User login
Heart-brain medicine: Update 2009
Last October, the 2009 Heart-Brain Summit—the fourth annual summit of this type presented by the Bakken Heart-Brain Institute—was held in Chicago and built on the the first three summits’ tradition of open-minded discussion, out-of-the-box thinking, scholarly activity, and engagement of attendees from varied backgrounds.
DEPRESSION AND HEART DISEASE: A WATERSHED YEAR, OR JUMPING THE GUN?
The year leading up to the 2009 summit may be remembered as a watershed period for the field of heart-brain medicine, in light of the American Heart Association’s (AHA’s) inclusion of the recommendation to screen patients with coronary artery disease (CAD) for depression in its science advisory on depression and CAD.1 As has been discussed at prior Heart-Brain Summits, there is incontrovertible evidence in the literature that CAD patients with depression have a worse prognosis than do their counterparts without depression.2–6 While the link is clear, the etiology or mechanism behind depression’s association with worse CAD outcomes is debated. Possible reasons for the association range from greater nonadherence with medical therapy7 to increased systemic inflammation related to the decreased vagal tone associated with depression.8 Furthermore, there is clear evidence that patients with depression and CAD can be treated for their depression safely with cognitive and pharmacologic therapy.5,9 What is lacking, however, is convincing data that the treatment of depression in patients with CAD leads to improved outcomes.10
The topic for the first half of the opening day of the 2009 summit was whether the AHA has gotten ahead of itself in its science advisory1 and whether we should require demonstrable benefits from the treatment of depression in CAD patients before screening for depression is recommended in all patients with CAD. This is a critically important question for the field as well as for the Bakken Heart-Brain Institute, which under our leadership has been advocating for a clinical trial to address this very issue. Cardiologists addressing this question were well reminded that logical therapeutic targets without proven end points have failed us in the past. For instance, it was a rational concept that the suppression of premature ventricular contractions in patients with a history of acute myocardial infarction would lead to decreased ventricular tachycardia and death. Unfortunately, when this concept was put to the test in a randomized clinical trial, increased death was observed in the treatment group.11 More recent examples—and perhaps more applicable to depression, given its chronic nature—come from recent clinical trials demonstrating that tight blood sugar control is associated with higher mortality than moderate blood sugar control in critically ill patients12 and that intensive blood pressure control does not yield greater reductions in cardiovascular events compared with moderate blood pressure control in patients with type 2 diabetes.13
So we are faced with a chronic disease state—depression—that is clearly linked to adverse outcomes and death in patients with CAD. In the context of this association, we also know the following:
- The AHA science advisory recommends that we screen all CAD patients for depression.
- Treating depression in heart disease patients is safe.
- There is no clear proof that treating depression will reverse the increased risk associated with depression in patients with CAD.
- There is a community of physicians who treat CAD patients who are skeptical about therapies that do not have outcomes data.
The summit’s first morning concluded with a debate on whether now is the time for a large-scale multicenter randomized trial, which raised several important issues:
- The limited effectiveness of treatment for depression (approximately 30% to 40%)
- The ethics of randomizing a patient with depression to placebo
- The required size of the trial, given the efficacy of antidepressant therapy
- Measures to define response to therapy
- The utility of surrogate markers for adverse events in CAD versus a mortality end point.
The discussion and presentations were excellent and animated. In the end, each attendee was left to reach his or her own conclusion. Personally, one of us (M.S.P.) was surprised to be left with the conclusion that we are not ready for a definitive clinical trial.
In the cardiovascular medicine literature we were faced with a similar situation regarding the management of patients with atrial fibrillation. In the AFFIRM trial, patients were randomized to conservative treatment (rate control and warfarin) or aggressive treatment (rate control, warfarin, and any and all therapies to convert to and maintain normal sinus rhythm).14 Ultimately there was no difference between the groups, with a trend toward improved outcomes in the conservatively treated patients. What we really learned was that our therapies to convert to and maintain normal sinus rhythm were inadequate, and that in the case of atrial fibrillation at least we could clearly identify which patients did not respond to therapy.14 These findings ultimately may have led the field astray, as we still do not know if we have efficacious therapies for the treatment of atrial fibrillation and whether patients would benefit.
STRATEGIES FOR MODULATING HEART-BRAIN INTERACTIONS
In line with the need for more effective strategies to modulate heart-brain interactions, the summit went on to review and discuss the role of biofeedback. If the effects of depression, post-traumatic stress disorder, and other psychological modulators of vagal tone are the mechanism of action for adverse outcomes in these patient populations, then methods to directly modulate vagal tone may prove efficacious.15 Within the Bakken Heart-Brain Institute we recently committed half a million dollars to fund a biofeedback program. The program’s goal is to investigate the efficacy of biofeedback in improving outcomes within and across several states of cardiovascular disease and chronic disease. We believe that rigorous and standardized delivery and quantification of the effects of biofeedback are critical in order to robustly determine the role of biofeedback in the treatment of patients with chronic disease.
The group of experts assembled at this year’s summit presented further evidence of the potential importance of biofeedback for the control and treatment of multiple disorders, including heart failure, epilepsy, and chronic headache. As the mechanisms underlying brain interactions with end-organ innervations and systemic inflammation are dissected, it is clear that this field of medicine will have greater impact on the outcomes of many patient populations.
CROSS-FERTILIZATION OF TREATMENT APPROACHES
The summit abounded with evidence and examples of how neurology, cardiology, and psychiatry continue to cross-fertilize one another and foster interdisciplinary innovation. We were fortunate to have Brian Litt, MD, from the University of Pennsylvania return for the 2009 summit to update us on the progress of detecting, mapping, and extinguishing early seizure activity before there is clinical evidence of a seizure. The lessons learned and clinical advancement of internal cardiac defibrillators offer insights and great hope for this potentially important advancement in the treatment of seizure disorders. Similarly, Irving Zucker, PhD, from the University of Nebraska reviewed how neuromodulation through the baroreceptors can be targeted to modulate arterial blood pressure. Clearly there is great potential for device-based therapies to augment the treatment of chronic hypertension and improve outcomes in clinical populations at risk.
A LOOK AHEAD
Many of the topics reviewed above are discussed in detail in the proceedings supplement that follows. We continue to be excited and gratified by the progress being made in the field of heart-brain medicine. The continuing commitment to the rigorous multidisciplinary approach that has served this field well to date will continue to advance our understanding of disease and improve outcomes in our patients. We hope you will join us September 23–24, 2010, at the Lou Ruvo Center for Brain Health in Las Vegas, Nevada, for the 2010 Heart-Brain Summit, our fifth annual gathering.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Frazier L, Vaughn WK, Willerson JT, Ballantyne CM, Boerwinkle E. Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events. Biol Res Nurs 2009; 11:163–173.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Davidson KW, Schwartz JE, Kirkland SA, et al Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Summers KM, Martin KE, Watson K. Impact and clinical management of depression in patients with coronary artery disease. Pharmacotherapy 2010; 30:304–322.
- Kendler KS, Gardner CO, Fiske A, Gatz M. Major depression and coronary artery disease in the Swedish Twin Registry. Arch Gen Psychiatry 2009; 66:857–863.
- Albert NM, Fonarow GC, Abraham WT, et al Depression and clinical outcomes in heart failure: an OPTIMIZE-HF analysis. Am J Med 2009; 122:366–373.
- Khawaja IS, Westermeyer JJ, Gajwani P, Feinstein RE. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont) 2009; 6:38–51.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. JAMA 2002; 288:701–709.
- Shapiro PA. Depression in coronary artery disease: does treatment help? Cleve Clin J Med 2008; 75( suppl 2):S5–S9.
- Echt DS, Liebson PR, Mitchell LB, et al Mortality and morbidity in patients receiving encainide, flecainide, or placebo: the Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991; 324:781–788.
- Finfer S, Chittock DR, Su SY, et al Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Cushman WC, Evans GW, Byington RP, et al Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Wyse DG, Waldo AL, DiMarco JP, et al A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Penn MS, Bakken EE. Heart-brain medicine: update 2008. Cleve Clin J Med 2009; 76( suppl 2):S5–S7.
Last October, the 2009 Heart-Brain Summit—the fourth annual summit of this type presented by the Bakken Heart-Brain Institute—was held in Chicago and built on the the first three summits’ tradition of open-minded discussion, out-of-the-box thinking, scholarly activity, and engagement of attendees from varied backgrounds.
DEPRESSION AND HEART DISEASE: A WATERSHED YEAR, OR JUMPING THE GUN?
The year leading up to the 2009 summit may be remembered as a watershed period for the field of heart-brain medicine, in light of the American Heart Association’s (AHA’s) inclusion of the recommendation to screen patients with coronary artery disease (CAD) for depression in its science advisory on depression and CAD.1 As has been discussed at prior Heart-Brain Summits, there is incontrovertible evidence in the literature that CAD patients with depression have a worse prognosis than do their counterparts without depression.2–6 While the link is clear, the etiology or mechanism behind depression’s association with worse CAD outcomes is debated. Possible reasons for the association range from greater nonadherence with medical therapy7 to increased systemic inflammation related to the decreased vagal tone associated with depression.8 Furthermore, there is clear evidence that patients with depression and CAD can be treated for their depression safely with cognitive and pharmacologic therapy.5,9 What is lacking, however, is convincing data that the treatment of depression in patients with CAD leads to improved outcomes.10
The topic for the first half of the opening day of the 2009 summit was whether the AHA has gotten ahead of itself in its science advisory1 and whether we should require demonstrable benefits from the treatment of depression in CAD patients before screening for depression is recommended in all patients with CAD. This is a critically important question for the field as well as for the Bakken Heart-Brain Institute, which under our leadership has been advocating for a clinical trial to address this very issue. Cardiologists addressing this question were well reminded that logical therapeutic targets without proven end points have failed us in the past. For instance, it was a rational concept that the suppression of premature ventricular contractions in patients with a history of acute myocardial infarction would lead to decreased ventricular tachycardia and death. Unfortunately, when this concept was put to the test in a randomized clinical trial, increased death was observed in the treatment group.11 More recent examples—and perhaps more applicable to depression, given its chronic nature—come from recent clinical trials demonstrating that tight blood sugar control is associated with higher mortality than moderate blood sugar control in critically ill patients12 and that intensive blood pressure control does not yield greater reductions in cardiovascular events compared with moderate blood pressure control in patients with type 2 diabetes.13
So we are faced with a chronic disease state—depression—that is clearly linked to adverse outcomes and death in patients with CAD. In the context of this association, we also know the following:
- The AHA science advisory recommends that we screen all CAD patients for depression.
- Treating depression in heart disease patients is safe.
- There is no clear proof that treating depression will reverse the increased risk associated with depression in patients with CAD.
- There is a community of physicians who treat CAD patients who are skeptical about therapies that do not have outcomes data.
The summit’s first morning concluded with a debate on whether now is the time for a large-scale multicenter randomized trial, which raised several important issues:
- The limited effectiveness of treatment for depression (approximately 30% to 40%)
- The ethics of randomizing a patient with depression to placebo
- The required size of the trial, given the efficacy of antidepressant therapy
- Measures to define response to therapy
- The utility of surrogate markers for adverse events in CAD versus a mortality end point.
The discussion and presentations were excellent and animated. In the end, each attendee was left to reach his or her own conclusion. Personally, one of us (M.S.P.) was surprised to be left with the conclusion that we are not ready for a definitive clinical trial.
In the cardiovascular medicine literature we were faced with a similar situation regarding the management of patients with atrial fibrillation. In the AFFIRM trial, patients were randomized to conservative treatment (rate control and warfarin) or aggressive treatment (rate control, warfarin, and any and all therapies to convert to and maintain normal sinus rhythm).14 Ultimately there was no difference between the groups, with a trend toward improved outcomes in the conservatively treated patients. What we really learned was that our therapies to convert to and maintain normal sinus rhythm were inadequate, and that in the case of atrial fibrillation at least we could clearly identify which patients did not respond to therapy.14 These findings ultimately may have led the field astray, as we still do not know if we have efficacious therapies for the treatment of atrial fibrillation and whether patients would benefit.
STRATEGIES FOR MODULATING HEART-BRAIN INTERACTIONS
In line with the need for more effective strategies to modulate heart-brain interactions, the summit went on to review and discuss the role of biofeedback. If the effects of depression, post-traumatic stress disorder, and other psychological modulators of vagal tone are the mechanism of action for adverse outcomes in these patient populations, then methods to directly modulate vagal tone may prove efficacious.15 Within the Bakken Heart-Brain Institute we recently committed half a million dollars to fund a biofeedback program. The program’s goal is to investigate the efficacy of biofeedback in improving outcomes within and across several states of cardiovascular disease and chronic disease. We believe that rigorous and standardized delivery and quantification of the effects of biofeedback are critical in order to robustly determine the role of biofeedback in the treatment of patients with chronic disease.
The group of experts assembled at this year’s summit presented further evidence of the potential importance of biofeedback for the control and treatment of multiple disorders, including heart failure, epilepsy, and chronic headache. As the mechanisms underlying brain interactions with end-organ innervations and systemic inflammation are dissected, it is clear that this field of medicine will have greater impact on the outcomes of many patient populations.
CROSS-FERTILIZATION OF TREATMENT APPROACHES
The summit abounded with evidence and examples of how neurology, cardiology, and psychiatry continue to cross-fertilize one another and foster interdisciplinary innovation. We were fortunate to have Brian Litt, MD, from the University of Pennsylvania return for the 2009 summit to update us on the progress of detecting, mapping, and extinguishing early seizure activity before there is clinical evidence of a seizure. The lessons learned and clinical advancement of internal cardiac defibrillators offer insights and great hope for this potentially important advancement in the treatment of seizure disorders. Similarly, Irving Zucker, PhD, from the University of Nebraska reviewed how neuromodulation through the baroreceptors can be targeted to modulate arterial blood pressure. Clearly there is great potential for device-based therapies to augment the treatment of chronic hypertension and improve outcomes in clinical populations at risk.
A LOOK AHEAD
Many of the topics reviewed above are discussed in detail in the proceedings supplement that follows. We continue to be excited and gratified by the progress being made in the field of heart-brain medicine. The continuing commitment to the rigorous multidisciplinary approach that has served this field well to date will continue to advance our understanding of disease and improve outcomes in our patients. We hope you will join us September 23–24, 2010, at the Lou Ruvo Center for Brain Health in Las Vegas, Nevada, for the 2010 Heart-Brain Summit, our fifth annual gathering.
Last October, the 2009 Heart-Brain Summit—the fourth annual summit of this type presented by the Bakken Heart-Brain Institute—was held in Chicago and built on the the first three summits’ tradition of open-minded discussion, out-of-the-box thinking, scholarly activity, and engagement of attendees from varied backgrounds.
DEPRESSION AND HEART DISEASE: A WATERSHED YEAR, OR JUMPING THE GUN?
The year leading up to the 2009 summit may be remembered as a watershed period for the field of heart-brain medicine, in light of the American Heart Association’s (AHA’s) inclusion of the recommendation to screen patients with coronary artery disease (CAD) for depression in its science advisory on depression and CAD.1 As has been discussed at prior Heart-Brain Summits, there is incontrovertible evidence in the literature that CAD patients with depression have a worse prognosis than do their counterparts without depression.2–6 While the link is clear, the etiology or mechanism behind depression’s association with worse CAD outcomes is debated. Possible reasons for the association range from greater nonadherence with medical therapy7 to increased systemic inflammation related to the decreased vagal tone associated with depression.8 Furthermore, there is clear evidence that patients with depression and CAD can be treated for their depression safely with cognitive and pharmacologic therapy.5,9 What is lacking, however, is convincing data that the treatment of depression in patients with CAD leads to improved outcomes.10
The topic for the first half of the opening day of the 2009 summit was whether the AHA has gotten ahead of itself in its science advisory1 and whether we should require demonstrable benefits from the treatment of depression in CAD patients before screening for depression is recommended in all patients with CAD. This is a critically important question for the field as well as for the Bakken Heart-Brain Institute, which under our leadership has been advocating for a clinical trial to address this very issue. Cardiologists addressing this question were well reminded that logical therapeutic targets without proven end points have failed us in the past. For instance, it was a rational concept that the suppression of premature ventricular contractions in patients with a history of acute myocardial infarction would lead to decreased ventricular tachycardia and death. Unfortunately, when this concept was put to the test in a randomized clinical trial, increased death was observed in the treatment group.11 More recent examples—and perhaps more applicable to depression, given its chronic nature—come from recent clinical trials demonstrating that tight blood sugar control is associated with higher mortality than moderate blood sugar control in critically ill patients12 and that intensive blood pressure control does not yield greater reductions in cardiovascular events compared with moderate blood pressure control in patients with type 2 diabetes.13
So we are faced with a chronic disease state—depression—that is clearly linked to adverse outcomes and death in patients with CAD. In the context of this association, we also know the following:
- The AHA science advisory recommends that we screen all CAD patients for depression.
- Treating depression in heart disease patients is safe.
- There is no clear proof that treating depression will reverse the increased risk associated with depression in patients with CAD.
- There is a community of physicians who treat CAD patients who are skeptical about therapies that do not have outcomes data.
The summit’s first morning concluded with a debate on whether now is the time for a large-scale multicenter randomized trial, which raised several important issues:
- The limited effectiveness of treatment for depression (approximately 30% to 40%)
- The ethics of randomizing a patient with depression to placebo
- The required size of the trial, given the efficacy of antidepressant therapy
- Measures to define response to therapy
- The utility of surrogate markers for adverse events in CAD versus a mortality end point.
The discussion and presentations were excellent and animated. In the end, each attendee was left to reach his or her own conclusion. Personally, one of us (M.S.P.) was surprised to be left with the conclusion that we are not ready for a definitive clinical trial.
In the cardiovascular medicine literature we were faced with a similar situation regarding the management of patients with atrial fibrillation. In the AFFIRM trial, patients were randomized to conservative treatment (rate control and warfarin) or aggressive treatment (rate control, warfarin, and any and all therapies to convert to and maintain normal sinus rhythm).14 Ultimately there was no difference between the groups, with a trend toward improved outcomes in the conservatively treated patients. What we really learned was that our therapies to convert to and maintain normal sinus rhythm were inadequate, and that in the case of atrial fibrillation at least we could clearly identify which patients did not respond to therapy.14 These findings ultimately may have led the field astray, as we still do not know if we have efficacious therapies for the treatment of atrial fibrillation and whether patients would benefit.
STRATEGIES FOR MODULATING HEART-BRAIN INTERACTIONS
In line with the need for more effective strategies to modulate heart-brain interactions, the summit went on to review and discuss the role of biofeedback. If the effects of depression, post-traumatic stress disorder, and other psychological modulators of vagal tone are the mechanism of action for adverse outcomes in these patient populations, then methods to directly modulate vagal tone may prove efficacious.15 Within the Bakken Heart-Brain Institute we recently committed half a million dollars to fund a biofeedback program. The program’s goal is to investigate the efficacy of biofeedback in improving outcomes within and across several states of cardiovascular disease and chronic disease. We believe that rigorous and standardized delivery and quantification of the effects of biofeedback are critical in order to robustly determine the role of biofeedback in the treatment of patients with chronic disease.
The group of experts assembled at this year’s summit presented further evidence of the potential importance of biofeedback for the control and treatment of multiple disorders, including heart failure, epilepsy, and chronic headache. As the mechanisms underlying brain interactions with end-organ innervations and systemic inflammation are dissected, it is clear that this field of medicine will have greater impact on the outcomes of many patient populations.
CROSS-FERTILIZATION OF TREATMENT APPROACHES
The summit abounded with evidence and examples of how neurology, cardiology, and psychiatry continue to cross-fertilize one another and foster interdisciplinary innovation. We were fortunate to have Brian Litt, MD, from the University of Pennsylvania return for the 2009 summit to update us on the progress of detecting, mapping, and extinguishing early seizure activity before there is clinical evidence of a seizure. The lessons learned and clinical advancement of internal cardiac defibrillators offer insights and great hope for this potentially important advancement in the treatment of seizure disorders. Similarly, Irving Zucker, PhD, from the University of Nebraska reviewed how neuromodulation through the baroreceptors can be targeted to modulate arterial blood pressure. Clearly there is great potential for device-based therapies to augment the treatment of chronic hypertension and improve outcomes in clinical populations at risk.
A LOOK AHEAD
Many of the topics reviewed above are discussed in detail in the proceedings supplement that follows. We continue to be excited and gratified by the progress being made in the field of heart-brain medicine. The continuing commitment to the rigorous multidisciplinary approach that has served this field well to date will continue to advance our understanding of disease and improve outcomes in our patients. We hope you will join us September 23–24, 2010, at the Lou Ruvo Center for Brain Health in Las Vegas, Nevada, for the 2010 Heart-Brain Summit, our fifth annual gathering.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Frazier L, Vaughn WK, Willerson JT, Ballantyne CM, Boerwinkle E. Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events. Biol Res Nurs 2009; 11:163–173.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Davidson KW, Schwartz JE, Kirkland SA, et al Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Summers KM, Martin KE, Watson K. Impact and clinical management of depression in patients with coronary artery disease. Pharmacotherapy 2010; 30:304–322.
- Kendler KS, Gardner CO, Fiske A, Gatz M. Major depression and coronary artery disease in the Swedish Twin Registry. Arch Gen Psychiatry 2009; 66:857–863.
- Albert NM, Fonarow GC, Abraham WT, et al Depression and clinical outcomes in heart failure: an OPTIMIZE-HF analysis. Am J Med 2009; 122:366–373.
- Khawaja IS, Westermeyer JJ, Gajwani P, Feinstein RE. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont) 2009; 6:38–51.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. JAMA 2002; 288:701–709.
- Shapiro PA. Depression in coronary artery disease: does treatment help? Cleve Clin J Med 2008; 75( suppl 2):S5–S9.
- Echt DS, Liebson PR, Mitchell LB, et al Mortality and morbidity in patients receiving encainide, flecainide, or placebo: the Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991; 324:781–788.
- Finfer S, Chittock DR, Su SY, et al Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Cushman WC, Evans GW, Byington RP, et al Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Wyse DG, Waldo AL, DiMarco JP, et al A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Penn MS, Bakken EE. Heart-brain medicine: update 2008. Cleve Clin J Med 2009; 76( suppl 2):S5–S7.
- Lichtman JH, Bigger JT, Blumenthal JA, et al Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research: endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Frazier L, Vaughn WK, Willerson JT, Ballantyne CM, Boerwinkle E. Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events. Biol Res Nurs 2009; 11:163–173.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Davidson KW, Schwartz JE, Kirkland SA, et al Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Summers KM, Martin KE, Watson K. Impact and clinical management of depression in patients with coronary artery disease. Pharmacotherapy 2010; 30:304–322.
- Kendler KS, Gardner CO, Fiske A, Gatz M. Major depression and coronary artery disease in the Swedish Twin Registry. Arch Gen Psychiatry 2009; 66:857–863.
- Albert NM, Fonarow GC, Abraham WT, et al Depression and clinical outcomes in heart failure: an OPTIMIZE-HF analysis. Am J Med 2009; 122:366–373.
- Khawaja IS, Westermeyer JJ, Gajwani P, Feinstein RE. Depression and coronary artery disease: the association, mechanisms, and therapeutic implications. Psychiatry (Edgmont) 2009; 6:38–51.
- Glassman AH, O’Connor CM, Califf RM, et al Sertraline treatment of major depression in patients with acute MI or unstable angina. Sertraline Antidepressant Heart Attack Randomized Trial (SADHART) Group. JAMA 2002; 288:701–709.
- Shapiro PA. Depression in coronary artery disease: does treatment help? Cleve Clin J Med 2008; 75( suppl 2):S5–S9.
- Echt DS, Liebson PR, Mitchell LB, et al Mortality and morbidity in patients receiving encainide, flecainide, or placebo: the Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991; 324:781–788.
- Finfer S, Chittock DR, Su SY, et al Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297.
- Cushman WC, Evans GW, Byington RP, et al Effects of intensive blood-pressure control in type 2 diabetes mellitus. N Engl J Med 2010; 362:1575–1585.
- Wyse DG, Waldo AL, DiMarco JP, et al A comparison of rate control and rhythm control in patients with atrial fibrillation. N Engl J Med 2002; 347:1825–1833.
- Penn MS, Bakken EE. Heart-brain medicine: update 2008. Cleve Clin J Med 2009; 76( suppl 2):S5–S7.
Multidisciplinary research in biofeedback
Heart-brain medicine: Update 2008
Investigators involved in heart-brain medicine are dedicated to defining the physiology associated with interactions of the neurological and cardiovascular systems. In 2004 the Bakken Heart-Brain Institute was founded at Cleveland Clinic because we believed that furthering our understanding of this physiology could lead to a better understanding of chronic disease, define novel therapies, and improve patient outcomes.
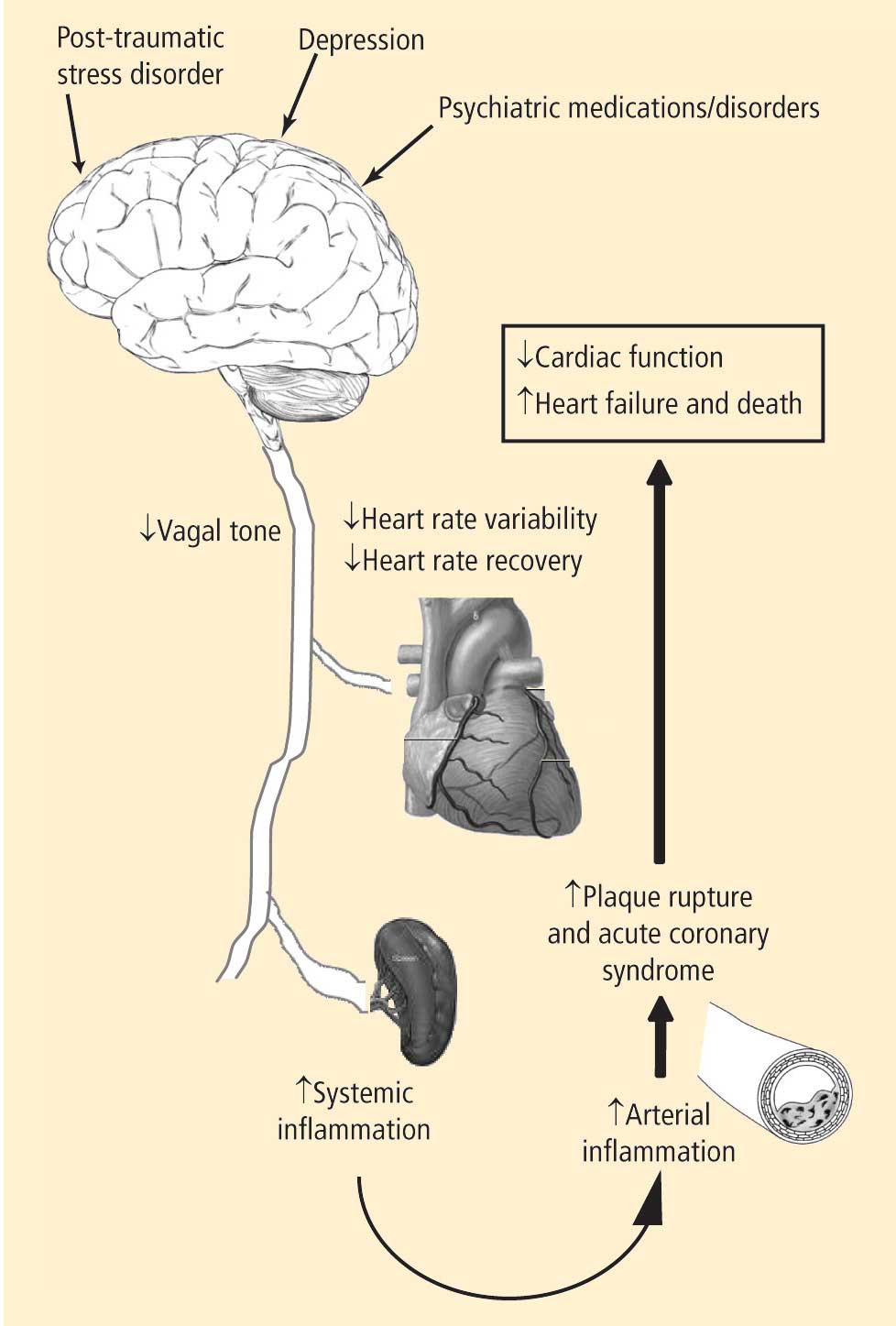
- Depression leads to decreased vagal tone
- Decreased vagal tone leads to increased inflammation
- Increased inflammation leads to acute coronary syndrome.
Speakers at the 2008 Summit offered insights into the physiology, clinical measures, and molecular pathways involved in linking the heart and the brain, including:
- Measures of heart rate variability in depression
- The utility of heart rate variability and heart rate recovery in quantifying vagal tone and outcome in patients with and without coronary artery disease
- Pathways of inflammation involved in acute coronary syndrome.
MOUNTING CLINICAL EVIDENCE LINKING DEPRESSION WITH CARDIAC OUTCOMES
The 2007 and 2008 Summits highlighted the link between depression and outcomes in patients with atherosclerosis (2007)1 and the potential associated mechanisms (2008). Just as exciting are the developments since last June: numerous papers have been published demonstrating this link in clinical populations, and depression screening has been included in recommendations from the American Heart Association on the treatment of patients with coronary artery disease—recommendations that are endorsed by the American Psychiatric Association.2
The studies published since June 2008 demonstrate clear links between depression and morbidity and mortality from cardiovascular causes. A recent paper from the Nurses’ Health Study showed that individuals with depression had a higher incidence of cardiovascular death.3 Notably, subjects in the Nurses’ Health Study had no clinical evidence of atherosclerotic heart disease at enrollment. In another recent study, depression was associated with worse outcomes in patients following coronary stenting.4 Finally, and most interestingly, depression was recently associated with endothelial dysfunction in patients with atypical angina and angiographically normal coronary arteries.5 Thus, regardless of the degree of underlying atherosclerosis, depression is associated with cardiovascular morbidity or mortality.
Less clear is the relationship between depression and inflammation as measured by surrogate inflammatory markers. An analysis of the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study suggested that increased inflammatory markers accounted for only a small portion of the risk of coronary heart disease associated with depression.6 Conversely, a recent analysis of patients with stable coronary artery disease demonstrated a strong correlation between major depressive disorders and highsensitivity C-reactive protein.7
Clearly, significant work has yet to be done to fully elucidate the molecular pathways that link depression and adverse outcomes in patients at risk for coronary artery disease. That said, it is very encouraging that professional societies are beginning to recognize the value and importance of heart-brain medicine in identifying novel strategies for improving patient outcomes.
STILL ELUSIVE: EVIDENCE THAT DEPRESSION THERAPY IMPROVES CARDIAC OUTCOMES
At the 2008 Summit there was clear enthusiasm among attendees and faculty for advances in our understanding of the pathways discussed above. Since then, as reviewed above, significant publications have furthered the link between heart and brain in the setting of atherosclerotic heart disease. That said, the missing piece—the demonstration that treating depression leads to improved outcomes in patients with coronary artery disease—remains missing.
Some advances in this regard have been made. A recent study from the Enhancing Recovery In Coronary Heart Disease (ENRICHD) clinical trial demonstrated that major depression in any patient who survived myocardial infarction decreased survival over 2.5 years.8 Interestingly, and perhaps critical for an event-driven treatment trial in the future, this analysis showed an even worse outcome in patients who experienced their initial episode of major depression after their myocardial infarction.8 The need, ethics, and design of clinical trials to determine whether treatment of depression leads to improved outcomes in patients with coronary artery disease will be a major topic of the 4th Annual Heart-Brain Summit, to be held in Chicago on October 15–16, 2009.
OTHER HIGHLIGHTS, INCLUDING ROLE OF THE HEALING ENVIRONMENT
While much of the early focus of the 2008 Heart-Brain Summit was on the interaction of depression, inflammation, and outcomes in patients with coronary artery disease, a significant portion of the Summit identified other disease states and opportunities. The disease states discussed can be divided into primary cardiac, primary psychiatric, and primary neurologic. Cardiac topics under continued investigation include the role of vagal tone on the inflammatory response that regulates left ventricular remodeling following acute myocardial infarction9 as well as the role of spinal stimulation for treatment of refractory myocardial ischemia. Psychiatric disorders of interest that have been shown to modulate vagal tone include post-traumatic stress disorder,10 which has also been shown to increase the risk for coronary heart disease.11,12 Neurologically, advances concerning the polyvagal theory of autonomic nervous system control and cardiac control were discussed.13,14
On the Summit’s final day, the discussions of neuropathways, inflammation, and cardiac control gave way to presentations on the role of the healing environment. Following discussions of how depression can have significant ramifications on systemic inflammation and acute coronary syndrome, it was interesting to review data on how the presence of family and the patient environment can improve patient outcomes.
Many of the topics touched on above are discussed in greater detail in the following pages of this proceedings of the 2008 Bakken Heart-Brain Summit. We are gratified to see the advancements in the field of heartbrain medicine over the past 5 years, and especially to see the recognition the discipline is receiving in our attempt to improve patient outcomes.
FAR MORE QUESTIONS REMAIN
Without a doubt there are more questions than answers at this time. That said, by continuing the rigorous multidisciplinary approach that has served this field well to date, many questions will be answered. We hope you will join us in Chicago on October 15–16, 2009, for the 4th Annual Heart-Brain Summit, which will be jointly hosted by the Society of Heart-Brain Medicine and the Bakken Heart-Brain Institute.
- Penn MS, Bakken EE. Heart-brain medicine: update 2007. Cleve Clin J Med 2008; 75( suppl 2):S3–S4.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research. Endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Whang W, Kubzansky LD, Kawachi I, et al. Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol 2009; 53:950–958.
- Frazier L, Vaughn W, Willerson J, Ballantyne C, Boerwinkle E Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events [published online ahead of print February 26, 2009]. Biol Res Nurs. doi:10.1177/1099800409332801.
- Kim JH, Kim JW, Ko YH, et al Coronary endothelial dysfunction associated with a depressive mood in patients with atypical angina but angiographically normal coronary artery [published online ahead of print March 7, 2009]. Int J Cardiol. doi:10.1016/j.ijcard.2009.02.004.
- Davidson KW, Schwartz JE, Kirkland SA, et al. Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Bankier B, Barajas J, Martinez-Rumayor A, Januzzi JL. Association between major depressive disorder and C-reactive protein levels in stable coronary heart disease patients. J Psychosom Res 2009; 66:189–194.
- Carney RM, Freedland KE, Steinmeyer B, et al History of depression and survival after acute myocardial infarction [published online ahead of print February 27, 2009]. Psychosom Med. doi:10.1097/PSY.0b013e31819b69e3.
- Vasilyev N, Williams T, Brennan ML, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005; 112:2812–2820.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
- Kubzansky LD, Koenen KC, Jones C, Eaton WW. A prospective study of posttraumatic stress disorder symptoms and coronary heart disease in women. Health Psychol 2009; 28:125–130.
- Kubzansky LD, Koenen KC, Spiro A, Vokonas PS, Sparrow D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch Gen Psychiatry 2007; 64:109–116.
- Porges SW. The polyvagal perspective. Biol Psychol 2007; 74:116–143.
- Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol 2001; 42:123–146.
Investigators involved in heart-brain medicine are dedicated to defining the physiology associated with interactions of the neurological and cardiovascular systems. In 2004 the Bakken Heart-Brain Institute was founded at Cleveland Clinic because we believed that furthering our understanding of this physiology could lead to a better understanding of chronic disease, define novel therapies, and improve patient outcomes.
- Depression leads to decreased vagal tone
- Decreased vagal tone leads to increased inflammation
- Increased inflammation leads to acute coronary syndrome.
Speakers at the 2008 Summit offered insights into the physiology, clinical measures, and molecular pathways involved in linking the heart and the brain, including:
- Measures of heart rate variability in depression
- The utility of heart rate variability and heart rate recovery in quantifying vagal tone and outcome in patients with and without coronary artery disease
- Pathways of inflammation involved in acute coronary syndrome.
MOUNTING CLINICAL EVIDENCE LINKING DEPRESSION WITH CARDIAC OUTCOMES
The 2007 and 2008 Summits highlighted the link between depression and outcomes in patients with atherosclerosis (2007)1 and the potential associated mechanisms (2008). Just as exciting are the developments since last June: numerous papers have been published demonstrating this link in clinical populations, and depression screening has been included in recommendations from the American Heart Association on the treatment of patients with coronary artery disease—recommendations that are endorsed by the American Psychiatric Association.2
The studies published since June 2008 demonstrate clear links between depression and morbidity and mortality from cardiovascular causes. A recent paper from the Nurses’ Health Study showed that individuals with depression had a higher incidence of cardiovascular death.3 Notably, subjects in the Nurses’ Health Study had no clinical evidence of atherosclerotic heart disease at enrollment. In another recent study, depression was associated with worse outcomes in patients following coronary stenting.4 Finally, and most interestingly, depression was recently associated with endothelial dysfunction in patients with atypical angina and angiographically normal coronary arteries.5 Thus, regardless of the degree of underlying atherosclerosis, depression is associated with cardiovascular morbidity or mortality.
Less clear is the relationship between depression and inflammation as measured by surrogate inflammatory markers. An analysis of the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study suggested that increased inflammatory markers accounted for only a small portion of the risk of coronary heart disease associated with depression.6 Conversely, a recent analysis of patients with stable coronary artery disease demonstrated a strong correlation between major depressive disorders and highsensitivity C-reactive protein.7
Clearly, significant work has yet to be done to fully elucidate the molecular pathways that link depression and adverse outcomes in patients at risk for coronary artery disease. That said, it is very encouraging that professional societies are beginning to recognize the value and importance of heart-brain medicine in identifying novel strategies for improving patient outcomes.
STILL ELUSIVE: EVIDENCE THAT DEPRESSION THERAPY IMPROVES CARDIAC OUTCOMES
At the 2008 Summit there was clear enthusiasm among attendees and faculty for advances in our understanding of the pathways discussed above. Since then, as reviewed above, significant publications have furthered the link between heart and brain in the setting of atherosclerotic heart disease. That said, the missing piece—the demonstration that treating depression leads to improved outcomes in patients with coronary artery disease—remains missing.
Some advances in this regard have been made. A recent study from the Enhancing Recovery In Coronary Heart Disease (ENRICHD) clinical trial demonstrated that major depression in any patient who survived myocardial infarction decreased survival over 2.5 years.8 Interestingly, and perhaps critical for an event-driven treatment trial in the future, this analysis showed an even worse outcome in patients who experienced their initial episode of major depression after their myocardial infarction.8 The need, ethics, and design of clinical trials to determine whether treatment of depression leads to improved outcomes in patients with coronary artery disease will be a major topic of the 4th Annual Heart-Brain Summit, to be held in Chicago on October 15–16, 2009.
OTHER HIGHLIGHTS, INCLUDING ROLE OF THE HEALING ENVIRONMENT
While much of the early focus of the 2008 Heart-Brain Summit was on the interaction of depression, inflammation, and outcomes in patients with coronary artery disease, a significant portion of the Summit identified other disease states and opportunities. The disease states discussed can be divided into primary cardiac, primary psychiatric, and primary neurologic. Cardiac topics under continued investigation include the role of vagal tone on the inflammatory response that regulates left ventricular remodeling following acute myocardial infarction9 as well as the role of spinal stimulation for treatment of refractory myocardial ischemia. Psychiatric disorders of interest that have been shown to modulate vagal tone include post-traumatic stress disorder,10 which has also been shown to increase the risk for coronary heart disease.11,12 Neurologically, advances concerning the polyvagal theory of autonomic nervous system control and cardiac control were discussed.13,14
On the Summit’s final day, the discussions of neuropathways, inflammation, and cardiac control gave way to presentations on the role of the healing environment. Following discussions of how depression can have significant ramifications on systemic inflammation and acute coronary syndrome, it was interesting to review data on how the presence of family and the patient environment can improve patient outcomes.
Many of the topics touched on above are discussed in greater detail in the following pages of this proceedings of the 2008 Bakken Heart-Brain Summit. We are gratified to see the advancements in the field of heartbrain medicine over the past 5 years, and especially to see the recognition the discipline is receiving in our attempt to improve patient outcomes.
FAR MORE QUESTIONS REMAIN
Without a doubt there are more questions than answers at this time. That said, by continuing the rigorous multidisciplinary approach that has served this field well to date, many questions will be answered. We hope you will join us in Chicago on October 15–16, 2009, for the 4th Annual Heart-Brain Summit, which will be jointly hosted by the Society of Heart-Brain Medicine and the Bakken Heart-Brain Institute.
Investigators involved in heart-brain medicine are dedicated to defining the physiology associated with interactions of the neurological and cardiovascular systems. In 2004 the Bakken Heart-Brain Institute was founded at Cleveland Clinic because we believed that furthering our understanding of this physiology could lead to a better understanding of chronic disease, define novel therapies, and improve patient outcomes.
- Depression leads to decreased vagal tone
- Decreased vagal tone leads to increased inflammation
- Increased inflammation leads to acute coronary syndrome.
Speakers at the 2008 Summit offered insights into the physiology, clinical measures, and molecular pathways involved in linking the heart and the brain, including:
- Measures of heart rate variability in depression
- The utility of heart rate variability and heart rate recovery in quantifying vagal tone and outcome in patients with and without coronary artery disease
- Pathways of inflammation involved in acute coronary syndrome.
MOUNTING CLINICAL EVIDENCE LINKING DEPRESSION WITH CARDIAC OUTCOMES
The 2007 and 2008 Summits highlighted the link between depression and outcomes in patients with atherosclerosis (2007)1 and the potential associated mechanisms (2008). Just as exciting are the developments since last June: numerous papers have been published demonstrating this link in clinical populations, and depression screening has been included in recommendations from the American Heart Association on the treatment of patients with coronary artery disease—recommendations that are endorsed by the American Psychiatric Association.2
The studies published since June 2008 demonstrate clear links between depression and morbidity and mortality from cardiovascular causes. A recent paper from the Nurses’ Health Study showed that individuals with depression had a higher incidence of cardiovascular death.3 Notably, subjects in the Nurses’ Health Study had no clinical evidence of atherosclerotic heart disease at enrollment. In another recent study, depression was associated with worse outcomes in patients following coronary stenting.4 Finally, and most interestingly, depression was recently associated with endothelial dysfunction in patients with atypical angina and angiographically normal coronary arteries.5 Thus, regardless of the degree of underlying atherosclerosis, depression is associated with cardiovascular morbidity or mortality.
Less clear is the relationship between depression and inflammation as measured by surrogate inflammatory markers. An analysis of the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study suggested that increased inflammatory markers accounted for only a small portion of the risk of coronary heart disease associated with depression.6 Conversely, a recent analysis of patients with stable coronary artery disease demonstrated a strong correlation between major depressive disorders and highsensitivity C-reactive protein.7
Clearly, significant work has yet to be done to fully elucidate the molecular pathways that link depression and adverse outcomes in patients at risk for coronary artery disease. That said, it is very encouraging that professional societies are beginning to recognize the value and importance of heart-brain medicine in identifying novel strategies for improving patient outcomes.
STILL ELUSIVE: EVIDENCE THAT DEPRESSION THERAPY IMPROVES CARDIAC OUTCOMES
At the 2008 Summit there was clear enthusiasm among attendees and faculty for advances in our understanding of the pathways discussed above. Since then, as reviewed above, significant publications have furthered the link between heart and brain in the setting of atherosclerotic heart disease. That said, the missing piece—the demonstration that treating depression leads to improved outcomes in patients with coronary artery disease—remains missing.
Some advances in this regard have been made. A recent study from the Enhancing Recovery In Coronary Heart Disease (ENRICHD) clinical trial demonstrated that major depression in any patient who survived myocardial infarction decreased survival over 2.5 years.8 Interestingly, and perhaps critical for an event-driven treatment trial in the future, this analysis showed an even worse outcome in patients who experienced their initial episode of major depression after their myocardial infarction.8 The need, ethics, and design of clinical trials to determine whether treatment of depression leads to improved outcomes in patients with coronary artery disease will be a major topic of the 4th Annual Heart-Brain Summit, to be held in Chicago on October 15–16, 2009.
OTHER HIGHLIGHTS, INCLUDING ROLE OF THE HEALING ENVIRONMENT
While much of the early focus of the 2008 Heart-Brain Summit was on the interaction of depression, inflammation, and outcomes in patients with coronary artery disease, a significant portion of the Summit identified other disease states and opportunities. The disease states discussed can be divided into primary cardiac, primary psychiatric, and primary neurologic. Cardiac topics under continued investigation include the role of vagal tone on the inflammatory response that regulates left ventricular remodeling following acute myocardial infarction9 as well as the role of spinal stimulation for treatment of refractory myocardial ischemia. Psychiatric disorders of interest that have been shown to modulate vagal tone include post-traumatic stress disorder,10 which has also been shown to increase the risk for coronary heart disease.11,12 Neurologically, advances concerning the polyvagal theory of autonomic nervous system control and cardiac control were discussed.13,14
On the Summit’s final day, the discussions of neuropathways, inflammation, and cardiac control gave way to presentations on the role of the healing environment. Following discussions of how depression can have significant ramifications on systemic inflammation and acute coronary syndrome, it was interesting to review data on how the presence of family and the patient environment can improve patient outcomes.
Many of the topics touched on above are discussed in greater detail in the following pages of this proceedings of the 2008 Bakken Heart-Brain Summit. We are gratified to see the advancements in the field of heartbrain medicine over the past 5 years, and especially to see the recognition the discipline is receiving in our attempt to improve patient outcomes.
FAR MORE QUESTIONS REMAIN
Without a doubt there are more questions than answers at this time. That said, by continuing the rigorous multidisciplinary approach that has served this field well to date, many questions will be answered. We hope you will join us in Chicago on October 15–16, 2009, for the 4th Annual Heart-Brain Summit, which will be jointly hosted by the Society of Heart-Brain Medicine and the Bakken Heart-Brain Institute.
- Penn MS, Bakken EE. Heart-brain medicine: update 2007. Cleve Clin J Med 2008; 75( suppl 2):S3–S4.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research. Endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Whang W, Kubzansky LD, Kawachi I, et al. Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol 2009; 53:950–958.
- Frazier L, Vaughn W, Willerson J, Ballantyne C, Boerwinkle E Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events [published online ahead of print February 26, 2009]. Biol Res Nurs. doi:10.1177/1099800409332801.
- Kim JH, Kim JW, Ko YH, et al Coronary endothelial dysfunction associated with a depressive mood in patients with atypical angina but angiographically normal coronary artery [published online ahead of print March 7, 2009]. Int J Cardiol. doi:10.1016/j.ijcard.2009.02.004.
- Davidson KW, Schwartz JE, Kirkland SA, et al. Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Bankier B, Barajas J, Martinez-Rumayor A, Januzzi JL. Association between major depressive disorder and C-reactive protein levels in stable coronary heart disease patients. J Psychosom Res 2009; 66:189–194.
- Carney RM, Freedland KE, Steinmeyer B, et al History of depression and survival after acute myocardial infarction [published online ahead of print February 27, 2009]. Psychosom Med. doi:10.1097/PSY.0b013e31819b69e3.
- Vasilyev N, Williams T, Brennan ML, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005; 112:2812–2820.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
- Kubzansky LD, Koenen KC, Jones C, Eaton WW. A prospective study of posttraumatic stress disorder symptoms and coronary heart disease in women. Health Psychol 2009; 28:125–130.
- Kubzansky LD, Koenen KC, Spiro A, Vokonas PS, Sparrow D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch Gen Psychiatry 2007; 64:109–116.
- Porges SW. The polyvagal perspective. Biol Psychol 2007; 74:116–143.
- Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol 2001; 42:123–146.
- Penn MS, Bakken EE. Heart-brain medicine: update 2007. Cleve Clin J Med 2008; 75( suppl 2):S3–S4.
- Lichtman JH, Bigger JT, Blumenthal JA, et al. Depression and coronary heart disease: recommendations for screening, referral, and treatment: a science advisory from the American Heart Association Prevention Committee of the Council on Cardiovascular Nursing, Council on Clinical Cardiology, Council on Epidemiology and Prevention, and Interdisciplinary Council on Quality of Care and Outcomes Research. Endorsed by the American Psychiatric Association. Circulation 2008; 118:1768–1775.
- Whang W, Kubzansky LD, Kawachi I, et al. Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses’ Health Study. J Am Coll Cardiol 2009; 53:950–958.
- Frazier L, Vaughn W, Willerson J, Ballantyne C, Boerwinkle E Inflammatory protein levels and depression screening after coronary stenting predict major adverse coronary events [published online ahead of print February 26, 2009]. Biol Res Nurs. doi:10.1177/1099800409332801.
- Kim JH, Kim JW, Ko YH, et al Coronary endothelial dysfunction associated with a depressive mood in patients with atypical angina but angiographically normal coronary artery [published online ahead of print March 7, 2009]. Int J Cardiol. doi:10.1016/j.ijcard.2009.02.004.
- Davidson KW, Schwartz JE, Kirkland SA, et al. Relation of inflammation to depression and incident coronary heart disease (from the Canadian Nova Scotia Health Survey [NSHS95] Prospective Population Study). Am J Cardiol 2009; 103:755–761.
- Bankier B, Barajas J, Martinez-Rumayor A, Januzzi JL. Association between major depressive disorder and C-reactive protein levels in stable coronary heart disease patients. J Psychosom Res 2009; 66:189–194.
- Carney RM, Freedland KE, Steinmeyer B, et al History of depression and survival after acute myocardial infarction [published online ahead of print February 27, 2009]. Psychosom Med. doi:10.1097/PSY.0b013e31819b69e3.
- Vasilyev N, Williams T, Brennan ML, et al. Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 2005; 112:2812–2820.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
- Kubzansky LD, Koenen KC, Jones C, Eaton WW. A prospective study of posttraumatic stress disorder symptoms and coronary heart disease in women. Health Psychol 2009; 28:125–130.
- Kubzansky LD, Koenen KC, Spiro A, Vokonas PS, Sparrow D. Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Arch Gen Psychiatry 2007; 64:109–116.
- Porges SW. The polyvagal perspective. Biol Psychol 2007; 74:116–143.
- Porges SW. The polyvagal theory: phylogenetic substrates of a social nervous system. Int J Psychophysiol 2001; 42:123–146.
Heart-brain medicine: Update 2007
Heart-brain medicine is dedicated to furthering our understanding of the interaction between the body’s neurologic and cardiovascular systems. As discussed previously,1 the advent of subspecialization in health care delivery has led to significant advances in the care of patients with acute disease or acute exacerbations of chronic disease. While these advances have led to improved outcomes, we were reminded several times this past year how difficult it is to further improve outcomes using the “silo”-based, highly subspecialized approach that has yielded results in the past.
The 2007 Bakken Heart-Brain Summit, held last June in Cleveland, further demonstrated real progress in our understanding of the importance of heart-brain interactions in health and disease. A series of presentations—highlighted by the Bakken Lecture given by Peter Shapiro, MD, an investigator with the SADHART trial—reviewed the effect of psychiatric disorders on the incidence of cardiovascular disease and its consequences. These presentations by leaders in the field (many of which are summarized in the pages that follow) offer irrefutable evidence of the following:
- Patients with depression and heart disease have worse outcomes than patients with heart disease without depression2
- Patients with depression have decreased vagal tone3
- Patients with coronary artery disease (CAD) can be safely treated with and respond to antidepressants.4
These data were complemented by a keynote presentation by Kevin Tracey, MD, whose elegant work over the past many years has demonstrated a link between vagal tone and inflammation.5 His most recent data have shown that the vagus has direct input into the inflammatory state of macrophages in the spleen. The effect is mediated via vagal innervation of the spleen and the α7 subunit of the nicotinic receptor expressed on the cell surface of the resident macrophages.6,7 The relevance of vagally mediated modulation of systemic inflammation has been shown in sepsis and more recently by our group in left ventricular remodeling following acute myocardial infarction.
‘RECONNECTING THE BODY’ TO IMPROVE OUTCOMES
The continuing emergence of the link between psychiatric and neurocontrol of systemic inflammation offers an undeveloped strategy for further improving outcomes in patients with cardiovascular disease. One of our interests in pursuing heart-brain medicine is to reconnect the body and exploit the physiologic interplay between the heart and brain to improve patient outcomes.1 Given the disappointments over the past year for new therapies like cholesteryl ester transfer protein inhibitors8 and vascular cell adhesion molecule (VCAM) inhibitors, strategies that have a singular organ or cellular target focus, now may be the time for exploiting multisystem approaches for modulating disease states such as CAD, congestive heart failure, and arrhythmia.
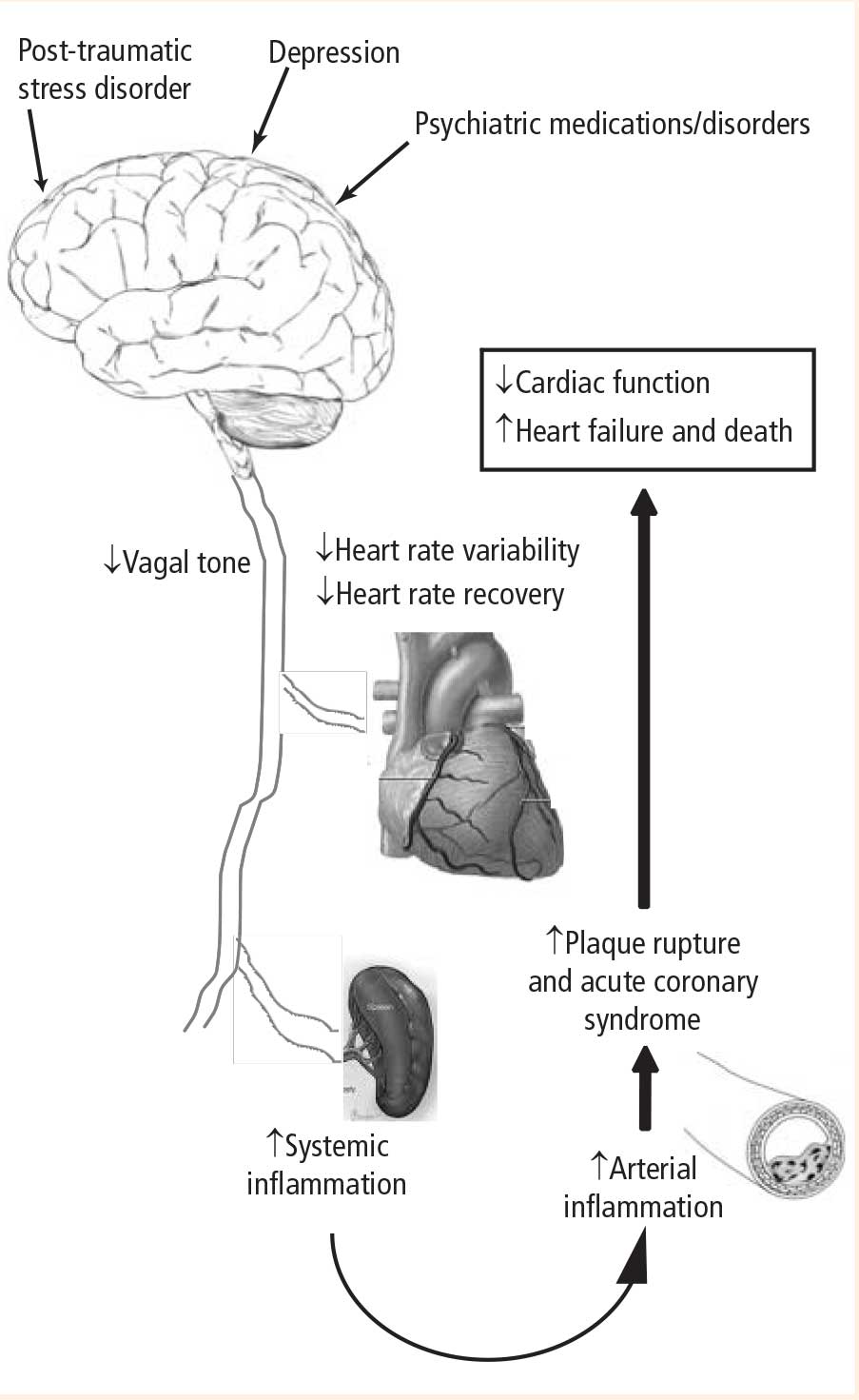
The potential consequences of these pathways are profound and include the following:
- A physiologic mechanism for the increased incidence of myocardial infarction observed with medications that have anticholinergic properties and potentially decrease autonomic tone
- Worse outcomes in patients with CAD and depression
- An increased incidence of CAD in patients with psychiatric disorders that in themselves may be associated with decreased vagal tone, as well as in patients on long-term drug therapies that alter parasympathetic tone
- Increased incidences of CAD, myocardial infarction, and death in patients with PTSD.
AN URGENT NEED FOR CLINICAL TRANSLATION
Clearly the underlying science of heart-brain medicine is fascinating and needs to be pursued vigorously. While the science is ongoing, the need to translate what we know to the bedside has never been greater, given the prevalence of CAD, chronic heart failure, and psychiatric and mood disorders, as well as the likelihood of an increasing incidence of PTSD in light of the Iraq war and terrorist threats.
Multiple studies have been performed to position the field for a trial to test whether treating depression leads to improved outcomes in patients with CAD. We know that patients with depression have decreased vagal tone based on decreased heart rate variability; we know that CAD can be safely treated with selective serotonin reuptake inhibitors; and we know that this patient population is more effectively treated with medications. There was a clear sentiment among faculty and attendees of the 2006 Bakken Heart-Brain Summit that the next step in the clinical science of heart disease and neurologic state is in fact a clinical trial to test the efficacy of this approach. Unfortunately, funding for such a trial from the pharmaceutical industry or government agencies is lacking. The Bakken Heart-Brain Institute is working diligently to secure private financing of such a trial from those with personal interests in moving this field forward. We hope to be able to commence such a trial in the near future. We believe the successful initiation of a multicenter trial not only will demonstrate new avenues for improving outcomes in millions of patients but will validate the concept and usher in a new age of cooperative medicine among multiple disciplines.
As we discussed last year,1 both the need for and the future of heart-brain medicine are great. The advances seen over the past year and those being pursued in basic and clinical science laboratories throughout the world are very exciting. We thank those colleagues who attended the 2007 Bakken Heart-Brain Summit, and we hope you can join us June 4–5, 2008, in Cleveland to continue this exciting pursuit.
- Penn MS, Bakken EE. Heart-brain medicine: where we go from here and why. Cleve Clin J Med 2007; 74 (Suppl 1):S4–S6.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Chambers AS, Allen JJ. Vagal tone as an indicator of treatment response in major depression. Psychophysiology 2002; 39:861–864.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Tracey KJ. Physiology and immunology of the cholinergic anti-inflammatory pathway. J Clin Invest 2007; 117:289–296.
- Huston JM, Ochani M, Rosas-Ballina M, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
Heart-brain medicine is dedicated to furthering our understanding of the interaction between the body’s neurologic and cardiovascular systems. As discussed previously,1 the advent of subspecialization in health care delivery has led to significant advances in the care of patients with acute disease or acute exacerbations of chronic disease. While these advances have led to improved outcomes, we were reminded several times this past year how difficult it is to further improve outcomes using the “silo”-based, highly subspecialized approach that has yielded results in the past.
The 2007 Bakken Heart-Brain Summit, held last June in Cleveland, further demonstrated real progress in our understanding of the importance of heart-brain interactions in health and disease. A series of presentations—highlighted by the Bakken Lecture given by Peter Shapiro, MD, an investigator with the SADHART trial—reviewed the effect of psychiatric disorders on the incidence of cardiovascular disease and its consequences. These presentations by leaders in the field (many of which are summarized in the pages that follow) offer irrefutable evidence of the following:
- Patients with depression and heart disease have worse outcomes than patients with heart disease without depression2
- Patients with depression have decreased vagal tone3
- Patients with coronary artery disease (CAD) can be safely treated with and respond to antidepressants.4
These data were complemented by a keynote presentation by Kevin Tracey, MD, whose elegant work over the past many years has demonstrated a link between vagal tone and inflammation.5 His most recent data have shown that the vagus has direct input into the inflammatory state of macrophages in the spleen. The effect is mediated via vagal innervation of the spleen and the α7 subunit of the nicotinic receptor expressed on the cell surface of the resident macrophages.6,7 The relevance of vagally mediated modulation of systemic inflammation has been shown in sepsis and more recently by our group in left ventricular remodeling following acute myocardial infarction.
‘RECONNECTING THE BODY’ TO IMPROVE OUTCOMES
The continuing emergence of the link between psychiatric and neurocontrol of systemic inflammation offers an undeveloped strategy for further improving outcomes in patients with cardiovascular disease. One of our interests in pursuing heart-brain medicine is to reconnect the body and exploit the physiologic interplay between the heart and brain to improve patient outcomes.1 Given the disappointments over the past year for new therapies like cholesteryl ester transfer protein inhibitors8 and vascular cell adhesion molecule (VCAM) inhibitors, strategies that have a singular organ or cellular target focus, now may be the time for exploiting multisystem approaches for modulating disease states such as CAD, congestive heart failure, and arrhythmia.
The potential consequences of these pathways are profound and include the following:
- A physiologic mechanism for the increased incidence of myocardial infarction observed with medications that have anticholinergic properties and potentially decrease autonomic tone
- Worse outcomes in patients with CAD and depression
- An increased incidence of CAD in patients with psychiatric disorders that in themselves may be associated with decreased vagal tone, as well as in patients on long-term drug therapies that alter parasympathetic tone
- Increased incidences of CAD, myocardial infarction, and death in patients with PTSD.
AN URGENT NEED FOR CLINICAL TRANSLATION
Clearly the underlying science of heart-brain medicine is fascinating and needs to be pursued vigorously. While the science is ongoing, the need to translate what we know to the bedside has never been greater, given the prevalence of CAD, chronic heart failure, and psychiatric and mood disorders, as well as the likelihood of an increasing incidence of PTSD in light of the Iraq war and terrorist threats.
Multiple studies have been performed to position the field for a trial to test whether treating depression leads to improved outcomes in patients with CAD. We know that patients with depression have decreased vagal tone based on decreased heart rate variability; we know that CAD can be safely treated with selective serotonin reuptake inhibitors; and we know that this patient population is more effectively treated with medications. There was a clear sentiment among faculty and attendees of the 2006 Bakken Heart-Brain Summit that the next step in the clinical science of heart disease and neurologic state is in fact a clinical trial to test the efficacy of this approach. Unfortunately, funding for such a trial from the pharmaceutical industry or government agencies is lacking. The Bakken Heart-Brain Institute is working diligently to secure private financing of such a trial from those with personal interests in moving this field forward. We hope to be able to commence such a trial in the near future. We believe the successful initiation of a multicenter trial not only will demonstrate new avenues for improving outcomes in millions of patients but will validate the concept and usher in a new age of cooperative medicine among multiple disciplines.
As we discussed last year,1 both the need for and the future of heart-brain medicine are great. The advances seen over the past year and those being pursued in basic and clinical science laboratories throughout the world are very exciting. We thank those colleagues who attended the 2007 Bakken Heart-Brain Summit, and we hope you can join us June 4–5, 2008, in Cleveland to continue this exciting pursuit.
Heart-brain medicine is dedicated to furthering our understanding of the interaction between the body’s neurologic and cardiovascular systems. As discussed previously,1 the advent of subspecialization in health care delivery has led to significant advances in the care of patients with acute disease or acute exacerbations of chronic disease. While these advances have led to improved outcomes, we were reminded several times this past year how difficult it is to further improve outcomes using the “silo”-based, highly subspecialized approach that has yielded results in the past.
The 2007 Bakken Heart-Brain Summit, held last June in Cleveland, further demonstrated real progress in our understanding of the importance of heart-brain interactions in health and disease. A series of presentations—highlighted by the Bakken Lecture given by Peter Shapiro, MD, an investigator with the SADHART trial—reviewed the effect of psychiatric disorders on the incidence of cardiovascular disease and its consequences. These presentations by leaders in the field (many of which are summarized in the pages that follow) offer irrefutable evidence of the following:
- Patients with depression and heart disease have worse outcomes than patients with heart disease without depression2
- Patients with depression have decreased vagal tone3
- Patients with coronary artery disease (CAD) can be safely treated with and respond to antidepressants.4
These data were complemented by a keynote presentation by Kevin Tracey, MD, whose elegant work over the past many years has demonstrated a link between vagal tone and inflammation.5 His most recent data have shown that the vagus has direct input into the inflammatory state of macrophages in the spleen. The effect is mediated via vagal innervation of the spleen and the α7 subunit of the nicotinic receptor expressed on the cell surface of the resident macrophages.6,7 The relevance of vagally mediated modulation of systemic inflammation has been shown in sepsis and more recently by our group in left ventricular remodeling following acute myocardial infarction.
‘RECONNECTING THE BODY’ TO IMPROVE OUTCOMES
The continuing emergence of the link between psychiatric and neurocontrol of systemic inflammation offers an undeveloped strategy for further improving outcomes in patients with cardiovascular disease. One of our interests in pursuing heart-brain medicine is to reconnect the body and exploit the physiologic interplay between the heart and brain to improve patient outcomes.1 Given the disappointments over the past year for new therapies like cholesteryl ester transfer protein inhibitors8 and vascular cell adhesion molecule (VCAM) inhibitors, strategies that have a singular organ or cellular target focus, now may be the time for exploiting multisystem approaches for modulating disease states such as CAD, congestive heart failure, and arrhythmia.
The potential consequences of these pathways are profound and include the following:
- A physiologic mechanism for the increased incidence of myocardial infarction observed with medications that have anticholinergic properties and potentially decrease autonomic tone
- Worse outcomes in patients with CAD and depression
- An increased incidence of CAD in patients with psychiatric disorders that in themselves may be associated with decreased vagal tone, as well as in patients on long-term drug therapies that alter parasympathetic tone
- Increased incidences of CAD, myocardial infarction, and death in patients with PTSD.
AN URGENT NEED FOR CLINICAL TRANSLATION
Clearly the underlying science of heart-brain medicine is fascinating and needs to be pursued vigorously. While the science is ongoing, the need to translate what we know to the bedside has never been greater, given the prevalence of CAD, chronic heart failure, and psychiatric and mood disorders, as well as the likelihood of an increasing incidence of PTSD in light of the Iraq war and terrorist threats.
Multiple studies have been performed to position the field for a trial to test whether treating depression leads to improved outcomes in patients with CAD. We know that patients with depression have decreased vagal tone based on decreased heart rate variability; we know that CAD can be safely treated with selective serotonin reuptake inhibitors; and we know that this patient population is more effectively treated with medications. There was a clear sentiment among faculty and attendees of the 2006 Bakken Heart-Brain Summit that the next step in the clinical science of heart disease and neurologic state is in fact a clinical trial to test the efficacy of this approach. Unfortunately, funding for such a trial from the pharmaceutical industry or government agencies is lacking. The Bakken Heart-Brain Institute is working diligently to secure private financing of such a trial from those with personal interests in moving this field forward. We hope to be able to commence such a trial in the near future. We believe the successful initiation of a multicenter trial not only will demonstrate new avenues for improving outcomes in millions of patients but will validate the concept and usher in a new age of cooperative medicine among multiple disciplines.
As we discussed last year,1 both the need for and the future of heart-brain medicine are great. The advances seen over the past year and those being pursued in basic and clinical science laboratories throughout the world are very exciting. We thank those colleagues who attended the 2007 Bakken Heart-Brain Summit, and we hope you can join us June 4–5, 2008, in Cleveland to continue this exciting pursuit.
- Penn MS, Bakken EE. Heart-brain medicine: where we go from here and why. Cleve Clin J Med 2007; 74 (Suppl 1):S4–S6.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Chambers AS, Allen JJ. Vagal tone as an indicator of treatment response in major depression. Psychophysiology 2002; 39:861–864.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Tracey KJ. Physiology and immunology of the cholinergic anti-inflammatory pathway. J Clin Invest 2007; 117:289–296.
- Huston JM, Ochani M, Rosas-Ballina M, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
- Penn MS, Bakken EE. Heart-brain medicine: where we go from here and why. Cleve Clin J Med 2007; 74 (Suppl 1):S4–S6.
- Connerney I, Shapiro PA, McLaughlin JS, Bagiella E, Sloan RP. Relation between depression after coronary artery bypass surgery and 12-month outcome: a prospective study. Lancet 2001; 358:1766–1771.
- Chambers AS, Allen JJ. Vagal tone as an indicator of treatment response in major depression. Psychophysiology 2002; 39:861–864.
- Glassman AH, O’Connor CM, Califf RM, et al. Sertraline treatment of major depression in patients with acute MI or unstable angina. JAMA 2002; 288:701–709.
- Tracey KJ. Physiology and immunology of the cholinergic anti-inflammatory pathway. J Clin Invest 2007; 117:289–296.
- Huston JM, Ochani M, Rosas-Ballina M, et al. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 2006; 203:1623–1628.
- Huston JM, Gallowitsch-Puerta M, Ochani M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med 2007; 35:2762–2768.
- Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med 2007; 357:2109–2122.
- Sack M, Hopper JW, Lamprecht F. Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: heart rate dynamics and individual differences in arousal regulation. Biol Psychiatry 2004; 55:284–290.
Preconditioning paradigms and pathways in the brain
The brain relies upon internal defense mechanisms for protection from injurious stimuli. Preconditioning is a phenomenon whereby low doses of these noxious insults shield the brain from future insults rather than inflicting damage. Preconditioning stimuli include but are not limited to transient global and focal ischemia,1–4 cortical spreading depression,5–7 brief episodes of seizure, exposure to anesthetic inhalants,8–10 low doses of endotoxin (lipopolysaccharide [LPS]),11,12 hypothermia and hyperthermia,13,14 and 3-nitropropionic acid treatment.15,16
Depending on the specific preconditioning stimulus, a state of neuronal tolerance can be established in at least two temporal profiles: one in which the trigger induces protection within minutes (rapid or acute tolerance),17 and one in which the protected state develops after a delay of several hours to days (delayed tolerance).4 Some preconditioning paradigms induce both phases of ischemic tolerance, while others can induce only the acute phase or only the delayed phase.18–21 The acute phase is most likely due to rapid posttranslational modifications of proteins.22,23 In contrast, the delayed phase is dependent on de novo protein synthesis.24,25
Preconditioning by ischemic tolerance was first identified in the heart by Murry et al,26 and was subsequently found to occur in the brain4,27 and a variety of organs including the liver, intestine, kidney, and lung. Preconditioning stimuli can be cross-tolerant, safeguarding against other types of injury. For example, endotoxin preconditioning can protect against subsequent ischemia and vice versa. Thus, there may be some overlapping mechanisms in preconditioning, and unraveling these pathways may uncover an arsenal of neuroprotective therapeutic targets. In this review, we will compare different preconditioning paradigms and discuss potential mechanisms in initiating brain ischemic tolerance.
PARADIGMS TO ESTABLISH PRECONDITIONING
Refinement of various preconditioning models is of great clinical significance. Cardiovascular or cerebrovascular surgery has a negative impact on brain function due to stoppage of blood flow during surgery. In fact, more than 25% of patients who receive coronary artery bypass surgery suffer from temporary or permanent memory loss.28,29 As a result, it is of premier importance to develop strategies to protect the brain either prior to vascular surgeries or in patients at high risk of stroke. While it would be dangerous and impractical to precondition at-risk patients with ischemia, the identification of underlying preconditioning mechanisms may lead to safer therapeutic factors that can be administered before surgery.
Ischemia
Global ischemic preconditioning in the brain is accomplished by occlusion of the bilateral common carotid arteries. In contrast, in focal ischemic preconditioning, occlusion of one side of the middle cerebral artery is induced for about 1 to 20 minutes, depending on methods and animal species.4,30–32 Twenty-four hours after ischemic preconditioning, stroke is induced in these animals. Preconditioning-induced neuroprotection is observed not only in terms of infarct volume but also in terms of neurological scores and behavior studies.
Lipopolysaccharide
Tolerance to ischemic injury can also be induced by a small dose of LPS injected into the peritoneal cavity. Dosages vary from 0.05 to 1 mg/kg body weight in small rodents such as mice and rats.11,33–36 This dose of LPS usually does not bring abnormal signs and symptoms to the animals. The ischemic protection yields a reduction of infarct volume of approximately 30%. This tolerant state can be sustained for about 1 week, with maximum protection occurring around 2 to 3 days after injection of LPS.
Hypoxia
A relatively convenient method for preconditioning animals is hypoxic exposure. Animals are put in a chamber in which oxygen and nitrogen proportions can be controlled. Oxygen concentration usually ranges from 8% to 13% with normobaric pressure. Exposure time ranges from 1 to 6 hours. Twenty-four to 72 hours later, transient or permanent focal stroke is induced in the animals.37–40 Hypoxia-preconditioned neuroprotection usually starts at 1 to 3 days with a significant reduction of infarct size. Hypoxic preconditioning has also been demonstrated for in vitro neuron culture models using oxygen-glucose deprivation injury.41
3-Nitropropionic acid
3-Nitropropionic acid (3-NP) is an irreversible inhibitor of succinate dehydrogenase, an enzyme required for oxidative phosphorylation and adenosine triphosphate production. When applied at low doses 1 to 4 days before ischemia, 3-NP can lead to ischemic tolerance in the forebrain of gerbils and rats.16,42,43 The dose ranges from 1 to 20 mg/kg body weight.16 Such treatment significantly improves neurological behavior and increases neuronal survival in the CA1 region of hippocampus. In addition, 3-NP preconditioning induces tolerance to hypoxia in hippocampal slice preparations.15,44
Hypothermia and hyperthermia
Hypothermia is a well-characterized protective procedure used during and after cerebral surgery. It is also reported that brief hypothermic or hyperthermic exposure can also lead to ischemic tolerance. The temperatures adopted range from 25°C to 32°C13,45,46 in hypothermia and from 42°C to 43°C in hyperthermia.14
Cortical spreading depression
Cortical spreading depression is defined as the electrophysiologic phenomenon of slowly propagating transient depolarization waves across the cortex. Usually 5 M of potassium chloride is infused into the cortex, or a cotton pad soaked with the solution is put on the surface of dura mater, which results in depolarization, firing of neurons, and cortical spreading depression. Cortical spreading depression induces a prolonged phase of ischemic tolerance that lasts 1 to 7 days.5,6,47,48
Anesthetics
Exposure to volatile anesthetics such as isoflurane and halothane within pharmacologic concentration ranges also confers delayed-phase ischemic tolerance of the brain.8–10,49
MOLECULAR PRECONDITIONING PATHWAYS
Mechanistically, cellular preconditioning can be subdivided into intrinsic neuronal pathways (preventing excitotoxic damage, signaling through anti-apoptotic molecules, and treatment by neurotrophic factors) or extrinsic nonneuronal pathways (peripheral cytokine production, microglial activation, and regulation of the cerebrovascular system). Several neuroprotective molecules are expressed and signal through multiple cell types both within and peripheral to the brain, so that assigning an exact source and paradigm for preconditioning pathways has proven difficult.
NMDA receptor activation and excitotoxicity protection
In neurons, ischemic tolerance is mediated largely by the activation of the N-methyl-d-aspartate (NMDA) glutamate receptors through increases in intracellular calcium.50–52 Although glutamate receptor activation is generally believed to be responsible for much of the neuronal damage caused by excitotoxicity, it appears to also be implicated in the establishment of preconditioning. One study demonstrated that exposure of cortical cell cultures to low levels of glutamate activated NMDA receptors in preconditioning.50 In addition, preconditioning by oxygen-glucose deprivation was blocked when an NMDA antagonist was applied. NMDA receptor activation can induce a tolerant state through rapid adaptation of the voltage-dependent calcium flux. In addition, activation of NMDA receptors leads to rapid release of brain-derived neurotrophic factor, which then binds to and activates its cognate receptor, receptor tyrosine kinase B. Both NMDA and tyrosine kinase B receptors activate nuclear factor–kappa B (NFκB), a transcription factor involved in protecting neurons against insults. In sublethal ischemic preconditioning, activation of NFκB and its translocation from the cytosol to the nucleus was required for the development of late cerebral protection against severe ischemia or epilepsy.53 Other key mediators involved in synaptic NMDA receptor–dependent neuroprotection are phosphatidylinositol 3-kinase (PI3K), Akt, and glycogen synthase kinase 3-beta.54
Preconditioning with cortical spreading depression results in the downregulation of the excitatory amino acid transporters EAAT1 and EAAT2 from cerebral cortex plasma membranes.55 Although these transporters are normally involved in glutamate uptake, it has been suggested that the influx of sodium that occurs during excitotoxicity may cause their reversal and result in additional glutamate release. Downregulating these transporters may thus contribute to ischemic tolerance.
Nitric oxide
Nitric oxide (NO) may play a key role as a mediator of the neuronal ischemic preconditioning response, either in conjunction with or independent of NMDA receptor activation. Both the inhibition of nitric oxide synthase (NOS) and the scavenging of NO during preconditioning significantly attenuated the induced neuronal tolerance, and neither endothelial NOS nor neuronal NOS knockout mice showed protection from rapid ischemic preconditioning.56,57 Treatment with the inducible NOS (iNOS) inhibitor aminoguanidine abolished the induced protection. The mechanisms responsible for NO-induced tolerance are not clear. Downregulation of the glutamate transporter GLT-1 might play a role.58 A common link to NMDA receptor activation and NO is p21ras (Ras). Preconditioning induces p21ras activation in an NMDA- and NO-dependent manner and leads to the downstream activation of Raf kinase, mitogen-activated protein kinases, and extracellular regulated kinase.59 Inhibition of these kinases attenuates subsequent protection from ischemia.60,61 Pharmacologic inhibition of Ras, as well as a dominant negative Ras mutant, blocked preconditioning, whereas a constitutively active form of Ras promoted neuroprotection against lethal insults. An important consideration regarding NO is also that preconditioning by volatile anesthetics appears to involve NO pathways.9
NO and reactive oxygen species (ROS) are also implicated in regulating the peripheral cerebrovascular system. Ischemia generated by occlusion of the middle cerebral artery causes defects in cerebrovascular function for not only the infarcted area but also the surrounding ischemic region. LPS preconditioning has been reported in some cases to increase this regional cerebral blood flow both before and after ischemia.1,21,36,62–64 LPS also improves microvascular perfusion.33,64 It was recently reported that LPS-stimulated cerebral blood flow is induced through reactive oxygen and nitrogen species (ROS or NO).1 Mouse knockouts of iNOS (NO production) or of the nox2 subunit of NADPH oxidase (ROS production) eliminated the LPS-upregulated cerebrovascular activity. Furthermore, blockage of these ROS and NO pathways reduced the preconditioning effect of LPS. Therefore, LPS may play a more direct role in preventing ischemic damage by increasing blood availability to the affected brain region.
Inflammatory cytokines and the innate immune system
LPS, a component of the gram-negative bacterial cell wall, can illicit a potent innate immune response. While this systemic inflammatory response can be destructive (at doses of 5 mg/kg),65 tolerable LPS doses of 0.05 to 1 mg/kg injected intraperitoneally render the brain,11 heart,66,67 liver,68,69 kidneys,70 and pancreas71 transiently resistant to subsequent ischemic injury. This preconditioning paradigm relies on the ability of a peripheral signal to cross into multiple organ systems. LPS injected into the gut can signal through peritoneal macrophages and circulating monocytes. Toll-like receptor 4 is a pattern-recognition receptor that binds to pathogen-associated molecular patterns in LPS and initiates a signaling cascade through the NFκB pathway. This pathway culminates in the expression and secretion of several proinflammatory cytokines to fight off the infection and anti-inflammatory cytokines to control the immune response.
The major output of LPS signaling is innate production of proinflammatory cytokines to fight infection and clear cellular debris. Central cytokines, including tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), can be neurodestructive if administered after ischemia. TNFα administration by cerebroventricular injection after ischemia augmented the extent of injury, and blockage of TNFα signaling proved neuroprotective.11,72,73 However, in LPS preconditioning, cytokine production precedes ischemia. Intracisternal injections of TNFα before middle cerebral artery occlusion (MCAO) were protective in reducing the infarct size of pretreated mice.74 Furthermore, intracisternal injection of ceramide analog, a downstream component of the TNFα signaling pathway, was also capable of reducing the MCAO infarct area.75 Preischemic treatment with IL-6 and IL-1 also reduced neuronal damage.76,77 TNFα knockout mice eliminated the LPS protective phenotype,72 demonstrating that cytokine production is a critical feature of LPS preconditioning in ischemia. Additionally, ischemic damage in the absence of LPS preconditioning was exacerbated in TNFα receptor 1 knockout mice.78,79 Consistently, TNFα protein levels are upregulated after LPS treatment but are downregulated following LPS-preconditioned MCAO.72 A unifying theme in LPS preconditioning comprises early activation of the innate immune system with ensuing suppression in ischemia.
As a potential mechanism, the initial inflammatory response induced by LPS appears to render the innate immune system hyporesponsive to subsequent insults such as ischemia. This may occur by persistence of anti-inflammatory cytokines produced by the primary insult. These molecules are expressed in tandem with proinflammatory cytokines to control the innate immune response, but may also play a role in delayed preconditioning. For instance, intravenous or intracerebroventricular IL-10 injection can reduce the infarct size with MCAO.80 Alternatively, several proinflammatory cytokine signaling pathways may be downregulated by negative feedback inhibition.20,81 This inhibition may occur extracellularly, using soluble cytokine receptors, decoy receptors, or receptor antagonists. For example, intravenous injection of IL-1 receptor antagonist can provide neuroprotection against ischemic injury from MCAO.82,83 Cytokine feedback inhibitors that act intracellularly are also induced with the innate immune response. Intracellular inhibition may involve direct downregulation of cytokine transcription (peroxisome proliferator-activated receptor gamma [PPAR-γ]) or inhibition of intracellular signaling pathways that promote cytokine production (suppressor of cytokine signaling [SOCS] and PI3K). Antisense mRNA knockdown of SOCS-3 exacerbates ischemic injury from MCAO.84 The MCAO infarction area is increased after treatment with PPAR-γ antagonists and decreased by PPAR-γ agonists.85,86 Administration of compounds that increase PI3K signaling is also capable of reducing ischemic damage.87 Thus, several defense mechanisms designed to suppress the innate immune response may play an active role in LPS ischemic preconditioning.
Role of microglia in ischemic preconditioning
Microglia represent the resident central nervous system (CNS) component of the innate immune system. Microglia and macrophages become activated with ischemia in the infarcted and surrounded area.88 Upon activation in ischemia, microglia will become phagocytic and secrete a multitude of noxious chemokines and cytokines.89 Accordingly, anti-inflammatory antibiotics such as doxycycline and minocycline reduce microglial activation and diminish the ischemic infarction area.90 Preconditioning the brain with LPS ameliorates microglial activation, neutrophil infiltration, and circulating monocyte activation following MCAO.35 However, primary ischemic damage is not correlated with CNS infiltration of peripheral leukocytes but rather with an increase in proliferating resident microglial cells.91 Alternatively, microglia can exhibit neuroprotective properties within the brain.92 In fact, greater ischemic damage from longer periods of MCAO is correlated with fewer proliferating microglia, suggesting a protective microglial role.91 Consistently, ablation of proliferating microglia increases the infarction area following MCAO.93 Therefore, microglia can be protective in ischemia, and preconditioning with LPS may render microglia more capable of reacting to ischemic conditions.
CONCLUSIONS
Preconditioning represents an adaptive response to prime the brain for protection against future injury. Elucidation of these endogenous cell survival pathways has significant clinical implications for preventing neuronal damage in susceptible patients. For this reason, understanding the underlying mechanisms in establishing a tolerant state will be a critical step in adapting preconditioning for safe patient applications. The field of ischemic research has made great strides in deciphering causative preconditioning factors but has been hampered by the complex, multifactorial nature of preconditioning paradigms. The study of tolerance is further complicated by the fact that signaling takes place both peripheral to and within the brain in multiple cell types. Future research will require the exploration of interactions between multiple pathways and roles of individual cell types in establishing ischemic tolerance. Only with a more thorough understanding of preconditioning mechanisms can we adapt these pathways for the most efficient and protective treatments.
- Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 2007; 27:7083–7093.
- Simon RP, Niiro M, Gwinn R. Prior ischemic stress protects against experimental stroke. Neurosci Lett 1993; 163:135–137.
- Chen J, Simon R. Ischemic tolerance in the brain. Neurology 1997; 48:306–311.
- Kitagawa K, Matsumoto M, Tagaya M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res 1990; 528:21–24.
- Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res 1995; 17:9–16.
- Taga K, Patel PM, Drummond JC, Cole DJ, Kelly PJ. Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology 1997; 87:918–925.
- Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab 1995; 15:721–727.
- Baughman VL, Hoffman WE, Miletich DJ, Albrecht RF, Thomas C. Neurologic outcome in rats following incomplete cerebral ischemia during halothane, isoflurane, or N2O. Anesthesiology 1988; 69:192–198.
- Kapinya KJ, Lowl D, Futterer C, et al. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 2002; 33:1889–1898.
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol Pharmacol 2004; 65:1172–1180.
- Tasaki K, Ruetzler CA, Ohtsuki T, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 1997; 748:267–270.
- Zimmermann C, Ginis I, Furuya K, et al. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res 2001; 895:59–65.
- Nishio S, Yunoki M, Chen ZF, Anzivino MJ, Lee KS. Ischemic tolerance in the rat neocortex following hypothermic preconditioning. J Neurosurg 2000; 93:845–851.
- Ota A, Ikeda T, Xia XY, Xia YX, Ikenoue T. Hypoxic-ischemic tolerance induced by hyperthermic pretreatment in newborn rats. J Soc Gynecol Investig 2000; 7:102–105.
- Riepe MW, Niemi WN, Megow D, Ludolph AC, Carpenter DO. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp Neurol 1996; 138:15–21.
- Sugino T, Nozaki K, Takagi Y, Hashimoto N. 3-Nitropropionic acid induces ischemic tolerance in gerbil hippocampus in vivo. Neurosci Lett 1999; 259:9–12.
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab 1997; 17:175–182.
- Stagliano NE, Perez-Pinzon MA, Moskowitz MA, Huang PL. Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 1999; 19:757–761.
- Perez-Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: role of the K+ ATP channel and protein kinase C. Neuroscience 1999; 89:453–459.
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 2004; 24:1288–1304.
- Hoyte LC, Papadakis M, Barber PA, Buchan AM. Improved regional cerebral blood flow is important for the protection seen in a mouse model of late phase ischemic preconditioning. Brain Res 2006; 1121:231–237.
- Nakase H, Heimann A, Uranishi R, Riepe MW, Kempski O. Early-onset tolerance in rat global cerebral ischemia induced by a mitochondrial inhibitor. Neurosci Lett 2000; 290:105–108.
- Meller R, Cameron JA, Torrey DJ, et al. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem 2006; 281:7429–7436.
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab 2002; 22:1283–1296.
- Stenzel-Poore MP, Stevens SL, Xiong Z, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003; 362:1028–1037.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74:1124–1136.
- Kitagawa K, Matsumoto M, Kuwabara K, et al. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res 1991; 561:203–211.
- Roach GW, Kanchuger M, Mangano CM, et al. Adverse cerebral outcomes after coronary bypass surgery. Multicenter Study of Perioperative Ischemia Research Group and the Ischemia Research and Education Foundation Investigators. N Engl J Med 1996; 335:1857–1863.
- Newman MF, Kirchner JL, Phillips-Bute B, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 2001; 344:395–402.
- Corbett D, Crooks P. Ischemic preconditioning: a long term survival study using behavioural and histological endpoints. Brain Res 1997; 760:129–136.
- Barone FC, White RF, Spera PA, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29:1937–1950.
- Wu C, Zhan RZ, Qi S, et al. A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods 2001; 107:101–106.
- Dawson DA, Furuya K, Gotoh J, et al. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. J Cereb Blood Flow Metab 1999; 19:616–623.
- Ahmed SH, He YY, Nassief A, et al. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 2000; 31:193–199.
- Rosenzweig HL, Lessov NS, Henshall DC, et al. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004; 35:2576–2581.
- Furuya K, Zhu L, Kawahara N, et al. Differences in infarct evolution between lipopolysaccharide-induced tolerant and nontolerant conditions to focal cerebral ischemia. J Neurosurg 2005; 103:715–723.
- Bernaudin M, Nedelec AS, Divoux D, et al. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab 2002; 22:393–403.
- Kulinskii VI, Minakina LN, Gavrilina TV. Neuroprotective effect of hypoxic preconditioning: phenomenon and mechanisms. Bull Exp Biol Med 2002; 133:202–204.
- Miller BA, Perez RS, Shah AR, et al. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 2001; 12:1663–1669.
- Prass K, Scharff A, Ruscher K, et al. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003; 34:1981–1986.
- Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol 2000; 278:C144–C153.
- Kuroiwa T, Yamada I, Endo S, Hakamata Y, Ito U. 3-Nitropropionic acid preconditioning ameliorates delayed neurological deterioration and infarction after transient focal cerebral ischemia in gerbils. Neurosci Lett 2000; 283:145–148.
- Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 2003; 34:1015–1020.
- Aketa S, Nakase H, Kamada Y, Hiramatsu K, Sakaki T. Chemical preconditioning with 3-nitropropionic acid in gerbil hippocampal slices: therapeutic window and the participation of adenosine receptor. Exp Neurol 2000; 166:385–391.
- Nishio S, Chen ZF, Yunoki M, et al. Hypothermia-induced ischemic tolerance. Ann N Y Acad Sci 1999; 890:26–41.
- Yunoki M, Nishio S, Ukita N, Anzivino MJ, Lee KS. Hypothermic preconditioning induces rapid tolerance to focal ischemic injury in the rat. Exp Neurol 2003; 181:291–300.
- Kawahara N, Ruetzler CA, Mies G, Klatzo I. Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp Neurol 1999; 158:27–36.
- Yanamoto H, Hashimoto N, Nagata I, Kikuchi H. Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res 1998; 784:239–249.
- McAuliffe JJ, Joseph B, Vorhees CV. Isoflurane-delayed preconditioning reduces immediate mortality and improves striatal function in adult mice after neonatal hypoxia-ischemia. Anesth Analg 2007; 104:1066–1077.
- Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 1999; 19:1657–1662.
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett 1992; 139:118–121.
- Kasischke K, Ludolph AC, Riepe MW. NMDA-antagonists reverse increased hypoxic tolerance by preceding chemical hypoxia. Neurosci Lett 1996; 214:175–178.
- Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-κB is a key event in brain tolerance. J Neurosci 2001; 21:4668–4677.
- Soriano FX, Papadia S, Hofmann F, et al. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci 2006; 26:4509–4518.
- Douen AG, Akiyama K, Hogan MJ, et al. Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebal cortex plasma membranes. J Neurochem 2000; 75:812–818.
- Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 2003; 34:1299–1303.
- Cho S, Park EM, Zhou P, et al. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab 2005; 25:493–501.
- Yamada T, Kawahara K, Kosugi T, Tanaka M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res 2006; 31:49–56.
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, et al. Requirement for nitric oxide activation of p21ras/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A 2000; 97:436–441.
- Kurkinen K, Keinanen R, Li W, Koistinaho J. Preconditioning with spreading depression activates specifically protein kinase Cδ. Neuroreport 2001; 12:269–273.
- Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience 1999; 93:81–88.
- Zhao L, Nowak TS Jr. CBF changes associated with focal ischemic preconditioning in the spontaneously hypertensive rat. J Cereb Blood Flow Metab 2006; 26:1128–1140.
- Nakamura H, Katsumata T, Nishiyama Y, et al. Effect of ischemic preconditioning on cerebral blood flow after subsequent lethal ischemia in gerbils. Life Sci 2006; 78:1713–1719.
- Bastide M, Gele P, Petrault O, et al. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab 2003; 23:399–405.
- Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007; 55:453–462.
- Eising GP, Mao L, Schmid-Schonbein GW, Engler RL, Ross J. Effects of induced tolerance to bacterial lipopolysaccharide on myocardial infarct size in rats. Cardiovasc Res 1996; 31:73–81.
- Zacharowski K, Otto M, Hafner G, et al. Endotoxin induces a second window of protection in the rat heart as determined by using pnitro-blue tetrazolium staining, cardiac troponin T release, and histology. Arterioscler Thromb Vasc Biol 1999; 19:2276–2280.
- Colletti LM, Remick DG, Campbell DA Jr. LPS pretreatment protects from hepatic ischemia/reperfusion. J Surg Res 1994; 57:337–343.
- Fernandez ED, Flohe S, Siemers F, et al. Endotoxin tolerance protects against local hepatic ischemia/reperfusion injury in the rat. J Endotoxin Res 2000; 6:321–328.
- Heemann U, Szabo A, Hamar P, et al. Lipopolysaccharide pretreatment protects from renal ischemia/reperfusion injury: possible connection to an interleukin-6-dependent pathway. Am J Pathol 2000; 156:287–293.
- Obermaier R, Drognitz O, Grub A, et al. Endotoxin preconditioning in pancreatic ischemia/reperfusion injury. Pancreas 2003; 27:e51–e56.
- Rosenzweig HL, Minami M, Lessov NS, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFα after stroke: a novel role for TNFα in LPS-ischemic tolerance. J Cereb Blood Flow Metab 2007; 27:1663–1674.
- Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997; 28:1233–1244.
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997; 17:483–490.
- Furuya K, Ginis I, Takeda H, et al. Cell permeable exogenous ceramide reduces infarct size in spontaneously hypertensive rats supporting in vitro studies that have implicated ceramide in induction of tolerance to ischemia. J Cereb Blood Flow Metab 2001; 21:226–232.
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18:176–179.
- Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J Cereb Blood Flow Metab 1996; 16:1137–1142.
- Bruce AJ, Boling W, Kindy MS, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996; 2:788–794.
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab 1998; 18:1283–1287.
- Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett 1998; 251:189–192.
- Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res 2004; 10:71–84.
- Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Pathol 1995; 147:1477–1486.
- Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol 1996; 138:206–213.
- Raghavendra RV, Bowen KK, Dhodda VK, et al. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem 2002; 83:1072–1086.
- Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARγ expression and activation after transient focal ischemia in rats. Eur J Neurosci 2006; 24:1653–1663.
- Zhao Y, Patzer A, Gohlke P, et al. The intracerebral application of the PPARγ-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci 2005; 22:278–282.
- Shioda N, Ishigami T, Han F, et al. Activation of phosphatidylinositol 3-kinase/protein kinase B pathway by a vanadyl compound mediates its neuroprotective effect in mouse brain ischemia. Neuroscience 2007; 148:221–229.
- Mabuchi T, Kitagawa K, Ohtsuki T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000; 31:1735–1743.
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol 2006; 84:49–59.
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 1998; 95:15769–15774.
- Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007; 27:1941–1953.
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002; 40:133–139.
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007; 27:2596–2605.
The brain relies upon internal defense mechanisms for protection from injurious stimuli. Preconditioning is a phenomenon whereby low doses of these noxious insults shield the brain from future insults rather than inflicting damage. Preconditioning stimuli include but are not limited to transient global and focal ischemia,1–4 cortical spreading depression,5–7 brief episodes of seizure, exposure to anesthetic inhalants,8–10 low doses of endotoxin (lipopolysaccharide [LPS]),11,12 hypothermia and hyperthermia,13,14 and 3-nitropropionic acid treatment.15,16
Depending on the specific preconditioning stimulus, a state of neuronal tolerance can be established in at least two temporal profiles: one in which the trigger induces protection within minutes (rapid or acute tolerance),17 and one in which the protected state develops after a delay of several hours to days (delayed tolerance).4 Some preconditioning paradigms induce both phases of ischemic tolerance, while others can induce only the acute phase or only the delayed phase.18–21 The acute phase is most likely due to rapid posttranslational modifications of proteins.22,23 In contrast, the delayed phase is dependent on de novo protein synthesis.24,25
Preconditioning by ischemic tolerance was first identified in the heart by Murry et al,26 and was subsequently found to occur in the brain4,27 and a variety of organs including the liver, intestine, kidney, and lung. Preconditioning stimuli can be cross-tolerant, safeguarding against other types of injury. For example, endotoxin preconditioning can protect against subsequent ischemia and vice versa. Thus, there may be some overlapping mechanisms in preconditioning, and unraveling these pathways may uncover an arsenal of neuroprotective therapeutic targets. In this review, we will compare different preconditioning paradigms and discuss potential mechanisms in initiating brain ischemic tolerance.
PARADIGMS TO ESTABLISH PRECONDITIONING
Refinement of various preconditioning models is of great clinical significance. Cardiovascular or cerebrovascular surgery has a negative impact on brain function due to stoppage of blood flow during surgery. In fact, more than 25% of patients who receive coronary artery bypass surgery suffer from temporary or permanent memory loss.28,29 As a result, it is of premier importance to develop strategies to protect the brain either prior to vascular surgeries or in patients at high risk of stroke. While it would be dangerous and impractical to precondition at-risk patients with ischemia, the identification of underlying preconditioning mechanisms may lead to safer therapeutic factors that can be administered before surgery.
Ischemia
Global ischemic preconditioning in the brain is accomplished by occlusion of the bilateral common carotid arteries. In contrast, in focal ischemic preconditioning, occlusion of one side of the middle cerebral artery is induced for about 1 to 20 minutes, depending on methods and animal species.4,30–32 Twenty-four hours after ischemic preconditioning, stroke is induced in these animals. Preconditioning-induced neuroprotection is observed not only in terms of infarct volume but also in terms of neurological scores and behavior studies.
Lipopolysaccharide
Tolerance to ischemic injury can also be induced by a small dose of LPS injected into the peritoneal cavity. Dosages vary from 0.05 to 1 mg/kg body weight in small rodents such as mice and rats.11,33–36 This dose of LPS usually does not bring abnormal signs and symptoms to the animals. The ischemic protection yields a reduction of infarct volume of approximately 30%. This tolerant state can be sustained for about 1 week, with maximum protection occurring around 2 to 3 days after injection of LPS.
Hypoxia
A relatively convenient method for preconditioning animals is hypoxic exposure. Animals are put in a chamber in which oxygen and nitrogen proportions can be controlled. Oxygen concentration usually ranges from 8% to 13% with normobaric pressure. Exposure time ranges from 1 to 6 hours. Twenty-four to 72 hours later, transient or permanent focal stroke is induced in the animals.37–40 Hypoxia-preconditioned neuroprotection usually starts at 1 to 3 days with a significant reduction of infarct size. Hypoxic preconditioning has also been demonstrated for in vitro neuron culture models using oxygen-glucose deprivation injury.41
3-Nitropropionic acid
3-Nitropropionic acid (3-NP) is an irreversible inhibitor of succinate dehydrogenase, an enzyme required for oxidative phosphorylation and adenosine triphosphate production. When applied at low doses 1 to 4 days before ischemia, 3-NP can lead to ischemic tolerance in the forebrain of gerbils and rats.16,42,43 The dose ranges from 1 to 20 mg/kg body weight.16 Such treatment significantly improves neurological behavior and increases neuronal survival in the CA1 region of hippocampus. In addition, 3-NP preconditioning induces tolerance to hypoxia in hippocampal slice preparations.15,44
Hypothermia and hyperthermia
Hypothermia is a well-characterized protective procedure used during and after cerebral surgery. It is also reported that brief hypothermic or hyperthermic exposure can also lead to ischemic tolerance. The temperatures adopted range from 25°C to 32°C13,45,46 in hypothermia and from 42°C to 43°C in hyperthermia.14
Cortical spreading depression
Cortical spreading depression is defined as the electrophysiologic phenomenon of slowly propagating transient depolarization waves across the cortex. Usually 5 M of potassium chloride is infused into the cortex, or a cotton pad soaked with the solution is put on the surface of dura mater, which results in depolarization, firing of neurons, and cortical spreading depression. Cortical spreading depression induces a prolonged phase of ischemic tolerance that lasts 1 to 7 days.5,6,47,48
Anesthetics
Exposure to volatile anesthetics such as isoflurane and halothane within pharmacologic concentration ranges also confers delayed-phase ischemic tolerance of the brain.8–10,49
MOLECULAR PRECONDITIONING PATHWAYS
Mechanistically, cellular preconditioning can be subdivided into intrinsic neuronal pathways (preventing excitotoxic damage, signaling through anti-apoptotic molecules, and treatment by neurotrophic factors) or extrinsic nonneuronal pathways (peripheral cytokine production, microglial activation, and regulation of the cerebrovascular system). Several neuroprotective molecules are expressed and signal through multiple cell types both within and peripheral to the brain, so that assigning an exact source and paradigm for preconditioning pathways has proven difficult.
NMDA receptor activation and excitotoxicity protection
In neurons, ischemic tolerance is mediated largely by the activation of the N-methyl-d-aspartate (NMDA) glutamate receptors through increases in intracellular calcium.50–52 Although glutamate receptor activation is generally believed to be responsible for much of the neuronal damage caused by excitotoxicity, it appears to also be implicated in the establishment of preconditioning. One study demonstrated that exposure of cortical cell cultures to low levels of glutamate activated NMDA receptors in preconditioning.50 In addition, preconditioning by oxygen-glucose deprivation was blocked when an NMDA antagonist was applied. NMDA receptor activation can induce a tolerant state through rapid adaptation of the voltage-dependent calcium flux. In addition, activation of NMDA receptors leads to rapid release of brain-derived neurotrophic factor, which then binds to and activates its cognate receptor, receptor tyrosine kinase B. Both NMDA and tyrosine kinase B receptors activate nuclear factor–kappa B (NFκB), a transcription factor involved in protecting neurons against insults. In sublethal ischemic preconditioning, activation of NFκB and its translocation from the cytosol to the nucleus was required for the development of late cerebral protection against severe ischemia or epilepsy.53 Other key mediators involved in synaptic NMDA receptor–dependent neuroprotection are phosphatidylinositol 3-kinase (PI3K), Akt, and glycogen synthase kinase 3-beta.54
Preconditioning with cortical spreading depression results in the downregulation of the excitatory amino acid transporters EAAT1 and EAAT2 from cerebral cortex plasma membranes.55 Although these transporters are normally involved in glutamate uptake, it has been suggested that the influx of sodium that occurs during excitotoxicity may cause their reversal and result in additional glutamate release. Downregulating these transporters may thus contribute to ischemic tolerance.
Nitric oxide
Nitric oxide (NO) may play a key role as a mediator of the neuronal ischemic preconditioning response, either in conjunction with or independent of NMDA receptor activation. Both the inhibition of nitric oxide synthase (NOS) and the scavenging of NO during preconditioning significantly attenuated the induced neuronal tolerance, and neither endothelial NOS nor neuronal NOS knockout mice showed protection from rapid ischemic preconditioning.56,57 Treatment with the inducible NOS (iNOS) inhibitor aminoguanidine abolished the induced protection. The mechanisms responsible for NO-induced tolerance are not clear. Downregulation of the glutamate transporter GLT-1 might play a role.58 A common link to NMDA receptor activation and NO is p21ras (Ras). Preconditioning induces p21ras activation in an NMDA- and NO-dependent manner and leads to the downstream activation of Raf kinase, mitogen-activated protein kinases, and extracellular regulated kinase.59 Inhibition of these kinases attenuates subsequent protection from ischemia.60,61 Pharmacologic inhibition of Ras, as well as a dominant negative Ras mutant, blocked preconditioning, whereas a constitutively active form of Ras promoted neuroprotection against lethal insults. An important consideration regarding NO is also that preconditioning by volatile anesthetics appears to involve NO pathways.9
NO and reactive oxygen species (ROS) are also implicated in regulating the peripheral cerebrovascular system. Ischemia generated by occlusion of the middle cerebral artery causes defects in cerebrovascular function for not only the infarcted area but also the surrounding ischemic region. LPS preconditioning has been reported in some cases to increase this regional cerebral blood flow both before and after ischemia.1,21,36,62–64 LPS also improves microvascular perfusion.33,64 It was recently reported that LPS-stimulated cerebral blood flow is induced through reactive oxygen and nitrogen species (ROS or NO).1 Mouse knockouts of iNOS (NO production) or of the nox2 subunit of NADPH oxidase (ROS production) eliminated the LPS-upregulated cerebrovascular activity. Furthermore, blockage of these ROS and NO pathways reduced the preconditioning effect of LPS. Therefore, LPS may play a more direct role in preventing ischemic damage by increasing blood availability to the affected brain region.
Inflammatory cytokines and the innate immune system
LPS, a component of the gram-negative bacterial cell wall, can illicit a potent innate immune response. While this systemic inflammatory response can be destructive (at doses of 5 mg/kg),65 tolerable LPS doses of 0.05 to 1 mg/kg injected intraperitoneally render the brain,11 heart,66,67 liver,68,69 kidneys,70 and pancreas71 transiently resistant to subsequent ischemic injury. This preconditioning paradigm relies on the ability of a peripheral signal to cross into multiple organ systems. LPS injected into the gut can signal through peritoneal macrophages and circulating monocytes. Toll-like receptor 4 is a pattern-recognition receptor that binds to pathogen-associated molecular patterns in LPS and initiates a signaling cascade through the NFκB pathway. This pathway culminates in the expression and secretion of several proinflammatory cytokines to fight off the infection and anti-inflammatory cytokines to control the immune response.
The major output of LPS signaling is innate production of proinflammatory cytokines to fight infection and clear cellular debris. Central cytokines, including tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), can be neurodestructive if administered after ischemia. TNFα administration by cerebroventricular injection after ischemia augmented the extent of injury, and blockage of TNFα signaling proved neuroprotective.11,72,73 However, in LPS preconditioning, cytokine production precedes ischemia. Intracisternal injections of TNFα before middle cerebral artery occlusion (MCAO) were protective in reducing the infarct size of pretreated mice.74 Furthermore, intracisternal injection of ceramide analog, a downstream component of the TNFα signaling pathway, was also capable of reducing the MCAO infarct area.75 Preischemic treatment with IL-6 and IL-1 also reduced neuronal damage.76,77 TNFα knockout mice eliminated the LPS protective phenotype,72 demonstrating that cytokine production is a critical feature of LPS preconditioning in ischemia. Additionally, ischemic damage in the absence of LPS preconditioning was exacerbated in TNFα receptor 1 knockout mice.78,79 Consistently, TNFα protein levels are upregulated after LPS treatment but are downregulated following LPS-preconditioned MCAO.72 A unifying theme in LPS preconditioning comprises early activation of the innate immune system with ensuing suppression in ischemia.
As a potential mechanism, the initial inflammatory response induced by LPS appears to render the innate immune system hyporesponsive to subsequent insults such as ischemia. This may occur by persistence of anti-inflammatory cytokines produced by the primary insult. These molecules are expressed in tandem with proinflammatory cytokines to control the innate immune response, but may also play a role in delayed preconditioning. For instance, intravenous or intracerebroventricular IL-10 injection can reduce the infarct size with MCAO.80 Alternatively, several proinflammatory cytokine signaling pathways may be downregulated by negative feedback inhibition.20,81 This inhibition may occur extracellularly, using soluble cytokine receptors, decoy receptors, or receptor antagonists. For example, intravenous injection of IL-1 receptor antagonist can provide neuroprotection against ischemic injury from MCAO.82,83 Cytokine feedback inhibitors that act intracellularly are also induced with the innate immune response. Intracellular inhibition may involve direct downregulation of cytokine transcription (peroxisome proliferator-activated receptor gamma [PPAR-γ]) or inhibition of intracellular signaling pathways that promote cytokine production (suppressor of cytokine signaling [SOCS] and PI3K). Antisense mRNA knockdown of SOCS-3 exacerbates ischemic injury from MCAO.84 The MCAO infarction area is increased after treatment with PPAR-γ antagonists and decreased by PPAR-γ agonists.85,86 Administration of compounds that increase PI3K signaling is also capable of reducing ischemic damage.87 Thus, several defense mechanisms designed to suppress the innate immune response may play an active role in LPS ischemic preconditioning.
Role of microglia in ischemic preconditioning
Microglia represent the resident central nervous system (CNS) component of the innate immune system. Microglia and macrophages become activated with ischemia in the infarcted and surrounded area.88 Upon activation in ischemia, microglia will become phagocytic and secrete a multitude of noxious chemokines and cytokines.89 Accordingly, anti-inflammatory antibiotics such as doxycycline and minocycline reduce microglial activation and diminish the ischemic infarction area.90 Preconditioning the brain with LPS ameliorates microglial activation, neutrophil infiltration, and circulating monocyte activation following MCAO.35 However, primary ischemic damage is not correlated with CNS infiltration of peripheral leukocytes but rather with an increase in proliferating resident microglial cells.91 Alternatively, microglia can exhibit neuroprotective properties within the brain.92 In fact, greater ischemic damage from longer periods of MCAO is correlated with fewer proliferating microglia, suggesting a protective microglial role.91 Consistently, ablation of proliferating microglia increases the infarction area following MCAO.93 Therefore, microglia can be protective in ischemia, and preconditioning with LPS may render microglia more capable of reacting to ischemic conditions.
CONCLUSIONS
Preconditioning represents an adaptive response to prime the brain for protection against future injury. Elucidation of these endogenous cell survival pathways has significant clinical implications for preventing neuronal damage in susceptible patients. For this reason, understanding the underlying mechanisms in establishing a tolerant state will be a critical step in adapting preconditioning for safe patient applications. The field of ischemic research has made great strides in deciphering causative preconditioning factors but has been hampered by the complex, multifactorial nature of preconditioning paradigms. The study of tolerance is further complicated by the fact that signaling takes place both peripheral to and within the brain in multiple cell types. Future research will require the exploration of interactions between multiple pathways and roles of individual cell types in establishing ischemic tolerance. Only with a more thorough understanding of preconditioning mechanisms can we adapt these pathways for the most efficient and protective treatments.
The brain relies upon internal defense mechanisms for protection from injurious stimuli. Preconditioning is a phenomenon whereby low doses of these noxious insults shield the brain from future insults rather than inflicting damage. Preconditioning stimuli include but are not limited to transient global and focal ischemia,1–4 cortical spreading depression,5–7 brief episodes of seizure, exposure to anesthetic inhalants,8–10 low doses of endotoxin (lipopolysaccharide [LPS]),11,12 hypothermia and hyperthermia,13,14 and 3-nitropropionic acid treatment.15,16
Depending on the specific preconditioning stimulus, a state of neuronal tolerance can be established in at least two temporal profiles: one in which the trigger induces protection within minutes (rapid or acute tolerance),17 and one in which the protected state develops after a delay of several hours to days (delayed tolerance).4 Some preconditioning paradigms induce both phases of ischemic tolerance, while others can induce only the acute phase or only the delayed phase.18–21 The acute phase is most likely due to rapid posttranslational modifications of proteins.22,23 In contrast, the delayed phase is dependent on de novo protein synthesis.24,25
Preconditioning by ischemic tolerance was first identified in the heart by Murry et al,26 and was subsequently found to occur in the brain4,27 and a variety of organs including the liver, intestine, kidney, and lung. Preconditioning stimuli can be cross-tolerant, safeguarding against other types of injury. For example, endotoxin preconditioning can protect against subsequent ischemia and vice versa. Thus, there may be some overlapping mechanisms in preconditioning, and unraveling these pathways may uncover an arsenal of neuroprotective therapeutic targets. In this review, we will compare different preconditioning paradigms and discuss potential mechanisms in initiating brain ischemic tolerance.
PARADIGMS TO ESTABLISH PRECONDITIONING
Refinement of various preconditioning models is of great clinical significance. Cardiovascular or cerebrovascular surgery has a negative impact on brain function due to stoppage of blood flow during surgery. In fact, more than 25% of patients who receive coronary artery bypass surgery suffer from temporary or permanent memory loss.28,29 As a result, it is of premier importance to develop strategies to protect the brain either prior to vascular surgeries or in patients at high risk of stroke. While it would be dangerous and impractical to precondition at-risk patients with ischemia, the identification of underlying preconditioning mechanisms may lead to safer therapeutic factors that can be administered before surgery.
Ischemia
Global ischemic preconditioning in the brain is accomplished by occlusion of the bilateral common carotid arteries. In contrast, in focal ischemic preconditioning, occlusion of one side of the middle cerebral artery is induced for about 1 to 20 minutes, depending on methods and animal species.4,30–32 Twenty-four hours after ischemic preconditioning, stroke is induced in these animals. Preconditioning-induced neuroprotection is observed not only in terms of infarct volume but also in terms of neurological scores and behavior studies.
Lipopolysaccharide
Tolerance to ischemic injury can also be induced by a small dose of LPS injected into the peritoneal cavity. Dosages vary from 0.05 to 1 mg/kg body weight in small rodents such as mice and rats.11,33–36 This dose of LPS usually does not bring abnormal signs and symptoms to the animals. The ischemic protection yields a reduction of infarct volume of approximately 30%. This tolerant state can be sustained for about 1 week, with maximum protection occurring around 2 to 3 days after injection of LPS.
Hypoxia
A relatively convenient method for preconditioning animals is hypoxic exposure. Animals are put in a chamber in which oxygen and nitrogen proportions can be controlled. Oxygen concentration usually ranges from 8% to 13% with normobaric pressure. Exposure time ranges from 1 to 6 hours. Twenty-four to 72 hours later, transient or permanent focal stroke is induced in the animals.37–40 Hypoxia-preconditioned neuroprotection usually starts at 1 to 3 days with a significant reduction of infarct size. Hypoxic preconditioning has also been demonstrated for in vitro neuron culture models using oxygen-glucose deprivation injury.41
3-Nitropropionic acid
3-Nitropropionic acid (3-NP) is an irreversible inhibitor of succinate dehydrogenase, an enzyme required for oxidative phosphorylation and adenosine triphosphate production. When applied at low doses 1 to 4 days before ischemia, 3-NP can lead to ischemic tolerance in the forebrain of gerbils and rats.16,42,43 The dose ranges from 1 to 20 mg/kg body weight.16 Such treatment significantly improves neurological behavior and increases neuronal survival in the CA1 region of hippocampus. In addition, 3-NP preconditioning induces tolerance to hypoxia in hippocampal slice preparations.15,44
Hypothermia and hyperthermia
Hypothermia is a well-characterized protective procedure used during and after cerebral surgery. It is also reported that brief hypothermic or hyperthermic exposure can also lead to ischemic tolerance. The temperatures adopted range from 25°C to 32°C13,45,46 in hypothermia and from 42°C to 43°C in hyperthermia.14
Cortical spreading depression
Cortical spreading depression is defined as the electrophysiologic phenomenon of slowly propagating transient depolarization waves across the cortex. Usually 5 M of potassium chloride is infused into the cortex, or a cotton pad soaked with the solution is put on the surface of dura mater, which results in depolarization, firing of neurons, and cortical spreading depression. Cortical spreading depression induces a prolonged phase of ischemic tolerance that lasts 1 to 7 days.5,6,47,48
Anesthetics
Exposure to volatile anesthetics such as isoflurane and halothane within pharmacologic concentration ranges also confers delayed-phase ischemic tolerance of the brain.8–10,49
MOLECULAR PRECONDITIONING PATHWAYS
Mechanistically, cellular preconditioning can be subdivided into intrinsic neuronal pathways (preventing excitotoxic damage, signaling through anti-apoptotic molecules, and treatment by neurotrophic factors) or extrinsic nonneuronal pathways (peripheral cytokine production, microglial activation, and regulation of the cerebrovascular system). Several neuroprotective molecules are expressed and signal through multiple cell types both within and peripheral to the brain, so that assigning an exact source and paradigm for preconditioning pathways has proven difficult.
NMDA receptor activation and excitotoxicity protection
In neurons, ischemic tolerance is mediated largely by the activation of the N-methyl-d-aspartate (NMDA) glutamate receptors through increases in intracellular calcium.50–52 Although glutamate receptor activation is generally believed to be responsible for much of the neuronal damage caused by excitotoxicity, it appears to also be implicated in the establishment of preconditioning. One study demonstrated that exposure of cortical cell cultures to low levels of glutamate activated NMDA receptors in preconditioning.50 In addition, preconditioning by oxygen-glucose deprivation was blocked when an NMDA antagonist was applied. NMDA receptor activation can induce a tolerant state through rapid adaptation of the voltage-dependent calcium flux. In addition, activation of NMDA receptors leads to rapid release of brain-derived neurotrophic factor, which then binds to and activates its cognate receptor, receptor tyrosine kinase B. Both NMDA and tyrosine kinase B receptors activate nuclear factor–kappa B (NFκB), a transcription factor involved in protecting neurons against insults. In sublethal ischemic preconditioning, activation of NFκB and its translocation from the cytosol to the nucleus was required for the development of late cerebral protection against severe ischemia or epilepsy.53 Other key mediators involved in synaptic NMDA receptor–dependent neuroprotection are phosphatidylinositol 3-kinase (PI3K), Akt, and glycogen synthase kinase 3-beta.54
Preconditioning with cortical spreading depression results in the downregulation of the excitatory amino acid transporters EAAT1 and EAAT2 from cerebral cortex plasma membranes.55 Although these transporters are normally involved in glutamate uptake, it has been suggested that the influx of sodium that occurs during excitotoxicity may cause their reversal and result in additional glutamate release. Downregulating these transporters may thus contribute to ischemic tolerance.
Nitric oxide
Nitric oxide (NO) may play a key role as a mediator of the neuronal ischemic preconditioning response, either in conjunction with or independent of NMDA receptor activation. Both the inhibition of nitric oxide synthase (NOS) and the scavenging of NO during preconditioning significantly attenuated the induced neuronal tolerance, and neither endothelial NOS nor neuronal NOS knockout mice showed protection from rapid ischemic preconditioning.56,57 Treatment with the inducible NOS (iNOS) inhibitor aminoguanidine abolished the induced protection. The mechanisms responsible for NO-induced tolerance are not clear. Downregulation of the glutamate transporter GLT-1 might play a role.58 A common link to NMDA receptor activation and NO is p21ras (Ras). Preconditioning induces p21ras activation in an NMDA- and NO-dependent manner and leads to the downstream activation of Raf kinase, mitogen-activated protein kinases, and extracellular regulated kinase.59 Inhibition of these kinases attenuates subsequent protection from ischemia.60,61 Pharmacologic inhibition of Ras, as well as a dominant negative Ras mutant, blocked preconditioning, whereas a constitutively active form of Ras promoted neuroprotection against lethal insults. An important consideration regarding NO is also that preconditioning by volatile anesthetics appears to involve NO pathways.9
NO and reactive oxygen species (ROS) are also implicated in regulating the peripheral cerebrovascular system. Ischemia generated by occlusion of the middle cerebral artery causes defects in cerebrovascular function for not only the infarcted area but also the surrounding ischemic region. LPS preconditioning has been reported in some cases to increase this regional cerebral blood flow both before and after ischemia.1,21,36,62–64 LPS also improves microvascular perfusion.33,64 It was recently reported that LPS-stimulated cerebral blood flow is induced through reactive oxygen and nitrogen species (ROS or NO).1 Mouse knockouts of iNOS (NO production) or of the nox2 subunit of NADPH oxidase (ROS production) eliminated the LPS-upregulated cerebrovascular activity. Furthermore, blockage of these ROS and NO pathways reduced the preconditioning effect of LPS. Therefore, LPS may play a more direct role in preventing ischemic damage by increasing blood availability to the affected brain region.
Inflammatory cytokines and the innate immune system
LPS, a component of the gram-negative bacterial cell wall, can illicit a potent innate immune response. While this systemic inflammatory response can be destructive (at doses of 5 mg/kg),65 tolerable LPS doses of 0.05 to 1 mg/kg injected intraperitoneally render the brain,11 heart,66,67 liver,68,69 kidneys,70 and pancreas71 transiently resistant to subsequent ischemic injury. This preconditioning paradigm relies on the ability of a peripheral signal to cross into multiple organ systems. LPS injected into the gut can signal through peritoneal macrophages and circulating monocytes. Toll-like receptor 4 is a pattern-recognition receptor that binds to pathogen-associated molecular patterns in LPS and initiates a signaling cascade through the NFκB pathway. This pathway culminates in the expression and secretion of several proinflammatory cytokines to fight off the infection and anti-inflammatory cytokines to control the immune response.
The major output of LPS signaling is innate production of proinflammatory cytokines to fight infection and clear cellular debris. Central cytokines, including tumor necrosis factor–alpha (TNFα), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β), can be neurodestructive if administered after ischemia. TNFα administration by cerebroventricular injection after ischemia augmented the extent of injury, and blockage of TNFα signaling proved neuroprotective.11,72,73 However, in LPS preconditioning, cytokine production precedes ischemia. Intracisternal injections of TNFα before middle cerebral artery occlusion (MCAO) were protective in reducing the infarct size of pretreated mice.74 Furthermore, intracisternal injection of ceramide analog, a downstream component of the TNFα signaling pathway, was also capable of reducing the MCAO infarct area.75 Preischemic treatment with IL-6 and IL-1 also reduced neuronal damage.76,77 TNFα knockout mice eliminated the LPS protective phenotype,72 demonstrating that cytokine production is a critical feature of LPS preconditioning in ischemia. Additionally, ischemic damage in the absence of LPS preconditioning was exacerbated in TNFα receptor 1 knockout mice.78,79 Consistently, TNFα protein levels are upregulated after LPS treatment but are downregulated following LPS-preconditioned MCAO.72 A unifying theme in LPS preconditioning comprises early activation of the innate immune system with ensuing suppression in ischemia.
As a potential mechanism, the initial inflammatory response induced by LPS appears to render the innate immune system hyporesponsive to subsequent insults such as ischemia. This may occur by persistence of anti-inflammatory cytokines produced by the primary insult. These molecules are expressed in tandem with proinflammatory cytokines to control the innate immune response, but may also play a role in delayed preconditioning. For instance, intravenous or intracerebroventricular IL-10 injection can reduce the infarct size with MCAO.80 Alternatively, several proinflammatory cytokine signaling pathways may be downregulated by negative feedback inhibition.20,81 This inhibition may occur extracellularly, using soluble cytokine receptors, decoy receptors, or receptor antagonists. For example, intravenous injection of IL-1 receptor antagonist can provide neuroprotection against ischemic injury from MCAO.82,83 Cytokine feedback inhibitors that act intracellularly are also induced with the innate immune response. Intracellular inhibition may involve direct downregulation of cytokine transcription (peroxisome proliferator-activated receptor gamma [PPAR-γ]) or inhibition of intracellular signaling pathways that promote cytokine production (suppressor of cytokine signaling [SOCS] and PI3K). Antisense mRNA knockdown of SOCS-3 exacerbates ischemic injury from MCAO.84 The MCAO infarction area is increased after treatment with PPAR-γ antagonists and decreased by PPAR-γ agonists.85,86 Administration of compounds that increase PI3K signaling is also capable of reducing ischemic damage.87 Thus, several defense mechanisms designed to suppress the innate immune response may play an active role in LPS ischemic preconditioning.
Role of microglia in ischemic preconditioning
Microglia represent the resident central nervous system (CNS) component of the innate immune system. Microglia and macrophages become activated with ischemia in the infarcted and surrounded area.88 Upon activation in ischemia, microglia will become phagocytic and secrete a multitude of noxious chemokines and cytokines.89 Accordingly, anti-inflammatory antibiotics such as doxycycline and minocycline reduce microglial activation and diminish the ischemic infarction area.90 Preconditioning the brain with LPS ameliorates microglial activation, neutrophil infiltration, and circulating monocyte activation following MCAO.35 However, primary ischemic damage is not correlated with CNS infiltration of peripheral leukocytes but rather with an increase in proliferating resident microglial cells.91 Alternatively, microglia can exhibit neuroprotective properties within the brain.92 In fact, greater ischemic damage from longer periods of MCAO is correlated with fewer proliferating microglia, suggesting a protective microglial role.91 Consistently, ablation of proliferating microglia increases the infarction area following MCAO.93 Therefore, microglia can be protective in ischemia, and preconditioning with LPS may render microglia more capable of reacting to ischemic conditions.
CONCLUSIONS
Preconditioning represents an adaptive response to prime the brain for protection against future injury. Elucidation of these endogenous cell survival pathways has significant clinical implications for preventing neuronal damage in susceptible patients. For this reason, understanding the underlying mechanisms in establishing a tolerant state will be a critical step in adapting preconditioning for safe patient applications. The field of ischemic research has made great strides in deciphering causative preconditioning factors but has been hampered by the complex, multifactorial nature of preconditioning paradigms. The study of tolerance is further complicated by the fact that signaling takes place both peripheral to and within the brain in multiple cell types. Future research will require the exploration of interactions between multiple pathways and roles of individual cell types in establishing ischemic tolerance. Only with a more thorough understanding of preconditioning mechanisms can we adapt these pathways for the most efficient and protective treatments.
- Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 2007; 27:7083–7093.
- Simon RP, Niiro M, Gwinn R. Prior ischemic stress protects against experimental stroke. Neurosci Lett 1993; 163:135–137.
- Chen J, Simon R. Ischemic tolerance in the brain. Neurology 1997; 48:306–311.
- Kitagawa K, Matsumoto M, Tagaya M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res 1990; 528:21–24.
- Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res 1995; 17:9–16.
- Taga K, Patel PM, Drummond JC, Cole DJ, Kelly PJ. Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology 1997; 87:918–925.
- Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab 1995; 15:721–727.
- Baughman VL, Hoffman WE, Miletich DJ, Albrecht RF, Thomas C. Neurologic outcome in rats following incomplete cerebral ischemia during halothane, isoflurane, or N2O. Anesthesiology 1988; 69:192–198.
- Kapinya KJ, Lowl D, Futterer C, et al. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 2002; 33:1889–1898.
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol Pharmacol 2004; 65:1172–1180.
- Tasaki K, Ruetzler CA, Ohtsuki T, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 1997; 748:267–270.
- Zimmermann C, Ginis I, Furuya K, et al. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res 2001; 895:59–65.
- Nishio S, Yunoki M, Chen ZF, Anzivino MJ, Lee KS. Ischemic tolerance in the rat neocortex following hypothermic preconditioning. J Neurosurg 2000; 93:845–851.
- Ota A, Ikeda T, Xia XY, Xia YX, Ikenoue T. Hypoxic-ischemic tolerance induced by hyperthermic pretreatment in newborn rats. J Soc Gynecol Investig 2000; 7:102–105.
- Riepe MW, Niemi WN, Megow D, Ludolph AC, Carpenter DO. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp Neurol 1996; 138:15–21.
- Sugino T, Nozaki K, Takagi Y, Hashimoto N. 3-Nitropropionic acid induces ischemic tolerance in gerbil hippocampus in vivo. Neurosci Lett 1999; 259:9–12.
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab 1997; 17:175–182.
- Stagliano NE, Perez-Pinzon MA, Moskowitz MA, Huang PL. Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 1999; 19:757–761.
- Perez-Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: role of the K+ ATP channel and protein kinase C. Neuroscience 1999; 89:453–459.
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 2004; 24:1288–1304.
- Hoyte LC, Papadakis M, Barber PA, Buchan AM. Improved regional cerebral blood flow is important for the protection seen in a mouse model of late phase ischemic preconditioning. Brain Res 2006; 1121:231–237.
- Nakase H, Heimann A, Uranishi R, Riepe MW, Kempski O. Early-onset tolerance in rat global cerebral ischemia induced by a mitochondrial inhibitor. Neurosci Lett 2000; 290:105–108.
- Meller R, Cameron JA, Torrey DJ, et al. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem 2006; 281:7429–7436.
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab 2002; 22:1283–1296.
- Stenzel-Poore MP, Stevens SL, Xiong Z, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003; 362:1028–1037.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74:1124–1136.
- Kitagawa K, Matsumoto M, Kuwabara K, et al. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res 1991; 561:203–211.
- Roach GW, Kanchuger M, Mangano CM, et al. Adverse cerebral outcomes after coronary bypass surgery. Multicenter Study of Perioperative Ischemia Research Group and the Ischemia Research and Education Foundation Investigators. N Engl J Med 1996; 335:1857–1863.
- Newman MF, Kirchner JL, Phillips-Bute B, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 2001; 344:395–402.
- Corbett D, Crooks P. Ischemic preconditioning: a long term survival study using behavioural and histological endpoints. Brain Res 1997; 760:129–136.
- Barone FC, White RF, Spera PA, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29:1937–1950.
- Wu C, Zhan RZ, Qi S, et al. A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods 2001; 107:101–106.
- Dawson DA, Furuya K, Gotoh J, et al. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. J Cereb Blood Flow Metab 1999; 19:616–623.
- Ahmed SH, He YY, Nassief A, et al. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 2000; 31:193–199.
- Rosenzweig HL, Lessov NS, Henshall DC, et al. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004; 35:2576–2581.
- Furuya K, Zhu L, Kawahara N, et al. Differences in infarct evolution between lipopolysaccharide-induced tolerant and nontolerant conditions to focal cerebral ischemia. J Neurosurg 2005; 103:715–723.
- Bernaudin M, Nedelec AS, Divoux D, et al. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab 2002; 22:393–403.
- Kulinskii VI, Minakina LN, Gavrilina TV. Neuroprotective effect of hypoxic preconditioning: phenomenon and mechanisms. Bull Exp Biol Med 2002; 133:202–204.
- Miller BA, Perez RS, Shah AR, et al. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 2001; 12:1663–1669.
- Prass K, Scharff A, Ruscher K, et al. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003; 34:1981–1986.
- Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol 2000; 278:C144–C153.
- Kuroiwa T, Yamada I, Endo S, Hakamata Y, Ito U. 3-Nitropropionic acid preconditioning ameliorates delayed neurological deterioration and infarction after transient focal cerebral ischemia in gerbils. Neurosci Lett 2000; 283:145–148.
- Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 2003; 34:1015–1020.
- Aketa S, Nakase H, Kamada Y, Hiramatsu K, Sakaki T. Chemical preconditioning with 3-nitropropionic acid in gerbil hippocampal slices: therapeutic window and the participation of adenosine receptor. Exp Neurol 2000; 166:385–391.
- Nishio S, Chen ZF, Yunoki M, et al. Hypothermia-induced ischemic tolerance. Ann N Y Acad Sci 1999; 890:26–41.
- Yunoki M, Nishio S, Ukita N, Anzivino MJ, Lee KS. Hypothermic preconditioning induces rapid tolerance to focal ischemic injury in the rat. Exp Neurol 2003; 181:291–300.
- Kawahara N, Ruetzler CA, Mies G, Klatzo I. Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp Neurol 1999; 158:27–36.
- Yanamoto H, Hashimoto N, Nagata I, Kikuchi H. Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res 1998; 784:239–249.
- McAuliffe JJ, Joseph B, Vorhees CV. Isoflurane-delayed preconditioning reduces immediate mortality and improves striatal function in adult mice after neonatal hypoxia-ischemia. Anesth Analg 2007; 104:1066–1077.
- Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 1999; 19:1657–1662.
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett 1992; 139:118–121.
- Kasischke K, Ludolph AC, Riepe MW. NMDA-antagonists reverse increased hypoxic tolerance by preceding chemical hypoxia. Neurosci Lett 1996; 214:175–178.
- Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-κB is a key event in brain tolerance. J Neurosci 2001; 21:4668–4677.
- Soriano FX, Papadia S, Hofmann F, et al. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci 2006; 26:4509–4518.
- Douen AG, Akiyama K, Hogan MJ, et al. Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebal cortex plasma membranes. J Neurochem 2000; 75:812–818.
- Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 2003; 34:1299–1303.
- Cho S, Park EM, Zhou P, et al. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab 2005; 25:493–501.
- Yamada T, Kawahara K, Kosugi T, Tanaka M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res 2006; 31:49–56.
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, et al. Requirement for nitric oxide activation of p21ras/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A 2000; 97:436–441.
- Kurkinen K, Keinanen R, Li W, Koistinaho J. Preconditioning with spreading depression activates specifically protein kinase Cδ. Neuroreport 2001; 12:269–273.
- Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience 1999; 93:81–88.
- Zhao L, Nowak TS Jr. CBF changes associated with focal ischemic preconditioning in the spontaneously hypertensive rat. J Cereb Blood Flow Metab 2006; 26:1128–1140.
- Nakamura H, Katsumata T, Nishiyama Y, et al. Effect of ischemic preconditioning on cerebral blood flow after subsequent lethal ischemia in gerbils. Life Sci 2006; 78:1713–1719.
- Bastide M, Gele P, Petrault O, et al. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab 2003; 23:399–405.
- Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007; 55:453–462.
- Eising GP, Mao L, Schmid-Schonbein GW, Engler RL, Ross J. Effects of induced tolerance to bacterial lipopolysaccharide on myocardial infarct size in rats. Cardiovasc Res 1996; 31:73–81.
- Zacharowski K, Otto M, Hafner G, et al. Endotoxin induces a second window of protection in the rat heart as determined by using pnitro-blue tetrazolium staining, cardiac troponin T release, and histology. Arterioscler Thromb Vasc Biol 1999; 19:2276–2280.
- Colletti LM, Remick DG, Campbell DA Jr. LPS pretreatment protects from hepatic ischemia/reperfusion. J Surg Res 1994; 57:337–343.
- Fernandez ED, Flohe S, Siemers F, et al. Endotoxin tolerance protects against local hepatic ischemia/reperfusion injury in the rat. J Endotoxin Res 2000; 6:321–328.
- Heemann U, Szabo A, Hamar P, et al. Lipopolysaccharide pretreatment protects from renal ischemia/reperfusion injury: possible connection to an interleukin-6-dependent pathway. Am J Pathol 2000; 156:287–293.
- Obermaier R, Drognitz O, Grub A, et al. Endotoxin preconditioning in pancreatic ischemia/reperfusion injury. Pancreas 2003; 27:e51–e56.
- Rosenzweig HL, Minami M, Lessov NS, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFα after stroke: a novel role for TNFα in LPS-ischemic tolerance. J Cereb Blood Flow Metab 2007; 27:1663–1674.
- Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997; 28:1233–1244.
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997; 17:483–490.
- Furuya K, Ginis I, Takeda H, et al. Cell permeable exogenous ceramide reduces infarct size in spontaneously hypertensive rats supporting in vitro studies that have implicated ceramide in induction of tolerance to ischemia. J Cereb Blood Flow Metab 2001; 21:226–232.
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18:176–179.
- Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J Cereb Blood Flow Metab 1996; 16:1137–1142.
- Bruce AJ, Boling W, Kindy MS, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996; 2:788–794.
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab 1998; 18:1283–1287.
- Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett 1998; 251:189–192.
- Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res 2004; 10:71–84.
- Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Pathol 1995; 147:1477–1486.
- Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol 1996; 138:206–213.
- Raghavendra RV, Bowen KK, Dhodda VK, et al. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem 2002; 83:1072–1086.
- Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARγ expression and activation after transient focal ischemia in rats. Eur J Neurosci 2006; 24:1653–1663.
- Zhao Y, Patzer A, Gohlke P, et al. The intracerebral application of the PPARγ-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci 2005; 22:278–282.
- Shioda N, Ishigami T, Han F, et al. Activation of phosphatidylinositol 3-kinase/protein kinase B pathway by a vanadyl compound mediates its neuroprotective effect in mouse brain ischemia. Neuroscience 2007; 148:221–229.
- Mabuchi T, Kitagawa K, Ohtsuki T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000; 31:1735–1743.
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol 2006; 84:49–59.
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 1998; 95:15769–15774.
- Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007; 27:1941–1953.
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002; 40:133–139.
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007; 27:2596–2605.
- Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 2007; 27:7083–7093.
- Simon RP, Niiro M, Gwinn R. Prior ischemic stress protects against experimental stroke. Neurosci Lett 1993; 163:135–137.
- Chen J, Simon R. Ischemic tolerance in the brain. Neurology 1997; 48:306–311.
- Kitagawa K, Matsumoto M, Tagaya M, et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res 1990; 528:21–24.
- Kawahara N, Ruetzler CA, Klatzo I. Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res 1995; 17:9–16.
- Taga K, Patel PM, Drummond JC, Cole DJ, Kelly PJ. Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology 1997; 87:918–925.
- Kobayashi S, Harris VA, Welsh FA. Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab 1995; 15:721–727.
- Baughman VL, Hoffman WE, Miletich DJ, Albrecht RF, Thomas C. Neurologic outcome in rats following incomplete cerebral ischemia during halothane, isoflurane, or N2O. Anesthesiology 1988; 69:192–198.
- Kapinya KJ, Lowl D, Futterer C, et al. Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 2002; 33:1889–1898.
- Zheng S, Zuo Z. Isoflurane preconditioning induces neuroprotection against ischemia via activation of P38 mitogen-activated protein kinases. Mol Pharmacol 2004; 65:1172–1180.
- Tasaki K, Ruetzler CA, Ohtsuki T, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res 1997; 748:267–270.
- Zimmermann C, Ginis I, Furuya K, et al. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res 2001; 895:59–65.
- Nishio S, Yunoki M, Chen ZF, Anzivino MJ, Lee KS. Ischemic tolerance in the rat neocortex following hypothermic preconditioning. J Neurosurg 2000; 93:845–851.
- Ota A, Ikeda T, Xia XY, Xia YX, Ikenoue T. Hypoxic-ischemic tolerance induced by hyperthermic pretreatment in newborn rats. J Soc Gynecol Investig 2000; 7:102–105.
- Riepe MW, Niemi WN, Megow D, Ludolph AC, Carpenter DO. Mitochondrial oxidation in rat hippocampus can be preconditioned by selective chemical inhibition of succinic dehydrogenase. Exp Neurol 1996; 138:15–21.
- Sugino T, Nozaki K, Takagi Y, Hashimoto N. 3-Nitropropionic acid induces ischemic tolerance in gerbil hippocampus in vivo. Neurosci Lett 1999; 259:9–12.
- Perez-Pinzon MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab 1997; 17:175–182.
- Stagliano NE, Perez-Pinzon MA, Moskowitz MA, Huang PL. Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 1999; 19:757–761.
- Perez-Pinzon MA, Born JG. Rapid preconditioning neuroprotection following anoxia in hippocampal slices: role of the K+ ATP channel and protein kinase C. Neuroscience 1999; 89:453–459.
- Kariko K, Weissman D, Welsh FA. Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 2004; 24:1288–1304.
- Hoyte LC, Papadakis M, Barber PA, Buchan AM. Improved regional cerebral blood flow is important for the protection seen in a mouse model of late phase ischemic preconditioning. Brain Res 2006; 1121:231–237.
- Nakase H, Heimann A, Uranishi R, Riepe MW, Kempski O. Early-onset tolerance in rat global cerebral ischemia induced by a mitochondrial inhibitor. Neurosci Lett 2000; 290:105–108.
- Meller R, Cameron JA, Torrey DJ, et al. Rapid degradation of Bim by the ubiquitin-proteasome pathway mediates short-term ischemic tolerance in cultured neurons. J Biol Chem 2006; 281:7429–7436.
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab 2002; 22:1283–1296.
- Stenzel-Poore MP, Stevens SL, Xiong Z, et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003; 362:1028–1037.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74:1124–1136.
- Kitagawa K, Matsumoto M, Kuwabara K, et al. ‘Ischemic tolerance’ phenomenon detected in various brain regions. Brain Res 1991; 561:203–211.
- Roach GW, Kanchuger M, Mangano CM, et al. Adverse cerebral outcomes after coronary bypass surgery. Multicenter Study of Perioperative Ischemia Research Group and the Ischemia Research and Education Foundation Investigators. N Engl J Med 1996; 335:1857–1863.
- Newman MF, Kirchner JL, Phillips-Bute B, et al. Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. N Engl J Med 2001; 344:395–402.
- Corbett D, Crooks P. Ischemic preconditioning: a long term survival study using behavioural and histological endpoints. Brain Res 1997; 760:129–136.
- Barone FC, White RF, Spera PA, et al. Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 1998; 29:1937–1950.
- Wu C, Zhan RZ, Qi S, et al. A forebrain ischemic preconditioning model established in C57Black/Crj6 mice. J Neurosci Methods 2001; 107:101–106.
- Dawson DA, Furuya K, Gotoh J, et al. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide-induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. J Cereb Blood Flow Metab 1999; 19:616–623.
- Ahmed SH, He YY, Nassief A, et al. Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 2000; 31:193–199.
- Rosenzweig HL, Lessov NS, Henshall DC, et al. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004; 35:2576–2581.
- Furuya K, Zhu L, Kawahara N, et al. Differences in infarct evolution between lipopolysaccharide-induced tolerant and nontolerant conditions to focal cerebral ischemia. J Neurosurg 2005; 103:715–723.
- Bernaudin M, Nedelec AS, Divoux D, et al. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J Cereb Blood Flow Metab 2002; 22:393–403.
- Kulinskii VI, Minakina LN, Gavrilina TV. Neuroprotective effect of hypoxic preconditioning: phenomenon and mechanisms. Bull Exp Biol Med 2002; 133:202–204.
- Miller BA, Perez RS, Shah AR, et al. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 2001; 12:1663–1669.
- Prass K, Scharff A, Ruscher K, et al. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003; 34:1981–1986.
- Liu J, Ginis I, Spatz M, Hallenbeck JM. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am J Physiol Cell Physiol 2000; 278:C144–C153.
- Kuroiwa T, Yamada I, Endo S, Hakamata Y, Ito U. 3-Nitropropionic acid preconditioning ameliorates delayed neurological deterioration and infarction after transient focal cerebral ischemia in gerbils. Neurosci Lett 2000; 283:145–148.
- Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 2003; 34:1015–1020.
- Aketa S, Nakase H, Kamada Y, Hiramatsu K, Sakaki T. Chemical preconditioning with 3-nitropropionic acid in gerbil hippocampal slices: therapeutic window and the participation of adenosine receptor. Exp Neurol 2000; 166:385–391.
- Nishio S, Chen ZF, Yunoki M, et al. Hypothermia-induced ischemic tolerance. Ann N Y Acad Sci 1999; 890:26–41.
- Yunoki M, Nishio S, Ukita N, Anzivino MJ, Lee KS. Hypothermic preconditioning induces rapid tolerance to focal ischemic injury in the rat. Exp Neurol 2003; 181:291–300.
- Kawahara N, Ruetzler CA, Mies G, Klatzo I. Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp Neurol 1999; 158:27–36.
- Yanamoto H, Hashimoto N, Nagata I, Kikuchi H. Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res 1998; 784:239–249.
- McAuliffe JJ, Joseph B, Vorhees CV. Isoflurane-delayed preconditioning reduces immediate mortality and improves striatal function in adult mice after neonatal hypoxia-ischemia. Anesth Analg 2007; 104:1066–1077.
- Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 1999; 19:1657–1662.
- Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett 1992; 139:118–121.
- Kasischke K, Ludolph AC, Riepe MW. NMDA-antagonists reverse increased hypoxic tolerance by preceding chemical hypoxia. Neurosci Lett 1996; 214:175–178.
- Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-κB is a key event in brain tolerance. J Neurosci 2001; 21:4668–4677.
- Soriano FX, Papadia S, Hofmann F, et al. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J Neurosci 2006; 26:4509–4518.
- Douen AG, Akiyama K, Hogan MJ, et al. Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebal cortex plasma membranes. J Neurochem 2000; 75:812–818.
- Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL. Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 2003; 34:1299–1303.
- Cho S, Park EM, Zhou P, et al. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab 2005; 25:493–501.
- Yamada T, Kawahara K, Kosugi T, Tanaka M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem Res 2006; 31:49–56.
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, et al. Requirement for nitric oxide activation of p21ras/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci U S A 2000; 97:436–441.
- Kurkinen K, Keinanen R, Li W, Koistinaho J. Preconditioning with spreading depression activates specifically protein kinase Cδ. Neuroreport 2001; 12:269–273.
- Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience 1999; 93:81–88.
- Zhao L, Nowak TS Jr. CBF changes associated with focal ischemic preconditioning in the spontaneously hypertensive rat. J Cereb Blood Flow Metab 2006; 26:1128–1140.
- Nakamura H, Katsumata T, Nishiyama Y, et al. Effect of ischemic preconditioning on cerebral blood flow after subsequent lethal ischemia in gerbils. Life Sci 2006; 78:1713–1719.
- Bastide M, Gele P, Petrault O, et al. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab 2003; 23:399–405.
- Qin L, Wu X, Block ML, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007; 55:453–462.
- Eising GP, Mao L, Schmid-Schonbein GW, Engler RL, Ross J. Effects of induced tolerance to bacterial lipopolysaccharide on myocardial infarct size in rats. Cardiovasc Res 1996; 31:73–81.
- Zacharowski K, Otto M, Hafner G, et al. Endotoxin induces a second window of protection in the rat heart as determined by using pnitro-blue tetrazolium staining, cardiac troponin T release, and histology. Arterioscler Thromb Vasc Biol 1999; 19:2276–2280.
- Colletti LM, Remick DG, Campbell DA Jr. LPS pretreatment protects from hepatic ischemia/reperfusion. J Surg Res 1994; 57:337–343.
- Fernandez ED, Flohe S, Siemers F, et al. Endotoxin tolerance protects against local hepatic ischemia/reperfusion injury in the rat. J Endotoxin Res 2000; 6:321–328.
- Heemann U, Szabo A, Hamar P, et al. Lipopolysaccharide pretreatment protects from renal ischemia/reperfusion injury: possible connection to an interleukin-6-dependent pathway. Am J Pathol 2000; 156:287–293.
- Obermaier R, Drognitz O, Grub A, et al. Endotoxin preconditioning in pancreatic ischemia/reperfusion injury. Pancreas 2003; 27:e51–e56.
- Rosenzweig HL, Minami M, Lessov NS, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFα after stroke: a novel role for TNFα in LPS-ischemic tolerance. J Cereb Blood Flow Metab 2007; 27:1663–1674.
- Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997; 28:1233–1244.
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab 1997; 17:483–490.
- Furuya K, Ginis I, Takeda H, et al. Cell permeable exogenous ceramide reduces infarct size in spontaneously hypertensive rats supporting in vitro studies that have implicated ceramide in induction of tolerance to ischemia. J Cereb Blood Flow Metab 2001; 21:226–232.
- Loddick SA, Turnbull AV, Rothwell NJ. Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 1998; 18:176–179.
- Ohtsuki T, Ruetzler CA, Tasaki K, Hallenbeck JM. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J Cereb Blood Flow Metab 1996; 16:1137–1142.
- Bruce AJ, Boling W, Kindy MS, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996; 2:788–794.
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab 1998; 18:1283–1287.
- Spera PA, Ellison JA, Feuerstein GZ, Barone FC. IL-10 reduces rat brain injury following focal stroke. Neurosci Lett 1998; 251:189–192.
- Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res 2004; 10:71–84.
- Garcia JH, Liu KF, Relton JK. Interleukin-1 receptor antagonist decreases the number of necrotic neurons in rats with middle cerebral artery occlusion. Am J Pathol 1995; 147:1477–1486.
- Relton JK, Martin D, Thompson RC, Russell DA. Peripheral administration of interleukin-1 receptor antagonist inhibits brain damage after focal cerebral ischemia in the rat. Exp Neurol 1996; 138:206–213.
- Raghavendra RV, Bowen KK, Dhodda VK, et al. Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem 2002; 83:1072–1086.
- Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARγ expression and activation after transient focal ischemia in rats. Eur J Neurosci 2006; 24:1653–1663.
- Zhao Y, Patzer A, Gohlke P, et al. The intracerebral application of the PPARγ-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci 2005; 22:278–282.
- Shioda N, Ishigami T, Han F, et al. Activation of phosphatidylinositol 3-kinase/protein kinase B pathway by a vanadyl compound mediates its neuroprotective effect in mouse brain ischemia. Neuroscience 2007; 148:221–229.
- Mabuchi T, Kitagawa K, Ohtsuki T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke 2000; 31:1735–1743.
- Lai AY, Todd KG. Microglia in cerebral ischemia: molecular actions and interactions. Can J Physiol Pharmacol 2006; 84:49–59.
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 1998; 95:15769–15774.
- Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 2007; 27:1941–1953.
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002; 40:133–139.
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 2007; 27:2596–2605.