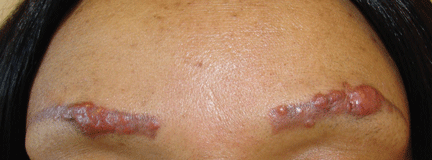
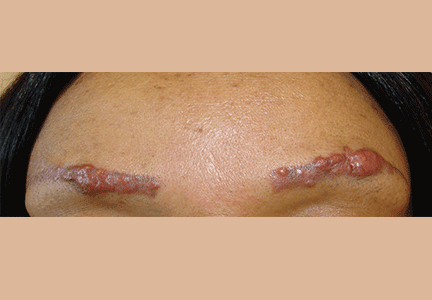
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
Figure 1. The patient had reddish papules and nodules on the eye-brows as well as on the lips. These areas had undergone cosmetic tattooing 10 years earlier.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
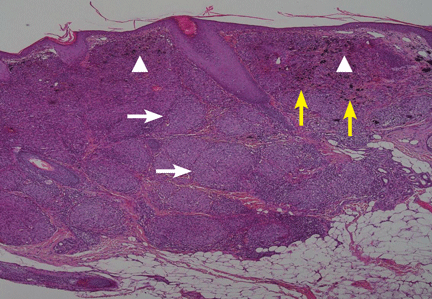
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Figure 2. Dermal noncaseating granulomas of epitheloid cells were seen (white arrows), with polarizable foreign material (yellow arrows)and pigment granules (arrowheads) (hematoxylin and eosin, x 10).
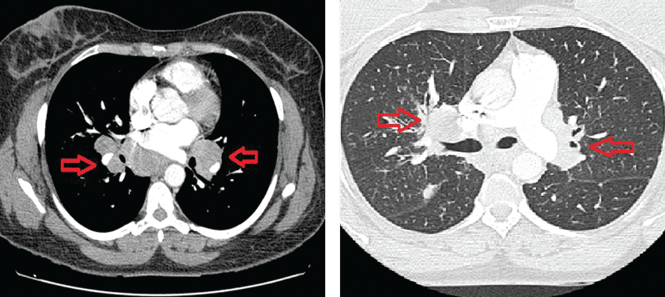
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Figure 3. Computed tomography without contrast (left) shows multiple areas of lymphadenopathy in the mediastinum and lung hila (red arrows). The “lung window” view (right) shows a perilymphatic micronodular interstitial pattern, with thickened and nodular bronchial walls; the red arrows indicate areas of hilar lymphadenopathy.
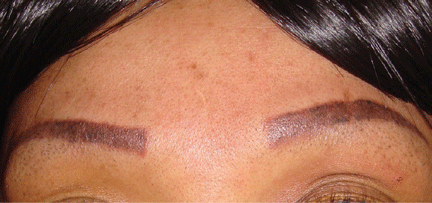
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
Figure 4. After treatment.
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
References
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
Laura Miguel-Gómez, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Basil Sido-Ahmed, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Carmen Moreno García Del Real, MD, PhD Department of Pathological Anatomy, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén-Olasolo, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Laura Miguel-Gómez, MD, Department of Dermatology, Ramón y Cajal Hospital, Carretera de Colmenar Viejo km 9.1, 28034 Madrid, Spain; e-mail: [email protected]
Laura Miguel-Gómez, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Basil Sido-Ahmed, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Carmen Moreno García Del Real, MD, PhD Department of Pathological Anatomy, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén-Olasolo, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Laura Miguel-Gómez, MD, Department of Dermatology, Ramón y Cajal Hospital, Carretera de Colmenar Viejo km 9.1, 28034 Madrid, Spain; e-mail: [email protected]
Author and Disclosure Information
Laura Miguel-Gómez, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Basil Sido-Ahmed, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Carmen Moreno García Del Real, MD, PhD Department of Pathological Anatomy, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén-Olasolo, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Laura Miguel-Gómez, MD, Department of Dermatology, Ramón y Cajal Hospital, Carretera de Colmenar Viejo km 9.1, 28034 Madrid, Spain; e-mail: [email protected]
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
Figure 1. The patient had reddish papules and nodules on the eye-brows as well as on the lips. These areas had undergone cosmetic tattooing 10 years earlier.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Figure 2. Dermal noncaseating granulomas of epitheloid cells were seen (white arrows), with polarizable foreign material (yellow arrows)and pigment granules (arrowheads) (hematoxylin and eosin, x 10).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Figure 3. Computed tomography without contrast (left) shows multiple areas of lymphadenopathy in the mediastinum and lung hila (red arrows). The “lung window” view (right) shows a perilymphatic micronodular interstitial pattern, with thickened and nodular bronchial walls; the red arrows indicate areas of hilar lymphadenopathy.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
Figure 4. After treatment.
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
A 42-year-old woman presented with painless lesions on her eyebrows that had been progressively growing for the past 3 months. Inspection and palpation revealed reddish papules and nodules on both eyebrows (Figure 1) and on the lips. The remainder of the physical examination was normal.
Figure 1. The patient had reddish papules and nodules on the eye-brows as well as on the lips. These areas had undergone cosmetic tattooing 10 years earlier.
The patient said she had undergone cosmetic tattooing of her eyebrows and lips 10 years previously. She had no other significant medical history.
Her complete blood cell count and biochemistry and immunologic test panels were normal except for a high angiotensin-converting enzyme level.
Suspecting that the lesions represented sarcoidal infiltration of the tattoos, we obtained a biopsy. Histopathologic study showed multiple dermal noncaseating granulomas of epithelioid cells, together with polarizable foreign material and pigmented granules (Figure 2).
Figure 2. Dermal noncaseating granulomas of epitheloid cells were seen (white arrows), with polarizable foreign material (yellow arrows)and pigment granules (arrowheads) (hematoxylin and eosin, x 10).
Thoracic multislice computed tomography revealed multiple areas of lymphadenopathy in the mediastinum and both lung hila, with a perilymphatic micronodular interstitial pattern and with thickened and nodular bronchial walls (Figure 3). The patient’s condition was diagnosed as systemic sarcoidosis presenting as sarcoidal infiltration of the tattooed areas.
Figure 3. Computed tomography without contrast (left) shows multiple areas of lymphadenopathy in the mediastinum and lung hila (red arrows). The “lung window” view (right) shows a perilymphatic micronodular interstitial pattern, with thickened and nodular bronchial walls; the red arrows indicate areas of hilar lymphadenopathy.
Treatment with oral prednisone 20 mg daily and monthly intralesional injections of triamcinolone 12 ng/mL in the eyebrows and lips resulted in clinical improvement after 3 months (Figure 4).
Figure 4. After treatment.
CUTANEOUS SARCOIDOSIS
Sarcoidosis is a multisystemic granulomatous disease characterized by hyperactivity of the cellular immune system. It usually appears between ages 25 and 35 and is more severe in African Americans.1 About one-third of patients with systemic sarcoidosis develop cutaneous lesions, and these may be the first sign of this disease.
Specific cutaneous lesions contain noncaseating granulomas. They manifest as reddish-brown papules, plaques, and macules and may appear over scars and tattoos.2,3 Other, nonspecific manifestations include erythema nodosum, erythema multiforme, nail clubbing, and Sweet syndrome.4
Diascopy and dermoscopy may be useful in diagnosing cutaneous sarcoidosis. The lesions typically have a characteristic yellowish-brown (“apple-jelly”) color.5
Sarcoidosis is a diagnosis of exclusion. It is suspected clinically, and histologic, radiologic, and analytical tests are indicated. Laboratory evaluation may show an elevated serum angiotensin-converting enzyme level, but this finding alone is not sensitive enough to be useful for diagnosis.6
The treatment and prognosis of skin sarcoidosis depend on the degree of systemic involvement. Localized cutaneous lesions can respond to intralesional corticosteroid injections and tacrolimus 0.1% ointment.7 However, if there is systemic involvement, low-dose prednisolone and weekly methotrexate should be started. This combination has few adverse effects.8 Recently, improvement of cutaneous and systemic sarcoidosis has been observed with agents that block tumor necrosis factor alpha.4
References
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
References
1. English JC 3rd, Patel PJ, Greer KE. Sarcoidosis. J Am Acad Dermatol 2001; 44:725–743.
2. Antonovich DD, Callen JP. Development of sarcoidosis in cosmetic tattoos. Arch Dermatol 2005; 141:869–872.
3. Anolik R, Mandal R, Franks AG Jr. Sarcoidal tattoo granuloma. Dermatol Online J 2010; 16:19.
4. Su Ö, Onsun N, Topukçu B, Özçelik HK, Çakıter AU, Büyükpınarbaşılı N. Disseminated scar sarcoidosis may predict pulmonary involvement in sarcoidosis. Acta Dermatovenerol Alp Pannonica Adriat 2013; 22:71–74.
5. Hunt RD, Gonzalez ME, Robinson M, Meehan SA, Franks AG Jr. Ulcerative sarcoidosis. Dermatol Online J 2012; 18:29.
6. Baughman RP, Culver DA, Judson MA. A concise review of pulmonary sarcoidosis. Am J Respir Crit Care Med 2011; 183:573–581.
7. Landers MC, Skokan M, Law S, Storrs FJ. Cutaneous and pulmonary sarcoidosis in association with tattoos. Cutis 2005; 75:44–48.
8. Nagai S, Yokomatsu T, Tanizawa K, et al. Treatment with methotrexate and low-dose corticosteroids in sarcoidosis patients with cardiac lesions. Intern Med 2014; 53:427–433.
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
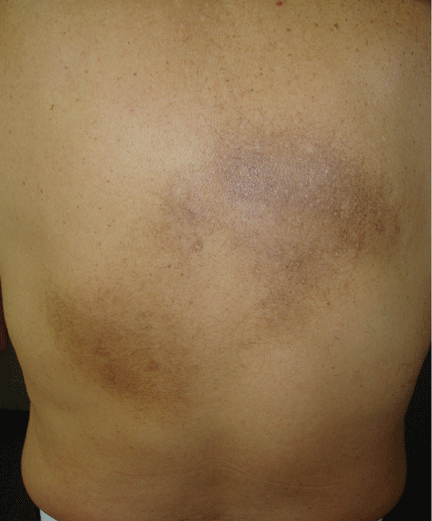
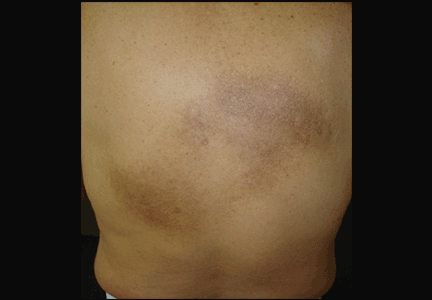
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
Figure 1. Two gray-brown ovoid patches on the back and on the right infrascapular region were associated with chronic itch.
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
References
Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed2011; 9:353–358.
Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol2011; 91:356–357.
Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol1995; 32:287–289.
Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol2005; 52:1085–1087.
Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol1999; 12:215–221.
Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol2010; 24:730–732.
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Mayte Truchuelo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Sergio Vañó-Galván, PhD, Department of Dermatology, Ramón y Cajal Hospital, University of Alcalá, Carretera Colmenar Viejo, km 9.100, 28034 Madrid, Spain; e-mail: [email protected]
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Mayte Truchuelo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Sergio Vañó-Galván, PhD, Department of Dermatology, Ramón y Cajal Hospital, University of Alcalá, Carretera Colmenar Viejo, km 9.100, 28034 Madrid, Spain; e-mail: [email protected]
Author and Disclosure Information
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Mayte Truchuelo, MD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Address: Sergio Vañó-Galván, PhD, Department of Dermatology, Ramón y Cajal Hospital, University of Alcalá, Carretera Colmenar Viejo, km 9.100, 28034 Madrid, Spain; e-mail: [email protected]
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
Figure 1. Two gray-brown ovoid patches on the back and on the right infrascapular region were associated with chronic itch.
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
A 47-year-old man had had a chronic itch on his back for 2 years. He had no history of trauma to the site, nor did he recall applying topical products to that area.
He was otherwise healthy. He worked as an electrician and said he occasionally experienced cervical and back pain while working.
An examination revealed two grayish-brown ovoid patches on the upper back, each 5 cm to 7 cm in diameter (Figure 1).
DIAGNOSIS: NOTALGIA PARESTHETICA
Figure 1. Two gray-brown ovoid patches on the back and on the right infrascapular region were associated with chronic itch.
Chronic, brown-gray, itching patches on the back in an adult patient are characteristic of notalgia paresthetica.
Conditions that may be included in the differential diagnosis but that do not match the presentation in this patient include the following:
Cutaneous sarcoidosis, which may exhibit several morphologies, but itching would be unusual
Chronic discoid lupus erythematosus, characterized by scarring and atrophic plaques, but mainly on the face and scalp
Contact dermatitis, an itchy eczematous condition, characterized by scaly erythematous plaques
Lichen amyloidosis, a variant of cutaneous amyloidosis characterized by the deposition of amyloid or amyloid-like proteins in the dermis, resulting in red-brown hyperkeratotic lichenoid papules, usually on the pretibial surfaces.
CAUSES AND MANAGEMENT
Notalgia paresthetica is a neuropathic syndrome of the skin of the middle of the back characterized by localized pruritus.1–3 Although common, it often goes undiagnosed.1,3,4 It tends to be chronic, with periodic remissions and exacerbations.
Notalgia paresthetica is thought to be a sensory neuropathy and may result from compression of the posterior rami of spinal nerve segments T2 to T6. Slight degenerative changes are often but not always observed, and their clinical significance is uncertain.1,2,4 The condition affects people of all races and both sexes, usually adults ages 40 to 80.
Clinically, it presents as localized pruritus on the back, usually within the dermatomes T2 to T6.5 Examination reveals a hyperpigmented patch, sometimes with excoriations.5
Diagnosis is based on clinical findings. Laboratory tests are not useful. Imaging is not needed, but magnetic resonance imaging and evaluation by an orthopedic surgeon are appropriate when there is chronic focal pain. Skin biopsy is usually not necessary, although it may be useful in some patients to exclude other conditions. When biopsy is done, macular amyloidosis or postinflammatory hyperpigmentation is seen.
Treatment is difficult. Topical steroids and oral antihistamines are usually ineffective,5 but topical capsaicin may provide temporary relief.3 The most recommended treatment in patients with notalgia paresthetica and underlying spinal disease is evaluation and conservative management of the spinal disease, including progressive exercise and rehabilitation.2 Other therapies include oxcarbazepine, gabapentin, transcutaneous electrical nerve stimulation, phototherapy,6 and botulinum toxin injection.
TREATMENT OF OUR PATIENT
In our patient, an orthopedic evaluation revealed cervicothoracic scoliosis. He underwent 6 months of conservative treatment under the care of his family physician and a dermatologist. Treatment consisted of exercise and rehabilitation for his scoliosis, and daily application of topical mometasone. The pain and itch gradual improved.
References
Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed2011; 9:353–358.
Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol2011; 91:356–357.
Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol1995; 32:287–289.
Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol2005; 52:1085–1087.
Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol1999; 12:215–221.
Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol2010; 24:730–732.
References
Pérez-Pérez LC. General features and treatment of notalgia paresthetica. Skinmed2011; 9:353–358.
Fleischer AB, Meade TJ, Fleischer AB. Notalgia paresthetica: successful treatment with exercises. Acta Derm Venereol2011; 91:356–357.
Wallengren J, Klinker M. Successful treatment of notalgia paresthetica with topical capsaicin: vehicle-controlled, double-blind, crossover study. J Am Acad Dermatol1995; 32:287–289.
Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol2005; 52:1085–1087.
Raison-Peyron N, Meunier L, Acevedo M, Meynadier J. Notalgia paresthetica: clinical, physiopathological and therapeutic aspects. A study of 12 cases. J Eur Acad Dermatol Venereol1999; 12:215–221.
Pérez-Pérez L, Allegue F, Fabeiro JM, Caeiro JL, Zulaica A. Notalgia paresthesica successfully treated with narrow-band UVB: report of five cases. J Eur Acad Dermatol Venereol2010; 24:730–732.
A 68-year-old man sought care in our emergency department for unilateral ptosis following superior and inferior right eyelid edema. The patient said that the edema had developed 3 days earlier and was getting worse each day; the ptosis was impairing his vision. The patient indicated that the edema was accompanied by mild burning in the right periocular region. His medical history included arterial hypertension, which was under control, and bilateral cataract surgery 5 years ago.
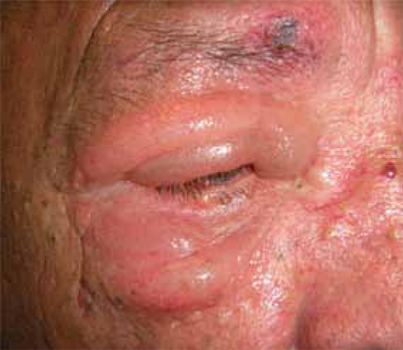
On examination, we noted superior and inferior nontender painless eyelid edema on the right eye, with no signs of acute inflammation (FIGURE). The patient also had right supraciliary folliculitis that was improving; the folliculitis had been treated 3 days earlier at a primary care facility.
We performed a complete ocular examination, including visual acuity (20/20 in both eyes) and found no other significant problems. Nor did the patient have a fever or any other systemic symptoms.
FIGURE 1 Edema of the upper and lower eyelid
The swelling was not accompanied by inflammation or fever, but it did impair the patient’s vision.
WHAT IS YOUR DIAGNOSIS? HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Noninflammatory eyelid edema
The fact that the patient’s eyelid was edematous (but not tender or red) and that he’d been scratching the area following a case of folliculitis prompted us to diagnose noninflammatory eyelid edema.
Noninflammatory eyelid edema—also called noninflammatory palpebral edema—is a relatively common disorder that usually occurs after some local irritation or microtrauma. In its early phase, patients start out with nonerythematous, painless edema of the upper eyelid, which eventually affects the lower eyelid.
FAST TRACK
Noninflammatory eyelid edema is a relatively common disorder that usually appears after some local irritation or microtrauma.
As was the case with our patient, this disorder is not associated with fever or any other systemic sign or symptom. Complete blood count and C-reactive protein will be negative, as no infection or systemic alteration is present.
Consider these infectious alternatives In cases like this one, be sure to differentiate between noninflammatory palpebral edema and other common but less benign ocular conditions. Among the infectious causes of unilateral edema to rule out:
Preseptal cellulitis is an infection that is localized between the skin and the orbital septum. This disorder usually appears as a tender erythematous eyelid edema with no proptosis and no pain with eye movement.1 It is usually accompanied by an elevated white blood cell (WBC) count. Blood cultures are rarely positive, but when they are, Haemophilus influenzae is the most frequent pathogen.2 Preseptal cellulitis is treated with broad-spectrum oral antibiotics (amoxicillin/ clavulanate) for at least 7 days.
Orbital cellulitis is an infection that affects the orbital extraocular structures. It can lead to blindness, so prompt diagnosis is critical.
Orbital cellulitis usually develops when a sinus infection spreads into the orbit.3 Suspect it in a patient with proptosis, orbital pain, tenderness, conjunctival chemosis, decreased vision, elevated intraocular pressure, and pain on eye movement. Also look for an elevated WBC count. Perform blood cultures to confirm the presence of a pathogen.
If you suspect orbital cellulitis, order an orbital computed tomography scan with contrast infusion, including axial and coronal views; it may reveal an infection of the soft tissue behind the orbital septum. Treat this condition with broad-spectrum intravenous antibiotics (ceftriaxone) for 1 week, followed by 2 weeks of oral antibiotics, completing a total of 21 days of antibiotics.
Hordeolum is a localized infection that occurs when a meibomian gland becomes blocked. It usually appears as a palpable subcutaneous nodule within the eyelid and requires treatment with warm compresses and a combination of antibiotics and steroid topical ointment (tobramycin and dexamethasone ophthalmic ointment 4 times a day for a week).4
Acute dacryoadenitis is a lacrimal gland infection that usually causes unilateral, painful swelling of the outer third of the upper lid. You may also see tearing and discharge.5 Look for an elevated WBC count; however, blood cultures are rarely positive. These patients must be referred to an ophthalmologist as treatment varies from symptomatic measures in viral cases (warm compresses and a nonsteroidal anti-inflammatory drug, such as ibuprofen) to hospitalization and oral or intravenous antibiotics (eg, amoxicillin/ clavulanate for at least 7 days), depending on the severity and origin of the infection.6
Ruling out other noninfectious disorders Noninfectious inflammatory diseases should also be part of the differential diagnosis, including:
Idiopathic orbital inflammation. Also known as orbital pseudotumor, this disorder is a nongranulomatous acute-to-subacute inflammatory disease with no systemic manifestations.7 It frequently involves the lacrimal gland and can cause abrupt pain, conjunctival edema, and lid edema. Other common signs are proptosis and ocular motility alterations, which the patient may report as diplopia and visual impairment.
This disorder can result from optic neuropathy, exudative retinal detachment, or uveitis. It is a diagnosis of exclusion and is often arrived at by seeing the patient respond to systemic corticosteroids. The specific drug and dose will vary based on disease severity.
Graves’ ophthalmopathy. This bilateral, asymmetric immunological disorder affects the conjunctiva, eyelids,8 extraocular muscles, lacrimal gland, and optic nerve. Although it is typically diagnosed clinically, thyroid hormone abnormalities (increased T4 and a drop in thyroid-stimulating hormone) help support the diagnosis. In the acute phase, it has to be treated with systemic steroids; if the condition becomes chronic, the complications may require surgery. Treatment of the underlying hyperthyroidism may, in some cases, worsen the ophthalmopathy.
One more consideration. Finally, consider a local allergic reaction in the eyelids,9 which can also cause painless palpebral edema. Expect to see erythematous edema and intense itching a few hours after exposure to the allergen. A cosmetic product is often the culprit. As you might expect, initial treatment calls for withdrawal of the offending product. Instruct the patient to apply a topical steroid ointment on the eyelid if the edema is intense.
FAST TRACK
The condition does not require any local or systemic treatment.
Good news for our patient Diagnosing noninflammatory eyelid edema requires keen observational skills and a knowledge of several alternative diagnoses. Fortunately for our patient, this benign condition did not require any local or systemic treatment. The edema resolved on its own in 6 days.
A 68-year-old man sought care in our emergency department for unilateral ptosis following superior and inferior right eyelid edema. The patient said that the edema had developed 3 days earlier and was getting worse each day; the ptosis was impairing his vision. The patient indicated that the edema was accompanied by mild burning in the right periocular region. His medical history included arterial hypertension, which was under control, and bilateral cataract surgery 5 years ago.
On examination, we noted superior and inferior nontender painless eyelid edema on the right eye, with no signs of acute inflammation (FIGURE). The patient also had right supraciliary folliculitis that was improving; the folliculitis had been treated 3 days earlier at a primary care facility.
We performed a complete ocular examination, including visual acuity (20/20 in both eyes) and found no other significant problems. Nor did the patient have a fever or any other systemic symptoms.
FIGURE 1 Edema of the upper and lower eyelid
The swelling was not accompanied by inflammation or fever, but it did impair the patient’s vision.
WHAT IS YOUR DIAGNOSIS? HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Noninflammatory eyelid edema
The fact that the patient’s eyelid was edematous (but not tender or red) and that he’d been scratching the area following a case of folliculitis prompted us to diagnose noninflammatory eyelid edema.
Noninflammatory eyelid edema—also called noninflammatory palpebral edema—is a relatively common disorder that usually occurs after some local irritation or microtrauma. In its early phase, patients start out with nonerythematous, painless edema of the upper eyelid, which eventually affects the lower eyelid.
FAST TRACK
Noninflammatory eyelid edema is a relatively common disorder that usually appears after some local irritation or microtrauma.
As was the case with our patient, this disorder is not associated with fever or any other systemic sign or symptom. Complete blood count and C-reactive protein will be negative, as no infection or systemic alteration is present.
Consider these infectious alternatives In cases like this one, be sure to differentiate between noninflammatory palpebral edema and other common but less benign ocular conditions. Among the infectious causes of unilateral edema to rule out:
Preseptal cellulitis is an infection that is localized between the skin and the orbital septum. This disorder usually appears as a tender erythematous eyelid edema with no proptosis and no pain with eye movement.1 It is usually accompanied by an elevated white blood cell (WBC) count. Blood cultures are rarely positive, but when they are, Haemophilus influenzae is the most frequent pathogen.2 Preseptal cellulitis is treated with broad-spectrum oral antibiotics (amoxicillin/ clavulanate) for at least 7 days.
Orbital cellulitis is an infection that affects the orbital extraocular structures. It can lead to blindness, so prompt diagnosis is critical.
Orbital cellulitis usually develops when a sinus infection spreads into the orbit.3 Suspect it in a patient with proptosis, orbital pain, tenderness, conjunctival chemosis, decreased vision, elevated intraocular pressure, and pain on eye movement. Also look for an elevated WBC count. Perform blood cultures to confirm the presence of a pathogen.
If you suspect orbital cellulitis, order an orbital computed tomography scan with contrast infusion, including axial and coronal views; it may reveal an infection of the soft tissue behind the orbital septum. Treat this condition with broad-spectrum intravenous antibiotics (ceftriaxone) for 1 week, followed by 2 weeks of oral antibiotics, completing a total of 21 days of antibiotics.
Hordeolum is a localized infection that occurs when a meibomian gland becomes blocked. It usually appears as a palpable subcutaneous nodule within the eyelid and requires treatment with warm compresses and a combination of antibiotics and steroid topical ointment (tobramycin and dexamethasone ophthalmic ointment 4 times a day for a week).4
Acute dacryoadenitis is a lacrimal gland infection that usually causes unilateral, painful swelling of the outer third of the upper lid. You may also see tearing and discharge.5 Look for an elevated WBC count; however, blood cultures are rarely positive. These patients must be referred to an ophthalmologist as treatment varies from symptomatic measures in viral cases (warm compresses and a nonsteroidal anti-inflammatory drug, such as ibuprofen) to hospitalization and oral or intravenous antibiotics (eg, amoxicillin/ clavulanate for at least 7 days), depending on the severity and origin of the infection.6
Ruling out other noninfectious disorders Noninfectious inflammatory diseases should also be part of the differential diagnosis, including:
Idiopathic orbital inflammation. Also known as orbital pseudotumor, this disorder is a nongranulomatous acute-to-subacute inflammatory disease with no systemic manifestations.7 It frequently involves the lacrimal gland and can cause abrupt pain, conjunctival edema, and lid edema. Other common signs are proptosis and ocular motility alterations, which the patient may report as diplopia and visual impairment.
This disorder can result from optic neuropathy, exudative retinal detachment, or uveitis. It is a diagnosis of exclusion and is often arrived at by seeing the patient respond to systemic corticosteroids. The specific drug and dose will vary based on disease severity.
Graves’ ophthalmopathy. This bilateral, asymmetric immunological disorder affects the conjunctiva, eyelids,8 extraocular muscles, lacrimal gland, and optic nerve. Although it is typically diagnosed clinically, thyroid hormone abnormalities (increased T4 and a drop in thyroid-stimulating hormone) help support the diagnosis. In the acute phase, it has to be treated with systemic steroids; if the condition becomes chronic, the complications may require surgery. Treatment of the underlying hyperthyroidism may, in some cases, worsen the ophthalmopathy.
One more consideration. Finally, consider a local allergic reaction in the eyelids,9 which can also cause painless palpebral edema. Expect to see erythematous edema and intense itching a few hours after exposure to the allergen. A cosmetic product is often the culprit. As you might expect, initial treatment calls for withdrawal of the offending product. Instruct the patient to apply a topical steroid ointment on the eyelid if the edema is intense.
FAST TRACK
The condition does not require any local or systemic treatment.
Good news for our patient Diagnosing noninflammatory eyelid edema requires keen observational skills and a knowledge of several alternative diagnoses. Fortunately for our patient, this benign condition did not require any local or systemic treatment. The edema resolved on its own in 6 days.
A 68-year-old man sought care in our emergency department for unilateral ptosis following superior and inferior right eyelid edema. The patient said that the edema had developed 3 days earlier and was getting worse each day; the ptosis was impairing his vision. The patient indicated that the edema was accompanied by mild burning in the right periocular region. His medical history included arterial hypertension, which was under control, and bilateral cataract surgery 5 years ago.
On examination, we noted superior and inferior nontender painless eyelid edema on the right eye, with no signs of acute inflammation (FIGURE). The patient also had right supraciliary folliculitis that was improving; the folliculitis had been treated 3 days earlier at a primary care facility.
We performed a complete ocular examination, including visual acuity (20/20 in both eyes) and found no other significant problems. Nor did the patient have a fever or any other systemic symptoms.
FIGURE 1 Edema of the upper and lower eyelid
The swelling was not accompanied by inflammation or fever, but it did impair the patient’s vision.
WHAT IS YOUR DIAGNOSIS? HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Noninflammatory eyelid edema
The fact that the patient’s eyelid was edematous (but not tender or red) and that he’d been scratching the area following a case of folliculitis prompted us to diagnose noninflammatory eyelid edema.
Noninflammatory eyelid edema—also called noninflammatory palpebral edema—is a relatively common disorder that usually occurs after some local irritation or microtrauma. In its early phase, patients start out with nonerythematous, painless edema of the upper eyelid, which eventually affects the lower eyelid.
FAST TRACK
Noninflammatory eyelid edema is a relatively common disorder that usually appears after some local irritation or microtrauma.
As was the case with our patient, this disorder is not associated with fever or any other systemic sign or symptom. Complete blood count and C-reactive protein will be negative, as no infection or systemic alteration is present.
Consider these infectious alternatives In cases like this one, be sure to differentiate between noninflammatory palpebral edema and other common but less benign ocular conditions. Among the infectious causes of unilateral edema to rule out:
Preseptal cellulitis is an infection that is localized between the skin and the orbital septum. This disorder usually appears as a tender erythematous eyelid edema with no proptosis and no pain with eye movement.1 It is usually accompanied by an elevated white blood cell (WBC) count. Blood cultures are rarely positive, but when they are, Haemophilus influenzae is the most frequent pathogen.2 Preseptal cellulitis is treated with broad-spectrum oral antibiotics (amoxicillin/ clavulanate) for at least 7 days.
Orbital cellulitis is an infection that affects the orbital extraocular structures. It can lead to blindness, so prompt diagnosis is critical.
Orbital cellulitis usually develops when a sinus infection spreads into the orbit.3 Suspect it in a patient with proptosis, orbital pain, tenderness, conjunctival chemosis, decreased vision, elevated intraocular pressure, and pain on eye movement. Also look for an elevated WBC count. Perform blood cultures to confirm the presence of a pathogen.
If you suspect orbital cellulitis, order an orbital computed tomography scan with contrast infusion, including axial and coronal views; it may reveal an infection of the soft tissue behind the orbital septum. Treat this condition with broad-spectrum intravenous antibiotics (ceftriaxone) for 1 week, followed by 2 weeks of oral antibiotics, completing a total of 21 days of antibiotics.
Hordeolum is a localized infection that occurs when a meibomian gland becomes blocked. It usually appears as a palpable subcutaneous nodule within the eyelid and requires treatment with warm compresses and a combination of antibiotics and steroid topical ointment (tobramycin and dexamethasone ophthalmic ointment 4 times a day for a week).4
Acute dacryoadenitis is a lacrimal gland infection that usually causes unilateral, painful swelling of the outer third of the upper lid. You may also see tearing and discharge.5 Look for an elevated WBC count; however, blood cultures are rarely positive. These patients must be referred to an ophthalmologist as treatment varies from symptomatic measures in viral cases (warm compresses and a nonsteroidal anti-inflammatory drug, such as ibuprofen) to hospitalization and oral or intravenous antibiotics (eg, amoxicillin/ clavulanate for at least 7 days), depending on the severity and origin of the infection.6
Ruling out other noninfectious disorders Noninfectious inflammatory diseases should also be part of the differential diagnosis, including:
Idiopathic orbital inflammation. Also known as orbital pseudotumor, this disorder is a nongranulomatous acute-to-subacute inflammatory disease with no systemic manifestations.7 It frequently involves the lacrimal gland and can cause abrupt pain, conjunctival edema, and lid edema. Other common signs are proptosis and ocular motility alterations, which the patient may report as diplopia and visual impairment.
This disorder can result from optic neuropathy, exudative retinal detachment, or uveitis. It is a diagnosis of exclusion and is often arrived at by seeing the patient respond to systemic corticosteroids. The specific drug and dose will vary based on disease severity.
Graves’ ophthalmopathy. This bilateral, asymmetric immunological disorder affects the conjunctiva, eyelids,8 extraocular muscles, lacrimal gland, and optic nerve. Although it is typically diagnosed clinically, thyroid hormone abnormalities (increased T4 and a drop in thyroid-stimulating hormone) help support the diagnosis. In the acute phase, it has to be treated with systemic steroids; if the condition becomes chronic, the complications may require surgery. Treatment of the underlying hyperthyroidism may, in some cases, worsen the ophthalmopathy.
One more consideration. Finally, consider a local allergic reaction in the eyelids,9 which can also cause painless palpebral edema. Expect to see erythematous edema and intense itching a few hours after exposure to the allergen. A cosmetic product is often the culprit. As you might expect, initial treatment calls for withdrawal of the offending product. Instruct the patient to apply a topical steroid ointment on the eyelid if the edema is intense.
FAST TRACK
The condition does not require any local or systemic treatment.
Good news for our patient Diagnosing noninflammatory eyelid edema requires keen observational skills and a knowledge of several alternative diagnoses. Fortunately for our patient, this benign condition did not require any local or systemic treatment. The edema resolved on its own in 6 days.
A 54-year-old woman presented with a 7-day history of odynophagia, pharyngeal swelling, and painful skin lesions on her left ear. She had been on antiretroviral therapy for human immunodeficiency virus infection but had not been fully compliant with the treatment.
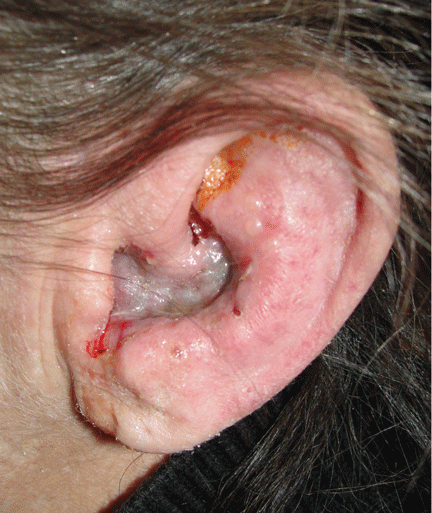
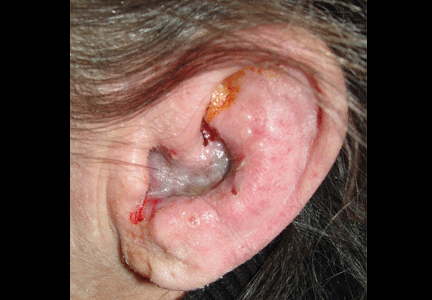
Figure 1. Vesicular eruption on the left concha and external auditory meatus.
On examination, she had painful erythematous vesicles and pustules on the left auricle and in the external auditory canal (Figure 1), as well as small vesicles and circumscribed erosions on the left anterior twothirds of her tongue (Figure 2) and left palate. Facial sensory function was normal; however, she had lagophthalmos, a flattened nasolabial fold, ptosis of the oral commissure, and a loss of the forehead wrinkles on the left side of her face—all signs of peripheral facial nerve paralysis.
Q: Which is the most likely diagnosis?
Ramsay Hunt syndrome
Herpes simplex
Contact dermatitis
Malignant external otitis
Erysipelas
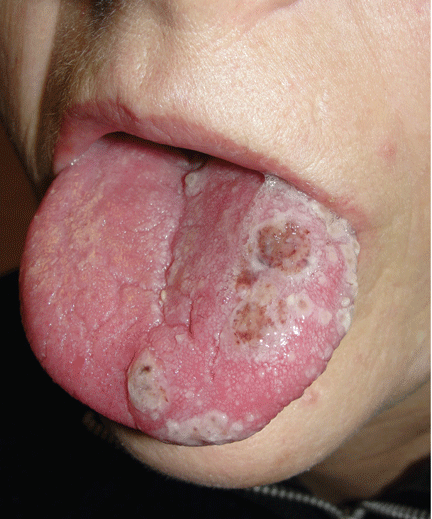
Figure 2. Multiple vesicles and pustules on the left side of the tongue and soft palate.
A: This patient had Ramsay Hunt syndrome, also known as herpes zoster oticus. It is a rare complication of herpes zoster in which the reactivation of latent varicella-zoster virus infection in the geniculate ganglion causes the triad of ipsilateral facial paralysis, ear pain, and vesicles in the auditory canal and auricle. Taste perception, hearing (eg, tinnitus, hyperacusis), and lacrimation can be affected.1
Ramsay Hunt syndrome is generally considered a polycranial neuropathy of cranial nerves VII (facial) and VIII (acoustic). In some cases other cranial neuropathies may be present and may involve cranial nerves V (trigeminal), IX (glossopharyngeal), and X (vagus). Vestibular disturbances such as vertigo are also often reported. It is more severe in patients with immune deficiency. Because the classic symptoms are not always present at the onset, the syndrome can be misdiagnosed.
DIAGNOSIS
Once the vesicular rash caused by herpes zoster has appeared, the diagnosis is usually readily apparent. The other main disease to consider in the differential diagnosis is herpes simplex. Herpes zoster infection is characterized by a painful sensory prodrome, dermatomal distribution, and lack of a history of a similar rash. However, if the patient has had a similar vesicular rash in the same location, then recurrent zosteriform herpes simplex should be considered. A noninfectious cause to consider is contact dermatitis. However, contact dermatitis usually produces intense itch rather than pain.
If the clinical presentation is uncertain, then viral culture, direct immunofluorescence testing, and a polymerase chain reaction assay is indicated to confirm the diagnosis. Polymerase chain reaction testing is the most sensitive test.3
TREATMENT
The rapid start of antiviral therapy is particularly critical in immunocompromised patients,4 even if the vesicles have been present for 72 hours. Immunocompromised patients with Ramsay Hunt syndrome and other forms of complicated herpes zoster infection should be hospitalized for intravenous acyclovir therapy.
Corticosteroids and oral acyclovir (10 mg/kg three times daily for 7 days) are commonly used in Ramsay Hunt syndrome. In a recent review,5 combination therapy with a corticosteroid and intravenous acyclovir did not show a benefit over corticosteroids alone in promoting resolution of facial neuropathy after 6 months.5 However, randomized clinical trials are needed to evaluate both therapies.
Although antiviral therapy reduces pain associated with acute neuritis, pain syndromes associated with herpes zoster can still be severe. Nonsteroidal antiinflammatory drugs and acetaminophen are useful for mild pain, either alone or in combination with a weak opioid analgesic (eg, tramadol, codeine). For moderate to severe pain that disturbs sleep, a stronger opioid analgesic (eg, oxycodone, morphine) may be necessary.6
Vestibular suppressants may be helpful if vestibular symptoms are severe. Temporary relief of otalgia may be achieved by applying a local anesthetic to the trigger point, if in the external auditory canal. Carbamazepine may be helpful, especially in cases of idiopathic geniculate neuralgia.7
OTHER CONSIDERATIONS
Once drug therapy is started, the patient should be seen at 2 weeks, 6 weeks, and 3 months to monitor the evolution of nerve paralysis.8
References
Mishell JH, Applebaum EL. Ramsay-Hunt syndrome in a patient with HIV infection. Otolaryngol Head Neck Surg1990; 102:177–179.
Adour KK. Otological complications of herpes zoster. Ann Neurol1994; 35(suppl):S62–S64.
Stránská R, Schuurman R, de Vos M, van Loon AM. Routine use of a highly automated and internally controlled real-time PCR assay for the diagnosis of herpes simplex and varicella-zoster virus infections. J Clin Virol2004; 30:39–44.
Miller GG, Dummer JS. Herpes simplex and varicella zoster viruses: forgotten but not gone. Am J Transplant2007; 7:741–747.
Uscategui T, Dorée C, Chamberlain IJ, Burton MJ. Antiviral therapy for Ramsay Hunt syndrome (herpes zoster oticus with facial palsy) in adults. Cochrane Database Syst Rev2008;(4):CD006851.
Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain2009; 142:209–217.
Edelsberg JS, Lord C, Oster G. Systematic review and meta-analysis of efficacy, safety, and tolerability data from randomized controlled trials of drugs used to treat postherpetic neuralgia. Ann Pharmacother2011; 45:1483–1490.
Ryu EW, Lee HY, Lee SY, Park MS, Yeo SG. Clinical manifestations and prognosis of patients with Ramsay Hunt syndrome. Am J Otolaryngol2012; 33:313–318.
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal University Hospital, Madrid, Spain
Angela Miguel-Morrondo, MD Department of Vascular Surgery, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology. Ramón y Cajal Hospital, Madrid, Spain
Address: Emiliano Grillo, MD, Department of Dermatology, Ramón y Cajal University Hospital, Carretera Colmenar km 9.100, 28034 Madrid, Spain; e-mail [email protected]
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal University Hospital, Madrid, Spain
Angela Miguel-Morrondo, MD Department of Vascular Surgery, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology. Ramón y Cajal Hospital, Madrid, Spain
Address: Emiliano Grillo, MD, Department of Dermatology, Ramón y Cajal University Hospital, Carretera Colmenar km 9.100, 28034 Madrid, Spain; e-mail [email protected]
Author and Disclosure Information
Emiliano Grillo, MD Department of Dermatology, Ramón y Cajal University Hospital, Madrid, Spain
Angela Miguel-Morrondo, MD Department of Vascular Surgery, Ramón y Cajal Hospital, Madrid, Spain
Sergio Vañó-Galván, MD, PhD Department of Dermatology, Ramón y Cajal Hospital, Madrid, Spain
Pedro Jaén, MD, PhD Department of Dermatology. Ramón y Cajal Hospital, Madrid, Spain
Address: Emiliano Grillo, MD, Department of Dermatology, Ramón y Cajal University Hospital, Carretera Colmenar km 9.100, 28034 Madrid, Spain; e-mail [email protected]
A 54-year-old woman presented with a 7-day history of odynophagia, pharyngeal swelling, and painful skin lesions on her left ear. She had been on antiretroviral therapy for human immunodeficiency virus infection but had not been fully compliant with the treatment.
Figure 1. Vesicular eruption on the left concha and external auditory meatus.
On examination, she had painful erythematous vesicles and pustules on the left auricle and in the external auditory canal (Figure 1), as well as small vesicles and circumscribed erosions on the left anterior twothirds of her tongue (Figure 2) and left palate. Facial sensory function was normal; however, she had lagophthalmos, a flattened nasolabial fold, ptosis of the oral commissure, and a loss of the forehead wrinkles on the left side of her face—all signs of peripheral facial nerve paralysis.
Q: Which is the most likely diagnosis?
Ramsay Hunt syndrome
Herpes simplex
Contact dermatitis
Malignant external otitis
Erysipelas
Figure 2. Multiple vesicles and pustules on the left side of the tongue and soft palate.
A: This patient had Ramsay Hunt syndrome, also known as herpes zoster oticus. It is a rare complication of herpes zoster in which the reactivation of latent varicella-zoster virus infection in the geniculate ganglion causes the triad of ipsilateral facial paralysis, ear pain, and vesicles in the auditory canal and auricle. Taste perception, hearing (eg, tinnitus, hyperacusis), and lacrimation can be affected.1
Ramsay Hunt syndrome is generally considered a polycranial neuropathy of cranial nerves VII (facial) and VIII (acoustic). In some cases other cranial neuropathies may be present and may involve cranial nerves V (trigeminal), IX (glossopharyngeal), and X (vagus). Vestibular disturbances such as vertigo are also often reported. It is more severe in patients with immune deficiency. Because the classic symptoms are not always present at the onset, the syndrome can be misdiagnosed.
DIAGNOSIS
Once the vesicular rash caused by herpes zoster has appeared, the diagnosis is usually readily apparent. The other main disease to consider in the differential diagnosis is herpes simplex. Herpes zoster infection is characterized by a painful sensory prodrome, dermatomal distribution, and lack of a history of a similar rash. However, if the patient has had a similar vesicular rash in the same location, then recurrent zosteriform herpes simplex should be considered. A noninfectious cause to consider is contact dermatitis. However, contact dermatitis usually produces intense itch rather than pain.
If the clinical presentation is uncertain, then viral culture, direct immunofluorescence testing, and a polymerase chain reaction assay is indicated to confirm the diagnosis. Polymerase chain reaction testing is the most sensitive test.3
TREATMENT
The rapid start of antiviral therapy is particularly critical in immunocompromised patients,4 even if the vesicles have been present for 72 hours. Immunocompromised patients with Ramsay Hunt syndrome and other forms of complicated herpes zoster infection should be hospitalized for intravenous acyclovir therapy.
Corticosteroids and oral acyclovir (10 mg/kg three times daily for 7 days) are commonly used in Ramsay Hunt syndrome. In a recent review,5 combination therapy with a corticosteroid and intravenous acyclovir did not show a benefit over corticosteroids alone in promoting resolution of facial neuropathy after 6 months.5 However, randomized clinical trials are needed to evaluate both therapies.
Although antiviral therapy reduces pain associated with acute neuritis, pain syndromes associated with herpes zoster can still be severe. Nonsteroidal antiinflammatory drugs and acetaminophen are useful for mild pain, either alone or in combination with a weak opioid analgesic (eg, tramadol, codeine). For moderate to severe pain that disturbs sleep, a stronger opioid analgesic (eg, oxycodone, morphine) may be necessary.6
Vestibular suppressants may be helpful if vestibular symptoms are severe. Temporary relief of otalgia may be achieved by applying a local anesthetic to the trigger point, if in the external auditory canal. Carbamazepine may be helpful, especially in cases of idiopathic geniculate neuralgia.7
OTHER CONSIDERATIONS
Once drug therapy is started, the patient should be seen at 2 weeks, 6 weeks, and 3 months to monitor the evolution of nerve paralysis.8
A 54-year-old woman presented with a 7-day history of odynophagia, pharyngeal swelling, and painful skin lesions on her left ear. She had been on antiretroviral therapy for human immunodeficiency virus infection but had not been fully compliant with the treatment.
Figure 1. Vesicular eruption on the left concha and external auditory meatus.
On examination, she had painful erythematous vesicles and pustules on the left auricle and in the external auditory canal (Figure 1), as well as small vesicles and circumscribed erosions on the left anterior twothirds of her tongue (Figure 2) and left palate. Facial sensory function was normal; however, she had lagophthalmos, a flattened nasolabial fold, ptosis of the oral commissure, and a loss of the forehead wrinkles on the left side of her face—all signs of peripheral facial nerve paralysis.
Q: Which is the most likely diagnosis?
Ramsay Hunt syndrome
Herpes simplex
Contact dermatitis
Malignant external otitis
Erysipelas
Figure 2. Multiple vesicles and pustules on the left side of the tongue and soft palate.
A: This patient had Ramsay Hunt syndrome, also known as herpes zoster oticus. It is a rare complication of herpes zoster in which the reactivation of latent varicella-zoster virus infection in the geniculate ganglion causes the triad of ipsilateral facial paralysis, ear pain, and vesicles in the auditory canal and auricle. Taste perception, hearing (eg, tinnitus, hyperacusis), and lacrimation can be affected.1
Ramsay Hunt syndrome is generally considered a polycranial neuropathy of cranial nerves VII (facial) and VIII (acoustic). In some cases other cranial neuropathies may be present and may involve cranial nerves V (trigeminal), IX (glossopharyngeal), and X (vagus). Vestibular disturbances such as vertigo are also often reported. It is more severe in patients with immune deficiency. Because the classic symptoms are not always present at the onset, the syndrome can be misdiagnosed.
DIAGNOSIS
Once the vesicular rash caused by herpes zoster has appeared, the diagnosis is usually readily apparent. The other main disease to consider in the differential diagnosis is herpes simplex. Herpes zoster infection is characterized by a painful sensory prodrome, dermatomal distribution, and lack of a history of a similar rash. However, if the patient has had a similar vesicular rash in the same location, then recurrent zosteriform herpes simplex should be considered. A noninfectious cause to consider is contact dermatitis. However, contact dermatitis usually produces intense itch rather than pain.
If the clinical presentation is uncertain, then viral culture, direct immunofluorescence testing, and a polymerase chain reaction assay is indicated to confirm the diagnosis. Polymerase chain reaction testing is the most sensitive test.3
TREATMENT
The rapid start of antiviral therapy is particularly critical in immunocompromised patients,4 even if the vesicles have been present for 72 hours. Immunocompromised patients with Ramsay Hunt syndrome and other forms of complicated herpes zoster infection should be hospitalized for intravenous acyclovir therapy.
Corticosteroids and oral acyclovir (10 mg/kg three times daily for 7 days) are commonly used in Ramsay Hunt syndrome. In a recent review,5 combination therapy with a corticosteroid and intravenous acyclovir did not show a benefit over corticosteroids alone in promoting resolution of facial neuropathy after 6 months.5 However, randomized clinical trials are needed to evaluate both therapies.
Although antiviral therapy reduces pain associated with acute neuritis, pain syndromes associated with herpes zoster can still be severe. Nonsteroidal antiinflammatory drugs and acetaminophen are useful for mild pain, either alone or in combination with a weak opioid analgesic (eg, tramadol, codeine). For moderate to severe pain that disturbs sleep, a stronger opioid analgesic (eg, oxycodone, morphine) may be necessary.6
Vestibular suppressants may be helpful if vestibular symptoms are severe. Temporary relief of otalgia may be achieved by applying a local anesthetic to the trigger point, if in the external auditory canal. Carbamazepine may be helpful, especially in cases of idiopathic geniculate neuralgia.7
OTHER CONSIDERATIONS
Once drug therapy is started, the patient should be seen at 2 weeks, 6 weeks, and 3 months to monitor the evolution of nerve paralysis.8
References
Mishell JH, Applebaum EL. Ramsay-Hunt syndrome in a patient with HIV infection. Otolaryngol Head Neck Surg1990; 102:177–179.
Adour KK. Otological complications of herpes zoster. Ann Neurol1994; 35(suppl):S62–S64.
Stránská R, Schuurman R, de Vos M, van Loon AM. Routine use of a highly automated and internally controlled real-time PCR assay for the diagnosis of herpes simplex and varicella-zoster virus infections. J Clin Virol2004; 30:39–44.
Miller GG, Dummer JS. Herpes simplex and varicella zoster viruses: forgotten but not gone. Am J Transplant2007; 7:741–747.
Uscategui T, Dorée C, Chamberlain IJ, Burton MJ. Antiviral therapy for Ramsay Hunt syndrome (herpes zoster oticus with facial palsy) in adults. Cochrane Database Syst Rev2008;(4):CD006851.
Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain2009; 142:209–217.
Edelsberg JS, Lord C, Oster G. Systematic review and meta-analysis of efficacy, safety, and tolerability data from randomized controlled trials of drugs used to treat postherpetic neuralgia. Ann Pharmacother2011; 45:1483–1490.
Ryu EW, Lee HY, Lee SY, Park MS, Yeo SG. Clinical manifestations and prognosis of patients with Ramsay Hunt syndrome. Am J Otolaryngol2012; 33:313–318.
References
Mishell JH, Applebaum EL. Ramsay-Hunt syndrome in a patient with HIV infection. Otolaryngol Head Neck Surg1990; 102:177–179.
Adour KK. Otological complications of herpes zoster. Ann Neurol1994; 35(suppl):S62–S64.
Stránská R, Schuurman R, de Vos M, van Loon AM. Routine use of a highly automated and internally controlled real-time PCR assay for the diagnosis of herpes simplex and varicella-zoster virus infections. J Clin Virol2004; 30:39–44.
Miller GG, Dummer JS. Herpes simplex and varicella zoster viruses: forgotten but not gone. Am J Transplant2007; 7:741–747.
Uscategui T, Dorée C, Chamberlain IJ, Burton MJ. Antiviral therapy for Ramsay Hunt syndrome (herpes zoster oticus with facial palsy) in adults. Cochrane Database Syst Rev2008;(4):CD006851.
Dworkin RH, Barbano RL, Tyring SK, et al. A randomized, placebo-controlled trial of oxycodone and of gabapentin for acute pain in herpes zoster. Pain2009; 142:209–217.
Edelsberg JS, Lord C, Oster G. Systematic review and meta-analysis of efficacy, safety, and tolerability data from randomized controlled trials of drugs used to treat postherpetic neuralgia. Ann Pharmacother2011; 45:1483–1490.
Ryu EW, Lee HY, Lee SY, Park MS, Yeo SG. Clinical manifestations and prognosis of patients with Ramsay Hunt syndrome. Am J Otolaryngol2012; 33:313–318.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
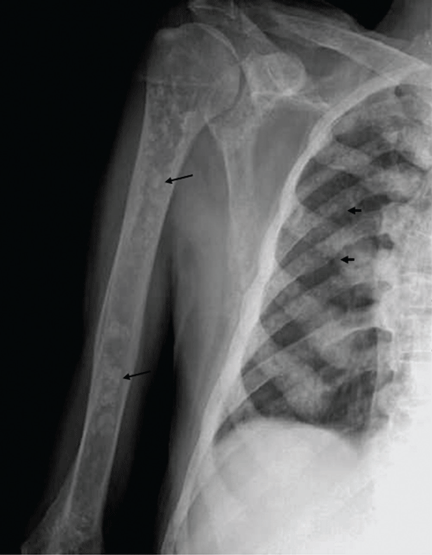
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
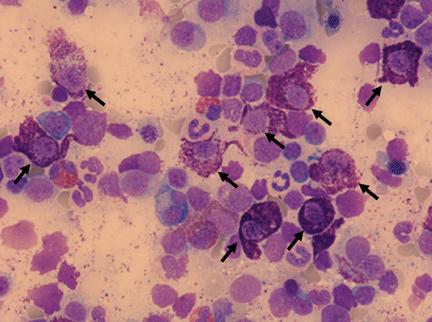
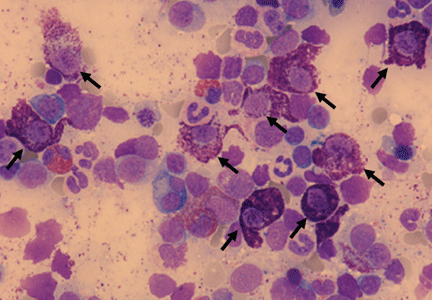
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
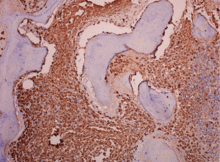
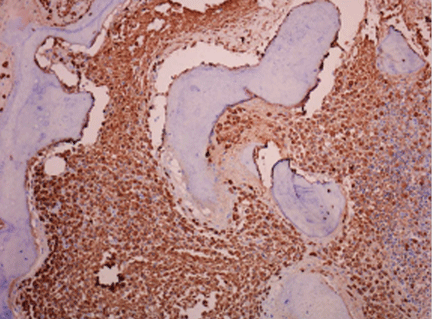
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
A 72-year-old man presented with abdominal cramping, diarrhea, intermittent flushing, asthenia, and a weight loss of 10 kg (22 lb) in the past 6 months. Physical examination revealed hepatosplenomegaly and an erythematous, maculopapular, confluent rash on the trunk (Figure 1) that displayed the Darier sign (redness, swelling, and itching in response to stroking in the involved area).
Laboratory analyses
Hemoglobin 9.8 g/dL (normal 13–17 g/dL)
White blood cell count 22.9 × 109/L (3.8–10)
Vitamin B12 1,730 pg/mL (220–900)
Serum tryptase 516 μg/L (5.5–13.5)
Beta-2 microglobulin 4.14 mg/L (1.39–2.11).
Radiologic evaluation
Figure 2. Radiologic evaluation showed diffuse osteosclerosis together with lytic and blastic areas (arrows).
Radiologic evaluation showed diffuse osteosclerosis with lytic and blastic areas (Figure 2).
Q: Which is the most likely diagnosis?
Carcinoid syndrome
Histiocytosis
Acute myeloblastic leukemia
Systemic mastocytosis
Chronic myeloblastic leukemia
A: The correct answer is systemic mastocytosis. The diagnosis was made according to the World Health Organization (WHO) diagnostic criteria for mastocytosis on the basis of the following findings in the bone marrow:
Figure 3. Bone marrow smear demonstrating increased numbers of abnormal mast cells (May-Grünwald-Giemsa stain, x 600).
Morphologically abnormal mast cells characterized by large size, spindle shape and poorly granulated cytoplasm (Figure 3) together with criteria for refractory cytopenia and multilineage dysplasia
Diffuse infiltration by tryptase-positive mast cells as assessed by immunohistochemical study (Figure 4)
Figure 4. Bone marrow study demonstrating a massive infiltrate of abnormal mast cells (tryptase stain, x 200).
One percent of mast cells that are immunophenotypically aberrant (CD25bright+), all of them showing an immature profile,1 associated with features of multilineage dysplasia2 as assessed by flow cytometry
The activating D816V KIT mutation, detected by peptide nucleic acid-mediated polymerase chain reaction clamping technique.3
MASTOCYTOSIS HAS SEVEN VARIANTS
Mastocytosis is a rare heterogeneous group of disorders characterized by proliferation and accumulation of abnormal mast cells in diverse organs and tissues, such as the skin, bone marrow, gastrointestinal tract, liver, spleen, or lymph nodes.4–6 The release of mast cell mediators causes a wide variety of symptoms, ranging from pruritus, flushing, abdominal cramping, and diarrhea to severe anaphylaxis with vascular collapse.7,8
The WHO defines seven variants6:
Cutaneous mastocytosis
Indolent systemic mastocytosis
Systemic mastocytosis with an associated (clonal) hematologic non-mast-cell disease (SM-AHNMD)
Aggressive systemic mastocytosis
Mast cell leukemia
Mast cell sarcoma
Extracutaneous mastocytoma.6
KIT mutation as a diagnostic criterion and prognostic factor
In most cases of systemic involvement, the clonal nature of the disease can be established by finding activating mutations of KIT, usually D816V, in lesions in the skin, bone marrow cells, or both.9 Apart from its value as a diagnostic criterion for systemic mastocytosis, KIT mutation has been reported to be strongly associated with progression of indolent systemic mastocytosis, including the development of myeloid malignancies, when the mutation is detected not only in mast cells but in all hematopoietic lineages.10
In cases of SM-AHNMD, a possible pathophysiologic relationship between the disorder in the mast cells and the disorder in other cells could be explained by a KIT mutation in early hematopoietic progenitor cells, which further evolve into phenotypically different subclones.
A rational management plan for mastocytosis must include carefully counselling the patient and care providers, avoiding factors that trigger acute release of mast cell mediators, and giving antimediator therapy such as oral cromolyn sodium (Gastrocrom), antihistamines, and leukotriene antagonists to relieve the symptoms caused by mast-cell-mediator release.11 In cases of SM-AHNMD, the clinical course and long-term prognosis are usually dominated by the concomitant hematologic malignancy, which should be treated as a separate entity.
CASE CONTINUED
Our patient’s bone marrow was analyzed for the KIT mutation in highly purified bone marrow cell subpopulations sorted by fluorescence-activated cell sorting. The mutation was detected in his mast cells, CD34+ cells, eosinophils, monocytes, neutrophils, lymphocytes, and nucleated erythroid precursors. According to the WHO recommendations, he had SMAHNMD, the associated hematologic disease being a myelodysplastic syndrome.
In view of his advanced age and concomitant myelodysplastic syndrome presenting with leukocytosis, we gave him hydroxyurea (Droxia; available in Spain as Hydrea) rather than other cytoreductive drugs as the first-line therapy. Additionally, we gave him corticosteroids in low doses, sodium cromolyn, and antihistamines to treat mastocytosis-related gastrointestinal symptoms. The patient was alive with stable disease 14 months after starting therapy.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.
References
Teodosio C, García-Montero AC, Jara-Acevedo M, et al. Mast cells from different molecular and prognostic subtypes of systemic mastocytosis display distinct immunophenotypes. J Allergy Clin Immunol2010; 125:719–726.
van de Loosdrecht AA, Alhan C, Béné MC, et al. Standardization of flow cytometry in myelodysplastic syndromes: report from the first European LeukemiaNet working conference on flow cytometry in myelodysplastic syndromes. Haematologica2009; 94:1124–1134.
Sotlar K, Escribano L, Landt O, et al. One-step detection of c-kit point mutations using peptide nucleic acid-mediated polymerase chain reaction clamping and hybridization probes. Am J Pathol2003; 162:737–746.
Valent P, Horny HP, Escribano L, et al. Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res2001; 25:603–625.
Valent P, Akin C, Escribano L, et al. Standards and standardization in mastocytosis: consensus statements on diagnostics, treatment recommendations and response criteria. Eur J Clin Invest2007; 37:435–453.
Horny HP, Metcalfe DD, Bennet JM, et al. Mastocytosis. In:Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008:54–63.
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am2006; 26:465–485.
González de Olano D, de la Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy2007; 37:1547–1555.
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood2006; 108:2366–2372.
Escribano L, Alvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol2009; 124:514–521.
Escribano L, Akin C, Castells M, Schwartz LB. Current options in the treatment of mast cell mediator-related symptoms in mastocytosis. Inflamm Allergy Drug Targets2006; 5:61–77.