User login
An infant girl presents with a growing pink-red leg nodule
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.

A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.

Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
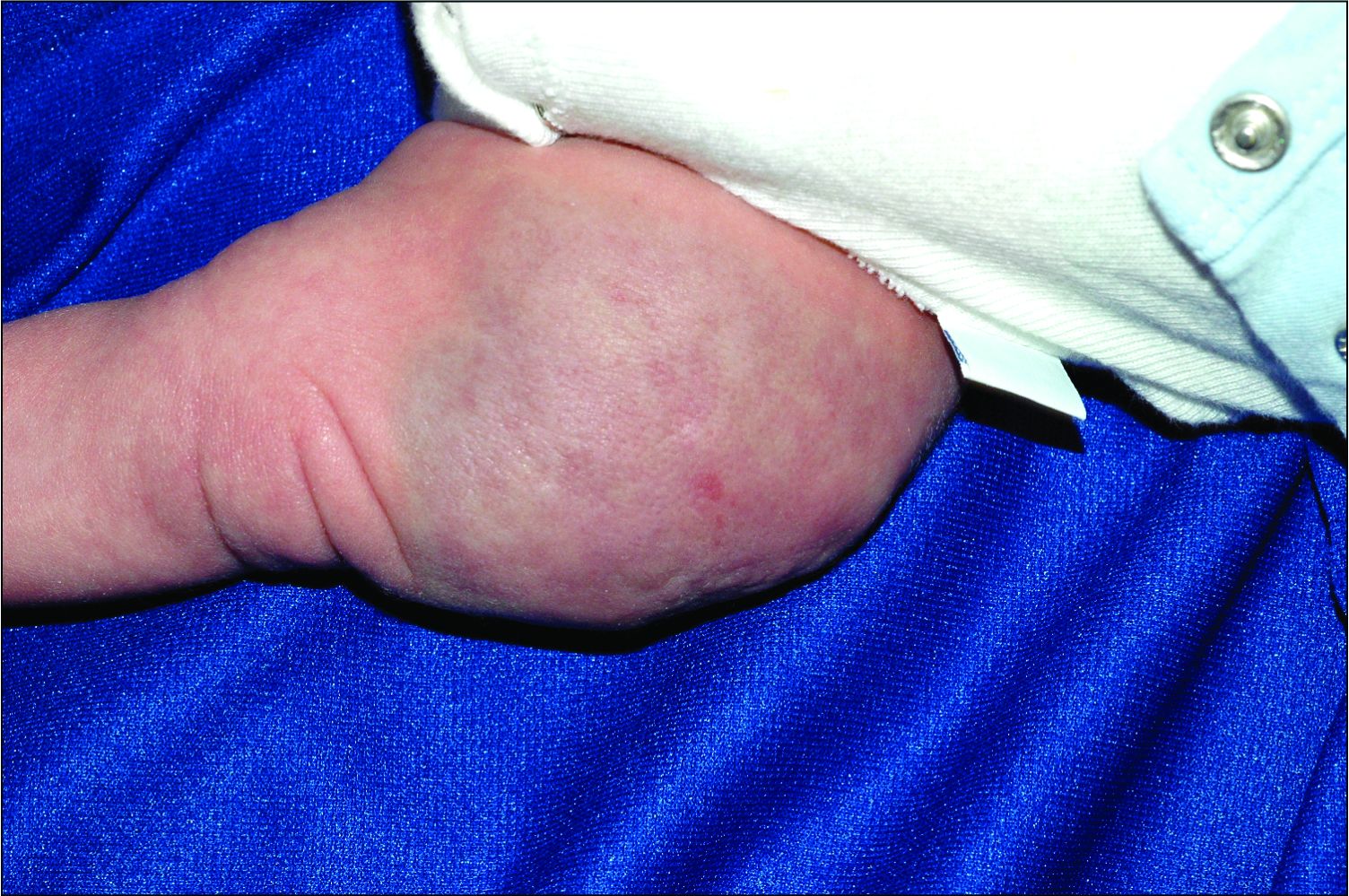
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
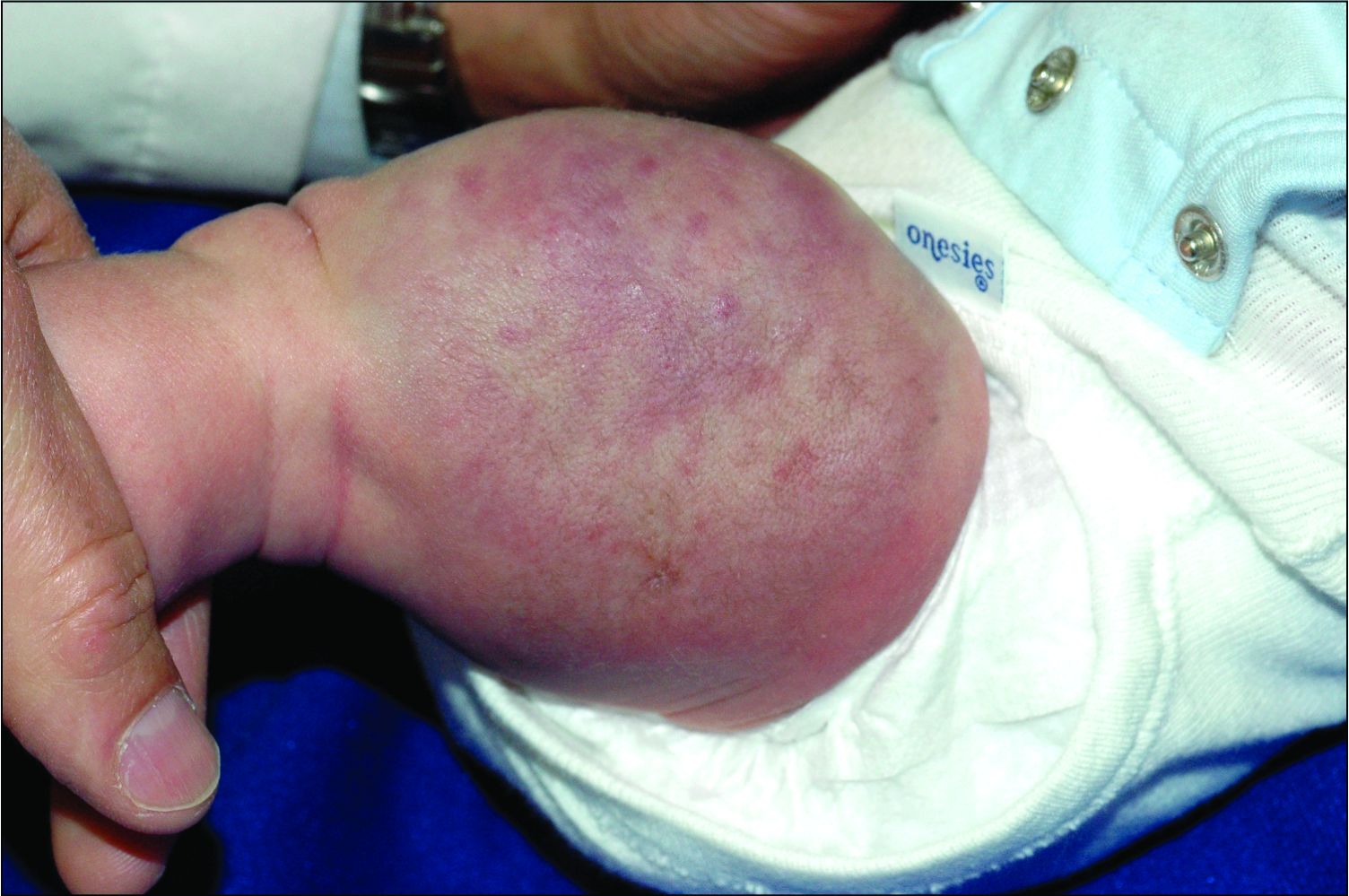
On physical exam, you see an infant with a mass of the left lower extremity. Close examination reveals an approximately 7 cm x 8 cm poorly defined mass with overlying central erythematous to violaceous color of the left anterior upper leg with a lumpy texture. The lesion is moderately firm and mildly tender on palpation.
A young girl presents with ‘itchy, rashy’ hands
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
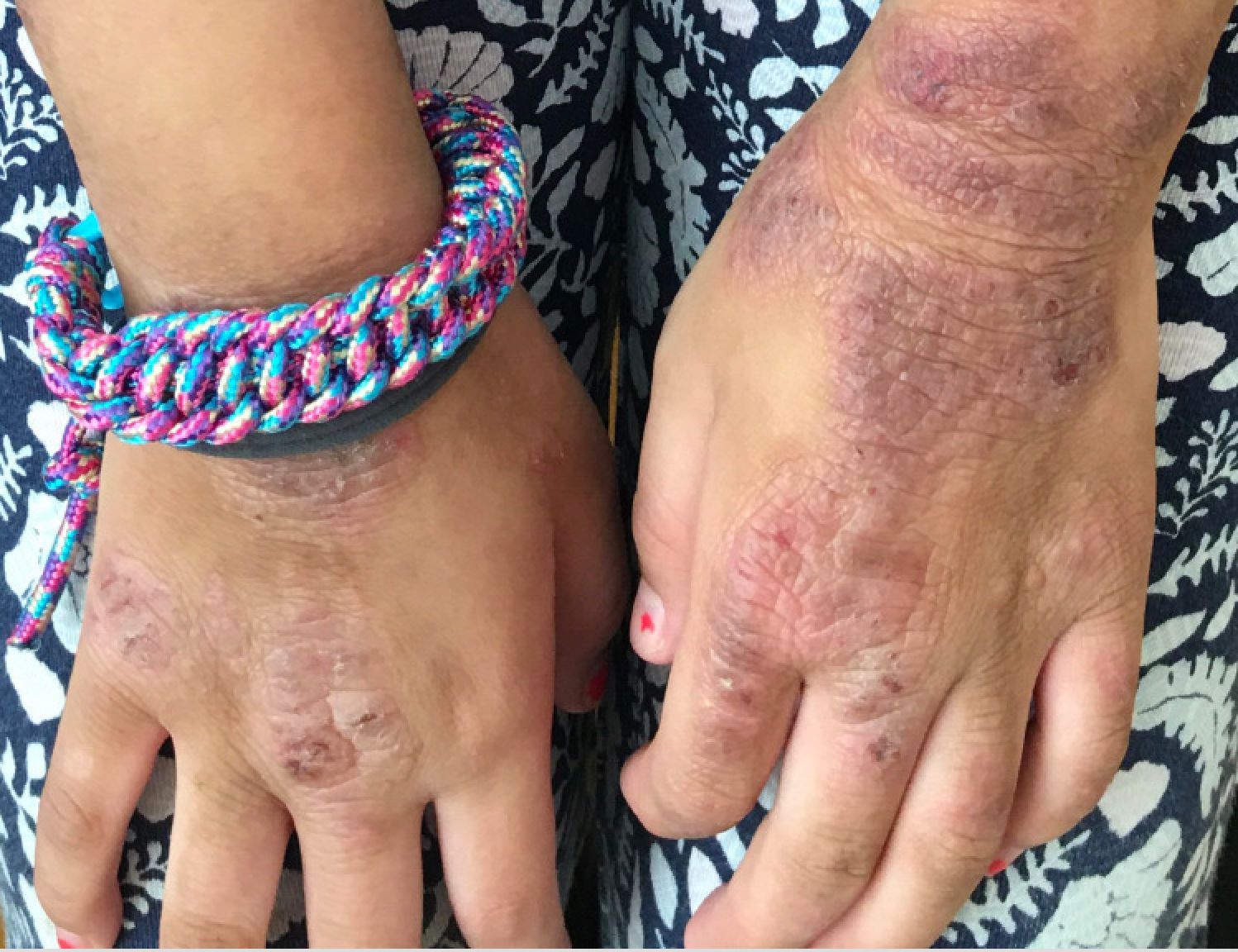
Examination findings of the bilateral hands and wrists demonstrate plaques of erythema, lichenification, and scale of the dorsal surfaces of the hands and digits. Closer inspection reveals fissuring and erythematous crust of the affected skin but normal nails. The rest of the skin exam is unremarkable.
An 11-year-old female with a 3-year history of alopecia
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at [email protected].
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
An 11-year-old female is seen in clinic with a 3-year history of alopecia. The patient recently immigrated to the United States from Afghanistan. Prior to immigrating, she was evaluated for "scarring alopecia" and had been treated with oral and topical steroids as well as oral and topical antifungals. When active, she had itching and tenderness. She is not actively losing any hair at this time, but she has not regrown any of her hair. The patient has no family members with alopecia. She reports some burning pain and itching of her scalp, and denies any muscle pain or weakness or sun sensitivity.
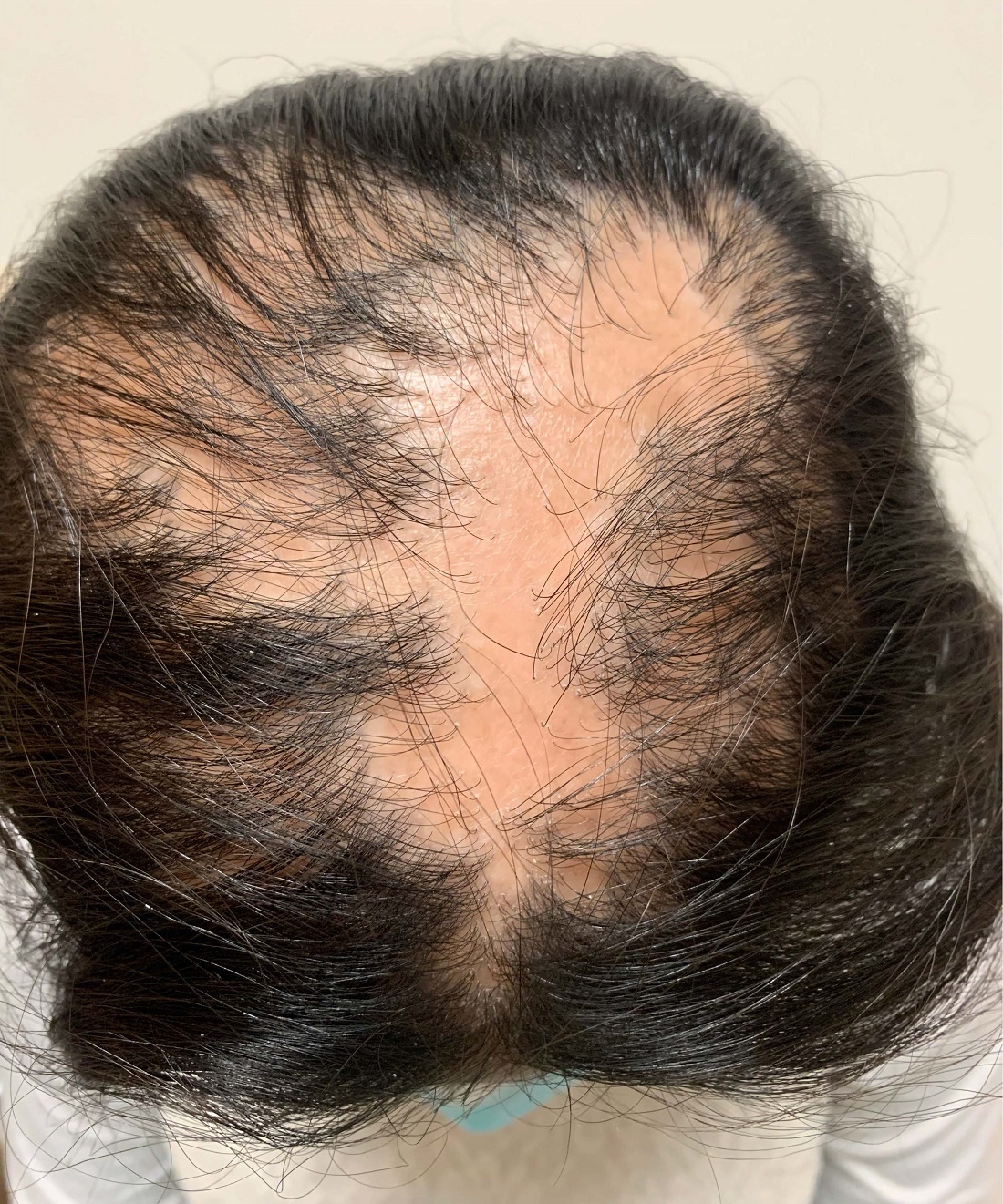
On physical exam, you see 50% loss of hair on the superior scalp with preservation of the anterior hair line. Patches of hair can be seen throughout, with segments of smooth-skinned alopecia, without pustules. There is a loss of the follicle pattern in scarred areas, and magnification or "dermoscopy" shows perifollicular erythema and scaling at the border of the affected scalp. Labs are all within normal limits. Bacterial and fungal cultures of the scalp do not grow organisms.