User login
A Fatal Case of Hemophagocytic Lymphohistiocytosis Secondary to Anti-MDA5–Positive Dermatomyositis
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
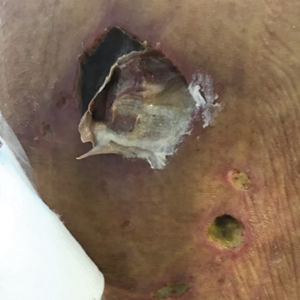
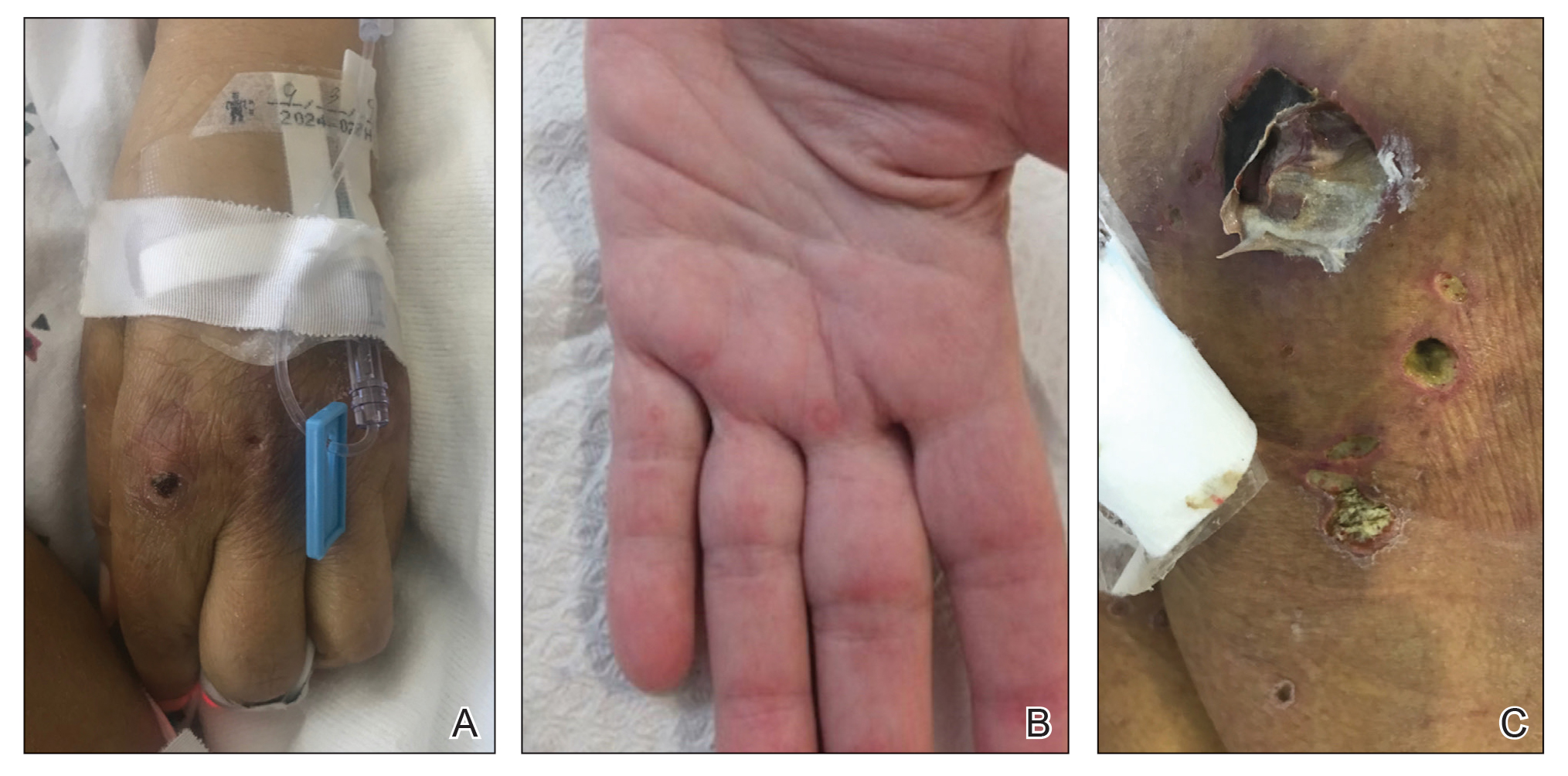
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
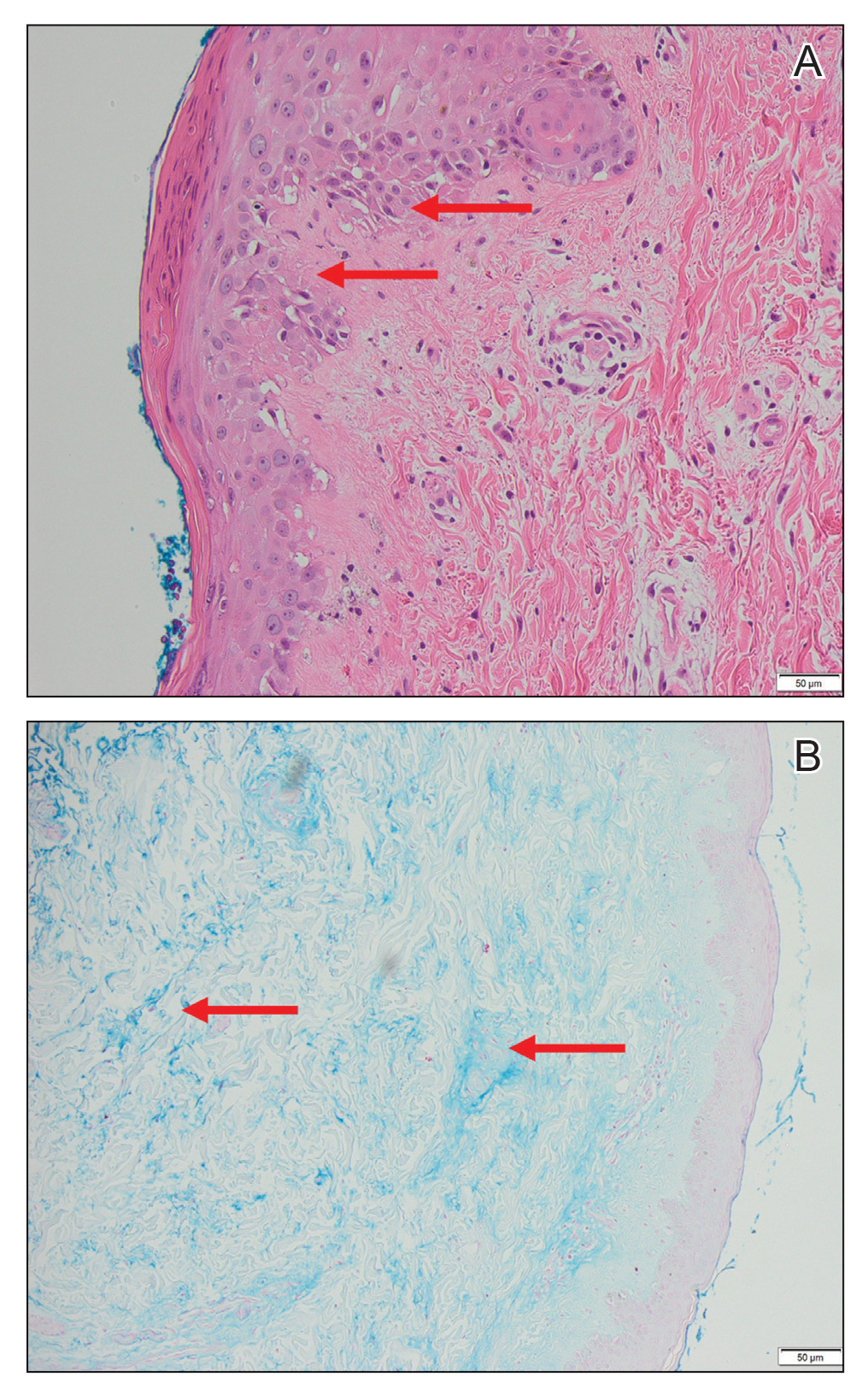
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
To the Editor:
Dermatomyositis (DM) is an idiopathic inflammatory myopathy characterized by bilateral, symmetrical, proximal muscle weakness and classic cutaneous manifestations.1 Patients with antibodies directed against melanoma differentiation–associated gene 5, MDA5, have a distinct presentation due to vasculopathy with more severe cutaneous ulcerations, palmar papules, alopecia, and an elevated risk of rapidly progressive interstitial lung disease.2 A ferritin level greater than 1600 ng/mL portends an increased risk for pulmonary disease and therefore can be of prognostic value.3 Further, patients with anti-MDA5 DM are at a lower risk of malignancy and are more likely to test negative for antinuclear antibodies in comparison to other patients with DM.2,4
Hemophagocytic lymphohistiocytosis (HLH), also known as hemophagocytic syndrome, is a potentially lethal condition whereby uncontrolled activation of histiocytes in the reticuloendothelial system causes hemophagocytosis and a hyperinflammatory state. Patients present with fever, splenomegaly, cytopenia, and hyperferritinemia.5 Autoimmune‐associated hemophagocytic syndrome (AAHS) describes HLH that develops in association with autoimmune conditions, most commonly systemic lupus erythematosus and adult-onset Still disease. Cases reported in association with DM exist but are few in number, and there is no standard-of-care treatment.6 We report a case of a woman with anti-MDA5 DM complicated by HLH and DM-associated liver injury.
A 50-year-old woman presented as a direct admit from the rheumatology clinic for diffuse muscle weakness of 8 months’ duration, 40-pound unintentional weight loss, pruritic rash, bilateral joint pains, dry eyes, dry mouth, and altered mental status. Four months prior, she presented to an outside hospital and was given a diagnosis of probable Sjögren syndrome and autoimmune hepatitis vs drug-induced liver injury. At that time, a workup was notable for antibodies against Sjögren syndrome–related antigen A, anti–smooth muscle antibodies, and transaminitis. Ultrasonography of the right upper quadrant revealed hepatic steatosis. The patient was started on oral prednisone and pilocarpine but had been off all medications for 1 month when she presented to our hospital.
On hospital admission, physical examination revealed a violaceous heliotrope rash; a v-sign on the chest; shawl sign; palmar papules with pits at the fingertips; and periungual erythema and ulcerations along the metacarpophalangeal joints, elbows, lateral feet, and upper eyelids (Figure 1). Laboratory workup showed the following results: white blood cell count, 4100/μL (reference range, 4000–11,000/μL); hemoglobin, 11.6 g/dL (reference range, 12–16 g/dL); platelet count, 100,000/μL (reference range, 150,000–450,000/μL); lactate dehydrogenase, 510 U/L (reference range, 80–225 U/L); alkaline phosphatase (ALP), 766 U/L (reference range, 30–120 U/L); alanine aminotransferase (ALT), 88 U/L (reference range, 10–40 U/L); aspartate aminotransferase (AST), 544 U/L (reference range, 10–40 U/L); total bilirubin, 4.2 mg/dL (reference range, 0.3–1.0 mg/dL); direct bilirubin, 3.7 mg/dL (reference range, 0.1–0.3 mg/dL); aldolase, 20.2 U/L (reference range, 1–7.5 U/L), creatine kinase, 180 U/L (reference range, 30–135 U/L); γ-glutamyltransferase (GGT), 2743 U/L (reference range, 8–40 U/L); high sensitivity C-reactive protein, 122.9 mg/L (low-risk reference range, <1.0 mg/L); triglycerides, 534 mg/dL (reference range, <150 mg/dL); ferritin, 3784 ng/mL (reference range, 24–307 ng/mL); antinuclear antibody, negative titer; antimitochondrial antibody, negative titer; soluble IL-2 receptor (CD25), 7000 U/mL (reference range, 189–846 U/mL); anti-Sjögren syndrome–related antigen A antibody, positive.
Magnetic resonance imaging of the shoulders showed diffuse soft-tissue edema. Computed tomography (CT) of the chest demonstrated parabronchial thickening and parenchymal bands suggestive of DM. An age-appropriate malignancy workup was negative, and results from a liver biopsy showed diffuse steatosis with no histologic evidence of autoimmune hepatitis. Punch biopsy results from a plaque on the left knee revealed vacuolar interface dermatitis with increased dermal mucin on colloidal iron staining, indicative of connective tissue disease (Figure 2). The patient was treated with intravenous (IV) methylprednisolone 250 mg twice daily for 2 days followed by oral prednisone 50 mg daily with IV immunoglobulin (IVIG) 0.4 mg/kg daily for 5 days. The patient’s symptoms improved, and she was discharged on oral prednisone 50 mg and mycophenolate mofetil 1000 mg twice daily with a plan for outpatient IVIG.
Two days after discharge, the patient was re-admitted for worsening muscle weakness; recalcitrant rash; new-onset hypophonia, dysphagia, and odynophagia; and intermittent fevers. Myositis panel results were positive for MDA5. Additionally, workup for HLH, which was initiated during the first hospital admission, revealed that she met 6 of 8 diagnostic criteria: intermittent fevers (maximum temperature, 38.2 °C), splenomegaly (12.6 cm on CT scan of abdomen), cytopenia in 2 cell lines (anemia, thrombocytopenia), hypertriglyceridemia, hyperferritinemia, and elevated IL-2 receptor (CD25). Based on these findings, the patient was diagnosed with anti-MDA5 DM associated with HLH.
The patient was started on IV methylprednisolone 1000 mg daily and received 1 rituximab infusion. Two days later, she experienced worsening fever with tachycardia, and a chest radiograph showed bibasilar infiltrates concerning for aspiration pneumonia, with sputum cultures growing Staphylococcus aureus. Due to the infection, the dosage of methylprednisolone was decreased to 16 mg 3 times daily and rituximab was stopped. The hematology department was consulted for the patient’s HLH, and due to her profound weakness and sepsis, the decision was made to hold initiation of etoposide, which, in addition to glucocorticoids, is considered first-line therapy for HLH. She subsequently experienced worsening hypoxia requiring intubation and received a second course of IVIG. Two days later, CT of the chest revealed progressive ground-glass opacities in the lower lobes of the lungs. The patient was then started on plasmapheresis every other day, hydroxychloroquine 200 mg daily, and IV methylprednisolone 1000 mg daily. Over the subsequent 6 days, she developed worsening renal failure, liver dysfunction, profound thrombocytopenia (13/μL), and acidemia. After extensive discussion with her family, the patient was transitioned to comfort care, and she died 33 days after the initial admission to our hospital.
Our case is a collection of several rare presentations: anti-MDA5 DM, with HLH and AAHS as complications of anti-MDA5 DM, and DM-associated liver injury. Anti-MDA5 DM is frequently refractory to conventional therapy, including high-dose glucocorticoids, cyclophosphamide, oral tacrolimus, and cyclosporine, and there currently is no single treatment algorithm.2 Lake and colleagues7 highlighted the importance of personalizing treatment of anti-MDA5 DM, as it can be one of the most aggressive rheumatologic diseases. We initially chose to treat our patient with high-dose methylprednisolone, IVIG, and rituximab. Kampylafka et al8 performed a retrospective analysis of the use of IVIG for DM as compared to standard therapy and demonstrated improved muscle and cutaneous involvement from a collection of 50 patients. Case reports have specifically revealed efficacy for the use of IVIG in patients with anti-MDA5 DM.9,10 Additionally, rituximab—an anti–B lymphocyte therapy—has been shown to be an effective supplemental therapy for cases of aggressive anti-MDA5 DM with associated interstitial lung disease, especially when conventional therapy has failed.11,12 Our patient’s sepsis secondary to S aureus pneumonia limited her to only receiving 1 dose of rituximab.
One promising treatment approach for anti-MDA5 DM recently published by Tsuji et al13 involves the use of combination therapy. In this prospective multicenter trial, patients were initially treated with a combination of high-dose glucocorticoids, oral tacrolimus, and IV cyclophosphamide. Plasmapheresis was then started for patients without symptomatic improvement. This method was compared to the more traditional step-up approach of high-dose steroids followed by another immunosuppressant. At 1-year follow-up, the combination therapy group demonstrated an 85% survival rate compared to 33% of historical controls.13
We suspect that our patient developed HLH and AAHS secondary to her underlying anti-MDA5 DM. Kumakura and Murakawa6 reported that among 116 cases of AAHS, 6.9% of cases were associated with DM, most commonly anti-Jo-1 DM. Hemophagocytic lymphohistiocytosis associated with anti-MDA5 DM has been described in only a few cases.14-16 The diagnosis of HLH is critical, as the treatments for HLH and DM differ. Both diseases manifest with hyperferritinemia—greater than 500 ng/mL in the case of HLH and 3784 ng/mL in our patient. Therefore, HLH can be easily overlooked. It is possible the rates of HLH associated with anti-MDA5 DM are higher than reported given their similar presentations.
Analogous to our case, Fujita et al15 reported a case of HLH associated with anti-MDA5 DM successfully treated with IV cyclophosphamide pulse therapy and plasmapheresis. The rationale for using plasmapheresis in anti-MDA5 DM is based on its success in patients with other antibody-mediated conditions such as Goodpasture syndrome and granulomatosis with polyangiitis.7 It is thought to expedite response to traditional treatment, and in the case described by Fujita et al,15 the patient received plasmapheresis 6 times total over the course of 9 days. The patient’s clinical symptoms, as well as platelet levels, liver enzymes, and ferritin value, improved.15 Our patient received 3 days of plasmapheresis with no improvement when the decision was made to discontinue plasmapheresis given her worsening clinical state.
Additionally, our patient had elevated hepatic enzymes (ALT, AST, ALP, GGT), and results of a liver biopsy demonstrated diffuse steatosis. We speculate her transaminitis was a complication of anti-MDA5 DM. Hepatocellular damage accompanying DM has been investigated in multiple studies and is most often defined as an elevated ALT.17-20 Improvement in ALT levels has been seen with DM treatment. However, investigators note that creatine kinase (CK) values often do not correlate with the resolution of the transaminitis, suggesting that CK denotes muscle damage whereas ALT represents separate liver damage.18-21
Nagashima et al22 highlighted that among 50 patients with DM without malignancy, only 20% presented with a transaminitis or elevated bilirubin. However, among those with liver injury, all were positive for antibodies against MDA5.22 The patients with anti-MDA5 DM liver dysfunction had higher ALT, ALP, and GGT levels compared to those without liver dysfunction. Similarly, in a retrospective review of 14 patients with anti-MDA5 DM, Gono and colleagues3 found elevated GGT levels and lower CK levels in comparison to patients with anti-aminoacyl-transfer RNA synthetase DM. Although liver enzymes can be elevated in patients with DM secondary to muscle damage, the authors argue that the specificity of GGT to the liver suggests intrinsic liver damage.3
The mechanism behind liver disease in anti-MDA5 DM is unclear, but it is hypothesized to be similar to nonalcoholic steatohepatitis.22 Other studies have revealed drug-induced hepatitis, hepatic congestion, nonspecific reactive hepatitis, metastatic liver tumor, primary biliary cholangitis, and autoimmune hepatitis as the etiology behind liver disease in their patients with DM.17-19 Liver biopsy results from patients with anti-MDA5 DM most commonly reveal hepatic steatosis, as seen in our patient, as well as hepatocyte ballooning and increased pigmented macrophages.22
We presented a case of anti-MDA5 DM complicated by HLH. Our patient had a fatal outcome despite aggressive treatment with high-dose methylprednisolone, IVIG, rituximab, and plasmapheresis. It is accepted that anti-MDA5 DM affects the lungs and skin, and our patient’s presentation also suggests liver involvement. In our case, onset of symptoms to fatality was approximately 1 year. It is essential to consider the diagnosis of HLH in all cases of anti-MDA5 DM given clinical disease overlap. Our patient could have benefited from earlier disease recognition and thus earlier aggressive therapy.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
1. Bohan A, Peter JB. Polymyositis and dermatomyositis. N Engl J Med. 1975;292:344-347.
2. Kurtzman DJB, Vleugels RA. Anti-melanoma differentiation-associated gene 5 (MDA5) dermatomyositis: a concise review with an emphasis on distinctive clinical features. J Am Acad Dermatol. 2018;78:776-785.
3. Gono T, Kawaguchi Y, Satoh T, et al. Clinical manifestation and prognostic factor in anti-melanoma differentiation-associated gene 5 antibody-associated interstitial lung disease as a complication of dermatomyositis. Rheumatology (Oxford). 2010;49:1713-1719.
4. Fiorentino D, Chung L, Zwerner J, et al. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J Am Acad Dermatol. 2011;65:25-34.
5. Sepulveda FE, de Saint Basile G. Hemophagocytic syndrome: primary forms and predisposing conditions. Curr Opin Immunol. 2017;49:20-26.
6. Kumakura S, Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum. 2014;66:2297-2307.
7. Lake M, George G, Summer R. Time to personalize the treatment of anti-MDA-5 associated lung disease. Ann Rheum Dis. 2019;78:E52.
8. Kampylafka EI, Kosmidis ML, Panagiotakos DB, et al. The effect of intravenous immunoglobulin (IVIG) treatment on patients with dermatomyositis: a 4-year follow-up study. Clin Exp Rheumatol. 2012;30:397-401.
9. Koguchi-Yoshioka H, Okiyama N, Iwamoto K, et al. Intravenous immunoglobulin contributes to the control of antimelanoma differentiation-associated protein 5 antibody-associated dermatomyositis with palmar violaceous macules/papules. Br J Dermatol. 2017;177:1442-1446.
10. Hamada-Ode K, Taniguchi Y, Kimata T, et al. High-dose intravenous immunoglobulin therapy for rapidly progressive interstitial pneumonitis accompanied by anti-melanoma differentiation-associated gene 5 antibody-positive amyopathic dermatomyositis. Eur J Rheumatol. 2015;2:83-85.
11. So H, Wong VTL, Lao VWN, et al. Rituximab for refractory rapidly progressive interstitial lung disease related to anti-MDA5 antibody-positive amyopathic dermatomyositis. Clin Rheumatol. 2018;37:1983-1989.
12. Koichi Y, Aya Y, Megumi U, et al. A case of anti-MDA5-positive rapidly progressive interstitial lung disease in a patient with clinically amyopathic dermatomyositis ameliorated by rituximab, in addition to standard immunosuppressive treatment. Mod Rheumatol. 2017;27:536-540.
13. Tsuji H, Nakashima R, Hosono Y, et al. Multicenter prospective study of the efficacy and safety of combined immunosuppressive therapy with high-dose glucocorticoid, tacrolimus, and cyclophosphamide in interstitial lung diseases accompanied by anti-melanoma differentiation-associated gene 5-positive dermatomyositis. Arthritis Rheumatol. 2020;72:488-498.
14. Honda M, Moriyama M, Kondo M, et al. Three cases of autoimmune-associated haemophagocytic syndrome in dermatomyositis with anti-MDA5 autoantibody. Scand J Rheumatol. 2020;49:244-246.
15. Fujita Y, Fukui S, Suzuki T, et al. Anti-MDA5 antibody-positive dermatomyositis complicated by autoimmune-associated hemophagocytic syndrome that was successfully treated with immunosuppressive therapy and plasmapheresis. Intern Med. 2018;57:3473-3478.
16. Gono T, Miyake K, Kawaguchi Y, et al. Hyperferritinaemia and macrophage activation in a patient with interstitial lung disease with clinically amyopathic DM. Rheumatology (Oxford). 2012;51:1336-1338.
17. Wada T, Abe G, Kudou, T, et al. Liver damage in patients with polymyositis and dermatomyositis. Kitasato Med Journal. 2016;46:40-46.
18. Takahashi A, Abe K, Yokokawa J, et al. Clinical features of liver dysfunction in collagen diseases. Hepatol Res. 2010;40:1092-1097.
19. Matsumoto T, Kobayashi S, Shimizu H, et al. The liver in collagen diseases: pathologic study of 160 cases with particular reference to hepatic arteritis, primary biliary cirrhosis, autoimmune hepatitis and nodular regenerative hyperplasia of the liver. Liver. 2000;20:366-373.
20. Shi Q, Niu J, Huang X, et al. Do muscle enzyme changes forecast liver injury in polymyositis/dermatomyositis patients treated with methylprednisolone and methotrexate? Ann Clin Lab Sci. 2016;46:266-269.
21. Noda S, Asano Y, Tamaki Z, et al. A case of dermatomyositis with “liver disease associated with rheumatoid diseases” positive for anti-liver-kidney microsome-1 antibody. Clin Rheumatol. 2010;29:941-943.
22. Nagashima T, Kamata Y, Iwamoto M, et al. Liver dysfunction in anti-melanoma differentiation-associated gene 5 antibody-positive patients with dermatomyositis. Rheumatol Int. 2019;39:901-909.
PRACTICE POINTS
- Anti-MDA5 (melanoma differentiation–associated gene 5 antibody)–positive dermatomyositis associated with hemophagocytic lymphohistiocytosis is a rare and aggressive condition associated with a poor prognosis, and there is no standard treatment.
- Dermatomyositis-associated liver injury is not well defined.