User login
Solitary Yellow Papule on the Upper Back in an Infant
The Diagnosis: Juvenile Xanthogranuloma
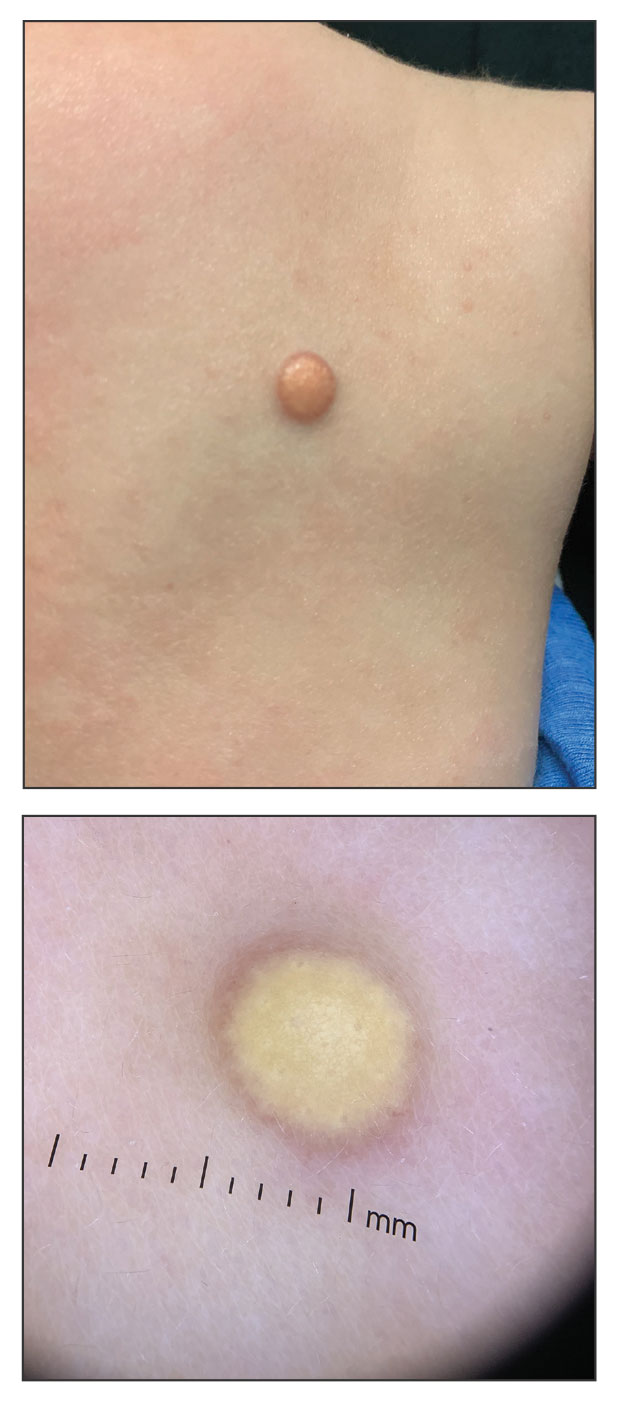
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
A 6-month-old male infant with a history of cradle cap and an infantile hemangioma on the left shoulder presented to the dermatology clinic for evaluation of a slow-growing yellow papule on the upper back of 3 months’ duration. The lesion initially was noted 2 months prior to the current presentation by the patient’s pediatrician, who recommended follow-up with dermatology after an unsuccessful attempt at incision and drainage. Physical examination revealed a 7-mm, yellow, dome-shaped papule with a red collarette on the right upper back. No axillary freckling, ocular findings, or other skin findings were found. The patient was born at term with no complications, and his mother reported that he was otherwise healthy. There were no developmental concerns or known allergies, and his family history was negative for any similar lesions. Dermoscopic examination of the lesion revealed a well-circumscribed, circular, yellow-orange papule with an erythematous border and setting-sun appearance.