User login
Multiple sclerosis (MS) is a disorder characterized by inflammation, demyelination, and degeneration of the central nervous system (CNS). The hallmark of the disorder is relapses and remissions of neurologic symptoms occurring early in the disease course, which are often associated with areas of CNS inflammation and myelin loss.1-3 The inciting cause for this inflammation is unknown but is believed to be multifactorial, with environmental and genetic influences creating an adaptive, T cell-mediated autoimmune response against the CNS.4 Separate from the acute attacks, progressive neurodegeneration can occur more chronically and is characterized by axonal loss and grey matter atrophy thought to be due to direct cytotoxic activity of the innate immune system as well as toxic intermediates, such as nitric oxide.4,5 Despite the multiple insults early on, neurologic disability typically becomes more apparent over time.6 The disability threshold theory argues that neurologic function compensates for brain tissue loss until a threshold of accumulated damage is exceeded.7
Background
The incidence of MS follows a geographic gradient; rates rise as the distance from the equator increases.8,9 This is thought to be due to the gradient of relative sun exposure and its role in the production of vitamin D, which plays an important role in immune regulation when converted to its active hormonal form. Multiple sclerosis is more prevalent in non-Hispanic white patients than it is in other racial groups, and women are affected nearly 2 to 3 times more often than are men.10 About 450,000 individuals in the U.S. and more than 2 million worldwide have MS.11-14
Multiple sclerosis is the most common cause of nontraumatic neurologic disability in young adults. It is typically diagnosed in the third and fourth decades of life, and those who are diagnosed after age 50 years often can recount neurologic symptoms that began years before. However, pediatric-onset and new-onset cases in the elderly have been reported. It has been estimated that up to 10% of patients with MS have onset before 18 years of age.15-17 Compared with adult-onset MS, pediatric-onset is associated with a longer period between initial attack and physical disability, although the average age of disability onset is about 10 years younger.17,18
Disease Courses
Relapsing-remitting MS (RRMS) is the most common disease course overall, and this pattern affects 97% of individuals with disease onset before age 18 years.15-17 The clinically isolated syndrome disease course leads to clinically definite MS in one-third of patients within 1 year and in one-half of patients within 2 years.19 In the majority of cases, the RRMS course transitions over time to secondary-progressive MS (SPMS), which is a disease pattern of progressively worsening disability with few neurologic relapses. Although inflammation is present at all stages, the difference is in the predominance of cell types involved.5 Why the shift from active to chronic inflammation occurs and how to prevent it remain central questions in MS research.4 Regardless, tentative evidence suggests that prevention of relapses may reduce disability accumulation and risk of conversion to progressive MS.20
A minority of patients with MS are diagnosed with primary-progressive MS (PPMS) at onset, which is characterized by a disease pattern that follows a relatively steady progression of neurologic symptoms over time, without clear relapses or remissions of these symptoms, though phases of stability or fluctuations in disability may still occur.21 It is typically diagnosed at an older age than is RRMS, and it is rare in children; suspicion of PPMS in this age group should prompt detailed assessment of alternative diagnoses.17,22 Primary-progressive MS is more equally distributed in men and women than is RRMS.
Regardless of onset type, disability progression seems to occur at the same rate among all patients with MS after a certain threshold is reached. The established assessment scale for disability progression in MS is the Kurtzke Expanded Disability Status scale (EDSS), which has a scoring range from 0 to 10. Data from several patient registries have shown that once EDSS step 4 is reached, progression thereafter occurs at a predictable rate that is similar across MS phenotypes.23 The time it takes patients to subsequently reach higher EDSS steps may be independent of preceding factors.23
MS Symptom Burden
The neurologic symptoms that patients experience are fluctuating and disabling throughout the disease course, irrespective of onset type. Typical MS symptoms include mobility impairment, changes in cognition and mood, pain and other sensation disturbances, bowel and bladder dysfunction, fatigue, and visual disturbances. The burden of these symptoms can significantly impact quality of life (QOL) for patients and their families. The symptom burden can pose a direct threat to a patient’s autonomy, necessitating adaptation to an unpredictable disease that requires frequent health care visits to many different health care providers (eg, neurologists; primary care providers; physiatrists; urologists; ophthalmologists; and speech, physical, and occupational therapists), periodic testing, and often costly medications.24
Compared with patients who have other chronic conditions, patients with MS experience diminished societal roles, along with decreased assessments in health, energy, and physical functions.25 These often lead to early exit from the workforce and limitations in household responsibilities, which further impact QOL.26 Including both direct and indirect costs of the disease, a patient with MS can expect a lifetime financial burden of nearly $1.2 million.27
Large population cohort studies in MS, along with MS registry studies of patients untreated with disease-modifying therapies, have shown reduced survival rates by an average of 7 to 14 years.23,28 Multiple sclerosis is the main cause of death in about 50% of cases (EDSS step 10), which is defined as “acute death due to brain stem involvement or to respiratory failure, or death consequent to the chronic bedridden state with terminal pneumonia, sepsis, uremia, or cardiorespiratory failure [and excluding] intercurrent causes of death.”23 For the remaining patients with MS, cause of death is similar to those of the general population, such as cardiovascular disease and cancer.23 However, the incidence of suicide is higher among patients with MS.23
All these factors underscore the importance of early diagnosis as well as early initiation of effective disease-modifying therapy. 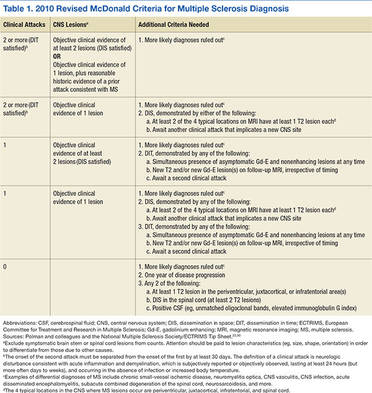
Disease-Modifying Therapies
The goal of MS disease-modifying therapy is to reduce the early clinical and subclinical disease activity that eventually contributes to long-term disability.31,32 There are currently 13 FDA-approved disease-modifying therapies for MS. These include 7 self-injecting therapies, 3 oral therapies, and 3 infusion therapies. These 13 medications have 8 different mechanisms of action (MOA) that target distinct areas of the immune-mediated disease process. They also differ in their frequencies and routes of administration in addition to their adverse effect (AE) profiles (Tables 2, 3, and 4).
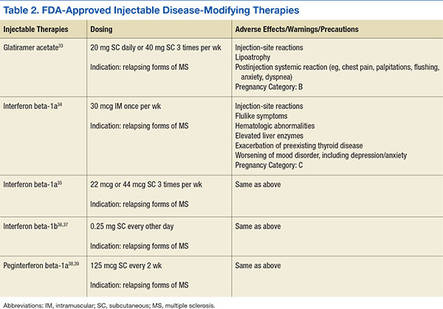
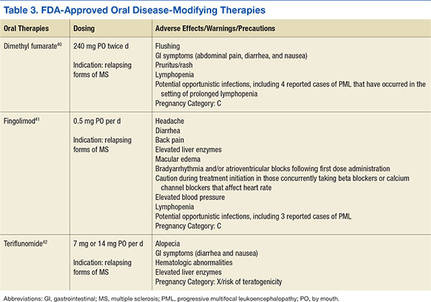
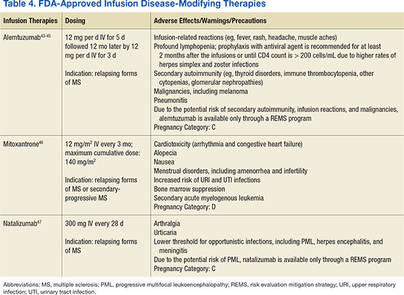
Treatment Considerations
In 1993, interferon beta-1b became the first FDA-approved MS medication. In the following 2 decades, there became 12 additional FDA-approved medications for MS, beginning with other injectables. The first infusion therapy was introduced in 2004, followed by various oral medications. The treatment landscape continues to change rapidly. This therapeutic revolution has occurred largely due to the improved understanding of the pathophysiology of MS and unquestionably has improved the prognosis and overall QOL for patients. The question is no longer how to treat MS but rather how to personalize and optimize treatment for each patient.20
Despite all available treatment options, none are curative, and none have been proven to offer neuroprotection or contribute to neural repair. To date, no studies have led to FDA-approved therapies for PPMS. Further, the efficacy of any of these medications varies from patient to patient. Due largely to the lack of biomarkers for disease activity and treatment response, drug efficacy continues to be measured according to the current gold standard, which is identification of gadolinium-enhancing lesions in the white matter on magnetic resonance imaging (MRI), combined with other markers of disease, including clinical relapse rate and confirmed disability progression.19 In general, the injectable therapies are expected to protect against about 20% to 35% of relapses; the oral agents, 50% to 55%; and the infusion therapies, > 60%.2
In conjunction with a medication’s efficacy rate and safety profile, the frequency and route of administration also must be considered. In general, MS medications are exceedingly expensive, some costing up to tens-of-thousands of dollars per year.48 All these factors have the real potential to negatively impact patient adherence. Nonadherence and gaps in treatment have been correlated with increased rates of relapses and progression of disability as well as negative MRI outcomes.49-53
When to Initiate Treatment
Once a patient is diagnosed, a common question is, when is the right time to initiate treatment? The primary target of the current MS medications is to decrease CNS inflammation (relapses). The ideal time to start treatment is as promptly as possible after confirmation of the diagnosis to combat the early inflammatory relapsing phase of the disease. There seems to be an early window in the disease course when achieving disease control can have a profound impact on long-term disability. Disease control is typically defined as decreasing relapses, slowing the accumulation of lesions visualized on MRI, and preventing the disability that occurs from both incomplete recovery after relapses and overall disease progression.54,55
Certain clinical indicators, such as higher relapse rates early in the disease course and MRI characteristics, including total lesion burden and the location of lesions within the CNS, seem to be associated with a higher risk of disease progression.56 These are potential prognostic indicators that can help tailor the choice of disease-modifying therapy for patients.57 Those with highly inflammatory and potentially aggressive disease at onset, for example, may benefit from early initiation of higher efficacy therapies, whereas those with more benign forms of MS at onset may fare well on lower efficacy therapies. In general, when it comes to currently available MS treatments, higher efficacy is often tied to riskier AE profiles, so the best medication may be the “least efficacious” one that can still control the disease.20
Hauser and colleagues suggested a treatment decision-making model that identifies the interferons, glatiramer acetate, dimethyl fumarate, and teriflunomide as acceptable first-line therapies; fingolimod and natalizumab as acceptable second-line options; and mitoxantrone and alemtuzumab as acceptable third-line therapeutic options.20 The authors generally agree with Hauser and colleagues’ model, and it is important to consider individual patient factors (eg, comorbidities, concurrent medications, life circumstances) and disease severity when deciding on a treatment plan.
Perhaps an even more difficult question is, when is the right time to switch therapies? There remains a dearth of either guidelines or comparative studies for treatment management decisions. Further, without reliable biomarkers, the clinical and pathologic heterogeneity of MS makes treatment difficult.4,19 In practice, there is general consensus that 1 year of treatment monitoring for effects on clinical and radiologic outcomes is an acceptable time frame to evaluate effectiveness of a disease-modifying treatment. If adherence is maintained and there is still evidence of clinical or MRI activity (suggesting a suboptimal response), an alternative therapy, particularly one with a different MOA, should be strongly considered. This highlights the importance of broad access to all available MS therapies to allow for early selection of a correct therapy that patients will remain adherent to and that controls their disease.
Conclusion
Multiple sclerosis remains a highly unpredictable disease, and relapses have the ability to produce a measurable and sustained impact on the level of disability.58 Still, the influence of reduced relapses on preventing disability in an individual patient remains unclear. Large, long-term, prospective cohort studies may clarify whether early treatment affects disease progression and disability.20 However, it is quite evident that effective relapse reduction decreases discomfort, reduces days lost from work and other important activities of daily life, and improves QOL.58,59
There is still much to learn about this unique disease, but emerging evidence in the medical literature highlights the importance of setting treatment goals that include targeting disease activity to achieve early and effective control. Attaining control with a MS medication seems to be a key component of slowing the physical and emotional disability that can accumulate, helping patients remain active and maintain the highest QOL possible for as long as possible.
1. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46(4):907-911.
2. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(pt 5):1175-1189.
3. Charil A, Filippi M. Inflammatory demyelination and neurodegeneration in early multiple sclerosis. J Neurol Sci. 2007;259(1-2):7-15.
4. Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65(3):239-248.
5. Grigoriadis N, van Pesch V; ParadigMS Group. A basic overview of multiple sclerosis immunopathology. Eur J Neurol. 2015;22(suppl 2):3-13.
6. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647-656.
7. Rudick RA, Lee JC, Simon J, Fisher E. Significance of T2 lesions in multiple sclerosis: a 13-year longitudinal study. Ann Neurol. 2006;60(2):236-242.
8. Alla S, Mason DF. Multiple sclerosis in New Zealand. J Clin Neurosci. 2014;21(8):1288-1291.
9. Simpson S Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(10):1132-1141.
10. Evans C, Beland SG, Kulaga S, et al. Incidence and prevalence of multiple sclerosis in the Americas: a systematic review. Neuroepidemiology. 2013;40(3):195-210.
11. Giesser BS. Diagnosis of multiple sclerosis. Neurol Clin. 2011;29(2):381-388.
12. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938-952.
13. Weinshenker BG. The natural history of multiple sclerosis. Neurol Clin. 1995;13(1):119-146.
14. Weinshenker BG. The natural history of multiple sclerosis: update 1998. Semin Neurol. 1998;18(3):301-307.
15. Simone IL, Carrara D, Tortorella C, Ceccarelli A, Livrea P. Early onset multiple sclerosis. Neurol Sci. 2000;21(4)(suppl 2):S861-S863.
16. Reinhardt K, Weiss S, Rosenbauer J, Gärtner J, von Kries R. Multiple sclerosis in children and adolescents: incidence and clinical picture--new insights from the nationwide German surveillance (2009-2011). Eur J Neurol. 2014;21(4):654-659.
17. Waldman A, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M, Banwell B. Multiple sclerosis in children: an update on clinical diagnosis, therapeutic strategies, and research. Lancet Neurol. 2014;13(9):936-948.
18. Renoux C, Vukusic S, Mikaeloff Y, et al; Adult Neurology Departments KIDMUS Study Group. Natural history of multiple sclerosis with childhood onset. N Engl J Med. 2007;356(25):2603-2613.
19. D'Ambrosio A, Pontecorvo S, Colastanti T, Zamboni S, Francia A, Margutti P. Peripheral blood biomarkers in multiple sclerosis. Autoimmun Rev. 2015;14(12):1097-1110.
20. Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol. 2013;74(3):317-327.
21. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
22. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
23. Hurwitz BJ. Analysis of current multiple sclerosis registries. Neurology. 2011;76(1)(suppl 1):S7-S13.
24. Boeije HR, Duijnstee MS, Grypdonck MH, Pool A. Encountering the downward phase: biographical work in people with multiple sclerosis living at home. Soc Sci Med. 2002;55(6):881-893.
25. Sprangers MA, de Regt EB, Andries F, et al. Which chronic conditions are associated with better or poorer quality of life? J Clin Epidemiol. 2000;53(9):895-907.
26. Julian LJ, Vella L, Vollmer T, Hadjimichael O, Mohr DC. Employment in multiple sclerosis. Exiting and re-entering the work force. J Neurol. 2008;255(9):1354-1360.
27. Trisolini M, Honeycutt A, Wiener J, Lesesne S. Global economic impact of multiple sclerosis. Multiple Sclerosis International Federation website. http://www.msif.org/wp-content/uploads/2014/09/Global_economic_impact_of_MS.pdf. Published May 2010. Accessed May 6, 2016
28. Scalfari A, Knappertz V, Cutter G, Goodin DS, Ashton R, Ebers GC. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
29. Gurevich M, Miron G, Achiron A. Optimizing multiple sclerosis diagnosis: gene expression and genomic association. Ann Clin Transl Neurol. 2015;2(3):271-277.
30. National Multiple Sclerosis Society, European Committee for Treatment and Research in Multiple Sclerosis. Tip sheet: 2010 revised McDonald diagnostic criteria for MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Paper-TipSheet_-2010-Revisions-to-the-McDonald-Criteria-for-the-Diagnosis-of-MS.pdf. Accessed April 22, 2016.
31. Freedman MS, Selchen D, Arnold DL, et al; Canadian Multiple Sclerosis Working Group. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013;40(3):307-323.
32. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
33. Copaxone [package insert]. Overland Park, KS: Teva Neuroscience, Inc; 2014.
34. Avonex [package insert]. Cambridge, MA: Biogen Idec Inc; 1996.
35. Rebif [package insert]. Rockland, MA: EMD Serono, Inc; New York, NY: Pfizer, Inc; 2012.
36. Betaseron [package insert]. Montville, NJ: Bayer HealthCare Pharmaceuticals Inc; 2012.
37. Extavia [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2014.
38. Plegridy [package insert]. Cambridge, MA: Biogen Idec Inc; 2013.
39. Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon ß-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657-665.
40. Tecfidera [package insert]. Cambridge, MA: Biogen Idec Inc; 2015.
41. Gilenya [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2016.
42. Aubagio [package insert]. Cambridge, MA: Genzyme Corp; 2012.
43. Lemtrada [package insert]. Cambridge, MA: Genzyme Corp; 2014.
44. Cohen JA, Coles AJ, Arnold DL, et al; CARE-MS I investigators. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819-1828.
45. Coles AJ, Twyman CL, Arnold DL, et al; CARE-MS II investigators. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829-1839.
46. Novantrone [package insert]. Rockland, MA: EMD Serono, Inc; 2008.
47. Tysabri [medication guide]. Cambridge, MA: Biogen Idec Inc; 2015.
48. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail. Neurology. 2015;84(21):2185-2192.
49. Cohen BA, Coyle PK, Leist T, Oleen-Burkey MA, Schwartz M, Zwibel H. Therapy Optimization in Multiple Sclerosis: a cohort study of therapy adherence and risk of relapse. Mult Scler Relat Disord. 2015;4(1):75-82.
50. Cohen B, Leist T, Coyle P, Zwibel H, Markowitz C, Tullman M. MS therapy adherence and relapse risk. Neurology. 2013;80(7) (suppl):P01.193.
51. Richert ND, Zierak MC, Bash CN, Lewis BK, McFarland HF, Frank JA. MRI and clinical activity in MS patients after terminating treatment with interferon beta-1b. Mult Scler. 2000;6(2):86-90.
52. Siger M, Durko A, Nicpan A, Konarska M, Grudziecka M, Selmaj K. Discontinuation of interferon beta therapy in multiple sclerosis patients with high pre-treatment disease activity leads to prompt return to previous disease activity. J Neurol Sci. 2011;303(1-2):50-52.
53. Wu X, Dastidar P, Kuusisto H, Ukkonen M, Huhtala H, Elovaara I. Increased disability and MRI lesions after discontinuation of IFN-beta-1a in secondary progressive MS. Acta Neurol Scand. 2005;112(4):242-247.
54. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133(pt 7):1914-1929.
55. Bates D. Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology. 2011;76(1)(suppl 1):S14-S25.
56. Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131(pt 3):808-817.
57. Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275(4):350-363.
58. Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003;61(11):1528-1532.
59. Kalb R. The emotional and psychological impact of multiple sclerosis relapses. J Neurol Sci. 2007;256(suppl 1):S29-S33.
Multiple sclerosis (MS) is a disorder characterized by inflammation, demyelination, and degeneration of the central nervous system (CNS). The hallmark of the disorder is relapses and remissions of neurologic symptoms occurring early in the disease course, which are often associated with areas of CNS inflammation and myelin loss.1-3 The inciting cause for this inflammation is unknown but is believed to be multifactorial, with environmental and genetic influences creating an adaptive, T cell-mediated autoimmune response against the CNS.4 Separate from the acute attacks, progressive neurodegeneration can occur more chronically and is characterized by axonal loss and grey matter atrophy thought to be due to direct cytotoxic activity of the innate immune system as well as toxic intermediates, such as nitric oxide.4,5 Despite the multiple insults early on, neurologic disability typically becomes more apparent over time.6 The disability threshold theory argues that neurologic function compensates for brain tissue loss until a threshold of accumulated damage is exceeded.7
Background
The incidence of MS follows a geographic gradient; rates rise as the distance from the equator increases.8,9 This is thought to be due to the gradient of relative sun exposure and its role in the production of vitamin D, which plays an important role in immune regulation when converted to its active hormonal form. Multiple sclerosis is more prevalent in non-Hispanic white patients than it is in other racial groups, and women are affected nearly 2 to 3 times more often than are men.10 About 450,000 individuals in the U.S. and more than 2 million worldwide have MS.11-14
Multiple sclerosis is the most common cause of nontraumatic neurologic disability in young adults. It is typically diagnosed in the third and fourth decades of life, and those who are diagnosed after age 50 years often can recount neurologic symptoms that began years before. However, pediatric-onset and new-onset cases in the elderly have been reported. It has been estimated that up to 10% of patients with MS have onset before 18 years of age.15-17 Compared with adult-onset MS, pediatric-onset is associated with a longer period between initial attack and physical disability, although the average age of disability onset is about 10 years younger.17,18
Disease Courses
Relapsing-remitting MS (RRMS) is the most common disease course overall, and this pattern affects 97% of individuals with disease onset before age 18 years.15-17 The clinically isolated syndrome disease course leads to clinically definite MS in one-third of patients within 1 year and in one-half of patients within 2 years.19 In the majority of cases, the RRMS course transitions over time to secondary-progressive MS (SPMS), which is a disease pattern of progressively worsening disability with few neurologic relapses. Although inflammation is present at all stages, the difference is in the predominance of cell types involved.5 Why the shift from active to chronic inflammation occurs and how to prevent it remain central questions in MS research.4 Regardless, tentative evidence suggests that prevention of relapses may reduce disability accumulation and risk of conversion to progressive MS.20
A minority of patients with MS are diagnosed with primary-progressive MS (PPMS) at onset, which is characterized by a disease pattern that follows a relatively steady progression of neurologic symptoms over time, without clear relapses or remissions of these symptoms, though phases of stability or fluctuations in disability may still occur.21 It is typically diagnosed at an older age than is RRMS, and it is rare in children; suspicion of PPMS in this age group should prompt detailed assessment of alternative diagnoses.17,22 Primary-progressive MS is more equally distributed in men and women than is RRMS.
Regardless of onset type, disability progression seems to occur at the same rate among all patients with MS after a certain threshold is reached. The established assessment scale for disability progression in MS is the Kurtzke Expanded Disability Status scale (EDSS), which has a scoring range from 0 to 10. Data from several patient registries have shown that once EDSS step 4 is reached, progression thereafter occurs at a predictable rate that is similar across MS phenotypes.23 The time it takes patients to subsequently reach higher EDSS steps may be independent of preceding factors.23
MS Symptom Burden
The neurologic symptoms that patients experience are fluctuating and disabling throughout the disease course, irrespective of onset type. Typical MS symptoms include mobility impairment, changes in cognition and mood, pain and other sensation disturbances, bowel and bladder dysfunction, fatigue, and visual disturbances. The burden of these symptoms can significantly impact quality of life (QOL) for patients and their families. The symptom burden can pose a direct threat to a patient’s autonomy, necessitating adaptation to an unpredictable disease that requires frequent health care visits to many different health care providers (eg, neurologists; primary care providers; physiatrists; urologists; ophthalmologists; and speech, physical, and occupational therapists), periodic testing, and often costly medications.24
Compared with patients who have other chronic conditions, patients with MS experience diminished societal roles, along with decreased assessments in health, energy, and physical functions.25 These often lead to early exit from the workforce and limitations in household responsibilities, which further impact QOL.26 Including both direct and indirect costs of the disease, a patient with MS can expect a lifetime financial burden of nearly $1.2 million.27
Large population cohort studies in MS, along with MS registry studies of patients untreated with disease-modifying therapies, have shown reduced survival rates by an average of 7 to 14 years.23,28 Multiple sclerosis is the main cause of death in about 50% of cases (EDSS step 10), which is defined as “acute death due to brain stem involvement or to respiratory failure, or death consequent to the chronic bedridden state with terminal pneumonia, sepsis, uremia, or cardiorespiratory failure [and excluding] intercurrent causes of death.”23 For the remaining patients with MS, cause of death is similar to those of the general population, such as cardiovascular disease and cancer.23 However, the incidence of suicide is higher among patients with MS.23
All these factors underscore the importance of early diagnosis as well as early initiation of effective disease-modifying therapy.
Disease-Modifying Therapies
The goal of MS disease-modifying therapy is to reduce the early clinical and subclinical disease activity that eventually contributes to long-term disability.31,32 There are currently 13 FDA-approved disease-modifying therapies for MS. These include 7 self-injecting therapies, 3 oral therapies, and 3 infusion therapies. These 13 medications have 8 different mechanisms of action (MOA) that target distinct areas of the immune-mediated disease process. They also differ in their frequencies and routes of administration in addition to their adverse effect (AE) profiles (Tables 2, 3, and 4).
Treatment Considerations
In 1993, interferon beta-1b became the first FDA-approved MS medication. In the following 2 decades, there became 12 additional FDA-approved medications for MS, beginning with other injectables. The first infusion therapy was introduced in 2004, followed by various oral medications. The treatment landscape continues to change rapidly. This therapeutic revolution has occurred largely due to the improved understanding of the pathophysiology of MS and unquestionably has improved the prognosis and overall QOL for patients. The question is no longer how to treat MS but rather how to personalize and optimize treatment for each patient.20
Despite all available treatment options, none are curative, and none have been proven to offer neuroprotection or contribute to neural repair. To date, no studies have led to FDA-approved therapies for PPMS. Further, the efficacy of any of these medications varies from patient to patient. Due largely to the lack of biomarkers for disease activity and treatment response, drug efficacy continues to be measured according to the current gold standard, which is identification of gadolinium-enhancing lesions in the white matter on magnetic resonance imaging (MRI), combined with other markers of disease, including clinical relapse rate and confirmed disability progression.19 In general, the injectable therapies are expected to protect against about 20% to 35% of relapses; the oral agents, 50% to 55%; and the infusion therapies, > 60%.2
In conjunction with a medication’s efficacy rate and safety profile, the frequency and route of administration also must be considered. In general, MS medications are exceedingly expensive, some costing up to tens-of-thousands of dollars per year.48 All these factors have the real potential to negatively impact patient adherence. Nonadherence and gaps in treatment have been correlated with increased rates of relapses and progression of disability as well as negative MRI outcomes.49-53
When to Initiate Treatment
Once a patient is diagnosed, a common question is, when is the right time to initiate treatment? The primary target of the current MS medications is to decrease CNS inflammation (relapses). The ideal time to start treatment is as promptly as possible after confirmation of the diagnosis to combat the early inflammatory relapsing phase of the disease. There seems to be an early window in the disease course when achieving disease control can have a profound impact on long-term disability. Disease control is typically defined as decreasing relapses, slowing the accumulation of lesions visualized on MRI, and preventing the disability that occurs from both incomplete recovery after relapses and overall disease progression.54,55
Certain clinical indicators, such as higher relapse rates early in the disease course and MRI characteristics, including total lesion burden and the location of lesions within the CNS, seem to be associated with a higher risk of disease progression.56 These are potential prognostic indicators that can help tailor the choice of disease-modifying therapy for patients.57 Those with highly inflammatory and potentially aggressive disease at onset, for example, may benefit from early initiation of higher efficacy therapies, whereas those with more benign forms of MS at onset may fare well on lower efficacy therapies. In general, when it comes to currently available MS treatments, higher efficacy is often tied to riskier AE profiles, so the best medication may be the “least efficacious” one that can still control the disease.20
Hauser and colleagues suggested a treatment decision-making model that identifies the interferons, glatiramer acetate, dimethyl fumarate, and teriflunomide as acceptable first-line therapies; fingolimod and natalizumab as acceptable second-line options; and mitoxantrone and alemtuzumab as acceptable third-line therapeutic options.20 The authors generally agree with Hauser and colleagues’ model, and it is important to consider individual patient factors (eg, comorbidities, concurrent medications, life circumstances) and disease severity when deciding on a treatment plan.
Perhaps an even more difficult question is, when is the right time to switch therapies? There remains a dearth of either guidelines or comparative studies for treatment management decisions. Further, without reliable biomarkers, the clinical and pathologic heterogeneity of MS makes treatment difficult.4,19 In practice, there is general consensus that 1 year of treatment monitoring for effects on clinical and radiologic outcomes is an acceptable time frame to evaluate effectiveness of a disease-modifying treatment. If adherence is maintained and there is still evidence of clinical or MRI activity (suggesting a suboptimal response), an alternative therapy, particularly one with a different MOA, should be strongly considered. This highlights the importance of broad access to all available MS therapies to allow for early selection of a correct therapy that patients will remain adherent to and that controls their disease.
Conclusion
Multiple sclerosis remains a highly unpredictable disease, and relapses have the ability to produce a measurable and sustained impact on the level of disability.58 Still, the influence of reduced relapses on preventing disability in an individual patient remains unclear. Large, long-term, prospective cohort studies may clarify whether early treatment affects disease progression and disability.20 However, it is quite evident that effective relapse reduction decreases discomfort, reduces days lost from work and other important activities of daily life, and improves QOL.58,59
There is still much to learn about this unique disease, but emerging evidence in the medical literature highlights the importance of setting treatment goals that include targeting disease activity to achieve early and effective control. Attaining control with a MS medication seems to be a key component of slowing the physical and emotional disability that can accumulate, helping patients remain active and maintain the highest QOL possible for as long as possible.
Multiple sclerosis (MS) is a disorder characterized by inflammation, demyelination, and degeneration of the central nervous system (CNS). The hallmark of the disorder is relapses and remissions of neurologic symptoms occurring early in the disease course, which are often associated with areas of CNS inflammation and myelin loss.1-3 The inciting cause for this inflammation is unknown but is believed to be multifactorial, with environmental and genetic influences creating an adaptive, T cell-mediated autoimmune response against the CNS.4 Separate from the acute attacks, progressive neurodegeneration can occur more chronically and is characterized by axonal loss and grey matter atrophy thought to be due to direct cytotoxic activity of the innate immune system as well as toxic intermediates, such as nitric oxide.4,5 Despite the multiple insults early on, neurologic disability typically becomes more apparent over time.6 The disability threshold theory argues that neurologic function compensates for brain tissue loss until a threshold of accumulated damage is exceeded.7
Background
The incidence of MS follows a geographic gradient; rates rise as the distance from the equator increases.8,9 This is thought to be due to the gradient of relative sun exposure and its role in the production of vitamin D, which plays an important role in immune regulation when converted to its active hormonal form. Multiple sclerosis is more prevalent in non-Hispanic white patients than it is in other racial groups, and women are affected nearly 2 to 3 times more often than are men.10 About 450,000 individuals in the U.S. and more than 2 million worldwide have MS.11-14
Multiple sclerosis is the most common cause of nontraumatic neurologic disability in young adults. It is typically diagnosed in the third and fourth decades of life, and those who are diagnosed after age 50 years often can recount neurologic symptoms that began years before. However, pediatric-onset and new-onset cases in the elderly have been reported. It has been estimated that up to 10% of patients with MS have onset before 18 years of age.15-17 Compared with adult-onset MS, pediatric-onset is associated with a longer period between initial attack and physical disability, although the average age of disability onset is about 10 years younger.17,18
Disease Courses
Relapsing-remitting MS (RRMS) is the most common disease course overall, and this pattern affects 97% of individuals with disease onset before age 18 years.15-17 The clinically isolated syndrome disease course leads to clinically definite MS in one-third of patients within 1 year and in one-half of patients within 2 years.19 In the majority of cases, the RRMS course transitions over time to secondary-progressive MS (SPMS), which is a disease pattern of progressively worsening disability with few neurologic relapses. Although inflammation is present at all stages, the difference is in the predominance of cell types involved.5 Why the shift from active to chronic inflammation occurs and how to prevent it remain central questions in MS research.4 Regardless, tentative evidence suggests that prevention of relapses may reduce disability accumulation and risk of conversion to progressive MS.20
A minority of patients with MS are diagnosed with primary-progressive MS (PPMS) at onset, which is characterized by a disease pattern that follows a relatively steady progression of neurologic symptoms over time, without clear relapses or remissions of these symptoms, though phases of stability or fluctuations in disability may still occur.21 It is typically diagnosed at an older age than is RRMS, and it is rare in children; suspicion of PPMS in this age group should prompt detailed assessment of alternative diagnoses.17,22 Primary-progressive MS is more equally distributed in men and women than is RRMS.
Regardless of onset type, disability progression seems to occur at the same rate among all patients with MS after a certain threshold is reached. The established assessment scale for disability progression in MS is the Kurtzke Expanded Disability Status scale (EDSS), which has a scoring range from 0 to 10. Data from several patient registries have shown that once EDSS step 4 is reached, progression thereafter occurs at a predictable rate that is similar across MS phenotypes.23 The time it takes patients to subsequently reach higher EDSS steps may be independent of preceding factors.23
MS Symptom Burden
The neurologic symptoms that patients experience are fluctuating and disabling throughout the disease course, irrespective of onset type. Typical MS symptoms include mobility impairment, changes in cognition and mood, pain and other sensation disturbances, bowel and bladder dysfunction, fatigue, and visual disturbances. The burden of these symptoms can significantly impact quality of life (QOL) for patients and their families. The symptom burden can pose a direct threat to a patient’s autonomy, necessitating adaptation to an unpredictable disease that requires frequent health care visits to many different health care providers (eg, neurologists; primary care providers; physiatrists; urologists; ophthalmologists; and speech, physical, and occupational therapists), periodic testing, and often costly medications.24
Compared with patients who have other chronic conditions, patients with MS experience diminished societal roles, along with decreased assessments in health, energy, and physical functions.25 These often lead to early exit from the workforce and limitations in household responsibilities, which further impact QOL.26 Including both direct and indirect costs of the disease, a patient with MS can expect a lifetime financial burden of nearly $1.2 million.27
Large population cohort studies in MS, along with MS registry studies of patients untreated with disease-modifying therapies, have shown reduced survival rates by an average of 7 to 14 years.23,28 Multiple sclerosis is the main cause of death in about 50% of cases (EDSS step 10), which is defined as “acute death due to brain stem involvement or to respiratory failure, or death consequent to the chronic bedridden state with terminal pneumonia, sepsis, uremia, or cardiorespiratory failure [and excluding] intercurrent causes of death.”23 For the remaining patients with MS, cause of death is similar to those of the general population, such as cardiovascular disease and cancer.23 However, the incidence of suicide is higher among patients with MS.23
All these factors underscore the importance of early diagnosis as well as early initiation of effective disease-modifying therapy.
Disease-Modifying Therapies
The goal of MS disease-modifying therapy is to reduce the early clinical and subclinical disease activity that eventually contributes to long-term disability.31,32 There are currently 13 FDA-approved disease-modifying therapies for MS. These include 7 self-injecting therapies, 3 oral therapies, and 3 infusion therapies. These 13 medications have 8 different mechanisms of action (MOA) that target distinct areas of the immune-mediated disease process. They also differ in their frequencies and routes of administration in addition to their adverse effect (AE) profiles (Tables 2, 3, and 4).
Treatment Considerations
In 1993, interferon beta-1b became the first FDA-approved MS medication. In the following 2 decades, there became 12 additional FDA-approved medications for MS, beginning with other injectables. The first infusion therapy was introduced in 2004, followed by various oral medications. The treatment landscape continues to change rapidly. This therapeutic revolution has occurred largely due to the improved understanding of the pathophysiology of MS and unquestionably has improved the prognosis and overall QOL for patients. The question is no longer how to treat MS but rather how to personalize and optimize treatment for each patient.20
Despite all available treatment options, none are curative, and none have been proven to offer neuroprotection or contribute to neural repair. To date, no studies have led to FDA-approved therapies for PPMS. Further, the efficacy of any of these medications varies from patient to patient. Due largely to the lack of biomarkers for disease activity and treatment response, drug efficacy continues to be measured according to the current gold standard, which is identification of gadolinium-enhancing lesions in the white matter on magnetic resonance imaging (MRI), combined with other markers of disease, including clinical relapse rate and confirmed disability progression.19 In general, the injectable therapies are expected to protect against about 20% to 35% of relapses; the oral agents, 50% to 55%; and the infusion therapies, > 60%.2
In conjunction with a medication’s efficacy rate and safety profile, the frequency and route of administration also must be considered. In general, MS medications are exceedingly expensive, some costing up to tens-of-thousands of dollars per year.48 All these factors have the real potential to negatively impact patient adherence. Nonadherence and gaps in treatment have been correlated with increased rates of relapses and progression of disability as well as negative MRI outcomes.49-53
When to Initiate Treatment
Once a patient is diagnosed, a common question is, when is the right time to initiate treatment? The primary target of the current MS medications is to decrease CNS inflammation (relapses). The ideal time to start treatment is as promptly as possible after confirmation of the diagnosis to combat the early inflammatory relapsing phase of the disease. There seems to be an early window in the disease course when achieving disease control can have a profound impact on long-term disability. Disease control is typically defined as decreasing relapses, slowing the accumulation of lesions visualized on MRI, and preventing the disability that occurs from both incomplete recovery after relapses and overall disease progression.54,55
Certain clinical indicators, such as higher relapse rates early in the disease course and MRI characteristics, including total lesion burden and the location of lesions within the CNS, seem to be associated with a higher risk of disease progression.56 These are potential prognostic indicators that can help tailor the choice of disease-modifying therapy for patients.57 Those with highly inflammatory and potentially aggressive disease at onset, for example, may benefit from early initiation of higher efficacy therapies, whereas those with more benign forms of MS at onset may fare well on lower efficacy therapies. In general, when it comes to currently available MS treatments, higher efficacy is often tied to riskier AE profiles, so the best medication may be the “least efficacious” one that can still control the disease.20
Hauser and colleagues suggested a treatment decision-making model that identifies the interferons, glatiramer acetate, dimethyl fumarate, and teriflunomide as acceptable first-line therapies; fingolimod and natalizumab as acceptable second-line options; and mitoxantrone and alemtuzumab as acceptable third-line therapeutic options.20 The authors generally agree with Hauser and colleagues’ model, and it is important to consider individual patient factors (eg, comorbidities, concurrent medications, life circumstances) and disease severity when deciding on a treatment plan.
Perhaps an even more difficult question is, when is the right time to switch therapies? There remains a dearth of either guidelines or comparative studies for treatment management decisions. Further, without reliable biomarkers, the clinical and pathologic heterogeneity of MS makes treatment difficult.4,19 In practice, there is general consensus that 1 year of treatment monitoring for effects on clinical and radiologic outcomes is an acceptable time frame to evaluate effectiveness of a disease-modifying treatment. If adherence is maintained and there is still evidence of clinical or MRI activity (suggesting a suboptimal response), an alternative therapy, particularly one with a different MOA, should be strongly considered. This highlights the importance of broad access to all available MS therapies to allow for early selection of a correct therapy that patients will remain adherent to and that controls their disease.
Conclusion
Multiple sclerosis remains a highly unpredictable disease, and relapses have the ability to produce a measurable and sustained impact on the level of disability.58 Still, the influence of reduced relapses on preventing disability in an individual patient remains unclear. Large, long-term, prospective cohort studies may clarify whether early treatment affects disease progression and disability.20 However, it is quite evident that effective relapse reduction decreases discomfort, reduces days lost from work and other important activities of daily life, and improves QOL.58,59
There is still much to learn about this unique disease, but emerging evidence in the medical literature highlights the importance of setting treatment goals that include targeting disease activity to achieve early and effective control. Attaining control with a MS medication seems to be a key component of slowing the physical and emotional disability that can accumulate, helping patients remain active and maintain the highest QOL possible for as long as possible.
1. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46(4):907-911.
2. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(pt 5):1175-1189.
3. Charil A, Filippi M. Inflammatory demyelination and neurodegeneration in early multiple sclerosis. J Neurol Sci. 2007;259(1-2):7-15.
4. Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65(3):239-248.
5. Grigoriadis N, van Pesch V; ParadigMS Group. A basic overview of multiple sclerosis immunopathology. Eur J Neurol. 2015;22(suppl 2):3-13.
6. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647-656.
7. Rudick RA, Lee JC, Simon J, Fisher E. Significance of T2 lesions in multiple sclerosis: a 13-year longitudinal study. Ann Neurol. 2006;60(2):236-242.
8. Alla S, Mason DF. Multiple sclerosis in New Zealand. J Clin Neurosci. 2014;21(8):1288-1291.
9. Simpson S Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(10):1132-1141.
10. Evans C, Beland SG, Kulaga S, et al. Incidence and prevalence of multiple sclerosis in the Americas: a systematic review. Neuroepidemiology. 2013;40(3):195-210.
11. Giesser BS. Diagnosis of multiple sclerosis. Neurol Clin. 2011;29(2):381-388.
12. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938-952.
13. Weinshenker BG. The natural history of multiple sclerosis. Neurol Clin. 1995;13(1):119-146.
14. Weinshenker BG. The natural history of multiple sclerosis: update 1998. Semin Neurol. 1998;18(3):301-307.
15. Simone IL, Carrara D, Tortorella C, Ceccarelli A, Livrea P. Early onset multiple sclerosis. Neurol Sci. 2000;21(4)(suppl 2):S861-S863.
16. Reinhardt K, Weiss S, Rosenbauer J, Gärtner J, von Kries R. Multiple sclerosis in children and adolescents: incidence and clinical picture--new insights from the nationwide German surveillance (2009-2011). Eur J Neurol. 2014;21(4):654-659.
17. Waldman A, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M, Banwell B. Multiple sclerosis in children: an update on clinical diagnosis, therapeutic strategies, and research. Lancet Neurol. 2014;13(9):936-948.
18. Renoux C, Vukusic S, Mikaeloff Y, et al; Adult Neurology Departments KIDMUS Study Group. Natural history of multiple sclerosis with childhood onset. N Engl J Med. 2007;356(25):2603-2613.
19. D'Ambrosio A, Pontecorvo S, Colastanti T, Zamboni S, Francia A, Margutti P. Peripheral blood biomarkers in multiple sclerosis. Autoimmun Rev. 2015;14(12):1097-1110.
20. Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol. 2013;74(3):317-327.
21. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
22. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
23. Hurwitz BJ. Analysis of current multiple sclerosis registries. Neurology. 2011;76(1)(suppl 1):S7-S13.
24. Boeije HR, Duijnstee MS, Grypdonck MH, Pool A. Encountering the downward phase: biographical work in people with multiple sclerosis living at home. Soc Sci Med. 2002;55(6):881-893.
25. Sprangers MA, de Regt EB, Andries F, et al. Which chronic conditions are associated with better or poorer quality of life? J Clin Epidemiol. 2000;53(9):895-907.
26. Julian LJ, Vella L, Vollmer T, Hadjimichael O, Mohr DC. Employment in multiple sclerosis. Exiting and re-entering the work force. J Neurol. 2008;255(9):1354-1360.
27. Trisolini M, Honeycutt A, Wiener J, Lesesne S. Global economic impact of multiple sclerosis. Multiple Sclerosis International Federation website. http://www.msif.org/wp-content/uploads/2014/09/Global_economic_impact_of_MS.pdf. Published May 2010. Accessed May 6, 2016
28. Scalfari A, Knappertz V, Cutter G, Goodin DS, Ashton R, Ebers GC. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
29. Gurevich M, Miron G, Achiron A. Optimizing multiple sclerosis diagnosis: gene expression and genomic association. Ann Clin Transl Neurol. 2015;2(3):271-277.
30. National Multiple Sclerosis Society, European Committee for Treatment and Research in Multiple Sclerosis. Tip sheet: 2010 revised McDonald diagnostic criteria for MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Paper-TipSheet_-2010-Revisions-to-the-McDonald-Criteria-for-the-Diagnosis-of-MS.pdf. Accessed April 22, 2016.
31. Freedman MS, Selchen D, Arnold DL, et al; Canadian Multiple Sclerosis Working Group. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013;40(3):307-323.
32. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
33. Copaxone [package insert]. Overland Park, KS: Teva Neuroscience, Inc; 2014.
34. Avonex [package insert]. Cambridge, MA: Biogen Idec Inc; 1996.
35. Rebif [package insert]. Rockland, MA: EMD Serono, Inc; New York, NY: Pfizer, Inc; 2012.
36. Betaseron [package insert]. Montville, NJ: Bayer HealthCare Pharmaceuticals Inc; 2012.
37. Extavia [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2014.
38. Plegridy [package insert]. Cambridge, MA: Biogen Idec Inc; 2013.
39. Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon ß-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657-665.
40. Tecfidera [package insert]. Cambridge, MA: Biogen Idec Inc; 2015.
41. Gilenya [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2016.
42. Aubagio [package insert]. Cambridge, MA: Genzyme Corp; 2012.
43. Lemtrada [package insert]. Cambridge, MA: Genzyme Corp; 2014.
44. Cohen JA, Coles AJ, Arnold DL, et al; CARE-MS I investigators. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819-1828.
45. Coles AJ, Twyman CL, Arnold DL, et al; CARE-MS II investigators. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829-1839.
46. Novantrone [package insert]. Rockland, MA: EMD Serono, Inc; 2008.
47. Tysabri [medication guide]. Cambridge, MA: Biogen Idec Inc; 2015.
48. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail. Neurology. 2015;84(21):2185-2192.
49. Cohen BA, Coyle PK, Leist T, Oleen-Burkey MA, Schwartz M, Zwibel H. Therapy Optimization in Multiple Sclerosis: a cohort study of therapy adherence and risk of relapse. Mult Scler Relat Disord. 2015;4(1):75-82.
50. Cohen B, Leist T, Coyle P, Zwibel H, Markowitz C, Tullman M. MS therapy adherence and relapse risk. Neurology. 2013;80(7) (suppl):P01.193.
51. Richert ND, Zierak MC, Bash CN, Lewis BK, McFarland HF, Frank JA. MRI and clinical activity in MS patients after terminating treatment with interferon beta-1b. Mult Scler. 2000;6(2):86-90.
52. Siger M, Durko A, Nicpan A, Konarska M, Grudziecka M, Selmaj K. Discontinuation of interferon beta therapy in multiple sclerosis patients with high pre-treatment disease activity leads to prompt return to previous disease activity. J Neurol Sci. 2011;303(1-2):50-52.
53. Wu X, Dastidar P, Kuusisto H, Ukkonen M, Huhtala H, Elovaara I. Increased disability and MRI lesions after discontinuation of IFN-beta-1a in secondary progressive MS. Acta Neurol Scand. 2005;112(4):242-247.
54. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133(pt 7):1914-1929.
55. Bates D. Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology. 2011;76(1)(suppl 1):S14-S25.
56. Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131(pt 3):808-817.
57. Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275(4):350-363.
58. Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003;61(11):1528-1532.
59. Kalb R. The emotional and psychological impact of multiple sclerosis relapses. J Neurol Sci. 2007;256(suppl 1):S29-S33.
1. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46(4):907-911.
2. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(pt 5):1175-1189.
3. Charil A, Filippi M. Inflammatory demyelination and neurodegeneration in early multiple sclerosis. J Neurol Sci. 2007;259(1-2):7-15.
4. Weiner HL. The challenge of multiple sclerosis: how do we cure a chronic heterogeneous disease? Ann Neurol. 2009;65(3):239-248.
5. Grigoriadis N, van Pesch V; ParadigMS Group. A basic overview of multiple sclerosis immunopathology. Eur J Neurol. 2015;22(suppl 2):3-13.
6. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647-656.
7. Rudick RA, Lee JC, Simon J, Fisher E. Significance of T2 lesions in multiple sclerosis: a 13-year longitudinal study. Ann Neurol. 2006;60(2):236-242.
8. Alla S, Mason DF. Multiple sclerosis in New Zealand. J Clin Neurosci. 2014;21(8):1288-1291.
9. Simpson S Jr, Blizzard L, Otahal P, Van der Mei I, Taylor B. Latitude is significantly associated with the prevalence of multiple sclerosis: a meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(10):1132-1141.
10. Evans C, Beland SG, Kulaga S, et al. Incidence and prevalence of multiple sclerosis in the Americas: a systematic review. Neuroepidemiology. 2013;40(3):195-210.
11. Giesser BS. Diagnosis of multiple sclerosis. Neurol Clin. 2011;29(2):381-388.
12. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343(13):938-952.
13. Weinshenker BG. The natural history of multiple sclerosis. Neurol Clin. 1995;13(1):119-146.
14. Weinshenker BG. The natural history of multiple sclerosis: update 1998. Semin Neurol. 1998;18(3):301-307.
15. Simone IL, Carrara D, Tortorella C, Ceccarelli A, Livrea P. Early onset multiple sclerosis. Neurol Sci. 2000;21(4)(suppl 2):S861-S863.
16. Reinhardt K, Weiss S, Rosenbauer J, Gärtner J, von Kries R. Multiple sclerosis in children and adolescents: incidence and clinical picture--new insights from the nationwide German surveillance (2009-2011). Eur J Neurol. 2014;21(4):654-659.
17. Waldman A, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M, Banwell B. Multiple sclerosis in children: an update on clinical diagnosis, therapeutic strategies, and research. Lancet Neurol. 2014;13(9):936-948.
18. Renoux C, Vukusic S, Mikaeloff Y, et al; Adult Neurology Departments KIDMUS Study Group. Natural history of multiple sclerosis with childhood onset. N Engl J Med. 2007;356(25):2603-2613.
19. D'Ambrosio A, Pontecorvo S, Colastanti T, Zamboni S, Francia A, Margutti P. Peripheral blood biomarkers in multiple sclerosis. Autoimmun Rev. 2015;14(12):1097-1110.
20. Hauser SL, Chan JR, Oksenberg JR. Multiple sclerosis: prospects and promise. Ann Neurol. 2013;74(3):317-327.
21. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278-286.
22. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292-302.
23. Hurwitz BJ. Analysis of current multiple sclerosis registries. Neurology. 2011;76(1)(suppl 1):S7-S13.
24. Boeije HR, Duijnstee MS, Grypdonck MH, Pool A. Encountering the downward phase: biographical work in people with multiple sclerosis living at home. Soc Sci Med. 2002;55(6):881-893.
25. Sprangers MA, de Regt EB, Andries F, et al. Which chronic conditions are associated with better or poorer quality of life? J Clin Epidemiol. 2000;53(9):895-907.
26. Julian LJ, Vella L, Vollmer T, Hadjimichael O, Mohr DC. Employment in multiple sclerosis. Exiting and re-entering the work force. J Neurol. 2008;255(9):1354-1360.
27. Trisolini M, Honeycutt A, Wiener J, Lesesne S. Global economic impact of multiple sclerosis. Multiple Sclerosis International Federation website. http://www.msif.org/wp-content/uploads/2014/09/Global_economic_impact_of_MS.pdf. Published May 2010. Accessed May 6, 2016
28. Scalfari A, Knappertz V, Cutter G, Goodin DS, Ashton R, Ebers GC. Mortality in patients with multiple sclerosis. Neurology. 2013;81(2):184-192.
29. Gurevich M, Miron G, Achiron A. Optimizing multiple sclerosis diagnosis: gene expression and genomic association. Ann Clin Transl Neurol. 2015;2(3):271-277.
30. National Multiple Sclerosis Society, European Committee for Treatment and Research in Multiple Sclerosis. Tip sheet: 2010 revised McDonald diagnostic criteria for MS. National Multiple Sclerosis Society website. http://www.nationalmssociety.org/NationalMSSociety/media/MSNationalFiles/Brochures/Paper-TipSheet_-2010-Revisions-to-the-McDonald-Criteria-for-the-Diagnosis-of-MS.pdf. Accessed April 22, 2016.
31. Freedman MS, Selchen D, Arnold DL, et al; Canadian Multiple Sclerosis Working Group. Treatment optimization in MS: Canadian MS Working Group updated recommendations. Can J Neurol Sci. 2013;40(3):307-323.
32. Gold R, Wolinsky JS, Amato MP, Comi G. Evolving expectations around early management of multiple sclerosis. Ther Adv Neurol Disord. 2010;3(6):351-367.
33. Copaxone [package insert]. Overland Park, KS: Teva Neuroscience, Inc; 2014.
34. Avonex [package insert]. Cambridge, MA: Biogen Idec Inc; 1996.
35. Rebif [package insert]. Rockland, MA: EMD Serono, Inc; New York, NY: Pfizer, Inc; 2012.
36. Betaseron [package insert]. Montville, NJ: Bayer HealthCare Pharmaceuticals Inc; 2012.
37. Extavia [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2014.
38. Plegridy [package insert]. Cambridge, MA: Biogen Idec Inc; 2013.
39. Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon ß-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 2014;13(7):657-665.
40. Tecfidera [package insert]. Cambridge, MA: Biogen Idec Inc; 2015.
41. Gilenya [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corp; 2016.
42. Aubagio [package insert]. Cambridge, MA: Genzyme Corp; 2012.
43. Lemtrada [package insert]. Cambridge, MA: Genzyme Corp; 2014.
44. Cohen JA, Coles AJ, Arnold DL, et al; CARE-MS I investigators. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819-1828.
45. Coles AJ, Twyman CL, Arnold DL, et al; CARE-MS II investigators. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829-1839.
46. Novantrone [package insert]. Rockland, MA: EMD Serono, Inc; 2008.
47. Tysabri [medication guide]. Cambridge, MA: Biogen Idec Inc; 2015.
48. Hartung DM, Bourdette DN, Ahmed SM, Whitham RH. The cost of multiple sclerosis drugs in the US and the pharmaceutical industry: too big to fail. Neurology. 2015;84(21):2185-2192.
49. Cohen BA, Coyle PK, Leist T, Oleen-Burkey MA, Schwartz M, Zwibel H. Therapy Optimization in Multiple Sclerosis: a cohort study of therapy adherence and risk of relapse. Mult Scler Relat Disord. 2015;4(1):75-82.
50. Cohen B, Leist T, Coyle P, Zwibel H, Markowitz C, Tullman M. MS therapy adherence and relapse risk. Neurology. 2013;80(7) (suppl):P01.193.
51. Richert ND, Zierak MC, Bash CN, Lewis BK, McFarland HF, Frank JA. MRI and clinical activity in MS patients after terminating treatment with interferon beta-1b. Mult Scler. 2000;6(2):86-90.
52. Siger M, Durko A, Nicpan A, Konarska M, Grudziecka M, Selmaj K. Discontinuation of interferon beta therapy in multiple sclerosis patients with high pre-treatment disease activity leads to prompt return to previous disease activity. J Neurol Sci. 2011;303(1-2):50-52.
53. Wu X, Dastidar P, Kuusisto H, Ukkonen M, Huhtala H, Elovaara I. Increased disability and MRI lesions after discontinuation of IFN-beta-1a in secondary progressive MS. Acta Neurol Scand. 2005;112(4):242-247.
54. Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133(pt 7):1914-1929.
55. Bates D. Treatment effects of immunomodulatory therapies at different stages of multiple sclerosis in short-term trials. Neurology. 2011;76(1)(suppl 1):S14-S25.
56. Fisniku LK, Brex PA, Altmann DR, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131(pt 3):808-817.
57. Cross AH, Naismith RT. Established and novel disease-modifying treatments in multiple sclerosis. J Intern Med. 2014;275(4):350-363.
58. Lublin FD, Baier M, Cutter G. Effect of relapses on development of residual deficit in multiple sclerosis. Neurology. 2003;61(11):1528-1532.
59. Kalb R. The emotional and psychological impact of multiple sclerosis relapses. J Neurol Sci. 2007;256(suppl 1):S29-S33.