User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Overlooked diagnoses
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
“Not all mood swings are bipolar disorder” (Current Psychiatry, February 2011, p. 38-52) is a highly relevant and helpful article with a glaring omission. There is no mention of the emotional lability and behavioral dyscontrol associated with abuse, trauma, and invalidation. “Mood swing” symptoms are prominent in developmental trauma disorder and complex posttraumatic stress disorder, although these diagnoses are not yet in the DSM. Unfortunately, the effects of abuse, trauma, and invalidation often are unrecognized in the differential diagnoses of these children and too often the “kneejerk” diagnoses of bipolar disorder, oppositional defiant disorder, and attention-deficit/hyperactivity disorder are inappropriately assigned, delaying the implementation of trauma theory-informed therapy.
Bradford B. Schwartz, MD
Private Practice
York, PA
The authors respond
We thank Dr. Schwartz for his comments regarding emotional lability and behavioral dyscontrol associated with children who have experienced trauma, abuse, and invalidation. An assessment for possible trauma always is part of the initial assessment of each child referred to our program. None of the patients discussed in our article had a history of abuse or trauma. Referrals to our pediatric mood disorders program initially are screened through the Cincinnati Children’s Hospital Psychiatric Intake and Response Center, which functions as triage, gathering psychiatric history, including assessing trauma, and children with a history of abuse and trauma are referred to other clinicians specializing in this area. But Dr. Schwartz’s point is well taken—trauma or abuse always should be part of the differential diagnosis of children and adolescents referred for mood swings.
Robert A. Kowatch, MD, PhD
Professor of Psychiatry and Pediatrics
Erin Monroe, CNS
Clinical Nurse Specialist
Division of Psychiatry
Sergio V. Delgado, MD
Associate Professor of Psychiatry
and Pediatrics
Cincinnati Children’s Hospital Medical Center
Cincinnati, OH
Polypharmacy subtypes: The necessary, the reasonable, the ridiculous, and the hazardous
You’ve heard about the 2 certainties in life: death and taxes. In psychiatric practice with complex and chronic patients, there is a third certainty: polypharmacy. It ranges from thoughtful to indiscriminate and seems to be entrenched in clinical practice, possibly reflecting practitioners’ desperation in trying to manage severely ill, treatment-resistant patients, usually in the absence of evidence-based guidelines.
I never fail to encounter polypharmacy in hospitals or clinics where I consult. I always wondered how the patient’s doctor knew which drug was exerting a therapeutic effect or which drug was causing side effects (parkinsonism, akathisia, sedation, orthostasis, dizziness, headache, blurry vision, etc. ). Over time, I came to categorize polypharmacy into 4 subtypes that span the spectrum from sensible to absurd. Here is my personal classification, which I trust that you, my readers, have witnessed as well.
Necessary polypharmacy. This variant of polypharmacy is evidence-based and proven in double-blind studies to be more effective than monotherapy. The most prominent example is adding an atypical antipsychotic to a mood stabilizer in bipolar mania. In fact, the superior efficacy of combination therapy in bipolar disorder is one of the oldest forms of rational polypharmacy, is supported by FDA trials, and is indicated whenever mood stabilizer monotherapy is not sufficient. For example, combining lithium and valproate is superior to either drug alone. Another example of FDA-approved combinations is combining small doses of an atypical antipsychotic to an antidepressant for treatment-resistant depression.
Reasonable polypharmacy. Although many of the combinations in this category are not FDA-approved, controlled studies support their use for suffering patients. Examples include:
- An atypical antipsychotic added to a selective serotonin reuptake inhibitor (SSRI) for obsessive-compulsive disorder (OCD) patients who do not improve on SSRI monotherapy.
- Modafinil added to clozapine in patients who suffer substantial and persistent daytime sedation or somnolence.
- Combining 2 antidepressants for major depressive disorder patients who partially respond to 1 antidepressant.
- Combining a mood stabilizer with an antidepressant for bipolar depression to prevent mood switching.
Ridiculous polypharmacy. The sky is the limit to the variations and degrees of ridiculous polypharmacy, but the theme is the same: an absurd concoction of psychotropic drugs across several classes, often including multiple agents from 1 or several classes. Here are examples I have seen in patient records:
- Two atypicals, an anticholinergic, a mood stabilizer, an antidepressant, and 2 benzodiazepines.
- Three antipsychotics (2 atypicals and 1 typical), 2 antidepressants, 3 sedative/hypnotics, and an anticonvulsant for weight control.
- This one takes the cake: 6 antipsychotics (2 typicals and 4 atypicals), plus an anticholinergic, 3 mood stabilizers, 2 antidepressants, 2 sleeping pills, a hypoglycemic agent, 2 antihypertensives, and a statin.
Hazardous polypharmacy. In this category, serious medical complications, toxic effects, or death may occur because of careless combinations of drugs that may interact to produce dangerous kinetic interactions or exacerbate a pre-existing medical condition. Examples include:
- Combining 1 psychotropic with another that may inhibit its metabolism (eg, prescribing fluvoxamine to a severely psychotic patient who developed OCD while receiving clozapine). There have been several toxic reactions and even death because fluvoxamine inhibits cytochrome 1A2, which metabolizes clozapine, thus increasing clozapine blood level by 400% to 500%.
- Combining 2 injectable drugs for agitation that may cause a serious medical complication. An example would be injecting a benzodiazepine such as lorazepam in a patient receiving olanzapine IM, which can cause severe respiratory depression or death.
- Combining several drugs, each of which may prolong the QTc interval, resulting in syncope or torsade de pointes.
Psychopharmacology can relieve the terrible anguish of psychosis, depression, or anxiety, but it also can carry iatrogenic risks if it is not based on scientific evidence. The practice of psychopharmacology requires the fully integrated skills of medical and psychiatric training to maximize benefit while avoiding harm. It also requires basic arithmetic skills: to consider subtracting drugs, not only adding them!

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest,visit http://www.facebook.com/CurrentPsychiatry, or go to our Web site at CurrentPsychiatry.com and click on the “Contact us” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest,visit http://www.facebook.com/CurrentPsychiatry, or go to our Web site at CurrentPsychiatry.com and click on the “Contact us” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest,visit http://www.facebook.com/CurrentPsychiatry, or go to our Web site at CurrentPsychiatry.com and click on the “Contact us” link.
You’ve heard about the 2 certainties in life: death and taxes. In psychiatric practice with complex and chronic patients, there is a third certainty: polypharmacy. It ranges from thoughtful to indiscriminate and seems to be entrenched in clinical practice, possibly reflecting practitioners’ desperation in trying to manage severely ill, treatment-resistant patients, usually in the absence of evidence-based guidelines.
I never fail to encounter polypharmacy in hospitals or clinics where I consult. I always wondered how the patient’s doctor knew which drug was exerting a therapeutic effect or which drug was causing side effects (parkinsonism, akathisia, sedation, orthostasis, dizziness, headache, blurry vision, etc. ). Over time, I came to categorize polypharmacy into 4 subtypes that span the spectrum from sensible to absurd. Here is my personal classification, which I trust that you, my readers, have witnessed as well.
Necessary polypharmacy. This variant of polypharmacy is evidence-based and proven in double-blind studies to be more effective than monotherapy. The most prominent example is adding an atypical antipsychotic to a mood stabilizer in bipolar mania. In fact, the superior efficacy of combination therapy in bipolar disorder is one of the oldest forms of rational polypharmacy, is supported by FDA trials, and is indicated whenever mood stabilizer monotherapy is not sufficient. For example, combining lithium and valproate is superior to either drug alone. Another example of FDA-approved combinations is combining small doses of an atypical antipsychotic to an antidepressant for treatment-resistant depression.
Reasonable polypharmacy. Although many of the combinations in this category are not FDA-approved, controlled studies support their use for suffering patients. Examples include:
- An atypical antipsychotic added to a selective serotonin reuptake inhibitor (SSRI) for obsessive-compulsive disorder (OCD) patients who do not improve on SSRI monotherapy.
- Modafinil added to clozapine in patients who suffer substantial and persistent daytime sedation or somnolence.
- Combining 2 antidepressants for major depressive disorder patients who partially respond to 1 antidepressant.
- Combining a mood stabilizer with an antidepressant for bipolar depression to prevent mood switching.
Ridiculous polypharmacy. The sky is the limit to the variations and degrees of ridiculous polypharmacy, but the theme is the same: an absurd concoction of psychotropic drugs across several classes, often including multiple agents from 1 or several classes. Here are examples I have seen in patient records:
- Two atypicals, an anticholinergic, a mood stabilizer, an antidepressant, and 2 benzodiazepines.
- Three antipsychotics (2 atypicals and 1 typical), 2 antidepressants, 3 sedative/hypnotics, and an anticonvulsant for weight control.
- This one takes the cake: 6 antipsychotics (2 typicals and 4 atypicals), plus an anticholinergic, 3 mood stabilizers, 2 antidepressants, 2 sleeping pills, a hypoglycemic agent, 2 antihypertensives, and a statin.
Hazardous polypharmacy. In this category, serious medical complications, toxic effects, or death may occur because of careless combinations of drugs that may interact to produce dangerous kinetic interactions or exacerbate a pre-existing medical condition. Examples include:
- Combining 1 psychotropic with another that may inhibit its metabolism (eg, prescribing fluvoxamine to a severely psychotic patient who developed OCD while receiving clozapine). There have been several toxic reactions and even death because fluvoxamine inhibits cytochrome 1A2, which metabolizes clozapine, thus increasing clozapine blood level by 400% to 500%.
- Combining 2 injectable drugs for agitation that may cause a serious medical complication. An example would be injecting a benzodiazepine such as lorazepam in a patient receiving olanzapine IM, which can cause severe respiratory depression or death.
- Combining several drugs, each of which may prolong the QTc interval, resulting in syncope or torsade de pointes.
Psychopharmacology can relieve the terrible anguish of psychosis, depression, or anxiety, but it also can carry iatrogenic risks if it is not based on scientific evidence. The practice of psychopharmacology requires the fully integrated skills of medical and psychiatric training to maximize benefit while avoiding harm. It also requires basic arithmetic skills: to consider subtracting drugs, not only adding them!
You’ve heard about the 2 certainties in life: death and taxes. In psychiatric practice with complex and chronic patients, there is a third certainty: polypharmacy. It ranges from thoughtful to indiscriminate and seems to be entrenched in clinical practice, possibly reflecting practitioners’ desperation in trying to manage severely ill, treatment-resistant patients, usually in the absence of evidence-based guidelines.
I never fail to encounter polypharmacy in hospitals or clinics where I consult. I always wondered how the patient’s doctor knew which drug was exerting a therapeutic effect or which drug was causing side effects (parkinsonism, akathisia, sedation, orthostasis, dizziness, headache, blurry vision, etc. ). Over time, I came to categorize polypharmacy into 4 subtypes that span the spectrum from sensible to absurd. Here is my personal classification, which I trust that you, my readers, have witnessed as well.
Necessary polypharmacy. This variant of polypharmacy is evidence-based and proven in double-blind studies to be more effective than monotherapy. The most prominent example is adding an atypical antipsychotic to a mood stabilizer in bipolar mania. In fact, the superior efficacy of combination therapy in bipolar disorder is one of the oldest forms of rational polypharmacy, is supported by FDA trials, and is indicated whenever mood stabilizer monotherapy is not sufficient. For example, combining lithium and valproate is superior to either drug alone. Another example of FDA-approved combinations is combining small doses of an atypical antipsychotic to an antidepressant for treatment-resistant depression.
Reasonable polypharmacy. Although many of the combinations in this category are not FDA-approved, controlled studies support their use for suffering patients. Examples include:
- An atypical antipsychotic added to a selective serotonin reuptake inhibitor (SSRI) for obsessive-compulsive disorder (OCD) patients who do not improve on SSRI monotherapy.
- Modafinil added to clozapine in patients who suffer substantial and persistent daytime sedation or somnolence.
- Combining 2 antidepressants for major depressive disorder patients who partially respond to 1 antidepressant.
- Combining a mood stabilizer with an antidepressant for bipolar depression to prevent mood switching.
Ridiculous polypharmacy. The sky is the limit to the variations and degrees of ridiculous polypharmacy, but the theme is the same: an absurd concoction of psychotropic drugs across several classes, often including multiple agents from 1 or several classes. Here are examples I have seen in patient records:
- Two atypicals, an anticholinergic, a mood stabilizer, an antidepressant, and 2 benzodiazepines.
- Three antipsychotics (2 atypicals and 1 typical), 2 antidepressants, 3 sedative/hypnotics, and an anticonvulsant for weight control.
- This one takes the cake: 6 antipsychotics (2 typicals and 4 atypicals), plus an anticholinergic, 3 mood stabilizers, 2 antidepressants, 2 sleeping pills, a hypoglycemic agent, 2 antihypertensives, and a statin.
Hazardous polypharmacy. In this category, serious medical complications, toxic effects, or death may occur because of careless combinations of drugs that may interact to produce dangerous kinetic interactions or exacerbate a pre-existing medical condition. Examples include:
- Combining 1 psychotropic with another that may inhibit its metabolism (eg, prescribing fluvoxamine to a severely psychotic patient who developed OCD while receiving clozapine). There have been several toxic reactions and even death because fluvoxamine inhibits cytochrome 1A2, which metabolizes clozapine, thus increasing clozapine blood level by 400% to 500%.
- Combining 2 injectable drugs for agitation that may cause a serious medical complication. An example would be injecting a benzodiazepine such as lorazepam in a patient receiving olanzapine IM, which can cause severe respiratory depression or death.
- Combining several drugs, each of which may prolong the QTc interval, resulting in syncope or torsade de pointes.
Psychopharmacology can relieve the terrible anguish of psychosis, depression, or anxiety, but it also can carry iatrogenic risks if it is not based on scientific evidence. The practice of psychopharmacology requires the fully integrated skills of medical and psychiatric training to maximize benefit while avoiding harm. It also requires basic arithmetic skills: to consider subtracting drugs, not only adding them!
Glutamate: New hope for schizophrenia treatment
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
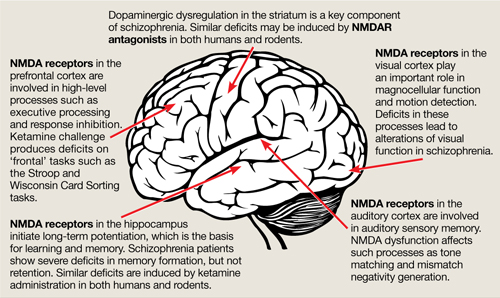
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6
NMDAR antagonists also reproduce the widespread neuropsychological abnormalities of schizophrenia (Figure).6 Ketamine infusion results in the severity and type of disorganized thinking seen in schizophrenia. Given the importance of neurocognitive dysfunction to the conceptualization of schizophrenia, these findings further support a glutamatergic model.
Sensory processing deficits
A key difference between dopaminergic and glutamatergic models is prediction of sensory processing deficits. Traditionally, dopaminergic models have viewed cognitive deficits of schizophrenia as being driven “top down” from higher order brain regions such as the prefrontal cortex, or from local dysfunction within regions such as the striatum.11 In contrast, glutamatergic models predict that deficits also should be observed within sensory brain regions, such as the primary auditory and visual cortex.
Because of the focus on higher-level brain dysfunction, little research on sensory processing deficits was performed until recently. It has become increasingly clear that:
- patients with schizophrenia show severe deficits in early auditory and visual processing
- these deficits significantly contribute to patterns of cognitive dysfunction and psychosocial impairment.12,13
In the auditory system, patients show deficits in pitch perception and, specifically, the ability to match tones after a brief delay. Schizophrenia patients show dysfunction in a specific part of the visual system called the magnocellular visual system. Deficits in these regions lead to impaired ability to detect emotion based on vocal intonation or facial expression, among other deficits.
In addition, reading ability—which was once thought to be normal in patients with schizophrenia—has been found to be severely disturbed.14 As in developmental dyslexia, impairments relate to dysfunction of underlying auditory and visual brain regions. Administering NMDAR antagonists to humans or animals causes deficits in the auditory and visual system similar to those seen in schizophrenia, which confirms the importance of NMDA dysfunction.
Glutamate-based treatments
Because NMDAR antagonists can induce schizophrenia symptoms, the most straightforward approach for treatment is to develop compounds that stimulate glutamate or NMDAR function (Table). The NMDAR contains modulatory sites that may be appropriate targets for drug development, including one that binds the amino acids glycine and D-serine and a redox site that is sensitive to brain glutathione levels. Reductions in brain D-serine and glutathione levels have been reported in schizophrenia, which suggests that impaired NMDAR regulation may contribute directly to brain dysfunction.15 Other treatment approaches being developed include targeting glycine transporters, which indirectly regulate brain levels of glycine, or metabotropic glutamate receptors, which modulate both pre-synaptic glutamate release and post-synaptic NMDAR function.
Table
Glutamatergic drugs in development
| Target | Proposed mechanism | Proposed agents | Phase of development |
|---|---|---|---|
| Glycine/D-serine receptor | Allosteric modulator of the NMDA receptor | Glycine, D-serine, D-alanine, D-cycloserine | Phase II |
| Glycine-type I transport inhibitor | Blocks the reuptake of glycine, akin to SSRIs’ action on serotonin | Sarcosine, RG1678 | Phase II/III |
| Metabotropic glutamate type 2/3 (mGluR2/3) | Blocks presynaptic glutamate release | LY-2140023 | Phase II |
| Redox sensitive site | Allosteric modulator of the NMDA receptor | N-acetylcysteine | Phase II |
| D-amino acid oxidase (DAAO) inhibitors | Inhibits the enzyme that metabolizes D-serine | Remains in preclinical stage | |
| Tetrahydrobiopterin (BH4) | Indirectly modulates glutamatergic system | Remains in preclinical stage | |
| NMDA: N-methyl-D-aspartate; SSRIs: selective serotonin reuptake inhibitors | |||
Glycine/D-serine site agonists. To date, most studies have used glutamatergic drugs adjunctive to antipsychotics and targeted the glycine/D-serine modulatory site, in part because glycine and D-serine are natural compounds and therefore FDA approval for their use could be obtained without the extensive preclinical development usually required for new chemical entities.16 Unfortunately, these agents are less potent than traditional pharmaceuticals, and delivering optimal doses may be impossible. Nevertheless, positive studies with these compounds have provided proof-of-concept for development of agents with higher affinity and specificity.
Studies have used glycine administered at doses up to 60 g/d, D-serine up to 8 g/d, or D-alanine approximately 6 g/d. For glycine, 60 g/d is the highest dose that can be given because of concerns about tolerability and replacement of other essential amino acids. D-serine originally was tested at approximately 2 g/d with promising results, but a recent open-label trial suggested that higher doses may be more efficacious.17 D-serine doses are limited by potential renal toxicity, as demonstrated in rodents studies.
Although not all studies of glycine/D-serine site agonists have been positive, a recent meta-analysis suggests significant improvement in negative symptoms across studies.18 Variability in statistical results across studies is related primarily to degree of placebo effect within individual trials, with a mean improvement in negative symptoms of approximately 15%. Glycine/D-serine site agonists seem to be less effective when combined with clozapine, possibly because clozapine may already enhance the glutamatergic system and increase synaptic glycine levels.6
One study that evaluated effects of open-label glycine in individuals with schizophrenia symptoms observed a large effect-size improvement, including early remission in 3 of 10 patients.19 These data—if confirmed by double-blind trials—would indicate that glycine/d-serine site agonists might have utility in treating the schizophrenia prodrome.
Glycine transport inhibitors. A potential indirect approach to raising glycine levels in the brain is using GlyT1-type glycine transport inhibitors (GTIs). GlyT1 transporters are co-localized in brain with NMDARs and modulate local glycine levels. Rather than binding directly to the NMDAR glycine binding site, GTIs increase glycine levels in the synapse by preventing its removal by GlyT1 transporters. Their function is analogous to using selective serotonin reuptake inhibitors to increase serotonin levels in patients with depression.6
Sarcosine (N-methylglycine) is a naturally occurring GlyT1 inhibitor that has been used in early clinical trials in Taiwan. Initial studies with sarcosine showed efficacy similar to—and in some cases better than—that of direct glycine/D-serine site agonists when added to first-generation or non-clozapine second-generation antipsychotics.18 Sarcosine also has been found to be effective for acute treatment of schizophrenia.20 At present, however, sarcosine is not available for experimental use in the United States because of toxicity considerations.
Using high-affinity GTIs for schizophrenia was first proposed in the mid-1990s,6 but such compounds are only now entering clinical efficacy studies. Most recently, phase II results were presented for RG1678, a compound developed by Hoffman LaRoche.21 The study targeted persistent negative symptoms in patients receiving chronic antipsychotic treatment. Adding RG1678, 10 mg and 30 mg, to antipsychotics led to significant improvement in persistent negative symptoms vs placebo. These promising results are being followed up in phase III studies.
Other glutamatergic options. Few compounds are available to modulate NMDARs at sites other than the glycine/D-serine site. One study administered N-acetylcysteine, a glutathione precursor, as a potential treatment for persistent negative symptoms.22 Encouraging clinical results were observed in this double-blind study, along with improvement in electrophysiologic measures, negative symptoms, and overall functioning, but the study was limited by relatively high rates of noncompletion. Preclinical studies have combined D-serine with an inhibitor of D-amino acid oxidase to prevent D-serine breakdown.23 In rodents, this approach produces a 30-fold increase in D-serine potency.
Tetrahydrobiopterin (BH4) is a cofactor for enzymes responsible for the synthesis of dopamine and other monoamines, and presynaptic release of dopamine and glutamate. Reductions in BH4 levels have been reported in schizophrenia, which suggests that this compound may be etiologically important.24 Researchers have initiated a study of this compound in schizophrenia.
Other schizophrenia models propose that the crucial issue is not NMDA blockade but subsequent dysregulation of presynaptic glutamate release. Type 2/3 metabotropic glutamate receptors (mGluR2/3) are located on presynaptic glutamate terminals and inhibit presynaptic glutamate release. mGluR2/3 agonists have been shown to reverse ketamine’s effects in humans and in animal models,25,26 which suggests a potential role in schizophrenia treatment.
The first mGluR2/3 agonist entered into monotherapy clinical efficacy trials for schizophrenia was LY-2140023. In an initial trial, this compound showed significant efficacy in improving positive and negative symptoms, comparable to that of olanzapine.27 However, a follow-up study failed because of a large placebo effect,28 which leaves the efficacy question unresolved.
In contrast to mGluR2/3, type 5 metabotropic receptors (mGluR5) are co-localized with NMDA receptors and potentiate activation. Thus, mGluR5 agonists also may be effective for treating schizophrenia. These compounds remain in preclinical development. Other approaches, such as stimulating specific types of GABA receptors to overcome glutamatergic deficits, remain promising but have not been tested in definitive clinical trials.
Related Resources
- Kantrowitz JT, Javitt DC. N-methyl-D-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010; 83(3-4): 108-121.
- Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT, Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
Drug Brand Names
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Ketamine • Ketalar
- Olanzapine • Zyprexa
Disclosures
Dr. Javitt receives grant/research support from Jazz Pharmaceuticals, Pfizer Inc., and Roche and is a consultant to AstraZeneca, Cypress, Eli Lilly and Company, NPS Pharmaceuticals, Sepracor, Solvay, Sunovion, and Takeda. He holds intellectual property rights for use of glycine, D-serine, and glycine transport inhibitors in treatment of schizophrenia and related disorders.
Dr. Kantrowitz receives grant/research support from Eli Lilly and Company, Jazz Pharmaceuticals, Pfizer Inc., Roche, and Sepracor.
Preparation of this manuscript was supported in part by National Institute of Health grants R01 DA03383, R37 MH49334, and P50 MH086385.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.
1. Fenton WS, McGlashan TH. Antecedents symptom progression, and long-term outcome of the deficit syndrome in schizophrenia. Am J Psychiatry. 1994;151(3):351-356.
2. Woodberry KA. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165(5):579-587.
3. Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull. 2009;35(3):549-562.
4. Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148(10):1301-1308.
5. Egan MF, Goldberg TE, Kolachana BS, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917-6922.
6. Kantrowitz JT, Javitt DC. Glutamatergic approaches to the conceptualization and treatment of schizophrenia. In: Kantrowitz JT Javitt DC, eds. Handbook of neurochemistry and molecular neurobiology: schizophrenia. 3rd ed. New York, NY: Springer; 2009.
7. Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51(3):199-214.
8. Krystal J, D’Souza DC, Mathalon D, et al. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology. 2003;169(3-4):215-233.
9. Malhotra AK, Pinals DA, Adler CM, et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology. 1997;17(3):141-150.
10. Lahti AC, Koffel B, LaPorte D, et al. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 1995;13(1):9-19.
11. Lesh TA, Niendam TA, Minzenberg MJ, et al. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36(1):316-338.
12. Leitman DI, Laukka P, Juslin PN, et al. Getting the cue: sensory contributions to auditory emotion recognition impairments in schizophrenia. Schizophr Bull. 2010;36(3):545-556.
13. Butler PD, Abeles IY, Weiskopf NG, et al. Sensory contributions to impaired emotion processing in schizophrenia. Schizophr Bull. 2009;35(6):1095-1107.
14. Revheim N, Butler PD, Schechter I, et al. Reading impairment and visual processing deficits in schizophrenia. Schizophr Res. 2006;87(1-3):238-245.
15. Hashimoto K, Fukushima T, Shimizu E, et al. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572-576.
16. Javitt DC, Balla A, Burch S, et al. Reversal of phencyclidine-induced dopaminergic dysregulation by N-methyl-D-aspartate receptor/glycine-site agonists. Neuropsychopharmacology. 2004;29(2):300-307.
17. Kantrowitz JT, Malhotra AK, Cornblatt B, et al. High dose D-serine in the treatment of schizophrenia. Schizophr Res. 2010;121(1-3):125-130.
18. Tsai GE, Lin PY. Strategies to enhance N-methyl-D-aspartate receptor-mediated neurotransmission in schizophrenia a critical review and meta-analysis. Curr Pharm Des. 2010;16(5):522-537.
19. Woods SW, Thomas L, Tully E, et al. Effects of oral glycine in the schizophrenia prodrome. Schizophr Res. 2004;70(suppl 1):79.-
20. Lane HY, Liu YC, Huang CL, et al. Sarcosine (N-methylglycine) treatment for acute schizophrenia: a randomized, double-blind study. Biol Psychiatry. 2008;63(1):9-12.
21. Umbricht D, Yoo K, Youssef E, et al. Glycine transporter type 1 (GLYT1) inhibitor RG1678: positive results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Neuropsychopharmacology. 2010;35:S320-S321.
22. Berk M, Copolov D, Dean O, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia—a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361-368.
23. Smith SM, Uslaner JM, Hutson PH. The therapeutic potential of d-amino acid oxidase (DAAO) inhibitors. Open Med Chem J. 2010;4:3-9.
24. Richardson MA, Read LL, Reilly MA, et al. Analysis of plasma biopterin levels in psychiatric disorders suggests a common BH4 deficit in schizophrenia and schizoaffective disorder. Neurochem Res. 2007;32(1):107-113.
25. Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281(5381):1349-1352.
26. Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl). 2005;179(1):303-309.
27. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13(9):1102-1107.
28. A multi-center, inpatient, phase 2, double-blind, placebo-controlled dose ranging study of LY2140023 in patients with DSM-IV schizophrenia. ClinicalTrials.gov identifier NCT00520923. Available at: http://clinicaltrials.gov/ct2/show/NCT00520923?intr=LY2140023&rank=1. Accessed February 23 2011.
Comorbid bipolar disorder and substance abuse: Evidence-based options
Discuss this article at www.facebook.com/CurrentPsychiatry
Among DSM axis I diagnoses, bipolar disorder (BD) has the highest rates of comorbid substance use disorders (SUDs).1-3 Approximately 60% of patients with bipolar I disorder have a lifetime diagnosis of an SUD.1 Excluding tobacco, alcohol is the substance most often abused by BD patients, followed by cannabis, amphetamines, and cocaine.1-3
BD patients with comorbid SUD usually exhibit more severe clinical presentations and poorer outcomes than their counterparts without SUDs. Compared with patients with BD alone, those with BD and SUD comorbidity (BD-SUD) experience earlier onset of mood symptoms; higher rates of anxiety disorders, suicide attempts, accidents, hospitalizations, and rapid cycling; more depressive episodes; and lower treatment compliance.4-9
Several treatment options are available for patients with BD-SUD, including psychotherapy modalities, medications primarily used to treat BD, and medications primarily used to treat SUDs. Evidence-based support for these treatments remains limited, and no treatment of choice has emerged. This article reviews evidence on the longer-term treatment of BD-SUD, including general strategies and specific psychosocial and pharmacologic interventions. Short-term treatment strategies, such as pharmacotherapy for detoxification, are outside the scope of this review.
General strategies
The causes of BD-SUD are complex. Evidence suggests that the presence of affective symptoms is associated with an increased risk for substance misuse. This should be kept in mind when treating a patient with BD-SUD because controlling mood symptoms probably will help control substance abuse. However, evidence also shows that SUDs may be independent of mood episodes. Therefore, treating only mood symptoms in the hope that doing so will control substance abuse may not be enough.
Because the negative impact of SUDs on BD outcome is well documented, inform patients that limiting their use of alcohol and/or drugs is vital to control their mood disorder. Efforts to educate, stimulate, and support patients to moderate or stop their alcohol and/or drug use are likely to result in positive changes.10 Therefore, treatment for BD-SUD should follow, in part, the same recommendations for treatment of SUDs in patients with no comorbid axis I disorders:
- identify the problem (ie, the existence of a comorbid SUD)
- share your concerns with your patient
- offer appropriate and specific treatments, such as detoxification and/or self-help and counseling programs.10
Because SUDs usually are chronic and relapsing conditions, periods of drug and/ or alcohol use should be expected and not considered a sign of treatment failure. In addition, integrating treatment for both conditions probably is better than managing each separately. Therefore, targeting BD symptoms with mood-stabilizing medications and substance abuse with nonpharmacologic modalities such as drug counseling likely will bring about the best results.
Compared with BD patients without comorbid SUD, BD-SUD patients have a 7-fold increased risk of antidepressantinduced mania.11 Therefore, antidepressants should be prescribed cautiously for patients with BD-SUD.
Integrated psychosocial therapy
BD-SUD patients may benefit from attending self-help programs such as Alcoholics Anonymous and Narcotics Anonymous, provided their mood is stable enough to allow them to participate. Other forms of psychotherapy for BD-SUD patients include standard group drug counseling and integrated group therapy that simultaneously addresses both conditions.
Integrated group therapy is based on the premise that changing maladaptive mood cognitions and behaviors will facilitate recovery from SUDs, and changing maladaptive substance use cognitions and behaviors will facilitate recovery from mood disorders.12 In a recent randomized controlled trial, 62 BD-SUD patients were blindly assigned to integrated group therapy or standard group drug counseling and followed for 3 months.12 Pharmacotherapy was prescribed as usual. Substance use decreased for both groups. However, compared with patients in the drug counseling group, those who participated in integrated group therapy spent fewer days using substances in general and alcohol in particular, fewer days using alcohol to intoxication, and had a shorter time from treatment initiation to the first abstinent month. There were no differences between groups in number of weeks in a mood episode.
Pharmacotherapy options
For a table that summarizes the dosages and indications of the medications used to treat BD-SUD that are described below, visit this article at CurrentPsychiatry.com.
Table
Medications used to treat substance use disorders in bipolar disorder patients*
| Drug | Dosages | FDA-approved indication(s) |
|---|---|---|
| Acamprosate | 1,998 mg/d | Maintenance of abstinence from alcohol in patients with alcohol dependence |
| Aripiprazole | 15 to 45 mg/d | Acute manic or mixed episode of bipolar disorder; augmentation therapy for major depressive disorder |
| Carbamazepine | 400 to 1,200 mg/d | Manic and mixed episodes associated with bipolar disorder |
| Disulfiram | 250 to 500 mg/d | Enforced sobriety in abstinent alcohol-dependence patients |
| Divalproex sodium | Initial dose: 750 mg/d Maximum dose: 60 mg/kg/d† | Manic episodes associated with bipolar disorder |
| Lamotrigine | 200 mg/d | Maintenance treatment of bipolar I disorder |
| Lithium | 900 to 1,800 mg/d for acute episodes 900 mg to 1,200 mg/d for maintenance‡. | Manic episodes associated with bipolar disorder; maintenance treatment of bipolar disorder |
| Naltrexone | 50 mg/d 380 mg/month | Alcohol dependence |
| Quetiapine | 300 mg/d for bipolar depression 400 to 800 mg/d for bipolar mania 400 to 800 mg/d for maintenance treatment of bipolar disorder | Depressive and acute manic episodes associated with bipolar I disorder; maintenance treatment of bipolar I disorder |
| Risperidone | 1 to 6 mg/d | Acute manic or mixed episodes associated with bipolar I disorder |
| * None of the medications cited in this table or the text have been specifically approved by the FDA for treating alcohol/drug abuse/dependence co-occurring with bipolar disorder †Dose should correspond to valproic acid therapeutic levels between 50 and 100 μg/mL ‡Dose should correspond to lithium therapeutic levels between 0.8 and 1.2 mEq/L for acute manic episode treatment and 0.6 and 1.0 mEq/L for maintenance treatment | ||
Lithium. Given its well-documented mood stabilizing effect, lithium would seem to be a reasonable option to treat BD-SUD patients, but scant evidence supports its role as an anti-alcohol or anti-drug medication (Table 1).13,14 Lithium’s efficacy was evaluated in a study of 25 adolescents suffering from mood disorders (mostly BD) and comorbid SUDs (mostly alcohol and cannabis) randomized to receive lithium or placebo for 6 weeks.13 Lithium was well tolerated and improved psychiatric symptoms. At week 3, patients receiving lithium produced fewer positive results on randomly administered urine drug screens than those receiving placebo.
However, lithium seems to have little efficacy in reducing cocaine use in cocaine-dependent patients with bipolar spectrum disorders.14 In an open-label study, 10 patients with a history of hypomania or cyclothymia received lithium monotherapy for 12 weeks. Although patients experienced improved mood symptoms and decreased cocaine use, the mean decrease was transitory and not statistically significant. Another factor that may limit lithium’s use for BD-SUD patients is that these patients are more likely to comply with valproate treatment than with lithium.15
Table 1
Lithium for BD patients with substance use disorders
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Geller et al, 199813 | Lithium vs placebo | Double-blind, placebo-controlled | Alcohol and cannabis use disorders | Decreased positive drug screen results |
| Nunes et al, 199014 | Lithium | Open label | Cocaine abuse | Nonsignificant decrease in cocaine use |
| BD: bipolar disorder | ||||
Anticonvulsants. In a double-blind, placebo-controlled study of 59 alcohol-dependent bipolar I disorder patients, lithium plus divalproex sodium was superior to lithium plus placebo in decreasing number of drinking days and number of drinks per day and in increasing periods of abstinence (Table 2).16-19 Divalproex sodium was well tolerated and liver function improved in the divalproex sodium group compared with the placebo group, which probably was a benefit of decreased alcohol consumption. In addition, there was a strong association between mood symptoms and alcohol use, which suggests that maximizing mood symptom treatment may decrease alcohol use. However, the divalproex sodium and placebo groups did not differ in measures of mood symptoms, which implies that divalproex sodium might exhibit a positive effect on drinking regardless of its mood-stabilizing properties.
Divalproex sodium also has been used to treat BD comorbid with cocaine dependence. In a small open-label study, 15 patients receiving divalproex sodium plus counseling for mood and substance use disorders were followed for 6 weeks.17 The 7 patients who completed the trial had significantly more cocaine-abstinent days, spent less money on cocaine, and experienced fewer manic and depressive symptoms. However, divalproex sodium’s effect on cocaine use cannot be determined solely from this study because there was no placebo control group.
Despite its widespread use as a mood stabilizer and potential use in alcohol detoxification, carbamazepine scarcely has been studied in BD-SUD patients. A double-blind, placebo-controlled study of 139 cocaine-dependent patients with BD or other affective disorders found that patients taking carbamazepine for 12 weeks experienced modest reductions in positive urine drug screens and increased time to cocaine use.18 They also reported less cocaine craving than patients taking placebo, and mood symptoms (mostly depressive) improved.
An open-label study used lamotrigine as adjunctive therapy or monotherapy for 62 cocaine-dependent BD patients followed for 36 weeks.19 There was some decrease in cocaine craving, money spent on cocaine, and rate of depressive and manic symptoms, but no effect on cocaine use. A placebo-controlled trial is necessary to confirm these modest effects.
No studies have evaluated the potential role of topiramate in treating BD-SUD, despite its FDA-approved indication for alcoholism treatment. Topiramate’s well-known safety and tolerability profile in BD patients make it an interesting option for those with co-occurring alcohol dependence.
Table 2
Studies suggest anticonvulsants may reduce alcohol, cocaine use in BD patients
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Salloum et al, 200516 | Divalproex sodium plus lithium vs placebo plus lithium | Double-blind, placebo-controlled | Alcohol dependence | Decreased number of drinking days and number of drinks per day and increased time of abstinence |
| Salloum et al, 200717 | Divalproex sodium | Open label | Cocaine dependence | Increased cocaine-abstinent days and decreased money spent on cocaine and cocaine use severity index |
| Brady et al,* 200218 | Carbamazepine vs placebo | Double-blind, placebo-controlled | Cocaine dependence | Decreased cocaine craving and cocaine use |
| Brown et al, 200619 | Lamotrigine | Open label | Cocaine dependence | Decreased cocaine craving and money spent on cocaine |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder | ||||
Atypical antipsychotics. In an open-label study, 16 weeks of quetiapine monotherapy effectively decreased alcohol consumption, alcohol craving, and psychotic and affective symptoms in 28 alcoholics with a variety of psychiatric diagnoses, including BD, schizoaffective disorder, and borderline personality disorder (Table 3).20-24 However, in a double-blind study of augmentation with quetiapine or placebo for 102 alcohol-dependent BD patients, no significant differences in alcohol use were found between groups.21
Quetiapine may be effective for treating BD patients with comorbid cocaine dependence. In an open-label study, 12 weeks of quetiapine augmentation in 17 cocaine-dependent BD patients was associated with decreased cocaine craving and improvement in depressive symptoms.22 In another open-label study, 80 BD patients with comorbid cocaine or amphetamine dependence were randomly assigned to receive quetiapine or risperidone as adjunctive therapy or monotherapy for 20 weeks.23 Both groups showed significantly decreased drug use and drug craving and improved mood. This study suggests that risperidone also may be an option for BD patients with comorbid cocaine or stimulant dependence.
A 20-week, open-label study of 20 BD-SUD patients found that switching patients from their previous antipsychotic to aripiprazole resulted in less cocaine craving, less alcohol craving, and less money spent on alcohol.24
Olanzapine has not been systematically studied in BD-SUD patients. Some case reports suggest that olanzapine may decrease cocaine craving and use in patients with schizoaffective disorder (bipolar type) and alcohol craving and use in BD patients with comorbid alcohol dependence.25
Table 3
Evidence of efficacy for antipsychotics for BD patients with SUDs
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Martinotti et al,* 200820 | Quetiapine | Open label | Alcohol dependence | Decreased alcohol consumption and alcohol craving |
| Brown et al, 200821 | Quetiapine vs placebo | Double-blind, placebo-controlled | Alcohol dependence | No difference between quetiapine and placebo in decreasing alcohol use and alcohol craving |
| Brown et al, 200222 | Quetiapine | Open label | Cocaine dependence | Decreased cocaine use and cocaine craving |
| Nejtek et al, 200823 | Risperidone vs quetiapine | Open label | Cocaine dependence and amphetamine dependence | Decreased drug use and drug craving |
| Brown et al, 200524 | Aripiprazole | Open label | Alcohol and cocaine dependence | Decreased alcohol and cocaine craving and money spent on alcohol |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder; SUDs: substance use disorders | ||||
SUD medications. Little evidence guides using medications indicated for treating SUDs—such as naltrexone, acamprosate, and disulfiram—as treatment for BD patients (Table 4).26-29 In an open-label trial of 34 BD patients with alcohol dependence, naltrexone was well tolerated and associated with decreased alcohol craving and use and modest improvement in manic and depressive symptoms.26
In a double-blind, placebo-controlled study, 50 alcohol-dependent BD patients treated with standard mood-stabilizing therapy and cognitive-behavioral therapy were randomized to receive add-on naltrexone, 50 mg/d, or placebo.27 Patients receiving naltrexone showed decreased alcohol consumption, although no measures were statistically significant. Effect sizes of alcohol use decrease and alcohol craving were moderate to large compared with placebo, which suggests that naltrexone may be effective for treating alcoholism in these patients.
Two other studies evaluated naltrexone and disulfiram in patients with BD or other mood disorders.28,29 Naltrexone was well tolerated, caused no serious adverse side effects, and was significantly more effective than placebo in decreasing drinking rates and increasing the number of abstinent days.28,29 Disulfiram was as effective as naltrexone, but the combination of both offered no advantage over use of either drug separately.
There are reports of a new-onset manic episode associated with naltrexone use in a patient with opioid dependence, and a manic episode triggered by naltrexone in a patient with BD with comorbid alcohol dependence.30,31 At both low and high doses, disulfiram is associated with induction of psychotic mania in alcoholic patients without a personal or family history of BD.32,33
We found no studies that evaluated treating BD patients who abused other substances, such as cannabis or opiates. We recommend that BD patients with these substance use disorders should be referred to treatment modalities that are condition-specific, such as psychotherapy for cannabis use disorders or methadone or naltrexone treatment for opiate dependence. More severe cases of comorbid SUD probably would benefit from a referral to or consultation with a SUD specialist.
Table 4
Naltrexone and disulfiram for BD patients with alcohol dependence
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Brown et al, 200626 | Naltrexone | Open label | Alcohol dependence | Decreased alcohol use and craving |
| Brown et al, 200927 | Naltrexone vs placebo | Double-blind, placebo-controlled | Alcohol dependence | Nonsignificant decrease in alcohol consumption |
| Petrakis et al, 200528 and 200729 | Naltrexone alone vs disulfiram alone vs naltrexone plus disulfiram | Double-blind, randomized, placebo-controlled | Alcohol dependence | More time in abstinence and decreased craving for both compounds |
| BD: bipolar disorder | ||||
Related Resource
- Tolliver BK. Bipolar disorder and substance abuse: Overcome the challenges of ‘dual diagnosis’ patients. Current Psychiatry. 2010; 9(8): 32-38.
Drug Brand Names
- Acamprosate • Campral
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, Equetro, others
- Disulfiram • Antabuse
- Divalproex sodium • Depakote,
- Depakote ER Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methadone • Dolophine
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Valproate • Depacon
Disclosures
Dr. Nery held a temporary work contract as a clinical research physician with Eli Lilly and Company Brazil from May 2009 to November 2009.
Dr. Soares was partly supported by National Institute of Health grants MH 68766, MH 69774, and RR 20571. He receives grant/research support from Bristol-Myers Squibb, Cephalon, GlaxoSmithKline, and Sunovion.
1. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
2. Kessler RC, Crum RM, Warner LA, et al. Lifetime cooccurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
3. Grant BF, Stinson FS, Hasin DS, et al. Prevalence, correlates, and comorbidity of bipolar I disorder and axis I and II disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2005;66(10):1205-1215.
4. Feinman JA, Dunner DL. The effect of alcohol and substance abuse on the course of bipolar affective disorder. J Affect Disord. 1996;37(1):43-49.
5. Cassidy F, Ahearn EP, Carroll BJ. Substance abuse in bipolar disorder. Bipolar Disord. 2001;3(4):181-188.
6. Frye MA, Altshuler LL, McElroy SL, et al. Gender differences in prevalence, risk, and clinical correlates of alcoholism comorbidity in bipolar disorder. Am J Psychiatry. 2003;160(5):883-889.
7. Khalsa HM, Salvatore P, Hennen J, et al. Suicidal events and accidents in 215 first-episode bipolar I disorder patients: predictive factors. J Affect Disord. 2008;106(1-2):179-184.
8. Baldessarini RJ, Perry R, Pike J. Factors associated with treatment nonadherence among US bipolar disorder patients. Hum Psychopharmacol. 2008;23(2):95-105.
9. Cardoso BM, Kauer Sant’Anna M, Dias VV, et al. The impact of co-morbid alcohol use disorder in bipolar patients. Alcohol. 2008;42(6):451-457.
10. Schuckit MA. Alcohol-use disorders. Lancet. 2009;373 (9662):492-501.
11. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795.
12. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
13. Geller B, Cooper TB, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry. 1998;37:171-178.
14. Nunes EV, McGrath PJ, Wager S, et al. Lithium treatment for cocaine abusers with bipolar spectrum disorders. Am J Psychiatry. 1990;147:655-657.
15. Weiss RD, Greenfield SF, Najavits LM, et al. Medication compliance among patients with bipolar disorder and substance use disorder. J Clin Psychiatry. 1998;59:172-174.
16. Salloum IM, Cornelius JR, Daley DC, et al. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
17. Salloum IM, Douaihy A, Cornelius JR, et al. Divalproex utility in bipolar disorder with co-occurring cocaine dependence: a pilot study. Addict Behav. 2007;32(2):410-405.
18. Brady KT, Sonne SC, Malcolm RJ, et al. Carbamazepine in the treatment of cocaine dependence: subtyping by affective disorder. Exp Clin Psychopharmacol. 2002;10:276-285.
19. Brown ES, Perantie DC, Dhanani N, et al. Lamotrigine for bipolar disorder and comorbid cocaine dependence: a replication and extension study. J Affect Disord. 2006;93(1-3):219-222.
20. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
21. Brown ES, Garza M, Carmody TJ. A randomized double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
22. Brown ES, Nejtek VA, Perantie DC, et al. Quetiapine in bipolar disorder and cocaine dependence. Bipolar Disord. 2002;4(6):406-411.
23. Nejtek VA, Avila M, Chen LA, et al. Do atypical antipsychotics effectively treat co-occurring bipolar disorder and stimulant dependence? A randomized, double-blind trial. J Clin Psychiatry. 2008;69(8):1257-1266.
24. Brown ES, Jeffress J, Liggin JD, et al. Switching outpatients with bipolar or schizoaffective disorders and substance abuse from their current antipsychotic to aripiprazole. J Clin Psychiatry. 2005;66:756-760.
25. Sattar SP, Grant K, Bhatia S, et al. Potential use of olanzapine in treatment of substance dependence disorders. J Clin Psychopharmacol. 2003;23:413-415.
26. Brown ES, Beard L, Dobbs L, et al. Naltrexone in patients with bipolar disorder and alcohol dependence. Depress Anxiety. 2006;23(8):492-495.
27. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebo-controlled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33:1863-1869.
28. Petrakis IL, Poling J, Levinson C, et al. and the VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
29. Petrakis I, Ralevski E, Nich C, et al. and the VA VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Sullivan MA, Nunes EV. New-onset mania and psychosis following heroin detoxification and naltrexone maintenance. Am J Addict. 2005;14(5):486-487.
31. Sonne SC, Brady KT. Naltrexone for individuals with comorbid bipolar disorder and alcohol dependence. J Clin Psychopharmacol. 2000;20(1):114-115.
32. Ceylan ME, Turkcan A, Mutlu E, et al. Manic episode with psychotic symptoms associated with high dose of disulfiram: a case report. J Clin Psychopharmacol. 2007;27(2):224-225.
33. Li MY, Shen YC. Manic episode with psychosis following a lower than recommended dosage regimen of disulfiram. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):311-312.
Discuss this article at www.facebook.com/CurrentPsychiatry
Among DSM axis I diagnoses, bipolar disorder (BD) has the highest rates of comorbid substance use disorders (SUDs).1-3 Approximately 60% of patients with bipolar I disorder have a lifetime diagnosis of an SUD.1 Excluding tobacco, alcohol is the substance most often abused by BD patients, followed by cannabis, amphetamines, and cocaine.1-3
BD patients with comorbid SUD usually exhibit more severe clinical presentations and poorer outcomes than their counterparts without SUDs. Compared with patients with BD alone, those with BD and SUD comorbidity (BD-SUD) experience earlier onset of mood symptoms; higher rates of anxiety disorders, suicide attempts, accidents, hospitalizations, and rapid cycling; more depressive episodes; and lower treatment compliance.4-9
Several treatment options are available for patients with BD-SUD, including psychotherapy modalities, medications primarily used to treat BD, and medications primarily used to treat SUDs. Evidence-based support for these treatments remains limited, and no treatment of choice has emerged. This article reviews evidence on the longer-term treatment of BD-SUD, including general strategies and specific psychosocial and pharmacologic interventions. Short-term treatment strategies, such as pharmacotherapy for detoxification, are outside the scope of this review.
General strategies
The causes of BD-SUD are complex. Evidence suggests that the presence of affective symptoms is associated with an increased risk for substance misuse. This should be kept in mind when treating a patient with BD-SUD because controlling mood symptoms probably will help control substance abuse. However, evidence also shows that SUDs may be independent of mood episodes. Therefore, treating only mood symptoms in the hope that doing so will control substance abuse may not be enough.
Because the negative impact of SUDs on BD outcome is well documented, inform patients that limiting their use of alcohol and/or drugs is vital to control their mood disorder. Efforts to educate, stimulate, and support patients to moderate or stop their alcohol and/or drug use are likely to result in positive changes.10 Therefore, treatment for BD-SUD should follow, in part, the same recommendations for treatment of SUDs in patients with no comorbid axis I disorders:
- identify the problem (ie, the existence of a comorbid SUD)
- share your concerns with your patient
- offer appropriate and specific treatments, such as detoxification and/or self-help and counseling programs.10
Because SUDs usually are chronic and relapsing conditions, periods of drug and/ or alcohol use should be expected and not considered a sign of treatment failure. In addition, integrating treatment for both conditions probably is better than managing each separately. Therefore, targeting BD symptoms with mood-stabilizing medications and substance abuse with nonpharmacologic modalities such as drug counseling likely will bring about the best results.
Compared with BD patients without comorbid SUD, BD-SUD patients have a 7-fold increased risk of antidepressantinduced mania.11 Therefore, antidepressants should be prescribed cautiously for patients with BD-SUD.
Integrated psychosocial therapy
BD-SUD patients may benefit from attending self-help programs such as Alcoholics Anonymous and Narcotics Anonymous, provided their mood is stable enough to allow them to participate. Other forms of psychotherapy for BD-SUD patients include standard group drug counseling and integrated group therapy that simultaneously addresses both conditions.
Integrated group therapy is based on the premise that changing maladaptive mood cognitions and behaviors will facilitate recovery from SUDs, and changing maladaptive substance use cognitions and behaviors will facilitate recovery from mood disorders.12 In a recent randomized controlled trial, 62 BD-SUD patients were blindly assigned to integrated group therapy or standard group drug counseling and followed for 3 months.12 Pharmacotherapy was prescribed as usual. Substance use decreased for both groups. However, compared with patients in the drug counseling group, those who participated in integrated group therapy spent fewer days using substances in general and alcohol in particular, fewer days using alcohol to intoxication, and had a shorter time from treatment initiation to the first abstinent month. There were no differences between groups in number of weeks in a mood episode.
Pharmacotherapy options
For a table that summarizes the dosages and indications of the medications used to treat BD-SUD that are described below, visit this article at CurrentPsychiatry.com.
Table
Medications used to treat substance use disorders in bipolar disorder patients*
| Drug | Dosages | FDA-approved indication(s) |
|---|---|---|
| Acamprosate | 1,998 mg/d | Maintenance of abstinence from alcohol in patients with alcohol dependence |
| Aripiprazole | 15 to 45 mg/d | Acute manic or mixed episode of bipolar disorder; augmentation therapy for major depressive disorder |
| Carbamazepine | 400 to 1,200 mg/d | Manic and mixed episodes associated with bipolar disorder |
| Disulfiram | 250 to 500 mg/d | Enforced sobriety in abstinent alcohol-dependence patients |
| Divalproex sodium | Initial dose: 750 mg/d Maximum dose: 60 mg/kg/d† | Manic episodes associated with bipolar disorder |
| Lamotrigine | 200 mg/d | Maintenance treatment of bipolar I disorder |
| Lithium | 900 to 1,800 mg/d for acute episodes 900 mg to 1,200 mg/d for maintenance‡. | Manic episodes associated with bipolar disorder; maintenance treatment of bipolar disorder |
| Naltrexone | 50 mg/d 380 mg/month | Alcohol dependence |
| Quetiapine | 300 mg/d for bipolar depression 400 to 800 mg/d for bipolar mania 400 to 800 mg/d for maintenance treatment of bipolar disorder | Depressive and acute manic episodes associated with bipolar I disorder; maintenance treatment of bipolar I disorder |
| Risperidone | 1 to 6 mg/d | Acute manic or mixed episodes associated with bipolar I disorder |
| * None of the medications cited in this table or the text have been specifically approved by the FDA for treating alcohol/drug abuse/dependence co-occurring with bipolar disorder †Dose should correspond to valproic acid therapeutic levels between 50 and 100 μg/mL ‡Dose should correspond to lithium therapeutic levels between 0.8 and 1.2 mEq/L for acute manic episode treatment and 0.6 and 1.0 mEq/L for maintenance treatment | ||
Lithium. Given its well-documented mood stabilizing effect, lithium would seem to be a reasonable option to treat BD-SUD patients, but scant evidence supports its role as an anti-alcohol or anti-drug medication (Table 1).13,14 Lithium’s efficacy was evaluated in a study of 25 adolescents suffering from mood disorders (mostly BD) and comorbid SUDs (mostly alcohol and cannabis) randomized to receive lithium or placebo for 6 weeks.13 Lithium was well tolerated and improved psychiatric symptoms. At week 3, patients receiving lithium produced fewer positive results on randomly administered urine drug screens than those receiving placebo.
However, lithium seems to have little efficacy in reducing cocaine use in cocaine-dependent patients with bipolar spectrum disorders.14 In an open-label study, 10 patients with a history of hypomania or cyclothymia received lithium monotherapy for 12 weeks. Although patients experienced improved mood symptoms and decreased cocaine use, the mean decrease was transitory and not statistically significant. Another factor that may limit lithium’s use for BD-SUD patients is that these patients are more likely to comply with valproate treatment than with lithium.15
Table 1
Lithium for BD patients with substance use disorders
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Geller et al, 199813 | Lithium vs placebo | Double-blind, placebo-controlled | Alcohol and cannabis use disorders | Decreased positive drug screen results |
| Nunes et al, 199014 | Lithium | Open label | Cocaine abuse | Nonsignificant decrease in cocaine use |
| BD: bipolar disorder | ||||
Anticonvulsants. In a double-blind, placebo-controlled study of 59 alcohol-dependent bipolar I disorder patients, lithium plus divalproex sodium was superior to lithium plus placebo in decreasing number of drinking days and number of drinks per day and in increasing periods of abstinence (Table 2).16-19 Divalproex sodium was well tolerated and liver function improved in the divalproex sodium group compared with the placebo group, which probably was a benefit of decreased alcohol consumption. In addition, there was a strong association between mood symptoms and alcohol use, which suggests that maximizing mood symptom treatment may decrease alcohol use. However, the divalproex sodium and placebo groups did not differ in measures of mood symptoms, which implies that divalproex sodium might exhibit a positive effect on drinking regardless of its mood-stabilizing properties.
Divalproex sodium also has been used to treat BD comorbid with cocaine dependence. In a small open-label study, 15 patients receiving divalproex sodium plus counseling for mood and substance use disorders were followed for 6 weeks.17 The 7 patients who completed the trial had significantly more cocaine-abstinent days, spent less money on cocaine, and experienced fewer manic and depressive symptoms. However, divalproex sodium’s effect on cocaine use cannot be determined solely from this study because there was no placebo control group.
Despite its widespread use as a mood stabilizer and potential use in alcohol detoxification, carbamazepine scarcely has been studied in BD-SUD patients. A double-blind, placebo-controlled study of 139 cocaine-dependent patients with BD or other affective disorders found that patients taking carbamazepine for 12 weeks experienced modest reductions in positive urine drug screens and increased time to cocaine use.18 They also reported less cocaine craving than patients taking placebo, and mood symptoms (mostly depressive) improved.
An open-label study used lamotrigine as adjunctive therapy or monotherapy for 62 cocaine-dependent BD patients followed for 36 weeks.19 There was some decrease in cocaine craving, money spent on cocaine, and rate of depressive and manic symptoms, but no effect on cocaine use. A placebo-controlled trial is necessary to confirm these modest effects.
No studies have evaluated the potential role of topiramate in treating BD-SUD, despite its FDA-approved indication for alcoholism treatment. Topiramate’s well-known safety and tolerability profile in BD patients make it an interesting option for those with co-occurring alcohol dependence.
Table 2
Studies suggest anticonvulsants may reduce alcohol, cocaine use in BD patients
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Salloum et al, 200516 | Divalproex sodium plus lithium vs placebo plus lithium | Double-blind, placebo-controlled | Alcohol dependence | Decreased number of drinking days and number of drinks per day and increased time of abstinence |
| Salloum et al, 200717 | Divalproex sodium | Open label | Cocaine dependence | Increased cocaine-abstinent days and decreased money spent on cocaine and cocaine use severity index |
| Brady et al,* 200218 | Carbamazepine vs placebo | Double-blind, placebo-controlled | Cocaine dependence | Decreased cocaine craving and cocaine use |
| Brown et al, 200619 | Lamotrigine | Open label | Cocaine dependence | Decreased cocaine craving and money spent on cocaine |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder | ||||
Atypical antipsychotics. In an open-label study, 16 weeks of quetiapine monotherapy effectively decreased alcohol consumption, alcohol craving, and psychotic and affective symptoms in 28 alcoholics with a variety of psychiatric diagnoses, including BD, schizoaffective disorder, and borderline personality disorder (Table 3).20-24 However, in a double-blind study of augmentation with quetiapine or placebo for 102 alcohol-dependent BD patients, no significant differences in alcohol use were found between groups.21
Quetiapine may be effective for treating BD patients with comorbid cocaine dependence. In an open-label study, 12 weeks of quetiapine augmentation in 17 cocaine-dependent BD patients was associated with decreased cocaine craving and improvement in depressive symptoms.22 In another open-label study, 80 BD patients with comorbid cocaine or amphetamine dependence were randomly assigned to receive quetiapine or risperidone as adjunctive therapy or monotherapy for 20 weeks.23 Both groups showed significantly decreased drug use and drug craving and improved mood. This study suggests that risperidone also may be an option for BD patients with comorbid cocaine or stimulant dependence.
A 20-week, open-label study of 20 BD-SUD patients found that switching patients from their previous antipsychotic to aripiprazole resulted in less cocaine craving, less alcohol craving, and less money spent on alcohol.24
Olanzapine has not been systematically studied in BD-SUD patients. Some case reports suggest that olanzapine may decrease cocaine craving and use in patients with schizoaffective disorder (bipolar type) and alcohol craving and use in BD patients with comorbid alcohol dependence.25
Table 3
Evidence of efficacy for antipsychotics for BD patients with SUDs
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Martinotti et al,* 200820 | Quetiapine | Open label | Alcohol dependence | Decreased alcohol consumption and alcohol craving |
| Brown et al, 200821 | Quetiapine vs placebo | Double-blind, placebo-controlled | Alcohol dependence | No difference between quetiapine and placebo in decreasing alcohol use and alcohol craving |
| Brown et al, 200222 | Quetiapine | Open label | Cocaine dependence | Decreased cocaine use and cocaine craving |
| Nejtek et al, 200823 | Risperidone vs quetiapine | Open label | Cocaine dependence and amphetamine dependence | Decreased drug use and drug craving |
| Brown et al, 200524 | Aripiprazole | Open label | Alcohol and cocaine dependence | Decreased alcohol and cocaine craving and money spent on alcohol |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder; SUDs: substance use disorders | ||||
SUD medications. Little evidence guides using medications indicated for treating SUDs—such as naltrexone, acamprosate, and disulfiram—as treatment for BD patients (Table 4).26-29 In an open-label trial of 34 BD patients with alcohol dependence, naltrexone was well tolerated and associated with decreased alcohol craving and use and modest improvement in manic and depressive symptoms.26
In a double-blind, placebo-controlled study, 50 alcohol-dependent BD patients treated with standard mood-stabilizing therapy and cognitive-behavioral therapy were randomized to receive add-on naltrexone, 50 mg/d, or placebo.27 Patients receiving naltrexone showed decreased alcohol consumption, although no measures were statistically significant. Effect sizes of alcohol use decrease and alcohol craving were moderate to large compared with placebo, which suggests that naltrexone may be effective for treating alcoholism in these patients.
Two other studies evaluated naltrexone and disulfiram in patients with BD or other mood disorders.28,29 Naltrexone was well tolerated, caused no serious adverse side effects, and was significantly more effective than placebo in decreasing drinking rates and increasing the number of abstinent days.28,29 Disulfiram was as effective as naltrexone, but the combination of both offered no advantage over use of either drug separately.
There are reports of a new-onset manic episode associated with naltrexone use in a patient with opioid dependence, and a manic episode triggered by naltrexone in a patient with BD with comorbid alcohol dependence.30,31 At both low and high doses, disulfiram is associated with induction of psychotic mania in alcoholic patients without a personal or family history of BD.32,33
We found no studies that evaluated treating BD patients who abused other substances, such as cannabis or opiates. We recommend that BD patients with these substance use disorders should be referred to treatment modalities that are condition-specific, such as psychotherapy for cannabis use disorders or methadone or naltrexone treatment for opiate dependence. More severe cases of comorbid SUD probably would benefit from a referral to or consultation with a SUD specialist.
Table 4
Naltrexone and disulfiram for BD patients with alcohol dependence
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Brown et al, 200626 | Naltrexone | Open label | Alcohol dependence | Decreased alcohol use and craving |
| Brown et al, 200927 | Naltrexone vs placebo | Double-blind, placebo-controlled | Alcohol dependence | Nonsignificant decrease in alcohol consumption |
| Petrakis et al, 200528 and 200729 | Naltrexone alone vs disulfiram alone vs naltrexone plus disulfiram | Double-blind, randomized, placebo-controlled | Alcohol dependence | More time in abstinence and decreased craving for both compounds |
| BD: bipolar disorder | ||||
Related Resource
- Tolliver BK. Bipolar disorder and substance abuse: Overcome the challenges of ‘dual diagnosis’ patients. Current Psychiatry. 2010; 9(8): 32-38.
Drug Brand Names
- Acamprosate • Campral
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, Equetro, others
- Disulfiram • Antabuse
- Divalproex sodium • Depakote,
- Depakote ER Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methadone • Dolophine
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Valproate • Depacon
Disclosures
Dr. Nery held a temporary work contract as a clinical research physician with Eli Lilly and Company Brazil from May 2009 to November 2009.
Dr. Soares was partly supported by National Institute of Health grants MH 68766, MH 69774, and RR 20571. He receives grant/research support from Bristol-Myers Squibb, Cephalon, GlaxoSmithKline, and Sunovion.
Discuss this article at www.facebook.com/CurrentPsychiatry
Among DSM axis I diagnoses, bipolar disorder (BD) has the highest rates of comorbid substance use disorders (SUDs).1-3 Approximately 60% of patients with bipolar I disorder have a lifetime diagnosis of an SUD.1 Excluding tobacco, alcohol is the substance most often abused by BD patients, followed by cannabis, amphetamines, and cocaine.1-3
BD patients with comorbid SUD usually exhibit more severe clinical presentations and poorer outcomes than their counterparts without SUDs. Compared with patients with BD alone, those with BD and SUD comorbidity (BD-SUD) experience earlier onset of mood symptoms; higher rates of anxiety disorders, suicide attempts, accidents, hospitalizations, and rapid cycling; more depressive episodes; and lower treatment compliance.4-9
Several treatment options are available for patients with BD-SUD, including psychotherapy modalities, medications primarily used to treat BD, and medications primarily used to treat SUDs. Evidence-based support for these treatments remains limited, and no treatment of choice has emerged. This article reviews evidence on the longer-term treatment of BD-SUD, including general strategies and specific psychosocial and pharmacologic interventions. Short-term treatment strategies, such as pharmacotherapy for detoxification, are outside the scope of this review.
General strategies
The causes of BD-SUD are complex. Evidence suggests that the presence of affective symptoms is associated with an increased risk for substance misuse. This should be kept in mind when treating a patient with BD-SUD because controlling mood symptoms probably will help control substance abuse. However, evidence also shows that SUDs may be independent of mood episodes. Therefore, treating only mood symptoms in the hope that doing so will control substance abuse may not be enough.
Because the negative impact of SUDs on BD outcome is well documented, inform patients that limiting their use of alcohol and/or drugs is vital to control their mood disorder. Efforts to educate, stimulate, and support patients to moderate or stop their alcohol and/or drug use are likely to result in positive changes.10 Therefore, treatment for BD-SUD should follow, in part, the same recommendations for treatment of SUDs in patients with no comorbid axis I disorders:
- identify the problem (ie, the existence of a comorbid SUD)
- share your concerns with your patient
- offer appropriate and specific treatments, such as detoxification and/or self-help and counseling programs.10
Because SUDs usually are chronic and relapsing conditions, periods of drug and/ or alcohol use should be expected and not considered a sign of treatment failure. In addition, integrating treatment for both conditions probably is better than managing each separately. Therefore, targeting BD symptoms with mood-stabilizing medications and substance abuse with nonpharmacologic modalities such as drug counseling likely will bring about the best results.
Compared with BD patients without comorbid SUD, BD-SUD patients have a 7-fold increased risk of antidepressantinduced mania.11 Therefore, antidepressants should be prescribed cautiously for patients with BD-SUD.
Integrated psychosocial therapy
BD-SUD patients may benefit from attending self-help programs such as Alcoholics Anonymous and Narcotics Anonymous, provided their mood is stable enough to allow them to participate. Other forms of psychotherapy for BD-SUD patients include standard group drug counseling and integrated group therapy that simultaneously addresses both conditions.
Integrated group therapy is based on the premise that changing maladaptive mood cognitions and behaviors will facilitate recovery from SUDs, and changing maladaptive substance use cognitions and behaviors will facilitate recovery from mood disorders.12 In a recent randomized controlled trial, 62 BD-SUD patients were blindly assigned to integrated group therapy or standard group drug counseling and followed for 3 months.12 Pharmacotherapy was prescribed as usual. Substance use decreased for both groups. However, compared with patients in the drug counseling group, those who participated in integrated group therapy spent fewer days using substances in general and alcohol in particular, fewer days using alcohol to intoxication, and had a shorter time from treatment initiation to the first abstinent month. There were no differences between groups in number of weeks in a mood episode.
Pharmacotherapy options
For a table that summarizes the dosages and indications of the medications used to treat BD-SUD that are described below, visit this article at CurrentPsychiatry.com.
Table
Medications used to treat substance use disorders in bipolar disorder patients*
| Drug | Dosages | FDA-approved indication(s) |
|---|---|---|
| Acamprosate | 1,998 mg/d | Maintenance of abstinence from alcohol in patients with alcohol dependence |
| Aripiprazole | 15 to 45 mg/d | Acute manic or mixed episode of bipolar disorder; augmentation therapy for major depressive disorder |
| Carbamazepine | 400 to 1,200 mg/d | Manic and mixed episodes associated with bipolar disorder |
| Disulfiram | 250 to 500 mg/d | Enforced sobriety in abstinent alcohol-dependence patients |
| Divalproex sodium | Initial dose: 750 mg/d Maximum dose: 60 mg/kg/d† | Manic episodes associated with bipolar disorder |
| Lamotrigine | 200 mg/d | Maintenance treatment of bipolar I disorder |
| Lithium | 900 to 1,800 mg/d for acute episodes 900 mg to 1,200 mg/d for maintenance‡. | Manic episodes associated with bipolar disorder; maintenance treatment of bipolar disorder |
| Naltrexone | 50 mg/d 380 mg/month | Alcohol dependence |
| Quetiapine | 300 mg/d for bipolar depression 400 to 800 mg/d for bipolar mania 400 to 800 mg/d for maintenance treatment of bipolar disorder | Depressive and acute manic episodes associated with bipolar I disorder; maintenance treatment of bipolar I disorder |
| Risperidone | 1 to 6 mg/d | Acute manic or mixed episodes associated with bipolar I disorder |
| * None of the medications cited in this table or the text have been specifically approved by the FDA for treating alcohol/drug abuse/dependence co-occurring with bipolar disorder †Dose should correspond to valproic acid therapeutic levels between 50 and 100 μg/mL ‡Dose should correspond to lithium therapeutic levels between 0.8 and 1.2 mEq/L for acute manic episode treatment and 0.6 and 1.0 mEq/L for maintenance treatment | ||
Lithium. Given its well-documented mood stabilizing effect, lithium would seem to be a reasonable option to treat BD-SUD patients, but scant evidence supports its role as an anti-alcohol or anti-drug medication (Table 1).13,14 Lithium’s efficacy was evaluated in a study of 25 adolescents suffering from mood disorders (mostly BD) and comorbid SUDs (mostly alcohol and cannabis) randomized to receive lithium or placebo for 6 weeks.13 Lithium was well tolerated and improved psychiatric symptoms. At week 3, patients receiving lithium produced fewer positive results on randomly administered urine drug screens than those receiving placebo.
However, lithium seems to have little efficacy in reducing cocaine use in cocaine-dependent patients with bipolar spectrum disorders.14 In an open-label study, 10 patients with a history of hypomania or cyclothymia received lithium monotherapy for 12 weeks. Although patients experienced improved mood symptoms and decreased cocaine use, the mean decrease was transitory and not statistically significant. Another factor that may limit lithium’s use for BD-SUD patients is that these patients are more likely to comply with valproate treatment than with lithium.15
Table 1
Lithium for BD patients with substance use disorders
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Geller et al, 199813 | Lithium vs placebo | Double-blind, placebo-controlled | Alcohol and cannabis use disorders | Decreased positive drug screen results |
| Nunes et al, 199014 | Lithium | Open label | Cocaine abuse | Nonsignificant decrease in cocaine use |
| BD: bipolar disorder | ||||
Anticonvulsants. In a double-blind, placebo-controlled study of 59 alcohol-dependent bipolar I disorder patients, lithium plus divalproex sodium was superior to lithium plus placebo in decreasing number of drinking days and number of drinks per day and in increasing periods of abstinence (Table 2).16-19 Divalproex sodium was well tolerated and liver function improved in the divalproex sodium group compared with the placebo group, which probably was a benefit of decreased alcohol consumption. In addition, there was a strong association between mood symptoms and alcohol use, which suggests that maximizing mood symptom treatment may decrease alcohol use. However, the divalproex sodium and placebo groups did not differ in measures of mood symptoms, which implies that divalproex sodium might exhibit a positive effect on drinking regardless of its mood-stabilizing properties.
Divalproex sodium also has been used to treat BD comorbid with cocaine dependence. In a small open-label study, 15 patients receiving divalproex sodium plus counseling for mood and substance use disorders were followed for 6 weeks.17 The 7 patients who completed the trial had significantly more cocaine-abstinent days, spent less money on cocaine, and experienced fewer manic and depressive symptoms. However, divalproex sodium’s effect on cocaine use cannot be determined solely from this study because there was no placebo control group.
Despite its widespread use as a mood stabilizer and potential use in alcohol detoxification, carbamazepine scarcely has been studied in BD-SUD patients. A double-blind, placebo-controlled study of 139 cocaine-dependent patients with BD or other affective disorders found that patients taking carbamazepine for 12 weeks experienced modest reductions in positive urine drug screens and increased time to cocaine use.18 They also reported less cocaine craving than patients taking placebo, and mood symptoms (mostly depressive) improved.
An open-label study used lamotrigine as adjunctive therapy or monotherapy for 62 cocaine-dependent BD patients followed for 36 weeks.19 There was some decrease in cocaine craving, money spent on cocaine, and rate of depressive and manic symptoms, but no effect on cocaine use. A placebo-controlled trial is necessary to confirm these modest effects.
No studies have evaluated the potential role of topiramate in treating BD-SUD, despite its FDA-approved indication for alcoholism treatment. Topiramate’s well-known safety and tolerability profile in BD patients make it an interesting option for those with co-occurring alcohol dependence.
Table 2
Studies suggest anticonvulsants may reduce alcohol, cocaine use in BD patients
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Salloum et al, 200516 | Divalproex sodium plus lithium vs placebo plus lithium | Double-blind, placebo-controlled | Alcohol dependence | Decreased number of drinking days and number of drinks per day and increased time of abstinence |
| Salloum et al, 200717 | Divalproex sodium | Open label | Cocaine dependence | Increased cocaine-abstinent days and decreased money spent on cocaine and cocaine use severity index |
| Brady et al,* 200218 | Carbamazepine vs placebo | Double-blind, placebo-controlled | Cocaine dependence | Decreased cocaine craving and cocaine use |
| Brown et al, 200619 | Lamotrigine | Open label | Cocaine dependence | Decreased cocaine craving and money spent on cocaine |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder | ||||
Atypical antipsychotics. In an open-label study, 16 weeks of quetiapine monotherapy effectively decreased alcohol consumption, alcohol craving, and psychotic and affective symptoms in 28 alcoholics with a variety of psychiatric diagnoses, including BD, schizoaffective disorder, and borderline personality disorder (Table 3).20-24 However, in a double-blind study of augmentation with quetiapine or placebo for 102 alcohol-dependent BD patients, no significant differences in alcohol use were found between groups.21
Quetiapine may be effective for treating BD patients with comorbid cocaine dependence. In an open-label study, 12 weeks of quetiapine augmentation in 17 cocaine-dependent BD patients was associated with decreased cocaine craving and improvement in depressive symptoms.22 In another open-label study, 80 BD patients with comorbid cocaine or amphetamine dependence were randomly assigned to receive quetiapine or risperidone as adjunctive therapy or monotherapy for 20 weeks.23 Both groups showed significantly decreased drug use and drug craving and improved mood. This study suggests that risperidone also may be an option for BD patients with comorbid cocaine or stimulant dependence.
A 20-week, open-label study of 20 BD-SUD patients found that switching patients from their previous antipsychotic to aripiprazole resulted in less cocaine craving, less alcohol craving, and less money spent on alcohol.24
Olanzapine has not been systematically studied in BD-SUD patients. Some case reports suggest that olanzapine may decrease cocaine craving and use in patients with schizoaffective disorder (bipolar type) and alcohol craving and use in BD patients with comorbid alcohol dependence.25
Table 3
Evidence of efficacy for antipsychotics for BD patients with SUDs
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Martinotti et al,* 200820 | Quetiapine | Open label | Alcohol dependence | Decreased alcohol consumption and alcohol craving |
| Brown et al, 200821 | Quetiapine vs placebo | Double-blind, placebo-controlled | Alcohol dependence | No difference between quetiapine and placebo in decreasing alcohol use and alcohol craving |
| Brown et al, 200222 | Quetiapine | Open label | Cocaine dependence | Decreased cocaine use and cocaine craving |
| Nejtek et al, 200823 | Risperidone vs quetiapine | Open label | Cocaine dependence and amphetamine dependence | Decreased drug use and drug craving |
| Brown et al, 200524 | Aripiprazole | Open label | Alcohol and cocaine dependence | Decreased alcohol and cocaine craving and money spent on alcohol |
| *Sample included, but was not limited to, patients with BD BD: bipolar disorder; SUDs: substance use disorders | ||||
SUD medications. Little evidence guides using medications indicated for treating SUDs—such as naltrexone, acamprosate, and disulfiram—as treatment for BD patients (Table 4).26-29 In an open-label trial of 34 BD patients with alcohol dependence, naltrexone was well tolerated and associated with decreased alcohol craving and use and modest improvement in manic and depressive symptoms.26
In a double-blind, placebo-controlled study, 50 alcohol-dependent BD patients treated with standard mood-stabilizing therapy and cognitive-behavioral therapy were randomized to receive add-on naltrexone, 50 mg/d, or placebo.27 Patients receiving naltrexone showed decreased alcohol consumption, although no measures were statistically significant. Effect sizes of alcohol use decrease and alcohol craving were moderate to large compared with placebo, which suggests that naltrexone may be effective for treating alcoholism in these patients.
Two other studies evaluated naltrexone and disulfiram in patients with BD or other mood disorders.28,29 Naltrexone was well tolerated, caused no serious adverse side effects, and was significantly more effective than placebo in decreasing drinking rates and increasing the number of abstinent days.28,29 Disulfiram was as effective as naltrexone, but the combination of both offered no advantage over use of either drug separately.
There are reports of a new-onset manic episode associated with naltrexone use in a patient with opioid dependence, and a manic episode triggered by naltrexone in a patient with BD with comorbid alcohol dependence.30,31 At both low and high doses, disulfiram is associated with induction of psychotic mania in alcoholic patients without a personal or family history of BD.32,33
We found no studies that evaluated treating BD patients who abused other substances, such as cannabis or opiates. We recommend that BD patients with these substance use disorders should be referred to treatment modalities that are condition-specific, such as psychotherapy for cannabis use disorders or methadone or naltrexone treatment for opiate dependence. More severe cases of comorbid SUD probably would benefit from a referral to or consultation with a SUD specialist.
Table 4
Naltrexone and disulfiram for BD patients with alcohol dependence
| Study | Intervention | Design | Substance use disorder | Results |
|---|---|---|---|---|
| Brown et al, 200626 | Naltrexone | Open label | Alcohol dependence | Decreased alcohol use and craving |
| Brown et al, 200927 | Naltrexone vs placebo | Double-blind, placebo-controlled | Alcohol dependence | Nonsignificant decrease in alcohol consumption |
| Petrakis et al, 200528 and 200729 | Naltrexone alone vs disulfiram alone vs naltrexone plus disulfiram | Double-blind, randomized, placebo-controlled | Alcohol dependence | More time in abstinence and decreased craving for both compounds |
| BD: bipolar disorder | ||||
Related Resource
- Tolliver BK. Bipolar disorder and substance abuse: Overcome the challenges of ‘dual diagnosis’ patients. Current Psychiatry. 2010; 9(8): 32-38.
Drug Brand Names
- Acamprosate • Campral
- Aripiprazole • Abilify
- Carbamazepine • Carbatrol, Equetro, others
- Disulfiram • Antabuse
- Divalproex sodium • Depakote,
- Depakote ER Lamotrigine • Lamictal
- Lithium • Eskalith, Lithobid
- Methadone • Dolophine
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Topiramate • Topamax
- Valproate • Depacon
Disclosures
Dr. Nery held a temporary work contract as a clinical research physician with Eli Lilly and Company Brazil from May 2009 to November 2009.
Dr. Soares was partly supported by National Institute of Health grants MH 68766, MH 69774, and RR 20571. He receives grant/research support from Bristol-Myers Squibb, Cephalon, GlaxoSmithKline, and Sunovion.
1. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
2. Kessler RC, Crum RM, Warner LA, et al. Lifetime cooccurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
3. Grant BF, Stinson FS, Hasin DS, et al. Prevalence, correlates, and comorbidity of bipolar I disorder and axis I and II disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2005;66(10):1205-1215.
4. Feinman JA, Dunner DL. The effect of alcohol and substance abuse on the course of bipolar affective disorder. J Affect Disord. 1996;37(1):43-49.
5. Cassidy F, Ahearn EP, Carroll BJ. Substance abuse in bipolar disorder. Bipolar Disord. 2001;3(4):181-188.
6. Frye MA, Altshuler LL, McElroy SL, et al. Gender differences in prevalence, risk, and clinical correlates of alcoholism comorbidity in bipolar disorder. Am J Psychiatry. 2003;160(5):883-889.
7. Khalsa HM, Salvatore P, Hennen J, et al. Suicidal events and accidents in 215 first-episode bipolar I disorder patients: predictive factors. J Affect Disord. 2008;106(1-2):179-184.
8. Baldessarini RJ, Perry R, Pike J. Factors associated with treatment nonadherence among US bipolar disorder patients. Hum Psychopharmacol. 2008;23(2):95-105.
9. Cardoso BM, Kauer Sant’Anna M, Dias VV, et al. The impact of co-morbid alcohol use disorder in bipolar patients. Alcohol. 2008;42(6):451-457.
10. Schuckit MA. Alcohol-use disorders. Lancet. 2009;373 (9662):492-501.
11. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795.
12. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
13. Geller B, Cooper TB, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry. 1998;37:171-178.
14. Nunes EV, McGrath PJ, Wager S, et al. Lithium treatment for cocaine abusers with bipolar spectrum disorders. Am J Psychiatry. 1990;147:655-657.
15. Weiss RD, Greenfield SF, Najavits LM, et al. Medication compliance among patients with bipolar disorder and substance use disorder. J Clin Psychiatry. 1998;59:172-174.
16. Salloum IM, Cornelius JR, Daley DC, et al. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
17. Salloum IM, Douaihy A, Cornelius JR, et al. Divalproex utility in bipolar disorder with co-occurring cocaine dependence: a pilot study. Addict Behav. 2007;32(2):410-405.
18. Brady KT, Sonne SC, Malcolm RJ, et al. Carbamazepine in the treatment of cocaine dependence: subtyping by affective disorder. Exp Clin Psychopharmacol. 2002;10:276-285.
19. Brown ES, Perantie DC, Dhanani N, et al. Lamotrigine for bipolar disorder and comorbid cocaine dependence: a replication and extension study. J Affect Disord. 2006;93(1-3):219-222.
20. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
21. Brown ES, Garza M, Carmody TJ. A randomized double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
22. Brown ES, Nejtek VA, Perantie DC, et al. Quetiapine in bipolar disorder and cocaine dependence. Bipolar Disord. 2002;4(6):406-411.
23. Nejtek VA, Avila M, Chen LA, et al. Do atypical antipsychotics effectively treat co-occurring bipolar disorder and stimulant dependence? A randomized, double-blind trial. J Clin Psychiatry. 2008;69(8):1257-1266.
24. Brown ES, Jeffress J, Liggin JD, et al. Switching outpatients with bipolar or schizoaffective disorders and substance abuse from their current antipsychotic to aripiprazole. J Clin Psychiatry. 2005;66:756-760.
25. Sattar SP, Grant K, Bhatia S, et al. Potential use of olanzapine in treatment of substance dependence disorders. J Clin Psychopharmacol. 2003;23:413-415.
26. Brown ES, Beard L, Dobbs L, et al. Naltrexone in patients with bipolar disorder and alcohol dependence. Depress Anxiety. 2006;23(8):492-495.
27. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebo-controlled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33:1863-1869.
28. Petrakis IL, Poling J, Levinson C, et al. and the VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
29. Petrakis I, Ralevski E, Nich C, et al. and the VA VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Sullivan MA, Nunes EV. New-onset mania and psychosis following heroin detoxification and naltrexone maintenance. Am J Addict. 2005;14(5):486-487.
31. Sonne SC, Brady KT. Naltrexone for individuals with comorbid bipolar disorder and alcohol dependence. J Clin Psychopharmacol. 2000;20(1):114-115.
32. Ceylan ME, Turkcan A, Mutlu E, et al. Manic episode with psychotic symptoms associated with high dose of disulfiram: a case report. J Clin Psychopharmacol. 2007;27(2):224-225.
33. Li MY, Shen YC. Manic episode with psychosis following a lower than recommended dosage regimen of disulfiram. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):311-312.
1. Regier DA, Farmer ME, Rae DS, et al. Comorbidity of mental disorders with alcohol and other drug abuse. Results from the Epidemiologic Catchment Area (ECA) Study. JAMA. 1990;264(19):2511-2518.
2. Kessler RC, Crum RM, Warner LA, et al. Lifetime cooccurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch Gen Psychiatry. 1997;54(4):313-321.
3. Grant BF, Stinson FS, Hasin DS, et al. Prevalence, correlates, and comorbidity of bipolar I disorder and axis I and II disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2005;66(10):1205-1215.
4. Feinman JA, Dunner DL. The effect of alcohol and substance abuse on the course of bipolar affective disorder. J Affect Disord. 1996;37(1):43-49.
5. Cassidy F, Ahearn EP, Carroll BJ. Substance abuse in bipolar disorder. Bipolar Disord. 2001;3(4):181-188.
6. Frye MA, Altshuler LL, McElroy SL, et al. Gender differences in prevalence, risk, and clinical correlates of alcoholism comorbidity in bipolar disorder. Am J Psychiatry. 2003;160(5):883-889.
7. Khalsa HM, Salvatore P, Hennen J, et al. Suicidal events and accidents in 215 first-episode bipolar I disorder patients: predictive factors. J Affect Disord. 2008;106(1-2):179-184.
8. Baldessarini RJ, Perry R, Pike J. Factors associated with treatment nonadherence among US bipolar disorder patients. Hum Psychopharmacol. 2008;23(2):95-105.
9. Cardoso BM, Kauer Sant’Anna M, Dias VV, et al. The impact of co-morbid alcohol use disorder in bipolar patients. Alcohol. 2008;42(6):451-457.
10. Schuckit MA. Alcohol-use disorders. Lancet. 2009;373 (9662):492-501.
11. Goldberg JF, Whiteside JE. The association between substance abuse and antidepressant-induced mania in bipolar disorder: a preliminary study. J Clin Psychiatry. 2002;63:791-795.
12. Weiss RD, Griffin ML, Kolodziej ME, et al. A randomized trial of integrated group therapy versus group drug counseling for patients with bipolar disorder and substance dependence. Am J Psychiatry. 2007;164(1):100-107.
13. Geller B, Cooper TB, Sun K, et al. Double-blind and placebo-controlled study of lithium for adolescent bipolar disorders with secondary substance dependency. J Am Acad Child Adolesc Psychiatry. 1998;37:171-178.
14. Nunes EV, McGrath PJ, Wager S, et al. Lithium treatment for cocaine abusers with bipolar spectrum disorders. Am J Psychiatry. 1990;147:655-657.
15. Weiss RD, Greenfield SF, Najavits LM, et al. Medication compliance among patients with bipolar disorder and substance use disorder. J Clin Psychiatry. 1998;59:172-174.
16. Salloum IM, Cornelius JR, Daley DC, et al. Efficacy of valproate maintenance in patients with bipolar disorder and alcoholism: a double-blind placebo-controlled study. Arch Gen Psychiatry. 2005;62(1):37-45.
17. Salloum IM, Douaihy A, Cornelius JR, et al. Divalproex utility in bipolar disorder with co-occurring cocaine dependence: a pilot study. Addict Behav. 2007;32(2):410-405.
18. Brady KT, Sonne SC, Malcolm RJ, et al. Carbamazepine in the treatment of cocaine dependence: subtyping by affective disorder. Exp Clin Psychopharmacol. 2002;10:276-285.
19. Brown ES, Perantie DC, Dhanani N, et al. Lamotrigine for bipolar disorder and comorbid cocaine dependence: a replication and extension study. J Affect Disord. 2006;93(1-3):219-222.
20. Martinotti G, Andreoli S, Di Nicola M, et al. Quetiapine decreases alcohol consumption, craving, and psychiatric symptoms in dually diagnosed alcoholics. Hum Psychopharmacol. 2008;23(5):417-424.
21. Brown ES, Garza M, Carmody TJ. A randomized double-blind, placebo-controlled add-on trial of quetiapine in outpatients with bipolar disorder and alcohol use disorders. J Clin Psychiatry. 2008;69(5):701-705.
22. Brown ES, Nejtek VA, Perantie DC, et al. Quetiapine in bipolar disorder and cocaine dependence. Bipolar Disord. 2002;4(6):406-411.
23. Nejtek VA, Avila M, Chen LA, et al. Do atypical antipsychotics effectively treat co-occurring bipolar disorder and stimulant dependence? A randomized, double-blind trial. J Clin Psychiatry. 2008;69(8):1257-1266.
24. Brown ES, Jeffress J, Liggin JD, et al. Switching outpatients with bipolar or schizoaffective disorders and substance abuse from their current antipsychotic to aripiprazole. J Clin Psychiatry. 2005;66:756-760.
25. Sattar SP, Grant K, Bhatia S, et al. Potential use of olanzapine in treatment of substance dependence disorders. J Clin Psychopharmacol. 2003;23:413-415.
26. Brown ES, Beard L, Dobbs L, et al. Naltrexone in patients with bipolar disorder and alcohol dependence. Depress Anxiety. 2006;23(8):492-495.
27. Brown ES, Carmody TJ, Schmitz JM, et al. A randomized, double-blind, placebo-controlled pilot study of naltrexone in outpatients with bipolar disorder and alcohol dependence. Alcohol Clin Exp Res. 2009;33:1863-1869.
28. Petrakis IL, Poling J, Levinson C, et al. and the VA New England VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and comorbid psychiatric disorders. Biol Psychiatry. 2005;57(10):1128-1137.
29. Petrakis I, Ralevski E, Nich C, et al. and the VA VISN I MIRECC Study Group. Naltrexone and disulfiram in patients with alcohol dependence and current depression. J Clin Psychopharmacol. 2007;27(2):160-165.
30. Sullivan MA, Nunes EV. New-onset mania and psychosis following heroin detoxification and naltrexone maintenance. Am J Addict. 2005;14(5):486-487.
31. Sonne SC, Brady KT. Naltrexone for individuals with comorbid bipolar disorder and alcohol dependence. J Clin Psychopharmacol. 2000;20(1):114-115.
32. Ceylan ME, Turkcan A, Mutlu E, et al. Manic episode with psychotic symptoms associated with high dose of disulfiram: a case report. J Clin Psychopharmacol. 2007;27(2):224-225.
33. Li MY, Shen YC. Manic episode with psychosis following a lower than recommended dosage regimen of disulfiram. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):311-312.
Subjective cognitive impairment: When to be concerned about ‘senior moments’
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
MS. F, age 66, requests genetic testing because she is concerned about mild memory difficulties, such as forgetting names and where she puts her keys or checkbook, and fears she may be developing Alzheimer’s disease (AD). Her mother and sister were diagnosed with AD in their early 60s. Ms. F has 20 years of education and reports no problems with driving, managing her finances, remembering to take her medications, or maintaining social activities, which her husband confirms.
Detailed questioning about anxiety and depressive symptoms reveals substantial worries about future cognitive decline and some concerns about her finances and her husband’s health. Ms. F says she occasionally feels down and has low energy but denies other depressive symptoms. She reports no sleep disturbances—including snoring and daytime sleepiness, which could indicate obstructive sleep apnea—which her husband confirms. Ms. F takes levothyroxine for hypothyroidism, atenolol for hypertension, aspirin and clopidogrel for coronary artery disease, and atorvastatin for hyperlipidemia. In addition, she provides a long list of over-the-counter (OTC) supplements—ginkgo, huperzine, ginseng, phosphatidylserine, B1, B12, folate, vitamin D, alpha-lipoic acid, and vinpocetine—that she takes to “protect” her brain from AD.
Subjective cognitive impairment (SCI) in older persons is a common condition with a largely unclear prognosis. Many older adults (age ≥65) express concern about mild cognitive problems—“senior moments”—such as word-finding difficulties and forgetfulness.1 Individuals may wonder if walking into a room only to forget why might be the first sign of dementia. Some older adults try to counteract these memory problems by engaging in brain exercises—including costly computer games—and taking OTC “brain-enhancing” vitamins, herbal remedies, and other supplements.
Although some clinicians may view SCI as benign, that is not always true (Table l).2-5 This article discusses the clinical significance of these mild cognitive complaints by examining:
- age-related cognitive decline (ARCD)
- SCI
- how SCI can be differentiated from more serious conditions, such as mild cognitive impairment (MCI) and early stages of AD and other dementias.
We also will discuss assessing and treating cognitive complaints. Although distinctions between SCI and ARCD may be controversial, evidence suggests clinicians need to adopt a more nuanced clinical approach.
Table 1
Why SCI should be taken seriously
| SCI may create emotional distress because patients are aware of decline in their ‘mental sharpness’ |
| SCI patients might consume unnecessary and potentially harmful OTC supplements touted to promote memory |
| Patients might limit their driving and financial management to avoid making mistakes |
| SCI might impair medication adherence2 |
| SCI may be an early sign of dementia3 |
| Patients’ worry about their self-perceived memory loss might predict dementia4 |
| SCI may predict nursing home placement5 |
| Addressing SCI gives health care providers an opportunity to address anxiety or depression that often accompany SCI |
| Evaluation of potential causes of SCI may uncover reversible conditions that can be treated |
| OTC: over-the-counter; SCI: subjective cognitive impairment |
‘Normal’ cognitive decline
ARCD is subtle decline in cognitive abilities, such as episodic memory, attention, and time needed to complete complex activities.6,7 Individuals with ARCD might not have subjective memory complaints or objective cognitive deficits, and their ability to live independently may not be compromised.7 The degree of decline in ARCD may be smaller than previously thought.8 Park9 summarizes 4 main mechanisms thought to underlie age-related declines in cognition:
- reduced speed of processing
- decreased working memory capabilities
- declining inhibitory control (eg, impaired complex attentional capabilities)
- sensory changes (eg, visual and auditory deficits).
ARCD traditionally is thought to result from predictable changes in the brain associated with aging, such as reduced brain volume in the hippocampus and frontal lobes, loss of myelin, loss of synapses, and cytoskeletal changes.7 However, not all older adults experience ARCD. Some remain highly functional in their later years and continue to actively engage in life well into very old age.6,9
Subjective cognitive impairment
One-quarter to one-half of community-dwelling older adults report subjective cognitive complaints, such as forgetfulness and word-finding difficulties.10 Patients with SCI do not show objective evidence of cognitive impairment on neuropsychological tests and their cognitive problems cause no functional decline.10
Preliminary evidence indicates that SCI may be a harbinger of further cognitive decline. Reisberg et al3 found that compared with patients without SCI, patients with SCI were 4.5 times more likely to develop MCI—cognitive difficulties that can be detected by cognitive tests, but do not cause functional decline—or dementia within 7 years.3 Studies also have suggested that SCI may be a pre-MCI stage of subsequent dementia.11-13 AD generally has a long (10 to 12 years) and progressive prodromal phase before dementia onset and is characterized by successive emergence of cognitive deficits, memory complaints, depressive symptoms, and functional impairment.14
In light of this research, we believe patients with SCI and other risk factors for AD, such as a family history of AD, may be at higher risk of further cognitive and functional decline compared with individuals with ARCD and no AD risk factors. Therefore, patients with SCI and other risk factors for AD (Table 2)15-19 may benefit from annual follow-up to determine if cognitive problems have progressed to MCI or AD.
SCI may be a response to subclinical alterations in neurobiology—a phenomenon known as reverse causality.20 Biomarkers, such as cerebrospinal fluid levels of ß-amyloid and phosphorylated tau, and amyloid imaging using positron emission tomography may help identify AD in SCI patients.21 In these patients, SCI is a misnomer because the cognitive impairment is real—not “subjective”—but current tests are not sensitive enough to detect the cognitive decline the patient has recognized. This group of patients should be differentiated from individuals who may perceive typical cognitive aging (ARCD) as pathologic and complain about it. In the future, biomarkers may help differentiate these 2 groups.
Table 2
Factors that increase SCI patients’ risk for dementia
| Family history of Alzheimer’s disease |
| Mild behavioral impairment |
| Slow gait |
| Depression |
| Rapid weight loss |
| Multiple subtle neurologic abnormalities |
| Vascular disease (eg, peripheral vascular disease, coronary artery disease, cerebrovascular disease) |
| SCI: subjective cognitive impairment Source: References 15-19 |
Mild cognitive impairment
MCI is similar to SCI because MCI patients may present with complaints of memory decline and other cognitive difficulties22 but neither condition is associated with significant impairment of daily activities.23 The key difference is that patients with MCI demonstrate impaired performance on objective cognitive tests whereas SCI patients do not.24 In our experience, office-based tests do not reliably differentiate the 2 conditions because many patients with SCI may show mild impairment in tests such as the Mini-Mental State Exam (MMSE)25 but comprehensive neuropsychological testing reveals no objective cognitive deficits. Neuropsychological testing is essential to reliably differentiate SCI from MCI.
The distinction between SCI and MCI is clinically relevant because evidence suggests that MCI patients have a near-term risk of developing dementia, particularly AD.22,23 In a longitudinal study of 76 individuals with MCI, 12% of patients progressed to AD each year compared with 1% to 2% of healthy older adults.26 Patients with MCI are at increased risk of delirium (especially during hospitalization), falls, medication errors, and difficulty managing their finances.24 Older adults with MCI also have increased mortality compared with older adults with normal cognitive functioning.22 Both SCI and MCI should be differentiated from mild dementia. Common dementias in older adults include:
- AD dementia
- Vascular dementia (may occur with or without AD)
- Lewy body dementia
- Frontotemporal dementia
- Parkinson’s disease dementia.
By definition, all dementia types are associated with impaired ability to perform daily activities and cognitive decline.27
Assessing cognitive complaints
Evaluation of older adults’ cognitive complaints should begin with a thorough history to elicit symptoms of anxiety, depression, physical complaints, and any associated functional decline; a physical exam; and a comprehensive mental status examination. This initial evaluation should be followed by routine and specific investigations as indicated (Table 3).22,24,28,29
In a 6-year study of 100 older adults with and without objective evidence of memory decline, both groups showed similar rates of cognitive complaints.30 Also, researchers found no relationship between individuals’ perception of their cognitive functioning and performance on neuropsychological testing. Mood, education level, and apolipoprotein E epsilon 4 genotype status also did not correlate with participants’ subjective cognitive complaints. These findings highlight the need for objective test data to determine whether older adults’ memory complaints reflect pathologic changes in cognition. After a thorough diagnostic workup, some patients complaining of memory decline will have no detectable evidence of cognitive dysfunction or an identifiable cause. However, others may have identifiable causes of memory impairment (Table 4)28,29,31,32—which could be treated—some will have MCI, and others may be in an early stage of dementia.
Table 3
Investigation of older adults with SCI
| Investigation | Rationale |
|---|---|
| Routine | |
| Neuropsychological testing | Delineation of cognitive syndromes (SCI vs MCI vs AD*) |
| Hematology (full blood count) | Screen for anemia |
| Biochemistry (electrolytes, renal function, liver function, thyroid function, B12, and folate) | Screen for treatable causes of cognitive complaints |
| For specific indication suggested by history, physical exam, or neuropsychological testing | |
| Neuroimaging | Generalized and regional imaging (eg, hippocampal atrophy, space occupying lesions) |
| Electroencephalography | Epilepsy/seizures (especially absence and complex partial) |
| Cardiac (eg, echocardiography) | May reveal cardiac arrhythmia or sources of emboli |
| Inflammatory markers (eg, ESR) | Screen for inflammatory processes |
| Treponemal serology | Tertiary syphilis |
| *Alzheimer’s disease and other dementias AD: Alzheimer’s disease; ESR: erythrocyte sedimentation rate; MCI: mild cognitive impairment; SCI: subjective cognitive impairment Source: References 22,24,28,29 | |
Table 4
Differential diagnosis of SCI
| Cause of cognitive impairment | Potential mechanism |
|---|---|
| ARCD | Allostatic load, ‘wear and tear’ from a lifetime of physiological or psychological stresses and adaptations |
| Anemia | Neuronal hypoxia |
| Alzheimer’s disease | Amyloid and/or tau-mediated neurotoxicity, neuroinflammation |
| Cerebrovascular disease | Neuronal ischemia and hypoxia, neuroinflammation |
| Vitamin deficiencies (eg, B1, B12, folate, D) | Impaired neuronal and neurotransmitter function |
| Inadequate protein intake | Impaired neuronal function |
| Anticholinergic drug use | Decreased cholinergic neurotransmission |
| Alcohol use | Direct neurotoxicity and indirect causes such as malnutrition or head injury |
| Depression, anxiety | Hippocampal dysfunction with or without atrophy |
| Obstructive sleep apnea | Neuronal hypoxia, neuroinflammation |
| Head injury | Neuronal and synaptic loss |
| ARCD: age-related cognitive decline; SCI: subjective cognitive impairment Source: References 28,29,31,32 | |
CASE CONTINUED: No measurable deficits
Ms. F’s medical history is remarkable for coronary artery disease, hypothyroidism, hypertension, hyperlipidemia, cataracts, arthritis, back surgery (secondary to spondylosis), and foot surgery. Ms. F denies a history of alcohol or illicit substance abuse. She smoked tobacco for 30 years (2 packs per day), but quit 5 years ago after her heart attack. Physical exam is unremarkable except for mild obesity (body mass index = 31 kg/m2).
Ms. F’s mental status exam reveals anxious mood and affect. Her recall is 2 out of 3 items. Her MMSE score is 29/30 (1 point lost on recall) and her Geriatric Depression Scale33 score is 2/15, indicating minimal depressive symptoms. On neuropsychological testing, Ms. F demonstrates high average intellectual abilities; compared with others her age, she performs within expectations on all measures. That is, she performs within the above-average to low-average range on measures of attention, working memory, speed of processing, expressive language, learning, memory, visual spatial abilities, executive functioning, and knowledge of basic health and safety information.
Enhancing neuroplasticity
We recommend neuroplasticity-based interventions to treat SCI and promote healthy brain aging.20,29 For a checklist clinicians can use to promote healthy brain aging and thus improve patients’ cognitive health see this article at CurrentPsychiatry. com. Table 51,29 lists cognitive strategies to improve memory and maintain cognitive vitality.
Enhancing brain plasticity and neurogenesis requires engaging older adults in demanding sensory, cognitive, and motor activities on an intensive basis.34 Therapeutic stimulation of neuroplasticity and neurogenesis might contribute to functional “repair” of the diseased adult brain before damage to whole neuronal networks has ensued.29 An important treatment component is reassuring patients with SCI that they do not have AD or MCI. Treating comorbid anxiety and depression and reversible causes of cognitive complaints is key to successful outcomes.
Table 5
Strategies to improve memory and maintain cognitive vitality
| Strategy | Description |
|---|---|
| Mindfulness | Focus on 1 task at a time rather than trying to multitask. Research shows that cognition is more efficient in this manner |
| Cognitive strategies | Use mnemonics (such as ROY G BIV to remember the colors of the rainbow). Make associations for information, such as when meeting someone new, relate their name to someone else you know well. Use cues such as memory notebooks to prompt information recall. Engage in learning new and challenging cognitive activities, such as a new language, a music instrument, or dance. Consider computer-based brain exercises |
| Rehearsal | Practice information you want to remember, such as repeating the information several times or writing it down |
| Be patient | Getting frustrated when you have memory difficulties makes it more challenging to remember information |
| Exercise (mental and physical) | Engage in mental activities, such as reading and crossword puzzles. Do something that you are interested in, rather than making it a chore. Research has demonstrated that physical exercise also aids memory |
| Diet | What is good for the heart is good for the brain. Fruits, vegetables, food rich in omega-3 fatty acids (eg, fatty fish such as salmon), whole grains, spices (eg, turmeric), and small amounts of tree nuts (eg, walnuts) are recommended as part of a balanced diet |
| Source: References 1,29 | |
CASE CONTINUED: Reassurance and risk reduction
Ms. F’s psychiatrist reassures her that she does not have AD. She receives genetic counseling and decides to forgo genetic testing. Her psychiatrist educates Ms. F about the risks of OTC supplements—especially increased risk of bleeding because she takes aspirin and clopidogrel—and lack of data supporting their use. Ms. F is counseled that a healthy lifestyle, including regular exercise, Mediterranean diet with increased intake of omega-3 fatty acids, learning new things, and being socially active, is the safest way to promote brain health. Over 3 months, Ms. F discontinues all supplements except the vitamins and omega-3, starts exercising, resumes piano lessons that she stopped 10 years ago, and becomes a vegetarian. She continues to have mild SCI but she says she is not bothered by it and feels satisfied that she is doing all she can to promote her brain health.
Related Resources
- Desai AK. Healthy brain aging: evidence based methods to preserve brain function and prevent dementia. Philadelphia, PA: W. B. Saunders; 2010.
- Doidge N. The brain that changes itself. New York, NY: Penguin Books; 2007.
- Vance DE, Roberson AJ, McGuinness TM, et al. How neuroplasticity and cognitive reserve protect cognitive functioning. J Psychosoc Nurs Ment Health Serv. 2010; 48: 1-8.
Brain Training Resources
- Weil A, Small G. The healthy brain kit. Boulder, CO: Sounds True, Inc.; 2007. Audio CDs, brain-training cards and workbooks.
- Posit Science. www.positscience.com.
- Sharp Brains. www.sharpbrains.com.
Drug Brand Names
- Atenolol • Tenormin
- Atorvastatin • Lipitor
- Clopidogrel • Plavix
- Levothyroxine • Levoxyl, Synthroid
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Featured Audio
Abhilash K. Desai, MD, discusses emerging research on biomarkers that may help clarify diagnosis.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
1. Small GW. What we need to know about age related memory loss. BMJ. 2002;324:1502-1505.
2. Hayes TL, Larimer N, Adami A, et al. Medication adherence in healthy elders. J Aging Health. 2009;21(4):567-580.
3. Reisberg B, Shulman MB, Torossian C, et al. Outcome over seven years of healthy adults with and without subjective cognitive impairment. Alzheimers Dement. 2010;6(1):11-24.
4. Jessen F, Wiese B, Bachmann C, et al. Prediction of dementia by subjective memory impairments: effects of severity and temporal association with cognitive impairment. Arch Gen Psychiatry. 2010;67:414-422.
5. Waldorff FB, Siersma V, Waldemar G. Association between subjective memory complaints and nursing home placement: a four-year follow-up. Int J Geriatr Psychiatry. 2009;24(6):602-609.
6. Salthouse TA. Selective review of cognitive aging. J Int Neuropsychol Soc. 2010;16:754-760.
7. Anderton B. Ageing of the brain. Mech Ageing Dev. 2002;23:811-817.
8. Salthouse TA. Influence of age on practice effects in longitudinal neurocognitive change. Neuropsychology. 2010;24(5):563-572.
9. Park D, Schwarz N. Cognitive aging: a primer. Philadelphia PA: Taylor and Francis Group; 2000.
10. Reisberg B, Shulman MB. Commentary on “a roadmap for the prevention of dementia II: Leon Thal Symposium 2008.” Subjective cognitive impairment as an antecedent of Alzheimer’s dementia: policy import. Alzheimers Dement. 2009;5:154-156.
11. Reisberg B, Gauthier S. Current evidence for subjective cognitive impairment (SCI) as the pre-mild cognitive impairment (MCI) stage of subsequently manifest Alzheimer’s disease. Int Psychogeriatr. 2008;20(1):1-16.
12. Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180-195.
13. Ramakers IH, Visser PJ, Aalten P, et al. Symptoms of preclinical dementia in general practice up to five years before dementia diagnosis. Dement Geriatr Cogn Disord. 2007;24(4):300-306.
14. Amieva H, Le Goff M, Millet X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008;64(5):492-498.
15. Taragano FE, Allegri RF, Krupitzki H, et al. Mild behavioral impairment and risk of dementia: a prospective cohort study of 358 patients. J Clin Psychiatry. 2009;70(4):584-592.
16. Jayadev S, Steinbart EJ, Chi YY, et al. Conjugal Alzheimer disease: risk in children when both parents have Alzheimer disease. Arch Neurol. 2008;65(3):373-378.
17. Hajjar I, Yang F, Sorond F, et al. A novel aging phenotype of slow gait, impaired executive function, and depressive symptoms: relationship to blood pressure and other cardiovascular risks. J Gerontol A Biol Sci Med Sci. 2009;64(9):994-1001.
18. Yamamoto N, Yamanaka G, Ishikawa M, et al. Cardio-ankle vascular index as a predictor of cognitive impairment in community-dwelling elderly people: four-year follow-up. Dement Geriatr Cogn Disord. 2009;28(2):153-158.
19. Inzitari M, Pozzi C, Ferrucci L, et al. Subtle neurological abnormalities as risk factors for cognitive and functional decline, cerebrovascular events, and mortality in older community-dwelling adults. Arch Intern Med. 2008;168(12):1270-1276.
20. Shineman DW, Salthouse TA, Launer LJ, et al. Therapeutics of cognitive aging. Ann N Y Acad Sci. 2010;1191(suppl 1):E1-E10.
21. Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118-1127.
22. Chertkow H, Massoud F, Nasreddine Z, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273-1285.
23. Rosenberg PB, Lyketsos C. Mild cognitive impairment: searching for the prodrome of Alzheimer’s disease. World Psychiatry. 2008;7(2):72-78.
24. Rosenberg PB, Johnston D, Lyketsos CG. A clinical approach to mild cognitive impairment. Am J Psychiatry. 2006;163(11):1884-1890.
25. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198.
26. Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56(3):303-308.
27. Diagnostic and statistical manual of mental disorders. 4th ed text rev. Washington, DC: American Psychiatric Association; 2000:135–180.
28. Malhotra R, Desai AK. Healthy brain aging: what has sleep got to do with it? Clin Geriatr Med. 2010;26:45-56.
29. Desai AK, Grossberg GT, Chibnall JT. Healthy brain aging: a road map. Clin Geriatr Med. 2010;26:1-16.
30. Weaver Cargin J, Collie A, Masters C, et al. The nature of cognitive complaints in healthy older adults with and without objective memory decline. J Clin Exp Neuropsychol. 2008;30:245-257.
31. Wilson RS, Arnold SE, Schneider JA, et al. Chronic distress, age-related neuropathology, and late-life dementia. Psychosom Med. 2007;69:47-53.
32. Deal JA, Carlson MC, Xue Q, et al. Anemia and 9-year domain-specific cognitive decline in community-dwelling older women: the Women’s Health and Aging Study II. J Am Geriatr Soc. 2009;57(9):1604-1611.
33. Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression scale: a preliminary report. J Psychiatr Res. 1983;17:37-49.
34. Mahncke HW, Bronstone A, Merzenich MM. Brain plasticity and functional losses in the aged: scientific bases for a novel intervention. Prog Brain Res. 2006;157:81-109.
Depression and suicide among physicians
Discuss this article at www.facebook.com/CurrentPsychiatry
Dr. G, a second-year surgical resident, becomes depressed when his girlfriend abruptly ends their relationship. His phone calls and e-mails seeking an explanation go unanswered. Having long struggled with his self-esteem, Dr. G interprets this rejection as confirmation of his self-criticism.
Because of his work schedule, Dr. G feels that there is no way to see a therapist or psychiatrist and believes that asking for time off to do so would adversely affect his evaluations. He feels too embarrassed and “weak” to disclose his breakup and depression to his colleagues and attending physicians and senses that fellow residents would resent having to “carry his load.” Dr. G has spent the past 2 years moonlighting at the local emergency room and thinks it would be humiliating to go there for psychiatric help. His work performance and attendance decline until eventually his residency director forces him to take a medical leave of absence.
Dr. G feels that his pain will never end. He writes goodbye letters to his family, makes arrangements for his possessions and funeral, and hangs himself from the balcony outside his apartment.
Although the rate of depression among physicians is comparable to that of the general population, physicians’ risk of suicide is markedly higher.1 Depression and other mood disorders may be under-recognized and inadequately treated in physicians because physicians might:
- be reluctant to seek treatment
- attempt to diagnose and treat themselves
- seek and receive “VIP treatment” from other health care providers.
This article examines physicians’ risk for depression and suicide, licensing concerns and other barriers to effective treatment, and what can be done to overcome such obstacles.
Not immune to depression
Rates of depression are higher in medical students and residents (15% to 30%) than in the general population.2-4 A longitudinal study of medical students at the University of California, San Francisco showed that students’ rates of depression when they enter medical school are similar to those of the general population, but students’ depression scores rise over time; approximately one-fourth of first- and second-year students were depressed.3 Fahrenkopf et al5 reported that 20% of 123 pediatric residents at 3 U.S. children’s hospitals were depressed. These depressed residents made 6.2 times more medication errors than did their non-depressed peers.5 For more information on physicians-in-training, see “Treating depression in medical residents“.
After completing residency, the risk of depression persists. The lifetime prevalence of depression among physicians is 13% in men and 20% in women6; these rates are comparable to those of the general population. Firth-Cozens7 found a range of factors that predict depression among general practitioners; relationships with senior doctors and patients were the main stressors (Table 1).7 Although these stressors increase depression risk, Vaillant et al8 showed that they did not increase suicide risk in physicians who did not have underlying psychological difficulties when they entered college. Certain personality traits common among physicians, such as self-criticism and perfectionism, may increase risk for depression and substance abuse.8
A depressed physician might enter a downward spiral. Feelings of hopelessness and worthlessness frequently lead to declining professional performance. Professional and personal relationships are strained as internal dysphoria manifests as irritability and anger. Spouses and partners can feel overwhelmed and bewildered by changes in the depressed person’s behavior, which may lead to separation or divorce. Patient care and the physician’s professional standing can be endangered. Signs that suggest a physician may be suffering from depression or another mental illness appear in Table 2.9
Table 1
Predictors of depression in physicians
| Difficult relationships with senior doctors, staff, and/or patients |
| Lack of sleep |
| Dealing with death |
| Making mistakes |
| Loneliness |
| 24-hour responsibility |
| Self-criticism |
| Source: Reference 7 |
Table 2
Manifestations of mental illness in physicians
| Severe irritability and anger, resulting in interpersonal conflict |
| Marked vacillations in energy, creativity, enthusiasm, confidence, and productivity |
| Erratic behavior at the office or hospital (ie, performing rounds at 3 am or not showing up until noon) |
| Inappropriate boundaries with patients, staff, or peers |
| Isolation and withdrawal |
| Increased errors in or inattention to chart work and patient calls |
| Personality change, mood swings |
| Impulsivity or irrationality in decision making or action |
| Inappropriate dress, change in hygiene |
| Sexually inappropriate comments or behavior |
| Diminished or heightened need for sleep |
| Frequent job changes and/or moves |
| Inconsistency in performance, absenteeism |
| Source: Adapted from reference 9 |
Increased suicide risk
A review of 14 studies found that the relative risk of suicide in physicians compared with the general population is between 1.1 and 3.4 for men and 2.5 to 5.7 for women.1 A retrospective study of English and Welsh doctors showed elevated suicide rates in female but not male physicians compared with the general population.10 There are no recent studies of suicide rates among U.S. physicians. A 1984-1995 study showed that white male physicians have a higher risk for suicide than other white male professionals.11 A survey of 4,500 women physicians found that female doctors are less likely to attempt suicide than the general female population6; however, their attempts more often are lethal, perhaps because they have greater knowledge of toxicology and access to lethal drugs.12
The relative rate of suicide among medical specialties is unknown. Studies had indicated higher rates of suicide among psychiatrists and anesthesiologists, but these trials were methodologically flawed.12
Silverman12 developed a profile of the physician at high risk for suicide: a workaholic white male age ≥50 or female age ≥45 who is divorced, single, or currently experiencing marital disruption and is suffering from depression. He or she has a substance abuse problem and a history of risk-taking (high-stakes gambling, etc.). Physicians with chronic pain or illness or with a recent change in occupational or financial status also are at risk. Recent increased work demands, personal losses, diminished autonomy, and access to lethal means (medications, firearms) complete the profile.
Protective factors that lower the risk of completed suicide include effective treatment, social and family support, resilience and coping skills, religious faith, and restricted access to lethal means.13,14
Barriers to treatment
Physicians often are hesitant to seek mental health treatment.15 They may fear social stigma and could have trouble finding a local provider who they trust but is not a colleague. Physicians might be concerned about confidentiality and fear recrimination by colleagues, facilities where they work, or licensing boards.16 Givens and Tjia3 found that only 22% of medical students who screened positive for depression sought help and only 42% of students with suicidal ideation received treatment. These students reported that time constraints, confidentiality concerns, stigma, cost, and fear that their illness will be documented on their academic record were major barriers to seeking mental health care.
Licensing concerns. Physicians may be required to disclose a mental health diagnosis or treatment history when applying for or renewing their medical license. Increasingly, medical boards are asking applicants if they have been treated for bipolar disorder, schizophrenia, paranoia, or other disorders.17 Credentialing bodies, clinics, and hospitals may make similar queries.
In an analysis of 51 medical licensing applications (50 states and the District of Columbia), Schroeder et al17 determined that 69% contained at least 1 question that was “likely impermissible” or “impermissible” in terms of compliance with the Americans with Disabilities Act (ADA). In 1993, a U.S. District Court found that the New Jersey State Board of Medical Examiners was in violation of the ADA because licensure application questions did not focus on current fitness to practice medicine but rather on information about a candidate’s status as a person with a disability (illness or diagnosis).18
In Alexander v Margolis,19 however, the court found that because patient safety is in question, medical licensing boards and credentialing bodies can solicit information about serious mental illness that could lead to impaired performance. Courts have ruled that questions regarding a history of treatment or hospitalization for bipolar disorder or schizophrenia and other psychotic disorders are permissible because they are considered “serious disorders” likely to interfere with a physician’s current ability to practice.20 In a 2008 review of all U.S. -affiliated medical licensing boards (N=54), Polfliet21 found that 7 specifically asked applicants about a history of bipolar disorder or schizophrenia, paranoia, and other psychotic disorders. Polfliet21 also found that state medical boards’ compliance with ADA guidelines was not uniform and some questions were “just as broad, and potentially discriminatory, as they were before enactment of the ADA.”
Worley22 reported a successful appeal to the Arkansas State Medical Board to revise its licensure questions following a cluster of medical student and physician suicides. The Board changed the question “Have you ever, or are you presently, being treated for a mental health condition?” to “Have you ever been advised or required by any licensing or privileging body to seek treatment for a physical or mental health condition?”
Providing inaccurate information on a medical licensure application may result in denial or revocation,23 but acknowledging a history of mental health or substance abuse treatment triggers a more in-depth inquiry by the medical board. The lack of distinction between diagnosis and impairment further stigmatizes physicians who seek care and impedes treatment.
Bipolar disorder. The trend in psychiatry toward diagnosing bipolar II disorder and “soft bipolarity” in patients previously diagnosed with and treated for major depression presents a new challenge. Despite no change in their history or functioning, a physician whose diagnosis is changed from depression to bipolar II disorder might be moved from a non-reportable to a board-reportable diagnostic category. With the evolving understanding of bipolar spectrum disorders, medical boards may need to revise their screening questions to ensure that they are seeking information about impairment, not simply the presence of a medical disorder.
Seeking special treatment
Self-treatment. Physicians may attempt to treat their mood disorder with self-prescribed medications before seeking consultation from a psychiatrist. Others use alcohol or illicit drugs to try to alleviate mood disorder symptoms. Self-diagnosis and treatment are not advisable because it is impossible to be objective. Professional boards and state medical boards discourage or prohibit self-prescribing because of the need for ongoing evaluation and monitoring for adverse reactions.
‘VIP’ treatment. When a physician comes to a colleague for help with a mental health issue, both parties might underestimate the severity of the crisis.24 Weintraub25 reported a case series of 12 “VIP” psychiatric inpatients, 10 of whom he described as “therapeutic failures, “including 2 who committed suicide and 3 who left the hospital against medical advice. He observed that improvement occurred only after patients lost their VIP status/treatment.
In a literature review, Groves et al26 found delays in pursuing diagnostic evaluation and treatment for physician patients. He described risks of VIP treatment (Table 3),26 including the physician’s ability to circumscribe the care regimen to obtain “special treatment, “which can create conflict among care providers and other patients. The ailing physician might have trouble relinquishing control. Care providers might not give physician patients necessary information about the illness or treatment because they make assumptions about the physician’s knowledge or fear causing narcissistic injury. Providers’ identification with their peers, deference to their background, and desire to preserve these patients’ autonomy may lead to interventions that are different from those they would provide to other patients.
Treating physicians might underestimate the patient’s suicide risk and tend to not hospitalize a physician patient who faces an imminent risk of self-harm. Similarly, a physician patient might know what key words to use to deny suicidal ideation or avoid hospitalization. Providers assessing physician patients should provide the same interventions they would give to nonphysician patients with the same history and suicide risk factors. To do otherwise is to risk a fatal outcome.
Physician health programs provide confidential treatment and assistance to physicians with mental illness and/or substance abuse problems. Some programs are affiliated with licensing boards, some are branches of the state medical societies, and others are independent of the licensing agencies. Directories of these programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Physician health programs aim to help impaired physicians receive treatment and rehabilitation without censure or licensure revocation, provided they comply with treatment and monitoring requirements.
Table 3
Risks of caring for ‘VIP’ patients
| Caregivers, family, and the patient may deny the possibility of alcohol or substance abuse |
| Caregivers may avoid or poorly handle discussions of death and ‘do not resuscitate’ orders |
| The patient may suffer from emotional isolation when protected from the normal hospital culture |
| The patient’s feelings of shame and fear in the sick role can go uncomforted |
| Caregivers may overlook neuropsychiatric symptoms because they do not wish to ‘insult’ the patient |
| Staff may neglect or poorly handle the patient’s toileting and hygiene |
| Ordinary clinical routine may be short-circuited |
| Caregivers may avoid discussing issues related to the patient’s sexuality |
| Source: Reference 26 |
- American Foundation for Suicide Prevention. www.afsp.org. 24-hour crisis line: 1-800-273-TALK (8255).
- Center for Patient and Professional Advocacy. www.mc.vanderbilt.edu/root/vumc.php?site=CPPA.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Federation of State Physician Health Programs, Inc. www.fsphp.org.
- National Alliance on Mental Illness. www.nami.org.
- Vanderbilt Center for Professional Health. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Lindeman S, Laara E, Hakko H, et al. A systematic review on gender-specific suicide mortality in medical doctors. Br J Psychiatry. 1996;168:274-279.
2. Zoccolillo M, Murphy GE, Wetzel RD. Depression among medical students. J Affect Disord. 1986;11(1):91-96.
3. Givens JL, Tjia J. Depressed medical students’ use of mental health services and barriers to use. Acad Med. 2002;77(9):918-921.
4. Shanafelt TD, Bradley KA, Wipf JE, et al. Burnout and self-reported patient care in an internal medicine residency program. Ann Intern Med. 2002;136(5):358-367.
5. Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
6. Frank E, Dingle AD. Self-reported depression and suicide attempts among U.S. women physicians. Am J Psychiatry. 1999;156:1887-94.
7. Firth-Cozens J. Individual and organizational predictors of depression in general practitioners. Br J Gen Practice. 1998;48:1647-1651.
8. Vaillant GE, Sobowale NC, McArthur C. Some psychological vulnerabilities of physicians. N Engl J Med. 1972;287:372-375.
9. Michalak EE, Yatham LN, Maxwell V, et al. The impact of bipolar disorder upon work functioning: a qualitative analysis. Bipolar Disord. 2007;9:126-143.
10. Hawton K, Clements A, Sakarovitch C, et al. Suicide in doctors: a study of risk according to gender, seniority and specialty in medical practitioners in England and Wales, 1979-1995. J Epidemiol Community Health. 2001;55:296-300.
11. Frank E, Biola H, Burnett CA. Mortality rates and causes among U.S. physicians. Am J Prev Med. 2000;19:155-159.
12. Silverman M. Physicians and suicide. In: The handbook of physician health: essential guide to understanding the health care needs of physicians. Goldman LS Myers M, Dickstein LJ, eds. Chicago, IL: American Medical Association; 2000:95–117.
13. Goldsmith SK, Pellmar TC, Kleinman AM, et al. eds. Reducing suicide: a national imperative. Washington, DC: National Academies Press; 2002.
14. Mann JJ. A current perspective of suicide and attempted suicide. Ann Intern Med. 2002;136:358-367.
15. Center CD, Davis M, Detre T, et al. Confronting depression and suicide in physicians: a consensus statement. JAMA. 2003;289(23):3161-3166.
16. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35(2):S106-116.
17. Schroeder R, Brazeau CM, Zackin F, et al. Do state medical board applications violate the Americans with Disabilities Act? Acad Med. 2009;84(6):776-781.
18. The Medical Society of New Jersey v Jacobs, No, 93-3670 (DNJ 1993)
19. Alexander v Margolis. 921 F Supp 482, 488 (WD Mich 1995).
20. Applicants v Texas State Board of Law examiners, WL 923404 (WD Tex 1994)
21. Polfliet SJ. A national analysis of medical licensure applications. J Am Acad Psychiatry Law. 2008;36(3):369-374.
22. Worley LL. Our fallen peers: a mandate for change. Acad Psychiatry. 2008;32(1):8-12.
23. Sansone RA, Wiederman MW, Sansone LA. Physician mental health and substance abuse. What are state medical licensure applications asking? Arch Fam Med. 1999;8(5):448-451.
24. Robbins GF, Macdonald MC, Pack GT. Delay in the diagnosis and treatment of physicians with cancer. Cancer. 1953;6(3):624-626.
25. Weintraub W. The VIP syndrome: a clinical study in hospital psychiatry. J Nerv Ment Dis. 1964;138:181-193.
26. Groves JE, Dunderdale BA, Stern TA. Celebrity patients VIPs, and potentates. Prim Care Companion J Clin Psychiatry. 2002;4(6):215-223.
Discuss this article at www.facebook.com/CurrentPsychiatry
Dr. G, a second-year surgical resident, becomes depressed when his girlfriend abruptly ends their relationship. His phone calls and e-mails seeking an explanation go unanswered. Having long struggled with his self-esteem, Dr. G interprets this rejection as confirmation of his self-criticism.
Because of his work schedule, Dr. G feels that there is no way to see a therapist or psychiatrist and believes that asking for time off to do so would adversely affect his evaluations. He feels too embarrassed and “weak” to disclose his breakup and depression to his colleagues and attending physicians and senses that fellow residents would resent having to “carry his load.” Dr. G has spent the past 2 years moonlighting at the local emergency room and thinks it would be humiliating to go there for psychiatric help. His work performance and attendance decline until eventually his residency director forces him to take a medical leave of absence.
Dr. G feels that his pain will never end. He writes goodbye letters to his family, makes arrangements for his possessions and funeral, and hangs himself from the balcony outside his apartment.
Although the rate of depression among physicians is comparable to that of the general population, physicians’ risk of suicide is markedly higher.1 Depression and other mood disorders may be under-recognized and inadequately treated in physicians because physicians might:
- be reluctant to seek treatment
- attempt to diagnose and treat themselves
- seek and receive “VIP treatment” from other health care providers.
This article examines physicians’ risk for depression and suicide, licensing concerns and other barriers to effective treatment, and what can be done to overcome such obstacles.
Not immune to depression
Rates of depression are higher in medical students and residents (15% to 30%) than in the general population.2-4 A longitudinal study of medical students at the University of California, San Francisco showed that students’ rates of depression when they enter medical school are similar to those of the general population, but students’ depression scores rise over time; approximately one-fourth of first- and second-year students were depressed.3 Fahrenkopf et al5 reported that 20% of 123 pediatric residents at 3 U.S. children’s hospitals were depressed. These depressed residents made 6.2 times more medication errors than did their non-depressed peers.5 For more information on physicians-in-training, see “Treating depression in medical residents“.
After completing residency, the risk of depression persists. The lifetime prevalence of depression among physicians is 13% in men and 20% in women6; these rates are comparable to those of the general population. Firth-Cozens7 found a range of factors that predict depression among general practitioners; relationships with senior doctors and patients were the main stressors (Table 1).7 Although these stressors increase depression risk, Vaillant et al8 showed that they did not increase suicide risk in physicians who did not have underlying psychological difficulties when they entered college. Certain personality traits common among physicians, such as self-criticism and perfectionism, may increase risk for depression and substance abuse.8
A depressed physician might enter a downward spiral. Feelings of hopelessness and worthlessness frequently lead to declining professional performance. Professional and personal relationships are strained as internal dysphoria manifests as irritability and anger. Spouses and partners can feel overwhelmed and bewildered by changes in the depressed person’s behavior, which may lead to separation or divorce. Patient care and the physician’s professional standing can be endangered. Signs that suggest a physician may be suffering from depression or another mental illness appear in Table 2.9
Table 1
Predictors of depression in physicians
| Difficult relationships with senior doctors, staff, and/or patients |
| Lack of sleep |
| Dealing with death |
| Making mistakes |
| Loneliness |
| 24-hour responsibility |
| Self-criticism |
| Source: Reference 7 |
Table 2
Manifestations of mental illness in physicians
| Severe irritability and anger, resulting in interpersonal conflict |
| Marked vacillations in energy, creativity, enthusiasm, confidence, and productivity |
| Erratic behavior at the office or hospital (ie, performing rounds at 3 am or not showing up until noon) |
| Inappropriate boundaries with patients, staff, or peers |
| Isolation and withdrawal |
| Increased errors in or inattention to chart work and patient calls |
| Personality change, mood swings |
| Impulsivity or irrationality in decision making or action |
| Inappropriate dress, change in hygiene |
| Sexually inappropriate comments or behavior |
| Diminished or heightened need for sleep |
| Frequent job changes and/or moves |
| Inconsistency in performance, absenteeism |
| Source: Adapted from reference 9 |
Increased suicide risk
A review of 14 studies found that the relative risk of suicide in physicians compared with the general population is between 1.1 and 3.4 for men and 2.5 to 5.7 for women.1 A retrospective study of English and Welsh doctors showed elevated suicide rates in female but not male physicians compared with the general population.10 There are no recent studies of suicide rates among U.S. physicians. A 1984-1995 study showed that white male physicians have a higher risk for suicide than other white male professionals.11 A survey of 4,500 women physicians found that female doctors are less likely to attempt suicide than the general female population6; however, their attempts more often are lethal, perhaps because they have greater knowledge of toxicology and access to lethal drugs.12
The relative rate of suicide among medical specialties is unknown. Studies had indicated higher rates of suicide among psychiatrists and anesthesiologists, but these trials were methodologically flawed.12
Silverman12 developed a profile of the physician at high risk for suicide: a workaholic white male age ≥50 or female age ≥45 who is divorced, single, or currently experiencing marital disruption and is suffering from depression. He or she has a substance abuse problem and a history of risk-taking (high-stakes gambling, etc.). Physicians with chronic pain or illness or with a recent change in occupational or financial status also are at risk. Recent increased work demands, personal losses, diminished autonomy, and access to lethal means (medications, firearms) complete the profile.
Protective factors that lower the risk of completed suicide include effective treatment, social and family support, resilience and coping skills, religious faith, and restricted access to lethal means.13,14
Barriers to treatment
Physicians often are hesitant to seek mental health treatment.15 They may fear social stigma and could have trouble finding a local provider who they trust but is not a colleague. Physicians might be concerned about confidentiality and fear recrimination by colleagues, facilities where they work, or licensing boards.16 Givens and Tjia3 found that only 22% of medical students who screened positive for depression sought help and only 42% of students with suicidal ideation received treatment. These students reported that time constraints, confidentiality concerns, stigma, cost, and fear that their illness will be documented on their academic record were major barriers to seeking mental health care.
Licensing concerns. Physicians may be required to disclose a mental health diagnosis or treatment history when applying for or renewing their medical license. Increasingly, medical boards are asking applicants if they have been treated for bipolar disorder, schizophrenia, paranoia, or other disorders.17 Credentialing bodies, clinics, and hospitals may make similar queries.
In an analysis of 51 medical licensing applications (50 states and the District of Columbia), Schroeder et al17 determined that 69% contained at least 1 question that was “likely impermissible” or “impermissible” in terms of compliance with the Americans with Disabilities Act (ADA). In 1993, a U.S. District Court found that the New Jersey State Board of Medical Examiners was in violation of the ADA because licensure application questions did not focus on current fitness to practice medicine but rather on information about a candidate’s status as a person with a disability (illness or diagnosis).18
In Alexander v Margolis,19 however, the court found that because patient safety is in question, medical licensing boards and credentialing bodies can solicit information about serious mental illness that could lead to impaired performance. Courts have ruled that questions regarding a history of treatment or hospitalization for bipolar disorder or schizophrenia and other psychotic disorders are permissible because they are considered “serious disorders” likely to interfere with a physician’s current ability to practice.20 In a 2008 review of all U.S. -affiliated medical licensing boards (N=54), Polfliet21 found that 7 specifically asked applicants about a history of bipolar disorder or schizophrenia, paranoia, and other psychotic disorders. Polfliet21 also found that state medical boards’ compliance with ADA guidelines was not uniform and some questions were “just as broad, and potentially discriminatory, as they were before enactment of the ADA.”
Worley22 reported a successful appeal to the Arkansas State Medical Board to revise its licensure questions following a cluster of medical student and physician suicides. The Board changed the question “Have you ever, or are you presently, being treated for a mental health condition?” to “Have you ever been advised or required by any licensing or privileging body to seek treatment for a physical or mental health condition?”
Providing inaccurate information on a medical licensure application may result in denial or revocation,23 but acknowledging a history of mental health or substance abuse treatment triggers a more in-depth inquiry by the medical board. The lack of distinction between diagnosis and impairment further stigmatizes physicians who seek care and impedes treatment.
Bipolar disorder. The trend in psychiatry toward diagnosing bipolar II disorder and “soft bipolarity” in patients previously diagnosed with and treated for major depression presents a new challenge. Despite no change in their history or functioning, a physician whose diagnosis is changed from depression to bipolar II disorder might be moved from a non-reportable to a board-reportable diagnostic category. With the evolving understanding of bipolar spectrum disorders, medical boards may need to revise their screening questions to ensure that they are seeking information about impairment, not simply the presence of a medical disorder.
Seeking special treatment
Self-treatment. Physicians may attempt to treat their mood disorder with self-prescribed medications before seeking consultation from a psychiatrist. Others use alcohol or illicit drugs to try to alleviate mood disorder symptoms. Self-diagnosis and treatment are not advisable because it is impossible to be objective. Professional boards and state medical boards discourage or prohibit self-prescribing because of the need for ongoing evaluation and monitoring for adverse reactions.
‘VIP’ treatment. When a physician comes to a colleague for help with a mental health issue, both parties might underestimate the severity of the crisis.24 Weintraub25 reported a case series of 12 “VIP” psychiatric inpatients, 10 of whom he described as “therapeutic failures, “including 2 who committed suicide and 3 who left the hospital against medical advice. He observed that improvement occurred only after patients lost their VIP status/treatment.
In a literature review, Groves et al26 found delays in pursuing diagnostic evaluation and treatment for physician patients. He described risks of VIP treatment (Table 3),26 including the physician’s ability to circumscribe the care regimen to obtain “special treatment, “which can create conflict among care providers and other patients. The ailing physician might have trouble relinquishing control. Care providers might not give physician patients necessary information about the illness or treatment because they make assumptions about the physician’s knowledge or fear causing narcissistic injury. Providers’ identification with their peers, deference to their background, and desire to preserve these patients’ autonomy may lead to interventions that are different from those they would provide to other patients.
Treating physicians might underestimate the patient’s suicide risk and tend to not hospitalize a physician patient who faces an imminent risk of self-harm. Similarly, a physician patient might know what key words to use to deny suicidal ideation or avoid hospitalization. Providers assessing physician patients should provide the same interventions they would give to nonphysician patients with the same history and suicide risk factors. To do otherwise is to risk a fatal outcome.
Physician health programs provide confidential treatment and assistance to physicians with mental illness and/or substance abuse problems. Some programs are affiliated with licensing boards, some are branches of the state medical societies, and others are independent of the licensing agencies. Directories of these programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Physician health programs aim to help impaired physicians receive treatment and rehabilitation without censure or licensure revocation, provided they comply with treatment and monitoring requirements.
Table 3
Risks of caring for ‘VIP’ patients
| Caregivers, family, and the patient may deny the possibility of alcohol or substance abuse |
| Caregivers may avoid or poorly handle discussions of death and ‘do not resuscitate’ orders |
| The patient may suffer from emotional isolation when protected from the normal hospital culture |
| The patient’s feelings of shame and fear in the sick role can go uncomforted |
| Caregivers may overlook neuropsychiatric symptoms because they do not wish to ‘insult’ the patient |
| Staff may neglect or poorly handle the patient’s toileting and hygiene |
| Ordinary clinical routine may be short-circuited |
| Caregivers may avoid discussing issues related to the patient’s sexuality |
| Source: Reference 26 |
- American Foundation for Suicide Prevention. www.afsp.org. 24-hour crisis line: 1-800-273-TALK (8255).
- Center for Patient and Professional Advocacy. www.mc.vanderbilt.edu/root/vumc.php?site=CPPA.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Federation of State Physician Health Programs, Inc. www.fsphp.org.
- National Alliance on Mental Illness. www.nami.org.
- Vanderbilt Center for Professional Health. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Dr. G, a second-year surgical resident, becomes depressed when his girlfriend abruptly ends their relationship. His phone calls and e-mails seeking an explanation go unanswered. Having long struggled with his self-esteem, Dr. G interprets this rejection as confirmation of his self-criticism.
Because of his work schedule, Dr. G feels that there is no way to see a therapist or psychiatrist and believes that asking for time off to do so would adversely affect his evaluations. He feels too embarrassed and “weak” to disclose his breakup and depression to his colleagues and attending physicians and senses that fellow residents would resent having to “carry his load.” Dr. G has spent the past 2 years moonlighting at the local emergency room and thinks it would be humiliating to go there for psychiatric help. His work performance and attendance decline until eventually his residency director forces him to take a medical leave of absence.
Dr. G feels that his pain will never end. He writes goodbye letters to his family, makes arrangements for his possessions and funeral, and hangs himself from the balcony outside his apartment.
Although the rate of depression among physicians is comparable to that of the general population, physicians’ risk of suicide is markedly higher.1 Depression and other mood disorders may be under-recognized and inadequately treated in physicians because physicians might:
- be reluctant to seek treatment
- attempt to diagnose and treat themselves
- seek and receive “VIP treatment” from other health care providers.
This article examines physicians’ risk for depression and suicide, licensing concerns and other barriers to effective treatment, and what can be done to overcome such obstacles.
Not immune to depression
Rates of depression are higher in medical students and residents (15% to 30%) than in the general population.2-4 A longitudinal study of medical students at the University of California, San Francisco showed that students’ rates of depression when they enter medical school are similar to those of the general population, but students’ depression scores rise over time; approximately one-fourth of first- and second-year students were depressed.3 Fahrenkopf et al5 reported that 20% of 123 pediatric residents at 3 U.S. children’s hospitals were depressed. These depressed residents made 6.2 times more medication errors than did their non-depressed peers.5 For more information on physicians-in-training, see “Treating depression in medical residents“.
After completing residency, the risk of depression persists. The lifetime prevalence of depression among physicians is 13% in men and 20% in women6; these rates are comparable to those of the general population. Firth-Cozens7 found a range of factors that predict depression among general practitioners; relationships with senior doctors and patients were the main stressors (Table 1).7 Although these stressors increase depression risk, Vaillant et al8 showed that they did not increase suicide risk in physicians who did not have underlying psychological difficulties when they entered college. Certain personality traits common among physicians, such as self-criticism and perfectionism, may increase risk for depression and substance abuse.8
A depressed physician might enter a downward spiral. Feelings of hopelessness and worthlessness frequently lead to declining professional performance. Professional and personal relationships are strained as internal dysphoria manifests as irritability and anger. Spouses and partners can feel overwhelmed and bewildered by changes in the depressed person’s behavior, which may lead to separation or divorce. Patient care and the physician’s professional standing can be endangered. Signs that suggest a physician may be suffering from depression or another mental illness appear in Table 2.9
Table 1
Predictors of depression in physicians
| Difficult relationships with senior doctors, staff, and/or patients |
| Lack of sleep |
| Dealing with death |
| Making mistakes |
| Loneliness |
| 24-hour responsibility |
| Self-criticism |
| Source: Reference 7 |
Table 2
Manifestations of mental illness in physicians
| Severe irritability and anger, resulting in interpersonal conflict |
| Marked vacillations in energy, creativity, enthusiasm, confidence, and productivity |
| Erratic behavior at the office or hospital (ie, performing rounds at 3 am or not showing up until noon) |
| Inappropriate boundaries with patients, staff, or peers |
| Isolation and withdrawal |
| Increased errors in or inattention to chart work and patient calls |
| Personality change, mood swings |
| Impulsivity or irrationality in decision making or action |
| Inappropriate dress, change in hygiene |
| Sexually inappropriate comments or behavior |
| Diminished or heightened need for sleep |
| Frequent job changes and/or moves |
| Inconsistency in performance, absenteeism |
| Source: Adapted from reference 9 |
Increased suicide risk
A review of 14 studies found that the relative risk of suicide in physicians compared with the general population is between 1.1 and 3.4 for men and 2.5 to 5.7 for women.1 A retrospective study of English and Welsh doctors showed elevated suicide rates in female but not male physicians compared with the general population.10 There are no recent studies of suicide rates among U.S. physicians. A 1984-1995 study showed that white male physicians have a higher risk for suicide than other white male professionals.11 A survey of 4,500 women physicians found that female doctors are less likely to attempt suicide than the general female population6; however, their attempts more often are lethal, perhaps because they have greater knowledge of toxicology and access to lethal drugs.12
The relative rate of suicide among medical specialties is unknown. Studies had indicated higher rates of suicide among psychiatrists and anesthesiologists, but these trials were methodologically flawed.12
Silverman12 developed a profile of the physician at high risk for suicide: a workaholic white male age ≥50 or female age ≥45 who is divorced, single, or currently experiencing marital disruption and is suffering from depression. He or she has a substance abuse problem and a history of risk-taking (high-stakes gambling, etc.). Physicians with chronic pain or illness or with a recent change in occupational or financial status also are at risk. Recent increased work demands, personal losses, diminished autonomy, and access to lethal means (medications, firearms) complete the profile.
Protective factors that lower the risk of completed suicide include effective treatment, social and family support, resilience and coping skills, religious faith, and restricted access to lethal means.13,14
Barriers to treatment
Physicians often are hesitant to seek mental health treatment.15 They may fear social stigma and could have trouble finding a local provider who they trust but is not a colleague. Physicians might be concerned about confidentiality and fear recrimination by colleagues, facilities where they work, or licensing boards.16 Givens and Tjia3 found that only 22% of medical students who screened positive for depression sought help and only 42% of students with suicidal ideation received treatment. These students reported that time constraints, confidentiality concerns, stigma, cost, and fear that their illness will be documented on their academic record were major barriers to seeking mental health care.
Licensing concerns. Physicians may be required to disclose a mental health diagnosis or treatment history when applying for or renewing their medical license. Increasingly, medical boards are asking applicants if they have been treated for bipolar disorder, schizophrenia, paranoia, or other disorders.17 Credentialing bodies, clinics, and hospitals may make similar queries.
In an analysis of 51 medical licensing applications (50 states and the District of Columbia), Schroeder et al17 determined that 69% contained at least 1 question that was “likely impermissible” or “impermissible” in terms of compliance with the Americans with Disabilities Act (ADA). In 1993, a U.S. District Court found that the New Jersey State Board of Medical Examiners was in violation of the ADA because licensure application questions did not focus on current fitness to practice medicine but rather on information about a candidate’s status as a person with a disability (illness or diagnosis).18
In Alexander v Margolis,19 however, the court found that because patient safety is in question, medical licensing boards and credentialing bodies can solicit information about serious mental illness that could lead to impaired performance. Courts have ruled that questions regarding a history of treatment or hospitalization for bipolar disorder or schizophrenia and other psychotic disorders are permissible because they are considered “serious disorders” likely to interfere with a physician’s current ability to practice.20 In a 2008 review of all U.S. -affiliated medical licensing boards (N=54), Polfliet21 found that 7 specifically asked applicants about a history of bipolar disorder or schizophrenia, paranoia, and other psychotic disorders. Polfliet21 also found that state medical boards’ compliance with ADA guidelines was not uniform and some questions were “just as broad, and potentially discriminatory, as they were before enactment of the ADA.”
Worley22 reported a successful appeal to the Arkansas State Medical Board to revise its licensure questions following a cluster of medical student and physician suicides. The Board changed the question “Have you ever, or are you presently, being treated for a mental health condition?” to “Have you ever been advised or required by any licensing or privileging body to seek treatment for a physical or mental health condition?”
Providing inaccurate information on a medical licensure application may result in denial or revocation,23 but acknowledging a history of mental health or substance abuse treatment triggers a more in-depth inquiry by the medical board. The lack of distinction between diagnosis and impairment further stigmatizes physicians who seek care and impedes treatment.
Bipolar disorder. The trend in psychiatry toward diagnosing bipolar II disorder and “soft bipolarity” in patients previously diagnosed with and treated for major depression presents a new challenge. Despite no change in their history or functioning, a physician whose diagnosis is changed from depression to bipolar II disorder might be moved from a non-reportable to a board-reportable diagnostic category. With the evolving understanding of bipolar spectrum disorders, medical boards may need to revise their screening questions to ensure that they are seeking information about impairment, not simply the presence of a medical disorder.
Seeking special treatment
Self-treatment. Physicians may attempt to treat their mood disorder with self-prescribed medications before seeking consultation from a psychiatrist. Others use alcohol or illicit drugs to try to alleviate mood disorder symptoms. Self-diagnosis and treatment are not advisable because it is impossible to be objective. Professional boards and state medical boards discourage or prohibit self-prescribing because of the need for ongoing evaluation and monitoring for adverse reactions.
‘VIP’ treatment. When a physician comes to a colleague for help with a mental health issue, both parties might underestimate the severity of the crisis.24 Weintraub25 reported a case series of 12 “VIP” psychiatric inpatients, 10 of whom he described as “therapeutic failures, “including 2 who committed suicide and 3 who left the hospital against medical advice. He observed that improvement occurred only after patients lost their VIP status/treatment.
In a literature review, Groves et al26 found delays in pursuing diagnostic evaluation and treatment for physician patients. He described risks of VIP treatment (Table 3),26 including the physician’s ability to circumscribe the care regimen to obtain “special treatment, “which can create conflict among care providers and other patients. The ailing physician might have trouble relinquishing control. Care providers might not give physician patients necessary information about the illness or treatment because they make assumptions about the physician’s knowledge or fear causing narcissistic injury. Providers’ identification with their peers, deference to their background, and desire to preserve these patients’ autonomy may lead to interventions that are different from those they would provide to other patients.
Treating physicians might underestimate the patient’s suicide risk and tend to not hospitalize a physician patient who faces an imminent risk of self-harm. Similarly, a physician patient might know what key words to use to deny suicidal ideation or avoid hospitalization. Providers assessing physician patients should provide the same interventions they would give to nonphysician patients with the same history and suicide risk factors. To do otherwise is to risk a fatal outcome.
Physician health programs provide confidential treatment and assistance to physicians with mental illness and/or substance abuse problems. Some programs are affiliated with licensing boards, some are branches of the state medical societies, and others are independent of the licensing agencies. Directories of these programs are available from the Federation of State Physician Health Programs and the Federation of State Medical Boards (see Related Resources). Physician health programs aim to help impaired physicians receive treatment and rehabilitation without censure or licensure revocation, provided they comply with treatment and monitoring requirements.
Table 3
Risks of caring for ‘VIP’ patients
| Caregivers, family, and the patient may deny the possibility of alcohol or substance abuse |
| Caregivers may avoid or poorly handle discussions of death and ‘do not resuscitate’ orders |
| The patient may suffer from emotional isolation when protected from the normal hospital culture |
| The patient’s feelings of shame and fear in the sick role can go uncomforted |
| Caregivers may overlook neuropsychiatric symptoms because they do not wish to ‘insult’ the patient |
| Staff may neglect or poorly handle the patient’s toileting and hygiene |
| Ordinary clinical routine may be short-circuited |
| Caregivers may avoid discussing issues related to the patient’s sexuality |
| Source: Reference 26 |
- American Foundation for Suicide Prevention. www.afsp.org. 24-hour crisis line: 1-800-273-TALK (8255).
- Center for Patient and Professional Advocacy. www.mc.vanderbilt.edu/root/vumc.php?site=CPPA.
- Depression and Bipolar Support Alliance. www.dbsalliance.org.
- Federation of State Physician Health Programs, Inc. www.fsphp.org.
- National Alliance on Mental Illness. www.nami.org.
- Vanderbilt Center for Professional Health. www.mc.vanderbilt.edu/cph.
- Vanderbilt Comprehensive Assessment Program. www.mc.vanderbilt.edu/root/vcap.
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Lindeman S, Laara E, Hakko H, et al. A systematic review on gender-specific suicide mortality in medical doctors. Br J Psychiatry. 1996;168:274-279.
2. Zoccolillo M, Murphy GE, Wetzel RD. Depression among medical students. J Affect Disord. 1986;11(1):91-96.
3. Givens JL, Tjia J. Depressed medical students’ use of mental health services and barriers to use. Acad Med. 2002;77(9):918-921.
4. Shanafelt TD, Bradley KA, Wipf JE, et al. Burnout and self-reported patient care in an internal medicine residency program. Ann Intern Med. 2002;136(5):358-367.
5. Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
6. Frank E, Dingle AD. Self-reported depression and suicide attempts among U.S. women physicians. Am J Psychiatry. 1999;156:1887-94.
7. Firth-Cozens J. Individual and organizational predictors of depression in general practitioners. Br J Gen Practice. 1998;48:1647-1651.
8. Vaillant GE, Sobowale NC, McArthur C. Some psychological vulnerabilities of physicians. N Engl J Med. 1972;287:372-375.
9. Michalak EE, Yatham LN, Maxwell V, et al. The impact of bipolar disorder upon work functioning: a qualitative analysis. Bipolar Disord. 2007;9:126-143.
10. Hawton K, Clements A, Sakarovitch C, et al. Suicide in doctors: a study of risk according to gender, seniority and specialty in medical practitioners in England and Wales, 1979-1995. J Epidemiol Community Health. 2001;55:296-300.
11. Frank E, Biola H, Burnett CA. Mortality rates and causes among U.S. physicians. Am J Prev Med. 2000;19:155-159.
12. Silverman M. Physicians and suicide. In: The handbook of physician health: essential guide to understanding the health care needs of physicians. Goldman LS Myers M, Dickstein LJ, eds. Chicago, IL: American Medical Association; 2000:95–117.
13. Goldsmith SK, Pellmar TC, Kleinman AM, et al. eds. Reducing suicide: a national imperative. Washington, DC: National Academies Press; 2002.
14. Mann JJ. A current perspective of suicide and attempted suicide. Ann Intern Med. 2002;136:358-367.
15. Center CD, Davis M, Detre T, et al. Confronting depression and suicide in physicians: a consensus statement. JAMA. 2003;289(23):3161-3166.
16. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35(2):S106-116.
17. Schroeder R, Brazeau CM, Zackin F, et al. Do state medical board applications violate the Americans with Disabilities Act? Acad Med. 2009;84(6):776-781.
18. The Medical Society of New Jersey v Jacobs, No, 93-3670 (DNJ 1993)
19. Alexander v Margolis. 921 F Supp 482, 488 (WD Mich 1995).
20. Applicants v Texas State Board of Law examiners, WL 923404 (WD Tex 1994)
21. Polfliet SJ. A national analysis of medical licensure applications. J Am Acad Psychiatry Law. 2008;36(3):369-374.
22. Worley LL. Our fallen peers: a mandate for change. Acad Psychiatry. 2008;32(1):8-12.
23. Sansone RA, Wiederman MW, Sansone LA. Physician mental health and substance abuse. What are state medical licensure applications asking? Arch Fam Med. 1999;8(5):448-451.
24. Robbins GF, Macdonald MC, Pack GT. Delay in the diagnosis and treatment of physicians with cancer. Cancer. 1953;6(3):624-626.
25. Weintraub W. The VIP syndrome: a clinical study in hospital psychiatry. J Nerv Ment Dis. 1964;138:181-193.
26. Groves JE, Dunderdale BA, Stern TA. Celebrity patients VIPs, and potentates. Prim Care Companion J Clin Psychiatry. 2002;4(6):215-223.
1. Lindeman S, Laara E, Hakko H, et al. A systematic review on gender-specific suicide mortality in medical doctors. Br J Psychiatry. 1996;168:274-279.
2. Zoccolillo M, Murphy GE, Wetzel RD. Depression among medical students. J Affect Disord. 1986;11(1):91-96.
3. Givens JL, Tjia J. Depressed medical students’ use of mental health services and barriers to use. Acad Med. 2002;77(9):918-921.
4. Shanafelt TD, Bradley KA, Wipf JE, et al. Burnout and self-reported patient care in an internal medicine residency program. Ann Intern Med. 2002;136(5):358-367.
5. Fahrenkopf AM, Sectish TC, Barger LK, et al. Rates of medication errors among depressed and burnt out residents: prospective cohort study. BMJ. 2008;336:488-491.
6. Frank E, Dingle AD. Self-reported depression and suicide attempts among U.S. women physicians. Am J Psychiatry. 1999;156:1887-94.
7. Firth-Cozens J. Individual and organizational predictors of depression in general practitioners. Br J Gen Practice. 1998;48:1647-1651.
8. Vaillant GE, Sobowale NC, McArthur C. Some psychological vulnerabilities of physicians. N Engl J Med. 1972;287:372-375.
9. Michalak EE, Yatham LN, Maxwell V, et al. The impact of bipolar disorder upon work functioning: a qualitative analysis. Bipolar Disord. 2007;9:126-143.
10. Hawton K, Clements A, Sakarovitch C, et al. Suicide in doctors: a study of risk according to gender, seniority and specialty in medical practitioners in England and Wales, 1979-1995. J Epidemiol Community Health. 2001;55:296-300.
11. Frank E, Biola H, Burnett CA. Mortality rates and causes among U.S. physicians. Am J Prev Med. 2000;19:155-159.
12. Silverman M. Physicians and suicide. In: The handbook of physician health: essential guide to understanding the health care needs of physicians. Goldman LS Myers M, Dickstein LJ, eds. Chicago, IL: American Medical Association; 2000:95–117.
13. Goldsmith SK, Pellmar TC, Kleinman AM, et al. eds. Reducing suicide: a national imperative. Washington, DC: National Academies Press; 2002.
14. Mann JJ. A current perspective of suicide and attempted suicide. Ann Intern Med. 2002;136:358-367.
15. Center CD, Davis M, Detre T, et al. Confronting depression and suicide in physicians: a consensus statement. JAMA. 2003;289(23):3161-3166.
16. Baldisseri MR. Impaired healthcare professional. Crit Care Med. 2007;35(2):S106-116.
17. Schroeder R, Brazeau CM, Zackin F, et al. Do state medical board applications violate the Americans with Disabilities Act? Acad Med. 2009;84(6):776-781.
18. The Medical Society of New Jersey v Jacobs, No, 93-3670 (DNJ 1993)
19. Alexander v Margolis. 921 F Supp 482, 488 (WD Mich 1995).
20. Applicants v Texas State Board of Law examiners, WL 923404 (WD Tex 1994)
21. Polfliet SJ. A national analysis of medical licensure applications. J Am Acad Psychiatry Law. 2008;36(3):369-374.
22. Worley LL. Our fallen peers: a mandate for change. Acad Psychiatry. 2008;32(1):8-12.
23. Sansone RA, Wiederman MW, Sansone LA. Physician mental health and substance abuse. What are state medical licensure applications asking? Arch Fam Med. 1999;8(5):448-451.
24. Robbins GF, Macdonald MC, Pack GT. Delay in the diagnosis and treatment of physicians with cancer. Cancer. 1953;6(3):624-626.
25. Weintraub W. The VIP syndrome: a clinical study in hospital psychiatry. J Nerv Ment Dis. 1964;138:181-193.
26. Groves JE, Dunderdale BA, Stern TA. Celebrity patients VIPs, and potentates. Prim Care Companion J Clin Psychiatry. 2002;4(6):215-223.
Teaching suicidal adolescents how to "walk the middle path"
The mysterious foreign accent
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Disruptive and withdrawn
Police bring Ms. D, age 33, to our psychiatric facility because of violent behavior at her group home. When confronted for allegedly stealing, she became upset, fought with a housemate, and spat. Six months before coming to our facility she was admitted to a private hospital for psychotic disorder, not otherwise specified (NOS) where she was mute, refused all food and medications, lay in her room, and covered her face with a sheet when someone tried to talk to her.
Ms. D denies having depressive symptoms, sleep disturbance, racing thoughts, thoughts of hurting herself or others, or auditory or visual hallucinations. She complains of poor appetite. Ms. D denies a history of mental illness and says she is not taking any medication. She is upset about being hospitalized and says she will not cooperate with treatment. We cannot obtain her complete psychiatric history but available records indicate that she has 1 previous psychiatric hospitalization for psychotic disorder NOS, and has received trials of haloperidol, lorazepam, diphenhydramine, escitalopram, ziprasidone, and benztropine. Her records do not indicate the dosages of these medications or how she responded to pharmacotherapy.
During her mental status exam, Ms. D is well dressed, covers her hair with a scarf, has no unusual body movements, and responds to questions appropriately. She describes her mood as “okay” but appears upset and anxious about being in the hospital. She exhibits no overt psychotic symptoms and does not appear to be responding to auditory hallucinations or having delusional thoughts. Her cognitive function is intact and her intelligence is judged to be average with impaired insight and judgment. However, she speaks with a distinct accent that sounds Jamaican; otherwise, her speech is articulate with normal rate and tone. When we ask about her accent, Ms. D, who is African American, does not disclose her ethnicity and seems to be unaware of her accent. We did not question the authenticity of her accent until after we obtained collateral information from her family.
The authors’ observations
Based on the available information, we make a provisional diagnosis of psychotic disorder NOS and Ms. D is admitted involuntarily because of concerns about her safety. She is reluctant to accept any treatment and receives an involuntary probate commitment for 90 days. At admission, Ms. D is evasive, guarded, secretive, and at times hostile. Her physical examination reveals no signs or symptoms of focal neurologic deficits. Laboratory testing, including urine toxicology, is unremarkable. She refuses an MRI. Later testing reveals a critical ammonia level of 143 μg/dL, warranting an axis III diagnosis of asymptomatic hyperammonemia.
HISTORY: Paranoia and delusions
Ms. D says she was born and raised in a southern state. She reports that she was born to an Egyptian mother who died during childbirth; her father, who is white, was an ambassador stationed abroad. Ms. D attended school until the 11thgrade and was married at age 19 to a Secret Service agent. She says she has a son who was kidnapped by her husband’s enemies, rescued by paying ransom, and currently lives with his grandfather. Ms. D is paranoid and fears that her life is in danger. She also believes that she has gluten sensitivity that could discolor and damage her hair, which is why she always keeps a scarf on her head for protection.
Through an Internet search, we find articles about Ms. D’s son’s kidnapping. The 7-year-old had been missing for weeks when police found him with his mother in safe condition in another state, after Ms. D called her mother to ask for money and a place to stay. The child was taken from Ms. D’s custody because of concerns for his safety. We also find Ms. D’s mother. Although Ms. D insists her mother is deceased, after some persuasion, she signs a release allowing us to talk to her.
Ms. D’s mother reports that her daughter’s psychiatric problems began when she was pregnant. At the time Ms. D did not have a foreign accent. She had started to “talk funny” when her psychiatric symptoms emerged after she married and became pregnant.
Foreign accent syndrome
A foreign accent can be acquired by normal phenomena, such as being immersed in a foreign language, or a pathological process,1 which can include psychiatric (functional) or neurologic illness (organic causes). Foreign accent syndrome (FAS) is a rare speech disorder characterized by the appearance of a new accent, different from the speaker’s native language, that is perceived as foreign by the listener and in most cases also by the speaker.2 Usually an FAS patient has had no exposure to the accent, although in some cases an old accent has re-emerged.3,4
FAS can result from lesions in brain areas involved in speech production, including precentral gyrus, premotor mid-frontal gyrus, left subcortical prerolandic gyrus, postrolandic gyri, and left parietal area.4 Most FAS cases are secondary to a structural lesion in the brain caused by stroke, traumatic brain injury, cerebral hemorrhage, or multiple sclerosis.2 There are a few cases in the literature of acquired foreign accent with psychogenic etiology in patients with schizophrenia and bipolar disorder with psychotic features.5
TREATMENT: Combination therapy
Based on Ms. D’s unstable mood, irritability, delusional beliefs, and paranoid ideas, we start divalproex, 500 mg/d titrated to 1, 750 mg/d, and risperidone, 3 mg in the morning and 4 mg at bedtime.
The unit psychologist evaluates Ms. D and provides individual psychotherapy, which is mainly supportive and psychoeducational. Ms. D gradually becomes cooperative and friendly. She is not willing to talk about her accent or its origin; however, as her psychiatric symptoms improve, her accent gradually diminishes. The accent never completely resolves, but reduces until it is barely noticeable.
The authors’ observations
Ms. D’s foreign accent was more prominent when she displayed positive psychotic symptoms, such as delusions and disorganized thinking, and gradually disappeared as her psychotic symptoms improved. Ms. D’s case was peculiar because her accent was 1 of the first symptoms before her psychosis fully manifested.
How are FAS and psychosis linked?
Language dysfunction in schizophrenia is common and characterized by derailment and disorganization. Severity of language dysfunction in schizophrenia is directly proportional to overall disease severity.6,7 Various hypotheses have suggested the origin of FAS. In patients with FAS secondary to a neurologic disorder, a lesion usually is found in the dominant brain hemisphere, but the cause is not clear in patients with psychosis who have normal MRI findings. One hypothesis by Reeves et al links development of FAS to the functional disconnection between the left dorsolateral prefrontal cortex (DLPFC) and the superior temporal gyrus (STG) during active psychosis.5 In normal speech production, electric impulses originate in the DLPFC and are transmitted to STG in Wernicke’s area. From there, information goes to Broca’s area, which activates the primary motor cortex to pronounce words. In healthy individuals, word generation activates the DLPFC and causes deactivation of the bilateral STG.8 In schizophrenia, the left STG fails to deactivate in the presence of activation of the left DLPFC.9 Interestingly, STG dysfunction is seen only during active phase of psychosis. Its absence in asymptomatic patients with schizophrenia and bipolar disorder10,11 suggest that a foreign accent-like syndrome may be linked to the functional disconnection between the left DLPFC and left STG dysfunction in patients with active psychosis.5
Performing functional neuroimaging, including positron-emission tomography, functional MRI, and single-photon emission computed tomography, of patients with FAS could shed more light on the possible link between FAS and psychosis. In a case report of a patient with bipolar disorder who developed FAS, MRI initially showed no structural lesion but a later functional imaging scan revealed a cerebral infarct in the left insular and anterior temporal cortex.2
One of the limitations in Ms. D’s case is the lack of neuroimaging studies. For the first few weeks of her hospitalization, it was difficult to communicate with Ms. D. She did not acknowledge her illness and would not cooperate with treatment. She was withdrawn and seemed to experience hysterical mutism, which she perceived as caused by extreme food allergies. Later, as her symptoms continued to improve with pharmacologic and psychotherapeutic interventions, neuroimaging was no longer clinically necessary.
OUTCOME: Accent disappears
As Ms. D improves, psychotherapy evolves to gently and carefully challenging her delusions and providing insight-oriented interventions and trauma therapy. As her delusions gradually start to loosen, Ms. D reveals she had been physically and emotionally abused by her husband.
At discharge after 90 days in the hospital, Ms. D’s symptoms are well managed and she no longer shows signs of a thought disorder. Her thinking is clear, rational, and logical. She demonstrates incredible insight and appreciation that she needs to stay in treatment and continue to take divalproex and risperidone. Her delusions appear to be completely resolved and she is focused on reuniting with her son. Many of her previous delusions appear to be related to trauma and partly dissociative.
Ms. D contacts the psychologist several months later to report she is doing well in the community, staying in treatment, and working on legal means to reunite with her son. No trace of any foreign accent is detectable in her voice.
Related Resources
- Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? Journal of Neurolinguistics. 2006; 19: 385-409.
- Tsuruga K, Kobayashi T, Hirai N, et al. Foreign accent syndrome in a case of dissociative (conversion) disorder. Seishin Shinkeigaku Zasshi. 2008; 110(2): 79-87.
Drug Brand Names
- Benztropine • Cogentin
- Diphenhydramine • Benadryl
- Divalproex • Depakote
- Escitalopram • Lexapro
- Haloperidol • Haldol
- Lorazepam • Ativan
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.
1. Miller N, Lowit A, O’Sullivan H. What makes acquired foreign accent syndrome foreign? J Neurolinguistics. 2006;19(5):385-409.
2. Poulin S, Macoir J, Paquet N, et al. Psychogenic or neurogenic origin of agrammatism and foreign accent syndrome in a bipolar patient: a case report. Ann Gen Psychiatry. 2007;6:1.-
3. Takayama Y, Sugishita M, Kido T, et al. A case of foreign accent syndrome without aphasia caused by a lesion of the left precentral gyrus. Neurology. 1993;43:1361-1363.
4. Roth EJ, Fink K, Cherney LR, et al. Reversion to a previously learned foreign accent after stroke. Arch Phys Med Rehabil. 1997;78:550-552.
5. Reeves RR, Burke RS, Parker JD. Characteristics of psychotic patients with foreign accent syndrome. J Neuropsychiatry Clin Neurosci. 2007;19:70-76.
6. Ceccherini-Nelli A, Crow TJ. Disintegration of the components of language as the path to a revision of Bleuler’s and Schneider’s concepts of schizophrenia: linguistic disturbances compared with first-rank symptoms in acute psychosis. Br J Psychiatry. 2003;182:233-240.
7. Harrow M, O’Connell EM, Herbener ES, et al. Disordered verbalizations in schizophrenia: a speech disturbance or thought disorder? Compr Psychiatry. 2003;44:353-359.
8. Friston KJ, Frith CD, Liddle PF, et al. Investigating a network of word generation with positron emission tomography. Proc R Soc Lond B Biol Sci. 1991;244:101-106.
9. Frith CD, Friston K, Herold S, et al. Regional brain activity in chronic schizophrenic patients during the performance of a verbal fluency task. Br J Psychiatry. 1995;167:343-349.
10. Spence SA, Liddle PF, Stefan MD, et al. Functional anatomy of verbal fluency in people with schizophrenia and those at genetic risk. Focal dysfunction and distributed disconnectivity reappraised. Br J Psychiatry. 2011;176:52-60.
11. Dye SM, Spence SA, Bench CJ, et al. No evidence for left superior temporal dysfunction in asymptomatic schizophrenia and bipolar disorder. PET study of verbal fluency. Br J Psychiatry. 1999;175:367-374.
Slippery slope
In the vast majority of instances, DSM defines psychotic disorders as manifesting with symptoms that, by definition, fly in the face of the physical constraints of reality. It is my opinion that the point of view Dr. Henry A. Nasrallah presents is boundless (“Are some nonpsychotic psychiatric disorders actually psychotic?” From the Editor, Current Psychiatry, November 2010, p. 16-19). Dr. Nasrallah’s hypothesis easily could extend to encompass circumstances such as over-reacting to being slighted by a friend or being offended by an inattentive store clerk, which may cause one to see things through (the distortion of) “grey (or perhaps rose) colored glasses. “ Although with time such perceptions may grow to take on psychotic proportions, this is a slippery slope upon which one must tread carefully, being vigilant not to fall prey to “pathologizing” thoughts and feelings associated with normal human angst.
Karen Fox, MD
Adult and Child Psychiatrist
The Upstate’s Golden Corner of Psychiatry
Seneca, SC
In the vast majority of instances, DSM defines psychotic disorders as manifesting with symptoms that, by definition, fly in the face of the physical constraints of reality. It is my opinion that the point of view Dr. Henry A. Nasrallah presents is boundless (“Are some nonpsychotic psychiatric disorders actually psychotic?” From the Editor, Current Psychiatry, November 2010, p. 16-19). Dr. Nasrallah’s hypothesis easily could extend to encompass circumstances such as over-reacting to being slighted by a friend or being offended by an inattentive store clerk, which may cause one to see things through (the distortion of) “grey (or perhaps rose) colored glasses. “ Although with time such perceptions may grow to take on psychotic proportions, this is a slippery slope upon which one must tread carefully, being vigilant not to fall prey to “pathologizing” thoughts and feelings associated with normal human angst.
Karen Fox, MD
Adult and Child Psychiatrist
The Upstate’s Golden Corner of Psychiatry
Seneca, SC
In the vast majority of instances, DSM defines psychotic disorders as manifesting with symptoms that, by definition, fly in the face of the physical constraints of reality. It is my opinion that the point of view Dr. Henry A. Nasrallah presents is boundless (“Are some nonpsychotic psychiatric disorders actually psychotic?” From the Editor, Current Psychiatry, November 2010, p. 16-19). Dr. Nasrallah’s hypothesis easily could extend to encompass circumstances such as over-reacting to being slighted by a friend or being offended by an inattentive store clerk, which may cause one to see things through (the distortion of) “grey (or perhaps rose) colored glasses. “ Although with time such perceptions may grow to take on psychotic proportions, this is a slippery slope upon which one must tread carefully, being vigilant not to fall prey to “pathologizing” thoughts and feelings associated with normal human angst.
Karen Fox, MD
Adult and Child Psychiatrist
The Upstate’s Golden Corner of Psychiatry
Seneca, SC
SGAs for delirium?
“Atypical antipsychotics for delirium: A reasonable alternative to haloperidol?” (Current Psychiatry, January 2011, p. 37-46) was an interesting article. I agree that low doses of haloperidol, (0. 5 to 3 mg), have low risk for causing acute dystonia, which is the major worry for its use in the ICU. However, it is my understanding that IV haloperidol has no risk of acute dystonia. If this is true, then reduced risk of acute dystonia may not be an advantage of second-generation antipsychotics.
Also, a possible obstacle to using ziprasidone is that the maximum IM dose is 40 mg/d, which may be inadequate for some patients. In my opinion, the FDA warnings have unfairly limited ziprasidone’s use, even though it has a favorable side effect profile in terms of weight gain and hypercholesterolemia. Its propensity to prolong the QTc interval is notable, but to my knowledge, this has never resulted in a death from torsade de pointes (TDP) in clinical trials. On the other hand, haloperidol has been linked to deaths from TDP.
In cases of extreme agitation in patients with delirium, I was wondering what the authors thought about using droperidol. I have found that it is perhaps one of the most sedating and calming agents one can use for delirium and agitation, but nursing staff and other psychiatrists are extremely reluctant to use it and sometimes request telemetry in addition to routine EKG before and after its administration. My feeling is that although the QTc prolongation associated with droperidol is real, it has resulted in the drug being underutilized and almost forgotten.
Corey Yilmaz, MD
Adult and Child Psychiatrist
Tolleson, AZ
Dr. Spiegel responds
Two studies could support Dr. Yilmaz’s statement that IV haloperidol has no risk of acute dystonia. In an early prospective study using IV haloperidol (mean dosage: 10 mg) primarily in delirium, acute dystonia did not occur after an average of 5 days of treatment.1 Furthermore, in a more recent prospective study using IV haloperidol (median dose: 10 mg) in patients with behavioral emergencies, acute dystonia was not reported; however, assessment occurred every 15 minutes for 1 hour.2 The former study included 4 patients and the latter 76 patients. In both studies, possible limitations include that dystonia could have developed beyond the evaluation periods (ie, 5 days and 1 hour), and the number of patients who received IV haloperidol was small. Therefore, while there were no reports of acute dystonia in these studies, it may be more prudent to state that IV haloperidol may have less risk of acute dystonia when compared with the oral formulation, but is not devoid of this risk.
Concerning ziprasidone and droperidol’s relationship with QT prolongation and TDP, as outlined in our article, I advocate for safety while using any psychotropic medication that can prolong QT interval or precipitate TDP. Nonetheless, 1 recent review reports that the FDA uses increases in QT interval as a proarrhythmic marker for TDP, because TDP is very uncommon and difficult to assess. Additionally, the review states there is general agreement by investigators that droperidol increases QT interval, and TDP is associated with an increase in the QT interval. But there is no established link between increased QT interval and incidence of TDP with droperidol administration.3
As with any treatment, a risk/benefit analysis should guide clinical decisions.
David R. Spiegel, MD
Associate Professor of Clinical Psychiatry and Behavioral Sciences
Director of Consultation-Liaison Services
Eastern Virginia Medical School
Norfolk, VA
1. Menza MMA, Murray GB, Holmes VF, et al. Decreased extrapyramidal symptoms with intravenous haloperidol. J Clin Psychiatry. 1987;48:278-280.
2. Hatta K, Nakamura M, Yoshida K, et al. A prospective naturalistic multicentre study of intravenous medications in behavioural emergencies: haloperidol versus flunitrazepam. Psychiatry Res. 2010;178:182-185.
3. Halloran K, Barash PG. Inside the black box: current policies and concerns with the United States Food and Drug Administration’s highest drug safety warning system. Curr Opin Anaesthesiol. 2010;23:423-427.
“Atypical antipsychotics for delirium: A reasonable alternative to haloperidol?” (Current Psychiatry, January 2011, p. 37-46) was an interesting article. I agree that low doses of haloperidol, (0. 5 to 3 mg), have low risk for causing acute dystonia, which is the major worry for its use in the ICU. However, it is my understanding that IV haloperidol has no risk of acute dystonia. If this is true, then reduced risk of acute dystonia may not be an advantage of second-generation antipsychotics.
Also, a possible obstacle to using ziprasidone is that the maximum IM dose is 40 mg/d, which may be inadequate for some patients. In my opinion, the FDA warnings have unfairly limited ziprasidone’s use, even though it has a favorable side effect profile in terms of weight gain and hypercholesterolemia. Its propensity to prolong the QTc interval is notable, but to my knowledge, this has never resulted in a death from torsade de pointes (TDP) in clinical trials. On the other hand, haloperidol has been linked to deaths from TDP.
In cases of extreme agitation in patients with delirium, I was wondering what the authors thought about using droperidol. I have found that it is perhaps one of the most sedating and calming agents one can use for delirium and agitation, but nursing staff and other psychiatrists are extremely reluctant to use it and sometimes request telemetry in addition to routine EKG before and after its administration. My feeling is that although the QTc prolongation associated with droperidol is real, it has resulted in the drug being underutilized and almost forgotten.
Corey Yilmaz, MD
Adult and Child Psychiatrist
Tolleson, AZ
Dr. Spiegel responds
Two studies could support Dr. Yilmaz’s statement that IV haloperidol has no risk of acute dystonia. In an early prospective study using IV haloperidol (mean dosage: 10 mg) primarily in delirium, acute dystonia did not occur after an average of 5 days of treatment.1 Furthermore, in a more recent prospective study using IV haloperidol (median dose: 10 mg) in patients with behavioral emergencies, acute dystonia was not reported; however, assessment occurred every 15 minutes for 1 hour.2 The former study included 4 patients and the latter 76 patients. In both studies, possible limitations include that dystonia could have developed beyond the evaluation periods (ie, 5 days and 1 hour), and the number of patients who received IV haloperidol was small. Therefore, while there were no reports of acute dystonia in these studies, it may be more prudent to state that IV haloperidol may have less risk of acute dystonia when compared with the oral formulation, but is not devoid of this risk.
Concerning ziprasidone and droperidol’s relationship with QT prolongation and TDP, as outlined in our article, I advocate for safety while using any psychotropic medication that can prolong QT interval or precipitate TDP. Nonetheless, 1 recent review reports that the FDA uses increases in QT interval as a proarrhythmic marker for TDP, because TDP is very uncommon and difficult to assess. Additionally, the review states there is general agreement by investigators that droperidol increases QT interval, and TDP is associated with an increase in the QT interval. But there is no established link between increased QT interval and incidence of TDP with droperidol administration.3
As with any treatment, a risk/benefit analysis should guide clinical decisions.
David R. Spiegel, MD
Associate Professor of Clinical Psychiatry and Behavioral Sciences
Director of Consultation-Liaison Services
Eastern Virginia Medical School
Norfolk, VA
“Atypical antipsychotics for delirium: A reasonable alternative to haloperidol?” (Current Psychiatry, January 2011, p. 37-46) was an interesting article. I agree that low doses of haloperidol, (0. 5 to 3 mg), have low risk for causing acute dystonia, which is the major worry for its use in the ICU. However, it is my understanding that IV haloperidol has no risk of acute dystonia. If this is true, then reduced risk of acute dystonia may not be an advantage of second-generation antipsychotics.
Also, a possible obstacle to using ziprasidone is that the maximum IM dose is 40 mg/d, which may be inadequate for some patients. In my opinion, the FDA warnings have unfairly limited ziprasidone’s use, even though it has a favorable side effect profile in terms of weight gain and hypercholesterolemia. Its propensity to prolong the QTc interval is notable, but to my knowledge, this has never resulted in a death from torsade de pointes (TDP) in clinical trials. On the other hand, haloperidol has been linked to deaths from TDP.
In cases of extreme agitation in patients with delirium, I was wondering what the authors thought about using droperidol. I have found that it is perhaps one of the most sedating and calming agents one can use for delirium and agitation, but nursing staff and other psychiatrists are extremely reluctant to use it and sometimes request telemetry in addition to routine EKG before and after its administration. My feeling is that although the QTc prolongation associated with droperidol is real, it has resulted in the drug being underutilized and almost forgotten.
Corey Yilmaz, MD
Adult and Child Psychiatrist
Tolleson, AZ
Dr. Spiegel responds
Two studies could support Dr. Yilmaz’s statement that IV haloperidol has no risk of acute dystonia. In an early prospective study using IV haloperidol (mean dosage: 10 mg) primarily in delirium, acute dystonia did not occur after an average of 5 days of treatment.1 Furthermore, in a more recent prospective study using IV haloperidol (median dose: 10 mg) in patients with behavioral emergencies, acute dystonia was not reported; however, assessment occurred every 15 minutes for 1 hour.2 The former study included 4 patients and the latter 76 patients. In both studies, possible limitations include that dystonia could have developed beyond the evaluation periods (ie, 5 days and 1 hour), and the number of patients who received IV haloperidol was small. Therefore, while there were no reports of acute dystonia in these studies, it may be more prudent to state that IV haloperidol may have less risk of acute dystonia when compared with the oral formulation, but is not devoid of this risk.
Concerning ziprasidone and droperidol’s relationship with QT prolongation and TDP, as outlined in our article, I advocate for safety while using any psychotropic medication that can prolong QT interval or precipitate TDP. Nonetheless, 1 recent review reports that the FDA uses increases in QT interval as a proarrhythmic marker for TDP, because TDP is very uncommon and difficult to assess. Additionally, the review states there is general agreement by investigators that droperidol increases QT interval, and TDP is associated with an increase in the QT interval. But there is no established link between increased QT interval and incidence of TDP with droperidol administration.3
As with any treatment, a risk/benefit analysis should guide clinical decisions.
David R. Spiegel, MD
Associate Professor of Clinical Psychiatry and Behavioral Sciences
Director of Consultation-Liaison Services
Eastern Virginia Medical School
Norfolk, VA
1. Menza MMA, Murray GB, Holmes VF, et al. Decreased extrapyramidal symptoms with intravenous haloperidol. J Clin Psychiatry. 1987;48:278-280.
2. Hatta K, Nakamura M, Yoshida K, et al. A prospective naturalistic multicentre study of intravenous medications in behavioural emergencies: haloperidol versus flunitrazepam. Psychiatry Res. 2010;178:182-185.
3. Halloran K, Barash PG. Inside the black box: current policies and concerns with the United States Food and Drug Administration’s highest drug safety warning system. Curr Opin Anaesthesiol. 2010;23:423-427.
1. Menza MMA, Murray GB, Holmes VF, et al. Decreased extrapyramidal symptoms with intravenous haloperidol. J Clin Psychiatry. 1987;48:278-280.
2. Hatta K, Nakamura M, Yoshida K, et al. A prospective naturalistic multicentre study of intravenous medications in behavioural emergencies: haloperidol versus flunitrazepam. Psychiatry Res. 2010;178:182-185.
3. Halloran K, Barash PG. Inside the black box: current policies and concerns with the United States Food and Drug Administration’s highest drug safety warning system. Curr Opin Anaesthesiol. 2010;23:423-427.