User login
The Diagnosis: Persistent Still Disease
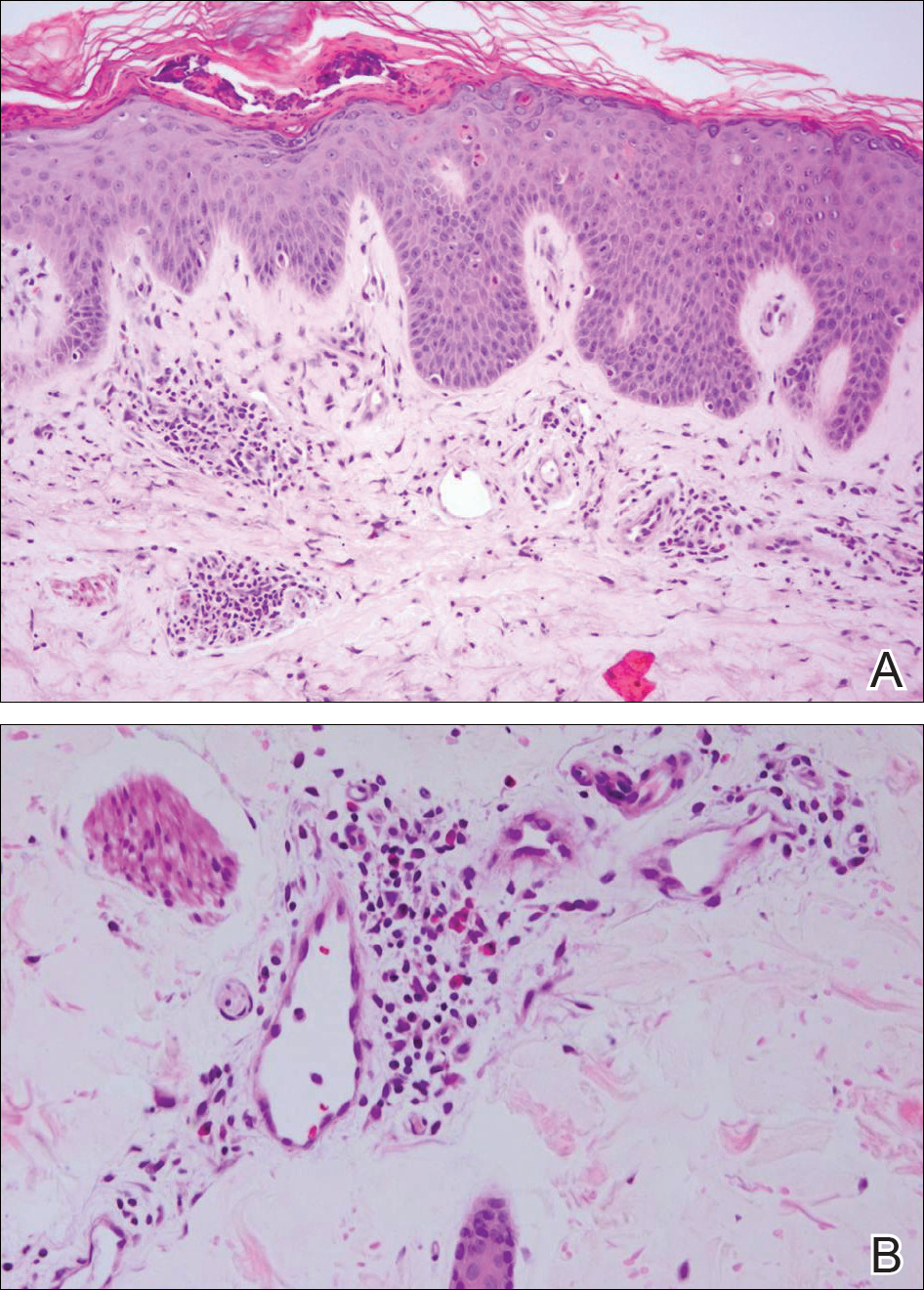
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
The Diagnosis: Persistent Still Disease
At the time of presentation, the patient had not taken systemic medications for a year. Laboratory studies revealed leukocytosis with neutrophilia and a serum ferritin level of 5493 ng/mL (reference range, 15-200 ng/mL). Rheumatoid factor and antinuclear antibody serologies were within reference range. Microbiologic workup was negative. Lymph node and bone marrow biopsies were negative for a lymphoproliferative disorder. Skin biopsies were performed on the back and forearm. Histologic evaluation revealed orthokeratosis, slight acanthosis, and dyskeratosis confined to the upper layers of the epidermis without evidence of interface dermatitis. There was a mixed perivascular infiltrate composed of lymphocytes and neutrophils with no attendant vasculitic change (Figure).
The patient was discharged on prednisone and seen for outpatient follow-up weeks later. Six weeks later, the cutaneous eruption remained unchanged. The patient was unable to start other systemic medications due to lack of insurance and ineligibility for the local patient-assistance program; he was subsequently lost to follow-up.
Adult-onset Still disease is a rare, systemic, inflammatory condition with a broad spectrum of clinical presentations.1-3 Still disease affects all age groups, and children with Still disease (<16 years) usually have a concurrent diagnosis of juvenile idiopathic arthritis (formerly known as juvenile rheumatoid arthritis).1,2,4 Still disease preferentially affects adolescents and adults aged 16 to 35 years, with more than 75% of new cases occurring in this age range.1 Worldwide, the incidence and prevalence of Still disease is disputed with no conclusive rates established.1,3
Still disease is characterized by 4 cardinal signs: high spiking fevers (temperature, ≥39°C); leukocytosis with a predominance of neutrophils (≥10,000 cells/mm3 with ≥80% neutrophils); arthralgia or arthritis; and an evanescent, nonpruritic, salmon-colored morbilliform eruption of the skin, typically on the trunk or extremities.2 Histologic evaluation of the classic Still disease eruption displays perivascular inflammation of the superficial dermis with infiltration by lymphocytes and histiocytes.3
In 1992, major and minor diagnostic criteria were established for adult-onset Still disease. For diagnosis, patients must meet 5 criteria, including 2 major criteria.5 Major criteria include arthralgia or arthritis present for more than 2 weeks, fever (temperature, >39°C) for at least 1 week, the classic Still disease morbilliform eruption (ie, salmon colored, evanescent, morbilliform), and leukocytosis with more than 80% neutrophils. Minor criteria include sore throat, lymphadenopathy and/or splenomegaly, negative rheumatoid factor and antinuclear antibody serologies, and abnormal liver function (defined as elevated transaminases).5 Although not included in the diagnostic criteria, there have been reports of elevated serum ferritin levels in patients with Still disease, a finding that potentially is useful in distinguishing between active and inactive rheumatic conditions.6,7
Several case reports have described persistent Still disease, a subtype of Still disease in which patients present with brown-red, persistent, pruritic macules, papules, and plaques that are widespread and oddly shaped.8,9 Histologically, this subtype is characterized by necrotic keratinocytes in the epidermis and dermal perivascular inflammation composed of neutrophils and lymphocytes.10 This histology differs from classic Still disease in that the latter typically does not have superficial epidermal dyskeratosis. Our case is consistent with reports of persistent Still disease.
Although the etiology of Still disease remains to be elucidated, HLA-B17, -B18, -B35, and -DR2 have been associated with the disease.3 Furthermore, helper T cell TH1, IL-2, IFN-γ, and tumor necrosis factor α have been implicated in disease pathology, enabling the use of newer targeted pharmacologic therapies. Canakinumab, an IL-1β inhibitor, has been found to improve arthritis, fever, and rash in patients with Still disease.11 These findings are particularly encouraging for patients who have not experienced improvement with traditional antirheumatic drugs, such as our patient who was not steroid responsive.3
Although a salmon-colored, evanescent, morbilliform eruption in the context of other systemic signs and symptoms readily evokes consideration of Still disease, the less common fixed cutaneous eruption seen in our case may evade accurate diagnosis. Our case aims to increase awareness of this unusual and rare subtype of the cutaneous eruption of Still disease, as a timely diagnosis may prevent potentially life-threatening sequelae including cardiopulmonary disease and respiratory failure.3,5,9
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
- Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still's disease [published online October 11, 2005]. Ann Rheum Dis. 2006;65:564-572.
- Fautrel B. Adult-onset Still disease. Best Pract Res Clin Rheumatol. 2008;22:773-792.
- Bagnari V, Colina M, Ciancio G, et al. Adult-onset Still's disease. Rheumatol Int. 2010;30:855-862.
- Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767-778.
- Yamaguchi M, Ohta A, Tsunematsu, T, et al. Preliminary criteria for classification of adult Still's disease. J Rheumatol. 1992;19:424-430.
- Van Reeth C, Le Moel G, Lasne Y, et al. Serum ferritin and isoferritins are tools for diagnosis of active adult Still's disease. J Rheumatol. 1994;21:890-895.
- Novak S, Anic F, Luke-Vrbanic TS. Extremely high serum ferritin levels as a main diagnostic tool of adult-onset Still's disease. Rheumatol Int. 2012;32:1091-1094.
- Fortna RR, Gudjonsson JE, Seidel G, et al. Persistent pruritic papules and plaques: a characteristic histopathologic presentation seen in a subset of patients with adult-onset and juvenile Still's disease. J Cutan Pathol. 2010;37:932-937.
- Yang CC, Lee JY, Liu MF, et al. Adult-onset Still's disease with persistent skin eruption and fatal respiratory failure in a Taiwanese woman. Eur J Dermatol. 2006;16:593-594.
- Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still's disease. J Am Acad Dermatol. 2005;52:1003-1008.
- Kontzias A, Efthimiou P. The use of canakinumab, a novel IL-1β long-acting inhibitor in refractory adult-onset Still's disease. Sem Arthritis Rheum. 2012;42:201-205.
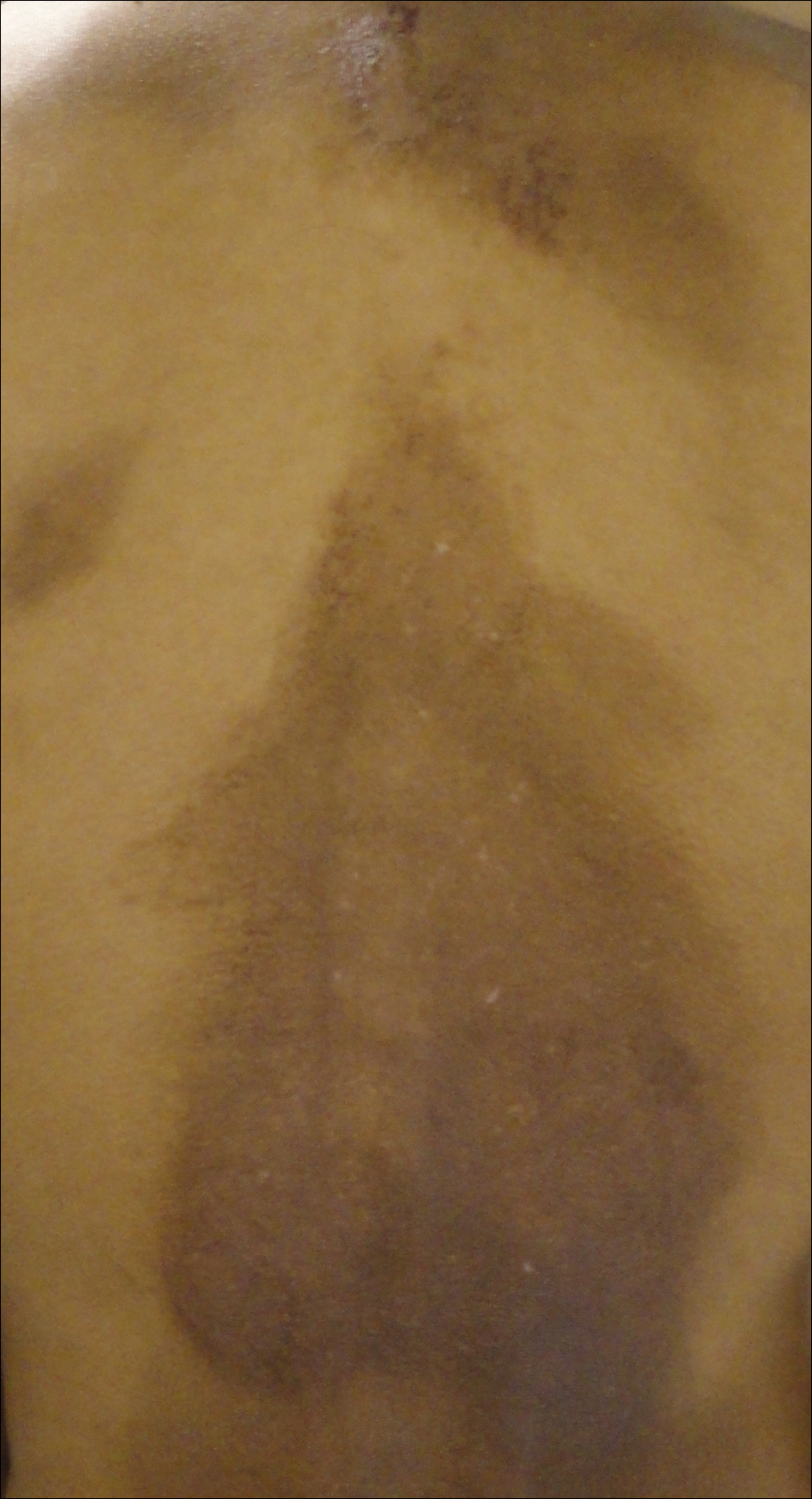
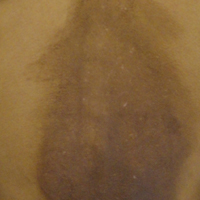
A 25-year-old Hispanic man with a history of juvenile idiopathic arthritis was admitted with a high-grade fever (temperature, >38.9°C) and diffuse nonlocalized abdominal pain of 2 days' duration. Physical examination revealed tachycardia, axillary lymphadenopathy, and hepatosplenomegaly. Cutaneous findings consisted of striking hyperpigmented patches on the chest and back, and hyperpigmented scaly lichenoid papules and plaques on the upper and lower extremities. The plaques on the lower extremities exhibited koebnerization. The patient reported that the eruption initially presented at 16 years of age as pruritic papules on the legs, which gradually spread to involve the arms, chest, and back. Prior treatments of juvenile idiopathic arthritis included prednisone, methotrexate, infliximab, and etanercept, though they were intermittent and temporary. Over time, the cutaneous eruption evolved into its current morphology and distribution, with periods of clearance observed while receiving systemic medications.