User login
The Diagnosis: Bullous Leukocytoclastic Vasculitis
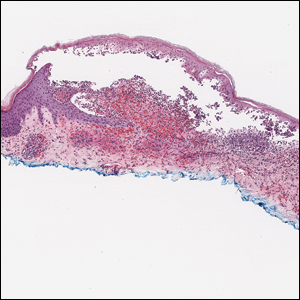
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
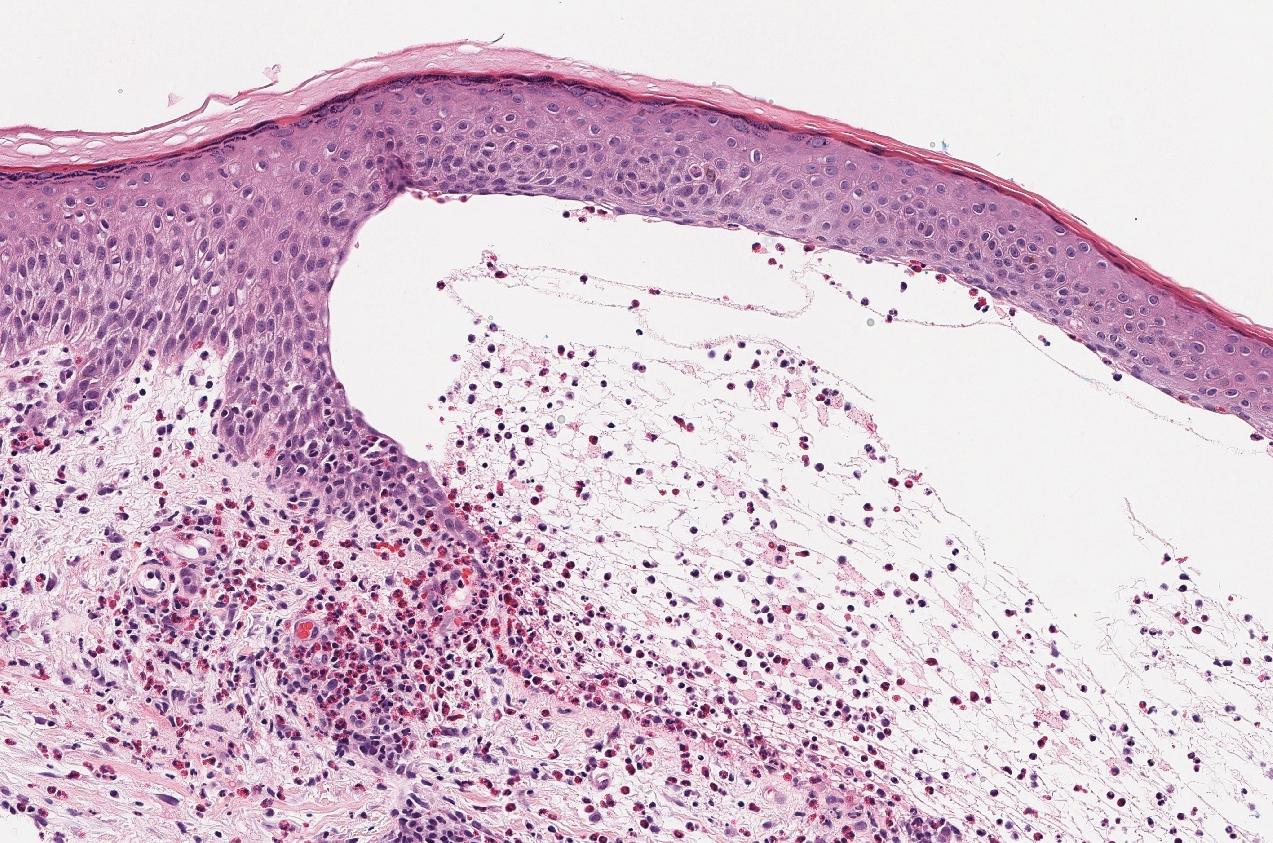
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
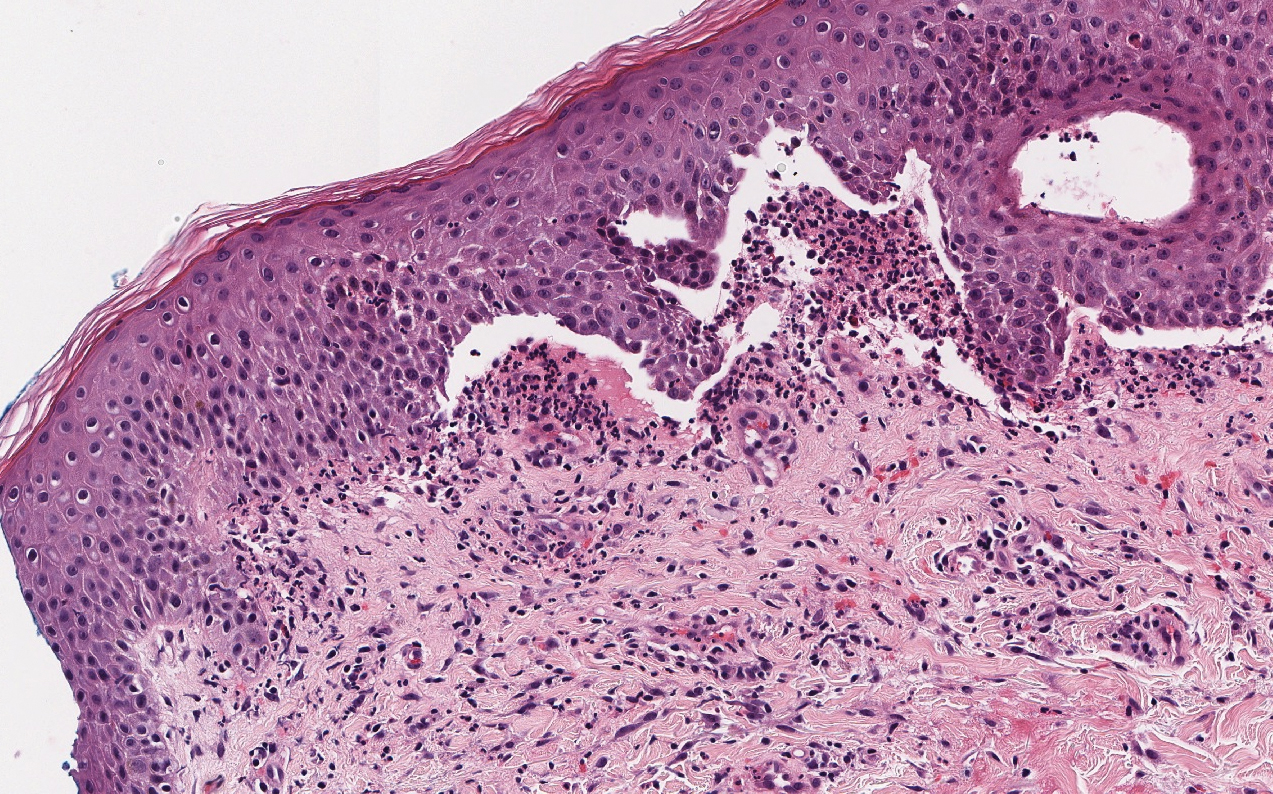
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
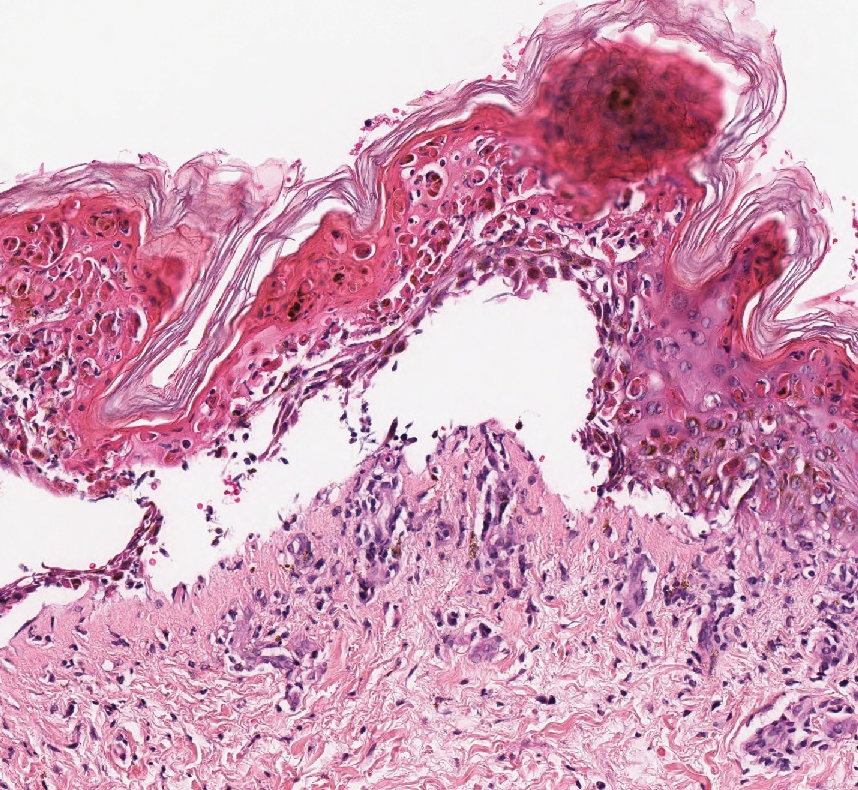
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.

Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
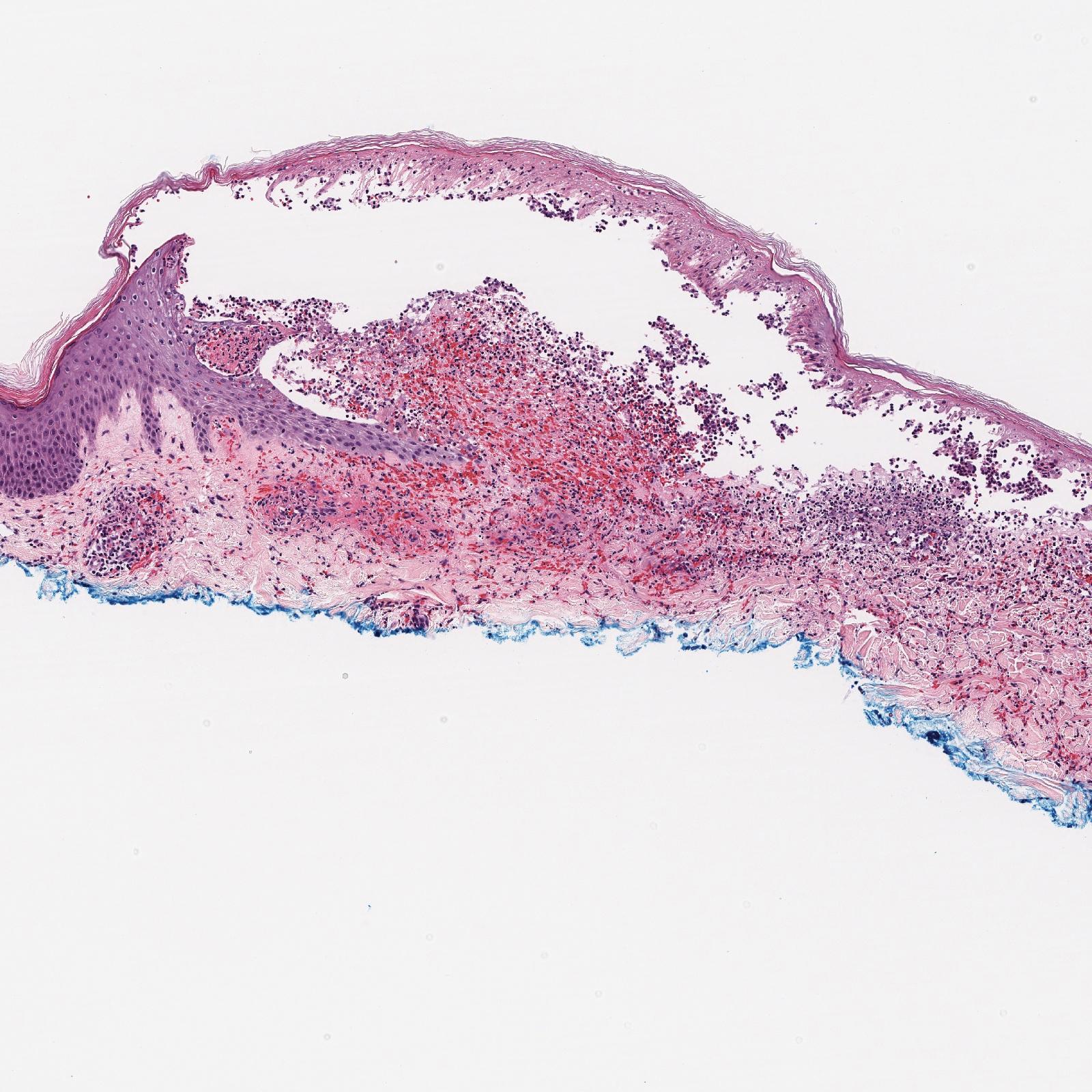
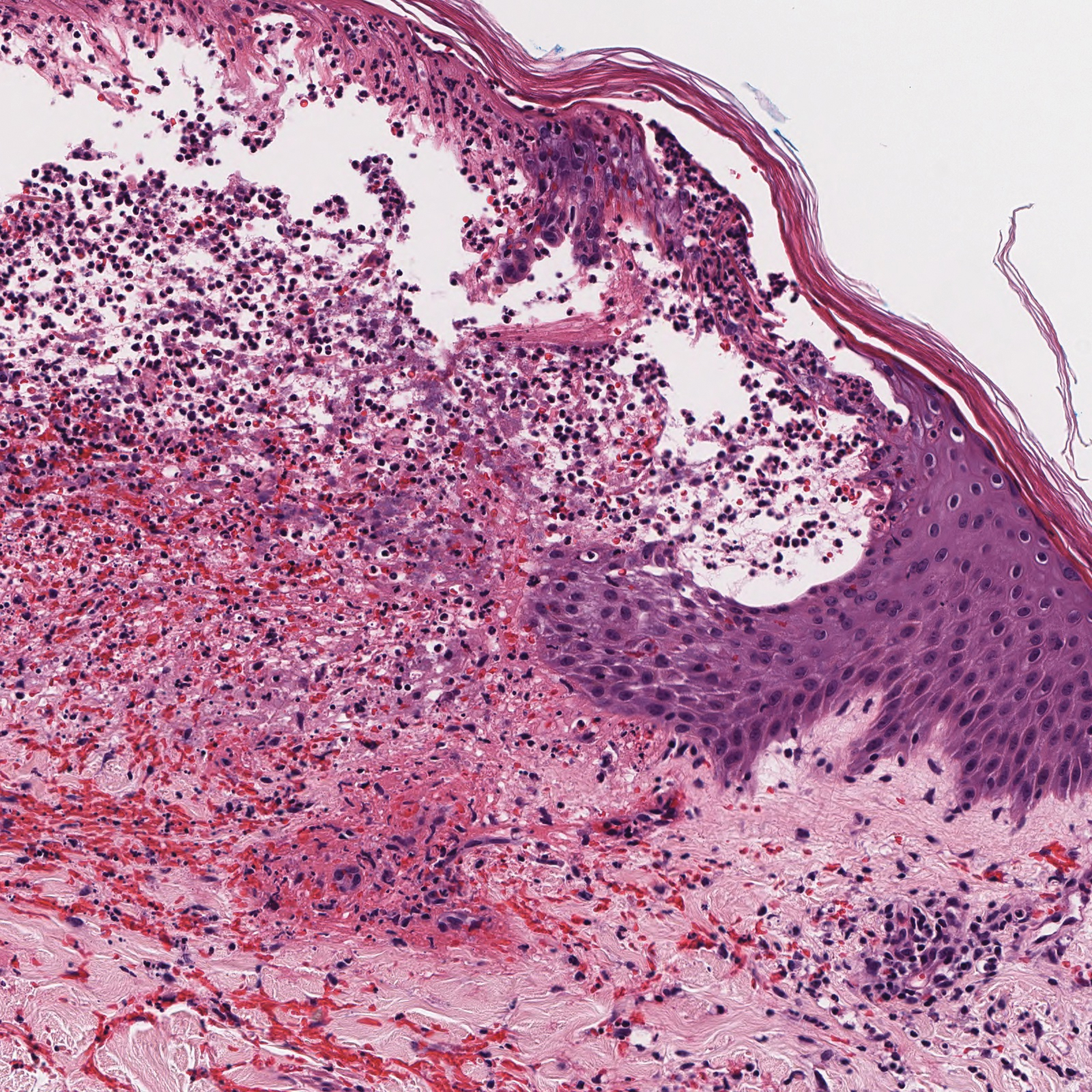
A 30-year-old woman with a medical history of uncontrolled type 2 diabetes mellitus and morbid obesity presented to the dermatology clinic with a painful blistering rash on the lower extremities with scattered red-purple papules of 1 week's duration. The rash began on the left dorsal foot. Physical examination showed nonblanching, 2- to 4-mm, violaceous papules with numerous vesiculopustular bullae on the lower extremities from the dorsal feet to the proximal knee. A shave biopsy with hematoxylin and eosin stain and a punch biopsy for direct immunofluorescence were performed.