User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Sponsored CME: Do drug companies influence the content?
Continuing medical education (CME) has grown into a thriving educational ‘business’ whose success is highly dependent on educational grants.
The notion of a “quid pro quo” has grown among observers because the pharmaceutical industry provides most funding for CME programs in psychiatry and other specialties. Evaluations completed at that end of CME programs sometimes reflect attendees’ perception that the content has been “slanted” in favor of the sponsor’s proprietary drug(s).
Congress weighs in. The issue of potential influence by pharmaceutical industry sponsors on the content of CME programs is heating up. Congress has decided to hold hearings to investigate allegations that drug companies may be using CME programs to skew doctors’ treatment decisions or to circumvent laws against promoting off-label uses of medications. Congress wants to investigate whether a conflict of interest exists when pharmaceutical companies sponsor CME programs, especially when the speakers have received research grants, speaking honoraria, or consulting fees from the pharmaceutical sponsors.
Realities of CME. CME is required for the license renewal of physicians and nurses in all states. It is rigorously regulated by the Accreditation Council for Continuing Medical Education (ACCME), whose parent is the American Medical Association. Several thousand CME providers (including all medical schools) solicit educational grants from sponsors and offer programs in the form of grand rounds at teaching institutions, symposia, or dinner programs, etc.
Most teaching institutions have practically no internal funds to cover CME program costs, such as administrative expenses, speakers’ travel and honoraria, refreshments and meals, venue charges, printing, parking, etc. Without grants from external sponsors, CME programs would shrink drastically, and the cost of CME credits for licensure renewal would skyrocket.
‘Hands-off’ policies. Over the past 3 years, the ACCME has tightened procedures for CME content development, and drug companies are complying with these “hands-off” requirements. All have adopted a similar process whereby a grants committee reviews applications and makes decisions devoid of marketing influences. As an applicant for CME grants, I find the process to have become more elaborate and the rate of funding lower than in the past.
Expert speakers. Most CME speakers are experts in psychopharmacology and have financial relationships with more than one pharmaceutical company. Because these companies produce drugs that are in vigorous competition, it would be difficult for the speakers to assume a conflict of interest. Only good science will stand the test of competing interests.
CME programs’ depth and scope might decline and learning objectives might not be met if the speakers were not researchers or experts in the published literature of psychopharmacology.
Balance, not bias. Many CME symposia are sponsored jointly by several competing pharmaceutical companies, which reduces the likelihood that content could be skewed in favor of any particular one. At the University of Cincinnati department of psychiatry, for example, no specific drug company ever sponsors our grand rounds, and no sponsor recommends any speaker. Rather, every week we simply express our appreciation for the support of several industry grant providers listed on a slide at the beginning of each grand rounds program. In post-meeting evaluations, attendees’ perception of bias in the presentations has been close to zero since we moved to multiple sponsorship.
Ongoing evaluation of educational content for balance is absolutely essential and is required of all major CME providers. Physicians and nurses would lose a valuable component of CME programs if excessive restrictions were to shackle the free exchange of the latest basic, clinical, and translational research data. That may include controversial issues such as emerging uses of drugs for other than their approved indications.
Discussions about off-label uses of FDA-approved medications are highly relevant to medical practice and can lead to a more critical, evidence-based approach to patient care—the ultimate goal of CME programs.

Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or visit currentpsychiatry.com and click on the “Contact Us” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or visit currentpsychiatry.com and click on the “Contact Us” link.
Henry A. Nasrallah, MD
Editor-in-Chief
To comment on this editorial or other topics of interest, contact Dr. Nasrallah at [email protected] or visit currentpsychiatry.com and click on the “Contact Us” link.
Continuing medical education (CME) has grown into a thriving educational ‘business’ whose success is highly dependent on educational grants.
The notion of a “quid pro quo” has grown among observers because the pharmaceutical industry provides most funding for CME programs in psychiatry and other specialties. Evaluations completed at that end of CME programs sometimes reflect attendees’ perception that the content has been “slanted” in favor of the sponsor’s proprietary drug(s).
Congress weighs in. The issue of potential influence by pharmaceutical industry sponsors on the content of CME programs is heating up. Congress has decided to hold hearings to investigate allegations that drug companies may be using CME programs to skew doctors’ treatment decisions or to circumvent laws against promoting off-label uses of medications. Congress wants to investigate whether a conflict of interest exists when pharmaceutical companies sponsor CME programs, especially when the speakers have received research grants, speaking honoraria, or consulting fees from the pharmaceutical sponsors.
Realities of CME. CME is required for the license renewal of physicians and nurses in all states. It is rigorously regulated by the Accreditation Council for Continuing Medical Education (ACCME), whose parent is the American Medical Association. Several thousand CME providers (including all medical schools) solicit educational grants from sponsors and offer programs in the form of grand rounds at teaching institutions, symposia, or dinner programs, etc.
Most teaching institutions have practically no internal funds to cover CME program costs, such as administrative expenses, speakers’ travel and honoraria, refreshments and meals, venue charges, printing, parking, etc. Without grants from external sponsors, CME programs would shrink drastically, and the cost of CME credits for licensure renewal would skyrocket.
‘Hands-off’ policies. Over the past 3 years, the ACCME has tightened procedures for CME content development, and drug companies are complying with these “hands-off” requirements. All have adopted a similar process whereby a grants committee reviews applications and makes decisions devoid of marketing influences. As an applicant for CME grants, I find the process to have become more elaborate and the rate of funding lower than in the past.
Expert speakers. Most CME speakers are experts in psychopharmacology and have financial relationships with more than one pharmaceutical company. Because these companies produce drugs that are in vigorous competition, it would be difficult for the speakers to assume a conflict of interest. Only good science will stand the test of competing interests.
CME programs’ depth and scope might decline and learning objectives might not be met if the speakers were not researchers or experts in the published literature of psychopharmacology.
Balance, not bias. Many CME symposia are sponsored jointly by several competing pharmaceutical companies, which reduces the likelihood that content could be skewed in favor of any particular one. At the University of Cincinnati department of psychiatry, for example, no specific drug company ever sponsors our grand rounds, and no sponsor recommends any speaker. Rather, every week we simply express our appreciation for the support of several industry grant providers listed on a slide at the beginning of each grand rounds program. In post-meeting evaluations, attendees’ perception of bias in the presentations has been close to zero since we moved to multiple sponsorship.
Ongoing evaluation of educational content for balance is absolutely essential and is required of all major CME providers. Physicians and nurses would lose a valuable component of CME programs if excessive restrictions were to shackle the free exchange of the latest basic, clinical, and translational research data. That may include controversial issues such as emerging uses of drugs for other than their approved indications.
Discussions about off-label uses of FDA-approved medications are highly relevant to medical practice and can lead to a more critical, evidence-based approach to patient care—the ultimate goal of CME programs.
Continuing medical education (CME) has grown into a thriving educational ‘business’ whose success is highly dependent on educational grants.
The notion of a “quid pro quo” has grown among observers because the pharmaceutical industry provides most funding for CME programs in psychiatry and other specialties. Evaluations completed at that end of CME programs sometimes reflect attendees’ perception that the content has been “slanted” in favor of the sponsor’s proprietary drug(s).
Congress weighs in. The issue of potential influence by pharmaceutical industry sponsors on the content of CME programs is heating up. Congress has decided to hold hearings to investigate allegations that drug companies may be using CME programs to skew doctors’ treatment decisions or to circumvent laws against promoting off-label uses of medications. Congress wants to investigate whether a conflict of interest exists when pharmaceutical companies sponsor CME programs, especially when the speakers have received research grants, speaking honoraria, or consulting fees from the pharmaceutical sponsors.
Realities of CME. CME is required for the license renewal of physicians and nurses in all states. It is rigorously regulated by the Accreditation Council for Continuing Medical Education (ACCME), whose parent is the American Medical Association. Several thousand CME providers (including all medical schools) solicit educational grants from sponsors and offer programs in the form of grand rounds at teaching institutions, symposia, or dinner programs, etc.
Most teaching institutions have practically no internal funds to cover CME program costs, such as administrative expenses, speakers’ travel and honoraria, refreshments and meals, venue charges, printing, parking, etc. Without grants from external sponsors, CME programs would shrink drastically, and the cost of CME credits for licensure renewal would skyrocket.
‘Hands-off’ policies. Over the past 3 years, the ACCME has tightened procedures for CME content development, and drug companies are complying with these “hands-off” requirements. All have adopted a similar process whereby a grants committee reviews applications and makes decisions devoid of marketing influences. As an applicant for CME grants, I find the process to have become more elaborate and the rate of funding lower than in the past.
Expert speakers. Most CME speakers are experts in psychopharmacology and have financial relationships with more than one pharmaceutical company. Because these companies produce drugs that are in vigorous competition, it would be difficult for the speakers to assume a conflict of interest. Only good science will stand the test of competing interests.
CME programs’ depth and scope might decline and learning objectives might not be met if the speakers were not researchers or experts in the published literature of psychopharmacology.
Balance, not bias. Many CME symposia are sponsored jointly by several competing pharmaceutical companies, which reduces the likelihood that content could be skewed in favor of any particular one. At the University of Cincinnati department of psychiatry, for example, no specific drug company ever sponsors our grand rounds, and no sponsor recommends any speaker. Rather, every week we simply express our appreciation for the support of several industry grant providers listed on a slide at the beginning of each grand rounds program. In post-meeting evaluations, attendees’ perception of bias in the presentations has been close to zero since we moved to multiple sponsorship.
Ongoing evaluation of educational content for balance is absolutely essential and is required of all major CME providers. Physicians and nurses would lose a valuable component of CME programs if excessive restrictions were to shackle the free exchange of the latest basic, clinical, and translational research data. That may include controversial issues such as emerging uses of drugs for other than their approved indications.
Discussions about off-label uses of FDA-approved medications are highly relevant to medical practice and can lead to a more critical, evidence-based approach to patient care—the ultimate goal of CME programs.
A life of drugs and ‘downtime’
CASE: Near-fatal combination
Inpatient psychiatry refers Mr. B, age 50, to our outpatient psychiatry clinic. Two weeks earlier, he tried to kill himself by sitting on a stepladder, tying a noose around his neck, and consuming large amounts of quetiapine, trazodone, and vodka. His wife found him unconscious on the floor with facial abrasions, empty pill bottles, and the noose lying next to him.
Emergency medical personnel brought Mr. B to the ER. His total Glasgow Coma Scale score of 3 indicated he was comatose. Pulse was 65 bpm (low-normal), and blood alcohol level was 106 mg/dL, suggesting he had ingested hazardous amounts of vodka. Quetiapine and trazodone blood levels were not measured.
Gastric lavage was unsuccessful because the orogastric tube became curled in the distal esophagus. Mr. B was successfully intubated and admitted to the intensive care unit. After 2 days, he was medically stable and regained consciousness, though he was delirious. He was transferred to inpatient psychiatry, where the attending psychiatrist diagnosed major depression and alcohol abuse disorder.
Before presentation, Mr. B had been taking venlafaxine, 75 mg/d, and mirtazapine, 30 mg at bedtime. His previous outpatient psychiatrist had added methylphenidate, 40 mg/d, to augment the antidepressants—which were not alleviating his depression—and the attending continued all 3 medications. Prior trials of sertraline, bupropion, trazodone, quetiapine, and aripiprazole were ineffective.
By the time Mr. B is transferred to us, his suicidal thoughts have remitted but he is still notably depressed. He is anergic, feels hopeless about the future, has markedly diminished self-worth, feels excessively guilty over past actions, is socially withdrawn, and shows a blunted, depressed affect. He also complains of insomnia despite taking mirtazapine at bedtime.
HISTORY: Depression and drugs
Mr. B says he has felt depressed on and off since his teens, and his current episode has been continuously severe for 1½ years. He began abusing alcohol and benzodiazepines during this episode but says he has been clean and sober for 2 weeks. He tried to kill himself 2 other times over 6 months by overdosing on alprazolam and was hospitalized after both attempts. He has no history of mania or psychosis.
Mr. B also abused opioids. In college, he was prescribed codeine for back pain after a sports injury. He experienced profound relief from depression after his first dose and soon began abusing codeine and other opioids for mood effects, including diphenoxylate/atropine and “cough syrup.” He says he has never used heroin.
Twenty years of illicit opioid use destroyed Mr. B’s occupational and social functioning, leaving him unable to work in his chosen field. During that period, he was frequently unemployed, socially isolated, and unable to sustain romantic relationships.
At age 40, Mr. B entered a methadone program, began working steadily, and got married. Five years later, he tapered off methadone and to our knowledge remained continuously opioid-free until presentation. Mr. B’s depression persisted while using opioids and worsened after stopping methadone. He also completed an 8-week residential substance abuse treatment program several months before presentation.
HISTORY: Family problems
Mr. B says he was emotionally abused as a child and described his father as excessively rageful. He says he entered a highly skilled profession to please his father but did not enjoy it and has not worked in the field since his early 30s. He has been unemployed for 1 year because his depression makes him feel “unworthy” to work.
The patient’s marriage of 10 years has been riddled with conflict. His depression, substance abuse, suicidality, and unemployment have fueled his wife’s resentment and anger.
The authors’ observations
Mr. B’s depression is challenging because of its severity and many possible causes and perpetuating factors. In addition to acute psychological stress and recent alcohol and benzodiazepine abuse, he has endured long-term opioid addiction. Although he had stayed opioid-free for 5 years, his past addiction contributed to his depression.
Whether Mr. B’s depression or opioid dependence came first is unclear. Either way, past opioid dependence can worsen depression prognosis.1 Opioid dependence might cause a withdrawal state that lasts years after acute withdrawal has subsided, although some researchers dispute this concept.2 According to Gold et al,3 long-term opioid use can cause endogenous opioid system derangements and depression after exogenous opioid use has ceased.
Depression is difficult to diagnose unambiguously in patients who have been using alcohol or anxiolytics because these CNS depressants’ effects might mimic depression. Patients whose symptoms suggest dual disorders commonly alternate between traditional psychiatric interventions and chemical dependence treatment.
As with Mr. B, a patient who abstains from 1 substance might start abusing another. This “replacement” is part of an “addiction interaction” theory that recognizes multiple substance and/or behavioral addictions in a patient.4 “Replacement” addiction indicates that substance abuse therapy is not adequately addressing some issues.
Coordinating concurrent depression and substance abuse treatment is critical. Although Mr. B’s ongoing psychosocial stress was addressed to varying degrees, endogenous opioid system derangements and/or prolonged opioid withdrawal may have been missed.
TREATMENT: Medication change
We discontinue methylphenidate because it is causing anxiety while leaving Mr. B’s depression unabated. Also, methylphenidate can be addictive.
Over several weeks, we titrate venlafaxine to 300 mg/d and continue mirtazapine, 30 mg at bedtime. We start weekly individual psychotherapy and encourage Mr. B to regularly attend Alcoholics Anonymous (AA) meetings, which he had been attending intermittently for years.
After 1 month, Mr. B’s depression improves marginally, but his depressed mood, anergia, and flat affect persist. He has not relapsed into alcohol or benzodiazepine dependence but reports occasional cravings for opioids and longs for the profound antidepressant effect they once gave him.
The authors’ observations
Sublingual buprenorphine is not FDA-approved to treat depression, although several small studies have described its antidepressant efficacy.5-7 How exogenous opioids reduce depressive symptoms is unknown, although some researchers believe that endogenous opioids:
- work with the mesolimbic dopaminergic system to mediate pleasure and reward
- modulate the mesolimbic system
- or have the same attenuating effect on both psychic and physical pain.
Buprenorphine also is a kappa receptor antagonist, which might explain its antidepressant efficacy.11 Whereas full mu agonism mediates euphoria, kappa receptor agonism results in dysphoria. By contrast, kappa receptor antagonism might cause a more stable, noneuphoric antidepressant effect.
Based on Mr. B’s clinical status, we ask him to consider sublingual buprenorphine/naloxone to treat depression and prevent relapse to opioid addiction.
The authors’ observations
Mr. B’s opioid addiction history and type of depression support buprenorphine augmentation. Whereas switching antidepressants or starting ECT would address only his persistent depression, buprenorphine also would target his opioid craving.
Numerous conventional psychotropics have not alleviated Mr. B’s depression, and changing antidepressants might nullify his small gains over the past month. We might consider ECT if buprenorphine does not reduce his depression.
Doctors need to obtain a waiver from the Drug Enforcement Administration (DEA) before using buprenorphine to treat opioid dependence—its approved indication (Box 1). This waiver is not necessary for off-label buprenorphine use. We needed the DEA waiver for Mr. B because we were using buprenorphine to treat opioid relapse prevention as well as depression. To prescribe buprenorphine without a DEA waiver, document that you are using the drug only for the off-label purpose.
The Drug Enforcement Administration (DEA) requires physicians to obtain a waiver to use buprenorphine to treat opioid dependence in outpatients. This waiver exempts outpatient practitioners from the DEA requirement that only specially licensed opioid treatment programs—such as methadone clinics—can dispense opioid medications.
To obtain the waiver, a physician must:
- show competency to use buprenorphine—usually by completing an 8-hour training course
- certify that he/she can conveniently refer patients for psychosocial treatment.
To receive DEA-approved buprenorphine training, in person or online, contact:
- American Society of Addiction Medicine. (888) 362-6784, www.asam.org/BuprenorphineCME.html
- American Academy of Addiction Psychiatry. (401) 524-3076, www.aaap.org/buprenorphine/buprenorphine.htm
- American Psychiatric Association. (703) 907-7300, www.psych.org/edu/bup_training.cfm
- American Osteopathic Academy of Addiction Medicine. (800) 621-1773, ext. 8163, www.aoaam.org.
For information on obtaining the waiver, visit www.buprenorphine.samhsa.gov.
Buprenorphine risks
Overdose. Buprenorphine can be abused by grinding and dissolving tablets, then injecting them intravenously. Doing this while under the influence of benzodiazepines or other sedatives can cause respiratory depression, leading to coma or death.
Combination buprenorphine/naloxone carries a much lower risk of IV overdose than buprenorphine alone because naloxone blocks mu opioid receptors. This formulation was created specifically to prevent buprenorphine misuse. Because naloxone is metabolized hepatically, it is not pharmacologically active when taken orally and will not block buprenorphine’s effect when buprenorphine/naloxone is taken as prescribed.
Physical dependence and withdrawal. Long-term buprenorphine use can cause physical dependence. Abrupt discontinuation or excessively high doses can precipitate withdrawal. How withdrawal is precipitated is unclear, although some believe the drug displaces itself from mu receptors when doses are too high. Myalgia, headache, abdominal discomfort, rhinorrhea, anxiety, and irritability are common buprenorphine withdrawal symptoms. The dosage at which the drug precipitates withdrawal varies with each patient’s tolerance for opioids.
When stopping buprenorphine therapy, taper the medication gradually to minimize withdrawal discomfort and relapse risk. Start tapering by 2 mg per month, then taper more rapidly or slowly based on the patient’s subjective experience.
TREATMENT: An opioid option
After discussing the risks and benefits with Mr. B and his wife, we add buprenorphine/naloxone, 8 mg/d, then increase it to 16 mg/d the next day. He tolerates the medication, and within 1 week his anergia disappears and he feels more motivated and productive. He reports no euphoria from buprenorphine but says it decreases his craving for alcohol, benzodiazepines, and opioids.
We continue buprenorphine/naloxone, 16 mg/d, and mirtazapine, 30 mg at bedtime, and reduce venlafaxine to 225 mg/d to mitigate sexual side effects. During weekly individual psychotherapy, we target Mr. B’s marital conflict and low self-esteem, and instruct him on overcoming life obstacles such as unemployment. He is looking for work and attends AA approximately 5 times a week.
Remember these 8 steps
- Address depression and substance abuse concurrently
- Communicate regularly with other providers about progress on depression and substance abuse issues
- Recommend and support involvement in 12-step programs such as AA
- Use medications for both depression—such as antidepressants—and relapse prevention—such as naltrexone, acamprosate, or buprenorphine/naloxone
- Explore family history of addiction and how this affected the patient developmentally. Find out if depression and substance abuse had common causes; this helps the patient realize that he/she did not become depressed or addicted by choice
- Ask about and discuss multiple addictions that were not initially reported
- Help the patient express, tolerate, and experience difficult feelings rather than avoid them
- Empathize with the patient; express understanding that factors out of the patient’s control caused depression and addiction
The authors’ observations
Considering the tumultuousness of Mr. B’s life, his willingness to enter psychotherapy and address underlying issues is significant. Adding buprenorphine to his antidepressant regimen helped stabilize his mood and make psychotherapy possible.
Psychotropics have not induced total remission of Mr. B’s depression, which is multifactorial and requires multimodal treatment. Still, we consider buprenorphine therapy at least partially successful—he has gone 6 months without attempting suicide or requiring psychiatric hospitalization.
Some clinicians consider buprenorphine’s potential for physical dependence a drawback to depression therapy. Physical dependence on a psychotropic does not necessarily outweigh its benefit in severe depression. Indeed, patients with depression can experience discontinuation symptoms from selective serotonin reuptake inhibitors and withdrawal from benzodiazepines.2,12
FOLLOW-UP: ‘Bup’ stigma
Mr. B feels stigmatized about buprenorphine use, partly because his wife shames him for his history of addiction and views buprenorphine as a constant reminder of his “failures.”
Mrs. B’s dysfunctional attitude leaves Mr. B too ashamed to tell his fellow AA members that he takes buprenorphine. His inability to share these feelings also diminishes his sense of belonging in the 12-step fellowship. Even so, he feels that buprenorphine has helped him tremendously and wants to continue taking it.
During psychotherapy, we address Mr. B’s buprenorphine-related stigma and pervasive shame stemming from his history of mental illness, addiction, inability to work in his chosen field, and past employment failures. We encourage him to overcome his shame by pointing out his strengths—such as the skills he can offer potential employers—and by emphasizing that he did not choose to become depressed and addicted.
The authors’ observations
Most patients addicted to opiates feel much less stigmatized by buprenorphine therapy than by methadone. Patients who feel shame while taking buprenorphine usually are reacting to past opioid addiction rather than current therapy. Mr. B’s buprenorphine-related shame stems from his personality structure.
Shame, however, could create negative expectations of buprenorphine therapy, and can lower some patients’ self-esteem to the point that they feel they do not deserve to get better. Some patients stop buprenorphine prematurely because they believe they have beaten the addiction, but this often leads to relapse to the previous opioid of choice.
Help patients work through the shame of past addiction and encourage them to view buprenorphine therapy as a positive step toward recovery (Box 2). As mental health professionals, we must not collude with society to shame people with past chemical addiction. Creatively yet responsibly broadening our perspective toward psychiatric intervention can help patients such as Mr. B receive optimal treatment.
Although members of a 12-step group might harbor an idiosyncratic position on medications or treatment, cooperation with professionals is the program’s mainstream stance. Ideally, combination pharmacotherapy, psychotherapy, and guidance for optimal use of support groups can provide a stable foundation for recovery from both psychiatric and addictive disorders.
Related resources
- U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Substance Abuse Treatment Knowledge Application Program, Treatment Improvement Protocol Series. www.kap.samhsa.gov/products/manuals/tips/index.htm.
- Acamprosate • Campral
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Bupropion • Wellbutrin
- Diphenoxylate/atropine • Lomotil
- Methadone • Dolophine
- Methylphenidate • Ritalin, Concerta
- Mirtazapine • Remeron
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
Dr. Roth is a speaker for Reckitt Benckiser.
Drs. Eiger and Tan report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nunes EV, Sullivan MA, Levin FR. Treatment of depression in patients with opiate dependence. Biol Psychiatry 2004;56:793-802.
2. Graham AW, Schultz TK, Mayo-Smith MF, et al, eds. Principles of addiction medicine. 3rd ed. Chevy Chase, MD: American Society of Addiction Medicine; 2003.
3. Gold MS, Pottash AL, Extein I, et al. Evidence for an endorphin dysfunction in methadone addicts: lack of ACTH response to naloxone. Drug Alcohol Depend 1981;8:257-62.
4. Carnes PJ, Murray RE, Charpentier L. Addiction interaction disorder. In: Coombs RH, ed. Handbook of addictive disorders: a practical guide to diagnosis and treatment. Hoboken, NJ: John Wiley & Sons 2004:31-59.
5. Kosten TR, Morgan C, Kosten TA. Depressive symptoms during buprenorphine treatment of opioid abusers. J Subst Abuse Treat. 1990;7:51-4.
6. Dean AJ, Bell J, Christie MJ, Mattick RP. Depressive symptoms during buprenorphine vs. methadone maintenance: findings from a randomized, controlled trial in opioid dependence. Eur Psychiatry. 2004;19:510-13.
7. Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15:49-57.
8. Jaffe JH, Jaffe AB. Neurobiology of opioids. In: Galanter M, Kleber HD, eds. Textbook of substance abuse treatment.. 3rd ed. Washington, DC: American Psychiatric Publishing; 2004:17-30.
9. Jones HE. Practical considerations for the clinical use of buprenorphine. NIDA Sci Pract Perspectives. 2004;2:4-20.
10. Geppert CM, Toney GB, Siracusano D, Thorius M. Outpatient buprenorphine treatment for opioid dependence. Fed Practitioner. 2005;22:9-40.
11. Mague SD, Pliakas AM, Todtenkopf MS, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323-30.
12. Van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt continuation. Eur J Clin Pharmacol. 2005;61:303-7.
CASE: Near-fatal combination
Inpatient psychiatry refers Mr. B, age 50, to our outpatient psychiatry clinic. Two weeks earlier, he tried to kill himself by sitting on a stepladder, tying a noose around his neck, and consuming large amounts of quetiapine, trazodone, and vodka. His wife found him unconscious on the floor with facial abrasions, empty pill bottles, and the noose lying next to him.
Emergency medical personnel brought Mr. B to the ER. His total Glasgow Coma Scale score of 3 indicated he was comatose. Pulse was 65 bpm (low-normal), and blood alcohol level was 106 mg/dL, suggesting he had ingested hazardous amounts of vodka. Quetiapine and trazodone blood levels were not measured.
Gastric lavage was unsuccessful because the orogastric tube became curled in the distal esophagus. Mr. B was successfully intubated and admitted to the intensive care unit. After 2 days, he was medically stable and regained consciousness, though he was delirious. He was transferred to inpatient psychiatry, where the attending psychiatrist diagnosed major depression and alcohol abuse disorder.
Before presentation, Mr. B had been taking venlafaxine, 75 mg/d, and mirtazapine, 30 mg at bedtime. His previous outpatient psychiatrist had added methylphenidate, 40 mg/d, to augment the antidepressants—which were not alleviating his depression—and the attending continued all 3 medications. Prior trials of sertraline, bupropion, trazodone, quetiapine, and aripiprazole were ineffective.
By the time Mr. B is transferred to us, his suicidal thoughts have remitted but he is still notably depressed. He is anergic, feels hopeless about the future, has markedly diminished self-worth, feels excessively guilty over past actions, is socially withdrawn, and shows a blunted, depressed affect. He also complains of insomnia despite taking mirtazapine at bedtime.
HISTORY: Depression and drugs
Mr. B says he has felt depressed on and off since his teens, and his current episode has been continuously severe for 1½ years. He began abusing alcohol and benzodiazepines during this episode but says he has been clean and sober for 2 weeks. He tried to kill himself 2 other times over 6 months by overdosing on alprazolam and was hospitalized after both attempts. He has no history of mania or psychosis.
Mr. B also abused opioids. In college, he was prescribed codeine for back pain after a sports injury. He experienced profound relief from depression after his first dose and soon began abusing codeine and other opioids for mood effects, including diphenoxylate/atropine and “cough syrup.” He says he has never used heroin.
Twenty years of illicit opioid use destroyed Mr. B’s occupational and social functioning, leaving him unable to work in his chosen field. During that period, he was frequently unemployed, socially isolated, and unable to sustain romantic relationships.
At age 40, Mr. B entered a methadone program, began working steadily, and got married. Five years later, he tapered off methadone and to our knowledge remained continuously opioid-free until presentation. Mr. B’s depression persisted while using opioids and worsened after stopping methadone. He also completed an 8-week residential substance abuse treatment program several months before presentation.
HISTORY: Family problems
Mr. B says he was emotionally abused as a child and described his father as excessively rageful. He says he entered a highly skilled profession to please his father but did not enjoy it and has not worked in the field since his early 30s. He has been unemployed for 1 year because his depression makes him feel “unworthy” to work.
The patient’s marriage of 10 years has been riddled with conflict. His depression, substance abuse, suicidality, and unemployment have fueled his wife’s resentment and anger.
The authors’ observations
Mr. B’s depression is challenging because of its severity and many possible causes and perpetuating factors. In addition to acute psychological stress and recent alcohol and benzodiazepine abuse, he has endured long-term opioid addiction. Although he had stayed opioid-free for 5 years, his past addiction contributed to his depression.
Whether Mr. B’s depression or opioid dependence came first is unclear. Either way, past opioid dependence can worsen depression prognosis.1 Opioid dependence might cause a withdrawal state that lasts years after acute withdrawal has subsided, although some researchers dispute this concept.2 According to Gold et al,3 long-term opioid use can cause endogenous opioid system derangements and depression after exogenous opioid use has ceased.
Depression is difficult to diagnose unambiguously in patients who have been using alcohol or anxiolytics because these CNS depressants’ effects might mimic depression. Patients whose symptoms suggest dual disorders commonly alternate between traditional psychiatric interventions and chemical dependence treatment.
As with Mr. B, a patient who abstains from 1 substance might start abusing another. This “replacement” is part of an “addiction interaction” theory that recognizes multiple substance and/or behavioral addictions in a patient.4 “Replacement” addiction indicates that substance abuse therapy is not adequately addressing some issues.
Coordinating concurrent depression and substance abuse treatment is critical. Although Mr. B’s ongoing psychosocial stress was addressed to varying degrees, endogenous opioid system derangements and/or prolonged opioid withdrawal may have been missed.
TREATMENT: Medication change
We discontinue methylphenidate because it is causing anxiety while leaving Mr. B’s depression unabated. Also, methylphenidate can be addictive.
Over several weeks, we titrate venlafaxine to 300 mg/d and continue mirtazapine, 30 mg at bedtime. We start weekly individual psychotherapy and encourage Mr. B to regularly attend Alcoholics Anonymous (AA) meetings, which he had been attending intermittently for years.
After 1 month, Mr. B’s depression improves marginally, but his depressed mood, anergia, and flat affect persist. He has not relapsed into alcohol or benzodiazepine dependence but reports occasional cravings for opioids and longs for the profound antidepressant effect they once gave him.
The authors’ observations
Sublingual buprenorphine is not FDA-approved to treat depression, although several small studies have described its antidepressant efficacy.5-7 How exogenous opioids reduce depressive symptoms is unknown, although some researchers believe that endogenous opioids:
- work with the mesolimbic dopaminergic system to mediate pleasure and reward
- modulate the mesolimbic system
- or have the same attenuating effect on both psychic and physical pain.
Buprenorphine also is a kappa receptor antagonist, which might explain its antidepressant efficacy.11 Whereas full mu agonism mediates euphoria, kappa receptor agonism results in dysphoria. By contrast, kappa receptor antagonism might cause a more stable, noneuphoric antidepressant effect.
Based on Mr. B’s clinical status, we ask him to consider sublingual buprenorphine/naloxone to treat depression and prevent relapse to opioid addiction.
The authors’ observations
Mr. B’s opioid addiction history and type of depression support buprenorphine augmentation. Whereas switching antidepressants or starting ECT would address only his persistent depression, buprenorphine also would target his opioid craving.
Numerous conventional psychotropics have not alleviated Mr. B’s depression, and changing antidepressants might nullify his small gains over the past month. We might consider ECT if buprenorphine does not reduce his depression.
Doctors need to obtain a waiver from the Drug Enforcement Administration (DEA) before using buprenorphine to treat opioid dependence—its approved indication (Box 1). This waiver is not necessary for off-label buprenorphine use. We needed the DEA waiver for Mr. B because we were using buprenorphine to treat opioid relapse prevention as well as depression. To prescribe buprenorphine without a DEA waiver, document that you are using the drug only for the off-label purpose.
The Drug Enforcement Administration (DEA) requires physicians to obtain a waiver to use buprenorphine to treat opioid dependence in outpatients. This waiver exempts outpatient practitioners from the DEA requirement that only specially licensed opioid treatment programs—such as methadone clinics—can dispense opioid medications.
To obtain the waiver, a physician must:
- show competency to use buprenorphine—usually by completing an 8-hour training course
- certify that he/she can conveniently refer patients for psychosocial treatment.
To receive DEA-approved buprenorphine training, in person or online, contact:
- American Society of Addiction Medicine. (888) 362-6784, www.asam.org/BuprenorphineCME.html
- American Academy of Addiction Psychiatry. (401) 524-3076, www.aaap.org/buprenorphine/buprenorphine.htm
- American Psychiatric Association. (703) 907-7300, www.psych.org/edu/bup_training.cfm
- American Osteopathic Academy of Addiction Medicine. (800) 621-1773, ext. 8163, www.aoaam.org.
For information on obtaining the waiver, visit www.buprenorphine.samhsa.gov.
Buprenorphine risks
Overdose. Buprenorphine can be abused by grinding and dissolving tablets, then injecting them intravenously. Doing this while under the influence of benzodiazepines or other sedatives can cause respiratory depression, leading to coma or death.
Combination buprenorphine/naloxone carries a much lower risk of IV overdose than buprenorphine alone because naloxone blocks mu opioid receptors. This formulation was created specifically to prevent buprenorphine misuse. Because naloxone is metabolized hepatically, it is not pharmacologically active when taken orally and will not block buprenorphine’s effect when buprenorphine/naloxone is taken as prescribed.
Physical dependence and withdrawal. Long-term buprenorphine use can cause physical dependence. Abrupt discontinuation or excessively high doses can precipitate withdrawal. How withdrawal is precipitated is unclear, although some believe the drug displaces itself from mu receptors when doses are too high. Myalgia, headache, abdominal discomfort, rhinorrhea, anxiety, and irritability are common buprenorphine withdrawal symptoms. The dosage at which the drug precipitates withdrawal varies with each patient’s tolerance for opioids.
When stopping buprenorphine therapy, taper the medication gradually to minimize withdrawal discomfort and relapse risk. Start tapering by 2 mg per month, then taper more rapidly or slowly based on the patient’s subjective experience.
TREATMENT: An opioid option
After discussing the risks and benefits with Mr. B and his wife, we add buprenorphine/naloxone, 8 mg/d, then increase it to 16 mg/d the next day. He tolerates the medication, and within 1 week his anergia disappears and he feels more motivated and productive. He reports no euphoria from buprenorphine but says it decreases his craving for alcohol, benzodiazepines, and opioids.
We continue buprenorphine/naloxone, 16 mg/d, and mirtazapine, 30 mg at bedtime, and reduce venlafaxine to 225 mg/d to mitigate sexual side effects. During weekly individual psychotherapy, we target Mr. B’s marital conflict and low self-esteem, and instruct him on overcoming life obstacles such as unemployment. He is looking for work and attends AA approximately 5 times a week.
Remember these 8 steps
- Address depression and substance abuse concurrently
- Communicate regularly with other providers about progress on depression and substance abuse issues
- Recommend and support involvement in 12-step programs such as AA
- Use medications for both depression—such as antidepressants—and relapse prevention—such as naltrexone, acamprosate, or buprenorphine/naloxone
- Explore family history of addiction and how this affected the patient developmentally. Find out if depression and substance abuse had common causes; this helps the patient realize that he/she did not become depressed or addicted by choice
- Ask about and discuss multiple addictions that were not initially reported
- Help the patient express, tolerate, and experience difficult feelings rather than avoid them
- Empathize with the patient; express understanding that factors out of the patient’s control caused depression and addiction
The authors’ observations
Considering the tumultuousness of Mr. B’s life, his willingness to enter psychotherapy and address underlying issues is significant. Adding buprenorphine to his antidepressant regimen helped stabilize his mood and make psychotherapy possible.
Psychotropics have not induced total remission of Mr. B’s depression, which is multifactorial and requires multimodal treatment. Still, we consider buprenorphine therapy at least partially successful—he has gone 6 months without attempting suicide or requiring psychiatric hospitalization.
Some clinicians consider buprenorphine’s potential for physical dependence a drawback to depression therapy. Physical dependence on a psychotropic does not necessarily outweigh its benefit in severe depression. Indeed, patients with depression can experience discontinuation symptoms from selective serotonin reuptake inhibitors and withdrawal from benzodiazepines.2,12
FOLLOW-UP: ‘Bup’ stigma
Mr. B feels stigmatized about buprenorphine use, partly because his wife shames him for his history of addiction and views buprenorphine as a constant reminder of his “failures.”
Mrs. B’s dysfunctional attitude leaves Mr. B too ashamed to tell his fellow AA members that he takes buprenorphine. His inability to share these feelings also diminishes his sense of belonging in the 12-step fellowship. Even so, he feels that buprenorphine has helped him tremendously and wants to continue taking it.
During psychotherapy, we address Mr. B’s buprenorphine-related stigma and pervasive shame stemming from his history of mental illness, addiction, inability to work in his chosen field, and past employment failures. We encourage him to overcome his shame by pointing out his strengths—such as the skills he can offer potential employers—and by emphasizing that he did not choose to become depressed and addicted.
The authors’ observations
Most patients addicted to opiates feel much less stigmatized by buprenorphine therapy than by methadone. Patients who feel shame while taking buprenorphine usually are reacting to past opioid addiction rather than current therapy. Mr. B’s buprenorphine-related shame stems from his personality structure.
Shame, however, could create negative expectations of buprenorphine therapy, and can lower some patients’ self-esteem to the point that they feel they do not deserve to get better. Some patients stop buprenorphine prematurely because they believe they have beaten the addiction, but this often leads to relapse to the previous opioid of choice.
Help patients work through the shame of past addiction and encourage them to view buprenorphine therapy as a positive step toward recovery (Box 2). As mental health professionals, we must not collude with society to shame people with past chemical addiction. Creatively yet responsibly broadening our perspective toward psychiatric intervention can help patients such as Mr. B receive optimal treatment.
Although members of a 12-step group might harbor an idiosyncratic position on medications or treatment, cooperation with professionals is the program’s mainstream stance. Ideally, combination pharmacotherapy, psychotherapy, and guidance for optimal use of support groups can provide a stable foundation for recovery from both psychiatric and addictive disorders.
Related resources
- U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Substance Abuse Treatment Knowledge Application Program, Treatment Improvement Protocol Series. www.kap.samhsa.gov/products/manuals/tips/index.htm.
- Acamprosate • Campral
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Bupropion • Wellbutrin
- Diphenoxylate/atropine • Lomotil
- Methadone • Dolophine
- Methylphenidate • Ritalin, Concerta
- Mirtazapine • Remeron
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
Dr. Roth is a speaker for Reckitt Benckiser.
Drs. Eiger and Tan report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
CASE: Near-fatal combination
Inpatient psychiatry refers Mr. B, age 50, to our outpatient psychiatry clinic. Two weeks earlier, he tried to kill himself by sitting on a stepladder, tying a noose around his neck, and consuming large amounts of quetiapine, trazodone, and vodka. His wife found him unconscious on the floor with facial abrasions, empty pill bottles, and the noose lying next to him.
Emergency medical personnel brought Mr. B to the ER. His total Glasgow Coma Scale score of 3 indicated he was comatose. Pulse was 65 bpm (low-normal), and blood alcohol level was 106 mg/dL, suggesting he had ingested hazardous amounts of vodka. Quetiapine and trazodone blood levels were not measured.
Gastric lavage was unsuccessful because the orogastric tube became curled in the distal esophagus. Mr. B was successfully intubated and admitted to the intensive care unit. After 2 days, he was medically stable and regained consciousness, though he was delirious. He was transferred to inpatient psychiatry, where the attending psychiatrist diagnosed major depression and alcohol abuse disorder.
Before presentation, Mr. B had been taking venlafaxine, 75 mg/d, and mirtazapine, 30 mg at bedtime. His previous outpatient psychiatrist had added methylphenidate, 40 mg/d, to augment the antidepressants—which were not alleviating his depression—and the attending continued all 3 medications. Prior trials of sertraline, bupropion, trazodone, quetiapine, and aripiprazole were ineffective.
By the time Mr. B is transferred to us, his suicidal thoughts have remitted but he is still notably depressed. He is anergic, feels hopeless about the future, has markedly diminished self-worth, feels excessively guilty over past actions, is socially withdrawn, and shows a blunted, depressed affect. He also complains of insomnia despite taking mirtazapine at bedtime.
HISTORY: Depression and drugs
Mr. B says he has felt depressed on and off since his teens, and his current episode has been continuously severe for 1½ years. He began abusing alcohol and benzodiazepines during this episode but says he has been clean and sober for 2 weeks. He tried to kill himself 2 other times over 6 months by overdosing on alprazolam and was hospitalized after both attempts. He has no history of mania or psychosis.
Mr. B also abused opioids. In college, he was prescribed codeine for back pain after a sports injury. He experienced profound relief from depression after his first dose and soon began abusing codeine and other opioids for mood effects, including diphenoxylate/atropine and “cough syrup.” He says he has never used heroin.
Twenty years of illicit opioid use destroyed Mr. B’s occupational and social functioning, leaving him unable to work in his chosen field. During that period, he was frequently unemployed, socially isolated, and unable to sustain romantic relationships.
At age 40, Mr. B entered a methadone program, began working steadily, and got married. Five years later, he tapered off methadone and to our knowledge remained continuously opioid-free until presentation. Mr. B’s depression persisted while using opioids and worsened after stopping methadone. He also completed an 8-week residential substance abuse treatment program several months before presentation.
HISTORY: Family problems
Mr. B says he was emotionally abused as a child and described his father as excessively rageful. He says he entered a highly skilled profession to please his father but did not enjoy it and has not worked in the field since his early 30s. He has been unemployed for 1 year because his depression makes him feel “unworthy” to work.
The patient’s marriage of 10 years has been riddled with conflict. His depression, substance abuse, suicidality, and unemployment have fueled his wife’s resentment and anger.
The authors’ observations
Mr. B’s depression is challenging because of its severity and many possible causes and perpetuating factors. In addition to acute psychological stress and recent alcohol and benzodiazepine abuse, he has endured long-term opioid addiction. Although he had stayed opioid-free for 5 years, his past addiction contributed to his depression.
Whether Mr. B’s depression or opioid dependence came first is unclear. Either way, past opioid dependence can worsen depression prognosis.1 Opioid dependence might cause a withdrawal state that lasts years after acute withdrawal has subsided, although some researchers dispute this concept.2 According to Gold et al,3 long-term opioid use can cause endogenous opioid system derangements and depression after exogenous opioid use has ceased.
Depression is difficult to diagnose unambiguously in patients who have been using alcohol or anxiolytics because these CNS depressants’ effects might mimic depression. Patients whose symptoms suggest dual disorders commonly alternate between traditional psychiatric interventions and chemical dependence treatment.
As with Mr. B, a patient who abstains from 1 substance might start abusing another. This “replacement” is part of an “addiction interaction” theory that recognizes multiple substance and/or behavioral addictions in a patient.4 “Replacement” addiction indicates that substance abuse therapy is not adequately addressing some issues.
Coordinating concurrent depression and substance abuse treatment is critical. Although Mr. B’s ongoing psychosocial stress was addressed to varying degrees, endogenous opioid system derangements and/or prolonged opioid withdrawal may have been missed.
TREATMENT: Medication change
We discontinue methylphenidate because it is causing anxiety while leaving Mr. B’s depression unabated. Also, methylphenidate can be addictive.
Over several weeks, we titrate venlafaxine to 300 mg/d and continue mirtazapine, 30 mg at bedtime. We start weekly individual psychotherapy and encourage Mr. B to regularly attend Alcoholics Anonymous (AA) meetings, which he had been attending intermittently for years.
After 1 month, Mr. B’s depression improves marginally, but his depressed mood, anergia, and flat affect persist. He has not relapsed into alcohol or benzodiazepine dependence but reports occasional cravings for opioids and longs for the profound antidepressant effect they once gave him.
The authors’ observations
Sublingual buprenorphine is not FDA-approved to treat depression, although several small studies have described its antidepressant efficacy.5-7 How exogenous opioids reduce depressive symptoms is unknown, although some researchers believe that endogenous opioids:
- work with the mesolimbic dopaminergic system to mediate pleasure and reward
- modulate the mesolimbic system
- or have the same attenuating effect on both psychic and physical pain.
Buprenorphine also is a kappa receptor antagonist, which might explain its antidepressant efficacy.11 Whereas full mu agonism mediates euphoria, kappa receptor agonism results in dysphoria. By contrast, kappa receptor antagonism might cause a more stable, noneuphoric antidepressant effect.
Based on Mr. B’s clinical status, we ask him to consider sublingual buprenorphine/naloxone to treat depression and prevent relapse to opioid addiction.
The authors’ observations
Mr. B’s opioid addiction history and type of depression support buprenorphine augmentation. Whereas switching antidepressants or starting ECT would address only his persistent depression, buprenorphine also would target his opioid craving.
Numerous conventional psychotropics have not alleviated Mr. B’s depression, and changing antidepressants might nullify his small gains over the past month. We might consider ECT if buprenorphine does not reduce his depression.
Doctors need to obtain a waiver from the Drug Enforcement Administration (DEA) before using buprenorphine to treat opioid dependence—its approved indication (Box 1). This waiver is not necessary for off-label buprenorphine use. We needed the DEA waiver for Mr. B because we were using buprenorphine to treat opioid relapse prevention as well as depression. To prescribe buprenorphine without a DEA waiver, document that you are using the drug only for the off-label purpose.
The Drug Enforcement Administration (DEA) requires physicians to obtain a waiver to use buprenorphine to treat opioid dependence in outpatients. This waiver exempts outpatient practitioners from the DEA requirement that only specially licensed opioid treatment programs—such as methadone clinics—can dispense opioid medications.
To obtain the waiver, a physician must:
- show competency to use buprenorphine—usually by completing an 8-hour training course
- certify that he/she can conveniently refer patients for psychosocial treatment.
To receive DEA-approved buprenorphine training, in person or online, contact:
- American Society of Addiction Medicine. (888) 362-6784, www.asam.org/BuprenorphineCME.html
- American Academy of Addiction Psychiatry. (401) 524-3076, www.aaap.org/buprenorphine/buprenorphine.htm
- American Psychiatric Association. (703) 907-7300, www.psych.org/edu/bup_training.cfm
- American Osteopathic Academy of Addiction Medicine. (800) 621-1773, ext. 8163, www.aoaam.org.
For information on obtaining the waiver, visit www.buprenorphine.samhsa.gov.
Buprenorphine risks
Overdose. Buprenorphine can be abused by grinding and dissolving tablets, then injecting them intravenously. Doing this while under the influence of benzodiazepines or other sedatives can cause respiratory depression, leading to coma or death.
Combination buprenorphine/naloxone carries a much lower risk of IV overdose than buprenorphine alone because naloxone blocks mu opioid receptors. This formulation was created specifically to prevent buprenorphine misuse. Because naloxone is metabolized hepatically, it is not pharmacologically active when taken orally and will not block buprenorphine’s effect when buprenorphine/naloxone is taken as prescribed.
Physical dependence and withdrawal. Long-term buprenorphine use can cause physical dependence. Abrupt discontinuation or excessively high doses can precipitate withdrawal. How withdrawal is precipitated is unclear, although some believe the drug displaces itself from mu receptors when doses are too high. Myalgia, headache, abdominal discomfort, rhinorrhea, anxiety, and irritability are common buprenorphine withdrawal symptoms. The dosage at which the drug precipitates withdrawal varies with each patient’s tolerance for opioids.
When stopping buprenorphine therapy, taper the medication gradually to minimize withdrawal discomfort and relapse risk. Start tapering by 2 mg per month, then taper more rapidly or slowly based on the patient’s subjective experience.
TREATMENT: An opioid option
After discussing the risks and benefits with Mr. B and his wife, we add buprenorphine/naloxone, 8 mg/d, then increase it to 16 mg/d the next day. He tolerates the medication, and within 1 week his anergia disappears and he feels more motivated and productive. He reports no euphoria from buprenorphine but says it decreases his craving for alcohol, benzodiazepines, and opioids.
We continue buprenorphine/naloxone, 16 mg/d, and mirtazapine, 30 mg at bedtime, and reduce venlafaxine to 225 mg/d to mitigate sexual side effects. During weekly individual psychotherapy, we target Mr. B’s marital conflict and low self-esteem, and instruct him on overcoming life obstacles such as unemployment. He is looking for work and attends AA approximately 5 times a week.
Remember these 8 steps
- Address depression and substance abuse concurrently
- Communicate regularly with other providers about progress on depression and substance abuse issues
- Recommend and support involvement in 12-step programs such as AA
- Use medications for both depression—such as antidepressants—and relapse prevention—such as naltrexone, acamprosate, or buprenorphine/naloxone
- Explore family history of addiction and how this affected the patient developmentally. Find out if depression and substance abuse had common causes; this helps the patient realize that he/she did not become depressed or addicted by choice
- Ask about and discuss multiple addictions that were not initially reported
- Help the patient express, tolerate, and experience difficult feelings rather than avoid them
- Empathize with the patient; express understanding that factors out of the patient’s control caused depression and addiction
The authors’ observations
Considering the tumultuousness of Mr. B’s life, his willingness to enter psychotherapy and address underlying issues is significant. Adding buprenorphine to his antidepressant regimen helped stabilize his mood and make psychotherapy possible.
Psychotropics have not induced total remission of Mr. B’s depression, which is multifactorial and requires multimodal treatment. Still, we consider buprenorphine therapy at least partially successful—he has gone 6 months without attempting suicide or requiring psychiatric hospitalization.
Some clinicians consider buprenorphine’s potential for physical dependence a drawback to depression therapy. Physical dependence on a psychotropic does not necessarily outweigh its benefit in severe depression. Indeed, patients with depression can experience discontinuation symptoms from selective serotonin reuptake inhibitors and withdrawal from benzodiazepines.2,12
FOLLOW-UP: ‘Bup’ stigma
Mr. B feels stigmatized about buprenorphine use, partly because his wife shames him for his history of addiction and views buprenorphine as a constant reminder of his “failures.”
Mrs. B’s dysfunctional attitude leaves Mr. B too ashamed to tell his fellow AA members that he takes buprenorphine. His inability to share these feelings also diminishes his sense of belonging in the 12-step fellowship. Even so, he feels that buprenorphine has helped him tremendously and wants to continue taking it.
During psychotherapy, we address Mr. B’s buprenorphine-related stigma and pervasive shame stemming from his history of mental illness, addiction, inability to work in his chosen field, and past employment failures. We encourage him to overcome his shame by pointing out his strengths—such as the skills he can offer potential employers—and by emphasizing that he did not choose to become depressed and addicted.
The authors’ observations
Most patients addicted to opiates feel much less stigmatized by buprenorphine therapy than by methadone. Patients who feel shame while taking buprenorphine usually are reacting to past opioid addiction rather than current therapy. Mr. B’s buprenorphine-related shame stems from his personality structure.
Shame, however, could create negative expectations of buprenorphine therapy, and can lower some patients’ self-esteem to the point that they feel they do not deserve to get better. Some patients stop buprenorphine prematurely because they believe they have beaten the addiction, but this often leads to relapse to the previous opioid of choice.
Help patients work through the shame of past addiction and encourage them to view buprenorphine therapy as a positive step toward recovery (Box 2). As mental health professionals, we must not collude with society to shame people with past chemical addiction. Creatively yet responsibly broadening our perspective toward psychiatric intervention can help patients such as Mr. B receive optimal treatment.
Although members of a 12-step group might harbor an idiosyncratic position on medications or treatment, cooperation with professionals is the program’s mainstream stance. Ideally, combination pharmacotherapy, psychotherapy, and guidance for optimal use of support groups can provide a stable foundation for recovery from both psychiatric and addictive disorders.
Related resources
- U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Substance Abuse Treatment Knowledge Application Program, Treatment Improvement Protocol Series. www.kap.samhsa.gov/products/manuals/tips/index.htm.
- Acamprosate • Campral
- Alprazolam • Xanax
- Aripiprazole • Abilify
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Bupropion • Wellbutrin
- Diphenoxylate/atropine • Lomotil
- Methadone • Dolophine
- Methylphenidate • Ritalin, Concerta
- Mirtazapine • Remeron
- Naltrexone • ReVia, Vivitrol
- Quetiapine • Seroquel
- Sertraline • Zoloft
- Trazodone • Desyrel
- Venlafaxine • Effexor
Dr. Roth is a speaker for Reckitt Benckiser.
Drs. Eiger and Tan report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Nunes EV, Sullivan MA, Levin FR. Treatment of depression in patients with opiate dependence. Biol Psychiatry 2004;56:793-802.
2. Graham AW, Schultz TK, Mayo-Smith MF, et al, eds. Principles of addiction medicine. 3rd ed. Chevy Chase, MD: American Society of Addiction Medicine; 2003.
3. Gold MS, Pottash AL, Extein I, et al. Evidence for an endorphin dysfunction in methadone addicts: lack of ACTH response to naloxone. Drug Alcohol Depend 1981;8:257-62.
4. Carnes PJ, Murray RE, Charpentier L. Addiction interaction disorder. In: Coombs RH, ed. Handbook of addictive disorders: a practical guide to diagnosis and treatment. Hoboken, NJ: John Wiley & Sons 2004:31-59.
5. Kosten TR, Morgan C, Kosten TA. Depressive symptoms during buprenorphine treatment of opioid abusers. J Subst Abuse Treat. 1990;7:51-4.
6. Dean AJ, Bell J, Christie MJ, Mattick RP. Depressive symptoms during buprenorphine vs. methadone maintenance: findings from a randomized, controlled trial in opioid dependence. Eur Psychiatry. 2004;19:510-13.
7. Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15:49-57.
8. Jaffe JH, Jaffe AB. Neurobiology of opioids. In: Galanter M, Kleber HD, eds. Textbook of substance abuse treatment.. 3rd ed. Washington, DC: American Psychiatric Publishing; 2004:17-30.
9. Jones HE. Practical considerations for the clinical use of buprenorphine. NIDA Sci Pract Perspectives. 2004;2:4-20.
10. Geppert CM, Toney GB, Siracusano D, Thorius M. Outpatient buprenorphine treatment for opioid dependence. Fed Practitioner. 2005;22:9-40.
11. Mague SD, Pliakas AM, Todtenkopf MS, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323-30.
12. Van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt continuation. Eur J Clin Pharmacol. 2005;61:303-7.
1. Nunes EV, Sullivan MA, Levin FR. Treatment of depression in patients with opiate dependence. Biol Psychiatry 2004;56:793-802.
2. Graham AW, Schultz TK, Mayo-Smith MF, et al, eds. Principles of addiction medicine. 3rd ed. Chevy Chase, MD: American Society of Addiction Medicine; 2003.
3. Gold MS, Pottash AL, Extein I, et al. Evidence for an endorphin dysfunction in methadone addicts: lack of ACTH response to naloxone. Drug Alcohol Depend 1981;8:257-62.
4. Carnes PJ, Murray RE, Charpentier L. Addiction interaction disorder. In: Coombs RH, ed. Handbook of addictive disorders: a practical guide to diagnosis and treatment. Hoboken, NJ: John Wiley & Sons 2004:31-59.
5. Kosten TR, Morgan C, Kosten TA. Depressive symptoms during buprenorphine treatment of opioid abusers. J Subst Abuse Treat. 1990;7:51-4.
6. Dean AJ, Bell J, Christie MJ, Mattick RP. Depressive symptoms during buprenorphine vs. methadone maintenance: findings from a randomized, controlled trial in opioid dependence. Eur Psychiatry. 2004;19:510-13.
7. Bodkin JA, Zornberg GL, Lukas SE, Cole JO. Buprenorphine treatment of refractory depression. J Clin Psychopharmacol. 1995;15:49-57.
8. Jaffe JH, Jaffe AB. Neurobiology of opioids. In: Galanter M, Kleber HD, eds. Textbook of substance abuse treatment.. 3rd ed. Washington, DC: American Psychiatric Publishing; 2004:17-30.
9. Jones HE. Practical considerations for the clinical use of buprenorphine. NIDA Sci Pract Perspectives. 2004;2:4-20.
10. Geppert CM, Toney GB, Siracusano D, Thorius M. Outpatient buprenorphine treatment for opioid dependence. Fed Practitioner. 2005;22:9-40.
11. Mague SD, Pliakas AM, Todtenkopf MS, et al. Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323-30.
12. Van Geffen EC, Hugtenburg JG, Heerdink ER, et al. Discontinuation symptoms in users of selective serotonin reuptake inhibitors in clinical practice: tapering versus abrupt continuation. Eur J Clin Pharmacol. 2005;61:303-7.
Can you interpret confidence intervals? It’s not that difficult
Number needed to treat (NNT) is a measure of clinical effect that has been called medicine’s “secret stat”(Box 1).1,2 By itself, however, the NNT provides no information about whether a trial result is probably true (statistical significance). If a NNT is statistically significant, the confidence interval (CI) can tell you the range of numbers within which the truth probably lies.
In the March 2007 issue of Current Psychiatry, we described how to use NNT to interpret and apply research data in daily practice.3 In this article, we explain the “secrets” of NNT and CI by providing sample calculations and several figures for visual learning. For illustration, we analyze data from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) in schizophrenia, this time focusing on phase 2E—the efficacy pathway in which patients were randomly assigned to open-label clozapine or a double-blinded second-generation antipsychotic (SGA).4
Confidence intervals: Is the NNT statistically significant?
To find out a NNT’s statistical significance, you can examine the CI. A 95% CI means that the truth lies between the interval’s lower and upper bounds with a 95% probability.
Calculating CI. Although formulas to calculate the CI appear complicated,5 they are easily inserted into a Microsoft Excel-brand spreadsheet. Useful alternatives are online calculators (seeRelated Resources), which can be downloaded to your hand-held device or pocket PC.
Time magazine recently declared NNT as medicine’s “secret stat.”1 NNT allows us to place a number on how often we would see a difference between 2 interventions.
In a handbook on essentials of evidence-based clinical practice, Guyatt et al2 define NNT as “the number of patients who must receive an intervention of therapy during a specific period of time to prevent 1 adverse outcome or produce 1 positive outcome.”
If a difference in therapeutic outcome is seen once in every 5 patients treated with 1 intervention vs another (NNT of 5), it will likely influence day-to-day practice. However, if a therapeutic difference occurs in 1 of every 100 patients (NNT of 100), the difference between 2 treatments is not usually of great concern (except, for example, in assessing immunization against a rare but very dangerous illness).
A 95% CI of 5 to 15 means we are dealing with a NNT that with 95% probability falls between 5 and 15. However, if the NNT is not statistically significant, it becomes more difficult to describe the CI.6 A non-statistically significant NNT would have a CI that includes a negative number and a positive number: When comparing intervention A with intervention B, A might be better than B or B might be better than A. One bound of the CI may be a NNT of 10 and the other may be –10. It would be tempting to describe the CI as –10 to 10, but this would be misleading.
Attributable risk. NNT is calculated by taking the reciprocal of the difference between 2 rates for a particular outcome (Box 2). This difference is known as the attributable risk (AR). We can calculate a 95% CI for the AR, and the AR is considered statistically significant if both ends of the 95% CI are positive or both ends are negative.
If the 95% CI includes zero, then the AR is considered not statistically significant.
An AR value of zero means the rates of the outcome of interest are the same for the 2 interventions (there is no difference). Translating this to NNT would mean that no matter how many patients you treat with 1 intervention versus the other, you will not see a difference on the outcome of interest. The NNT would be “infinite” (represented by the symbol “∞”). Mathematically, if we tried to calculate the NNT when AR was zero, we would be trying to calculate the reciprocal of zero.
CI in CATIE’s efficacy phase
What do NNT and CI calculations tell us about data from clinical trials such as CATIE for schizophrenia? In CATIE, 1,493 patients were randomly assigned to 1 of 5 antipsychotics—perphenazine, olanzapine, quetiapine, risperidone, or ziprasidone—for up to 18 months. Patients who received an SGA and discontinued phase 1 before 18 months could participate in phase 2:
- Those who discontinued because of poor symptom control were expected to enter the efficacy arm (2E) and receive open-label clozapine (n = 49) or an SGA not taken in phase 1 (n = 50).
- Those who discontinued phase 1 because of poor tolerability (n = 444) were expected to enter the tolerability arm (2T), and receive an SGA they had not taken in phase 1.
The investigator could choose which arm a patient entered, but many more patients entered 2T than 2E (perhaps because they were reluctant to enter a pathway in which they might receive clozapine). Those in phase 2E who were randomly assigned to clozapine knew they were receiving clozapine and that clozapine was a treatment for patients who did not have successful outcomes with other antipsychotic(s). This design may have influenced whether or not patients remained in the study.
In phase 2E, time until treatment discontinuation for any reason was statistically significantly longer for clozapine (median 10.5 months) than for quetiapine (median 3.3 months) or risperidone (median 2.8 months) but not statistically significantly longer than for olanzapine (median 2.7 months).
What is the NNT for an outcome for drug A versus drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
Attributable risk (AR) = fA-fB
NNT = 1/AR
(By convention, we round up the NNT to the next higher whole number.)
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to see a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75-0.55 = 0.20
- NNT = 1/0.20 = 5
What happens if response rates are reversed?
- Difference in response rates = 0.55–0.75 = -0.20
- NNT = 1/(–0.20) = -5
Here the NNT is –5, meaning a disadvantage for drug B, or a number needed to harm (NNH) of +5
What happens if response rates are identical?
- Difference in response rates = 0.75-0.75 = 0
- NNT = 1/0 = "infinity" (∞)
A NNT of 8 means it would take an infinite number of patients on drug A vs drug B to see a difference (in other words, no difference). This is by definition the "weakest" possible effect size.
What happens if the response rate is 100% for one intervention and zero for the other?
- Difference in response rates = 1.00–0 = 1.00
- NNT = 1/1 = 1
Theoretically, this is the "strongest" possible effect size.
Thus all possible values of NNT range from 1 to ∞, or –1 to –∞ it is not possible for a NNT to be zero.
Time to discontinuation because of inadequate therapeutic effect was significantly longer for clozapine than for olanzapine, quetiapine, or risperi-done.4 These statements give us the rank order of the tested medications’ performance and some idea of the size of the differences. We do not know, however, how often these differences will affect day-to-day patient treatment.
The question becomes “how many patients do I need to treat with clozapine instead of [olanzapine, quetiapine, or risperidone] before I see 1 extra success (defined as remaining on the medication)?” Similar questions can be asked about other outcomes, such as adverse events. NNT can convert the study results to a common language: numbers of patients.
Advantages for clozapine. NNTs for outcomes in CATIE phase 2E are shown in the Table. From the conventional analysis,4 we knew that patients randomly assigned to clozapine were more likely to stay on clozapine than patients assigned to other SGAs. The NNT comparing clozapine with quetiapine is 3, which means for every 3 patients treated with clozapine instead of quetiapine, 1 extra patient remained on the drug. A NNT of 3 is a medium to large effect size,7 similar to that seen when antidepressant treatment is compared with placebo in terms of reducing depressive symptoms by at least 50% among patients with major depressive disorder.8
The NNT comparing clozapine with risperidone was 4 and that for olanzapine was 7. The difference in all-cause discontinuation between clozapine and olanzapine was not statistically significant, however, perhaps because of a small sample size. The effectiveness analysis included
only 45 patients assigned to clozapine, 14 to quetiapine, 14 to risperidone, and 17 to olanzapine—far fewer than the 183 to 333 subjects in each arm in the phase-1 effectiveness analyses.9
Disadvantages for clozapine can be seen as “negative” NNT values in the Table. A negative NNT can be interpreted as a number needed to harm (NNH).
Tolerability. Discontinuation because of poor tolerability showed a disadvantage when clozapine was compared with risperidone, with a NNT of –9 (in other words, a NNH of 9). This means that for every 9 patients receiving clozapine instead of risperidone, 1 extra patient would discontinue because of poor tolerability.
Anticholinergic effects. Another statistically significant disadvantage is seen when clozapine was compared with olanzapine on the occurrence of urinary hesitancy, dry mouth, or constipation, with a NNT for clozapine of –5 (NNH 5). The comparison of clozapine with risperidone on this outcome, which yielded a NNT of –8, was not statistically significant. Clozapine vs quetiapine on this measure also was not statistically significant but showed an advantage for clozapine (disadvantage for quetiapine), with a NNT of 4.
Sialorrhea is a common adverse event attributed to clozapine. Here the NNTs for clozapine compared with olanzapine, risperidone, and quetiapine were –5, –5, and –4, respectively. The comparison with risperidone was not statistically significant.
Table
Using NNTs to compare clozapine’s effects in CATIE phase 2E
| Comparison | Clozapine vs olanzapine | Clozapine vs risperidone | Clozapine vs quetiapine |
|---|---|---|---|
| All cause discontinuation | 7 | 4* | 3* |
| Discontinuation because of poor efficacy | 5 | 4* | 4* |
| Discontinuation because of poor tolerability | –20 | –9* | 10 |
| Urinary hesitancy, dry mouth, constipation | –5* | –8 | 4 |
| Sialorrhea | –5* | –5 | –4* |
| *Statistically significant p<0.05 | |||
Interpreting the CI
The CI width is affected by the variability of the estimate and the sample size, not the true population effect size. This means that a larger sample size might decrease the CI width. Sometimes, narrowing the CI width will change a nonsignificant result to statistically significant. When researchers design a study, a large sample size helps minimize the chance of not finding a statistically significant difference if a true difference exists.
A CI that includes ∞ indicates a NNT that is not statistically significant, but low CI boundaries (close to 1 or –1) can suggest potentially important results and the need for more studies to provide additional data. The study might have been “under-powered” with an inadequate sample size.
NNTs for all-cause discontinuation and their CIs when comparing clozapine with olanzapine, risperidone, or quetiapine in CATIE phase 2E are shown in Figure 1. The figure’s y-axis is centered on zero, but because a NNT must fall between 1 and ∞ (or –1 to –∞), we “grayed out” the interval around zero.
CI is easy to interpret for a statistically significant NNT. For NNT values that are not statistically significant, the CI contains 2 ranges of numbers. For the comparison of clozapine vs olanzapine, the 2 ranges are 3 to ∞ and –10 to –∞. The NNT of 7 falls within the range of 3 to ∞, but the 95% confidence interval also includes the range of –10 to –∞.
It may be easier to visualize and understand the CI by reformatting the figure so that it is centered on ∞ (Figure 2). Any CI that “crosses” ∞ represents a result that is not statistically significant. In Figure 1 and Figure 2 we also can examine the “width” of the CI. The comparison of clozapine vs quetiapine yields a NNT with a narrower CI than the comparison of clozapine vs risperidone. A narrow CI implies greater precision of our estimate of NNT and potentially its clinical importance.
Figure 1
CATIE Phase 2E: What was the advantage for clozapine?
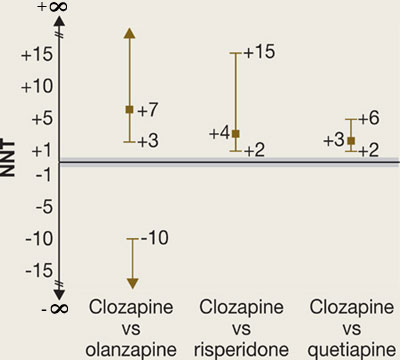
NNTs for all-cause discontinuation and 95% confidence intervals in comparing clozapine with other SGAs. The y-axis is centered on zero, but because a NNT must fall between 1 and infinity (∞) (or –1 to –∞), the interval around zero is ‘grayed out.’
Figure 2
CATIE Phase 2E: What was the advantage for clozapine (revised)?
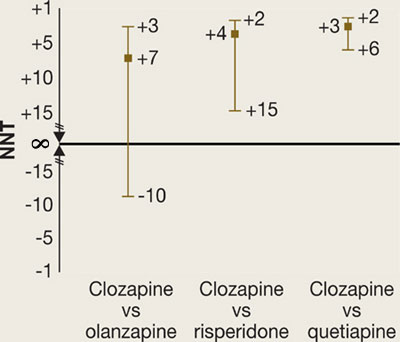
NNTs for all-cause discontinuation and 95% confidence intervals (CI) in comparing clozapine with other SGAs. Figure 2 shows Figure 1 reformatted to center on infinity (∞). Any CI that ‘crosses’ ∞ represents a result that is not statistically significant.Related Resources
- Confidence interval calculator. www.cebm.utoronto.ca/practise/ca/statscal.
- Guyatt G, Rennie D. Users’ guides to the medical literature: a manual for evidence-based clinical practice. Chicago, IL: AMA Press; 2001.
- Straus SE, Richardson WS, Glasziou P, et al. Evidence-based medicine: how to practice and teach EBM. 3rd ed. Edinburgh, UK: Elsevier/Churchill Livingstone; 2005.
Drug brand names
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceutica, Jazz Pharmaceuticals, and Pfizer Inc.
1. Lemonick MD. Medicine’s secret stat. Time. February 15, 2007. Available at: http://www.time.com/time/printout/0,8816,1590464,00.html. Accessed February 20, 2007.
2. Guyatt G, Cook D, Devereaux PJ, et al. Therapy. In: Guyatt G, Rennie D, eds. Users’ guides to the medical literature, chapter 1B1. Chicago, IL: AMA Press, 2002.
3. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
4. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-10.
5. Citrome L, Stroup TS. Schizophrenia, Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-40.
6. Altman DG. Confidence intervals for the number needed to treat. BMJ. 1998;317(7168):1309-12.
7. Kraemer HC, Kupfer DJ. Size of treatment effects and their importance to clinical research and practice. Biol Psychiatry. 2006;59(11):990-6.
8. Pinson L, Gray GE. Number needed to treat: an underused measure of treatment effect. Psychiatr Serv. 2003;54(2):145-6,154.
9. Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-23.
Number needed to treat (NNT) is a measure of clinical effect that has been called medicine’s “secret stat”(Box 1).1,2 By itself, however, the NNT provides no information about whether a trial result is probably true (statistical significance). If a NNT is statistically significant, the confidence interval (CI) can tell you the range of numbers within which the truth probably lies.
In the March 2007 issue of Current Psychiatry, we described how to use NNT to interpret and apply research data in daily practice.3 In this article, we explain the “secrets” of NNT and CI by providing sample calculations and several figures for visual learning. For illustration, we analyze data from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) in schizophrenia, this time focusing on phase 2E—the efficacy pathway in which patients were randomly assigned to open-label clozapine or a double-blinded second-generation antipsychotic (SGA).4
Confidence intervals: Is the NNT statistically significant?
To find out a NNT’s statistical significance, you can examine the CI. A 95% CI means that the truth lies between the interval’s lower and upper bounds with a 95% probability.
Calculating CI. Although formulas to calculate the CI appear complicated,5 they are easily inserted into a Microsoft Excel-brand spreadsheet. Useful alternatives are online calculators (seeRelated Resources), which can be downloaded to your hand-held device or pocket PC.
Time magazine recently declared NNT as medicine’s “secret stat.”1 NNT allows us to place a number on how often we would see a difference between 2 interventions.
In a handbook on essentials of evidence-based clinical practice, Guyatt et al2 define NNT as “the number of patients who must receive an intervention of therapy during a specific period of time to prevent 1 adverse outcome or produce 1 positive outcome.”
If a difference in therapeutic outcome is seen once in every 5 patients treated with 1 intervention vs another (NNT of 5), it will likely influence day-to-day practice. However, if a therapeutic difference occurs in 1 of every 100 patients (NNT of 100), the difference between 2 treatments is not usually of great concern (except, for example, in assessing immunization against a rare but very dangerous illness).
A 95% CI of 5 to 15 means we are dealing with a NNT that with 95% probability falls between 5 and 15. However, if the NNT is not statistically significant, it becomes more difficult to describe the CI.6 A non-statistically significant NNT would have a CI that includes a negative number and a positive number: When comparing intervention A with intervention B, A might be better than B or B might be better than A. One bound of the CI may be a NNT of 10 and the other may be –10. It would be tempting to describe the CI as –10 to 10, but this would be misleading.
Attributable risk. NNT is calculated by taking the reciprocal of the difference between 2 rates for a particular outcome (Box 2). This difference is known as the attributable risk (AR). We can calculate a 95% CI for the AR, and the AR is considered statistically significant if both ends of the 95% CI are positive or both ends are negative.
If the 95% CI includes zero, then the AR is considered not statistically significant.
An AR value of zero means the rates of the outcome of interest are the same for the 2 interventions (there is no difference). Translating this to NNT would mean that no matter how many patients you treat with 1 intervention versus the other, you will not see a difference on the outcome of interest. The NNT would be “infinite” (represented by the symbol “∞”). Mathematically, if we tried to calculate the NNT when AR was zero, we would be trying to calculate the reciprocal of zero.
CI in CATIE’s efficacy phase
What do NNT and CI calculations tell us about data from clinical trials such as CATIE for schizophrenia? In CATIE, 1,493 patients were randomly assigned to 1 of 5 antipsychotics—perphenazine, olanzapine, quetiapine, risperidone, or ziprasidone—for up to 18 months. Patients who received an SGA and discontinued phase 1 before 18 months could participate in phase 2:
- Those who discontinued because of poor symptom control were expected to enter the efficacy arm (2E) and receive open-label clozapine (n = 49) or an SGA not taken in phase 1 (n = 50).
- Those who discontinued phase 1 because of poor tolerability (n = 444) were expected to enter the tolerability arm (2T), and receive an SGA they had not taken in phase 1.
The investigator could choose which arm a patient entered, but many more patients entered 2T than 2E (perhaps because they were reluctant to enter a pathway in which they might receive clozapine). Those in phase 2E who were randomly assigned to clozapine knew they were receiving clozapine and that clozapine was a treatment for patients who did not have successful outcomes with other antipsychotic(s). This design may have influenced whether or not patients remained in the study.
In phase 2E, time until treatment discontinuation for any reason was statistically significantly longer for clozapine (median 10.5 months) than for quetiapine (median 3.3 months) or risperidone (median 2.8 months) but not statistically significantly longer than for olanzapine (median 2.7 months).
What is the NNT for an outcome for drug A versus drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
Attributable risk (AR) = fA-fB
NNT = 1/AR
(By convention, we round up the NNT to the next higher whole number.)
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to see a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75-0.55 = 0.20
- NNT = 1/0.20 = 5
What happens if response rates are reversed?
- Difference in response rates = 0.55–0.75 = -0.20
- NNT = 1/(–0.20) = -5
Here the NNT is –5, meaning a disadvantage for drug B, or a number needed to harm (NNH) of +5
What happens if response rates are identical?
- Difference in response rates = 0.75-0.75 = 0
- NNT = 1/0 = "infinity" (∞)
A NNT of 8 means it would take an infinite number of patients on drug A vs drug B to see a difference (in other words, no difference). This is by definition the "weakest" possible effect size.
What happens if the response rate is 100% for one intervention and zero for the other?
- Difference in response rates = 1.00–0 = 1.00
- NNT = 1/1 = 1
Theoretically, this is the "strongest" possible effect size.
Thus all possible values of NNT range from 1 to ∞, or –1 to –∞ it is not possible for a NNT to be zero.
Time to discontinuation because of inadequate therapeutic effect was significantly longer for clozapine than for olanzapine, quetiapine, or risperi-done.4 These statements give us the rank order of the tested medications’ performance and some idea of the size of the differences. We do not know, however, how often these differences will affect day-to-day patient treatment.
The question becomes “how many patients do I need to treat with clozapine instead of [olanzapine, quetiapine, or risperidone] before I see 1 extra success (defined as remaining on the medication)?” Similar questions can be asked about other outcomes, such as adverse events. NNT can convert the study results to a common language: numbers of patients.
Advantages for clozapine. NNTs for outcomes in CATIE phase 2E are shown in the Table. From the conventional analysis,4 we knew that patients randomly assigned to clozapine were more likely to stay on clozapine than patients assigned to other SGAs. The NNT comparing clozapine with quetiapine is 3, which means for every 3 patients treated with clozapine instead of quetiapine, 1 extra patient remained on the drug. A NNT of 3 is a medium to large effect size,7 similar to that seen when antidepressant treatment is compared with placebo in terms of reducing depressive symptoms by at least 50% among patients with major depressive disorder.8
The NNT comparing clozapine with risperidone was 4 and that for olanzapine was 7. The difference in all-cause discontinuation between clozapine and olanzapine was not statistically significant, however, perhaps because of a small sample size. The effectiveness analysis included
only 45 patients assigned to clozapine, 14 to quetiapine, 14 to risperidone, and 17 to olanzapine—far fewer than the 183 to 333 subjects in each arm in the phase-1 effectiveness analyses.9
Disadvantages for clozapine can be seen as “negative” NNT values in the Table. A negative NNT can be interpreted as a number needed to harm (NNH).
Tolerability. Discontinuation because of poor tolerability showed a disadvantage when clozapine was compared with risperidone, with a NNT of –9 (in other words, a NNH of 9). This means that for every 9 patients receiving clozapine instead of risperidone, 1 extra patient would discontinue because of poor tolerability.
Anticholinergic effects. Another statistically significant disadvantage is seen when clozapine was compared with olanzapine on the occurrence of urinary hesitancy, dry mouth, or constipation, with a NNT for clozapine of –5 (NNH 5). The comparison of clozapine with risperidone on this outcome, which yielded a NNT of –8, was not statistically significant. Clozapine vs quetiapine on this measure also was not statistically significant but showed an advantage for clozapine (disadvantage for quetiapine), with a NNT of 4.
Sialorrhea is a common adverse event attributed to clozapine. Here the NNTs for clozapine compared with olanzapine, risperidone, and quetiapine were –5, –5, and –4, respectively. The comparison with risperidone was not statistically significant.
Table
Using NNTs to compare clozapine’s effects in CATIE phase 2E
| Comparison | Clozapine vs olanzapine | Clozapine vs risperidone | Clozapine vs quetiapine |
|---|---|---|---|
| All cause discontinuation | 7 | 4* | 3* |
| Discontinuation because of poor efficacy | 5 | 4* | 4* |
| Discontinuation because of poor tolerability | –20 | –9* | 10 |
| Urinary hesitancy, dry mouth, constipation | –5* | –8 | 4 |
| Sialorrhea | –5* | –5 | –4* |
| *Statistically significant p<0.05 | |||
Interpreting the CI
The CI width is affected by the variability of the estimate and the sample size, not the true population effect size. This means that a larger sample size might decrease the CI width. Sometimes, narrowing the CI width will change a nonsignificant result to statistically significant. When researchers design a study, a large sample size helps minimize the chance of not finding a statistically significant difference if a true difference exists.
A CI that includes ∞ indicates a NNT that is not statistically significant, but low CI boundaries (close to 1 or –1) can suggest potentially important results and the need for more studies to provide additional data. The study might have been “under-powered” with an inadequate sample size.
NNTs for all-cause discontinuation and their CIs when comparing clozapine with olanzapine, risperidone, or quetiapine in CATIE phase 2E are shown in Figure 1. The figure’s y-axis is centered on zero, but because a NNT must fall between 1 and ∞ (or –1 to –∞), we “grayed out” the interval around zero.
CI is easy to interpret for a statistically significant NNT. For NNT values that are not statistically significant, the CI contains 2 ranges of numbers. For the comparison of clozapine vs olanzapine, the 2 ranges are 3 to ∞ and –10 to –∞. The NNT of 7 falls within the range of 3 to ∞, but the 95% confidence interval also includes the range of –10 to –∞.
It may be easier to visualize and understand the CI by reformatting the figure so that it is centered on ∞ (Figure 2). Any CI that “crosses” ∞ represents a result that is not statistically significant. In Figure 1 and Figure 2 we also can examine the “width” of the CI. The comparison of clozapine vs quetiapine yields a NNT with a narrower CI than the comparison of clozapine vs risperidone. A narrow CI implies greater precision of our estimate of NNT and potentially its clinical importance.
Figure 1
CATIE Phase 2E: What was the advantage for clozapine?
NNTs for all-cause discontinuation and 95% confidence intervals in comparing clozapine with other SGAs. The y-axis is centered on zero, but because a NNT must fall between 1 and infinity (∞) (or –1 to –∞), the interval around zero is ‘grayed out.’
Figure 2
CATIE Phase 2E: What was the advantage for clozapine (revised)?
NNTs for all-cause discontinuation and 95% confidence intervals (CI) in comparing clozapine with other SGAs. Figure 2 shows Figure 1 reformatted to center on infinity (∞). Any CI that ‘crosses’ ∞ represents a result that is not statistically significant.Related Resources
- Confidence interval calculator. www.cebm.utoronto.ca/practise/ca/statscal.
- Guyatt G, Rennie D. Users’ guides to the medical literature: a manual for evidence-based clinical practice. Chicago, IL: AMA Press; 2001.
- Straus SE, Richardson WS, Glasziou P, et al. Evidence-based medicine: how to practice and teach EBM. 3rd ed. Edinburgh, UK: Elsevier/Churchill Livingstone; 2005.
Drug brand names
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceutica, Jazz Pharmaceuticals, and Pfizer Inc.
Number needed to treat (NNT) is a measure of clinical effect that has been called medicine’s “secret stat”(Box 1).1,2 By itself, however, the NNT provides no information about whether a trial result is probably true (statistical significance). If a NNT is statistically significant, the confidence interval (CI) can tell you the range of numbers within which the truth probably lies.
In the March 2007 issue of Current Psychiatry, we described how to use NNT to interpret and apply research data in daily practice.3 In this article, we explain the “secrets” of NNT and CI by providing sample calculations and several figures for visual learning. For illustration, we analyze data from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) in schizophrenia, this time focusing on phase 2E—the efficacy pathway in which patients were randomly assigned to open-label clozapine or a double-blinded second-generation antipsychotic (SGA).4
Confidence intervals: Is the NNT statistically significant?
To find out a NNT’s statistical significance, you can examine the CI. A 95% CI means that the truth lies between the interval’s lower and upper bounds with a 95% probability.
Calculating CI. Although formulas to calculate the CI appear complicated,5 they are easily inserted into a Microsoft Excel-brand spreadsheet. Useful alternatives are online calculators (seeRelated Resources), which can be downloaded to your hand-held device or pocket PC.
Time magazine recently declared NNT as medicine’s “secret stat.”1 NNT allows us to place a number on how often we would see a difference between 2 interventions.
In a handbook on essentials of evidence-based clinical practice, Guyatt et al2 define NNT as “the number of patients who must receive an intervention of therapy during a specific period of time to prevent 1 adverse outcome or produce 1 positive outcome.”
If a difference in therapeutic outcome is seen once in every 5 patients treated with 1 intervention vs another (NNT of 5), it will likely influence day-to-day practice. However, if a therapeutic difference occurs in 1 of every 100 patients (NNT of 100), the difference between 2 treatments is not usually of great concern (except, for example, in assessing immunization against a rare but very dangerous illness).
A 95% CI of 5 to 15 means we are dealing with a NNT that with 95% probability falls between 5 and 15. However, if the NNT is not statistically significant, it becomes more difficult to describe the CI.6 A non-statistically significant NNT would have a CI that includes a negative number and a positive number: When comparing intervention A with intervention B, A might be better than B or B might be better than A. One bound of the CI may be a NNT of 10 and the other may be –10. It would be tempting to describe the CI as –10 to 10, but this would be misleading.
Attributable risk. NNT is calculated by taking the reciprocal of the difference between 2 rates for a particular outcome (Box 2). This difference is known as the attributable risk (AR). We can calculate a 95% CI for the AR, and the AR is considered statistically significant if both ends of the 95% CI are positive or both ends are negative.
If the 95% CI includes zero, then the AR is considered not statistically significant.
An AR value of zero means the rates of the outcome of interest are the same for the 2 interventions (there is no difference). Translating this to NNT would mean that no matter how many patients you treat with 1 intervention versus the other, you will not see a difference on the outcome of interest. The NNT would be “infinite” (represented by the symbol “∞”). Mathematically, if we tried to calculate the NNT when AR was zero, we would be trying to calculate the reciprocal of zero.
CI in CATIE’s efficacy phase
What do NNT and CI calculations tell us about data from clinical trials such as CATIE for schizophrenia? In CATIE, 1,493 patients were randomly assigned to 1 of 5 antipsychotics—perphenazine, olanzapine, quetiapine, risperidone, or ziprasidone—for up to 18 months. Patients who received an SGA and discontinued phase 1 before 18 months could participate in phase 2:
- Those who discontinued because of poor symptom control were expected to enter the efficacy arm (2E) and receive open-label clozapine (n = 49) or an SGA not taken in phase 1 (n = 50).
- Those who discontinued phase 1 because of poor tolerability (n = 444) were expected to enter the tolerability arm (2T), and receive an SGA they had not taken in phase 1.
The investigator could choose which arm a patient entered, but many more patients entered 2T than 2E (perhaps because they were reluctant to enter a pathway in which they might receive clozapine). Those in phase 2E who were randomly assigned to clozapine knew they were receiving clozapine and that clozapine was a treatment for patients who did not have successful outcomes with other antipsychotic(s). This design may have influenced whether or not patients remained in the study.
In phase 2E, time until treatment discontinuation for any reason was statistically significantly longer for clozapine (median 10.5 months) than for quetiapine (median 3.3 months) or risperidone (median 2.8 months) but not statistically significantly longer than for olanzapine (median 2.7 months).
What is the NNT for an outcome for drug A versus drug B?
fA = frequency of outcome for drug A
fB = frequency of outcome for drug B
Attributable risk (AR) = fA-fB
NNT = 1/AR
(By convention, we round up the NNT to the next higher whole number.)
For example, let’s say drugs A and B are used to treat depression, and they result in 6-week response rates of 55% and 75%, respectively. The NNT to see a difference between drug B and drug A in terms of responders at 6 weeks can be calculated as follows:
- Difference in response rates = 0.75-0.55 = 0.20
- NNT = 1/0.20 = 5
What happens if response rates are reversed?
- Difference in response rates = 0.55–0.75 = -0.20
- NNT = 1/(–0.20) = -5
Here the NNT is –5, meaning a disadvantage for drug B, or a number needed to harm (NNH) of +5
What happens if response rates are identical?
- Difference in response rates = 0.75-0.75 = 0
- NNT = 1/0 = "infinity" (∞)
A NNT of 8 means it would take an infinite number of patients on drug A vs drug B to see a difference (in other words, no difference). This is by definition the "weakest" possible effect size.
What happens if the response rate is 100% for one intervention and zero for the other?
- Difference in response rates = 1.00–0 = 1.00
- NNT = 1/1 = 1
Theoretically, this is the "strongest" possible effect size.
Thus all possible values of NNT range from 1 to ∞, or –1 to –∞ it is not possible for a NNT to be zero.
Time to discontinuation because of inadequate therapeutic effect was significantly longer for clozapine than for olanzapine, quetiapine, or risperi-done.4 These statements give us the rank order of the tested medications’ performance and some idea of the size of the differences. We do not know, however, how often these differences will affect day-to-day patient treatment.
The question becomes “how many patients do I need to treat with clozapine instead of [olanzapine, quetiapine, or risperidone] before I see 1 extra success (defined as remaining on the medication)?” Similar questions can be asked about other outcomes, such as adverse events. NNT can convert the study results to a common language: numbers of patients.
Advantages for clozapine. NNTs for outcomes in CATIE phase 2E are shown in the Table. From the conventional analysis,4 we knew that patients randomly assigned to clozapine were more likely to stay on clozapine than patients assigned to other SGAs. The NNT comparing clozapine with quetiapine is 3, which means for every 3 patients treated with clozapine instead of quetiapine, 1 extra patient remained on the drug. A NNT of 3 is a medium to large effect size,7 similar to that seen when antidepressant treatment is compared with placebo in terms of reducing depressive symptoms by at least 50% among patients with major depressive disorder.8
The NNT comparing clozapine with risperidone was 4 and that for olanzapine was 7. The difference in all-cause discontinuation between clozapine and olanzapine was not statistically significant, however, perhaps because of a small sample size. The effectiveness analysis included
only 45 patients assigned to clozapine, 14 to quetiapine, 14 to risperidone, and 17 to olanzapine—far fewer than the 183 to 333 subjects in each arm in the phase-1 effectiveness analyses.9
Disadvantages for clozapine can be seen as “negative” NNT values in the Table. A negative NNT can be interpreted as a number needed to harm (NNH).
Tolerability. Discontinuation because of poor tolerability showed a disadvantage when clozapine was compared with risperidone, with a NNT of –9 (in other words, a NNH of 9). This means that for every 9 patients receiving clozapine instead of risperidone, 1 extra patient would discontinue because of poor tolerability.
Anticholinergic effects. Another statistically significant disadvantage is seen when clozapine was compared with olanzapine on the occurrence of urinary hesitancy, dry mouth, or constipation, with a NNT for clozapine of –5 (NNH 5). The comparison of clozapine with risperidone on this outcome, which yielded a NNT of –8, was not statistically significant. Clozapine vs quetiapine on this measure also was not statistically significant but showed an advantage for clozapine (disadvantage for quetiapine), with a NNT of 4.
Sialorrhea is a common adverse event attributed to clozapine. Here the NNTs for clozapine compared with olanzapine, risperidone, and quetiapine were –5, –5, and –4, respectively. The comparison with risperidone was not statistically significant.
Table
Using NNTs to compare clozapine’s effects in CATIE phase 2E
| Comparison | Clozapine vs olanzapine | Clozapine vs risperidone | Clozapine vs quetiapine |
|---|---|---|---|
| All cause discontinuation | 7 | 4* | 3* |
| Discontinuation because of poor efficacy | 5 | 4* | 4* |
| Discontinuation because of poor tolerability | –20 | –9* | 10 |
| Urinary hesitancy, dry mouth, constipation | –5* | –8 | 4 |
| Sialorrhea | –5* | –5 | –4* |
| *Statistically significant p<0.05 | |||
Interpreting the CI
The CI width is affected by the variability of the estimate and the sample size, not the true population effect size. This means that a larger sample size might decrease the CI width. Sometimes, narrowing the CI width will change a nonsignificant result to statistically significant. When researchers design a study, a large sample size helps minimize the chance of not finding a statistically significant difference if a true difference exists.
A CI that includes ∞ indicates a NNT that is not statistically significant, but low CI boundaries (close to 1 or –1) can suggest potentially important results and the need for more studies to provide additional data. The study might have been “under-powered” with an inadequate sample size.
NNTs for all-cause discontinuation and their CIs when comparing clozapine with olanzapine, risperidone, or quetiapine in CATIE phase 2E are shown in Figure 1. The figure’s y-axis is centered on zero, but because a NNT must fall between 1 and ∞ (or –1 to –∞), we “grayed out” the interval around zero.
CI is easy to interpret for a statistically significant NNT. For NNT values that are not statistically significant, the CI contains 2 ranges of numbers. For the comparison of clozapine vs olanzapine, the 2 ranges are 3 to ∞ and –10 to –∞. The NNT of 7 falls within the range of 3 to ∞, but the 95% confidence interval also includes the range of –10 to –∞.
It may be easier to visualize and understand the CI by reformatting the figure so that it is centered on ∞ (Figure 2). Any CI that “crosses” ∞ represents a result that is not statistically significant. In Figure 1 and Figure 2 we also can examine the “width” of the CI. The comparison of clozapine vs quetiapine yields a NNT with a narrower CI than the comparison of clozapine vs risperidone. A narrow CI implies greater precision of our estimate of NNT and potentially its clinical importance.
Figure 1
CATIE Phase 2E: What was the advantage for clozapine?
NNTs for all-cause discontinuation and 95% confidence intervals in comparing clozapine with other SGAs. The y-axis is centered on zero, but because a NNT must fall between 1 and infinity (∞) (or –1 to –∞), the interval around zero is ‘grayed out.’
Figure 2
CATIE Phase 2E: What was the advantage for clozapine (revised)?
NNTs for all-cause discontinuation and 95% confidence intervals (CI) in comparing clozapine with other SGAs. Figure 2 shows Figure 1 reformatted to center on infinity (∞). Any CI that ‘crosses’ ∞ represents a result that is not statistically significant.Related Resources
- Confidence interval calculator. www.cebm.utoronto.ca/practise/ca/statscal.
- Guyatt G, Rennie D. Users’ guides to the medical literature: a manual for evidence-based clinical practice. Chicago, IL: AMA Press; 2001.
- Straus SE, Richardson WS, Glasziou P, et al. Evidence-based medicine: how to practice and teach EBM. 3rd ed. Edinburgh, UK: Elsevier/Churchill Livingstone; 2005.
Drug brand names
- Clozapine • Clozaril
- Olanzapine • Zyprexa
- Perphenazine • Trilafon
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ziprasidone • Geodon
Disclosures
Dr. Citrome is a consultant for, has received honoraria from, or has conducted clinical research supported by Abbott Laboratories, AstraZeneca Pharmaceuticals, Barr Laboratories, Bristol-Myers Squibb, Eli Lilly and Company, Forest Research Institute, GlaxoSmithKline, Janssen Pharmaceutica, Jazz Pharmaceuticals, and Pfizer Inc.
1. Lemonick MD. Medicine’s secret stat. Time. February 15, 2007. Available at: http://www.time.com/time/printout/0,8816,1590464,00.html. Accessed February 20, 2007.
2. Guyatt G, Cook D, Devereaux PJ, et al. Therapy. In: Guyatt G, Rennie D, eds. Users’ guides to the medical literature, chapter 1B1. Chicago, IL: AMA Press, 2002.
3. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
4. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-10.
5. Citrome L, Stroup TS. Schizophrenia, Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-40.
6. Altman DG. Confidence intervals for the number needed to treat. BMJ. 1998;317(7168):1309-12.
7. Kraemer HC, Kupfer DJ. Size of treatment effects and their importance to clinical research and practice. Biol Psychiatry. 2006;59(11):990-6.
8. Pinson L, Gray GE. Number needed to treat: an underused measure of treatment effect. Psychiatr Serv. 2003;54(2):145-6,154.
9. Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-23.
1. Lemonick MD. Medicine’s secret stat. Time. February 15, 2007. Available at: http://www.time.com/time/printout/0,8816,1590464,00.html. Accessed February 20, 2007.
2. Guyatt G, Cook D, Devereaux PJ, et al. Therapy. In: Guyatt G, Rennie D, eds. Users’ guides to the medical literature, chapter 1B1. Chicago, IL: AMA Press, 2002.
3. Citrome L. Dissecting clinical trials with ‘number needed to treat.’ Current Psychiatry. 2007;6(3):66-71.
4. McEvoy JP, Lieberman JA, Stroup TS, et al. Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry. 2006;163(4):600-10.
5. Citrome L, Stroup TS. Schizophrenia, Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), and number needed to treat: how can CATIE inform clinicians? Int J Clin Pract. 2006;60(8):933-40.
6. Altman DG. Confidence intervals for the number needed to treat. BMJ. 1998;317(7168):1309-12.
7. Kraemer HC, Kupfer DJ. Size of treatment effects and their importance to clinical research and practice. Biol Psychiatry. 2006;59(11):990-6.
8. Pinson L, Gray GE. Number needed to treat: an underused measure of treatment effect. Psychiatr Serv. 2003;54(2):145-6,154.
9. Lieberman JA, Stroup TS, McEvoy JP, et al. Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-23.
Which to treat first: Comorbid anxiety or alcohol disorder?
Men and women with anxiety disorders are at three times the general population’s risk of being alcohol-dependent (Table),1 and those who seek treatment for an anxiety disorder are at even higher risk of alcohol disorder.2,3 This comorbidity can complicate treatment attempts if either disorder remains unaddressed, leading to increased relapse risk and multiple treatment episodes.
Based on our research and clinical work in helping patients with comorbid alcohol dependence and anxiety disorders,2,4-6 this article describes:
- potential relationships between anxiety disorders and alcohol disorder
- pros and cons of 3 approaches to treating this comorbidity
- how to identify and address alcohol disorder in patients with anxiety disorders, depending on available resources.
Table
Comorbidity rates of anxiety disorders and alcohol dependence*
| Anxiety disorder | Odds ratio for having alcohol dependence | |
|---|---|---|
| Men | Women | |
| Any | 3.2 | 3.3 |
| Panic disorder | 3.8 | 3.7 |
| Social phobia | 2.6 | 3.6 |
| Generalized anxiety disorder | 3.6 | 3.4 |
| Specific phobia | 2.8 | 2.9 |
| * Numbers indicate odds of having alcohol dependence when the anxiety disorder is present vs absent. | ||
| Source: 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), reference 1 | ||
3 comorbidity models
The most common understanding of this comorbidity (Figure 1) is that having an anxiety disorder predisposes one to develop an alcohol or substance use disorder via self-medication—using alcohol or drugs to modulate anxiety and negative affect.7-9 However, substance use disorder experts have argued that the social, occupational, and physiologic effects of substance use can generate new anxiety symptoms in vulnerable individuals.10 In other words, physiologic and/or environmental disruptions from chronic alcohol or substance use could promote conditions and circumstances in which anxiety symptoms are more likely to emerge or worsen.
Although DSM-IV-TR does not delve into the causes of mental disorders, it states that substance use can cause or “induce” an anxiety syndrome with symptoms that resemble or are identical to those of the various anxiety syndromes that are not related to substance disorder (Box).11,12
Alternatively, the idea that a third factor can serve as a common cause for both conditions fits with the view that substance use disorder and anxiety disorder can be phenotypic expressions of a common underlying genetic/physiologic liability.13
Finally, these models are not mutually exclusive. Anxiety symptoms or substance use could cause or aggravate the other.
Figure 1
Hypothetical models of comorbidity
Which comes first?
Anxiety disorder typically begins before a substance use disorder in comorbid cases, although some studies have reported the opposite pattern or roughly simultaneous onset of both disorders.2
Using a prospective method in college students, we found that the risk of developing alcohol dependence for the first time as a junior or senior more than tripled among students who had an anxiety diagnosis as a freshman.14 We also found, however, that students who were alcohol dependent as freshman were 4 times more likely than other freshman to develop an anxiety disorder for the first time within the next 6 years.
In short, having either an anxiety or alcohol disorder earlier in life appears to increase the probability of developing the other later. This finding supports the idea that the types of associations that link pathologic anxiety and substance use vary among individuals and, perhaps, within individuals over time.
3 treatment approaches
Treating 1 of the comorbid conditions—anxiety or alcohol disorder—does not tend to resolve the other.3,15 This suggests that therapies aimed at treating a single disorder are not satisfactory for treating comorbid cases. Possible multi-focused approaches include:
- serial (or sequential) approach—treating comorbid disorders one at a time
- parallel approach—providing simultaneous but separate treatments for each comorbidity
- integrated approach—providing one treatment that focuses on both comorbid disorders, especially as they interact with one another.
Each has strengths and weaknesses, and the approach you choose for your patient may depend on clinical circumstances and available resources.
Serial treatment has the structure to empirically evaluate whether the initially untreated comorbid condition is resolved by treating the other condition. For example, you could treat an anxiety disorder as usual and refer the patient for alcohol disorder treatment only if drinking remains an active problem following anxiety treatment. This approach also allows you to use well-established treatment systems, programs, and specialists as usual.
One disadvantage to the serial approach, however, is that the initially untreated comorbid disorder could undermine the resolution of the treated disorder. We found, for example, that treating either the anxiety or the alcohol disorder alone fails to resolve the comorbid condition and might leave a patient vulnerable to relapse before serial treatment can be completed.3,15
When implementing a serial treatment, it is not always clear which disorder to treat first. Distinguishing comorbid disorders as “primary” or “secondary” often is done inconsistently and imprecisely, so treatment decisions based on these terms can be erroneous. Using the order of disorder onset also is an unreliable guide to which disorder is in priority need of treatment.7,16
Based on our experience and research, where and why the comorbid patient presents for treatment should be factored heavily in these treatment decisions. For example, individuals seeking anxiety treatment who have a comorbid alcohol use disorder typically possess little insight into their drinking problem and a frank resistance to clinician-driven attempts to modify their drinking behavior. We would expect a similar reaction from patients presenting for alcohol disorder treatment who are told they must first obtain psychiatric treatment for anxiety symptoms. The serial approach often necessitates that patients be treated initially for the problem for which they present and then referred afterward for the comorbid condition as needed.
In an open-label pilot treatment study of 5 subjects with social anxiety disorder and a co-occurring alcohol use disorder (C.L.R., S.W.B., unpublished data, 2007), we first treated the anxiety disorder with the selective serotonin reuptake inhibitor paroxetine, up to 60 mg/d. After 6 weeks, we addressed the comorbid alcohol problem using a brief alcohol intervention. This approach met with little or no resistance to reduce drinking—all 5 subjects successfully decreased their alcohol consumption, and none dropped out of treatment. A controlled follow-up trial is planned to provide empiric support for serial treatment of anxiety and alcohol use disorders in mental health treatment settings.
DSM-IV-TR describes an anxiety disorder as independent from a coexisting substance use disorder only if the anxiety disorder:
- began distinctly before the substance use disorder
- or persisted during periods of extended abstinence (>1 month) from substance use/abuse.
Otherwise, substance abuse is presumed to have induced the anxiety disorder. This perspective implies that no specific treatment beyond drug/alcohol abstinence is required to resolve a substance-induced anxiety disorder.
In a large community-based sample, Grant et al11 found that <0.5% of individuals with comorbid anxiety and substance abuse met the strictly defined DSM-IV-TR criteria for a substance-induced anxiety disorder. Cases in which a comorbid anxiety disorder resolved during periods of substance abuse abstinence were especially rare. This observation suggests that substance-induced anxiety syndrome as defined by DSM-IV-TR is very rare in clinical practice.
DSM-IV-TR diagnostic criteria do not recognize an “anxiety-induced substance use disorder,” in which pathologic anxiety might induce a substance use disorder. Conceptually, however, this idea is as reasonable as substance-induced anxiety disorder and fits within the self-medication model.12
Parallel treatments, which can mitigate some disadvantages of the serial approach, increasingly are being used in chemical dependency treatment settings, where it is common to have psychiatric consultations. Based on our experience, however, this approach is far less common in psychiatric treatment settings, where clinicians do not routinely treat (or sometimes even assess for) comorbid alcohol or drug disorder in anxious and depressed patients. Also, the parallel approach often requires coordinating the times, locations, and strategies of treatments systems and clinicians, which can lead to problems:
- Substance use disorder treatment expertise is not always available for patients in mental health treatment.
- Clinicians from disparate systems may not fully understand the impact of the comorbid disorder and the culture of the parallel treatment system.
- Practitioners (or patients) might see medications or cognitive-behavioral therapy (CBT) exercises for anxiety as contradicting core tenants of the parallel treatment approach.
- It is not certain that standard treatments validated in non-comorbid patients would have the same therapeutic benefits when administered in a parallel treatment.5
Integrated treatments seek to address both comorbid disorders in a single treatment program. Our group has found, for example, that a CBT program aimed at treating comorbid anxiety could be successfully integrated with a standard alcohol disorder treatment.17
Several factors limit the use of integrated treatments, however:
- Few such programs exist.
- Treatment providers in mental health and addiction settings typically are not cross-trained.
- Personnel and other institutional supports are often lacking for integrated treatment programs.
Integrated treatment plan
Our CBT-based integrated approach to alcoholism treatment in patients with a comorbid anxiety disorder incorporates 3 components:
- psychoeducation
- cognitive restructuring
- cue exposure.
Psychoeducation. The goal of psychoeducation is to explain the biopsychosocial model of anxiety disorder, alcohol disorders, and their interactions. This information is the general platform on which the specific treatment program is established in the next phase of the treatment.
In addition to providing basic epidemiologic facts, we use simple language and graphics to emphasize the vicious cycle that can emerge between drinking and anxiety, wherein the more one uses alcohol to manage anxiety in the short run the more anxiety there is to manage in the long run.
We introduce the role of cognitions, thoughts, beliefs, and expectations in how individuals react to situations to produce anxiety and drinking urges. Finally, we teach patients a standard paced diaphragmatic breathing exercise designed to minimize hyperventilation commonly identified among individuals with anxiety disorders.
Cognitive restructuring. We teach patients about thinking patterns that contribute to initiating and maintaining anxiety and panic. We also teach patients how to recognize and restructure cognitions that promote alcohol use as a means of coping with anxiety, such as focusing on alcohol’s short-term calming effects instead of its longer-term anxiogenic effects. This phase requires clinical expertise in CBT skills; a wide range of resource materials is available to walk the patient (and clinician) through cognitive restructuring exercises (see Related Resources).
Cue exposure involves systematic therapist-guided exposure to fear-provoking situations and sensations with the goal of decoupling them from anxiety-inducing thoughts about catastrophic outcomes.
Exposures are used:
- for reality testing
- to allow patients to practice new anxiety management skills
- to increase patients’ sense that they can successfully cope in feared situations (“self-efficacy”).
We expand this approach to include alcohol-relevant cues associated with anxiety states. Exposures—imaginal and in vivo—incorporate this information to help patients decouple anxiety feelings from drinking urges and to practice alternate coping strategies.
Pilot data for integrated Tx
After 4 months of participating in an integrated CBT program, 32 alcoholism treatment patients with panic disorder were significantly less likely to meet criteria for panic disorder, compared with 17 patients who received standard chemical dependency treatment without the CBT program (M.G.K., C.D., B.F., unpublished data, 2007). Before treatment, both groups averaged approximately 2.5 panic attacks per week. At follow-up the group that received CBT averaged <0.5 panic attacks per week, whereas the control group averaged approximately 2 panic attacks per week.
Overall, there was a positive effect for CBT treatment in terms of relapse to full alcohol dependence—10% in the treatment group met this criteria vs 35% in the control group. Integrated CBT treatment was more effective in reducing relapse risk among patients who reported the strongest baseline expectations that alcohol consumption helps to control their anxiety symptoms: 0% in the treatment group relapsed to full alcohol dependence vs. 57% in the control group. For comorbid cases that had the weakest anxiety-reduction expectancies, 21% in the CBT group met the relapse criterion compared with about 20% in the control group.
In summary, an integrated CBT program for comorbid panic disorder appears to provide the greatest added value to standard alcoholism treatment among patients who expect alcohol to relieve their anxiety symptoms.
Treatment in a psychiatric setting
Our group is in the process of generating a database upon which to make empirically based treatment recommendations. Until then, we can offer treatment recommendations based upon experience and the limited data available.
When planning psychiatric treatment for a patient with an anxiety disorder, start by assessing the patient’s alcohol use (Figure 2). The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools (see Related Resources) to help you judge whether a patient’s alcohol use exceeds recommended limits (for example, 7 drinks per week for women and 14 per week for men).
Teach individuals whose drinking is excessive and/or regular (especially deliberate drinking aimed at coping with anxiety) about the risk associated with alcohol use and potential interference of alcohol/drugs with successful anxiety treatment. Suggest that patients reduce their drinking, and solicit their input into what would be a reasonable goal, such as those suggested in the NIAAA clinical guidelines (see Related Resources). Also advise patients to refrain from drinking/using before or during anxiety exposures so they can obtain the maximum benefits of treatment.
Individuals who are severely alcohol dependent or fail to meet their reduced drinking goals may require additional treatment. Options include:
- referral to a specialized addiction treatment setting
- pharmacotherapy with FDA-approved medications for treating alcohol dependence, such as naltrexone, acamprosate, or disulfiram.
Figure 2
Identifying and addressing alcohol use in patients with anxiety disorder
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools to help you judge whether a patient’s alcohol use exceeds recommended limits (seeRelated Resources).Related resources
- Al’Absi, M, ed. Stress and addiction: Biological and psychological mechanisms. 1st ed. New York: Elsevier-Academic Press; 2007.
- Beck AT, Wright FD, Newman CF, Liese BS. Cognitive therapy of substance abuse. New York: Guilford Press; 1993.
- National Institute on Alcohol Abuse and Alcoholism. Alcoholism screening tools. http://pubs.niaaa.nih.gov/publications/arh28-2/78-79.htm.
- National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005 ed. Rockville, MD: U.S. Department of Health and Human Services; 2005. NIH Publication No. 07-3769. http://pubs.niaaa. nih.gov/publications/Practitioner/CliniciansGuide2005/ clinicians_guide.htm.
- Padesky CA, Greenberger D. Mind over mood. New York: Guilford Press; 1995.
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • Depade, ReVia
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Eric Maurer for his help in preparing this manuscript.
This work was supported, in part, by the following grants: NIAAA Grant R01 AA0105069 (MGK), R01 AA013379 (CLR), P50 AA010761 (CLR, SWB).
1. National Institute on Alcohol Abuse and Alcoholism. Alcohol use and alcohol use disorders in the United States: main findings from the 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Bethesda, MD; National Institutes of Health; 2006. NIH Publication No. 05-5737.
2. Kushner, Sher KJ, Beitman BD. The relation between alcohol problems and the anxiety disorders. Am J Psychiatry 1990;147:685-95.
3. Kushner MG, Abrams K, Thuras P, et al. Follow-up study of anxiety disorder and alcohol dependence in comorbid alcoholism treatment patients. Alcohol Clin Exp Res 2005;29:1432-43.
4. Kushner MG, Abrams K, Borchardt C. Anxiety disorders co-occurring with alcohol or drug use disorders: a review of major perspectives and findings. Clin Psychol Rev 2000;20:149-71.
5. Randall CL, Thomas S, Thevos AK. Concurrent alcoholism and social anxiety disorder: a first step toward developing effective treatments. Alcohol Clin Exp Res 2001;25:210-20.
6. Thomas SE, Thevos AK, Randall CL. Alcoholics with and without social phobia: a comparison of substance use and psychiatric variables. J Stud Alcohol 1999;60(4):472-9.
7. Conger JJ. Reinforcement theory and the dynamics of alcoholism. Q J Stud Alcohol 1956;17:296-305.
8. Sher KG, Levenson RW. Risk for alcoholism and individual differences in the stress-response dampening effect of alcohol. J Abnorm Psychol 1982;91:350-68.
9. Carrigan MH, Randall CL. Self-medication in social phobia: a review of the alcohol literature. Addict Behav 2003;28(2):269-84.
10. Schuckit MA, Hesselbrock V. Alcohol dependence and anxiety disorders: what is the relationship? Am J Psychiatry 1994;151:1723-34.
11. Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders. Arch Gen Psychiatry 2004;61:807-16.
12. Thomas SE, Randall CL, Carrigan MH. Drinking to cope in socially anxious individuals: a controlled study. Alcohol Clin Exp Res 2003;27(12):1937-43.
13. Merikangas KR, Metha RL, Molnar BE, et al. Comorbidity of substance use disorders with mood and anxiety disorders: results of the International Consortium in Psychiatry Epidemiology. Addict Behav 1996;23(6):893-907.
14. Kushner MG, Sher KJ, Erickson DJ. Prospective analysis of the relation between DSM-III anxiety disorders and alcohol use disorders. Am J Psychiatry 1990;156:723-32.
15. Book SW, Thomas SE, Randall PK, Randall CL. Paroxetine reduces social anxiety in individuals with a co-occurring alcohol use disorder. J Anxiety Disord. In press.
16. Kadden RM, Kranzler HR, Rounsaville BJ. Validity of the distinction between “substance-induced” and “independent” depression and anxiety disorders. Am J Addict 1995;4:107-17.
17. Kushner MG, Donahue C, Sletten S. Cognitive behavioral treatment of comorbid anxiety disorder in alcoholism treatment patients: presentation of a prototype program and future directions. J Mental Health 2006;15:697-708.
Men and women with anxiety disorders are at three times the general population’s risk of being alcohol-dependent (Table),1 and those who seek treatment for an anxiety disorder are at even higher risk of alcohol disorder.2,3 This comorbidity can complicate treatment attempts if either disorder remains unaddressed, leading to increased relapse risk and multiple treatment episodes.
Based on our research and clinical work in helping patients with comorbid alcohol dependence and anxiety disorders,2,4-6 this article describes:
- potential relationships between anxiety disorders and alcohol disorder
- pros and cons of 3 approaches to treating this comorbidity
- how to identify and address alcohol disorder in patients with anxiety disorders, depending on available resources.
Table
Comorbidity rates of anxiety disorders and alcohol dependence*
| Anxiety disorder | Odds ratio for having alcohol dependence | |
|---|---|---|
| Men | Women | |
| Any | 3.2 | 3.3 |
| Panic disorder | 3.8 | 3.7 |
| Social phobia | 2.6 | 3.6 |
| Generalized anxiety disorder | 3.6 | 3.4 |
| Specific phobia | 2.8 | 2.9 |
| * Numbers indicate odds of having alcohol dependence when the anxiety disorder is present vs absent. | ||
| Source: 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), reference 1 | ||
3 comorbidity models
The most common understanding of this comorbidity (Figure 1) is that having an anxiety disorder predisposes one to develop an alcohol or substance use disorder via self-medication—using alcohol or drugs to modulate anxiety and negative affect.7-9 However, substance use disorder experts have argued that the social, occupational, and physiologic effects of substance use can generate new anxiety symptoms in vulnerable individuals.10 In other words, physiologic and/or environmental disruptions from chronic alcohol or substance use could promote conditions and circumstances in which anxiety symptoms are more likely to emerge or worsen.
Although DSM-IV-TR does not delve into the causes of mental disorders, it states that substance use can cause or “induce” an anxiety syndrome with symptoms that resemble or are identical to those of the various anxiety syndromes that are not related to substance disorder (Box).11,12
Alternatively, the idea that a third factor can serve as a common cause for both conditions fits with the view that substance use disorder and anxiety disorder can be phenotypic expressions of a common underlying genetic/physiologic liability.13
Finally, these models are not mutually exclusive. Anxiety symptoms or substance use could cause or aggravate the other.
Figure 1
Hypothetical models of comorbidity
Which comes first?
Anxiety disorder typically begins before a substance use disorder in comorbid cases, although some studies have reported the opposite pattern or roughly simultaneous onset of both disorders.2
Using a prospective method in college students, we found that the risk of developing alcohol dependence for the first time as a junior or senior more than tripled among students who had an anxiety diagnosis as a freshman.14 We also found, however, that students who were alcohol dependent as freshman were 4 times more likely than other freshman to develop an anxiety disorder for the first time within the next 6 years.
In short, having either an anxiety or alcohol disorder earlier in life appears to increase the probability of developing the other later. This finding supports the idea that the types of associations that link pathologic anxiety and substance use vary among individuals and, perhaps, within individuals over time.
3 treatment approaches
Treating 1 of the comorbid conditions—anxiety or alcohol disorder—does not tend to resolve the other.3,15 This suggests that therapies aimed at treating a single disorder are not satisfactory for treating comorbid cases. Possible multi-focused approaches include:
- serial (or sequential) approach—treating comorbid disorders one at a time
- parallel approach—providing simultaneous but separate treatments for each comorbidity
- integrated approach—providing one treatment that focuses on both comorbid disorders, especially as they interact with one another.
Each has strengths and weaknesses, and the approach you choose for your patient may depend on clinical circumstances and available resources.
Serial treatment has the structure to empirically evaluate whether the initially untreated comorbid condition is resolved by treating the other condition. For example, you could treat an anxiety disorder as usual and refer the patient for alcohol disorder treatment only if drinking remains an active problem following anxiety treatment. This approach also allows you to use well-established treatment systems, programs, and specialists as usual.
One disadvantage to the serial approach, however, is that the initially untreated comorbid disorder could undermine the resolution of the treated disorder. We found, for example, that treating either the anxiety or the alcohol disorder alone fails to resolve the comorbid condition and might leave a patient vulnerable to relapse before serial treatment can be completed.3,15
When implementing a serial treatment, it is not always clear which disorder to treat first. Distinguishing comorbid disorders as “primary” or “secondary” often is done inconsistently and imprecisely, so treatment decisions based on these terms can be erroneous. Using the order of disorder onset also is an unreliable guide to which disorder is in priority need of treatment.7,16
Based on our experience and research, where and why the comorbid patient presents for treatment should be factored heavily in these treatment decisions. For example, individuals seeking anxiety treatment who have a comorbid alcohol use disorder typically possess little insight into their drinking problem and a frank resistance to clinician-driven attempts to modify their drinking behavior. We would expect a similar reaction from patients presenting for alcohol disorder treatment who are told they must first obtain psychiatric treatment for anxiety symptoms. The serial approach often necessitates that patients be treated initially for the problem for which they present and then referred afterward for the comorbid condition as needed.
In an open-label pilot treatment study of 5 subjects with social anxiety disorder and a co-occurring alcohol use disorder (C.L.R., S.W.B., unpublished data, 2007), we first treated the anxiety disorder with the selective serotonin reuptake inhibitor paroxetine, up to 60 mg/d. After 6 weeks, we addressed the comorbid alcohol problem using a brief alcohol intervention. This approach met with little or no resistance to reduce drinking—all 5 subjects successfully decreased their alcohol consumption, and none dropped out of treatment. A controlled follow-up trial is planned to provide empiric support for serial treatment of anxiety and alcohol use disorders in mental health treatment settings.
DSM-IV-TR describes an anxiety disorder as independent from a coexisting substance use disorder only if the anxiety disorder:
- began distinctly before the substance use disorder
- or persisted during periods of extended abstinence (>1 month) from substance use/abuse.
Otherwise, substance abuse is presumed to have induced the anxiety disorder. This perspective implies that no specific treatment beyond drug/alcohol abstinence is required to resolve a substance-induced anxiety disorder.
In a large community-based sample, Grant et al11 found that <0.5% of individuals with comorbid anxiety and substance abuse met the strictly defined DSM-IV-TR criteria for a substance-induced anxiety disorder. Cases in which a comorbid anxiety disorder resolved during periods of substance abuse abstinence were especially rare. This observation suggests that substance-induced anxiety syndrome as defined by DSM-IV-TR is very rare in clinical practice.
DSM-IV-TR diagnostic criteria do not recognize an “anxiety-induced substance use disorder,” in which pathologic anxiety might induce a substance use disorder. Conceptually, however, this idea is as reasonable as substance-induced anxiety disorder and fits within the self-medication model.12
Parallel treatments, which can mitigate some disadvantages of the serial approach, increasingly are being used in chemical dependency treatment settings, where it is common to have psychiatric consultations. Based on our experience, however, this approach is far less common in psychiatric treatment settings, where clinicians do not routinely treat (or sometimes even assess for) comorbid alcohol or drug disorder in anxious and depressed patients. Also, the parallel approach often requires coordinating the times, locations, and strategies of treatments systems and clinicians, which can lead to problems:
- Substance use disorder treatment expertise is not always available for patients in mental health treatment.
- Clinicians from disparate systems may not fully understand the impact of the comorbid disorder and the culture of the parallel treatment system.
- Practitioners (or patients) might see medications or cognitive-behavioral therapy (CBT) exercises for anxiety as contradicting core tenants of the parallel treatment approach.
- It is not certain that standard treatments validated in non-comorbid patients would have the same therapeutic benefits when administered in a parallel treatment.5
Integrated treatments seek to address both comorbid disorders in a single treatment program. Our group has found, for example, that a CBT program aimed at treating comorbid anxiety could be successfully integrated with a standard alcohol disorder treatment.17
Several factors limit the use of integrated treatments, however:
- Few such programs exist.
- Treatment providers in mental health and addiction settings typically are not cross-trained.
- Personnel and other institutional supports are often lacking for integrated treatment programs.
Integrated treatment plan
Our CBT-based integrated approach to alcoholism treatment in patients with a comorbid anxiety disorder incorporates 3 components:
- psychoeducation
- cognitive restructuring
- cue exposure.
Psychoeducation. The goal of psychoeducation is to explain the biopsychosocial model of anxiety disorder, alcohol disorders, and their interactions. This information is the general platform on which the specific treatment program is established in the next phase of the treatment.
In addition to providing basic epidemiologic facts, we use simple language and graphics to emphasize the vicious cycle that can emerge between drinking and anxiety, wherein the more one uses alcohol to manage anxiety in the short run the more anxiety there is to manage in the long run.
We introduce the role of cognitions, thoughts, beliefs, and expectations in how individuals react to situations to produce anxiety and drinking urges. Finally, we teach patients a standard paced diaphragmatic breathing exercise designed to minimize hyperventilation commonly identified among individuals with anxiety disorders.
Cognitive restructuring. We teach patients about thinking patterns that contribute to initiating and maintaining anxiety and panic. We also teach patients how to recognize and restructure cognitions that promote alcohol use as a means of coping with anxiety, such as focusing on alcohol’s short-term calming effects instead of its longer-term anxiogenic effects. This phase requires clinical expertise in CBT skills; a wide range of resource materials is available to walk the patient (and clinician) through cognitive restructuring exercises (see Related Resources).
Cue exposure involves systematic therapist-guided exposure to fear-provoking situations and sensations with the goal of decoupling them from anxiety-inducing thoughts about catastrophic outcomes.
Exposures are used:
- for reality testing
- to allow patients to practice new anxiety management skills
- to increase patients’ sense that they can successfully cope in feared situations (“self-efficacy”).
We expand this approach to include alcohol-relevant cues associated with anxiety states. Exposures—imaginal and in vivo—incorporate this information to help patients decouple anxiety feelings from drinking urges and to practice alternate coping strategies.
Pilot data for integrated Tx
After 4 months of participating in an integrated CBT program, 32 alcoholism treatment patients with panic disorder were significantly less likely to meet criteria for panic disorder, compared with 17 patients who received standard chemical dependency treatment without the CBT program (M.G.K., C.D., B.F., unpublished data, 2007). Before treatment, both groups averaged approximately 2.5 panic attacks per week. At follow-up the group that received CBT averaged <0.5 panic attacks per week, whereas the control group averaged approximately 2 panic attacks per week.
Overall, there was a positive effect for CBT treatment in terms of relapse to full alcohol dependence—10% in the treatment group met this criteria vs 35% in the control group. Integrated CBT treatment was more effective in reducing relapse risk among patients who reported the strongest baseline expectations that alcohol consumption helps to control their anxiety symptoms: 0% in the treatment group relapsed to full alcohol dependence vs. 57% in the control group. For comorbid cases that had the weakest anxiety-reduction expectancies, 21% in the CBT group met the relapse criterion compared with about 20% in the control group.
In summary, an integrated CBT program for comorbid panic disorder appears to provide the greatest added value to standard alcoholism treatment among patients who expect alcohol to relieve their anxiety symptoms.
Treatment in a psychiatric setting
Our group is in the process of generating a database upon which to make empirically based treatment recommendations. Until then, we can offer treatment recommendations based upon experience and the limited data available.
When planning psychiatric treatment for a patient with an anxiety disorder, start by assessing the patient’s alcohol use (Figure 2). The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools (see Related Resources) to help you judge whether a patient’s alcohol use exceeds recommended limits (for example, 7 drinks per week for women and 14 per week for men).
Teach individuals whose drinking is excessive and/or regular (especially deliberate drinking aimed at coping with anxiety) about the risk associated with alcohol use and potential interference of alcohol/drugs with successful anxiety treatment. Suggest that patients reduce their drinking, and solicit their input into what would be a reasonable goal, such as those suggested in the NIAAA clinical guidelines (see Related Resources). Also advise patients to refrain from drinking/using before or during anxiety exposures so they can obtain the maximum benefits of treatment.
Individuals who are severely alcohol dependent or fail to meet their reduced drinking goals may require additional treatment. Options include:
- referral to a specialized addiction treatment setting
- pharmacotherapy with FDA-approved medications for treating alcohol dependence, such as naltrexone, acamprosate, or disulfiram.
Figure 2
Identifying and addressing alcohol use in patients with anxiety disorder
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools to help you judge whether a patient’s alcohol use exceeds recommended limits (seeRelated Resources).Related resources
- Al’Absi, M, ed. Stress and addiction: Biological and psychological mechanisms. 1st ed. New York: Elsevier-Academic Press; 2007.
- Beck AT, Wright FD, Newman CF, Liese BS. Cognitive therapy of substance abuse. New York: Guilford Press; 1993.
- National Institute on Alcohol Abuse and Alcoholism. Alcoholism screening tools. http://pubs.niaaa.nih.gov/publications/arh28-2/78-79.htm.
- National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005 ed. Rockville, MD: U.S. Department of Health and Human Services; 2005. NIH Publication No. 07-3769. http://pubs.niaaa. nih.gov/publications/Practitioner/CliniciansGuide2005/ clinicians_guide.htm.
- Padesky CA, Greenberger D. Mind over mood. New York: Guilford Press; 1995.
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • Depade, ReVia
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Eric Maurer for his help in preparing this manuscript.
This work was supported, in part, by the following grants: NIAAA Grant R01 AA0105069 (MGK), R01 AA013379 (CLR), P50 AA010761 (CLR, SWB).
Men and women with anxiety disorders are at three times the general population’s risk of being alcohol-dependent (Table),1 and those who seek treatment for an anxiety disorder are at even higher risk of alcohol disorder.2,3 This comorbidity can complicate treatment attempts if either disorder remains unaddressed, leading to increased relapse risk and multiple treatment episodes.
Based on our research and clinical work in helping patients with comorbid alcohol dependence and anxiety disorders,2,4-6 this article describes:
- potential relationships between anxiety disorders and alcohol disorder
- pros and cons of 3 approaches to treating this comorbidity
- how to identify and address alcohol disorder in patients with anxiety disorders, depending on available resources.
Table
Comorbidity rates of anxiety disorders and alcohol dependence*
| Anxiety disorder | Odds ratio for having alcohol dependence | |
|---|---|---|
| Men | Women | |
| Any | 3.2 | 3.3 |
| Panic disorder | 3.8 | 3.7 |
| Social phobia | 2.6 | 3.6 |
| Generalized anxiety disorder | 3.6 | 3.4 |
| Specific phobia | 2.8 | 2.9 |
| * Numbers indicate odds of having alcohol dependence when the anxiety disorder is present vs absent. | ||
| Source: 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC), reference 1 | ||
3 comorbidity models
The most common understanding of this comorbidity (Figure 1) is that having an anxiety disorder predisposes one to develop an alcohol or substance use disorder via self-medication—using alcohol or drugs to modulate anxiety and negative affect.7-9 However, substance use disorder experts have argued that the social, occupational, and physiologic effects of substance use can generate new anxiety symptoms in vulnerable individuals.10 In other words, physiologic and/or environmental disruptions from chronic alcohol or substance use could promote conditions and circumstances in which anxiety symptoms are more likely to emerge or worsen.
Although DSM-IV-TR does not delve into the causes of mental disorders, it states that substance use can cause or “induce” an anxiety syndrome with symptoms that resemble or are identical to those of the various anxiety syndromes that are not related to substance disorder (Box).11,12
Alternatively, the idea that a third factor can serve as a common cause for both conditions fits with the view that substance use disorder and anxiety disorder can be phenotypic expressions of a common underlying genetic/physiologic liability.13
Finally, these models are not mutually exclusive. Anxiety symptoms or substance use could cause or aggravate the other.
Figure 1
Hypothetical models of comorbidity
Which comes first?
Anxiety disorder typically begins before a substance use disorder in comorbid cases, although some studies have reported the opposite pattern or roughly simultaneous onset of both disorders.2
Using a prospective method in college students, we found that the risk of developing alcohol dependence for the first time as a junior or senior more than tripled among students who had an anxiety diagnosis as a freshman.14 We also found, however, that students who were alcohol dependent as freshman were 4 times more likely than other freshman to develop an anxiety disorder for the first time within the next 6 years.
In short, having either an anxiety or alcohol disorder earlier in life appears to increase the probability of developing the other later. This finding supports the idea that the types of associations that link pathologic anxiety and substance use vary among individuals and, perhaps, within individuals over time.
3 treatment approaches
Treating 1 of the comorbid conditions—anxiety or alcohol disorder—does not tend to resolve the other.3,15 This suggests that therapies aimed at treating a single disorder are not satisfactory for treating comorbid cases. Possible multi-focused approaches include:
- serial (or sequential) approach—treating comorbid disorders one at a time
- parallel approach—providing simultaneous but separate treatments for each comorbidity
- integrated approach—providing one treatment that focuses on both comorbid disorders, especially as they interact with one another.
Each has strengths and weaknesses, and the approach you choose for your patient may depend on clinical circumstances and available resources.
Serial treatment has the structure to empirically evaluate whether the initially untreated comorbid condition is resolved by treating the other condition. For example, you could treat an anxiety disorder as usual and refer the patient for alcohol disorder treatment only if drinking remains an active problem following anxiety treatment. This approach also allows you to use well-established treatment systems, programs, and specialists as usual.
One disadvantage to the serial approach, however, is that the initially untreated comorbid disorder could undermine the resolution of the treated disorder. We found, for example, that treating either the anxiety or the alcohol disorder alone fails to resolve the comorbid condition and might leave a patient vulnerable to relapse before serial treatment can be completed.3,15
When implementing a serial treatment, it is not always clear which disorder to treat first. Distinguishing comorbid disorders as “primary” or “secondary” often is done inconsistently and imprecisely, so treatment decisions based on these terms can be erroneous. Using the order of disorder onset also is an unreliable guide to which disorder is in priority need of treatment.7,16
Based on our experience and research, where and why the comorbid patient presents for treatment should be factored heavily in these treatment decisions. For example, individuals seeking anxiety treatment who have a comorbid alcohol use disorder typically possess little insight into their drinking problem and a frank resistance to clinician-driven attempts to modify their drinking behavior. We would expect a similar reaction from patients presenting for alcohol disorder treatment who are told they must first obtain psychiatric treatment for anxiety symptoms. The serial approach often necessitates that patients be treated initially for the problem for which they present and then referred afterward for the comorbid condition as needed.
In an open-label pilot treatment study of 5 subjects with social anxiety disorder and a co-occurring alcohol use disorder (C.L.R., S.W.B., unpublished data, 2007), we first treated the anxiety disorder with the selective serotonin reuptake inhibitor paroxetine, up to 60 mg/d. After 6 weeks, we addressed the comorbid alcohol problem using a brief alcohol intervention. This approach met with little or no resistance to reduce drinking—all 5 subjects successfully decreased their alcohol consumption, and none dropped out of treatment. A controlled follow-up trial is planned to provide empiric support for serial treatment of anxiety and alcohol use disorders in mental health treatment settings.
DSM-IV-TR describes an anxiety disorder as independent from a coexisting substance use disorder only if the anxiety disorder:
- began distinctly before the substance use disorder
- or persisted during periods of extended abstinence (>1 month) from substance use/abuse.
Otherwise, substance abuse is presumed to have induced the anxiety disorder. This perspective implies that no specific treatment beyond drug/alcohol abstinence is required to resolve a substance-induced anxiety disorder.
In a large community-based sample, Grant et al11 found that <0.5% of individuals with comorbid anxiety and substance abuse met the strictly defined DSM-IV-TR criteria for a substance-induced anxiety disorder. Cases in which a comorbid anxiety disorder resolved during periods of substance abuse abstinence were especially rare. This observation suggests that substance-induced anxiety syndrome as defined by DSM-IV-TR is very rare in clinical practice.
DSM-IV-TR diagnostic criteria do not recognize an “anxiety-induced substance use disorder,” in which pathologic anxiety might induce a substance use disorder. Conceptually, however, this idea is as reasonable as substance-induced anxiety disorder and fits within the self-medication model.12
Parallel treatments, which can mitigate some disadvantages of the serial approach, increasingly are being used in chemical dependency treatment settings, where it is common to have psychiatric consultations. Based on our experience, however, this approach is far less common in psychiatric treatment settings, where clinicians do not routinely treat (or sometimes even assess for) comorbid alcohol or drug disorder in anxious and depressed patients. Also, the parallel approach often requires coordinating the times, locations, and strategies of treatments systems and clinicians, which can lead to problems:
- Substance use disorder treatment expertise is not always available for patients in mental health treatment.
- Clinicians from disparate systems may not fully understand the impact of the comorbid disorder and the culture of the parallel treatment system.
- Practitioners (or patients) might see medications or cognitive-behavioral therapy (CBT) exercises for anxiety as contradicting core tenants of the parallel treatment approach.
- It is not certain that standard treatments validated in non-comorbid patients would have the same therapeutic benefits when administered in a parallel treatment.5
Integrated treatments seek to address both comorbid disorders in a single treatment program. Our group has found, for example, that a CBT program aimed at treating comorbid anxiety could be successfully integrated with a standard alcohol disorder treatment.17
Several factors limit the use of integrated treatments, however:
- Few such programs exist.
- Treatment providers in mental health and addiction settings typically are not cross-trained.
- Personnel and other institutional supports are often lacking for integrated treatment programs.
Integrated treatment plan
Our CBT-based integrated approach to alcoholism treatment in patients with a comorbid anxiety disorder incorporates 3 components:
- psychoeducation
- cognitive restructuring
- cue exposure.
Psychoeducation. The goal of psychoeducation is to explain the biopsychosocial model of anxiety disorder, alcohol disorders, and their interactions. This information is the general platform on which the specific treatment program is established in the next phase of the treatment.
In addition to providing basic epidemiologic facts, we use simple language and graphics to emphasize the vicious cycle that can emerge between drinking and anxiety, wherein the more one uses alcohol to manage anxiety in the short run the more anxiety there is to manage in the long run.
We introduce the role of cognitions, thoughts, beliefs, and expectations in how individuals react to situations to produce anxiety and drinking urges. Finally, we teach patients a standard paced diaphragmatic breathing exercise designed to minimize hyperventilation commonly identified among individuals with anxiety disorders.
Cognitive restructuring. We teach patients about thinking patterns that contribute to initiating and maintaining anxiety and panic. We also teach patients how to recognize and restructure cognitions that promote alcohol use as a means of coping with anxiety, such as focusing on alcohol’s short-term calming effects instead of its longer-term anxiogenic effects. This phase requires clinical expertise in CBT skills; a wide range of resource materials is available to walk the patient (and clinician) through cognitive restructuring exercises (see Related Resources).
Cue exposure involves systematic therapist-guided exposure to fear-provoking situations and sensations with the goal of decoupling them from anxiety-inducing thoughts about catastrophic outcomes.
Exposures are used:
- for reality testing
- to allow patients to practice new anxiety management skills
- to increase patients’ sense that they can successfully cope in feared situations (“self-efficacy”).
We expand this approach to include alcohol-relevant cues associated with anxiety states. Exposures—imaginal and in vivo—incorporate this information to help patients decouple anxiety feelings from drinking urges and to practice alternate coping strategies.
Pilot data for integrated Tx
After 4 months of participating in an integrated CBT program, 32 alcoholism treatment patients with panic disorder were significantly less likely to meet criteria for panic disorder, compared with 17 patients who received standard chemical dependency treatment without the CBT program (M.G.K., C.D., B.F., unpublished data, 2007). Before treatment, both groups averaged approximately 2.5 panic attacks per week. At follow-up the group that received CBT averaged <0.5 panic attacks per week, whereas the control group averaged approximately 2 panic attacks per week.
Overall, there was a positive effect for CBT treatment in terms of relapse to full alcohol dependence—10% in the treatment group met this criteria vs 35% in the control group. Integrated CBT treatment was more effective in reducing relapse risk among patients who reported the strongest baseline expectations that alcohol consumption helps to control their anxiety symptoms: 0% in the treatment group relapsed to full alcohol dependence vs. 57% in the control group. For comorbid cases that had the weakest anxiety-reduction expectancies, 21% in the CBT group met the relapse criterion compared with about 20% in the control group.
In summary, an integrated CBT program for comorbid panic disorder appears to provide the greatest added value to standard alcoholism treatment among patients who expect alcohol to relieve their anxiety symptoms.
Treatment in a psychiatric setting
Our group is in the process of generating a database upon which to make empirically based treatment recommendations. Until then, we can offer treatment recommendations based upon experience and the limited data available.
When planning psychiatric treatment for a patient with an anxiety disorder, start by assessing the patient’s alcohol use (Figure 2). The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools (see Related Resources) to help you judge whether a patient’s alcohol use exceeds recommended limits (for example, 7 drinks per week for women and 14 per week for men).
Teach individuals whose drinking is excessive and/or regular (especially deliberate drinking aimed at coping with anxiety) about the risk associated with alcohol use and potential interference of alcohol/drugs with successful anxiety treatment. Suggest that patients reduce their drinking, and solicit their input into what would be a reasonable goal, such as those suggested in the NIAAA clinical guidelines (see Related Resources). Also advise patients to refrain from drinking/using before or during anxiety exposures so they can obtain the maximum benefits of treatment.
Individuals who are severely alcohol dependent or fail to meet their reduced drinking goals may require additional treatment. Options include:
- referral to a specialized addiction treatment setting
- pharmacotherapy with FDA-approved medications for treating alcohol dependence, such as naltrexone, acamprosate, or disulfiram.
Figure 2
Identifying and addressing alcohol use in patients with anxiety disorder
The National Institute on Alcohol Abuse and Alcoholism (NIAAA) offers assessment tools to help you judge whether a patient’s alcohol use exceeds recommended limits (seeRelated Resources).Related resources
- Al’Absi, M, ed. Stress and addiction: Biological and psychological mechanisms. 1st ed. New York: Elsevier-Academic Press; 2007.
- Beck AT, Wright FD, Newman CF, Liese BS. Cognitive therapy of substance abuse. New York: Guilford Press; 1993.
- National Institute on Alcohol Abuse and Alcoholism. Alcoholism screening tools. http://pubs.niaaa.nih.gov/publications/arh28-2/78-79.htm.
- National Institute on Alcohol Abuse and Alcoholism. Helping patients who drink too much: a clinician’s guide. 2005 ed. Rockville, MD: U.S. Department of Health and Human Services; 2005. NIH Publication No. 07-3769. http://pubs.niaaa. nih.gov/publications/Practitioner/CliniciansGuide2005/ clinicians_guide.htm.
- Padesky CA, Greenberger D. Mind over mood. New York: Guilford Press; 1995.
Drug brand names
- Acamprosate • Campral
- Disulfiram • Antabuse
- Naltrexone • Depade, ReVia
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Acknowledgements
The authors thank Eric Maurer for his help in preparing this manuscript.
This work was supported, in part, by the following grants: NIAAA Grant R01 AA0105069 (MGK), R01 AA013379 (CLR), P50 AA010761 (CLR, SWB).
1. National Institute on Alcohol Abuse and Alcoholism. Alcohol use and alcohol use disorders in the United States: main findings from the 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Bethesda, MD; National Institutes of Health; 2006. NIH Publication No. 05-5737.
2. Kushner, Sher KJ, Beitman BD. The relation between alcohol problems and the anxiety disorders. Am J Psychiatry 1990;147:685-95.
3. Kushner MG, Abrams K, Thuras P, et al. Follow-up study of anxiety disorder and alcohol dependence in comorbid alcoholism treatment patients. Alcohol Clin Exp Res 2005;29:1432-43.
4. Kushner MG, Abrams K, Borchardt C. Anxiety disorders co-occurring with alcohol or drug use disorders: a review of major perspectives and findings. Clin Psychol Rev 2000;20:149-71.
5. Randall CL, Thomas S, Thevos AK. Concurrent alcoholism and social anxiety disorder: a first step toward developing effective treatments. Alcohol Clin Exp Res 2001;25:210-20.
6. Thomas SE, Thevos AK, Randall CL. Alcoholics with and without social phobia: a comparison of substance use and psychiatric variables. J Stud Alcohol 1999;60(4):472-9.
7. Conger JJ. Reinforcement theory and the dynamics of alcoholism. Q J Stud Alcohol 1956;17:296-305.
8. Sher KG, Levenson RW. Risk for alcoholism and individual differences in the stress-response dampening effect of alcohol. J Abnorm Psychol 1982;91:350-68.
9. Carrigan MH, Randall CL. Self-medication in social phobia: a review of the alcohol literature. Addict Behav 2003;28(2):269-84.
10. Schuckit MA, Hesselbrock V. Alcohol dependence and anxiety disorders: what is the relationship? Am J Psychiatry 1994;151:1723-34.
11. Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders. Arch Gen Psychiatry 2004;61:807-16.
12. Thomas SE, Randall CL, Carrigan MH. Drinking to cope in socially anxious individuals: a controlled study. Alcohol Clin Exp Res 2003;27(12):1937-43.
13. Merikangas KR, Metha RL, Molnar BE, et al. Comorbidity of substance use disorders with mood and anxiety disorders: results of the International Consortium in Psychiatry Epidemiology. Addict Behav 1996;23(6):893-907.
14. Kushner MG, Sher KJ, Erickson DJ. Prospective analysis of the relation between DSM-III anxiety disorders and alcohol use disorders. Am J Psychiatry 1990;156:723-32.
15. Book SW, Thomas SE, Randall PK, Randall CL. Paroxetine reduces social anxiety in individuals with a co-occurring alcohol use disorder. J Anxiety Disord. In press.
16. Kadden RM, Kranzler HR, Rounsaville BJ. Validity of the distinction between “substance-induced” and “independent” depression and anxiety disorders. Am J Addict 1995;4:107-17.
17. Kushner MG, Donahue C, Sletten S. Cognitive behavioral treatment of comorbid anxiety disorder in alcoholism treatment patients: presentation of a prototype program and future directions. J Mental Health 2006;15:697-708.
1. National Institute on Alcohol Abuse and Alcoholism. Alcohol use and alcohol use disorders in the United States: main findings from the 2001-2002 National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Bethesda, MD; National Institutes of Health; 2006. NIH Publication No. 05-5737.
2. Kushner, Sher KJ, Beitman BD. The relation between alcohol problems and the anxiety disorders. Am J Psychiatry 1990;147:685-95.
3. Kushner MG, Abrams K, Thuras P, et al. Follow-up study of anxiety disorder and alcohol dependence in comorbid alcoholism treatment patients. Alcohol Clin Exp Res 2005;29:1432-43.
4. Kushner MG, Abrams K, Borchardt C. Anxiety disorders co-occurring with alcohol or drug use disorders: a review of major perspectives and findings. Clin Psychol Rev 2000;20:149-71.
5. Randall CL, Thomas S, Thevos AK. Concurrent alcoholism and social anxiety disorder: a first step toward developing effective treatments. Alcohol Clin Exp Res 2001;25:210-20.
6. Thomas SE, Thevos AK, Randall CL. Alcoholics with and without social phobia: a comparison of substance use and psychiatric variables. J Stud Alcohol 1999;60(4):472-9.
7. Conger JJ. Reinforcement theory and the dynamics of alcoholism. Q J Stud Alcohol 1956;17:296-305.
8. Sher KG, Levenson RW. Risk for alcoholism and individual differences in the stress-response dampening effect of alcohol. J Abnorm Psychol 1982;91:350-68.
9. Carrigan MH, Randall CL. Self-medication in social phobia: a review of the alcohol literature. Addict Behav 2003;28(2):269-84.
10. Schuckit MA, Hesselbrock V. Alcohol dependence and anxiety disorders: what is the relationship? Am J Psychiatry 1994;151:1723-34.
11. Grant BF, Stinson FS, Dawson DA, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders. Arch Gen Psychiatry 2004;61:807-16.
12. Thomas SE, Randall CL, Carrigan MH. Drinking to cope in socially anxious individuals: a controlled study. Alcohol Clin Exp Res 2003;27(12):1937-43.
13. Merikangas KR, Metha RL, Molnar BE, et al. Comorbidity of substance use disorders with mood and anxiety disorders: results of the International Consortium in Psychiatry Epidemiology. Addict Behav 1996;23(6):893-907.
14. Kushner MG, Sher KJ, Erickson DJ. Prospective analysis of the relation between DSM-III anxiety disorders and alcohol use disorders. Am J Psychiatry 1990;156:723-32.
15. Book SW, Thomas SE, Randall PK, Randall CL. Paroxetine reduces social anxiety in individuals with a co-occurring alcohol use disorder. J Anxiety Disord. In press.
16. Kadden RM, Kranzler HR, Rounsaville BJ. Validity of the distinction between “substance-induced” and “independent” depression and anxiety disorders. Am J Addict 1995;4:107-17.
17. Kushner MG, Donahue C, Sletten S. Cognitive behavioral treatment of comorbid anxiety disorder in alcoholism treatment patients: presentation of a prototype program and future directions. J Mental Health 2006;15:697-708.
Sedation with antipsychotics: Manage, don't accept adverse 'calming' effect
Manage, don’t accept adverse ‘calming’ effect
Sedation is a frequent side effect of antipsychotics, especially at relatively high doses. Antipsychotics’ sedative effects can reduce agitation in acute psychosis and promote sleep in insomnia, but long-term sedation may:
- interfere with schizophrenia patients’ efforts to go to work or school or engage in normal socialization
- prevent improvement from psychosocial training, psychiatric rehabilitation, and other treatments.
This article discusses how to manage acute psychosis without oversedation and ways to address persistent sedation and chronic insomnia with less-sedating antipsychotics or adjunctive medications.
Neurobiology or psychopharmacology?
Many patients experience only mild, transient somnolence at the beginning of antipsychotic treatment, and most develop some tolerance to the sedating effects with continued administration. Others may have persistent daytime sedation that interferes with normal functioning.
Sedation is especially common in elderly patients receiving antipsychotics. Compared with younger patients, older patients receiving the same doses of the same medications become more heavily sedated for longer periods of time. The resulting sedation can impair arousal levels during the day and increase the risk of falls.
Sedation can occur with first-generation antipsychotics (FGAs) and second-generation antipsychotics (SGAs), but it is seen more commonly and tends to be more severe with low-potency FGAs than with SGAs. Clinical challenges come with:
- distinguishing between sedation and negative symptoms of schizophrenia such as avolition, amotivation, withdrawal, and anhedonia
- determining whether an individual’s cognitive impairment is related to the antipsychotic’s sedative properties.
Because the treatments are different, it is important to try to distinguish negative symptoms and/or cognitive impairment related to schizophrenia’s neurobiology from sedation related to the antipsychotic. Ask patients if they nap during the day or just lie around, and if they want to do things but can’t:
- If they want to do things but feel too tired, this likely is sedation caused by the antipsychotic. Treatment might be a dose reduction.
- If they are not interested in doing things, the likely cause is negative symptoms. Treatment might be a medication such as a selective serotonin reuptake inhibitor.
- If they want to do things but cannot organize themselves to be able to do them, this likely is cognitive impairment. Treatment might be cognitive training or remediation.
Efficacy and sedation
Antipsychotics are thought to exert their effect by antagonism of postsynaptic dopamine D2 and serotonin 5HT2A receptors and possibly other receptors in the brain. Four SGAs—risperidone, olanzapine, quetiapine, and ziprasidone—act as dopamine D2 and 5HT2A antagonists. Aripiprazole is a dopamine D2 partial agonist, serotonin 5HT1A partial agonist, and serotonin 5HT2A antagonist.1 Efficacy data comparing SGAs with each other and with FGAs vary, but all 5 of these SGAs have been shown to be effective antipsychotics.2-4 They also generally cause less sedation than FGAs.
Patients with acute exacerbation of psychosis often have insomnia and frequently report paranoia that “something” will happen to them while they sleep. When treating agitated patients, many clinicians consider calming effects and true antipsychotic effects to be one in the same, which is not correct. All available antipsychotics are, on average, equally effective in treating acute psychotic symptoms but vary considerably in the amount of sedation they produce. Studies of short-acting injectable SGAs, such as ziprasidone and aripiprazole, have shown that agitation and acute symptoms can be controlled without significant sedation.5,6
Antipsychotic effects are not immediate and historically were thought to occur over several weeks. Recent meta-analyses suggest, however, that some antipsychotic effects are evident within the first week of treatment.7,8 To avoid overmedication, therefore, it’s important to separate calming effects from antipsychotic effects.
Recommendations. Choose the initial antipsychotic based on its effectiveness in treating the underlying disease, rather than relying on side effects—such as sedation—to control disease manifestations. Without sedation, patients are better able to engage in therapy; participate in family, social, school, and work activities; and increase their chances of recovery.
Initiate the antipsychotic at or titrate to a reasonable, not overly high dose—such as:
- olanzapine, 10 to 20 mg/d
- risperidone, 3 to 6 mg/d
- ziprasidone, 100 to 140 mg/d
- quetiapine, 400 to 600 mg/d
- aripiprazole, 15 mg/d.
Continue the patient on that dose, and use a nonantipsychotic such as a benzodiazepine to help control insomnia, anxiety, and agitation. Two to 4 weeks is generally adequate, but some patients may need the adjunctive therapy for several months. If you initiate a more sedating antipsychotic acutely, switching to a less sedating agent when the patient is stable and the illness is in remission may support adherence and improve outcomes.
Dosages and sedation. Not all FGAs have the same sedative effect, nor do all SGAs (Table 1).9 In general, the high-milligram, low-potency FGAs—such as chlorpromazine—produce more sedation than the low-milligram, high-potency FGAs—such as haloperidol and fluphenazine.9 This principle tends to hold true for the SGAs as well. For example, the high-potency, low-dose SGA risperidone is less sedating than the lower-potency, high-dose SGAs quetiapine and clozapine.
Dose does not always determine sedation, however. Olanzapine, which is commonly dosed at 15 to 30 mg/d, is more sedating than ziprasidone, for which the usual range is 80 to 160 mg/d.3,10-12
Table 1
Antipsychotics’ potency, dosages, and sedative properties
| Medication | Relative potency (mg)* | Common dosage (mg/d) | Sedation |
|---|---|---|---|
| First-generation antipsychotics | |||
| Chlorpromazine | 100.0 | 300 to 600 | Moderate |
| Fluphenazine | 1.0 to 2.0 | 4 to 20 | Mild |
| Haloperidol | 2.0 | 5 to 20 | Mild |
| Second-generation antipsychotics | |||
| Aripiprazole | 7.5 | 15 to 30 | Mild |
| Clozapine | 50.0 | 250 to 500 | Marked |
| Olanzapine | 4.0 | 15 to 30 | Moderate |
| Quetiapine | 80.0 | 300 to 800 | Moderate |
| Risperidone | 1.0 | 2 to 6 | Mild |
| Ziprasidone | 20.0 | 80 to 160 | Mild |
| * Approximate dose equivalent to 100 mg of chlorpromazine | |||
| Source: Data from reference 9. | |||
Mechanism and sedation
The mechanisms of antipsychotics’ therapeutic and sedative properties appear to be different.13 The degree of sedation shows little relationship with the various antipsychotics’ potency at the dopamine D2 receptor, which suggests that dopamine D2 receptor antagonism is not involved in causing sedation.
Instead, the degree of sedation may be associated with each antipsychotic’s affinity for the histamine H1 receptor, which is highly variable (Table 2).1,14,15 In general:
- agents that are more potent histamine H1 antagonists—such as olanzapine and clozapine14—produce more sedation
- agents that are weaker H1 antagonists—such as risperidone, ziprasidone, and aripiprazole—produce less sedation.
Although dosage and affinity for histamine H1 receptors play important roles in the sedative effect of a medication, what ultimately determines sedative effect is the combination of histamine H1 affinity and the amount of drug reaching the histamine H1 receptors in the CNS.
For example, the SGA quetiapine—which has moderate affinity for histamine H1 receptors14—also has relatively low affinity for dopamine D2 receptors. Higher dosages of quetiapine are therefore required to produce antipsychotic effects compared with other SGAs—such as risperidone, ziprasidone, and aripiprazole—that have higher affinities for the dopamine D2 receptors.
Because of higher dosing, higher amounts of quetiapine are assumed to be reaching the histamine H1 receptors in the CNS. This is why quetiapine causes more sedation in clinical use than does risperidone, even though risperidone has greater affinity for the histamine H1 receptor.
Table 2
Antipsychotics’ sedative effects by the numbers: Equilibrium dissociation constants at brain receptors*
| Receptors | |||
|---|---|---|---|
| Dopamine D2 | Serotonin 5-HT2A | Histamine H1 | |
| FGA | |||
| Haloperidol | 2.60 | 61.00 | 260.0 |
| SGAs | |||
| Aripiprazole | 0.34 | 3.40 | 61.00 |
| Clozapine | 210.00 | 2.60 | 3.10 |
| Olanzapine | 20.00 | 1.50 | 0.10 |
| Risperidone | 3.80 | 0.15 | 5.20 |
| Quetiapine | 770.00 | 31.00 | 19.00 |
| Ziprasidone | 2.60 | 0.12 | 4.60 |
| * Lower numbers are equivalent to higher receptor binding affinity. Each antipsychotic’s sedative effect is determined by histamine H1 affinity and the amount of drug that reaches H1 receptors in the CNS—which in turn is affected by the agent’s dosage and dopamine D2 receptor affinity. | |||
| FGA: first-generation antipsychotic; SGAs: second-generation antipsychotics | |||
| Source: References 1,14,15 | |||
Sleep patterns in mental illness
Sleep disturbances—including changes in sleep patterns, insomnia, and excessive sleeping—occur frequently in patients with psychiatric disorders. The sleep process itself (Table 3) is disrupted in patients with schizophrenia.
A study that examined sleep patterns in 40 patients with schizophrenia found longer sleep latency, more frequent arousals, and increased periods of wakefulness after sleep onset compared with controls without a psychiatric disorder. Ratings of sleep efficiency—ratio of sleep time to time in bed—were:
- 95% in the control group
- 78% in antipsychotic-naïve patients with schizophrenia
- 72% in patients with chronic schizophrenia.15
A study of sleep in 19 patients with schizophrenia and 13 nonpsychiatric controls16 found individuals with schizophrenia had:
- increased duration of stage 1 sleep
- decreased duration of stages 3 and 4 (slow-wave) sleep
- 83% total sleep efficiency, compared with 95% in nonpsychiatric controls.
Because of these differences in sleep patterns, patients with schizophrenia often experience inadequate sleep.
Antipsychotic effects on sleep patterns. Your choice of an antipsychotic also can affect the patient’s sleep. In a study of sleep measures in patients with schizophrenia treated with risperidone or haloperidol, Yamashita et al17 reported a significant difference in the time each group spent in slow-wave sleep (27% with risperidone vs 20% with haloperidol). The authors suggested that risperidone might lengthen the amount of slow-wave sleep because of its higher affinity for serotonin 5-HT2 receptors compared with haloperidol.
Salin-Pascual et al18 found that olanzapine improved total sleep time and sleep efficacy, reduced stage 1 sleep, and significantly enhanced stage 2 and slow-wave (delta) sleep.
Serotonin 5-HT2 receptors have been reported to be involved in controlling sleep quality.19 Similar to risperidone and olanzapine, the other SGAs also have a higher affinity than haloperidol for serotonin 5-HT2 receptors (Table 2). Thus, although antipsychotics’ sedative effects may adversely affect patients, SGAs may have the potential to improve sleep quality in individuals with schizophrenia. SGAs increase slow-wave sleep, and patients feel more rested after awakening.
Table 3
Normal sleep architecture, which can be disordered in schizophrenia
| Nonrapid eye movement (NREM) sleep | |
| Stage 1 | Drowsiness; represents transition between waking and sleeping |
| Stage 2 | Sleep deepens |
| Stages 3 and 4 | Deepest levels of NREM sleep subjects are most difficult to arouse (slow-wave or delta sleep) |
| Rapid eye movement (REM) sleep | |
| Vivid dreams; pulse and respiration rates are higher and more variable than during NREM sleep | |
| During the second half of the night, slow-wave sleep decreases compared with the first half of the night, but REM periods become more frequent and prolonged. | |
Managing excessive sedation
Take steps to minimize bothersome sedation in patients taking antipsychotics (Figure). To reduce daytime sedation, instruct the patient to take all or most of the antipsychotic dose at bedtime. Also rule out medical conditions that can produce fatigue and sedation, such as hypothyroidism, obstructive sleep apnea (OSA), and restless legs syndrome.
Figure
Managing sedation in patients with schizophrenia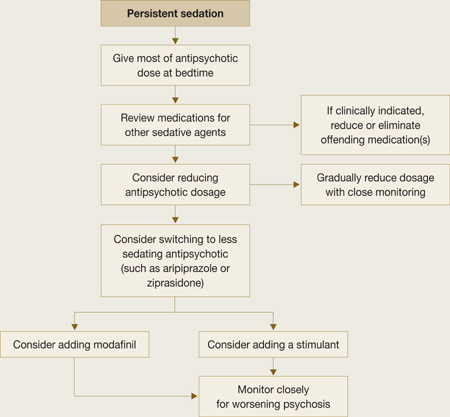
Review the patient’s medication list to determine if other potentially sedating medications can be reduced or eliminated. Psychotropics that can cause sedation include:
- antidepressants such as the tricyclics and mirtazapine
- mood stabilizers (particularly valproic acid, but also carbamazepine, lithium, and lamotrigine).
Also consider gradually reducing the patient’s antipsychotic dose, and closely monitor for worsening of psychosis.
If sedation persists despite these interventions, consider switching the patient to a less sedating antipsychotic such as ziprasidone or aripiprazole. If these efforts also are ineffective, caffeine or off-label bupropion—75 to 100 mg once in the morning or up to twice daily—might help the patient feel more alert. Many patients taking antipsychotics drink several cups of coffee every morning to feel less sedated.
Stimulants. A consensus guideline on treating schizophrenia20 recommends prescribing amphetamine-related stimulants for patients who are persistently sedated, but this practice is highly controversial. Many stimulants increase dopamine release in the CNS, which theoretically can worsen psychosis. A clinician could be held liable for the actions of patients medicated with stimulants.
Modafinil is a nonamphetamine CNS stimulant approved for use in disorders of excessive sleep such as narcolepsy, OSA, shift work sleep disorder, and fatigue related to multiple sclerosis. Its mechanism of action in promoting wakefulness in these disorders is not fully understood; it may activate histaminergic projections in the frontal cortex from the tuberomammillary nucleus, which plays a major role in maintaining wakefulness.21
Modafinil, 200 mg in the morning, has been reported to reduce total sleep time without adverse effects in 3 patients experiencing sedation associated with antipsychotics.22 A later double-blind, placebo-controlled trial by Sevy et al23 found that modafinil and placebo were associated with similar, significant improvement in fatigue over time. Narendran24 reported a case in which modafinil might have exacerbated psychosis in a patient with schizophrenia who was taking 200 mg qid.
Managing chronic insomnia
Schizophrenia patients with chronic insomnia usually require education about appropriate sleep habits, combined with additional treatments.
Sleep hygiene. Instruct patients to:
- Wake up at the same time every day, regardless of when they went to sleep.
- Maintain a consistent bedtime.
- Exercise regularly, preferably in the late afternoon but not within 2 to 4 hours of bedtime.
- Perform relaxing activities before bed.
- Keep the bedroom quiet and cool (extreme temperatures compromise sleep).
- Do not watch the clock at night.
- Avoid caffeine and nicotine for at least 6 hours before bedtime.
- Drink alcohol only in moderation, and avoid use for at least 4 hours before bedtime.
- Avoid napping; it may interfere with the ability to fall asleep at night.
Medications. The consensus guideline on treating schizophrenia20 offers the option of switching the patient with chronic insomnia to one of the more sedating antipsychotics, such as olanzapine, quetiapine, or clozapine. Sedation alone should not be the reason to switch to clozapine, however.
You could consider adding a bedtime sedative to the patient’s medications (Table 4). FDA-approved sedatives include nonbenzodiazepines such as zolpidem, zolpidem extended-release, zaleplon, or eszopiclone, and the melatonin receptor agonist ramelteon. Although not approved as sedatives, some antidepressants such as trazodone or mirtazapine and antihistamines such as diphenhydramine and hydroxyzine are used to promote sleep. Benzodiazepines can be helpful but require caution when prescribed to patients with comorbid substance abuse disorders.
Sedatives have been studied extensively in general populations with insomnia but not in patients receiving antipsychotics. Combining antipsychotics and sedatives can produce daytime drowsiness and sedation.
Table 4
Sedatives to treat insomnia in patients with schizophrenia
| Medication | Common bedtime dose range* |
|---|---|
| Benzodiazepines | |
| Estazolam | 1 to 2 mg |
| Flurazepam | 15 to 30 mg |
| Temazepam | 7.5 to 30 mg |
| Triazolam | 0.125 to 0.25 mg |
| Benzodiazepine agonists | |
| Eszopiclone | 2 to 3 mg |
| Zaleplon | 5 to 10 mg |
| Zolpidem | 5 to 10 mg |
| Zolpidem CR | 6.25 to 12.5 mg |
| Melatonin receptor agonist | |
| Ramelteon | 8 mg |
| H1 antihistamines | |
| Diphenhydramine | 25 to 50 mg |
| Hydroxyzine | 50 to 100 mg |
| Antidepressants | |
| Mirtazapine | 15 to 30 mg |
| Trazodone | 50 to 200 mg |
| * No dosage adjustment is required in this patient population | |
Related resources
- Miller DD. Atypical antipsychotics: sleep, sedation, and efficacy. Prim Care Companion J Clin Psychiatry 2004;6(suppl 2):3-7.
- Benca RM, Diagnosis and treatment of chronic insomnia: a review. Psychiatr Serv 2005;56(3):332-43.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Diphenhydramine • Benadryl
- Estazolam • ProSom
- Eszopiclone • Lunesta
- Fluphenazine • Prolixin
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Hydroxyzine • Vistaril, Atarax
- Lamotrigine • Lamictal
- Mirtazapine • Remeron
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Ramelteon • Rozerem
- Risperidone • Risperdal
- Temazepam • Restor
- Trazodone • Desyrel
- Triazolam • Halcion
- Valproic acid • Depakote
- Zaleplon • Sonata
- Ziprasidone • Geodon
- Zolpidem • Ambien, Ambien CR
Disclosure
Dr. Miller receives research support from Pfizer Inc. and has received honoraria from AstraZeneca, Bristol-Myers Squibb, Janssen Pharmaceutica, and Pfizer Inc.
1. McQuade R, Burris KD, Jordan S, et al. Aripiprazole: a dopamine-serotonin system stabilizer [abstract no. P.3.W.080]. Int J Neuropsychopharmacol 2002;5(suppl 1):S176.-
2. Davis JM, Chen N, Glick ID. A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry 2003;60(6):553-64.
3. Gardner DM, Baldessarini RJ, Waraich P. Modern antipsychotic drugs: a critical overview. Can Med Assoc J 2005;172(13):1703-11.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
5. Andrezina R, Josiassen RC, Marcus RN, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacol (Berl) 2006;188(3):281-92.
6. Brook S, Lucey JV, Gunn KP. Intramuscular ziprasidone compared with intramuscular haloperidol in the treatment of acute psychosis. Ziprasidone I.M. Study Group. J Clin Psychiatry 2000;61(12):933-41.
7. Agid O, Kapur S, Arenovich T, Zipursky RB. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 2003;60(12):1228-35.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry 2005;57(12):1543-9.
9. Jibson MD, Tandon R. An overview of antipsychotic medications. CNS News 2001;3:49-54.
10. Collaborative Working Group on Clinical Trial Evaluations. Treatment of special populations with the atypical antipsychotics. J Clin Psychiatry 1998;59(suppl 12):46-52.
11. El-Sayeh HG, Morganti C. Aripiprazole for schizophrenia. Cochrane Database Syst Rev 2004(2);CD004578.-
12. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
13. Tandon R. Safety and tolerability: how do newer generation ”atypical” antipsychotics compare? Psychiatr Q 2002;73(4):297-311.
14. Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci 2000;68(1):29-39.
15. Tandon R, Shipley JE, Taylor S, et al. Electroencephalographic sleep abnormalities in schizophrenia. Relationship to positive/negative symptoms and prior neuroleptic treatment. Arch Gen Psychiatry 1992;49(3):185-94.
16. Benson KL, Zarcone VP Jr. Rapid eye movement sleep eye movements in schizophrenia and depression. Arch Gen Psychiatry 1993;50(6):474-82.
17. Yamashita H, Morinobu S, Yamawaki S, et al. Effect of risperidone on sleep in schizophrenia: a comparison with haloperidol. Psychiatry Res 2002;109(2):137-42.
18. Salin-Pascual RJ, Herrera-Estrella M, Galicia-Polo L, Laurrabaquio MR. Olanzapine acute administration in schizophrenic patients increases delta sleep and sleep efficiency. Biol Psychiatry 1999;46(1):141-3.
19. Idzikowski C, Mills FJ, Glennard R. 5-Hydroxytryptamine-2 antagonist increases human slow wave sleep. Brain Res 1986;378(1):164-8.
20. Expert Consensus Guideline Series Treatment of schizophrenia. J Clin Psychiatry 1999;60(suppl 11):1-80.
21. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
22. Makela EH, Miller K, Cutlip WD, 2nd. Three case reports of modafinil use in treating sedation induced by antipsychotic medications. J Clin Psychiatry 2003;64(4):485-6.
23. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
24. Narendran R, Young CM, Valenti AM, et al. Is psychosis exacerbated by modafinil? Arch Gen Psychiatry 2002;59(3):292-3.
Manage, don’t accept adverse ‘calming’ effect
Sedation is a frequent side effect of antipsychotics, especially at relatively high doses. Antipsychotics’ sedative effects can reduce agitation in acute psychosis and promote sleep in insomnia, but long-term sedation may:
- interfere with schizophrenia patients’ efforts to go to work or school or engage in normal socialization
- prevent improvement from psychosocial training, psychiatric rehabilitation, and other treatments.
This article discusses how to manage acute psychosis without oversedation and ways to address persistent sedation and chronic insomnia with less-sedating antipsychotics or adjunctive medications.
Neurobiology or psychopharmacology?
Many patients experience only mild, transient somnolence at the beginning of antipsychotic treatment, and most develop some tolerance to the sedating effects with continued administration. Others may have persistent daytime sedation that interferes with normal functioning.
Sedation is especially common in elderly patients receiving antipsychotics. Compared with younger patients, older patients receiving the same doses of the same medications become more heavily sedated for longer periods of time. The resulting sedation can impair arousal levels during the day and increase the risk of falls.
Sedation can occur with first-generation antipsychotics (FGAs) and second-generation antipsychotics (SGAs), but it is seen more commonly and tends to be more severe with low-potency FGAs than with SGAs. Clinical challenges come with:
- distinguishing between sedation and negative symptoms of schizophrenia such as avolition, amotivation, withdrawal, and anhedonia
- determining whether an individual’s cognitive impairment is related to the antipsychotic’s sedative properties.
Because the treatments are different, it is important to try to distinguish negative symptoms and/or cognitive impairment related to schizophrenia’s neurobiology from sedation related to the antipsychotic. Ask patients if they nap during the day or just lie around, and if they want to do things but can’t:
- If they want to do things but feel too tired, this likely is sedation caused by the antipsychotic. Treatment might be a dose reduction.
- If they are not interested in doing things, the likely cause is negative symptoms. Treatment might be a medication such as a selective serotonin reuptake inhibitor.
- If they want to do things but cannot organize themselves to be able to do them, this likely is cognitive impairment. Treatment might be cognitive training or remediation.
Efficacy and sedation
Antipsychotics are thought to exert their effect by antagonism of postsynaptic dopamine D2 and serotonin 5HT2A receptors and possibly other receptors in the brain. Four SGAs—risperidone, olanzapine, quetiapine, and ziprasidone—act as dopamine D2 and 5HT2A antagonists. Aripiprazole is a dopamine D2 partial agonist, serotonin 5HT1A partial agonist, and serotonin 5HT2A antagonist.1 Efficacy data comparing SGAs with each other and with FGAs vary, but all 5 of these SGAs have been shown to be effective antipsychotics.2-4 They also generally cause less sedation than FGAs.
Patients with acute exacerbation of psychosis often have insomnia and frequently report paranoia that “something” will happen to them while they sleep. When treating agitated patients, many clinicians consider calming effects and true antipsychotic effects to be one in the same, which is not correct. All available antipsychotics are, on average, equally effective in treating acute psychotic symptoms but vary considerably in the amount of sedation they produce. Studies of short-acting injectable SGAs, such as ziprasidone and aripiprazole, have shown that agitation and acute symptoms can be controlled without significant sedation.5,6
Antipsychotic effects are not immediate and historically were thought to occur over several weeks. Recent meta-analyses suggest, however, that some antipsychotic effects are evident within the first week of treatment.7,8 To avoid overmedication, therefore, it’s important to separate calming effects from antipsychotic effects.
Recommendations. Choose the initial antipsychotic based on its effectiveness in treating the underlying disease, rather than relying on side effects—such as sedation—to control disease manifestations. Without sedation, patients are better able to engage in therapy; participate in family, social, school, and work activities; and increase their chances of recovery.
Initiate the antipsychotic at or titrate to a reasonable, not overly high dose—such as:
- olanzapine, 10 to 20 mg/d
- risperidone, 3 to 6 mg/d
- ziprasidone, 100 to 140 mg/d
- quetiapine, 400 to 600 mg/d
- aripiprazole, 15 mg/d.
Continue the patient on that dose, and use a nonantipsychotic such as a benzodiazepine to help control insomnia, anxiety, and agitation. Two to 4 weeks is generally adequate, but some patients may need the adjunctive therapy for several months. If you initiate a more sedating antipsychotic acutely, switching to a less sedating agent when the patient is stable and the illness is in remission may support adherence and improve outcomes.
Dosages and sedation. Not all FGAs have the same sedative effect, nor do all SGAs (Table 1).9 In general, the high-milligram, low-potency FGAs—such as chlorpromazine—produce more sedation than the low-milligram, high-potency FGAs—such as haloperidol and fluphenazine.9 This principle tends to hold true for the SGAs as well. For example, the high-potency, low-dose SGA risperidone is less sedating than the lower-potency, high-dose SGAs quetiapine and clozapine.
Dose does not always determine sedation, however. Olanzapine, which is commonly dosed at 15 to 30 mg/d, is more sedating than ziprasidone, for which the usual range is 80 to 160 mg/d.3,10-12
Table 1
Antipsychotics’ potency, dosages, and sedative properties
| Medication | Relative potency (mg)* | Common dosage (mg/d) | Sedation |
|---|---|---|---|
| First-generation antipsychotics | |||
| Chlorpromazine | 100.0 | 300 to 600 | Moderate |
| Fluphenazine | 1.0 to 2.0 | 4 to 20 | Mild |
| Haloperidol | 2.0 | 5 to 20 | Mild |
| Second-generation antipsychotics | |||
| Aripiprazole | 7.5 | 15 to 30 | Mild |
| Clozapine | 50.0 | 250 to 500 | Marked |
| Olanzapine | 4.0 | 15 to 30 | Moderate |
| Quetiapine | 80.0 | 300 to 800 | Moderate |
| Risperidone | 1.0 | 2 to 6 | Mild |
| Ziprasidone | 20.0 | 80 to 160 | Mild |
| * Approximate dose equivalent to 100 mg of chlorpromazine | |||
| Source: Data from reference 9. | |||
Mechanism and sedation
The mechanisms of antipsychotics’ therapeutic and sedative properties appear to be different.13 The degree of sedation shows little relationship with the various antipsychotics’ potency at the dopamine D2 receptor, which suggests that dopamine D2 receptor antagonism is not involved in causing sedation.
Instead, the degree of sedation may be associated with each antipsychotic’s affinity for the histamine H1 receptor, which is highly variable (Table 2).1,14,15 In general:
- agents that are more potent histamine H1 antagonists—such as olanzapine and clozapine14—produce more sedation
- agents that are weaker H1 antagonists—such as risperidone, ziprasidone, and aripiprazole—produce less sedation.
Although dosage and affinity for histamine H1 receptors play important roles in the sedative effect of a medication, what ultimately determines sedative effect is the combination of histamine H1 affinity and the amount of drug reaching the histamine H1 receptors in the CNS.
For example, the SGA quetiapine—which has moderate affinity for histamine H1 receptors14—also has relatively low affinity for dopamine D2 receptors. Higher dosages of quetiapine are therefore required to produce antipsychotic effects compared with other SGAs—such as risperidone, ziprasidone, and aripiprazole—that have higher affinities for the dopamine D2 receptors.
Because of higher dosing, higher amounts of quetiapine are assumed to be reaching the histamine H1 receptors in the CNS. This is why quetiapine causes more sedation in clinical use than does risperidone, even though risperidone has greater affinity for the histamine H1 receptor.
Table 2
Antipsychotics’ sedative effects by the numbers: Equilibrium dissociation constants at brain receptors*
| Receptors | |||
|---|---|---|---|
| Dopamine D2 | Serotonin 5-HT2A | Histamine H1 | |
| FGA | |||
| Haloperidol | 2.60 | 61.00 | 260.0 |
| SGAs | |||
| Aripiprazole | 0.34 | 3.40 | 61.00 |
| Clozapine | 210.00 | 2.60 | 3.10 |
| Olanzapine | 20.00 | 1.50 | 0.10 |
| Risperidone | 3.80 | 0.15 | 5.20 |
| Quetiapine | 770.00 | 31.00 | 19.00 |
| Ziprasidone | 2.60 | 0.12 | 4.60 |
| * Lower numbers are equivalent to higher receptor binding affinity. Each antipsychotic’s sedative effect is determined by histamine H1 affinity and the amount of drug that reaches H1 receptors in the CNS—which in turn is affected by the agent’s dosage and dopamine D2 receptor affinity. | |||
| FGA: first-generation antipsychotic; SGAs: second-generation antipsychotics | |||
| Source: References 1,14,15 | |||
Sleep patterns in mental illness
Sleep disturbances—including changes in sleep patterns, insomnia, and excessive sleeping—occur frequently in patients with psychiatric disorders. The sleep process itself (Table 3) is disrupted in patients with schizophrenia.
A study that examined sleep patterns in 40 patients with schizophrenia found longer sleep latency, more frequent arousals, and increased periods of wakefulness after sleep onset compared with controls without a psychiatric disorder. Ratings of sleep efficiency—ratio of sleep time to time in bed—were:
- 95% in the control group
- 78% in antipsychotic-naïve patients with schizophrenia
- 72% in patients with chronic schizophrenia.15
A study of sleep in 19 patients with schizophrenia and 13 nonpsychiatric controls16 found individuals with schizophrenia had:
- increased duration of stage 1 sleep
- decreased duration of stages 3 and 4 (slow-wave) sleep
- 83% total sleep efficiency, compared with 95% in nonpsychiatric controls.
Because of these differences in sleep patterns, patients with schizophrenia often experience inadequate sleep.
Antipsychotic effects on sleep patterns. Your choice of an antipsychotic also can affect the patient’s sleep. In a study of sleep measures in patients with schizophrenia treated with risperidone or haloperidol, Yamashita et al17 reported a significant difference in the time each group spent in slow-wave sleep (27% with risperidone vs 20% with haloperidol). The authors suggested that risperidone might lengthen the amount of slow-wave sleep because of its higher affinity for serotonin 5-HT2 receptors compared with haloperidol.
Salin-Pascual et al18 found that olanzapine improved total sleep time and sleep efficacy, reduced stage 1 sleep, and significantly enhanced stage 2 and slow-wave (delta) sleep.
Serotonin 5-HT2 receptors have been reported to be involved in controlling sleep quality.19 Similar to risperidone and olanzapine, the other SGAs also have a higher affinity than haloperidol for serotonin 5-HT2 receptors (Table 2). Thus, although antipsychotics’ sedative effects may adversely affect patients, SGAs may have the potential to improve sleep quality in individuals with schizophrenia. SGAs increase slow-wave sleep, and patients feel more rested after awakening.
Table 3
Normal sleep architecture, which can be disordered in schizophrenia
| Nonrapid eye movement (NREM) sleep | |
| Stage 1 | Drowsiness; represents transition between waking and sleeping |
| Stage 2 | Sleep deepens |
| Stages 3 and 4 | Deepest levels of NREM sleep subjects are most difficult to arouse (slow-wave or delta sleep) |
| Rapid eye movement (REM) sleep | |
| Vivid dreams; pulse and respiration rates are higher and more variable than during NREM sleep | |
| During the second half of the night, slow-wave sleep decreases compared with the first half of the night, but REM periods become more frequent and prolonged. | |
Managing excessive sedation
Take steps to minimize bothersome sedation in patients taking antipsychotics (Figure). To reduce daytime sedation, instruct the patient to take all or most of the antipsychotic dose at bedtime. Also rule out medical conditions that can produce fatigue and sedation, such as hypothyroidism, obstructive sleep apnea (OSA), and restless legs syndrome.
Figure
Managing sedation in patients with schizophrenia
Review the patient’s medication list to determine if other potentially sedating medications can be reduced or eliminated. Psychotropics that can cause sedation include:
- antidepressants such as the tricyclics and mirtazapine
- mood stabilizers (particularly valproic acid, but also carbamazepine, lithium, and lamotrigine).
Also consider gradually reducing the patient’s antipsychotic dose, and closely monitor for worsening of psychosis.
If sedation persists despite these interventions, consider switching the patient to a less sedating antipsychotic such as ziprasidone or aripiprazole. If these efforts also are ineffective, caffeine or off-label bupropion—75 to 100 mg once in the morning or up to twice daily—might help the patient feel more alert. Many patients taking antipsychotics drink several cups of coffee every morning to feel less sedated.
Stimulants. A consensus guideline on treating schizophrenia20 recommends prescribing amphetamine-related stimulants for patients who are persistently sedated, but this practice is highly controversial. Many stimulants increase dopamine release in the CNS, which theoretically can worsen psychosis. A clinician could be held liable for the actions of patients medicated with stimulants.
Modafinil is a nonamphetamine CNS stimulant approved for use in disorders of excessive sleep such as narcolepsy, OSA, shift work sleep disorder, and fatigue related to multiple sclerosis. Its mechanism of action in promoting wakefulness in these disorders is not fully understood; it may activate histaminergic projections in the frontal cortex from the tuberomammillary nucleus, which plays a major role in maintaining wakefulness.21
Modafinil, 200 mg in the morning, has been reported to reduce total sleep time without adverse effects in 3 patients experiencing sedation associated with antipsychotics.22 A later double-blind, placebo-controlled trial by Sevy et al23 found that modafinil and placebo were associated with similar, significant improvement in fatigue over time. Narendran24 reported a case in which modafinil might have exacerbated psychosis in a patient with schizophrenia who was taking 200 mg qid.
Managing chronic insomnia
Schizophrenia patients with chronic insomnia usually require education about appropriate sleep habits, combined with additional treatments.
Sleep hygiene. Instruct patients to:
- Wake up at the same time every day, regardless of when they went to sleep.
- Maintain a consistent bedtime.
- Exercise regularly, preferably in the late afternoon but not within 2 to 4 hours of bedtime.
- Perform relaxing activities before bed.
- Keep the bedroom quiet and cool (extreme temperatures compromise sleep).
- Do not watch the clock at night.
- Avoid caffeine and nicotine for at least 6 hours before bedtime.
- Drink alcohol only in moderation, and avoid use for at least 4 hours before bedtime.
- Avoid napping; it may interfere with the ability to fall asleep at night.
Medications. The consensus guideline on treating schizophrenia20 offers the option of switching the patient with chronic insomnia to one of the more sedating antipsychotics, such as olanzapine, quetiapine, or clozapine. Sedation alone should not be the reason to switch to clozapine, however.
You could consider adding a bedtime sedative to the patient’s medications (Table 4). FDA-approved sedatives include nonbenzodiazepines such as zolpidem, zolpidem extended-release, zaleplon, or eszopiclone, and the melatonin receptor agonist ramelteon. Although not approved as sedatives, some antidepressants such as trazodone or mirtazapine and antihistamines such as diphenhydramine and hydroxyzine are used to promote sleep. Benzodiazepines can be helpful but require caution when prescribed to patients with comorbid substance abuse disorders.
Sedatives have been studied extensively in general populations with insomnia but not in patients receiving antipsychotics. Combining antipsychotics and sedatives can produce daytime drowsiness and sedation.
Table 4
Sedatives to treat insomnia in patients with schizophrenia
| Medication | Common bedtime dose range* |
|---|---|
| Benzodiazepines | |
| Estazolam | 1 to 2 mg |
| Flurazepam | 15 to 30 mg |
| Temazepam | 7.5 to 30 mg |
| Triazolam | 0.125 to 0.25 mg |
| Benzodiazepine agonists | |
| Eszopiclone | 2 to 3 mg |
| Zaleplon | 5 to 10 mg |
| Zolpidem | 5 to 10 mg |
| Zolpidem CR | 6.25 to 12.5 mg |
| Melatonin receptor agonist | |
| Ramelteon | 8 mg |
| H1 antihistamines | |
| Diphenhydramine | 25 to 50 mg |
| Hydroxyzine | 50 to 100 mg |
| Antidepressants | |
| Mirtazapine | 15 to 30 mg |
| Trazodone | 50 to 200 mg |
| * No dosage adjustment is required in this patient population | |
Related resources
- Miller DD. Atypical antipsychotics: sleep, sedation, and efficacy. Prim Care Companion J Clin Psychiatry 2004;6(suppl 2):3-7.
- Benca RM, Diagnosis and treatment of chronic insomnia: a review. Psychiatr Serv 2005;56(3):332-43.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Diphenhydramine • Benadryl
- Estazolam • ProSom
- Eszopiclone • Lunesta
- Fluphenazine • Prolixin
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Hydroxyzine • Vistaril, Atarax
- Lamotrigine • Lamictal
- Mirtazapine • Remeron
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Ramelteon • Rozerem
- Risperidone • Risperdal
- Temazepam • Restor
- Trazodone • Desyrel
- Triazolam • Halcion
- Valproic acid • Depakote
- Zaleplon • Sonata
- Ziprasidone • Geodon
- Zolpidem • Ambien, Ambien CR
Disclosure
Dr. Miller receives research support from Pfizer Inc. and has received honoraria from AstraZeneca, Bristol-Myers Squibb, Janssen Pharmaceutica, and Pfizer Inc.
Manage, don’t accept adverse ‘calming’ effect
Sedation is a frequent side effect of antipsychotics, especially at relatively high doses. Antipsychotics’ sedative effects can reduce agitation in acute psychosis and promote sleep in insomnia, but long-term sedation may:
- interfere with schizophrenia patients’ efforts to go to work or school or engage in normal socialization
- prevent improvement from psychosocial training, psychiatric rehabilitation, and other treatments.
This article discusses how to manage acute psychosis without oversedation and ways to address persistent sedation and chronic insomnia with less-sedating antipsychotics or adjunctive medications.
Neurobiology or psychopharmacology?
Many patients experience only mild, transient somnolence at the beginning of antipsychotic treatment, and most develop some tolerance to the sedating effects with continued administration. Others may have persistent daytime sedation that interferes with normal functioning.
Sedation is especially common in elderly patients receiving antipsychotics. Compared with younger patients, older patients receiving the same doses of the same medications become more heavily sedated for longer periods of time. The resulting sedation can impair arousal levels during the day and increase the risk of falls.
Sedation can occur with first-generation antipsychotics (FGAs) and second-generation antipsychotics (SGAs), but it is seen more commonly and tends to be more severe with low-potency FGAs than with SGAs. Clinical challenges come with:
- distinguishing between sedation and negative symptoms of schizophrenia such as avolition, amotivation, withdrawal, and anhedonia
- determining whether an individual’s cognitive impairment is related to the antipsychotic’s sedative properties.
Because the treatments are different, it is important to try to distinguish negative symptoms and/or cognitive impairment related to schizophrenia’s neurobiology from sedation related to the antipsychotic. Ask patients if they nap during the day or just lie around, and if they want to do things but can’t:
- If they want to do things but feel too tired, this likely is sedation caused by the antipsychotic. Treatment might be a dose reduction.
- If they are not interested in doing things, the likely cause is negative symptoms. Treatment might be a medication such as a selective serotonin reuptake inhibitor.
- If they want to do things but cannot organize themselves to be able to do them, this likely is cognitive impairment. Treatment might be cognitive training or remediation.
Efficacy and sedation
Antipsychotics are thought to exert their effect by antagonism of postsynaptic dopamine D2 and serotonin 5HT2A receptors and possibly other receptors in the brain. Four SGAs—risperidone, olanzapine, quetiapine, and ziprasidone—act as dopamine D2 and 5HT2A antagonists. Aripiprazole is a dopamine D2 partial agonist, serotonin 5HT1A partial agonist, and serotonin 5HT2A antagonist.1 Efficacy data comparing SGAs with each other and with FGAs vary, but all 5 of these SGAs have been shown to be effective antipsychotics.2-4 They also generally cause less sedation than FGAs.
Patients with acute exacerbation of psychosis often have insomnia and frequently report paranoia that “something” will happen to them while they sleep. When treating agitated patients, many clinicians consider calming effects and true antipsychotic effects to be one in the same, which is not correct. All available antipsychotics are, on average, equally effective in treating acute psychotic symptoms but vary considerably in the amount of sedation they produce. Studies of short-acting injectable SGAs, such as ziprasidone and aripiprazole, have shown that agitation and acute symptoms can be controlled without significant sedation.5,6
Antipsychotic effects are not immediate and historically were thought to occur over several weeks. Recent meta-analyses suggest, however, that some antipsychotic effects are evident within the first week of treatment.7,8 To avoid overmedication, therefore, it’s important to separate calming effects from antipsychotic effects.
Recommendations. Choose the initial antipsychotic based on its effectiveness in treating the underlying disease, rather than relying on side effects—such as sedation—to control disease manifestations. Without sedation, patients are better able to engage in therapy; participate in family, social, school, and work activities; and increase their chances of recovery.
Initiate the antipsychotic at or titrate to a reasonable, not overly high dose—such as:
- olanzapine, 10 to 20 mg/d
- risperidone, 3 to 6 mg/d
- ziprasidone, 100 to 140 mg/d
- quetiapine, 400 to 600 mg/d
- aripiprazole, 15 mg/d.
Continue the patient on that dose, and use a nonantipsychotic such as a benzodiazepine to help control insomnia, anxiety, and agitation. Two to 4 weeks is generally adequate, but some patients may need the adjunctive therapy for several months. If you initiate a more sedating antipsychotic acutely, switching to a less sedating agent when the patient is stable and the illness is in remission may support adherence and improve outcomes.
Dosages and sedation. Not all FGAs have the same sedative effect, nor do all SGAs (Table 1).9 In general, the high-milligram, low-potency FGAs—such as chlorpromazine—produce more sedation than the low-milligram, high-potency FGAs—such as haloperidol and fluphenazine.9 This principle tends to hold true for the SGAs as well. For example, the high-potency, low-dose SGA risperidone is less sedating than the lower-potency, high-dose SGAs quetiapine and clozapine.
Dose does not always determine sedation, however. Olanzapine, which is commonly dosed at 15 to 30 mg/d, is more sedating than ziprasidone, for which the usual range is 80 to 160 mg/d.3,10-12
Table 1
Antipsychotics’ potency, dosages, and sedative properties
| Medication | Relative potency (mg)* | Common dosage (mg/d) | Sedation |
|---|---|---|---|
| First-generation antipsychotics | |||
| Chlorpromazine | 100.0 | 300 to 600 | Moderate |
| Fluphenazine | 1.0 to 2.0 | 4 to 20 | Mild |
| Haloperidol | 2.0 | 5 to 20 | Mild |
| Second-generation antipsychotics | |||
| Aripiprazole | 7.5 | 15 to 30 | Mild |
| Clozapine | 50.0 | 250 to 500 | Marked |
| Olanzapine | 4.0 | 15 to 30 | Moderate |
| Quetiapine | 80.0 | 300 to 800 | Moderate |
| Risperidone | 1.0 | 2 to 6 | Mild |
| Ziprasidone | 20.0 | 80 to 160 | Mild |
| * Approximate dose equivalent to 100 mg of chlorpromazine | |||
| Source: Data from reference 9. | |||
Mechanism and sedation
The mechanisms of antipsychotics’ therapeutic and sedative properties appear to be different.13 The degree of sedation shows little relationship with the various antipsychotics’ potency at the dopamine D2 receptor, which suggests that dopamine D2 receptor antagonism is not involved in causing sedation.
Instead, the degree of sedation may be associated with each antipsychotic’s affinity for the histamine H1 receptor, which is highly variable (Table 2).1,14,15 In general:
- agents that are more potent histamine H1 antagonists—such as olanzapine and clozapine14—produce more sedation
- agents that are weaker H1 antagonists—such as risperidone, ziprasidone, and aripiprazole—produce less sedation.
Although dosage and affinity for histamine H1 receptors play important roles in the sedative effect of a medication, what ultimately determines sedative effect is the combination of histamine H1 affinity and the amount of drug reaching the histamine H1 receptors in the CNS.
For example, the SGA quetiapine—which has moderate affinity for histamine H1 receptors14—also has relatively low affinity for dopamine D2 receptors. Higher dosages of quetiapine are therefore required to produce antipsychotic effects compared with other SGAs—such as risperidone, ziprasidone, and aripiprazole—that have higher affinities for the dopamine D2 receptors.
Because of higher dosing, higher amounts of quetiapine are assumed to be reaching the histamine H1 receptors in the CNS. This is why quetiapine causes more sedation in clinical use than does risperidone, even though risperidone has greater affinity for the histamine H1 receptor.
Table 2
Antipsychotics’ sedative effects by the numbers: Equilibrium dissociation constants at brain receptors*
| Receptors | |||
|---|---|---|---|
| Dopamine D2 | Serotonin 5-HT2A | Histamine H1 | |
| FGA | |||
| Haloperidol | 2.60 | 61.00 | 260.0 |
| SGAs | |||
| Aripiprazole | 0.34 | 3.40 | 61.00 |
| Clozapine | 210.00 | 2.60 | 3.10 |
| Olanzapine | 20.00 | 1.50 | 0.10 |
| Risperidone | 3.80 | 0.15 | 5.20 |
| Quetiapine | 770.00 | 31.00 | 19.00 |
| Ziprasidone | 2.60 | 0.12 | 4.60 |
| * Lower numbers are equivalent to higher receptor binding affinity. Each antipsychotic’s sedative effect is determined by histamine H1 affinity and the amount of drug that reaches H1 receptors in the CNS—which in turn is affected by the agent’s dosage and dopamine D2 receptor affinity. | |||
| FGA: first-generation antipsychotic; SGAs: second-generation antipsychotics | |||
| Source: References 1,14,15 | |||
Sleep patterns in mental illness
Sleep disturbances—including changes in sleep patterns, insomnia, and excessive sleeping—occur frequently in patients with psychiatric disorders. The sleep process itself (Table 3) is disrupted in patients with schizophrenia.
A study that examined sleep patterns in 40 patients with schizophrenia found longer sleep latency, more frequent arousals, and increased periods of wakefulness after sleep onset compared with controls without a psychiatric disorder. Ratings of sleep efficiency—ratio of sleep time to time in bed—were:
- 95% in the control group
- 78% in antipsychotic-naïve patients with schizophrenia
- 72% in patients with chronic schizophrenia.15
A study of sleep in 19 patients with schizophrenia and 13 nonpsychiatric controls16 found individuals with schizophrenia had:
- increased duration of stage 1 sleep
- decreased duration of stages 3 and 4 (slow-wave) sleep
- 83% total sleep efficiency, compared with 95% in nonpsychiatric controls.
Because of these differences in sleep patterns, patients with schizophrenia often experience inadequate sleep.
Antipsychotic effects on sleep patterns. Your choice of an antipsychotic also can affect the patient’s sleep. In a study of sleep measures in patients with schizophrenia treated with risperidone or haloperidol, Yamashita et al17 reported a significant difference in the time each group spent in slow-wave sleep (27% with risperidone vs 20% with haloperidol). The authors suggested that risperidone might lengthen the amount of slow-wave sleep because of its higher affinity for serotonin 5-HT2 receptors compared with haloperidol.
Salin-Pascual et al18 found that olanzapine improved total sleep time and sleep efficacy, reduced stage 1 sleep, and significantly enhanced stage 2 and slow-wave (delta) sleep.
Serotonin 5-HT2 receptors have been reported to be involved in controlling sleep quality.19 Similar to risperidone and olanzapine, the other SGAs also have a higher affinity than haloperidol for serotonin 5-HT2 receptors (Table 2). Thus, although antipsychotics’ sedative effects may adversely affect patients, SGAs may have the potential to improve sleep quality in individuals with schizophrenia. SGAs increase slow-wave sleep, and patients feel more rested after awakening.
Table 3
Normal sleep architecture, which can be disordered in schizophrenia
| Nonrapid eye movement (NREM) sleep | |
| Stage 1 | Drowsiness; represents transition between waking and sleeping |
| Stage 2 | Sleep deepens |
| Stages 3 and 4 | Deepest levels of NREM sleep subjects are most difficult to arouse (slow-wave or delta sleep) |
| Rapid eye movement (REM) sleep | |
| Vivid dreams; pulse and respiration rates are higher and more variable than during NREM sleep | |
| During the second half of the night, slow-wave sleep decreases compared with the first half of the night, but REM periods become more frequent and prolonged. | |
Managing excessive sedation
Take steps to minimize bothersome sedation in patients taking antipsychotics (Figure). To reduce daytime sedation, instruct the patient to take all or most of the antipsychotic dose at bedtime. Also rule out medical conditions that can produce fatigue and sedation, such as hypothyroidism, obstructive sleep apnea (OSA), and restless legs syndrome.
Figure
Managing sedation in patients with schizophrenia
Review the patient’s medication list to determine if other potentially sedating medications can be reduced or eliminated. Psychotropics that can cause sedation include:
- antidepressants such as the tricyclics and mirtazapine
- mood stabilizers (particularly valproic acid, but also carbamazepine, lithium, and lamotrigine).
Also consider gradually reducing the patient’s antipsychotic dose, and closely monitor for worsening of psychosis.
If sedation persists despite these interventions, consider switching the patient to a less sedating antipsychotic such as ziprasidone or aripiprazole. If these efforts also are ineffective, caffeine or off-label bupropion—75 to 100 mg once in the morning or up to twice daily—might help the patient feel more alert. Many patients taking antipsychotics drink several cups of coffee every morning to feel less sedated.
Stimulants. A consensus guideline on treating schizophrenia20 recommends prescribing amphetamine-related stimulants for patients who are persistently sedated, but this practice is highly controversial. Many stimulants increase dopamine release in the CNS, which theoretically can worsen psychosis. A clinician could be held liable for the actions of patients medicated with stimulants.
Modafinil is a nonamphetamine CNS stimulant approved for use in disorders of excessive sleep such as narcolepsy, OSA, shift work sleep disorder, and fatigue related to multiple sclerosis. Its mechanism of action in promoting wakefulness in these disorders is not fully understood; it may activate histaminergic projections in the frontal cortex from the tuberomammillary nucleus, which plays a major role in maintaining wakefulness.21
Modafinil, 200 mg in the morning, has been reported to reduce total sleep time without adverse effects in 3 patients experiencing sedation associated with antipsychotics.22 A later double-blind, placebo-controlled trial by Sevy et al23 found that modafinil and placebo were associated with similar, significant improvement in fatigue over time. Narendran24 reported a case in which modafinil might have exacerbated psychosis in a patient with schizophrenia who was taking 200 mg qid.
Managing chronic insomnia
Schizophrenia patients with chronic insomnia usually require education about appropriate sleep habits, combined with additional treatments.
Sleep hygiene. Instruct patients to:
- Wake up at the same time every day, regardless of when they went to sleep.
- Maintain a consistent bedtime.
- Exercise regularly, preferably in the late afternoon but not within 2 to 4 hours of bedtime.
- Perform relaxing activities before bed.
- Keep the bedroom quiet and cool (extreme temperatures compromise sleep).
- Do not watch the clock at night.
- Avoid caffeine and nicotine for at least 6 hours before bedtime.
- Drink alcohol only in moderation, and avoid use for at least 4 hours before bedtime.
- Avoid napping; it may interfere with the ability to fall asleep at night.
Medications. The consensus guideline on treating schizophrenia20 offers the option of switching the patient with chronic insomnia to one of the more sedating antipsychotics, such as olanzapine, quetiapine, or clozapine. Sedation alone should not be the reason to switch to clozapine, however.
You could consider adding a bedtime sedative to the patient’s medications (Table 4). FDA-approved sedatives include nonbenzodiazepines such as zolpidem, zolpidem extended-release, zaleplon, or eszopiclone, and the melatonin receptor agonist ramelteon. Although not approved as sedatives, some antidepressants such as trazodone or mirtazapine and antihistamines such as diphenhydramine and hydroxyzine are used to promote sleep. Benzodiazepines can be helpful but require caution when prescribed to patients with comorbid substance abuse disorders.
Sedatives have been studied extensively in general populations with insomnia but not in patients receiving antipsychotics. Combining antipsychotics and sedatives can produce daytime drowsiness and sedation.
Table 4
Sedatives to treat insomnia in patients with schizophrenia
| Medication | Common bedtime dose range* |
|---|---|
| Benzodiazepines | |
| Estazolam | 1 to 2 mg |
| Flurazepam | 15 to 30 mg |
| Temazepam | 7.5 to 30 mg |
| Triazolam | 0.125 to 0.25 mg |
| Benzodiazepine agonists | |
| Eszopiclone | 2 to 3 mg |
| Zaleplon | 5 to 10 mg |
| Zolpidem | 5 to 10 mg |
| Zolpidem CR | 6.25 to 12.5 mg |
| Melatonin receptor agonist | |
| Ramelteon | 8 mg |
| H1 antihistamines | |
| Diphenhydramine | 25 to 50 mg |
| Hydroxyzine | 50 to 100 mg |
| Antidepressants | |
| Mirtazapine | 15 to 30 mg |
| Trazodone | 50 to 200 mg |
| * No dosage adjustment is required in this patient population | |
Related resources
- Miller DD. Atypical antipsychotics: sleep, sedation, and efficacy. Prim Care Companion J Clin Psychiatry 2004;6(suppl 2):3-7.
- Benca RM, Diagnosis and treatment of chronic insomnia: a review. Psychiatr Serv 2005;56(3):332-43.
Drug Brand Names
- Aripiprazole • Abilify
- Bupropion • Wellbutrin
- Carbamazepine • Tegretol
- Chlorpromazine • Thorazine
- Clozapine • Clozaril
- Diphenhydramine • Benadryl
- Estazolam • ProSom
- Eszopiclone • Lunesta
- Fluphenazine • Prolixin
- Flurazepam • Dalmane
- Haloperidol • Haldol
- Hydroxyzine • Vistaril, Atarax
- Lamotrigine • Lamictal
- Mirtazapine • Remeron
- Modafinil • Provigil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Ramelteon • Rozerem
- Risperidone • Risperdal
- Temazepam • Restor
- Trazodone • Desyrel
- Triazolam • Halcion
- Valproic acid • Depakote
- Zaleplon • Sonata
- Ziprasidone • Geodon
- Zolpidem • Ambien, Ambien CR
Disclosure
Dr. Miller receives research support from Pfizer Inc. and has received honoraria from AstraZeneca, Bristol-Myers Squibb, Janssen Pharmaceutica, and Pfizer Inc.
1. McQuade R, Burris KD, Jordan S, et al. Aripiprazole: a dopamine-serotonin system stabilizer [abstract no. P.3.W.080]. Int J Neuropsychopharmacol 2002;5(suppl 1):S176.-
2. Davis JM, Chen N, Glick ID. A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry 2003;60(6):553-64.
3. Gardner DM, Baldessarini RJ, Waraich P. Modern antipsychotic drugs: a critical overview. Can Med Assoc J 2005;172(13):1703-11.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
5. Andrezina R, Josiassen RC, Marcus RN, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacol (Berl) 2006;188(3):281-92.
6. Brook S, Lucey JV, Gunn KP. Intramuscular ziprasidone compared with intramuscular haloperidol in the treatment of acute psychosis. Ziprasidone I.M. Study Group. J Clin Psychiatry 2000;61(12):933-41.
7. Agid O, Kapur S, Arenovich T, Zipursky RB. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 2003;60(12):1228-35.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry 2005;57(12):1543-9.
9. Jibson MD, Tandon R. An overview of antipsychotic medications. CNS News 2001;3:49-54.
10. Collaborative Working Group on Clinical Trial Evaluations. Treatment of special populations with the atypical antipsychotics. J Clin Psychiatry 1998;59(suppl 12):46-52.
11. El-Sayeh HG, Morganti C. Aripiprazole for schizophrenia. Cochrane Database Syst Rev 2004(2);CD004578.-
12. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
13. Tandon R. Safety and tolerability: how do newer generation ”atypical” antipsychotics compare? Psychiatr Q 2002;73(4):297-311.
14. Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci 2000;68(1):29-39.
15. Tandon R, Shipley JE, Taylor S, et al. Electroencephalographic sleep abnormalities in schizophrenia. Relationship to positive/negative symptoms and prior neuroleptic treatment. Arch Gen Psychiatry 1992;49(3):185-94.
16. Benson KL, Zarcone VP Jr. Rapid eye movement sleep eye movements in schizophrenia and depression. Arch Gen Psychiatry 1993;50(6):474-82.
17. Yamashita H, Morinobu S, Yamawaki S, et al. Effect of risperidone on sleep in schizophrenia: a comparison with haloperidol. Psychiatry Res 2002;109(2):137-42.
18. Salin-Pascual RJ, Herrera-Estrella M, Galicia-Polo L, Laurrabaquio MR. Olanzapine acute administration in schizophrenic patients increases delta sleep and sleep efficiency. Biol Psychiatry 1999;46(1):141-3.
19. Idzikowski C, Mills FJ, Glennard R. 5-Hydroxytryptamine-2 antagonist increases human slow wave sleep. Brain Res 1986;378(1):164-8.
20. Expert Consensus Guideline Series Treatment of schizophrenia. J Clin Psychiatry 1999;60(suppl 11):1-80.
21. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
22. Makela EH, Miller K, Cutlip WD, 2nd. Three case reports of modafinil use in treating sedation induced by antipsychotic medications. J Clin Psychiatry 2003;64(4):485-6.
23. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
24. Narendran R, Young CM, Valenti AM, et al. Is psychosis exacerbated by modafinil? Arch Gen Psychiatry 2002;59(3):292-3.
1. McQuade R, Burris KD, Jordan S, et al. Aripiprazole: a dopamine-serotonin system stabilizer [abstract no. P.3.W.080]. Int J Neuropsychopharmacol 2002;5(suppl 1):S176.-
2. Davis JM, Chen N, Glick ID. A meta-analysis of the efficacy of second-generation antipsychotics. Arch Gen Psychiatry 2003;60(6):553-64.
3. Gardner DM, Baldessarini RJ, Waraich P. Modern antipsychotic drugs: a critical overview. Can Med Assoc J 2005;172(13):1703-11.
4. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 2005;353(12):1209-23.
5. Andrezina R, Josiassen RC, Marcus RN, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacol (Berl) 2006;188(3):281-92.
6. Brook S, Lucey JV, Gunn KP. Intramuscular ziprasidone compared with intramuscular haloperidol in the treatment of acute psychosis. Ziprasidone I.M. Study Group. J Clin Psychiatry 2000;61(12):933-41.
7. Agid O, Kapur S, Arenovich T, Zipursky RB. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 2003;60(12):1228-35.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry 2005;57(12):1543-9.
9. Jibson MD, Tandon R. An overview of antipsychotic medications. CNS News 2001;3:49-54.
10. Collaborative Working Group on Clinical Trial Evaluations. Treatment of special populations with the atypical antipsychotics. J Clin Psychiatry 1998;59(suppl 12):46-52.
11. El-Sayeh HG, Morganti C. Aripiprazole for schizophrenia. Cochrane Database Syst Rev 2004(2);CD004578.-
12. Tandon R, Jibson MD. Efficacy of newer generation antipsychotics in the treatment of schizophrenia. Psychoneuroendocrinol 2003;28(suppl 1):9-26.
13. Tandon R. Safety and tolerability: how do newer generation ”atypical” antipsychotics compare? Psychiatr Q 2002;73(4):297-311.
14. Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci 2000;68(1):29-39.
15. Tandon R, Shipley JE, Taylor S, et al. Electroencephalographic sleep abnormalities in schizophrenia. Relationship to positive/negative symptoms and prior neuroleptic treatment. Arch Gen Psychiatry 1992;49(3):185-94.
16. Benson KL, Zarcone VP Jr. Rapid eye movement sleep eye movements in schizophrenia and depression. Arch Gen Psychiatry 1993;50(6):474-82.
17. Yamashita H, Morinobu S, Yamawaki S, et al. Effect of risperidone on sleep in schizophrenia: a comparison with haloperidol. Psychiatry Res 2002;109(2):137-42.
18. Salin-Pascual RJ, Herrera-Estrella M, Galicia-Polo L, Laurrabaquio MR. Olanzapine acute administration in schizophrenic patients increases delta sleep and sleep efficiency. Biol Psychiatry 1999;46(1):141-3.
19. Idzikowski C, Mills FJ, Glennard R. 5-Hydroxytryptamine-2 antagonist increases human slow wave sleep. Brain Res 1986;378(1):164-8.
20. Expert Consensus Guideline Series Treatment of schizophrenia. J Clin Psychiatry 1999;60(suppl 11):1-80.
21. Scammell TE, Estabrooke IV, McCarthy MT, et al. Hypothalamic arousal regions are activated during modafinil-induced wakefulness. J Neurosci 2000;20(22):8620-8.
22. Makela EH, Miller K, Cutlip WD, 2nd. Three case reports of modafinil use in treating sedation induced by antipsychotic medications. J Clin Psychiatry 2003;64(4):485-6.
23. Sevy S, Rosenthal MH, Alvir J, et al. Double-blind, placebo-controlled study of modafinil for fatigue and cognition in schizophrenia patients treated with psychotropic medications. J Clin Psychiatry 2005;66(7):839-43.
24. Narendran R, Young CM, Valenti AM, et al. Is psychosis exacerbated by modafinil? Arch Gen Psychiatry 2002;59(3):292-3.
Neurocognitive impairment: Feigned, exaggerated, or real?
Mrs. M, age 27, suffered a head injury in a motor vehicle accident 9 months ago. She is referred to you by a neurologist with complaints of persistent headache and diffculties with memory and attention “worse now than right after the accident.” She tried to return to work 3 months after the accident but could not concentrate enough to be productive.
Review of medical records shows that she had minimal, if any, loss of consciousness at the accident scene, and she followed commands at the emergency room without apparent difficulty. Neurologic exam and head CT were normal. She is cooperative and fully oriented but appears upset about the difficulties she has experienced and occasionally complains of headache.
Three days later you receive a signed release of information from her attorney, requesting all records related to her examination.
In cases such as Mrs. M’s, the differential diagnosis often comes down to a somatoform disorder vs factitious disorder vs malingering, a decision that rarely seems as clear-cut as one might believe when reading the DSM-IV-TR. Particularly in litigation- or compensation-related situations, clinicians must make 2 fundamental judgments:
- Is the patient intentionally generating the symptoms?
- Are the symptoms plausibly related to neurologic injury or illness?
This article describes how symptom validity testing (SVT) as part of a comprehensive neuropsychological evaluation can help answer these questions. Inconsistencies in the way patients perform on SVT (Table)18-30 can provide “red flags” to possible embellishment of neurocognitive symptoms. We also offer recently developed guidelines for diagnosing malingering of neurocognitive dysfunction that may be more helpful than the DSM-IV-TR criteria.
Table
Performance consistencies in patients
who fail symptom validity testing (SVT)
| Consistency | Comment |
|---|---|
| 25% to 40% of patients seeking some form compensation for their injuries or illness fail SVT | This appears to hold true not only for ‘brain’ cases but also for ‘pain’ cases |
| Deficits are not exaggerated in a constant manner across tests of different abilities | Deficits most likely to be exaggerated are concentration, memory, weakness, and processing speed; may be due to assumptions about what ‘brain damage’ looks like |
| Patients failing SVT report greater levels of emotional distress, psychological maladjustment, and severity of neurocognitive difficulties on self-report measures | Patterns of exaggerated responses are not the same as those exaggerating psychopathology |
| Very few patients who fail SVT score significantly below chance | Below-chance responding is an insensitive criterion for identifying suboptimal effort, but this level of performance is quite specific; short of confession, below-chance performance on SVT is closest to an evidentiary ‘gold standard’ for malingering |
| Not all SVTs are created equal | Sensitivity and specificity vary, and measures may disagree when more than one is administered |
| Coaching makes a difference | Malingering subjects who are told which tests to look for and how to approach them are more difficult to discriminate from genuine patients |
| Invalid effort does not rule out a genuine neurologic injury or illness | Exaggeration can coexist with neurologically driven neurocognitive deficits; neuropsychologists who do forensic work encounter patients with documented injuries who fail SVT, sometimes in blatantly obvious or absurd ways |
| Source: References 1-13 | |
Why ‘gut feelings’ are fallible
Differential diagnosis of neurocognitive impairment is challenging. Some patients have normal neurologic examinations in all respects but cognition, such as those with early Alzheimer’s disease or recent concussion. Others may show significant neurobehavioral changes but normal results on neuroimaging (such as the rare patient in a coma after a traumatic brain injury whose head CT is read as normal). Thus, the absence of findings other than impaired cognition in a neurologic exam is not proof that a disorder is driven primarily by psychiatric or behavioral issues.
- rely on their training and intuition
- refer for psychological evaluation
- rely on traditional malingering measures in standard psychological tests, such as the Minnesota Multiphasic Personality Inventory-2 (MMPI-2).
The problem with this approach is its high error rate. Health care professionals do not discriminate poor effort from genuine neurocognitive impairment very effectively. Diagnostic algorithms routinely outperform clinical judgment, particularly when diagnostic parameters are relatively well understood.19
Although discerning conscious intent often remains more art than science, neuropsychologists have developed cross-validated techniques to identify implausible cognitive performances that suggest embellished symptoms. Thus, relying on clinical judgment is accepting an error rate that can be reduced by using other approaches.
‘Let the psychologist figure it out.’ The success of this approach depends on the psychologist’s methodology. The psychologist’s gut instinct is no more accurate than that of the psychiatrist or neurologist.
Relying on traditional scales. Measures of malingering in psychological testing can be quite effective for identifying exaggerated psychopathology,20-22 but exaggerated psychopathology differs from exaggerated neurocognitive symptoms.23 Embellished psychopathology is not the same as embellished “brain damage,” and they are not detected equally well by the same techniques.
Validity scales on the MMPI and MMPI-2 do a poor job of detecting patients known to be exaggerating neurocognitive impairment23 (although the more recently developed Lees-Haley “Fake Bad Scale” has shown promise).24 Thus, the clinician who feels confident that a patient has not exaggerated neurocognitive complaints because he or she scored in the normal range on the MMPI-2 validity scales (or other measures shown to help identify exaggeration of psychopathology) has drawn a conclusion based on scales that likely are inadequate for this purpose.
3 ways to measure patient effort
Using SVT is the most effective way to determine the validity of a patient’s effort on a neuropsychological test battery. SVT using 3 approaches has been shown to reliably discriminate patients who are putting forth valid effort from those who are not:
- forced-choice testing
- unusual patterns of responses within established neurocognitive tests
- unusual patterns of variability on the same test given on different occasions.
On some validated forced-choice SVTs, patients with moderate to severe traumatic brain injuries perform at ≥90% accuracy; thus, a far lower performance from a mildly injured patient raises a red flag that some-thing exceptional is occurring that demands an explanation.
Patterns within established tests. As empiric evidence about SVTs grows, we understand more about how neurologically impaired patients perform—and do not perform—on these tests. These patterns can then be used to examine the extent to which they discriminate between patients who are exaggerating and those who are not. Cross-validated techniques are available for the Wechsler Adult Intelligence Scale, 3rd edition, and the California Verbal Learning Test, among others.25,26
Patterns across different evaluations. Variation in test results is expected when a patient takes the same test on different dates. Along with having previously seen the test, other patient factors may include fatigue or inattention. When a patient is recovering from a brain injury or illness, additional variation is expected because of recovery or progression over time.
Some abilities—and test scores—are more stable than others, however, even in patients with genuine neurologic damage. At least one method that analyzes data from different administrations of the Halstead Reitan Neuropsychological Battery uses this insight,27 although this method has yet to be cross-validated.
What have we learned?
Cross-validated techniques have demonstrated that effort has a significant effect on neurocognitive test scores, often greater than the effect of the neurologic condition being studied.28,29 For example, you will be more accurate predicting a patient’s overall performance on a neuropsychological test battery on the basis of their performance on the Word Memory Test (one type of SVT) than on how long he or she was in a coma after a head injury until the coma has persisted for >6 days.30
SVTs are not ‘malingering tests.’ A malingering patient simulates or exaggerates symptoms with the conscious intention of deceiving someone. An SVT does a good job identifying exaggerated symptoms, but it has little (and, in most cases, nothing) to say about the extent to which this exaggeration is conscious or intentional.
For instance, patients with somatoform disorders tend to fail SVT at a higher rate than general medical populations.6,31 Our group32 recently reported that approximately one-half of patients diagnosed with psychogenic nonepileptic seizures at an epilepsy center fail SVTs. It is unlikely, however, that all—or even most—of these patients were malingering.
SVTs do not reveal motivation or intention—they merely state the extent to which the effort put into testing provides a valid estimate of neurocognitive function.
Alternative guidelines have been suggested to guide decisions about when to diagnose a patient as malingering neurocognitive deficits.33 See the original publication for a full explication of the criteria.
Clinical recommendations
Particularly when someone with a mild brain injury is seeking compensation, keep in mind that 25% to 40% of these patients perform in such a way on SVT that the validity of their cognitive performances should be questioned. It does not necessarily mean they are malingering; rather, they are performing in a way that cannot be explained by established brain-behavior relationships in the absence of obvious severe neurologic injury or illness.
Ask for SVT when you refer cases such as this for neuropsychological or psychological evaluation. SVT can provide an empirically based foundation on which to formulate an opinion, particularly about the severity of reported cognitive symptoms. Your opinion about the intentionality of symptoms likely will rely primarily on other information (such as the consistency of complaints with behavior during the assessment or presence of primary or secondary gain), but SVT provides a valuable tool with which to examine the validity of cognitive complaints.
Related resources
- Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
- National Academy of Neuropsychology. Position paper: Symptom validity testing: practice issues and medical necessity. http://nanonline.org/paio/svt.shtm.
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bianchini KJ, Greve KW, Glynn G. On the diagnosis of malingered pain-related disability: lessons from cognitive malingering research. Spine 2005;5:404-17.
2. Gervais RO, Green P, Allen LM, Iverson GL. Effects of coaching on symptom validity testing in chronic pain patients presenting for disability assessments. J Forensic Neuropsychol 2001;2:1.-
3. Meyers JE, Diep A. Assessment of malingering in chronic pain patients using neuropsychological tests. Applied Neuropsychol 2000;7:133-9.
4. Williamson DJ, Rohling ML, Green P, Allen L. Evaluating effort with the Word Memory Test and Category Test–or not: inconsistencies in a forensic sample. J Forensic Neuropsychol 2003;3:19-44.
5. Gouvier WD, Prestholdt P, Warner M. A survey of common misperceptions about head injury and recovery. Arch Clin Neuropsychology 1988;3:331-43.
6. Boone KB, Lu P. Impact of somatoform symptomatology on credibility of cognitive performance. Clin Neuropsychol 1999;13:414-9.
7. Greiffenstein MF, Baker WJ, Axelrod BN, et al. The Fake Bad Scale and MMPI-2 F-family in detection of implausible psychological trauma claims. Clin Neuropsychol 2004;18:573-90.
8. Larrabee GJ. Detection of symptom exaggeration with the MMPI-2 in litigants with malingered neurocognitive dysfunction. Clin Neuropsychol 2003;17:54.-
9. Tan JE, Slick DJ, Strauss E, Hultsch DF. How’d they do it? Malingering strategies on symptom validity tests. Clin Neuropsychol 2002;16:495.-
10. Iverson GL, Binder LM. Detecting exaggeration and malingering in neuropsychological assessment. J Head Trauma Rehabil 2000;15:829-58.
11. Sweet JJ. Malingering: differential diagnosis. In: Sweet JJ, ed. Forensic neuropsychology: fundamentals and practice. New York: Swets & Zeitlinger, 1999:255-85.
12. Coleman RD, Rapport LJ, Millis SR, et al. Effects of coaching on detection of malingering on the California Verbal Learning Test. J Clin Exp Neuropsychol 1998;20:201.-
13. Rapport LJ, Farchione TJ, Coleman RD, Axelrod BN. Effects of coaching on malingered motor function profiles. J Clin Exp Neuropsychol 1998;20:89-97.
14. Carone DA, Benedict RHB, Munschauer FE, et al. Interpreting patient/informant discrepancies of reported cognitive symptoms in MS. J Int Neuropsychol Soc 2005;11:574.-
15. Meador KJ, Loring DW, Vahle VJ, et al. Subjective perception of cognitive effects of antiepileptic drugs is more related to mood than to objective performance. Epilepsia 2005;46:261-2.
16. Sawrie SM, Martin RC, Kuzniecky R, et al. Subjective versus objective memory change after temporal lobe epilepsy surgery. Neurology 1999;53:1511.-
17. Davis DA, Mazmanian PE, Fordis M, et al. Accuracy of physician self-assessment compared with observed measures of competence: a systematic review. JAMA 2006;296:1094-102.
18. Faust D. The detection of deception. Neurol Clin 1995;13:255-65.
19. Swets JA, Dawes RM, Monahan J. Psychological science can improve diagnostic decisions. Psychol Sci Public Interest 2000;1:1-26.
20. Knoll J, Resnick PJ. The detection of malingered post-traumatic stress disorder. Psychiatr Clin North Am 2006;29:629-47.
21. Morey LC, Lanier VW. Operating characteristics of six response distortion indicators for the Personality Assessment Inventory. Assessment 1998;5:203-14.
22. Rogers R, Sewell KW, Salekin RT. A meta-analysis of malingering on the MMPI-2. Assessment 1994;1:227-37.
23. Larrabee GJ. Somatic malingering on the MMPI and MMPI-2 in personal injury litigants. Clin Neuropsychol 1998;12:179.-
24. Nelson NW, Sweet JJ, Demakis GJ. Meta-analysis of the MMPI-2 Fake Bad Scale: utility in forensic practice. Clin Neuropsychol 2006;20:39-58.
25. Curtis KL, Greve KW, Bianchini KJ, Brennan A. California Verbal Learning Test indicators of malingered neurocognitive dysfunction: sensitivity and specificity in traumatic brain injury. Assessment 2006;13:46.-
26. Greve KW, Bianchini KJ, Mathias CW, et al. Detecting malingered performance on the Wechsler Adult Intelligence Scale: validation of Mittenberg’s approach in traumatic brain injury. Arch Clin Neuropsychol 2003;18:245.-
27. Reitan RM, Wolfson D. The question of validity of neuropsychological test scores among head-injured litigants: development of a dissimulation index. Arch Clin Neuropsychology 1996;11:573-80.
28. Green P, Lees-Haley PR, Allen LM. The Word Memory Test and the validity of neuropsychological test scores. J Forensic Neuropsychol 2002;2:97.-
29. Vickery CD, Berry DTR, Hanlon IT, et al. Detection of inadequate effort on neuropsychological testing: a meta-analytic review of selected procedures. Arch Clin Neuropsychology 2002;16:45-73.
30. Iverson GL, Viljoen JL. Practical and ethical issues regarding assessment of exaggeration, poor effort, and malingering in neuropsychology. Presented at First International Conference of Symptom, Diagnostic, and Disability Validity, 2002 Toronto, Ontario, Canada.
31. Mittenberg W, Patton C, Canyock EM, Condit DC. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol 2002;24:1094.-
32. Drane DL, Williamson DJ, Stroup ES, et al. Cognitive impairment is not equal in patients with epileptic and psychogenic nonepileptic seizures. Epilepsia 2006;47(11):1879-86.
33. Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
Mrs. M, age 27, suffered a head injury in a motor vehicle accident 9 months ago. She is referred to you by a neurologist with complaints of persistent headache and diffculties with memory and attention “worse now than right after the accident.” She tried to return to work 3 months after the accident but could not concentrate enough to be productive.
Review of medical records shows that she had minimal, if any, loss of consciousness at the accident scene, and she followed commands at the emergency room without apparent difficulty. Neurologic exam and head CT were normal. She is cooperative and fully oriented but appears upset about the difficulties she has experienced and occasionally complains of headache.
Three days later you receive a signed release of information from her attorney, requesting all records related to her examination.
In cases such as Mrs. M’s, the differential diagnosis often comes down to a somatoform disorder vs factitious disorder vs malingering, a decision that rarely seems as clear-cut as one might believe when reading the DSM-IV-TR. Particularly in litigation- or compensation-related situations, clinicians must make 2 fundamental judgments:
- Is the patient intentionally generating the symptoms?
- Are the symptoms plausibly related to neurologic injury or illness?
This article describes how symptom validity testing (SVT) as part of a comprehensive neuropsychological evaluation can help answer these questions. Inconsistencies in the way patients perform on SVT (Table)18-30 can provide “red flags” to possible embellishment of neurocognitive symptoms. We also offer recently developed guidelines for diagnosing malingering of neurocognitive dysfunction that may be more helpful than the DSM-IV-TR criteria.
Table
Performance consistencies in patients
who fail symptom validity testing (SVT)
| Consistency | Comment |
|---|---|
| 25% to 40% of patients seeking some form compensation for their injuries or illness fail SVT | This appears to hold true not only for ‘brain’ cases but also for ‘pain’ cases |
| Deficits are not exaggerated in a constant manner across tests of different abilities | Deficits most likely to be exaggerated are concentration, memory, weakness, and processing speed; may be due to assumptions about what ‘brain damage’ looks like |
| Patients failing SVT report greater levels of emotional distress, psychological maladjustment, and severity of neurocognitive difficulties on self-report measures | Patterns of exaggerated responses are not the same as those exaggerating psychopathology |
| Very few patients who fail SVT score significantly below chance | Below-chance responding is an insensitive criterion for identifying suboptimal effort, but this level of performance is quite specific; short of confession, below-chance performance on SVT is closest to an evidentiary ‘gold standard’ for malingering |
| Not all SVTs are created equal | Sensitivity and specificity vary, and measures may disagree when more than one is administered |
| Coaching makes a difference | Malingering subjects who are told which tests to look for and how to approach them are more difficult to discriminate from genuine patients |
| Invalid effort does not rule out a genuine neurologic injury or illness | Exaggeration can coexist with neurologically driven neurocognitive deficits; neuropsychologists who do forensic work encounter patients with documented injuries who fail SVT, sometimes in blatantly obvious or absurd ways |
| Source: References 1-13 | |
Why ‘gut feelings’ are fallible
Differential diagnosis of neurocognitive impairment is challenging. Some patients have normal neurologic examinations in all respects but cognition, such as those with early Alzheimer’s disease or recent concussion. Others may show significant neurobehavioral changes but normal results on neuroimaging (such as the rare patient in a coma after a traumatic brain injury whose head CT is read as normal). Thus, the absence of findings other than impaired cognition in a neurologic exam is not proof that a disorder is driven primarily by psychiatric or behavioral issues.
- rely on their training and intuition
- refer for psychological evaluation
- rely on traditional malingering measures in standard psychological tests, such as the Minnesota Multiphasic Personality Inventory-2 (MMPI-2).
The problem with this approach is its high error rate. Health care professionals do not discriminate poor effort from genuine neurocognitive impairment very effectively. Diagnostic algorithms routinely outperform clinical judgment, particularly when diagnostic parameters are relatively well understood.19
Although discerning conscious intent often remains more art than science, neuropsychologists have developed cross-validated techniques to identify implausible cognitive performances that suggest embellished symptoms. Thus, relying on clinical judgment is accepting an error rate that can be reduced by using other approaches.
‘Let the psychologist figure it out.’ The success of this approach depends on the psychologist’s methodology. The psychologist’s gut instinct is no more accurate than that of the psychiatrist or neurologist.
Relying on traditional scales. Measures of malingering in psychological testing can be quite effective for identifying exaggerated psychopathology,20-22 but exaggerated psychopathology differs from exaggerated neurocognitive symptoms.23 Embellished psychopathology is not the same as embellished “brain damage,” and they are not detected equally well by the same techniques.
Validity scales on the MMPI and MMPI-2 do a poor job of detecting patients known to be exaggerating neurocognitive impairment23 (although the more recently developed Lees-Haley “Fake Bad Scale” has shown promise).24 Thus, the clinician who feels confident that a patient has not exaggerated neurocognitive complaints because he or she scored in the normal range on the MMPI-2 validity scales (or other measures shown to help identify exaggeration of psychopathology) has drawn a conclusion based on scales that likely are inadequate for this purpose.
3 ways to measure patient effort
Using SVT is the most effective way to determine the validity of a patient’s effort on a neuropsychological test battery. SVT using 3 approaches has been shown to reliably discriminate patients who are putting forth valid effort from those who are not:
- forced-choice testing
- unusual patterns of responses within established neurocognitive tests
- unusual patterns of variability on the same test given on different occasions.
On some validated forced-choice SVTs, patients with moderate to severe traumatic brain injuries perform at ≥90% accuracy; thus, a far lower performance from a mildly injured patient raises a red flag that some-thing exceptional is occurring that demands an explanation.
Patterns within established tests. As empiric evidence about SVTs grows, we understand more about how neurologically impaired patients perform—and do not perform—on these tests. These patterns can then be used to examine the extent to which they discriminate between patients who are exaggerating and those who are not. Cross-validated techniques are available for the Wechsler Adult Intelligence Scale, 3rd edition, and the California Verbal Learning Test, among others.25,26
Patterns across different evaluations. Variation in test results is expected when a patient takes the same test on different dates. Along with having previously seen the test, other patient factors may include fatigue or inattention. When a patient is recovering from a brain injury or illness, additional variation is expected because of recovery or progression over time.
Some abilities—and test scores—are more stable than others, however, even in patients with genuine neurologic damage. At least one method that analyzes data from different administrations of the Halstead Reitan Neuropsychological Battery uses this insight,27 although this method has yet to be cross-validated.
What have we learned?
Cross-validated techniques have demonstrated that effort has a significant effect on neurocognitive test scores, often greater than the effect of the neurologic condition being studied.28,29 For example, you will be more accurate predicting a patient’s overall performance on a neuropsychological test battery on the basis of their performance on the Word Memory Test (one type of SVT) than on how long he or she was in a coma after a head injury until the coma has persisted for >6 days.30
SVTs are not ‘malingering tests.’ A malingering patient simulates or exaggerates symptoms with the conscious intention of deceiving someone. An SVT does a good job identifying exaggerated symptoms, but it has little (and, in most cases, nothing) to say about the extent to which this exaggeration is conscious or intentional.
For instance, patients with somatoform disorders tend to fail SVT at a higher rate than general medical populations.6,31 Our group32 recently reported that approximately one-half of patients diagnosed with psychogenic nonepileptic seizures at an epilepsy center fail SVTs. It is unlikely, however, that all—or even most—of these patients were malingering.
SVTs do not reveal motivation or intention—they merely state the extent to which the effort put into testing provides a valid estimate of neurocognitive function.
Alternative guidelines have been suggested to guide decisions about when to diagnose a patient as malingering neurocognitive deficits.33 See the original publication for a full explication of the criteria.
Clinical recommendations
Particularly when someone with a mild brain injury is seeking compensation, keep in mind that 25% to 40% of these patients perform in such a way on SVT that the validity of their cognitive performances should be questioned. It does not necessarily mean they are malingering; rather, they are performing in a way that cannot be explained by established brain-behavior relationships in the absence of obvious severe neurologic injury or illness.
Ask for SVT when you refer cases such as this for neuropsychological or psychological evaluation. SVT can provide an empirically based foundation on which to formulate an opinion, particularly about the severity of reported cognitive symptoms. Your opinion about the intentionality of symptoms likely will rely primarily on other information (such as the consistency of complaints with behavior during the assessment or presence of primary or secondary gain), but SVT provides a valuable tool with which to examine the validity of cognitive complaints.
Related resources
- Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
- National Academy of Neuropsychology. Position paper: Symptom validity testing: practice issues and medical necessity. http://nanonline.org/paio/svt.shtm.
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mrs. M, age 27, suffered a head injury in a motor vehicle accident 9 months ago. She is referred to you by a neurologist with complaints of persistent headache and diffculties with memory and attention “worse now than right after the accident.” She tried to return to work 3 months after the accident but could not concentrate enough to be productive.
Review of medical records shows that she had minimal, if any, loss of consciousness at the accident scene, and she followed commands at the emergency room without apparent difficulty. Neurologic exam and head CT were normal. She is cooperative and fully oriented but appears upset about the difficulties she has experienced and occasionally complains of headache.
Three days later you receive a signed release of information from her attorney, requesting all records related to her examination.
In cases such as Mrs. M’s, the differential diagnosis often comes down to a somatoform disorder vs factitious disorder vs malingering, a decision that rarely seems as clear-cut as one might believe when reading the DSM-IV-TR. Particularly in litigation- or compensation-related situations, clinicians must make 2 fundamental judgments:
- Is the patient intentionally generating the symptoms?
- Are the symptoms plausibly related to neurologic injury or illness?
This article describes how symptom validity testing (SVT) as part of a comprehensive neuropsychological evaluation can help answer these questions. Inconsistencies in the way patients perform on SVT (Table)18-30 can provide “red flags” to possible embellishment of neurocognitive symptoms. We also offer recently developed guidelines for diagnosing malingering of neurocognitive dysfunction that may be more helpful than the DSM-IV-TR criteria.
Table
Performance consistencies in patients
who fail symptom validity testing (SVT)
| Consistency | Comment |
|---|---|
| 25% to 40% of patients seeking some form compensation for their injuries or illness fail SVT | This appears to hold true not only for ‘brain’ cases but also for ‘pain’ cases |
| Deficits are not exaggerated in a constant manner across tests of different abilities | Deficits most likely to be exaggerated are concentration, memory, weakness, and processing speed; may be due to assumptions about what ‘brain damage’ looks like |
| Patients failing SVT report greater levels of emotional distress, psychological maladjustment, and severity of neurocognitive difficulties on self-report measures | Patterns of exaggerated responses are not the same as those exaggerating psychopathology |
| Very few patients who fail SVT score significantly below chance | Below-chance responding is an insensitive criterion for identifying suboptimal effort, but this level of performance is quite specific; short of confession, below-chance performance on SVT is closest to an evidentiary ‘gold standard’ for malingering |
| Not all SVTs are created equal | Sensitivity and specificity vary, and measures may disagree when more than one is administered |
| Coaching makes a difference | Malingering subjects who are told which tests to look for and how to approach them are more difficult to discriminate from genuine patients |
| Invalid effort does not rule out a genuine neurologic injury or illness | Exaggeration can coexist with neurologically driven neurocognitive deficits; neuropsychologists who do forensic work encounter patients with documented injuries who fail SVT, sometimes in blatantly obvious or absurd ways |
| Source: References 1-13 | |
Why ‘gut feelings’ are fallible
Differential diagnosis of neurocognitive impairment is challenging. Some patients have normal neurologic examinations in all respects but cognition, such as those with early Alzheimer’s disease or recent concussion. Others may show significant neurobehavioral changes but normal results on neuroimaging (such as the rare patient in a coma after a traumatic brain injury whose head CT is read as normal). Thus, the absence of findings other than impaired cognition in a neurologic exam is not proof that a disorder is driven primarily by psychiatric or behavioral issues.
- rely on their training and intuition
- refer for psychological evaluation
- rely on traditional malingering measures in standard psychological tests, such as the Minnesota Multiphasic Personality Inventory-2 (MMPI-2).
The problem with this approach is its high error rate. Health care professionals do not discriminate poor effort from genuine neurocognitive impairment very effectively. Diagnostic algorithms routinely outperform clinical judgment, particularly when diagnostic parameters are relatively well understood.19
Although discerning conscious intent often remains more art than science, neuropsychologists have developed cross-validated techniques to identify implausible cognitive performances that suggest embellished symptoms. Thus, relying on clinical judgment is accepting an error rate that can be reduced by using other approaches.
‘Let the psychologist figure it out.’ The success of this approach depends on the psychologist’s methodology. The psychologist’s gut instinct is no more accurate than that of the psychiatrist or neurologist.
Relying on traditional scales. Measures of malingering in psychological testing can be quite effective for identifying exaggerated psychopathology,20-22 but exaggerated psychopathology differs from exaggerated neurocognitive symptoms.23 Embellished psychopathology is not the same as embellished “brain damage,” and they are not detected equally well by the same techniques.
Validity scales on the MMPI and MMPI-2 do a poor job of detecting patients known to be exaggerating neurocognitive impairment23 (although the more recently developed Lees-Haley “Fake Bad Scale” has shown promise).24 Thus, the clinician who feels confident that a patient has not exaggerated neurocognitive complaints because he or she scored in the normal range on the MMPI-2 validity scales (or other measures shown to help identify exaggeration of psychopathology) has drawn a conclusion based on scales that likely are inadequate for this purpose.
3 ways to measure patient effort
Using SVT is the most effective way to determine the validity of a patient’s effort on a neuropsychological test battery. SVT using 3 approaches has been shown to reliably discriminate patients who are putting forth valid effort from those who are not:
- forced-choice testing
- unusual patterns of responses within established neurocognitive tests
- unusual patterns of variability on the same test given on different occasions.
On some validated forced-choice SVTs, patients with moderate to severe traumatic brain injuries perform at ≥90% accuracy; thus, a far lower performance from a mildly injured patient raises a red flag that some-thing exceptional is occurring that demands an explanation.
Patterns within established tests. As empiric evidence about SVTs grows, we understand more about how neurologically impaired patients perform—and do not perform—on these tests. These patterns can then be used to examine the extent to which they discriminate between patients who are exaggerating and those who are not. Cross-validated techniques are available for the Wechsler Adult Intelligence Scale, 3rd edition, and the California Verbal Learning Test, among others.25,26
Patterns across different evaluations. Variation in test results is expected when a patient takes the same test on different dates. Along with having previously seen the test, other patient factors may include fatigue or inattention. When a patient is recovering from a brain injury or illness, additional variation is expected because of recovery or progression over time.
Some abilities—and test scores—are more stable than others, however, even in patients with genuine neurologic damage. At least one method that analyzes data from different administrations of the Halstead Reitan Neuropsychological Battery uses this insight,27 although this method has yet to be cross-validated.
What have we learned?
Cross-validated techniques have demonstrated that effort has a significant effect on neurocognitive test scores, often greater than the effect of the neurologic condition being studied.28,29 For example, you will be more accurate predicting a patient’s overall performance on a neuropsychological test battery on the basis of their performance on the Word Memory Test (one type of SVT) than on how long he or she was in a coma after a head injury until the coma has persisted for >6 days.30
SVTs are not ‘malingering tests.’ A malingering patient simulates or exaggerates symptoms with the conscious intention of deceiving someone. An SVT does a good job identifying exaggerated symptoms, but it has little (and, in most cases, nothing) to say about the extent to which this exaggeration is conscious or intentional.
For instance, patients with somatoform disorders tend to fail SVT at a higher rate than general medical populations.6,31 Our group32 recently reported that approximately one-half of patients diagnosed with psychogenic nonepileptic seizures at an epilepsy center fail SVTs. It is unlikely, however, that all—or even most—of these patients were malingering.
SVTs do not reveal motivation or intention—they merely state the extent to which the effort put into testing provides a valid estimate of neurocognitive function.
Alternative guidelines have been suggested to guide decisions about when to diagnose a patient as malingering neurocognitive deficits.33 See the original publication for a full explication of the criteria.
Clinical recommendations
Particularly when someone with a mild brain injury is seeking compensation, keep in mind that 25% to 40% of these patients perform in such a way on SVT that the validity of their cognitive performances should be questioned. It does not necessarily mean they are malingering; rather, they are performing in a way that cannot be explained by established brain-behavior relationships in the absence of obvious severe neurologic injury or illness.
Ask for SVT when you refer cases such as this for neuropsychological or psychological evaluation. SVT can provide an empirically based foundation on which to formulate an opinion, particularly about the severity of reported cognitive symptoms. Your opinion about the intentionality of symptoms likely will rely primarily on other information (such as the consistency of complaints with behavior during the assessment or presence of primary or secondary gain), but SVT provides a valuable tool with which to examine the validity of cognitive complaints.
Related resources
- Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
- National Academy of Neuropsychology. Position paper: Symptom validity testing: practice issues and medical necessity. http://nanonline.org/paio/svt.shtm.
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bianchini KJ, Greve KW, Glynn G. On the diagnosis of malingered pain-related disability: lessons from cognitive malingering research. Spine 2005;5:404-17.
2. Gervais RO, Green P, Allen LM, Iverson GL. Effects of coaching on symptom validity testing in chronic pain patients presenting for disability assessments. J Forensic Neuropsychol 2001;2:1.-
3. Meyers JE, Diep A. Assessment of malingering in chronic pain patients using neuropsychological tests. Applied Neuropsychol 2000;7:133-9.
4. Williamson DJ, Rohling ML, Green P, Allen L. Evaluating effort with the Word Memory Test and Category Test–or not: inconsistencies in a forensic sample. J Forensic Neuropsychol 2003;3:19-44.
5. Gouvier WD, Prestholdt P, Warner M. A survey of common misperceptions about head injury and recovery. Arch Clin Neuropsychology 1988;3:331-43.
6. Boone KB, Lu P. Impact of somatoform symptomatology on credibility of cognitive performance. Clin Neuropsychol 1999;13:414-9.
7. Greiffenstein MF, Baker WJ, Axelrod BN, et al. The Fake Bad Scale and MMPI-2 F-family in detection of implausible psychological trauma claims. Clin Neuropsychol 2004;18:573-90.
8. Larrabee GJ. Detection of symptom exaggeration with the MMPI-2 in litigants with malingered neurocognitive dysfunction. Clin Neuropsychol 2003;17:54.-
9. Tan JE, Slick DJ, Strauss E, Hultsch DF. How’d they do it? Malingering strategies on symptom validity tests. Clin Neuropsychol 2002;16:495.-
10. Iverson GL, Binder LM. Detecting exaggeration and malingering in neuropsychological assessment. J Head Trauma Rehabil 2000;15:829-58.
11. Sweet JJ. Malingering: differential diagnosis. In: Sweet JJ, ed. Forensic neuropsychology: fundamentals and practice. New York: Swets & Zeitlinger, 1999:255-85.
12. Coleman RD, Rapport LJ, Millis SR, et al. Effects of coaching on detection of malingering on the California Verbal Learning Test. J Clin Exp Neuropsychol 1998;20:201.-
13. Rapport LJ, Farchione TJ, Coleman RD, Axelrod BN. Effects of coaching on malingered motor function profiles. J Clin Exp Neuropsychol 1998;20:89-97.
14. Carone DA, Benedict RHB, Munschauer FE, et al. Interpreting patient/informant discrepancies of reported cognitive symptoms in MS. J Int Neuropsychol Soc 2005;11:574.-
15. Meador KJ, Loring DW, Vahle VJ, et al. Subjective perception of cognitive effects of antiepileptic drugs is more related to mood than to objective performance. Epilepsia 2005;46:261-2.
16. Sawrie SM, Martin RC, Kuzniecky R, et al. Subjective versus objective memory change after temporal lobe epilepsy surgery. Neurology 1999;53:1511.-
17. Davis DA, Mazmanian PE, Fordis M, et al. Accuracy of physician self-assessment compared with observed measures of competence: a systematic review. JAMA 2006;296:1094-102.
18. Faust D. The detection of deception. Neurol Clin 1995;13:255-65.
19. Swets JA, Dawes RM, Monahan J. Psychological science can improve diagnostic decisions. Psychol Sci Public Interest 2000;1:1-26.
20. Knoll J, Resnick PJ. The detection of malingered post-traumatic stress disorder. Psychiatr Clin North Am 2006;29:629-47.
21. Morey LC, Lanier VW. Operating characteristics of six response distortion indicators for the Personality Assessment Inventory. Assessment 1998;5:203-14.
22. Rogers R, Sewell KW, Salekin RT. A meta-analysis of malingering on the MMPI-2. Assessment 1994;1:227-37.
23. Larrabee GJ. Somatic malingering on the MMPI and MMPI-2 in personal injury litigants. Clin Neuropsychol 1998;12:179.-
24. Nelson NW, Sweet JJ, Demakis GJ. Meta-analysis of the MMPI-2 Fake Bad Scale: utility in forensic practice. Clin Neuropsychol 2006;20:39-58.
25. Curtis KL, Greve KW, Bianchini KJ, Brennan A. California Verbal Learning Test indicators of malingered neurocognitive dysfunction: sensitivity and specificity in traumatic brain injury. Assessment 2006;13:46.-
26. Greve KW, Bianchini KJ, Mathias CW, et al. Detecting malingered performance on the Wechsler Adult Intelligence Scale: validation of Mittenberg’s approach in traumatic brain injury. Arch Clin Neuropsychol 2003;18:245.-
27. Reitan RM, Wolfson D. The question of validity of neuropsychological test scores among head-injured litigants: development of a dissimulation index. Arch Clin Neuropsychology 1996;11:573-80.
28. Green P, Lees-Haley PR, Allen LM. The Word Memory Test and the validity of neuropsychological test scores. J Forensic Neuropsychol 2002;2:97.-
29. Vickery CD, Berry DTR, Hanlon IT, et al. Detection of inadequate effort on neuropsychological testing: a meta-analytic review of selected procedures. Arch Clin Neuropsychology 2002;16:45-73.
30. Iverson GL, Viljoen JL. Practical and ethical issues regarding assessment of exaggeration, poor effort, and malingering in neuropsychology. Presented at First International Conference of Symptom, Diagnostic, and Disability Validity, 2002 Toronto, Ontario, Canada.
31. Mittenberg W, Patton C, Canyock EM, Condit DC. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol 2002;24:1094.-
32. Drane DL, Williamson DJ, Stroup ES, et al. Cognitive impairment is not equal in patients with epileptic and psychogenic nonepileptic seizures. Epilepsia 2006;47(11):1879-86.
33. Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
1. Bianchini KJ, Greve KW, Glynn G. On the diagnosis of malingered pain-related disability: lessons from cognitive malingering research. Spine 2005;5:404-17.
2. Gervais RO, Green P, Allen LM, Iverson GL. Effects of coaching on symptom validity testing in chronic pain patients presenting for disability assessments. J Forensic Neuropsychol 2001;2:1.-
3. Meyers JE, Diep A. Assessment of malingering in chronic pain patients using neuropsychological tests. Applied Neuropsychol 2000;7:133-9.
4. Williamson DJ, Rohling ML, Green P, Allen L. Evaluating effort with the Word Memory Test and Category Test–or not: inconsistencies in a forensic sample. J Forensic Neuropsychol 2003;3:19-44.
5. Gouvier WD, Prestholdt P, Warner M. A survey of common misperceptions about head injury and recovery. Arch Clin Neuropsychology 1988;3:331-43.
6. Boone KB, Lu P. Impact of somatoform symptomatology on credibility of cognitive performance. Clin Neuropsychol 1999;13:414-9.
7. Greiffenstein MF, Baker WJ, Axelrod BN, et al. The Fake Bad Scale and MMPI-2 F-family in detection of implausible psychological trauma claims. Clin Neuropsychol 2004;18:573-90.
8. Larrabee GJ. Detection of symptom exaggeration with the MMPI-2 in litigants with malingered neurocognitive dysfunction. Clin Neuropsychol 2003;17:54.-
9. Tan JE, Slick DJ, Strauss E, Hultsch DF. How’d they do it? Malingering strategies on symptom validity tests. Clin Neuropsychol 2002;16:495.-
10. Iverson GL, Binder LM. Detecting exaggeration and malingering in neuropsychological assessment. J Head Trauma Rehabil 2000;15:829-58.
11. Sweet JJ. Malingering: differential diagnosis. In: Sweet JJ, ed. Forensic neuropsychology: fundamentals and practice. New York: Swets & Zeitlinger, 1999:255-85.
12. Coleman RD, Rapport LJ, Millis SR, et al. Effects of coaching on detection of malingering on the California Verbal Learning Test. J Clin Exp Neuropsychol 1998;20:201.-
13. Rapport LJ, Farchione TJ, Coleman RD, Axelrod BN. Effects of coaching on malingered motor function profiles. J Clin Exp Neuropsychol 1998;20:89-97.
14. Carone DA, Benedict RHB, Munschauer FE, et al. Interpreting patient/informant discrepancies of reported cognitive symptoms in MS. J Int Neuropsychol Soc 2005;11:574.-
15. Meador KJ, Loring DW, Vahle VJ, et al. Subjective perception of cognitive effects of antiepileptic drugs is more related to mood than to objective performance. Epilepsia 2005;46:261-2.
16. Sawrie SM, Martin RC, Kuzniecky R, et al. Subjective versus objective memory change after temporal lobe epilepsy surgery. Neurology 1999;53:1511.-
17. Davis DA, Mazmanian PE, Fordis M, et al. Accuracy of physician self-assessment compared with observed measures of competence: a systematic review. JAMA 2006;296:1094-102.
18. Faust D. The detection of deception. Neurol Clin 1995;13:255-65.
19. Swets JA, Dawes RM, Monahan J. Psychological science can improve diagnostic decisions. Psychol Sci Public Interest 2000;1:1-26.
20. Knoll J, Resnick PJ. The detection of malingered post-traumatic stress disorder. Psychiatr Clin North Am 2006;29:629-47.
21. Morey LC, Lanier VW. Operating characteristics of six response distortion indicators for the Personality Assessment Inventory. Assessment 1998;5:203-14.
22. Rogers R, Sewell KW, Salekin RT. A meta-analysis of malingering on the MMPI-2. Assessment 1994;1:227-37.
23. Larrabee GJ. Somatic malingering on the MMPI and MMPI-2 in personal injury litigants. Clin Neuropsychol 1998;12:179.-
24. Nelson NW, Sweet JJ, Demakis GJ. Meta-analysis of the MMPI-2 Fake Bad Scale: utility in forensic practice. Clin Neuropsychol 2006;20:39-58.
25. Curtis KL, Greve KW, Bianchini KJ, Brennan A. California Verbal Learning Test indicators of malingered neurocognitive dysfunction: sensitivity and specificity in traumatic brain injury. Assessment 2006;13:46.-
26. Greve KW, Bianchini KJ, Mathias CW, et al. Detecting malingered performance on the Wechsler Adult Intelligence Scale: validation of Mittenberg’s approach in traumatic brain injury. Arch Clin Neuropsychol 2003;18:245.-
27. Reitan RM, Wolfson D. The question of validity of neuropsychological test scores among head-injured litigants: development of a dissimulation index. Arch Clin Neuropsychology 1996;11:573-80.
28. Green P, Lees-Haley PR, Allen LM. The Word Memory Test and the validity of neuropsychological test scores. J Forensic Neuropsychol 2002;2:97.-
29. Vickery CD, Berry DTR, Hanlon IT, et al. Detection of inadequate effort on neuropsychological testing: a meta-analytic review of selected procedures. Arch Clin Neuropsychology 2002;16:45-73.
30. Iverson GL, Viljoen JL. Practical and ethical issues regarding assessment of exaggeration, poor effort, and malingering in neuropsychology. Presented at First International Conference of Symptom, Diagnostic, and Disability Validity, 2002 Toronto, Ontario, Canada.
31. Mittenberg W, Patton C, Canyock EM, Condit DC. Base rates of malingering and symptom exaggeration. J Clin Exp Neuropsychol 2002;24:1094.-
32. Drane DL, Williamson DJ, Stroup ES, et al. Cognitive impairment is not equal in patients with epileptic and psychogenic nonepileptic seizures. Epilepsia 2006;47(11):1879-86.
33. Slick DJ, Sherman EMS, Iverson GL. Diagnostic criteria for malingering cognitive dysfunction: proposed standards for clinical practice and research. Clin Neuropsychol 1999;13:545-61.
IM aripiprazole for acute agitation
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
In recent clinical trials, a new intramuscular (IM) form of the second-generation antipsychotic (SGA) aripiprazole has controlled agitation in adults with schizophrenia or bipolar mania without causing significant side effects (Table 1).1-3
Table 1
IM aripiprazole: Fast facts
| Brand name: Abilify |
| Class: Second-generation antipsychotic |
| Indication: Acute agitation associated with schizophrenia or type I bipolar disorder (mixed or manic episodes) |
| Manufacturer: Otsuka America Pharmaceutical (marketed in collaboration with Bristol-Myers Squibb) |
| Dosing forms: 1.3-mL vial of clear, aqueous solution containing 9.75 mg of active drug |
| Recommended dosage: 9.75 mg every 2 hours as needed; do not exceed 30 mg across 24 hours |
Clinical implications
Rapid intervention is critical to protecting the patient and caregivers when violent and/or destructive behavior accompanies agitation. IM aripiprazole substantially reduced agitation within 45 to 60 minutes of dosing in randomized, double-blind, placebo-controlled studies.1-3
How it works
Whereas other SGAs have relatively little effect on D2 (dopamine) receptors and relatively high 5-HT2A (serotonin) receptor affinities, aripiprazole appears to work via partial D2 receptor agonism. The medication:
- blocks D2 receptors in brain regions where dopamine is overactive in schizophrenia, such as the mesolimbic pathway. This produces an antipsychotic effect.
- maintains or moderately boosts dopamine activity as needed in regions such as the nigrostriatal pathway. This reduces the risk of motor side effects and might improve negative and cognitive schizophrenia symptoms.
Aripiprazole is a partial 5-HT1A receptor agonist and—like other SGAs—a 5-HT2A receptor antagonist. These receptor subtypes have been implicated in antipsychotic action. In particular, partial 5-HT1A receptor agonism is thought to help:
- reduce anxiety
- lessen depressive, negative, and cognitive symptoms
- decrease extrapyramidal symptom (EPS) liability.4
Aripiprazole also has moderate affinity for histaminic and alpha-adrenergic receptors and no appreciable effect on cholinergic muscarinic receptors.5-8
Pharmacokinetics
IM aripiprazole’s activity has been attributed to its parent drug and to a lesser extent its major metabolite, dehydroaripiprazole. Both moieties act on D2 receptors, and dehydroaripiprazole accounts for 40% of the parent drug’s exposure in plasma.
Mean elimination half-lives for aripiprazole and dehydroaripiprazole are approximately 75 and 94 hours, respectively, allowing for daily administration. Both active moieties reach steady-state concentration within 14 days of dosing. Because aripiprazole accumulation is predictable after a single dose and its pharmacokinetics are dose-proportional at steady state, higher doses are not always more effective and could increase side-effect risk.
Aripiprazole is metabolized mainly through the liver by cytochrome P-450 2D6 and 3A4 isozymes. This requires careful monitoring when prescribing the drug concomitantly with:
- agents that induce CYP 3A4—such as carbamazepine—which could diminish aripiprazole’s effectiveness by increasing its clearance and decreasing aripiprazole blood levels
- CYP 3A4 inhibitors such as ketoconazole or CYP 2D6 inhibitors such as quinidine, fluoxetine, or paroxetine, which can inhibit aripiprazole elimination9 and increase the risk of adverse events.
Similarly, aripiprazole could be efficacious at lower-than-therapeutic dosages when taken with medications that raise aripiprazole blood levels.
Efficacy
In 3 randomized, placebo-controlled, double-blind trials, IM aripiprazole reduced agitation in inpatients with schizophrenia, schizoaffective disorder, or type I bipolar disorder with manic or mixed episodes, with or without psychotic features.
In each trial, IM aripiprazole was as effective as comparable dosages of haloperidol or lorazepam IM preparations. Patients were moderately to severely agitated based on Positive and Negative Syndrome Scale Excited Component (PANSS-EC) assessments, which gauged impulse control, tension, hostility, uncooperativeness, and excitement.
Patients could receive up to 3 injections within 24 hours but had to wait ≥2 hours for the second injection so that investigators could record follow-up PANSS-EC scores. Clinical Global Impression of Improvement (CGI-I) scale scores were a key secondary measure.
Examination of population subsets in the studies showed no differential response based on age, race, or gender.
Tran-Johnson et al1followed 357 patients with schizophreniform disorders, schizophrenia, or schizoaffective disorders.
Two hours after initial injection, mean PANSS-EC scores decreased approximately 3 points with placebo and 4 to 6.5 points among patients receiving 7.5 mg of IM haloperidol or 5.25, 9.75, or 15 mg of IM aripiprazole. Agitation improved significantly after 45 minutes among patients receiving 9.75 mg of IM aripiprazole, compared with 105 minutes in the IM haloperidol group.
Prevalence of EPS across 24 hours with haloperidol was 19.3%, compared with an average 5.2% among all IM aripiprazole groups, suggesting that IM aripiprazole carries a substantially lower EPS risk.
Andrezina et al2followed 448 patients with schizophrenia or schizoaffective disorder. Two hours after injection, patients in both treatment groups showed much greater improvement compared with placebo based on mean PANSS-EC score decreases and mean CGI-I scores (Table 2).
Prevalence of EPS was 1.7% with IM aripiprazole, 2.3% with placebo, and 12.6% with IM haloperidol. Prevalence of EPS-related adverse events was 0% with IM aripiprazole, 1.6% with placebo, and 16.5% with IM haloperidol.
Zimbroff et al3 gave IM aripiprazole, 9.75 or 15 mg; IM lorazepam, 2 mg; or placebo to 301 patients with type I bipolar disorder with manic or mixed episodes.
Two hours later, all 3 treatment groups showed significantly greater agitation improvement as shown by PANSS-EC scores, compared with placebo (Table 3).
Across 2 hours, oversedation—defined as an Agitation-Calmness Evaluation Scale score of 8 or 9—was less prevalent among patients receiving IM aripiprazole, 9.75 mg (6.7%), compared with IM aripiprazole, 15 mg (17.3%), or IM lorazepam (19.1%).
Table 2
Agitation, symptom improvement 2 hours after aripiprazole or haloperidol injection
| Assessment scale | IM aripiprazole, 9.75 mg | IM haloperidol, 6.5 mg | Placebo |
|---|---|---|---|
| PANSS-EC mean score decrease (P | 7.27 | 7.75 | 4.78 |
| CGI-I mean score (P | 2.42 | 2.37 | 3.10 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component; CGI-I: Clinical Global Impression of Improvement | |||
| Source: Adapted from reference 2 | |||
Table 3
Agitation improvement 2 hours after aripiprazole or lorazepam injection
| IM preparation | PANSS-EC mean score decrease |
|---|---|
| Aripiprazole, 9.75 mg | 8.7 |
| Aripiprazole, 15 mg | 8.7 |
| Lorazepam, 2 mg | 9.6 |
| Placebo | 5.6 |
| PANSS-EC: Positive and Negative Syndrome Scale Excited Component | |
| Source: Adapted from reference 3 | |
Safety and tolerability
IM aripiprazole was well tolerated in clinical trials and did not cause excessive sedation10 or injection-site pain.1-3
Most frequently reported adverse events were headache (12% with IM aripiprazole vs 7% with placebo), nausea (9% vs 3%), dizziness (8% vs 5%), and somnolence (7% vs 4%).
Prevalence of akathisia or dystonia among all IM aripiprazole groups in the 3 trials was 2% and
No clinically significant ECG abnormalities were reported among the aripiprazole groups.1-3,11
Dosing
Start at 9.75 mg every 2 hours as needed, but do not exceed 30 mg/d across 24 hours. Controlled studies have not evaluated efficacy or safety of more-frequent injections or safety of total daily doses >30 mg.
Try a lower dose (5.25 mg) for patients who are elderly or small in body size or have reacted adversely to other antipsychotics. If necessary, give another 5.25 mg in 2 hours. If the patient is still agitated 2 hours after the second dose, consider a third dose at 9.75 mg. Again, do not exceed 30 mg over 24 hours. Obtain lower doses by administering a portion of the vial.
Transitioning to oral Tx
If IM aripiprazole reduces psychotic symptoms as well as acute behaviors, switch the patient to oral aripiprazole once the risk of violence has diminished.12 If psychosis does not improve with IM aripiprazole, weigh clinical factors before choosing an oral antipsychotic.
Only one controlled trial12 has examined transitioning from IM aripiprazole to an oral antipsychotic. In the randomized study, 448 patients receiving IM aripiprazole, 9.75 mg; IM haloperidol, 6.5 mg; or placebo for agitation secondary to schizophrenia or schizoaffective disorder were transitioned to the oral preparation of the drug they were receiving: aripiprazole, 10 to 15 mg/d, or haloperidol, 7.5 to 10 mg/d. Placebo-group patients transitioned to oral aripiprazole.
Over 4 days, both oral treatments provided continued efficacy, suggesting that:
- patients receiving IM aripiprazole can be conveniently switched to the oral preparation
- IM and oral aripiprazole are equally safe.
Oral and IM aripiprazole doses are equivalent and the pharmacokinetics are comparable. For example, a patient receiving 20 mg/d of IM aripiprazole can take 20 mg of oral aripiprazole within 24 hours of the last injection.
Related resources
- Allen MH, Currier GW, Carpenter D, et al. Expert consensus guidelines: treatment of behavioral emergencies. J Psychiatr Pract 2005;11(suppl 1):5-108.
- Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
Drug brand names
- Aripiprazole IM • Abilify
- Carbamazepine • Tegretol, others
- Fluoxetine • Prozac
- Haloperidol • Haldol
- Ketoconazole • Nizoral
- Lorazepam • Ativan
- Paroxetine • Paxil
- Quinidine • Quinaglute
Disclosure
Dr. Josiassen was principal investigator and Dr. Shaughnessy a co-investigator on a pre-approval clinical trial of IM aripiprazole. Both have conducted sponsor- and investigator-initiated studies for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Janssen, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceuticals, Otsuka Maryland Research Institute, Pfizer, and Yamanuchi.
Acknowledgments
This article was supported in part by the Arthur P. Noyes Research Foundation.
The authors thank Margit Kacso, Cara Bendler, Dawn Filmyer, and Jon Weinstein for their technical and editorial assistance in preparing this article.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
1. Tran-Johnson TK, Sack DA, Marcus RN, et al. Efficacy and safety of intramuscular aripiprazole inpatients with acute agitation: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2007;68:111-9.
2. Andrezina R, Josiassen RC, Marcus R, et al. Intramuscular aripiprazole for the treatment of acute agitation in patients with schizophrenia or schizoaffective disorder: a double-blind, placebo-controlled comparison with intramuscular haloperidol. Psychopharmacology (Berl) 2006;188:281-92.
3. Zimbroff DL, Marcus RN, Manos G, et al. Management of acute agitation in patients with bipolar disorder: efficacy and safety of intramuscular aripiprazole. J Clin Psychopharmacol 2007;27:171-6.
4. Stark AD, Jordan S, Allers KA, et al. Interaction of the novel antipsychotic aripiprazole with HT1A and 5-HT2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacol 2007;190:373-82.
5. Burris KD, Molski TF, Xu C, et al. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302:381-9.
6. Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology 2003;28:1400-11.
7. Jordan S, Koprivica V, Dunn R, et al. In vivo effects of aripiprazole on cortical and striatal dopaminergic and serotonergic function. Eur J Pharmacol 2004;483:45-53.
8. Hirose T, Uwahodo Y, Yamada S, et al. Mechanism of action of aripiprazole predicts clinical efficacy and a favourable side-effect profile. J Psychopharmacol 2004;18:375-83.
9. Physicians’ desk reference 61st ed. Montvale, NJ: Thomson PDR; 2007.
10. Currier GW, Crandall D, Archibald D, et al. Intramuscular (IM) aripiprazole controls agitation in patients with schizophrenia or bipolar I disorder without excessive sedation. Paper presented at: Annual Meeting of the American Psychiatric Association; May 20-25, 2006; Toronto, Canada.
11. Currier GW, Citrome LL, Zimbroff DL, et al. Intramuscular aripiprazole in the control of agitation. J Psychiatr Pract 2007;13:159-69.
12. Daniel DG, Currier GW, Zimbroff DL, et al. Efficacy and safety of oral aripiprazole compared with haloperidol in patients transitioning from acute treatment with intramuscular formulations. J Psychiatr Pract 2007;13:170-7.
Dr. Josiassen is chief scientific officer, Arthur P. Noyes Research Foundation, Norristown, PA, and adjunct professor of psychiatry, University of Pennsylvania, Philadelphia.
Dr. Shaughnessy is associate professor of psychiatry, Drexel University College of Medicine, Philadelphia, and medical consultant, Arthur P. Noyes Research Foundation.
Dangerous duo: Antiepileptics plus herbals
Antiepileptic drugs (AEDs)—one of the most frequently prescribed medication classes—are used to manage seizures, epilepsy, pain syndromes, migraines, and psychiatric disorders such as bipolar disorder, anxiety, schizophrenia, and depression. When prescribing AEDs and monitoring patient response, consider possible adverse interactions with complementary and alternative medicines (CAM).
Approximately 40% of Americans use herbals or botanicals,1 whose pharmacokinetics, efficacy, or safety have not been rigorously studied. When used concurrently, these alternative remedies may reduce AEDs’ efficacy, increase the risk of seizures, or cause other neurologic adverse effects.
Common agents. In the United States, the most commonly used herbals and botanicals are garlic, ginkgo biloba, soy, melatonin, kava kava, St. John’s Wort, saw palmetto, and ginseng.2 Many first- and second-generation AEDs are known to interact with herbals and botanicals. All first-generation AEDs (such as carbamazepine, valproic acid, phenytoin, phenobarbital, and primidone) are cytochrome P-450 inducers or inhibitors, which means they have the potential to interact with other drugs that undergo hepatic metabolism. Because these interactions are unpredictable, it is important to carefully question your patient about the clinical effect of a prescribed AED.
Moreover, some botanicals—such as black cohosh, water hemlock, ephedra, kava kava, yohimbine, guarana, and ginkgo seeds—are known to induce seizures, which could negate an AED’s efficacy.
Communication. When managing psychiatric patients taking AEDs, maintain open communication regarding CAM. If a patient does not show clinical response to an AED or reports an adverse effect, gently inquire about his or her use of herbal remedies. Maintain a nonjudgmental tone when a patient reports using alternative remedies. Several studies have shown that patients often are reluctant to share this information with their physicians2-4 because they fear the physician may have a negative opinion about CAM.
The key to any patient inquiry regarding herbals is to identify why the patient initially chose the CAM. Doing so might reveal that the patient is not happy with the prescribed therapy, in which case you might be able to lower the risk of an adverse drug interaction by switching to another AED or persuading the patient to discontinue the herbal remedy.
Referneces
1. Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA 1998;280(18):1569-75.
2. Sirven JI, Drazkowski JF, Zimmerman RS, et al. Complementary/alternative medicine for epilepsy in Arizona. Neurology 2003;61(4):576-7.
3. Astin JA. Why patients use alternative medicine: results of a national study. JAMA 1998;279(19):1548-53.
4. National Center for Complementary and Alternative Medicine. What is CAM? Available at: http://nccam.nih.gov/health/whatiscam. Accessed June 7, 2007.
Dr. Sirven is associate professor of neurology, Mayo Clinic College of Medicine, Phoenix, AZ.
Antiepileptic drugs (AEDs)—one of the most frequently prescribed medication classes—are used to manage seizures, epilepsy, pain syndromes, migraines, and psychiatric disorders such as bipolar disorder, anxiety, schizophrenia, and depression. When prescribing AEDs and monitoring patient response, consider possible adverse interactions with complementary and alternative medicines (CAM).
Approximately 40% of Americans use herbals or botanicals,1 whose pharmacokinetics, efficacy, or safety have not been rigorously studied. When used concurrently, these alternative remedies may reduce AEDs’ efficacy, increase the risk of seizures, or cause other neurologic adverse effects.
Common agents. In the United States, the most commonly used herbals and botanicals are garlic, ginkgo biloba, soy, melatonin, kava kava, St. John’s Wort, saw palmetto, and ginseng.2 Many first- and second-generation AEDs are known to interact with herbals and botanicals. All first-generation AEDs (such as carbamazepine, valproic acid, phenytoin, phenobarbital, and primidone) are cytochrome P-450 inducers or inhibitors, which means they have the potential to interact with other drugs that undergo hepatic metabolism. Because these interactions are unpredictable, it is important to carefully question your patient about the clinical effect of a prescribed AED.
Moreover, some botanicals—such as black cohosh, water hemlock, ephedra, kava kava, yohimbine, guarana, and ginkgo seeds—are known to induce seizures, which could negate an AED’s efficacy.
Communication. When managing psychiatric patients taking AEDs, maintain open communication regarding CAM. If a patient does not show clinical response to an AED or reports an adverse effect, gently inquire about his or her use of herbal remedies. Maintain a nonjudgmental tone when a patient reports using alternative remedies. Several studies have shown that patients often are reluctant to share this information with their physicians2-4 because they fear the physician may have a negative opinion about CAM.
The key to any patient inquiry regarding herbals is to identify why the patient initially chose the CAM. Doing so might reveal that the patient is not happy with the prescribed therapy, in which case you might be able to lower the risk of an adverse drug interaction by switching to another AED or persuading the patient to discontinue the herbal remedy.
Antiepileptic drugs (AEDs)—one of the most frequently prescribed medication classes—are used to manage seizures, epilepsy, pain syndromes, migraines, and psychiatric disorders such as bipolar disorder, anxiety, schizophrenia, and depression. When prescribing AEDs and monitoring patient response, consider possible adverse interactions with complementary and alternative medicines (CAM).
Approximately 40% of Americans use herbals or botanicals,1 whose pharmacokinetics, efficacy, or safety have not been rigorously studied. When used concurrently, these alternative remedies may reduce AEDs’ efficacy, increase the risk of seizures, or cause other neurologic adverse effects.
Common agents. In the United States, the most commonly used herbals and botanicals are garlic, ginkgo biloba, soy, melatonin, kava kava, St. John’s Wort, saw palmetto, and ginseng.2 Many first- and second-generation AEDs are known to interact with herbals and botanicals. All first-generation AEDs (such as carbamazepine, valproic acid, phenytoin, phenobarbital, and primidone) are cytochrome P-450 inducers or inhibitors, which means they have the potential to interact with other drugs that undergo hepatic metabolism. Because these interactions are unpredictable, it is important to carefully question your patient about the clinical effect of a prescribed AED.
Moreover, some botanicals—such as black cohosh, water hemlock, ephedra, kava kava, yohimbine, guarana, and ginkgo seeds—are known to induce seizures, which could negate an AED’s efficacy.
Communication. When managing psychiatric patients taking AEDs, maintain open communication regarding CAM. If a patient does not show clinical response to an AED or reports an adverse effect, gently inquire about his or her use of herbal remedies. Maintain a nonjudgmental tone when a patient reports using alternative remedies. Several studies have shown that patients often are reluctant to share this information with their physicians2-4 because they fear the physician may have a negative opinion about CAM.
The key to any patient inquiry regarding herbals is to identify why the patient initially chose the CAM. Doing so might reveal that the patient is not happy with the prescribed therapy, in which case you might be able to lower the risk of an adverse drug interaction by switching to another AED or persuading the patient to discontinue the herbal remedy.
Referneces
1. Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA 1998;280(18):1569-75.
2. Sirven JI, Drazkowski JF, Zimmerman RS, et al. Complementary/alternative medicine for epilepsy in Arizona. Neurology 2003;61(4):576-7.
3. Astin JA. Why patients use alternative medicine: results of a national study. JAMA 1998;279(19):1548-53.
4. National Center for Complementary and Alternative Medicine. What is CAM? Available at: http://nccam.nih.gov/health/whatiscam. Accessed June 7, 2007.
Dr. Sirven is associate professor of neurology, Mayo Clinic College of Medicine, Phoenix, AZ.
Referneces
1. Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990-1997: results of a follow-up national survey. JAMA 1998;280(18):1569-75.
2. Sirven JI, Drazkowski JF, Zimmerman RS, et al. Complementary/alternative medicine for epilepsy in Arizona. Neurology 2003;61(4):576-7.
3. Astin JA. Why patients use alternative medicine: results of a national study. JAMA 1998;279(19):1548-53.
4. National Center for Complementary and Alternative Medicine. What is CAM? Available at: http://nccam.nih.gov/health/whatiscam. Accessed June 7, 2007.
Dr. Sirven is associate professor of neurology, Mayo Clinic College of Medicine, Phoenix, AZ.
Parkinson’s symptoms or depression? Look for clinical signs
Many depressive symptoms are seen in the normal course of Parkinson’s disease (PD) (Table 1).1 As a result, depression—the most common neuropsychiatric disturbance in PD—is difficult to assess in PD and easily can go undetected and untreated.2
Making the diagnosis is important, however, because depression causes PD patients suffering and may accelerate decline in motor and cognitive function, activities of daily living, and quality of life.3 In the absence of specific guidelines (Box),4 we provide evidence to help you sort through the overlapping symptoms to find clinical signs that differentiate depression from PD symptoms.5,6
Table 1
Symptoms of depression that occur in or mimic those in the natural course of PD
| Psychomotor retardation (bradykinesia) |
| Depressed or emotionless appearance (‘masked facies,’ stooped posture) |
| Agitation (dyskinesias) |
| Decreased interest and enjoyment (apathy and decreased initiative) |
| Impaired memory and concentration |
| Fatigue or decreased energy |
| Impaired sleep |
| Weight and appetite changes |
| Physical complaints |
| Source: Adapted from reference 1 |
DSM-IV-TR depression criteria
Approximately 20% of PD patients meet DSM-IV-TR criteria for major depression, and another 20% meet criteria for dysthymia.5 By DSM-IV-TR criteria,6 diagnosis of a major depressive episode requires ≥5 of 9 symptoms, of which at least 1 is depressed mood or loss of interest or pleasure. Because these symptoms must be present during the same 2-week period and represent a change in functioning, this diagnosis has an acute quality.
Dysthymia—also frequently called “chronic depression”—is characterized by a mostly depressed mood for 2 years, accompanied by ≥2 of 6 symptoms: appetite changes, sleep changes, low energy/fatigue, low self-esteem, poor concentration/indecisiveness, and hopelessness.6
All of these depression symptoms may overlap with those of PD.
1 Mood. In mid-stage and late PD, mood often fluctuates in concert with daily periods of increased rigidity and tremor (“off” periods) interspersed with improved motor functioning (“on” periods).7 Thus, when evaluating the PD patient:
- take a detailed history of motor fluctuations and their associations with mood symptoms
- also evaluate mood during “on” periods.
- behavioral (lack of effort)
- cognitive (loss of interest/concern)
- affective (decreased emotional response or “flat” affect).
3 Weight changes. Patients with PD tend to have lower body weight than matched subjects. As a result, weight loss in the course of PD can be confused with weight loss associated with depression.
Weight loss appears to start 2 to 4 years before a PD diagnosis and continues thereafter. Despite the weight loss, PD patients report higher energy intake after the diagnosis compared with individuals without PD.11 A related, not necessarily contradictory finding is that a higher premorbid body mass index (BMI) seems to be associated with an increased risk of developing PD.12
In general, dopaminergic treatment of PD seems to be associated with weight loss.13 However, weight gain has been reported after pramipexole treatment, which the authors of the study attributed to limbic D3 receptor stimulation.14
4 Sleep and excessive daytime sleepiness. Sleep disturbances are very common in individuals with PD.15 A community study found that two-thirds of PD patients complained of sleep problems, with sleep fragmentation and early awakening being the most common complaints.16 Initial insomnia was less common, and a surprisingly high number of PD patients reported symptoms that suggested obstructive sleep apnea, periodic limb movements of sleep, and REM sleep behavior disorder.17
Excessive daytime sleepiness has been associated with PD and with the medications used to treat it. Give special consideration to diagnosing sleep attacks—abrupt, unavoidable transitions from wakefulness to sleep—which are reported in up to 30% of PD patients taking dopaminergic agonists. These attacks can occur during critical activities, such as driving,18 and likely are a class effect of dopamine replacement therapies.19
5 Psychomotor retardation as a core symptom of PD is clinically indistinguishable from that seen in severe depression.
6 Fatigue. Most studies of fatigue in PD do not define whether the term applies to prolonged mental exhaustion or lack of physical endurance. In any case, one-third to one-half of PD patients report fatigue, and many consider it one of the most disabling symptoms—worse in this regard than motor symptoms.20 Fatigue is more than twice as common in PD patients as in healthy controls and is associated with depression, dementia, disease severity, disease duration, levodopa dose, and use of sleep medications.21
7 Feeling worthless/excessive or inappropriate guilt. DSM-IV-TR defines this symptom as not merely self-reproach or guilt about being sick.6 Guilt or self-blame seem to be less common in PD depression compared with dysphoria, pessimism, and somatic symptoms.22 Nonetheless, feelings of decreased self-worth are common in PD patients, especially as the illness limits work, productivity at home, and social activities.
8 Concentration and decision-making. PD patients show cognitive changes such as difficulty in changing tasks and impaired executive function (planning, sequencing, and executing). In tasks of divided attention—such as “multitasking”—PD patients have difficulty filtering out nonrelevant information.23 Difficulties with memory, attention, and language also have been observed in PD and often are exacerbated by depression.24 These cognitive changes affect PD patients’ ability to concentrate, maintain focus, and engage in effective decision making.25
Attention problems in PD are compounded by dementia, which affects at least 20% to 40% of PD patients26 and perhaps considerably more.27
To study depression in PD patients, the NINDS/NIMH Work Group on Depression in Parkinson’s Disease4 recommended that researchers use DSM-IV-TR criteria for depression and count all overlapping depressive symptoms toward a depression diagnosis.
Unfortunately, this provisional recommendation—intended only to “provide a common starting point for clinical research in PD-associated depression”4—is not evidence-based, and its specificity and sensitivity are unknown. If you follow this recommendation in clinical practice, you might overdiagnose depression in PD patients by including false positives and nonsignificant cases.
Until these issues are clarified, we recommend that you focus on the most specific symptoms, such as mood, when assessing depression in PD patients.
Features of depression in PD
The specificity and clinical usefulness of individual depression symptoms in PD is variable. Some symptoms seem to be as common in nondepressed as in depressed PD patients (Table 2).29
Distinguishing characteristics. Using Hamilton Depression Rating Scale (HAM-D) and Montgomery-Åsburg Depression Rating Scale items, a study of nondemented PD patients found the presence of suicidal thoughts to be the most reliable discriminator between depressed and nondepressed patients. Other symptoms with good discriminating reliability for depression in PD were (in descending order):
- feelings of guilt
- psychic anxiety
- reduced appetite
- depressed mood
- reduction of work and interest.
Symptom profile. The most recent studies comparing depression symptoms in PD patients with those in non-PD populations seem to indicate:
- the profile of depression in PD is not different from that of other elderly depressed populations
- or PD patients show more cognitive symptoms, which is not surprising considering PD’s cognitive involvement.31
Psychiatric comorbidities. A relatively high association with anxiety, cognitive impairment, and psychosis also complicates depression’s picture in PD.32 Often this relationship seems to be bidirectional, with the comorbidities increasing the risk for depression and vice versa.
Table 2
Frequency of depressive symptoms in PD
| Effect | Symptoms |
|---|---|
| Significantly higher frequency in PD patients with depression | Worrying, brooding, loss of interest, hopelessness, suicidal tendencies, social withdrawal, self-depreciation, ideas of reference, anxiety symptoms, loss of appetite, initial and middle insomnia, loss of libido |
| No significant differences in frequency compared with PD patients without depression | Anergia, motor retardation, early morning awakening |
| PD: Parkinson’s disease | |
| Source: Reference 29 | |
Recommendations
As we have seen, depression’s somatic and cognitive symptoms and PD’s motor, somatic, and cognitive features overlap substantially. How, then, should clinicians handle symptoms that can be attributed to either depression or PD? Several approaches are possible (Table 3),33 and each has strengths and weaknesses.
An exclusionary approach may be indicated for research, whereas an inclusive approach may be better suited to clinical settings. As mentioned, the National Institute of Neurological Disorders and Stroke/National Institute of Mental Health Work Group on Depression in Parkinson’s Disease4 supports an inclusive approach when evaluating depression symptoms. This group (Box) also recommends eliminating the DSM-IV-TR general exclusion criterion “due to the effects of a medical condition” applied to the diagnosis of depression.4
As we have seen, however, most DSM-IV-TR depressive symptoms overlap with PD symptoms. The false-positive results likely to occur with an inclusive definition of depression might discourage clinicians from screening PD patients for depression.
In clinical practice, finding recent changes in these overlapping symptoms might point to depression. Therefore, try to establish recent changes—associated with depression—in a PD patient’s somatic or cognitive symptoms, such as weight loss, lack of interest, impaired concentration, or decreased energy. This may be difficult, however, given:
- the subjective nature of many of these symptoms
- the decreased reporting ability of patients with cognitive deterioration
- medical comorbidities in PD that also could produce the referred symptoms.
1. Mood. Try to differentiate pervasive depressed mood from mood fluctuations associated with motor fluctuations and poorly controlled motor symptoms. Start with simple, open-ended questions and progress toward precise estimates.
2. Interest. Depressive loss of interest may be more acute and fluctuating than apathy. Also, selective loss of interest in some areas—such as social life, work, or hobbies—as opposed to the pervasive character of apathy, may suggest depression.
When evaluating interest in PD patients, consider that they may be avoiding activities that interest them out of fear that motor impairment may cause poor performance or social embarrassment.
3. Weight/appetite. Appetite may be a better indicator of depression than weight changes, as weight loss seems to be common in PD patients. Keep in mind, however, that the GI side effects of dopaminergic medications may limit what patients can eat.
4. Insomnia/hypersomnia. Insomnia associated with PD is usually characterized by sleep maintenance problems (middle insomnia or “broken” sleep). Thus, initial and terminal insomnia are probably better indicators of the presence of depression.
5. Agitation/retardation. Psychomotor retardation is common in PD, but acute exacerbations associated with depression may be noticed. Also note that depression-associated anxiety may exacerbate dyskinesias.
Table 3
4 options for diagnosing depression in PD patients
| Approach | Definition | Comment |
|---|---|---|
| Inclusive | Count all depressive symptoms toward a depression diagnosis | Recommended by NINDS/NIMH Work Group on Depression in Parkinson’s Disease, but may result in overdiagnosis of depression in PD patients |
| Exclusive | Ignore any depressive symptoms that could otherwise be explained | May be indicated for research |
| Etiologic exclusive | Ignore symptoms that likely are the result of the medical illness | The NINDS/NIMH Work Group on Depression in Parkinson’s Disease recommends avoiding attributing symptoms to a particular cause |
| Substitutive | Replace the most confusing diagnostic features with others that are less controversial | Theoretically the best approach, but establishing this approach as evidence-based would require substantial research |
| PD: Parkinson’s disease; NINDS/NIMH: National Institute of Neurological Disorders and Stroke/National Institute of Mental Health | ||
| Source: References 4,33 | ||
7. Worthlessness/guilt. PD is an incapacitating illness that causes work, family, and social dysfunction. To count as a depression criterion, worthlessness and guilty feelings need to be excessive or inappropriate and relatively constant and not merely self-reproach or guilt about being sick.
8. Diminished ability to think and concentrate is another a symptom that is difficult to ascribe to either depression or PD. A recent change in the context of mood symptoms might point to depression.
9. Recurrent thoughts of death. As mentioned, suicide seems to be less common in patients with PD than in the general population, but suicidal ideation—when found—is highly specific. Fear of dying from PD is not considered a depressive criterion, however.
Related resources
- Menza M, Marsh L, eds. Psychiatric issues in Parkinson’s disease: a practical guide. New York: Taylor and Francis; 2006.
- Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
- Parkinson’s Disease Foundation: www.pdf.org.
- Michael J. Fox Foundation: www.michaeljfox.org.
Drug brand names
- Levodopa • Dopar, Larodopa
- Pramipexole • Mirapex
Dr. Marin reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Menza receives research support from the National Institutes of Health, Astra-Zeneca, Bristol-Myers Squibb, Cephalon, Forest Laboratories, GlaxoSmithKline, Janssen, Lilly, Merck, Pfizer, sanofi-aventis, Sepracor, Takeda, and Wyeth. He is a consultant for the National Institutes of Health, GlaxoSmithKline, Kyowa, Lilly Research Laboratories, Ono, Pfizer, Sepracor, and Takeda. He is a speaker for Bristol-Myers Squibb, Lilly Research Laboratories, Sepracor, sanofi-aventis, and Takeda.
Dr. Dobkin receives research grants from Takeda.
1. Marsh L. Neuropsychiatric aspects of Parkinson’s disease. Psychosomatics 2000;41:15-23.
2. Weintraub D, Moberg PJ, Duda JE, et al. Recognition and treatment of depression in Parkinson’s disease. J Geriatr Psychiatry Neurol 2003;16:178-83.
3. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992;55:377-82.
4. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
5. Cummings JL. Depression and Parkinson’s disease. Am J Psychiatry 1992;149:443-54.
6. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
7. Schrag A, Jahanshahi M, Quinn N. What contributes to quality of life in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 2000;69:308-12.
8. Kirsch-Darrow L, Fernandez HF, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33-8.
9. Pluck GC, Brown RG. Apathy in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2002;73:636-42.
10. Isella V, Iurlaro S, Piolti R, et al. Physical anhedonia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2003;74(9):1308-11.
11. Chen H, Zhang SM, Hernan MA, et al. Weight loss in Parkinson’s disease. Ann Neurol 2003;53:676-9.
12. Hu G, Jousilahti P, Nissinen A, et al. Body mass index and the risk for Parkinson’s disease. Neurology 2006;67:1955-9.
13. Palhagen S, Lorefait B, Carlsson M, et al. Does L-dopa treatment contribute to reduction in body weight in elderly patients with Parkinson’s disease? Acta Neurol Scand 2005;111:12-20.
14. Kumru H, Santamaria J, Valldeoriola F, et al. Increase in body weight after pramipexole treatment in Parkinson’s disease. Mov Disord 2006;21:1972-4.
15. Lees A, Blackburn N, Campbell V. The nighttime problems of Parkinson’s disease. Clin Neuropharmacol 1988;11:512-9.
16. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson’s disease. Mov Disord 1998;13:895-9.
17. Oerlemans WGH, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson’s disease: a self-reported, community-based study. Sleep Med 2002;3:147-9.
18. Frucht S, Rogers JD, Geen P, et al. Falling asleep at the wheel: motor vehicle mishaps in persons taking pramipexole and ropinirole. Neurology 2003;61:40-5.
19. Homann CN, Wenzel K, Suppan A, et al. Sleep attacks—facts and fiction: a critical review. Adv Neurol 2003;91:335-41
20. Friedman JH, Brown RG, Comella C, et al. Fatigue in Parkinson’s disease: a review. Mov Disord 2007;22(3):297-308.
21. Karlsen K, Larsen JP, Tandberg E, et al. Fatigue in patients with Parkinson’s disease. Mov Disord 1999;14(2):237-41.
22. Brown RG, MacCarthy B, Gotham AM, et al. Depression and disability in Parkinson’s disease: a follow-up of 132 cases. Psychol Med 1988;18(1):49-55.
23. Zgaljardic DJ, Borod JC, Foldi NS, et al. A review of the cognitive and behavioral sequelae of Parkinson’s disease: relationship to frontostriatal circuitry. Cogn Behav Neurol 2003;16(4):193-210.
24. Kuzis G, Sabe L, Tiberti C, et al. Cognitive functions in major depression and Parkinson’s disease. Arch Neurol 1997;54(8):982-6.
25. Mimura M, Oeda R, Kawamura M. Impaired decision-making in Parkinson’s disease. Parkinsonism Relat Disord 2006;12:169-75.
26. Marder K, Jacobs DM. Dementia. In: Factor SA, Weiner WJ, eds. Parkinson’s disease: diagnosis and clinical management. New York: Demos Medical Publishing; 2002.
27. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60(3):387-92.
28. Myslobodsky M, Lalonde FM, Hicks L. Are patients with Parkinson’s disease suicidal? J Geriatr Pscyhiatr Neurol 2001;14(3)120:4.-
29. Starkstein SE, Preziosi TJ, Forrester AW, et al. Specificity of affective and autonomic symptoms of depression in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1990;53:869-73.
30. Leentjens AFG, Marinus J, Van Hilten JJ, et al. The contribution of somatic symptoms to the diagnosis of depressive disorder in Parkinson’s disease: a discriminant analytic approach. J Neuropsychiatry Clin Neurosci 2003;15(1):74-7.
31. Erdal KJ. Depressive symptom patterns in patients with Parkinson’s disease and other older adults. J Clin Psychol 2001;57(12):1559-69.
32. Marsh L, Williams JR, Rocco M, et al. Psychiatric comorbidities in patients with Parkinson disease and psychosis. Neurology 2004;63(2):293-300.
33. Koenig HG, George LG, Peterson BL, et al. Depression in medically ill hospitalized older adults: prevalence, characteristics, and course of symptoms according to six diagnostic schemes. Am J Psychiatry 1997;154:1376-83.
Many depressive symptoms are seen in the normal course of Parkinson’s disease (PD) (Table 1).1 As a result, depression—the most common neuropsychiatric disturbance in PD—is difficult to assess in PD and easily can go undetected and untreated.2
Making the diagnosis is important, however, because depression causes PD patients suffering and may accelerate decline in motor and cognitive function, activities of daily living, and quality of life.3 In the absence of specific guidelines (Box),4 we provide evidence to help you sort through the overlapping symptoms to find clinical signs that differentiate depression from PD symptoms.5,6
Table 1
Symptoms of depression that occur in or mimic those in the natural course of PD
| Psychomotor retardation (bradykinesia) |
| Depressed or emotionless appearance (‘masked facies,’ stooped posture) |
| Agitation (dyskinesias) |
| Decreased interest and enjoyment (apathy and decreased initiative) |
| Impaired memory and concentration |
| Fatigue or decreased energy |
| Impaired sleep |
| Weight and appetite changes |
| Physical complaints |
| Source: Adapted from reference 1 |
DSM-IV-TR depression criteria
Approximately 20% of PD patients meet DSM-IV-TR criteria for major depression, and another 20% meet criteria for dysthymia.5 By DSM-IV-TR criteria,6 diagnosis of a major depressive episode requires ≥5 of 9 symptoms, of which at least 1 is depressed mood or loss of interest or pleasure. Because these symptoms must be present during the same 2-week period and represent a change in functioning, this diagnosis has an acute quality.
Dysthymia—also frequently called “chronic depression”—is characterized by a mostly depressed mood for 2 years, accompanied by ≥2 of 6 symptoms: appetite changes, sleep changes, low energy/fatigue, low self-esteem, poor concentration/indecisiveness, and hopelessness.6
All of these depression symptoms may overlap with those of PD.
1 Mood. In mid-stage and late PD, mood often fluctuates in concert with daily periods of increased rigidity and tremor (“off” periods) interspersed with improved motor functioning (“on” periods).7 Thus, when evaluating the PD patient:
- take a detailed history of motor fluctuations and their associations with mood symptoms
- also evaluate mood during “on” periods.
- behavioral (lack of effort)
- cognitive (loss of interest/concern)
- affective (decreased emotional response or “flat” affect).
3 Weight changes. Patients with PD tend to have lower body weight than matched subjects. As a result, weight loss in the course of PD can be confused with weight loss associated with depression.
Weight loss appears to start 2 to 4 years before a PD diagnosis and continues thereafter. Despite the weight loss, PD patients report higher energy intake after the diagnosis compared with individuals without PD.11 A related, not necessarily contradictory finding is that a higher premorbid body mass index (BMI) seems to be associated with an increased risk of developing PD.12
In general, dopaminergic treatment of PD seems to be associated with weight loss.13 However, weight gain has been reported after pramipexole treatment, which the authors of the study attributed to limbic D3 receptor stimulation.14
4 Sleep and excessive daytime sleepiness. Sleep disturbances are very common in individuals with PD.15 A community study found that two-thirds of PD patients complained of sleep problems, with sleep fragmentation and early awakening being the most common complaints.16 Initial insomnia was less common, and a surprisingly high number of PD patients reported symptoms that suggested obstructive sleep apnea, periodic limb movements of sleep, and REM sleep behavior disorder.17
Excessive daytime sleepiness has been associated with PD and with the medications used to treat it. Give special consideration to diagnosing sleep attacks—abrupt, unavoidable transitions from wakefulness to sleep—which are reported in up to 30% of PD patients taking dopaminergic agonists. These attacks can occur during critical activities, such as driving,18 and likely are a class effect of dopamine replacement therapies.19
5 Psychomotor retardation as a core symptom of PD is clinically indistinguishable from that seen in severe depression.
6 Fatigue. Most studies of fatigue in PD do not define whether the term applies to prolonged mental exhaustion or lack of physical endurance. In any case, one-third to one-half of PD patients report fatigue, and many consider it one of the most disabling symptoms—worse in this regard than motor symptoms.20 Fatigue is more than twice as common in PD patients as in healthy controls and is associated with depression, dementia, disease severity, disease duration, levodopa dose, and use of sleep medications.21
7 Feeling worthless/excessive or inappropriate guilt. DSM-IV-TR defines this symptom as not merely self-reproach or guilt about being sick.6 Guilt or self-blame seem to be less common in PD depression compared with dysphoria, pessimism, and somatic symptoms.22 Nonetheless, feelings of decreased self-worth are common in PD patients, especially as the illness limits work, productivity at home, and social activities.
8 Concentration and decision-making. PD patients show cognitive changes such as difficulty in changing tasks and impaired executive function (planning, sequencing, and executing). In tasks of divided attention—such as “multitasking”—PD patients have difficulty filtering out nonrelevant information.23 Difficulties with memory, attention, and language also have been observed in PD and often are exacerbated by depression.24 These cognitive changes affect PD patients’ ability to concentrate, maintain focus, and engage in effective decision making.25
Attention problems in PD are compounded by dementia, which affects at least 20% to 40% of PD patients26 and perhaps considerably more.27
To study depression in PD patients, the NINDS/NIMH Work Group on Depression in Parkinson’s Disease4 recommended that researchers use DSM-IV-TR criteria for depression and count all overlapping depressive symptoms toward a depression diagnosis.
Unfortunately, this provisional recommendation—intended only to “provide a common starting point for clinical research in PD-associated depression”4—is not evidence-based, and its specificity and sensitivity are unknown. If you follow this recommendation in clinical practice, you might overdiagnose depression in PD patients by including false positives and nonsignificant cases.
Until these issues are clarified, we recommend that you focus on the most specific symptoms, such as mood, when assessing depression in PD patients.
Features of depression in PD
The specificity and clinical usefulness of individual depression symptoms in PD is variable. Some symptoms seem to be as common in nondepressed as in depressed PD patients (Table 2).29
Distinguishing characteristics. Using Hamilton Depression Rating Scale (HAM-D) and Montgomery-Åsburg Depression Rating Scale items, a study of nondemented PD patients found the presence of suicidal thoughts to be the most reliable discriminator between depressed and nondepressed patients. Other symptoms with good discriminating reliability for depression in PD were (in descending order):
- feelings of guilt
- psychic anxiety
- reduced appetite
- depressed mood
- reduction of work and interest.
Symptom profile. The most recent studies comparing depression symptoms in PD patients with those in non-PD populations seem to indicate:
- the profile of depression in PD is not different from that of other elderly depressed populations
- or PD patients show more cognitive symptoms, which is not surprising considering PD’s cognitive involvement.31
Psychiatric comorbidities. A relatively high association with anxiety, cognitive impairment, and psychosis also complicates depression’s picture in PD.32 Often this relationship seems to be bidirectional, with the comorbidities increasing the risk for depression and vice versa.
Table 2
Frequency of depressive symptoms in PD
| Effect | Symptoms |
|---|---|
| Significantly higher frequency in PD patients with depression | Worrying, brooding, loss of interest, hopelessness, suicidal tendencies, social withdrawal, self-depreciation, ideas of reference, anxiety symptoms, loss of appetite, initial and middle insomnia, loss of libido |
| No significant differences in frequency compared with PD patients without depression | Anergia, motor retardation, early morning awakening |
| PD: Parkinson’s disease | |
| Source: Reference 29 | |
Recommendations
As we have seen, depression’s somatic and cognitive symptoms and PD’s motor, somatic, and cognitive features overlap substantially. How, then, should clinicians handle symptoms that can be attributed to either depression or PD? Several approaches are possible (Table 3),33 and each has strengths and weaknesses.
An exclusionary approach may be indicated for research, whereas an inclusive approach may be better suited to clinical settings. As mentioned, the National Institute of Neurological Disorders and Stroke/National Institute of Mental Health Work Group on Depression in Parkinson’s Disease4 supports an inclusive approach when evaluating depression symptoms. This group (Box) also recommends eliminating the DSM-IV-TR general exclusion criterion “due to the effects of a medical condition” applied to the diagnosis of depression.4
As we have seen, however, most DSM-IV-TR depressive symptoms overlap with PD symptoms. The false-positive results likely to occur with an inclusive definition of depression might discourage clinicians from screening PD patients for depression.
In clinical practice, finding recent changes in these overlapping symptoms might point to depression. Therefore, try to establish recent changes—associated with depression—in a PD patient’s somatic or cognitive symptoms, such as weight loss, lack of interest, impaired concentration, or decreased energy. This may be difficult, however, given:
- the subjective nature of many of these symptoms
- the decreased reporting ability of patients with cognitive deterioration
- medical comorbidities in PD that also could produce the referred symptoms.
1. Mood. Try to differentiate pervasive depressed mood from mood fluctuations associated with motor fluctuations and poorly controlled motor symptoms. Start with simple, open-ended questions and progress toward precise estimates.
2. Interest. Depressive loss of interest may be more acute and fluctuating than apathy. Also, selective loss of interest in some areas—such as social life, work, or hobbies—as opposed to the pervasive character of apathy, may suggest depression.
When evaluating interest in PD patients, consider that they may be avoiding activities that interest them out of fear that motor impairment may cause poor performance or social embarrassment.
3. Weight/appetite. Appetite may be a better indicator of depression than weight changes, as weight loss seems to be common in PD patients. Keep in mind, however, that the GI side effects of dopaminergic medications may limit what patients can eat.
4. Insomnia/hypersomnia. Insomnia associated with PD is usually characterized by sleep maintenance problems (middle insomnia or “broken” sleep). Thus, initial and terminal insomnia are probably better indicators of the presence of depression.
5. Agitation/retardation. Psychomotor retardation is common in PD, but acute exacerbations associated with depression may be noticed. Also note that depression-associated anxiety may exacerbate dyskinesias.
Table 3
4 options for diagnosing depression in PD patients
| Approach | Definition | Comment |
|---|---|---|
| Inclusive | Count all depressive symptoms toward a depression diagnosis | Recommended by NINDS/NIMH Work Group on Depression in Parkinson’s Disease, but may result in overdiagnosis of depression in PD patients |
| Exclusive | Ignore any depressive symptoms that could otherwise be explained | May be indicated for research |
| Etiologic exclusive | Ignore symptoms that likely are the result of the medical illness | The NINDS/NIMH Work Group on Depression in Parkinson’s Disease recommends avoiding attributing symptoms to a particular cause |
| Substitutive | Replace the most confusing diagnostic features with others that are less controversial | Theoretically the best approach, but establishing this approach as evidence-based would require substantial research |
| PD: Parkinson’s disease; NINDS/NIMH: National Institute of Neurological Disorders and Stroke/National Institute of Mental Health | ||
| Source: References 4,33 | ||
7. Worthlessness/guilt. PD is an incapacitating illness that causes work, family, and social dysfunction. To count as a depression criterion, worthlessness and guilty feelings need to be excessive or inappropriate and relatively constant and not merely self-reproach or guilt about being sick.
8. Diminished ability to think and concentrate is another a symptom that is difficult to ascribe to either depression or PD. A recent change in the context of mood symptoms might point to depression.
9. Recurrent thoughts of death. As mentioned, suicide seems to be less common in patients with PD than in the general population, but suicidal ideation—when found—is highly specific. Fear of dying from PD is not considered a depressive criterion, however.
Related resources
- Menza M, Marsh L, eds. Psychiatric issues in Parkinson’s disease: a practical guide. New York: Taylor and Francis; 2006.
- Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
- Parkinson’s Disease Foundation: www.pdf.org.
- Michael J. Fox Foundation: www.michaeljfox.org.
Drug brand names
- Levodopa • Dopar, Larodopa
- Pramipexole • Mirapex
Dr. Marin reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Menza receives research support from the National Institutes of Health, Astra-Zeneca, Bristol-Myers Squibb, Cephalon, Forest Laboratories, GlaxoSmithKline, Janssen, Lilly, Merck, Pfizer, sanofi-aventis, Sepracor, Takeda, and Wyeth. He is a consultant for the National Institutes of Health, GlaxoSmithKline, Kyowa, Lilly Research Laboratories, Ono, Pfizer, Sepracor, and Takeda. He is a speaker for Bristol-Myers Squibb, Lilly Research Laboratories, Sepracor, sanofi-aventis, and Takeda.
Dr. Dobkin receives research grants from Takeda.
Many depressive symptoms are seen in the normal course of Parkinson’s disease (PD) (Table 1).1 As a result, depression—the most common neuropsychiatric disturbance in PD—is difficult to assess in PD and easily can go undetected and untreated.2
Making the diagnosis is important, however, because depression causes PD patients suffering and may accelerate decline in motor and cognitive function, activities of daily living, and quality of life.3 In the absence of specific guidelines (Box),4 we provide evidence to help you sort through the overlapping symptoms to find clinical signs that differentiate depression from PD symptoms.5,6
Table 1
Symptoms of depression that occur in or mimic those in the natural course of PD
| Psychomotor retardation (bradykinesia) |
| Depressed or emotionless appearance (‘masked facies,’ stooped posture) |
| Agitation (dyskinesias) |
| Decreased interest and enjoyment (apathy and decreased initiative) |
| Impaired memory and concentration |
| Fatigue or decreased energy |
| Impaired sleep |
| Weight and appetite changes |
| Physical complaints |
| Source: Adapted from reference 1 |
DSM-IV-TR depression criteria
Approximately 20% of PD patients meet DSM-IV-TR criteria for major depression, and another 20% meet criteria for dysthymia.5 By DSM-IV-TR criteria,6 diagnosis of a major depressive episode requires ≥5 of 9 symptoms, of which at least 1 is depressed mood or loss of interest or pleasure. Because these symptoms must be present during the same 2-week period and represent a change in functioning, this diagnosis has an acute quality.
Dysthymia—also frequently called “chronic depression”—is characterized by a mostly depressed mood for 2 years, accompanied by ≥2 of 6 symptoms: appetite changes, sleep changes, low energy/fatigue, low self-esteem, poor concentration/indecisiveness, and hopelessness.6
All of these depression symptoms may overlap with those of PD.
1 Mood. In mid-stage and late PD, mood often fluctuates in concert with daily periods of increased rigidity and tremor (“off” periods) interspersed with improved motor functioning (“on” periods).7 Thus, when evaluating the PD patient:
- take a detailed history of motor fluctuations and their associations with mood symptoms
- also evaluate mood during “on” periods.
- behavioral (lack of effort)
- cognitive (loss of interest/concern)
- affective (decreased emotional response or “flat” affect).
3 Weight changes. Patients with PD tend to have lower body weight than matched subjects. As a result, weight loss in the course of PD can be confused with weight loss associated with depression.
Weight loss appears to start 2 to 4 years before a PD diagnosis and continues thereafter. Despite the weight loss, PD patients report higher energy intake after the diagnosis compared with individuals without PD.11 A related, not necessarily contradictory finding is that a higher premorbid body mass index (BMI) seems to be associated with an increased risk of developing PD.12
In general, dopaminergic treatment of PD seems to be associated with weight loss.13 However, weight gain has been reported after pramipexole treatment, which the authors of the study attributed to limbic D3 receptor stimulation.14
4 Sleep and excessive daytime sleepiness. Sleep disturbances are very common in individuals with PD.15 A community study found that two-thirds of PD patients complained of sleep problems, with sleep fragmentation and early awakening being the most common complaints.16 Initial insomnia was less common, and a surprisingly high number of PD patients reported symptoms that suggested obstructive sleep apnea, periodic limb movements of sleep, and REM sleep behavior disorder.17
Excessive daytime sleepiness has been associated with PD and with the medications used to treat it. Give special consideration to diagnosing sleep attacks—abrupt, unavoidable transitions from wakefulness to sleep—which are reported in up to 30% of PD patients taking dopaminergic agonists. These attacks can occur during critical activities, such as driving,18 and likely are a class effect of dopamine replacement therapies.19
5 Psychomotor retardation as a core symptom of PD is clinically indistinguishable from that seen in severe depression.
6 Fatigue. Most studies of fatigue in PD do not define whether the term applies to prolonged mental exhaustion or lack of physical endurance. In any case, one-third to one-half of PD patients report fatigue, and many consider it one of the most disabling symptoms—worse in this regard than motor symptoms.20 Fatigue is more than twice as common in PD patients as in healthy controls and is associated with depression, dementia, disease severity, disease duration, levodopa dose, and use of sleep medications.21
7 Feeling worthless/excessive or inappropriate guilt. DSM-IV-TR defines this symptom as not merely self-reproach or guilt about being sick.6 Guilt or self-blame seem to be less common in PD depression compared with dysphoria, pessimism, and somatic symptoms.22 Nonetheless, feelings of decreased self-worth are common in PD patients, especially as the illness limits work, productivity at home, and social activities.
8 Concentration and decision-making. PD patients show cognitive changes such as difficulty in changing tasks and impaired executive function (planning, sequencing, and executing). In tasks of divided attention—such as “multitasking”—PD patients have difficulty filtering out nonrelevant information.23 Difficulties with memory, attention, and language also have been observed in PD and often are exacerbated by depression.24 These cognitive changes affect PD patients’ ability to concentrate, maintain focus, and engage in effective decision making.25
Attention problems in PD are compounded by dementia, which affects at least 20% to 40% of PD patients26 and perhaps considerably more.27
To study depression in PD patients, the NINDS/NIMH Work Group on Depression in Parkinson’s Disease4 recommended that researchers use DSM-IV-TR criteria for depression and count all overlapping depressive symptoms toward a depression diagnosis.
Unfortunately, this provisional recommendation—intended only to “provide a common starting point for clinical research in PD-associated depression”4—is not evidence-based, and its specificity and sensitivity are unknown. If you follow this recommendation in clinical practice, you might overdiagnose depression in PD patients by including false positives and nonsignificant cases.
Until these issues are clarified, we recommend that you focus on the most specific symptoms, such as mood, when assessing depression in PD patients.
Features of depression in PD
The specificity and clinical usefulness of individual depression symptoms in PD is variable. Some symptoms seem to be as common in nondepressed as in depressed PD patients (Table 2).29
Distinguishing characteristics. Using Hamilton Depression Rating Scale (HAM-D) and Montgomery-Åsburg Depression Rating Scale items, a study of nondemented PD patients found the presence of suicidal thoughts to be the most reliable discriminator between depressed and nondepressed patients. Other symptoms with good discriminating reliability for depression in PD were (in descending order):
- feelings of guilt
- psychic anxiety
- reduced appetite
- depressed mood
- reduction of work and interest.
Symptom profile. The most recent studies comparing depression symptoms in PD patients with those in non-PD populations seem to indicate:
- the profile of depression in PD is not different from that of other elderly depressed populations
- or PD patients show more cognitive symptoms, which is not surprising considering PD’s cognitive involvement.31
Psychiatric comorbidities. A relatively high association with anxiety, cognitive impairment, and psychosis also complicates depression’s picture in PD.32 Often this relationship seems to be bidirectional, with the comorbidities increasing the risk for depression and vice versa.
Table 2
Frequency of depressive symptoms in PD
| Effect | Symptoms |
|---|---|
| Significantly higher frequency in PD patients with depression | Worrying, brooding, loss of interest, hopelessness, suicidal tendencies, social withdrawal, self-depreciation, ideas of reference, anxiety symptoms, loss of appetite, initial and middle insomnia, loss of libido |
| No significant differences in frequency compared with PD patients without depression | Anergia, motor retardation, early morning awakening |
| PD: Parkinson’s disease | |
| Source: Reference 29 | |
Recommendations
As we have seen, depression’s somatic and cognitive symptoms and PD’s motor, somatic, and cognitive features overlap substantially. How, then, should clinicians handle symptoms that can be attributed to either depression or PD? Several approaches are possible (Table 3),33 and each has strengths and weaknesses.
An exclusionary approach may be indicated for research, whereas an inclusive approach may be better suited to clinical settings. As mentioned, the National Institute of Neurological Disorders and Stroke/National Institute of Mental Health Work Group on Depression in Parkinson’s Disease4 supports an inclusive approach when evaluating depression symptoms. This group (Box) also recommends eliminating the DSM-IV-TR general exclusion criterion “due to the effects of a medical condition” applied to the diagnosis of depression.4
As we have seen, however, most DSM-IV-TR depressive symptoms overlap with PD symptoms. The false-positive results likely to occur with an inclusive definition of depression might discourage clinicians from screening PD patients for depression.
In clinical practice, finding recent changes in these overlapping symptoms might point to depression. Therefore, try to establish recent changes—associated with depression—in a PD patient’s somatic or cognitive symptoms, such as weight loss, lack of interest, impaired concentration, or decreased energy. This may be difficult, however, given:
- the subjective nature of many of these symptoms
- the decreased reporting ability of patients with cognitive deterioration
- medical comorbidities in PD that also could produce the referred symptoms.
1. Mood. Try to differentiate pervasive depressed mood from mood fluctuations associated with motor fluctuations and poorly controlled motor symptoms. Start with simple, open-ended questions and progress toward precise estimates.
2. Interest. Depressive loss of interest may be more acute and fluctuating than apathy. Also, selective loss of interest in some areas—such as social life, work, or hobbies—as opposed to the pervasive character of apathy, may suggest depression.
When evaluating interest in PD patients, consider that they may be avoiding activities that interest them out of fear that motor impairment may cause poor performance or social embarrassment.
3. Weight/appetite. Appetite may be a better indicator of depression than weight changes, as weight loss seems to be common in PD patients. Keep in mind, however, that the GI side effects of dopaminergic medications may limit what patients can eat.
4. Insomnia/hypersomnia. Insomnia associated with PD is usually characterized by sleep maintenance problems (middle insomnia or “broken” sleep). Thus, initial and terminal insomnia are probably better indicators of the presence of depression.
5. Agitation/retardation. Psychomotor retardation is common in PD, but acute exacerbations associated with depression may be noticed. Also note that depression-associated anxiety may exacerbate dyskinesias.
Table 3
4 options for diagnosing depression in PD patients
| Approach | Definition | Comment |
|---|---|---|
| Inclusive | Count all depressive symptoms toward a depression diagnosis | Recommended by NINDS/NIMH Work Group on Depression in Parkinson’s Disease, but may result in overdiagnosis of depression in PD patients |
| Exclusive | Ignore any depressive symptoms that could otherwise be explained | May be indicated for research |
| Etiologic exclusive | Ignore symptoms that likely are the result of the medical illness | The NINDS/NIMH Work Group on Depression in Parkinson’s Disease recommends avoiding attributing symptoms to a particular cause |
| Substitutive | Replace the most confusing diagnostic features with others that are less controversial | Theoretically the best approach, but establishing this approach as evidence-based would require substantial research |
| PD: Parkinson’s disease; NINDS/NIMH: National Institute of Neurological Disorders and Stroke/National Institute of Mental Health | ||
| Source: References 4,33 | ||
7. Worthlessness/guilt. PD is an incapacitating illness that causes work, family, and social dysfunction. To count as a depression criterion, worthlessness and guilty feelings need to be excessive or inappropriate and relatively constant and not merely self-reproach or guilt about being sick.
8. Diminished ability to think and concentrate is another a symptom that is difficult to ascribe to either depression or PD. A recent change in the context of mood symptoms might point to depression.
9. Recurrent thoughts of death. As mentioned, suicide seems to be less common in patients with PD than in the general population, but suicidal ideation—when found—is highly specific. Fear of dying from PD is not considered a depressive criterion, however.
Related resources
- Menza M, Marsh L, eds. Psychiatric issues in Parkinson’s disease: a practical guide. New York: Taylor and Francis; 2006.
- Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
- Parkinson’s Disease Foundation: www.pdf.org.
- Michael J. Fox Foundation: www.michaeljfox.org.
Drug brand names
- Levodopa • Dopar, Larodopa
- Pramipexole • Mirapex
Dr. Marin reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Menza receives research support from the National Institutes of Health, Astra-Zeneca, Bristol-Myers Squibb, Cephalon, Forest Laboratories, GlaxoSmithKline, Janssen, Lilly, Merck, Pfizer, sanofi-aventis, Sepracor, Takeda, and Wyeth. He is a consultant for the National Institutes of Health, GlaxoSmithKline, Kyowa, Lilly Research Laboratories, Ono, Pfizer, Sepracor, and Takeda. He is a speaker for Bristol-Myers Squibb, Lilly Research Laboratories, Sepracor, sanofi-aventis, and Takeda.
Dr. Dobkin receives research grants from Takeda.
1. Marsh L. Neuropsychiatric aspects of Parkinson’s disease. Psychosomatics 2000;41:15-23.
2. Weintraub D, Moberg PJ, Duda JE, et al. Recognition and treatment of depression in Parkinson’s disease. J Geriatr Psychiatry Neurol 2003;16:178-83.
3. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992;55:377-82.
4. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
5. Cummings JL. Depression and Parkinson’s disease. Am J Psychiatry 1992;149:443-54.
6. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
7. Schrag A, Jahanshahi M, Quinn N. What contributes to quality of life in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 2000;69:308-12.
8. Kirsch-Darrow L, Fernandez HF, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33-8.
9. Pluck GC, Brown RG. Apathy in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2002;73:636-42.
10. Isella V, Iurlaro S, Piolti R, et al. Physical anhedonia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2003;74(9):1308-11.
11. Chen H, Zhang SM, Hernan MA, et al. Weight loss in Parkinson’s disease. Ann Neurol 2003;53:676-9.
12. Hu G, Jousilahti P, Nissinen A, et al. Body mass index and the risk for Parkinson’s disease. Neurology 2006;67:1955-9.
13. Palhagen S, Lorefait B, Carlsson M, et al. Does L-dopa treatment contribute to reduction in body weight in elderly patients with Parkinson’s disease? Acta Neurol Scand 2005;111:12-20.
14. Kumru H, Santamaria J, Valldeoriola F, et al. Increase in body weight after pramipexole treatment in Parkinson’s disease. Mov Disord 2006;21:1972-4.
15. Lees A, Blackburn N, Campbell V. The nighttime problems of Parkinson’s disease. Clin Neuropharmacol 1988;11:512-9.
16. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson’s disease. Mov Disord 1998;13:895-9.
17. Oerlemans WGH, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson’s disease: a self-reported, community-based study. Sleep Med 2002;3:147-9.
18. Frucht S, Rogers JD, Geen P, et al. Falling asleep at the wheel: motor vehicle mishaps in persons taking pramipexole and ropinirole. Neurology 2003;61:40-5.
19. Homann CN, Wenzel K, Suppan A, et al. Sleep attacks—facts and fiction: a critical review. Adv Neurol 2003;91:335-41
20. Friedman JH, Brown RG, Comella C, et al. Fatigue in Parkinson’s disease: a review. Mov Disord 2007;22(3):297-308.
21. Karlsen K, Larsen JP, Tandberg E, et al. Fatigue in patients with Parkinson’s disease. Mov Disord 1999;14(2):237-41.
22. Brown RG, MacCarthy B, Gotham AM, et al. Depression and disability in Parkinson’s disease: a follow-up of 132 cases. Psychol Med 1988;18(1):49-55.
23. Zgaljardic DJ, Borod JC, Foldi NS, et al. A review of the cognitive and behavioral sequelae of Parkinson’s disease: relationship to frontostriatal circuitry. Cogn Behav Neurol 2003;16(4):193-210.
24. Kuzis G, Sabe L, Tiberti C, et al. Cognitive functions in major depression and Parkinson’s disease. Arch Neurol 1997;54(8):982-6.
25. Mimura M, Oeda R, Kawamura M. Impaired decision-making in Parkinson’s disease. Parkinsonism Relat Disord 2006;12:169-75.
26. Marder K, Jacobs DM. Dementia. In: Factor SA, Weiner WJ, eds. Parkinson’s disease: diagnosis and clinical management. New York: Demos Medical Publishing; 2002.
27. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60(3):387-92.
28. Myslobodsky M, Lalonde FM, Hicks L. Are patients with Parkinson’s disease suicidal? J Geriatr Pscyhiatr Neurol 2001;14(3)120:4.-
29. Starkstein SE, Preziosi TJ, Forrester AW, et al. Specificity of affective and autonomic symptoms of depression in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1990;53:869-73.
30. Leentjens AFG, Marinus J, Van Hilten JJ, et al. The contribution of somatic symptoms to the diagnosis of depressive disorder in Parkinson’s disease: a discriminant analytic approach. J Neuropsychiatry Clin Neurosci 2003;15(1):74-7.
31. Erdal KJ. Depressive symptom patterns in patients with Parkinson’s disease and other older adults. J Clin Psychol 2001;57(12):1559-69.
32. Marsh L, Williams JR, Rocco M, et al. Psychiatric comorbidities in patients with Parkinson disease and psychosis. Neurology 2004;63(2):293-300.
33. Koenig HG, George LG, Peterson BL, et al. Depression in medically ill hospitalized older adults: prevalence, characteristics, and course of symptoms according to six diagnostic schemes. Am J Psychiatry 1997;154:1376-83.
1. Marsh L. Neuropsychiatric aspects of Parkinson’s disease. Psychosomatics 2000;41:15-23.
2. Weintraub D, Moberg PJ, Duda JE, et al. Recognition and treatment of depression in Parkinson’s disease. J Geriatr Psychiatry Neurol 2003;16:178-83.
3. Starkstein SE, Mayberg HS, Leiguarda R, et al. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992;55:377-82.
4. Marsh L, McDonald WM, Cummings J, et al. Provisional diagnostic criteria for depression in Parkinson’s disease: report of an NINDS/NIMH work group. Mov Disord 2006;21:148-58.
5. Cummings JL. Depression and Parkinson’s disease. Am J Psychiatry 1992;149:443-54.
6. Diagnostic and statistical manual of disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
7. Schrag A, Jahanshahi M, Quinn N. What contributes to quality of life in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry 2000;69:308-12.
8. Kirsch-Darrow L, Fernandez HF, Marsiske M, et al. Dissociating apathy and depression in Parkinson disease. Neurology 2006;67:33-8.
9. Pluck GC, Brown RG. Apathy in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2002;73:636-42.
10. Isella V, Iurlaro S, Piolti R, et al. Physical anhedonia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2003;74(9):1308-11.
11. Chen H, Zhang SM, Hernan MA, et al. Weight loss in Parkinson’s disease. Ann Neurol 2003;53:676-9.
12. Hu G, Jousilahti P, Nissinen A, et al. Body mass index and the risk for Parkinson’s disease. Neurology 2006;67:1955-9.
13. Palhagen S, Lorefait B, Carlsson M, et al. Does L-dopa treatment contribute to reduction in body weight in elderly patients with Parkinson’s disease? Acta Neurol Scand 2005;111:12-20.
14. Kumru H, Santamaria J, Valldeoriola F, et al. Increase in body weight after pramipexole treatment in Parkinson’s disease. Mov Disord 2006;21:1972-4.
15. Lees A, Blackburn N, Campbell V. The nighttime problems of Parkinson’s disease. Clin Neuropharmacol 1988;11:512-9.
16. Tandberg E, Larsen JP, Karlsen K. A community-based study of sleep disorders in patients with Parkinson’s disease. Mov Disord 1998;13:895-9.
17. Oerlemans WGH, de Weerd AW. The prevalence of sleep disorders in patients with Parkinson’s disease: a self-reported, community-based study. Sleep Med 2002;3:147-9.
18. Frucht S, Rogers JD, Geen P, et al. Falling asleep at the wheel: motor vehicle mishaps in persons taking pramipexole and ropinirole. Neurology 2003;61:40-5.
19. Homann CN, Wenzel K, Suppan A, et al. Sleep attacks—facts and fiction: a critical review. Adv Neurol 2003;91:335-41
20. Friedman JH, Brown RG, Comella C, et al. Fatigue in Parkinson’s disease: a review. Mov Disord 2007;22(3):297-308.
21. Karlsen K, Larsen JP, Tandberg E, et al. Fatigue in patients with Parkinson’s disease. Mov Disord 1999;14(2):237-41.
22. Brown RG, MacCarthy B, Gotham AM, et al. Depression and disability in Parkinson’s disease: a follow-up of 132 cases. Psychol Med 1988;18(1):49-55.
23. Zgaljardic DJ, Borod JC, Foldi NS, et al. A review of the cognitive and behavioral sequelae of Parkinson’s disease: relationship to frontostriatal circuitry. Cogn Behav Neurol 2003;16(4):193-210.
24. Kuzis G, Sabe L, Tiberti C, et al. Cognitive functions in major depression and Parkinson’s disease. Arch Neurol 1997;54(8):982-6.
25. Mimura M, Oeda R, Kawamura M. Impaired decision-making in Parkinson’s disease. Parkinsonism Relat Disord 2006;12:169-75.
26. Marder K, Jacobs DM. Dementia. In: Factor SA, Weiner WJ, eds. Parkinson’s disease: diagnosis and clinical management. New York: Demos Medical Publishing; 2002.
27. Aarsland D, Andersen K, Larsen JP, et al. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60(3):387-92.
28. Myslobodsky M, Lalonde FM, Hicks L. Are patients with Parkinson’s disease suicidal? J Geriatr Pscyhiatr Neurol 2001;14(3)120:4.-
29. Starkstein SE, Preziosi TJ, Forrester AW, et al. Specificity of affective and autonomic symptoms of depression in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1990;53:869-73.
30. Leentjens AFG, Marinus J, Van Hilten JJ, et al. The contribution of somatic symptoms to the diagnosis of depressive disorder in Parkinson’s disease: a discriminant analytic approach. J Neuropsychiatry Clin Neurosci 2003;15(1):74-7.
31. Erdal KJ. Depressive symptom patterns in patients with Parkinson’s disease and other older adults. J Clin Psychol 2001;57(12):1559-69.
32. Marsh L, Williams JR, Rocco M, et al. Psychiatric comorbidities in patients with Parkinson disease and psychosis. Neurology 2004;63(2):293-300.
33. Koenig HG, George LG, Peterson BL, et al. Depression in medically ill hospitalized older adults: prevalence, characteristics, and course of symptoms according to six diagnostic schemes. Am J Psychiatry 1997;154:1376-83.
DAMA dispute
As one who has been practicing acute inpatient psychiatry for approximately 20 years, I have to take issue with “‘I want to leave now’: Handling discharge against medical advice” (Current Psychiatry, May 2007).
Psychiatrists’ power to involuntarily confine citizens needs to be exercised with the greatest care and sensitivity. Every case where a voluntary inpatient requests discharge needs to be evaluated in a careful and individualized manner that often has little to do with the items listed in the article as disqualifiers for discharge against medical advice (DAMA).
Specifically, I have seen numerous cases where patients with delusions, dementia, and even acute psychosis have been discharged despite the treatment team’s wish that they stay longer because the patients did not meet criteria for involuntary hospitalization in New York. I suspect laws in other states also would have mandated these patients’ discharge. The same situation has occurred with patients expressing homicidal or suicidal ideation. Many patients have chronic suicidal “ideation” but do not intend to act upon these thoughts.
I don’t think the table of patient characteristics that are risk factors for DAMA has much clinical value. In fact I would venture to say that a large percentage of DAMA patients have every one of the factors listed.
This is not to say that the decision to discharge these patients is made without great deliberation. The point is that looking at a few isolated symptoms often is a misleading oversimplification. The decision is a complex process focusing on acute suicidal, violent, or criminal potential that cannot be operationalized.
I agree with the authors that “DAMA does not absolve the physician of responsibility for poor out-comes.” The psychiatrist needs to carefully document the factors that lead to the decision to discharge a patient. The documentation needs to reflect that the DAMA decision was necessary given the state’s statutes.
Bennett Cohen, MD
New York, NY
As one who has been practicing acute inpatient psychiatry for approximately 20 years, I have to take issue with “‘I want to leave now’: Handling discharge against medical advice” (Current Psychiatry, May 2007).
Psychiatrists’ power to involuntarily confine citizens needs to be exercised with the greatest care and sensitivity. Every case where a voluntary inpatient requests discharge needs to be evaluated in a careful and individualized manner that often has little to do with the items listed in the article as disqualifiers for discharge against medical advice (DAMA).
Specifically, I have seen numerous cases where patients with delusions, dementia, and even acute psychosis have been discharged despite the treatment team’s wish that they stay longer because the patients did not meet criteria for involuntary hospitalization in New York. I suspect laws in other states also would have mandated these patients’ discharge. The same situation has occurred with patients expressing homicidal or suicidal ideation. Many patients have chronic suicidal “ideation” but do not intend to act upon these thoughts.
I don’t think the table of patient characteristics that are risk factors for DAMA has much clinical value. In fact I would venture to say that a large percentage of DAMA patients have every one of the factors listed.
This is not to say that the decision to discharge these patients is made without great deliberation. The point is that looking at a few isolated symptoms often is a misleading oversimplification. The decision is a complex process focusing on acute suicidal, violent, or criminal potential that cannot be operationalized.
I agree with the authors that “DAMA does not absolve the physician of responsibility for poor out-comes.” The psychiatrist needs to carefully document the factors that lead to the decision to discharge a patient. The documentation needs to reflect that the DAMA decision was necessary given the state’s statutes.
Bennett Cohen, MD
New York, NY
As one who has been practicing acute inpatient psychiatry for approximately 20 years, I have to take issue with “‘I want to leave now’: Handling discharge against medical advice” (Current Psychiatry, May 2007).
Psychiatrists’ power to involuntarily confine citizens needs to be exercised with the greatest care and sensitivity. Every case where a voluntary inpatient requests discharge needs to be evaluated in a careful and individualized manner that often has little to do with the items listed in the article as disqualifiers for discharge against medical advice (DAMA).
Specifically, I have seen numerous cases where patients with delusions, dementia, and even acute psychosis have been discharged despite the treatment team’s wish that they stay longer because the patients did not meet criteria for involuntary hospitalization in New York. I suspect laws in other states also would have mandated these patients’ discharge. The same situation has occurred with patients expressing homicidal or suicidal ideation. Many patients have chronic suicidal “ideation” but do not intend to act upon these thoughts.
I don’t think the table of patient characteristics that are risk factors for DAMA has much clinical value. In fact I would venture to say that a large percentage of DAMA patients have every one of the factors listed.
This is not to say that the decision to discharge these patients is made without great deliberation. The point is that looking at a few isolated symptoms often is a misleading oversimplification. The decision is a complex process focusing on acute suicidal, violent, or criminal potential that cannot be operationalized.
I agree with the authors that “DAMA does not absolve the physician of responsibility for poor out-comes.” The psychiatrist needs to carefully document the factors that lead to the decision to discharge a patient. The documentation needs to reflect that the DAMA decision was necessary given the state’s statutes.
Bennett Cohen, MD
New York, NY