User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Self-deception : A double-edged trait
Consider these common human tales:
- A prominent politician who made his reputation combating prostitution loses his job after being discovered to have consorted with many "escorts." He believed he would never be caught.
- A sociopathic man charms a young woman and convinces her he will love her forever. She is infatuated with him. He dumps her a month later.
- A gambler is "convinced" his next bet will win back his previous losses and ends up losing his shirt again.
- Voters elect a politician who promises to solve all their problems but are disillusioned a few years later when things have barely changed.
- A woman with severe chronic fibromyalgia seeks the help of a shaman in her village in Haiti. He pain amazingly disappears for a few days before recurring.
The human brain has been both blessed and cursed during its evolutionary journey by developing the capacity for self-deception. Unlike other living things, humans are capable of massive self-deception—as these tales show.
Advantage: survival
Self-deception's upside is obvious, with established survival value. Hope, optimism, and self-confidence in dark times are antidotes to capitulation, despair, and inaction. Infatuation helps perpetuate the human species, and "eternal love" leads to other obligatory self-deceptions such as "till death do us part." Sometimes self-deception helps communities survive by promoting altruism, charity, and compassion for strangers.
For us in the health professions—especially psychiatry—self-deception's benefits for patients are well recognized: a remarkable healing capacity, an almost magical placebo effect from drug therapy or psychotherapy, and the advantages of positive transference toward the physician. Without self-deception, our patients could not respond to support and reassurance or resist hopelessness and the urge to give up and end their lives.
Disadvantage: suffering
But self-deception has a serious downside as well, from hubris and arrogance that end badly to blind faith and gullibility that lead to joining cults and "drinking the Kool-Aid," from unshakable belief in astrology or fanatical pursuit of a cause to believing in nothing and wasting one's life with nihilism.
The biology of self-deception also may represent the foundation of psychopathology, such as:
- unremitting panic and anxiety associated with a firm belief in impending doom
- bizarre, fixed, false beliefs of schizophrenia
- grandiose delusions of bipolar mania
- melancholia's profound and inconsolable sorrow, futility, and worthlessness
- pervasive belief in one's repulsiveness by attractive women with dysmorphic body disorder
- distorted conviction of obesity in skin-and-bones teenagers with anorexia nervosa
- unshakable parasitosis of a delusional disorder
- tortured and agonizing obsessions to perform meaningless rituals.
Psychiatric disorders are extremes of self-deception gone awry across complex neural pathways, encompassing emotions, thoughts, behavior, and cognitions. Human adaptation to stress or serious illness is often enhanced by the blissful escape of self-deception, but its curse can destroy lives and cause untold suffering.
So, are nonhuman creatures spared the double-edged sword of self-deception? If so, then why do dogs have unshakable loyalty, even when their owners abuse them? Maybe self-deception is not uniquely human after all.

Henry A. Nasrallah, MD
Editor-in-Chief To comment on this editorial or other topics of interest, contact Dr. Nasrallah or click here.
Henry A. Nasrallah, MD
Editor-in-Chief To comment on this editorial or other topics of interest, contact Dr. Nasrallah or click here.
Henry A. Nasrallah, MD
Editor-in-Chief To comment on this editorial or other topics of interest, contact Dr. Nasrallah or click here.
Consider these common human tales:
- A prominent politician who made his reputation combating prostitution loses his job after being discovered to have consorted with many "escorts." He believed he would never be caught.
- A sociopathic man charms a young woman and convinces her he will love her forever. She is infatuated with him. He dumps her a month later.
- A gambler is "convinced" his next bet will win back his previous losses and ends up losing his shirt again.
- Voters elect a politician who promises to solve all their problems but are disillusioned a few years later when things have barely changed.
- A woman with severe chronic fibromyalgia seeks the help of a shaman in her village in Haiti. He pain amazingly disappears for a few days before recurring.
The human brain has been both blessed and cursed during its evolutionary journey by developing the capacity for self-deception. Unlike other living things, humans are capable of massive self-deception—as these tales show.
Advantage: survival
Self-deception's upside is obvious, with established survival value. Hope, optimism, and self-confidence in dark times are antidotes to capitulation, despair, and inaction. Infatuation helps perpetuate the human species, and "eternal love" leads to other obligatory self-deceptions such as "till death do us part." Sometimes self-deception helps communities survive by promoting altruism, charity, and compassion for strangers.
For us in the health professions—especially psychiatry—self-deception's benefits for patients are well recognized: a remarkable healing capacity, an almost magical placebo effect from drug therapy or psychotherapy, and the advantages of positive transference toward the physician. Without self-deception, our patients could not respond to support and reassurance or resist hopelessness and the urge to give up and end their lives.
Disadvantage: suffering
But self-deception has a serious downside as well, from hubris and arrogance that end badly to blind faith and gullibility that lead to joining cults and "drinking the Kool-Aid," from unshakable belief in astrology or fanatical pursuit of a cause to believing in nothing and wasting one's life with nihilism.
The biology of self-deception also may represent the foundation of psychopathology, such as:
- unremitting panic and anxiety associated with a firm belief in impending doom
- bizarre, fixed, false beliefs of schizophrenia
- grandiose delusions of bipolar mania
- melancholia's profound and inconsolable sorrow, futility, and worthlessness
- pervasive belief in one's repulsiveness by attractive women with dysmorphic body disorder
- distorted conviction of obesity in skin-and-bones teenagers with anorexia nervosa
- unshakable parasitosis of a delusional disorder
- tortured and agonizing obsessions to perform meaningless rituals.
Psychiatric disorders are extremes of self-deception gone awry across complex neural pathways, encompassing emotions, thoughts, behavior, and cognitions. Human adaptation to stress or serious illness is often enhanced by the blissful escape of self-deception, but its curse can destroy lives and cause untold suffering.
So, are nonhuman creatures spared the double-edged sword of self-deception? If so, then why do dogs have unshakable loyalty, even when their owners abuse them? Maybe self-deception is not uniquely human after all.
Consider these common human tales:
- A prominent politician who made his reputation combating prostitution loses his job after being discovered to have consorted with many "escorts." He believed he would never be caught.
- A sociopathic man charms a young woman and convinces her he will love her forever. She is infatuated with him. He dumps her a month later.
- A gambler is "convinced" his next bet will win back his previous losses and ends up losing his shirt again.
- Voters elect a politician who promises to solve all their problems but are disillusioned a few years later when things have barely changed.
- A woman with severe chronic fibromyalgia seeks the help of a shaman in her village in Haiti. He pain amazingly disappears for a few days before recurring.
The human brain has been both blessed and cursed during its evolutionary journey by developing the capacity for self-deception. Unlike other living things, humans are capable of massive self-deception—as these tales show.
Advantage: survival
Self-deception's upside is obvious, with established survival value. Hope, optimism, and self-confidence in dark times are antidotes to capitulation, despair, and inaction. Infatuation helps perpetuate the human species, and "eternal love" leads to other obligatory self-deceptions such as "till death do us part." Sometimes self-deception helps communities survive by promoting altruism, charity, and compassion for strangers.
For us in the health professions—especially psychiatry—self-deception's benefits for patients are well recognized: a remarkable healing capacity, an almost magical placebo effect from drug therapy or psychotherapy, and the advantages of positive transference toward the physician. Without self-deception, our patients could not respond to support and reassurance or resist hopelessness and the urge to give up and end their lives.
Disadvantage: suffering
But self-deception has a serious downside as well, from hubris and arrogance that end badly to blind faith and gullibility that lead to joining cults and "drinking the Kool-Aid," from unshakable belief in astrology or fanatical pursuit of a cause to believing in nothing and wasting one's life with nihilism.
The biology of self-deception also may represent the foundation of psychopathology, such as:
- unremitting panic and anxiety associated with a firm belief in impending doom
- bizarre, fixed, false beliefs of schizophrenia
- grandiose delusions of bipolar mania
- melancholia's profound and inconsolable sorrow, futility, and worthlessness
- pervasive belief in one's repulsiveness by attractive women with dysmorphic body disorder
- distorted conviction of obesity in skin-and-bones teenagers with anorexia nervosa
- unshakable parasitosis of a delusional disorder
- tortured and agonizing obsessions to perform meaningless rituals.
Psychiatric disorders are extremes of self-deception gone awry across complex neural pathways, encompassing emotions, thoughts, behavior, and cognitions. Human adaptation to stress or serious illness is often enhanced by the blissful escape of self-deception, but its curse can destroy lives and cause untold suffering.
So, are nonhuman creatures spared the double-edged sword of self-deception? If so, then why do dogs have unshakable loyalty, even when their owners abuse them? Maybe self-deception is not uniquely human after all.
‘I’ve been abducted by aliens’
CASE: ’I’m not crazy’
Ms. S, age 55, presents for treatment because she is feeling depressed and anxious. Her symptoms include decreased concentration, intermittent irritability, hoarding, and difficulty starting and completing tasks. She also has chronic sleep difficulties that often keep her awake until dawn.
Fatigue, lack of focus, and poor comprehension and motivation have left her unemployed. She and her teenage daughter live with Ms. S’s elderly mother. Ms. S feels tremendous guilt because she cannot be the mother and daughter she wants to be.
Initially, I (PK) diagnose Ms. S with major depressive disorder and prescribe sertraline, 100 mg/d, which improves her mood and energy. However, her inability to stay organized results in her being “let go” from job training.
Ms. S reports similar difficulties in school as a child. I determine that she meets DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD). Adding methylphenidate, 10 mg bid, improves her concentration and ability to complete tasks. It also reduces the impulsivity that has disrupted her relationships.
Despite a strong desire to normalize her sleep schedule, Ms. S continues to have difficulty falling asleep, so I add melatonin, 3 to 6 mg at bedtime. Her sleeping pattern is improved, but still variable. She also tries quetiapine, 25 mg at bedtime, but soon discontinues it due to intolerance.
As our rapport strengthens, Ms. S reveals that she has had multiple encounters with aliens beginning at age 3. Although she has not had an “alien experience” for about 5 years, she does not feel safe sleeping at night and instead sleeps during the day. Her efforts to stay awake at night strain her relationship with her mother.
The authors’ observations
Approximately 1% of the U.S. population report alien abduction experiences (AAE)—an umbrella term that includes alleged contact with aliens ranging from sightings to abductions.1 Patients rarely report AAE to mental health professionals. In our society, claiming to be an “abductee” implies that one might be insane. A survey of 398 Canadian students that assessed attitudes, beliefs, and experiences regarding alien abductions found that 79% of respondents believed they would have mostly negative consequences—such as being laughed at or socially isolated—if they claimed to have encountered aliens.1
Persons who have AAE may attend support groups of fellow “abductees” to accumulate behavior-consonant information (hearing other people’s abduction stories) and reduce dissonance by being surrounded by others who share a questionable belief.2 A survey of “abductees” found that 88% report at least some positive aspects of the experience, such as a sense of importance or feeling as though they were chosen to bridge communication between extraterrestrials and humans.3
- high levels of psychic energy
- self-sufficiency
- resourcefulness
- a tendency to question authority and to be exposed to situational conflicts.1
After Ms. S reveals her alien experiences, I reassure her in a nonjudgmental manner that we will explore her experiences and determine ways to help her cope with them.
HISTORY: Terrifying experiences
Ms. S elaborates on her alien experiences, relating a particularly terrifying example from her teen years. She was lying awake in bed, looking at the ceiling, where she saw a jeweled spider with a drill. As the spider descended from the ceiling and spread its legs, she recalled a noise like a dentist’s drill. As the spider neared her face, it grew larger and larger. Terrified, Ms. S was unable to scream for help or move anything except her eyes as the spider clamped its legs around her head and bored into her skull. She reported that although she could feel the drill go in, it wasn’t painful.
Other experiences included giving birth, undergoing examinations or probes, and communicating with aliens. Although she is very distressed by most memories, she feels she benefited from others. For example, as a child, Ms. S’s math skills improved dramatically after an AAE episode; she believes this was a gift from the aliens. Ms. S’s AAE memories are as vivid to her as memories of her college graduation. She had been reluctant to discuss these events with anyone outside her family out of fear of being perceived as “crazy.”
Ms. S says she was a shy child who had difficulty making friends. She was plagued with fatigue and worry about family members. She believed that aliens might attack her sisters and felt obligated to stay awake at night to protect them. Aside from alien experiences, Ms. S reports a happy childhood.
She has always been an avid reader. At age 8 or 9, after reading a book on alien abduction, she concluded that she had been abducted. Later, she joined a group of professed alien abductees. She feels accepted and validated by this group and has a forum for discussing her experiences without fear of ridicule or rejection.
Ms. S remains frightened by things that remind her of aliens. Although she wrote a summary of her alien experiences, she cannot draw a picture of an alien, and thoughts or images of the prototypical “grey” alien trigger panic. She also feels somewhat “different,” nervous, and distant from others.
The authors’ observations
Reviewing AAE literature led me to consider several diagnoses, including:
- psychosis
- seizures
- false memory (sexual abuse, trauma)
- narcolepsy
- sleep paralysis.
Electroencephalography (during drowsiness) revealed abnormal activity (occurrences of widely scattered bursts of nonspecific, round, sharply contoured slow waves in the left frontal region) only in the F7 electrode. In the absence of clinical symptoms and when found in a single lead, this is considered a normal variant.
Diagnostic testing ruled out hallucinosis related to seizures. I also ruled out false memory related to sexual abuse or trauma, which is commonly found in patients who present with AAE.
Collaborative information from relatives did not uncover a history of psychosis. She and family members reported, however, that Ms. S’s father and 1 sister had periodic sleep disturbances with associated hallucinations. I began to suspect sleep paralysis.
The authors’ observations
Full-body paralysis normally accompanies rapid eye movement (REM) sleep, which occurs several times a night.4 Sleep paralysis is a transient state that occurs when an individual becomes conscious of this immobility, typically while falling asleep or awakening.5 These experiences can be accompanied by hypnagogic (while falling asleep) or hypnopompic (while awakening) hallucinations. An estimated 30% of the population has had at least one sleep paralysis episode.6 In one study, 5% of sleep paralysis patients had episodes that were accompanied by hallucinations.7
Although individuals cannot make gross body movements during sleep paralysis, they can open their eyes and are able to report events that occurred around them during the episode.8 Patients interpret sleep paralysis experiences in subjective terms. Common descriptions include intense fear, breathing difficulties, feeling of bodily pressure—especially on the chest—and sensations of floating, flying, or falling (Table 1).7,9
During sleep paralysis episodes, individuals typically sense a threatening presence.6 Patients have reported beastly and demonic figures of doom: devils, demons, witches, aliens, and even cinematic villains such as Darth Vader and Freddy Kruger.6 Others have described this presence in terms of alien visitations or abductions.
Table 1
4 types of sleep paralysis-related hallucinations
| Intruder | Vague sense of a threatening presence accompanied by visual, auditory, and tactile hallucinations—noises, footsteps, gibbering voices, humanoid apparitions, and sensation of being touched or grabbed |
| Incubus | Breathing difficulties, feelings of suffocation, bodily pressure (particularly on the chest, as if someone were sitting or standing on it), pain, and thoughts of impending death |
| Vestibular-motor | Sensations of floating (levitation), flying, and falling |
| Other | Out-of-body experiences, autoscopy (seeing oneself from an external point), and fictive motor movements, ranging from simple arm movements to sitting up to apparent locomotion through the environment |
| Source: References 7,9 | |
A Harvard University study of 11 individuals who reported alien abductions found that all participants experienced a similar sequence of events:
- They suspected abduction after sleep episodes characterized by awakening, full-body paralysis, intense fear, and a feeling of a presence. Several reported tactile or visual sensations strikingly similar to descriptions of sleep paralysis, such as levitating, being touched, and seeing shadowy figures.
- They sought explanations for what they perceived as anomalous experiences.
- They “recovered” abduction memories in therapy (with the help of techniques such as hypnosis) or spontaneously (after reading books or seeing movies or television shows depicting similar episodes).4
Ms. S reported no daytime sleep attacks, cataplexy, or rapid onset of dreaming. Because her reported AAEs were spread out and the last occurred approximately 5 years ago, I decided against conducting a sleep study because it likely would be low yield and costly. I reached a diagnosis of sleep paralysis-familial type, chronic based on:
- an absence of organic or psychiatric dysfunction
- a familial pattern of sleep disturbances
- the temporal pattern and description of her symptoms (Table 2).11
Table 2
Diagnostic criteria for sleep paralysis
| A. Patient complains of inability to move the trunk or limbs at sleep onset or upon awakening |
| B. Brief episodes of partial or complete skeletal muscle paralysis |
| C. Episodes can be associated with hypnagogic (preceding sleep) hallucinations or dreamlike mentation |
D. Polysomnographic monitoring demonstrates at least 1 of the following:
|
| E. Symptoms are not associated with other medical or mental disorders, such as hysteria or hypokalemic paralysis |
| Minimal criteria are A plus B plus E |
| Note: If symptoms are associated with a familial history, the diagnosis is sleep paralysis-familial type. If symptoms are not associated with a familial history, the diagnosis is sleep paralysis-isolated type |
| Severity criteria Mild: Moderate: >1 episode per month but Severe: ≥1 episode per week |
| Duration criteria Acute: ≤1 month Subacute: >1 month but Chronic: ≥6 months |
| REM: rapid eye movement |
| Source: Reference 11 |
TREATMENT: Reassurance, therapy
Effective treatment for Ms. S required helping her to understand that an organic condition was the foundation of her experiences. I began by conveying the sleep paralysis diagnosis and my understanding of the occupational and personal consequences that this condition had had for her. I explained the physiology of sleep paralysis and that memories or hallucinations (dreamlike mentation) are preserved in an extremely vivid fashion because her eyes are open. I acknowledged the realistic character of her experiences and the resulting symptoms of posttraumatic stress disorder (PTSD).
I refer Ms. S to a therapist for psychotherapy. The therapist begins by using trauma informed techniques to address Ms. S’s PTSD. As she improves, her therapy evolves into a combination of narrative and supportive psychotherapy, and then family systems therapy to address issues with her daughter and mother.
In a follow-up visit 1 year after beginning treatment, Ms. S cites multiple improvements, with no recurrence of sleep paralysis episodes. She continues to take sertraline, which relieves her depression and anxiety, and methylphenidate to improve her attention and concentration. She has taken on more responsibility at home, cleaning, preparing meals, helping her daughter choose a college, and attending to her mother’s health issues. Ms. S still has difficulties with her sleep patterns, and her new psychiatrist is exploring the possibility of a bipolar component to her mood disorder.
The authors’ observations
Like other traumas, AAE can induce symptoms of acute or chronic PTSD. The various psychoses, personality disorders, and dissociative disorders that could account for abduction experiences are characterized by delusions, so conduct ongoing assessment for these conditions in patients who report AAE. However, evidence suggests that serious psychopathology is no more common among “abductees” than among the general population.12
Persons reporting AAE exhibit physiologic reactivity as profound as that of survivors of combat or sexual assault.13 This reactivity confirms that the emotional power of the memory is as evocative and problematic as the physiologic reactions attributable to genuine (documented) traumatic events. Because patients have difficulty differentiating these hallucinations from actual events, they experience emotional pain and suffering. Fifty-seven percent of sleep paralysis patients who report AAE attempt suicide.14
There are no FDA-approved medications for treating sleep paralysis. Pharmacotherapy can be used to address psychiatric symptoms such as the depression and anxiety Ms. S exhibited.
Related resources
- American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine; 2001:166-9.
- Cheyne JA. Sleep paralysis and associated hypnagogic and hypnopompic experiences. http://watarts.uwaterloo.ca/~acheyne/S_P.html.
- Methylphenidate • Ritalin
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Patry AL, Pelletier LG. Extraterrestrial beliefs and experiences: an application of the theory of reasoned action. J Soc Psychol 2001;141(2):199-217.
2. Newman LS, Baumeister RF. Toward an explanation of the UFO abduction phenomenon: hypnotic elaboration, extraterrestrial sadomasochism, and spurious memories. Psychol Inq 1996;7(2):99-126.
3. Bader CD. Supernatural support groups: who are the UFO abductees and ritual-abuse survivors? J Sci Study Relig 2003;42(4):669-78.
4. Clancy SA, McNally RJ, Schacter DL, et al. Memory distortion in people reporting abduction by aliens. J Abnorm Psychol 2002;111(3):455-61.
5. Girard TA, Cheyne JA. Individual differences in lateralization of hallucinations associated with sleep paralysis. Laterality 2004;9(1):93-111.
6. Cheyne JA. The ominous numinous: sensed presence and “other” hallucinations. Journal of Consciousness Studies 2001;8(5-7):133-50.
7. Cheyne JA, Newby-Clark IR, Rueffer SD. Relations among hypnagogic and hypnopompic experiences associated with sleep paralysis. J Sleep Res 1999;8:313-7.
8. Cheyne JA, Rueffer SD, Newby-Clark IR. Hypnagogic and hypnopompic hallucinations during sleep paralysis: neurological and cultural construction of the night-mare. Conscious Cogn 1999;8(3):319-37.
9. Cheyne JA. Sleep paralysis and the structure of waking-nightmare hallucinations. Dreaming 2003;13(3):163-79.
10. Spanos NP, Cross PA, Dickson K, et al. Close encounters: an examination of UFO experiences. J Abnorm Psychol 1993;102(4):624-32.
11. American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine. 2001;166-9.
12. Holden KJ, French CC. Alien abduction experiences: some clues from neuropsychology and neuropsychiatry. Cognit Neuropsychiatry 2002;7(3):163-78.
13. McNally RJ. Applying biological data in forensic and policy arenas. Ann N Y Acad Sci 2006;1071:267-76.
14. Stone-Carmen J. A descriptive study of people reporting abduction by unidentified flying objects (UFOs). In: Pritchard A, Pritchard DE, Mack JE, et al, eds. Alien discussions: proceedings of the abduction study conference held at MIT. Cambridge, MA: North Cambridge Press; 1994:309-15
CASE: ’I’m not crazy’
Ms. S, age 55, presents for treatment because she is feeling depressed and anxious. Her symptoms include decreased concentration, intermittent irritability, hoarding, and difficulty starting and completing tasks. She also has chronic sleep difficulties that often keep her awake until dawn.
Fatigue, lack of focus, and poor comprehension and motivation have left her unemployed. She and her teenage daughter live with Ms. S’s elderly mother. Ms. S feels tremendous guilt because she cannot be the mother and daughter she wants to be.
Initially, I (PK) diagnose Ms. S with major depressive disorder and prescribe sertraline, 100 mg/d, which improves her mood and energy. However, her inability to stay organized results in her being “let go” from job training.
Ms. S reports similar difficulties in school as a child. I determine that she meets DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD). Adding methylphenidate, 10 mg bid, improves her concentration and ability to complete tasks. It also reduces the impulsivity that has disrupted her relationships.
Despite a strong desire to normalize her sleep schedule, Ms. S continues to have difficulty falling asleep, so I add melatonin, 3 to 6 mg at bedtime. Her sleeping pattern is improved, but still variable. She also tries quetiapine, 25 mg at bedtime, but soon discontinues it due to intolerance.
As our rapport strengthens, Ms. S reveals that she has had multiple encounters with aliens beginning at age 3. Although she has not had an “alien experience” for about 5 years, she does not feel safe sleeping at night and instead sleeps during the day. Her efforts to stay awake at night strain her relationship with her mother.
The authors’ observations
Approximately 1% of the U.S. population report alien abduction experiences (AAE)—an umbrella term that includes alleged contact with aliens ranging from sightings to abductions.1 Patients rarely report AAE to mental health professionals. In our society, claiming to be an “abductee” implies that one might be insane. A survey of 398 Canadian students that assessed attitudes, beliefs, and experiences regarding alien abductions found that 79% of respondents believed they would have mostly negative consequences—such as being laughed at or socially isolated—if they claimed to have encountered aliens.1
Persons who have AAE may attend support groups of fellow “abductees” to accumulate behavior-consonant information (hearing other people’s abduction stories) and reduce dissonance by being surrounded by others who share a questionable belief.2 A survey of “abductees” found that 88% report at least some positive aspects of the experience, such as a sense of importance or feeling as though they were chosen to bridge communication between extraterrestrials and humans.3
- high levels of psychic energy
- self-sufficiency
- resourcefulness
- a tendency to question authority and to be exposed to situational conflicts.1
After Ms. S reveals her alien experiences, I reassure her in a nonjudgmental manner that we will explore her experiences and determine ways to help her cope with them.
HISTORY: Terrifying experiences
Ms. S elaborates on her alien experiences, relating a particularly terrifying example from her teen years. She was lying awake in bed, looking at the ceiling, where she saw a jeweled spider with a drill. As the spider descended from the ceiling and spread its legs, she recalled a noise like a dentist’s drill. As the spider neared her face, it grew larger and larger. Terrified, Ms. S was unable to scream for help or move anything except her eyes as the spider clamped its legs around her head and bored into her skull. She reported that although she could feel the drill go in, it wasn’t painful.
Other experiences included giving birth, undergoing examinations or probes, and communicating with aliens. Although she is very distressed by most memories, she feels she benefited from others. For example, as a child, Ms. S’s math skills improved dramatically after an AAE episode; she believes this was a gift from the aliens. Ms. S’s AAE memories are as vivid to her as memories of her college graduation. She had been reluctant to discuss these events with anyone outside her family out of fear of being perceived as “crazy.”
Ms. S says she was a shy child who had difficulty making friends. She was plagued with fatigue and worry about family members. She believed that aliens might attack her sisters and felt obligated to stay awake at night to protect them. Aside from alien experiences, Ms. S reports a happy childhood.
She has always been an avid reader. At age 8 or 9, after reading a book on alien abduction, she concluded that she had been abducted. Later, she joined a group of professed alien abductees. She feels accepted and validated by this group and has a forum for discussing her experiences without fear of ridicule or rejection.
Ms. S remains frightened by things that remind her of aliens. Although she wrote a summary of her alien experiences, she cannot draw a picture of an alien, and thoughts or images of the prototypical “grey” alien trigger panic. She also feels somewhat “different,” nervous, and distant from others.
The authors’ observations
Reviewing AAE literature led me to consider several diagnoses, including:
- psychosis
- seizures
- false memory (sexual abuse, trauma)
- narcolepsy
- sleep paralysis.
Electroencephalography (during drowsiness) revealed abnormal activity (occurrences of widely scattered bursts of nonspecific, round, sharply contoured slow waves in the left frontal region) only in the F7 electrode. In the absence of clinical symptoms and when found in a single lead, this is considered a normal variant.
Diagnostic testing ruled out hallucinosis related to seizures. I also ruled out false memory related to sexual abuse or trauma, which is commonly found in patients who present with AAE.
Collaborative information from relatives did not uncover a history of psychosis. She and family members reported, however, that Ms. S’s father and 1 sister had periodic sleep disturbances with associated hallucinations. I began to suspect sleep paralysis.
The authors’ observations
Full-body paralysis normally accompanies rapid eye movement (REM) sleep, which occurs several times a night.4 Sleep paralysis is a transient state that occurs when an individual becomes conscious of this immobility, typically while falling asleep or awakening.5 These experiences can be accompanied by hypnagogic (while falling asleep) or hypnopompic (while awakening) hallucinations. An estimated 30% of the population has had at least one sleep paralysis episode.6 In one study, 5% of sleep paralysis patients had episodes that were accompanied by hallucinations.7
Although individuals cannot make gross body movements during sleep paralysis, they can open their eyes and are able to report events that occurred around them during the episode.8 Patients interpret sleep paralysis experiences in subjective terms. Common descriptions include intense fear, breathing difficulties, feeling of bodily pressure—especially on the chest—and sensations of floating, flying, or falling (Table 1).7,9
During sleep paralysis episodes, individuals typically sense a threatening presence.6 Patients have reported beastly and demonic figures of doom: devils, demons, witches, aliens, and even cinematic villains such as Darth Vader and Freddy Kruger.6 Others have described this presence in terms of alien visitations or abductions.
Table 1
4 types of sleep paralysis-related hallucinations
| Intruder | Vague sense of a threatening presence accompanied by visual, auditory, and tactile hallucinations—noises, footsteps, gibbering voices, humanoid apparitions, and sensation of being touched or grabbed |
| Incubus | Breathing difficulties, feelings of suffocation, bodily pressure (particularly on the chest, as if someone were sitting or standing on it), pain, and thoughts of impending death |
| Vestibular-motor | Sensations of floating (levitation), flying, and falling |
| Other | Out-of-body experiences, autoscopy (seeing oneself from an external point), and fictive motor movements, ranging from simple arm movements to sitting up to apparent locomotion through the environment |
| Source: References 7,9 | |
A Harvard University study of 11 individuals who reported alien abductions found that all participants experienced a similar sequence of events:
- They suspected abduction after sleep episodes characterized by awakening, full-body paralysis, intense fear, and a feeling of a presence. Several reported tactile or visual sensations strikingly similar to descriptions of sleep paralysis, such as levitating, being touched, and seeing shadowy figures.
- They sought explanations for what they perceived as anomalous experiences.
- They “recovered” abduction memories in therapy (with the help of techniques such as hypnosis) or spontaneously (after reading books or seeing movies or television shows depicting similar episodes).4
Ms. S reported no daytime sleep attacks, cataplexy, or rapid onset of dreaming. Because her reported AAEs were spread out and the last occurred approximately 5 years ago, I decided against conducting a sleep study because it likely would be low yield and costly. I reached a diagnosis of sleep paralysis-familial type, chronic based on:
- an absence of organic or psychiatric dysfunction
- a familial pattern of sleep disturbances
- the temporal pattern and description of her symptoms (Table 2).11
Table 2
Diagnostic criteria for sleep paralysis
| A. Patient complains of inability to move the trunk or limbs at sleep onset or upon awakening |
| B. Brief episodes of partial or complete skeletal muscle paralysis |
| C. Episodes can be associated with hypnagogic (preceding sleep) hallucinations or dreamlike mentation |
D. Polysomnographic monitoring demonstrates at least 1 of the following:
|
| E. Symptoms are not associated with other medical or mental disorders, such as hysteria or hypokalemic paralysis |
| Minimal criteria are A plus B plus E |
| Note: If symptoms are associated with a familial history, the diagnosis is sleep paralysis-familial type. If symptoms are not associated with a familial history, the diagnosis is sleep paralysis-isolated type |
| Severity criteria Mild: Moderate: >1 episode per month but Severe: ≥1 episode per week |
| Duration criteria Acute: ≤1 month Subacute: >1 month but Chronic: ≥6 months |
| REM: rapid eye movement |
| Source: Reference 11 |
TREATMENT: Reassurance, therapy
Effective treatment for Ms. S required helping her to understand that an organic condition was the foundation of her experiences. I began by conveying the sleep paralysis diagnosis and my understanding of the occupational and personal consequences that this condition had had for her. I explained the physiology of sleep paralysis and that memories or hallucinations (dreamlike mentation) are preserved in an extremely vivid fashion because her eyes are open. I acknowledged the realistic character of her experiences and the resulting symptoms of posttraumatic stress disorder (PTSD).
I refer Ms. S to a therapist for psychotherapy. The therapist begins by using trauma informed techniques to address Ms. S’s PTSD. As she improves, her therapy evolves into a combination of narrative and supportive psychotherapy, and then family systems therapy to address issues with her daughter and mother.
In a follow-up visit 1 year after beginning treatment, Ms. S cites multiple improvements, with no recurrence of sleep paralysis episodes. She continues to take sertraline, which relieves her depression and anxiety, and methylphenidate to improve her attention and concentration. She has taken on more responsibility at home, cleaning, preparing meals, helping her daughter choose a college, and attending to her mother’s health issues. Ms. S still has difficulties with her sleep patterns, and her new psychiatrist is exploring the possibility of a bipolar component to her mood disorder.
The authors’ observations
Like other traumas, AAE can induce symptoms of acute or chronic PTSD. The various psychoses, personality disorders, and dissociative disorders that could account for abduction experiences are characterized by delusions, so conduct ongoing assessment for these conditions in patients who report AAE. However, evidence suggests that serious psychopathology is no more common among “abductees” than among the general population.12
Persons reporting AAE exhibit physiologic reactivity as profound as that of survivors of combat or sexual assault.13 This reactivity confirms that the emotional power of the memory is as evocative and problematic as the physiologic reactions attributable to genuine (documented) traumatic events. Because patients have difficulty differentiating these hallucinations from actual events, they experience emotional pain and suffering. Fifty-seven percent of sleep paralysis patients who report AAE attempt suicide.14
There are no FDA-approved medications for treating sleep paralysis. Pharmacotherapy can be used to address psychiatric symptoms such as the depression and anxiety Ms. S exhibited.
Related resources
- American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine; 2001:166-9.
- Cheyne JA. Sleep paralysis and associated hypnagogic and hypnopompic experiences. http://watarts.uwaterloo.ca/~acheyne/S_P.html.
- Methylphenidate • Ritalin
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: ’I’m not crazy’
Ms. S, age 55, presents for treatment because she is feeling depressed and anxious. Her symptoms include decreased concentration, intermittent irritability, hoarding, and difficulty starting and completing tasks. She also has chronic sleep difficulties that often keep her awake until dawn.
Fatigue, lack of focus, and poor comprehension and motivation have left her unemployed. She and her teenage daughter live with Ms. S’s elderly mother. Ms. S feels tremendous guilt because she cannot be the mother and daughter she wants to be.
Initially, I (PK) diagnose Ms. S with major depressive disorder and prescribe sertraline, 100 mg/d, which improves her mood and energy. However, her inability to stay organized results in her being “let go” from job training.
Ms. S reports similar difficulties in school as a child. I determine that she meets DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD). Adding methylphenidate, 10 mg bid, improves her concentration and ability to complete tasks. It also reduces the impulsivity that has disrupted her relationships.
Despite a strong desire to normalize her sleep schedule, Ms. S continues to have difficulty falling asleep, so I add melatonin, 3 to 6 mg at bedtime. Her sleeping pattern is improved, but still variable. She also tries quetiapine, 25 mg at bedtime, but soon discontinues it due to intolerance.
As our rapport strengthens, Ms. S reveals that she has had multiple encounters with aliens beginning at age 3. Although she has not had an “alien experience” for about 5 years, she does not feel safe sleeping at night and instead sleeps during the day. Her efforts to stay awake at night strain her relationship with her mother.
The authors’ observations
Approximately 1% of the U.S. population report alien abduction experiences (AAE)—an umbrella term that includes alleged contact with aliens ranging from sightings to abductions.1 Patients rarely report AAE to mental health professionals. In our society, claiming to be an “abductee” implies that one might be insane. A survey of 398 Canadian students that assessed attitudes, beliefs, and experiences regarding alien abductions found that 79% of respondents believed they would have mostly negative consequences—such as being laughed at or socially isolated—if they claimed to have encountered aliens.1
Persons who have AAE may attend support groups of fellow “abductees” to accumulate behavior-consonant information (hearing other people’s abduction stories) and reduce dissonance by being surrounded by others who share a questionable belief.2 A survey of “abductees” found that 88% report at least some positive aspects of the experience, such as a sense of importance or feeling as though they were chosen to bridge communication between extraterrestrials and humans.3
- high levels of psychic energy
- self-sufficiency
- resourcefulness
- a tendency to question authority and to be exposed to situational conflicts.1
After Ms. S reveals her alien experiences, I reassure her in a nonjudgmental manner that we will explore her experiences and determine ways to help her cope with them.
HISTORY: Terrifying experiences
Ms. S elaborates on her alien experiences, relating a particularly terrifying example from her teen years. She was lying awake in bed, looking at the ceiling, where she saw a jeweled spider with a drill. As the spider descended from the ceiling and spread its legs, she recalled a noise like a dentist’s drill. As the spider neared her face, it grew larger and larger. Terrified, Ms. S was unable to scream for help or move anything except her eyes as the spider clamped its legs around her head and bored into her skull. She reported that although she could feel the drill go in, it wasn’t painful.
Other experiences included giving birth, undergoing examinations or probes, and communicating with aliens. Although she is very distressed by most memories, she feels she benefited from others. For example, as a child, Ms. S’s math skills improved dramatically after an AAE episode; she believes this was a gift from the aliens. Ms. S’s AAE memories are as vivid to her as memories of her college graduation. She had been reluctant to discuss these events with anyone outside her family out of fear of being perceived as “crazy.”
Ms. S says she was a shy child who had difficulty making friends. She was plagued with fatigue and worry about family members. She believed that aliens might attack her sisters and felt obligated to stay awake at night to protect them. Aside from alien experiences, Ms. S reports a happy childhood.
She has always been an avid reader. At age 8 or 9, after reading a book on alien abduction, she concluded that she had been abducted. Later, she joined a group of professed alien abductees. She feels accepted and validated by this group and has a forum for discussing her experiences without fear of ridicule or rejection.
Ms. S remains frightened by things that remind her of aliens. Although she wrote a summary of her alien experiences, she cannot draw a picture of an alien, and thoughts or images of the prototypical “grey” alien trigger panic. She also feels somewhat “different,” nervous, and distant from others.
The authors’ observations
Reviewing AAE literature led me to consider several diagnoses, including:
- psychosis
- seizures
- false memory (sexual abuse, trauma)
- narcolepsy
- sleep paralysis.
Electroencephalography (during drowsiness) revealed abnormal activity (occurrences of widely scattered bursts of nonspecific, round, sharply contoured slow waves in the left frontal region) only in the F7 electrode. In the absence of clinical symptoms and when found in a single lead, this is considered a normal variant.
Diagnostic testing ruled out hallucinosis related to seizures. I also ruled out false memory related to sexual abuse or trauma, which is commonly found in patients who present with AAE.
Collaborative information from relatives did not uncover a history of psychosis. She and family members reported, however, that Ms. S’s father and 1 sister had periodic sleep disturbances with associated hallucinations. I began to suspect sleep paralysis.
The authors’ observations
Full-body paralysis normally accompanies rapid eye movement (REM) sleep, which occurs several times a night.4 Sleep paralysis is a transient state that occurs when an individual becomes conscious of this immobility, typically while falling asleep or awakening.5 These experiences can be accompanied by hypnagogic (while falling asleep) or hypnopompic (while awakening) hallucinations. An estimated 30% of the population has had at least one sleep paralysis episode.6 In one study, 5% of sleep paralysis patients had episodes that were accompanied by hallucinations.7
Although individuals cannot make gross body movements during sleep paralysis, they can open their eyes and are able to report events that occurred around them during the episode.8 Patients interpret sleep paralysis experiences in subjective terms. Common descriptions include intense fear, breathing difficulties, feeling of bodily pressure—especially on the chest—and sensations of floating, flying, or falling (Table 1).7,9
During sleep paralysis episodes, individuals typically sense a threatening presence.6 Patients have reported beastly and demonic figures of doom: devils, demons, witches, aliens, and even cinematic villains such as Darth Vader and Freddy Kruger.6 Others have described this presence in terms of alien visitations or abductions.
Table 1
4 types of sleep paralysis-related hallucinations
| Intruder | Vague sense of a threatening presence accompanied by visual, auditory, and tactile hallucinations—noises, footsteps, gibbering voices, humanoid apparitions, and sensation of being touched or grabbed |
| Incubus | Breathing difficulties, feelings of suffocation, bodily pressure (particularly on the chest, as if someone were sitting or standing on it), pain, and thoughts of impending death |
| Vestibular-motor | Sensations of floating (levitation), flying, and falling |
| Other | Out-of-body experiences, autoscopy (seeing oneself from an external point), and fictive motor movements, ranging from simple arm movements to sitting up to apparent locomotion through the environment |
| Source: References 7,9 | |
A Harvard University study of 11 individuals who reported alien abductions found that all participants experienced a similar sequence of events:
- They suspected abduction after sleep episodes characterized by awakening, full-body paralysis, intense fear, and a feeling of a presence. Several reported tactile or visual sensations strikingly similar to descriptions of sleep paralysis, such as levitating, being touched, and seeing shadowy figures.
- They sought explanations for what they perceived as anomalous experiences.
- They “recovered” abduction memories in therapy (with the help of techniques such as hypnosis) or spontaneously (after reading books or seeing movies or television shows depicting similar episodes).4
Ms. S reported no daytime sleep attacks, cataplexy, or rapid onset of dreaming. Because her reported AAEs were spread out and the last occurred approximately 5 years ago, I decided against conducting a sleep study because it likely would be low yield and costly. I reached a diagnosis of sleep paralysis-familial type, chronic based on:
- an absence of organic or psychiatric dysfunction
- a familial pattern of sleep disturbances
- the temporal pattern and description of her symptoms (Table 2).11
Table 2
Diagnostic criteria for sleep paralysis
| A. Patient complains of inability to move the trunk or limbs at sleep onset or upon awakening |
| B. Brief episodes of partial or complete skeletal muscle paralysis |
| C. Episodes can be associated with hypnagogic (preceding sleep) hallucinations or dreamlike mentation |
D. Polysomnographic monitoring demonstrates at least 1 of the following:
|
| E. Symptoms are not associated with other medical or mental disorders, such as hysteria or hypokalemic paralysis |
| Minimal criteria are A plus B plus E |
| Note: If symptoms are associated with a familial history, the diagnosis is sleep paralysis-familial type. If symptoms are not associated with a familial history, the diagnosis is sleep paralysis-isolated type |
| Severity criteria Mild: Moderate: >1 episode per month but Severe: ≥1 episode per week |
| Duration criteria Acute: ≤1 month Subacute: >1 month but Chronic: ≥6 months |
| REM: rapid eye movement |
| Source: Reference 11 |
TREATMENT: Reassurance, therapy
Effective treatment for Ms. S required helping her to understand that an organic condition was the foundation of her experiences. I began by conveying the sleep paralysis diagnosis and my understanding of the occupational and personal consequences that this condition had had for her. I explained the physiology of sleep paralysis and that memories or hallucinations (dreamlike mentation) are preserved in an extremely vivid fashion because her eyes are open. I acknowledged the realistic character of her experiences and the resulting symptoms of posttraumatic stress disorder (PTSD).
I refer Ms. S to a therapist for psychotherapy. The therapist begins by using trauma informed techniques to address Ms. S’s PTSD. As she improves, her therapy evolves into a combination of narrative and supportive psychotherapy, and then family systems therapy to address issues with her daughter and mother.
In a follow-up visit 1 year after beginning treatment, Ms. S cites multiple improvements, with no recurrence of sleep paralysis episodes. She continues to take sertraline, which relieves her depression and anxiety, and methylphenidate to improve her attention and concentration. She has taken on more responsibility at home, cleaning, preparing meals, helping her daughter choose a college, and attending to her mother’s health issues. Ms. S still has difficulties with her sleep patterns, and her new psychiatrist is exploring the possibility of a bipolar component to her mood disorder.
The authors’ observations
Like other traumas, AAE can induce symptoms of acute or chronic PTSD. The various psychoses, personality disorders, and dissociative disorders that could account for abduction experiences are characterized by delusions, so conduct ongoing assessment for these conditions in patients who report AAE. However, evidence suggests that serious psychopathology is no more common among “abductees” than among the general population.12
Persons reporting AAE exhibit physiologic reactivity as profound as that of survivors of combat or sexual assault.13 This reactivity confirms that the emotional power of the memory is as evocative and problematic as the physiologic reactions attributable to genuine (documented) traumatic events. Because patients have difficulty differentiating these hallucinations from actual events, they experience emotional pain and suffering. Fifty-seven percent of sleep paralysis patients who report AAE attempt suicide.14
There are no FDA-approved medications for treating sleep paralysis. Pharmacotherapy can be used to address psychiatric symptoms such as the depression and anxiety Ms. S exhibited.
Related resources
- American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine; 2001:166-9.
- Cheyne JA. Sleep paralysis and associated hypnagogic and hypnopompic experiences. http://watarts.uwaterloo.ca/~acheyne/S_P.html.
- Methylphenidate • Ritalin
- Quetiapine • Seroquel
- Sertraline • Zoloft
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Patry AL, Pelletier LG. Extraterrestrial beliefs and experiences: an application of the theory of reasoned action. J Soc Psychol 2001;141(2):199-217.
2. Newman LS, Baumeister RF. Toward an explanation of the UFO abduction phenomenon: hypnotic elaboration, extraterrestrial sadomasochism, and spurious memories. Psychol Inq 1996;7(2):99-126.
3. Bader CD. Supernatural support groups: who are the UFO abductees and ritual-abuse survivors? J Sci Study Relig 2003;42(4):669-78.
4. Clancy SA, McNally RJ, Schacter DL, et al. Memory distortion in people reporting abduction by aliens. J Abnorm Psychol 2002;111(3):455-61.
5. Girard TA, Cheyne JA. Individual differences in lateralization of hallucinations associated with sleep paralysis. Laterality 2004;9(1):93-111.
6. Cheyne JA. The ominous numinous: sensed presence and “other” hallucinations. Journal of Consciousness Studies 2001;8(5-7):133-50.
7. Cheyne JA, Newby-Clark IR, Rueffer SD. Relations among hypnagogic and hypnopompic experiences associated with sleep paralysis. J Sleep Res 1999;8:313-7.
8. Cheyne JA, Rueffer SD, Newby-Clark IR. Hypnagogic and hypnopompic hallucinations during sleep paralysis: neurological and cultural construction of the night-mare. Conscious Cogn 1999;8(3):319-37.
9. Cheyne JA. Sleep paralysis and the structure of waking-nightmare hallucinations. Dreaming 2003;13(3):163-79.
10. Spanos NP, Cross PA, Dickson K, et al. Close encounters: an examination of UFO experiences. J Abnorm Psychol 1993;102(4):624-32.
11. American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine. 2001;166-9.
12. Holden KJ, French CC. Alien abduction experiences: some clues from neuropsychology and neuropsychiatry. Cognit Neuropsychiatry 2002;7(3):163-78.
13. McNally RJ. Applying biological data in forensic and policy arenas. Ann N Y Acad Sci 2006;1071:267-76.
14. Stone-Carmen J. A descriptive study of people reporting abduction by unidentified flying objects (UFOs). In: Pritchard A, Pritchard DE, Mack JE, et al, eds. Alien discussions: proceedings of the abduction study conference held at MIT. Cambridge, MA: North Cambridge Press; 1994:309-15
1. Patry AL, Pelletier LG. Extraterrestrial beliefs and experiences: an application of the theory of reasoned action. J Soc Psychol 2001;141(2):199-217.
2. Newman LS, Baumeister RF. Toward an explanation of the UFO abduction phenomenon: hypnotic elaboration, extraterrestrial sadomasochism, and spurious memories. Psychol Inq 1996;7(2):99-126.
3. Bader CD. Supernatural support groups: who are the UFO abductees and ritual-abuse survivors? J Sci Study Relig 2003;42(4):669-78.
4. Clancy SA, McNally RJ, Schacter DL, et al. Memory distortion in people reporting abduction by aliens. J Abnorm Psychol 2002;111(3):455-61.
5. Girard TA, Cheyne JA. Individual differences in lateralization of hallucinations associated with sleep paralysis. Laterality 2004;9(1):93-111.
6. Cheyne JA. The ominous numinous: sensed presence and “other” hallucinations. Journal of Consciousness Studies 2001;8(5-7):133-50.
7. Cheyne JA, Newby-Clark IR, Rueffer SD. Relations among hypnagogic and hypnopompic experiences associated with sleep paralysis. J Sleep Res 1999;8:313-7.
8. Cheyne JA, Rueffer SD, Newby-Clark IR. Hypnagogic and hypnopompic hallucinations during sleep paralysis: neurological and cultural construction of the night-mare. Conscious Cogn 1999;8(3):319-37.
9. Cheyne JA. Sleep paralysis and the structure of waking-nightmare hallucinations. Dreaming 2003;13(3):163-79.
10. Spanos NP, Cross PA, Dickson K, et al. Close encounters: an examination of UFO experiences. J Abnorm Psychol 1993;102(4):624-32.
11. American Academy of Sleep Medicine. International classification of sleep disorders, revised: diagnostic and coding manual. Chicago, IL: American Academy of Sleep Medicine. 2001;166-9.
12. Holden KJ, French CC. Alien abduction experiences: some clues from neuropsychology and neuropsychiatry. Cognit Neuropsychiatry 2002;7(3):163-78.
13. McNally RJ. Applying biological data in forensic and policy arenas. Ann N Y Acad Sci 2006;1071:267-76.
14. Stone-Carmen J. A descriptive study of people reporting abduction by unidentified flying objects (UFOs). In: Pritchard A, Pritchard DE, Mack JE, et al, eds. Alien discussions: proceedings of the abduction study conference held at MIT. Cambridge, MA: North Cambridge Press; 1994:309-15
Extended-release fluvoxamine for social anxiety disorder and OCD
Fluvoxamine extended-release formulation was FDA-approved to treat generalized social anxiety disorder (GSAD) and obsessive-compulsive disorder (OCD) because it demonstrated efficacy in reducing anxiety symptoms of these disorders in 3 clinical trials. The new formulation may benefit patients unable to tolerate the existing immediate-release form.
Clinical implications
Like other selective serotonin reuptake inhibitors (SSRIs), fluvoxamine alleviates symptoms of GSAD and OCD. The extended-release formulation allows the medication to be administered once daily (Table 1) and, according to the manufacturer, may reduce side effects and improve tolerability.
Many clinicians have prescribed immediate-release fluvoxamine once daily, and the efficacy and tolerability of the immediate- and extended-release formulations have not been compared in head-to-head trials. In addition, no studies have examined the efficacy of extended-release fluvoxamine in treating other psychiatric conditions.
Table 1
Extended-release fluvoxamine: Fast facts
| Brand name: Luvox CR |
| Class: Selective serotonin reuptake inhibitor |
| Indication: Generalized social anxiety disorder and obsessive-compulsive disorder |
| Approval date: February 29, 2008 |
| Availability date: March 2008 |
| Manufacturer: Jazz Pharmaceuticals |
| Dosing forms: 100 mg and 150 mg extended-release capsules |
| Recommended dose: Starting dose: 100 mg/d. Titrate in 50-mg/week increments until maximum therapeutic benefit is achieved. Maximum recommended dose: 300 mg/d |
How it works
Decreased serotonin levels are associated with GSAD and OCD. Fluvoxamine’s therapeutic effect is thought to be mediated through its specific serotonin reuptake inhibition in the CNS.1
The drug acts primarily on serotonin 2C receptors, with no reported significant affinity for histaminergic, adrenergic, muscarinic, or dopaminergic receptors.1 Fluvoxamine’s 1-sigma receptor antagonism is unique among SSRIs, and researchers have suggested that this may make fluvoxamine more effective than other SSRIs in treating anxious or delusional depression.2
The extended-release formulation uses a spheroidal oral drug absorption system, a proprietary technology that limits peak-to-trough variance for 24 hours.1 The manufacturer postulates that decreased plasma concentration variability will improve fluvoxamine’s tolerability.1
Pharmacokinetics
In a single-dose crossover study of 28 healthy subjects, the mean maximum concentration of drug (Cmax) for extended-release fluvoxamine was 38% lower than that of the immediate-release formulation, which may reduce the risk of adverse effects.1 Its relative bioavailability was 84%, and mean plasma half-life was 16.3 hours in male and female volunteers.1
Fluvoxamine is extensively metabolized in the liver, primarily through oxidative demethylation and deamination.1 Nine metabolites constitute 85% of the urinary excretion product; the main metabolite is fluvoxamine acid.1 Approximately 2% of fluvoxamine is excreted unchanged in urine. Administering extended-release fluvoxamine capsules with food does not appear to affect the drug’s absorption.1
Fluvoxamine is a potent inhibitor of the cytochrome P450 (CYP) 1A2 isoenzyme and also is believed to significantly inhibit CYP3A4, CYP2C9, CYP3A4, and CYP2C19. It is a relatively weak inhibitor of CYP2D6.1
Efficacy
The FDA based its approval of extended-release fluvoxamine on data from 3 clinical trials with positive outcomes: 2 for GSAD and 1 for OCD (Table 2).1,3-6
GSAD trials. In the first GSAD study—a randomized, double-blind, placebo-controlled, multicenter trial of 300 subjects with GSAD—participants were randomly assigned to receive extended-release fluvoxamine or placebo for 12 weeks.3 The extended-release fluvoxamine group started at 100 mg administered at night, with dosages titrated at 50 mg/week based on efficacy and tolerability to a maximum of 300 mg/d.1 Subjects in the extended-release fluvoxamine group demonstrated a statistically significant change in Liebowitz Social Anxiety Scale (LSAS) scores from baseline compared with those receiving placebo (P=0.02). Researchers observed similar results in secondary measures.
In an extension of this study, 112 subjects who demonstrated at least minimal improvement from extended-release fluvoxamine by week 12 continued the same dosing regimen for an additional 12 weeks. Investigators found the drug’s beneficial effects persisted to 24 weeks, although the magnitude of the effect decreased.4
A separate study using the same dosing regimen enrolled 279 adult patients in a 12-week, multicenter, randomized, placebo-controlled trial.5 The fluvoxamine-treated group showed statistically and clinically significant improvement:
- by week 4 on the LSAS and the Clinical Global Impression-Improvement (CGI-I) scale
- by week 6 on the Sheehan Disability Scale, Clinical Global Impression-Severity scale (CGI-S) and Patient Global Impression of Improvement (PGI) scale.5
OCD trial. Hollander et al6 conducted a 12-week, double-blind, placebo-controlled, flexible-dose, parallel multicenter trial of 253 adult patients with OCD.6 Compared with those receiving placebo, subjects treated with extended-release fluvoxamine, 100 to 300 mg/d, showed a statistically significant decrease in score on the Yale-Brown Obsessive Compulsive Scale (P=0.001).6 Analysis of the CGIS and CGI-I also revealed statistically significant improvement compared with placebo. The effect appeared to begin at week 2.
As did the GSAD studies, this study compared extended-release fluvoxamine with placebo and not with the immediate-release formulation. Although no additional studies have examined the efficacy of extended-release fluvoxamine in treating OCD and the drug has not been evaluated in pediatric patients, the manufacturer notes that the immediate-release formulation has been evaluated in 2 studies with adult OCD patients and 1 pediatric OCD study, all of which had positive results.1
Table 2
Fluvoxamine extended-release: What the evidence says
| Study | Measures used | Results |
|---|---|---|
| Generalized social anxiety disorder | ||
| Westenberg et al (2004)3 | LSAS, CGI-S, CGI-I, SDS, PGI | Fluvoxamine was significantly more effective than placebo in decreasing LSAS total score (primary measure) starting at week 4 and in improving SDS, CGI-S, and CGI-I (secondary measures) |
| Stein et al (2003)4 | LSAS, CGI-S, CGI-I, SDS, PGI | Severity of illness on the CGI-S scale and disability on the SDS were significantly lower in the fluvoxamine group than in the placebo group; fluvoxamine-treated subjects had a numerically greater decrease in LSAS total scores than subjects treated with placebo |
| Davidson et al (2004)5 | LSAS, CGI-G, SDS, CGI-S, PGI | Fluvoxamine produced statistically and clinically significant improvements in symptoms starting at week 4 on the LSAS and CGI-I and at week 6 on the SDS, CGI-S, and PGI |
| Obsessive-compulsive disorder | ||
| Hollander et al (2003)6 | YBOCS, CGI-S, CGI-I | Fluvoxamine was significantly more effective than placebo in decreasing YBOCS total score beginning at week 2 and in improving CGI-S and CGI-I scores |
| LSAS: Liebowitz Social Anxiety Scale; SDS: Sheehan Disability Scale; CGI-S: Clinical Global Impression-Severity of illness; CGI-I: Clinical Global Impression-Improvement; PGI: Patient Global Impression of Improvement; YBOCS: Yale-Brown Obsessive Compulsive Scale | ||
Tolerability
In the 3 published trials of extended-release fluvoxamine, adverse event rates were similar and consistent with earlier studies of the immediate-release formulation. 1 The manufacturer considered adverse events likely to be drug-related if they had an incidence ≥5% and at least twice that of placebo (Table 3). 1, 3- 6
Adverse events caused 26% of patients in the GSAD studies and 19% in the OCD trial to discontinue treatment. No deaths, life-threatening adverse events, or suicide attempts were reported.3-6 No statistically significant differences in weight gain or loss, vital signs, laboratory findings, or ECG changes were found between patients treated with extended-release fluvoxamine and those receiving placebo.1
Table 3
Extended-release fluvoxamine: Adverse events*
| Study | Adverse events |
|---|---|
| Both GSAD and OCD studies | Abnormal ejaculation, anorexia, anorgasmia, asthenia, diarrhea, nausea, somnolence, sweating, tremor |
| GSAD studies only | Dyspepsia, dizziness, insomnia, yawning |
| OCD study only | Accidental injury, anxiety, decreased libido, myalgia, pharyngitis, emesis |
| * Includes events with an incidence ≥5% and at least twice that of placebo GSAD: generalized social anxiety disorder; OCD: obsessive-compulsive disorder | |
| Source: References 3-6 | |
Contraindications
Immediate- and extended-release fluvoxamine have the same active ingredient and therefore the same contraindications. Coadministration of alosetron, pimozide, thioridazine, or tizanidine, is contraindicated, as is using monoamine oxidase (MAO) inhibitors with extended-release fluvoxamine or within 14 days of discontinuing fluvoxamine treatment. Extended-release fluvoxamine has the same warnings that all SSRIs share regarding clinical worsening and suicide risk, administration to bipolar patients, neuroleptic malignant syndrome, serotonin syndrome, and possible increases in coagulation.1,2
The FDA classifies extended-release fluvoxamine as pregnancy category C.1 The drug is not contraindicated for lactating mothers, but because fluvoxamine is secreted in breast milk discuss with breast-feeding patients the benefits and risks of continuing fluvoxamine therapy.1 Infants exposed to immediate-release fluvoxamine in late pregnancy have developed serious adverse reactions, including respiratory distress, cyanosis, apnea, and seizures.1
Dosing
The recommended starting dose of extended-release fluvoxamine is 100 mg once daily, with or without food.1 The dose can be titrated in 50-mg/week increments as tolerated to achieve maximum therapeutic benefit, to the maximum recommended dose of 300 mg/d. Unlike immediate-release fluvoxamine, which is occasionally split into twice-daily doses, extended-release fluvoxamine must be administered only once daily, even at high doses.1,2
Related resource
- Luvox CR prescribing information. www.jazzpharmaceuticals.com/content/news/documents/LUVOX_CR.pdf.
Drug brand names
- Alosetron • Lotronex
- Fluvoxamine • Luvox
- Fluvoxamine extended-release • Luvox CR
- Pimozide • Orap
- Thioridazine • Mellaril
- Tizanidine • Zanaflex
Disclosures
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives research/grant support from Forest Laboratories and is a consultant to Jazz Pharmaceuticals.
1. Luvox CR [package insert]. Palo Alto, CA: Jazz Pharmaceuticals; 2008.
2. Stahl SM. Essential psychopharmacology: the prescriber’s guide. Revised and updated edition. New York, NY: Cambridge University Press; 2006.
3. Westenberg HG, Stein DJ, Yang H, et al. A double-blind placebo-controlled study of controlled release fluvoxamine for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(1):49-55.
4. Stein DJ, Westenberg HG, Yang H, et al. Fluvoxamine CR in the long-term treatment of social anxiety disorder: the 12- to 24-week extension phase of a multicentre, randomized, placebo-controlled trial. Int J Neuropsychopharmacol 2003;6(4):317-23.
5. Davidson J, Yaryura-Tobias J, DuPont R, et al. Fluvoxamine-controlled release formulation for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(2):118-25.
6. Hollander E, Koran LM, Goodman WK, et al. A double-blind, placebo-controlled study of the efficacy and safety of controlled-release fluvoxamine in patients with obsessive-compulsive disorder. J Clin Psychiatry 2003;64(6):640-7.
Fluvoxamine extended-release formulation was FDA-approved to treat generalized social anxiety disorder (GSAD) and obsessive-compulsive disorder (OCD) because it demonstrated efficacy in reducing anxiety symptoms of these disorders in 3 clinical trials. The new formulation may benefit patients unable to tolerate the existing immediate-release form.
Clinical implications
Like other selective serotonin reuptake inhibitors (SSRIs), fluvoxamine alleviates symptoms of GSAD and OCD. The extended-release formulation allows the medication to be administered once daily (Table 1) and, according to the manufacturer, may reduce side effects and improve tolerability.
Many clinicians have prescribed immediate-release fluvoxamine once daily, and the efficacy and tolerability of the immediate- and extended-release formulations have not been compared in head-to-head trials. In addition, no studies have examined the efficacy of extended-release fluvoxamine in treating other psychiatric conditions.
Table 1
Extended-release fluvoxamine: Fast facts
| Brand name: Luvox CR |
| Class: Selective serotonin reuptake inhibitor |
| Indication: Generalized social anxiety disorder and obsessive-compulsive disorder |
| Approval date: February 29, 2008 |
| Availability date: March 2008 |
| Manufacturer: Jazz Pharmaceuticals |
| Dosing forms: 100 mg and 150 mg extended-release capsules |
| Recommended dose: Starting dose: 100 mg/d. Titrate in 50-mg/week increments until maximum therapeutic benefit is achieved. Maximum recommended dose: 300 mg/d |
How it works
Decreased serotonin levels are associated with GSAD and OCD. Fluvoxamine’s therapeutic effect is thought to be mediated through its specific serotonin reuptake inhibition in the CNS.1
The drug acts primarily on serotonin 2C receptors, with no reported significant affinity for histaminergic, adrenergic, muscarinic, or dopaminergic receptors.1 Fluvoxamine’s 1-sigma receptor antagonism is unique among SSRIs, and researchers have suggested that this may make fluvoxamine more effective than other SSRIs in treating anxious or delusional depression.2
The extended-release formulation uses a spheroidal oral drug absorption system, a proprietary technology that limits peak-to-trough variance for 24 hours.1 The manufacturer postulates that decreased plasma concentration variability will improve fluvoxamine’s tolerability.1
Pharmacokinetics
In a single-dose crossover study of 28 healthy subjects, the mean maximum concentration of drug (Cmax) for extended-release fluvoxamine was 38% lower than that of the immediate-release formulation, which may reduce the risk of adverse effects.1 Its relative bioavailability was 84%, and mean plasma half-life was 16.3 hours in male and female volunteers.1
Fluvoxamine is extensively metabolized in the liver, primarily through oxidative demethylation and deamination.1 Nine metabolites constitute 85% of the urinary excretion product; the main metabolite is fluvoxamine acid.1 Approximately 2% of fluvoxamine is excreted unchanged in urine. Administering extended-release fluvoxamine capsules with food does not appear to affect the drug’s absorption.1
Fluvoxamine is a potent inhibitor of the cytochrome P450 (CYP) 1A2 isoenzyme and also is believed to significantly inhibit CYP3A4, CYP2C9, CYP3A4, and CYP2C19. It is a relatively weak inhibitor of CYP2D6.1
Efficacy
The FDA based its approval of extended-release fluvoxamine on data from 3 clinical trials with positive outcomes: 2 for GSAD and 1 for OCD (Table 2).1,3-6
GSAD trials. In the first GSAD study—a randomized, double-blind, placebo-controlled, multicenter trial of 300 subjects with GSAD—participants were randomly assigned to receive extended-release fluvoxamine or placebo for 12 weeks.3 The extended-release fluvoxamine group started at 100 mg administered at night, with dosages titrated at 50 mg/week based on efficacy and tolerability to a maximum of 300 mg/d.1 Subjects in the extended-release fluvoxamine group demonstrated a statistically significant change in Liebowitz Social Anxiety Scale (LSAS) scores from baseline compared with those receiving placebo (P=0.02). Researchers observed similar results in secondary measures.
In an extension of this study, 112 subjects who demonstrated at least minimal improvement from extended-release fluvoxamine by week 12 continued the same dosing regimen for an additional 12 weeks. Investigators found the drug’s beneficial effects persisted to 24 weeks, although the magnitude of the effect decreased.4
A separate study using the same dosing regimen enrolled 279 adult patients in a 12-week, multicenter, randomized, placebo-controlled trial.5 The fluvoxamine-treated group showed statistically and clinically significant improvement:
- by week 4 on the LSAS and the Clinical Global Impression-Improvement (CGI-I) scale
- by week 6 on the Sheehan Disability Scale, Clinical Global Impression-Severity scale (CGI-S) and Patient Global Impression of Improvement (PGI) scale.5
OCD trial. Hollander et al6 conducted a 12-week, double-blind, placebo-controlled, flexible-dose, parallel multicenter trial of 253 adult patients with OCD.6 Compared with those receiving placebo, subjects treated with extended-release fluvoxamine, 100 to 300 mg/d, showed a statistically significant decrease in score on the Yale-Brown Obsessive Compulsive Scale (P=0.001).6 Analysis of the CGIS and CGI-I also revealed statistically significant improvement compared with placebo. The effect appeared to begin at week 2.
As did the GSAD studies, this study compared extended-release fluvoxamine with placebo and not with the immediate-release formulation. Although no additional studies have examined the efficacy of extended-release fluvoxamine in treating OCD and the drug has not been evaluated in pediatric patients, the manufacturer notes that the immediate-release formulation has been evaluated in 2 studies with adult OCD patients and 1 pediatric OCD study, all of which had positive results.1
Table 2
Fluvoxamine extended-release: What the evidence says
| Study | Measures used | Results |
|---|---|---|
| Generalized social anxiety disorder | ||
| Westenberg et al (2004)3 | LSAS, CGI-S, CGI-I, SDS, PGI | Fluvoxamine was significantly more effective than placebo in decreasing LSAS total score (primary measure) starting at week 4 and in improving SDS, CGI-S, and CGI-I (secondary measures) |
| Stein et al (2003)4 | LSAS, CGI-S, CGI-I, SDS, PGI | Severity of illness on the CGI-S scale and disability on the SDS were significantly lower in the fluvoxamine group than in the placebo group; fluvoxamine-treated subjects had a numerically greater decrease in LSAS total scores than subjects treated with placebo |
| Davidson et al (2004)5 | LSAS, CGI-G, SDS, CGI-S, PGI | Fluvoxamine produced statistically and clinically significant improvements in symptoms starting at week 4 on the LSAS and CGI-I and at week 6 on the SDS, CGI-S, and PGI |
| Obsessive-compulsive disorder | ||
| Hollander et al (2003)6 | YBOCS, CGI-S, CGI-I | Fluvoxamine was significantly more effective than placebo in decreasing YBOCS total score beginning at week 2 and in improving CGI-S and CGI-I scores |
| LSAS: Liebowitz Social Anxiety Scale; SDS: Sheehan Disability Scale; CGI-S: Clinical Global Impression-Severity of illness; CGI-I: Clinical Global Impression-Improvement; PGI: Patient Global Impression of Improvement; YBOCS: Yale-Brown Obsessive Compulsive Scale | ||
Tolerability
In the 3 published trials of extended-release fluvoxamine, adverse event rates were similar and consistent with earlier studies of the immediate-release formulation. 1 The manufacturer considered adverse events likely to be drug-related if they had an incidence ≥5% and at least twice that of placebo (Table 3). 1, 3- 6
Adverse events caused 26% of patients in the GSAD studies and 19% in the OCD trial to discontinue treatment. No deaths, life-threatening adverse events, or suicide attempts were reported.3-6 No statistically significant differences in weight gain or loss, vital signs, laboratory findings, or ECG changes were found between patients treated with extended-release fluvoxamine and those receiving placebo.1
Table 3
Extended-release fluvoxamine: Adverse events*
| Study | Adverse events |
|---|---|
| Both GSAD and OCD studies | Abnormal ejaculation, anorexia, anorgasmia, asthenia, diarrhea, nausea, somnolence, sweating, tremor |
| GSAD studies only | Dyspepsia, dizziness, insomnia, yawning |
| OCD study only | Accidental injury, anxiety, decreased libido, myalgia, pharyngitis, emesis |
| * Includes events with an incidence ≥5% and at least twice that of placebo GSAD: generalized social anxiety disorder; OCD: obsessive-compulsive disorder | |
| Source: References 3-6 | |
Contraindications
Immediate- and extended-release fluvoxamine have the same active ingredient and therefore the same contraindications. Coadministration of alosetron, pimozide, thioridazine, or tizanidine, is contraindicated, as is using monoamine oxidase (MAO) inhibitors with extended-release fluvoxamine or within 14 days of discontinuing fluvoxamine treatment. Extended-release fluvoxamine has the same warnings that all SSRIs share regarding clinical worsening and suicide risk, administration to bipolar patients, neuroleptic malignant syndrome, serotonin syndrome, and possible increases in coagulation.1,2
The FDA classifies extended-release fluvoxamine as pregnancy category C.1 The drug is not contraindicated for lactating mothers, but because fluvoxamine is secreted in breast milk discuss with breast-feeding patients the benefits and risks of continuing fluvoxamine therapy.1 Infants exposed to immediate-release fluvoxamine in late pregnancy have developed serious adverse reactions, including respiratory distress, cyanosis, apnea, and seizures.1
Dosing
The recommended starting dose of extended-release fluvoxamine is 100 mg once daily, with or without food.1 The dose can be titrated in 50-mg/week increments as tolerated to achieve maximum therapeutic benefit, to the maximum recommended dose of 300 mg/d. Unlike immediate-release fluvoxamine, which is occasionally split into twice-daily doses, extended-release fluvoxamine must be administered only once daily, even at high doses.1,2
Related resource
- Luvox CR prescribing information. www.jazzpharmaceuticals.com/content/news/documents/LUVOX_CR.pdf.
Drug brand names
- Alosetron • Lotronex
- Fluvoxamine • Luvox
- Fluvoxamine extended-release • Luvox CR
- Pimozide • Orap
- Thioridazine • Mellaril
- Tizanidine • Zanaflex
Disclosures
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives research/grant support from Forest Laboratories and is a consultant to Jazz Pharmaceuticals.
Fluvoxamine extended-release formulation was FDA-approved to treat generalized social anxiety disorder (GSAD) and obsessive-compulsive disorder (OCD) because it demonstrated efficacy in reducing anxiety symptoms of these disorders in 3 clinical trials. The new formulation may benefit patients unable to tolerate the existing immediate-release form.
Clinical implications
Like other selective serotonin reuptake inhibitors (SSRIs), fluvoxamine alleviates symptoms of GSAD and OCD. The extended-release formulation allows the medication to be administered once daily (Table 1) and, according to the manufacturer, may reduce side effects and improve tolerability.
Many clinicians have prescribed immediate-release fluvoxamine once daily, and the efficacy and tolerability of the immediate- and extended-release formulations have not been compared in head-to-head trials. In addition, no studies have examined the efficacy of extended-release fluvoxamine in treating other psychiatric conditions.
Table 1
Extended-release fluvoxamine: Fast facts
| Brand name: Luvox CR |
| Class: Selective serotonin reuptake inhibitor |
| Indication: Generalized social anxiety disorder and obsessive-compulsive disorder |
| Approval date: February 29, 2008 |
| Availability date: March 2008 |
| Manufacturer: Jazz Pharmaceuticals |
| Dosing forms: 100 mg and 150 mg extended-release capsules |
| Recommended dose: Starting dose: 100 mg/d. Titrate in 50-mg/week increments until maximum therapeutic benefit is achieved. Maximum recommended dose: 300 mg/d |
How it works
Decreased serotonin levels are associated with GSAD and OCD. Fluvoxamine’s therapeutic effect is thought to be mediated through its specific serotonin reuptake inhibition in the CNS.1
The drug acts primarily on serotonin 2C receptors, with no reported significant affinity for histaminergic, adrenergic, muscarinic, or dopaminergic receptors.1 Fluvoxamine’s 1-sigma receptor antagonism is unique among SSRIs, and researchers have suggested that this may make fluvoxamine more effective than other SSRIs in treating anxious or delusional depression.2
The extended-release formulation uses a spheroidal oral drug absorption system, a proprietary technology that limits peak-to-trough variance for 24 hours.1 The manufacturer postulates that decreased plasma concentration variability will improve fluvoxamine’s tolerability.1
Pharmacokinetics
In a single-dose crossover study of 28 healthy subjects, the mean maximum concentration of drug (Cmax) for extended-release fluvoxamine was 38% lower than that of the immediate-release formulation, which may reduce the risk of adverse effects.1 Its relative bioavailability was 84%, and mean plasma half-life was 16.3 hours in male and female volunteers.1
Fluvoxamine is extensively metabolized in the liver, primarily through oxidative demethylation and deamination.1 Nine metabolites constitute 85% of the urinary excretion product; the main metabolite is fluvoxamine acid.1 Approximately 2% of fluvoxamine is excreted unchanged in urine. Administering extended-release fluvoxamine capsules with food does not appear to affect the drug’s absorption.1
Fluvoxamine is a potent inhibitor of the cytochrome P450 (CYP) 1A2 isoenzyme and also is believed to significantly inhibit CYP3A4, CYP2C9, CYP3A4, and CYP2C19. It is a relatively weak inhibitor of CYP2D6.1
Efficacy
The FDA based its approval of extended-release fluvoxamine on data from 3 clinical trials with positive outcomes: 2 for GSAD and 1 for OCD (Table 2).1,3-6
GSAD trials. In the first GSAD study—a randomized, double-blind, placebo-controlled, multicenter trial of 300 subjects with GSAD—participants were randomly assigned to receive extended-release fluvoxamine or placebo for 12 weeks.3 The extended-release fluvoxamine group started at 100 mg administered at night, with dosages titrated at 50 mg/week based on efficacy and tolerability to a maximum of 300 mg/d.1 Subjects in the extended-release fluvoxamine group demonstrated a statistically significant change in Liebowitz Social Anxiety Scale (LSAS) scores from baseline compared with those receiving placebo (P=0.02). Researchers observed similar results in secondary measures.
In an extension of this study, 112 subjects who demonstrated at least minimal improvement from extended-release fluvoxamine by week 12 continued the same dosing regimen for an additional 12 weeks. Investigators found the drug’s beneficial effects persisted to 24 weeks, although the magnitude of the effect decreased.4
A separate study using the same dosing regimen enrolled 279 adult patients in a 12-week, multicenter, randomized, placebo-controlled trial.5 The fluvoxamine-treated group showed statistically and clinically significant improvement:
- by week 4 on the LSAS and the Clinical Global Impression-Improvement (CGI-I) scale
- by week 6 on the Sheehan Disability Scale, Clinical Global Impression-Severity scale (CGI-S) and Patient Global Impression of Improvement (PGI) scale.5
OCD trial. Hollander et al6 conducted a 12-week, double-blind, placebo-controlled, flexible-dose, parallel multicenter trial of 253 adult patients with OCD.6 Compared with those receiving placebo, subjects treated with extended-release fluvoxamine, 100 to 300 mg/d, showed a statistically significant decrease in score on the Yale-Brown Obsessive Compulsive Scale (P=0.001).6 Analysis of the CGIS and CGI-I also revealed statistically significant improvement compared with placebo. The effect appeared to begin at week 2.
As did the GSAD studies, this study compared extended-release fluvoxamine with placebo and not with the immediate-release formulation. Although no additional studies have examined the efficacy of extended-release fluvoxamine in treating OCD and the drug has not been evaluated in pediatric patients, the manufacturer notes that the immediate-release formulation has been evaluated in 2 studies with adult OCD patients and 1 pediatric OCD study, all of which had positive results.1
Table 2
Fluvoxamine extended-release: What the evidence says
| Study | Measures used | Results |
|---|---|---|
| Generalized social anxiety disorder | ||
| Westenberg et al (2004)3 | LSAS, CGI-S, CGI-I, SDS, PGI | Fluvoxamine was significantly more effective than placebo in decreasing LSAS total score (primary measure) starting at week 4 and in improving SDS, CGI-S, and CGI-I (secondary measures) |
| Stein et al (2003)4 | LSAS, CGI-S, CGI-I, SDS, PGI | Severity of illness on the CGI-S scale and disability on the SDS were significantly lower in the fluvoxamine group than in the placebo group; fluvoxamine-treated subjects had a numerically greater decrease in LSAS total scores than subjects treated with placebo |
| Davidson et al (2004)5 | LSAS, CGI-G, SDS, CGI-S, PGI | Fluvoxamine produced statistically and clinically significant improvements in symptoms starting at week 4 on the LSAS and CGI-I and at week 6 on the SDS, CGI-S, and PGI |
| Obsessive-compulsive disorder | ||
| Hollander et al (2003)6 | YBOCS, CGI-S, CGI-I | Fluvoxamine was significantly more effective than placebo in decreasing YBOCS total score beginning at week 2 and in improving CGI-S and CGI-I scores |
| LSAS: Liebowitz Social Anxiety Scale; SDS: Sheehan Disability Scale; CGI-S: Clinical Global Impression-Severity of illness; CGI-I: Clinical Global Impression-Improvement; PGI: Patient Global Impression of Improvement; YBOCS: Yale-Brown Obsessive Compulsive Scale | ||
Tolerability
In the 3 published trials of extended-release fluvoxamine, adverse event rates were similar and consistent with earlier studies of the immediate-release formulation. 1 The manufacturer considered adverse events likely to be drug-related if they had an incidence ≥5% and at least twice that of placebo (Table 3). 1, 3- 6
Adverse events caused 26% of patients in the GSAD studies and 19% in the OCD trial to discontinue treatment. No deaths, life-threatening adverse events, or suicide attempts were reported.3-6 No statistically significant differences in weight gain or loss, vital signs, laboratory findings, or ECG changes were found between patients treated with extended-release fluvoxamine and those receiving placebo.1
Table 3
Extended-release fluvoxamine: Adverse events*
| Study | Adverse events |
|---|---|
| Both GSAD and OCD studies | Abnormal ejaculation, anorexia, anorgasmia, asthenia, diarrhea, nausea, somnolence, sweating, tremor |
| GSAD studies only | Dyspepsia, dizziness, insomnia, yawning |
| OCD study only | Accidental injury, anxiety, decreased libido, myalgia, pharyngitis, emesis |
| * Includes events with an incidence ≥5% and at least twice that of placebo GSAD: generalized social anxiety disorder; OCD: obsessive-compulsive disorder | |
| Source: References 3-6 | |
Contraindications
Immediate- and extended-release fluvoxamine have the same active ingredient and therefore the same contraindications. Coadministration of alosetron, pimozide, thioridazine, or tizanidine, is contraindicated, as is using monoamine oxidase (MAO) inhibitors with extended-release fluvoxamine or within 14 days of discontinuing fluvoxamine treatment. Extended-release fluvoxamine has the same warnings that all SSRIs share regarding clinical worsening and suicide risk, administration to bipolar patients, neuroleptic malignant syndrome, serotonin syndrome, and possible increases in coagulation.1,2
The FDA classifies extended-release fluvoxamine as pregnancy category C.1 The drug is not contraindicated for lactating mothers, but because fluvoxamine is secreted in breast milk discuss with breast-feeding patients the benefits and risks of continuing fluvoxamine therapy.1 Infants exposed to immediate-release fluvoxamine in late pregnancy have developed serious adverse reactions, including respiratory distress, cyanosis, apnea, and seizures.1
Dosing
The recommended starting dose of extended-release fluvoxamine is 100 mg once daily, with or without food.1 The dose can be titrated in 50-mg/week increments as tolerated to achieve maximum therapeutic benefit, to the maximum recommended dose of 300 mg/d. Unlike immediate-release fluvoxamine, which is occasionally split into twice-daily doses, extended-release fluvoxamine must be administered only once daily, even at high doses.1,2
Related resource
- Luvox CR prescribing information. www.jazzpharmaceuticals.com/content/news/documents/LUVOX_CR.pdf.
Drug brand names
- Alosetron • Lotronex
- Fluvoxamine • Luvox
- Fluvoxamine extended-release • Luvox CR
- Pimozide • Orap
- Thioridazine • Mellaril
- Tizanidine • Zanaflex
Disclosures
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives research/grant support from Forest Laboratories and is a consultant to Jazz Pharmaceuticals.
1. Luvox CR [package insert]. Palo Alto, CA: Jazz Pharmaceuticals; 2008.
2. Stahl SM. Essential psychopharmacology: the prescriber’s guide. Revised and updated edition. New York, NY: Cambridge University Press; 2006.
3. Westenberg HG, Stein DJ, Yang H, et al. A double-blind placebo-controlled study of controlled release fluvoxamine for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(1):49-55.
4. Stein DJ, Westenberg HG, Yang H, et al. Fluvoxamine CR in the long-term treatment of social anxiety disorder: the 12- to 24-week extension phase of a multicentre, randomized, placebo-controlled trial. Int J Neuropsychopharmacol 2003;6(4):317-23.
5. Davidson J, Yaryura-Tobias J, DuPont R, et al. Fluvoxamine-controlled release formulation for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(2):118-25.
6. Hollander E, Koran LM, Goodman WK, et al. A double-blind, placebo-controlled study of the efficacy and safety of controlled-release fluvoxamine in patients with obsessive-compulsive disorder. J Clin Psychiatry 2003;64(6):640-7.
1. Luvox CR [package insert]. Palo Alto, CA: Jazz Pharmaceuticals; 2008.
2. Stahl SM. Essential psychopharmacology: the prescriber’s guide. Revised and updated edition. New York, NY: Cambridge University Press; 2006.
3. Westenberg HG, Stein DJ, Yang H, et al. A double-blind placebo-controlled study of controlled release fluvoxamine for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(1):49-55.
4. Stein DJ, Westenberg HG, Yang H, et al. Fluvoxamine CR in the long-term treatment of social anxiety disorder: the 12- to 24-week extension phase of a multicentre, randomized, placebo-controlled trial. Int J Neuropsychopharmacol 2003;6(4):317-23.
5. Davidson J, Yaryura-Tobias J, DuPont R, et al. Fluvoxamine-controlled release formulation for the treatment of generalized social anxiety disorder. J Clin Psychopharmacol 2004;24(2):118-25.
6. Hollander E, Koran LM, Goodman WK, et al. A double-blind, placebo-controlled study of the efficacy and safety of controlled-release fluvoxamine in patients with obsessive-compulsive disorder. J Clin Psychiatry 2003;64(6):640-7.
STAT: 7 tips for the psychiatric ER
Psychiatric emergency rooms (ERs) often are the first stop for patients experiencing severe psychiatric symptoms. Following these strategies can help as you assess and treat a variety of patients and create a modicum of calm out of the chaos.
‘Heal’ borderlines
Although patients diagnosed with borderline personality disorder often present treatment challenges, a supportive psychotherapeutic approach based on empathic listening often can be helpful. Allowing these patients to feel understood in the midst of an interpersonal crisis may be enough to help them navigate their predicament in a healthier way.
Beware of shift changes
Patients arriving during a staff shift change might not receive the time and attention necessary for a comprehensive psychiatric evaluation. Resist pressure to speed up the workflow, and do not leave patients waiting to be seen by the oncoming shift. Working only by the clock may result in a rushed and inadequate assessment and a suboptimal treatment plan.
Sleeping it off
Patients often arrive intoxicated and might not be able to adequately participate in a psychiatric assessment. Talk to intoxicated patients briefly, get an adequate medical history, ensure their safety and monitoring, and then let them sleep in the ER. Re-evaluation in the morning often yields dramatically different mental status findings.
Be familiar with social services
Although some ERs employ staff members who specialize in coordinating social services, be familiar with available homeless shelters, travelers’ aid societies, halfway houses, and safe homes. Armed with this information, you can refer to appropriate agencies patients with problems that are more social than psychiatric.
Know your staff
Psychiatric ERs are staffed by a variety of mental health professionals, and individual team members’ experience, training, and knowledge can vary greatly. It is your responsibility to be familiar with the strengths and weaknesses of these workers to guard against having to repeat tasks.
Feed your patients
Providing patients with food is a straight-forward way to demonstrate you care about their needs and want to help. Though it is important that patients do not view psychiatric ERs as places to come to get a hot meal, generously dispensing food often helps lay the groundwork for a therapeutic relationship.
Be generous with thiamine
Many ER patients are undernourished or abuse alcohol and therefore are at risk for thiamine deficiency. The sequelae of thiamine deficiency, including Wernicke’s encephalopathy and Korsakoff’s syndrome, are serious and in some cases irreversible.
Psychiatric emergency rooms (ERs) often are the first stop for patients experiencing severe psychiatric symptoms. Following these strategies can help as you assess and treat a variety of patients and create a modicum of calm out of the chaos.
‘Heal’ borderlines
Although patients diagnosed with borderline personality disorder often present treatment challenges, a supportive psychotherapeutic approach based on empathic listening often can be helpful. Allowing these patients to feel understood in the midst of an interpersonal crisis may be enough to help them navigate their predicament in a healthier way.
Beware of shift changes
Patients arriving during a staff shift change might not receive the time and attention necessary for a comprehensive psychiatric evaluation. Resist pressure to speed up the workflow, and do not leave patients waiting to be seen by the oncoming shift. Working only by the clock may result in a rushed and inadequate assessment and a suboptimal treatment plan.
Sleeping it off
Patients often arrive intoxicated and might not be able to adequately participate in a psychiatric assessment. Talk to intoxicated patients briefly, get an adequate medical history, ensure their safety and monitoring, and then let them sleep in the ER. Re-evaluation in the morning often yields dramatically different mental status findings.
Be familiar with social services
Although some ERs employ staff members who specialize in coordinating social services, be familiar with available homeless shelters, travelers’ aid societies, halfway houses, and safe homes. Armed with this information, you can refer to appropriate agencies patients with problems that are more social than psychiatric.
Know your staff
Psychiatric ERs are staffed by a variety of mental health professionals, and individual team members’ experience, training, and knowledge can vary greatly. It is your responsibility to be familiar with the strengths and weaknesses of these workers to guard against having to repeat tasks.
Feed your patients
Providing patients with food is a straight-forward way to demonstrate you care about their needs and want to help. Though it is important that patients do not view psychiatric ERs as places to come to get a hot meal, generously dispensing food often helps lay the groundwork for a therapeutic relationship.
Be generous with thiamine
Many ER patients are undernourished or abuse alcohol and therefore are at risk for thiamine deficiency. The sequelae of thiamine deficiency, including Wernicke’s encephalopathy and Korsakoff’s syndrome, are serious and in some cases irreversible.
Psychiatric emergency rooms (ERs) often are the first stop for patients experiencing severe psychiatric symptoms. Following these strategies can help as you assess and treat a variety of patients and create a modicum of calm out of the chaos.
‘Heal’ borderlines
Although patients diagnosed with borderline personality disorder often present treatment challenges, a supportive psychotherapeutic approach based on empathic listening often can be helpful. Allowing these patients to feel understood in the midst of an interpersonal crisis may be enough to help them navigate their predicament in a healthier way.
Beware of shift changes
Patients arriving during a staff shift change might not receive the time and attention necessary for a comprehensive psychiatric evaluation. Resist pressure to speed up the workflow, and do not leave patients waiting to be seen by the oncoming shift. Working only by the clock may result in a rushed and inadequate assessment and a suboptimal treatment plan.
Sleeping it off
Patients often arrive intoxicated and might not be able to adequately participate in a psychiatric assessment. Talk to intoxicated patients briefly, get an adequate medical history, ensure their safety and monitoring, and then let them sleep in the ER. Re-evaluation in the morning often yields dramatically different mental status findings.
Be familiar with social services
Although some ERs employ staff members who specialize in coordinating social services, be familiar with available homeless shelters, travelers’ aid societies, halfway houses, and safe homes. Armed with this information, you can refer to appropriate agencies patients with problems that are more social than psychiatric.
Know your staff
Psychiatric ERs are staffed by a variety of mental health professionals, and individual team members’ experience, training, and knowledge can vary greatly. It is your responsibility to be familiar with the strengths and weaknesses of these workers to guard against having to repeat tasks.
Feed your patients
Providing patients with food is a straight-forward way to demonstrate you care about their needs and want to help. Though it is important that patients do not view psychiatric ERs as places to come to get a hot meal, generously dispensing food often helps lay the groundwork for a therapeutic relationship.
Be generous with thiamine
Many ER patients are undernourished or abuse alcohol and therefore are at risk for thiamine deficiency. The sequelae of thiamine deficiency, including Wernicke’s encephalopathy and Korsakoff’s syndrome, are serious and in some cases irreversible.
Did Internet-purchased diet pills cause serotonin syndrome?
Ms. G, age 28, presents to a tertiary care hospital with altered mental status. Six weeks ago she started taking phentermine, 37.5 mg/d, to lose weight. Her body mass index is 24 kg/m2 (normal range), and she obtained the stimulant agent via the Internet. Her family reports Ms. G was very busy in the past week, staying up until 2 AM cleaning. They say she also was irritable with her 5-year-old son.
Two days ago, Ms. G complained of fatigue and nausea without emesis. She went to bed early and did not awaken the next morning. Her sister found her in bed, minimally responsive to verbal stimuli, and brought her to the hospital.
Patients have used phentermine as a weight-reducing agent since the FDA approved this amphetamine-like compound in 1960.1 Phentermine’s mechanism of action is thought to involve dopaminergic, noradrenergic, and serotonergic effects.2 Stimulation of norepinephrine (NE) release is its most potent effect, followed by NE reuptake inhibition, stimulation of dopamine (DA) release, DA reuptake inhibition, stimulation of serotonin (5-HT) release, and 5-HT reuptake inhibition (weak).3
Because phentermine could in theory cause serotonin syndrome,4 its use is contraindicated with monoamine oxidase inhibitors (MAOIs) and not recommended with selective serotonin reuptake inhibitors (SSRIs).5 One case report describes an interaction between fluoxetine and phentermine that appears consistent with serotonin syndrome.6 We are aware of no case reports of serotonin syndrome caused by phentermine alone.
This article reports the case of Ms. G, who presented with probable serotonin syndrome associated with phentermine use and subsequently developed a rapid-onset, superimposed neuroleptic malignant syndrome (NMS). We hypothesize that phentermine use may increase NMS risk through adverse drug events and discuss potential pathophysiologic mechanisms and treatment implications.
Serotonin syndrome vs NMS
Serotonin syndrome is an infrequent and potentially life-threatening adverse drug reaction that presumably results from excess serotonin activity (Box 1).7-10 NMS also is an infrequent and potentially life-threatening neurologic emergency (Box 2).11-18 Similarities between disorders of increased serotonergic activity and disorders of low dopaminergic activity (Table 1) suggest both may result from an imbalance between the serotonergic and dopaminergic systems, which have reciprocal relationships in the CNS.19
Differentiating between serotonin syndrome and NMS is further complicated when both antipsychotics and serotonergic agents may be implicated.20 Clinical trials are not feasible because NMS and serotonin syndrome rarely occur.
Sternbach7 first summarized serotonin syndrome’s clinical presentation in a review of 38 cases. The most frequent clinical features include changes in mental status, restlessness, myoclonus, hyperreflexia, diaphoresis, shivering, and tremor (Table 1).
The clinical syndrome varies in scope and intensity. Animal models suggest the pathophysiologic mechanism involves brainstem and spinal cord inundation with serotonin, acting on 5-HT1A and 5-HT2A receptors. Recent evidence supports a greater role for 5-HT2A receptors.8
Primary treatment calls for discontinuing the suspected serotonergic agent and instituting supportive measures. Case reports also suggest using serotonin receptor antagonists—such as cyproheptadine, methysergide, chlorpromazine, or propranolol—to clinically manage serotonin syndrome, although empiric support is limited.9
The syndrome often improves within 24 hours of primary treatment, although confusion sometimes last for days and death has been reported.10
NMS—characterized by fever, extrapyramidal rigidity, and disturbances of autonomic function and consciousness—was first described with the use of haloperidol.11 Risk factors include catatonia, disorganized presentation, and dehydration.12 NMS is thought to result from deficient compensatory mechanisms following blockade of dopaminergic regulation of muscle tone and autonomic function.13
Although possibly idiosyncratic, the reaction has been associated with:
- intramuscular, higher total dose, or abruptly increasing doses of antipsychotics14
- withdrawal of dopaminergic agents, such as those used to treat Parkinson’s disease.15
Akin to serotonin syndrome, managing NMS focuses on removing the offending agent(s) and providing supportive care. Severe cases require intensive monitoring, aggressive IV hydration, and respiratory support. Dopaminergics such as bromocriptine16 and skeletal muscle relaxants such as dantrolene17 also have been used to manage NMS.
Unlike serotonin syndrome, NMS often resolves slowly (typically >1 week). NMS’ mortality rate of 11% to 38% appears to be declining in recent years, perhaps because it is being recognized more rapidly.18
Signs and symptoms of NMS vs serotonin syndrome
| NMS | Serotonin syndrome | |
|---|---|---|
| Onset | Insidious, days to weeks | Acute (minutes to hours) |
| Resolution | Slow, often >1 week | Improvement or resolution often within 24 hours |
| Autonomic | Fever, tachycardia, diaphoresis, elevated or labile blood pressure, sialorrhea, tachypnea, incontinence | Diaphoresis, shivering, fever, tachycardia, hypertension, mydriasis |
| Gastrointestinal | Dysphagia, elevated transaminases | Diarrhea, nausea, vomiting, elevated ammonia and transaminases |
| Neuromuscular | Rigidity, bradykinesia, dysarthria, dyskinesias, coarse tremor, ataxia, opisthotonos, oculogyric crisis, rhabdomyolysis | Clonus, myoclonus, hyperreflexia, ataxia, incoordination, rigidity, tremor |
| Psychiatric | Altered mental status, stupor, somnolence, mutism | Altered mental status, agitation, hypomania, hyperactivity, restlessness, somnolence (less common) |
| Other | Leukocytosis, elevated creatine kinase (significant), elevated serum creatinine, proteinuria, renal failure, disseminated intravascular coagulation | Leukocytosis (rarely >20K cells/mm3), elevated creatine kinase (less common), disseminated intravascular coagulation, metabolic acidosis |
| NMS: neuroleptic malignant syndrome | ||
| Note: Classically reported symptoms are italicized | ||
CASE CONTINUED: Fever follows haloperidol
Initial workup. Ms. G has no significant medical or psychiatric history. She has no history of seizures, head trauma, changes in mental status, recent travel, tick bites, or mosquito bites. Family history is relevant only for a maternal aunt with a history of 1 seizure. Ms. G is employed and lives with her husband and son. She is not taking other medications, herbal supplements, or vitamins and does not use tobacco, alcohol, caffeine, or illicit drugs.
On admission, she is somnolent and arousable only to painful stimuli. Temperature is 36.7°C, blood pressure 89/58 mm Hg, heart rate 73 bpm, and respirations 21/minute. She does not talk but is cooperative to physical examination, which is otherwise unremarkable.
Neurologic exam also is unremarkable, with no evidence of meningeal irritation, abnormal reflexes, or muscle tone. Serum ammonia (51 µmol/L; normal range 7 to 42 µmol/L) is slightly elevated. Liver function tests, electrolytes, blood urea nitrogen, creatinine, complete blood counts, urinalysis, urine culture, and blood cultures are unremarkable. Ethanol, salicylate, and acetaminophen levels are negative. Evaluation reveals a positive urine drug screen only for amphetamines, attributed to use of phentermine. Chest radiography and head CT are unremarkable.
Electroencephalography (EEG) 17 hours after admission reveals left anterior temporal spikes suggestive of seizure activity lasting 50 seconds. The patient is described as stuporous but arousable during EEG, and diffuse delta slow waves are superimposed on an alpha rhythm with intermittent diffuse delta bursts. Brain MRI is unremarkable.
Despite no clinical evidence of seizure, Ms. G is transferred to the cardiac telemetry ward to monitor for potential side effects from IV phenytoin loading, at which time (24 hours after admission) she is found to have intermittent sinus tachycardia ≤140 bpm.
Antipsychotic therapy. Thirty hours after admission—after phenytoin loading and normalized EEG—Ms. G shows periodic episodes of sudden startling, with repetitive leg shaking. Continuous ankle clonus is present bilaterally. She complains of severe paresthesias in her legs and is unable to urinate on her own.
NMS signs emerge. Forty-eight hours after admission, Ms. G becomes febrile (38.3°C) and shows tachycardia, with heart rate consistently >130 bpm. Her vital signs did not normalize before the fever developed. She remains somnolent and continues to have spastic lower leg and ankle clonus. She shows no seizure activity on video EEG monitoring during later episodes of repetitive leg shaking, approximately 60 hours after admission.
Ms. G receives empiric vancomycin, ceftriaxone, ampicillin, and acyclovir for possible infectious encephalitis, and lumbar puncture is done emergently. Further laboratory tests reveal creatine kinase (CK) elevation (17,282 U/L, from 270 on admission), leukocytosis (white blood cell count 16.1K/mm3, from 7.2K on admission), and elevated transaminases (AST 199 U/L, up from 21 on admission; ALT 84 U/L, up from 19 on admission).
She is transferred to the ICU with a preliminary diagnosis of NMS. Again, continuous EEG monitoring does not show seizure activity. CSF specimen is negative for infection (negative cultures, negative herpes simplex virus PCR, protein 31 mg/dl, glucose 75 mg/dl). She is started on dantrolene, bromocriptine, and levodopa but shows no initial improvement.
Intubation. On hospital day 8, the patient is intubated to protect her airway and placed in a pentobarbital coma for 2 days, with no improvement. On hospital day 9, cyproheptadine, 24 mg/d, is added for possible serotonin syndrome, and continued for 9 days.
On day 11, the addition of IV diazepam, 10 mg per hour, is followed by gradual improvement in rigidity. Ms. G remains on continuous EEG, with no evidence of seizure activity before diazepam was added or after it is tapered off by day 23.
Discharge. Ms. G is extubated on hospital day 18. On day 23 she can follow commands but is not fully oriented, and levodopa, phenytoin, bromocriptine, and dantrolene are tapered off. She is discharged to a rehabilitation facility, where she again requires phenytoin for a witnessed seizure, attributed to anticonvulsant withdrawal.
On follow-up phone interviews 4 and 18 months after hospitalization, Ms. G says she remains seizure-free without taking anticonvulsants. She reports a subjective, interval improvement in cognitive function, which has since returned to baseline.
Evidence for serotonin syndrome
This case involves a young woman with a several-week history of phentermine use for weight reduction who presented with confusion, sedation, mutism, and nausea. She was initially found to have an abnormal EEG, for which she was loaded with the anticonvulsant phenytoin. However, she continued to exhibit altered mental status, myoclonus, and hyperreflexia along with autonomic dysregulation—such as urinary retention and tachycardia—despite a negative EEG on continuous monitoring.
On retrospective review, we believe she likely was experiencing serotonin toxicity from phentermine. She later developed NMS within several hours of receiving the antipsychotic haloperidol.
Even though phentermine is thought to have a relatively weak serotonergic effect,3 it has been shown to markedly increase serotonin efflux in the rat hypothalamus (to a greater degree than the SSRI fluoxetine).23 Although Ms. G did not report having consumed foods or supplements that could have interfered with phentermine’s metabolism, such use could have contributed to or prolonged a serotonin syndrome.20 Phentermine misuse also cannot be ruled out.
Excess phentermine or concomitant use of other serotonergic agents may have precipitated serotonin syndrome. Ms. G’s hyperactivity a few days before she complained of fatigue and somnolence may represent:
- a sign of phentermine intoxication or overuse
- a harbinger of serotonin syndrome, because these symptoms were followed by overt serotonin syndrome signs such as confusion, disorientation, myoclonus, and autonomic dysfunction.
Evidence for NMS
Ms. G received haloperidol because her agitation obstructed urgent evaluation. After several doses, she rapidly developed signs and symptoms highly consistent with NMS. Onset was rapid compared with the typically described, more insidious NMS evolution of 24 to 72 hours, however.25 Rapid NMS onset may have been precipitated in 2 ways:
- dopaminergic (phentermine) withdrawal combined with dopamine antagonist challenge (haloperidol)25,26
- background serotonin syndrome caused by amphetamine (phentermine) predisposing the patient to develop NMS.27
Although phentermine-induced sympathetic hyperactivity also could have predisposed Ms. G to NMS,31 we think this is unlikely because phentermine was discontinued 3 to 4 days before she developed NMS. Nonetheless, sympathetic hyperactivity secondary to phentermine or serotonin syndrome may increase the risk of developing NMS.
Treatment strategy
Because serotonin syndrome and NMS share many clinical findings, differentiating between the 2 syndromes may be difficult, especially when the patient’s medication history does not implicate a specific agent. A detailed history and physical may help distinguish the syndromes. Clonus may be particularly specific and is important in the diagnosis of serotonin syndrome.32 If you are unable to differentiate between serotonin syndrome and NMS in a patient with this acute neurotoxic abnormal behavior syndrome,33 consider a common treatment strategy (Table 2).19,25
In Ms. G’s case, she probably should not have received bromocriptine for NMS,20 given the potential role of serotonin syndrome in precipitating her symptoms.
Case reports support our hypotheses of an increased predilection for NMS with dopaminergic withdrawal or serotonin syndrome. Growing evidence supports the use of chlorpromazine for serotonin syndrome,34 but consider its use contraindicated in patients with NMS.
Table 2
Serotonin syndrome or NMS?
When in doubt, follow 4 management principles
| Avoid serotonin agonists and dopamine antagonists when a patient presents with features of serotonin syndrome or neuroleptic malignant syndrome (NMS) and the diagnosis is unclear20 |
| Provide supportive care with monitoring, cooling blankets as needed, and hydration |
| Avoid using antipsychotics for agitation, when possible; benzodiazepines may be preferable, although their use in NMS is controversial25 |
| Avoid using bromocriptine, given its contraindication in serotonin syndrome, but consider cyproheptadine for the serotonin syndrome component and dantrolene for skeletal muscle rigidity20 |
Related resources
- Carbone JR. The neuroleptic malignant and serotonin syndromes. Emerg Med Clin North Am 2000;18:317-25.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999;13:100-9.
- Neuroleptic Malignant Syndrome Information Service (NMSIS). www.nmsis.org. NMS hotline for medical professionals (toll-free 888-667-8367) handles calls on NMS, serotonin syndrome, heat stroke, malignant catatonia, and other drug-induced heat-related disorders.
This paper was among those entered in the 2007 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s competition was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
- Acyclovir • Zovirax
- Ampicillin • various
- Bromocriptine • Parlodel
- Ceftriaxone • Rocephin
- Chlorpromazine • Thorazine
- Cyproheptadine • Periactin
- Dantrolene • Dantrium
- Diazepam • Valium
- Dextroamphetamine • Dexedrine
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Levodopa • various
- Methysergide • Sansert
- Pentobarbital • Nembutal
- Phentermine • various
- Phenytoin • Dilantin
- Propranolol • Inderal
- Vancomycin • various
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mosby’s Drug Consult, 16th ed. St. Louis, MO: Mosby; 2006.
2. McEvoy G. AHFS drug information. Bethesda, MD: American Society of Health-System Pharmacists; 2006.
3. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001;39:32-41.
4. Ener RA, Meglathery SB, Van Decker WA, Gallagher RM. Serotonin syndrome and other serotonergic disorders. Pain Med 2003;4:63-74.
5. Phentermine hydrochloride. Physicians’ Desk Reference, 53rd ed. Montvale, NJ: Medical Economics; 1999:3055-6.
6. Bostwick JM, Brown TM. A toxic reaction from combining fluoxetine and phentermine. J Clin Psychopharmacol 1996;16:189-90.
7. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-13.
8. Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001;890:23-31.
9. Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000;79:201-9.
10. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005;352:1112-20.
11. Delay J, Pichot P, Lemperiere T, et al. [A non-phenothiazine and non-reserpine major neuroleptic, haloperidol, in the treatment of psychoses.] Ann Med Psychol (Paris). 1960;118:145-52.
12. Berardi D, Dell’Atti M, Amore M, et al. Clinical risk factors for neuroleptic malignant syndrome. Hum Psychopharmacol 2002;17:99-102.
13. Pearlman CA. Neuroleptic malignant syndrome: a review of the literature. J Clin Psychopharmacol 1986;6:257-73.
14. Viejo LF, Morales V, Punal P, et al. Risk factors in neuroleptic malignant syndrome. A case-control study. Acta Psychiatr Scand 2003;107:45-9.
15. Ikebe S, Harada T, Hashimoto T, et al. Prevention and treatment of malignant syndrome in Parkinson’s disease: a consensus statement of the Malignant Syndrome Research Group. Parkinsonism Relat Disord 2003;9(suppl 1):S47-9.
16. Mueller PS, Vester JW, Fermaglich J. Neuroleptic malignant syndrome. Successful treatment with bromocriptine. JAMA 1983;249:386-8.
17. Coons DJ, Hillman FJ, Marshall RW. Treatment of neuroleptic malignant syndrome with dantrolene sodium: a case report. Am J Psychiatry 1982;139:944-5.
18. Spivak B, Maline DI, Kozyrev VN, et al. Frequency of neuroleptic malignant syndrome in a large psychiatric hospital in Moscow. Eur Psychiatry 2000;15:330-3.
19. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother 1998;32:692-8.
20. Kaufman KR, Levitt MJ, Schiltz JF, Sunderram J. Neuroleptic malignant syndrome and serotonin syndrome in the critical care setting: case analysis. Ann Clin Psychiatry 2006;18:201-4.
21. Spencer DC, Hwang J, Morrell MJ. Fenfluramine-phentermine (Fen-Phen) and seizures: evidence for an association. Epilepsy Behav 2000;1(6):448-52.
22. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin 1997;13(4):763-83.
23. Tao R, Fray A, Aspley S, et al. Effects on serotonin in rat hypothalamus of D-fenfluramine, aminorex, phentermine and fluoxetine. Eur J Pharmacol 2002;445(1-2):69-81.
24. Gillman PK. Serotonin syndrome: history and risk. Fundam Clin Pharmacol 1998;12:482-91.
25. Bhanushali MJ, Tuite PJ. The evaluation and management of patients with neuroleptic malignant syndrome. Neurol Clin 2004;22:389-411.
26. Ebadi M, Pfeiffer RF, Murrin LC. Pathogenesis and treatment of neuroleptic malignant syndrome. Gen Pharmacol 1990;21:367-86.
27. Green AR, Cross AJ, Goodwin GM. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or “Ecstasy”). Psychopharmacology (Berl) 1995;119:247-60.
28. Chayasirisobhon S, Cullis P, Veeramasuneni RR. Occurrence of neuroleptic malignant syndrome in a narcoleptic patient. Hosp Community Psychiatry 1983;34:548-50.
29. Serrano-Dueñas M. Neuroleptic malignant syndrome-like, or—dopaminergic malignant syndrome—due to levodopa therapy withdrawal. Clinical features in 11 patients. Parkinsonism Relat Disord 2003;9:175-8.
30. Kline SS, Mauro LS, Scala-Barnett DM, Zick D. Serotonin syndrome versus neuroleptic malignant syndrome as a cause of death. Clin Pharm 1989;8(7):510-14.
31. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
32. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003;96:635-42.
33. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
34. Gillman PK. Serotonin syndrome treated with chlorpromazine. J Clin Psychopharmacol 1997;17:128-9.
Ms. G, age 28, presents to a tertiary care hospital with altered mental status. Six weeks ago she started taking phentermine, 37.5 mg/d, to lose weight. Her body mass index is 24 kg/m2 (normal range), and she obtained the stimulant agent via the Internet. Her family reports Ms. G was very busy in the past week, staying up until 2 AM cleaning. They say she also was irritable with her 5-year-old son.
Two days ago, Ms. G complained of fatigue and nausea without emesis. She went to bed early and did not awaken the next morning. Her sister found her in bed, minimally responsive to verbal stimuli, and brought her to the hospital.
Patients have used phentermine as a weight-reducing agent since the FDA approved this amphetamine-like compound in 1960.1 Phentermine’s mechanism of action is thought to involve dopaminergic, noradrenergic, and serotonergic effects.2 Stimulation of norepinephrine (NE) release is its most potent effect, followed by NE reuptake inhibition, stimulation of dopamine (DA) release, DA reuptake inhibition, stimulation of serotonin (5-HT) release, and 5-HT reuptake inhibition (weak).3
Because phentermine could in theory cause serotonin syndrome,4 its use is contraindicated with monoamine oxidase inhibitors (MAOIs) and not recommended with selective serotonin reuptake inhibitors (SSRIs).5 One case report describes an interaction between fluoxetine and phentermine that appears consistent with serotonin syndrome.6 We are aware of no case reports of serotonin syndrome caused by phentermine alone.
This article reports the case of Ms. G, who presented with probable serotonin syndrome associated with phentermine use and subsequently developed a rapid-onset, superimposed neuroleptic malignant syndrome (NMS). We hypothesize that phentermine use may increase NMS risk through adverse drug events and discuss potential pathophysiologic mechanisms and treatment implications.
Serotonin syndrome vs NMS
Serotonin syndrome is an infrequent and potentially life-threatening adverse drug reaction that presumably results from excess serotonin activity (Box 1).7-10 NMS also is an infrequent and potentially life-threatening neurologic emergency (Box 2).11-18 Similarities between disorders of increased serotonergic activity and disorders of low dopaminergic activity (Table 1) suggest both may result from an imbalance between the serotonergic and dopaminergic systems, which have reciprocal relationships in the CNS.19
Differentiating between serotonin syndrome and NMS is further complicated when both antipsychotics and serotonergic agents may be implicated.20 Clinical trials are not feasible because NMS and serotonin syndrome rarely occur.
Sternbach7 first summarized serotonin syndrome’s clinical presentation in a review of 38 cases. The most frequent clinical features include changes in mental status, restlessness, myoclonus, hyperreflexia, diaphoresis, shivering, and tremor (Table 1).
The clinical syndrome varies in scope and intensity. Animal models suggest the pathophysiologic mechanism involves brainstem and spinal cord inundation with serotonin, acting on 5-HT1A and 5-HT2A receptors. Recent evidence supports a greater role for 5-HT2A receptors.8
Primary treatment calls for discontinuing the suspected serotonergic agent and instituting supportive measures. Case reports also suggest using serotonin receptor antagonists—such as cyproheptadine, methysergide, chlorpromazine, or propranolol—to clinically manage serotonin syndrome, although empiric support is limited.9
The syndrome often improves within 24 hours of primary treatment, although confusion sometimes last for days and death has been reported.10
NMS—characterized by fever, extrapyramidal rigidity, and disturbances of autonomic function and consciousness—was first described with the use of haloperidol.11 Risk factors include catatonia, disorganized presentation, and dehydration.12 NMS is thought to result from deficient compensatory mechanisms following blockade of dopaminergic regulation of muscle tone and autonomic function.13
Although possibly idiosyncratic, the reaction has been associated with:
- intramuscular, higher total dose, or abruptly increasing doses of antipsychotics14
- withdrawal of dopaminergic agents, such as those used to treat Parkinson’s disease.15
Akin to serotonin syndrome, managing NMS focuses on removing the offending agent(s) and providing supportive care. Severe cases require intensive monitoring, aggressive IV hydration, and respiratory support. Dopaminergics such as bromocriptine16 and skeletal muscle relaxants such as dantrolene17 also have been used to manage NMS.
Unlike serotonin syndrome, NMS often resolves slowly (typically >1 week). NMS’ mortality rate of 11% to 38% appears to be declining in recent years, perhaps because it is being recognized more rapidly.18
Signs and symptoms of NMS vs serotonin syndrome
| NMS | Serotonin syndrome | |
|---|---|---|
| Onset | Insidious, days to weeks | Acute (minutes to hours) |
| Resolution | Slow, often >1 week | Improvement or resolution often within 24 hours |
| Autonomic | Fever, tachycardia, diaphoresis, elevated or labile blood pressure, sialorrhea, tachypnea, incontinence | Diaphoresis, shivering, fever, tachycardia, hypertension, mydriasis |
| Gastrointestinal | Dysphagia, elevated transaminases | Diarrhea, nausea, vomiting, elevated ammonia and transaminases |
| Neuromuscular | Rigidity, bradykinesia, dysarthria, dyskinesias, coarse tremor, ataxia, opisthotonos, oculogyric crisis, rhabdomyolysis | Clonus, myoclonus, hyperreflexia, ataxia, incoordination, rigidity, tremor |
| Psychiatric | Altered mental status, stupor, somnolence, mutism | Altered mental status, agitation, hypomania, hyperactivity, restlessness, somnolence (less common) |
| Other | Leukocytosis, elevated creatine kinase (significant), elevated serum creatinine, proteinuria, renal failure, disseminated intravascular coagulation | Leukocytosis (rarely >20K cells/mm3), elevated creatine kinase (less common), disseminated intravascular coagulation, metabolic acidosis |
| NMS: neuroleptic malignant syndrome | ||
| Note: Classically reported symptoms are italicized | ||
CASE CONTINUED: Fever follows haloperidol
Initial workup. Ms. G has no significant medical or psychiatric history. She has no history of seizures, head trauma, changes in mental status, recent travel, tick bites, or mosquito bites. Family history is relevant only for a maternal aunt with a history of 1 seizure. Ms. G is employed and lives with her husband and son. She is not taking other medications, herbal supplements, or vitamins and does not use tobacco, alcohol, caffeine, or illicit drugs.
On admission, she is somnolent and arousable only to painful stimuli. Temperature is 36.7°C, blood pressure 89/58 mm Hg, heart rate 73 bpm, and respirations 21/minute. She does not talk but is cooperative to physical examination, which is otherwise unremarkable.
Neurologic exam also is unremarkable, with no evidence of meningeal irritation, abnormal reflexes, or muscle tone. Serum ammonia (51 µmol/L; normal range 7 to 42 µmol/L) is slightly elevated. Liver function tests, electrolytes, blood urea nitrogen, creatinine, complete blood counts, urinalysis, urine culture, and blood cultures are unremarkable. Ethanol, salicylate, and acetaminophen levels are negative. Evaluation reveals a positive urine drug screen only for amphetamines, attributed to use of phentermine. Chest radiography and head CT are unremarkable.
Electroencephalography (EEG) 17 hours after admission reveals left anterior temporal spikes suggestive of seizure activity lasting 50 seconds. The patient is described as stuporous but arousable during EEG, and diffuse delta slow waves are superimposed on an alpha rhythm with intermittent diffuse delta bursts. Brain MRI is unremarkable.
Despite no clinical evidence of seizure, Ms. G is transferred to the cardiac telemetry ward to monitor for potential side effects from IV phenytoin loading, at which time (24 hours after admission) she is found to have intermittent sinus tachycardia ≤140 bpm.
Antipsychotic therapy. Thirty hours after admission—after phenytoin loading and normalized EEG—Ms. G shows periodic episodes of sudden startling, with repetitive leg shaking. Continuous ankle clonus is present bilaterally. She complains of severe paresthesias in her legs and is unable to urinate on her own.
NMS signs emerge. Forty-eight hours after admission, Ms. G becomes febrile (38.3°C) and shows tachycardia, with heart rate consistently >130 bpm. Her vital signs did not normalize before the fever developed. She remains somnolent and continues to have spastic lower leg and ankle clonus. She shows no seizure activity on video EEG monitoring during later episodes of repetitive leg shaking, approximately 60 hours after admission.
Ms. G receives empiric vancomycin, ceftriaxone, ampicillin, and acyclovir for possible infectious encephalitis, and lumbar puncture is done emergently. Further laboratory tests reveal creatine kinase (CK) elevation (17,282 U/L, from 270 on admission), leukocytosis (white blood cell count 16.1K/mm3, from 7.2K on admission), and elevated transaminases (AST 199 U/L, up from 21 on admission; ALT 84 U/L, up from 19 on admission).
She is transferred to the ICU with a preliminary diagnosis of NMS. Again, continuous EEG monitoring does not show seizure activity. CSF specimen is negative for infection (negative cultures, negative herpes simplex virus PCR, protein 31 mg/dl, glucose 75 mg/dl). She is started on dantrolene, bromocriptine, and levodopa but shows no initial improvement.
Intubation. On hospital day 8, the patient is intubated to protect her airway and placed in a pentobarbital coma for 2 days, with no improvement. On hospital day 9, cyproheptadine, 24 mg/d, is added for possible serotonin syndrome, and continued for 9 days.
On day 11, the addition of IV diazepam, 10 mg per hour, is followed by gradual improvement in rigidity. Ms. G remains on continuous EEG, with no evidence of seizure activity before diazepam was added or after it is tapered off by day 23.
Discharge. Ms. G is extubated on hospital day 18. On day 23 she can follow commands but is not fully oriented, and levodopa, phenytoin, bromocriptine, and dantrolene are tapered off. She is discharged to a rehabilitation facility, where she again requires phenytoin for a witnessed seizure, attributed to anticonvulsant withdrawal.
On follow-up phone interviews 4 and 18 months after hospitalization, Ms. G says she remains seizure-free without taking anticonvulsants. She reports a subjective, interval improvement in cognitive function, which has since returned to baseline.
Evidence for serotonin syndrome
This case involves a young woman with a several-week history of phentermine use for weight reduction who presented with confusion, sedation, mutism, and nausea. She was initially found to have an abnormal EEG, for which she was loaded with the anticonvulsant phenytoin. However, she continued to exhibit altered mental status, myoclonus, and hyperreflexia along with autonomic dysregulation—such as urinary retention and tachycardia—despite a negative EEG on continuous monitoring.
On retrospective review, we believe she likely was experiencing serotonin toxicity from phentermine. She later developed NMS within several hours of receiving the antipsychotic haloperidol.
Even though phentermine is thought to have a relatively weak serotonergic effect,3 it has been shown to markedly increase serotonin efflux in the rat hypothalamus (to a greater degree than the SSRI fluoxetine).23 Although Ms. G did not report having consumed foods or supplements that could have interfered with phentermine’s metabolism, such use could have contributed to or prolonged a serotonin syndrome.20 Phentermine misuse also cannot be ruled out.
Excess phentermine or concomitant use of other serotonergic agents may have precipitated serotonin syndrome. Ms. G’s hyperactivity a few days before she complained of fatigue and somnolence may represent:
- a sign of phentermine intoxication or overuse
- a harbinger of serotonin syndrome, because these symptoms were followed by overt serotonin syndrome signs such as confusion, disorientation, myoclonus, and autonomic dysfunction.
Evidence for NMS
Ms. G received haloperidol because her agitation obstructed urgent evaluation. After several doses, she rapidly developed signs and symptoms highly consistent with NMS. Onset was rapid compared with the typically described, more insidious NMS evolution of 24 to 72 hours, however.25 Rapid NMS onset may have been precipitated in 2 ways:
- dopaminergic (phentermine) withdrawal combined with dopamine antagonist challenge (haloperidol)25,26
- background serotonin syndrome caused by amphetamine (phentermine) predisposing the patient to develop NMS.27
Although phentermine-induced sympathetic hyperactivity also could have predisposed Ms. G to NMS,31 we think this is unlikely because phentermine was discontinued 3 to 4 days before she developed NMS. Nonetheless, sympathetic hyperactivity secondary to phentermine or serotonin syndrome may increase the risk of developing NMS.
Treatment strategy
Because serotonin syndrome and NMS share many clinical findings, differentiating between the 2 syndromes may be difficult, especially when the patient’s medication history does not implicate a specific agent. A detailed history and physical may help distinguish the syndromes. Clonus may be particularly specific and is important in the diagnosis of serotonin syndrome.32 If you are unable to differentiate between serotonin syndrome and NMS in a patient with this acute neurotoxic abnormal behavior syndrome,33 consider a common treatment strategy (Table 2).19,25
In Ms. G’s case, she probably should not have received bromocriptine for NMS,20 given the potential role of serotonin syndrome in precipitating her symptoms.
Case reports support our hypotheses of an increased predilection for NMS with dopaminergic withdrawal or serotonin syndrome. Growing evidence supports the use of chlorpromazine for serotonin syndrome,34 but consider its use contraindicated in patients with NMS.
Table 2
Serotonin syndrome or NMS?
When in doubt, follow 4 management principles
| Avoid serotonin agonists and dopamine antagonists when a patient presents with features of serotonin syndrome or neuroleptic malignant syndrome (NMS) and the diagnosis is unclear20 |
| Provide supportive care with monitoring, cooling blankets as needed, and hydration |
| Avoid using antipsychotics for agitation, when possible; benzodiazepines may be preferable, although their use in NMS is controversial25 |
| Avoid using bromocriptine, given its contraindication in serotonin syndrome, but consider cyproheptadine for the serotonin syndrome component and dantrolene for skeletal muscle rigidity20 |
Related resources
- Carbone JR. The neuroleptic malignant and serotonin syndromes. Emerg Med Clin North Am 2000;18:317-25.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999;13:100-9.
- Neuroleptic Malignant Syndrome Information Service (NMSIS). www.nmsis.org. NMS hotline for medical professionals (toll-free 888-667-8367) handles calls on NMS, serotonin syndrome, heat stroke, malignant catatonia, and other drug-induced heat-related disorders.
This paper was among those entered in the 2007 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s competition was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
- Acyclovir • Zovirax
- Ampicillin • various
- Bromocriptine • Parlodel
- Ceftriaxone • Rocephin
- Chlorpromazine • Thorazine
- Cyproheptadine • Periactin
- Dantrolene • Dantrium
- Diazepam • Valium
- Dextroamphetamine • Dexedrine
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Levodopa • various
- Methysergide • Sansert
- Pentobarbital • Nembutal
- Phentermine • various
- Phenytoin • Dilantin
- Propranolol • Inderal
- Vancomycin • various
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. G, age 28, presents to a tertiary care hospital with altered mental status. Six weeks ago she started taking phentermine, 37.5 mg/d, to lose weight. Her body mass index is 24 kg/m2 (normal range), and she obtained the stimulant agent via the Internet. Her family reports Ms. G was very busy in the past week, staying up until 2 AM cleaning. They say she also was irritable with her 5-year-old son.
Two days ago, Ms. G complained of fatigue and nausea without emesis. She went to bed early and did not awaken the next morning. Her sister found her in bed, minimally responsive to verbal stimuli, and brought her to the hospital.
Patients have used phentermine as a weight-reducing agent since the FDA approved this amphetamine-like compound in 1960.1 Phentermine’s mechanism of action is thought to involve dopaminergic, noradrenergic, and serotonergic effects.2 Stimulation of norepinephrine (NE) release is its most potent effect, followed by NE reuptake inhibition, stimulation of dopamine (DA) release, DA reuptake inhibition, stimulation of serotonin (5-HT) release, and 5-HT reuptake inhibition (weak).3
Because phentermine could in theory cause serotonin syndrome,4 its use is contraindicated with monoamine oxidase inhibitors (MAOIs) and not recommended with selective serotonin reuptake inhibitors (SSRIs).5 One case report describes an interaction between fluoxetine and phentermine that appears consistent with serotonin syndrome.6 We are aware of no case reports of serotonin syndrome caused by phentermine alone.
This article reports the case of Ms. G, who presented with probable serotonin syndrome associated with phentermine use and subsequently developed a rapid-onset, superimposed neuroleptic malignant syndrome (NMS). We hypothesize that phentermine use may increase NMS risk through adverse drug events and discuss potential pathophysiologic mechanisms and treatment implications.
Serotonin syndrome vs NMS
Serotonin syndrome is an infrequent and potentially life-threatening adverse drug reaction that presumably results from excess serotonin activity (Box 1).7-10 NMS also is an infrequent and potentially life-threatening neurologic emergency (Box 2).11-18 Similarities between disorders of increased serotonergic activity and disorders of low dopaminergic activity (Table 1) suggest both may result from an imbalance between the serotonergic and dopaminergic systems, which have reciprocal relationships in the CNS.19
Differentiating between serotonin syndrome and NMS is further complicated when both antipsychotics and serotonergic agents may be implicated.20 Clinical trials are not feasible because NMS and serotonin syndrome rarely occur.
Sternbach7 first summarized serotonin syndrome’s clinical presentation in a review of 38 cases. The most frequent clinical features include changes in mental status, restlessness, myoclonus, hyperreflexia, diaphoresis, shivering, and tremor (Table 1).
The clinical syndrome varies in scope and intensity. Animal models suggest the pathophysiologic mechanism involves brainstem and spinal cord inundation with serotonin, acting on 5-HT1A and 5-HT2A receptors. Recent evidence supports a greater role for 5-HT2A receptors.8
Primary treatment calls for discontinuing the suspected serotonergic agent and instituting supportive measures. Case reports also suggest using serotonin receptor antagonists—such as cyproheptadine, methysergide, chlorpromazine, or propranolol—to clinically manage serotonin syndrome, although empiric support is limited.9
The syndrome often improves within 24 hours of primary treatment, although confusion sometimes last for days and death has been reported.10
NMS—characterized by fever, extrapyramidal rigidity, and disturbances of autonomic function and consciousness—was first described with the use of haloperidol.11 Risk factors include catatonia, disorganized presentation, and dehydration.12 NMS is thought to result from deficient compensatory mechanisms following blockade of dopaminergic regulation of muscle tone and autonomic function.13
Although possibly idiosyncratic, the reaction has been associated with:
- intramuscular, higher total dose, or abruptly increasing doses of antipsychotics14
- withdrawal of dopaminergic agents, such as those used to treat Parkinson’s disease.15
Akin to serotonin syndrome, managing NMS focuses on removing the offending agent(s) and providing supportive care. Severe cases require intensive monitoring, aggressive IV hydration, and respiratory support. Dopaminergics such as bromocriptine16 and skeletal muscle relaxants such as dantrolene17 also have been used to manage NMS.
Unlike serotonin syndrome, NMS often resolves slowly (typically >1 week). NMS’ mortality rate of 11% to 38% appears to be declining in recent years, perhaps because it is being recognized more rapidly.18
Signs and symptoms of NMS vs serotonin syndrome
| NMS | Serotonin syndrome | |
|---|---|---|
| Onset | Insidious, days to weeks | Acute (minutes to hours) |
| Resolution | Slow, often >1 week | Improvement or resolution often within 24 hours |
| Autonomic | Fever, tachycardia, diaphoresis, elevated or labile blood pressure, sialorrhea, tachypnea, incontinence | Diaphoresis, shivering, fever, tachycardia, hypertension, mydriasis |
| Gastrointestinal | Dysphagia, elevated transaminases | Diarrhea, nausea, vomiting, elevated ammonia and transaminases |
| Neuromuscular | Rigidity, bradykinesia, dysarthria, dyskinesias, coarse tremor, ataxia, opisthotonos, oculogyric crisis, rhabdomyolysis | Clonus, myoclonus, hyperreflexia, ataxia, incoordination, rigidity, tremor |
| Psychiatric | Altered mental status, stupor, somnolence, mutism | Altered mental status, agitation, hypomania, hyperactivity, restlessness, somnolence (less common) |
| Other | Leukocytosis, elevated creatine kinase (significant), elevated serum creatinine, proteinuria, renal failure, disseminated intravascular coagulation | Leukocytosis (rarely >20K cells/mm3), elevated creatine kinase (less common), disseminated intravascular coagulation, metabolic acidosis |
| NMS: neuroleptic malignant syndrome | ||
| Note: Classically reported symptoms are italicized | ||
CASE CONTINUED: Fever follows haloperidol
Initial workup. Ms. G has no significant medical or psychiatric history. She has no history of seizures, head trauma, changes in mental status, recent travel, tick bites, or mosquito bites. Family history is relevant only for a maternal aunt with a history of 1 seizure. Ms. G is employed and lives with her husband and son. She is not taking other medications, herbal supplements, or vitamins and does not use tobacco, alcohol, caffeine, or illicit drugs.
On admission, she is somnolent and arousable only to painful stimuli. Temperature is 36.7°C, blood pressure 89/58 mm Hg, heart rate 73 bpm, and respirations 21/minute. She does not talk but is cooperative to physical examination, which is otherwise unremarkable.
Neurologic exam also is unremarkable, with no evidence of meningeal irritation, abnormal reflexes, or muscle tone. Serum ammonia (51 µmol/L; normal range 7 to 42 µmol/L) is slightly elevated. Liver function tests, electrolytes, blood urea nitrogen, creatinine, complete blood counts, urinalysis, urine culture, and blood cultures are unremarkable. Ethanol, salicylate, and acetaminophen levels are negative. Evaluation reveals a positive urine drug screen only for amphetamines, attributed to use of phentermine. Chest radiography and head CT are unremarkable.
Electroencephalography (EEG) 17 hours after admission reveals left anterior temporal spikes suggestive of seizure activity lasting 50 seconds. The patient is described as stuporous but arousable during EEG, and diffuse delta slow waves are superimposed on an alpha rhythm with intermittent diffuse delta bursts. Brain MRI is unremarkable.
Despite no clinical evidence of seizure, Ms. G is transferred to the cardiac telemetry ward to monitor for potential side effects from IV phenytoin loading, at which time (24 hours after admission) she is found to have intermittent sinus tachycardia ≤140 bpm.
Antipsychotic therapy. Thirty hours after admission—after phenytoin loading and normalized EEG—Ms. G shows periodic episodes of sudden startling, with repetitive leg shaking. Continuous ankle clonus is present bilaterally. She complains of severe paresthesias in her legs and is unable to urinate on her own.
NMS signs emerge. Forty-eight hours after admission, Ms. G becomes febrile (38.3°C) and shows tachycardia, with heart rate consistently >130 bpm. Her vital signs did not normalize before the fever developed. She remains somnolent and continues to have spastic lower leg and ankle clonus. She shows no seizure activity on video EEG monitoring during later episodes of repetitive leg shaking, approximately 60 hours after admission.
Ms. G receives empiric vancomycin, ceftriaxone, ampicillin, and acyclovir for possible infectious encephalitis, and lumbar puncture is done emergently. Further laboratory tests reveal creatine kinase (CK) elevation (17,282 U/L, from 270 on admission), leukocytosis (white blood cell count 16.1K/mm3, from 7.2K on admission), and elevated transaminases (AST 199 U/L, up from 21 on admission; ALT 84 U/L, up from 19 on admission).
She is transferred to the ICU with a preliminary diagnosis of NMS. Again, continuous EEG monitoring does not show seizure activity. CSF specimen is negative for infection (negative cultures, negative herpes simplex virus PCR, protein 31 mg/dl, glucose 75 mg/dl). She is started on dantrolene, bromocriptine, and levodopa but shows no initial improvement.
Intubation. On hospital day 8, the patient is intubated to protect her airway and placed in a pentobarbital coma for 2 days, with no improvement. On hospital day 9, cyproheptadine, 24 mg/d, is added for possible serotonin syndrome, and continued for 9 days.
On day 11, the addition of IV diazepam, 10 mg per hour, is followed by gradual improvement in rigidity. Ms. G remains on continuous EEG, with no evidence of seizure activity before diazepam was added or after it is tapered off by day 23.
Discharge. Ms. G is extubated on hospital day 18. On day 23 she can follow commands but is not fully oriented, and levodopa, phenytoin, bromocriptine, and dantrolene are tapered off. She is discharged to a rehabilitation facility, where she again requires phenytoin for a witnessed seizure, attributed to anticonvulsant withdrawal.
On follow-up phone interviews 4 and 18 months after hospitalization, Ms. G says she remains seizure-free without taking anticonvulsants. She reports a subjective, interval improvement in cognitive function, which has since returned to baseline.
Evidence for serotonin syndrome
This case involves a young woman with a several-week history of phentermine use for weight reduction who presented with confusion, sedation, mutism, and nausea. She was initially found to have an abnormal EEG, for which she was loaded with the anticonvulsant phenytoin. However, she continued to exhibit altered mental status, myoclonus, and hyperreflexia along with autonomic dysregulation—such as urinary retention and tachycardia—despite a negative EEG on continuous monitoring.
On retrospective review, we believe she likely was experiencing serotonin toxicity from phentermine. She later developed NMS within several hours of receiving the antipsychotic haloperidol.
Even though phentermine is thought to have a relatively weak serotonergic effect,3 it has been shown to markedly increase serotonin efflux in the rat hypothalamus (to a greater degree than the SSRI fluoxetine).23 Although Ms. G did not report having consumed foods or supplements that could have interfered with phentermine’s metabolism, such use could have contributed to or prolonged a serotonin syndrome.20 Phentermine misuse also cannot be ruled out.
Excess phentermine or concomitant use of other serotonergic agents may have precipitated serotonin syndrome. Ms. G’s hyperactivity a few days before she complained of fatigue and somnolence may represent:
- a sign of phentermine intoxication or overuse
- a harbinger of serotonin syndrome, because these symptoms were followed by overt serotonin syndrome signs such as confusion, disorientation, myoclonus, and autonomic dysfunction.
Evidence for NMS
Ms. G received haloperidol because her agitation obstructed urgent evaluation. After several doses, she rapidly developed signs and symptoms highly consistent with NMS. Onset was rapid compared with the typically described, more insidious NMS evolution of 24 to 72 hours, however.25 Rapid NMS onset may have been precipitated in 2 ways:
- dopaminergic (phentermine) withdrawal combined with dopamine antagonist challenge (haloperidol)25,26
- background serotonin syndrome caused by amphetamine (phentermine) predisposing the patient to develop NMS.27
Although phentermine-induced sympathetic hyperactivity also could have predisposed Ms. G to NMS,31 we think this is unlikely because phentermine was discontinued 3 to 4 days before she developed NMS. Nonetheless, sympathetic hyperactivity secondary to phentermine or serotonin syndrome may increase the risk of developing NMS.
Treatment strategy
Because serotonin syndrome and NMS share many clinical findings, differentiating between the 2 syndromes may be difficult, especially when the patient’s medication history does not implicate a specific agent. A detailed history and physical may help distinguish the syndromes. Clonus may be particularly specific and is important in the diagnosis of serotonin syndrome.32 If you are unable to differentiate between serotonin syndrome and NMS in a patient with this acute neurotoxic abnormal behavior syndrome,33 consider a common treatment strategy (Table 2).19,25
In Ms. G’s case, she probably should not have received bromocriptine for NMS,20 given the potential role of serotonin syndrome in precipitating her symptoms.
Case reports support our hypotheses of an increased predilection for NMS with dopaminergic withdrawal or serotonin syndrome. Growing evidence supports the use of chlorpromazine for serotonin syndrome,34 but consider its use contraindicated in patients with NMS.
Table 2
Serotonin syndrome or NMS?
When in doubt, follow 4 management principles
| Avoid serotonin agonists and dopamine antagonists when a patient presents with features of serotonin syndrome or neuroleptic malignant syndrome (NMS) and the diagnosis is unclear20 |
| Provide supportive care with monitoring, cooling blankets as needed, and hydration |
| Avoid using antipsychotics for agitation, when possible; benzodiazepines may be preferable, although their use in NMS is controversial25 |
| Avoid using bromocriptine, given its contraindication in serotonin syndrome, but consider cyproheptadine for the serotonin syndrome component and dantrolene for skeletal muscle rigidity20 |
Related resources
- Carbone JR. The neuroleptic malignant and serotonin syndromes. Emerg Med Clin North Am 2000;18:317-25.
- Gillman PK. The serotonin syndrome and its treatment. J Psychopharmacol 1999;13:100-9.
- Neuroleptic Malignant Syndrome Information Service (NMSIS). www.nmsis.org. NMS hotline for medical professionals (toll-free 888-667-8367) handles calls on NMS, serotonin syndrome, heat stroke, malignant catatonia, and other drug-induced heat-related disorders.
This paper was among those entered in the 2007 Promising New Investigators competition sponsored by the Neuroleptic Malignant Syndrome Information Service (NMSIS). The theme of this year’s competition was “New insights on psychotropic drug safety and side effects.”
Current Psychiatry is honored to publish this peer-reviewed, evidence-based article on a clinically important topic for practicing psychiatrists.
- Acyclovir • Zovirax
- Ampicillin • various
- Bromocriptine • Parlodel
- Ceftriaxone • Rocephin
- Chlorpromazine • Thorazine
- Cyproheptadine • Periactin
- Dantrolene • Dantrium
- Diazepam • Valium
- Dextroamphetamine • Dexedrine
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Levodopa • various
- Methysergide • Sansert
- Pentobarbital • Nembutal
- Phentermine • various
- Phenytoin • Dilantin
- Propranolol • Inderal
- Vancomycin • various
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Mosby’s Drug Consult, 16th ed. St. Louis, MO: Mosby; 2006.
2. McEvoy G. AHFS drug information. Bethesda, MD: American Society of Health-System Pharmacists; 2006.
3. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001;39:32-41.
4. Ener RA, Meglathery SB, Van Decker WA, Gallagher RM. Serotonin syndrome and other serotonergic disorders. Pain Med 2003;4:63-74.
5. Phentermine hydrochloride. Physicians’ Desk Reference, 53rd ed. Montvale, NJ: Medical Economics; 1999:3055-6.
6. Bostwick JM, Brown TM. A toxic reaction from combining fluoxetine and phentermine. J Clin Psychopharmacol 1996;16:189-90.
7. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-13.
8. Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001;890:23-31.
9. Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000;79:201-9.
10. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005;352:1112-20.
11. Delay J, Pichot P, Lemperiere T, et al. [A non-phenothiazine and non-reserpine major neuroleptic, haloperidol, in the treatment of psychoses.] Ann Med Psychol (Paris). 1960;118:145-52.
12. Berardi D, Dell’Atti M, Amore M, et al. Clinical risk factors for neuroleptic malignant syndrome. Hum Psychopharmacol 2002;17:99-102.
13. Pearlman CA. Neuroleptic malignant syndrome: a review of the literature. J Clin Psychopharmacol 1986;6:257-73.
14. Viejo LF, Morales V, Punal P, et al. Risk factors in neuroleptic malignant syndrome. A case-control study. Acta Psychiatr Scand 2003;107:45-9.
15. Ikebe S, Harada T, Hashimoto T, et al. Prevention and treatment of malignant syndrome in Parkinson’s disease: a consensus statement of the Malignant Syndrome Research Group. Parkinsonism Relat Disord 2003;9(suppl 1):S47-9.
16. Mueller PS, Vester JW, Fermaglich J. Neuroleptic malignant syndrome. Successful treatment with bromocriptine. JAMA 1983;249:386-8.
17. Coons DJ, Hillman FJ, Marshall RW. Treatment of neuroleptic malignant syndrome with dantrolene sodium: a case report. Am J Psychiatry 1982;139:944-5.
18. Spivak B, Maline DI, Kozyrev VN, et al. Frequency of neuroleptic malignant syndrome in a large psychiatric hospital in Moscow. Eur Psychiatry 2000;15:330-3.
19. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother 1998;32:692-8.
20. Kaufman KR, Levitt MJ, Schiltz JF, Sunderram J. Neuroleptic malignant syndrome and serotonin syndrome in the critical care setting: case analysis. Ann Clin Psychiatry 2006;18:201-4.
21. Spencer DC, Hwang J, Morrell MJ. Fenfluramine-phentermine (Fen-Phen) and seizures: evidence for an association. Epilepsy Behav 2000;1(6):448-52.
22. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin 1997;13(4):763-83.
23. Tao R, Fray A, Aspley S, et al. Effects on serotonin in rat hypothalamus of D-fenfluramine, aminorex, phentermine and fluoxetine. Eur J Pharmacol 2002;445(1-2):69-81.
24. Gillman PK. Serotonin syndrome: history and risk. Fundam Clin Pharmacol 1998;12:482-91.
25. Bhanushali MJ, Tuite PJ. The evaluation and management of patients with neuroleptic malignant syndrome. Neurol Clin 2004;22:389-411.
26. Ebadi M, Pfeiffer RF, Murrin LC. Pathogenesis and treatment of neuroleptic malignant syndrome. Gen Pharmacol 1990;21:367-86.
27. Green AR, Cross AJ, Goodwin GM. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or “Ecstasy”). Psychopharmacology (Berl) 1995;119:247-60.
28. Chayasirisobhon S, Cullis P, Veeramasuneni RR. Occurrence of neuroleptic malignant syndrome in a narcoleptic patient. Hosp Community Psychiatry 1983;34:548-50.
29. Serrano-Dueñas M. Neuroleptic malignant syndrome-like, or—dopaminergic malignant syndrome—due to levodopa therapy withdrawal. Clinical features in 11 patients. Parkinsonism Relat Disord 2003;9:175-8.
30. Kline SS, Mauro LS, Scala-Barnett DM, Zick D. Serotonin syndrome versus neuroleptic malignant syndrome as a cause of death. Clin Pharm 1989;8(7):510-14.
31. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
32. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003;96:635-42.
33. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
34. Gillman PK. Serotonin syndrome treated with chlorpromazine. J Clin Psychopharmacol 1997;17:128-9.
1. Mosby’s Drug Consult, 16th ed. St. Louis, MO: Mosby; 2006.
2. McEvoy G. AHFS drug information. Bethesda, MD: American Society of Health-System Pharmacists; 2006.
3. Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001;39:32-41.
4. Ener RA, Meglathery SB, Van Decker WA, Gallagher RM. Serotonin syndrome and other serotonergic disorders. Pain Med 2003;4:63-74.
5. Phentermine hydrochloride. Physicians’ Desk Reference, 53rd ed. Montvale, NJ: Medical Economics; 1999:3055-6.
6. Bostwick JM, Brown TM. A toxic reaction from combining fluoxetine and phentermine. J Clin Psychopharmacol 1996;16:189-90.
7. Sternbach H. The serotonin syndrome. Am J Psychiatry 1991;148(6):705-13.
8. Nisijima K, Yoshino T, Yui K, Katoh S. Potent serotonin (5-HT)(2A) receptor antagonists completely prevent the development of hyperthermia in an animal model of the 5-HT syndrome. Brain Res 2001;890:23-31.
9. Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome. Presentation of 2 cases and review of the literature. Medicine (Baltimore) 2000;79:201-9.
10. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005;352:1112-20.
11. Delay J, Pichot P, Lemperiere T, et al. [A non-phenothiazine and non-reserpine major neuroleptic, haloperidol, in the treatment of psychoses.] Ann Med Psychol (Paris). 1960;118:145-52.
12. Berardi D, Dell’Atti M, Amore M, et al. Clinical risk factors for neuroleptic malignant syndrome. Hum Psychopharmacol 2002;17:99-102.
13. Pearlman CA. Neuroleptic malignant syndrome: a review of the literature. J Clin Psychopharmacol 1986;6:257-73.
14. Viejo LF, Morales V, Punal P, et al. Risk factors in neuroleptic malignant syndrome. A case-control study. Acta Psychiatr Scand 2003;107:45-9.
15. Ikebe S, Harada T, Hashimoto T, et al. Prevention and treatment of malignant syndrome in Parkinson’s disease: a consensus statement of the Malignant Syndrome Research Group. Parkinsonism Relat Disord 2003;9(suppl 1):S47-9.
16. Mueller PS, Vester JW, Fermaglich J. Neuroleptic malignant syndrome. Successful treatment with bromocriptine. JAMA 1983;249:386-8.
17. Coons DJ, Hillman FJ, Marshall RW. Treatment of neuroleptic malignant syndrome with dantrolene sodium: a case report. Am J Psychiatry 1982;139:944-5.
18. Spivak B, Maline DI, Kozyrev VN, et al. Frequency of neuroleptic malignant syndrome in a large psychiatric hospital in Moscow. Eur Psychiatry 2000;15:330-3.
19. Gerber PE, Lynd LD. Selective serotonin-reuptake inhibitor-induced movement disorders. Ann Pharmacother 1998;32:692-8.
20. Kaufman KR, Levitt MJ, Schiltz JF, Sunderram J. Neuroleptic malignant syndrome and serotonin syndrome in the critical care setting: case analysis. Ann Clin Psychiatry 2006;18:201-4.
21. Spencer DC, Hwang J, Morrell MJ. Fenfluramine-phentermine (Fen-Phen) and seizures: evidence for an association. Epilepsy Behav 2000;1(6):448-52.
22. Mills KC. Serotonin syndrome. A clinical update. Crit Care Clin 1997;13(4):763-83.
23. Tao R, Fray A, Aspley S, et al. Effects on serotonin in rat hypothalamus of D-fenfluramine, aminorex, phentermine and fluoxetine. Eur J Pharmacol 2002;445(1-2):69-81.
24. Gillman PK. Serotonin syndrome: history and risk. Fundam Clin Pharmacol 1998;12:482-91.
25. Bhanushali MJ, Tuite PJ. The evaluation and management of patients with neuroleptic malignant syndrome. Neurol Clin 2004;22:389-411.
26. Ebadi M, Pfeiffer RF, Murrin LC. Pathogenesis and treatment of neuroleptic malignant syndrome. Gen Pharmacol 1990;21:367-86.
27. Green AR, Cross AJ, Goodwin GM. Review of the pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA or “Ecstasy”). Psychopharmacology (Berl) 1995;119:247-60.
28. Chayasirisobhon S, Cullis P, Veeramasuneni RR. Occurrence of neuroleptic malignant syndrome in a narcoleptic patient. Hosp Community Psychiatry 1983;34:548-50.
29. Serrano-Dueñas M. Neuroleptic malignant syndrome-like, or—dopaminergic malignant syndrome—due to levodopa therapy withdrawal. Clinical features in 11 patients. Parkinsonism Relat Disord 2003;9:175-8.
30. Kline SS, Mauro LS, Scala-Barnett DM, Zick D. Serotonin syndrome versus neuroleptic malignant syndrome as a cause of death. Clin Pharm 1989;8(7):510-14.
31. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
32. Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003;96:635-42.
33. Fink M. Toxic serotonin syndrome or neuroleptic malignant syndrome? Pharmacopsychiatry 1996;29:159-61.
34. Gillman PK. Serotonin syndrome treated with chlorpromazine. J Clin Psychopharmacol 1997;17:128-9.
Antipsychotic combinations: Blind step or logical?
In a perfect world, every treatment decision would fall under the protective umbrella of evidence-based medicine. The reality is that up to 30% of schizophrenia patients respond poorly to antipsychotic monotherapy,1 and addressing their chronic debilitating illness requires clinicians to step outside the realm of evidence.
This does not have to be a blind step, however. Guided by logic, you can apply knowledge of receptor binding profiles, adverse effects, and kinetic considerations when choosing antipsychotic polypharmacy. This article offers evidence to answer 2 questions:
- What clinical evidence and/or pharmacologic rationale support using >1 antipsychotic?
- When might it be appropriate to use 2 antipsychotics in patients with treatment-resistant psychosis?
Antipsychotic polypharmacy defined
“Polypharmacy” can carry a negative connotation, but not all forms are bad. In some circumstances, antipsychotic polypharmacy may be necessary to provide optimum benefit and prevent harm to the patient and/or staff.
Short-term polypharmacy often occurs when switching patients from 1 antipsychotic to another. This “crossover phase” is justified to provide a smooth transition between the 2 agents, as abrupt antipsychotic discontinuation may cause a rebound worsening of psychosis. Other short-term antipsychotic polypharmacy strategies may be necessary in inpatient settings, particularly for a patient who is acutely psychotic or aggressive.
The use of a first-generation antipsychotic (FGA) to lead in a second-generation antipsychotic (SGA) is a justifiable treatment strategy. In addition, sedative antipsychotics such as quetiapine often are used during initial treatment of acutely ill patients and subsequently withdrawn.
As our understanding of psychosis’ pathophysiology of improves, more options will come for treatment-resistant cases. Changes in the glutamatergic system, for example, have been implicated in schizophrenia’s pathophysiology.15
Lamotrigine—a second-generation anticonvulsant with antiglutamatergic activity—has been studied as augmentation to antipsychotics in patients with schizophrenia. Several randomized, controlled trials suggested clinical benefit from adjunctive lamotrigine,16-18 but 2 recent multicenter, randomized, double-blind trials failed to support that finding.19
Although not adequately studied, other possible augmentation options may include GABA agonists, COX-2 inhibitors, and selective serotonin reuptake inhibitors.20
Experience-based treatment?
Antipsychotic polypharmacy is prevalent (reported in up to 25% of outpatients and 50% of inpatients with schizophrenia),2-7 costly for patients and insurers,8 and likely to be associated with increased risk of adverse effects and drug-drug interactions. Despite what is known, a wide gap exists between the science and clinical practice of combination antipsychotic therapy in schizophrenia (Table 1).
Clinical trials. The efficacy and safety of antipsychotic combinations in schizophrenia (with options including FGA + FGA, FGA + SGA, and SGA + SGA) has not been studied adequately in well-controlled, systematic trials. Four short-term—6 to 26 weeks—randomized, double-blind, controlled studies9-12 have examined antipsychotic polypharmacy (clozapine + risperidone) in patients with schizophrenia:
- In 3 studies,9,11,12 adding risperidone to clozapine did not significantly improve positive or negative symptoms.
- In all 4 studies, clozapine + risperidone was associated with increased sedation, akathisia, hyperprolactinemia, and elevated fasting blood glucose.
Not all combinations make pharmacologic sense, however, such as adding haloperidol to aripiprazole. Haloperidol’s pharmacologic binding profile (potent D2 blockade) may cancel out any benefits with regard to extrapyramidal symptoms and hyperprolactinemia from aripiprazole’s receptor binding profile (D2 agonist/antagonist). In theory, any displacement of antipsychotic medication from D2 receptors because of competing inhibition may increase risk of symptom exacerbation.
Mortality risk? Two independent, longitudinal cohort studies have found antipsychotic polypharmacy to be a statistically significant predictor of reduced survival.21,22 Although these studies have identified a possible association, additional research is required to determine whether increased mortality in schizophrenia is attributable to the disorder, comorbid medical conditions, antipsychotic medications, or a complex interaction of factors.
Treatment guidelines—such as the Texas Medication Algorithm Project’s updated treatment algorithm for schizophrenia23—reflect the paucity of controlled studies of antipsychotic combinations. The expert consensus panel that developed the TMAP algorithm recommends clozapine augmentation with an FGA or SGA, or electroconvulsive therapy after adequate trials of antipsychotic monotherapy, including clozapine. The panel recommends reserving other antipsychotic combinations as a last-line strategy (see Related Resources).
Table 1
Take-home points about antipsychotic polypharmacy
| Long-term antipsychotic polypharmacy is common, even in schizophrenia patients without treatment-refractory psychosis |
| Controlled clinical trials do not support antipsychotic polypharmacy; many clinicians use this strategy, however, so it may have perceived value |
| Which antipsychotic combinations are best—in terms of efficacy and safety—is unclear |
| Controlled trials of combination antipsychotic therapy are difficult to conduct, which limits the availability of evidence to inform clinical practice |
| Whenever you initiate antipsychotic polypharmacy, document your rationale and the alternatives you considered |
‘Sensible’ pharmacology
Despite the lack of supporting evidence, many clinicians apparently are using antipsychotic polypharmacy for schizophrenia patients with treatment-resistant psychosis. Moreover, reports that up to one-fourth of outpatients and one-half of inpatients may receive antipsychotic polypharmacy2-7 suggest that this approach is not being reserved for treatment-resistant psychosis. Rather, it is being used in non-treatment-refractory schizophrenia patients as well—a practice Stahl labeled a “dirty little secret.”24
Before you consider using antipsychotic polypharmacy for a schizophrenia patient, we suggest that you answer a series of questions to rationalize your decision (Table 2). These questions seem intuitive, but they represent appropriate clinical practice and may support the use of multiple antipsychotics in selected patients.
Which combination? If you determine that a patient is an appropriate candidate for antipsychotic polypharmacy, think about the pharmacologic profiles of available agents. Administering 2 antipsychotics may augment pharmacologic activity, provide an additive effect, or worsen your patient’s symptoms.
Although data from well-controlled studies of clozapine + risperidone do not support its efficacy,9-12 this combination is rational from a pharmacologic perspective. Clozapine shows a lower D2 receptor occupancy (16% to 68%) than that of risperidone (63% to 89%),25 so risperidone’s additional D2 receptor occupancy may enhance a patient’s response to clozapine. Table 3 lists other potentially “sensible” antipsychoticantipsychotic combinations.
Table 2
Questions to consider before initiating antipsychotic polypharmacy
| Ask yourself, ‘Have I… |
|---|
| Determined if my patient is taking the prescribed medication correctly or even at all? |
| Allowed for an adequate trial—dosage and duration—of antipsychotic monotherapy? |
| Maximized the dosage of the current antipsychotic? |
| Tried at least 2 to 3 trials of a first-generation and/or second-generation antipsychotic? |
| Tried an adequate trial of clozapine? |
| Re-evaluated my patient’s diagnosis? |
| Considered tolerability and safety issues associated with adding another antipsychotic? |
| Considered drug-drug interactions that may occur as a result of adding another antipsychotic? |
| Considered nonpharmacologic alternatives, including psychosocial interventions? |
| Augmented with a nonantipsychotic medication, such as valproic acid? |
| Considered my patient’s ability to pay for an additional antipsychotic? |
| Considered whether I can monitor my patient more closely while he/she is on multiple antipsychotics? |
Theoretically beneficial antipsychotic combinations
| Antipsychotic #1 | Antipsychotic #2 | Theoretical pharmacologic benefit | Theoretical safety/tolerability concerns |
|---|---|---|---|
| Clozapine | Olanzapine | Additional D2 receptor occupancy | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Quetiapine | D2 agonist/antagonist in addition to ‘fast on/fast off’ D2 blockade; unique 5HT activity | Sedation |
| Quetiapine | Olanzapine | Differing D2 blockade properties with minimal increase in EPS risk; 2 agents with structural similarity to clozapine | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Loxapine | D2 agonist/antagonist plus a typical antipsychotic that has atypical properties at low doses; 2 agents thought to not potentiate weight gain | Orthostasis, sedation |
| D2: dopamine; 5HT: serotonergic; EPS: extrapyramidal symptoms | |||
Safety/tolerability
Reduced dosages. Combining antipsychotics may allow you to increase treatment efficacy and improve patient tolerability. Lower dosages of 2 antipsychotics may cause fewer side effects than a high dosage of 1 antipsychotic.
For example, case reports and retrospective studies26,27 suggest that adding aripiprazole to clozapine may improve antipsychotic efficacy and reduce metabolic adverse events in treatment-resistant patients. In these cases, clozapine dosages were lower than those usually used in patients with schizophrenia.
Metabolic effects. Carefully weigh the propensity of some antipsychotics to induce weight gain, hyperlipidemia, or glucose dysregulation if you plan to use these agents as part of a polypharmacy regimen. Among SGAs, clozapine and olanzapine are associated with the highest risks of metabolic adverse effects, followed by quetiapine and risperidone. Aripiprazole and ziprasidone are less likely than other SGAs to cause these effects.28
A recent study found a higher incidence of metabolic syndrome in patients receiving antipsychotic polypharmacy. The increased incidence was linked to demographics and clinical risk factors, however, and was not independently associated with the use of multiple antipsychotics.29
Because evidence is scarce and inconclusive, the risk of metabolic adverse events is unknown when antipsychotics are combined. Exercise caution when combining antipsychotics—particularly those known to cause adverse metabolic effects—in case the risk is additive.
Tardive dyskinesia (TD). SGAs are associated with a lower incidence of TD compared with FGAs, but adding an FGA to an SGA may increase the patient’s TD risk.30 Also assess patients regularly (as often as weekly during acute treatment and every 6 to 12 months during maintenance treatment)31 for extrapyramidal symptoms, including akathisia. Administer appropriate rating scales (such as the Abnormal Involuntary Movements Scale [AIMS], Barnes Akathisia Rating Scale [BARS], or Simpson-Angus Rating Scale [SARS]), and treat these adverse events as clinically indicated.
Other adverse effects. The concurrent use of 2 antipsychotics may amplify side effects that are generally considered mild, such as sedation. For example, risperidone and ziprasidone are considered to cause low to moderate sedation. This combination may result in an additive sedative effect that could negatively impact the patient’s psychosocial functioning.
Anticholinergic effects may also be potentiated, especially if a particular combination of antipsychotics warrants anticholinergic medication use for extrapyramidal symptoms.
- Texas Medication Algorithm Project (TMAP). Schizophrenia antipsychotic treatment algorithm. www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1-56.
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Dr. Gibson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. He receives research support from the PhRMA Foundation.
Dr. Patel receives grant support from Takeda Pharmaceuticals and is a consultant for Eli Lilly and Company and Shire Pharmaceuticals.
Dr. Lauriello receives grant support from Eli Lilly and Company and serves as a consultant to Eli Lilly and Company and Vanda Pharmaceuticals.
Dr. Buckley receives grant support from AstraZeneca, National Institute of Mental Health, Pfizer Inc., Solvay, and Wyeth and is a consultant for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen Pharmaceutica, Lundbeck, Pfizer Inc., Solvay, and Wyeth. He receives honoraria from Bristol Myers-Squibb, Janssen Pharmaceutica, Lundbeck, and Pfizer Inc.
1. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
2. Chakos MH, Glick ID, Miller AL, et al. Baseline use of concomitant psychotropic medications to treat schizophrenia in the CATIE trial. Psychiatr Serv 2006;57(8):1094-101.
3. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv 2003;54(8):1086.-
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv 2003;54(1):55-9.
5. Paton C, Lelliott P, Harrington M, et al. Patterns of antipsychotic and anticholinergic prescribing for hospital inpatients. J Psychopharmacol 2003;17(2):223-9.
6. Tempier RP, Pawliuk NH. Conventional, atypical, and combination antipsychotic prescriptions: a 2-year comparison. J Clin Psychiatry 2003;64(6):673-9.
7. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf 2003;12(1):41-8.
8. Valuck RJ, Morrato EH, Dodd S, et al. How expensive is antipsychotic polypharmacy? Experience from five US state Medicaid programs. Curr Med Res Opin 2007;23(10):2567-76.
9. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res 2007;92(1-3):90-4.
10. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
11. Honer WG, Thornton AE, Chen EY, et al. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med 2006;354(5):472-82.
12. Anil Yağcioğlu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
13. Chan J, Sweeting M. Combination therapy with nonclozapine atypical antipsychotic medication: a review of current evidence. J Psychopharmacol 2007;21(6):657-64.
14. Lerner V, Libov I, Kotler M, Strous RD. Combination of “atypical” antipsychotic medication in the management of treatment-resistant schizophrenia and schizoaffective disorder. Prog Neuropsychopharmacol Biol Psychiatry 2004;28(1):89-98.
15. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 2001;158(9):1367-77.
16. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry 2004;56(6):441-6.
17. Tiihonen J, Hallikainen T, Ryynänen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry 2003;54(11):1241-8.
18. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res 2007;93(1-3):109-16.
19. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol 2007;27(6):582-9.
20. Nasrallah HA. Innovative polypharmacy: when dopamine blockade is not enough [editorial]. Current Psychiatry 2007;6(11):17-18.
21. Waddington JL, Youssef HA, Kinsella A. Mortality in schizophrenia. Antipsychotic polypharmacy and absence of adjunctive anticholinergics over the course of a 10-year prospective study. Br J Psychiatry 1998;173:325-9.
22. Joukamaa M, Heliovaara M, Knekt P, et al. Schizophrenia, neuroleptic medication and mortality. Br J Psychiatry 2006;188:122-7.
23. Moore TA, Buchanan RW, Buckley PF, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2006 update. J Clin Psychiatry 2007;68(11):1751-62.
24. Stahl SM. Antipsychotic polypharmacy, part 1: therapeutic option or dirty little secret? J Clin Psychiatry 1999;60(7):425-6.
25. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry 1999;156(2):286-93.
26. Lim S, Pralea C, Schnitt J, et al. Possible increased efficacy of low-dosed clozapine when combined with aripiprazole. J Clin Psychiatry 2004;65(9):1284-5.
27. Karunakaran K, Tungaraza TE, Harborne GC. Is clozapine-aripiprazole combination a useful regimen in the management of treatment-resistant schizophrenia? J Psychopharmacol 2007;21(4):453-6.
28. Haddad PM, Sharma SG. Adverse effects of atypical antipsychotics: differential risk and clinical implications. CNS Drugs 2007;21(11):911-36.
29. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk of metabolic syndrome? Schizophr Res 2007;89(1-3):91-100.
30. Haddad PM, Dursun SM. Neurologic complications of psychiatric drugs: clinical features and management. Hum Psychopharmacol 2008;23(suppl 1):15-26.
31. Marder SR, Essock S, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161(8):1334-49.
In a perfect world, every treatment decision would fall under the protective umbrella of evidence-based medicine. The reality is that up to 30% of schizophrenia patients respond poorly to antipsychotic monotherapy,1 and addressing their chronic debilitating illness requires clinicians to step outside the realm of evidence.
This does not have to be a blind step, however. Guided by logic, you can apply knowledge of receptor binding profiles, adverse effects, and kinetic considerations when choosing antipsychotic polypharmacy. This article offers evidence to answer 2 questions:
- What clinical evidence and/or pharmacologic rationale support using >1 antipsychotic?
- When might it be appropriate to use 2 antipsychotics in patients with treatment-resistant psychosis?
Antipsychotic polypharmacy defined
“Polypharmacy” can carry a negative connotation, but not all forms are bad. In some circumstances, antipsychotic polypharmacy may be necessary to provide optimum benefit and prevent harm to the patient and/or staff.
Short-term polypharmacy often occurs when switching patients from 1 antipsychotic to another. This “crossover phase” is justified to provide a smooth transition between the 2 agents, as abrupt antipsychotic discontinuation may cause a rebound worsening of psychosis. Other short-term antipsychotic polypharmacy strategies may be necessary in inpatient settings, particularly for a patient who is acutely psychotic or aggressive.
The use of a first-generation antipsychotic (FGA) to lead in a second-generation antipsychotic (SGA) is a justifiable treatment strategy. In addition, sedative antipsychotics such as quetiapine often are used during initial treatment of acutely ill patients and subsequently withdrawn.
As our understanding of psychosis’ pathophysiology of improves, more options will come for treatment-resistant cases. Changes in the glutamatergic system, for example, have been implicated in schizophrenia’s pathophysiology.15
Lamotrigine—a second-generation anticonvulsant with antiglutamatergic activity—has been studied as augmentation to antipsychotics in patients with schizophrenia. Several randomized, controlled trials suggested clinical benefit from adjunctive lamotrigine,16-18 but 2 recent multicenter, randomized, double-blind trials failed to support that finding.19
Although not adequately studied, other possible augmentation options may include GABA agonists, COX-2 inhibitors, and selective serotonin reuptake inhibitors.20
Experience-based treatment?
Antipsychotic polypharmacy is prevalent (reported in up to 25% of outpatients and 50% of inpatients with schizophrenia),2-7 costly for patients and insurers,8 and likely to be associated with increased risk of adverse effects and drug-drug interactions. Despite what is known, a wide gap exists between the science and clinical practice of combination antipsychotic therapy in schizophrenia (Table 1).
Clinical trials. The efficacy and safety of antipsychotic combinations in schizophrenia (with options including FGA + FGA, FGA + SGA, and SGA + SGA) has not been studied adequately in well-controlled, systematic trials. Four short-term—6 to 26 weeks—randomized, double-blind, controlled studies9-12 have examined antipsychotic polypharmacy (clozapine + risperidone) in patients with schizophrenia:
- In 3 studies,9,11,12 adding risperidone to clozapine did not significantly improve positive or negative symptoms.
- In all 4 studies, clozapine + risperidone was associated with increased sedation, akathisia, hyperprolactinemia, and elevated fasting blood glucose.
Not all combinations make pharmacologic sense, however, such as adding haloperidol to aripiprazole. Haloperidol’s pharmacologic binding profile (potent D2 blockade) may cancel out any benefits with regard to extrapyramidal symptoms and hyperprolactinemia from aripiprazole’s receptor binding profile (D2 agonist/antagonist). In theory, any displacement of antipsychotic medication from D2 receptors because of competing inhibition may increase risk of symptom exacerbation.
Mortality risk? Two independent, longitudinal cohort studies have found antipsychotic polypharmacy to be a statistically significant predictor of reduced survival.21,22 Although these studies have identified a possible association, additional research is required to determine whether increased mortality in schizophrenia is attributable to the disorder, comorbid medical conditions, antipsychotic medications, or a complex interaction of factors.
Treatment guidelines—such as the Texas Medication Algorithm Project’s updated treatment algorithm for schizophrenia23—reflect the paucity of controlled studies of antipsychotic combinations. The expert consensus panel that developed the TMAP algorithm recommends clozapine augmentation with an FGA or SGA, or electroconvulsive therapy after adequate trials of antipsychotic monotherapy, including clozapine. The panel recommends reserving other antipsychotic combinations as a last-line strategy (see Related Resources).
Table 1
Take-home points about antipsychotic polypharmacy
| Long-term antipsychotic polypharmacy is common, even in schizophrenia patients without treatment-refractory psychosis |
| Controlled clinical trials do not support antipsychotic polypharmacy; many clinicians use this strategy, however, so it may have perceived value |
| Which antipsychotic combinations are best—in terms of efficacy and safety—is unclear |
| Controlled trials of combination antipsychotic therapy are difficult to conduct, which limits the availability of evidence to inform clinical practice |
| Whenever you initiate antipsychotic polypharmacy, document your rationale and the alternatives you considered |
‘Sensible’ pharmacology
Despite the lack of supporting evidence, many clinicians apparently are using antipsychotic polypharmacy for schizophrenia patients with treatment-resistant psychosis. Moreover, reports that up to one-fourth of outpatients and one-half of inpatients may receive antipsychotic polypharmacy2-7 suggest that this approach is not being reserved for treatment-resistant psychosis. Rather, it is being used in non-treatment-refractory schizophrenia patients as well—a practice Stahl labeled a “dirty little secret.”24
Before you consider using antipsychotic polypharmacy for a schizophrenia patient, we suggest that you answer a series of questions to rationalize your decision (Table 2). These questions seem intuitive, but they represent appropriate clinical practice and may support the use of multiple antipsychotics in selected patients.
Which combination? If you determine that a patient is an appropriate candidate for antipsychotic polypharmacy, think about the pharmacologic profiles of available agents. Administering 2 antipsychotics may augment pharmacologic activity, provide an additive effect, or worsen your patient’s symptoms.
Although data from well-controlled studies of clozapine + risperidone do not support its efficacy,9-12 this combination is rational from a pharmacologic perspective. Clozapine shows a lower D2 receptor occupancy (16% to 68%) than that of risperidone (63% to 89%),25 so risperidone’s additional D2 receptor occupancy may enhance a patient’s response to clozapine. Table 3 lists other potentially “sensible” antipsychoticantipsychotic combinations.
Table 2
Questions to consider before initiating antipsychotic polypharmacy
| Ask yourself, ‘Have I… |
|---|
| Determined if my patient is taking the prescribed medication correctly or even at all? |
| Allowed for an adequate trial—dosage and duration—of antipsychotic monotherapy? |
| Maximized the dosage of the current antipsychotic? |
| Tried at least 2 to 3 trials of a first-generation and/or second-generation antipsychotic? |
| Tried an adequate trial of clozapine? |
| Re-evaluated my patient’s diagnosis? |
| Considered tolerability and safety issues associated with adding another antipsychotic? |
| Considered drug-drug interactions that may occur as a result of adding another antipsychotic? |
| Considered nonpharmacologic alternatives, including psychosocial interventions? |
| Augmented with a nonantipsychotic medication, such as valproic acid? |
| Considered my patient’s ability to pay for an additional antipsychotic? |
| Considered whether I can monitor my patient more closely while he/she is on multiple antipsychotics? |
Theoretically beneficial antipsychotic combinations
| Antipsychotic #1 | Antipsychotic #2 | Theoretical pharmacologic benefit | Theoretical safety/tolerability concerns |
|---|---|---|---|
| Clozapine | Olanzapine | Additional D2 receptor occupancy | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Quetiapine | D2 agonist/antagonist in addition to ‘fast on/fast off’ D2 blockade; unique 5HT activity | Sedation |
| Quetiapine | Olanzapine | Differing D2 blockade properties with minimal increase in EPS risk; 2 agents with structural similarity to clozapine | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Loxapine | D2 agonist/antagonist plus a typical antipsychotic that has atypical properties at low doses; 2 agents thought to not potentiate weight gain | Orthostasis, sedation |
| D2: dopamine; 5HT: serotonergic; EPS: extrapyramidal symptoms | |||
Safety/tolerability
Reduced dosages. Combining antipsychotics may allow you to increase treatment efficacy and improve patient tolerability. Lower dosages of 2 antipsychotics may cause fewer side effects than a high dosage of 1 antipsychotic.
For example, case reports and retrospective studies26,27 suggest that adding aripiprazole to clozapine may improve antipsychotic efficacy and reduce metabolic adverse events in treatment-resistant patients. In these cases, clozapine dosages were lower than those usually used in patients with schizophrenia.
Metabolic effects. Carefully weigh the propensity of some antipsychotics to induce weight gain, hyperlipidemia, or glucose dysregulation if you plan to use these agents as part of a polypharmacy regimen. Among SGAs, clozapine and olanzapine are associated with the highest risks of metabolic adverse effects, followed by quetiapine and risperidone. Aripiprazole and ziprasidone are less likely than other SGAs to cause these effects.28
A recent study found a higher incidence of metabolic syndrome in patients receiving antipsychotic polypharmacy. The increased incidence was linked to demographics and clinical risk factors, however, and was not independently associated with the use of multiple antipsychotics.29
Because evidence is scarce and inconclusive, the risk of metabolic adverse events is unknown when antipsychotics are combined. Exercise caution when combining antipsychotics—particularly those known to cause adverse metabolic effects—in case the risk is additive.
Tardive dyskinesia (TD). SGAs are associated with a lower incidence of TD compared with FGAs, but adding an FGA to an SGA may increase the patient’s TD risk.30 Also assess patients regularly (as often as weekly during acute treatment and every 6 to 12 months during maintenance treatment)31 for extrapyramidal symptoms, including akathisia. Administer appropriate rating scales (such as the Abnormal Involuntary Movements Scale [AIMS], Barnes Akathisia Rating Scale [BARS], or Simpson-Angus Rating Scale [SARS]), and treat these adverse events as clinically indicated.
Other adverse effects. The concurrent use of 2 antipsychotics may amplify side effects that are generally considered mild, such as sedation. For example, risperidone and ziprasidone are considered to cause low to moderate sedation. This combination may result in an additive sedative effect that could negatively impact the patient’s psychosocial functioning.
Anticholinergic effects may also be potentiated, especially if a particular combination of antipsychotics warrants anticholinergic medication use for extrapyramidal symptoms.
- Texas Medication Algorithm Project (TMAP). Schizophrenia antipsychotic treatment algorithm. www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1-56.
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Dr. Gibson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. He receives research support from the PhRMA Foundation.
Dr. Patel receives grant support from Takeda Pharmaceuticals and is a consultant for Eli Lilly and Company and Shire Pharmaceuticals.
Dr. Lauriello receives grant support from Eli Lilly and Company and serves as a consultant to Eli Lilly and Company and Vanda Pharmaceuticals.
Dr. Buckley receives grant support from AstraZeneca, National Institute of Mental Health, Pfizer Inc., Solvay, and Wyeth and is a consultant for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen Pharmaceutica, Lundbeck, Pfizer Inc., Solvay, and Wyeth. He receives honoraria from Bristol Myers-Squibb, Janssen Pharmaceutica, Lundbeck, and Pfizer Inc.
In a perfect world, every treatment decision would fall under the protective umbrella of evidence-based medicine. The reality is that up to 30% of schizophrenia patients respond poorly to antipsychotic monotherapy,1 and addressing their chronic debilitating illness requires clinicians to step outside the realm of evidence.
This does not have to be a blind step, however. Guided by logic, you can apply knowledge of receptor binding profiles, adverse effects, and kinetic considerations when choosing antipsychotic polypharmacy. This article offers evidence to answer 2 questions:
- What clinical evidence and/or pharmacologic rationale support using >1 antipsychotic?
- When might it be appropriate to use 2 antipsychotics in patients with treatment-resistant psychosis?
Antipsychotic polypharmacy defined
“Polypharmacy” can carry a negative connotation, but not all forms are bad. In some circumstances, antipsychotic polypharmacy may be necessary to provide optimum benefit and prevent harm to the patient and/or staff.
Short-term polypharmacy often occurs when switching patients from 1 antipsychotic to another. This “crossover phase” is justified to provide a smooth transition between the 2 agents, as abrupt antipsychotic discontinuation may cause a rebound worsening of psychosis. Other short-term antipsychotic polypharmacy strategies may be necessary in inpatient settings, particularly for a patient who is acutely psychotic or aggressive.
The use of a first-generation antipsychotic (FGA) to lead in a second-generation antipsychotic (SGA) is a justifiable treatment strategy. In addition, sedative antipsychotics such as quetiapine often are used during initial treatment of acutely ill patients and subsequently withdrawn.
As our understanding of psychosis’ pathophysiology of improves, more options will come for treatment-resistant cases. Changes in the glutamatergic system, for example, have been implicated in schizophrenia’s pathophysiology.15
Lamotrigine—a second-generation anticonvulsant with antiglutamatergic activity—has been studied as augmentation to antipsychotics in patients with schizophrenia. Several randomized, controlled trials suggested clinical benefit from adjunctive lamotrigine,16-18 but 2 recent multicenter, randomized, double-blind trials failed to support that finding.19
Although not adequately studied, other possible augmentation options may include GABA agonists, COX-2 inhibitors, and selective serotonin reuptake inhibitors.20
Experience-based treatment?
Antipsychotic polypharmacy is prevalent (reported in up to 25% of outpatients and 50% of inpatients with schizophrenia),2-7 costly for patients and insurers,8 and likely to be associated with increased risk of adverse effects and drug-drug interactions. Despite what is known, a wide gap exists between the science and clinical practice of combination antipsychotic therapy in schizophrenia (Table 1).
Clinical trials. The efficacy and safety of antipsychotic combinations in schizophrenia (with options including FGA + FGA, FGA + SGA, and SGA + SGA) has not been studied adequately in well-controlled, systematic trials. Four short-term—6 to 26 weeks—randomized, double-blind, controlled studies9-12 have examined antipsychotic polypharmacy (clozapine + risperidone) in patients with schizophrenia:
- In 3 studies,9,11,12 adding risperidone to clozapine did not significantly improve positive or negative symptoms.
- In all 4 studies, clozapine + risperidone was associated with increased sedation, akathisia, hyperprolactinemia, and elevated fasting blood glucose.
Not all combinations make pharmacologic sense, however, such as adding haloperidol to aripiprazole. Haloperidol’s pharmacologic binding profile (potent D2 blockade) may cancel out any benefits with regard to extrapyramidal symptoms and hyperprolactinemia from aripiprazole’s receptor binding profile (D2 agonist/antagonist). In theory, any displacement of antipsychotic medication from D2 receptors because of competing inhibition may increase risk of symptom exacerbation.
Mortality risk? Two independent, longitudinal cohort studies have found antipsychotic polypharmacy to be a statistically significant predictor of reduced survival.21,22 Although these studies have identified a possible association, additional research is required to determine whether increased mortality in schizophrenia is attributable to the disorder, comorbid medical conditions, antipsychotic medications, or a complex interaction of factors.
Treatment guidelines—such as the Texas Medication Algorithm Project’s updated treatment algorithm for schizophrenia23—reflect the paucity of controlled studies of antipsychotic combinations. The expert consensus panel that developed the TMAP algorithm recommends clozapine augmentation with an FGA or SGA, or electroconvulsive therapy after adequate trials of antipsychotic monotherapy, including clozapine. The panel recommends reserving other antipsychotic combinations as a last-line strategy (see Related Resources).
Table 1
Take-home points about antipsychotic polypharmacy
| Long-term antipsychotic polypharmacy is common, even in schizophrenia patients without treatment-refractory psychosis |
| Controlled clinical trials do not support antipsychotic polypharmacy; many clinicians use this strategy, however, so it may have perceived value |
| Which antipsychotic combinations are best—in terms of efficacy and safety—is unclear |
| Controlled trials of combination antipsychotic therapy are difficult to conduct, which limits the availability of evidence to inform clinical practice |
| Whenever you initiate antipsychotic polypharmacy, document your rationale and the alternatives you considered |
‘Sensible’ pharmacology
Despite the lack of supporting evidence, many clinicians apparently are using antipsychotic polypharmacy for schizophrenia patients with treatment-resistant psychosis. Moreover, reports that up to one-fourth of outpatients and one-half of inpatients may receive antipsychotic polypharmacy2-7 suggest that this approach is not being reserved for treatment-resistant psychosis. Rather, it is being used in non-treatment-refractory schizophrenia patients as well—a practice Stahl labeled a “dirty little secret.”24
Before you consider using antipsychotic polypharmacy for a schizophrenia patient, we suggest that you answer a series of questions to rationalize your decision (Table 2). These questions seem intuitive, but they represent appropriate clinical practice and may support the use of multiple antipsychotics in selected patients.
Which combination? If you determine that a patient is an appropriate candidate for antipsychotic polypharmacy, think about the pharmacologic profiles of available agents. Administering 2 antipsychotics may augment pharmacologic activity, provide an additive effect, or worsen your patient’s symptoms.
Although data from well-controlled studies of clozapine + risperidone do not support its efficacy,9-12 this combination is rational from a pharmacologic perspective. Clozapine shows a lower D2 receptor occupancy (16% to 68%) than that of risperidone (63% to 89%),25 so risperidone’s additional D2 receptor occupancy may enhance a patient’s response to clozapine. Table 3 lists other potentially “sensible” antipsychoticantipsychotic combinations.
Table 2
Questions to consider before initiating antipsychotic polypharmacy
| Ask yourself, ‘Have I… |
|---|
| Determined if my patient is taking the prescribed medication correctly or even at all? |
| Allowed for an adequate trial—dosage and duration—of antipsychotic monotherapy? |
| Maximized the dosage of the current antipsychotic? |
| Tried at least 2 to 3 trials of a first-generation and/or second-generation antipsychotic? |
| Tried an adequate trial of clozapine? |
| Re-evaluated my patient’s diagnosis? |
| Considered tolerability and safety issues associated with adding another antipsychotic? |
| Considered drug-drug interactions that may occur as a result of adding another antipsychotic? |
| Considered nonpharmacologic alternatives, including psychosocial interventions? |
| Augmented with a nonantipsychotic medication, such as valproic acid? |
| Considered my patient’s ability to pay for an additional antipsychotic? |
| Considered whether I can monitor my patient more closely while he/she is on multiple antipsychotics? |
Theoretically beneficial antipsychotic combinations
| Antipsychotic #1 | Antipsychotic #2 | Theoretical pharmacologic benefit | Theoretical safety/tolerability concerns |
|---|---|---|---|
| Clozapine | Olanzapine | Additional D2 receptor occupancy | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Quetiapine | D2 agonist/antagonist in addition to ‘fast on/fast off’ D2 blockade; unique 5HT activity | Sedation |
| Quetiapine | Olanzapine | Differing D2 blockade properties with minimal increase in EPS risk; 2 agents with structural similarity to clozapine | Anticholinergic effects, metabolic adverse events, orthostasis, sedation |
| Aripiprazole | Loxapine | D2 agonist/antagonist plus a typical antipsychotic that has atypical properties at low doses; 2 agents thought to not potentiate weight gain | Orthostasis, sedation |
| D2: dopamine; 5HT: serotonergic; EPS: extrapyramidal symptoms | |||
Safety/tolerability
Reduced dosages. Combining antipsychotics may allow you to increase treatment efficacy and improve patient tolerability. Lower dosages of 2 antipsychotics may cause fewer side effects than a high dosage of 1 antipsychotic.
For example, case reports and retrospective studies26,27 suggest that adding aripiprazole to clozapine may improve antipsychotic efficacy and reduce metabolic adverse events in treatment-resistant patients. In these cases, clozapine dosages were lower than those usually used in patients with schizophrenia.
Metabolic effects. Carefully weigh the propensity of some antipsychotics to induce weight gain, hyperlipidemia, or glucose dysregulation if you plan to use these agents as part of a polypharmacy regimen. Among SGAs, clozapine and olanzapine are associated with the highest risks of metabolic adverse effects, followed by quetiapine and risperidone. Aripiprazole and ziprasidone are less likely than other SGAs to cause these effects.28
A recent study found a higher incidence of metabolic syndrome in patients receiving antipsychotic polypharmacy. The increased incidence was linked to demographics and clinical risk factors, however, and was not independently associated with the use of multiple antipsychotics.29
Because evidence is scarce and inconclusive, the risk of metabolic adverse events is unknown when antipsychotics are combined. Exercise caution when combining antipsychotics—particularly those known to cause adverse metabolic effects—in case the risk is additive.
Tardive dyskinesia (TD). SGAs are associated with a lower incidence of TD compared with FGAs, but adding an FGA to an SGA may increase the patient’s TD risk.30 Also assess patients regularly (as often as weekly during acute treatment and every 6 to 12 months during maintenance treatment)31 for extrapyramidal symptoms, including akathisia. Administer appropriate rating scales (such as the Abnormal Involuntary Movements Scale [AIMS], Barnes Akathisia Rating Scale [BARS], or Simpson-Angus Rating Scale [SARS]), and treat these adverse events as clinically indicated.
Other adverse effects. The concurrent use of 2 antipsychotics may amplify side effects that are generally considered mild, such as sedation. For example, risperidone and ziprasidone are considered to cause low to moderate sedation. This combination may result in an additive sedative effect that could negatively impact the patient’s psychosocial functioning.
Anticholinergic effects may also be potentiated, especially if a particular combination of antipsychotics warrants anticholinergic medication use for extrapyramidal symptoms.
- Texas Medication Algorithm Project (TMAP). Schizophrenia antipsychotic treatment algorithm. www.dshs.state.tx.us/mhprograms/TIMA.shtm.
- Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, 2nd ed. Am J Psychiatry 2004;161(suppl):1-56.
- Aripiprazole • Abilify
- Clozapine • Clozaril
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Loxapine • Loxitane
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Valproic acid • Depakene
- Ziprasidone • Geodon
Dr. Gibson reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. He receives research support from the PhRMA Foundation.
Dr. Patel receives grant support from Takeda Pharmaceuticals and is a consultant for Eli Lilly and Company and Shire Pharmaceuticals.
Dr. Lauriello receives grant support from Eli Lilly and Company and serves as a consultant to Eli Lilly and Company and Vanda Pharmaceuticals.
Dr. Buckley receives grant support from AstraZeneca, National Institute of Mental Health, Pfizer Inc., Solvay, and Wyeth and is a consultant for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen Pharmaceutica, Lundbeck, Pfizer Inc., Solvay, and Wyeth. He receives honoraria from Bristol Myers-Squibb, Janssen Pharmaceutica, Lundbeck, and Pfizer Inc.
1. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
2. Chakos MH, Glick ID, Miller AL, et al. Baseline use of concomitant psychotropic medications to treat schizophrenia in the CATIE trial. Psychiatr Serv 2006;57(8):1094-101.
3. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv 2003;54(8):1086.-
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv 2003;54(1):55-9.
5. Paton C, Lelliott P, Harrington M, et al. Patterns of antipsychotic and anticholinergic prescribing for hospital inpatients. J Psychopharmacol 2003;17(2):223-9.
6. Tempier RP, Pawliuk NH. Conventional, atypical, and combination antipsychotic prescriptions: a 2-year comparison. J Clin Psychiatry 2003;64(6):673-9.
7. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf 2003;12(1):41-8.
8. Valuck RJ, Morrato EH, Dodd S, et al. How expensive is antipsychotic polypharmacy? Experience from five US state Medicaid programs. Curr Med Res Opin 2007;23(10):2567-76.
9. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res 2007;92(1-3):90-4.
10. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
11. Honer WG, Thornton AE, Chen EY, et al. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med 2006;354(5):472-82.
12. Anil Yağcioğlu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
13. Chan J, Sweeting M. Combination therapy with nonclozapine atypical antipsychotic medication: a review of current evidence. J Psychopharmacol 2007;21(6):657-64.
14. Lerner V, Libov I, Kotler M, Strous RD. Combination of “atypical” antipsychotic medication in the management of treatment-resistant schizophrenia and schizoaffective disorder. Prog Neuropsychopharmacol Biol Psychiatry 2004;28(1):89-98.
15. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 2001;158(9):1367-77.
16. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry 2004;56(6):441-6.
17. Tiihonen J, Hallikainen T, Ryynänen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry 2003;54(11):1241-8.
18. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res 2007;93(1-3):109-16.
19. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol 2007;27(6):582-9.
20. Nasrallah HA. Innovative polypharmacy: when dopamine blockade is not enough [editorial]. Current Psychiatry 2007;6(11):17-18.
21. Waddington JL, Youssef HA, Kinsella A. Mortality in schizophrenia. Antipsychotic polypharmacy and absence of adjunctive anticholinergics over the course of a 10-year prospective study. Br J Psychiatry 1998;173:325-9.
22. Joukamaa M, Heliovaara M, Knekt P, et al. Schizophrenia, neuroleptic medication and mortality. Br J Psychiatry 2006;188:122-7.
23. Moore TA, Buchanan RW, Buckley PF, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2006 update. J Clin Psychiatry 2007;68(11):1751-62.
24. Stahl SM. Antipsychotic polypharmacy, part 1: therapeutic option or dirty little secret? J Clin Psychiatry 1999;60(7):425-6.
25. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry 1999;156(2):286-93.
26. Lim S, Pralea C, Schnitt J, et al. Possible increased efficacy of low-dosed clozapine when combined with aripiprazole. J Clin Psychiatry 2004;65(9):1284-5.
27. Karunakaran K, Tungaraza TE, Harborne GC. Is clozapine-aripiprazole combination a useful regimen in the management of treatment-resistant schizophrenia? J Psychopharmacol 2007;21(4):453-6.
28. Haddad PM, Sharma SG. Adverse effects of atypical antipsychotics: differential risk and clinical implications. CNS Drugs 2007;21(11):911-36.
29. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk of metabolic syndrome? Schizophr Res 2007;89(1-3):91-100.
30. Haddad PM, Dursun SM. Neurologic complications of psychiatric drugs: clinical features and management. Hum Psychopharmacol 2008;23(suppl 1):15-26.
31. Marder SR, Essock S, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161(8):1334-49.
1. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
2. Chakos MH, Glick ID, Miller AL, et al. Baseline use of concomitant psychotropic medications to treat schizophrenia in the CATIE trial. Psychiatr Serv 2006;57(8):1094-101.
3. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv 2003;54(8):1086.-
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv 2003;54(1):55-9.
5. Paton C, Lelliott P, Harrington M, et al. Patterns of antipsychotic and anticholinergic prescribing for hospital inpatients. J Psychopharmacol 2003;17(2):223-9.
6. Tempier RP, Pawliuk NH. Conventional, atypical, and combination antipsychotic prescriptions: a 2-year comparison. J Clin Psychiatry 2003;64(6):673-9.
7. Jaffe AB, Levine J. Antipsychotic medication coprescribing in a large state hospital system. Pharmacoepidemiol Drug Saf 2003;12(1):41-8.
8. Valuck RJ, Morrato EH, Dodd S, et al. How expensive is antipsychotic polypharmacy? Experience from five US state Medicaid programs. Curr Med Res Opin 2007;23(10):2567-76.
9. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res 2007;92(1-3):90-4.
10. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 2005;162(1):130-6.
11. Honer WG, Thornton AE, Chen EY, et al. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med 2006;354(5):472-82.
12. Anil Yağcioğlu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry 2005;66(1):63-72.
13. Chan J, Sweeting M. Combination therapy with nonclozapine atypical antipsychotic medication: a review of current evidence. J Psychopharmacol 2007;21(6):657-64.
14. Lerner V, Libov I, Kotler M, Strous RD. Combination of “atypical” antipsychotic medication in the management of treatment-resistant schizophrenia and schizoaffective disorder. Prog Neuropsychopharmacol Biol Psychiatry 2004;28(1):89-98.
15. Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 2001;158(9):1367-77.
16. Kremer I, Vass A, Gorelik I, et al. Placebo-controlled trial of lamotrigine added to conventional and atypical antipsychotics in schizophrenia. Biol Psychiatry 2004;56(6):441-6.
17. Tiihonen J, Hallikainen T, Ryynänen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry 2003;54(11):1241-8.
18. Zoccali R, Muscatello MR, Bruno A, et al. The effect of lamotrigine augmentation of clozapine in a sample of treatment-resistant schizophrenic patients: a double-blind, placebo-controlled study. Schizophr Res 2007;93(1-3):109-16.
19. Goff DC, Keefe R, Citrome L, et al. Lamotrigine as add-on therapy in schizophrenia: results of 2 placebo-controlled trials. J Clin Psychopharmacol 2007;27(6):582-9.
20. Nasrallah HA. Innovative polypharmacy: when dopamine blockade is not enough [editorial]. Current Psychiatry 2007;6(11):17-18.
21. Waddington JL, Youssef HA, Kinsella A. Mortality in schizophrenia. Antipsychotic polypharmacy and absence of adjunctive anticholinergics over the course of a 10-year prospective study. Br J Psychiatry 1998;173:325-9.
22. Joukamaa M, Heliovaara M, Knekt P, et al. Schizophrenia, neuroleptic medication and mortality. Br J Psychiatry 2006;188:122-7.
23. Moore TA, Buchanan RW, Buckley PF, et al. The Texas Medication Algorithm Project antipsychotic algorithm for schizophrenia: 2006 update. J Clin Psychiatry 2007;68(11):1751-62.
24. Stahl SM. Antipsychotic polypharmacy, part 1: therapeutic option or dirty little secret? J Clin Psychiatry 1999;60(7):425-6.
25. Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry 1999;156(2):286-93.
26. Lim S, Pralea C, Schnitt J, et al. Possible increased efficacy of low-dosed clozapine when combined with aripiprazole. J Clin Psychiatry 2004;65(9):1284-5.
27. Karunakaran K, Tungaraza TE, Harborne GC. Is clozapine-aripiprazole combination a useful regimen in the management of treatment-resistant schizophrenia? J Psychopharmacol 2007;21(4):453-6.
28. Haddad PM, Sharma SG. Adverse effects of atypical antipsychotics: differential risk and clinical implications. CNS Drugs 2007;21(11):911-36.
29. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk of metabolic syndrome? Schizophr Res 2007;89(1-3):91-100.
30. Haddad PM, Dursun SM. Neurologic complications of psychiatric drugs: clinical features and management. Hum Psychopharmacol 2008;23(suppl 1):15-26.
31. Marder SR, Essock S, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004;161(8):1334-49.
‘Morning sickness’ in pregnancy loses psychogenic stigma
Nausea and vomiting in pregnancy (NVP) is a misunderstood disorder associated with stress, anxiety, and depression. Prejudice toward women is thought to have guided the historical psychoanalytic concept of NVP as psychogenic, but this view is being replaced by newer biologic theories.
This article examines the evidence for psychological and organic causes of NVP to inform psychiatrists treating pregnant patients. We review guidelines for pharmacologic treatment of NVP and discuss potentially useful psychotherapies and alternative approaches.
Definitive cause unknown
“Morning sickness” affects 50% to 80% of pregnant women, occurring so commonly that NVP is often considered normal.1,2 Approximately 0.5% to 2% of women experience the most severe NVP—hyperemesis gravidarum (HG)3—characterized by intractable vomiting, weight loss, and electrolyte imbalance that can lead to hospitalization.
Without modern supportive care, HG can be lethal; although Charlotte Brontë’s death certificate states she died of “phthisis” (tuberculosis), the author of Jane Eyre is popularly believed to have succumbed to HG.4,5
The search for effective NVP treatments has been disappointing, partly because no cause has been identified. After other conditions that may lead to nausea and vomiting are ruled out (Table 1), NVP medical management is supportive. Correcting dehydration and encouraging dietary and lifestyle changes (Table 2)6 are important adjuncts to step-wise pharmacologic treatment recommended by the American College of Obstetrics and Gynecology (Algorithm).7
Algorithm Pharmacologic treatment of nausea and vomiting in pregnancy*
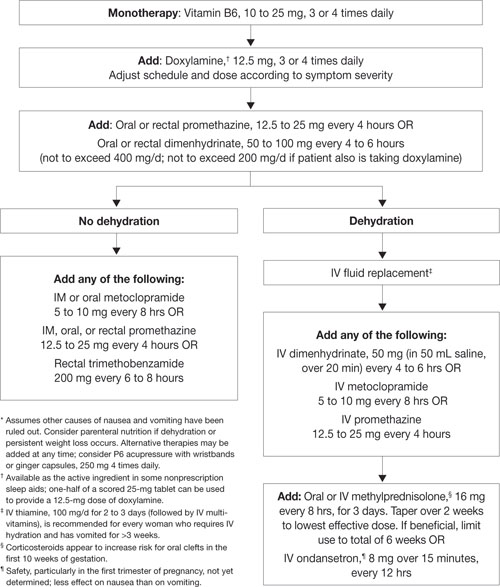
Source: Adapted and reprinted with permission from Canadian Family Physician. Levichek Z, Atanackovic G, Oepkes D, et al. Nausea and vomiting of pregnancy. Evidence-based treatment algorithm. Can Fam Physician 2002;48:267-77Table 1
Medical causes of nausea and vomiting in pregnancy
| Possible cause | How to rule it out |
|---|---|
| Appendicitis | History; do physical, order imaging |
| Hepatitis | Jaundice; order liver function tests, antibody studies, imaging |
| Pancreatitis | History of alcohol use, abdominal pain; check amylase and lipase level |
| Gastrointestinal obstruction | History of surgeries; order imaging |
| Peptic ulcer disease | History; order upper GI series/endoscopy |
| Thyroid disease | Thyroid function tests |
| Urinary tract infection | Urinalysis, culture-sensitivity |
| Trophoblastic disease | Check hCG,* order ultrasound |
| * Elevated human chorionic gonadotropin (hCG) has shown evidence of an association with NVP20 | |
Table 2
Advice for patients: Strategies to manage NVP
| Correct dehydration |
| Drink small amount of fluids frequently |
| Dietary changes |
| Eat frequent, small meals |
| Avoid high-fat foods |
| Snack before getting out of bed and before going to sleep |
| Don’t force yourself to eat |
| Use candy and salty snacks to combat nausea |
| Avoid strong odors and scents; try cold foods, which may have less odor than hot foods |
| Take advantage of good days or good hours of the day for eating |
| Lifestyle changes |
| Get out of bed slowly |
| Lie down when nauseated |
| Avoid stressful situations |
| NVP: nausea and vomiting in pregnancy Source: Reference 6 |
Prejudice vs evidence
Psychological factors. Historically, psychological factors have been blamed for NVP, but support comes from a few poorly designed studies or case reports.3,4
Psychoanalytically, pregnancy and childbirth are significant events in a woman’s life and a rich environment for conflict that could lead to physical expression of symptoms. Freud believed pregnancy and childbirth involve the unconscious substitution of the penis with the child.8 Later writers viewed motherhood as woman’s most powerful wish and the primary organizer of her sexual drive and personality.8
NVP has been considered a conversion or somatization disorder in which symptoms are a “hysterical” expression of unconscious conflict. A psychoanalytic view contends that women who experience NVP are ambivalent about the pregnancy and seek to reject it.1,9 Vomiting, in this view, represents an oral abortion attempt.10 Others claim NVP is a rejection of femininity3 or that symptoms in women with overly attached maternal relationships mask unconscious aggressive feelings toward their mothers.11
Robertson11 proposed an association between NVP and a woman’s view of sexual experiences and her ability to achieve orgasm. He interviewed 100 women and found that 40 of 57 with NVP had “disturbed sexual functioning” or were “frigid” (defined as experiencing coitus as undesirable and unaccompanied by orgasm).
Higgins12 in 1887 proposed that the cause of NVP “is sexual intercourse, the husband too eager for it and the wife too adverse.” Additionally, NVP and HG have been associated with infantile, childish, immature, and hysterical personalities.12-14
Psychiatric comorbidities. Attempts have been made to associate NVP with other psychiatric disorders such as depression, bipolar disorder, schizophrenia, and anxiety disorders, including posttraumatic stress disorder (PTSD). No definitive association has been found between NVP and depressive illness, bipolar disorder, or schizophrenia15 or the use of antidepressants before or during early pregnancy.16 Studies reporting an association with depression have not established a cause-effect relationship.17
Seng18 reported increased NVP risk in women with PTSD. High levels of stress, anxiety, and depression found in women with NVP are thought to result from—rather than cause—NVP’s physical symptoms, however.3,19,20
Psychosocial stressors have been implicated, with higher NVP rates reported in unmarried women, those with unwanted or unplanned pregnancies, immigrants, and those living in crowded situations.21 NVP also is more frequent among women who experience emotionally disturbing events or interpersonal, economic, or occupational difficulties during pregnancy.22 Physical symptoms may provide secondary gain in attention and sympathy and a time-out from stressful home events.14
These psychosocial theories are poorly supported by data, but some clinicians may still believe NVP has a psychogenic cause. Lennane and Lennane23 proposed in 1973 that this perception may result from gender bias because:
- most conditions believed to have psychogenic causes affect women more than men
- the belief that NVP is psychogenic has been perpetuated primarily by male authors.
They argued that sexual prejudice may prevent women from receiving necessary symptomatic treatment and impede research into the cause of NVP.23
Gender bias continues to be found in the diagnosis of women with physical complaints. In 2006, Chiaramonte and Friend24 found strong, consistent gender bias among medical students and residents when evaluating women who reported coronary disease symptoms during stressful life events.
Organic theories
Organic theories view NVP as multifactorial, with contributions from evolution and multiple organ systems. Endocrine, vestibular, gastrointestinal, and CNS contributions have been described, but none have solved NVP’s etiologic mystery.
Evolutionary. NVP may provide an evolutionary advantage by protecting the embryo and mother. This theory states that potential toxins are present in many foods, especially if eaten in large quantities. NVP prevents the pregnant woman from eating very much and harming the embryo. Below-average miscarriage rates are seen in women with NVP.1,2,25
And because a woman’s immune system is depressed during pregnancy, NVP may be advantageous for the mother by limiting her ingestion of potential toxins.25
Endocrine. Human chorionic gonadotropin (hCG), estrogens, progesterone, and leptin, as well as adrenal cortex insufficiencies have been investigated for a role in NVP. Only hCG has shown clear evidence of an association, and some researchers believe it is the most likely cause of NVP.20
NVP rates are higher in pregnancies with elevated hCG. Molar and multiple-gestation pregnancies—each associated with elevated hCG—are complicated more frequently with the severest form of NVP.20,26 Conversely, NVP is less common in women who smoke, which is associated with lower hCG.26
During pregnancy, actions of hCG stimulate the thyroid. Hyperstimulation, leading to transient hyperthyroidism, has been implicated in NVP development.20,26 Symptom severity and the degree of thyroid stimulating hormone (TSH) suppression are closely correlated.26
Elevated hCG levels, hypersensitive TSH receptors, and the presence of a hyperactive hCG isoform have been proposed.20
Gastrointestinal disorders are believed to be involved in the pathogenesis of persistent NVP. Women with NVP usually lack structural or mucosal abnormalities and have normal endoscopic upper GI evaluations. They may, however, have disorders of the stomach’s neuromuscular function. Severe cases of gastric dysrhythmias and abnormalities of gastric tone may lead to gastroparesis.27
Stomach motility in pregnancy is influenced by neurohormonal changes, specifically in estrogen and thyroid hormones. Gastric motility abnormalities—evaluated by electrogastrography (EGG)—have been associated with NVP symptoms and normal EGGs with the absence of symptoms. Some women who had NVP and abnormal EGGs were retested after delivery when symptom-free and found to have normal myoelectric EGG patterns.27
Helicobacter pylori also may be involved in NVP, and at least 1 study found active H pylori infection and HG to be highly correlated. Pregnancy is not believed to predispose to H pylori infection, but active infection compounded by pregnancy’s hormonal changes may exacerbate NVP.28
NVP and motion sickness share many features, suggesting that NVP treatment could be targeted if a vestibular disorder could be discovered.29 Abnormal electroencephalography—particularly generalized slowing—that is not present in asymptomatic pregnant women has been reported in women with NVP.30
CNS contributions. Persistent NVP may be a learned behavior,24 a view based on findings of anticipatory nausea and vomiting in chemotherapy patients. Through conditioning, a pregnant woman may associate her physical symptoms with elements in her life that maintain the cycle of nausea and vomiting.31
Treating psychological symptoms
Brief psychotherapy to identify and correct sources of anxiety in pregnancy may alleviate a patient’s nausea and vomiting.32
- Progressive muscle relaxation training, often combined with guided imagery, can decrease nausea and vomiting associated with chemotherapy and may prevent anticipatory symptoms by decreasing anxiety.
- Systematic desensitization is successful in most chemotherapy patients who try it. In this technique, relaxation is counter-conditioned as a response to stimuli known to elicit symptoms.31
Hypnosis allows patients to achieve a physiologic state incompatible with nausea and vomiting31 and can terminate vomiting after 1 to 3 sessions.3
Medication. Similar to ondansetron, the antidepressant mirtazapine exhibits an antiemetic effect by blocking the 5-HT3 receptor. In treatment-resistant cases, mirtazapine, 30 mg/d, has been reported to ameliorate NVP symptoms, usually within 24 hours. Patients were able to return to normal diets and discontinue treatment after 6 to 10 days. Mirtazapine appears to be safe during pregnancy, based on animal studies using 17 and 20 times the maximum recommended human dose.33
For patients with anxiety symptoms, consider other medications—including selective serotonin reuptake inhibitors and benzodiazepines—only after counseling the patient about potential risks and benefits to her and the fetus.34
Alternative treatments. In traditional Indian medicine, a mixture of powdered ginger and honey is given to women with NVP. At least 2 studies demonstrate ginger’s efficacy.35
In traditional Chinese medicine, stimulating the Neiguan point (P6) on the wrist is believed to relieve nausea and vomiting. Although results are inconclusive, studies suggest that P6 stimulation can help control NVP.36 The FDA has approved wristbands that stimulate the P6 site, either electrically or by acupressure (Figure).36
Figure Wristband to manage nausea and vomiting in pregnancy

The FDA-cleared BioBand acupressure wristband may help control nausea and vomiting in pregnancy by stimulating the Neiguan point (P6) on the wrist.
Source: Reference 36Consider thiamine supplementation for women with severe symptoms, as Wernicke’s encephalopathy is a rare complication of prolonged NVP.19,37
Related resources
- Motherisk Program at The Hospital for Sick Children, Toronto, Ontario, Canada. Website with information on vomiting during pregnancy: www.motherisk.org/women/morningSickness.jsp. Nausea and vomiting of pregnancy (NVP) forum. www.motherisk.org/women/forum.jsp.
- BioBand acupuncture wristband. www.BioBands.com.
- Koren G, Bishai R, eds. Nausea and vomiting of pregnancy: state of the art 2000. Toronto, Ontario, Canada: The Motherisk Program; 2000.
Drug brand names
- Dimenhydrinate • Dramamine
- Doxylamine • Unisom
- Methylprednisolone • Medrol
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Ondansetron • Zofran
- Promethazine • Phenergan
- Trimethobenzamide • Tigan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis 1990;178:655-9.
2. Davis M. Nausea and vomiting of pregnancy: an evidence-based review. J Perinat Neonatal Nurs 2004;18:312-28.
3. Buckwalter JG, Simpson SW. Psychological factors in the etiology and treatment of severe nausea and vomiting in pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S210-4.
4. Bogen JT. Neurosis: a Ms-diagnosis. Perspect Biol Med 1994;37(2):263-74.
5. Weiss G. The death of Charlotte Brontë. Obstet Gynecol 1991;78(4):705-8.
6. Lester EP, Notman MT. Pregnancy, developmental crisis and object relations: psychoanalytic considerations. Int J Psychoanal 1986;67(pt 3):357-66.
7. Sheehan P. Hyperemesis gravidarum—assessment and management. Aust Fam Physician 2007;36:698-701.
8. American College of Obstetrics and Gynecology. ACOG practice bulletin: nausea and vomiting of pregnancy. Obstet Gynecol 2004;103:803-14.
9. Iancu I, Kotler M, Spivak B, et al. Psychiatric aspects of hyperemesis gravidarum. Psychother Psychosom 1994;61:143-9.
10. Munch S. Chicken or the egg? The biological-psychological controversy surrounding hyperemesis gravidarum. Soc Sci Med 2002;55:1267-78.
11. Robertson GG. Nausea and vomiting of pregnancy: a study in psychosomatic and social medicine. Lancet 1946;336-45.
12. Fairweather DV. Nausea and vomiting in pregnancy. Am J Obstet Gynecol 1968;102:135-75.
13. Katon WJ, Ries RK, Bokan JA, Kleinman A. Hyperemesis gravidarum: a biopsychosocial perspective. Int J Psychiatry Med 1980;10:151-62.
14. Simpson SW, Goodwin TM, Robins SB, et al. Psychological factors and hyperemesis gravidarum. J Womens Health Gend Based Med 2001;10:471-7.
15. Majerus PW, Guze SB, Delong WB, Robins E. Psychologic factors and psychiatric disease in hyperemesis gravidarum: a follow-up study of 69 vomiters and 66 controls. Am J Psychiatry 1960;117:421-8.
16. Bozzo P, Koren G, Nava-Ocampo AA, Einarson A. The incidence of nausea and vomiting of pregnancy (NVP): a comparison between depressed women treated with antidepressants and non-depressed women. Clin Invest Med 2006;29(6):347-50.
17. Markl GE, Strunz-Lehner C, Egen-Lappe V, et al. The association of psychosocial factors with nausea and vomiting during pregnancy. J Psychosom Obstet Gynaecol 2007;1-6.
18. Seng JS, Oakley DJ, Sampselle CM, et al. Posttraumatic stress disorder and pregnancy complications. Obstet Gynecol 2001;97(1):17-22.
19. Ismail SK, Kenny L. Review on hyperemesis gravidarum. Best Pract Res Clin Gastroenterol 2007;21:755-69.
20. Verberg MF, Gillott DJ, Al-Fardan N, Grudzinskas JG. Hyperemesis gravidarum, a literature review. Hum Reprod Update 2005;11:527-39.
21. Deuchar N. Nausea and vomiting in pregnancy: a review of the problem with particular regard to psychological and social aspects. Br J Obstet Gynaecol 1995;102(1):6-8.
22. Iatrakis GM, Sakellaropoulos GG, Kourkoubas AH, Kabounia SE. Vomiting and nausea in the first 12 weeks of pregnancy. Psychother Psychosom 1988;49(1):22-4.
23. Lennane KJ, Lennane RJ. Alleged psychogenic disorders in women—a possible manifestation of sexual prejudice. N Engl J Med 1973;288(6):288-92.
24. Chiaramonte GR, Friend R. Medical students’ and residents’ gender bias in the diagnosis, treatment, and interpretation of coronary heart disease symptoms. Health Psychol 2006;25:255-66.
25. Sherman PW, Flaxman SM. Nausea and vomiting of pregnancy in an evolutionary perspective. Am J Obstet Gynecol 2002;186(5 suppl):S190-7.
26. Goodwin TM. Nausea and vomiting of pregnancy: an obstetric syndrome. Am J Obstet Gynecol 2002;186(5 suppl):S184-9.
27. Koch KL. Gastrointestinal factors in nausea and vomiting of pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S198-203.
28. Golberg D, Szilagyi A, Graves L. Hyperemesis gravidarum and Helicobacter pylori infection: a systematic review. Obstet Gynecol 2007;110:695-703.
29. Black FO. Maternal susceptibility to nausea and vomiting of pregnancy: is the vestibular system involved? Am J Obstet Gynecol 2002;186(5 suppl):S204-9.
30. Vaknin Z, Halperin R, Schneider D, et al. Hyperemesis gravidarum and nonspecific abnormal EEG findings: a preliminary report. J Reprod Med 2006;51:623-7.
31. Matteson S, Roscoe J, Hickok J, Morrow GR. The role of behavioral conditioning in the development of nausea. Am J Obstet Gynecol 2002;186(5 suppl):S239-43.
32. Zechnich R, Hammer T. Brief psychotherapy for hyperemesis gravidarum. Am Fam Physician 1982;26:179-81.
33. Guclu S, Gol M, Dogan E, Saygili U. Mirtazapine use in resistant hyperemesis gravidarum: report of three cases and review of the literature. Arch Gynecol Obstet 2005;272:298-300.
34. Raphael DB, Ross J, Brizendine L. Treating anxiety during pregnancy: Just how safe are SSRIs? Current Psychiatry 2008;7(2):39-52.
35. Niebyl JR, Goodwin TM. Overview of nausea and vomiting of pregnancy with an emphasis on vitamins and ginger. Am J Obstet Gynecol 2002;186(5 suppl):S253-5.
36. Roscoe JA, Matteson SE. Acupressure and acustimulation bands for control of nausea: a brief review. Am J Obstet Gynecol 2002;186(5 suppl):S244-7.
37. Chiossi G, Neri I, Cavazzuti M, et al. Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet Gynecol Surv 2006;61:255-68.
Nausea and vomiting in pregnancy (NVP) is a misunderstood disorder associated with stress, anxiety, and depression. Prejudice toward women is thought to have guided the historical psychoanalytic concept of NVP as psychogenic, but this view is being replaced by newer biologic theories.
This article examines the evidence for psychological and organic causes of NVP to inform psychiatrists treating pregnant patients. We review guidelines for pharmacologic treatment of NVP and discuss potentially useful psychotherapies and alternative approaches.
Definitive cause unknown
“Morning sickness” affects 50% to 80% of pregnant women, occurring so commonly that NVP is often considered normal.1,2 Approximately 0.5% to 2% of women experience the most severe NVP—hyperemesis gravidarum (HG)3—characterized by intractable vomiting, weight loss, and electrolyte imbalance that can lead to hospitalization.
Without modern supportive care, HG can be lethal; although Charlotte Brontë’s death certificate states she died of “phthisis” (tuberculosis), the author of Jane Eyre is popularly believed to have succumbed to HG.4,5
The search for effective NVP treatments has been disappointing, partly because no cause has been identified. After other conditions that may lead to nausea and vomiting are ruled out (Table 1), NVP medical management is supportive. Correcting dehydration and encouraging dietary and lifestyle changes (Table 2)6 are important adjuncts to step-wise pharmacologic treatment recommended by the American College of Obstetrics and Gynecology (Algorithm).7
Algorithm Pharmacologic treatment of nausea and vomiting in pregnancy*
Source: Adapted and reprinted with permission from Canadian Family Physician. Levichek Z, Atanackovic G, Oepkes D, et al. Nausea and vomiting of pregnancy. Evidence-based treatment algorithm. Can Fam Physician 2002;48:267-77Table 1
Medical causes of nausea and vomiting in pregnancy
| Possible cause | How to rule it out |
|---|---|
| Appendicitis | History; do physical, order imaging |
| Hepatitis | Jaundice; order liver function tests, antibody studies, imaging |
| Pancreatitis | History of alcohol use, abdominal pain; check amylase and lipase level |
| Gastrointestinal obstruction | History of surgeries; order imaging |
| Peptic ulcer disease | History; order upper GI series/endoscopy |
| Thyroid disease | Thyroid function tests |
| Urinary tract infection | Urinalysis, culture-sensitivity |
| Trophoblastic disease | Check hCG,* order ultrasound |
| * Elevated human chorionic gonadotropin (hCG) has shown evidence of an association with NVP20 | |
Table 2
Advice for patients: Strategies to manage NVP
| Correct dehydration |
| Drink small amount of fluids frequently |
| Dietary changes |
| Eat frequent, small meals |
| Avoid high-fat foods |
| Snack before getting out of bed and before going to sleep |
| Don’t force yourself to eat |
| Use candy and salty snacks to combat nausea |
| Avoid strong odors and scents; try cold foods, which may have less odor than hot foods |
| Take advantage of good days or good hours of the day for eating |
| Lifestyle changes |
| Get out of bed slowly |
| Lie down when nauseated |
| Avoid stressful situations |
| NVP: nausea and vomiting in pregnancy Source: Reference 6 |
Prejudice vs evidence
Psychological factors. Historically, psychological factors have been blamed for NVP, but support comes from a few poorly designed studies or case reports.3,4
Psychoanalytically, pregnancy and childbirth are significant events in a woman’s life and a rich environment for conflict that could lead to physical expression of symptoms. Freud believed pregnancy and childbirth involve the unconscious substitution of the penis with the child.8 Later writers viewed motherhood as woman’s most powerful wish and the primary organizer of her sexual drive and personality.8
NVP has been considered a conversion or somatization disorder in which symptoms are a “hysterical” expression of unconscious conflict. A psychoanalytic view contends that women who experience NVP are ambivalent about the pregnancy and seek to reject it.1,9 Vomiting, in this view, represents an oral abortion attempt.10 Others claim NVP is a rejection of femininity3 or that symptoms in women with overly attached maternal relationships mask unconscious aggressive feelings toward their mothers.11
Robertson11 proposed an association between NVP and a woman’s view of sexual experiences and her ability to achieve orgasm. He interviewed 100 women and found that 40 of 57 with NVP had “disturbed sexual functioning” or were “frigid” (defined as experiencing coitus as undesirable and unaccompanied by orgasm).
Higgins12 in 1887 proposed that the cause of NVP “is sexual intercourse, the husband too eager for it and the wife too adverse.” Additionally, NVP and HG have been associated with infantile, childish, immature, and hysterical personalities.12-14
Psychiatric comorbidities. Attempts have been made to associate NVP with other psychiatric disorders such as depression, bipolar disorder, schizophrenia, and anxiety disorders, including posttraumatic stress disorder (PTSD). No definitive association has been found between NVP and depressive illness, bipolar disorder, or schizophrenia15 or the use of antidepressants before or during early pregnancy.16 Studies reporting an association with depression have not established a cause-effect relationship.17
Seng18 reported increased NVP risk in women with PTSD. High levels of stress, anxiety, and depression found in women with NVP are thought to result from—rather than cause—NVP’s physical symptoms, however.3,19,20
Psychosocial stressors have been implicated, with higher NVP rates reported in unmarried women, those with unwanted or unplanned pregnancies, immigrants, and those living in crowded situations.21 NVP also is more frequent among women who experience emotionally disturbing events or interpersonal, economic, or occupational difficulties during pregnancy.22 Physical symptoms may provide secondary gain in attention and sympathy and a time-out from stressful home events.14
These psychosocial theories are poorly supported by data, but some clinicians may still believe NVP has a psychogenic cause. Lennane and Lennane23 proposed in 1973 that this perception may result from gender bias because:
- most conditions believed to have psychogenic causes affect women more than men
- the belief that NVP is psychogenic has been perpetuated primarily by male authors.
They argued that sexual prejudice may prevent women from receiving necessary symptomatic treatment and impede research into the cause of NVP.23
Gender bias continues to be found in the diagnosis of women with physical complaints. In 2006, Chiaramonte and Friend24 found strong, consistent gender bias among medical students and residents when evaluating women who reported coronary disease symptoms during stressful life events.
Organic theories
Organic theories view NVP as multifactorial, with contributions from evolution and multiple organ systems. Endocrine, vestibular, gastrointestinal, and CNS contributions have been described, but none have solved NVP’s etiologic mystery.
Evolutionary. NVP may provide an evolutionary advantage by protecting the embryo and mother. This theory states that potential toxins are present in many foods, especially if eaten in large quantities. NVP prevents the pregnant woman from eating very much and harming the embryo. Below-average miscarriage rates are seen in women with NVP.1,2,25
And because a woman’s immune system is depressed during pregnancy, NVP may be advantageous for the mother by limiting her ingestion of potential toxins.25
Endocrine. Human chorionic gonadotropin (hCG), estrogens, progesterone, and leptin, as well as adrenal cortex insufficiencies have been investigated for a role in NVP. Only hCG has shown clear evidence of an association, and some researchers believe it is the most likely cause of NVP.20
NVP rates are higher in pregnancies with elevated hCG. Molar and multiple-gestation pregnancies—each associated with elevated hCG—are complicated more frequently with the severest form of NVP.20,26 Conversely, NVP is less common in women who smoke, which is associated with lower hCG.26
During pregnancy, actions of hCG stimulate the thyroid. Hyperstimulation, leading to transient hyperthyroidism, has been implicated in NVP development.20,26 Symptom severity and the degree of thyroid stimulating hormone (TSH) suppression are closely correlated.26
Elevated hCG levels, hypersensitive TSH receptors, and the presence of a hyperactive hCG isoform have been proposed.20
Gastrointestinal disorders are believed to be involved in the pathogenesis of persistent NVP. Women with NVP usually lack structural or mucosal abnormalities and have normal endoscopic upper GI evaluations. They may, however, have disorders of the stomach’s neuromuscular function. Severe cases of gastric dysrhythmias and abnormalities of gastric tone may lead to gastroparesis.27
Stomach motility in pregnancy is influenced by neurohormonal changes, specifically in estrogen and thyroid hormones. Gastric motility abnormalities—evaluated by electrogastrography (EGG)—have been associated with NVP symptoms and normal EGGs with the absence of symptoms. Some women who had NVP and abnormal EGGs were retested after delivery when symptom-free and found to have normal myoelectric EGG patterns.27
Helicobacter pylori also may be involved in NVP, and at least 1 study found active H pylori infection and HG to be highly correlated. Pregnancy is not believed to predispose to H pylori infection, but active infection compounded by pregnancy’s hormonal changes may exacerbate NVP.28
NVP and motion sickness share many features, suggesting that NVP treatment could be targeted if a vestibular disorder could be discovered.29 Abnormal electroencephalography—particularly generalized slowing—that is not present in asymptomatic pregnant women has been reported in women with NVP.30
CNS contributions. Persistent NVP may be a learned behavior,24 a view based on findings of anticipatory nausea and vomiting in chemotherapy patients. Through conditioning, a pregnant woman may associate her physical symptoms with elements in her life that maintain the cycle of nausea and vomiting.31
Treating psychological symptoms
Brief psychotherapy to identify and correct sources of anxiety in pregnancy may alleviate a patient’s nausea and vomiting.32
- Progressive muscle relaxation training, often combined with guided imagery, can decrease nausea and vomiting associated with chemotherapy and may prevent anticipatory symptoms by decreasing anxiety.
- Systematic desensitization is successful in most chemotherapy patients who try it. In this technique, relaxation is counter-conditioned as a response to stimuli known to elicit symptoms.31
Hypnosis allows patients to achieve a physiologic state incompatible with nausea and vomiting31 and can terminate vomiting after 1 to 3 sessions.3
Medication. Similar to ondansetron, the antidepressant mirtazapine exhibits an antiemetic effect by blocking the 5-HT3 receptor. In treatment-resistant cases, mirtazapine, 30 mg/d, has been reported to ameliorate NVP symptoms, usually within 24 hours. Patients were able to return to normal diets and discontinue treatment after 6 to 10 days. Mirtazapine appears to be safe during pregnancy, based on animal studies using 17 and 20 times the maximum recommended human dose.33
For patients with anxiety symptoms, consider other medications—including selective serotonin reuptake inhibitors and benzodiazepines—only after counseling the patient about potential risks and benefits to her and the fetus.34
Alternative treatments. In traditional Indian medicine, a mixture of powdered ginger and honey is given to women with NVP. At least 2 studies demonstrate ginger’s efficacy.35
In traditional Chinese medicine, stimulating the Neiguan point (P6) on the wrist is believed to relieve nausea and vomiting. Although results are inconclusive, studies suggest that P6 stimulation can help control NVP.36 The FDA has approved wristbands that stimulate the P6 site, either electrically or by acupressure (Figure).36
Figure Wristband to manage nausea and vomiting in pregnancy
The FDA-cleared BioBand acupressure wristband may help control nausea and vomiting in pregnancy by stimulating the Neiguan point (P6) on the wrist.
Source: Reference 36Consider thiamine supplementation for women with severe symptoms, as Wernicke’s encephalopathy is a rare complication of prolonged NVP.19,37
Related resources
- Motherisk Program at The Hospital for Sick Children, Toronto, Ontario, Canada. Website with information on vomiting during pregnancy: www.motherisk.org/women/morningSickness.jsp. Nausea and vomiting of pregnancy (NVP) forum. www.motherisk.org/women/forum.jsp.
- BioBand acupuncture wristband. www.BioBands.com.
- Koren G, Bishai R, eds. Nausea and vomiting of pregnancy: state of the art 2000. Toronto, Ontario, Canada: The Motherisk Program; 2000.
Drug brand names
- Dimenhydrinate • Dramamine
- Doxylamine • Unisom
- Methylprednisolone • Medrol
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Ondansetron • Zofran
- Promethazine • Phenergan
- Trimethobenzamide • Tigan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Nausea and vomiting in pregnancy (NVP) is a misunderstood disorder associated with stress, anxiety, and depression. Prejudice toward women is thought to have guided the historical psychoanalytic concept of NVP as psychogenic, but this view is being replaced by newer biologic theories.
This article examines the evidence for psychological and organic causes of NVP to inform psychiatrists treating pregnant patients. We review guidelines for pharmacologic treatment of NVP and discuss potentially useful psychotherapies and alternative approaches.
Definitive cause unknown
“Morning sickness” affects 50% to 80% of pregnant women, occurring so commonly that NVP is often considered normal.1,2 Approximately 0.5% to 2% of women experience the most severe NVP—hyperemesis gravidarum (HG)3—characterized by intractable vomiting, weight loss, and electrolyte imbalance that can lead to hospitalization.
Without modern supportive care, HG can be lethal; although Charlotte Brontë’s death certificate states she died of “phthisis” (tuberculosis), the author of Jane Eyre is popularly believed to have succumbed to HG.4,5
The search for effective NVP treatments has been disappointing, partly because no cause has been identified. After other conditions that may lead to nausea and vomiting are ruled out (Table 1), NVP medical management is supportive. Correcting dehydration and encouraging dietary and lifestyle changes (Table 2)6 are important adjuncts to step-wise pharmacologic treatment recommended by the American College of Obstetrics and Gynecology (Algorithm).7
Algorithm Pharmacologic treatment of nausea and vomiting in pregnancy*
Source: Adapted and reprinted with permission from Canadian Family Physician. Levichek Z, Atanackovic G, Oepkes D, et al. Nausea and vomiting of pregnancy. Evidence-based treatment algorithm. Can Fam Physician 2002;48:267-77Table 1
Medical causes of nausea and vomiting in pregnancy
| Possible cause | How to rule it out |
|---|---|
| Appendicitis | History; do physical, order imaging |
| Hepatitis | Jaundice; order liver function tests, antibody studies, imaging |
| Pancreatitis | History of alcohol use, abdominal pain; check amylase and lipase level |
| Gastrointestinal obstruction | History of surgeries; order imaging |
| Peptic ulcer disease | History; order upper GI series/endoscopy |
| Thyroid disease | Thyroid function tests |
| Urinary tract infection | Urinalysis, culture-sensitivity |
| Trophoblastic disease | Check hCG,* order ultrasound |
| * Elevated human chorionic gonadotropin (hCG) has shown evidence of an association with NVP20 | |
Table 2
Advice for patients: Strategies to manage NVP
| Correct dehydration |
| Drink small amount of fluids frequently |
| Dietary changes |
| Eat frequent, small meals |
| Avoid high-fat foods |
| Snack before getting out of bed and before going to sleep |
| Don’t force yourself to eat |
| Use candy and salty snacks to combat nausea |
| Avoid strong odors and scents; try cold foods, which may have less odor than hot foods |
| Take advantage of good days or good hours of the day for eating |
| Lifestyle changes |
| Get out of bed slowly |
| Lie down when nauseated |
| Avoid stressful situations |
| NVP: nausea and vomiting in pregnancy Source: Reference 6 |
Prejudice vs evidence
Psychological factors. Historically, psychological factors have been blamed for NVP, but support comes from a few poorly designed studies or case reports.3,4
Psychoanalytically, pregnancy and childbirth are significant events in a woman’s life and a rich environment for conflict that could lead to physical expression of symptoms. Freud believed pregnancy and childbirth involve the unconscious substitution of the penis with the child.8 Later writers viewed motherhood as woman’s most powerful wish and the primary organizer of her sexual drive and personality.8
NVP has been considered a conversion or somatization disorder in which symptoms are a “hysterical” expression of unconscious conflict. A psychoanalytic view contends that women who experience NVP are ambivalent about the pregnancy and seek to reject it.1,9 Vomiting, in this view, represents an oral abortion attempt.10 Others claim NVP is a rejection of femininity3 or that symptoms in women with overly attached maternal relationships mask unconscious aggressive feelings toward their mothers.11
Robertson11 proposed an association between NVP and a woman’s view of sexual experiences and her ability to achieve orgasm. He interviewed 100 women and found that 40 of 57 with NVP had “disturbed sexual functioning” or were “frigid” (defined as experiencing coitus as undesirable and unaccompanied by orgasm).
Higgins12 in 1887 proposed that the cause of NVP “is sexual intercourse, the husband too eager for it and the wife too adverse.” Additionally, NVP and HG have been associated with infantile, childish, immature, and hysterical personalities.12-14
Psychiatric comorbidities. Attempts have been made to associate NVP with other psychiatric disorders such as depression, bipolar disorder, schizophrenia, and anxiety disorders, including posttraumatic stress disorder (PTSD). No definitive association has been found between NVP and depressive illness, bipolar disorder, or schizophrenia15 or the use of antidepressants before or during early pregnancy.16 Studies reporting an association with depression have not established a cause-effect relationship.17
Seng18 reported increased NVP risk in women with PTSD. High levels of stress, anxiety, and depression found in women with NVP are thought to result from—rather than cause—NVP’s physical symptoms, however.3,19,20
Psychosocial stressors have been implicated, with higher NVP rates reported in unmarried women, those with unwanted or unplanned pregnancies, immigrants, and those living in crowded situations.21 NVP also is more frequent among women who experience emotionally disturbing events or interpersonal, economic, or occupational difficulties during pregnancy.22 Physical symptoms may provide secondary gain in attention and sympathy and a time-out from stressful home events.14
These psychosocial theories are poorly supported by data, but some clinicians may still believe NVP has a psychogenic cause. Lennane and Lennane23 proposed in 1973 that this perception may result from gender bias because:
- most conditions believed to have psychogenic causes affect women more than men
- the belief that NVP is psychogenic has been perpetuated primarily by male authors.
They argued that sexual prejudice may prevent women from receiving necessary symptomatic treatment and impede research into the cause of NVP.23
Gender bias continues to be found in the diagnosis of women with physical complaints. In 2006, Chiaramonte and Friend24 found strong, consistent gender bias among medical students and residents when evaluating women who reported coronary disease symptoms during stressful life events.
Organic theories
Organic theories view NVP as multifactorial, with contributions from evolution and multiple organ systems. Endocrine, vestibular, gastrointestinal, and CNS contributions have been described, but none have solved NVP’s etiologic mystery.
Evolutionary. NVP may provide an evolutionary advantage by protecting the embryo and mother. This theory states that potential toxins are present in many foods, especially if eaten in large quantities. NVP prevents the pregnant woman from eating very much and harming the embryo. Below-average miscarriage rates are seen in women with NVP.1,2,25
And because a woman’s immune system is depressed during pregnancy, NVP may be advantageous for the mother by limiting her ingestion of potential toxins.25
Endocrine. Human chorionic gonadotropin (hCG), estrogens, progesterone, and leptin, as well as adrenal cortex insufficiencies have been investigated for a role in NVP. Only hCG has shown clear evidence of an association, and some researchers believe it is the most likely cause of NVP.20
NVP rates are higher in pregnancies with elevated hCG. Molar and multiple-gestation pregnancies—each associated with elevated hCG—are complicated more frequently with the severest form of NVP.20,26 Conversely, NVP is less common in women who smoke, which is associated with lower hCG.26
During pregnancy, actions of hCG stimulate the thyroid. Hyperstimulation, leading to transient hyperthyroidism, has been implicated in NVP development.20,26 Symptom severity and the degree of thyroid stimulating hormone (TSH) suppression are closely correlated.26
Elevated hCG levels, hypersensitive TSH receptors, and the presence of a hyperactive hCG isoform have been proposed.20
Gastrointestinal disorders are believed to be involved in the pathogenesis of persistent NVP. Women with NVP usually lack structural or mucosal abnormalities and have normal endoscopic upper GI evaluations. They may, however, have disorders of the stomach’s neuromuscular function. Severe cases of gastric dysrhythmias and abnormalities of gastric tone may lead to gastroparesis.27
Stomach motility in pregnancy is influenced by neurohormonal changes, specifically in estrogen and thyroid hormones. Gastric motility abnormalities—evaluated by electrogastrography (EGG)—have been associated with NVP symptoms and normal EGGs with the absence of symptoms. Some women who had NVP and abnormal EGGs were retested after delivery when symptom-free and found to have normal myoelectric EGG patterns.27
Helicobacter pylori also may be involved in NVP, and at least 1 study found active H pylori infection and HG to be highly correlated. Pregnancy is not believed to predispose to H pylori infection, but active infection compounded by pregnancy’s hormonal changes may exacerbate NVP.28
NVP and motion sickness share many features, suggesting that NVP treatment could be targeted if a vestibular disorder could be discovered.29 Abnormal electroencephalography—particularly generalized slowing—that is not present in asymptomatic pregnant women has been reported in women with NVP.30
CNS contributions. Persistent NVP may be a learned behavior,24 a view based on findings of anticipatory nausea and vomiting in chemotherapy patients. Through conditioning, a pregnant woman may associate her physical symptoms with elements in her life that maintain the cycle of nausea and vomiting.31
Treating psychological symptoms
Brief psychotherapy to identify and correct sources of anxiety in pregnancy may alleviate a patient’s nausea and vomiting.32
- Progressive muscle relaxation training, often combined with guided imagery, can decrease nausea and vomiting associated with chemotherapy and may prevent anticipatory symptoms by decreasing anxiety.
- Systematic desensitization is successful in most chemotherapy patients who try it. In this technique, relaxation is counter-conditioned as a response to stimuli known to elicit symptoms.31
Hypnosis allows patients to achieve a physiologic state incompatible with nausea and vomiting31 and can terminate vomiting after 1 to 3 sessions.3
Medication. Similar to ondansetron, the antidepressant mirtazapine exhibits an antiemetic effect by blocking the 5-HT3 receptor. In treatment-resistant cases, mirtazapine, 30 mg/d, has been reported to ameliorate NVP symptoms, usually within 24 hours. Patients were able to return to normal diets and discontinue treatment after 6 to 10 days. Mirtazapine appears to be safe during pregnancy, based on animal studies using 17 and 20 times the maximum recommended human dose.33
For patients with anxiety symptoms, consider other medications—including selective serotonin reuptake inhibitors and benzodiazepines—only after counseling the patient about potential risks and benefits to her and the fetus.34
Alternative treatments. In traditional Indian medicine, a mixture of powdered ginger and honey is given to women with NVP. At least 2 studies demonstrate ginger’s efficacy.35
In traditional Chinese medicine, stimulating the Neiguan point (P6) on the wrist is believed to relieve nausea and vomiting. Although results are inconclusive, studies suggest that P6 stimulation can help control NVP.36 The FDA has approved wristbands that stimulate the P6 site, either electrically or by acupressure (Figure).36
Figure Wristband to manage nausea and vomiting in pregnancy
The FDA-cleared BioBand acupressure wristband may help control nausea and vomiting in pregnancy by stimulating the Neiguan point (P6) on the wrist.
Source: Reference 36Consider thiamine supplementation for women with severe symptoms, as Wernicke’s encephalopathy is a rare complication of prolonged NVP.19,37
Related resources
- Motherisk Program at The Hospital for Sick Children, Toronto, Ontario, Canada. Website with information on vomiting during pregnancy: www.motherisk.org/women/morningSickness.jsp. Nausea and vomiting of pregnancy (NVP) forum. www.motherisk.org/women/forum.jsp.
- BioBand acupuncture wristband. www.BioBands.com.
- Koren G, Bishai R, eds. Nausea and vomiting of pregnancy: state of the art 2000. Toronto, Ontario, Canada: The Motherisk Program; 2000.
Drug brand names
- Dimenhydrinate • Dramamine
- Doxylamine • Unisom
- Methylprednisolone • Medrol
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Ondansetron • Zofran
- Promethazine • Phenergan
- Trimethobenzamide • Tigan
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis 1990;178:655-9.
2. Davis M. Nausea and vomiting of pregnancy: an evidence-based review. J Perinat Neonatal Nurs 2004;18:312-28.
3. Buckwalter JG, Simpson SW. Psychological factors in the etiology and treatment of severe nausea and vomiting in pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S210-4.
4. Bogen JT. Neurosis: a Ms-diagnosis. Perspect Biol Med 1994;37(2):263-74.
5. Weiss G. The death of Charlotte Brontë. Obstet Gynecol 1991;78(4):705-8.
6. Lester EP, Notman MT. Pregnancy, developmental crisis and object relations: psychoanalytic considerations. Int J Psychoanal 1986;67(pt 3):357-66.
7. Sheehan P. Hyperemesis gravidarum—assessment and management. Aust Fam Physician 2007;36:698-701.
8. American College of Obstetrics and Gynecology. ACOG practice bulletin: nausea and vomiting of pregnancy. Obstet Gynecol 2004;103:803-14.
9. Iancu I, Kotler M, Spivak B, et al. Psychiatric aspects of hyperemesis gravidarum. Psychother Psychosom 1994;61:143-9.
10. Munch S. Chicken or the egg? The biological-psychological controversy surrounding hyperemesis gravidarum. Soc Sci Med 2002;55:1267-78.
11. Robertson GG. Nausea and vomiting of pregnancy: a study in psychosomatic and social medicine. Lancet 1946;336-45.
12. Fairweather DV. Nausea and vomiting in pregnancy. Am J Obstet Gynecol 1968;102:135-75.
13. Katon WJ, Ries RK, Bokan JA, Kleinman A. Hyperemesis gravidarum: a biopsychosocial perspective. Int J Psychiatry Med 1980;10:151-62.
14. Simpson SW, Goodwin TM, Robins SB, et al. Psychological factors and hyperemesis gravidarum. J Womens Health Gend Based Med 2001;10:471-7.
15. Majerus PW, Guze SB, Delong WB, Robins E. Psychologic factors and psychiatric disease in hyperemesis gravidarum: a follow-up study of 69 vomiters and 66 controls. Am J Psychiatry 1960;117:421-8.
16. Bozzo P, Koren G, Nava-Ocampo AA, Einarson A. The incidence of nausea and vomiting of pregnancy (NVP): a comparison between depressed women treated with antidepressants and non-depressed women. Clin Invest Med 2006;29(6):347-50.
17. Markl GE, Strunz-Lehner C, Egen-Lappe V, et al. The association of psychosocial factors with nausea and vomiting during pregnancy. J Psychosom Obstet Gynaecol 2007;1-6.
18. Seng JS, Oakley DJ, Sampselle CM, et al. Posttraumatic stress disorder and pregnancy complications. Obstet Gynecol 2001;97(1):17-22.
19. Ismail SK, Kenny L. Review on hyperemesis gravidarum. Best Pract Res Clin Gastroenterol 2007;21:755-69.
20. Verberg MF, Gillott DJ, Al-Fardan N, Grudzinskas JG. Hyperemesis gravidarum, a literature review. Hum Reprod Update 2005;11:527-39.
21. Deuchar N. Nausea and vomiting in pregnancy: a review of the problem with particular regard to psychological and social aspects. Br J Obstet Gynaecol 1995;102(1):6-8.
22. Iatrakis GM, Sakellaropoulos GG, Kourkoubas AH, Kabounia SE. Vomiting and nausea in the first 12 weeks of pregnancy. Psychother Psychosom 1988;49(1):22-4.
23. Lennane KJ, Lennane RJ. Alleged psychogenic disorders in women—a possible manifestation of sexual prejudice. N Engl J Med 1973;288(6):288-92.
24. Chiaramonte GR, Friend R. Medical students’ and residents’ gender bias in the diagnosis, treatment, and interpretation of coronary heart disease symptoms. Health Psychol 2006;25:255-66.
25. Sherman PW, Flaxman SM. Nausea and vomiting of pregnancy in an evolutionary perspective. Am J Obstet Gynecol 2002;186(5 suppl):S190-7.
26. Goodwin TM. Nausea and vomiting of pregnancy: an obstetric syndrome. Am J Obstet Gynecol 2002;186(5 suppl):S184-9.
27. Koch KL. Gastrointestinal factors in nausea and vomiting of pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S198-203.
28. Golberg D, Szilagyi A, Graves L. Hyperemesis gravidarum and Helicobacter pylori infection: a systematic review. Obstet Gynecol 2007;110:695-703.
29. Black FO. Maternal susceptibility to nausea and vomiting of pregnancy: is the vestibular system involved? Am J Obstet Gynecol 2002;186(5 suppl):S204-9.
30. Vaknin Z, Halperin R, Schneider D, et al. Hyperemesis gravidarum and nonspecific abnormal EEG findings: a preliminary report. J Reprod Med 2006;51:623-7.
31. Matteson S, Roscoe J, Hickok J, Morrow GR. The role of behavioral conditioning in the development of nausea. Am J Obstet Gynecol 2002;186(5 suppl):S239-43.
32. Zechnich R, Hammer T. Brief psychotherapy for hyperemesis gravidarum. Am Fam Physician 1982;26:179-81.
33. Guclu S, Gol M, Dogan E, Saygili U. Mirtazapine use in resistant hyperemesis gravidarum: report of three cases and review of the literature. Arch Gynecol Obstet 2005;272:298-300.
34. Raphael DB, Ross J, Brizendine L. Treating anxiety during pregnancy: Just how safe are SSRIs? Current Psychiatry 2008;7(2):39-52.
35. Niebyl JR, Goodwin TM. Overview of nausea and vomiting of pregnancy with an emphasis on vitamins and ginger. Am J Obstet Gynecol 2002;186(5 suppl):S253-5.
36. Roscoe JA, Matteson SE. Acupressure and acustimulation bands for control of nausea: a brief review. Am J Obstet Gynecol 2002;186(5 suppl):S244-7.
37. Chiossi G, Neri I, Cavazzuti M, et al. Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet Gynecol Surv 2006;61:255-68.
1. el-Mallakh RS, Liebowitz NR, Hale MS. Hyperemesis gravidarum as conversion disorder. J Nerv Ment Dis 1990;178:655-9.
2. Davis M. Nausea and vomiting of pregnancy: an evidence-based review. J Perinat Neonatal Nurs 2004;18:312-28.
3. Buckwalter JG, Simpson SW. Psychological factors in the etiology and treatment of severe nausea and vomiting in pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S210-4.
4. Bogen JT. Neurosis: a Ms-diagnosis. Perspect Biol Med 1994;37(2):263-74.
5. Weiss G. The death of Charlotte Brontë. Obstet Gynecol 1991;78(4):705-8.
6. Lester EP, Notman MT. Pregnancy, developmental crisis and object relations: psychoanalytic considerations. Int J Psychoanal 1986;67(pt 3):357-66.
7. Sheehan P. Hyperemesis gravidarum—assessment and management. Aust Fam Physician 2007;36:698-701.
8. American College of Obstetrics and Gynecology. ACOG practice bulletin: nausea and vomiting of pregnancy. Obstet Gynecol 2004;103:803-14.
9. Iancu I, Kotler M, Spivak B, et al. Psychiatric aspects of hyperemesis gravidarum. Psychother Psychosom 1994;61:143-9.
10. Munch S. Chicken or the egg? The biological-psychological controversy surrounding hyperemesis gravidarum. Soc Sci Med 2002;55:1267-78.
11. Robertson GG. Nausea and vomiting of pregnancy: a study in psychosomatic and social medicine. Lancet 1946;336-45.
12. Fairweather DV. Nausea and vomiting in pregnancy. Am J Obstet Gynecol 1968;102:135-75.
13. Katon WJ, Ries RK, Bokan JA, Kleinman A. Hyperemesis gravidarum: a biopsychosocial perspective. Int J Psychiatry Med 1980;10:151-62.
14. Simpson SW, Goodwin TM, Robins SB, et al. Psychological factors and hyperemesis gravidarum. J Womens Health Gend Based Med 2001;10:471-7.
15. Majerus PW, Guze SB, Delong WB, Robins E. Psychologic factors and psychiatric disease in hyperemesis gravidarum: a follow-up study of 69 vomiters and 66 controls. Am J Psychiatry 1960;117:421-8.
16. Bozzo P, Koren G, Nava-Ocampo AA, Einarson A. The incidence of nausea and vomiting of pregnancy (NVP): a comparison between depressed women treated with antidepressants and non-depressed women. Clin Invest Med 2006;29(6):347-50.
17. Markl GE, Strunz-Lehner C, Egen-Lappe V, et al. The association of psychosocial factors with nausea and vomiting during pregnancy. J Psychosom Obstet Gynaecol 2007;1-6.
18. Seng JS, Oakley DJ, Sampselle CM, et al. Posttraumatic stress disorder and pregnancy complications. Obstet Gynecol 2001;97(1):17-22.
19. Ismail SK, Kenny L. Review on hyperemesis gravidarum. Best Pract Res Clin Gastroenterol 2007;21:755-69.
20. Verberg MF, Gillott DJ, Al-Fardan N, Grudzinskas JG. Hyperemesis gravidarum, a literature review. Hum Reprod Update 2005;11:527-39.
21. Deuchar N. Nausea and vomiting in pregnancy: a review of the problem with particular regard to psychological and social aspects. Br J Obstet Gynaecol 1995;102(1):6-8.
22. Iatrakis GM, Sakellaropoulos GG, Kourkoubas AH, Kabounia SE. Vomiting and nausea in the first 12 weeks of pregnancy. Psychother Psychosom 1988;49(1):22-4.
23. Lennane KJ, Lennane RJ. Alleged psychogenic disorders in women—a possible manifestation of sexual prejudice. N Engl J Med 1973;288(6):288-92.
24. Chiaramonte GR, Friend R. Medical students’ and residents’ gender bias in the diagnosis, treatment, and interpretation of coronary heart disease symptoms. Health Psychol 2006;25:255-66.
25. Sherman PW, Flaxman SM. Nausea and vomiting of pregnancy in an evolutionary perspective. Am J Obstet Gynecol 2002;186(5 suppl):S190-7.
26. Goodwin TM. Nausea and vomiting of pregnancy: an obstetric syndrome. Am J Obstet Gynecol 2002;186(5 suppl):S184-9.
27. Koch KL. Gastrointestinal factors in nausea and vomiting of pregnancy. Am J Obstet Gynecol 2002;186(5 suppl):S198-203.
28. Golberg D, Szilagyi A, Graves L. Hyperemesis gravidarum and Helicobacter pylori infection: a systematic review. Obstet Gynecol 2007;110:695-703.
29. Black FO. Maternal susceptibility to nausea and vomiting of pregnancy: is the vestibular system involved? Am J Obstet Gynecol 2002;186(5 suppl):S204-9.
30. Vaknin Z, Halperin R, Schneider D, et al. Hyperemesis gravidarum and nonspecific abnormal EEG findings: a preliminary report. J Reprod Med 2006;51:623-7.
31. Matteson S, Roscoe J, Hickok J, Morrow GR. The role of behavioral conditioning in the development of nausea. Am J Obstet Gynecol 2002;186(5 suppl):S239-43.
32. Zechnich R, Hammer T. Brief psychotherapy for hyperemesis gravidarum. Am Fam Physician 1982;26:179-81.
33. Guclu S, Gol M, Dogan E, Saygili U. Mirtazapine use in resistant hyperemesis gravidarum: report of three cases and review of the literature. Arch Gynecol Obstet 2005;272:298-300.
34. Raphael DB, Ross J, Brizendine L. Treating anxiety during pregnancy: Just how safe are SSRIs? Current Psychiatry 2008;7(2):39-52.
35. Niebyl JR, Goodwin TM. Overview of nausea and vomiting of pregnancy with an emphasis on vitamins and ginger. Am J Obstet Gynecol 2002;186(5 suppl):S253-5.
36. Roscoe JA, Matteson SE. Acupressure and acustimulation bands for control of nausea: a brief review. Am J Obstet Gynecol 2002;186(5 suppl):S244-7.
37. Chiossi G, Neri I, Cavazzuti M, et al. Hyperemesis gravidarum complicated by Wernicke encephalopathy: background, case report, and review of the literature. Obstet Gynecol Surv 2006;61:255-68.
Your vote, please: What’s ailing psychiatry?
Psychiatry has become a mature neuroscience discipline and medical specialty, helping millions of Americans suffering from mental illness. Neurobiologic and psychosocial research into causes of and treatments for psychiatric brain disorders is booming, and molecular genetic discoveries promise unprecedented insights into the neural mechanisms of thought, feeling, emotional, behavioral, and cognitive disturbances.
As psychiatry’s promise grows, however, so do its frustrations. I list here 17 challenges psychiatrists face, and I would like to know how important you feel each challenge is to our profession.
Please visit the HTML version of this article to rank each challenge from 10 (highest importance) to 1 (lowest importance). To add other challenges not on my list, simply use the comments field at the bottom of the survey. We will share the results with you in Current Psychiatry, and you can compare your opinions with those of your colleagues.
- Still-elusive pathophysiology of psychiatric disorders, despite the abundance of new knowledge.
- Lack of an objective diagnostic schema, including laboratory tests to confirm diagnoses.
- High rates of off-label psychotropic use because >80% of DSM-IV-TR psychiatric disorders have no approved medications and practitioners have few options.
- Tightening regulations, suffocating litigation, and a demoralized drug-discovery industry that have blunted innovation and threaten drug development.
- Limited evidence-based psychotherapy research, compared with controlled FDA trials for drug therapy or medical devices.
- Persistent stigma of mental illness interfering with prevention and early intervention.
- Lack of full parity for mental illness with physical illness.
- Excessive, harmful intrusion of the courts into psychiatric care, more than any other medical specialty.
- A “broken” public mental health system with red tape, inadequate funding, nonevidence-based treatments, homelessness, drug abuse, and lack of primary care.
- Far less private philanthropy bestowed on psychiatry compared with cancer, cardiology, or other medical specialities.
- Shrinking funds for psychiatric research because of National Institutes of Health belt-tightening and dwindling of unrestricted pharmaceutical industry research grants.
- Atrociously low compensation for psychiatrists, with hourly earnings a fraction of those for procedural specialists (such as surgeons or radiologists), although the years of training are similar.
- Unreimbursed time spent on paperwork or phone calls related to patient care.
- Little time to read journals or attend continuing medical education meetings to keep up with rapid advances in psychiatry.
- Lack of primary care for the seriously mentally ill because of barriers to collaborative care between psychiatrists and primary care providers.
- Shortage of psychiatric beds, both acute and longterm, and a ridiculously brief length of stay for psychotic or suicidal patients.
- Too few academic psychiatrists to train and mentor young clinicians, researchers, and educators who represent the cutting edge of the speciality.
I am very interested in knowing your opinions. Ranking the importance of these challenges may be a sobering task, but problem solving always starts by assessing the challenges and developing strategies to address them in a targeted, systematic manner.
Finally, it is interesting that while there is a serious national shortage of psychiatrists, there is certainly no shortage of challenges. Psychiatrists, patients, families, and advocates should combine their efforts to find effective solutions, one challenge at a time.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or click here
Psychiatry has become a mature neuroscience discipline and medical specialty, helping millions of Americans suffering from mental illness. Neurobiologic and psychosocial research into causes of and treatments for psychiatric brain disorders is booming, and molecular genetic discoveries promise unprecedented insights into the neural mechanisms of thought, feeling, emotional, behavioral, and cognitive disturbances.
As psychiatry’s promise grows, however, so do its frustrations. I list here 17 challenges psychiatrists face, and I would like to know how important you feel each challenge is to our profession.
Please visit the HTML version of this article to rank each challenge from 10 (highest importance) to 1 (lowest importance). To add other challenges not on my list, simply use the comments field at the bottom of the survey. We will share the results with you in Current Psychiatry, and you can compare your opinions with those of your colleagues.
- Still-elusive pathophysiology of psychiatric disorders, despite the abundance of new knowledge.
- Lack of an objective diagnostic schema, including laboratory tests to confirm diagnoses.
- High rates of off-label psychotropic use because >80% of DSM-IV-TR psychiatric disorders have no approved medications and practitioners have few options.
- Tightening regulations, suffocating litigation, and a demoralized drug-discovery industry that have blunted innovation and threaten drug development.
- Limited evidence-based psychotherapy research, compared with controlled FDA trials for drug therapy or medical devices.
- Persistent stigma of mental illness interfering with prevention and early intervention.
- Lack of full parity for mental illness with physical illness.
- Excessive, harmful intrusion of the courts into psychiatric care, more than any other medical specialty.
- A “broken” public mental health system with red tape, inadequate funding, nonevidence-based treatments, homelessness, drug abuse, and lack of primary care.
- Far less private philanthropy bestowed on psychiatry compared with cancer, cardiology, or other medical specialities.
- Shrinking funds for psychiatric research because of National Institutes of Health belt-tightening and dwindling of unrestricted pharmaceutical industry research grants.
- Atrociously low compensation for psychiatrists, with hourly earnings a fraction of those for procedural specialists (such as surgeons or radiologists), although the years of training are similar.
- Unreimbursed time spent on paperwork or phone calls related to patient care.
- Little time to read journals or attend continuing medical education meetings to keep up with rapid advances in psychiatry.
- Lack of primary care for the seriously mentally ill because of barriers to collaborative care between psychiatrists and primary care providers.
- Shortage of psychiatric beds, both acute and longterm, and a ridiculously brief length of stay for psychotic or suicidal patients.
- Too few academic psychiatrists to train and mentor young clinicians, researchers, and educators who represent the cutting edge of the speciality.
I am very interested in knowing your opinions. Ranking the importance of these challenges may be a sobering task, but problem solving always starts by assessing the challenges and developing strategies to address them in a targeted, systematic manner.
Finally, it is interesting that while there is a serious national shortage of psychiatrists, there is certainly no shortage of challenges. Psychiatrists, patients, families, and advocates should combine their efforts to find effective solutions, one challenge at a time.
Psychiatry has become a mature neuroscience discipline and medical specialty, helping millions of Americans suffering from mental illness. Neurobiologic and psychosocial research into causes of and treatments for psychiatric brain disorders is booming, and molecular genetic discoveries promise unprecedented insights into the neural mechanisms of thought, feeling, emotional, behavioral, and cognitive disturbances.
As psychiatry’s promise grows, however, so do its frustrations. I list here 17 challenges psychiatrists face, and I would like to know how important you feel each challenge is to our profession.
Please visit the HTML version of this article to rank each challenge from 10 (highest importance) to 1 (lowest importance). To add other challenges not on my list, simply use the comments field at the bottom of the survey. We will share the results with you in Current Psychiatry, and you can compare your opinions with those of your colleagues.
- Still-elusive pathophysiology of psychiatric disorders, despite the abundance of new knowledge.
- Lack of an objective diagnostic schema, including laboratory tests to confirm diagnoses.
- High rates of off-label psychotropic use because >80% of DSM-IV-TR psychiatric disorders have no approved medications and practitioners have few options.
- Tightening regulations, suffocating litigation, and a demoralized drug-discovery industry that have blunted innovation and threaten drug development.
- Limited evidence-based psychotherapy research, compared with controlled FDA trials for drug therapy or medical devices.
- Persistent stigma of mental illness interfering with prevention and early intervention.
- Lack of full parity for mental illness with physical illness.
- Excessive, harmful intrusion of the courts into psychiatric care, more than any other medical specialty.
- A “broken” public mental health system with red tape, inadequate funding, nonevidence-based treatments, homelessness, drug abuse, and lack of primary care.
- Far less private philanthropy bestowed on psychiatry compared with cancer, cardiology, or other medical specialities.
- Shrinking funds for psychiatric research because of National Institutes of Health belt-tightening and dwindling of unrestricted pharmaceutical industry research grants.
- Atrociously low compensation for psychiatrists, with hourly earnings a fraction of those for procedural specialists (such as surgeons or radiologists), although the years of training are similar.
- Unreimbursed time spent on paperwork or phone calls related to patient care.
- Little time to read journals or attend continuing medical education meetings to keep up with rapid advances in psychiatry.
- Lack of primary care for the seriously mentally ill because of barriers to collaborative care between psychiatrists and primary care providers.
- Shortage of psychiatric beds, both acute and longterm, and a ridiculously brief length of stay for psychotic or suicidal patients.
- Too few academic psychiatrists to train and mentor young clinicians, researchers, and educators who represent the cutting edge of the speciality.
I am very interested in knowing your opinions. Ranking the importance of these challenges may be a sobering task, but problem solving always starts by assessing the challenges and developing strategies to address them in a targeted, systematic manner.
Finally, it is interesting that while there is a serious national shortage of psychiatrists, there is certainly no shortage of challenges. Psychiatrists, patients, families, and advocates should combine their efforts to find effective solutions, one challenge at a time.
Malpractice minute
Could a patient’s violent act
have been prevented?
THE PATIENT. A man under outpatient care of the state’s regional behavioral health authority was diagnosed with schizophrenia, paranoid type.
CASE FACTS. The patient killed his developmentally disabled niece, age 26.
THE VICTIM’S FAMILY’S CLAIM. The death would not have occurred if the patient had been civilly committed or heavily medicated.
THE BEHAVIORAL HEALTH AUTHORITY’S DEFENSE. The violent act was unforeseeable, and the patient was compliant with treatment. The victim’s mother should not have left the disabled woman alone with the patient.
Submit your verdict and find out how the court ruled. Click on “Have more to say about this topic?” to comment.
Cases are selected by Current Psychiatry from Medical Malpractice Verdicts, Settlements & Experts, with permission of its editor, Lewis Laska of Nashville, TN (www.verdictslaska.com). Information may be incomplete in some instances, but these cases represent clinical situations that typically result in litigation.
Could a patient’s violent act
have been prevented?
THE PATIENT. A man under outpatient care of the state’s regional behavioral health authority was diagnosed with schizophrenia, paranoid type.
CASE FACTS. The patient killed his developmentally disabled niece, age 26.
THE VICTIM’S FAMILY’S CLAIM. The death would not have occurred if the patient had been civilly committed or heavily medicated.
THE BEHAVIORAL HEALTH AUTHORITY’S DEFENSE. The violent act was unforeseeable, and the patient was compliant with treatment. The victim’s mother should not have left the disabled woman alone with the patient.
Submit your verdict and find out how the court ruled. Click on “Have more to say about this topic?” to comment.
Could a patient’s violent act
have been prevented?
THE PATIENT. A man under outpatient care of the state’s regional behavioral health authority was diagnosed with schizophrenia, paranoid type.
CASE FACTS. The patient killed his developmentally disabled niece, age 26.
THE VICTIM’S FAMILY’S CLAIM. The death would not have occurred if the patient had been civilly committed or heavily medicated.
THE BEHAVIORAL HEALTH AUTHORITY’S DEFENSE. The violent act was unforeseeable, and the patient was compliant with treatment. The victim’s mother should not have left the disabled woman alone with the patient.
Submit your verdict and find out how the court ruled. Click on “Have more to say about this topic?” to comment.
Cases are selected by Current Psychiatry from Medical Malpractice Verdicts, Settlements & Experts, with permission of its editor, Lewis Laska of Nashville, TN (www.verdictslaska.com). Information may be incomplete in some instances, but these cases represent clinical situations that typically result in litigation.
Cases are selected by Current Psychiatry from Medical Malpractice Verdicts, Settlements & Experts, with permission of its editor, Lewis Laska of Nashville, TN (www.verdictslaska.com). Information may be incomplete in some instances, but these cases represent clinical situations that typically result in litigation.
Desvenlafaxine for depression
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
Compared with other antidepressants, desvenlafaxine might have more predictable effects and a lower risk of drug-drug interactions because of the way it is metabolized. The FDA approved this selective serotonin-norepinephrine reuptake inhibitor (SNRI)—a major active metabolite of venlafaxine—for treating major depressive disorder (MDD, Table 1). In clinical trials, desvenlafaxine was more effective than placebo in improving patients’ scores on scales of depressive symptoms and overall improvement.1
Table 1
Desvenlafaxine: Fast facts
| Brand name: Pristiq |
| Class: Serotonin-norepinephrine reuptake inhibitor |
| Indication: Major depressive disorder |
| Approval date: February 2008 |
| Availability date: May 2008 |
| Manufacturer: Wyeth Pharmaceuticals |
| Dosing forms: 50- and 100-mg tablets |
| Recommended dose: 50 to 100 mg/d |
Clinical implications
Unlike other SNRIs (venlafaxine and duloxetine), desvenlafaxine does not depend on cytochrome P450 (CYP) 2D6 for bio-transformation. As a result plasma concentrations vary less among individual patients, which should result in more predictable efficacy and tolerability. In addition, unlike bupropion, duloxetine, fluoxetine, and paroxetine, desvenlafaxine does not affect the functional activity of CYP 2D6. This translates into a lower risk of drug-drug interactions and more predictable effects on coadministered drugs that are cleared by CYP 2D6.
How it works
Serotonin, norepinephrine, and dopamine in the CNS are involved in mood and neurovegetative functions that are disturbed in patients with MDD. Desvenlafaxine selectively inhibits serotonin and norepinephrine reuptake pumps, therefore increasing serotonin and norepinephrine concentration in the synaptic cleft.2 The drug has weak binding affinity for the dopamine transporter and does not cause substantial changes in extracellular dopamine concentration. Decreased presynaptic serotonin and norepinephrine uptake increases the synaptic concentration of these neurotransmitters. These effects are thought to be responsible for desvenlafaxine’s antidepressant efficacy.
Pharmacokinetics
Desvenlafaxine’s single-dose pharmacokinetics are linear and dose-proportional over the recommended 50 to 100 mg/d dosing range. The half-life is approximately 11 hours. Steady-state plasma concentration is achieved in 4 to 5 days with once-daily dosing.
Food does not affect intestinal absorption. Bioavailability after oral administration is 80%, and time to reach maximum concentration (Tmax) is 7.5 hours. Plasma protein binding is 30% and is independent of desvenlafaxine concentration.1
Desvenlafaxine is excreted renally:
- unchanged (45% at 72 hours after administration)
- as desvenlafaxine-glucuronide
- as N-desvenlafaxine-glucuronide.
Desvenlafaxine-glucuronide is the final metabolite of conjugation reaction with glucuronic acid. N-desvenlafaxine-glucuronide is an end product of a 2-step metabolic reaction that starts with oxidation by CYP 3A4 to produce N-desvenlafaxine, which is conjugated with glucuronic acid to create N-desvenlafaxine-glucuronide. As a result of these metabolic and elimination pathways, dosing adjustment is recommended for patients with severe renal impairment or who are taking a CYP 3A4 inhibitor.
Dosing
Desvenlafaxine is available as 50-mg and 100-mg tablets. The recommended dosage is 50 mg/d, and the maximum recommended dosage in patients with hepatic impairment is 100 mg/d.
No dosing adjustment is necessary for patients with moderate renal impairment. The recommend regimen for those with severe renal impairment or end-stage renal disease is 50 mg every other day.
Instruct patients to take desvenlafaxine at approximately the same time each day, with or without food. Tell them not to discontinue the drug abruptly and to immediately report any adverse effects (AEs).
Efficacy
Desvenlafaxine’s antidepressant efficacy was established in four 8-week, randomized, double-blind, placebo-controlled, fixed-dose (50 mg to 400 mg once daily) studies in adult outpatients who met DSM-IV-TR criteria for MDD.1,2,4
In the first study,3 461 patients received desvenlafaxine, 100 mg, 200 mg, or 400 mg, or placebo. In the second study,4 369 patients received 200 mg or 400 mg or placebo. In 2 additional studies, a total of 930 patients received 50 mg or 100 mg or placebo.1
All studies used the 17-item Hamilton Rating Scale for Depression (HAM-D17) to measure depressive symptom improvement and the Clinical Global Impressions-Improvement (CGI-I) scale to measure overall improvement. Desvenlafaxine was more effective than placebo in HAM-D17 score improvement in all 4 studies and in CGI-I score improvement in 3 studies.
In studies comparing 50 mg/d with 100 mg/d, doses >50 mg/d provided no additional benefit. Higher starting fixed doses were associated with more frequent AEs and discontinuation.
Gender or age had no effect on treatment outcome. There was no difference in safety in elderly vs younger patients. Data are insufficient to establish a relationship between race and desvenlafaxine responsiveness. Desvenlafaxine’s safety and effectiveness in children and adolescents was not evaluated, and the drug is not approved for these patients.
Tolerability and safety
Desvenlafaxine’s tolerability is comparable to that of other SNRIs. In premarketing studies, 12% of patients receiving desvenlafaxine (50 mg/d to 400 mg/d) discontinued treatment because of AEs, compared with 3% in the placebo group. The discontinuation rate in patients receiving 100 mg/d was 8.7% compared with 4.1% in patients taking 50 mg/d.
AEs generally occur during the first week of treatment. In the 8-week trials, the most common AEs were nausea and dizziness (Table 2). In a long-term study (up to 9 months), the most common AE was vomiting. Although the recommended starting dose is 50 mg/d, to avoid AEs consider beginning with every-other-day dosing.
Table 2
Desvenlafaxine trials: Rates of adverse effects
| Desvenlafaxine dose | |||
|---|---|---|---|
| Adverse effect | 50 mg/d | 100 mg/d | Placebo |
| Nausea | 22% | 26% | 10% |
| Dizziness | 13% | 10% | 5% |
| Insomnia | 9% | 12% | 6% |
| Hyperhidrosis | 10% | 11% | 4% |
| Constipation | 9% | 9% | 4% |
| Somnolence | 4% | 9% | 4% |
| Decreased appetite | 5% | 8% | 2% |
| Erectile dysfunction | 3% | 6% | 1% |
| Decreased libido | 4% | 5% | 1% |
| Anxiety | 3% | 5% | 2% |
| Source: Reference 1 | |||
Abruptly discontinuing desvenlafaxine can cause withdrawal symptoms, including dizziness, nausea, headache, irritability, insomnia, diarrhea, anxiety, abnormal dreams, fatigue, and hyperhidrosis. The frequency of withdrawal symptoms is higher with longer treatment duration. Gradually reducing the dose by administering 50 mg of desvenlafaxine less often can reduce withdrawal symptoms.
Clinical issues
All SNRIs and selective serotonin reuptake inhibitors (SSRIs) have a “black-box” warning about the potential for clinical worsening and increased suicidality early in treatment. Closely monitor patients for suicidal ideation/behaviors during the first months of treatment and with dose changes.
When taken in the third trimester of pregnancy, SNRIs and SSRIs can cause serious neonatal complications—including respiratory distress, cyanosis, apnea, and seizures—that may require longer hospitalization, respiratory support, or tube feeding for the infant. Carefully consider risks and benefits of third-trimester antidepressant use.5 Desvenlafaxine is excreted in breast milk and may cause AEs in infants who are breast-fed.
In clinical trials, patients taking desvenlafaxine experienced increased cholesterol, triglycerides, and blood pressure. Monitor these parameters closely in patients taking desvenlafaxine, and use the drug with caution in patients with cerebrovascular and cardiovascular disease.
Other concerns in patients taking desvenlafaxine include:
- Antidepressant medications can trigger hypomania or mania in patients with bipolar disorder.
- Patients—particularly those who are elderly or taking diuretics—may develop hyponatremia as a result of syndrome of in-appropriate antidiuretic hormone.
- Patients with an increased risk of glaucoma need to be monitored because of the drug’s effect on blood pressure.
Drug interactions. Coadministering desvenlafaxine with serotonergic medications— such as triptans, other antidepressants, and tramadol—can cause serotonin syndrome, a potentially life-threatening condition characterized by mental status changes, autonomic instability, neuromuscular aberrations, and gastrointestinal symptoms. Concomitant use of desvenlafaxine and blood-thinning medications such as warfarin, aspirin, and nonsteroidal anti-inflammatory drugs may result in abnormal bleeding. Patients taking a potent CYP 3A4 inhibitor such as ketoconazole may have increased desvenlafaxine concentration.
Contraindications
Do not prescribe desvenlafaxine to patients who are:
- hypersensitive to venlafaxine chloride, desvenlafaxine succinate, or any parts of the desvenlafaxine formulation
- taking a monoamine oxidase inhibitor (MAOI), or have discontinued an MAOI within 14 days.
Patients who stop taking desvenlafaxine should wait 7 days before starting an MAOI.
Drug brand names
- Bupropion • Wellbutrin
- Desvenlafaxine • Pristiq
- Duloxetine • Cymbalta
- Fluoxetine • Prozac
- Ketoconazole • Nizoral
- Paroxetine • Prozac
- Tramadol • Ultram
- Venlafaxine • Effexor
- Warfarin • Coumadin
Disclosures
Dr. Lincoln reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has in the past year received research/ grant support from and served as a speaker for Wyeth Pharmaceuticals. Previously, he has received research/grant support from or served as a speaker for or consultant to Abbott Laboratories, AstraZeneca, Aventis, Biovail, Boehringer Ingleheim, Bristol-Myers Squibb, Eisai, Eli Lilly and Company, GlaxoSmithKline, Hoffman LaRoche, Janssen, L.P., Johnson & Johnson, Lundbeck, Merck, Novartis, Organon, Otsuka, Pfizer Inc., Solvay, Somerset, Sumitomo, and Yamanouchi.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.
1. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals; 2008.
2. Deecher DC, Beyer CE, Johnston G, et al. Desvenlafaxine succinate: a new serotonin and norepinephrine reuptake inhibitor. J Pharmacol Exp Ther 2006;318(2):657-65.
3. DeMartinis NA, Yeung PP, Entsuah R, Manley AL. A double-blind, placebo-controlled study of the efficacy and safety of desvenlafaxine succinate in the treatment of major depressive disorder. J Clin Psychiatry 2007;68(5):677-88.
4. Septien-Velez L, Pitrosky B, Padmanabhan SK, et al. A randomized, double-blind, placebo-controlled trial of desvenlafaxine succinate in the treatment of major depressive disorder. Int Clin Psychopharmacol 2007;22(6):338-47.
5. Lennestål R, Källén B. Delivery outcome in relation to maternal use of some recently introduced antidepressants. J Clin Psychopharmacol 2007;27(6):607-13.
Dr. Lincoln is a clinical instructor and Dr. Preskorn is a professor of psychiatry, University of Kansas Medical Center, Wichita. Dr. Preskorn also is the psychopharmacology Section Editor of CURRENT PSYCHIATRY and president and CEO of Clinical Research Institute, Wichita.