User login
Nutritional Dermatoses in the Hospitalized Patient
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
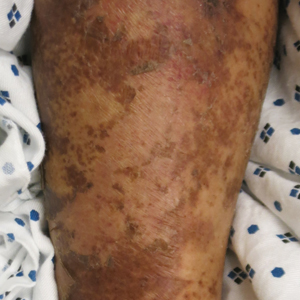
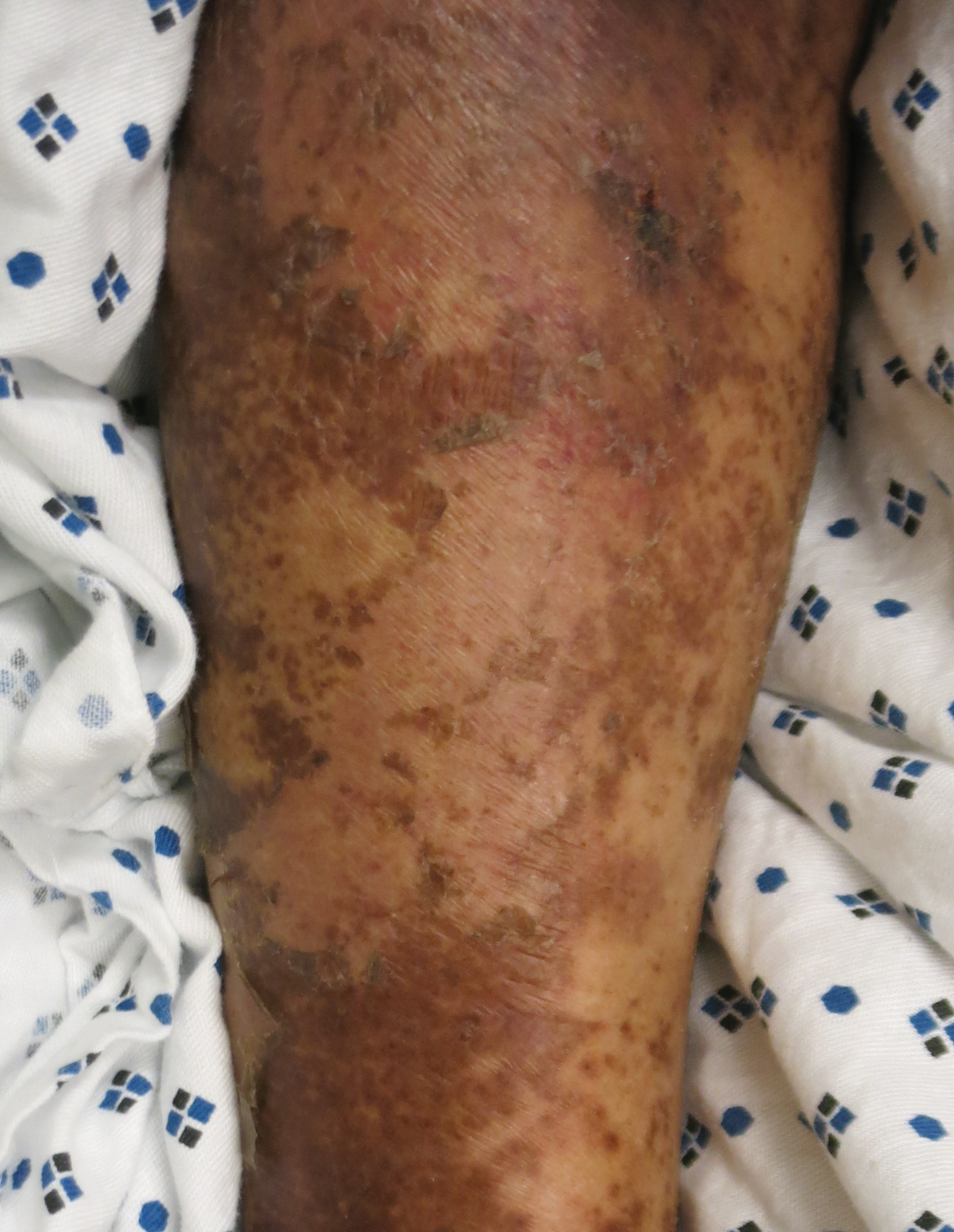
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
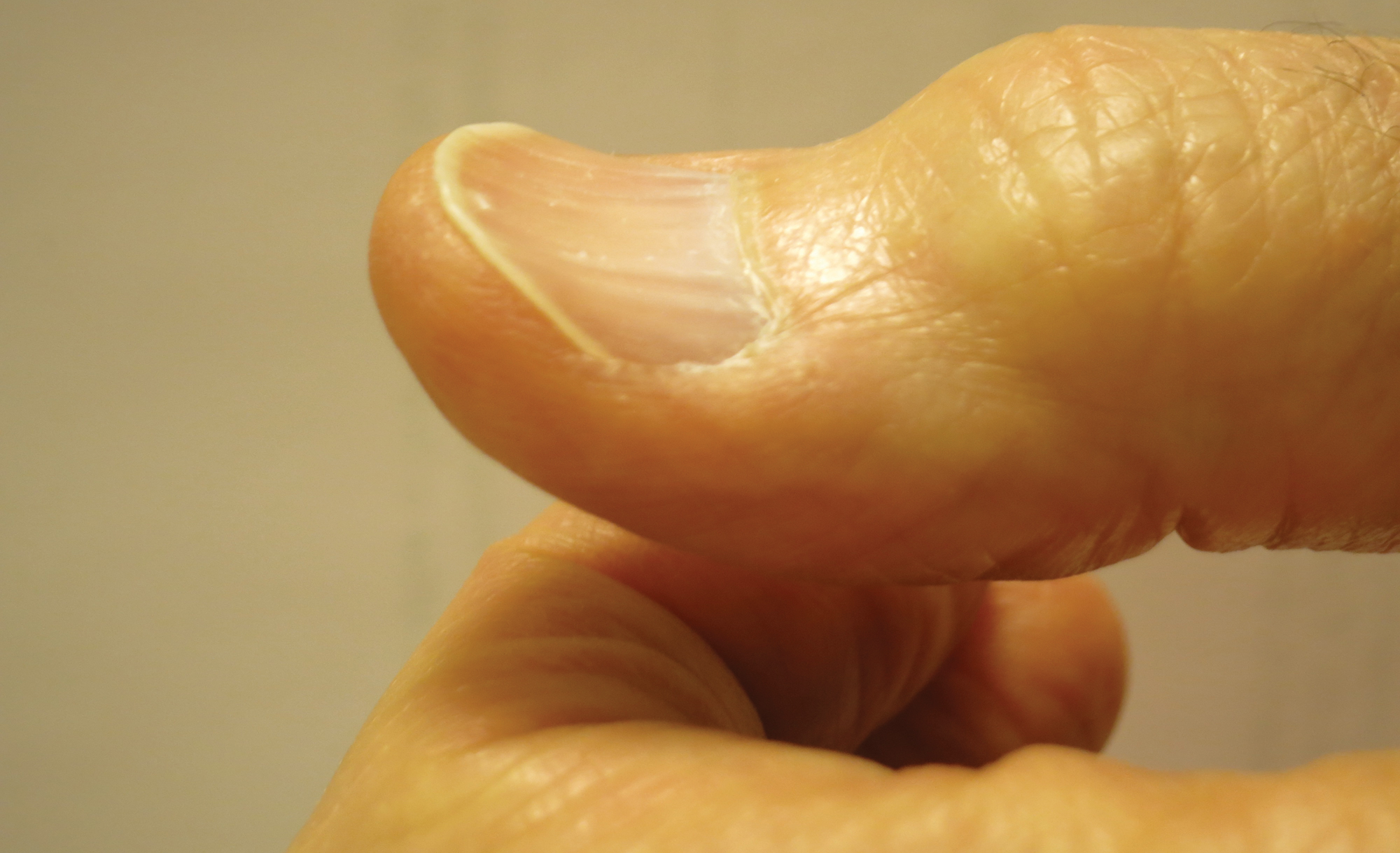
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
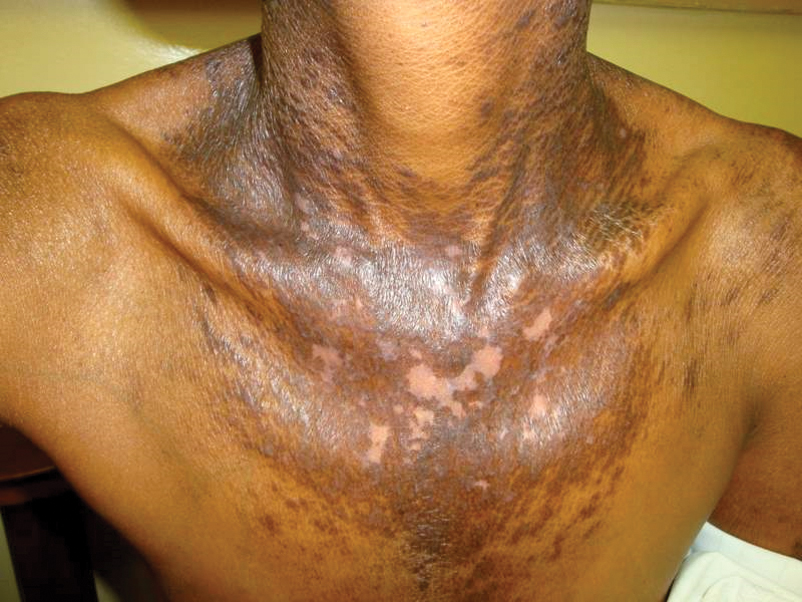
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
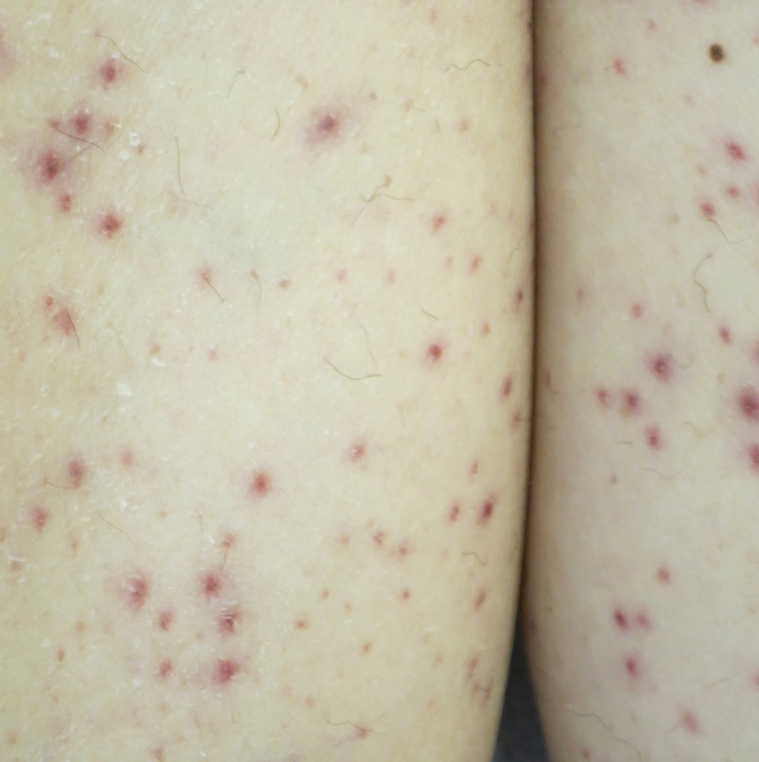
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
Practice Points
- Nutritional deficiencies are common in hospitalized patients and often go unrecognized.
- Awareness of the risk factors predisposing patients to nutritional deficiencies and the cutaneous manifestations associated with undernutrition can promote early diagnosis.
- When investigating cutaneous findings, undernutrition should be considered in patients with chronic infections, malabsorptive states, psychiatric illness, and strict dietary practices, as well as in those using certain medications.
- Prompt nutritional supplementation can prevent patient morbidity and mortality and reverse skin disease.