User login
Nail Unit Squamous Cell Carcinoma: Updates on Diagnosis, Surgical Approach, and the Use of Mohs Micrographic Surgery
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.


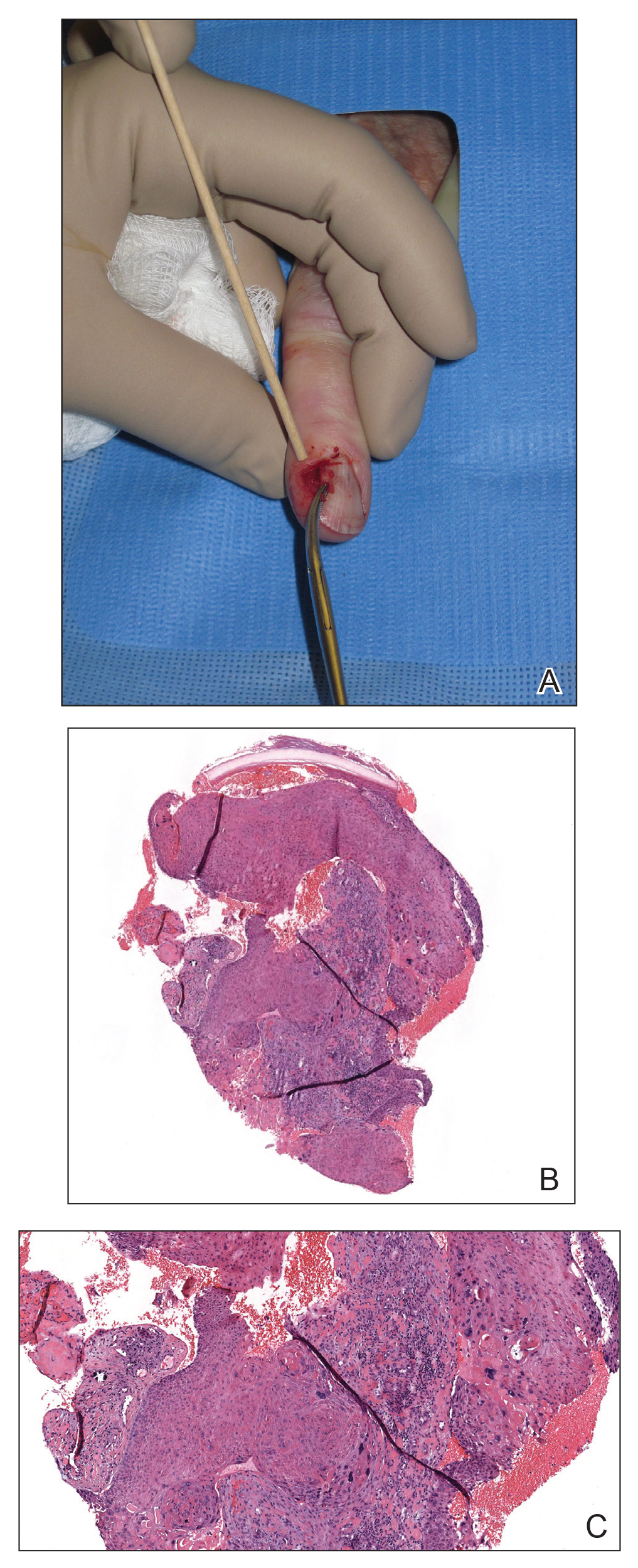
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
Nail unit squamous cell carcinoma (NSCC) is a malignant neoplasm that can arise from any part of the nail unit. Diagnosis often is delayed due to its clinical presentation mimicking benign conditions such as onychomycosis, warts, and paronychia. Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion. It is imperative for dermatologists who are early in their training to recognize this entity and refer for treatment. Many approaches have been used to treat NSCC, including wide local excision, digital amputation, cryotherapy, topical modalities, and recently Mohs micrographic surgery (MMS). This article provides an overview of the clinical presentation and diagnosis of NSCC, the role of human papillomavirus (HPV) in NSCC pathogenesis, and the evidence supporting surgical management.
NSCC Clinical Presentation and Diagnosis
Nail unit squamous cell carcinoma is a malignant neoplasm that can arise from any part of the nail unit including the nail bed, matrix, groove, and nail fold.1 Although NSCC is the most common malignant nail neoplasm, its diagnosis often is delayed partly due to the clinical presentation of NSCC mimicking benign conditions such as onychomycosis, warts, and paronychia.2,3 Nail unit SCC most commonly is mistaken for verruca vulgaris, and thus it is important to exclude malignancy in nonresolving verrucae of the fingernails or toenails. Another reason for a delay in the diagnosis is the painless and often asymptomatic presentation of this tumor, which keeps patients from seeking care.4 While evaluating a subungual lesion, dermatologists should keep in mind red flags that would prompt a biopsy to rule out NSCC (Table 1), including chronic nonhealing lesions, nail plate nodularity, known history of infection with HPV types 16 and 18, history of radiation or arsenic exposure, and immunosuppression. Table 2 lists the differential diagnosis of a persisting or nonhealing subungual tumor.
Nail unit SCC has a low rate of metastasis; however, a delayed diagnosis often can result in local destruction and bone invasion.5 Based on several reports, NSCC more commonly is found in middle-aged and older individuals, has a male predilection, and more often is seen on fingernails than toenails.1,2,6 Figure A shows an example of the clinical presentation of NSCC affecting the right thumb.
Although there often is a delay in the presentation and biopsy of NSCC, no correlation has been observed between time to biopsy and rate of disease invasion and recurrence.7 Nevertheless, Starace et al7 noted that a low threshold for biopsy of nail unit lesions is necessary. It is recommended to perform a deep shave or a nail matrix biopsy, especially if matrical involvement is suspected.8 Patients should be closely followed after a diagnosis of NSCC is made, especially if they are immunocompromised or have genetic skin cancer syndromes, as multiple NSCCs can occur in the same individual.9 For instance, one report discussed a patient with xeroderma pigmentosum who developed 3 separate NSCCs. Interestingly, in this patient, the authors suspected HPV as a cause for the field cancerization, as 2 of 3 NSCCs were noted on initial histopathology to have arisen from verrucae.10
Histologic Features
A biopsy from an NSCC tumor shows features similar to cutaneous SCC in the affected areas (ie, nail bed, nail matrix, nail groove, nail fold). Characteristic histologic findings include tongues or whorls of atypical squamous epithelium that invade deeply into the dermis.11 The cells appear as atypical keratinocytes, exhibit distinct intracellular bridges, and possess hyperchromatic and pleomorphic nuclei with dyskeratosis and keratin pearls within the dermis.12 Immunoperoxidase staining for cytokeratin AE1/AE3 can be helpful to confirm the diagnosis and assess whether the depth of invasion involves the bone.13 Figures B and C demonstrate the histopathology of NSCC biopsied from the tumor shown in Figure A.
Role of HPV in NSCC Pathogenesis
There is no clear pathogenic etiology for NSCC; however, there have been some reports of HPV as a risk factor. Shimizu et al14 reviewed 136 cases of HPV-associated NSCC and found that half of the cases were associated with high-risk HPV. They also found that 24% of the patients with NSCC had a history of other HPV-associated diseases. As such, the authors hypothesized that there is a possibility for genitodigital HPV transmission and that NSCC could be a reservoir for sexually transmitted high-risk HPV.14 Other risk factors are radiation exposure, chemical insult, and chronic trauma.15 The higher propensity for fingernails likely is reflective of the role of UV light exposure and infection with HPV in the development of these tumors.14,15
Several nonsurgical approaches have been suggested to treat NSCC, including topical agents, cryotherapy, CO2 laser, and photodynamic therapy.3,16 Unfortunately, there are no large case series to demonstrate the cure rate or effectiveness of these methods.17 In one study, the authors did not recommend use of photodynamic therapy or topical modalities such as imiquimod cream 5% or fluorouracil cream 5% as first-line treatments of NSCC due to the difficulty in ensuring complete treatment of the sulci of the lateral and proximal nail folds.18
More evidence in the literature supports surgical approaches, including wide local excision, MMS, and digital amputation. Clinicians should consider relapse rates and the impact on digital functioning when choosing a surgical approach.
For wide local excisions, the most common approach is en bloc excision of the nail unit including the lateral nail folds, the proximal nail fold, and the distal nail fold. The excision starts with a transverse incision on the base of the distal phalanx, which is then prolonged laterally and distally to the distal nail fold down to the bone. After the incision is made to the depth of the bone, the matrical horns are destroyed by electrocoagulation, and the defect is closed either by a full-thickness skin graft or secondary intent.19
Topin-Ruiz et al19 followed patients with biopsy-proven NSCC without bone invasion who underwent en bloc excision followed by full-thickness skin graft. In their consecutive series of 55 patients with 5 years of follow-up, the rate of recurrence was only 4%. There was a low rate of complications including graft infection, delayed wound healing, and severe pain in a small percentage of patients. They also reported a high patient satisfaction rate.19 Due to the low recurrence rate, this study suggested that total excision of the nail unit followed by a full-thickness skin graft is a safe and efficient treatment of NSCC without bone involvement. Similarly, in another case series, wide local excision of the entire nail apparatus had a relapse rate of only 5%, in contrast to partial excision of the nail unit with a relapse of 56%.20 These studies suggest that wide nail unit excision is an acceptable and effective approach; however, in cases in which invasion cannot be ruled out, histologic clearance would be a reasonable approach.21 As such, several case series demonstrated the merits of MMS for NSCC. de Berker et al22 reported 8 patients with NSCC treated using slow MMS and showed tumor clearance after a mean of 3 stages over a mean period of 6.9 days. In all cases, the wounds were allowed to heal by secondary intention, and the distal phalanx was preserved. During a mean follow-up period of 3.1 years, no recurrence was seen, and involved digits remained functional.22
Other studies tested the efficacy of MMS for NSCC. Young et al23 reported the outcomes of 14 NSCC cases treated with MMS. In their case series, they found that the mean number of MMS surgical stages required to achieve histologic clearance was 2, while the mean number of tissue sections was 4.23 All cases were allowed to heal by secondary intent with excellent outcomes, except for 1 patient who received primary closure of a small defect. They reported a 78% cure rate with an average time to recurrence of 47 months.23 In a series of 42 cases of NSCC treated with MMS, Gou et al17 noted a cure rate close to 93%. In their study, recurrences were observed in only 3 patients (7.1%). These recurrent cases were then successfully treated with another round of MMS.17 This study’s cure rate was comparable to the cure rate of MMS for SCC in other cutaneous areas. Goldminz and Bennett24 demonstrated a cure rate of 92% in their case series of 25 patients. Two patients developed recurrent disease and were treated again with MMS resulting in no subsequent recurrence. In this study, the authors allowed all defects to heal by secondary intention and found that there were excellent cosmetic and functional outcomes.24 Dika et al25 evaluated the long-term effectiveness of MMS in the treatment of NSCC, in particular its ability to reduce the number of digital amputations. Fifteen patients diagnosed with NSCC were treated with MMS as the first-line surgical approach and were followed for 2 to 5 years. They found that in utilizing MMS, they were able to avoid amputations in 13 of 15 cases with no recurrence in any of these tumors. Two cases, however, still required amputation of the distal phalanx.25
Although these studies suggest that MMS achieves a high cure rate ranging from 78% to 93%, it is not yet clear in the literature whether MMS is superior to wide local excision. More studies and clinical trials comparing these 2 surgical approaches should be performed to identify which surgical approach would be the gold standard for NSCC and which select cases would benefit from MMS as first-line treatment.
Final Thoughts
Nail unit SCC is one of the most common nail unit malignancies and can mimic several benign entities. Dermatologists who are early in their training should consider biopsy of subungual lesions with certain red flags (Table 1). It is important to diagnose NSCC for early intervention. Referral for wide local excision or MMS would be ideal. There are data in the literature supporting both surgical approaches as being effective; however, there are no trials comparing both approaches. Distal amputation should be considered as a last resort when wide local excision is not reasonable or when MMS fails to achieve clear margins, thereby reducing unnecessary amputations and patient morbidity.17
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
- Dika E, Starace M, Patrizi A, et al. Squamous cell carcinoma of the nail unit: a clinical histopathologic study and a proposal for classification. Dermatol Surg. 2019;45:365-370.
- Lee TM, Jo G, Kim M, et al. Squamous cell carcinoma of the nail unit: a retrospective review of 19 cases in Asia and comparative review of Western literature. Int J Dermatol. 2019;58:428-432.
- Tambe SA, Patil PD, Saple DG, et al. Squamous cell carcinoma of the nail bed: the great mimicker. J Cutan Aesthet Surg. 2017;10:59-60.
- Perrin C. Tumors of the nail unit. a review. part II: acquired localized longitudinal pachyonychia and masked nail tumors. Am J Dermatopathol. 2013;35:693-712.
- Li PF, Zhu N, Lu H. Squamous cell carcinoma of the nail bed: a case report. World J Clin Cases. 2019;7:3590-3594.
- Kaul S, Singal A, Grover C, et al. Clinical and histological spectrum of nail psoriasis: a cross-sectional study. J Cutan Pathol. 2018;45:824-830.
- Starace M, Alessandrini A, Dika E, et al. Squamous cell carcinoma of the nail unit. Dermatol Pract Concept. 2018;8:238-244.
- Kelly KJ, Kalani AD, Storrs S, et al. Subungual squamous cell carcinoma of the toe: working toward a standardized therapeutic approach. J Surg Educ. 2008;65:297-301.
- Ormerod E, De Berker D. Nail unit squamous cell carcinoma in people with immunosuppression. Br J Dermatol. 2015;173:701-712.
- Ventéjou S, Bagny K, Waldmeyer J, et al. Skin cancers in patients of skin phototype V or VI with xeroderma pigmentosum type C (XP-C): a retrospective study. Ann Dermatol Venereol. 2019;146:192-203.
- Mikhail GR. Subungual epidermoid carcinoma. J Am Acad Dermatol. 1984;11:291-298.
- Lecerf P, Richert B, Theunis A, et al. A retrospective study of squamous cell carcinoma of the nail unit diagnosed in a Belgian general hospital over a 15-year period. J Am Acad Dermatol. 2013;69:253-261.
- Kurokawa I, Senba Y, Kakeda M, et al. Cytokeratin expression in subungual squamous cell carcinoma. J Int Med Res. 2006;34:441-443.
- Shimizu A, Kuriyama Y, Hasegawa M, et al. Nail squamous cell carcinoma: a hidden high-risk human papillomavirus reservoir for sexually transmitted infections. J Am Acad Dermatol. 2019;81:1358-1370.
- Tang N, Maloney ME, Clark AH, et al. A retrospective study of nail squamous cell carcinoma at 2 institutions. Dermatol Surg. 2016;42(suppl 1):S8-S17.
- An Q, Zheng S, Zhang L, et al. Subungual squamous cell carcinoma treated by topical photodynamic therapy. Chin Med J (Engl). 2020;133:881-882.
- Gou D, Nijhawan RI, Srivastava D. Mohs micrographic surgery as the standard of care for nail unit squamous cell carcinoma. Dermatol Surg. 2020;46:725-732.
- Dika E, Fanti PA, Patrizi A, et al. Mohs surgery for squamous cell carcinoma of the nail unit: 10 years of experience. Dermatol Surg. 2015;41:1015-1019.
- Topin-Ruiz S, Surinach C, Dalle S, et al. Surgical treatment of subungual squamous cell carcinoma by wide excision of the nail unit and skin graft reconstruction: an evaluation of treatment efficiency and outcomes. JAMA Dermatol. 2017;153:442-448.
- Dalle S, Depape L, Phan A, et al. Squamous cell carcinoma of the nail apparatus: clinicopathological study of 35 cases. Br J Dermatol. 2007;156:871-874.
- Zaiac MN, Weiss E. Mohs micrographic surgery of the nail unit and squamous cell carcinoma. Dermatol Surg. 2001;27:246-251.
- de Berker DA, Dahl MG, Malcolm AJ, et al. Micrographic surgery for subungual squamous cell carcinoma. Br J Plast Surg. 1996;49:414-419.
- Young LC, Tuxen AJ, Goodman G. Mohs’ micrographic surgery as treatment for squamous dysplasia of the nail unit. Australas J Dermatol. 2012;53:123-127.
- Goldminz D, Bennett RG. Mohs micrographic surgery of the nail unit. J Dermatol Surg Oncol. 1992;18:721-726.
- Dika E, Piraccini BM, Balestri R, et al. Mohs surgery for squamous cell carcinoma of the nail: report of 15 cases. our experience and a long-term follow-up. Br J Dermatol. 2012;167:1310-1314.
Resident Pearls
- The diagnosis of nail unit squamous cell carcinoma often is delayed due to its clinical presentation, which frequently mimics benign nail conditions.
- Treatment includes wide local excision, Mohs micrographic surgery, digital amputation, cryotherapy, and topical modalities.

PD-1 Signaling in Extramammary Paget Disease
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.

Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.
Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.
Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
Resident Pearls
- Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts, while secondary EMPD is an extension of malignant cells from an underlying internal neoplasm.
- Surgical margin clearance in EMPD often is problematic, with high recurrence rates indicating the need for additional treatment modalities.
- Programmed cell death receptor 1 (PD-1) signaling can serve as a potential target in EMPD. Further studies and clinical trials are needed to test the efficacy of PD-1 inhibitors in unresectable or invasive/metastatic EMPD.
The DNA Mismatch Repair System in Sebaceous Tumors: An Update on the Genetics and Workup of Muir-Torre Syndrome
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
Resident Pearls
- When patients present with a solitary sebaceous tumor, there is a high likelihood they have Muir-Torre syndrome (MTS) and thus are at a high risk to develop visceral malignancies.
- It is important to perform further testing using immunohistochemistry for DNA mismatch repair proteins and microsatellite instability gene analysis in some cases to confirm the diagnosis of MTS and to perform the appropriate cancer screening tests.