User login
Flat-topped papules on the neck, arms, and trunk
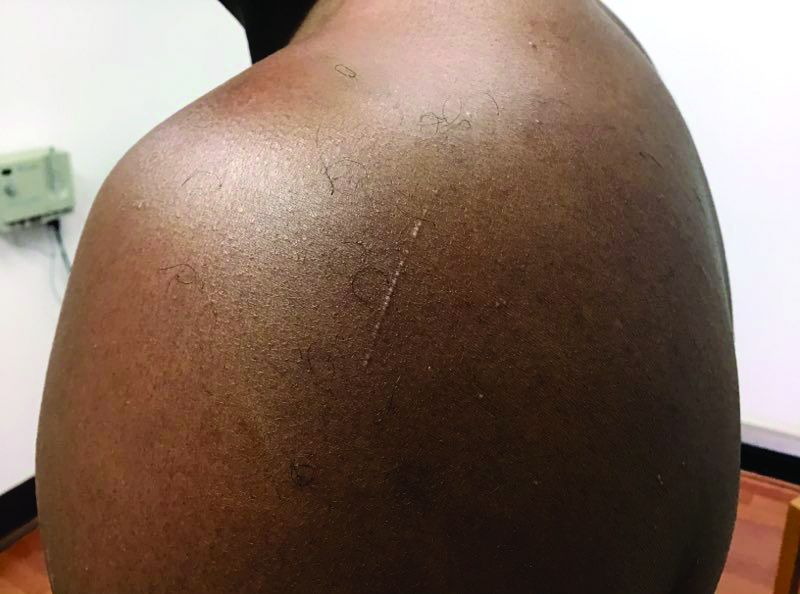
that typically occur on the trunk, extremities, or genitalia of children and young adults. However, it can affect people of all ages and all areas of skin. Lichen nitidus lesions may emerge in areas of trauma, often in a linear arrangement, called the Koebner phenomenon. It is not thought to be associated with any systemic disease. Nail involvement may be present. Oral lesions are not commonly seen. The diagnosis of LN is often a clinical one.
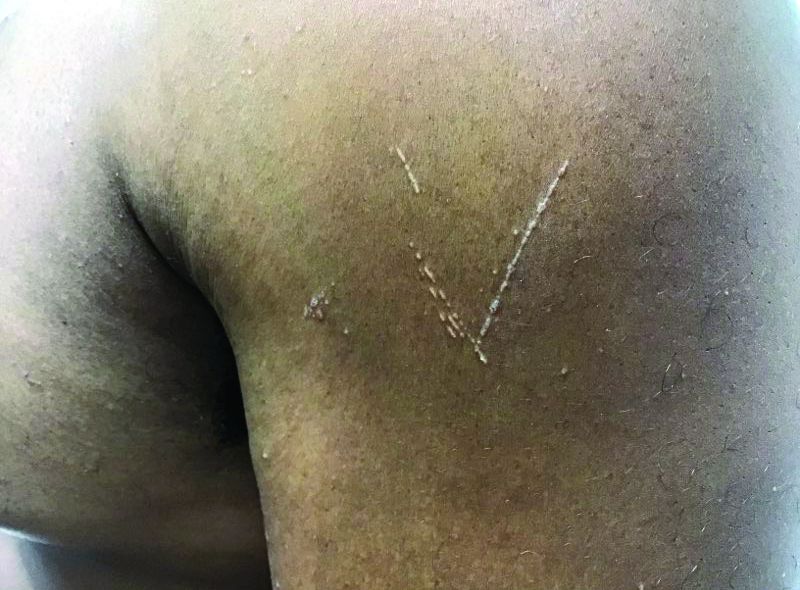
Histopathology for this patient showed a focally dense lymphohistiocytic infiltrate with multinucleate giant cells in the papillary dermis, associated with overlying epidermal atrophy and adjacent elongated rete ridges surrounding the infiltrate in a characteristic “ball and claw” pattern. These findings were consistent with a diagnosis of lichen nitidus.
The differential diagnosis includes lichen planus (LP). In LP, lesions tend to be larger and more violaceous. They tend to favor wrists, lower extremities, and genitalia. Oral and nail involvement are common. Histologically, a band-like lichenoid infiltrate in the dermis is present. Granulomatous inflammation and giant cells are absent. Direct immunofluorescence is positive for globular deposits of IgG, IgA, IgM and/or complement at the dermal-epidermal junction.
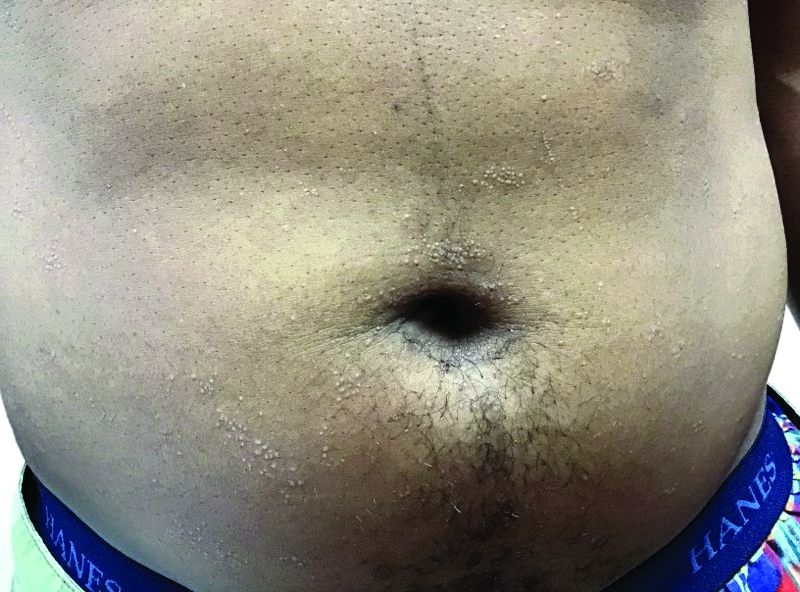
A hepatitis panel was drawn for this patient and was negative. Treatment for lichen nitidus is only needed if symptomatic because lesions will generally resolve spontaneously. Lesions may take months or years to resolve. For significant pruritus, topical corticosteroids or antihistamines may be used. Topical emollients are recommended. Topical tacrolimus has been reported to improve lesions. Oral steroids and light therapy have been reported to improve generalized lichen nitidus not responding to topical treatments.

The case and these photos were submitted byMs. Swartz of Nova Southeastern University, Ft. Lauderdale, Fla.; Dr. Chen and Dr. Walder of Bay Harbor Islands, Fla.; and Dr. Winslow of Pompano Beach, Fla. Donna Bilu Martin, MD, editor of this column, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].
that typically occur on the trunk, extremities, or genitalia of children and young adults. However, it can affect people of all ages and all areas of skin. Lichen nitidus lesions may emerge in areas of trauma, often in a linear arrangement, called the Koebner phenomenon. It is not thought to be associated with any systemic disease. Nail involvement may be present. Oral lesions are not commonly seen. The diagnosis of LN is often a clinical one.
Histopathology for this patient showed a focally dense lymphohistiocytic infiltrate with multinucleate giant cells in the papillary dermis, associated with overlying epidermal atrophy and adjacent elongated rete ridges surrounding the infiltrate in a characteristic “ball and claw” pattern. These findings were consistent with a diagnosis of lichen nitidus.
The differential diagnosis includes lichen planus (LP). In LP, lesions tend to be larger and more violaceous. They tend to favor wrists, lower extremities, and genitalia. Oral and nail involvement are common. Histologically, a band-like lichenoid infiltrate in the dermis is present. Granulomatous inflammation and giant cells are absent. Direct immunofluorescence is positive for globular deposits of IgG, IgA, IgM and/or complement at the dermal-epidermal junction.
A hepatitis panel was drawn for this patient and was negative. Treatment for lichen nitidus is only needed if symptomatic because lesions will generally resolve spontaneously. Lesions may take months or years to resolve. For significant pruritus, topical corticosteroids or antihistamines may be used. Topical emollients are recommended. Topical tacrolimus has been reported to improve lesions. Oral steroids and light therapy have been reported to improve generalized lichen nitidus not responding to topical treatments.
The case and these photos were submitted byMs. Swartz of Nova Southeastern University, Ft. Lauderdale, Fla.; Dr. Chen and Dr. Walder of Bay Harbor Islands, Fla.; and Dr. Winslow of Pompano Beach, Fla. Donna Bilu Martin, MD, editor of this column, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].
that typically occur on the trunk, extremities, or genitalia of children and young adults. However, it can affect people of all ages and all areas of skin. Lichen nitidus lesions may emerge in areas of trauma, often in a linear arrangement, called the Koebner phenomenon. It is not thought to be associated with any systemic disease. Nail involvement may be present. Oral lesions are not commonly seen. The diagnosis of LN is often a clinical one.
Histopathology for this patient showed a focally dense lymphohistiocytic infiltrate with multinucleate giant cells in the papillary dermis, associated with overlying epidermal atrophy and adjacent elongated rete ridges surrounding the infiltrate in a characteristic “ball and claw” pattern. These findings were consistent with a diagnosis of lichen nitidus.
The differential diagnosis includes lichen planus (LP). In LP, lesions tend to be larger and more violaceous. They tend to favor wrists, lower extremities, and genitalia. Oral and nail involvement are common. Histologically, a band-like lichenoid infiltrate in the dermis is present. Granulomatous inflammation and giant cells are absent. Direct immunofluorescence is positive for globular deposits of IgG, IgA, IgM and/or complement at the dermal-epidermal junction.
A hepatitis panel was drawn for this patient and was negative. Treatment for lichen nitidus is only needed if symptomatic because lesions will generally resolve spontaneously. Lesions may take months or years to resolve. For significant pruritus, topical corticosteroids or antihistamines may be used. Topical emollients are recommended. Topical tacrolimus has been reported to improve lesions. Oral steroids and light therapy have been reported to improve generalized lichen nitidus not responding to topical treatments.
The case and these photos were submitted byMs. Swartz of Nova Southeastern University, Ft. Lauderdale, Fla.; Dr. Chen and Dr. Walder of Bay Harbor Islands, Fla.; and Dr. Winslow of Pompano Beach, Fla. Donna Bilu Martin, MD, editor of this column, is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at MDedge.com/Dermatology. To submit a case for possible publication, send an email to [email protected].

What Do We Know About Opioid-Induced Hyperalgesia?
From the Massachusetts General Hospital Center for Pain Medicine, Boston, MA.
Abstract
- Objective: To review evidence from clinical and preclinical studies related to the phenomenon of opioid-induced hyperalgesia (OIH) and discuss issues relevant to clinical diagnosis and management.
- Methods: Literature review.
- Results: OIH is defined as a state of nociceptive sensitization caused by exposure to opioids such that a patient receiving opioids to treat pain could become more sensitive to painful stimuli. Interest in understanding OIH has grown over years and multiple mechanisms have been proposed. Both OIH and opioid tolerance can reduce opioid analgesic efficacy, complicating clinical management of chronic pain. When a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful in differentiating between tolerance and OIH. It is unclear whether certain types of opioids or routes of administration are more likely to lead to OIH.
- Conclusion: Clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While OIH has been well documented over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined.
Opioids, which produce analgesia through a primarily inhibitory effect on the nociceptive system, have been used for decades for the clinical management of moderate to severe pain. Opioid analgesics act on 3 major classes of opioid receptors, including the µ, k, δ (mu, kappa, and delta) receptors. Activation of opioid receptors not only produces analgesia but also other effects, such as euphoria, respiratory depression, decreased gastrointestinal motility, and cardiovascular effects. Exposure to opioids, however, can also lead to the development of opioid tolerance and opioid-induced hyperalgesia (OIH). Both opioid tolerance and OIH can decrease opioid analgesic efficacy, making chronic pain management a challenge. OIH is a state of nociceptive sensitization caused by exposure to opioids, such that a patient receiving opioids for the treatment of pain could actually become more sensitive to painful stimulation, resulting in a paradoxical adverse response to opioid therapy. In this article, we will review evidence from preclinical and clinical studies and discuss issues relevant to clinical diagnosis and management of OIH.
Evidence of OIH in Animal Studies
In early 1990s, an original preclinical study showed that there was a progressive reduction in baseline nociceptive threshold by using a foot withdraw test in rats receiving repeated intrathecal morphine administration (10-20 mg) over a 7-day period [1]. A number of animal studies later also provided similar data. A reduced baseline nociceptive threshold was observed in animals receiving subcutaneous fentanyl boluses using the Randall-Sellitto test, in which a constantly increasing pressure was applied to a rat’s hind paw. The decreased baseline nociceptive threshold lasted 5 days after cessation of 4 fentanyl bolus injections [2]. In another study, a reduced baseline nociceptive threshold was detected in animals with repeated heroin administration [3]. In other studies, rats exposed to morphine also developed a latent sensitization of visceral pain with a shift of the morphine dose-response curve to the right [4]; exposure to methadone also induced hyperalgesia in rats, which was not prevented by a weak NMDA receptor antagonist (memantine) [5]; and a partial µ-receptor agonist buprenophine produced a dose-related OIH as well [6].
These results indicate that a progressive and lasting reduction of baseline nociceptive threshold, which was referred to as OIH, can result from repeated opioid administration [7–9]. However, different from previous preclinical observations in which a large dose of intrathecal morphine was given, these studies resulted in hyperalgesic response [10,11] in a clinically relevant opioid dose. Of interest is that OIH was observed in animals even when there was continuous opioid infusion via an implanted osmotic pump, suggesting the involvement of active cellular mechanisms in the process [12]. Therefore, prolonged opioid treatment results in not only loss of the opioid analgesic effect (anti-nociceptive effect or desensitization) but also activation of a hyperalgesic effect (a pro-nociceptive effect with reduced nociceptive threshold or increased sensitization). Although both opioid tolerance and OIH are initiated by opioid administration, two opposing cellular mechanisms (ie, desensitization versus sensitization) may be involved in the process. Subsequently, many studies explored the neural and cellular mechanisms underlying the development of OIH and their interaction with the mechanism of opioid tolerance.
Proposed Cellular Mechanisms of OIH
A significant number of recent studies have explored the neurobiological basis of OIH, revealing a divergent range of cellular elements contributory to OIH. These mechanisms include (1) N-methyl-D-aspartate (NMDA) receptor and related intracellular pathways; (2) involvement of G-protein coupled receptors including 5-HT receptors and neurokinin-1 receptors; (3) nitrix oxide and nitric oxide sunthase; (4) TRPV1 receptors; (5) calcium channels; and (6) miscellaneous mechanisms including sex differences [7–9,13–41].
In summary, an increasing number of preclinical studies in the area of OIH indicates that there is enormous interest in understanding the cellular mechanisms of OIH, and the current evidence points to a progressive sensitization process within the central nervous system that involves a constellation of cellular elements such as NMDA receptors, similar to those contributory to the mechanisms of pathological pain.
Evidence of OIH in Human Studies
In animal studies, changes in baseline nociceptive thresholds can be measured in a controlled setting. It is, however, difficult to assess changes in pain threshold in clinical environment following opioid administration [9]. It is often a challenge to distinguish opioid pharmacologic tolerance from OIH because the outcome of opioid therapy is based primarily on subjective pain scores. In the face of these challenges, an increasing number of clinical anecdotal case reports and studies suggest that OIH is likely to be a significant factor in clinical opioid therapy [47–54].
In a study of 1620 patients in which remifentanil was used for general anesthesia, the incidence of postoperative remifentanil-induced hyperalgesia was 16.1%. This study found that age younger than 16 years, male sex, operation duration longer than 2 hours, and remifentanil dose greater than 30 mg/kga were associated with higher rates of OIH [55]. On the other hand, heroin or other opioid addicts not only demonstrated OIH but also had prolonged symptoms of OIH after detoxification from opioids for at least 1 month [56]. In chronic pain patients without opioid dependence, significantly lower pain threshold and tolerance as assessed by pressure pain stimulation were detected [57]. It appears that the sensitivity of detecting OIH in the clinical setting may be influenced by the modality of sensory stimulation [58].
In a prospective preliminary study of 6 patients with chronic low back pain, hyperalgesic response was detected after 1 month of oral morphine therapy using a cold pressor test but not a heat pain test [59]. In another prospective randomized, placebo-controlled, 2-way crossover study in healthy human volunteers, the development of OIH was quantified as changes in the average radius of the area of secondary hyperalgesia generated by electrical pain stimulation. A 23.6% increase in the area of secondary hyperalgesia over baseline was detected following the remifentanil infusion. The same study showed that endogenous opioids did not seem to have an effect on OIH because a single bolus of naloxone did not change the size of secondary hyperalgesia [60].
OIH Prevention Studies
Currently, efforts have also been made to see whether OIH can be prevented with different approaches in human subjects. The following is a brief summary of these studies. In a study of adolescents undergoing scoliosis surgery, treatment with morphine (150 mg/kg) prior to commencing remifentanil infusion did not prevent the development of remifentanil-induced hyperalgesia [61]. In another study, propofol infusion alone with remifentanil both delayed and attenuated remifentanil-induced hyperalgesia [53]. In yet another study, intraoperative 70% N2O administration appeared to reduce postoperative OIH following an intraoperative remifentanil-propofol anesthesia regimen [62].
In a study of 15 healthy male volunteers, preventive administration of parecoxib significantly diminished OIH after withdrawal from remifentanil. In contrast, parecoxib given together with remifentanil did not prevent OIH, suggesting that pre-treatment, not parallel treatment, with opioid may be required to prevent OIH [63]. Other NSAIDs administered preemptively also appear to prevent remifentanil-induced hyperalgesia [64].
Another study investigated the effect of intra-operative magnesium sulfate administration in patients undergoing robot-assisted laparoscopic prostatectomy. Magnesium sulfate administration reduced postoperative opioid consumption and OIH in subjects receiving intra-operative remifentanil-based anesthesia [65,66]. Intra-operative adenosine infusion also prevented acute opioid tolerance and remifentanil-induced hyperalgesia [67]. Continuous intra-operative infusion of ketamine, an NMDA receptor antagonist, significant lowered postoperative VAS and morphine use in gynecologic surgery patients [68]. Also, in a randomized, double-blind, placebo-controlled study of 90 patients who underwent total abdominal hysterectomy, cumulative morphine consumption was significantly greater in subjects with fentanyl alone than those with saline alone, ketamine alone, ketamine with fentanyl, or fentanyl with lornoxicam at 3, 6, and 12 hours postoperatively [69].
Finally, in a double-blind, randomized, placebo-controlled study of 40 patients undergoing elective shoulder surgery, clonidine was given intra-operatively in a remifentanil/propofol-based anesthesia. The results showed that clonidine did not reduce postoperative morphine consumption and pain score in these patients [70]. However, dexmedetomidine, another α2 receptor agonist, substantially reduced baseline opioid doses in hospitalized patients with OIH [71].
Quantitative Sensory Testing and OIH
Currently, diagnostic tools for OIH are still being developed. Many clinical studies have used quantitative sensory testing (QST) as a tool to assess OIH [72,73]. In a recent study, QST was used to compare pain threshold, pain tolerance, and the degree of temporal summation of pain in response to thermal stimulation among 3 groups of subjects: Group 1 (no pain and no opioid), Group 2 (chronic pain but no opioid therapy), and Group 3 (both chronic pain and opioid therapy). Group 3 subjects displayed a decreased heat pain threshold and exacerbated temporal summation of pain to thermal stimulation as compared with both group 1 and group 2 subjects. There were no differences in cold or warm sensation among all 3 groups. Among clinical factors, daily opioid dose consistently correlated with the decreased heat pain threshold and exacerbated temporal summation of second pain in group 3 subjects [72]. Another study investigated the sensitivity to cold pain and the magnitude of diffuse noxious inhibitory control (DNIC) using QST in subjects with or without opioid therapy. Pain threshold, intensity and tolerance in response to the cold pressor (1°C) were measured. They found that oral opioid use did not result in abnormal sensitivity to cold pain but altered pain modulation as detected by DNIC [74].
Opioid Regimen and OIH
Opioid regimen features, including type of opioid and dose, may influence the development of OIH. Anecdotal clinical observations have suggested that degree of OIH may vary according to opioid regimen [75]. Although the exact relationship between the dose regimen and the development of OIH remains to be determined, it is conceivable that OIH would be more likely to develop in patients receiving high opioid doses with a prolonged treatment course, although OIH has been demonstrated in patients receiving a short course of highly potent opioid analgesics [76]. Moreover, patients with a pathological pain condition (eg, neuropathic pain) treated with opioid therapy may be more susceptible to developing opioid-induced pain, because both pathological pain and OIH may share a common cellular mechanism [77].
If OIH develops following exposure to one opioid, can switching to a different opioid diminish OIH [78]?If cross-pain sensitivity does not develop between different opioids, switching to a different opioid would be justified, a similar rationale to that for opioid rotation to overcome opioid tolerance. This issue remains to be addressed.
OIH and Pre-emptive Analgesia
There is an ongoing debate about the clinical effectiveness of pre-emptive analgesia in pain management. However, use of opioid analgesic as the sole agent for pre-emptive analgesia may not be desirable for several reasons. First, a large dose of intra-operative opioids could activate a pro-nociceptive mechanism leading to the development of postoperative OIH [79]. This may confound the assessment of postoperative pain and counteract the opioid analgesic effect. Second, pre-emptive analgesia calls for pre-emptive inhibition of neuroplastic changes mediated through multiple cellular mechanisms such as the central glutamatergic system. Paradoxically, opioid administration could activate the central glutamatergic system as discussed above. Third, the neural mechanism of opioid tolerance and OIH may interact with that of pathological pain and pathological pain could be exacerbated following opioid administration [80,81]. This issue needs to be investigated in future studies.
Clinical Implications and Management of OIH
Until recently, a decreased opioid analgesic effect associated with opioid therapy was often recognized as the presence of pharmacologic opioid tolerance (ie, desensitization of the responsiveness of the opioid receptor and its cellular mechanism) and/or a worsening of the clinical pain condition. Therefore, opioid dose escalation appeared to be a logical approach to regain analgesic effectiveness. This practice should be reconsidered in light of the information on OIH. In the clinical setting, apparent opioid tolerance may result from pharmacological tolerance, worsening pain condition due to disease progression, and/or OIH. Below are some factors to consider in forming a differential diagnosis in the clinical setting [82].
First, the quality, location, and distribution pattern of the pain related to OIH would be different from a pre-existing pain condition. Because opioid analgesics are often administered systemically, changes in pain quality would be diffuse as compared with the pre-existing pain condition. Since the mechanism of OIH is similar to that of pathological pain, such as neuropathic pain, changes in pain threshold, tolerability, and distribution patterns seen in OIH would be similar to those seen in neuropathic pain patients. Quantitative sensory testing may be a useful tool to detect such changes.
Second, OIH would possibly exacerbate a pre-existing pain condition. Overall pain intensity (VAS) would be conceivably increased above the level of pre-existing pain in the absence of disease progression. Opioid dose escalation could only transiently and minimally reduce pain intensity in such a setting, with a subsequent increase in pain intensity due to OIH.
Third, when a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful to differentiate between tolerance and OIH. In an undertreated, worsening pain condition due to disease progress and/or pharma-cologic opioid tolerance, improved pain control may well be seen after a trial of opioid dose escalation. On the other hand, opioid dose escalation may exacerbate pain due to OIH, while a supervised opioid tapering may reduce OIH and improve clinical pain management. In this regard, if a patient is on a low opioid dose regimen and complains of unsatisfactory pain relief, a trial of opioid dose escalation may be appropriate; if a patient is already on a high dose of opioid analgesics, further dose escalation is rarely justified and may exacerbate OIH.
It is important to remember that the clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While any opioid dose escalation may transiently increase the analgesic effect, albeit by a small degree in many cases, the real issue is whether the same dose escalation may also exacerbate OIH, which could quickly overtake the transient increase in the opioid analgesic effect. Therefore, clinical judgment is fundamentally important and all clinical conditions related to opioid therapy need to be taken into consideration in the decision making process.
Summary
Interest in understanding OIH has grown over the last decade. Many discussions and reviews have centered around several key issues: (1) opioids not only produce analgesia through their anti-nociceptive effect, but also induce hyperalgesia via a pro-nociceptive effect; (2) opioid tolerance itself may be part of sensitization of a pro-nociceptive process; (3) the onset of OIH may be later than that of opioid tolerance and OIH may be a dose-related process, although OIH has been reported following acute and chronic opioid exposure at both high and low doses; (4) it is unclear whether a certain type of opioid and route of administration may be more likely to lead to clinical presentation of OIH; and (5) although opioid tolerance, OIH, and opioid withdrawal may share some common factors and mechanisms, the mechanism underlying each of these phenomena remains unclear [83].
While OIH has been well documented and investigated over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined [84]. In addition, opioid tolerance should be differentiated from OIH, although both have a similar clinical presentation with regard to change in pain intensity [85]. Clinically, OIH should be considered when the adjustment of opioid dose is contemplated if prior opioid dose escalation fails to provide the expected analgesic effect and there is unexplainable pain exacerbation following an initial period of effective opioid analgesia. Although in some cases increasing opioid dose leads to some improvement in pain management, in other cases less opioid may be more effective in pain reduction. This goal may be accomplished by initiating a trial of opioid tapering, opioid rotation, adding adjunctive medications, or combining opioid with a clinically available NMDA receptor antagonist. Continuing opioid therapy with endless dose escalation in the absence of clinical evidence of improved pain management is neither scientifically sound nor clinically justified.
Corresponding author: Lucy L. Chen, MD, MGH Center for Pain Medicine, WACC #340, 15 Parkman St., Boston, MA 02114.
Financial disclosures: None.
1. Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: Roles of excitatory amino acid receptors and protein kinase C. J Neurosci 1994;14:2301–12.
2. Celerier E, Rivat C, Jun Y, et al. Long-lasting hyperalgesia induced by fentanyl in rats: Preventive effect of ketamine. Anesthesiology 2000;92:465–72.
3. Laulin JP, Maurette P, Corcuff JB, et al. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg 2002;94:1263–9.
4. Lian B, Vera-Portocarrero L, King T, et al. Opioid-induced latent sensitization in a model of non-inflammatory viscerosomatic hypersensitivity. Brain Res 2010;1358:64–70.
5. Hay JL, Kaboutari J, White JM, et al. Model of methadone-induced hyperalgesia in rats and effect of memantine. Eur J Pharmacol 2010;626:229–33.
6. Wala EP, Holtman JR Jr. Buprenorphine-induced hyperalgesia in the rat. Eur J Pharmacol. 2011;651:89–95.
7. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: Implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–23.
8. Mao J, Sung B, Ji RR, Lim G. Neuronal apoptosis associated with morphine tolerance: Evidence for an opioid-induced neurotoxic mechanism. J Neurosci 2002;22:7650–61.
9. Mao J. Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep 2006;10:67–70.
10. Woolf CJ. Intrathecal high dose morphine produces hyperalgesia in the rat. Brain Res 1981;209:491–5.
11. Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: Clinical and theoretic implications. Anesthesiology 1986;64:590–7.
12. Vanderah TW, Ossipov MH, Lai J, et al. Mechanisms of opioid-induced pain and antinociceptive tolerance: Descending facilitation and spinal dynorphin. Pain 2001;92:5–9.
13. Zhao M, Joo DT. Enhancement of spinal N-methyl-D-aspartate receptor function by remifentanil action at delta-opioid receptors as a mechanism for acute opioid-induced hyperalgesia or tolerance. Anesthesiology 2008;109:308–17.
14. Ghelardini C, Galeotti N, Vivoli E, et al. Molecular interaction in the mouse PAG between NMDA and opioid receptors in morphine-induced acute thermal nociception. J Neurochem 2008;105:91–100.
15. Chen Y, Yang C, Wang ZJ. Ca2+/calmodulin-dependent protein kinase II alpha is required for the initiation and maintenance of opioid-induced hyperalgesia. J Neurosci 2010;30:38–46.
16. Van Elstraete AC, Sitbon P, Mazoit JX, et al. Protective effect of prior administration of magnesium on delayed hyperalgesia induced by fentanyl in rats. Can J Anaesth 2006;53:1180–5.
17. Gu X, Wu X, Liu Y, et al. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor 2B subunit in spinal cord contributes to remifentanil-induced postoperative hyperalgesia: the preventive effect of ketamine. Mol Pain 2009;5:76.
18. Minville V, Fourcade O, Girolami JP, Tack I. Opioid-induced hyperalgesia in a mice model of orthopaedic pain: Preventive effect of ketamine. Br J Anaesth 2010;104:231–8.
19. Cui W, Li Y, Li S, et al. Systemic lidocaine inhibits remifentanil-induced hyperalgesia via the inhibition of cPKCgamma membrane translocation in spinal dorsal horn of rats. J Neurosurg Anesthesiol 2009;21:318–25.
20. Gupta LK, Gupta R, Tripathi CD. N-methyl-D-aspartate receptor modulators block hyperalgesia induced by acute low-dose morphine. Clin Exp Pharmacol Physiol 2011;38:592–7.
21. Bianchi E, Galeotti N, Menicacci C, Ghelardini C. Contribution of G inhibitory protein alpha subunits in paradoxical hyperalgesia elicited by exceedingly low doses of morphine in mice. Life Sci 2011;89:918–25.
22. Ruiz-Medina J, Ledent C, Valverde O. GPR3 orphan receptor is involved in neuropathic pain after peripheral nerve injury and regulates morphine-induced antinociception. Neuropharmacology 2011;61:43–50.
23. Bianchi E, Norcini M, Smrcka A, Ghelardini C. Supraspinal gbetagamma-dependent stimulation of PLCbeta originating from G inhibitory protein-mu opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J Neurochem 2009;111:171–80.
24. Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology 2011;114:1180–9.
25. Rivat C, Vera-Portocarrero LP, Ibrahim MM, et al. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci 2009;29:727–37.
26. Vera-Portocarrero LP, Zhang ET, King T, et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain 2007;129:35–45.
27. Simonin F, Schmitt M, Laulin JP, et al. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A 2006;103:466–71.
28. Celerier E, Gonzalez JR, Maldonado R, et al. Opioid-induced hyperalgesia in a murine model of postoperative pain: Role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology 2006;104:546–55.
29. Vardanyan A, Wang R, Vanderah TW, et al. TRPV1 receptor in expression of opioid-induced hyperalgesia. J Pain 2009;10:243–52.
30. Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 2010;30:4460–6.
31. Esmaeili-Mahani S, Shimokawa N, Javan M, et al. Low-dose morphine induces hyperalgesia through activation of G alphas, protein kinase C, and L-type ca 2+ channels in rats. J Neurosci Res 2008;86:471–9.
32. Esmaeili-Mahani S, Fereidoni M, Javan M, et al. Nifedipine suppresses morphine-induced thermal hyperalgesia: Evidence for the role of corticosterone. Eur J Pharmacol 2007;567:95–101.
33. Van Elstraete AC, Sitbon P, Mazoit JX, Benhamou D. Gabapentin prevents delayed and long-lasting hyperalgesia induced by fentanyl in rats. Anesthesiology 2008;108:484–94.
34. Van Elstraete AC, Sitbon P, Benhamou D, Mazoit JX. The median effective dose of ketamine and gabapentin in opioid-induced hyperalgesia in rats: An isobolographic analysis of their interaction. Anesth Analg 2011;113:634–40.
35. Wei X, Wei W. Role of gabapentin in preventing fentanyl- and morphine-withdrawal-induced hyperalgesia in rats. J Anesth 2012;26:236–41.
36. Bessiere B, Richebe P, Laboureyras E, Laulin JP, Contarino A, Simonnet G. Nitrous oxide (N2O) prevents latent pain sensitization and long-term anxiety-like behavior in pain and opioid-experienced rats. Neuropharmacology 2007;53:733–40.
37. Hamlin AS, McNally GP, Osborne PB. Induction of c-fos and zif268 in the nociceptive amygdala parallel abstinence hyperalgesia in rats briefly exposed to morphine. Neuropharmacology 2007;53:330–43.
38. Doyle T, Bryant L, Muscoli C, et al. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett 2010;483:85–9.
39. Liang DY, Liao G, Wang J, et al. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology 2006;104:1054–62.
40. Liang DY, Liao G, Lighthall GK, Peltz G, Clark DJ. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenet Genomics 2006;16:825–35.
41. Wilson NM, Jung H, Ripsch MS, et al. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav Immun 2011;25:565–73.
42. Terashvili M, Wu HE, Schwasinger E, Tseng LF. Paradoxical hyperalgesia induced by mu-opioid receptor agonist endomorphin-2, but not endomorphin-1, microinjected into the centromedial amygdala of the rat. Eur J Pharmacol 2007;554:137–44.
43. Juni A, Klein G, Pintar JE, Kest B. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 2007;147:439–44.
44. Waxman AR, Arout C, Caldwell M, et al. Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 2009;462:68–72.
45. Juni A, Cai M, Stankova M, et al. Sex-specific mediation of opioid-induced hyperalgesia by the melanocortin-1 receptor. Anesthesiology 2010;112:181–8.
46. Juni A, Klein G, Kowalczyk B, et al. Sex differences in hyperalgesia during morphine infusion: Effect of gonadectomy and estrogen treatment. Neuropharmacology 2008;54:1264–70.
47. Forero M, Chan PS, Restrepo-Garces CE. Successful reversal of hyperalgesia/myoclonus complex with low-dose ketamine infusion. Pain Pract 2012;12:154–8.
48. Vorobeychik Y, Chen L, Bush MC, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med 2008;9:724–7.
49. Cortinas Saenz M, Geronimo Pardo M, Cortinas Saenz ML, et al. Acute opiate tolerance and postoperative hyperalgesia after a brief infusion of remifentanil managed with multimodal analgesia. Rev Esp Anestesiol Reanim 2008;55:40–2.
50. Siniscalchi A, Piraccini E, Miklosova Z, et al. Opioid-induced hyperalgesia and rapid opioid detoxification after tacrolimus administration. Anesth Analg 2008;106:645–6.
51. Okon TR, George ML. Fentanyl-induced neurotoxicity and paradoxic pain. J Pain Symptom Manage 2008;35:327–33.
52. Axelrod DJ, Reville B. Using methadone to treat opioid-induced hyperalgesia and refractory pain. J Opioid Manag 2007;3:113–4.
53. Singler B, Troster A, Manering N, et al. Modulation of remifentanil-induced postinfusion hyperalgesia by propofol. Anesth Analg 2007;104:1397–403.
54. Hallett BR, Chalkiadis GA. Suspected opioid-induced hyperalgesia in an infant. Br J Anaesth 2012;108:116–8.
55. Ma JF, Huang ZL, Li J, et al. Cohort study of remifentanil-induced hyperalgesia in postoperative patients. Zhonghua Yi Xue Za Zhi 2011;91:977–9.
56. Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain perception: New evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend 2006;82:218–23.
57. Fishbain DA, Lewis JE, Gao J. Are psychoactive substance (opioid)-dependent chronic pain patients hyperalgesic? Pain Pract 2011;11:337–43.
58. Hay JL, White JM, Bochner F, et al. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain 2009;10:316–22.
59. Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: A preliminary prospective study. J Pain 2006;7:43–8.
60. Chu LF, Dairmont J, Zamora AK, et al. The endogenous opioid system is not involved in modulation of opioid-induced hyperalgesia. J Pain 2011;12:108–15.
61. McDonnell C, Zaarour C, Hull R, et al. Pre-treatment with morphine does not prevent the development of remifentanil-induced hyperalgesia. Can J Anaesth 2008;55:813–8.
62. Echevarria G, Elgueta F, Fierro C, et al. Nitrous oxide (N(2)O) reduces postoperative opioid-induced hyperalgesia after remifentanil-propofol anaesthesia in humans. Br J Anaesth 2011;107:959–65.
63. Troster A, Sittl R, Singler B, et al. Modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by parecoxib in humans. Anesthesiology 2006;105:1016–23.
64. Tuncer S, Yalcin N, Reisli R, Alper Y. The effects of lornoxicam in preventing remifentanil-induced postoperative hyperalgesia. Agri 2009;2:161–7.
65. Lee C, Song YK, Jeong HM, Park SN. The effects of magnesium sulfate infiltration on perioperative opioid consumption and opioid-induced hyperalgesia in patients undergoing robot-assisted laparoscopic prostatectomy with remifentanil-based anesthesia. Korean J Anesthesiol 2011;61:244–50.
66. Song JW, Lee YW, Yoon KB, et al. Magnesium sulfate prevents remifentanil-induced postoperative hyperalgesia in patients undergoing thyroidectomy. Anesth Analg 2011;113:390–7.
67. Lee C, Song YK, Lee JH, Ha SM. The effects of intraoperative adenosine infusion on acute opioid tolerance and opioid induced hyperalgesia induced by remifentanil in adult patients undergoing tonsillectomy. Korean J Pain 2011;24:7–12.
68. Hong BH, Lee WY, Kim YH, et al. Effects of intraoperative low dose ketamine on remifentanil-induced hyperalgesia in gynecologic surgery with sevoflurane anesthesia. Korean J Anesthesiol 2011;61:238–43.
69. Xuerong Y, Yuguang H, Xia J, Hailan W. Ketamine and lornoxicam for preventing a fentanyl-induced increase in postoperative morphine requirement. Anesth Analg 2008;107:2032–7.
70. Schlimp CJ, Pipam W, Wolrab C, Ohner C, Kager HI, Likar R. Clonidine for remifentanil-induced hyperalgesia: A double-blind randomized, placebo-controlled study of clonidine under intra-operative use of remifentanil in elective surgery of the shoulder. Schmerz 2011;25:290–5.
71. Belgrade M, Hall S. Dexmedetomidine infusion for the management of opioid-induced hyperalgesia. Pain Med 2010;11:1819–26.
72. Chen L, Malarick C, Seefeld L, et al. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain 2009;143:65–70.
73. Bannister K, Dickenson AH. Opioid hyperalgesia. Curr Opin Support Palliat Care 2010;4:1–5.
74. Ram KC, Eisenberg E, Haddad M, Pud D. Oral opioid use alters DNIC but not cold pain perception in patients with chronic pain - new perspective of opioid-induced hyperalgesia. Pain 2008;139:431–8.
75. Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: Effect of long-acting maintenance agent. Drug Alcohol Depend 2001;63:139–46.
76. Vinik HR, Kissin I. Rapid development of tolerance to analgesia during remifentanil infusion in humans. Anesth Analg 1998;86:1307–11.
77. Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: A current view of their possible interactions. Pain 1995;62:259–74.
78. Sjogren P, Jensen NH, Jensen TS. Disappearance of morphine-induced hyperalgesia after discontinuing or substituting morphine with other opioid agonists. Pain 1994;59:313–6.
79. Guignard B, Bossard AE, Coste C, et al. Acute opioid tolerance: Intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology 2000;93:409–17.
80. Angst MS, Clark JD. Opioid-induced hyperalgesia: A qualitative systematic review. Anesthesiology 2006;104:570–87.
81. Baron MJ, McDonald PW. Significant pain reduction in chronic pain patients after detoxification from high-dose opioids. J Opioid Manag 2006;2:277–82
82. Mao J. Opioid-induced abnormal pain sensitivity: Implications in clinical opioid therapy. Pain 2002;100:213–7.
83. Low Y, Clarke CF, Huh BK. Opioid-induced hyperalgesia: A review of epidemiology, mechanisms and management. Singapore Med J 2012;53:357–60.
84. Fishbain DA, Cole B, Lewis JE, et al. Do opioids induce hyperalgesia in humans? An evidence-based structured review. Pain Med 2009:829–39.
85. Chu LF, D’Arcy N, Brady C, et al. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: a double-blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain 2012;153:1583–92.
From the Massachusetts General Hospital Center for Pain Medicine, Boston, MA.
Abstract
- Objective: To review evidence from clinical and preclinical studies related to the phenomenon of opioid-induced hyperalgesia (OIH) and discuss issues relevant to clinical diagnosis and management.
- Methods: Literature review.
- Results: OIH is defined as a state of nociceptive sensitization caused by exposure to opioids such that a patient receiving opioids to treat pain could become more sensitive to painful stimuli. Interest in understanding OIH has grown over years and multiple mechanisms have been proposed. Both OIH and opioid tolerance can reduce opioid analgesic efficacy, complicating clinical management of chronic pain. When a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful in differentiating between tolerance and OIH. It is unclear whether certain types of opioids or routes of administration are more likely to lead to OIH.
- Conclusion: Clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While OIH has been well documented over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined.
Opioids, which produce analgesia through a primarily inhibitory effect on the nociceptive system, have been used for decades for the clinical management of moderate to severe pain. Opioid analgesics act on 3 major classes of opioid receptors, including the µ, k, δ (mu, kappa, and delta) receptors. Activation of opioid receptors not only produces analgesia but also other effects, such as euphoria, respiratory depression, decreased gastrointestinal motility, and cardiovascular effects. Exposure to opioids, however, can also lead to the development of opioid tolerance and opioid-induced hyperalgesia (OIH). Both opioid tolerance and OIH can decrease opioid analgesic efficacy, making chronic pain management a challenge. OIH is a state of nociceptive sensitization caused by exposure to opioids, such that a patient receiving opioids for the treatment of pain could actually become more sensitive to painful stimulation, resulting in a paradoxical adverse response to opioid therapy. In this article, we will review evidence from preclinical and clinical studies and discuss issues relevant to clinical diagnosis and management of OIH.
Evidence of OIH in Animal Studies
In early 1990s, an original preclinical study showed that there was a progressive reduction in baseline nociceptive threshold by using a foot withdraw test in rats receiving repeated intrathecal morphine administration (10-20 mg) over a 7-day period [1]. A number of animal studies later also provided similar data. A reduced baseline nociceptive threshold was observed in animals receiving subcutaneous fentanyl boluses using the Randall-Sellitto test, in which a constantly increasing pressure was applied to a rat’s hind paw. The decreased baseline nociceptive threshold lasted 5 days after cessation of 4 fentanyl bolus injections [2]. In another study, a reduced baseline nociceptive threshold was detected in animals with repeated heroin administration [3]. In other studies, rats exposed to morphine also developed a latent sensitization of visceral pain with a shift of the morphine dose-response curve to the right [4]; exposure to methadone also induced hyperalgesia in rats, which was not prevented by a weak NMDA receptor antagonist (memantine) [5]; and a partial µ-receptor agonist buprenophine produced a dose-related OIH as well [6].
These results indicate that a progressive and lasting reduction of baseline nociceptive threshold, which was referred to as OIH, can result from repeated opioid administration [7–9]. However, different from previous preclinical observations in which a large dose of intrathecal morphine was given, these studies resulted in hyperalgesic response [10,11] in a clinically relevant opioid dose. Of interest is that OIH was observed in animals even when there was continuous opioid infusion via an implanted osmotic pump, suggesting the involvement of active cellular mechanisms in the process [12]. Therefore, prolonged opioid treatment results in not only loss of the opioid analgesic effect (anti-nociceptive effect or desensitization) but also activation of a hyperalgesic effect (a pro-nociceptive effect with reduced nociceptive threshold or increased sensitization). Although both opioid tolerance and OIH are initiated by opioid administration, two opposing cellular mechanisms (ie, desensitization versus sensitization) may be involved in the process. Subsequently, many studies explored the neural and cellular mechanisms underlying the development of OIH and their interaction with the mechanism of opioid tolerance.
Proposed Cellular Mechanisms of OIH
A significant number of recent studies have explored the neurobiological basis of OIH, revealing a divergent range of cellular elements contributory to OIH. These mechanisms include (1) N-methyl-D-aspartate (NMDA) receptor and related intracellular pathways; (2) involvement of G-protein coupled receptors including 5-HT receptors and neurokinin-1 receptors; (3) nitrix oxide and nitric oxide sunthase; (4) TRPV1 receptors; (5) calcium channels; and (6) miscellaneous mechanisms including sex differences [7–9,13–41].
In summary, an increasing number of preclinical studies in the area of OIH indicates that there is enormous interest in understanding the cellular mechanisms of OIH, and the current evidence points to a progressive sensitization process within the central nervous system that involves a constellation of cellular elements such as NMDA receptors, similar to those contributory to the mechanisms of pathological pain.
Evidence of OIH in Human Studies
In animal studies, changes in baseline nociceptive thresholds can be measured in a controlled setting. It is, however, difficult to assess changes in pain threshold in clinical environment following opioid administration [9]. It is often a challenge to distinguish opioid pharmacologic tolerance from OIH because the outcome of opioid therapy is based primarily on subjective pain scores. In the face of these challenges, an increasing number of clinical anecdotal case reports and studies suggest that OIH is likely to be a significant factor in clinical opioid therapy [47–54].
In a study of 1620 patients in which remifentanil was used for general anesthesia, the incidence of postoperative remifentanil-induced hyperalgesia was 16.1%. This study found that age younger than 16 years, male sex, operation duration longer than 2 hours, and remifentanil dose greater than 30 mg/kga were associated with higher rates of OIH [55]. On the other hand, heroin or other opioid addicts not only demonstrated OIH but also had prolonged symptoms of OIH after detoxification from opioids for at least 1 month [56]. In chronic pain patients without opioid dependence, significantly lower pain threshold and tolerance as assessed by pressure pain stimulation were detected [57]. It appears that the sensitivity of detecting OIH in the clinical setting may be influenced by the modality of sensory stimulation [58].
In a prospective preliminary study of 6 patients with chronic low back pain, hyperalgesic response was detected after 1 month of oral morphine therapy using a cold pressor test but not a heat pain test [59]. In another prospective randomized, placebo-controlled, 2-way crossover study in healthy human volunteers, the development of OIH was quantified as changes in the average radius of the area of secondary hyperalgesia generated by electrical pain stimulation. A 23.6% increase in the area of secondary hyperalgesia over baseline was detected following the remifentanil infusion. The same study showed that endogenous opioids did not seem to have an effect on OIH because a single bolus of naloxone did not change the size of secondary hyperalgesia [60].
OIH Prevention Studies
Currently, efforts have also been made to see whether OIH can be prevented with different approaches in human subjects. The following is a brief summary of these studies. In a study of adolescents undergoing scoliosis surgery, treatment with morphine (150 mg/kg) prior to commencing remifentanil infusion did not prevent the development of remifentanil-induced hyperalgesia [61]. In another study, propofol infusion alone with remifentanil both delayed and attenuated remifentanil-induced hyperalgesia [53]. In yet another study, intraoperative 70% N2O administration appeared to reduce postoperative OIH following an intraoperative remifentanil-propofol anesthesia regimen [62].
In a study of 15 healthy male volunteers, preventive administration of parecoxib significantly diminished OIH after withdrawal from remifentanil. In contrast, parecoxib given together with remifentanil did not prevent OIH, suggesting that pre-treatment, not parallel treatment, with opioid may be required to prevent OIH [63]. Other NSAIDs administered preemptively also appear to prevent remifentanil-induced hyperalgesia [64].
Another study investigated the effect of intra-operative magnesium sulfate administration in patients undergoing robot-assisted laparoscopic prostatectomy. Magnesium sulfate administration reduced postoperative opioid consumption and OIH in subjects receiving intra-operative remifentanil-based anesthesia [65,66]. Intra-operative adenosine infusion also prevented acute opioid tolerance and remifentanil-induced hyperalgesia [67]. Continuous intra-operative infusion of ketamine, an NMDA receptor antagonist, significant lowered postoperative VAS and morphine use in gynecologic surgery patients [68]. Also, in a randomized, double-blind, placebo-controlled study of 90 patients who underwent total abdominal hysterectomy, cumulative morphine consumption was significantly greater in subjects with fentanyl alone than those with saline alone, ketamine alone, ketamine with fentanyl, or fentanyl with lornoxicam at 3, 6, and 12 hours postoperatively [69].
Finally, in a double-blind, randomized, placebo-controlled study of 40 patients undergoing elective shoulder surgery, clonidine was given intra-operatively in a remifentanil/propofol-based anesthesia. The results showed that clonidine did not reduce postoperative morphine consumption and pain score in these patients [70]. However, dexmedetomidine, another α2 receptor agonist, substantially reduced baseline opioid doses in hospitalized patients with OIH [71].
Quantitative Sensory Testing and OIH
Currently, diagnostic tools for OIH are still being developed. Many clinical studies have used quantitative sensory testing (QST) as a tool to assess OIH [72,73]. In a recent study, QST was used to compare pain threshold, pain tolerance, and the degree of temporal summation of pain in response to thermal stimulation among 3 groups of subjects: Group 1 (no pain and no opioid), Group 2 (chronic pain but no opioid therapy), and Group 3 (both chronic pain and opioid therapy). Group 3 subjects displayed a decreased heat pain threshold and exacerbated temporal summation of pain to thermal stimulation as compared with both group 1 and group 2 subjects. There were no differences in cold or warm sensation among all 3 groups. Among clinical factors, daily opioid dose consistently correlated with the decreased heat pain threshold and exacerbated temporal summation of second pain in group 3 subjects [72]. Another study investigated the sensitivity to cold pain and the magnitude of diffuse noxious inhibitory control (DNIC) using QST in subjects with or without opioid therapy. Pain threshold, intensity and tolerance in response to the cold pressor (1°C) were measured. They found that oral opioid use did not result in abnormal sensitivity to cold pain but altered pain modulation as detected by DNIC [74].
Opioid Regimen and OIH
Opioid regimen features, including type of opioid and dose, may influence the development of OIH. Anecdotal clinical observations have suggested that degree of OIH may vary according to opioid regimen [75]. Although the exact relationship between the dose regimen and the development of OIH remains to be determined, it is conceivable that OIH would be more likely to develop in patients receiving high opioid doses with a prolonged treatment course, although OIH has been demonstrated in patients receiving a short course of highly potent opioid analgesics [76]. Moreover, patients with a pathological pain condition (eg, neuropathic pain) treated with opioid therapy may be more susceptible to developing opioid-induced pain, because both pathological pain and OIH may share a common cellular mechanism [77].
If OIH develops following exposure to one opioid, can switching to a different opioid diminish OIH [78]?If cross-pain sensitivity does not develop between different opioids, switching to a different opioid would be justified, a similar rationale to that for opioid rotation to overcome opioid tolerance. This issue remains to be addressed.
OIH and Pre-emptive Analgesia
There is an ongoing debate about the clinical effectiveness of pre-emptive analgesia in pain management. However, use of opioid analgesic as the sole agent for pre-emptive analgesia may not be desirable for several reasons. First, a large dose of intra-operative opioids could activate a pro-nociceptive mechanism leading to the development of postoperative OIH [79]. This may confound the assessment of postoperative pain and counteract the opioid analgesic effect. Second, pre-emptive analgesia calls for pre-emptive inhibition of neuroplastic changes mediated through multiple cellular mechanisms such as the central glutamatergic system. Paradoxically, opioid administration could activate the central glutamatergic system as discussed above. Third, the neural mechanism of opioid tolerance and OIH may interact with that of pathological pain and pathological pain could be exacerbated following opioid administration [80,81]. This issue needs to be investigated in future studies.
Clinical Implications and Management of OIH
Until recently, a decreased opioid analgesic effect associated with opioid therapy was often recognized as the presence of pharmacologic opioid tolerance (ie, desensitization of the responsiveness of the opioid receptor and its cellular mechanism) and/or a worsening of the clinical pain condition. Therefore, opioid dose escalation appeared to be a logical approach to regain analgesic effectiveness. This practice should be reconsidered in light of the information on OIH. In the clinical setting, apparent opioid tolerance may result from pharmacological tolerance, worsening pain condition due to disease progression, and/or OIH. Below are some factors to consider in forming a differential diagnosis in the clinical setting [82].
First, the quality, location, and distribution pattern of the pain related to OIH would be different from a pre-existing pain condition. Because opioid analgesics are often administered systemically, changes in pain quality would be diffuse as compared with the pre-existing pain condition. Since the mechanism of OIH is similar to that of pathological pain, such as neuropathic pain, changes in pain threshold, tolerability, and distribution patterns seen in OIH would be similar to those seen in neuropathic pain patients. Quantitative sensory testing may be a useful tool to detect such changes.
Second, OIH would possibly exacerbate a pre-existing pain condition. Overall pain intensity (VAS) would be conceivably increased above the level of pre-existing pain in the absence of disease progression. Opioid dose escalation could only transiently and minimally reduce pain intensity in such a setting, with a subsequent increase in pain intensity due to OIH.
Third, when a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful to differentiate between tolerance and OIH. In an undertreated, worsening pain condition due to disease progress and/or pharma-cologic opioid tolerance, improved pain control may well be seen after a trial of opioid dose escalation. On the other hand, opioid dose escalation may exacerbate pain due to OIH, while a supervised opioid tapering may reduce OIH and improve clinical pain management. In this regard, if a patient is on a low opioid dose regimen and complains of unsatisfactory pain relief, a trial of opioid dose escalation may be appropriate; if a patient is already on a high dose of opioid analgesics, further dose escalation is rarely justified and may exacerbate OIH.
It is important to remember that the clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While any opioid dose escalation may transiently increase the analgesic effect, albeit by a small degree in many cases, the real issue is whether the same dose escalation may also exacerbate OIH, which could quickly overtake the transient increase in the opioid analgesic effect. Therefore, clinical judgment is fundamentally important and all clinical conditions related to opioid therapy need to be taken into consideration in the decision making process.
Summary
Interest in understanding OIH has grown over the last decade. Many discussions and reviews have centered around several key issues: (1) opioids not only produce analgesia through their anti-nociceptive effect, but also induce hyperalgesia via a pro-nociceptive effect; (2) opioid tolerance itself may be part of sensitization of a pro-nociceptive process; (3) the onset of OIH may be later than that of opioid tolerance and OIH may be a dose-related process, although OIH has been reported following acute and chronic opioid exposure at both high and low doses; (4) it is unclear whether a certain type of opioid and route of administration may be more likely to lead to clinical presentation of OIH; and (5) although opioid tolerance, OIH, and opioid withdrawal may share some common factors and mechanisms, the mechanism underlying each of these phenomena remains unclear [83].
While OIH has been well documented and investigated over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined [84]. In addition, opioid tolerance should be differentiated from OIH, although both have a similar clinical presentation with regard to change in pain intensity [85]. Clinically, OIH should be considered when the adjustment of opioid dose is contemplated if prior opioid dose escalation fails to provide the expected analgesic effect and there is unexplainable pain exacerbation following an initial period of effective opioid analgesia. Although in some cases increasing opioid dose leads to some improvement in pain management, in other cases less opioid may be more effective in pain reduction. This goal may be accomplished by initiating a trial of opioid tapering, opioid rotation, adding adjunctive medications, or combining opioid with a clinically available NMDA receptor antagonist. Continuing opioid therapy with endless dose escalation in the absence of clinical evidence of improved pain management is neither scientifically sound nor clinically justified.
Corresponding author: Lucy L. Chen, MD, MGH Center for Pain Medicine, WACC #340, 15 Parkman St., Boston, MA 02114.
Financial disclosures: None.
From the Massachusetts General Hospital Center for Pain Medicine, Boston, MA.
Abstract
- Objective: To review evidence from clinical and preclinical studies related to the phenomenon of opioid-induced hyperalgesia (OIH) and discuss issues relevant to clinical diagnosis and management.
- Methods: Literature review.
- Results: OIH is defined as a state of nociceptive sensitization caused by exposure to opioids such that a patient receiving opioids to treat pain could become more sensitive to painful stimuli. Interest in understanding OIH has grown over years and multiple mechanisms have been proposed. Both OIH and opioid tolerance can reduce opioid analgesic efficacy, complicating clinical management of chronic pain. When a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful in differentiating between tolerance and OIH. It is unclear whether certain types of opioids or routes of administration are more likely to lead to OIH.
- Conclusion: Clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While OIH has been well documented over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined.
Opioids, which produce analgesia through a primarily inhibitory effect on the nociceptive system, have been used for decades for the clinical management of moderate to severe pain. Opioid analgesics act on 3 major classes of opioid receptors, including the µ, k, δ (mu, kappa, and delta) receptors. Activation of opioid receptors not only produces analgesia but also other effects, such as euphoria, respiratory depression, decreased gastrointestinal motility, and cardiovascular effects. Exposure to opioids, however, can also lead to the development of opioid tolerance and opioid-induced hyperalgesia (OIH). Both opioid tolerance and OIH can decrease opioid analgesic efficacy, making chronic pain management a challenge. OIH is a state of nociceptive sensitization caused by exposure to opioids, such that a patient receiving opioids for the treatment of pain could actually become more sensitive to painful stimulation, resulting in a paradoxical adverse response to opioid therapy. In this article, we will review evidence from preclinical and clinical studies and discuss issues relevant to clinical diagnosis and management of OIH.
Evidence of OIH in Animal Studies
In early 1990s, an original preclinical study showed that there was a progressive reduction in baseline nociceptive threshold by using a foot withdraw test in rats receiving repeated intrathecal morphine administration (10-20 mg) over a 7-day period [1]. A number of animal studies later also provided similar data. A reduced baseline nociceptive threshold was observed in animals receiving subcutaneous fentanyl boluses using the Randall-Sellitto test, in which a constantly increasing pressure was applied to a rat’s hind paw. The decreased baseline nociceptive threshold lasted 5 days after cessation of 4 fentanyl bolus injections [2]. In another study, a reduced baseline nociceptive threshold was detected in animals with repeated heroin administration [3]. In other studies, rats exposed to morphine also developed a latent sensitization of visceral pain with a shift of the morphine dose-response curve to the right [4]; exposure to methadone also induced hyperalgesia in rats, which was not prevented by a weak NMDA receptor antagonist (memantine) [5]; and a partial µ-receptor agonist buprenophine produced a dose-related OIH as well [6].
These results indicate that a progressive and lasting reduction of baseline nociceptive threshold, which was referred to as OIH, can result from repeated opioid administration [7–9]. However, different from previous preclinical observations in which a large dose of intrathecal morphine was given, these studies resulted in hyperalgesic response [10,11] in a clinically relevant opioid dose. Of interest is that OIH was observed in animals even when there was continuous opioid infusion via an implanted osmotic pump, suggesting the involvement of active cellular mechanisms in the process [12]. Therefore, prolonged opioid treatment results in not only loss of the opioid analgesic effect (anti-nociceptive effect or desensitization) but also activation of a hyperalgesic effect (a pro-nociceptive effect with reduced nociceptive threshold or increased sensitization). Although both opioid tolerance and OIH are initiated by opioid administration, two opposing cellular mechanisms (ie, desensitization versus sensitization) may be involved in the process. Subsequently, many studies explored the neural and cellular mechanisms underlying the development of OIH and their interaction with the mechanism of opioid tolerance.
Proposed Cellular Mechanisms of OIH
A significant number of recent studies have explored the neurobiological basis of OIH, revealing a divergent range of cellular elements contributory to OIH. These mechanisms include (1) N-methyl-D-aspartate (NMDA) receptor and related intracellular pathways; (2) involvement of G-protein coupled receptors including 5-HT receptors and neurokinin-1 receptors; (3) nitrix oxide and nitric oxide sunthase; (4) TRPV1 receptors; (5) calcium channels; and (6) miscellaneous mechanisms including sex differences [7–9,13–41].
In summary, an increasing number of preclinical studies in the area of OIH indicates that there is enormous interest in understanding the cellular mechanisms of OIH, and the current evidence points to a progressive sensitization process within the central nervous system that involves a constellation of cellular elements such as NMDA receptors, similar to those contributory to the mechanisms of pathological pain.
Evidence of OIH in Human Studies
In animal studies, changes in baseline nociceptive thresholds can be measured in a controlled setting. It is, however, difficult to assess changes in pain threshold in clinical environment following opioid administration [9]. It is often a challenge to distinguish opioid pharmacologic tolerance from OIH because the outcome of opioid therapy is based primarily on subjective pain scores. In the face of these challenges, an increasing number of clinical anecdotal case reports and studies suggest that OIH is likely to be a significant factor in clinical opioid therapy [47–54].
In a study of 1620 patients in which remifentanil was used for general anesthesia, the incidence of postoperative remifentanil-induced hyperalgesia was 16.1%. This study found that age younger than 16 years, male sex, operation duration longer than 2 hours, and remifentanil dose greater than 30 mg/kga were associated with higher rates of OIH [55]. On the other hand, heroin or other opioid addicts not only demonstrated OIH but also had prolonged symptoms of OIH after detoxification from opioids for at least 1 month [56]. In chronic pain patients without opioid dependence, significantly lower pain threshold and tolerance as assessed by pressure pain stimulation were detected [57]. It appears that the sensitivity of detecting OIH in the clinical setting may be influenced by the modality of sensory stimulation [58].
In a prospective preliminary study of 6 patients with chronic low back pain, hyperalgesic response was detected after 1 month of oral morphine therapy using a cold pressor test but not a heat pain test [59]. In another prospective randomized, placebo-controlled, 2-way crossover study in healthy human volunteers, the development of OIH was quantified as changes in the average radius of the area of secondary hyperalgesia generated by electrical pain stimulation. A 23.6% increase in the area of secondary hyperalgesia over baseline was detected following the remifentanil infusion. The same study showed that endogenous opioids did not seem to have an effect on OIH because a single bolus of naloxone did not change the size of secondary hyperalgesia [60].
OIH Prevention Studies
Currently, efforts have also been made to see whether OIH can be prevented with different approaches in human subjects. The following is a brief summary of these studies. In a study of adolescents undergoing scoliosis surgery, treatment with morphine (150 mg/kg) prior to commencing remifentanil infusion did not prevent the development of remifentanil-induced hyperalgesia [61]. In another study, propofol infusion alone with remifentanil both delayed and attenuated remifentanil-induced hyperalgesia [53]. In yet another study, intraoperative 70% N2O administration appeared to reduce postoperative OIH following an intraoperative remifentanil-propofol anesthesia regimen [62].
In a study of 15 healthy male volunteers, preventive administration of parecoxib significantly diminished OIH after withdrawal from remifentanil. In contrast, parecoxib given together with remifentanil did not prevent OIH, suggesting that pre-treatment, not parallel treatment, with opioid may be required to prevent OIH [63]. Other NSAIDs administered preemptively also appear to prevent remifentanil-induced hyperalgesia [64].
Another study investigated the effect of intra-operative magnesium sulfate administration in patients undergoing robot-assisted laparoscopic prostatectomy. Magnesium sulfate administration reduced postoperative opioid consumption and OIH in subjects receiving intra-operative remifentanil-based anesthesia [65,66]. Intra-operative adenosine infusion also prevented acute opioid tolerance and remifentanil-induced hyperalgesia [67]. Continuous intra-operative infusion of ketamine, an NMDA receptor antagonist, significant lowered postoperative VAS and morphine use in gynecologic surgery patients [68]. Also, in a randomized, double-blind, placebo-controlled study of 90 patients who underwent total abdominal hysterectomy, cumulative morphine consumption was significantly greater in subjects with fentanyl alone than those with saline alone, ketamine alone, ketamine with fentanyl, or fentanyl with lornoxicam at 3, 6, and 12 hours postoperatively [69].
Finally, in a double-blind, randomized, placebo-controlled study of 40 patients undergoing elective shoulder surgery, clonidine was given intra-operatively in a remifentanil/propofol-based anesthesia. The results showed that clonidine did not reduce postoperative morphine consumption and pain score in these patients [70]. However, dexmedetomidine, another α2 receptor agonist, substantially reduced baseline opioid doses in hospitalized patients with OIH [71].
Quantitative Sensory Testing and OIH
Currently, diagnostic tools for OIH are still being developed. Many clinical studies have used quantitative sensory testing (QST) as a tool to assess OIH [72,73]. In a recent study, QST was used to compare pain threshold, pain tolerance, and the degree of temporal summation of pain in response to thermal stimulation among 3 groups of subjects: Group 1 (no pain and no opioid), Group 2 (chronic pain but no opioid therapy), and Group 3 (both chronic pain and opioid therapy). Group 3 subjects displayed a decreased heat pain threshold and exacerbated temporal summation of pain to thermal stimulation as compared with both group 1 and group 2 subjects. There were no differences in cold or warm sensation among all 3 groups. Among clinical factors, daily opioid dose consistently correlated with the decreased heat pain threshold and exacerbated temporal summation of second pain in group 3 subjects [72]. Another study investigated the sensitivity to cold pain and the magnitude of diffuse noxious inhibitory control (DNIC) using QST in subjects with or without opioid therapy. Pain threshold, intensity and tolerance in response to the cold pressor (1°C) were measured. They found that oral opioid use did not result in abnormal sensitivity to cold pain but altered pain modulation as detected by DNIC [74].
Opioid Regimen and OIH
Opioid regimen features, including type of opioid and dose, may influence the development of OIH. Anecdotal clinical observations have suggested that degree of OIH may vary according to opioid regimen [75]. Although the exact relationship between the dose regimen and the development of OIH remains to be determined, it is conceivable that OIH would be more likely to develop in patients receiving high opioid doses with a prolonged treatment course, although OIH has been demonstrated in patients receiving a short course of highly potent opioid analgesics [76]. Moreover, patients with a pathological pain condition (eg, neuropathic pain) treated with opioid therapy may be more susceptible to developing opioid-induced pain, because both pathological pain and OIH may share a common cellular mechanism [77].
If OIH develops following exposure to one opioid, can switching to a different opioid diminish OIH [78]?If cross-pain sensitivity does not develop between different opioids, switching to a different opioid would be justified, a similar rationale to that for opioid rotation to overcome opioid tolerance. This issue remains to be addressed.
OIH and Pre-emptive Analgesia
There is an ongoing debate about the clinical effectiveness of pre-emptive analgesia in pain management. However, use of opioid analgesic as the sole agent for pre-emptive analgesia may not be desirable for several reasons. First, a large dose of intra-operative opioids could activate a pro-nociceptive mechanism leading to the development of postoperative OIH [79]. This may confound the assessment of postoperative pain and counteract the opioid analgesic effect. Second, pre-emptive analgesia calls for pre-emptive inhibition of neuroplastic changes mediated through multiple cellular mechanisms such as the central glutamatergic system. Paradoxically, opioid administration could activate the central glutamatergic system as discussed above. Third, the neural mechanism of opioid tolerance and OIH may interact with that of pathological pain and pathological pain could be exacerbated following opioid administration [80,81]. This issue needs to be investigated in future studies.
Clinical Implications and Management of OIH
Until recently, a decreased opioid analgesic effect associated with opioid therapy was often recognized as the presence of pharmacologic opioid tolerance (ie, desensitization of the responsiveness of the opioid receptor and its cellular mechanism) and/or a worsening of the clinical pain condition. Therefore, opioid dose escalation appeared to be a logical approach to regain analgesic effectiveness. This practice should be reconsidered in light of the information on OIH. In the clinical setting, apparent opioid tolerance may result from pharmacological tolerance, worsening pain condition due to disease progression, and/or OIH. Below are some factors to consider in forming a differential diagnosis in the clinical setting [82].
First, the quality, location, and distribution pattern of the pain related to OIH would be different from a pre-existing pain condition. Because opioid analgesics are often administered systemically, changes in pain quality would be diffuse as compared with the pre-existing pain condition. Since the mechanism of OIH is similar to that of pathological pain, such as neuropathic pain, changes in pain threshold, tolerability, and distribution patterns seen in OIH would be similar to those seen in neuropathic pain patients. Quantitative sensory testing may be a useful tool to detect such changes.
Second, OIH would possibly exacerbate a pre-existing pain condition. Overall pain intensity (VAS) would be conceivably increased above the level of pre-existing pain in the absence of disease progression. Opioid dose escalation could only transiently and minimally reduce pain intensity in such a setting, with a subsequent increase in pain intensity due to OIH.
Third, when a diagnosis is uncertain, a trial of opioid dose escalation or tapering may be helpful to differentiate between tolerance and OIH. In an undertreated, worsening pain condition due to disease progress and/or pharma-cologic opioid tolerance, improved pain control may well be seen after a trial of opioid dose escalation. On the other hand, opioid dose escalation may exacerbate pain due to OIH, while a supervised opioid tapering may reduce OIH and improve clinical pain management. In this regard, if a patient is on a low opioid dose regimen and complains of unsatisfactory pain relief, a trial of opioid dose escalation may be appropriate; if a patient is already on a high dose of opioid analgesics, further dose escalation is rarely justified and may exacerbate OIH.
It is important to remember that the clinical outcome of opioid therapy is a dynamic balance among the opioid analgesic effect, OIH, and worsening pain due to disease progression. While any opioid dose escalation may transiently increase the analgesic effect, albeit by a small degree in many cases, the real issue is whether the same dose escalation may also exacerbate OIH, which could quickly overtake the transient increase in the opioid analgesic effect. Therefore, clinical judgment is fundamentally important and all clinical conditions related to opioid therapy need to be taken into consideration in the decision making process.
Summary
Interest in understanding OIH has grown over the last decade. Many discussions and reviews have centered around several key issues: (1) opioids not only produce analgesia through their anti-nociceptive effect, but also induce hyperalgesia via a pro-nociceptive effect; (2) opioid tolerance itself may be part of sensitization of a pro-nociceptive process; (3) the onset of OIH may be later than that of opioid tolerance and OIH may be a dose-related process, although OIH has been reported following acute and chronic opioid exposure at both high and low doses; (4) it is unclear whether a certain type of opioid and route of administration may be more likely to lead to clinical presentation of OIH; and (5) although opioid tolerance, OIH, and opioid withdrawal may share some common factors and mechanisms, the mechanism underlying each of these phenomena remains unclear [83].
While OIH has been well documented and investigated over nearly 2 decades, its exact clinical characteristics and underlying mechanisms have yet to be fully determined [84]. In addition, opioid tolerance should be differentiated from OIH, although both have a similar clinical presentation with regard to change in pain intensity [85]. Clinically, OIH should be considered when the adjustment of opioid dose is contemplated if prior opioid dose escalation fails to provide the expected analgesic effect and there is unexplainable pain exacerbation following an initial period of effective opioid analgesia. Although in some cases increasing opioid dose leads to some improvement in pain management, in other cases less opioid may be more effective in pain reduction. This goal may be accomplished by initiating a trial of opioid tapering, opioid rotation, adding adjunctive medications, or combining opioid with a clinically available NMDA receptor antagonist. Continuing opioid therapy with endless dose escalation in the absence of clinical evidence of improved pain management is neither scientifically sound nor clinically justified.
Corresponding author: Lucy L. Chen, MD, MGH Center for Pain Medicine, WACC #340, 15 Parkman St., Boston, MA 02114.
Financial disclosures: None.
1. Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: Roles of excitatory amino acid receptors and protein kinase C. J Neurosci 1994;14:2301–12.
2. Celerier E, Rivat C, Jun Y, et al. Long-lasting hyperalgesia induced by fentanyl in rats: Preventive effect of ketamine. Anesthesiology 2000;92:465–72.
3. Laulin JP, Maurette P, Corcuff JB, et al. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg 2002;94:1263–9.
4. Lian B, Vera-Portocarrero L, King T, et al. Opioid-induced latent sensitization in a model of non-inflammatory viscerosomatic hypersensitivity. Brain Res 2010;1358:64–70.
5. Hay JL, Kaboutari J, White JM, et al. Model of methadone-induced hyperalgesia in rats and effect of memantine. Eur J Pharmacol 2010;626:229–33.
6. Wala EP, Holtman JR Jr. Buprenorphine-induced hyperalgesia in the rat. Eur J Pharmacol. 2011;651:89–95.
7. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: Implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–23.
8. Mao J, Sung B, Ji RR, Lim G. Neuronal apoptosis associated with morphine tolerance: Evidence for an opioid-induced neurotoxic mechanism. J Neurosci 2002;22:7650–61.
9. Mao J. Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep 2006;10:67–70.
10. Woolf CJ. Intrathecal high dose morphine produces hyperalgesia in the rat. Brain Res 1981;209:491–5.
11. Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: Clinical and theoretic implications. Anesthesiology 1986;64:590–7.
12. Vanderah TW, Ossipov MH, Lai J, et al. Mechanisms of opioid-induced pain and antinociceptive tolerance: Descending facilitation and spinal dynorphin. Pain 2001;92:5–9.
13. Zhao M, Joo DT. Enhancement of spinal N-methyl-D-aspartate receptor function by remifentanil action at delta-opioid receptors as a mechanism for acute opioid-induced hyperalgesia or tolerance. Anesthesiology 2008;109:308–17.
14. Ghelardini C, Galeotti N, Vivoli E, et al. Molecular interaction in the mouse PAG between NMDA and opioid receptors in morphine-induced acute thermal nociception. J Neurochem 2008;105:91–100.
15. Chen Y, Yang C, Wang ZJ. Ca2+/calmodulin-dependent protein kinase II alpha is required for the initiation and maintenance of opioid-induced hyperalgesia. J Neurosci 2010;30:38–46.
16. Van Elstraete AC, Sitbon P, Mazoit JX, et al. Protective effect of prior administration of magnesium on delayed hyperalgesia induced by fentanyl in rats. Can J Anaesth 2006;53:1180–5.
17. Gu X, Wu X, Liu Y, et al. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor 2B subunit in spinal cord contributes to remifentanil-induced postoperative hyperalgesia: the preventive effect of ketamine. Mol Pain 2009;5:76.
18. Minville V, Fourcade O, Girolami JP, Tack I. Opioid-induced hyperalgesia in a mice model of orthopaedic pain: Preventive effect of ketamine. Br J Anaesth 2010;104:231–8.
19. Cui W, Li Y, Li S, et al. Systemic lidocaine inhibits remifentanil-induced hyperalgesia via the inhibition of cPKCgamma membrane translocation in spinal dorsal horn of rats. J Neurosurg Anesthesiol 2009;21:318–25.
20. Gupta LK, Gupta R, Tripathi CD. N-methyl-D-aspartate receptor modulators block hyperalgesia induced by acute low-dose morphine. Clin Exp Pharmacol Physiol 2011;38:592–7.
21. Bianchi E, Galeotti N, Menicacci C, Ghelardini C. Contribution of G inhibitory protein alpha subunits in paradoxical hyperalgesia elicited by exceedingly low doses of morphine in mice. Life Sci 2011;89:918–25.
22. Ruiz-Medina J, Ledent C, Valverde O. GPR3 orphan receptor is involved in neuropathic pain after peripheral nerve injury and regulates morphine-induced antinociception. Neuropharmacology 2011;61:43–50.
23. Bianchi E, Norcini M, Smrcka A, Ghelardini C. Supraspinal gbetagamma-dependent stimulation of PLCbeta originating from G inhibitory protein-mu opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J Neurochem 2009;111:171–80.
24. Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology 2011;114:1180–9.
25. Rivat C, Vera-Portocarrero LP, Ibrahim MM, et al. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci 2009;29:727–37.
26. Vera-Portocarrero LP, Zhang ET, King T, et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain 2007;129:35–45.
27. Simonin F, Schmitt M, Laulin JP, et al. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A 2006;103:466–71.
28. Celerier E, Gonzalez JR, Maldonado R, et al. Opioid-induced hyperalgesia in a murine model of postoperative pain: Role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology 2006;104:546–55.
29. Vardanyan A, Wang R, Vanderah TW, et al. TRPV1 receptor in expression of opioid-induced hyperalgesia. J Pain 2009;10:243–52.
30. Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 2010;30:4460–6.
31. Esmaeili-Mahani S, Shimokawa N, Javan M, et al. Low-dose morphine induces hyperalgesia through activation of G alphas, protein kinase C, and L-type ca 2+ channels in rats. J Neurosci Res 2008;86:471–9.
32. Esmaeili-Mahani S, Fereidoni M, Javan M, et al. Nifedipine suppresses morphine-induced thermal hyperalgesia: Evidence for the role of corticosterone. Eur J Pharmacol 2007;567:95–101.
33. Van Elstraete AC, Sitbon P, Mazoit JX, Benhamou D. Gabapentin prevents delayed and long-lasting hyperalgesia induced by fentanyl in rats. Anesthesiology 2008;108:484–94.
34. Van Elstraete AC, Sitbon P, Benhamou D, Mazoit JX. The median effective dose of ketamine and gabapentin in opioid-induced hyperalgesia in rats: An isobolographic analysis of their interaction. Anesth Analg 2011;113:634–40.
35. Wei X, Wei W. Role of gabapentin in preventing fentanyl- and morphine-withdrawal-induced hyperalgesia in rats. J Anesth 2012;26:236–41.
36. Bessiere B, Richebe P, Laboureyras E, Laulin JP, Contarino A, Simonnet G. Nitrous oxide (N2O) prevents latent pain sensitization and long-term anxiety-like behavior in pain and opioid-experienced rats. Neuropharmacology 2007;53:733–40.
37. Hamlin AS, McNally GP, Osborne PB. Induction of c-fos and zif268 in the nociceptive amygdala parallel abstinence hyperalgesia in rats briefly exposed to morphine. Neuropharmacology 2007;53:330–43.
38. Doyle T, Bryant L, Muscoli C, et al. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett 2010;483:85–9.
39. Liang DY, Liao G, Wang J, et al. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology 2006;104:1054–62.
40. Liang DY, Liao G, Lighthall GK, Peltz G, Clark DJ. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenet Genomics 2006;16:825–35.
41. Wilson NM, Jung H, Ripsch MS, et al. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav Immun 2011;25:565–73.
42. Terashvili M, Wu HE, Schwasinger E, Tseng LF. Paradoxical hyperalgesia induced by mu-opioid receptor agonist endomorphin-2, but not endomorphin-1, microinjected into the centromedial amygdala of the rat. Eur J Pharmacol 2007;554:137–44.
43. Juni A, Klein G, Pintar JE, Kest B. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 2007;147:439–44.
44. Waxman AR, Arout C, Caldwell M, et al. Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 2009;462:68–72.
45. Juni A, Cai M, Stankova M, et al. Sex-specific mediation of opioid-induced hyperalgesia by the melanocortin-1 receptor. Anesthesiology 2010;112:181–8.
46. Juni A, Klein G, Kowalczyk B, et al. Sex differences in hyperalgesia during morphine infusion: Effect of gonadectomy and estrogen treatment. Neuropharmacology 2008;54:1264–70.
47. Forero M, Chan PS, Restrepo-Garces CE. Successful reversal of hyperalgesia/myoclonus complex with low-dose ketamine infusion. Pain Pract 2012;12:154–8.
48. Vorobeychik Y, Chen L, Bush MC, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med 2008;9:724–7.
49. Cortinas Saenz M, Geronimo Pardo M, Cortinas Saenz ML, et al. Acute opiate tolerance and postoperative hyperalgesia after a brief infusion of remifentanil managed with multimodal analgesia. Rev Esp Anestesiol Reanim 2008;55:40–2.
50. Siniscalchi A, Piraccini E, Miklosova Z, et al. Opioid-induced hyperalgesia and rapid opioid detoxification after tacrolimus administration. Anesth Analg 2008;106:645–6.
51. Okon TR, George ML. Fentanyl-induced neurotoxicity and paradoxic pain. J Pain Symptom Manage 2008;35:327–33.
52. Axelrod DJ, Reville B. Using methadone to treat opioid-induced hyperalgesia and refractory pain. J Opioid Manag 2007;3:113–4.
53. Singler B, Troster A, Manering N, et al. Modulation of remifentanil-induced postinfusion hyperalgesia by propofol. Anesth Analg 2007;104:1397–403.
54. Hallett BR, Chalkiadis GA. Suspected opioid-induced hyperalgesia in an infant. Br J Anaesth 2012;108:116–8.
55. Ma JF, Huang ZL, Li J, et al. Cohort study of remifentanil-induced hyperalgesia in postoperative patients. Zhonghua Yi Xue Za Zhi 2011;91:977–9.
56. Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain perception: New evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend 2006;82:218–23.
57. Fishbain DA, Lewis JE, Gao J. Are psychoactive substance (opioid)-dependent chronic pain patients hyperalgesic? Pain Pract 2011;11:337–43.
58. Hay JL, White JM, Bochner F, et al. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain 2009;10:316–22.
59. Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: A preliminary prospective study. J Pain 2006;7:43–8.
60. Chu LF, Dairmont J, Zamora AK, et al. The endogenous opioid system is not involved in modulation of opioid-induced hyperalgesia. J Pain 2011;12:108–15.
61. McDonnell C, Zaarour C, Hull R, et al. Pre-treatment with morphine does not prevent the development of remifentanil-induced hyperalgesia. Can J Anaesth 2008;55:813–8.
62. Echevarria G, Elgueta F, Fierro C, et al. Nitrous oxide (N(2)O) reduces postoperative opioid-induced hyperalgesia after remifentanil-propofol anaesthesia in humans. Br J Anaesth 2011;107:959–65.
63. Troster A, Sittl R, Singler B, et al. Modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by parecoxib in humans. Anesthesiology 2006;105:1016–23.
64. Tuncer S, Yalcin N, Reisli R, Alper Y. The effects of lornoxicam in preventing remifentanil-induced postoperative hyperalgesia. Agri 2009;2:161–7.
65. Lee C, Song YK, Jeong HM, Park SN. The effects of magnesium sulfate infiltration on perioperative opioid consumption and opioid-induced hyperalgesia in patients undergoing robot-assisted laparoscopic prostatectomy with remifentanil-based anesthesia. Korean J Anesthesiol 2011;61:244–50.
66. Song JW, Lee YW, Yoon KB, et al. Magnesium sulfate prevents remifentanil-induced postoperative hyperalgesia in patients undergoing thyroidectomy. Anesth Analg 2011;113:390–7.
67. Lee C, Song YK, Lee JH, Ha SM. The effects of intraoperative adenosine infusion on acute opioid tolerance and opioid induced hyperalgesia induced by remifentanil in adult patients undergoing tonsillectomy. Korean J Pain 2011;24:7–12.
68. Hong BH, Lee WY, Kim YH, et al. Effects of intraoperative low dose ketamine on remifentanil-induced hyperalgesia in gynecologic surgery with sevoflurane anesthesia. Korean J Anesthesiol 2011;61:238–43.
69. Xuerong Y, Yuguang H, Xia J, Hailan W. Ketamine and lornoxicam for preventing a fentanyl-induced increase in postoperative morphine requirement. Anesth Analg 2008;107:2032–7.
70. Schlimp CJ, Pipam W, Wolrab C, Ohner C, Kager HI, Likar R. Clonidine for remifentanil-induced hyperalgesia: A double-blind randomized, placebo-controlled study of clonidine under intra-operative use of remifentanil in elective surgery of the shoulder. Schmerz 2011;25:290–5.
71. Belgrade M, Hall S. Dexmedetomidine infusion for the management of opioid-induced hyperalgesia. Pain Med 2010;11:1819–26.
72. Chen L, Malarick C, Seefeld L, et al. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain 2009;143:65–70.
73. Bannister K, Dickenson AH. Opioid hyperalgesia. Curr Opin Support Palliat Care 2010;4:1–5.
74. Ram KC, Eisenberg E, Haddad M, Pud D. Oral opioid use alters DNIC but not cold pain perception in patients with chronic pain - new perspective of opioid-induced hyperalgesia. Pain 2008;139:431–8.
75. Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: Effect of long-acting maintenance agent. Drug Alcohol Depend 2001;63:139–46.
76. Vinik HR, Kissin I. Rapid development of tolerance to analgesia during remifentanil infusion in humans. Anesth Analg 1998;86:1307–11.
77. Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: A current view of their possible interactions. Pain 1995;62:259–74.
78. Sjogren P, Jensen NH, Jensen TS. Disappearance of morphine-induced hyperalgesia after discontinuing or substituting morphine with other opioid agonists. Pain 1994;59:313–6.
79. Guignard B, Bossard AE, Coste C, et al. Acute opioid tolerance: Intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology 2000;93:409–17.
80. Angst MS, Clark JD. Opioid-induced hyperalgesia: A qualitative systematic review. Anesthesiology 2006;104:570–87.
81. Baron MJ, McDonald PW. Significant pain reduction in chronic pain patients after detoxification from high-dose opioids. J Opioid Manag 2006;2:277–82
82. Mao J. Opioid-induced abnormal pain sensitivity: Implications in clinical opioid therapy. Pain 2002;100:213–7.
83. Low Y, Clarke CF, Huh BK. Opioid-induced hyperalgesia: A review of epidemiology, mechanisms and management. Singapore Med J 2012;53:357–60.
84. Fishbain DA, Cole B, Lewis JE, et al. Do opioids induce hyperalgesia in humans? An evidence-based structured review. Pain Med 2009:829–39.
85. Chu LF, D’Arcy N, Brady C, et al. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: a double-blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain 2012;153:1583–92.
1. Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: Roles of excitatory amino acid receptors and protein kinase C. J Neurosci 1994;14:2301–12.
2. Celerier E, Rivat C, Jun Y, et al. Long-lasting hyperalgesia induced by fentanyl in rats: Preventive effect of ketamine. Anesthesiology 2000;92:465–72.
3. Laulin JP, Maurette P, Corcuff JB, et al. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg 2002;94:1263–9.
4. Lian B, Vera-Portocarrero L, King T, et al. Opioid-induced latent sensitization in a model of non-inflammatory viscerosomatic hypersensitivity. Brain Res 2010;1358:64–70.
5. Hay JL, Kaboutari J, White JM, et al. Model of methadone-induced hyperalgesia in rats and effect of memantine. Eur J Pharmacol 2010;626:229–33.
6. Wala EP, Holtman JR Jr. Buprenorphine-induced hyperalgesia in the rat. Eur J Pharmacol. 2011;651:89–95.
7. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: Implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–23.
8. Mao J, Sung B, Ji RR, Lim G. Neuronal apoptosis associated with morphine tolerance: Evidence for an opioid-induced neurotoxic mechanism. J Neurosci 2002;22:7650–61.
9. Mao J. Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep 2006;10:67–70.
10. Woolf CJ. Intrathecal high dose morphine produces hyperalgesia in the rat. Brain Res 1981;209:491–5.
11. Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: Clinical and theoretic implications. Anesthesiology 1986;64:590–7.
12. Vanderah TW, Ossipov MH, Lai J, et al. Mechanisms of opioid-induced pain and antinociceptive tolerance: Descending facilitation and spinal dynorphin. Pain 2001;92:5–9.
13. Zhao M, Joo DT. Enhancement of spinal N-methyl-D-aspartate receptor function by remifentanil action at delta-opioid receptors as a mechanism for acute opioid-induced hyperalgesia or tolerance. Anesthesiology 2008;109:308–17.
14. Ghelardini C, Galeotti N, Vivoli E, et al. Molecular interaction in the mouse PAG between NMDA and opioid receptors in morphine-induced acute thermal nociception. J Neurochem 2008;105:91–100.
15. Chen Y, Yang C, Wang ZJ. Ca2+/calmodulin-dependent protein kinase II alpha is required for the initiation and maintenance of opioid-induced hyperalgesia. J Neurosci 2010;30:38–46.
16. Van Elstraete AC, Sitbon P, Mazoit JX, et al. Protective effect of prior administration of magnesium on delayed hyperalgesia induced by fentanyl in rats. Can J Anaesth 2006;53:1180–5.
17. Gu X, Wu X, Liu Y, et al. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor 2B subunit in spinal cord contributes to remifentanil-induced postoperative hyperalgesia: the preventive effect of ketamine. Mol Pain 2009;5:76.
18. Minville V, Fourcade O, Girolami JP, Tack I. Opioid-induced hyperalgesia in a mice model of orthopaedic pain: Preventive effect of ketamine. Br J Anaesth 2010;104:231–8.
19. Cui W, Li Y, Li S, et al. Systemic lidocaine inhibits remifentanil-induced hyperalgesia via the inhibition of cPKCgamma membrane translocation in spinal dorsal horn of rats. J Neurosurg Anesthesiol 2009;21:318–25.
20. Gupta LK, Gupta R, Tripathi CD. N-methyl-D-aspartate receptor modulators block hyperalgesia induced by acute low-dose morphine. Clin Exp Pharmacol Physiol 2011;38:592–7.
21. Bianchi E, Galeotti N, Menicacci C, Ghelardini C. Contribution of G inhibitory protein alpha subunits in paradoxical hyperalgesia elicited by exceedingly low doses of morphine in mice. Life Sci 2011;89:918–25.
22. Ruiz-Medina J, Ledent C, Valverde O. GPR3 orphan receptor is involved in neuropathic pain after peripheral nerve injury and regulates morphine-induced antinociception. Neuropharmacology 2011;61:43–50.
23. Bianchi E, Norcini M, Smrcka A, Ghelardini C. Supraspinal gbetagamma-dependent stimulation of PLCbeta originating from G inhibitory protein-mu opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J Neurochem 2009;111:171–80.
24. Liang DY, Li X, Clark JD. 5-hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology 2011;114:1180–9.
25. Rivat C, Vera-Portocarrero LP, Ibrahim MM, et al. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci 2009;29:727–37.
26. Vera-Portocarrero LP, Zhang ET, King T, et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain 2007;129:35–45.
27. Simonin F, Schmitt M, Laulin JP, et al. RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc Natl Acad Sci U S A 2006;103:466–71.
28. Celerier E, Gonzalez JR, Maldonado R, et al. Opioid-induced hyperalgesia in a murine model of postoperative pain: Role of nitric oxide generated from the inducible nitric oxide synthase. Anesthesiology 2006;104:546–55.
29. Vardanyan A, Wang R, Vanderah TW, et al. TRPV1 receptor in expression of opioid-induced hyperalgesia. J Pain 2009;10:243–52.
30. Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci 2010;30:4460–6.
31. Esmaeili-Mahani S, Shimokawa N, Javan M, et al. Low-dose morphine induces hyperalgesia through activation of G alphas, protein kinase C, and L-type ca 2+ channels in rats. J Neurosci Res 2008;86:471–9.
32. Esmaeili-Mahani S, Fereidoni M, Javan M, et al. Nifedipine suppresses morphine-induced thermal hyperalgesia: Evidence for the role of corticosterone. Eur J Pharmacol 2007;567:95–101.
33. Van Elstraete AC, Sitbon P, Mazoit JX, Benhamou D. Gabapentin prevents delayed and long-lasting hyperalgesia induced by fentanyl in rats. Anesthesiology 2008;108:484–94.
34. Van Elstraete AC, Sitbon P, Benhamou D, Mazoit JX. The median effective dose of ketamine and gabapentin in opioid-induced hyperalgesia in rats: An isobolographic analysis of their interaction. Anesth Analg 2011;113:634–40.
35. Wei X, Wei W. Role of gabapentin in preventing fentanyl- and morphine-withdrawal-induced hyperalgesia in rats. J Anesth 2012;26:236–41.
36. Bessiere B, Richebe P, Laboureyras E, Laulin JP, Contarino A, Simonnet G. Nitrous oxide (N2O) prevents latent pain sensitization and long-term anxiety-like behavior in pain and opioid-experienced rats. Neuropharmacology 2007;53:733–40.
37. Hamlin AS, McNally GP, Osborne PB. Induction of c-fos and zif268 in the nociceptive amygdala parallel abstinence hyperalgesia in rats briefly exposed to morphine. Neuropharmacology 2007;53:330–43.
38. Doyle T, Bryant L, Muscoli C, et al. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett 2010;483:85–9.
39. Liang DY, Liao G, Wang J, et al. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology 2006;104:1054–62.
40. Liang DY, Liao G, Lighthall GK, Peltz G, Clark DJ. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenet Genomics 2006;16:825–35.
41. Wilson NM, Jung H, Ripsch MS, et al. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav Immun 2011;25:565–73.
42. Terashvili M, Wu HE, Schwasinger E, Tseng LF. Paradoxical hyperalgesia induced by mu-opioid receptor agonist endomorphin-2, but not endomorphin-1, microinjected into the centromedial amygdala of the rat. Eur J Pharmacol 2007;554:137–44.
43. Juni A, Klein G, Pintar JE, Kest B. Nociception increases during opioid infusion in opioid receptor triple knock-out mice. Neuroscience 2007;147:439–44.
44. Waxman AR, Arout C, Caldwell M, et al. Acute and chronic fentanyl administration causes hyperalgesia independently of opioid receptor activity in mice. Neurosci Lett 2009;462:68–72.
45. Juni A, Cai M, Stankova M, et al. Sex-specific mediation of opioid-induced hyperalgesia by the melanocortin-1 receptor. Anesthesiology 2010;112:181–8.
46. Juni A, Klein G, Kowalczyk B, et al. Sex differences in hyperalgesia during morphine infusion: Effect of gonadectomy and estrogen treatment. Neuropharmacology 2008;54:1264–70.
47. Forero M, Chan PS, Restrepo-Garces CE. Successful reversal of hyperalgesia/myoclonus complex with low-dose ketamine infusion. Pain Pract 2012;12:154–8.
48. Vorobeychik Y, Chen L, Bush MC, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med 2008;9:724–7.
49. Cortinas Saenz M, Geronimo Pardo M, Cortinas Saenz ML, et al. Acute opiate tolerance and postoperative hyperalgesia after a brief infusion of remifentanil managed with multimodal analgesia. Rev Esp Anestesiol Reanim 2008;55:40–2.
50. Siniscalchi A, Piraccini E, Miklosova Z, et al. Opioid-induced hyperalgesia and rapid opioid detoxification after tacrolimus administration. Anesth Analg 2008;106:645–6.
51. Okon TR, George ML. Fentanyl-induced neurotoxicity and paradoxic pain. J Pain Symptom Manage 2008;35:327–33.
52. Axelrod DJ, Reville B. Using methadone to treat opioid-induced hyperalgesia and refractory pain. J Opioid Manag 2007;3:113–4.
53. Singler B, Troster A, Manering N, et al. Modulation of remifentanil-induced postinfusion hyperalgesia by propofol. Anesth Analg 2007;104:1397–403.
54. Hallett BR, Chalkiadis GA. Suspected opioid-induced hyperalgesia in an infant. Br J Anaesth 2012;108:116–8.
55. Ma JF, Huang ZL, Li J, et al. Cohort study of remifentanil-induced hyperalgesia in postoperative patients. Zhonghua Yi Xue Za Zhi 2011;91:977–9.
56. Pud D, Cohen D, Lawental E, Eisenberg E. Opioids and abnormal pain perception: New evidence from a study of chronic opioid addicts and healthy subjects. Drug Alcohol Depend 2006;82:218–23.
57. Fishbain DA, Lewis JE, Gao J. Are psychoactive substance (opioid)-dependent chronic pain patients hyperalgesic? Pain Pract 2011;11:337–43.
58. Hay JL, White JM, Bochner F, et al. Hyperalgesia in opioid-managed chronic pain and opioid-dependent patients. J Pain 2009;10:316–22.
59. Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: A preliminary prospective study. J Pain 2006;7:43–8.
60. Chu LF, Dairmont J, Zamora AK, et al. The endogenous opioid system is not involved in modulation of opioid-induced hyperalgesia. J Pain 2011;12:108–15.
61. McDonnell C, Zaarour C, Hull R, et al. Pre-treatment with morphine does not prevent the development of remifentanil-induced hyperalgesia. Can J Anaesth 2008;55:813–8.
62. Echevarria G, Elgueta F, Fierro C, et al. Nitrous oxide (N(2)O) reduces postoperative opioid-induced hyperalgesia after remifentanil-propofol anaesthesia in humans. Br J Anaesth 2011;107:959–65.
63. Troster A, Sittl R, Singler B, et al. Modulation of remifentanil-induced analgesia and postinfusion hyperalgesia by parecoxib in humans. Anesthesiology 2006;105:1016–23.
64. Tuncer S, Yalcin N, Reisli R, Alper Y. The effects of lornoxicam in preventing remifentanil-induced postoperative hyperalgesia. Agri 2009;2:161–7.
65. Lee C, Song YK, Jeong HM, Park SN. The effects of magnesium sulfate infiltration on perioperative opioid consumption and opioid-induced hyperalgesia in patients undergoing robot-assisted laparoscopic prostatectomy with remifentanil-based anesthesia. Korean J Anesthesiol 2011;61:244–50.
66. Song JW, Lee YW, Yoon KB, et al. Magnesium sulfate prevents remifentanil-induced postoperative hyperalgesia in patients undergoing thyroidectomy. Anesth Analg 2011;113:390–7.
67. Lee C, Song YK, Lee JH, Ha SM. The effects of intraoperative adenosine infusion on acute opioid tolerance and opioid induced hyperalgesia induced by remifentanil in adult patients undergoing tonsillectomy. Korean J Pain 2011;24:7–12.
68. Hong BH, Lee WY, Kim YH, et al. Effects of intraoperative low dose ketamine on remifentanil-induced hyperalgesia in gynecologic surgery with sevoflurane anesthesia. Korean J Anesthesiol 2011;61:238–43.
69. Xuerong Y, Yuguang H, Xia J, Hailan W. Ketamine and lornoxicam for preventing a fentanyl-induced increase in postoperative morphine requirement. Anesth Analg 2008;107:2032–7.
70. Schlimp CJ, Pipam W, Wolrab C, Ohner C, Kager HI, Likar R. Clonidine for remifentanil-induced hyperalgesia: A double-blind randomized, placebo-controlled study of clonidine under intra-operative use of remifentanil in elective surgery of the shoulder. Schmerz 2011;25:290–5.
71. Belgrade M, Hall S. Dexmedetomidine infusion for the management of opioid-induced hyperalgesia. Pain Med 2010;11:1819–26.
72. Chen L, Malarick C, Seefeld L, et al. Altered quantitative sensory testing outcome in subjects with opioid therapy. Pain 2009;143:65–70.
73. Bannister K, Dickenson AH. Opioid hyperalgesia. Curr Opin Support Palliat Care 2010;4:1–5.
74. Ram KC, Eisenberg E, Haddad M, Pud D. Oral opioid use alters DNIC but not cold pain perception in patients with chronic pain - new perspective of opioid-induced hyperalgesia. Pain 2008;139:431–8.
75. Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: Effect of long-acting maintenance agent. Drug Alcohol Depend 2001;63:139–46.
76. Vinik HR, Kissin I. Rapid development of tolerance to analgesia during remifentanil infusion in humans. Anesth Analg 1998;86:1307–11.
77. Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: A current view of their possible interactions. Pain 1995;62:259–74.
78. Sjogren P, Jensen NH, Jensen TS. Disappearance of morphine-induced hyperalgesia after discontinuing or substituting morphine with other opioid agonists. Pain 1994;59:313–6.
79. Guignard B, Bossard AE, Coste C, et al. Acute opioid tolerance: Intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology 2000;93:409–17.
80. Angst MS, Clark JD. Opioid-induced hyperalgesia: A qualitative systematic review. Anesthesiology 2006;104:570–87.
81. Baron MJ, McDonald PW. Significant pain reduction in chronic pain patients after detoxification from high-dose opioids. J Opioid Manag 2006;2:277–82
82. Mao J. Opioid-induced abnormal pain sensitivity: Implications in clinical opioid therapy. Pain 2002;100:213–7.
83. Low Y, Clarke CF, Huh BK. Opioid-induced hyperalgesia: A review of epidemiology, mechanisms and management. Singapore Med J 2012;53:357–60.
84. Fishbain DA, Cole B, Lewis JE, et al. Do opioids induce hyperalgesia in humans? An evidence-based structured review. Pain Med 2009:829–39.
85. Chu LF, D’Arcy N, Brady C, et al. Analgesic tolerance without demonstrable opioid-induced hyperalgesia: a double-blinded, randomized, placebo-controlled trial of sustained-release morphine for treatment of chronic nonradicular low-back pain. Pain 2012;153:1583–92.