User login
Research and Reviews for the Practicing Oncologist
Emerging biosimilars market presents opportunities and challenges
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
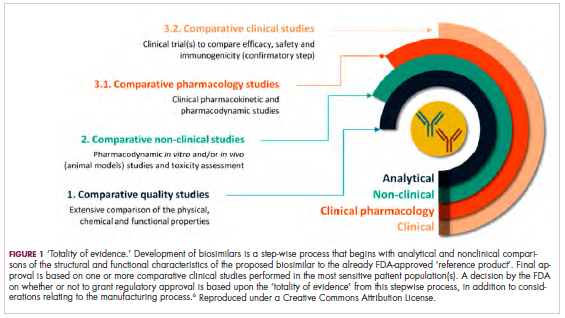
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6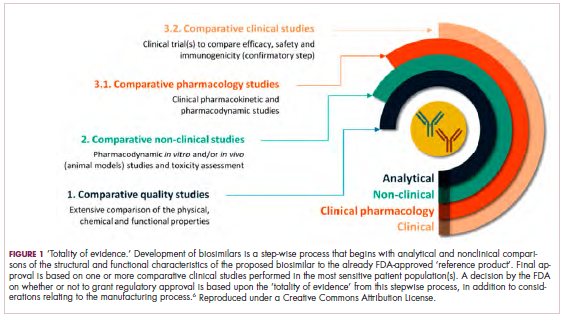
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10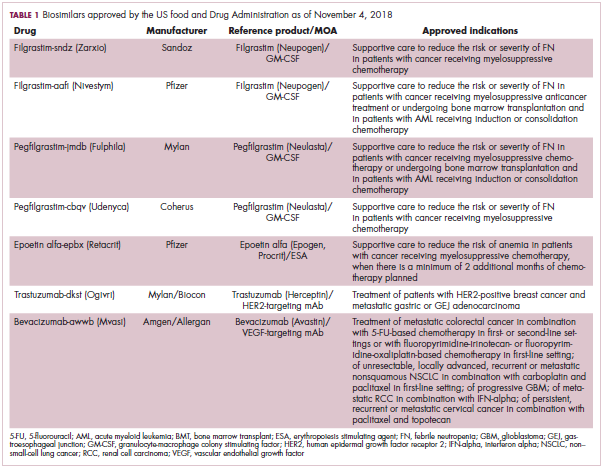
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22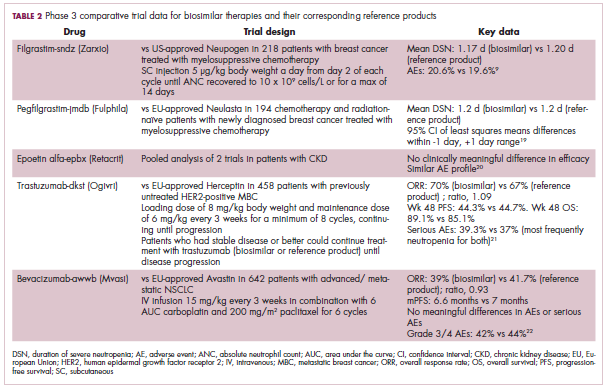
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.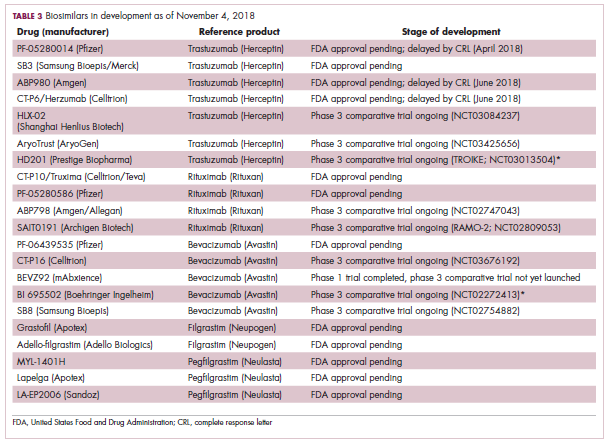
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
The development of biologic therapies has led to some of the most significant advances in the treatment of cancer, but these drugs are also very expensive. As patents for the biologics begin to expire, the development of biosimilars has the potential to dramatically cut therapy costs thereby making the therapies more readily accessible to patients. Here, we discuss biosimilar development and the challenges that need to be overcome to create a robust market.
Biosimilar, not generic
Biologic therapies are derived from living organisms and include the targeted monoclonal antibodies (mAbs) and cell-based therapies that have revolutionized the treatment of certain cancer types. Yet, their greater complexity makes them more difficult to manufacture, store, and administer, making them a costly therapeutic option that ultimately drives up health care costs. According to a 2011 drug expenditure analysis, biologic therapies accounted for more than half of the total expenditure on anticancer drugs in the US health care system.1,2
Generally, when drug patents expire, other companies can develop their own identical generic versions to increase competition in the marketplace and drive down costs. However, the paradigm for generic development cannot be applied to biologic therapies because the way in which they are manufactured makes it impossible to generate an identical copy.
Instead, the Biologics Price Competition and Innovation Act, a provision of the Patient Protection and Affordable Care Act, has allowed for submission of an application for “licensure of a biologic product based on its similarity to a licensed biologic product”.3
These “biosimilars” have been positioned as game-changers in oncology, with the potential to reduce costs and improve access to biologic therapies. With the patents on several blockbuster cancer biologics already expired or due to expire by 2020, an increasing number of biosimilars are being developed.4
Totality of evidence
Biosimilars require more rigorous testing than generics, but they don’t require the same type of scientific data that the original biologic products, termed “reference products,” did. Therefore, they are governed by legislation unique to them and approved by different regulatory pathways. The US Food and Drug Administration (FDA) has established a unique shortened regulatory pathway for their approval, known as the 351(k) pathway. So whereas the pathway for reference products is geared toward demonstrating patient benefit, biosimilars are required instead to show equivalence to the reference product.5
Biosimilars are produced through reverse engineering the reference product. Then, through a stepwise process, to generate what the FDA calls a “totality of evidence,” biosimilar manufacturers must demonstrate structural and functional similarities (through comparative quality studies) and comparable pharmacokinetics and pharmacodynamics (through comparative nonclinical and clinical studies) to the reference product. Final approval is based on 1 or more comparative clinical studies performed in the most sensitive patient population(s) (Figure 1).6
The primary endpoint of biosimilar clinical trials is chosen to detect clinically relevant differences and may not be the same as that used in pivotal trials of the reference product. Endpoints such as progression-free survival (PFS) and overall survival (OS) may not be feasible or sensitive enough to demonstrate biosimilarity.
Clinical trials of biosimilars should also be carried out in the most sensitive patient population, so that any potential differences can be attributed to the drug and not the patient population itself. If the reference product is approved across several different indications and there is sufficient scientific evidence to allow it, including the demonstration that the mechanism of action of the drug is the same across all indications, the FDA can extend the approval of the biosimilar to all of these indications without the need for individual clinical trials through a process known as extrapolation.
Biosimilar manufacturers must also provide evidence of the composition of their formulation and of quality control in their manufacturing processes, to ensure that biosimilarity can be maintained from batch to batch. As with the reference product, even small changes in the manufacturing process can have serious ramifications for clinical efficacy and safety.7,8
A flurry of approvals
The first biosimilar approvals in oncology in the United States came in the supportive care niche (Table 1). Filgrastim-sndz (Zarxio), approved in March 2015, is a biosimilar of the granulocyte-macrophage colony stimulating factor (G-CSF) analog filgrastim (Neupogen). Owing to its mechanism of action in stimulating the production of neutrophils in the bone marrow, filgrastim is used to help reduce the risk or severity of neutropenia in patients undergoing myelosuppressive chemotherapy regimens.
Filgrastim-sndz was approved for use across all 5 indications for which the reference product is approved, based on the totality of evidence, which included results from the key phase 3 PIONEER study.9 Market entry was initially delayed by lawsuits filed by Amgen, the maker of the reference product, but the biosimilar was subsequently cleared by the US Court of Appeals for the Federal Circuit. The wholesale acquisition cost (WAC) for a 300µg syringe is $324.30 for filgrastim and $275.66 figrastim-sndz, representing a 15% reduction on the reference product.10
In 2018, the FDA approved a second filgrastim biosimilar, filgrastim-aafi (Nivestym),11 in addition to 2 biosimilars of the pegylated form of filgrastim, pegfilgrastim-jmdb (Fulphila)12 and pegfilgrastim-cbqv (Udenyca)13 – these forms of filgrastim have been modified by the addition of polyethylene glycol polymer chains that help to increase circulation time.
Approval for the 2 pegfilgrastm biosimilars was originally delayed by complete response letters (CRLs) from the FDA. For pegfilgrastim-jmdb, the CRL was reported to be related to a pending update of the Biologic’s License Application (BLA) to include information regarding facility requalification activities that had been taken after the addition of plant modifications. The CRL for pegfilgrastim-cbqv requested that the company provide additional manufacturing information and reanalyze a subset of samples with a revised immunogenicity assay.
Once the CRL concerns were addressed, regulatory approval was awarded and Mylan recently confirmed that pegfilgrastim-jmdb has been launched in the US marketplace at a WAC that reflects a 33% discount over the reference product.14
Approval data for filgrastim-aafi and pegfilgrastim-cbqv have not yet been published, however the respective manufacturers reported that approval was based on totality of evidence demonstrating a high degree of similarity to the reference products. Filgrastim-aafi was approved for all of the indications of the reference product and launched in the US on October 1, 2018 at a 30% discounted WAC.15
Epoetin alfa-epbx (Retacrit), a biosimilar of epoetin alfa, was also approved in 2018. It is a recombinant analog of erythropoietin (EPO), which stimulates the production of blood cells and has proved useful for the treatment of anemia, including in cancer patients receiving myelosuppressive chemotherapy. Approval of the biosimilar followed earlier receipt of a CRL from the FDA citing concerns relating to the manufacturing facility, which the company addressed. Pfizer has said that it expects to launch the biosimilar this year (2018), but a WAC has not been disclosed.16The FDA also recently approved the first biosimilars for the treatment of cancer. Trastuzumab-dkst (Ogivri) and bevacizumab-awwb (Mvasi) were approved in the second half of 2017 for the same indications as their respective reference products, which are mAbs directed at the human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor, respectively.17,18
Approval data for bevacizumab-awwb included a comparative clinical trial in patients with advanced/metastatic non–small-cell lung cancer (NSCLC), which was considered the most sensitive patient population. The BLA for trastuzumab-dkst included data from the phase 3 comparative HERiTAge clinical trial, in which the biosimilar was compared with the reference product, both in combination with docetaxel or paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Neither biosimilar has been launched on the US market yet because the patents for their reference products do not expire until 2019, so it is not clear what the price discount will be for these drugs (Table 2).9,19-22
Biosimilars in development
While numerous other biosimilars of filgrastim and pegfilgrastim are in development, the major focus has been on the development of more biosimilars to treat cancer (Table 3). BLAs have been submitted for 4 biosimilars of trastuzumab and 1 bevacizumab biosimilar. Approval for several of the trastuzumab biosimilars has been delayed by CRLs from the FDA, mostly regarding issues with the manufacturing process or facility. Several other trastuzumab and bevacizumab biosimilars are in late-stage clinical trials.
The results of several phase 3 comparative clinical trials were recently published or reported at annual conferences. Pfizer’s PF-05280014 was compared with the European Union (EU)–approved trastuzumab, both in combination with paclitaxel, in patients with previously untreated HER2-positive metastatic breast cancer. Data reported at the European Society for Medical Oncology congress in 2017 demonstrated equivalence between the reference product and biosimilar in overall response rate (ORR).23
Another recently published trial compared this biosimilar to EU-trastuzumab, both in combination with carboplatin and docetaxel, as neoadjuvant treatment for patients with resectable HER2-positive breast cancer. Among 226 patients randomized to receive 8 mg/kg in cycle 1 and 6 mg/kg thereafter of the biosimilar or reference product, every 3 weeks for 6 cycles, the pathologic complete response (pCR) rates were 47% and 50%, respectively.24
The results of a phase 3 study comparing Samsung Bioepis/Merck’s joint offering SB3 were recently published. A total of 875 patients were randomized 1:1 to receive SB3 or reference trastuzumab in combination with chemotherapy (4 cycles docetaxel followed by 4 cycles 5-fluorouracil/epirubicin/cyclophosphamide) prior to surgery, followed by 10 cycles of adjuvant SB3 or trastuzumab reference. Rates of event-free survival (EFS) were comparable between the 2 groups at 12 months (93.7% vs 96.1%, respectively).25
Amgen’s ABP980 was evaluated in the phase 3 LILAC trial, which measured the effect of the biosimilar on pCR in women with HER2-positive early breast cancer compared with reference trastuzumab. After 4 cycles of run-in anthracycline-based chemotherapy, ABP980 or reference trastuzumab were administered in combination with paclitaxel. This was followed by surgery and then ABP980 or reference trastuzumab in the adjuvant setting for up to 1 year, with the option to continue on the same drug as the neoadjuvant setting or to switch to the other. Among 696 assessable patients, the pCR rates were 48% and 42%, respectively.26
Most advanced in clinical testing among the upcoming bevacizumab biosimilars is Pfizer’s PF-06439535, for which the results of a phase 3 comparative trial were presented at the 2018 annual meeting of the American Society for Clinical Oncology. PF-06439535 was compared with the EU-approved bevacizumab, both in combination with paclitaxel and carboplatin, as first-line therapy for patients with advanced non-squamous NSCLC. Among 719 patients, the primary endpoint of ORR was 45.3% and 44.6%, respectively.27
Biosimilars of a third blockbuster cancer drug, the CD20-targeting mAb rituximab (Rituxan) are also in development and FDA approval is pending for 2. The patent for Rituxan expired in 2016, so these drugs could hit the market as soon as they are approved.
In a race to the finish for the first US-approved rituximab biosimilar, Celltrion-Teva’s CT-P10 (Truxima) seems most likely to come first; the Oncologic Drugs Advisory Committee voted unanimously in October 2018 to recommend its approval. Phase 3 comparative data were recently published; patients with newly diagnosed advanced-stage follicular lymphoma were randomized to receive intravenous infusions of 375 mg/m2 CT-P10 or reference rituximab, both in combination with cyclophosphamide, vincristine, and prednisone, on day 1 of 8 21-day cycles. The ORRs were identical (92.6%) for both drugs, pharmacokinetics data also suggested bioequivalence, and the incidence of AEs was also comparable (83% vs 80%).28
Biosimilars of the epidermal growth factor receptor (EGFR)-targeting mAb cetuximab are also listed in the pipeline for several biosimilar developers, but there is no indication of their developmental status as yet and no clinical trials are ongoing in the US.
Sorrento is developing STI-001, a cetuximab biosimilar, and reported that a phase 3 trial had been completed. Instead of a comparison with the reference product, however, the trial compared STI-001 in combination with irinotecan with irinotecan alone. They reported significantly higher ORR, PFS, and OS with the biosimilar compared with irinotecan alone, and a significant increase over historical data with the reference product, as well as fewer side effects and immunogenicity, which they attribute to its manufacture in a different cell line. However, no data has been published and no trials are ongoing in the United States, so the status of its development remains unclear.29
Challenges to a robust market
It is an exciting time for biosimilars, with many approvals and drugs being brought to market in the US in the past several years and more poised to follow suit as patents expire. Yet many challenges remain around the growth of a robust biosimilars market.
Several surveys conducted in recent years have demonstrated suboptimal knowledge of all aspects of biosimilars and highlighted the need for evidence-based education across specialties.30,31 In response, the FDA recently announced that it was launching an educational campaign to further understanding of biosimilars, including naming conventions (Figure 2).32,33 Numerous other medical professional societies have produced or are in the process of producing biosimilar guidelines.
Educational outreach by the FDA forms part of their 4-step plan to aid biosimilar development, which also aims to improve the efficiency of biosimilar development and approval, to provide regulatory clarity for manufacturers, to facilitate public understanding and acceptance, and to support a competitive marketplace.
Among the most critical educational gaps is confusion over the issue of interchangeability. Once approved by the FDA, generic drugs are considered interchangeable with the brand name drug and can be substituted at the pharmacy level without referring to the prescribing physician. This is not the case for biosimilars; owing to their more complex nature, biosimilars require a separate designation for interchangeability and none of those approved so far have been given this designation by the FDA.
There has been some confusion about what will be required to demonstrate interchangeability, and the FDA recently produced draft guidance, saying that essentially it should be proven that switching out the reference product for a biosimilar does not increase risk in terms of diminished efficacy or safety. Several companies are beginning to incorporate a switching component into their clinical trials of biosimilars.
Continued postmarketing and real-world studies will also be particularly important for biosimilars to increase confidence in prescribing them by demonstrating their continued efficacy and safety in the long-term. Several real-world studies are now ongoing, including the MONITOR-GCSF trial of filgrastim biosimilars.
Another major barrier to the development of a thriving biosimilars market that achieves the goals of reduced costs and increased access is the financial burden of their development. They are vastly more costly to develop and produce than generics. Added to litigation costs, this can limit their ability to compete in terms of price, which has been reflected in the lower-than-anticipated cost savings with some approved biosimilars thus far.
Experts have suggested that there might be much to learn from the European market, where biosimilars have been available for more than a decade and over time have reached even higher-than-expected savings. With high financial stakes and an increasingly important role in the treatment of cancer, the need to iron out the kinks is more pressing than ever.7,8,34,35
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
. Abraham J. Developing oncology biosimilars: an essential approach for the future. Semin Oncol. 2013;40 Suppl 1:S5-24.
2. Doloresco F, Fominaya C, Schumock GT, et al. Projecting future drug expenditures: 2011. Am J Health Syst Pharm. 2011;68(10):921-932.
3. Prepared by the Office of the Legislative Counsel. HHS website. Compilation of the Patient Protection and Affordable Care Act [as amended through May 1, 2010] including Patient Protection and Affordable Care Act health-related portions of the Health Care and Education Reconciliation Act of 2010. https://www.hhs.gov/sites/default/files/ppacacon.pdf. Released June 9, 2010. Accessed November 7, 2018.
4. Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3-3.
5. Hung A, Vu Q, Mostovoy L. A systematic review of US biosimilar approvals: what evidence does the FDA require and how are manufacturers responding? J Manag Care Spec Pharm. 2017;23(12):1234-1244.
6. Uifălean A, Ilieş M, Nicoară R, Rus LM, Hegheş SC, Iuga C-A. Concepts and challenges of biosimilars in breast cancer: the emergence of trastuzumab biosimilars. Pharmaceutics. 2018;10(4):E168.
7. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev. 2016;46:73-79.
8. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3(5):596-610.
9. Blackwell K, Semiglazov V, Krasnozhon D, et al. Comparison of EP2006, a filgrastim biosimilar, to the reference: a phase III, randomized, double-blind clinical study in the prevention of severe neutropenia in patients with breast cancer receiving myelosuppressive chemotherapy. Ann Oncol. 2015;26(9):1948-1953.
10. FDA News. Sandoz launches Zarxio at 15 percent lower price than Neupogen. https://www.fdanews.com/articles/173036-sandoz-launches-zarxio-at-15-percent-lower-price-than-neupogen. Released September 11, 2015. Accessed November 7, 2018.
11. Pfizer. US FDA approves Pfizer's biosimilar Nivestym (filgrastim-aafi). https://www.pfizer.com/news/press-release/press-release-detail/u_s_fda_approves_pfizer_s_biosimilar_nivestym_filgrastim_aafi-0. Released July 2o, 2018. Accessed November 7, 2018.
12. United States Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm609805.htm. Released on June 4, 2018. Accessed November 7, 2018.
13. Coherus Biosciences. US FDA approves Udenyca (pegfilgrastim-cbqv). http://investors.coherus.com/news-releases/news-release-details/us-fda-approves-udenycatm-pegfilgrastim-cbqv. Released November 2, 2018. Accessed November 7, 2018.
14. The Center for Biosimilars. Mylan confirms that it has launched Fulphila in the United States. https://www.centerforbiosimilars.com/news/mylan-confirms-that-it-has-launched-fulphila-in-the-united-states. Released July 30, 2018. Accessed November 7, 2018.
15. The Center for Biosimilars. Pfizer launches biosimilar filgrastim, Nivestym, at a substantial discount. https://www.centerforbiosimilars.com/news/pfizer-launches-biosimilar-filgrastim-nivestym-at-a-substantial-discount. Released October 3, 2018. Accessed November 7, 2018.
16. The Center for Biosimilars. FDA approves Pfizer's epoetin alfa biosimilar, Retacrit. https://www.centerforbiosimilars.com/news/fda-approves-pfizers-epoetin-alfa-biosimilar-retacrit. Released May 15, 2018. Accessed November 7, 2018.
17. United States Food and Drug Administration. FDA approves Ogivri as a biosimilar to Herceptin. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm587404.htm. Last updated December 1, 2017. Accessed November 7, 2018.
18. United States Food and Drug Administration. FDA approves first biosimilar for the treatment of cancer. 2017; https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm576112.htm. Last updated March 26, 2018. Accessed November 7, 2018.
19. Waller CF, Blakeley C, Pennella E, et al. Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL-1401H vs EU-neulasta in the prophylaxis of chemotherapy-induced neutropenia. Ann Oncol. 2016;27(suppl_6):14330.
20. US Food and Drug Administration. 'Epoetin Hospira,' a proposed biosimilar to US-licensed Epogen/Procrit. 2017. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM559962.pdf. Accessed November 7, 2018.
21. Manikhas A, Pennella EJ, Bondarenko I, et al. Biosimilar trastuzumab-dkst monotherapy versus trastuzumab monotherapy after combination therapy: toxicity, efficacy, and immunogenicity from the phase 3 Heritage trial. J Clin Oncol. 2018;36(15_suppl):110.
22. Thatcher N, Thomas M, Paz-Ares L, et al. Randomized, double-blind, phase 3 study evaluating efficacy and safety of ABP 215 compared with bevacizumab in patients with non-squamous NSCLC. J Clin Oncol. 2016;34(15_suppl):9095.
23. Pegram M, Tan-Chiu E, Freyman A, et al. A randomized, double-blind study of PF-05280014 (a potential trastuzumab biosimilar) vs trastuzumab, both in combination with paclitaxel, as first-line therapy. Ann Oncol. 2017;28(suppl_5):v74-v108.
24. Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119(3):266-273.
25. Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safety, immunogenicity and survival results. Eur J Cancer. 2018;93:19-27.
26. von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2018;19(7):987-998.
27. Socinski MA, Pawel JV, Kasahara K, et al. A comparative clinical study of PF-06439535, a candidate bevacizumab biosimilar, and reference bevacizumab, in patients with advanced non-squamous non-small cell lung cancer. J Clin Oncol. 2018;36(15_suppl):109-109.
28. Kim WS, Buske C, Ogura M, et al. Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: a randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4(8):e362-e373.
29. PRNewsire. Sorrento announces positive data from phase 3 studies of biosimilar antibodies, STI-001 and STI-002. https://www.prnewswire.com/news-releases/sorrento-announces-positive-data-from-phase-3-studies-of-biosimilar-antibodies-sti-001-and-sti-002-300202054.html. Released January 11, 2016. Accessed November 7, 2018.
30. Molinari AL, Gewanter HL, Loaiza-Bonilla A, Reilly M, Kennedy B, Charles D. Global survey of physicians' attitudes toward biologic and biosimilar therapies. J Clin Oncol. 2016;34(15_suppl):e18025-e18025.
31. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160-2172.
32. Tomaszewski D. Biosimilar naming conventions: pharmacist perceptions and impact on confidence in dispensing biologics. J Manag Care Spec Pharm. 2016;22(8):919-926.
33. US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. https://www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Released January 2017. Accessed November 7, 2018.
34. Lyman GH. Emerging opportunities and challenges of biosimilars in oncology practice. J Clin Oncol Pract. 2017;13(9_suppl):7s-9s.
35. Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4(2):241-247.
Intravascular large B-cell lymphoma: an elusive diagnosis with challenging management
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.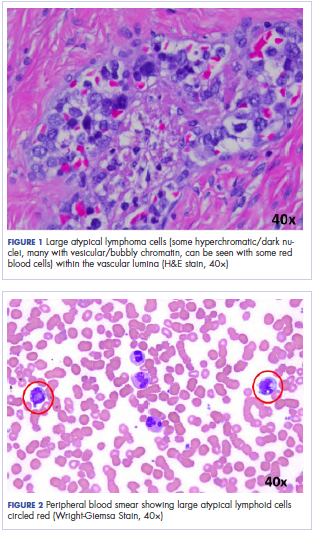
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.
Discussion
Clinical presentation
The clinical manifestation of this disease is highly variable, and virtually any organ can be involved. Besides causing constitutional symptoms, including fatigue, B symptoms, and decline in performance status, heterogeneity of the clinical presentation depends on the organ system involved. One of the exceptional features of this disease is the difference in clinical presentation based on the geographical origin of the patient.2-4
Western-variant IVBCL has a higher frequency of CNS and skin involvement, whereas Asian-variant IVBCL shows preferential involvement of bone marrow with hemophagocytosis, hepatosplenomegaly, and thrombocytopenia. However, these 2 clinical variants have no difference in clinical outcome, except with the cutaneous-variant kind.24 A retrospective case series of 38 Western-variant IVBCL cases showed that 55% of patients had B symptoms with poor performance status.3 Brain and skin were the organs that were most frequently involved, with 68% of patients having involvement of at least 1 of those organs. Ten patients in this case series had disease that was exclusively limited to the skin and described as a “cutaneous variant” of IVBCL.3
Similarly, a retrospective case series of 96 cases of Asian-variant IVBCL showed B symptoms in 76% of patients, with predominant bone marrow involvement in 75% of patients, accompanied by hemophagocytosis in 66% and hepatosplenomegaly and anemia/thrombocytopenia in 77% and 84% of the patients, respectively.4 This difference in clinical presentation might have existed as a result of ethnic difference associated with production of inflammatory cytokines, including interferon gamma, tumor necrosis factor-alpha, interlukin-1 beta, and soluble interlukin-2 receptor, with levels of soluble interlukin-2 receptor found to be significantly higher in Asian patients than non-Asian patients.2
Diagnosis
Involved organ biopsy is mandatory for establishing the diagnosis of IVBCL. Laboratory findings are nonspecific, with the most common abnormality being increased serum LDH and beta-2 microglobulin levels observed in 80% to 90% or more of patients. Despite its intravascular growth pattern, IVBCL was associated with peripheral blood involvement in only 5% to 9% of patients.1
Staging
Clinical staging work-up suggested for IVBCL patients by International Extranodal lymphoma study group in 2005 included physical examination (with emphasis on nervous system and skin), routine blood studies, peripheral blood smear, total body CT scan with contrast or PET–CT scan, MRI brain with contrast, CSF cytology, and bone marrow or organ biopsy.1 The role of fluorodeoxyglucose-PET scan is controversial but can be helpful to detect unexpected locations for biopsy and to assess treatment response.5,6
Morphology and immunophenotyping
In general, IVBCL histopathology shows large neoplastic lymphoid cells with large nuclei along with one or more nucleoli and scant cytoplasm within blood vessel lumen. Immunophenotypically, IVBCL cells mostly express nongerminal B-cell–associated markers with CD79a (100%), CD20 (96%), MUM-IRF4 (95%), CD5 (38%), and CD10 (12%) expressions. IVBCL cells have been demonstrated to lack cell surface protein CD29 and CD54 critical to transvascular migration. Similarly, aberrant expression of proteins such as CD11a and CXCR3 allows lymphoma cells to be attracted to endothelial cells, which might explain their intravascular confinement.7
Genetics
No pathognomic cytogenetic abnormalities have been reported in IVBCL to date, and the genetic features of this disease are not yet completely understood.2,7
Management
IVBCL is considered a stage IV disseminated disease with an International Prognostic Index score of high-intermediate to high in most cases. Half of the patients with IVBCL who were treated with anthracycline-based chemotherapy relapsed and died within 18 months of diagnosis. One third of the relapses involved the CNS, thereby highlighting the importance of prophylactic CNS-directed Intrathecal therapy in an induction treatment regimen.2-4 Ferreri and colleagues reported in their case series response rates of about 60%, with an overall survival (OS) of 3 years of 30% in patients who were treated with anthracycline-based chemotherapy. A multivariate analysis of the entire series showed cutaneous variant of the disease to be an independent favorable prognostic factor for OS.3
In the Murase and colleagues case series, the authors reported 67% response rates and a median OS of 13 months with CHOP (cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, prednisone) or CHOP-like regimens. Multivariate analysis showed older age, thrombocytopenia, and absence of anthracycline-based chemotherapy to be an independent negative prognostic factor for OS.4 Another retrospective analysis by Shimada and colleagues of 106 patients with IVBCL showed improved outcome with the addition of rituximab to CHOP-based chemotherapy (R-CHOP). Complete response rate (CR), 2-year progression-free survival, and OS were significantly higher for patients in rituximab-chemotherapy group than for those in the chemotherapy-alone group (CR, 82% vs 51%, respectively, P = .001; PFS, 56% vs 27%; OS, 66% vs 46%, P = .001), thereby establishing rituximab with CHOP-based therapy as induction therapy for IVBCL patients.8
The role of high-dose chemotherapy followed by ASCT could also be used as consolidation therapy to improve clinical outcomes as reported in 7 patients, showing durable remission after transplant in these 2 case series.3,4 Another retrospective analysis of 6 patients with IVBCL who were treated with 6 cycles of R-CHOP as induction therapy and consolidated with ASCT reported all patients to be alive and in complete remission after a median follow-up of 56 months.9 Based on the retrospective case series data by Kato and colleagues and considering that more than 80% of the patients with IVBCL were in the high-risk International Prognostic Index group, ASCT in first remission might be a useful treatment option for durable remission; however, because the median age for the diagnosis of IVBCL is about 70 years, ASCT may not be a realistic option for all patients.
Conclusions
IVBCL is a rare, aggressive, and distinct type of DLBCL with complex constellations of symptoms requiring strong clinical suspicion to establish this challenging diagnosis. Rituximab with anthracycline-based therapy along with prophylactic CNS-directed therapy followed by consolidative ASCT may lead to long-term remission. More research is needed into the genetic features of this disease to better understand its pathogenesis and potential targets for treatment.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25(21):3168-3173.
2. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10(9):895-902.
3. Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’. Br J Haematol. 2004;127(2):173-183.
4. Murase T, Yamaguchi M, Suzuki R, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007;109(2):478-485.
5. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. Mayo Clin Proc. 2010;85(8):e56-e57.
6. Shimada K, Kosugi H, Shimada S, et al. Evaluation of organ involvement in intravascular large B-cell lymphoma by 18F-fluorodeoxyglucose positron emission tomography. Int J Hematol. 2008;88(2):149-153.
7. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Arch Pathol Lab Med. 2012;136(3):333-338.
8. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26(19):3189-3195.
9. Kato K, Ohno Y, Kamimura T, et al. Long-term remission after high-dose chemotherapy followed by auto-SCT as consolidation for intravascular large B-cell lymphoma. Bone Marrow Transplant. 2014;49(12):1543-1544.
Elevated liver function tests in a patient on palbociclib and fulvestrant
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
About 12.4% of women in the United States will be diagnosed with breast cancer at some point in their lifetime.1 A percentage of these women will develop metastatic disease and are estimated to have a 5-year survival rate of 22%.2 There have been meaningful improvements in su
However, endocrine resistance inevitably occurs, and a great deal of research has been focused on developing strategies to combat resistance. One mechanism of endocrine resistance is though the Cyclin-dependent kinases 4 and 6 (CDK4/6) complexes. Among the most promising of the strategies to prevent resistance are the CDK4/6 inhibitors. There are now 3 approved CDK4/6 inhibitor drugs that can be used in combination with endocrine therapy, 1 of which can also be used as a single agent. When used in combination with endocrine therapy, the use of CDK 4/6 inhibitors has significantly improved progression-free survival (PFS) in patients with hormone-sensitive HER2-negative metastatic breast cancer by inhibiting cellular division and growth.3 In postmenopausal women, endocrine therapy plus CDK4/6 inhibitors are the preferred first-line regimen for metastatic disease.
Since the approval of palbociclib by the US Food and Drug Administration in 2015, the most common hematologic lab abnormalities are anemia, leukopenia, neutropenia, and thrombocytopenia. The most common nonhematologic adverse events (AEs) are fatigue, infection, nausea, and stomatitis. Hepatic toxicity has not been commonly observed. We report here the case of a 57-year-old woman on palbociclib and fulvestrant who developed significant elevation of liver function tests after starting palbociclib, suggesting a possible drug-induced liver injury from palbociclib.
Case presentation and summary
A 57-year-old woman with history of hypothyroidism and hypertension presented in May 2016 with a lump in her right breast and back pain. The lump was biopsied and revealed invasive ductal carcinoma, moderately differentiated, estrogen receptor (ER) positive 100%, progesterone receptor (PR) positive 95%, and HER2 negative. A positron emission tomography (PET)–computed tomography (CT) scan and magnetic resonance imaging showed bone metastasis at several vertebral levels, and the results of a bone biopsy confirmed metastatic adenocarcinoma of breast origin, ER positive 60%, PR positive 40%, and HER2 negative. No liver lesions were seen on imaging, but there was suggestion of fatty liver. She was started on letrozole 2.5 mg daily in July 2016 while undergoing kyphoplasty and subsequent radiation. A restaging PET scan revealed progression of disease on letrozole, with possible new rib lesion and progression in the breast. No liver disease was noted. Therapy was changed to fulvestrant and palbociclib. Fulvestrant was started in March 2017 with standard dosing of 500 mg intramuscular on days 1, 15, and 29, and then once a month thereafter. Her first cycle of palbociclib was started on April 5, dosed at 125 mg by mouth daily for 21 days, followed by 7 days off, repeated every 28 days (all dates hereinafter fell within 2017, unless otherwise stipulated).
Labs checked on April 28 and May 26 were unremarkable. A restaging CT scan of the chest, abdomen, and pelvis was done on June 21 after completion of 3 cycles of fulvestrant and palbociclib. There was no evidence of liver metastases, only the fatty infiltration of the liver that had been seen previously. On June 23, 2017, lab results showed a transaminitis with an alanine aminotransferase (ALT) level of 446 IU/L (reference range 10-33 IU/L) and aspartate aminotransferase (AST) level of 183 IU/L (reference range 0-32 IU/L).
The patient’s liver enzyme levels continued to increase and peaked on July 3 at ALT >700 IU/L and AST at 421 IU/L. Her total bilirubin and alkaline phosphatase levels remained within normal limits. She had received her final dose of fulvestrant on May 31 and had taken her last dose of palbociclib on June 20, 2017. She had no history of elevated liver enzymes or liver disease, although the initial PET scan done at diagnosis had suggested hepatic steatosis. She said she had not recently used antibiotics, alcohol, or over-the-counter medications or supplements. There was no family history of liver problems, inflammatory bowel disease, or gastrointestinal malignancy. The only other medications she had taken recently were denosumab, levothyroxine for hypothyroidism, and amlodipine for hypertension. She was seen by hepatology for evaluation of acute hepatitis. Other etiologies for her elevated liver enzymes were ruled out, and she was diagnosed with a drug-induced liver injury from one of her anticancer medications. Her treatments with fulvestrant and palbociclib were held, and the results of her liver function tests normalized by September 2017.
Fulvestrant was restarted on August 24, and her lab results remained normal through November of that year, when restaging scans showed progression with new axillary adenopathy suspicious for metastasis. Imaging also showed a 1.6-cm hepatic lesion suggestive of a focal area of fat deposition or atypical hemangioma without definitive evidence of metastasis. Follow-up imaging was recommended. She was therefore rechallenged with palbociclib at a reduced dose of 100 mg by mouth daily and received the first dose on November 30. On December 8, repeat labs again showed elevated liver function tests (ALT, 285 IU/L; AST, 112 IU/L). Treatment with palbociclib was discontinued on December 10. Because the patient was not able to tolerate palbociclib, and fulvestrant alone was not controlling the disease, she was started on an alternate endocrine therapy with tamoxifen on December 26. The patient’s liver function tests normalized again by January 2018.
Discussion
The use of targeted therapies has changed the landscape of oncologic treatments. Several studies have evaluated the safety and efficacy of palbociclib in combination with endocrine therapy. The Palbociclib Ongoing Trials in the Management of Breast Cancer (PALOMA)-1 study, an open-label, randomized, phase-2 trial involving patients with newly diagnosed metastatic hormone sensitive HER2-negative breast cancer, demonstrated that palbociclib in combination with letrozole was associated with significantly longer PFS than letrozole alone.4 These results were later confirmed in the larger PALOMA-2 study, a randomized, double-blind, phase-3 trial that evaluated 666 postmenopausal patients with no prior systemic therapy. In that study, median PFS for the palbociclib–letrozole group was 24.8 months, compared with 14.5 months for the letrozole-alone group (hazard ratio [HR] for disease progression or death, 0.58 [0.46–0.72], P < .001).5 The most recent PALOMA-3 study, a phase-3 trial involving 521 patients with advanced hormone receptor–positive, HER2-negative breast cancer that had progressed during initial endocrine therapy, evaluated the efficacy of combined palbociclib and fulvestrant in a randomized, double-blind, placebo-controlled, parallel-group trial. The result was that the palbociclib–fulvestrant combination resulted in longer median PFS of 9.2 months, compared with 3.8 months with fulvestrant alone (P < .001).6
These trials also monitored the number of AEs as secondary aims. The most commonly reported AEs in the PALOMA trials for those patients in the palbociclib group were hematologic, with neutropenia being the most common, followed by leukopenia, anemia, and thrombocytopenia. The most common nonhematologic AEs reported in the palbociclib-fulvestrant group were fatigue, nausea, and headache. Elevated liver function tests were a rare but reported AE in 7.2% of the palbociclib-treated patients in the PALOMA-1 study.7 In the PALOMA-2 study, ALT and AST elevations were reported as AEs (all grades) in 9.9% and 9.7% of palbociclib-treated patients, respectively.5 In the PALOMA-3 study, there was 1 fatal serious AE of hepatic failure with grade 5 disease progression in the palbociclib group; however, the patient’s medical history included progressive liver metastasis and disease progression.6 A pooled safety analysis conducted across all PALOMA studies demonstrated that grade 3/4 AST and ALT elevations occurred in 3.3% and 2.3% of palbociclib-treated patients, respectively, again highlighting a reported but rare occurrence.8
The patient described in the present case report started on combination fulvestrant and palbociclib after her disease showed progression on letrozole. She developed an increase in transaminases after completing 3 cycles of palbociclib. Liver function tests increased nearly 12 weeks after beginning her first cycle of the CDK 4/6 inhibitor. Staging scans of the patient demonstrated fatty liver. It is not known if her fatty liver contributed to her transminitis; however, her baseline labs showed normal liver function tests, and they did not increase until after therapy with fulvestrant–palbociclib was started. It might have been that her fatty liver caused her to be at higher risk of transaminitis with administration of palbociclib, although we cannot be certain. Her lab results remained normal while she was on fulvestrant alone, and the liver function test results increased only after palbociclib was started, making this drug the more likely culprit.
Both events of increased liver enzymes occurred within a week of the last palbociclib dose; however, we note that hepatotoxicity developed at a faster rate when the patient was rechallenged with palbociclib at a lower dose, with elevated liver function tests increasing 1 week after restarting treatment as opposed to the first episode that occurred after 3 cycles of the palbociclib. After discontinuation of the medication, liver function tests again normalized, suggesting that palbociclib was most likely the causative agent. In addition, the degree of elevated liver enzymes was less severe on re-exposure at the lower dose of 100 mg, which raises the possibility that there could be a dose-dependent association between palbociclib and hepatotoxicity. There have been few case reports of increased liver enzymes associated with palbociclib, and it is only recently that this association has been more recognized. A meta-analysis by Zaw and colleagues has demonstrated that CDK 4/6 inhibitor–based regimens are associated with a higher risk of elevated AST and ALT; however, their relation with dose dependence was not described. In particular, they found that CDK 4/6 inhibitors increased the risk of high-grade, elevated ALT with a relative risk of 4.33 (95% confidence interval, 2.15-8.71; P < .0001). The meta-analysis also included other CDK 4/6 inhibitors such as abemaciclib and ribociclib, which have been more commonly associated with liver toxicity than palbociclib has.9 Our case report highlights the specific association between palbociclib and elevated liver enzymes.
In conclusion, this case report illustrates that our patient’s elevated liver enzymes were likely related to palbociclib. This is further supported by the fact that this AE occurred twice, both times after palbociclib exposure. In each instance, liver enzymes normalized after discontinuation of palbociclib. One cannot entirely rule out that fulvestrant might have been the culprit medication, but the patient’s normal hepatic panel for several months after starting fulvestrant suggests that is less likely. This case report is indicative of an uncommon complication in the treatment of metastatic breast cancer, one that is starting to gain more recognition, and we must think of palbociclib as a possible cause of drug-induced liver injury when targeted CDK 4/6–based regimens are used.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
1. Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975-2014. Bethesda, MD: National Cancer Institute; 2017. https://seer.cancer.gov/csr/1975_2014/. Accessed April 3, 2018.
2. American Cancer Society. Breast cancer survival rates. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/breast-cancer-survival-rates.html. Accessed April 3, 2018.
3. Wolff AC. CDK4 and CDK6 inhibition in breast cancer - a new standard. N Engl J Med. 2016; 375(20):1993-1994.
4. Finn RS, Crown JP, Lang I, et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomized phase 2 study. Lancet Oncol. 2015;16(1):25-35.
5. Finn RS, Martin M, Rugo, HS et. al. Palbociclib and letrozole in advanced breast cancer. New Engl J Med. 2016;375:1925-1936
6. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomized controlled trial. Lancet Oncol. 2016;17(4):425-439.
7. Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373(3):209-219.
8. Dieras V, Rugo HS, Schnell P, et al. Long-term pooled safety analysis of palbociclib in combination with endocrine therapy for HR+/HR- advanced breast cancer [published online July 18, 2018]. Natl Cancer Inst. 2018;111.
9. Zaw M, Thein KZ, Tun A, et al. A systematic review and meta-analysis of randomized controlled trials to evaluate the risk of gastrointestinal and hepatic toxicities in patients with hormone receptor positive HER2-negative breast cancer treated with CKD 4/6 inhibitors. J Clin Oncol. 2017;35(suppl 31):209.
The challenge of managing a cetuximab rash
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.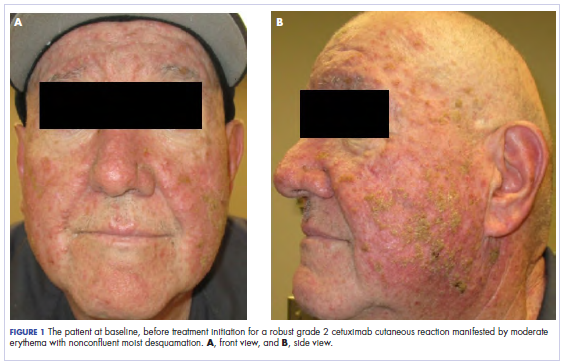
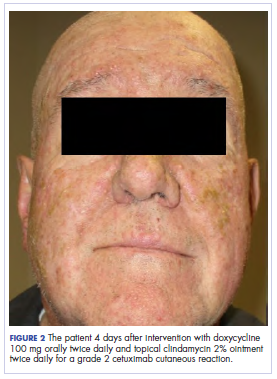
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.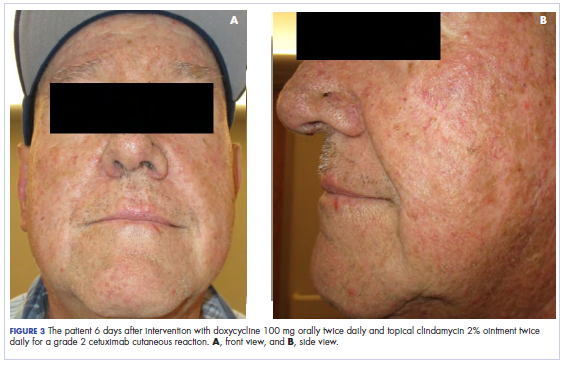
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.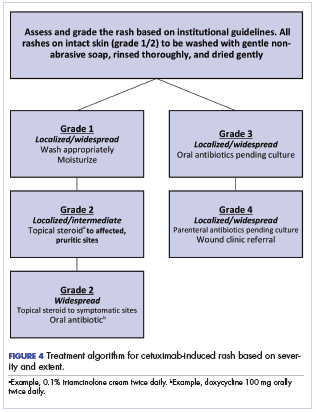
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
Epidermal growth factor receptor antibodies (EGFR) such as cetuximab have been approved for use as first-line management as well as salvage therapy for head and neck and colorectal cancers. Among the most common expected toxicity is a cutaneous eruption described as acneiform. The presence of a rash has been postulated to predict a more favorable treatment outcome for cancers of the head and neck1 but not for colorectum.2 With more severe drug reactions, patients may require a treatment break, which has been shown to reduce locoregional control and survival, particularly in patients with head and neck cancer.3 This has prompted clinicians to affect rapid therapy to reverse the drug eruption. Given the controversy around rapid and effective reversal of this drug reaction, this report aims to address the current status of clinical management using an actual patient vignette.
Case presentation and summary
The patient was a 57-year-old white man who had been diagnosed with stage 4 T4N0M1 grade 3 cutaneous squamous cell carcinoma (SCC) of the right postauricular soft tissues, with erosion into the right mastoid and biopsy-proven metastatic disease involving the contralateral left supraclavicular fossa and bilateral lungs. His disease became chemotherapy-refractory, and he was referred for palliative local therapy to the base of skull. Because of the size of the tumor (4 cm × 5 cm), he was considered for sensitizing chemotherapy, but cisplatin was not appropriate because of chronic hearing loss.4 The patient was recommended sensitizing doses of cetuximab. This EGFR antibody has been shown to offer similar benefits to those seen with cisplatin in the definitive management of head and neck SCC.5
The standard loading dose of cetuximab was given at 400 mg/m2 intravenously (IV). The following week, the sensitizing dose of 250 mg/m2 IV was given along with daily radiotherapy to the target volumes. The weekly dose of cetuximab continued at 250 mg/m2. The radiotherapy prescription was for 6,000 cGy in 200 cGy daily fractions, encompassing the gross tumor volume as identified on a computed-tomographic scan with 3-mm cuts. We used a noncoplanar arc radiotherapy beam arrangement because it inherently spreads the dose over a larger volume of normal tissue while conformally delivering its largest dose to the gross tumor volume. As such, a volume of the patient’s oropharynx and oral cavity was included within the radiotherapy dose penumbra. After receiving 3 weekly doses of cetuximab (1 loading dose and 2 weekly sensitizing doses) and 2,000 cGy of radiotherapy, the patient developed a robust grade 2 cutaneous eruption delimited to the face, with few scattered lesions on the upper anterior chest. He was seen in the medical oncology department and was prescribed doxycycline 100 mg orally twice daily and topical clindamycin 2% ointment twice daily.
In the radiation oncology clinic, his drug therapy was manipulated. His cetuximab cutaneous reaction was a grade 2, manifested by moderate erythema with nonconfluent moist desquamation. Because of concern that the patient would develop oral candida, which would further delay his therapy, the oral and topical antibiotics were discontinued, as was the oral prednisone. He was prescribed triamcinolone cream 0.1% to be applied to the facial and few chest wall areas twice daily and an oncology mouth rinse to address early nonconfluent mucositis. The accompanying images show the extent of the patient’s cetuximab cutaneous reaction at baseline before treatment initiation (Figure 1), at 4 days after the intervention (Figure 2), and again at 6 days after the intervention (Figure 3). The patient consented to having his photographs taken and understood that they would be used for educational and research publication purposes.
As can be seen from the photographs, the patient’s rash began to dry and peel by day 4 after the intervention, and there were no new eruptions. The pruritus that accompanied the rash had entirely resolved. By day 6, the rash had completely subsided. Because of the response to the topical steroid, the patient continued cetuximab without a dose modification. He was recommended to continue with the triamcinolone cream until the chemoradiotherapy course concluded.
Discussion
A cetuximab-induced rash is common. In a 2011 meta-analysis quantifying grades 1 to 4 in severity, about 75% of patients treated with an EGFR inhibitor experienced a rash. Most of the rashes were lower than grade 3, and the drug was either dose-reduced or temporarily held, but it was not generally discontinued.6 Of note is that in a nonselected survey of medical oncologists who were prescribing cetuximab, 76% reported holding the drug owing to rash severity, 60% reported dose reductions for a drug rash, and 32% reported changing the drug because of rash severity.7
In the initial pharmaceutical registration trial, 76% to 88% of patients who received cetuximab developed a rash, 17% of which were at least grade 3. The pharma recommendations for managing the drug rash include a drug delay for up to 2 weeks for a rash of grade 3 or less and to terminate use of the drug if there is no clinical improvement after 2 weeks.8 Biopsies of the rash confirm a suppurative inflammatory reaction separate from an infectious acne reaction,9 resulting in a recommendation to treat with topical steroid therapy. In some circumstances, the drug reaction can become infected or involve the paronychia, often related to Staphylococcus aureus.10 Despite what would otherwise be a problem addressed by anti-inflammatory medical therapy, the clinical appearance of the rash marked by pustules, coupled with the relative immunosuppressed state of a cancer patient, has prompted medical oncologists to prescribe antibiotic therapy.
To address the many single-institutional reports on management of the EGFR rash, several guidelines have been published. The earliest guideline – after a report that concurrent cetuximab and radiotherapy was superior to radiotherapy alone in locally advanced head and neck cancer, which documented a 23% incidence of at least grade 3 cutaneous toxicity in the cetuximab arm1 – attempted to score the severity of the rash according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events (CTCAE). Under those criteria, the authors defined grade 2 toxicity as moderate to brisk erythema with patchy moist desquamation, mostly confined to skin folds and creases. Grade 3 toxicity was described as moist desquamation other than skin folds and creases with bleeding induced by minor trauma, and grade 4 skin toxicity was defined as skin necrosis or ulceration of full thickness dermis with spontaneous bleeding from the involved site. The authors went on to describe a grade-related treatment algorithm that included gently washing the skin, keeping it dry, and using topical anti-inflammatory agents, including steroids. Antibiotics should be used in the presence of a suspected infection after culturing the area, and grade 4 toxicity should be referred to a wound care center.11
In a consensus statement from the National Comprehensive Cancer Network, the authors noted that most management recommendations were anecdotal. They recommended against the use of astringents and other drying agents because they exacerbate pain. The ultimate choice of topical steroids or antibiotics was based entirely on subjective judgement given the absence of prospective data.12
A Spanish consensus conference report argued against any prophylaxis against a skin reaction, other than keeping the skin clean and dry.13 The authors of the report recommended against washing the affected skin more than twice a day to avoid excess drying, and they advocated for moisturizers and debridement of skin crusting with hydrogels to reduce superinfection and bleeding.13 The authors also noted that some guidelines have suggested that topical steroids might exacerbate a skin rash,14 but they concluded that topical steroids are beneficial as long as they are used for less than 2 weeks. Any use of antibiotics should be based on clear evidence of an infection.13
In the first modification of the NCI’s CTCAE rash grading scale, an international panel addressed the increasing number of reports in the literature suggesting that the previous toxicity scale was possibly inadequate in its recommendations for appropriate treatment. The initial scale had defined only the skin reaction and not what therapy should be administered; therefore, in the update, the descriptions for grades 1 and 2 toxicity remained unchanged, but oral antibiotics were recommended for grade 3 lesion, and parenteral antibiotics with skin grafting were required with grade 4 toxicity.15
An Asian expert panel suggested modifying the bioradiation dermatitis scale, defining a grade 3 dermatitis as >50% moist desquamation of the involved field with formation of confluent lesions because of treatment. They recommended both topical and oral therapy, wound care, and possible hospitalization in severe cases. The panel suggested topical and systemic steroids and antibiotics.16
Finally, in an Italian consensus report, the members again modified the skin toxicity grading and were notably more aggressive in terms of their management recommendations. They defined grade 2 toxicity as pustules or papules covering 10% to 30% of the body surface area, with potential pruritus or tenderness. They also noted the psychosocial impact of skin toxicities on patients and the limits to their activities of daily living. They recommended vitamin K1 (menadione) cream, topical antibiotics, topical intermediate potency steroids, and oral antibiotic therapy for up to 4 weeks for grade 2 toxicity. Despite this aggressive treatment course, the authors admitted that the utility of topical steroids and antibiotics was unknown. They defined grade 3 toxicity as pustules or papules covering more than 30% of the body surface area, with signs of possible pruritus and tenderness. Activities of daily living and self-care were affected, and there was evidence of a superinfection. The panel suggested use of antibiotics pending culture results, oral prednisone, antihistamines, and oral analgesics. Topical therapy was not included.17 It is noteworthy that only the Italian panel recommended the use of vitamin K1 cream. In a prospective randomized, double-blinded, placebo-controlled phase 2 trial of 30 patients, menadione exhibited no clinical benefit in terms of reducing the severity of cetuximab skin lesions.18
Figure 4 illustrates our institutional approach to treating cetuximab rash based on a combination of the Spanish and NCI approaches.
The ultimate choice of therapy to manage a cetuximab rash must be patient and treatment specific. Our institutional approach, like that of the Spanish series,13 is to avoid chemoprophylaxis against a rash; rather, we recommend daily washing of the skin with a gentle soap followed by thorough rinsing and adequate, nonaggressive drying. Moisturizing the intact skin has been shown to reduce exfoliation, and we have incorporated that approach into our regimen.19
In our patient, whose head and neck radiotherapy tumor volume included a portion of the oral cavity and oropharynx, systemic antibiotic and steroid therapy would likely lead to further complications with the development of oral candidiasis. Therefore, while the severity of the reaction remained a grade 2, it seemed appropriate to treat with topical intermediate potency steroids and skin cleansing only. If the reaction had become more severe, then cultures would have been obtained to guide our decision on antibiotic therapy. Our patient’s response to topical steroids was predictable and effective, and he was able to proceed with his course of cancer therapy.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
1. Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21-28.
2. Sommeijer DW, Karapetis CS, Zalcberg JR, et al. The relationship between rash, tumor mutation KRAS status and clinical and quality of life outcomes in patients with advanced colorectal cancer treated with cetuximab in the NCIC CTG/AGITG CO.17. Acta Oncol. 2014;53(7):877-884.
3. Vahabzadeh-Hagh AM, Rwigema JM, Nabili V, Wang MB, Lorentz WC. Predictors of prolongation in radiation treatment time in a veteran population treated with chemoradiation for oropharyngeal cancer. Acta Otolaryngol. 2018;138(1):80-84.
4. Waissbluth S, Peleva E, Daniel SJ. Platinum-induced ototoxicity: a prevailing ototoxicity criteria. Eur Arch Otorhinlaryngol. 2017;274(3):1187-1196.
5. Huang J, Zhang J, Shi C, Liu L, Wei Y. Survival, recurrence and toxicity of HNSCC in comparison of a radiotherapy combination with cisplatin versus cetuximab: a meta-analysis. BMC cancer. 2016;16(1):689-713.
6. Mittman N, Seung SJ. Rash rates with EGFR inhibitors: meta-analysis. Curr Oncol. 2011;18(2):e54-e63.
7. Boone SL, Rademaker A, Liu D, Pfeiffer C, Mauro DJ, Lacouture ME. Impact and management of skin toxicity associated with anti-epidermal growth factor receptor therapy: survey results. Oncology. 2007;72(3-4):152-159.
8. Erbitux (cetuximab). Ask Lilly website. www.erbitux.com/hcp/index.html. Updated July 3, 2018. Accessed November 27.
9. Busam KJ, Capodieci P, Motzer R, Kiehn T, Phelan D, Halpern AC. Cutaneous side-effects in cancer patients treated with antiepidermal growth factor receptor antibody C225. Br J Dermatol. 2001;144(6):1169-1176.
10. Agero AL, Dusza SW, Benvenuto-Andrade C, Busam KJ, Myskowski P, Halpern AC. Dermatologic side effects associated with the epidermal growth factor receptor inhibitors. J Am Acad Dermatol. 2006;55:657-670, 2006.
11. Bernier J, Bonner J, Vermorken JB, et al. Consensus guidelines for the management of radiation dermatitis and coexisting acne-like rash in patients receiving radiotherapy plus EGFR inhibitors for the treatment of squamous cell carcinoma of the head and neck. Ann Oncol. 2008;19(1):142-149.
12. Burtness B, Anadkat M, Basti S, et al. NCCN task force report: management of dermatologic and other toxicities associated with EGFR inhibition in patients with cancer. J Natl Compr Canc Netw. 2009;7(suppl 1):S5-S21.
13.
14. Li T, Perez-Soler R. Skin toxicity associated with epidermal growth factor receptor inhibitors. Target Oncol. 2009;4(2):107-119.
15. Bernier J, Russi EG, Homey B, et al. Management of radiation dermatitis in patients receiving cetuximab and radiotherapy for locally advanced squamous cell carcinoma of the head and neck: proposals for a revised grading system and consensus management guidelines. Ann Oncol. 2011;22(10):2191-2200.
16. Zhu G, Lin JC, Kim SB, Bernier J, et al. Asian expert recommendation on management of skin and mucosal effects of radiation, with or without the addition of cetuximab or chemotherapy, in treatment of head and neck squamous cell carcinoma. BMC Cancer. 2016;16:42-62.
17. Pinto C, Barone CA, Girolomoni G, et al. Management of skin reactions during cetuximab treatment in association with chemotherapy or radiotherapy: update of the Italian expert recommendations. Am J Clin Oncol. 2016;39(4):407-415.
18. Eriksen JG, Kaalund I, Clemmensen O, Overgaard J, Pfeiffer P. Placebo-controlled phase II study of vitamin K3 cream for the treatment of cetuximab-induced rash. Support Care Cancer. 2017;25(7):2179-2185.
19. Watanabe S, Nakamura M, Takahashi H, et al. Dermopathy associated with cetuximab and panitumumab: investigation of the usefulness of moisturizers in its management. Clin Cosmet Investig Dermatol. 2017;10:353-361.
Comparing risk models guiding growth factor use in chemotherapy
Chemotherapy-induced neutropenia (CIN) and its corollary febrile neutropenia (FN) are well recognized, and they are serious consequences of many agents used in the treatment of malignancy. FN in particular has been associated with a considerable risk of morbidity and mortality, namely sepsis with multiorgan failure and eventual death. 1 The mainstay of prophylaxis for patients who are deemed to be at high risk for CIN and FN is colony-stimulating factors (CSF). These agents have been shown to significantly decrease FN-related mortality, and therefore their use is potentially life-saving. 2 However, CSF are not cheap, with the cost of peg-filgrastim as much as US $6195.99 per cycle of chemotherapy. 3 Therefore, not only do FN and CIN pose significant risk to patients, they also carry a high burden of cost to the patient and health care system both in treatment and prophylaxis. 4 As such, it is prudent for oncologists to accurately identify high-risk patients and judiciously use CSF in an evidence-based manner.
However, this has proven to be difficult because of the extent of variability between patients and the heterogeneity of the various risk models in the literature. Currently, there are 2 widely used guidelines, 1 developed by the National Comprehensive Cancer Network (NCCN) and another by the American Society of Clinical Oncology (ASCO). Both guidelines suggest the use of prophylactic CSF if the chemotherapy regimen has an FN risk of more than 20% (high risk). If the chemotherapy is deemed to be of intermediate risk (10%-20% FN risk), then patient-specific factors need to be considered. 5,6
In lung cancer, the NCCN lists only topotecan for small cell carcinomas as being high risk for FN, and therefore it is the only regimen that would warrant definitive use of prophylactic CSF. 5 The most recent ASCO guidelines do not list chemotherapy regimens that are high risk for FN. 6 For intermediate-risk regimens, the NCCN states that CSF prophylaxis should be considered if the patient has had previous chemotherapy or radiation therapy, persistent neutropenia, bone marrow involvement by tumor, recent surgery or open wounds, liver dysfunction (total bilirubin, >2.0 mg/dL), or renal dysfunction (creatinine clearance, <50 mL/min), or is older than 65 years. 5
ASCO guidelines state that in intermediate-risk chemotherapy regimens, the following factors are to be considered: age >65 years, advanced disease, previous chemotherapy or radiation therapy, pre-existing neutropenia or marrow involvement by tumor, infection, open wounds or recent surgery, poor performance status or nutritional status, poor renal function, liver dysfunction (most notably bilirubin elevation), cardiovascular disease, multiple comorbid conditions, and HIV infection. However, in the ASCO guidelines, there is no suggestion as to whether CSF should be administered if patients have one of these risk factors, only to “consider these factors when estimating patients’ overall risk of febrile neutropenia.” 6
There is some uncertainty with the NCCN and ASCO guidelines as to whether prophylactic CSF should be given to these intermediate-risk patients. There are suggestions but no definitive guidelines. In our study, we looked at lung cancer patients treated with intermediate-risk chemotherapy regimens and applied 2 different risk models created by Hosmer 7 and Bozcuk 8 and their respective colleagues (Hosmer and Bozcuk hereinafter). Our goal was to assess the efficacy differences between the 2 risk models and to compare their outcomes and recommendations with the NCCN and ASCO guidelines. This was done to showcase the tools available to a clinical oncologist who must decide whether to prescribe prophylactic CSF in these more challenging clinical situations.
Methods
Study population
This was a cross-sectional, retrospective study looking at male and female patients aged 18 to 75 years who were treated in the hematology–oncology offices of Drexel University in Philadelphia, Pennsylvania, from 2005 through 2016, who had a diagnosis of lung cancer and were, at some point during their disease, treated with chemotherapy. By using ICD-10 codes for any type of lung cancer, we identified 242 patients. Of those, 106 patients were excluded because they had never received chemotherapy, 16 were excluded either because of miscoding of the type of cancer or because they never actually had cancer, and 61 were excluded either because chemotherapy had not been delivered at our institution or because there were insufficient data to apply the 2 risk models. Of the remaining 59 patients, 16 were excluded because they had received prophylactic CSF with their first cycle of chemotherapy, leaving a total of 43 patients to whom the various risk models and guidelines could be applied (Table 1). If any of the 43 patients were found to be neutropenic, they were given growth factor shortly thereafter.
Chemotherapy for these 43 patients consisted of either a platinum doublet (cisplatin or carboplatin with either etoposide, pemetrexed, gemcitabine, or paclitaxel) or monotherapy with either paclitaxel, abraxane, navelbine, or pemetrexed. Of the 43 patients, 32 had platinum-based doublets, and 11 had monotherapy with one of the listed agents (Table 1).
Formal patient consent was not required because this was a retrospective study.
Defining CIN and FN
Neutropenia was defined as an absolute neutrophil count (ANC) of less than 1500 neutrophils per microliter. The levels of neutropenia were defined as mild (ANC, 1000-1500 neutrophils/μL), moderate (ANC, 500-1000 neutrophils/μL), and severe (ANC, <500 neutrophils/μL). The NCCN guidelines define FN as a single temperature of >38.3°C orally or >38.0°C over 1 hour, with an associated ANC of <500 or <1000 with a predicted decline to <500 over the next 48 hours. 5
Risk models
It should be noted that the Hosmer and Bozcuk calculators were powered to detect occurrence of FN. 7,8 However, we also applied them for the risk of any CIN. In scoring for the Hosmer calculator, points are given to each risk factor and are added together to give a final risk score. This risk score correlates to a percentage of predicted FN. The score for the Hosmer calculator is from minus 18 to plus 19, in which a score of 13 or higher correlates to a 15% predicted risk of FN, and a score of 0 or less correlates to a 1.6% risk of FN. 7 For the Bozcuk calculator, a nomogram is used to calculate risk. Individual points are given to each risk factor and are then summed to give a total that correlates to a risk of FN. The score range for the Bozcuk calculator is 0 to 300, with a score of greater than 190 correlating to a greater than 90% risk of FN, and a score of 0 correlating to a 0% predicted risk of FN. 8
For sensitivity and specificity threshold values, Hosmer reported using a risk score of 10 or above as being a reasonable value for the use of prophylactic CSF. They reported this score would predict an FN risk of about 10%, sensitivity of 24%, and specificity of 93% in detecting FN. 7 Bozcuk reported that using 110 as a cutoff value would correlate to about a 50% FN risk, sensitivity of 100%, and specificity of 49%. However, they did not suggest that value be applied as a threshold for the use of prophylactic CSF as Hosmer did. 8 Despite that, we used the thresholds of 10 and 110 for sensitivity and specificity analyses.
Regarding the current cycle of chemotherapy, the Hosmer calculator looked only at the first cycle, whereas the Bozcuk calculator looked at any cycle of chemotherapy. 7,8 In our study, we used the cycle correlating to the lowest ANC nadir the patient achieved. For example, if a patient achieved a nadir of 1,000 in cycle 1 but 200 in cycle 2, then we used the cycle 2 data to complete the calculators.
With respect to the NCCN and ASCO guidelines, we evaluated our cohort of 43 patients for the risk factors listed in the respective guidelines. If a patient had 1 or more of the risk factors, they were deemed to be high risk and therefore were recommended to receive CSF.
Results
General data
Of the 43 patients studied, 21 developed some level of CIN. Nine patients developed severe CIN, 4 developed moderate CIN, and 8 developed mild CIN. Of the severely neutropenic patients, 4 developed FN. None of the 16 patients who received prophylactic CSF developed FN, although 2 developed severe neutropenia despite CSF administration. Nadirs of ANC were seen on average during cycle 3 of chemotherapy. In all, 15 of the 43 patients achieved lowest ANC nadir during cycle 1.
Risk models
The Bozcuk calculator. A total of 22 patients had risk scores above the calculator’s threshold value of 110. Of those 22 patients, 7 developed severe CIN, 5 developed either mild or moderate CIN, and 3 developed FN. Of the remaining 21 patients who had risk scores of below 110, 2 developed severe CIN, 7 developed mild or moderate CIN, and 1 developed FN. Sensitivity and specificity values are shown in Table 2.
The Hosmer calculator. A total of 26 patients had risk scores above the calculator’s threshold value of 10. Of those 26 patients, 8 developed severe CIN, 4 developed either mild or moderate CIN, and 4 developed FN. Of the remaining 17 patients who had risk scores of less than 10, 1 developed severe CIN, 8 developed mild or moderate CIN, and none developed FN. Sensitivity and specificity values are listed in Table 2.
Current guidelines
NCCN guidelines. If one were to use the NCCN guidelines on our cohort of 43 patients, 25 would have been recommended to receive prophylactic CSF. Of those 25, 6 developed severe CIN (2 with FN), 2 moderate CIN, and 5 mild CIN. Of the 18 patients who would not have been recommended to receive CSF, 3 developed severe CIN (with 2 FN), 2 moderate CIN, and 3 mild CIN. Sensitivity and specificity values are listed in Table 2.
ASCO guidelines. Using the ASCO guidelines on our cohort of 43 patients, 38 had 1 or more of the high-risk features, and, therefore, CSF would have been considered for them. Of those 38 patients, 8 developed severe CIN (4 with FN), 4 developed moderate CIN, and 7 developed mild CIN. Of the 5 patients who would not have received CSF, 1 developed severe CIN and 1 mild CIN. Sensitivity and specificity values are listed in Table 2.
Discussion
In our study, we looked at 2 CIN risk models and compared them with the current NCCN and ASCO guidelines. The models were created to predict risk of FN, but we also looked at their predictive value for any level of CIN. To this end, we found that the Hosmer and Bozcuk calculators both were acceptable for predicting risk of severe CIN and FN. Because of the small number of patients in this study, differences in sensitivities and specificities cannot be quantitatively compared. Nevertheless, qualitatively, it can be said that both calculators were accurate in assigning high-risk scores to patients who developed severe CIN or FN. However, both calculators had many patients with high-risk scores who never developed CIN.
When comparing the 2 risk models with the NCCN and ASCO guidelines, the ASCO guidelines tended to be more liberal in their consideration of CSF use, whereas the NCCN guidelines tended to be more conservative and more similar to the 2 risk models we tested. The NCCN guidelines suggested not giving prophylactic CSF to 2 of our patients who developed FN and to not give CSF to an additional patient who developed severe CIN. The ASCO guidelines suggested considering using CSF for most of our patients, with only 5 patients not to be considered for CSF administration.
The differences in efficacy between the current guidelines and the 2 risk models may be indicative of the fact that the risk models are more accurate in assigning risk in older patients who are clinically more complicated. In our patients, the chemotherapies used were all considered to be intermediate risk, so patient-specific factors were used to guide the administration of CSF. However, because many our patients had at least 1 of the risk factors listed by the NCCN or ASCO, they were automatically deemed to be high risk and to receive prophylactic CSF.
Consequently, the Hosmer and Bozcuk calculators may be of greatest utility in more clinically complicated patients and those who have more comorbidities. The best approach may be a combination of either the NCCN or ASCO guidelines with 1 of the calculators, in our opinion the Hosmer system, for these complicated patients. Likely, the 2 risk models would not be as useful for chemotherapies deemed to have a high risk for FN because, in those situations, the efficacy and benefit of prophylactic CSF are clear. 9 Rather, their use could be beneficial in the grayer areas in which the risk is intermediate and decision-making is more difficult.
Limitations
There were several limitations in our study. First, the size of the cohort was small, and, therefore, the data that we gathered was limited in its scope. However, the goal of this study was to help provide guidance to oncologists in real-world settings about the validity and use of the available risk calculators. A further study should compare the calculators and guidelines in a much larger cohort to see if present results still hold true.
The second possible limitation of the study was our application of the Hosmer calculator because our patient population did not fit the criteria for inclusion in their original study. Hosmer had included only the first cycle of chemotherapy, whereas we included all cycles of chemotherapy. However, despite that, the calculator still performed well and could predict severe CIN and FN even with later cycles of chemotherapy. Therefore, we suggest using this calculator in any cycle of chemotherapy rather than just the first. This would expand its scope and utility in clinical practice.
Conclusions
This article provides oncologists with a comparison of 2 CIN risk models with the currently available NCCN and ASCO guidelines for use in patients with lung cancer. We prefer the Hosmer calculator over the Bozcuk calculator because of its simplicity of use and the accuracy of results. We anticipate that it may be useful and practical as an adjunct tool to the NCCN or ASCO guidelines in patients receiving intermediate-risk chemotherapy regimens. Larger studies combining the calculators and determining accuracy need to be completed to prove this hypothesis.
1. Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer. 2006;106(10):2258-2266.
2. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007;25(21):3158-3167.
3. Good Rx, Inc. Peg-filgrastim. https://www.goodrx.com/neulasta. Accessed September 2018.
4. Schilling MB, Parks C, Deeter RG. Costs and outcomes associated with hospitalized cancer patients with neutropenic complications: a retrospective study. Exp Ther Med. 2011;2(5):859-866.
5. National Comprehensive Cancer Network. Myeloid growth factors. In: NCCN Clinical Practice Guidelines in Oncology. Plymouth Meeting, PA: National Comprehensive Cancer Network; 2018.
6. Smith T, Bohlke K, Lyman GH, et al. Recommendations for the use of WBC growth factors: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2015;33(28):3199-3212.
7. Hosmer W, Malin J, Wong M. Development and validation of a prediction model for the risk of developing febrile neutropenia in the first cycle of chemotherapy among elderly patients with breast, lung, colorectal, and prostate cancer. Support Care Cancer. 2011;19(3):333-341.
8. Bozcuk H, Yıldız M, Artaç M, et al. A prospectively validated nomogram for predicting the risk of chemotherapy-induced febrile neutropenia: a multicenter study. Support Care Cancer. 2015;23(6):1759-1767.
9. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol. 2005;23(6):1178-1184.
Chemotherapy-induced neutropenia (CIN) and its corollary febrile neutropenia (FN) are well recognized, and they are serious consequences of many agents used in the treatment of malignancy. FN in particular has been associated with a considerable risk of morbidity and mortality, namely sepsis with multiorgan failure and eventual death. 1 The mainstay of prophylaxis for patients who are deemed to be at high risk for CIN and FN is colony-stimulating factors (CSF). These agents have been shown to significantly decrease FN-related mortality, and therefore their use is potentially life-saving. 2 However, CSF are not cheap, with the cost of peg-filgrastim as much as US $6195.99 per cycle of chemotherapy. 3 Therefore, not only do FN and CIN pose significant risk to patients, they also carry a high burden of cost to the patient and health care system both in treatment and prophylaxis. 4 As such, it is prudent for oncologists to accurately identify high-risk patients and judiciously use CSF in an evidence-based manner.
However, this has proven to be difficult because of the extent of variability between patients and the heterogeneity of the various risk models in the literature. Currently, there are 2 widely used guidelines, 1 developed by the National Comprehensive Cancer Network (NCCN) and another by the American Society of Clinical Oncology (ASCO). Both guidelines suggest the use of prophylactic CSF if the chemotherapy regimen has an FN risk of more than 20% (high risk). If the chemotherapy is deemed to be of intermediate risk (10%-20% FN risk), then patient-specific factors need to be considered. 5,6
In lung cancer, the NCCN lists only topotecan for small cell carcinomas as being high risk for FN, and therefore it is the only regimen that would warrant definitive use of prophylactic CSF. 5 The most recent ASCO guidelines do not list chemotherapy regimens that are high risk for FN. 6 For intermediate-risk regimens, the NCCN states that CSF prophylaxis should be considered if the patient has had previous chemotherapy or radiation therapy, persistent neutropenia, bone marrow involvement by tumor, recent surgery or open wounds, liver dysfunction (total bilirubin, >2.0 mg/dL), or renal dysfunction (creatinine clearance, <50 mL/min), or is older than 65 years. 5
ASCO guidelines state that in intermediate-risk chemotherapy regimens, the following factors are to be considered: age >65 years, advanced disease, previous chemotherapy or radiation therapy, pre-existing neutropenia or marrow involvement by tumor, infection, open wounds or recent surgery, poor performance status or nutritional status, poor renal function, liver dysfunction (most notably bilirubin elevation), cardiovascular disease, multiple comorbid conditions, and HIV infection. However, in the ASCO guidelines, there is no suggestion as to whether CSF should be administered if patients have one of these risk factors, only to “consider these factors when estimating patients’ overall risk of febrile neutropenia.” 6
There is some uncertainty with the NCCN and ASCO guidelines as to whether prophylactic CSF should be given to these intermediate-risk patients. There are suggestions but no definitive guidelines. In our study, we looked at lung cancer patients treated with intermediate-risk chemotherapy regimens and applied 2 different risk models created by Hosmer 7 and Bozcuk 8 and their respective colleagues (Hosmer and Bozcuk hereinafter). Our goal was to assess the efficacy differences between the 2 risk models and to compare their outcomes and recommendations with the NCCN and ASCO guidelines. This was done to showcase the tools available to a clinical oncologist who must decide whether to prescribe prophylactic CSF in these more challenging clinical situations.
Methods
Study population
This was a cross-sectional, retrospective study looking at male and female patients aged 18 to 75 years who were treated in the hematology–oncology offices of Drexel University in Philadelphia, Pennsylvania, from 2005 through 2016, who had a diagnosis of lung cancer and were, at some point during their disease, treated with chemotherapy. By using ICD-10 codes for any type of lung cancer, we identified 242 patients. Of those, 106 patients were excluded because they had never received chemotherapy, 16 were excluded either because of miscoding of the type of cancer or because they never actually had cancer, and 61 were excluded either because chemotherapy had not been delivered at our institution or because there were insufficient data to apply the 2 risk models. Of the remaining 59 patients, 16 were excluded because they had received prophylactic CSF with their first cycle of chemotherapy, leaving a total of 43 patients to whom the various risk models and guidelines could be applied (Table 1). If any of the 43 patients were found to be neutropenic, they were given growth factor shortly thereafter.
Chemotherapy for these 43 patients consisted of either a platinum doublet (cisplatin or carboplatin with either etoposide, pemetrexed, gemcitabine, or paclitaxel) or monotherapy with either paclitaxel, abraxane, navelbine, or pemetrexed. Of the 43 patients, 32 had platinum-based doublets, and 11 had monotherapy with one of the listed agents (Table 1).
Formal patient consent was not required because this was a retrospective study.
Defining CIN and FN
Neutropenia was defined as an absolute neutrophil count (ANC) of less than 1500 neutrophils per microliter. The levels of neutropenia were defined as mild (ANC, 1000-1500 neutrophils/μL), moderate (ANC, 500-1000 neutrophils/μL), and severe (ANC, <500 neutrophils/μL). The NCCN guidelines define FN as a single temperature of >38.3°C orally or >38.0°C over 1 hour, with an associated ANC of <500 or <1000 with a predicted decline to <500 over the next 48 hours. 5
Risk models
It should be noted that the Hosmer and Bozcuk calculators were powered to detect occurrence of FN. 7,8 However, we also applied them for the risk of any CIN. In scoring for the Hosmer calculator, points are given to each risk factor and are added together to give a final risk score. This risk score correlates to a percentage of predicted FN. The score for the Hosmer calculator is from minus 18 to plus 19, in which a score of 13 or higher correlates to a 15% predicted risk of FN, and a score of 0 or less correlates to a 1.6% risk of FN. 7 For the Bozcuk calculator, a nomogram is used to calculate risk. Individual points are given to each risk factor and are then summed to give a total that correlates to a risk of FN. The score range for the Bozcuk calculator is 0 to 300, with a score of greater than 190 correlating to a greater than 90% risk of FN, and a score of 0 correlating to a 0% predicted risk of FN. 8
For sensitivity and specificity threshold values, Hosmer reported using a risk score of 10 or above as being a reasonable value for the use of prophylactic CSF. They reported this score would predict an FN risk of about 10%, sensitivity of 24%, and specificity of 93% in detecting FN. 7 Bozcuk reported that using 110 as a cutoff value would correlate to about a 50% FN risk, sensitivity of 100%, and specificity of 49%. However, they did not suggest that value be applied as a threshold for the use of prophylactic CSF as Hosmer did. 8 Despite that, we used the thresholds of 10 and 110 for sensitivity and specificity analyses.
Regarding the current cycle of chemotherapy, the Hosmer calculator looked only at the first cycle, whereas the Bozcuk calculator looked at any cycle of chemotherapy. 7,8 In our study, we used the cycle correlating to the lowest ANC nadir the patient achieved. For example, if a patient achieved a nadir of 1,000 in cycle 1 but 200 in cycle 2, then we used the cycle 2 data to complete the calculators.
With respect to the NCCN and ASCO guidelines, we evaluated our cohort of 43 patients for the risk factors listed in the respective guidelines. If a patient had 1 or more of the risk factors, they were deemed to be high risk and therefore were recommended to receive CSF.
Results
General data
Of the 43 patients studied, 21 developed some level of CIN. Nine patients developed severe CIN, 4 developed moderate CIN, and 8 developed mild CIN. Of the severely neutropenic patients, 4 developed FN. None of the 16 patients who received prophylactic CSF developed FN, although 2 developed severe neutropenia despite CSF administration. Nadirs of ANC were seen on average during cycle 3 of chemotherapy. In all, 15 of the 43 patients achieved lowest ANC nadir during cycle 1.
Risk models
The Bozcuk calculator. A total of 22 patients had risk scores above the calculator’s threshold value of 110. Of those 22 patients, 7 developed severe CIN, 5 developed either mild or moderate CIN, and 3 developed FN. Of the remaining 21 patients who had risk scores of below 110, 2 developed severe CIN, 7 developed mild or moderate CIN, and 1 developed FN. Sensitivity and specificity values are shown in Table 2.
The Hosmer calculator. A total of 26 patients had risk scores above the calculator’s threshold value of 10. Of those 26 patients, 8 developed severe CIN, 4 developed either mild or moderate CIN, and 4 developed FN. Of the remaining 17 patients who had risk scores of less than 10, 1 developed severe CIN, 8 developed mild or moderate CIN, and none developed FN. Sensitivity and specificity values are listed in Table 2.
Current guidelines
NCCN guidelines. If one were to use the NCCN guidelines on our cohort of 43 patients, 25 would have been recommended to receive prophylactic CSF. Of those 25, 6 developed severe CIN (2 with FN), 2 moderate CIN, and 5 mild CIN. Of the 18 patients who would not have been recommended to receive CSF, 3 developed severe CIN (with 2 FN), 2 moderate CIN, and 3 mild CIN. Sensitivity and specificity values are listed in Table 2.
ASCO guidelines. Using the ASCO guidelines on our cohort of 43 patients, 38 had 1 or more of the high-risk features, and, therefore, CSF would have been considered for them. Of those 38 patients, 8 developed severe CIN (4 with FN), 4 developed moderate CIN, and 7 developed mild CIN. Of the 5 patients who would not have received CSF, 1 developed severe CIN and 1 mild CIN. Sensitivity and specificity values are listed in Table 2.
Discussion
In our study, we looked at 2 CIN risk models and compared them with the current NCCN and ASCO guidelines. The models were created to predict risk of FN, but we also looked at their predictive value for any level of CIN. To this end, we found that the Hosmer and Bozcuk calculators both were acceptable for predicting risk of severe CIN and FN. Because of the small number of patients in this study, differences in sensitivities and specificities cannot be quantitatively compared. Nevertheless, qualitatively, it can be said that both calculators were accurate in assigning high-risk scores to patients who developed severe CIN or FN. However, both calculators had many patients with high-risk scores who never developed CIN.
When comparing the 2 risk models with the NCCN and ASCO guidelines, the ASCO guidelines tended to be more liberal in their consideration of CSF use, whereas the NCCN guidelines tended to be more conservative and more similar to the 2 risk models we tested. The NCCN guidelines suggested not giving prophylactic CSF to 2 of our patients who developed FN and to not give CSF to an additional patient who developed severe CIN. The ASCO guidelines suggested considering using CSF for most of our patients, with only 5 patients not to be considered for CSF administration.
The differences in efficacy between the current guidelines and the 2 risk models may be indicative of the fact that the risk models are more accurate in assigning risk in older patients who are clinically more complicated. In our patients, the chemotherapies used were all considered to be intermediate risk, so patient-specific factors were used to guide the administration of CSF. However, because many our patients had at least 1 of the risk factors listed by the NCCN or ASCO, they were automatically deemed to be high risk and to receive prophylactic CSF.
Consequently, the Hosmer and Bozcuk calculators may be of greatest utility in more clinically complicated patients and those who have more comorbidities. The best approach may be a combination of either the NCCN or ASCO guidelines with 1 of the calculators, in our opinion the Hosmer system, for these complicated patients. Likely, the 2 risk models would not be as useful for chemotherapies deemed to have a high risk for FN because, in those situations, the efficacy and benefit of prophylactic CSF are clear. 9 Rather, their use could be beneficial in the grayer areas in which the risk is intermediate and decision-making is more difficult.
Limitations
There were several limitations in our study. First, the size of the cohort was small, and, therefore, the data that we gathered was limited in its scope. However, the goal of this study was to help provide guidance to oncologists in real-world settings about the validity and use of the available risk calculators. A further study should compare the calculators and guidelines in a much larger cohort to see if present results still hold true.
The second possible limitation of the study was our application of the Hosmer calculator because our patient population did not fit the criteria for inclusion in their original study. Hosmer had included only the first cycle of chemotherapy, whereas we included all cycles of chemotherapy. However, despite that, the calculator still performed well and could predict severe CIN and FN even with later cycles of chemotherapy. Therefore, we suggest using this calculator in any cycle of chemotherapy rather than just the first. This would expand its scope and utility in clinical practice.
Conclusions
This article provides oncologists with a comparison of 2 CIN risk models with the currently available NCCN and ASCO guidelines for use in patients with lung cancer. We prefer the Hosmer calculator over the Bozcuk calculator because of its simplicity of use and the accuracy of results. We anticipate that it may be useful and practical as an adjunct tool to the NCCN or ASCO guidelines in patients receiving intermediate-risk chemotherapy regimens. Larger studies combining the calculators and determining accuracy need to be completed to prove this hypothesis.
Chemotherapy-induced neutropenia (CIN) and its corollary febrile neutropenia (FN) are well recognized, and they are serious consequences of many agents used in the treatment of malignancy. FN in particular has been associated with a considerable risk of morbidity and mortality, namely sepsis with multiorgan failure and eventual death. 1 The mainstay of prophylaxis for patients who are deemed to be at high risk for CIN and FN is colony-stimulating factors (CSF). These agents have been shown to significantly decrease FN-related mortality, and therefore their use is potentially life-saving. 2 However, CSF are not cheap, with the cost of peg-filgrastim as much as US $6195.99 per cycle of chemotherapy. 3 Therefore, not only do FN and CIN pose significant risk to patients, they also carry a high burden of cost to the patient and health care system both in treatment and prophylaxis. 4 As such, it is prudent for oncologists to accurately identify high-risk patients and judiciously use CSF in an evidence-based manner.
However, this has proven to be difficult because of the extent of variability between patients and the heterogeneity of the various risk models in the literature. Currently, there are 2 widely used guidelines, 1 developed by the National Comprehensive Cancer Network (NCCN) and another by the American Society of Clinical Oncology (ASCO). Both guidelines suggest the use of prophylactic CSF if the chemotherapy regimen has an FN risk of more than 20% (high risk). If the chemotherapy is deemed to be of intermediate risk (10%-20% FN risk), then patient-specific factors need to be considered. 5,6
In lung cancer, the NCCN lists only topotecan for small cell carcinomas as being high risk for FN, and therefore it is the only regimen that would warrant definitive use of prophylactic CSF. 5 The most recent ASCO guidelines do not list chemotherapy regimens that are high risk for FN. 6 For intermediate-risk regimens, the NCCN states that CSF prophylaxis should be considered if the patient has had previous chemotherapy or radiation therapy, persistent neutropenia, bone marrow involvement by tumor, recent surgery or open wounds, liver dysfunction (total bilirubin, >2.0 mg/dL), or renal dysfunction (creatinine clearance, <50 mL/min), or is older than 65 years. 5
ASCO guidelines state that in intermediate-risk chemotherapy regimens, the following factors are to be considered: age >65 years, advanced disease, previous chemotherapy or radiation therapy, pre-existing neutropenia or marrow involvement by tumor, infection, open wounds or recent surgery, poor performance status or nutritional status, poor renal function, liver dysfunction (most notably bilirubin elevation), cardiovascular disease, multiple comorbid conditions, and HIV infection. However, in the ASCO guidelines, there is no suggestion as to whether CSF should be administered if patients have one of these risk factors, only to “consider these factors when estimating patients’ overall risk of febrile neutropenia.” 6
There is some uncertainty with the NCCN and ASCO guidelines as to whether prophylactic CSF should be given to these intermediate-risk patients. There are suggestions but no definitive guidelines. In our study, we looked at lung cancer patients treated with intermediate-risk chemotherapy regimens and applied 2 different risk models created by Hosmer 7 and Bozcuk 8 and their respective colleagues (Hosmer and Bozcuk hereinafter). Our goal was to assess the efficacy differences between the 2 risk models and to compare their outcomes and recommendations with the NCCN and ASCO guidelines. This was done to showcase the tools available to a clinical oncologist who must decide whether to prescribe prophylactic CSF in these more challenging clinical situations.
Methods
Study population
This was a cross-sectional, retrospective study looking at male and female patients aged 18 to 75 years who were treated in the hematology–oncology offices of Drexel University in Philadelphia, Pennsylvania, from 2005 through 2016, who had a diagnosis of lung cancer and were, at some point during their disease, treated with chemotherapy. By using ICD-10 codes for any type of lung cancer, we identified 242 patients. Of those, 106 patients were excluded because they had never received chemotherapy, 16 were excluded either because of miscoding of the type of cancer or because they never actually had cancer, and 61 were excluded either because chemotherapy had not been delivered at our institution or because there were insufficient data to apply the 2 risk models. Of the remaining 59 patients, 16 were excluded because they had received prophylactic CSF with their first cycle of chemotherapy, leaving a total of 43 patients to whom the various risk models and guidelines could be applied (Table 1). If any of the 43 patients were found to be neutropenic, they were given growth factor shortly thereafter.
Chemotherapy for these 43 patients consisted of either a platinum doublet (cisplatin or carboplatin with either etoposide, pemetrexed, gemcitabine, or paclitaxel) or monotherapy with either paclitaxel, abraxane, navelbine, or pemetrexed. Of the 43 patients, 32 had platinum-based doublets, and 11 had monotherapy with one of the listed agents (Table 1).
Formal patient consent was not required because this was a retrospective study.
Defining CIN and FN
Neutropenia was defined as an absolute neutrophil count (ANC) of less than 1500 neutrophils per microliter. The levels of neutropenia were defined as mild (ANC, 1000-1500 neutrophils/μL), moderate (ANC, 500-1000 neutrophils/μL), and severe (ANC, <500 neutrophils/μL). The NCCN guidelines define FN as a single temperature of >38.3°C orally or >38.0°C over 1 hour, with an associated ANC of <500 or <1000 with a predicted decline to <500 over the next 48 hours. 5
Risk models
It should be noted that the Hosmer and Bozcuk calculators were powered to detect occurrence of FN. 7,8 However, we also applied them for the risk of any CIN. In scoring for the Hosmer calculator, points are given to each risk factor and are added together to give a final risk score. This risk score correlates to a percentage of predicted FN. The score for the Hosmer calculator is from minus 18 to plus 19, in which a score of 13 or higher correlates to a 15% predicted risk of FN, and a score of 0 or less correlates to a 1.6% risk of FN. 7 For the Bozcuk calculator, a nomogram is used to calculate risk. Individual points are given to each risk factor and are then summed to give a total that correlates to a risk of FN. The score range for the Bozcuk calculator is 0 to 300, with a score of greater than 190 correlating to a greater than 90% risk of FN, and a score of 0 correlating to a 0% predicted risk of FN. 8
For sensitivity and specificity threshold values, Hosmer reported using a risk score of 10 or above as being a reasonable value for the use of prophylactic CSF. They reported this score would predict an FN risk of about 10%, sensitivity of 24%, and specificity of 93% in detecting FN. 7 Bozcuk reported that using 110 as a cutoff value would correlate to about a 50% FN risk, sensitivity of 100%, and specificity of 49%. However, they did not suggest that value be applied as a threshold for the use of prophylactic CSF as Hosmer did. 8 Despite that, we used the thresholds of 10 and 110 for sensitivity and specificity analyses.
Regarding the current cycle of chemotherapy, the Hosmer calculator looked only at the first cycle, whereas the Bozcuk calculator looked at any cycle of chemotherapy. 7,8 In our study, we used the cycle correlating to the lowest ANC nadir the patient achieved. For example, if a patient achieved a nadir of 1,000 in cycle 1 but 200 in cycle 2, then we used the cycle 2 data to complete the calculators.
With respect to the NCCN and ASCO guidelines, we evaluated our cohort of 43 patients for the risk factors listed in the respective guidelines. If a patient had 1 or more of the risk factors, they were deemed to be high risk and therefore were recommended to receive CSF.
Results
General data
Of the 43 patients studied, 21 developed some level of CIN. Nine patients developed severe CIN, 4 developed moderate CIN, and 8 developed mild CIN. Of the severely neutropenic patients, 4 developed FN. None of the 16 patients who received prophylactic CSF developed FN, although 2 developed severe neutropenia despite CSF administration. Nadirs of ANC were seen on average during cycle 3 of chemotherapy. In all, 15 of the 43 patients achieved lowest ANC nadir during cycle 1.
Risk models
The Bozcuk calculator. A total of 22 patients had risk scores above the calculator’s threshold value of 110. Of those 22 patients, 7 developed severe CIN, 5 developed either mild or moderate CIN, and 3 developed FN. Of the remaining 21 patients who had risk scores of below 110, 2 developed severe CIN, 7 developed mild or moderate CIN, and 1 developed FN. Sensitivity and specificity values are shown in Table 2.
The Hosmer calculator. A total of 26 patients had risk scores above the calculator’s threshold value of 10. Of those 26 patients, 8 developed severe CIN, 4 developed either mild or moderate CIN, and 4 developed FN. Of the remaining 17 patients who had risk scores of less than 10, 1 developed severe CIN, 8 developed mild or moderate CIN, and none developed FN. Sensitivity and specificity values are listed in Table 2.
Current guidelines
NCCN guidelines. If one were to use the NCCN guidelines on our cohort of 43 patients, 25 would have been recommended to receive prophylactic CSF. Of those 25, 6 developed severe CIN (2 with FN), 2 moderate CIN, and 5 mild CIN. Of the 18 patients who would not have been recommended to receive CSF, 3 developed severe CIN (with 2 FN), 2 moderate CIN, and 3 mild CIN. Sensitivity and specificity values are listed in Table 2.
ASCO guidelines. Using the ASCO guidelines on our cohort of 43 patients, 38 had 1 or more of the high-risk features, and, therefore, CSF would have been considered for them. Of those 38 patients, 8 developed severe CIN (4 with FN), 4 developed moderate CIN, and 7 developed mild CIN. Of the 5 patients who would not have received CSF, 1 developed severe CIN and 1 mild CIN. Sensitivity and specificity values are listed in Table 2.
Discussion
In our study, we looked at 2 CIN risk models and compared them with the current NCCN and ASCO guidelines. The models were created to predict risk of FN, but we also looked at their predictive value for any level of CIN. To this end, we found that the Hosmer and Bozcuk calculators both were acceptable for predicting risk of severe CIN and FN. Because of the small number of patients in this study, differences in sensitivities and specificities cannot be quantitatively compared. Nevertheless, qualitatively, it can be said that both calculators were accurate in assigning high-risk scores to patients who developed severe CIN or FN. However, both calculators had many patients with high-risk scores who never developed CIN.
When comparing the 2 risk models with the NCCN and ASCO guidelines, the ASCO guidelines tended to be more liberal in their consideration of CSF use, whereas the NCCN guidelines tended to be more conservative and more similar to the 2 risk models we tested. The NCCN guidelines suggested not giving prophylactic CSF to 2 of our patients who developed FN and to not give CSF to an additional patient who developed severe CIN. The ASCO guidelines suggested considering using CSF for most of our patients, with only 5 patients not to be considered for CSF administration.
The differences in efficacy between the current guidelines and the 2 risk models may be indicative of the fact that the risk models are more accurate in assigning risk in older patients who are clinically more complicated. In our patients, the chemotherapies used were all considered to be intermediate risk, so patient-specific factors were used to guide the administration of CSF. However, because many our patients had at least 1 of the risk factors listed by the NCCN or ASCO, they were automatically deemed to be high risk and to receive prophylactic CSF.
Consequently, the Hosmer and Bozcuk calculators may be of greatest utility in more clinically complicated patients and those who have more comorbidities. The best approach may be a combination of either the NCCN or ASCO guidelines with 1 of the calculators, in our opinion the Hosmer system, for these complicated patients. Likely, the 2 risk models would not be as useful for chemotherapies deemed to have a high risk for FN because, in those situations, the efficacy and benefit of prophylactic CSF are clear. 9 Rather, their use could be beneficial in the grayer areas in which the risk is intermediate and decision-making is more difficult.
Limitations
There were several limitations in our study. First, the size of the cohort was small, and, therefore, the data that we gathered was limited in its scope. However, the goal of this study was to help provide guidance to oncologists in real-world settings about the validity and use of the available risk calculators. A further study should compare the calculators and guidelines in a much larger cohort to see if present results still hold true.
The second possible limitation of the study was our application of the Hosmer calculator because our patient population did not fit the criteria for inclusion in their original study. Hosmer had included only the first cycle of chemotherapy, whereas we included all cycles of chemotherapy. However, despite that, the calculator still performed well and could predict severe CIN and FN even with later cycles of chemotherapy. Therefore, we suggest using this calculator in any cycle of chemotherapy rather than just the first. This would expand its scope and utility in clinical practice.
Conclusions
This article provides oncologists with a comparison of 2 CIN risk models with the currently available NCCN and ASCO guidelines for use in patients with lung cancer. We prefer the Hosmer calculator over the Bozcuk calculator because of its simplicity of use and the accuracy of results. We anticipate that it may be useful and practical as an adjunct tool to the NCCN or ASCO guidelines in patients receiving intermediate-risk chemotherapy regimens. Larger studies combining the calculators and determining accuracy need to be completed to prove this hypothesis.
1. Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer. 2006;106(10):2258-2266.
2. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007;25(21):3158-3167.
3. Good Rx, Inc. Peg-filgrastim. https://www.goodrx.com/neulasta. Accessed September 2018.
4. Schilling MB, Parks C, Deeter RG. Costs and outcomes associated with hospitalized cancer patients with neutropenic complications: a retrospective study. Exp Ther Med. 2011;2(5):859-866.
5. National Comprehensive Cancer Network. Myeloid growth factors. In: NCCN Clinical Practice Guidelines in Oncology. Plymouth Meeting, PA: National Comprehensive Cancer Network; 2018.
6. Smith T, Bohlke K, Lyman GH, et al. Recommendations for the use of WBC growth factors: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2015;33(28):3199-3212.
7. Hosmer W, Malin J, Wong M. Development and validation of a prediction model for the risk of developing febrile neutropenia in the first cycle of chemotherapy among elderly patients with breast, lung, colorectal, and prostate cancer. Support Care Cancer. 2011;19(3):333-341.
8. Bozcuk H, Yıldız M, Artaç M, et al. A prospectively validated nomogram for predicting the risk of chemotherapy-induced febrile neutropenia: a multicenter study. Support Care Cancer. 2015;23(6):1759-1767.
9. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol. 2005;23(6):1178-1184.
1. Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in adult cancer patients. Cancer. 2006;106(10):2258-2266.
2. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007;25(21):3158-3167.
3. Good Rx, Inc. Peg-filgrastim. https://www.goodrx.com/neulasta. Accessed September 2018.
4. Schilling MB, Parks C, Deeter RG. Costs and outcomes associated with hospitalized cancer patients with neutropenic complications: a retrospective study. Exp Ther Med. 2011;2(5):859-866.
5. National Comprehensive Cancer Network. Myeloid growth factors. In: NCCN Clinical Practice Guidelines in Oncology. Plymouth Meeting, PA: National Comprehensive Cancer Network; 2018.
6. Smith T, Bohlke K, Lyman GH, et al. Recommendations for the use of WBC growth factors: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2015;33(28):3199-3212.
7. Hosmer W, Malin J, Wong M. Development and validation of a prediction model for the risk of developing febrile neutropenia in the first cycle of chemotherapy among elderly patients with breast, lung, colorectal, and prostate cancer. Support Care Cancer. 2011;19(3):333-341.
8. Bozcuk H, Yıldız M, Artaç M, et al. A prospectively validated nomogram for predicting the risk of chemotherapy-induced febrile neutropenia: a multicenter study. Support Care Cancer. 2015;23(6):1759-1767.
9. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double-blind, placebo-controlled phase III study. J Clin Oncol. 2005;23(6):1178-1184.
Mortality outcomes in hospitalized oncology patients after rapid response team activation
Cancer is the second leading cause of death in the United States, exceeded only by heart disease.1 Despite the overall decline in cancer death rates from 2000 through 2014, physicians struggle to accurately predict disease progression and mortality in patients with cancer who are within 6 months of death.2-8 This prognostic uncertainty makes clinical decision making difficult for patients, families, and health care providers. On a health care system level, an insight into end-of-life prognostication could also have substantial financial implications. In 2013, $74 billion was spent on cancer-related health care in the United States.9 Studies have shown that from 5% to 6% of Medicare beneficiaries with cancer consumed up to 30% of the annual Medicare payments, with a staggering 78% of costs being from acute care in the final 30 days of life.10
Rapid response teams (RRTs) were first introduced in 1995 and are now widely used at many hospitals to identify and provide critical care at the bedside of deteriorating patients outside of the intensive care unit (ICU) to prevent morbidity and mortality.11-15 Although not the original aim, RRTs are commonly activated on patients at the end of life and have therefore come to play an important role in end-of-life care.11,16 RRT activation in the oncology population is of special interest because the activation may predict higher inpatient mortality.17 In addition, RRT activation can serve as a sentinel event that fosters discussion on goals of care, change in code status, and initiation of palliative care or hospice use, particularly when also accompanied by an upgrade in level of care.11,18 As such, the ability to predict mortality after an RRT event, both inpatient and at 100 days after the event, could be of great help in deciding whether to pursue further treatments or, alternatively, palliative or hospice care.
To that end, the purpose of this study was to identify baseline patient characteristics, causes of deterioration leading to the RRT event, and vital signs and laboratory abnormalities in the peri-RRT period –
Methods and materials
A retrospective study was performed at a single, 900+ bed academic center in the northeastern United States during a 2-year study period from October 2014 through November 2016. The Institutional Review Board at Thomas Jefferson University Hospital in Philadelphia, Pennsylvania, reviewed and approved the study.
Through our institution’s RRT database, all consecutive RRT activations during the study period involving hospitalized oncology patients were reviewed. We included patients 18 years or older with a cancer diagnosis, including solid tumor and hematologic malignancy, as well as those who were status post–bone marrow transplantation (BMT), who required rapid response activation while hospitalized at our institution. We excluded patients who activated rapid response while they were in the ICU, including the BMT unit, those on the surgical floors, and those with RRT activation at other hospitals before transfer to our institution. Data for both in-hospital mortality as well as 100-day mortality for all admitted oncology patients was obtained from a separate electronic health record database at our institution from a similar time period.
Our goal was to identify patient characteristics, reasons for the RRT activation, and vital sign and laboratory abnormalities in the peri-RRT period that were associated with increased mortality, both inpatient and at 100 days after RRT activation. Our institution’s RRT database and electronic health records were accessed for data collection. Primary outcome variables for this study were inpatient and 100-day mortality post-RRT activation. We investigated the following predictor variables: age, sex, cancer diagnosis, code status at the time of RRT activation, duration from hospital admission to RRT event, length of hospital stay, time of the day the RRT event occurred (daytime vs nighttime), change in level of care (telemetry upgrade and ICU transfer), previous ICU treatment during the same hospital stay, hospice discharge, reasons cited for the RRT event (increased work of breathing, hypotension, tachyarrhythmia, change in mental status, stroke, gastrointestinal bleed, and seizure), peri-RRT lactate level, international normalized ratio (INR), hemoglobin, positive blood cultures, peri-RRT blood product administration, and scores for systemic inflammatory response syndrome (SIRS) and quick sequential organ failure assessment (qSOFA) in the 24 hours preceding the RRT activation. The SIRS includes abnormal temperature (>38°C or <36°C), heart rate of >90 bpm, increased respiratory rate of >20 times/min, and abnormal white blood cell count (>12,000 cells/mm3, <4,000/mm3, or >10% bands). Its score ranges from 0 to 4, based on the number of SIRS criteria documented. The qSOFA includes hypotension (systolic blood pressure of ≤100 mmHg), increased respiratory rate of ≥22 times/min, and altered mentation and ranges from 0 to 3 based on the number of qSOFA score documented.
Descriptive statistics were generated, and we then conducted bivariate analysis using chi-square tests or Fisher exact tests for categorical variables and simple logistic regression for continuous variables. Multivariable logistic regression models were performed to identify predictors of inpatient and 100-day mortality. Regression models were fit separately for subsets defined by the type of cancer diagnosis. Variables with P < .2 were included in the models, and backward selection method was performed, keeping variables with P < .2. The results are presented as odds ratios (OR) and 95% confidence intervals (CI). C-statistics were used to measure goodness of fit for the models. A c-statistic value of 0.5 indicates the model is not better than random chance; a value higher than 0.7 indicates moderate accuracy, whereas a value higher than 0.8 indicates strong accuracy. P < .05 was considered significant. All analyses were conducted using SAS version 9.4 (SAS Institute Inc, Cary, NC).
Results
A total of 179 hospitalized oncology patients had an RRT activation during the 2-year study period during October 2014 through November 2016. During that time, 4,654 medical oncology patients were admitted to the hospital, resulting in a rate of RRT activation of 38.4 events per 1,000 admissions. In all, 179 patients were included in the analyses for inpatient mortality, and 175 patients were included for 100-day mortality post-RRT. Patients with unknown mortality status (n = 4) at 100 days after RRT were excluded from the analyses.
The average age of the study patients was 62.3 years (standard deviation [SD], 13.3; Table 1). They comprised equal proportions of men (52%) and women (48%). Just more than half (52%) of the patients carried a diagnosis of solid malignancy, 39% of hematologic malignancy, and 9% status post-BMT. Most of the patients were full code (80%) at the time of RRT activation. The average number of days from admission to RRT event was 9.5 days (SD, 12.1). Equal proportions of RRT events took place during the daytime (52%) and nighttime (48%), and more than half of the study patients (56%) were transferred to the ICU within 24 hours of the RRT activation. Of all the study patients, 11.7% were discharged to hospice after the RRT event, and 53% required RRT evaluation for increased work of breathing. Forty-nine percent of the total study patients had peri-RRT lactate levels ≥2 mmol/L (reference range, 0.5-2.0 mmol/L), and 58% had peri-RRT INR levels ≥1.2 (reference range, 0.85-1.15). The average SIRS score was 2.8 (SD, 1.1), and the qSOFA score was 1.4 (SD, 0.8) in the 24 hours preceding the RRT activation.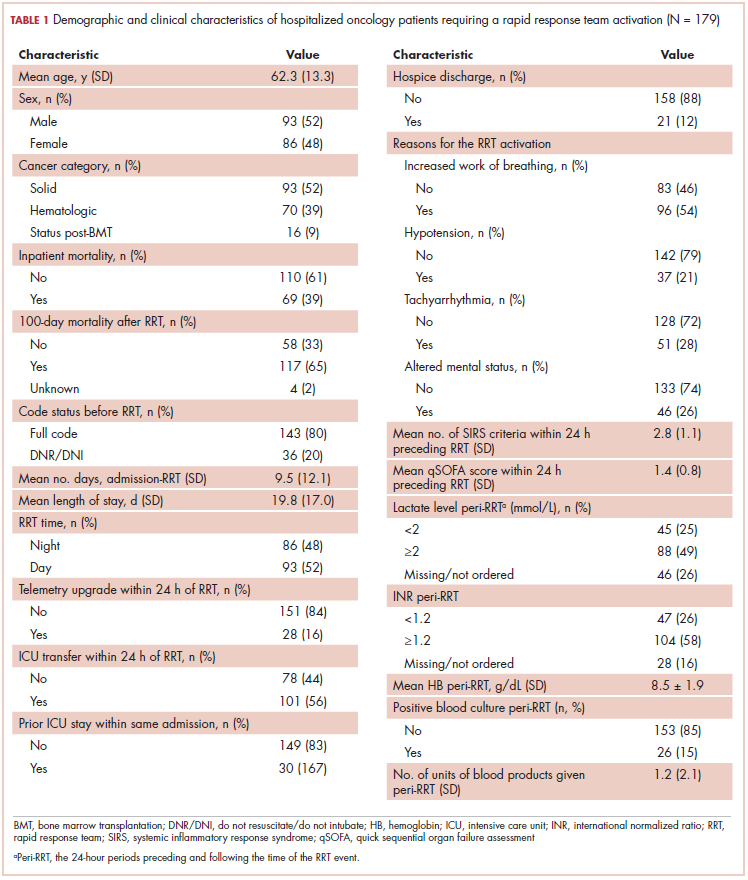
Over the 2-year study period, the inpatient mortality rate for all admitted oncology patients was 2.3% (108 deaths in 4,654 oncology inpatients), according to claims data. By comparison, of the 179 patients who required an RRT activation, 39% did not survive to discharge. When those patients were categorized based on their cancer type, 43% of the solid malignancy patients died within the same hospital stay after an RRT event, 35% of the hematologic malignancy patients died, and 25% of the status post-BMT patients died. Of the 175 patients with known mortality status at 100 days after RRT, 65% of total patients had died within that time compared with only 15.7% (347 deaths in 2,217 patients) of all admitted patients with cancer who did not experience an RRT event. When categorized based on their cancer type, significantly more patients (78%) with solid tumors had died within 100 days after RRT activation, whereas only 55% of those with a hematologic malignancy and 50% of those who were post-BMT died within the same time period.
Tables 2 and 3 present major findings from regression models with a moderate to strong level of prediction. The characteristics associated with increased odds of inpatient mortality among solid tumor patients after an RRT event were female sex (OR, 4.91; 95% CI, 1.45-16.6), increased work of breathing as the reason for the RRT activation (OR, 5.53; 95% CI, 1.69-18.1), having no lactate level ordered (OR, 5.12; 95% CI, 1.05-25.1), each unit increase in SIRS score (OR, 1.92; 95% CI, 1.01-3.66), each unit increase in qSOFA score (OR, 3.32; 95% CI, 1.45-7.56), and each unit increase in peri-RRT blood products being given (OR, 1.74; 95% CI, 1.03-2.94). Among hematologic malignancy patients, ICU transfer within 24 hours of the RRT (OR, 3.85; 95% CI, 1.14-13.0) was associated with increased inpatient mortality, whereas having no lactate level ordered (OR, 0.09; 95% CI, 0.01-0.96) was associated with lower odds of inpatient mortality.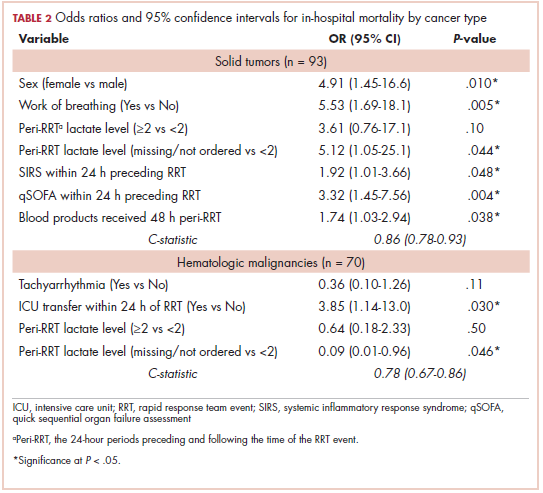
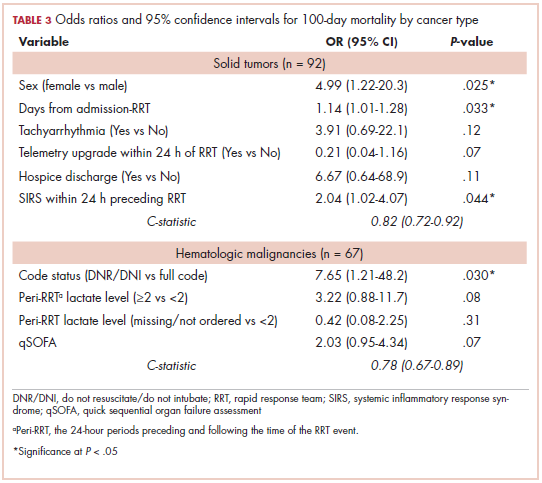
The characteristics associated with increased odds of 100-day mortality in patients with solid tumors were female sex (OR, 4.99; 95% CI, 1.22-20.3), increase in each day from admission to RRT event (OR, 1.14; 95% CI, 1.01-1.18), and each unit increase in SIRS score (OR, 2.04; 95% CI, 1.02-4.07). For hematologic malignancy patients, being do not resuscitate (DNR) or do not intubate (DNI) (OR, 7.65; 95% CI, 1.21-48.2) was associated with increased odds of 100-day mortality.
Discussion
The results of the study highlight the very high mortality rates associated with oncology patients requiring RRT activations, with 39% of patients dying within the same hospital stay and 65% dying within 100 days of the RRT event. These results are particularly notable when contrasted with the 2.3% inpatient and 15.7% 100-day postdischarge mortality rates in the total oncology patient population over a similar time period. The inpatient mortality rate after an RRT activation in our study closely resembled the rate reported by Austin and colleagues, which was 33% (hospital mortality in oncology patients cited during the time was 48.2 deaths per 1,000 patient admissions).17 Of note in our study is that solid tumor patients had higher mortality than the hematologic malignancy patients; 43% died within the same hospital stay and 78% died within 100 days, compared with 35% and 55%, respectively, in patients with hematologic malignancies. The poor prognosis of oncology patients requiring an RRT evaluation must be conveyed to the patients and families and taken into consideration by health care team to determine the most appropriate course of care subsequent to RRT activation.
Our finding that female sex is significantly and strongly associated with increased inpatient and 100-day mortality in patients with solid tumors was unexpected. The cause for this disparity remains elusive. We noted that, in our study, the following types of malignancies were more common in women than men (comparison of women vs men shown in parentheses): lung (53% vs 47%), colon (60% vs 40%), acute lymphoblastic leukemia (83% vs 17%), diffuse large B-cell lymphoma (64% vs 36%), and multiple myeloma (58% vs 42%). Whether these types of cancers are more clinically aggressive and associated with earlier mortality post-RRT could not be ascertained from our data. Gender bias in clinicians’ bedside determination of severity of illness may also play some role in this substantial mortality gap.
Among all the causes for RRT activation, increased work of breathing was the only variable associated with increased inpatient mortality in solid tumor patients. In a study by Austin and colleagues, decreased oxygen saturation was the most common reason for the RRT evaluation, though it did not reach statistical significance as a predictor of inpatient mortality.17 SIRS and qSOFA scores in the 24 hours preceding the RRT event along with peri-RRT blood product administration were all significant predictors of inpatient mortality among patients with solid tumors but were not so for those with hematologic malignancies. It is interesting to note that low hemoglobin was found to be associated with inpatient mortality in a study on 456 hospitalized patients with solid tumors (there was no data on RRT evaluation in their dataset).13 The fact that these well-validated measurements of illness severity correlate positively with RRT activation and increased mortality is intuitive and lends external credibility to other findings in this study.
In patients with hematologic malignancies, ICU transfers within 24 hours of the RRT activation were associated with 4-fold increased odds of inpatient death. This was not shown to be the case in patients with solid tumors. This should be explored in future studies because it could be crucial in conducting goals-of-care discussions in terminally ill cancer patients. The study also showed that patients with hematologic malignancies who were DNR or DNI were associated with almost 8-fold increased odds of 100-day mortality. This argues for a fair predictive ability of the care teams in this particular subgroup. Conversely, hospice referral is underused; of the patients that died at 100 days after the RRT event, only 16.2% were referred to hospice at the time of discharge.
Limitations
Limitations of the study include its retrospective nature at a single medical center on a small group of study participants. Variables such as lactate dehydrogenase level and Eastern Conference Oncology Group Performance Status, which have been found to be predictive of increased mortality in hospitalized oncology patients,19 were not consistently available for analysis in the data set. We had 4 patients whose mortality status was not known at 100 days and were excluded from the study. Because of a lack of documentation, we were also not able to reliably collect the data on patients with multiple RRT events. This presumably would be associated with increased mortality on its own. We only included the data associated with the earliest RRT activation in our electronic health records.
In addition, it is important to note that 26% and 16% of the study patients had missing lactate and INR values, respectively. Given the small size of the study and the unclear significance of the missing lactate and INR, we opted to include the patients with the missing data for final analyses of the regression models. The significance of a care team not ordering a lactate level is perhaps associated with the reason for RRT activation (ie, the patient seemed to be less ill) and perhaps could be associated with non–sepsis-related RRT events.
Conclusions
This study reports on the outcomes of oncology patients admitted to the hospital whose clinical deterioration required activation of a rapid response team. Female sex, increased qSOFA and SIRS scores in the 24 hours preceding the RRT event, and the need for blood product administrations around the time of the RRT event correlated with increased inpatient mortality. Hospitalized oncology patients’ d undestood and response evaluation if perPatientoutcomes, both regarding inpatient and 100-day mortality, demonstrated surprisingly poor survival, with solid malignancy patients bearing significantly higher burden of both inpatient mortality and mortality at 100 days after the RRT event. The findings from the study could help patients, families, and providers make informed decisions regarding advance care and end-of-life planning for terminally ill cancer patients.
The Cancer Center Support Grant 5P30CA056036-17 and the Biostatistics Shared Resource and Thomas Jefferson University Hospital’s Rapid Response Team (RRT) committee.
1. National Center for Health Statistics. Health, United States, 2016: with Chartbook on long-term trends in health. Hyattsville, MD: National Center for Health Statistics; 2017.
2. Lambden J, Zhang B, Friedlander R, Prigerson HG. Accuracy of oncologists’ life-expectancy estimates recalled by their advanced cancer patients: correlates and outcomes. J Palliat Med. 2016;19(12):1296-1303.
3. Maltoni M, Caraceni A, Brunelli C, et al. Prognostic factors in advanced cancer patients: evidence-based clinical recommendations—a study by the Steering Committee of the European Association for Palliative Care. J Clin Oncol. 2005;23(25):6240-6248.
4. Viganó A, Bruera E, Jhangri GS, Newman SC, Fields AL, Suarez-Almazor ME. Clinical survival predictors in patients with advanced cancer. Arch Intern Med. 2000;160(6):861-868.
5. Hauser CA, Stockler MR, Tattersall MH. Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review. Support Care Cancer. 2006;14(10):999-1011.
6. Al-Zahrani AS, El-Kashif AT, Mohammad AA, Elsamany S, Alsirafy SA. Prediction of in-hospital mortality of patients with advanced cancer using the Chuang Prognostic Score. Am J Hosp Palliat Med. 2013;30(7):707-711.
7. Hui D, Kilgore K, Fellman B, et al. Development and cross-validation of the in-hospital mortality prediction in advanced cancer patients score: a preliminary study. J Palliat Med. 2012;15(8):902-909.
8. Shouval R, Labopin M, Bondi O, et al. Prediction of allogeneic hematopoietic stem-cell transplantation mortality 100 days after transplantation using a machine learning algorithm: a European group for blood and marrow transplantation acute leukemia working party retrospective data mining study. J Clin Oncol. 2015;33(28):3144-3151.
9. Agency for Healthcare Research and Quality. Total expenses and percent distribution for selected conditions by type of service: United States, 2013. Medical Expenditure Panel Survey website. https://meps.ahrq.gov/mepsweb/data_stats/tables_compendia_hh_interactive.jsp?_SERVICE=MEPSSocket0&_PROGRAM=MEPSPGM.TC.SAS&File=HCFY2013&Table=HCFY2012_CNDXP_C&_Debug=. Accessed November 10, 2018.
10. McCall N. Utilization and costs of Medicare services by beneficiaries in their last year of life. Med Care. 1984;22(4):329-342.
11. Jones D, Moran J, Winters B, Welch J. The rapid response system and end-of-life care. Curr Opin Crit Care. 2013;19(6):616-623.
12. Solomon RS, Corwin GS, Barclay DC, Quddusi SF, Dannenberg MD. Effectiveness of rapid response teams on rates of in‐hospital cardiopulmonary arrest and mortality: a systematic review and meta‐analysis. J Hosp Med. 2016;11(6):438-445.
13. Jung B, Daurat A, De Jong A, et al. Rapid response team and hospital mortality in hospitalized patients. Intensive Care Med. 2016;42(4):494-504.
14. Sulistio M, Franco M, Vo A, Poon P, William L. Hospital rapid response team and patients with life-limiting illness: a multicentre retrospective cohort study. Palliat Med. 2015;29(4):302-309.
15. Wang J, Hahn SS, Kline M, Cohen RI. Early in-hospital clinical deterioration is not predicted by severity of illness, functional status, or comorbidity. Int J Gen Med. 2017;10:329-334.
16. Dargin JM, Mackey CG, Lei Y, Liesching TN. Resource utilization and end‐of‐life care in a US hospital following medical emergency team‐implemented do not resuscitate orders. J Hosp Med. 2014;9(6):372-378.
17. Austin CA, Hanzaker C, Stafford R, et al. Utilization of rapid response resources and outcomes in a comprehensive cancer center. Crit Care Med. 2014;42(4):905-909.
18. Smith RL, Hayashi VN, Lee YI, Navarro-Mariazeta L, Felner K. The medical emergency team call: a sentinel event that triggers goals of care discussion. Crit Care Med. 2014;42(2):322-327.
19. Bozcuk H, Koyuncu E, Yildiz M, et al. A simple and accurate prediction model to estimate the intrahospital mortality risk of hospitalised cancer patients. Int J Clin Pract. 2004;58(11):1014-1019.
Cancer is the second leading cause of death in the United States, exceeded only by heart disease.1 Despite the overall decline in cancer death rates from 2000 through 2014, physicians struggle to accurately predict disease progression and mortality in patients with cancer who are within 6 months of death.2-8 This prognostic uncertainty makes clinical decision making difficult for patients, families, and health care providers. On a health care system level, an insight into end-of-life prognostication could also have substantial financial implications. In 2013, $74 billion was spent on cancer-related health care in the United States.9 Studies have shown that from 5% to 6% of Medicare beneficiaries with cancer consumed up to 30% of the annual Medicare payments, with a staggering 78% of costs being from acute care in the final 30 days of life.10
Rapid response teams (RRTs) were first introduced in 1995 and are now widely used at many hospitals to identify and provide critical care at the bedside of deteriorating patients outside of the intensive care unit (ICU) to prevent morbidity and mortality.11-15 Although not the original aim, RRTs are commonly activated on patients at the end of life and have therefore come to play an important role in end-of-life care.11,16 RRT activation in the oncology population is of special interest because the activation may predict higher inpatient mortality.17 In addition, RRT activation can serve as a sentinel event that fosters discussion on goals of care, change in code status, and initiation of palliative care or hospice use, particularly when also accompanied by an upgrade in level of care.11,18 As such, the ability to predict mortality after an RRT event, both inpatient and at 100 days after the event, could be of great help in deciding whether to pursue further treatments or, alternatively, palliative or hospice care.
To that end, the purpose of this study was to identify baseline patient characteristics, causes of deterioration leading to the RRT event, and vital signs and laboratory abnormalities in the peri-RRT period –
Methods and materials
A retrospective study was performed at a single, 900+ bed academic center in the northeastern United States during a 2-year study period from October 2014 through November 2016. The Institutional Review Board at Thomas Jefferson University Hospital in Philadelphia, Pennsylvania, reviewed and approved the study.
Through our institution’s RRT database, all consecutive RRT activations during the study period involving hospitalized oncology patients were reviewed. We included patients 18 years or older with a cancer diagnosis, including solid tumor and hematologic malignancy, as well as those who were status post–bone marrow transplantation (BMT), who required rapid response activation while hospitalized at our institution. We excluded patients who activated rapid response while they were in the ICU, including the BMT unit, those on the surgical floors, and those with RRT activation at other hospitals before transfer to our institution. Data for both in-hospital mortality as well as 100-day mortality for all admitted oncology patients was obtained from a separate electronic health record database at our institution from a similar time period.
Our goal was to identify patient characteristics, reasons for the RRT activation, and vital sign and laboratory abnormalities in the peri-RRT period that were associated with increased mortality, both inpatient and at 100 days after RRT activation. Our institution’s RRT database and electronic health records were accessed for data collection. Primary outcome variables for this study were inpatient and 100-day mortality post-RRT activation. We investigated the following predictor variables: age, sex, cancer diagnosis, code status at the time of RRT activation, duration from hospital admission to RRT event, length of hospital stay, time of the day the RRT event occurred (daytime vs nighttime), change in level of care (telemetry upgrade and ICU transfer), previous ICU treatment during the same hospital stay, hospice discharge, reasons cited for the RRT event (increased work of breathing, hypotension, tachyarrhythmia, change in mental status, stroke, gastrointestinal bleed, and seizure), peri-RRT lactate level, international normalized ratio (INR), hemoglobin, positive blood cultures, peri-RRT blood product administration, and scores for systemic inflammatory response syndrome (SIRS) and quick sequential organ failure assessment (qSOFA) in the 24 hours preceding the RRT activation. The SIRS includes abnormal temperature (>38°C or <36°C), heart rate of >90 bpm, increased respiratory rate of >20 times/min, and abnormal white blood cell count (>12,000 cells/mm3, <4,000/mm3, or >10% bands). Its score ranges from 0 to 4, based on the number of SIRS criteria documented. The qSOFA includes hypotension (systolic blood pressure of ≤100 mmHg), increased respiratory rate of ≥22 times/min, and altered mentation and ranges from 0 to 3 based on the number of qSOFA score documented.
Descriptive statistics were generated, and we then conducted bivariate analysis using chi-square tests or Fisher exact tests for categorical variables and simple logistic regression for continuous variables. Multivariable logistic regression models were performed to identify predictors of inpatient and 100-day mortality. Regression models were fit separately for subsets defined by the type of cancer diagnosis. Variables with P < .2 were included in the models, and backward selection method was performed, keeping variables with P < .2. The results are presented as odds ratios (OR) and 95% confidence intervals (CI). C-statistics were used to measure goodness of fit for the models. A c-statistic value of 0.5 indicates the model is not better than random chance; a value higher than 0.7 indicates moderate accuracy, whereas a value higher than 0.8 indicates strong accuracy. P < .05 was considered significant. All analyses were conducted using SAS version 9.4 (SAS Institute Inc, Cary, NC).
Results
A total of 179 hospitalized oncology patients had an RRT activation during the 2-year study period during October 2014 through November 2016. During that time, 4,654 medical oncology patients were admitted to the hospital, resulting in a rate of RRT activation of 38.4 events per 1,000 admissions. In all, 179 patients were included in the analyses for inpatient mortality, and 175 patients were included for 100-day mortality post-RRT. Patients with unknown mortality status (n = 4) at 100 days after RRT were excluded from the analyses.
The average age of the study patients was 62.3 years (standard deviation [SD], 13.3; Table 1). They comprised equal proportions of men (52%) and women (48%). Just more than half (52%) of the patients carried a diagnosis of solid malignancy, 39% of hematologic malignancy, and 9% status post-BMT. Most of the patients were full code (80%) at the time of RRT activation. The average number of days from admission to RRT event was 9.5 days (SD, 12.1). Equal proportions of RRT events took place during the daytime (52%) and nighttime (48%), and more than half of the study patients (56%) were transferred to the ICU within 24 hours of the RRT activation. Of all the study patients, 11.7% were discharged to hospice after the RRT event, and 53% required RRT evaluation for increased work of breathing. Forty-nine percent of the total study patients had peri-RRT lactate levels ≥2 mmol/L (reference range, 0.5-2.0 mmol/L), and 58% had peri-RRT INR levels ≥1.2 (reference range, 0.85-1.15). The average SIRS score was 2.8 (SD, 1.1), and the qSOFA score was 1.4 (SD, 0.8) in the 24 hours preceding the RRT activation.
Over the 2-year study period, the inpatient mortality rate for all admitted oncology patients was 2.3% (108 deaths in 4,654 oncology inpatients), according to claims data. By comparison, of the 179 patients who required an RRT activation, 39% did not survive to discharge. When those patients were categorized based on their cancer type, 43% of the solid malignancy patients died within the same hospital stay after an RRT event, 35% of the hematologic malignancy patients died, and 25% of the status post-BMT patients died. Of the 175 patients with known mortality status at 100 days after RRT, 65% of total patients had died within that time compared with only 15.7% (347 deaths in 2,217 patients) of all admitted patients with cancer who did not experience an RRT event. When categorized based on their cancer type, significantly more patients (78%) with solid tumors had died within 100 days after RRT activation, whereas only 55% of those with a hematologic malignancy and 50% of those who were post-BMT died within the same time period.
Tables 2 and 3 present major findings from regression models with a moderate to strong level of prediction. The characteristics associated with increased odds of inpatient mortality among solid tumor patients after an RRT event were female sex (OR, 4.91; 95% CI, 1.45-16.6), increased work of breathing as the reason for the RRT activation (OR, 5.53; 95% CI, 1.69-18.1), having no lactate level ordered (OR, 5.12; 95% CI, 1.05-25.1), each unit increase in SIRS score (OR, 1.92; 95% CI, 1.01-3.66), each unit increase in qSOFA score (OR, 3.32; 95% CI, 1.45-7.56), and each unit increase in peri-RRT blood products being given (OR, 1.74; 95% CI, 1.03-2.94). Among hematologic malignancy patients, ICU transfer within 24 hours of the RRT (OR, 3.85; 95% CI, 1.14-13.0) was associated with increased inpatient mortality, whereas having no lactate level ordered (OR, 0.09; 95% CI, 0.01-0.96) was associated with lower odds of inpatient mortality.
The characteristics associated with increased odds of 100-day mortality in patients with solid tumors were female sex (OR, 4.99; 95% CI, 1.22-20.3), increase in each day from admission to RRT event (OR, 1.14; 95% CI, 1.01-1.18), and each unit increase in SIRS score (OR, 2.04; 95% CI, 1.02-4.07). For hematologic malignancy patients, being do not resuscitate (DNR) or do not intubate (DNI) (OR, 7.65; 95% CI, 1.21-48.2) was associated with increased odds of 100-day mortality.
Discussion
The results of the study highlight the very high mortality rates associated with oncology patients requiring RRT activations, with 39% of patients dying within the same hospital stay and 65% dying within 100 days of the RRT event. These results are particularly notable when contrasted with the 2.3% inpatient and 15.7% 100-day postdischarge mortality rates in the total oncology patient population over a similar time period. The inpatient mortality rate after an RRT activation in our study closely resembled the rate reported by Austin and colleagues, which was 33% (hospital mortality in oncology patients cited during the time was 48.2 deaths per 1,000 patient admissions).17 Of note in our study is that solid tumor patients had higher mortality than the hematologic malignancy patients; 43% died within the same hospital stay and 78% died within 100 days, compared with 35% and 55%, respectively, in patients with hematologic malignancies. The poor prognosis of oncology patients requiring an RRT evaluation must be conveyed to the patients and families and taken into consideration by health care team to determine the most appropriate course of care subsequent to RRT activation.
Our finding that female sex is significantly and strongly associated with increased inpatient and 100-day mortality in patients with solid tumors was unexpected. The cause for this disparity remains elusive. We noted that, in our study, the following types of malignancies were more common in women than men (comparison of women vs men shown in parentheses): lung (53% vs 47%), colon (60% vs 40%), acute lymphoblastic leukemia (83% vs 17%), diffuse large B-cell lymphoma (64% vs 36%), and multiple myeloma (58% vs 42%). Whether these types of cancers are more clinically aggressive and associated with earlier mortality post-RRT could not be ascertained from our data. Gender bias in clinicians’ bedside determination of severity of illness may also play some role in this substantial mortality gap.
Among all the causes for RRT activation, increased work of breathing was the only variable associated with increased inpatient mortality in solid tumor patients. In a study by Austin and colleagues, decreased oxygen saturation was the most common reason for the RRT evaluation, though it did not reach statistical significance as a predictor of inpatient mortality.17 SIRS and qSOFA scores in the 24 hours preceding the RRT event along with peri-RRT blood product administration were all significant predictors of inpatient mortality among patients with solid tumors but were not so for those with hematologic malignancies. It is interesting to note that low hemoglobin was found to be associated with inpatient mortality in a study on 456 hospitalized patients with solid tumors (there was no data on RRT evaluation in their dataset).13 The fact that these well-validated measurements of illness severity correlate positively with RRT activation and increased mortality is intuitive and lends external credibility to other findings in this study.
In patients with hematologic malignancies, ICU transfers within 24 hours of the RRT activation were associated with 4-fold increased odds of inpatient death. This was not shown to be the case in patients with solid tumors. This should be explored in future studies because it could be crucial in conducting goals-of-care discussions in terminally ill cancer patients. The study also showed that patients with hematologic malignancies who were DNR or DNI were associated with almost 8-fold increased odds of 100-day mortality. This argues for a fair predictive ability of the care teams in this particular subgroup. Conversely, hospice referral is underused; of the patients that died at 100 days after the RRT event, only 16.2% were referred to hospice at the time of discharge.
Limitations
Limitations of the study include its retrospective nature at a single medical center on a small group of study participants. Variables such as lactate dehydrogenase level and Eastern Conference Oncology Group Performance Status, which have been found to be predictive of increased mortality in hospitalized oncology patients,19 were not consistently available for analysis in the data set. We had 4 patients whose mortality status was not known at 100 days and were excluded from the study. Because of a lack of documentation, we were also not able to reliably collect the data on patients with multiple RRT events. This presumably would be associated with increased mortality on its own. We only included the data associated with the earliest RRT activation in our electronic health records.
In addition, it is important to note that 26% and 16% of the study patients had missing lactate and INR values, respectively. Given the small size of the study and the unclear significance of the missing lactate and INR, we opted to include the patients with the missing data for final analyses of the regression models. The significance of a care team not ordering a lactate level is perhaps associated with the reason for RRT activation (ie, the patient seemed to be less ill) and perhaps could be associated with non–sepsis-related RRT events.
Conclusions
This study reports on the outcomes of oncology patients admitted to the hospital whose clinical deterioration required activation of a rapid response team. Female sex, increased qSOFA and SIRS scores in the 24 hours preceding the RRT event, and the need for blood product administrations around the time of the RRT event correlated with increased inpatient mortality. Hospitalized oncology patients’ d undestood and response evaluation if perPatientoutcomes, both regarding inpatient and 100-day mortality, demonstrated surprisingly poor survival, with solid malignancy patients bearing significantly higher burden of both inpatient mortality and mortality at 100 days after the RRT event. The findings from the study could help patients, families, and providers make informed decisions regarding advance care and end-of-life planning for terminally ill cancer patients.
The Cancer Center Support Grant 5P30CA056036-17 and the Biostatistics Shared Resource and Thomas Jefferson University Hospital’s Rapid Response Team (RRT) committee.
Cancer is the second leading cause of death in the United States, exceeded only by heart disease.1 Despite the overall decline in cancer death rates from 2000 through 2014, physicians struggle to accurately predict disease progression and mortality in patients with cancer who are within 6 months of death.2-8 This prognostic uncertainty makes clinical decision making difficult for patients, families, and health care providers. On a health care system level, an insight into end-of-life prognostication could also have substantial financial implications. In 2013, $74 billion was spent on cancer-related health care in the United States.9 Studies have shown that from 5% to 6% of Medicare beneficiaries with cancer consumed up to 30% of the annual Medicare payments, with a staggering 78% of costs being from acute care in the final 30 days of life.10
Rapid response teams (RRTs) were first introduced in 1995 and are now widely used at many hospitals to identify and provide critical care at the bedside of deteriorating patients outside of the intensive care unit (ICU) to prevent morbidity and mortality.11-15 Although not the original aim, RRTs are commonly activated on patients at the end of life and have therefore come to play an important role in end-of-life care.11,16 RRT activation in the oncology population is of special interest because the activation may predict higher inpatient mortality.17 In addition, RRT activation can serve as a sentinel event that fosters discussion on goals of care, change in code status, and initiation of palliative care or hospice use, particularly when also accompanied by an upgrade in level of care.11,18 As such, the ability to predict mortality after an RRT event, both inpatient and at 100 days after the event, could be of great help in deciding whether to pursue further treatments or, alternatively, palliative or hospice care.
To that end, the purpose of this study was to identify baseline patient characteristics, causes of deterioration leading to the RRT event, and vital signs and laboratory abnormalities in the peri-RRT period –
Methods and materials
A retrospective study was performed at a single, 900+ bed academic center in the northeastern United States during a 2-year study period from October 2014 through November 2016. The Institutional Review Board at Thomas Jefferson University Hospital in Philadelphia, Pennsylvania, reviewed and approved the study.
Through our institution’s RRT database, all consecutive RRT activations during the study period involving hospitalized oncology patients were reviewed. We included patients 18 years or older with a cancer diagnosis, including solid tumor and hematologic malignancy, as well as those who were status post–bone marrow transplantation (BMT), who required rapid response activation while hospitalized at our institution. We excluded patients who activated rapid response while they were in the ICU, including the BMT unit, those on the surgical floors, and those with RRT activation at other hospitals before transfer to our institution. Data for both in-hospital mortality as well as 100-day mortality for all admitted oncology patients was obtained from a separate electronic health record database at our institution from a similar time period.
Our goal was to identify patient characteristics, reasons for the RRT activation, and vital sign and laboratory abnormalities in the peri-RRT period that were associated with increased mortality, both inpatient and at 100 days after RRT activation. Our institution’s RRT database and electronic health records were accessed for data collection. Primary outcome variables for this study were inpatient and 100-day mortality post-RRT activation. We investigated the following predictor variables: age, sex, cancer diagnosis, code status at the time of RRT activation, duration from hospital admission to RRT event, length of hospital stay, time of the day the RRT event occurred (daytime vs nighttime), change in level of care (telemetry upgrade and ICU transfer), previous ICU treatment during the same hospital stay, hospice discharge, reasons cited for the RRT event (increased work of breathing, hypotension, tachyarrhythmia, change in mental status, stroke, gastrointestinal bleed, and seizure), peri-RRT lactate level, international normalized ratio (INR), hemoglobin, positive blood cultures, peri-RRT blood product administration, and scores for systemic inflammatory response syndrome (SIRS) and quick sequential organ failure assessment (qSOFA) in the 24 hours preceding the RRT activation. The SIRS includes abnormal temperature (>38°C or <36°C), heart rate of >90 bpm, increased respiratory rate of >20 times/min, and abnormal white blood cell count (>12,000 cells/mm3, <4,000/mm3, or >10% bands). Its score ranges from 0 to 4, based on the number of SIRS criteria documented. The qSOFA includes hypotension (systolic blood pressure of ≤100 mmHg), increased respiratory rate of ≥22 times/min, and altered mentation and ranges from 0 to 3 based on the number of qSOFA score documented.
Descriptive statistics were generated, and we then conducted bivariate analysis using chi-square tests or Fisher exact tests for categorical variables and simple logistic regression for continuous variables. Multivariable logistic regression models were performed to identify predictors of inpatient and 100-day mortality. Regression models were fit separately for subsets defined by the type of cancer diagnosis. Variables with P < .2 were included in the models, and backward selection method was performed, keeping variables with P < .2. The results are presented as odds ratios (OR) and 95% confidence intervals (CI). C-statistics were used to measure goodness of fit for the models. A c-statistic value of 0.5 indicates the model is not better than random chance; a value higher than 0.7 indicates moderate accuracy, whereas a value higher than 0.8 indicates strong accuracy. P < .05 was considered significant. All analyses were conducted using SAS version 9.4 (SAS Institute Inc, Cary, NC).
Results
A total of 179 hospitalized oncology patients had an RRT activation during the 2-year study period during October 2014 through November 2016. During that time, 4,654 medical oncology patients were admitted to the hospital, resulting in a rate of RRT activation of 38.4 events per 1,000 admissions. In all, 179 patients were included in the analyses for inpatient mortality, and 175 patients were included for 100-day mortality post-RRT. Patients with unknown mortality status (n = 4) at 100 days after RRT were excluded from the analyses.
The average age of the study patients was 62.3 years (standard deviation [SD], 13.3; Table 1). They comprised equal proportions of men (52%) and women (48%). Just more than half (52%) of the patients carried a diagnosis of solid malignancy, 39% of hematologic malignancy, and 9% status post-BMT. Most of the patients were full code (80%) at the time of RRT activation. The average number of days from admission to RRT event was 9.5 days (SD, 12.1). Equal proportions of RRT events took place during the daytime (52%) and nighttime (48%), and more than half of the study patients (56%) were transferred to the ICU within 24 hours of the RRT activation. Of all the study patients, 11.7% were discharged to hospice after the RRT event, and 53% required RRT evaluation for increased work of breathing. Forty-nine percent of the total study patients had peri-RRT lactate levels ≥2 mmol/L (reference range, 0.5-2.0 mmol/L), and 58% had peri-RRT INR levels ≥1.2 (reference range, 0.85-1.15). The average SIRS score was 2.8 (SD, 1.1), and the qSOFA score was 1.4 (SD, 0.8) in the 24 hours preceding the RRT activation.
Over the 2-year study period, the inpatient mortality rate for all admitted oncology patients was 2.3% (108 deaths in 4,654 oncology inpatients), according to claims data. By comparison, of the 179 patients who required an RRT activation, 39% did not survive to discharge. When those patients were categorized based on their cancer type, 43% of the solid malignancy patients died within the same hospital stay after an RRT event, 35% of the hematologic malignancy patients died, and 25% of the status post-BMT patients died. Of the 175 patients with known mortality status at 100 days after RRT, 65% of total patients had died within that time compared with only 15.7% (347 deaths in 2,217 patients) of all admitted patients with cancer who did not experience an RRT event. When categorized based on their cancer type, significantly more patients (78%) with solid tumors had died within 100 days after RRT activation, whereas only 55% of those with a hematologic malignancy and 50% of those who were post-BMT died within the same time period.
Tables 2 and 3 present major findings from regression models with a moderate to strong level of prediction. The characteristics associated with increased odds of inpatient mortality among solid tumor patients after an RRT event were female sex (OR, 4.91; 95% CI, 1.45-16.6), increased work of breathing as the reason for the RRT activation (OR, 5.53; 95% CI, 1.69-18.1), having no lactate level ordered (OR, 5.12; 95% CI, 1.05-25.1), each unit increase in SIRS score (OR, 1.92; 95% CI, 1.01-3.66), each unit increase in qSOFA score (OR, 3.32; 95% CI, 1.45-7.56), and each unit increase in peri-RRT blood products being given (OR, 1.74; 95% CI, 1.03-2.94). Among hematologic malignancy patients, ICU transfer within 24 hours of the RRT (OR, 3.85; 95% CI, 1.14-13.0) was associated with increased inpatient mortality, whereas having no lactate level ordered (OR, 0.09; 95% CI, 0.01-0.96) was associated with lower odds of inpatient mortality.
The characteristics associated with increased odds of 100-day mortality in patients with solid tumors were female sex (OR, 4.99; 95% CI, 1.22-20.3), increase in each day from admission to RRT event (OR, 1.14; 95% CI, 1.01-1.18), and each unit increase in SIRS score (OR, 2.04; 95% CI, 1.02-4.07). For hematologic malignancy patients, being do not resuscitate (DNR) or do not intubate (DNI) (OR, 7.65; 95% CI, 1.21-48.2) was associated with increased odds of 100-day mortality.
Discussion
The results of the study highlight the very high mortality rates associated with oncology patients requiring RRT activations, with 39% of patients dying within the same hospital stay and 65% dying within 100 days of the RRT event. These results are particularly notable when contrasted with the 2.3% inpatient and 15.7% 100-day postdischarge mortality rates in the total oncology patient population over a similar time period. The inpatient mortality rate after an RRT activation in our study closely resembled the rate reported by Austin and colleagues, which was 33% (hospital mortality in oncology patients cited during the time was 48.2 deaths per 1,000 patient admissions).17 Of note in our study is that solid tumor patients had higher mortality than the hematologic malignancy patients; 43% died within the same hospital stay and 78% died within 100 days, compared with 35% and 55%, respectively, in patients with hematologic malignancies. The poor prognosis of oncology patients requiring an RRT evaluation must be conveyed to the patients and families and taken into consideration by health care team to determine the most appropriate course of care subsequent to RRT activation.
Our finding that female sex is significantly and strongly associated with increased inpatient and 100-day mortality in patients with solid tumors was unexpected. The cause for this disparity remains elusive. We noted that, in our study, the following types of malignancies were more common in women than men (comparison of women vs men shown in parentheses): lung (53% vs 47%), colon (60% vs 40%), acute lymphoblastic leukemia (83% vs 17%), diffuse large B-cell lymphoma (64% vs 36%), and multiple myeloma (58% vs 42%). Whether these types of cancers are more clinically aggressive and associated with earlier mortality post-RRT could not be ascertained from our data. Gender bias in clinicians’ bedside determination of severity of illness may also play some role in this substantial mortality gap.
Among all the causes for RRT activation, increased work of breathing was the only variable associated with increased inpatient mortality in solid tumor patients. In a study by Austin and colleagues, decreased oxygen saturation was the most common reason for the RRT evaluation, though it did not reach statistical significance as a predictor of inpatient mortality.17 SIRS and qSOFA scores in the 24 hours preceding the RRT event along with peri-RRT blood product administration were all significant predictors of inpatient mortality among patients with solid tumors but were not so for those with hematologic malignancies. It is interesting to note that low hemoglobin was found to be associated with inpatient mortality in a study on 456 hospitalized patients with solid tumors (there was no data on RRT evaluation in their dataset).13 The fact that these well-validated measurements of illness severity correlate positively with RRT activation and increased mortality is intuitive and lends external credibility to other findings in this study.
In patients with hematologic malignancies, ICU transfers within 24 hours of the RRT activation were associated with 4-fold increased odds of inpatient death. This was not shown to be the case in patients with solid tumors. This should be explored in future studies because it could be crucial in conducting goals-of-care discussions in terminally ill cancer patients. The study also showed that patients with hematologic malignancies who were DNR or DNI were associated with almost 8-fold increased odds of 100-day mortality. This argues for a fair predictive ability of the care teams in this particular subgroup. Conversely, hospice referral is underused; of the patients that died at 100 days after the RRT event, only 16.2% were referred to hospice at the time of discharge.
Limitations
Limitations of the study include its retrospective nature at a single medical center on a small group of study participants. Variables such as lactate dehydrogenase level and Eastern Conference Oncology Group Performance Status, which have been found to be predictive of increased mortality in hospitalized oncology patients,19 were not consistently available for analysis in the data set. We had 4 patients whose mortality status was not known at 100 days and were excluded from the study. Because of a lack of documentation, we were also not able to reliably collect the data on patients with multiple RRT events. This presumably would be associated with increased mortality on its own. We only included the data associated with the earliest RRT activation in our electronic health records.
In addition, it is important to note that 26% and 16% of the study patients had missing lactate and INR values, respectively. Given the small size of the study and the unclear significance of the missing lactate and INR, we opted to include the patients with the missing data for final analyses of the regression models. The significance of a care team not ordering a lactate level is perhaps associated with the reason for RRT activation (ie, the patient seemed to be less ill) and perhaps could be associated with non–sepsis-related RRT events.
Conclusions
This study reports on the outcomes of oncology patients admitted to the hospital whose clinical deterioration required activation of a rapid response team. Female sex, increased qSOFA and SIRS scores in the 24 hours preceding the RRT event, and the need for blood product administrations around the time of the RRT event correlated with increased inpatient mortality. Hospitalized oncology patients’ d undestood and response evaluation if perPatientoutcomes, both regarding inpatient and 100-day mortality, demonstrated surprisingly poor survival, with solid malignancy patients bearing significantly higher burden of both inpatient mortality and mortality at 100 days after the RRT event. The findings from the study could help patients, families, and providers make informed decisions regarding advance care and end-of-life planning for terminally ill cancer patients.
The Cancer Center Support Grant 5P30CA056036-17 and the Biostatistics Shared Resource and Thomas Jefferson University Hospital’s Rapid Response Team (RRT) committee.
1. National Center for Health Statistics. Health, United States, 2016: with Chartbook on long-term trends in health. Hyattsville, MD: National Center for Health Statistics; 2017.
2. Lambden J, Zhang B, Friedlander R, Prigerson HG. Accuracy of oncologists’ life-expectancy estimates recalled by their advanced cancer patients: correlates and outcomes. J Palliat Med. 2016;19(12):1296-1303.
3. Maltoni M, Caraceni A, Brunelli C, et al. Prognostic factors in advanced cancer patients: evidence-based clinical recommendations—a study by the Steering Committee of the European Association for Palliative Care. J Clin Oncol. 2005;23(25):6240-6248.
4. Viganó A, Bruera E, Jhangri GS, Newman SC, Fields AL, Suarez-Almazor ME. Clinical survival predictors in patients with advanced cancer. Arch Intern Med. 2000;160(6):861-868.
5. Hauser CA, Stockler MR, Tattersall MH. Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review. Support Care Cancer. 2006;14(10):999-1011.
6. Al-Zahrani AS, El-Kashif AT, Mohammad AA, Elsamany S, Alsirafy SA. Prediction of in-hospital mortality of patients with advanced cancer using the Chuang Prognostic Score. Am J Hosp Palliat Med. 2013;30(7):707-711.
7. Hui D, Kilgore K, Fellman B, et al. Development and cross-validation of the in-hospital mortality prediction in advanced cancer patients score: a preliminary study. J Palliat Med. 2012;15(8):902-909.
8. Shouval R, Labopin M, Bondi O, et al. Prediction of allogeneic hematopoietic stem-cell transplantation mortality 100 days after transplantation using a machine learning algorithm: a European group for blood and marrow transplantation acute leukemia working party retrospective data mining study. J Clin Oncol. 2015;33(28):3144-3151.
9. Agency for Healthcare Research and Quality. Total expenses and percent distribution for selected conditions by type of service: United States, 2013. Medical Expenditure Panel Survey website. https://meps.ahrq.gov/mepsweb/data_stats/tables_compendia_hh_interactive.jsp?_SERVICE=MEPSSocket0&_PROGRAM=MEPSPGM.TC.SAS&File=HCFY2013&Table=HCFY2012_CNDXP_C&_Debug=. Accessed November 10, 2018.
10. McCall N. Utilization and costs of Medicare services by beneficiaries in their last year of life. Med Care. 1984;22(4):329-342.
11. Jones D, Moran J, Winters B, Welch J. The rapid response system and end-of-life care. Curr Opin Crit Care. 2013;19(6):616-623.
12. Solomon RS, Corwin GS, Barclay DC, Quddusi SF, Dannenberg MD. Effectiveness of rapid response teams on rates of in‐hospital cardiopulmonary arrest and mortality: a systematic review and meta‐analysis. J Hosp Med. 2016;11(6):438-445.
13. Jung B, Daurat A, De Jong A, et al. Rapid response team and hospital mortality in hospitalized patients. Intensive Care Med. 2016;42(4):494-504.
14. Sulistio M, Franco M, Vo A, Poon P, William L. Hospital rapid response team and patients with life-limiting illness: a multicentre retrospective cohort study. Palliat Med. 2015;29(4):302-309.
15. Wang J, Hahn SS, Kline M, Cohen RI. Early in-hospital clinical deterioration is not predicted by severity of illness, functional status, or comorbidity. Int J Gen Med. 2017;10:329-334.
16. Dargin JM, Mackey CG, Lei Y, Liesching TN. Resource utilization and end‐of‐life care in a US hospital following medical emergency team‐implemented do not resuscitate orders. J Hosp Med. 2014;9(6):372-378.
17. Austin CA, Hanzaker C, Stafford R, et al. Utilization of rapid response resources and outcomes in a comprehensive cancer center. Crit Care Med. 2014;42(4):905-909.
18. Smith RL, Hayashi VN, Lee YI, Navarro-Mariazeta L, Felner K. The medical emergency team call: a sentinel event that triggers goals of care discussion. Crit Care Med. 2014;42(2):322-327.
19. Bozcuk H, Koyuncu E, Yildiz M, et al. A simple and accurate prediction model to estimate the intrahospital mortality risk of hospitalised cancer patients. Int J Clin Pract. 2004;58(11):1014-1019.
1. National Center for Health Statistics. Health, United States, 2016: with Chartbook on long-term trends in health. Hyattsville, MD: National Center for Health Statistics; 2017.
2. Lambden J, Zhang B, Friedlander R, Prigerson HG. Accuracy of oncologists’ life-expectancy estimates recalled by their advanced cancer patients: correlates and outcomes. J Palliat Med. 2016;19(12):1296-1303.
3. Maltoni M, Caraceni A, Brunelli C, et al. Prognostic factors in advanced cancer patients: evidence-based clinical recommendations—a study by the Steering Committee of the European Association for Palliative Care. J Clin Oncol. 2005;23(25):6240-6248.
4. Viganó A, Bruera E, Jhangri GS, Newman SC, Fields AL, Suarez-Almazor ME. Clinical survival predictors in patients with advanced cancer. Arch Intern Med. 2000;160(6):861-868.
5. Hauser CA, Stockler MR, Tattersall MH. Prognostic factors in patients with recently diagnosed incurable cancer: a systematic review. Support Care Cancer. 2006;14(10):999-1011.
6. Al-Zahrani AS, El-Kashif AT, Mohammad AA, Elsamany S, Alsirafy SA. Prediction of in-hospital mortality of patients with advanced cancer using the Chuang Prognostic Score. Am J Hosp Palliat Med. 2013;30(7):707-711.
7. Hui D, Kilgore K, Fellman B, et al. Development and cross-validation of the in-hospital mortality prediction in advanced cancer patients score: a preliminary study. J Palliat Med. 2012;15(8):902-909.
8. Shouval R, Labopin M, Bondi O, et al. Prediction of allogeneic hematopoietic stem-cell transplantation mortality 100 days after transplantation using a machine learning algorithm: a European group for blood and marrow transplantation acute leukemia working party retrospective data mining study. J Clin Oncol. 2015;33(28):3144-3151.
9. Agency for Healthcare Research and Quality. Total expenses and percent distribution for selected conditions by type of service: United States, 2013. Medical Expenditure Panel Survey website. https://meps.ahrq.gov/mepsweb/data_stats/tables_compendia_hh_interactive.jsp?_SERVICE=MEPSSocket0&_PROGRAM=MEPSPGM.TC.SAS&File=HCFY2013&Table=HCFY2012_CNDXP_C&_Debug=. Accessed November 10, 2018.
10. McCall N. Utilization and costs of Medicare services by beneficiaries in their last year of life. Med Care. 1984;22(4):329-342.
11. Jones D, Moran J, Winters B, Welch J. The rapid response system and end-of-life care. Curr Opin Crit Care. 2013;19(6):616-623.
12. Solomon RS, Corwin GS, Barclay DC, Quddusi SF, Dannenberg MD. Effectiveness of rapid response teams on rates of in‐hospital cardiopulmonary arrest and mortality: a systematic review and meta‐analysis. J Hosp Med. 2016;11(6):438-445.
13. Jung B, Daurat A, De Jong A, et al. Rapid response team and hospital mortality in hospitalized patients. Intensive Care Med. 2016;42(4):494-504.
14. Sulistio M, Franco M, Vo A, Poon P, William L. Hospital rapid response team and patients with life-limiting illness: a multicentre retrospective cohort study. Palliat Med. 2015;29(4):302-309.
15. Wang J, Hahn SS, Kline M, Cohen RI. Early in-hospital clinical deterioration is not predicted by severity of illness, functional status, or comorbidity. Int J Gen Med. 2017;10:329-334.
16. Dargin JM, Mackey CG, Lei Y, Liesching TN. Resource utilization and end‐of‐life care in a US hospital following medical emergency team‐implemented do not resuscitate orders. J Hosp Med. 2014;9(6):372-378.
17. Austin CA, Hanzaker C, Stafford R, et al. Utilization of rapid response resources and outcomes in a comprehensive cancer center. Crit Care Med. 2014;42(4):905-909.
18. Smith RL, Hayashi VN, Lee YI, Navarro-Mariazeta L, Felner K. The medical emergency team call: a sentinel event that triggers goals of care discussion. Crit Care Med. 2014;42(2):322-327.
19. Bozcuk H, Koyuncu E, Yildiz M, et al. A simple and accurate prediction model to estimate the intrahospital mortality risk of hospitalised cancer patients. Int J Clin Pract. 2004;58(11):1014-1019.
Effectiveness of duloxetine in treatment of painful chemotherapy-induced peripheral neuropathy: a systematic review
Chemotherapy-induced peripheral neuropathy (CIPN) is a serious side effect that can be dose limiting and affect patient quality of life for prolonged time,1 with an overall incidence of about 38% in patients who are treated with multiple chemotherapeutic agents. 2
The most common antineoplastic agents causing peripheral neuropathy are oxaliplatin, cisplatin, taxanes, Vinca alkaloids, bortezomib, and thalidomide.3,8,9
Different components of the nervous system are targets of various chemotherapeutic agents, from dorsal root ganglion (DRG) cells to the distal axon. The DRG is the most vulnerable to neurotoxicity because it is less protected by the nervous system blood barrier, hence the predominance of sensory symptoms in CIPN.10 The pathogenesis of CIPN is not fully understood, and it is most probably multifaceted and not always related to the antineoplastic mechanism. Findings from experimental studies have shown an accumulation of chemotherapeutic compounds in the cell bodies of the DRG, resulting in decreased cellular metabolism and axoplasmic transport. Another proposed mechanism is the induction of apoptosis in sensory neuron of the posterior spinal ganglion after binding to DNA strands.7,11
Opioids had been used for managing pain in patients with cancer, but their addictive side effects limit use in the treatment of chronic pain,12 so several drugs called coanalgesics have been introduced as a treatment
The imbalance of 5HT and NE in the pain inhibitory pathways may contribute to the peripheral neuropathic pain.20 Duloxetine hydrochloride is a 5HT–NE reuptake inhibitor used to treat depression and generalized anxiety disorder.21 Duloxetine effect in decreasing pain transmission through increasing synaptic concentrations of NE and 5HT, which results in blocking input signals to the dorsal horn neurons in the spinal cord.12
Methods
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA statement) guidelines during the preparation of this systematic review.22
Inclusion criteria
Trial or study type. Articles publishing findings from randomized controlled trials, nonrandomized controlled trials, retrospective studies, and single-arm studies of duloxetine in the treatment of CIPN were included in our review.
Intervention. The intervention was duloxetine with all doses, either administered alone or with other antineuropathic drugs.
Comparator. The comparator was placebo (control group) or other antineuropathic drugs or no control group.
Population. The population included cancer patients with painful CIPN.
Outcome. At least one of the following outcomes was used for pain assessment:
Exclusion criteria
Studies in a non-English language, animal studies, studies whose full-text article was not available, and thesis and conference papers were not included.
Objective and study design
The objective of this systematic review was to systematically assess the effectiveness of duloxetine in the treatment of pain in patients with CIPN.
Information sources and search
Medical electronic databases. PubMed and Scopus, from inception to January 2018, were searched using the following search queries: (((duloxetine) AND chemotherapy induced peripheral neuropathy)) OR ((((chemotherapy) AND (neuropathic pain OR peripheral neuropathy))) AND duloxetine).
Selection of studies. The authors selected eligible studies. The screening of search results was performed in the following 2 steps:
n Screen title and abstracts against the selection criteria. Articles that were unclear from their title or abstract were reviewed against the selection criteria through the full text.
n Retrieve and screen full-text articles of eligible abstracts for eligibility to systematic review.
Data extraction
Two authors extracted the following data independently: sample size, mean age, chemotherapeutic drug, duloxetine dosage, and outcomes for pain assessment using at least one score from VAS, BPI-SF, neuropathic pain score using the NCI-CTCAE v3.0 and v4.0, or FACT-Tax, and other secondary outcomes. The data was exported from the online forms as a Microsoft Excel sheet.
Statistical analysis
We calculated the mean age and associated standard deviations (SDs) for all patients by using the pooled mean and pooled SD equation, according to Cochrane handbook of systematic reviews of interventions 5.1.0 (updated March 2011).23 When data are expressed as median and interquartile range, we used Hozo and colleagues’ BMC Research Methodology equation to calculate or estimate the mean and SD.24
Data are expressed as means with SD (unless stated otherwise); statistical results were considered significant when the P-value was less than .05. Data analysis was performed using the SPSS Statistical Package, version 23 (IBM Corp., Armonk, NY).
Synthesis of data and analysis
Because of heterogeneity and low sample size of studies, no statistically justified analyses could be performed on the provided data. Instead, a descriptive analysis of published studies was performed.
Summary measures
The search strings, the list of relevant reviews, the data coding, and the quality criteria that were used can be requested from the corresponding author.
Results
Selection of articles
The 
Study characteristics
Characteristics of the included studies and patient outcome are summarized in Table 1 and Table 2. A total of 5 studies from 2012 through 2017 were included in the descriptive analysis and systematic review. In all, 4 trials were prospective studies, and 1 trial was retrospective; among all trials, 2 studies were single arm and 3 were placebo-controlled and/or crossover.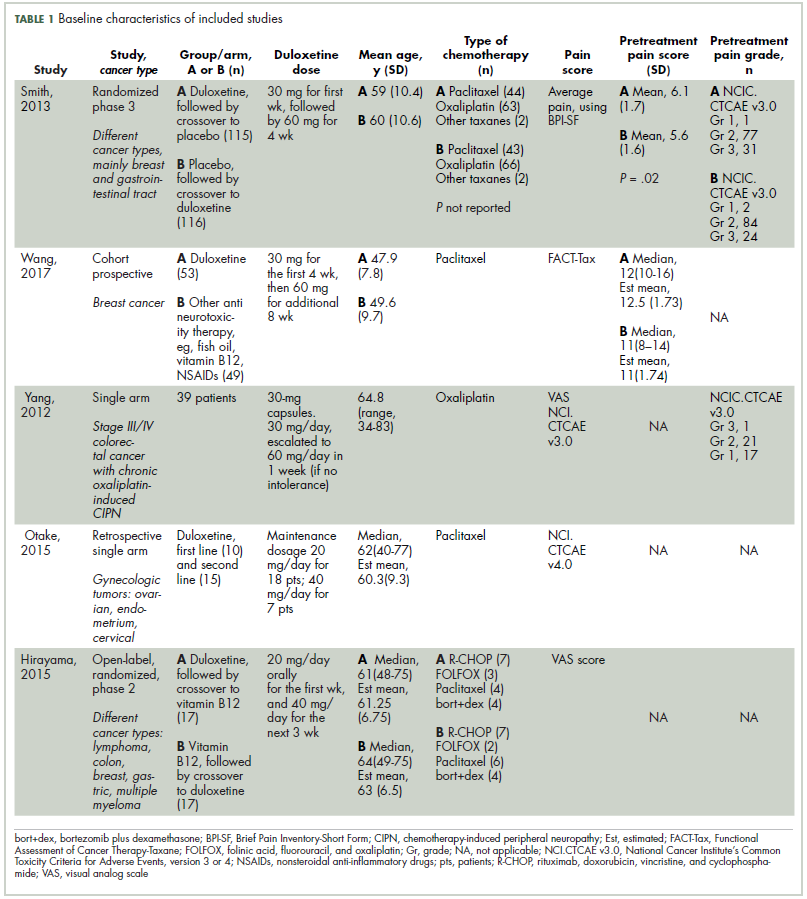

There were 431 participants in the total 5 studies included in this systematic review. The number of patients per study ranged from 25 to 231. Patients were mostly older, with mean sample ages ranging from 47.9 to 63 years, and the pooled mean age for all participants in the total 5 studies was 57.7 years.
In all included studies, duloxetine was given in varying doses of 20 mg, 30 mg, 40 mg, or 60 mg. Also, different therapeutic regimens of duloxetine were used, including placebo control or crossover with vitamin B12; 80% of the studies used escalation of doses over time (only 1 trial used fixed doses for each group of patients in the study). Escalation of duloxetine by doubling the dose was done in all 4 studies, with duloxetine doses of 30 mg and 60 mg used in 75% of those studies (3 out of 4 studies).
Comparator drug was used in 4 studies (1 was single arm) in our review analysis. The comparator drug was placebo in 1 study only, and the remaining 3 studies used other antineurotoxicity or antineuropathic pain therapy, mainly vitamin B12 (as only comparator in 1 study), fish oil, pregabalin, selective 5HT reuptake inhibitors, and nonsteroidal anti-inflammatory agents.
Regarding CIPN, the chemotherapeutic agents used in the studies were as follows (after exclusion of 11 patients who never received treatment in 1 study): 224 patients (52.9%) were on paclitaxel, 168 (39.7%) on oxaliplatin, 14 (3.30%) on R-CHOP, 8 (1.89%) on combined bortezomib–dexamethasone, 5 (1.18%) on FOLFOX, and 4 (0.94%) on other taxanes.
Improvement in pain scores was the primary and/or secondary endpoint in the included studies (Table 2). Pain was assessed by 6 different scores, including the VAS, BPI-SF, neuropathic pain score using NCI-CTCAE v3.0 and v4.0, and FACT-Tax, with all reported once except the VAS score, which was reported in 2 studies. Only 1 study, by Yang and colleagues,25 measured pain by 2 scores (the VAS and NCI-CTCAE v3.0), with the rest of the studies assessing pain by just 1 of the aforementioned scores. The pretreatment pain score was reported in only 2 studies, by Smith and colleagues and Wang and colleagues, using BPI-SF and FACT-Tax scores, respectively, with total respective mean scores of 5.8 (SD, 1.7) and 11.77 (SD, 1.73).17,26
Secondary endpoints were related mainly to pain score, drug adverse effect, and assessment of quality of life (Table 2). In the study by Yang and colleagues,25 9 patients (28.1%) discontinued duloxetine because of intolerable adverse events, with dizziness or giddiness as the most common cause (44.4% of patients who discontinued treatment). Studies by Otake and colleagues12 and Hirayama and colleagues2 reported duloxetine adverse events that were very mild and usually well tolerated in collectively 22 patients, with fatigue (n = 6) and somnolence (n = 5) as the most reported adverse effects. Wang and colleagues17 reported nonneuropathic adverse events that were attributed to chemotherapy and were mild and similar in both study groups.
Discussion
To our knowledge, this is the first systematic review to discuss the effectiveness of duloxetine specifically in treatment of pain in CIPN. The administration of chemotherapeutic agents such as paclitaxel, cisplatin, oxaliplatin, and vincristine was accompanied by CIPN. The currently available treatment options for CIPN are limited. To date, no drug has been approved specifically for treatment of pain in CIPN.12
In our review, we included cancer patients with CIPN and associated pain. Several previous studies8,27,28 discussed tingling and numbness as a common adverse effect in CIPN, and usually about 20% to 42% of patients develop chronic pain.
Six different pain assessment scores were reported in the total 5 studies in our review, with VAS and NCI-CTCAE scores reported in more than 1 study. This reflects the major challenges facing the assessment of CIPN, as various scales and tools are available for pain assessment but without a standardized approach for CIPN that can be precisely implemented.8 Several other challenges regarding pain scores were observed, with Smith and colleagues as the only authors to report both pretreatment pain score and grade, while the rest of the studies failed to report either pain score or grade, or even both.
Another difficulty observed in our review was the variability in study participants in both population size and type of cancer treated. The population size in largest study included in our review was 231 patients and the smallest was 25 patients; collectively, there were only 431 patients in the included studies. Although the type of primary cancer varied in between studies, gynecologic malignancies comprised most cases (215 patients), followed by gastrointestinal tumors, and few cases of hematologic and genitourinary malignancies were reported. Similar results were observed by Geber and colleagues in their large study screening pain in cancer patients, in which gynecologic malignancies were diagnosed in 28 patients out of 61 with CIPN, representing the highest percentage (45.9%) of malignancy type in that study.26
In the study by Otake and colleagues12 examining duloxetine for CIPN in patients with gynecologic cancer, the authors concluded that duloxetine dosage either 20 mg/day or 40 mg/day was not associated with the effectiveness of duloxetine treatment by either univariate or multivariate analysis. Previous authors have provided an explanation for the difference in duloxetine response among CIPN patients and attributed those differences to the underlying pain mechanisms.14,29 In other words, pain in those patients is both peripheral nociceptive and central neuropathic, and it is likely to be caused by mixed mechanisms.
Another variation observed among CIPN patients in our review was the chemotherapeutic agents used. That was noted by Smith and colleagues,26 who reported that patients with cancer who received platinum therapies (oxaliplatin) experienced more benefit from duloxetine in terms of pain improvement than those who received taxanes (P = .13). We found no other published studies on the response to duloxetine among different chemotherapeutic agents used. However, 2 studies of duloxetine response in patients with other pain-related disorders (painful diabetic peripheral neuropathy and fibromyalgia) showed significant improvement in pain symptoms compared with placebo. In a study of pain in chemotherapy-induced neuropathy (CIN) by Geber and colleagues,29 the preexisting pain medication was not reported, but the authors concluded that treatment for CIN-related neuropathic pain differs from that for nonneuropathic (ie, musculoskeletal) pain, with the former being treated mainly with pharmacotherapy and the latter with physiotherapy and behavioral exercises. They asserted that different pain patterns could help flag neuropathic or musculoskeletal pain so that the selected treatments would be more specific. Differences in pain improvement related to duloxetine may be attributed to the underlying pain mechanism, and whether it is mixed or centrally or peripherally related was also discussed by Geber and colleagues.29
In the study by
Findings from studies on the effect of duloxetine in treatment of pain in diabetic peripheral neuropathy have shown that duloxetine at a dose of 60 mg/day effectively improves pain in 43% to 68% of patients.15,16,30 Similarly, in our review, the study by Yang and colleagues25 showed a 63% subjective reduction in pain severity by VAS score in CIPN patients but lower improvement of 47.4% by NCI-CTCAE v3.0; this can be attributed to the simplistic 4-grade rating scale of the latter.
During our analysis of studies, we noticed that no diagnostic criteria were implemented for diagnosis or inclusion of CIPN patients in any of the included studies, and this represents a major challenge in any analysis of studies with neuropathic pain patients. In 2016, Finnerup and colleagues updated the previous 2008 grading system for diagnosis of neuropathic pain, which is intended to determine the level of certainty with which the pain in question is neuropathic.31 The system defines the diagnostic certainty into 3 levels: Possible, Probable, and Definite. Although the number of studies used the grading system during the inclusion of neuropathic pain patients increased from 5% in 2009 to 30% in 2014, still more than two-thirds of studies do not use a standardized system for diagnosis and/or inclusion of neuropathic pain in patients.
Strength and limitations
The first strength of this review is that it identifies gaps in our current knowledge about duloxetine in the treatment of pain in cancer patients with CIPN. Second, we collected all available articles from inception until January 2018. Third, this review can serve as a model for future studies investigating the effectiveness of duloxetine in treatment of CIPN.
There are also limitations to this review that should be discussed. First, the studies vary greatly in samples, methodologies, and outcomes measured. Second, the diagnostic criteria for CIPN and the pain assessment tools vary among the studies. Third, there is also variability in the duloxetine doses and administration regimens among the studies, and some articles did not report the precise outcome in pain scores. Furthermore, the articles reviewed included retrospective, single-arm, or nonrandomized controlled studies with relatively small numbers of participants.
To improve the results, more placebo-controlled or head-to-head trials (with other agents used in treatment of CIPN) with large sample sizes would be needed.
Conclusions
Our purpose was to describe the effectiveness of duloxetine in improving pain scores among CIPN patients
Acknowledgments
That authors express their sincere gratitude to Nahla A Merghany and Sarah M Abd Elfadel for helping them retrieve all the relevant articles for this review.
1. Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13(1):27-46.
2. Hirayama Y, Ishitani K, Sato Y, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. Int J Clin Oncol. 2015;20(5):866-871.
3. Stubblefield MD, McNeely ML, Alfano CM, Mayer DK. A prospective surveillance model for physical rehabilitation of women with breast cancer: chemotherapy-induced peripheral neuropathy. Cancer. 2012;118(suppl 8):2250-2260.
4. Park SB, Goldstein D, Krishnan AV, et al. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin. 2013;63(6):419-437.
5. Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer Manag Res. 2014;6(1):135-147.
6. Bakitas MA. Background noise: the experience of chemotherapy-induced peripheral neuropathy. Nurs Res. 2007;56(5):323-331.
7. Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: a comprehensive survey. Cancer Treat Rev. 2014;40(7):872-882.
8. Hausheer FH, Schilsky RL, Bain S, Berghorn EJ, Lieberman F. Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin Oncol. 2006;33(1):15-49.
9. Park SB, Krishnan AV, Lin CS, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15(29):3081-3094.
10. Caponero R, Montarroyos ES, Tahamtani SMM. Post-chemotherapy neuropathy. Rev Dor. Sao Paulo. 2016;17(suppl 1):S56-S58.
11. Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: an unresolved issue. Neurologia. 2010;25(2):116-131.
12. Otake A, Yoshino K, Ueda Y, et al. Usefulness of duloxetine for paclitaxel-induced peripheral neuropathy treatment in gynecological cancer patients. Anticancer Res. 2015;35(1):359-363.
13. Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967.
14. Smith EM, Pang H, Ye C, et al. Predictors of duloxetine response in patients with oxaliplatin-induced painful chemotherapy-induced peripheral neuropathy (CIPN): a secondary analysis of randomised controlled trial – CALGB/alliance 170601 [published online November 25, 2015]. Eur J Cancer Care (Engl). 2017;26(2). doi:10.1111/ecc.12421
15. Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs placebo in patients with painful diabetic neuropathy. Pain. 2005;116(1-2):109-118.
16. Raskin J, Pritchett YL, Wang F, et al. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med. 2005;6(5):346-356.
17. Wang J, Li Q, Xu B, Zhang T, Chen S, Luo Y. Efficacy and safety of duloxetine in Chinese breast cancer patients with paclitaxel-induced peripheral neuropathy. Chin J Cancer Res. 2017;29(5):411-418.
18. Irving G, Tanenberg RJ, Raskin J, Risser RC, Malcolm S. Comparative safety and tolerability of duloxetine vs pregabalin vs duloxetine plus gabapentin in patients with diabetic peripheral neuropathic pain. Int J Clin Pract. 2014;68(9):1130-1140.
19. Esin E, Yalcin S. Neuropathic cancer pain: what we are dealing with? How to manage it? Onco Targets Ther. 2014;7:599-618.
20. Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends Pharmacol Sci. 2004;25(12):613-617.
21. Mancini M, Perna G, Rossi A, Petralia A. Use of duloxetine in patients with an anxiety disorder, or with comorbid anxiety and major depressive disorder: a review of the literature. Expert Opin Pharmacother. 2010;11(7):1167-1181.
22. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264-269.
23. Higgins JPT, Green S, eds. Cochrane handbook for systematic reviews of interventions. Version 5.1.0. http://handbook-5-1.cochrane.org/. Updated March 2011. Accessed November 19, 2018.
24. Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol. 2005;5(1):13.
25. Yang YH, Lin JK, Chen WS, et al. Duloxetine improves oxaliplatin-induced neuropathy in patients with colorectal cancer: an open-label pilot study. Support Care Cancer. 2012;20(7):1491-1497.
26. Smith EM, Pang H, Cirrincione C, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359-1367.
27. Dworkin RH. An overview of neuropathic pain: syndromes, symptoms, signs, and several mechanisms. Clin J Pain. 2002;18(6):343-349.
28. Cavenagh J, Good P, Ravenscroft P. Neuropathic pain: are we out of the woods yet? Intern Med J. 2006;36(4):251-255.
29. Geber C, Breimhorst M, Burbach B, et al. Pain in chemotherapy-induced neuropathy—more than neuropathic? Pain. 2013;154(12):2877-2887.
30. Wernicke JF, Pritchett YL, D’Souza DN, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology. 2006;67(8):1411–1420.
31. Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. 2016;157(8):1599-1606.
Chemotherapy-induced peripheral neuropathy (CIPN) is a serious side effect that can be dose limiting and affect patient quality of life for prolonged time,1 with an overall incidence of about 38% in patients who are treated with multiple chemotherapeutic agents. 2
The most common antineoplastic agents causing peripheral neuropathy are oxaliplatin, cisplatin, taxanes, Vinca alkaloids, bortezomib, and thalidomide.3,8,9
Different components of the nervous system are targets of various chemotherapeutic agents, from dorsal root ganglion (DRG) cells to the distal axon. The DRG is the most vulnerable to neurotoxicity because it is less protected by the nervous system blood barrier, hence the predominance of sensory symptoms in CIPN.10 The pathogenesis of CIPN is not fully understood, and it is most probably multifaceted and not always related to the antineoplastic mechanism. Findings from experimental studies have shown an accumulation of chemotherapeutic compounds in the cell bodies of the DRG, resulting in decreased cellular metabolism and axoplasmic transport. Another proposed mechanism is the induction of apoptosis in sensory neuron of the posterior spinal ganglion after binding to DNA strands.7,11
Opioids had been used for managing pain in patients with cancer, but their addictive side effects limit use in the treatment of chronic pain,12 so several drugs called coanalgesics have been introduced as a treatment
The imbalance of 5HT and NE in the pain inhibitory pathways may contribute to the peripheral neuropathic pain.20 Duloxetine hydrochloride is a 5HT–NE reuptake inhibitor used to treat depression and generalized anxiety disorder.21 Duloxetine effect in decreasing pain transmission through increasing synaptic concentrations of NE and 5HT, which results in blocking input signals to the dorsal horn neurons in the spinal cord.12
Methods
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA statement) guidelines during the preparation of this systematic review.22
Inclusion criteria
Trial or study type. Articles publishing findings from randomized controlled trials, nonrandomized controlled trials, retrospective studies, and single-arm studies of duloxetine in the treatment of CIPN were included in our review.
Intervention. The intervention was duloxetine with all doses, either administered alone or with other antineuropathic drugs.
Comparator. The comparator was placebo (control group) or other antineuropathic drugs or no control group.
Population. The population included cancer patients with painful CIPN.
Outcome. At least one of the following outcomes was used for pain assessment:
Exclusion criteria
Studies in a non-English language, animal studies, studies whose full-text article was not available, and thesis and conference papers were not included.
Objective and study design
The objective of this systematic review was to systematically assess the effectiveness of duloxetine in the treatment of pain in patients with CIPN.
Information sources and search
Medical electronic databases. PubMed and Scopus, from inception to January 2018, were searched using the following search queries: (((duloxetine) AND chemotherapy induced peripheral neuropathy)) OR ((((chemotherapy) AND (neuropathic pain OR peripheral neuropathy))) AND duloxetine).
Selection of studies. The authors selected eligible studies. The screening of search results was performed in the following 2 steps:
n Screen title and abstracts against the selection criteria. Articles that were unclear from their title or abstract were reviewed against the selection criteria through the full text.
n Retrieve and screen full-text articles of eligible abstracts for eligibility to systematic review.
Data extraction
Two authors extracted the following data independently: sample size, mean age, chemotherapeutic drug, duloxetine dosage, and outcomes for pain assessment using at least one score from VAS, BPI-SF, neuropathic pain score using the NCI-CTCAE v3.0 and v4.0, or FACT-Tax, and other secondary outcomes. The data was exported from the online forms as a Microsoft Excel sheet.
Statistical analysis
We calculated the mean age and associated standard deviations (SDs) for all patients by using the pooled mean and pooled SD equation, according to Cochrane handbook of systematic reviews of interventions 5.1.0 (updated March 2011).23 When data are expressed as median and interquartile range, we used Hozo and colleagues’ BMC Research Methodology equation to calculate or estimate the mean and SD.24
Data are expressed as means with SD (unless stated otherwise); statistical results were considered significant when the P-value was less than .05. Data analysis was performed using the SPSS Statistical Package, version 23 (IBM Corp., Armonk, NY).
Synthesis of data and analysis
Because of heterogeneity and low sample size of studies, no statistically justified analyses could be performed on the provided data. Instead, a descriptive analysis of published studies was performed.
Summary measures
The search strings, the list of relevant reviews, the data coding, and the quality criteria that were used can be requested from the corresponding author.
Results
Selection of articles
The
Study characteristics
Characteristics of the included studies and patient outcome are summarized in Table 1 and Table 2. A total of 5 studies from 2012 through 2017 were included in the descriptive analysis and systematic review. In all, 4 trials were prospective studies, and 1 trial was retrospective; among all trials, 2 studies were single arm and 3 were placebo-controlled and/or crossover.
There were 431 participants in the total 5 studies included in this systematic review. The number of patients per study ranged from 25 to 231. Patients were mostly older, with mean sample ages ranging from 47.9 to 63 years, and the pooled mean age for all participants in the total 5 studies was 57.7 years.
In all included studies, duloxetine was given in varying doses of 20 mg, 30 mg, 40 mg, or 60 mg. Also, different therapeutic regimens of duloxetine were used, including placebo control or crossover with vitamin B12; 80% of the studies used escalation of doses over time (only 1 trial used fixed doses for each group of patients in the study). Escalation of duloxetine by doubling the dose was done in all 4 studies, with duloxetine doses of 30 mg and 60 mg used in 75% of those studies (3 out of 4 studies).
Comparator drug was used in 4 studies (1 was single arm) in our review analysis. The comparator drug was placebo in 1 study only, and the remaining 3 studies used other antineurotoxicity or antineuropathic pain therapy, mainly vitamin B12 (as only comparator in 1 study), fish oil, pregabalin, selective 5HT reuptake inhibitors, and nonsteroidal anti-inflammatory agents.
Regarding CIPN, the chemotherapeutic agents used in the studies were as follows (after exclusion of 11 patients who never received treatment in 1 study): 224 patients (52.9%) were on paclitaxel, 168 (39.7%) on oxaliplatin, 14 (3.30%) on R-CHOP, 8 (1.89%) on combined bortezomib–dexamethasone, 5 (1.18%) on FOLFOX, and 4 (0.94%) on other taxanes.
Improvement in pain scores was the primary and/or secondary endpoint in the included studies (Table 2). Pain was assessed by 6 different scores, including the VAS, BPI-SF, neuropathic pain score using NCI-CTCAE v3.0 and v4.0, and FACT-Tax, with all reported once except the VAS score, which was reported in 2 studies. Only 1 study, by Yang and colleagues,25 measured pain by 2 scores (the VAS and NCI-CTCAE v3.0), with the rest of the studies assessing pain by just 1 of the aforementioned scores. The pretreatment pain score was reported in only 2 studies, by Smith and colleagues and Wang and colleagues, using BPI-SF and FACT-Tax scores, respectively, with total respective mean scores of 5.8 (SD, 1.7) and 11.77 (SD, 1.73).17,26
Secondary endpoints were related mainly to pain score, drug adverse effect, and assessment of quality of life (Table 2). In the study by Yang and colleagues,25 9 patients (28.1%) discontinued duloxetine because of intolerable adverse events, with dizziness or giddiness as the most common cause (44.4% of patients who discontinued treatment). Studies by Otake and colleagues12 and Hirayama and colleagues2 reported duloxetine adverse events that were very mild and usually well tolerated in collectively 22 patients, with fatigue (n = 6) and somnolence (n = 5) as the most reported adverse effects. Wang and colleagues17 reported nonneuropathic adverse events that were attributed to chemotherapy and were mild and similar in both study groups.
Discussion
To our knowledge, this is the first systematic review to discuss the effectiveness of duloxetine specifically in treatment of pain in CIPN. The administration of chemotherapeutic agents such as paclitaxel, cisplatin, oxaliplatin, and vincristine was accompanied by CIPN. The currently available treatment options for CIPN are limited. To date, no drug has been approved specifically for treatment of pain in CIPN.12
In our review, we included cancer patients with CIPN and associated pain. Several previous studies8,27,28 discussed tingling and numbness as a common adverse effect in CIPN, and usually about 20% to 42% of patients develop chronic pain.
Six different pain assessment scores were reported in the total 5 studies in our review, with VAS and NCI-CTCAE scores reported in more than 1 study. This reflects the major challenges facing the assessment of CIPN, as various scales and tools are available for pain assessment but without a standardized approach for CIPN that can be precisely implemented.8 Several other challenges regarding pain scores were observed, with Smith and colleagues as the only authors to report both pretreatment pain score and grade, while the rest of the studies failed to report either pain score or grade, or even both.
Another difficulty observed in our review was the variability in study participants in both population size and type of cancer treated. The population size in largest study included in our review was 231 patients and the smallest was 25 patients; collectively, there were only 431 patients in the included studies. Although the type of primary cancer varied in between studies, gynecologic malignancies comprised most cases (215 patients), followed by gastrointestinal tumors, and few cases of hematologic and genitourinary malignancies were reported. Similar results were observed by Geber and colleagues in their large study screening pain in cancer patients, in which gynecologic malignancies were diagnosed in 28 patients out of 61 with CIPN, representing the highest percentage (45.9%) of malignancy type in that study.26
In the study by Otake and colleagues12 examining duloxetine for CIPN in patients with gynecologic cancer, the authors concluded that duloxetine dosage either 20 mg/day or 40 mg/day was not associated with the effectiveness of duloxetine treatment by either univariate or multivariate analysis. Previous authors have provided an explanation for the difference in duloxetine response among CIPN patients and attributed those differences to the underlying pain mechanisms.14,29 In other words, pain in those patients is both peripheral nociceptive and central neuropathic, and it is likely to be caused by mixed mechanisms.
Another variation observed among CIPN patients in our review was the chemotherapeutic agents used. That was noted by Smith and colleagues,26 who reported that patients with cancer who received platinum therapies (oxaliplatin) experienced more benefit from duloxetine in terms of pain improvement than those who received taxanes (P = .13). We found no other published studies on the response to duloxetine among different chemotherapeutic agents used. However, 2 studies of duloxetine response in patients with other pain-related disorders (painful diabetic peripheral neuropathy and fibromyalgia) showed significant improvement in pain symptoms compared with placebo. In a study of pain in chemotherapy-induced neuropathy (CIN) by Geber and colleagues,29 the preexisting pain medication was not reported, but the authors concluded that treatment for CIN-related neuropathic pain differs from that for nonneuropathic (ie, musculoskeletal) pain, with the former being treated mainly with pharmacotherapy and the latter with physiotherapy and behavioral exercises. They asserted that different pain patterns could help flag neuropathic or musculoskeletal pain so that the selected treatments would be more specific. Differences in pain improvement related to duloxetine may be attributed to the underlying pain mechanism, and whether it is mixed or centrally or peripherally related was also discussed by Geber and colleagues.29
In the study by
Findings from studies on the effect of duloxetine in treatment of pain in diabetic peripheral neuropathy have shown that duloxetine at a dose of 60 mg/day effectively improves pain in 43% to 68% of patients.15,16,30 Similarly, in our review, the study by Yang and colleagues25 showed a 63% subjective reduction in pain severity by VAS score in CIPN patients but lower improvement of 47.4% by NCI-CTCAE v3.0; this can be attributed to the simplistic 4-grade rating scale of the latter.
During our analysis of studies, we noticed that no diagnostic criteria were implemented for diagnosis or inclusion of CIPN patients in any of the included studies, and this represents a major challenge in any analysis of studies with neuropathic pain patients. In 2016, Finnerup and colleagues updated the previous 2008 grading system for diagnosis of neuropathic pain, which is intended to determine the level of certainty with which the pain in question is neuropathic.31 The system defines the diagnostic certainty into 3 levels: Possible, Probable, and Definite. Although the number of studies used the grading system during the inclusion of neuropathic pain patients increased from 5% in 2009 to 30% in 2014, still more than two-thirds of studies do not use a standardized system for diagnosis and/or inclusion of neuropathic pain in patients.
Strength and limitations
The first strength of this review is that it identifies gaps in our current knowledge about duloxetine in the treatment of pain in cancer patients with CIPN. Second, we collected all available articles from inception until January 2018. Third, this review can serve as a model for future studies investigating the effectiveness of duloxetine in treatment of CIPN.
There are also limitations to this review that should be discussed. First, the studies vary greatly in samples, methodologies, and outcomes measured. Second, the diagnostic criteria for CIPN and the pain assessment tools vary among the studies. Third, there is also variability in the duloxetine doses and administration regimens among the studies, and some articles did not report the precise outcome in pain scores. Furthermore, the articles reviewed included retrospective, single-arm, or nonrandomized controlled studies with relatively small numbers of participants.
To improve the results, more placebo-controlled or head-to-head trials (with other agents used in treatment of CIPN) with large sample sizes would be needed.
Conclusions
Our purpose was to describe the effectiveness of duloxetine in improving pain scores among CIPN patients
Acknowledgments
That authors express their sincere gratitude to Nahla A Merghany and Sarah M Abd Elfadel for helping them retrieve all the relevant articles for this review.
Chemotherapy-induced peripheral neuropathy (CIPN) is a serious side effect that can be dose limiting and affect patient quality of life for prolonged time,1 with an overall incidence of about 38% in patients who are treated with multiple chemotherapeutic agents. 2
The most common antineoplastic agents causing peripheral neuropathy are oxaliplatin, cisplatin, taxanes, Vinca alkaloids, bortezomib, and thalidomide.3,8,9
Different components of the nervous system are targets of various chemotherapeutic agents, from dorsal root ganglion (DRG) cells to the distal axon. The DRG is the most vulnerable to neurotoxicity because it is less protected by the nervous system blood barrier, hence the predominance of sensory symptoms in CIPN.10 The pathogenesis of CIPN is not fully understood, and it is most probably multifaceted and not always related to the antineoplastic mechanism. Findings from experimental studies have shown an accumulation of chemotherapeutic compounds in the cell bodies of the DRG, resulting in decreased cellular metabolism and axoplasmic transport. Another proposed mechanism is the induction of apoptosis in sensory neuron of the posterior spinal ganglion after binding to DNA strands.7,11
Opioids had been used for managing pain in patients with cancer, but their addictive side effects limit use in the treatment of chronic pain,12 so several drugs called coanalgesics have been introduced as a treatment
The imbalance of 5HT and NE in the pain inhibitory pathways may contribute to the peripheral neuropathic pain.20 Duloxetine hydrochloride is a 5HT–NE reuptake inhibitor used to treat depression and generalized anxiety disorder.21 Duloxetine effect in decreasing pain transmission through increasing synaptic concentrations of NE and 5HT, which results in blocking input signals to the dorsal horn neurons in the spinal cord.12
Methods
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA statement) guidelines during the preparation of this systematic review.22
Inclusion criteria
Trial or study type. Articles publishing findings from randomized controlled trials, nonrandomized controlled trials, retrospective studies, and single-arm studies of duloxetine in the treatment of CIPN were included in our review.
Intervention. The intervention was duloxetine with all doses, either administered alone or with other antineuropathic drugs.
Comparator. The comparator was placebo (control group) or other antineuropathic drugs or no control group.
Population. The population included cancer patients with painful CIPN.
Outcome. At least one of the following outcomes was used for pain assessment:
Exclusion criteria
Studies in a non-English language, animal studies, studies whose full-text article was not available, and thesis and conference papers were not included.
Objective and study design
The objective of this systematic review was to systematically assess the effectiveness of duloxetine in the treatment of pain in patients with CIPN.
Information sources and search
Medical electronic databases. PubMed and Scopus, from inception to January 2018, were searched using the following search queries: (((duloxetine) AND chemotherapy induced peripheral neuropathy)) OR ((((chemotherapy) AND (neuropathic pain OR peripheral neuropathy))) AND duloxetine).
Selection of studies. The authors selected eligible studies. The screening of search results was performed in the following 2 steps:
n Screen title and abstracts against the selection criteria. Articles that were unclear from their title or abstract were reviewed against the selection criteria through the full text.
n Retrieve and screen full-text articles of eligible abstracts for eligibility to systematic review.
Data extraction
Two authors extracted the following data independently: sample size, mean age, chemotherapeutic drug, duloxetine dosage, and outcomes for pain assessment using at least one score from VAS, BPI-SF, neuropathic pain score using the NCI-CTCAE v3.0 and v4.0, or FACT-Tax, and other secondary outcomes. The data was exported from the online forms as a Microsoft Excel sheet.
Statistical analysis
We calculated the mean age and associated standard deviations (SDs) for all patients by using the pooled mean and pooled SD equation, according to Cochrane handbook of systematic reviews of interventions 5.1.0 (updated March 2011).23 When data are expressed as median and interquartile range, we used Hozo and colleagues’ BMC Research Methodology equation to calculate or estimate the mean and SD.24
Data are expressed as means with SD (unless stated otherwise); statistical results were considered significant when the P-value was less than .05. Data analysis was performed using the SPSS Statistical Package, version 23 (IBM Corp., Armonk, NY).
Synthesis of data and analysis
Because of heterogeneity and low sample size of studies, no statistically justified analyses could be performed on the provided data. Instead, a descriptive analysis of published studies was performed.
Summary measures
The search strings, the list of relevant reviews, the data coding, and the quality criteria that were used can be requested from the corresponding author.
Results
Selection of articles
The
Study characteristics
Characteristics of the included studies and patient outcome are summarized in Table 1 and Table 2. A total of 5 studies from 2012 through 2017 were included in the descriptive analysis and systematic review. In all, 4 trials were prospective studies, and 1 trial was retrospective; among all trials, 2 studies were single arm and 3 were placebo-controlled and/or crossover.
There were 431 participants in the total 5 studies included in this systematic review. The number of patients per study ranged from 25 to 231. Patients were mostly older, with mean sample ages ranging from 47.9 to 63 years, and the pooled mean age for all participants in the total 5 studies was 57.7 years.
In all included studies, duloxetine was given in varying doses of 20 mg, 30 mg, 40 mg, or 60 mg. Also, different therapeutic regimens of duloxetine were used, including placebo control or crossover with vitamin B12; 80% of the studies used escalation of doses over time (only 1 trial used fixed doses for each group of patients in the study). Escalation of duloxetine by doubling the dose was done in all 4 studies, with duloxetine doses of 30 mg and 60 mg used in 75% of those studies (3 out of 4 studies).
Comparator drug was used in 4 studies (1 was single arm) in our review analysis. The comparator drug was placebo in 1 study only, and the remaining 3 studies used other antineurotoxicity or antineuropathic pain therapy, mainly vitamin B12 (as only comparator in 1 study), fish oil, pregabalin, selective 5HT reuptake inhibitors, and nonsteroidal anti-inflammatory agents.
Regarding CIPN, the chemotherapeutic agents used in the studies were as follows (after exclusion of 11 patients who never received treatment in 1 study): 224 patients (52.9%) were on paclitaxel, 168 (39.7%) on oxaliplatin, 14 (3.30%) on R-CHOP, 8 (1.89%) on combined bortezomib–dexamethasone, 5 (1.18%) on FOLFOX, and 4 (0.94%) on other taxanes.
Improvement in pain scores was the primary and/or secondary endpoint in the included studies (Table 2). Pain was assessed by 6 different scores, including the VAS, BPI-SF, neuropathic pain score using NCI-CTCAE v3.0 and v4.0, and FACT-Tax, with all reported once except the VAS score, which was reported in 2 studies. Only 1 study, by Yang and colleagues,25 measured pain by 2 scores (the VAS and NCI-CTCAE v3.0), with the rest of the studies assessing pain by just 1 of the aforementioned scores. The pretreatment pain score was reported in only 2 studies, by Smith and colleagues and Wang and colleagues, using BPI-SF and FACT-Tax scores, respectively, with total respective mean scores of 5.8 (SD, 1.7) and 11.77 (SD, 1.73).17,26
Secondary endpoints were related mainly to pain score, drug adverse effect, and assessment of quality of life (Table 2). In the study by Yang and colleagues,25 9 patients (28.1%) discontinued duloxetine because of intolerable adverse events, with dizziness or giddiness as the most common cause (44.4% of patients who discontinued treatment). Studies by Otake and colleagues12 and Hirayama and colleagues2 reported duloxetine adverse events that were very mild and usually well tolerated in collectively 22 patients, with fatigue (n = 6) and somnolence (n = 5) as the most reported adverse effects. Wang and colleagues17 reported nonneuropathic adverse events that were attributed to chemotherapy and were mild and similar in both study groups.
Discussion
To our knowledge, this is the first systematic review to discuss the effectiveness of duloxetine specifically in treatment of pain in CIPN. The administration of chemotherapeutic agents such as paclitaxel, cisplatin, oxaliplatin, and vincristine was accompanied by CIPN. The currently available treatment options for CIPN are limited. To date, no drug has been approved specifically for treatment of pain in CIPN.12
In our review, we included cancer patients with CIPN and associated pain. Several previous studies8,27,28 discussed tingling and numbness as a common adverse effect in CIPN, and usually about 20% to 42% of patients develop chronic pain.
Six different pain assessment scores were reported in the total 5 studies in our review, with VAS and NCI-CTCAE scores reported in more than 1 study. This reflects the major challenges facing the assessment of CIPN, as various scales and tools are available for pain assessment but without a standardized approach for CIPN that can be precisely implemented.8 Several other challenges regarding pain scores were observed, with Smith and colleagues as the only authors to report both pretreatment pain score and grade, while the rest of the studies failed to report either pain score or grade, or even both.
Another difficulty observed in our review was the variability in study participants in both population size and type of cancer treated. The population size in largest study included in our review was 231 patients and the smallest was 25 patients; collectively, there were only 431 patients in the included studies. Although the type of primary cancer varied in between studies, gynecologic malignancies comprised most cases (215 patients), followed by gastrointestinal tumors, and few cases of hematologic and genitourinary malignancies were reported. Similar results were observed by Geber and colleagues in their large study screening pain in cancer patients, in which gynecologic malignancies were diagnosed in 28 patients out of 61 with CIPN, representing the highest percentage (45.9%) of malignancy type in that study.26
In the study by Otake and colleagues12 examining duloxetine for CIPN in patients with gynecologic cancer, the authors concluded that duloxetine dosage either 20 mg/day or 40 mg/day was not associated with the effectiveness of duloxetine treatment by either univariate or multivariate analysis. Previous authors have provided an explanation for the difference in duloxetine response among CIPN patients and attributed those differences to the underlying pain mechanisms.14,29 In other words, pain in those patients is both peripheral nociceptive and central neuropathic, and it is likely to be caused by mixed mechanisms.
Another variation observed among CIPN patients in our review was the chemotherapeutic agents used. That was noted by Smith and colleagues,26 who reported that patients with cancer who received platinum therapies (oxaliplatin) experienced more benefit from duloxetine in terms of pain improvement than those who received taxanes (P = .13). We found no other published studies on the response to duloxetine among different chemotherapeutic agents used. However, 2 studies of duloxetine response in patients with other pain-related disorders (painful diabetic peripheral neuropathy and fibromyalgia) showed significant improvement in pain symptoms compared with placebo. In a study of pain in chemotherapy-induced neuropathy (CIN) by Geber and colleagues,29 the preexisting pain medication was not reported, but the authors concluded that treatment for CIN-related neuropathic pain differs from that for nonneuropathic (ie, musculoskeletal) pain, with the former being treated mainly with pharmacotherapy and the latter with physiotherapy and behavioral exercises. They asserted that different pain patterns could help flag neuropathic or musculoskeletal pain so that the selected treatments would be more specific. Differences in pain improvement related to duloxetine may be attributed to the underlying pain mechanism, and whether it is mixed or centrally or peripherally related was also discussed by Geber and colleagues.29
In the study by
Findings from studies on the effect of duloxetine in treatment of pain in diabetic peripheral neuropathy have shown that duloxetine at a dose of 60 mg/day effectively improves pain in 43% to 68% of patients.15,16,30 Similarly, in our review, the study by Yang and colleagues25 showed a 63% subjective reduction in pain severity by VAS score in CIPN patients but lower improvement of 47.4% by NCI-CTCAE v3.0; this can be attributed to the simplistic 4-grade rating scale of the latter.
During our analysis of studies, we noticed that no diagnostic criteria were implemented for diagnosis or inclusion of CIPN patients in any of the included studies, and this represents a major challenge in any analysis of studies with neuropathic pain patients. In 2016, Finnerup and colleagues updated the previous 2008 grading system for diagnosis of neuropathic pain, which is intended to determine the level of certainty with which the pain in question is neuropathic.31 The system defines the diagnostic certainty into 3 levels: Possible, Probable, and Definite. Although the number of studies used the grading system during the inclusion of neuropathic pain patients increased from 5% in 2009 to 30% in 2014, still more than two-thirds of studies do not use a standardized system for diagnosis and/or inclusion of neuropathic pain in patients.
Strength and limitations
The first strength of this review is that it identifies gaps in our current knowledge about duloxetine in the treatment of pain in cancer patients with CIPN. Second, we collected all available articles from inception until January 2018. Third, this review can serve as a model for future studies investigating the effectiveness of duloxetine in treatment of CIPN.
There are also limitations to this review that should be discussed. First, the studies vary greatly in samples, methodologies, and outcomes measured. Second, the diagnostic criteria for CIPN and the pain assessment tools vary among the studies. Third, there is also variability in the duloxetine doses and administration regimens among the studies, and some articles did not report the precise outcome in pain scores. Furthermore, the articles reviewed included retrospective, single-arm, or nonrandomized controlled studies with relatively small numbers of participants.
To improve the results, more placebo-controlled or head-to-head trials (with other agents used in treatment of CIPN) with large sample sizes would be needed.
Conclusions
Our purpose was to describe the effectiveness of duloxetine in improving pain scores among CIPN patients
Acknowledgments
That authors express their sincere gratitude to Nahla A Merghany and Sarah M Abd Elfadel for helping them retrieve all the relevant articles for this review.
1. Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13(1):27-46.
2. Hirayama Y, Ishitani K, Sato Y, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. Int J Clin Oncol. 2015;20(5):866-871.
3. Stubblefield MD, McNeely ML, Alfano CM, Mayer DK. A prospective surveillance model for physical rehabilitation of women with breast cancer: chemotherapy-induced peripheral neuropathy. Cancer. 2012;118(suppl 8):2250-2260.
4. Park SB, Goldstein D, Krishnan AV, et al. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin. 2013;63(6):419-437.
5. Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer Manag Res. 2014;6(1):135-147.
6. Bakitas MA. Background noise: the experience of chemotherapy-induced peripheral neuropathy. Nurs Res. 2007;56(5):323-331.
7. Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: a comprehensive survey. Cancer Treat Rev. 2014;40(7):872-882.
8. Hausheer FH, Schilsky RL, Bain S, Berghorn EJ, Lieberman F. Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin Oncol. 2006;33(1):15-49.
9. Park SB, Krishnan AV, Lin CS, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15(29):3081-3094.
10. Caponero R, Montarroyos ES, Tahamtani SMM. Post-chemotherapy neuropathy. Rev Dor. Sao Paulo. 2016;17(suppl 1):S56-S58.
11. Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: an unresolved issue. Neurologia. 2010;25(2):116-131.
12. Otake A, Yoshino K, Ueda Y, et al. Usefulness of duloxetine for paclitaxel-induced peripheral neuropathy treatment in gynecological cancer patients. Anticancer Res. 2015;35(1):359-363.
13. Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967.
14. Smith EM, Pang H, Ye C, et al. Predictors of duloxetine response in patients with oxaliplatin-induced painful chemotherapy-induced peripheral neuropathy (CIPN): a secondary analysis of randomised controlled trial – CALGB/alliance 170601 [published online November 25, 2015]. Eur J Cancer Care (Engl). 2017;26(2). doi:10.1111/ecc.12421
15. Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs placebo in patients with painful diabetic neuropathy. Pain. 2005;116(1-2):109-118.
16. Raskin J, Pritchett YL, Wang F, et al. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med. 2005;6(5):346-356.
17. Wang J, Li Q, Xu B, Zhang T, Chen S, Luo Y. Efficacy and safety of duloxetine in Chinese breast cancer patients with paclitaxel-induced peripheral neuropathy. Chin J Cancer Res. 2017;29(5):411-418.
18. Irving G, Tanenberg RJ, Raskin J, Risser RC, Malcolm S. Comparative safety and tolerability of duloxetine vs pregabalin vs duloxetine plus gabapentin in patients with diabetic peripheral neuropathic pain. Int J Clin Pract. 2014;68(9):1130-1140.
19. Esin E, Yalcin S. Neuropathic cancer pain: what we are dealing with? How to manage it? Onco Targets Ther. 2014;7:599-618.
20. Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends Pharmacol Sci. 2004;25(12):613-617.
21. Mancini M, Perna G, Rossi A, Petralia A. Use of duloxetine in patients with an anxiety disorder, or with comorbid anxiety and major depressive disorder: a review of the literature. Expert Opin Pharmacother. 2010;11(7):1167-1181.
22. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264-269.
23. Higgins JPT, Green S, eds. Cochrane handbook for systematic reviews of interventions. Version 5.1.0. http://handbook-5-1.cochrane.org/. Updated March 2011. Accessed November 19, 2018.
24. Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol. 2005;5(1):13.
25. Yang YH, Lin JK, Chen WS, et al. Duloxetine improves oxaliplatin-induced neuropathy in patients with colorectal cancer: an open-label pilot study. Support Care Cancer. 2012;20(7):1491-1497.
26. Smith EM, Pang H, Cirrincione C, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359-1367.
27. Dworkin RH. An overview of neuropathic pain: syndromes, symptoms, signs, and several mechanisms. Clin J Pain. 2002;18(6):343-349.
28. Cavenagh J, Good P, Ravenscroft P. Neuropathic pain: are we out of the woods yet? Intern Med J. 2006;36(4):251-255.
29. Geber C, Breimhorst M, Burbach B, et al. Pain in chemotherapy-induced neuropathy—more than neuropathic? Pain. 2013;154(12):2877-2887.
30. Wernicke JF, Pritchett YL, D’Souza DN, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology. 2006;67(8):1411–1420.
31. Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. 2016;157(8):1599-1606.
1. Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13(1):27-46.
2. Hirayama Y, Ishitani K, Sato Y, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. Int J Clin Oncol. 2015;20(5):866-871.
3. Stubblefield MD, McNeely ML, Alfano CM, Mayer DK. A prospective surveillance model for physical rehabilitation of women with breast cancer: chemotherapy-induced peripheral neuropathy. Cancer. 2012;118(suppl 8):2250-2260.
4. Park SB, Goldstein D, Krishnan AV, et al. Chemotherapy-induced peripheral neurotoxicity: a critical analysis. CA Cancer J Clin. 2013;63(6):419-437.
5. Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer Manag Res. 2014;6(1):135-147.
6. Bakitas MA. Background noise: the experience of chemotherapy-induced peripheral neuropathy. Nurs Res. 2007;56(5):323-331.
7. Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy: a comprehensive survey. Cancer Treat Rev. 2014;40(7):872-882.
8. Hausheer FH, Schilsky RL, Bain S, Berghorn EJ, Lieberman F. Diagnosis, management, and evaluation of chemotherapy-induced peripheral neuropathy. Semin Oncol. 2006;33(1):15-49.
9. Park SB, Krishnan AV, Lin CS, Goldstein D, Friedlander M, Kiernan MC. Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Curr Med Chem. 2008;15(29):3081-3094.
10. Caponero R, Montarroyos ES, Tahamtani SMM. Post-chemotherapy neuropathy. Rev Dor. Sao Paulo. 2016;17(suppl 1):S56-S58.
11. Velasco R, Bruna J. Chemotherapy-induced peripheral neuropathy: an unresolved issue. Neurologia. 2010;25(2):116-131.
12. Otake A, Yoshino K, Ueda Y, et al. Usefulness of duloxetine for paclitaxel-induced peripheral neuropathy treatment in gynecological cancer patients. Anticancer Res. 2015;35(1):359-363.
13. Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941-1967.
14. Smith EM, Pang H, Ye C, et al. Predictors of duloxetine response in patients with oxaliplatin-induced painful chemotherapy-induced peripheral neuropathy (CIPN): a secondary analysis of randomised controlled trial – CALGB/alliance 170601 [published online November 25, 2015]. Eur J Cancer Care (Engl). 2017;26(2). doi:10.1111/ecc.12421
15. Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetine vs placebo in patients with painful diabetic neuropathy. Pain. 2005;116(1-2):109-118.
16. Raskin J, Pritchett YL, Wang F, et al. A double-blind, randomized multicenter trial comparing duloxetine with placebo in the management of diabetic peripheral neuropathic pain. Pain Med. 2005;6(5):346-356.
17. Wang J, Li Q, Xu B, Zhang T, Chen S, Luo Y. Efficacy and safety of duloxetine in Chinese breast cancer patients with paclitaxel-induced peripheral neuropathy. Chin J Cancer Res. 2017;29(5):411-418.
18. Irving G, Tanenberg RJ, Raskin J, Risser RC, Malcolm S. Comparative safety and tolerability of duloxetine vs pregabalin vs duloxetine plus gabapentin in patients with diabetic peripheral neuropathic pain. Int J Clin Pract. 2014;68(9):1130-1140.
19. Esin E, Yalcin S. Neuropathic cancer pain: what we are dealing with? How to manage it? Onco Targets Ther. 2014;7:599-618.
20. Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends Pharmacol Sci. 2004;25(12):613-617.
21. Mancini M, Perna G, Rossi A, Petralia A. Use of duloxetine in patients with an anxiety disorder, or with comorbid anxiety and major depressive disorder: a review of the literature. Expert Opin Pharmacother. 2010;11(7):1167-1181.
22. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med. 2009;151(4):264-269.
23. Higgins JPT, Green S, eds. Cochrane handbook for systematic reviews of interventions. Version 5.1.0. http://handbook-5-1.cochrane.org/. Updated March 2011. Accessed November 19, 2018.
24. Hozo SP, Djulbegovic B, Hozo I. Estimating the mean and variance from the median, range, and the size of a sample. BMC Med Res Methodol. 2005;5(1):13.
25. Yang YH, Lin JK, Chen WS, et al. Duloxetine improves oxaliplatin-induced neuropathy in patients with colorectal cancer: an open-label pilot study. Support Care Cancer. 2012;20(7):1491-1497.
26. Smith EM, Pang H, Cirrincione C, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359-1367.
27. Dworkin RH. An overview of neuropathic pain: syndromes, symptoms, signs, and several mechanisms. Clin J Pain. 2002;18(6):343-349.
28. Cavenagh J, Good P, Ravenscroft P. Neuropathic pain: are we out of the woods yet? Intern Med J. 2006;36(4):251-255.
29. Geber C, Breimhorst M, Burbach B, et al. Pain in chemotherapy-induced neuropathy—more than neuropathic? Pain. 2013;154(12):2877-2887.
30. Wernicke JF, Pritchett YL, D’Souza DN, et al. A randomized controlled trial of duloxetine in diabetic peripheral neuropathic pain. Neurology. 2006;67(8):1411–1420.
31. Finnerup NB, Haroutounian S, Kamerman P, et al. Neuropathic pain: an updated grading system for research and clinical practice. 2016;157(8):1599-1606.
Development, implementation, and evaluation of a prostate cancer supportive care program
Prostate cancer is the most common malignancy diagnosed in Canadian men. An estimated 21,300 Canadian men were diagnosed with the disease in 2017, representing 21% of all new cancer cases.1 There are about 176,000 men living with prostate cancer in Canada.1 In the United States, there were 2,778,630 survivors of prostate cancer as of 2012 and that population is expected to increase by more than 1 million (40%) to 3,922,600 by 2022.2
Although 96% of men diagnosed with prostate cancer now survive longer than 5 years3, many will suffer from treatment-related sequelae that can have a profound effect on quality of life for themselves and their partners.4,5 Impacts include sexual, urinary, and bowel dysfunctions6 owing to treatment of the primary tumor as well as reduced muscle and bone mass, osteoporosis, fatigue, obesity, and glucose intolerance or diabetes7 owing to androgen-deprivation therapy (ADT). Many men also suffer from psychological issues such as depression, anxiety, anger and irritability, sense of isolation, grief, and loss of masculinity.8,9 The psychological impacts also continue well beyond the completion of treatment and can be significant for both patients and their partners.5,8
With posttreatment longevity and the associated complex sequelae, prostate cancer is being viewed increasingly as a chronic disease whose effects must be managed for many years after the completion of primary treatment. Supportive care that “[manages] symptoms and side effects, enables adaptation and coping, optimizes understanding and informed decision-making, and minimizes decrements in functioning”10 is becoming recognized as a critical component of direct oncologic care before, during, and after treatment. Health care professionals, scientists, governments, and patient advocates are increasingly calling for the development of comprehensive supportive care programs improve the quality of life for people diagnosed with cancer. A common model for survivorship care is a general program for all cancer survivors that provides disease- and patient-specific care plans. These care plans outline patients’ prior therapies, potential side effects, recommendations for monitoring (for side effects or relapse of cancer), and advice on how patients can maintain a healthy lifestyle.11 However, there are few survivorship programs for men with prostate cancer and their partners, and the evidence base around best practices for these programs is scant.12 Furthermore, up to 87% of men with a prostate cancer diagnosis report specific and significant unmet supportive care needs,10,13 with sexuality-related and psychological issues10,14 being the areas of greatest concern.
To address the complex supportive care needs of men with prostate cancer in British Columbia, Canada, the Vancouver Prostate Centre (VPC) and Department of Urologic Sciences at the University of British Columbia developed the multidisciplinary Prostate Cancer Supportive Care (PCSC) Program. The program aims to address the challenges of decision-making and coping faced by men with prostate cancer and their partners and family members along the entire disease trajectory. Services are provided at no cost to participants. Here, we outline the guiding principles for the PCSC program and its scope, delivery, and evaluation. We provide information on the more than 1,200 patients who have participated in the program since its inception in January of 2013, the rates of participation across the different program modules, and a selection of patient satisfaction measures. We also discuss successes and limitations and ongoing research and evaluation efforts, providing lessons learned to support the development of other supportive care programs in Canada and internationally.
Program description
Guiding principles
The PCSC Program is a clinical, educational, and research-based program, with 4 guiding principles: it is comprehensive, patient- and partner-centered, evidence-based, and supports new research. The program serves patients, partners, and families along the entire disease trajectory, recognizing that cancer is a family disease, affecting both the individual and social network, and that the psychological stress associated with a diagnosis of prostate cancer is borne heavily by partners. It has been designed, implemented, and refined with the best available evidence and with the intention to undergo consistent and repeated evaluation. Finally, it was designed to provide opportunities for targeted research efforts, supporting the growth of the evidence base in this area.
Patient entry and module descriptions
Patients can be referred to the program by a physician or other allied health professional. They may also self-refer, having been made aware of the program through our website, a variety of print materials, or by word of mouth. On referral, the program coordinator collects patients’ basic clinical and demographic data, assesses health literacy and lifestyle factors, and provides them with information on the program modules. As of December 2015, the program consisted of 6 distinct modules, each focusing on different elements of the disease trajectory or on addressing specific physical or mental health concerns. Modules are led by licensed health professionals with experience in oncology. No elements of the program are mandatory, and participants are free to pick and choose the components that are most relevant to them and their partners.
Introduction to prostate cancer and primary treatment options. This is a group-based module that focuses on educating newly diagnosed patients (and those going on or off active surveillance) on the basic biology of prostate cancer, the primary treatment options for localized disease, and the main side effects associated with the treatments. It also includes information about the other services offered by the program and any ongoing research studies. The session is held twice a month in the early evening and is run collectively by a urologist, radiation oncologist, patient representative, and program coordinator. It includes a brief one-on-one discussion between each patient and their partner or family member and the urologist and radiation oncologist to address any remaining questions. A copy of the patient’s biopsy report is on hand for the physician(s). Attendance of this session has been shown to significantly reduce pretreatment distress in both patients and their partners.15
Managing sexual function and intimacy. Sexual intimacy is tied to overall health outcomes, relationship satisfaction, and quality of life.16 Primary therapy for prostate cancer can be associated with substantial side-effects (eg, erectile dysfunction, incontinence, altered libido, penile shortening) that negatively affect sexual intimacy and have an impact on the patient individually as well as the sexual relationship he has with his partner.17
The program’s Sexual Health Service (SHS) provides patients and partners with information on the impact of treatment on sexual health.18 The SHS offers educational sessions led by a sexual rehabilitation nurse and clinical psychologist with a specialization in sexual health. Sessions focus on the impact of prostate cancer treatments on sexual function and therapeutic modalities, promote an understanding of the barriers to sexual adaptation posttreatment, and present options for sexual activity that are not solely dependent on the ability to achieve an erection. Once participants have attended an educational session, they are offered individual consultations with the sexual health nurse every 3 to 6 months for 2 years or longer, depending on the patient’s or couple’s needs. They are referred to the SHS’s sexual medicine physician if further medical intervention is warranted. The sexual health nurse works with the patient and partner to develop an individualized Sexual Health Rehabilitation Action Plan (SHRAP), which assists the couple in sexual adaptation going forward. The SHRAP is a tool devised by the sexual health nurse based on her clinical experience with couples affected by prostate cancer.
Couples who have been evaluated within the SHS are also invited to attend a second workshop on intimacy that is offered quarterly. Workshop participants discuss the impact of sexual changes on relationships, and strategies on how to enhance intimacy and sexual communication are presented. A resource package is provided to each couple to help re-establish and/or strengthen their various dimensions of intimacy.
Lifestyle management. The lifestyle management modules include separate nutrition and physical activity or exercise components. Referral to the smoking cessation program in the Vancouver Coastal Health Authority is made at program registration, if appropriate. The nutrition group-based education session is delivered by a registered dietitian from the British Columbia Cancer Agency who specializes in prostate cancer. The session focuses on evidence-based recommendations on diet after a diagnosis of prostate cancer, the use of dietary supplements, body weight and health, and practical nutrition tips. The exercise session is delivered by an exercise physiologist who specializes in working with cancer patients. It covers the value of exercise in terms of safety, prevention and reduction of treatment side effects (including from ADT), treatment prehabilitation and recovery, advanced cancer management, and long-term survival. A one-on-one exercise counseling clinic is also offered and aims to increase exercise adoption and long-term adherence in line with Canadian Physical Activity guidelines and exercise oncology guidelines,19,20 with follow-up appointments at 3, 6, and 12 months to help patients stay motivated and ensure they are exercising correctly. The individual consultations with the exercise physiologist include physical measures, exercise volume, treatment side effects, and coconstructed goal setting with an individualized formal exercise regimen (exercise prescription).
Adapting to ADT. This is an educational module offered to patients with metastatic prostate cancer who are starting hormone therapy treatments that lower serum testosterone into the castrate range. This program was one of several available through TrueNTH, a portfolio of projects funded by the Movember Foundation, through Prostate Cancer Canada. The session is delivered by a patient facilitator and focuses on strategies to recognize and adapt to the side effects of ADT21 while maintaining a good quality of life and strong intimate relationships with partners.22,23
Pelvic-floor physical therapy for urinary incontinence. This module includes a group-based and individualized education session for patients (either pre- or posttreatment) focused on reducing the effects of surgery and/or radiation therapy on urinary and sexual continence as well as on how to cope with these symptoms and minimize the effect they have on quality of life.24 The session is conducted by a physical therapist who is certified as a pelvic-floor specialist. Supervised pelvic-floor re-education and/or exercise has been shown to successfully reduce the degree of incontinence in this population.25 The module therefore also includes 3 one-on-one clinical appointments for patients who are still experiencing bother from incontinence 12 weeks after a radical prostatectomy or postradiation treatment.
Psycho-oncology. In recognition of the emotional and psychological burden associated with prostate cancer and the important role partners play in facilitating treatment of these psychological and/or psychosocial issues, the program offers appointments with a registered clinical counselor to address acute emotional distress. These are usually 1-hour appointments offered to both patients and partners, either separately or together. Appointments can be attended in person or conducted by telephone. When appropriate, patients are referred for further long-term individual support or couple support with their partners. A group therapy workshop was also initiated in 2016. In this program, men participate in a guided autobiographical life review through a process that focuses on developing a cohesive working group, learning strategic communication skills, and understanding and learning how to manage difficult emotions and transitional life stressors associated with prostate cancer. It also focuses on processing and integrating critical events that contribute to the men’s identity and psychological function and involves the consolidation of the personal learning that occurs. Postgroup referral plans are developed on an individual basis as needed.
Methods
Data
We obtained sociodemographic, diagnostic, and treatment information as well as clinic visit records for all PCSC Program registrants from the electronic medical record held at the VPC. Clinical variables included age at diagnosis, Gleason score, and primary treatment modality (including active surveillance and ADT use). The Gleason score determines the aggressiveness of a patient’s prostate cancer based on biopsy results. A score of 6 or less indicates that the disease is likely to grow slowly. A grade of 7 is considered intermediate risk (with primary score of 3 and secondary 4 being lower risk than those with a primary score of 4 and secondary of 3). A Gleason score of 8 or higher indicates aggressive disease that is poorly differentiated or high grade. Sociodemographic characteristics included age, travel distance to the clinic, and income quintile. We obtained attendance records for the modular education sessions from the program’s database. Patients who did not have any medical visits at the VPC (referred to henceforth as non-VPC patients) did not have a clinic record, so we excluded them from the subset of the analyses that depended on specific clinical variables.
All patients and partners who participate in any PCSC Program education sessions (introduction to prostate cancer, sexual health, nutrition, exercise, ADT, and pelvic-floor physical therapy) are asked to complete voluntary, anonymous feedback forms. These forms assess participant satisfaction using a series of Likert-based and Boolean response items as well as qualitative commentary. They include questions that assess the timing, structure, and content of each session.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. For this type of study, formal consent is not required.
Statistical approach
Descriptive statistics were used to analyze participant characteristics, program participation rates, and participant satisfaction. For each module’s education session, we compared the overall satisfaction between patients and partners using t tests. We also compared the level of satisfaction across the different modules using a 1-way analysis of variance. For the sexual health and pelvic-floor physical therapy sessions, we compared satisfaction between participants who attended the education sessions before to those who attended following their primary treatment using t tests. We provide the eta squared (for analyses of variance) and Cohen d (for t tests) to provide an effect size estimate of any significant differences observed.
Results
Participants
From the program’s founding in January of 2013 to December 31, 2016, a total of 1,269 patients registered (an average of 317 patients a year). Of those, 1,026 (80.9%) had at least 1 prostate cancer–related visit at the VPC. The remaining 243 (19.1%) were non-VPC patients (Figure). Overall, 1,062 men (83.4%) who registered with the program went on to attend at least 1 education session or clinic appointment.
Average age among male program participants was 67.7 years, and age at diagnosis was 62.5 years (Table 1). In all, 273 men (31.7%) had Gleason 3+4, and 117 (13.7%) had Gleason 4+3. Most of the participants (76.9%) elected to undergo radical prostatectomy for primary treatment. Ninety-five men (8.9%) received at least some ADT treatment as an adjunct to radiation or to treat recurrent disease. Participants traveled an average of 83.1 km (51.6 miles; median, 6.9 km and 10.5 miles, respectively) to attend the program; 10% of participants traveled further than 112 km (70 mi) to the clinic. One hundred and four (10.9%) and 301 (31.5%) of registrants were in the lowest and highest income quintiles respectively. Four hundred and ninety-seven (46.8%) attended at lesson 1 session or clinic appointment with a partner or family member.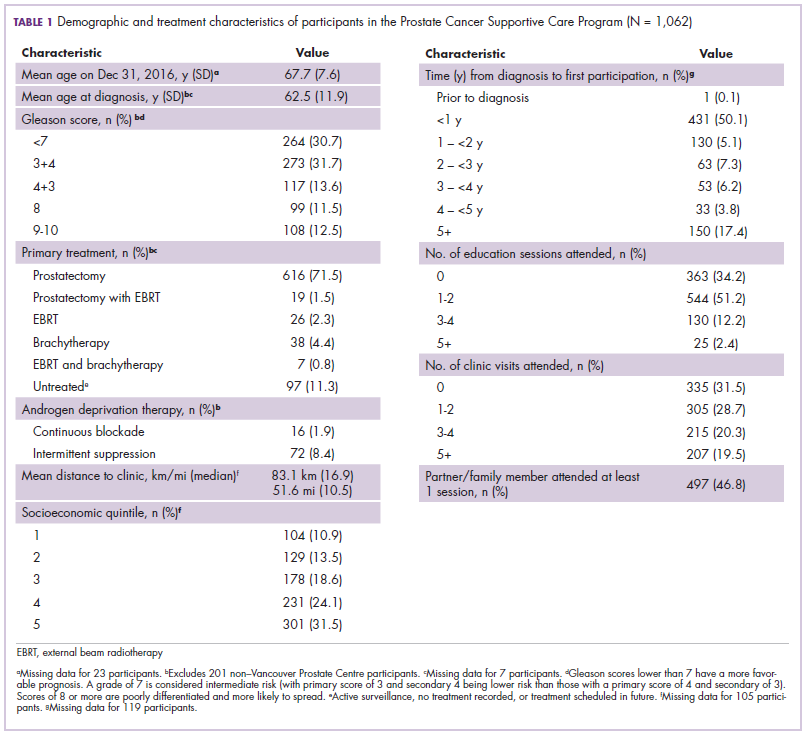
Program participation
Of the 1,062 men who participated in the program, 867 (80.1%) were patients of the VPC, and 205 (19.1%) were non-VPC patients. The education sessions for the introduction to prostate cancer and sexual health modules had the largest numbers of participants (309 and 265, respectively; Table 2); however, pelvic-floor physical therapy had the highest participation rate per quarter (25 patients). The clinical services offered within the sexual health module had the larger number of participants and highest participation rate per quarter (590 total patients, 42/quarter). Timing of program participation was highly variable, ranging from 6 days to 18.5 years after diagnosis (SD, 1,301 days). More than half of participants attended a session or clinic visit within the first year of their diagnosis. A total of 17% of patients who registered did not attend any part of the program.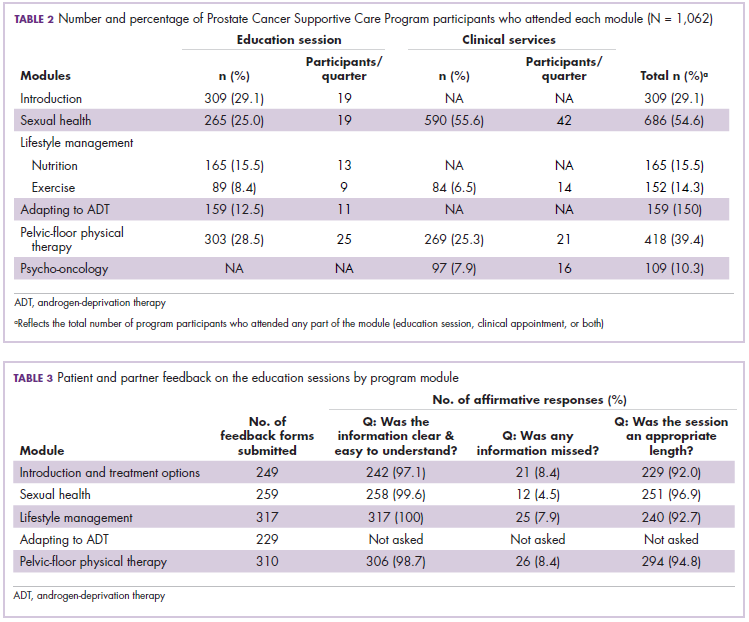
Satisfaction
Most patients and partners said that they found the information presented at the modular education sessions comprehensive, clear, and easy to understand (Table 3). Although the overall average satisfaction score varied significantly across sessions, ranging from 3.5 (out of a possible 4) for pelvic-floor physical therapy to 3.8 for introduction to prostate cancer (F = 3.8, P < .001), the effect size of this difference was small (η2 = .039; Table 4A). We found no difference in the level of satisfaction between patients and partners, with the exception of the sexual health module, which was rated better by partners than by patients (patients: 3.6, partners: 3.8; t = 2.0; P = .03); however, the effect size of this difference was again small (Cohen d = .29). A total of 86% of patients found the inclusion of their partners at the sessions useful. For both pelvic-floor physical therapy and sexual health, attendees were more satisfied if they attended before treatment initiation rather than after completion (Table 4B).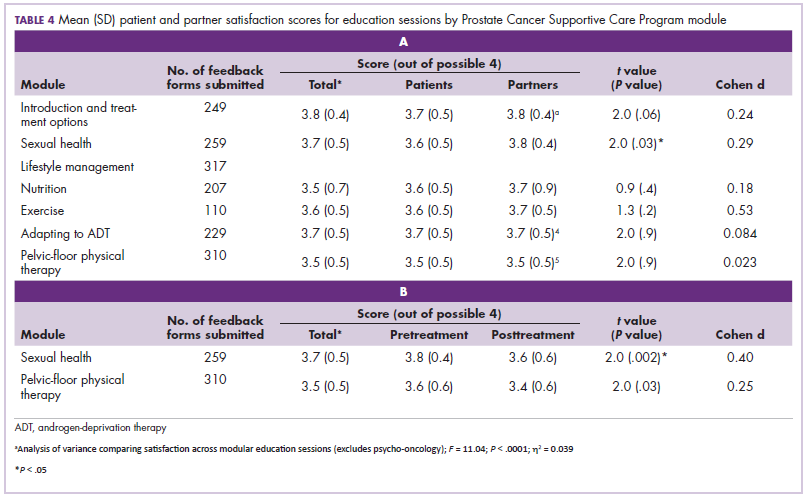
Discussion
The purpose of this descriptive analysis was to outline a comprehensive, multidisciplinary supportive care program for men with prostate cancer and to present initial data on the population that has used the program and their satisfaction with the services provided. Within the first 5 years of the PCSC Program, 1,269 patients registered to participate. However, nearly 1 in 6 men who registered for the program did not subsequently attend any education sessions or use any clinical services offered, despite the fact that all services were free of charge. It is possible that nonparticipation may be related to men on active surveillance choosing not to engage with the program until they are faced with making a treatment decision, which may not happen until several years after an initial positive biopsy.26 This and other factors that affect a patient’s decision not to participate will be investigated in a future study. There is existing evidence documenting high levels of distress and anxiety for patients and their partners resulting from decision-making around prostate cancer treatment,27,28 and many face both decisional conflict and subsequent regret.15,29 Further work to help patients access the program could include defining a prehabilitation program for which patients can sign up that automatically selects the education sessions and clinical services most relevant to them.
The number of attendees varied across the 6 education sessions, with introduction to prostate cancer and sexual health being the best attended. This is consistent with the literature concerning the specific unmet supportive care and information needs in this population10,13 and with the value that men have placed on taking an active role in the decisions around their prostate cancer treatment.30 It is also possible that attendance varied because modules were introduced in a stepwise fashion and were offered on different schedules. Patients and partners both reported a high degree of satisfaction with all of the modules’ education sessions, reporting that the length, content, and delivery were appropriate.
Since 2013, a wide research portfolio has grown alongside the program. It has acted as a recruitment site for multicenter national studies and has attracted funding for several in-house research projects and evaluations. In addition, the VPC has implemented clinic-wide electronic collection of several patient-reported outcome measures using iPads. Patients have the option of contributing their data to Canadian (PC360o) and Global (TrueNTH Global Registry – Prostate Cancer Outcomes) registries for prostate cancer. The program has also created educational opportunities by supporting postdoctoral fellows. It has also provided a rich environment for urology and radiation oncology residents and fellows to participate in a multidisciplinary supportive care team, ensuring that the next generation of surgeons and oncologists recognize the importance of this approach to care.
Limitations
This is a brief descriptive study that relies on a mixture of anonymized survey and clinical chart data. Because the program’s patient feedback forms are anonymous, we are not able to link satisfaction scores to differences in sociodemographic, clinical, or prognostic factors. We also have not directly measured clinical, psychological, or quality of life outcomes; however, all 3 will be included in future studies of the program. An additional limitation is that not all program modules were offered for the entirety of the study duration and are offered at different frequencies. Thus, some modules have disproportionally higher participation rates than others. Lastly, we are missing clinical information for 16% of our participants who are not patients at the VPC.
The program is offered within an academic and teaching hospital in a major metropolitan center and depends on the work of a large interdisciplinary team. Cancer programs that are not embedded within a similar environment, such as those located in smaller rural communities, may not have access to the specialized clinical professionals who run our program, affecting its direct generalizability to these locations. Other specialists, such as palliative care teams, could be well positioned to provide support in locations that do not have a similar level of resource available. Furthermore, some program elements will be adapted to be delivered using telemedicine technology, which is an additional approach to improving access for patients who are beyond the reach of a tertiary care facility.
Conclusions
There is a growing need to provide consistent and comprehensive supportive care to patients with prostate cancer and their partners and families throughout the disease and treatment journey. The PCSC Program uses a multidisciplinary, evidenced-based, disease-focused approach to support informed treatment decision-making and address the physical, psychological, and psychosocial effects of prostate cancer diagnosis and treatment. We proactively collect data on disease, personal demographic details, and symptoms or quality of life, supporting opportunities to partner with researchers with the goal of further improving quality of life based on evidenced-based practices. Going forward, we will conduct detailed examinations of the costs and benefits (in terms of symptom management and quality of life) of the PCSC Program, further contributing to the development of evidence-based best practices for supportive care for men with prostate cancer and their families.
Acknowledgments
The authors express their gratitude to the urologists and radiation oncologists who referred their patients to the program and participated in delivering education sessions, including Dr Martin Gleave, Dr Peter Black, Dr Alan So, Dr Scott Tyldesley, and Dr Mira Keyes. They also thank Dr Richard Wassersug for his contributions to the initial program design and implementation. They thank the patients and their families for participating, and all of their current or past staff and collaborators. Lastly, they thank the funders of the program: the Specialist Services Committee, the BC Ministry of Health, the Prostate Cancer Foundation of BC, and philanthropic donors. They acknowledge Vancouver Coastal Health Research Institute and the University of British Columbia for their institutional support.
1. Cancer Research UK. Prostate cancer statistics. http://www.cancer.ca/en/cancer-information/cancer-type/prostate/statistics/?region=sk. Published 2015. Accessed June 22, 2017.
2. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220-241.
3. Canadian Cancer Society. Canadian cancer statistics special topic: predictions of the future burden of cancer in Canada. Ottawa, Canada: Public Health Agency of Canada; 2015.
4. Roth AJ, Weinberger MI, Nelson CJ. Prostate cancer: psychosocial implications and management. Future Oncol. 2008;4(4):561-568.
5. Couper J, Bloch S, Love A, Macvean M, Duchesne GM, Kissane D. Psychosocial adjustment of female partners of men with prostate cancer: a review of the literature. Psychooncology 2006;15(11):937-953.
6. Wilt TJ, MacDonald R, Rutks I, Shamliyan TA, Taylor BC, Kane RL. Systematic review: comparative effectiveness and harms of treatments for clinically localized prostate cancer. Ann Intern Med. 2008;148(6):435-448.
7. Galvão DA, Spry NA, Taaffe DR, et al. Changes in muscle, fat and bone mass after 36 weeks of maximal androgen blockade for prostate cancer. BJU Int. 2008;102(1):44-47.
8. Watts S, Leydon G, Birch B, et al. Depression and anxiety in prostate cancer: a systematic review and meta-analysis of prevalence rates. BMJ Open. 2014;4(3):e003901.
9. Zaider T, Manne S, Nelson C, Mulhall J, Kissane D. Loss of masculine identity, marital affection, and sexual bother in men with localized prostate cancer. J Sex Med. 2012;9(10):2724-2732.
10. Ream E, Quennell A, Fincham L, et al. Supportive care needs of men living with prostate cancer in England: a survey. Br J Cancer. 2008;98(12):1903-1909.
11. Howell D, Hack TF, Oliver TK, et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
12. Halpern MT, Viswanathan M, Evans TS, Birken SA, Basch E, Mayer DK. Models of cancer survivorship care: overview and summary of current evidence. J Oncol Pract. 2015;11(1):e19-e27.
13. Smith DP, Supramaniam R, King MT, Ward J, Berry M, Armstrong BK. Age, health, and education determine supportive care needs of men younger than 70 years with prostate cancer. J Clin Oncol. 2007;25(18):2560-2566.
14. Northouse LL, Mood DW, Montie JE, et al. Living with prostate cancer: patients’ and spouses’ psychosocial status and quality of life. J Clin Oncol. 2007;25(27):4171-4177.
15. Hedden L, Wassersug R, Mahovlich S, et al. Evaluating an educational intervention to alleviate distress amongst men with newly diagnosed prostate cancer and their partners. BJU Int. 2017;120(5B):E21-E29.
16. Bradley EB, Bissonette EA, Theodorescu D. Determinants of long-term quality of life and voiding function of patients treated with radical prostatectomy or permanent brachytherapy for prostate cancer. BJU Int. 2004;94(7):1003-1009.
17. Ramsey SD, Zeliadt SB, Blough DK, et al. Impact of prostate cancer on sexual relationships: a longitudinal perspective on intimate partners’ experiences. J Sex Med. 2013;10(12):3135-3143.
18. Wittmann D, Carolan M, Given B, et al. Exploring the role of the partner in couples’ sexual recovery after surgery for prostate cancer. Support Care Cancer. 2014;22(9):2509-2515.
19. Schmitz KH, Courneya KS, Matthews C, et al. American college of sports medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42(7):1409-1426.
20. Rock CL, Doyle C, Demark-Wahnefried W, et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J Clin. 2012;62(4):243-274.
21. Elliott S, Latini DM, Walker LM, Wassersug R, Robinson JW; ADT Suvivorship Working Group. Androgen deprivation therapy for prostate cancer: recommendations to improve patient and partner quality of life. J Sex Med. 2010;7(9):2996-3010.
22. Wassersug RJ, Walker LM, Robinson JW. Androgen deprivation therapy: an essential guide for prostate cancer patients and their loved ones. New York, NY: Demos Health; 2014.
23. Wibowo E, Walker LM, Wilyman S, et al. Androgen deprivation therapy educational program: a Canadian True NTH initiative. J Clin Oncol. 2016;34(suppl 3):243.
24. Stanford JL, Feng Z, Hamilton AS, et al. Urinary and sexual function after radical prostatectomy for clinically localized prostate cancer. JAMA. 2000;283(3):354-360.
25. Overgård M, Angelsen A, Lydersen S, Mørkved S. Does physiotherapist-guided pelvic floor muscle training reduce urinary incontinence after radical prostatectomy? A randomised controlled trial. Eur Urol. 2008;54(2):438-448.
26. Godtman RA, Holmberg E, Khatami A, Pihl CG, Stranne J, Hugosson J. Long-term results of active surveillance in the Göteborg randomized, population-based prostate cancer screening trial. Eur Urol. 2016;70(5):760-766.
27. Cohen H, Britten N. Who decides about prostate cancer treatment? A qualitative study. Fam Pract. 2003;20(6):724-729.
28. Denberg TD, Melhado TV, Steiner JF. Patient treatment preferences in localized prostate carcinoma: the influence of emotion, misconception, and anecdote. Cancer. 2006;107(3):620-630.
29. Morris BB, Farnan L, Song L, et al. Treatment decisional regret among men with prostate cancer: racial differences and influential factors in the North Carolina health access and prostate cancer treatment project (HCaP-NC). Cancer. 2015;121(12):2029-2035.
30. Feldman-Stewart D, Capirci C, Brennenstuhl S, et al. Information for decision making by patients with early-stage prostate cancer: a comparison across 9 countries. Med Decis Making. 2011;31(5):754-766.
Prostate cancer is the most common malignancy diagnosed in Canadian men. An estimated 21,300 Canadian men were diagnosed with the disease in 2017, representing 21% of all new cancer cases.1 There are about 176,000 men living with prostate cancer in Canada.1 In the United States, there were 2,778,630 survivors of prostate cancer as of 2012 and that population is expected to increase by more than 1 million (40%) to 3,922,600 by 2022.2
Although 96% of men diagnosed with prostate cancer now survive longer than 5 years3, many will suffer from treatment-related sequelae that can have a profound effect on quality of life for themselves and their partners.4,5 Impacts include sexual, urinary, and bowel dysfunctions6 owing to treatment of the primary tumor as well as reduced muscle and bone mass, osteoporosis, fatigue, obesity, and glucose intolerance or diabetes7 owing to androgen-deprivation therapy (ADT). Many men also suffer from psychological issues such as depression, anxiety, anger and irritability, sense of isolation, grief, and loss of masculinity.8,9 The psychological impacts also continue well beyond the completion of treatment and can be significant for both patients and their partners.5,8
With posttreatment longevity and the associated complex sequelae, prostate cancer is being viewed increasingly as a chronic disease whose effects must be managed for many years after the completion of primary treatment. Supportive care that “[manages] symptoms and side effects, enables adaptation and coping, optimizes understanding and informed decision-making, and minimizes decrements in functioning”10 is becoming recognized as a critical component of direct oncologic care before, during, and after treatment. Health care professionals, scientists, governments, and patient advocates are increasingly calling for the development of comprehensive supportive care programs improve the quality of life for people diagnosed with cancer. A common model for survivorship care is a general program for all cancer survivors that provides disease- and patient-specific care plans. These care plans outline patients’ prior therapies, potential side effects, recommendations for monitoring (for side effects or relapse of cancer), and advice on how patients can maintain a healthy lifestyle.11 However, there are few survivorship programs for men with prostate cancer and their partners, and the evidence base around best practices for these programs is scant.12 Furthermore, up to 87% of men with a prostate cancer diagnosis report specific and significant unmet supportive care needs,10,13 with sexuality-related and psychological issues10,14 being the areas of greatest concern.
To address the complex supportive care needs of men with prostate cancer in British Columbia, Canada, the Vancouver Prostate Centre (VPC) and Department of Urologic Sciences at the University of British Columbia developed the multidisciplinary Prostate Cancer Supportive Care (PCSC) Program. The program aims to address the challenges of decision-making and coping faced by men with prostate cancer and their partners and family members along the entire disease trajectory. Services are provided at no cost to participants. Here, we outline the guiding principles for the PCSC program and its scope, delivery, and evaluation. We provide information on the more than 1,200 patients who have participated in the program since its inception in January of 2013, the rates of participation across the different program modules, and a selection of patient satisfaction measures. We also discuss successes and limitations and ongoing research and evaluation efforts, providing lessons learned to support the development of other supportive care programs in Canada and internationally.
Program description
Guiding principles
The PCSC Program is a clinical, educational, and research-based program, with 4 guiding principles: it is comprehensive, patient- and partner-centered, evidence-based, and supports new research. The program serves patients, partners, and families along the entire disease trajectory, recognizing that cancer is a family disease, affecting both the individual and social network, and that the psychological stress associated with a diagnosis of prostate cancer is borne heavily by partners. It has been designed, implemented, and refined with the best available evidence and with the intention to undergo consistent and repeated evaluation. Finally, it was designed to provide opportunities for targeted research efforts, supporting the growth of the evidence base in this area.
Patient entry and module descriptions
Patients can be referred to the program by a physician or other allied health professional. They may also self-refer, having been made aware of the program through our website, a variety of print materials, or by word of mouth. On referral, the program coordinator collects patients’ basic clinical and demographic data, assesses health literacy and lifestyle factors, and provides them with information on the program modules. As of December 2015, the program consisted of 6 distinct modules, each focusing on different elements of the disease trajectory or on addressing specific physical or mental health concerns. Modules are led by licensed health professionals with experience in oncology. No elements of the program are mandatory, and participants are free to pick and choose the components that are most relevant to them and their partners.
Introduction to prostate cancer and primary treatment options. This is a group-based module that focuses on educating newly diagnosed patients (and those going on or off active surveillance) on the basic biology of prostate cancer, the primary treatment options for localized disease, and the main side effects associated with the treatments. It also includes information about the other services offered by the program and any ongoing research studies. The session is held twice a month in the early evening and is run collectively by a urologist, radiation oncologist, patient representative, and program coordinator. It includes a brief one-on-one discussion between each patient and their partner or family member and the urologist and radiation oncologist to address any remaining questions. A copy of the patient’s biopsy report is on hand for the physician(s). Attendance of this session has been shown to significantly reduce pretreatment distress in both patients and their partners.15
Managing sexual function and intimacy. Sexual intimacy is tied to overall health outcomes, relationship satisfaction, and quality of life.16 Primary therapy for prostate cancer can be associated with substantial side-effects (eg, erectile dysfunction, incontinence, altered libido, penile shortening) that negatively affect sexual intimacy and have an impact on the patient individually as well as the sexual relationship he has with his partner.17
The program’s Sexual Health Service (SHS) provides patients and partners with information on the impact of treatment on sexual health.18 The SHS offers educational sessions led by a sexual rehabilitation nurse and clinical psychologist with a specialization in sexual health. Sessions focus on the impact of prostate cancer treatments on sexual function and therapeutic modalities, promote an understanding of the barriers to sexual adaptation posttreatment, and present options for sexual activity that are not solely dependent on the ability to achieve an erection. Once participants have attended an educational session, they are offered individual consultations with the sexual health nurse every 3 to 6 months for 2 years or longer, depending on the patient’s or couple’s needs. They are referred to the SHS’s sexual medicine physician if further medical intervention is warranted. The sexual health nurse works with the patient and partner to develop an individualized Sexual Health Rehabilitation Action Plan (SHRAP), which assists the couple in sexual adaptation going forward. The SHRAP is a tool devised by the sexual health nurse based on her clinical experience with couples affected by prostate cancer.
Couples who have been evaluated within the SHS are also invited to attend a second workshop on intimacy that is offered quarterly. Workshop participants discuss the impact of sexual changes on relationships, and strategies on how to enhance intimacy and sexual communication are presented. A resource package is provided to each couple to help re-establish and/or strengthen their various dimensions of intimacy.
Lifestyle management. The lifestyle management modules include separate nutrition and physical activity or exercise components. Referral to the smoking cessation program in the Vancouver Coastal Health Authority is made at program registration, if appropriate. The nutrition group-based education session is delivered by a registered dietitian from the British Columbia Cancer Agency who specializes in prostate cancer. The session focuses on evidence-based recommendations on diet after a diagnosis of prostate cancer, the use of dietary supplements, body weight and health, and practical nutrition tips. The exercise session is delivered by an exercise physiologist who specializes in working with cancer patients. It covers the value of exercise in terms of safety, prevention and reduction of treatment side effects (including from ADT), treatment prehabilitation and recovery, advanced cancer management, and long-term survival. A one-on-one exercise counseling clinic is also offered and aims to increase exercise adoption and long-term adherence in line with Canadian Physical Activity guidelines and exercise oncology guidelines,19,20 with follow-up appointments at 3, 6, and 12 months to help patients stay motivated and ensure they are exercising correctly. The individual consultations with the exercise physiologist include physical measures, exercise volume, treatment side effects, and coconstructed goal setting with an individualized formal exercise regimen (exercise prescription).
Adapting to ADT. This is an educational module offered to patients with metastatic prostate cancer who are starting hormone therapy treatments that lower serum testosterone into the castrate range. This program was one of several available through TrueNTH, a portfolio of projects funded by the Movember Foundation, through Prostate Cancer Canada. The session is delivered by a patient facilitator and focuses on strategies to recognize and adapt to the side effects of ADT21 while maintaining a good quality of life and strong intimate relationships with partners.22,23
Pelvic-floor physical therapy for urinary incontinence. This module includes a group-based and individualized education session for patients (either pre- or posttreatment) focused on reducing the effects of surgery and/or radiation therapy on urinary and sexual continence as well as on how to cope with these symptoms and minimize the effect they have on quality of life.24 The session is conducted by a physical therapist who is certified as a pelvic-floor specialist. Supervised pelvic-floor re-education and/or exercise has been shown to successfully reduce the degree of incontinence in this population.25 The module therefore also includes 3 one-on-one clinical appointments for patients who are still experiencing bother from incontinence 12 weeks after a radical prostatectomy or postradiation treatment.
Psycho-oncology. In recognition of the emotional and psychological burden associated with prostate cancer and the important role partners play in facilitating treatment of these psychological and/or psychosocial issues, the program offers appointments with a registered clinical counselor to address acute emotional distress. These are usually 1-hour appointments offered to both patients and partners, either separately or together. Appointments can be attended in person or conducted by telephone. When appropriate, patients are referred for further long-term individual support or couple support with their partners. A group therapy workshop was also initiated in 2016. In this program, men participate in a guided autobiographical life review through a process that focuses on developing a cohesive working group, learning strategic communication skills, and understanding and learning how to manage difficult emotions and transitional life stressors associated with prostate cancer. It also focuses on processing and integrating critical events that contribute to the men’s identity and psychological function and involves the consolidation of the personal learning that occurs. Postgroup referral plans are developed on an individual basis as needed.
Methods
Data
We obtained sociodemographic, diagnostic, and treatment information as well as clinic visit records for all PCSC Program registrants from the electronic medical record held at the VPC. Clinical variables included age at diagnosis, Gleason score, and primary treatment modality (including active surveillance and ADT use). The Gleason score determines the aggressiveness of a patient’s prostate cancer based on biopsy results. A score of 6 or less indicates that the disease is likely to grow slowly. A grade of 7 is considered intermediate risk (with primary score of 3 and secondary 4 being lower risk than those with a primary score of 4 and secondary of 3). A Gleason score of 8 or higher indicates aggressive disease that is poorly differentiated or high grade. Sociodemographic characteristics included age, travel distance to the clinic, and income quintile. We obtained attendance records for the modular education sessions from the program’s database. Patients who did not have any medical visits at the VPC (referred to henceforth as non-VPC patients) did not have a clinic record, so we excluded them from the subset of the analyses that depended on specific clinical variables.
All patients and partners who participate in any PCSC Program education sessions (introduction to prostate cancer, sexual health, nutrition, exercise, ADT, and pelvic-floor physical therapy) are asked to complete voluntary, anonymous feedback forms. These forms assess participant satisfaction using a series of Likert-based and Boolean response items as well as qualitative commentary. They include questions that assess the timing, structure, and content of each session.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. For this type of study, formal consent is not required.
Statistical approach
Descriptive statistics were used to analyze participant characteristics, program participation rates, and participant satisfaction. For each module’s education session, we compared the overall satisfaction between patients and partners using t tests. We also compared the level of satisfaction across the different modules using a 1-way analysis of variance. For the sexual health and pelvic-floor physical therapy sessions, we compared satisfaction between participants who attended the education sessions before to those who attended following their primary treatment using t tests. We provide the eta squared (for analyses of variance) and Cohen d (for t tests) to provide an effect size estimate of any significant differences observed.
Results
Participants
From the program’s founding in January of 2013 to December 31, 2016, a total of 1,269 patients registered (an average of 317 patients a year). Of those, 1,026 (80.9%) had at least 1 prostate cancer–related visit at the VPC. The remaining 243 (19.1%) were non-VPC patients (Figure). Overall, 1,062 men (83.4%) who registered with the program went on to attend at least 1 education session or clinic appointment.
Average age among male program participants was 67.7 years, and age at diagnosis was 62.5 years (Table 1). In all, 273 men (31.7%) had Gleason 3+4, and 117 (13.7%) had Gleason 4+3. Most of the participants (76.9%) elected to undergo radical prostatectomy for primary treatment. Ninety-five men (8.9%) received at least some ADT treatment as an adjunct to radiation or to treat recurrent disease. Participants traveled an average of 83.1 km (51.6 miles; median, 6.9 km and 10.5 miles, respectively) to attend the program; 10% of participants traveled further than 112 km (70 mi) to the clinic. One hundred and four (10.9%) and 301 (31.5%) of registrants were in the lowest and highest income quintiles respectively. Four hundred and ninety-seven (46.8%) attended at lesson 1 session or clinic appointment with a partner or family member.
Program participation
Of the 1,062 men who participated in the program, 867 (80.1%) were patients of the VPC, and 205 (19.1%) were non-VPC patients. The education sessions for the introduction to prostate cancer and sexual health modules had the largest numbers of participants (309 and 265, respectively; Table 2); however, pelvic-floor physical therapy had the highest participation rate per quarter (25 patients). The clinical services offered within the sexual health module had the larger number of participants and highest participation rate per quarter (590 total patients, 42/quarter). Timing of program participation was highly variable, ranging from 6 days to 18.5 years after diagnosis (SD, 1,301 days). More than half of participants attended a session or clinic visit within the first year of their diagnosis. A total of 17% of patients who registered did not attend any part of the program.
Satisfaction
Most patients and partners said that they found the information presented at the modular education sessions comprehensive, clear, and easy to understand (Table 3). Although the overall average satisfaction score varied significantly across sessions, ranging from 3.5 (out of a possible 4) for pelvic-floor physical therapy to 3.8 for introduction to prostate cancer (F = 3.8, P < .001), the effect size of this difference was small (η2 = .039; Table 4A). We found no difference in the level of satisfaction between patients and partners, with the exception of the sexual health module, which was rated better by partners than by patients (patients: 3.6, partners: 3.8; t = 2.0; P = .03); however, the effect size of this difference was again small (Cohen d = .29). A total of 86% of patients found the inclusion of their partners at the sessions useful. For both pelvic-floor physical therapy and sexual health, attendees were more satisfied if they attended before treatment initiation rather than after completion (Table 4B).
Discussion
The purpose of this descriptive analysis was to outline a comprehensive, multidisciplinary supportive care program for men with prostate cancer and to present initial data on the population that has used the program and their satisfaction with the services provided. Within the first 5 years of the PCSC Program, 1,269 patients registered to participate. However, nearly 1 in 6 men who registered for the program did not subsequently attend any education sessions or use any clinical services offered, despite the fact that all services were free of charge. It is possible that nonparticipation may be related to men on active surveillance choosing not to engage with the program until they are faced with making a treatment decision, which may not happen until several years after an initial positive biopsy.26 This and other factors that affect a patient’s decision not to participate will be investigated in a future study. There is existing evidence documenting high levels of distress and anxiety for patients and their partners resulting from decision-making around prostate cancer treatment,27,28 and many face both decisional conflict and subsequent regret.15,29 Further work to help patients access the program could include defining a prehabilitation program for which patients can sign up that automatically selects the education sessions and clinical services most relevant to them.
The number of attendees varied across the 6 education sessions, with introduction to prostate cancer and sexual health being the best attended. This is consistent with the literature concerning the specific unmet supportive care and information needs in this population10,13 and with the value that men have placed on taking an active role in the decisions around their prostate cancer treatment.30 It is also possible that attendance varied because modules were introduced in a stepwise fashion and were offered on different schedules. Patients and partners both reported a high degree of satisfaction with all of the modules’ education sessions, reporting that the length, content, and delivery were appropriate.
Since 2013, a wide research portfolio has grown alongside the program. It has acted as a recruitment site for multicenter national studies and has attracted funding for several in-house research projects and evaluations. In addition, the VPC has implemented clinic-wide electronic collection of several patient-reported outcome measures using iPads. Patients have the option of contributing their data to Canadian (PC360o) and Global (TrueNTH Global Registry – Prostate Cancer Outcomes) registries for prostate cancer. The program has also created educational opportunities by supporting postdoctoral fellows. It has also provided a rich environment for urology and radiation oncology residents and fellows to participate in a multidisciplinary supportive care team, ensuring that the next generation of surgeons and oncologists recognize the importance of this approach to care.
Limitations
This is a brief descriptive study that relies on a mixture of anonymized survey and clinical chart data. Because the program’s patient feedback forms are anonymous, we are not able to link satisfaction scores to differences in sociodemographic, clinical, or prognostic factors. We also have not directly measured clinical, psychological, or quality of life outcomes; however, all 3 will be included in future studies of the program. An additional limitation is that not all program modules were offered for the entirety of the study duration and are offered at different frequencies. Thus, some modules have disproportionally higher participation rates than others. Lastly, we are missing clinical information for 16% of our participants who are not patients at the VPC.
The program is offered within an academic and teaching hospital in a major metropolitan center and depends on the work of a large interdisciplinary team. Cancer programs that are not embedded within a similar environment, such as those located in smaller rural communities, may not have access to the specialized clinical professionals who run our program, affecting its direct generalizability to these locations. Other specialists, such as palliative care teams, could be well positioned to provide support in locations that do not have a similar level of resource available. Furthermore, some program elements will be adapted to be delivered using telemedicine technology, which is an additional approach to improving access for patients who are beyond the reach of a tertiary care facility.
Conclusions
There is a growing need to provide consistent and comprehensive supportive care to patients with prostate cancer and their partners and families throughout the disease and treatment journey. The PCSC Program uses a multidisciplinary, evidenced-based, disease-focused approach to support informed treatment decision-making and address the physical, psychological, and psychosocial effects of prostate cancer diagnosis and treatment. We proactively collect data on disease, personal demographic details, and symptoms or quality of life, supporting opportunities to partner with researchers with the goal of further improving quality of life based on evidenced-based practices. Going forward, we will conduct detailed examinations of the costs and benefits (in terms of symptom management and quality of life) of the PCSC Program, further contributing to the development of evidence-based best practices for supportive care for men with prostate cancer and their families.
Acknowledgments
The authors express their gratitude to the urologists and radiation oncologists who referred their patients to the program and participated in delivering education sessions, including Dr Martin Gleave, Dr Peter Black, Dr Alan So, Dr Scott Tyldesley, and Dr Mira Keyes. They also thank Dr Richard Wassersug for his contributions to the initial program design and implementation. They thank the patients and their families for participating, and all of their current or past staff and collaborators. Lastly, they thank the funders of the program: the Specialist Services Committee, the BC Ministry of Health, the Prostate Cancer Foundation of BC, and philanthropic donors. They acknowledge Vancouver Coastal Health Research Institute and the University of British Columbia for their institutional support.
Prostate cancer is the most common malignancy diagnosed in Canadian men. An estimated 21,300 Canadian men were diagnosed with the disease in 2017, representing 21% of all new cancer cases.1 There are about 176,000 men living with prostate cancer in Canada.1 In the United States, there were 2,778,630 survivors of prostate cancer as of 2012 and that population is expected to increase by more than 1 million (40%) to 3,922,600 by 2022.2
Although 96% of men diagnosed with prostate cancer now survive longer than 5 years3, many will suffer from treatment-related sequelae that can have a profound effect on quality of life for themselves and their partners.4,5 Impacts include sexual, urinary, and bowel dysfunctions6 owing to treatment of the primary tumor as well as reduced muscle and bone mass, osteoporosis, fatigue, obesity, and glucose intolerance or diabetes7 owing to androgen-deprivation therapy (ADT). Many men also suffer from psychological issues such as depression, anxiety, anger and irritability, sense of isolation, grief, and loss of masculinity.8,9 The psychological impacts also continue well beyond the completion of treatment and can be significant for both patients and their partners.5,8
With posttreatment longevity and the associated complex sequelae, prostate cancer is being viewed increasingly as a chronic disease whose effects must be managed for many years after the completion of primary treatment. Supportive care that “[manages] symptoms and side effects, enables adaptation and coping, optimizes understanding and informed decision-making, and minimizes decrements in functioning”10 is becoming recognized as a critical component of direct oncologic care before, during, and after treatment. Health care professionals, scientists, governments, and patient advocates are increasingly calling for the development of comprehensive supportive care programs improve the quality of life for people diagnosed with cancer. A common model for survivorship care is a general program for all cancer survivors that provides disease- and patient-specific care plans. These care plans outline patients’ prior therapies, potential side effects, recommendations for monitoring (for side effects or relapse of cancer), and advice on how patients can maintain a healthy lifestyle.11 However, there are few survivorship programs for men with prostate cancer and their partners, and the evidence base around best practices for these programs is scant.12 Furthermore, up to 87% of men with a prostate cancer diagnosis report specific and significant unmet supportive care needs,10,13 with sexuality-related and psychological issues10,14 being the areas of greatest concern.
To address the complex supportive care needs of men with prostate cancer in British Columbia, Canada, the Vancouver Prostate Centre (VPC) and Department of Urologic Sciences at the University of British Columbia developed the multidisciplinary Prostate Cancer Supportive Care (PCSC) Program. The program aims to address the challenges of decision-making and coping faced by men with prostate cancer and their partners and family members along the entire disease trajectory. Services are provided at no cost to participants. Here, we outline the guiding principles for the PCSC program and its scope, delivery, and evaluation. We provide information on the more than 1,200 patients who have participated in the program since its inception in January of 2013, the rates of participation across the different program modules, and a selection of patient satisfaction measures. We also discuss successes and limitations and ongoing research and evaluation efforts, providing lessons learned to support the development of other supportive care programs in Canada and internationally.
Program description
Guiding principles
The PCSC Program is a clinical, educational, and research-based program, with 4 guiding principles: it is comprehensive, patient- and partner-centered, evidence-based, and supports new research. The program serves patients, partners, and families along the entire disease trajectory, recognizing that cancer is a family disease, affecting both the individual and social network, and that the psychological stress associated with a diagnosis of prostate cancer is borne heavily by partners. It has been designed, implemented, and refined with the best available evidence and with the intention to undergo consistent and repeated evaluation. Finally, it was designed to provide opportunities for targeted research efforts, supporting the growth of the evidence base in this area.
Patient entry and module descriptions
Patients can be referred to the program by a physician or other allied health professional. They may also self-refer, having been made aware of the program through our website, a variety of print materials, or by word of mouth. On referral, the program coordinator collects patients’ basic clinical and demographic data, assesses health literacy and lifestyle factors, and provides them with information on the program modules. As of December 2015, the program consisted of 6 distinct modules, each focusing on different elements of the disease trajectory or on addressing specific physical or mental health concerns. Modules are led by licensed health professionals with experience in oncology. No elements of the program are mandatory, and participants are free to pick and choose the components that are most relevant to them and their partners.
Introduction to prostate cancer and primary treatment options. This is a group-based module that focuses on educating newly diagnosed patients (and those going on or off active surveillance) on the basic biology of prostate cancer, the primary treatment options for localized disease, and the main side effects associated with the treatments. It also includes information about the other services offered by the program and any ongoing research studies. The session is held twice a month in the early evening and is run collectively by a urologist, radiation oncologist, patient representative, and program coordinator. It includes a brief one-on-one discussion between each patient and their partner or family member and the urologist and radiation oncologist to address any remaining questions. A copy of the patient’s biopsy report is on hand for the physician(s). Attendance of this session has been shown to significantly reduce pretreatment distress in both patients and their partners.15
Managing sexual function and intimacy. Sexual intimacy is tied to overall health outcomes, relationship satisfaction, and quality of life.16 Primary therapy for prostate cancer can be associated with substantial side-effects (eg, erectile dysfunction, incontinence, altered libido, penile shortening) that negatively affect sexual intimacy and have an impact on the patient individually as well as the sexual relationship he has with his partner.17
The program’s Sexual Health Service (SHS) provides patients and partners with information on the impact of treatment on sexual health.18 The SHS offers educational sessions led by a sexual rehabilitation nurse and clinical psychologist with a specialization in sexual health. Sessions focus on the impact of prostate cancer treatments on sexual function and therapeutic modalities, promote an understanding of the barriers to sexual adaptation posttreatment, and present options for sexual activity that are not solely dependent on the ability to achieve an erection. Once participants have attended an educational session, they are offered individual consultations with the sexual health nurse every 3 to 6 months for 2 years or longer, depending on the patient’s or couple’s needs. They are referred to the SHS’s sexual medicine physician if further medical intervention is warranted. The sexual health nurse works with the patient and partner to develop an individualized Sexual Health Rehabilitation Action Plan (SHRAP), which assists the couple in sexual adaptation going forward. The SHRAP is a tool devised by the sexual health nurse based on her clinical experience with couples affected by prostate cancer.
Couples who have been evaluated within the SHS are also invited to attend a second workshop on intimacy that is offered quarterly. Workshop participants discuss the impact of sexual changes on relationships, and strategies on how to enhance intimacy and sexual communication are presented. A resource package is provided to each couple to help re-establish and/or strengthen their various dimensions of intimacy.
Lifestyle management. The lifestyle management modules include separate nutrition and physical activity or exercise components. Referral to the smoking cessation program in the Vancouver Coastal Health Authority is made at program registration, if appropriate. The nutrition group-based education session is delivered by a registered dietitian from the British Columbia Cancer Agency who specializes in prostate cancer. The session focuses on evidence-based recommendations on diet after a diagnosis of prostate cancer, the use of dietary supplements, body weight and health, and practical nutrition tips. The exercise session is delivered by an exercise physiologist who specializes in working with cancer patients. It covers the value of exercise in terms of safety, prevention and reduction of treatment side effects (including from ADT), treatment prehabilitation and recovery, advanced cancer management, and long-term survival. A one-on-one exercise counseling clinic is also offered and aims to increase exercise adoption and long-term adherence in line with Canadian Physical Activity guidelines and exercise oncology guidelines,19,20 with follow-up appointments at 3, 6, and 12 months to help patients stay motivated and ensure they are exercising correctly. The individual consultations with the exercise physiologist include physical measures, exercise volume, treatment side effects, and coconstructed goal setting with an individualized formal exercise regimen (exercise prescription).
Adapting to ADT. This is an educational module offered to patients with metastatic prostate cancer who are starting hormone therapy treatments that lower serum testosterone into the castrate range. This program was one of several available through TrueNTH, a portfolio of projects funded by the Movember Foundation, through Prostate Cancer Canada. The session is delivered by a patient facilitator and focuses on strategies to recognize and adapt to the side effects of ADT21 while maintaining a good quality of life and strong intimate relationships with partners.22,23
Pelvic-floor physical therapy for urinary incontinence. This module includes a group-based and individualized education session for patients (either pre- or posttreatment) focused on reducing the effects of surgery and/or radiation therapy on urinary and sexual continence as well as on how to cope with these symptoms and minimize the effect they have on quality of life.24 The session is conducted by a physical therapist who is certified as a pelvic-floor specialist. Supervised pelvic-floor re-education and/or exercise has been shown to successfully reduce the degree of incontinence in this population.25 The module therefore also includes 3 one-on-one clinical appointments for patients who are still experiencing bother from incontinence 12 weeks after a radical prostatectomy or postradiation treatment.
Psycho-oncology. In recognition of the emotional and psychological burden associated with prostate cancer and the important role partners play in facilitating treatment of these psychological and/or psychosocial issues, the program offers appointments with a registered clinical counselor to address acute emotional distress. These are usually 1-hour appointments offered to both patients and partners, either separately or together. Appointments can be attended in person or conducted by telephone. When appropriate, patients are referred for further long-term individual support or couple support with their partners. A group therapy workshop was also initiated in 2016. In this program, men participate in a guided autobiographical life review through a process that focuses on developing a cohesive working group, learning strategic communication skills, and understanding and learning how to manage difficult emotions and transitional life stressors associated with prostate cancer. It also focuses on processing and integrating critical events that contribute to the men’s identity and psychological function and involves the consolidation of the personal learning that occurs. Postgroup referral plans are developed on an individual basis as needed.
Methods
Data
We obtained sociodemographic, diagnostic, and treatment information as well as clinic visit records for all PCSC Program registrants from the electronic medical record held at the VPC. Clinical variables included age at diagnosis, Gleason score, and primary treatment modality (including active surveillance and ADT use). The Gleason score determines the aggressiveness of a patient’s prostate cancer based on biopsy results. A score of 6 or less indicates that the disease is likely to grow slowly. A grade of 7 is considered intermediate risk (with primary score of 3 and secondary 4 being lower risk than those with a primary score of 4 and secondary of 3). A Gleason score of 8 or higher indicates aggressive disease that is poorly differentiated or high grade. Sociodemographic characteristics included age, travel distance to the clinic, and income quintile. We obtained attendance records for the modular education sessions from the program’s database. Patients who did not have any medical visits at the VPC (referred to henceforth as non-VPC patients) did not have a clinic record, so we excluded them from the subset of the analyses that depended on specific clinical variables.
All patients and partners who participate in any PCSC Program education sessions (introduction to prostate cancer, sexual health, nutrition, exercise, ADT, and pelvic-floor physical therapy) are asked to complete voluntary, anonymous feedback forms. These forms assess participant satisfaction using a series of Likert-based and Boolean response items as well as qualitative commentary. They include questions that assess the timing, structure, and content of each session.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. For this type of study, formal consent is not required.
Statistical approach
Descriptive statistics were used to analyze participant characteristics, program participation rates, and participant satisfaction. For each module’s education session, we compared the overall satisfaction between patients and partners using t tests. We also compared the level of satisfaction across the different modules using a 1-way analysis of variance. For the sexual health and pelvic-floor physical therapy sessions, we compared satisfaction between participants who attended the education sessions before to those who attended following their primary treatment using t tests. We provide the eta squared (for analyses of variance) and Cohen d (for t tests) to provide an effect size estimate of any significant differences observed.
Results
Participants
From the program’s founding in January of 2013 to December 31, 2016, a total of 1,269 patients registered (an average of 317 patients a year). Of those, 1,026 (80.9%) had at least 1 prostate cancer–related visit at the VPC. The remaining 243 (19.1%) were non-VPC patients (Figure). Overall, 1,062 men (83.4%) who registered with the program went on to attend at least 1 education session or clinic appointment.
Average age among male program participants was 67.7 years, and age at diagnosis was 62.5 years (Table 1). In all, 273 men (31.7%) had Gleason 3+4, and 117 (13.7%) had Gleason 4+3. Most of the participants (76.9%) elected to undergo radical prostatectomy for primary treatment. Ninety-five men (8.9%) received at least some ADT treatment as an adjunct to radiation or to treat recurrent disease. Participants traveled an average of 83.1 km (51.6 miles; median, 6.9 km and 10.5 miles, respectively) to attend the program; 10% of participants traveled further than 112 km (70 mi) to the clinic. One hundred and four (10.9%) and 301 (31.5%) of registrants were in the lowest and highest income quintiles respectively. Four hundred and ninety-seven (46.8%) attended at lesson 1 session or clinic appointment with a partner or family member.
Program participation
Of the 1,062 men who participated in the program, 867 (80.1%) were patients of the VPC, and 205 (19.1%) were non-VPC patients. The education sessions for the introduction to prostate cancer and sexual health modules had the largest numbers of participants (309 and 265, respectively; Table 2); however, pelvic-floor physical therapy had the highest participation rate per quarter (25 patients). The clinical services offered within the sexual health module had the larger number of participants and highest participation rate per quarter (590 total patients, 42/quarter). Timing of program participation was highly variable, ranging from 6 days to 18.5 years after diagnosis (SD, 1,301 days). More than half of participants attended a session or clinic visit within the first year of their diagnosis. A total of 17% of patients who registered did not attend any part of the program.
Satisfaction
Most patients and partners said that they found the information presented at the modular education sessions comprehensive, clear, and easy to understand (Table 3). Although the overall average satisfaction score varied significantly across sessions, ranging from 3.5 (out of a possible 4) for pelvic-floor physical therapy to 3.8 for introduction to prostate cancer (F = 3.8, P < .001), the effect size of this difference was small (η2 = .039; Table 4A). We found no difference in the level of satisfaction between patients and partners, with the exception of the sexual health module, which was rated better by partners than by patients (patients: 3.6, partners: 3.8; t = 2.0; P = .03); however, the effect size of this difference was again small (Cohen d = .29). A total of 86% of patients found the inclusion of their partners at the sessions useful. For both pelvic-floor physical therapy and sexual health, attendees were more satisfied if they attended before treatment initiation rather than after completion (Table 4B).
Discussion
The purpose of this descriptive analysis was to outline a comprehensive, multidisciplinary supportive care program for men with prostate cancer and to present initial data on the population that has used the program and their satisfaction with the services provided. Within the first 5 years of the PCSC Program, 1,269 patients registered to participate. However, nearly 1 in 6 men who registered for the program did not subsequently attend any education sessions or use any clinical services offered, despite the fact that all services were free of charge. It is possible that nonparticipation may be related to men on active surveillance choosing not to engage with the program until they are faced with making a treatment decision, which may not happen until several years after an initial positive biopsy.26 This and other factors that affect a patient’s decision not to participate will be investigated in a future study. There is existing evidence documenting high levels of distress and anxiety for patients and their partners resulting from decision-making around prostate cancer treatment,27,28 and many face both decisional conflict and subsequent regret.15,29 Further work to help patients access the program could include defining a prehabilitation program for which patients can sign up that automatically selects the education sessions and clinical services most relevant to them.
The number of attendees varied across the 6 education sessions, with introduction to prostate cancer and sexual health being the best attended. This is consistent with the literature concerning the specific unmet supportive care and information needs in this population10,13 and with the value that men have placed on taking an active role in the decisions around their prostate cancer treatment.30 It is also possible that attendance varied because modules were introduced in a stepwise fashion and were offered on different schedules. Patients and partners both reported a high degree of satisfaction with all of the modules’ education sessions, reporting that the length, content, and delivery were appropriate.
Since 2013, a wide research portfolio has grown alongside the program. It has acted as a recruitment site for multicenter national studies and has attracted funding for several in-house research projects and evaluations. In addition, the VPC has implemented clinic-wide electronic collection of several patient-reported outcome measures using iPads. Patients have the option of contributing their data to Canadian (PC360o) and Global (TrueNTH Global Registry – Prostate Cancer Outcomes) registries for prostate cancer. The program has also created educational opportunities by supporting postdoctoral fellows. It has also provided a rich environment for urology and radiation oncology residents and fellows to participate in a multidisciplinary supportive care team, ensuring that the next generation of surgeons and oncologists recognize the importance of this approach to care.
Limitations
This is a brief descriptive study that relies on a mixture of anonymized survey and clinical chart data. Because the program’s patient feedback forms are anonymous, we are not able to link satisfaction scores to differences in sociodemographic, clinical, or prognostic factors. We also have not directly measured clinical, psychological, or quality of life outcomes; however, all 3 will be included in future studies of the program. An additional limitation is that not all program modules were offered for the entirety of the study duration and are offered at different frequencies. Thus, some modules have disproportionally higher participation rates than others. Lastly, we are missing clinical information for 16% of our participants who are not patients at the VPC.
The program is offered within an academic and teaching hospital in a major metropolitan center and depends on the work of a large interdisciplinary team. Cancer programs that are not embedded within a similar environment, such as those located in smaller rural communities, may not have access to the specialized clinical professionals who run our program, affecting its direct generalizability to these locations. Other specialists, such as palliative care teams, could be well positioned to provide support in locations that do not have a similar level of resource available. Furthermore, some program elements will be adapted to be delivered using telemedicine technology, which is an additional approach to improving access for patients who are beyond the reach of a tertiary care facility.
Conclusions
There is a growing need to provide consistent and comprehensive supportive care to patients with prostate cancer and their partners and families throughout the disease and treatment journey. The PCSC Program uses a multidisciplinary, evidenced-based, disease-focused approach to support informed treatment decision-making and address the physical, psychological, and psychosocial effects of prostate cancer diagnosis and treatment. We proactively collect data on disease, personal demographic details, and symptoms or quality of life, supporting opportunities to partner with researchers with the goal of further improving quality of life based on evidenced-based practices. Going forward, we will conduct detailed examinations of the costs and benefits (in terms of symptom management and quality of life) of the PCSC Program, further contributing to the development of evidence-based best practices for supportive care for men with prostate cancer and their families.
Acknowledgments
The authors express their gratitude to the urologists and radiation oncologists who referred their patients to the program and participated in delivering education sessions, including Dr Martin Gleave, Dr Peter Black, Dr Alan So, Dr Scott Tyldesley, and Dr Mira Keyes. They also thank Dr Richard Wassersug for his contributions to the initial program design and implementation. They thank the patients and their families for participating, and all of their current or past staff and collaborators. Lastly, they thank the funders of the program: the Specialist Services Committee, the BC Ministry of Health, the Prostate Cancer Foundation of BC, and philanthropic donors. They acknowledge Vancouver Coastal Health Research Institute and the University of British Columbia for their institutional support.
1. Cancer Research UK. Prostate cancer statistics. http://www.cancer.ca/en/cancer-information/cancer-type/prostate/statistics/?region=sk. Published 2015. Accessed June 22, 2017.
2. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220-241.
3. Canadian Cancer Society. Canadian cancer statistics special topic: predictions of the future burden of cancer in Canada. Ottawa, Canada: Public Health Agency of Canada; 2015.
4. Roth AJ, Weinberger MI, Nelson CJ. Prostate cancer: psychosocial implications and management. Future Oncol. 2008;4(4):561-568.
5. Couper J, Bloch S, Love A, Macvean M, Duchesne GM, Kissane D. Psychosocial adjustment of female partners of men with prostate cancer: a review of the literature. Psychooncology 2006;15(11):937-953.
6. Wilt TJ, MacDonald R, Rutks I, Shamliyan TA, Taylor BC, Kane RL. Systematic review: comparative effectiveness and harms of treatments for clinically localized prostate cancer. Ann Intern Med. 2008;148(6):435-448.
7. Galvão DA, Spry NA, Taaffe DR, et al. Changes in muscle, fat and bone mass after 36 weeks of maximal androgen blockade for prostate cancer. BJU Int. 2008;102(1):44-47.
8. Watts S, Leydon G, Birch B, et al. Depression and anxiety in prostate cancer: a systematic review and meta-analysis of prevalence rates. BMJ Open. 2014;4(3):e003901.
9. Zaider T, Manne S, Nelson C, Mulhall J, Kissane D. Loss of masculine identity, marital affection, and sexual bother in men with localized prostate cancer. J Sex Med. 2012;9(10):2724-2732.
10. Ream E, Quennell A, Fincham L, et al. Supportive care needs of men living with prostate cancer in England: a survey. Br J Cancer. 2008;98(12):1903-1909.
11. Howell D, Hack TF, Oliver TK, et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
12. Halpern MT, Viswanathan M, Evans TS, Birken SA, Basch E, Mayer DK. Models of cancer survivorship care: overview and summary of current evidence. J Oncol Pract. 2015;11(1):e19-e27.
13. Smith DP, Supramaniam R, King MT, Ward J, Berry M, Armstrong BK. Age, health, and education determine supportive care needs of men younger than 70 years with prostate cancer. J Clin Oncol. 2007;25(18):2560-2566.
14. Northouse LL, Mood DW, Montie JE, et al. Living with prostate cancer: patients’ and spouses’ psychosocial status and quality of life. J Clin Oncol. 2007;25(27):4171-4177.
15. Hedden L, Wassersug R, Mahovlich S, et al. Evaluating an educational intervention to alleviate distress amongst men with newly diagnosed prostate cancer and their partners. BJU Int. 2017;120(5B):E21-E29.
16. Bradley EB, Bissonette EA, Theodorescu D. Determinants of long-term quality of life and voiding function of patients treated with radical prostatectomy or permanent brachytherapy for prostate cancer. BJU Int. 2004;94(7):1003-1009.
17. Ramsey SD, Zeliadt SB, Blough DK, et al. Impact of prostate cancer on sexual relationships: a longitudinal perspective on intimate partners’ experiences. J Sex Med. 2013;10(12):3135-3143.
18. Wittmann D, Carolan M, Given B, et al. Exploring the role of the partner in couples’ sexual recovery after surgery for prostate cancer. Support Care Cancer. 2014;22(9):2509-2515.
19. Schmitz KH, Courneya KS, Matthews C, et al. American college of sports medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42(7):1409-1426.
20. Rock CL, Doyle C, Demark-Wahnefried W, et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J Clin. 2012;62(4):243-274.
21. Elliott S, Latini DM, Walker LM, Wassersug R, Robinson JW; ADT Suvivorship Working Group. Androgen deprivation therapy for prostate cancer: recommendations to improve patient and partner quality of life. J Sex Med. 2010;7(9):2996-3010.
22. Wassersug RJ, Walker LM, Robinson JW. Androgen deprivation therapy: an essential guide for prostate cancer patients and their loved ones. New York, NY: Demos Health; 2014.
23. Wibowo E, Walker LM, Wilyman S, et al. Androgen deprivation therapy educational program: a Canadian True NTH initiative. J Clin Oncol. 2016;34(suppl 3):243.
24. Stanford JL, Feng Z, Hamilton AS, et al. Urinary and sexual function after radical prostatectomy for clinically localized prostate cancer. JAMA. 2000;283(3):354-360.
25. Overgård M, Angelsen A, Lydersen S, Mørkved S. Does physiotherapist-guided pelvic floor muscle training reduce urinary incontinence after radical prostatectomy? A randomised controlled trial. Eur Urol. 2008;54(2):438-448.
26. Godtman RA, Holmberg E, Khatami A, Pihl CG, Stranne J, Hugosson J. Long-term results of active surveillance in the Göteborg randomized, population-based prostate cancer screening trial. Eur Urol. 2016;70(5):760-766.
27. Cohen H, Britten N. Who decides about prostate cancer treatment? A qualitative study. Fam Pract. 2003;20(6):724-729.
28. Denberg TD, Melhado TV, Steiner JF. Patient treatment preferences in localized prostate carcinoma: the influence of emotion, misconception, and anecdote. Cancer. 2006;107(3):620-630.
29. Morris BB, Farnan L, Song L, et al. Treatment decisional regret among men with prostate cancer: racial differences and influential factors in the North Carolina health access and prostate cancer treatment project (HCaP-NC). Cancer. 2015;121(12):2029-2035.
30. Feldman-Stewart D, Capirci C, Brennenstuhl S, et al. Information for decision making by patients with early-stage prostate cancer: a comparison across 9 countries. Med Decis Making. 2011;31(5):754-766.
1. Cancer Research UK. Prostate cancer statistics. http://www.cancer.ca/en/cancer-information/cancer-type/prostate/statistics/?region=sk. Published 2015. Accessed June 22, 2017.
2. Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220-241.
3. Canadian Cancer Society. Canadian cancer statistics special topic: predictions of the future burden of cancer in Canada. Ottawa, Canada: Public Health Agency of Canada; 2015.
4. Roth AJ, Weinberger MI, Nelson CJ. Prostate cancer: psychosocial implications and management. Future Oncol. 2008;4(4):561-568.
5. Couper J, Bloch S, Love A, Macvean M, Duchesne GM, Kissane D. Psychosocial adjustment of female partners of men with prostate cancer: a review of the literature. Psychooncology 2006;15(11):937-953.
6. Wilt TJ, MacDonald R, Rutks I, Shamliyan TA, Taylor BC, Kane RL. Systematic review: comparative effectiveness and harms of treatments for clinically localized prostate cancer. Ann Intern Med. 2008;148(6):435-448.
7. Galvão DA, Spry NA, Taaffe DR, et al. Changes in muscle, fat and bone mass after 36 weeks of maximal androgen blockade for prostate cancer. BJU Int. 2008;102(1):44-47.
8. Watts S, Leydon G, Birch B, et al. Depression and anxiety in prostate cancer: a systematic review and meta-analysis of prevalence rates. BMJ Open. 2014;4(3):e003901.
9. Zaider T, Manne S, Nelson C, Mulhall J, Kissane D. Loss of masculine identity, marital affection, and sexual bother in men with localized prostate cancer. J Sex Med. 2012;9(10):2724-2732.
10. Ream E, Quennell A, Fincham L, et al. Supportive care needs of men living with prostate cancer in England: a survey. Br J Cancer. 2008;98(12):1903-1909.
11. Howell D, Hack TF, Oliver TK, et al. Models of care for post-treatment follow-up of adult cancer survivors: a systematic review and quality appraisal of the evidence. J Cancer Surviv. 2012;6(4):359-371.
12. Halpern MT, Viswanathan M, Evans TS, Birken SA, Basch E, Mayer DK. Models of cancer survivorship care: overview and summary of current evidence. J Oncol Pract. 2015;11(1):e19-e27.
13. Smith DP, Supramaniam R, King MT, Ward J, Berry M, Armstrong BK. Age, health, and education determine supportive care needs of men younger than 70 years with prostate cancer. J Clin Oncol. 2007;25(18):2560-2566.
14. Northouse LL, Mood DW, Montie JE, et al. Living with prostate cancer: patients’ and spouses’ psychosocial status and quality of life. J Clin Oncol. 2007;25(27):4171-4177.
15. Hedden L, Wassersug R, Mahovlich S, et al. Evaluating an educational intervention to alleviate distress amongst men with newly diagnosed prostate cancer and their partners. BJU Int. 2017;120(5B):E21-E29.
16. Bradley EB, Bissonette EA, Theodorescu D. Determinants of long-term quality of life and voiding function of patients treated with radical prostatectomy or permanent brachytherapy for prostate cancer. BJU Int. 2004;94(7):1003-1009.
17. Ramsey SD, Zeliadt SB, Blough DK, et al. Impact of prostate cancer on sexual relationships: a longitudinal perspective on intimate partners’ experiences. J Sex Med. 2013;10(12):3135-3143.
18. Wittmann D, Carolan M, Given B, et al. Exploring the role of the partner in couples’ sexual recovery after surgery for prostate cancer. Support Care Cancer. 2014;22(9):2509-2515.
19. Schmitz KH, Courneya KS, Matthews C, et al. American college of sports medicine roundtable on exercise guidelines for cancer survivors. Med Sci Sports Exerc. 2010;42(7):1409-1426.
20. Rock CL, Doyle C, Demark-Wahnefried W, et al. Nutrition and physical activity guidelines for cancer survivors. CA Cancer J Clin. 2012;62(4):243-274.
21. Elliott S, Latini DM, Walker LM, Wassersug R, Robinson JW; ADT Suvivorship Working Group. Androgen deprivation therapy for prostate cancer: recommendations to improve patient and partner quality of life. J Sex Med. 2010;7(9):2996-3010.
22. Wassersug RJ, Walker LM, Robinson JW. Androgen deprivation therapy: an essential guide for prostate cancer patients and their loved ones. New York, NY: Demos Health; 2014.
23. Wibowo E, Walker LM, Wilyman S, et al. Androgen deprivation therapy educational program: a Canadian True NTH initiative. J Clin Oncol. 2016;34(suppl 3):243.
24. Stanford JL, Feng Z, Hamilton AS, et al. Urinary and sexual function after radical prostatectomy for clinically localized prostate cancer. JAMA. 2000;283(3):354-360.
25. Overgård M, Angelsen A, Lydersen S, Mørkved S. Does physiotherapist-guided pelvic floor muscle training reduce urinary incontinence after radical prostatectomy? A randomised controlled trial. Eur Urol. 2008;54(2):438-448.
26. Godtman RA, Holmberg E, Khatami A, Pihl CG, Stranne J, Hugosson J. Long-term results of active surveillance in the Göteborg randomized, population-based prostate cancer screening trial. Eur Urol. 2016;70(5):760-766.
27. Cohen H, Britten N. Who decides about prostate cancer treatment? A qualitative study. Fam Pract. 2003;20(6):724-729.
28. Denberg TD, Melhado TV, Steiner JF. Patient treatment preferences in localized prostate carcinoma: the influence of emotion, misconception, and anecdote. Cancer. 2006;107(3):620-630.
29. Morris BB, Farnan L, Song L, et al. Treatment decisional regret among men with prostate cancer: racial differences and influential factors in the North Carolina health access and prostate cancer treatment project (HCaP-NC). Cancer. 2015;121(12):2029-2035.
30. Feldman-Stewart D, Capirci C, Brennenstuhl S, et al. Information for decision making by patients with early-stage prostate cancer: a comparison across 9 countries. Med Decis Making. 2011;31(5):754-766.
Paradigm-changing osimertinib approval in front-line for advanced NSCLC
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
The US Food and Drug Administration awarded regulatory approval this spring to the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib for the treatment of patients with exon 19 deletion- or exon21 L858R mutation-positive advanced non–small-cell lung cancer (NSCLC) not previously treated for advanced disease.
Osimertinib is designed to target both sensitizing and resistant mutant forms of EGFR, but not the wildtype protein, in an effort to improve safety and efficacy compared with other standard of care (SoC) EGFR inhibitors. It was previously approved in the second-line setting in NSCLC following failure of prior EGFR inhibitor therapy in 2015. The current approval represents a paradigm shift in the front-line treatment of advanced NSCLC, reinforcing the role of osimertinib, which has been recommended in this setting by the National Comprehensive Cancer Network Guidelines in Oncology for more than a year.
Approval was based on the phase 3, multicenter, international, randomized, double-blind, active-controlled FLAURA trial. A total of 556 patients were randomized 1:1 to receive an oral daily dose of 80 mg osimertinib or gefitinib 250 mg or erlotinib 150 mg. The trial was conducted during December 2014 through March 2016 at 132 sites in 29 countries.
Eligible patients were aged 18 or over and had locally advanced or metastatic NSCLC, had not previously received treatment for advanced disease, were eligible for first-line treatment with erlotinib or gefitinib, had locally or centrally confirmed EGFR exon 19 deletion or L858R mutations alone or concurrently with other EGFR mutations, and a World Health Organization Performance Status of 0 (fully active, able to carry on all predisease performance without restriction) or 1 (restricted in strenuous activity but ambulatory and able to carry out light work), and a minimum life expectancy of 12 weeks.
Patients with central nervous system metastases were eligible if their condition was neurologically stable. Patients who had previous definitive treatment or glucocorticoid therapy had to have completed it at least 2 weeks before the start of the trial. Patients were excluded from the trial if they had any previous treatment with any systemic anticancer therapy for advanced NSCLC, had major surgery within 4 weeks of the first dose of the study drug, had radiation therapy to more than 30% of the bone marrow or a wide field of radiation within 4 weeks of the first dose of the study drug, or were currently receiving potent inhibitors or inducers of cytochrome P450 3A4.
Osimertinib cut the risk of disease progression or death by more than 50% compared with standard TKI therapy. The estimated median progression-free survival (PFS) was 18.9 months with osimertinib, compared with 10.2 months for erlotinib or gefitinib (hazard ratio [HR]: 0.46; P < .0001). PFS benefit extended across all prespecified subgroups, including patients with CNS metastases (median PFS: 15.2 months vs 9.6 months; HR: 0.47; P = .0009). Confirmed overall response rate was 77% and 69% in the study and SoC groups, respectively, and estimated duration of response (DoR) was 17.6 months and 9.6 months. At the time of analysis, there were too few deaths to compare overall survival.
The most common adverse events (AEs) experienced by patients treated with osimertinib were diarrhea, rash, dry skin, nail toxicity, stomatitis, and reduced appetite. Serious AEs occurred in 4% of patients treated with osimertinib, most commonly involving pneumonia, interstitial lung disease/pneumonitis, and pulmonary embolism (PE). The rate of grade 3/4 AEs was 33.7% in the osimertinib group and 44.8% in the SoC group. Patients treated with osimertinib were less likely to discontinue treatment due to AEs (13.3% vs 18.1% of those receiving SoC).
Osimertinib is marketed as Tagrisso by AstraZeneca and the recommended dose is 80 mg orally once daily, with or without food. The prescribing information details warnings and precautions relating to interstitial lung disease and pneumonitis, QTc interval prolongation, cardiomyopathy, keratitis, and embryofetal toxicity.
Treatment with osimertinib should be withheld in patients presenting with worsening of respiratory symptoms indicative of ILD and permanently discontinued if ILD is confirmed. Electrocardiograms and electrolytes should be monitored periodically in patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities or in patients taking medications known to prolong QTc interval. Treatment should be permanently discontinued in those who develop QTc interval prolongation with signs and symptoms of life-threatening arrhythmia.
Cardiac monitoring, including assessment of left ventricular ejection fraction should be performed at baseline and throughout treatment in patients with cardiac risk factors and treatment should be permanently discontinued in patients who develop symptomatic congestive heart failure. Patients with signs and symptoms of keratitis should be referred to an ophthalmologist. Osimertinib can cause fetal harm and patients should be advised of the potential risk and the need for effective contraception use during treatment and for 6 weeks after the final dose is administered.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.
1. US Food and Drug Administration Website. FDA approves osimertinib for first-line treatment of metastatic NSCLC with most common EGFR mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm605113.htm. Last updated April 18, 2018. Accessed October 6, 2018.
2. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-
3. Tagrisso (osimertinib tablets) for oral use. Prescribing information. AstraZeneca. https://www.azpicentral.com/tagrisso/tagrisso.pdf#page=1. August 2018. Accessed October 6, 2018.

BRAF-MEK inhibitor combo approved for adjuvant melanoma therapy
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
On April 30, 2018, the US Food and Drug Administration expanded the indication for the combined use of dabrafenib and trametinib to include adjuvant treatment of BRAF-mutant melanoma following complete surgical resection. Dabrafenib is an inhibitor of the BRAF kinase, and trametinib is an inhibitor of the MEK kinase, both of which are components of the mitogen-activated protein kinase (MAPK) signaling pathway. The 2 drugs are already approved as both single agents and in combination for the treatment of BRAF-mutated metastatic melanoma.
The current approval was based on data from a phase 3, international, multicenter, randomized, double-blind, placebo-controlled trial. The COMBI-AD trial was carried out from January 2013 through December 2014 at 169 sites in 26 countries. A total of 870 patients with stage III melanoma and BRAF V600E/K mutations and pathologic involvement of regional lymph nodes following complete resection were randomly assigned to receive dabrafenib 150 mg twice daily in combination with trametinib 2 mg once daily, or 2 matched placebos for up to 1 year. Randomization was stratified according to BRAF mutation status (V600E or V600K) and disease stage (IIIA, IIIB or IIIC).
Eligible patients were aged 18 years or older and had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (on a scale of 1-5, with higher scores indicating greater disability). Patients who had undergone previous systemic anticancer therapy or radiotherapy were excluded from the study.
The primary endpoint was relapse-free survival (RFS), defined as the time from randomization to disease recurrence or death from any cause. Secondary endpoints included overall survival (OS), distant metastasis-free survival (DMFS), freedom from relapse (FFR), and safety. Clinical examination and imaging by computed tomography, magnetic resonance imaging, or both was performed every 3 months for the first 2 years and then every 6 months until disease recurrence or trial completion.
As of the data cut-off, the combination of dabrafenib and trametinib reduced the risk of disease recurrence or death by 53% compared with placebo (hazard ratio [HR], 0.47; P < .001). Median RFS was not yet reached in the combination arm, compared with 16.6 months in the placebo arm. The RFS benefit was observed across all prespecified subgroups, and the combination was also found to improve OS, DMFS, and FFR.
The most common adverse events (AEs) included pyrexia, fatigue, nausea, rash, vomiting, diarrhea, chills, and myalgia. Overall, 97% of patients experienced an AE, 41% experienced a grade 3/4 AE, and 26% had an AE that led to treatment discontinuation. In patients treated with placebo, those numbers were 88%, 14%, and 3%, respectively.
The separate prescribing information for dabrafenib and trametinib detail warnings and precautions relating to their combined use, including the need to confirm BRAF status before starting therapy (because use in BRAF wildtype tumors can promote tumor cell proliferation), new primary malignancies, hemorrhage, cardiomyopathy, uveitis, serious febrile reactions, serious skin toxicity, hyperglycemia, glucose-6-phosphate dehydrogenase (G6PD) deficiency, colitis and gastrointestinal perforation, venous thromboembolism, ocular toxicities, interstitial lung disease, and embryofetal toxicity.
Dermatologic evaluations should be completed before starting therapy, every 2 months during and for up to 6 months after completion of therapy, and patients should be monitored closely for the signs and symptoms of noncutaneous primary malignancies. Treatment should be discontinued for all grade 4 hemorrhagic events and for any grade 3 events that do not improve, and withheld for grade 3 events until they resolve, at which point treatment can be resumed at the next lowest dose as described in the prescribing information.
Left ventricular ejection fraction (LVEF) values should be assessed before initiating therapy, after 1 month, and then at intervals of 2-3 months. Treatment should be withheld for up to 4 weeks if absolute LVEF values decrease by 10% and are less than the lower limit of normal (LLN) and it should be permanently discontinued for symptomatic cardiomyopathy or persistent, asymptomatic left ventricular dysfunction of >20% from baseline that is below LLN and does not resolve within 4 weeks.
Treatment should be withheld for fevers higher than 104°F or for serious febrile reactions or fever accompanied by hypotension, rigors or chills, dehydration, or renal failure. Serum creatinine levels should be monitored, along with other evidence of renal function, during, and after severe pyrexia. Antipyretics should be administered as secondary prophylaxis when treatment is resumed if the patient had previous episodes of severe febrile reaction or if fever was associated with complications. Corticosteroids should be administered for at least 5 days for second or subsequent pyrexia if the body temperature dose not return to baseline within 3 days of fever onset or for pyrexia associated with complications and no evidence of active infection.
Treatment should also be withheld for intolerable or severe skin toxicity and resumed at a lower dose as per guidelines in patients who improve or recover within 3 weeks. Serum glucose levels should be monitored at the start of treatment and as clinically appropriate in patients with pre-existing diabetes or hyperglycemia. Patients with G6PD deficiency should be monitored closely for signs of hemolytic anemia.
Patients should be monitored closely for signs and symptoms of colitis and gastrointestinal
Ophthalmological evaluations should be performed periodically and within 24 hours of patient-reported loss of vision or other visual disturbances. Treatment should be permanently discontinued in patients with documented retinal vein occlusion and withheld for retinal pigment epithelial detachment. Treatment should also be withheld in patients presenting with new or progressive pulmonary symptoms and findings and permanently discontinued for treatment-related interstitial lung disease or pneumonitis.
Both dabrafenib and trametinib can cause fetal harm and patients should be warned of this risk and the need for adequate contraceptive measures. Dabrafenib and trametinib are marketed as Tafinlar and Mekinist by Novartis.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.
1. US Food and Drug Administration Website. FDA approves dabrafenib plus trametinib for adjuvant treatment of melanoma with BRAF V600E or V600K mutations. https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm606165.htm. Last updated April 30, 2018. Accessed October 6, 2018.
2. Long GV, Hauschild A, Santinami M, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1913-1823.
3. Tafinlar (dabrafenib) capsules, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/tafinlar.pdf. May 2018. Accessed October 6, 2018.
4. Mekinist (trametinib) tablets, for oral use. Prescribing information. Novartis. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/mekinist.pdf. May 2018. Accessed October 6th, 2018.
