User login
Research and Reviews for the Practicing Oncologist
New myeloma drugs improve response and extend survival
In this interview, Dr David Henry, the Editor-in-Chief of The Journal of Community and Supportive Oncology, and Dr Ken Anderson, the Kraft Family Professor of Medicine at Harvard Medical School and an international thought leader and investigator in myeloma, discuss three cases of patients with myeloma that are indicative of the remarkable therapeutic advances in oncology in general, and in myeloma in particular.
Listen to the podcast below
In this interview, Dr David Henry, the Editor-in-Chief of The Journal of Community and Supportive Oncology, and Dr Ken Anderson, the Kraft Family Professor of Medicine at Harvard Medical School and an international thought leader and investigator in myeloma, discuss three cases of patients with myeloma that are indicative of the remarkable therapeutic advances in oncology in general, and in myeloma in particular.
Listen to the podcast below
In this interview, Dr David Henry, the Editor-in-Chief of The Journal of Community and Supportive Oncology, and Dr Ken Anderson, the Kraft Family Professor of Medicine at Harvard Medical School and an international thought leader and investigator in myeloma, discuss three cases of patients with myeloma that are indicative of the remarkable therapeutic advances in oncology in general, and in myeloma in particular.
Listen to the podcast below
First CAR T-cell therapy approvals bolster booming immunotherapy market
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
There were a number of landmark approvals by the US Food and Drug Administration (FDA) in 2017 for cancer therapies, among them, the approval of the first two chimeric antigen receptor (CAR) T-cell therapies for cancer: tisagenlecleucel (in August) and axicabtagene ciloluecel (in October).1 CAR T-cells are a type of adoptive cell therapy or immunotherapy, in which the patient’s own immune cells are genetically engineered to target a tumor-associated antigen, in this case CD19. In tisagenlecleucel, CD19 proteins on B cells are targeted in the treatment of B-cell precursor acute lymphoblastic leukemia. Axicabtagene ciloluecel, the second anti-CD19 CAR T-cell therapy, was approved for the treatment of refractory, aggressive B-cell non-Hodgkin lymphoma.
Tisagenlecleucel
Tisagenlecleucel was approved for the treatment of pediatric patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia (ALL) whose disease is refractory to treatment or who have relapsed after second-line therapy or beyond.2 Approval was based on the pivotal ELIANA trial, a single-arm, global phase 2 trial conducted at 25 centers worldwide during April 2015 through April 2017. Patients were eligible for enrollment if they had relapsed or refractory B-cell ALL and were at least 3 years of age at screening and no older than 21 years of age at diagnosis, had at least 5% lymphoblasts in the bone marrow at screening, had tumor expression of CD19, had adequate organ function, and a Karnofsky (adult) or Lansky (child) Performance Status of ≥50 (with the worst allowable score, 50, indicating a patient who requires considerable assistance and frequent medical care [Karnofsky] and lying around much of the day, but gets dressed; no active playing but participates in all quiet play and activities [Lansky]). Exclusion criteria included previous receipt of anti-CD19 therapy, concomitant genetic syndromes associated with bone marrow failure, previous malignancy, and/or active or latent hepatitis B or C virus (HBV/HCV) infection.
The overall remission rate (ORR) was evaluated in 75 patients who were given a single dose of tisagenlecleucel (a median weight-adjusted dose of 3.1 x 106 transduced viable T cells per kg of body weight) within 14 days of completing a lymphodepleting chemotherapy regimen. The confirmed ORR after at least 3 months of follow-up, as assessed by independent central review, was 81%, which included 60% of patients in complete remission (CR) and 21% in complete remission with incomplete hematologic recovery, all of whom were negative for minimal residual disease.
The most common adverse events (AEs) associated with tisagenlecleucel treatment were cytokine release syndrome (CRS), hypogammaglobulinemia, infection, pyrexia, decreased appetite, headache, encephalopathy, hypotension, bleeding episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, and delirium. AEs were of grade 3/4 severity in 84% of patients.3
To combat serious safety issues, including CRS and neurologic toxicities, the FDA approved tisagenlecleucel with a Risk Evaluation and Mitigation Strategy (REMS) that, in part, requires health care providers who administer the drug to be trained in their management. It also requires the facility where treatment is administered to have immediate, onsite access to the drug tocilizumab, which was approved in conjunction with tisagenlecleucel for the treatment of patients who experience CRS.
In addition to information about the REMS, the prescribing information details warnings and precautions relating to several other common toxicities. These include hypersensitivity reactions, serious infections, prolonged cytopenias, and hypogammaglobulinemia.
Patients should be monitored for signs and symptoms of infection and treated appropriately. Viral reactivation can occur after tisagenlecleucel treatment, so patients should be screened for HBV, HCV, and human immunodeficiency virus before collection of cells.
The administration of myeloid growth factors is not recommended during the first 3 weeks after infusion or until CRS has resolved. Immunoglobulin levels should be monitored after treatment and hypogammaglobulinemia managed using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement according to standard guidelines.
Patients treated with tisagenlecleucel should also be monitored for life for secondary malignancies, should not be treated with live vaccines from 2 weeks before the start of lymphodepleting chemotherapy until immune recovery after tisagenlecleucel infusion, and should be aware of the potential for neurological events to impact their ability to drive and use dangerous machinery.4
Tisagenlecleucel is marketed as Kymriah by Novartis Pharmaceuticals. The recommended dose is 1 infusion of 0.2-5 x 106 CAR-positive viable T cells per kilogram of body weight intravenously (for patients ≤50kg) and 0.1-2.5 x 108 cells/kg (for patients >50kg), administered 2-14 days after lymphodepleting chemotherapy.
Axicabtagene ciloleucel
Axicabtagene ciloleucel was approved for the treatment of adult patients with certain types of relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma.5 It is not indicated for the treatment of patients with primary central nervous system lymphoma.
Approval followed positive results from the phase 2 single-arm, multicenter ZUMA-1 trial.6 Patients were included if they were aged 18 years of age and older, had histologically confirmed aggressive B-cell non-Hodgkin lymphoma that was chemotherapy refractory, had received adequate previous therapy, had at least 1 measurable lesion, had completed radiation or systemic therapy at least 2 weeks before, had resolved toxicities related to previous therapy, and had an Eastern Cooperative Oncology Group Performance Status of 0 (asymptomatic) or 1 (symptomatic), an absolute neutrophil count of ≥1000/µL, a platelet count of ≥50,000/µL, and adequate hepatic, renal and cardiac function. They were treated with a single infusion of axicabtagene ciloleucel after lymphodepleting chemotherapy.
Patients who had received previous CD19-targeted therapy, who had concomitant genetic syndromes associated with bone marrow failure, who had previous malignancy, and who had active or latent HBV/HCV infection were among those excluded from the study.
Patients were enrolled in 2 cohorts; those with DLBCL (n = 77) and those with PMBCL or transformed follicular lymphoma (n = 24). The primary endpoint was objective response rate, and after a primary analysis at a minimum of 6 months follow-up, the objective response rate was 82%, with a CR rate of 52%. Among patients who achieved CR, the median duration of response was not reached after a median follow-up of 7.9 months.
A subsequent updated analysis was performed when 108 patients had been followed for a minimum of 1 year. The objective response rate was 82%, and the CR rate was 58%, with some patients having CR in the absence of additional therapies as late as 15 months after treatment. At this updated analysis, 42% of patients continued to have a response, 40% of whom remained in CR.
The most common grade 3 or higher AEs included febrile neutropenia, fever, CRS, encephalopathy, infections, hypotension, and hypoxia. Serious AEs occurred in 52% of patients and included CRS, neurologic toxicity, prolonged cytopenias, and serious infections. Grade 3 or higher CRS or neurologic toxicities occurred in 13% and 28% of patients, respectively. Three patients died during treatment.
To mitigate the risk of CRS and neurologic toxicity, axicabtagene ciloleucel is approved with an REMS that requires appropriate certification and training before hospitals are cleared to administer the therapy.
Other warnings and precautions in the prescribing information relate to serious infections (monitor for signs and symptoms and treat appropriately), prolonged cytopenias (monitor blood counts), hypogammaglobulinemia (monitor immunoglobulin levels and manage appropriately), secondary malignancies (life-long monitoring), and the potential effects of neurologic events on a patient’s ability to drive and operate dangerous machinery (avoid for at least 8 weeks after infusion).7
Axicabtagene ciloleucel is marketed as Yescarta by Kite Pharma Inc. The recommended dose is a single intravenous infusion with a target of 2 x 106 CAR-positive viable T cells per kilogram of body weight, preceded by fludarabine and cyclophosphamide lymphodepleting chemotherapy.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
1. Bosserman LD. Cancer care in 2017: the promise of more cures with the challenges of an unstable health care system. JCSO 2017;15(6):e283-e290.
2. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA News Release. August 30, 2017. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/
ucm574154.htm. Accessed March 31, 2018.
3. Maude S.L, Laetsch T.W, Buechner S, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018;378:439-48.
4. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Novartis Pharmaceuticals Corporation, August, 2017. https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.
com/files/kymriah.pdf. Accessed March 31, 2018.
5. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA News Release. October 18, 2017. https://www.fda.gov/Drugs/
InformationOnDrugs/ApprovedDrugs/ucm581296.htm. Accessed March 31, 2018.
6. Neelapu, S.S, Locke F.L, Bartlett, L.J, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531-44.
7. Kymriah (tisagenlecleucel) suspension for intravenous use. Prescribing information. Kite Pharma Inc. October 2017. https://www.yescarta.com/wp-content/uploads/yescarta-pi.pdf. Accessed March 31, 2018.
An unusual case of primary cardiac prosthetic valve-associated lymphoma
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
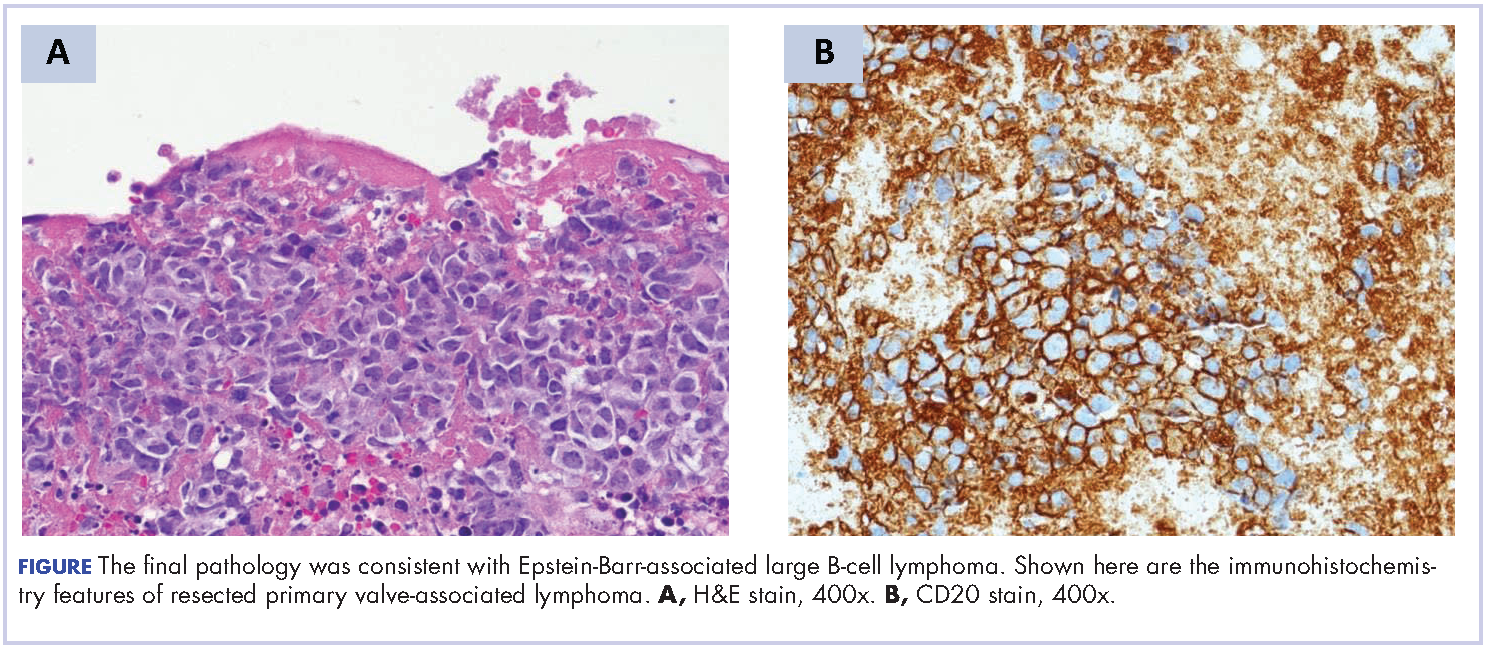
Discussion
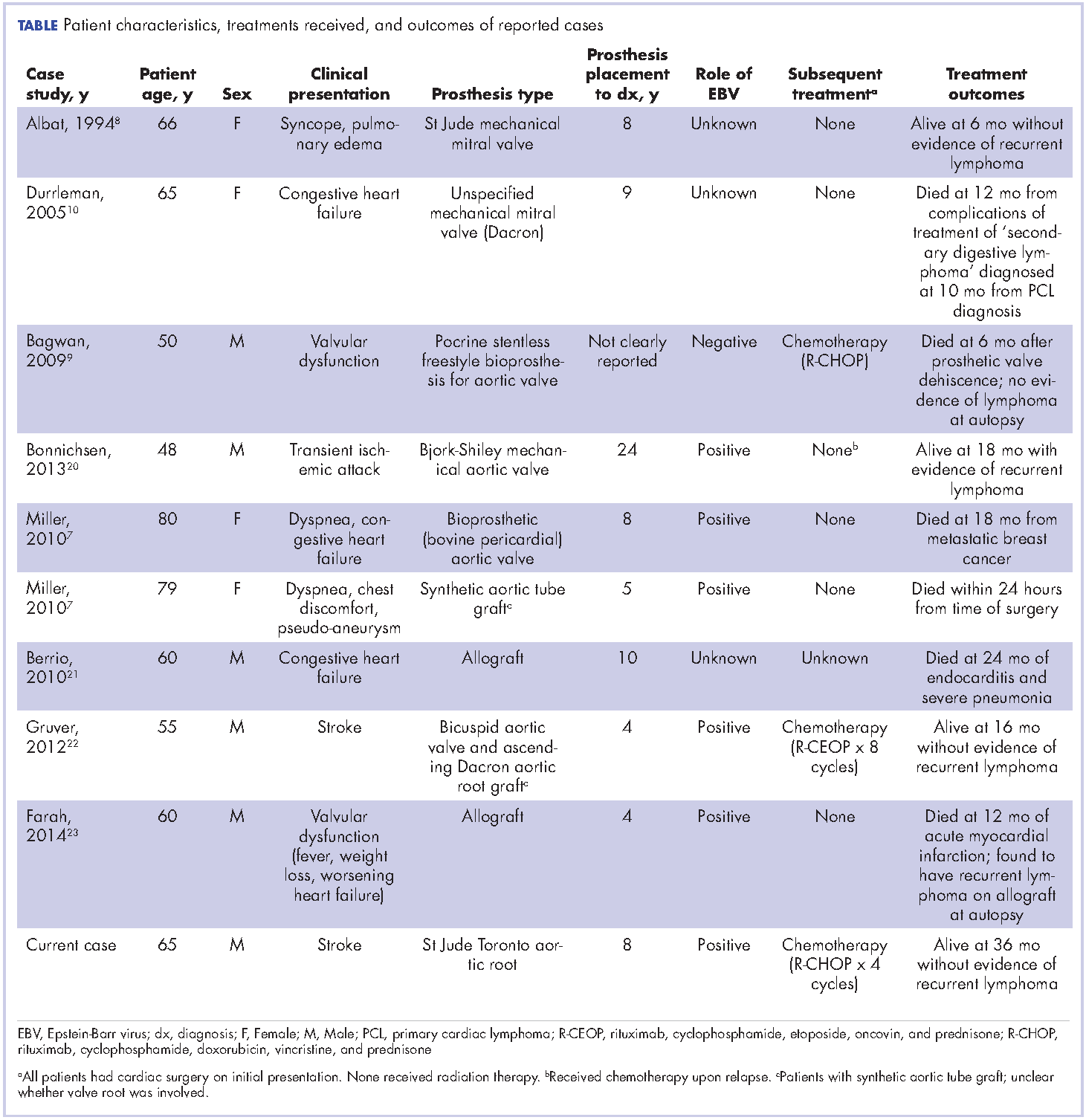
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
Primary cardiac tumors are extremely rare neoplasms with an incidence of less than 0.4%.1-3 Primary cardiac lymphoma (PCL), the majority of which is non-Hodgkin lymphoma, accounts for around 2% of cardiac tumors and less than 0.5% of extranodal lymphomas.1,4-6 Primary lymphoma involving cardiac valves has been described in few case reports and small case series owing to its rarity.7-10 Most cases of PCL present with manifestations of congestive heart failure or cardiac arrhythmias,11 whereas primary valve-associated lymphoma (PV-AL) is usually diagnosed incidentally during valve repair or replacement. The pathophysiology remains unclear, but a few cases have been associated with Epstein Barr virus (EBV).7 Cases previously described in the literature carried an overall poor prognosis and to date there is no standardized treatment approach. We provide here an unusual case of primary prosthetic valve-associated cardiac large B-cell lymphoma, which was successfully treated with adjuvant chemotherapy after valve repair and which resulted in an excellent long-term outcome.
Case presentation and summary
The patient presented in 2012 as a 65-year-old man with a history of ascending aortic aneurysm with secondary aortic insufficiency who in 2004 had undergone composite valve replacement of the aortic valve (AV) root and ascending aorta with a St Jude Toronto root. In June 2011, he was found to have a right parietal intraparenchymal hemorrhage that was thought to be a thromboembolic hemorrhagic ischemic stroke. In March 2012, he had routine follow-up brain magnetic resonance imaging that incidentally showed a left frontal ischemic stroke with hemorrhagic conversion. In June 2012, he was found to have first degree atrioventricular block with episodic runs of supraventricular tachycardia.
In September 2012, transthoracic echocardiography was done for further evaluation of possible recurrent cryptogenic strokes. The results showed a hypo-echogenic mass within the proximal ascending aortic root, but this was not confirmed on transesophageal echocardiography. A chest computed-tomography (CT) scan was therefore performed, and it showed aneurysmal dilatation of the aortic root with an irregular marginal filling defect just above the AV suggestive of intraluminal thrombus. The patient was placed on full anticoagulation with warfarin and referred for cardiothoracic surgery to consider graft and valve replacement. However, 3 weeks later and before the surgery, the patient developed a third thromboembolic ischemic event (transient ischemic attack). The recurrent strokes were attributed to thromboembolic events secondary to prosthetic AV thrombosis.
A repeat transthoracic echocardiography was significant for an abnormal AV bioprosthesis with associated thrombus extending to the ascending aorta. Surgical excision and replacement of the AV conduit explant were performed in November 2012. The final pathology was consistent with EBV-associated large B-cell lymphoma (Figure). The initial staging evaluation, including a CT and positron-emission tomography scan and bone marrow biopsy, was negative for any systemic disease. The patient received 4 cycles of R-CHOP-21 (rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2 , vincristine 2 mg, and prednisone 100 mg) every 3 weeks in an “adjuvant” setting (because patient had no evidence of disease when given the systemic chemotherapy). The patient tolerated chemotherapy well without significant complications, and he is now over 36 months post-treatment without evidence of recurrent disease.
Discussion
Cardiac lymphoma limited only to prosthetic valves is rare, but it has been reported increasingly over the past few years. Until 2010, only six cases of PV-AL had been reported in the literature.7 Including our case, we identified four additional PubMed-indexed cases (using a PubMed search through February 2015). The patient characteristics and treatments received for all identified cases are described in the accompanying Table. The pathology from all of the cases revealed non-Hodgkin lymphoma of large B-cell subtype. PV-AL predominated among men (60%) and older patients with a median age of 62.5 years at diagnosis (range, 48-80 years). Patients had a median duration of 8 years (range, 4-24 years) from date of prosthesis placement to date of lymphoma diagnosis. The three most common presenting manifestations were valvular dysfunction, stroke, and congestive heart failure. All of the patients had surgical intervention on initial presentation. However, management after surgery was not uniform, with only 3 patients reported to have received systemic chemotherapy (Table). None of the patients received adjuvant radiation therapy. Calculated from date of diagnosis, survival duration ranged from less than a month7 to more than 36 months (as reported in our case).
The pathophysiology of PV-AL is not well understood given the rarity of the condition. Similar to other prosthetic-related neoplasms (metallic implants, breast implants),12-14 it has been hypothesized that chronic inflammation and EBV infection may play an essential role in the pathogenesis of this entity. Further, it has been suggested that Dacron, which is used in composite cardiac valve replacements, is carcinogenic and may play a role in some cases.7,15 PV-AL should be highly considered in the differential diagnosis of a suspicious prosthetic valve mass. Various imaging modalities, including echocardiography, CT, and magnetic resonance imaging have been described to have a role in the preoperative evaluation of cardiac tumors by assessing the cardiac function and defining the location and extent of the cardiac tumors.16-19
Given the rarity of this disease entity, there is no standardized approach for treatment. Surgical resection along with repair or replacement of primary involved prosthetic valve is essential for initial treatment. However, there is no consensus about the best approach for subsequent therapy. We cannot be conclusive about the optimum treatment, because of the limited number of published cases, but based on our reading of those cases, it would seem that early surgical intervention and “adjuvant” systemic therapy may have influenced prognosis. We speculate that poor outcomes in the first 6 months were most likely related to primary cardiopulmonary deterioration, whereas later poor outcomes were more likely to be attributable to recurrent lymphoma, particularly for patients who received suboptimal systemic chemotherapy treatment after surgery. All 3 patients who received chemotherapy had no evidence of recurrent disease at last follow-up. Of the 4 patients who received no chemotherapy and survived longer than 6 months (all except 1 died; Table), 2 had recurrent valve lymphoma, 1 had secondary systemic lymphoma, and 1 died of metastatic breast cancer. Those outcomes are in contrast to the 2 out of 3 patients who received adjuvant chemotherapy and who were reported to be alive at 16 and 36 months after diagnosis.
In conclusion, cardiac PV-AL is an increasingly recognized entity that warrants greater awareness among health care providers for early diagnosis and timely surgical intervention. Most of the cases are large B-cell lymphoma. Similar to patients with limited-stage DLBCL, fit patients should be highly considered for “adjuvant” systemic chemotherapy to optimize long-term outcomes. Reporting of similar cases is highly encouraged to better define this rare iatrogenic malignancy.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
1. Hudzik B, Miszalski-Jamka K, Glowacki J, et al. Malignant tumors of the heart. Cancer epidemiol. 2015;39(5):665-672.
2. Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC, eds. Pathology and genetics of tumours of the lung, pleura, thymus and heart. Lyon, France: IARC Press; 2004.
3. Reynen K. Frequency of primary tumors of the heart. Am J Cardiol. 1996;77(1):107.
4. Neragi-Miandoab S, Kim J, Vlahakes GJ. Malignant tumours of the heart: a review of tumour type, diagnosis and therapy. Clin Oncol. 2007;19(10):748-756.
5. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6(4):219-228.
6. Burke A, Virmani R. Tumors of the heart and great vessels. In: Atlas of tumor pathology, 3rd Series, Fascicle 16. Washington, DC: Armed Forces Institute of Pathology, 1996.
7. Miller DV, Firchau DJ, McClure RF, Kurtin PJ, Feldman AL. Epstein-Barr virus-associated diffuse large B-cell lymphoma arising on cardiac prostheses. Am J Surg Pathol. 2010;34(3):377-384.
8. Albat B, Messner-Pellenc P, Thevenet A. Surgical treatment for primary lymphoma of the heart simulating prosthetic mitral valve thrombosis. J Thoracic Cardiovasc Surg. 1994;108(1):188-189.
9. Bagwan IN, Desai S, Wotherspoon A, Sheppard MN. Unusual presentation of primary cardiac lymphoma. Interact Cardiovasc Thorac Surg. 2009;9(1):127-129.
10. Durrleman NM, El-Hamamsy I, Demaria RG, Carrier M, Perrault LP, Albat B. Cardiac lymphoma following mitral valve replacement. Ann Thorac Surg. 2005;79(3):1040-1042.
11. Petrich A, Cho SI, Billett H. Primary cardiac lymphoma: an analysis of presentation, treatment, and outcome patterns. Cancer. 2011;117(3):581-589.
12. Cheuk W, Chan AC, Chan JK, Lau GT, Chan VN, Yiu HH. Metallic implant-associated lymphoma: a distinct subgroup of large B-cell lymphoma related to pyothorax-associated lymphoma? Am J Surg Pathol. 2005;29(6):832-836.
13. Roden AC, Macon WR, Keeney GL, Myers JL, Feldman AL, Dogan A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: an indolent T-cell lymphoproliferative disorder. Mod Pathol. 2008;21(4):455-463.
14. de Jong D, Vasmel WL, de Boer JP, et al. Anaplastic large-cell lymphoma in women with breast implants. JAMA. 2008;300(17):2030-2035.
15. Durrleman N, El Hamamsy I, Demaria R, Carrier M, Perrault LP, Albat B. Is Dacron carcinogenic? Apropos of a case and review of the literature [In French]. Arch Mal Coeur Vaiss. 2004 Mar;97(3):267-270.16. Peters PJ, Reinhardt S. The echocardiographic evaluation of intracardiac masses: a review. J Am Soc Echocard. 2006;19(2):230-240.
17. Gulati G, Sharma S, Kothari SS, Juneja R, Saxena A, Talwar KK. Comparison of echo and MRI in the imaging evaluation of intracardiac masses. Cardiovasc Intervent Radiol. 2004;27(5):459-469.
18. Krombach GA, Spuentrup E, Buecker A, et al. Heart tumors: magnetic resonance imaging and multislice spiral CT [In German]. RoFo. 2005;177(9):1205-1218.
19. Hoey ET, Mankad K, Puppala S, Gopalan D, Sivananthan MU. MRI and CT appearances of cardiac tumours in adults. Clin Radiol. 2009;64(12):1214-1230.
20. Bonnichsen CR, Dearani JA, Maleszewski JJ, Colgan JP, Williamson EE, Ammash NM. Recurrent Epstein-Barr virus-associated diffuse large B-cell lymphoma in an ascending aorta graft. Circulation. 2013;128(13):1481-1483.
21. Berrio G, Suryadevara A, Singh NK, Wesly OH. Diffuse large B-cell lymphoma in an aortic valve allograft. Tex Heart Inst J. 2010;37(4):492-493.
22. Gruver AM, Huba MA, Dogan A, Hsi ED. Fibrin-associated large B-cell lymphoma: part of the spectrum of cardiac lymphomas. Am J Surg Pathol. 2012;36(10):1527-1537.
23. Farah FJ, Chiles CD. Recurrent primary cardiac lymphoma on aortic valve allograft: implications for therapy. Tex Heart Inst J. 2014;41(5):543-546.
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.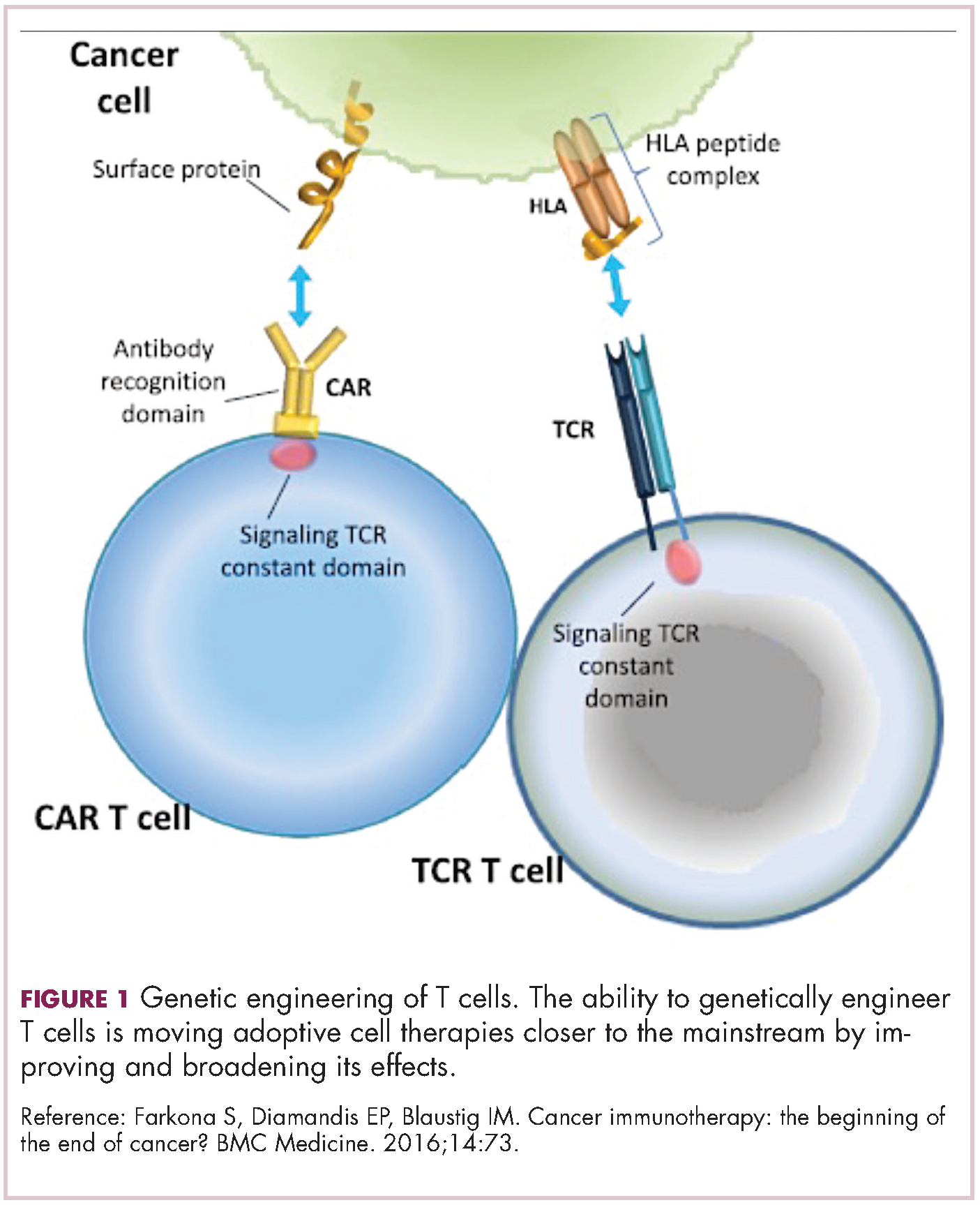
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17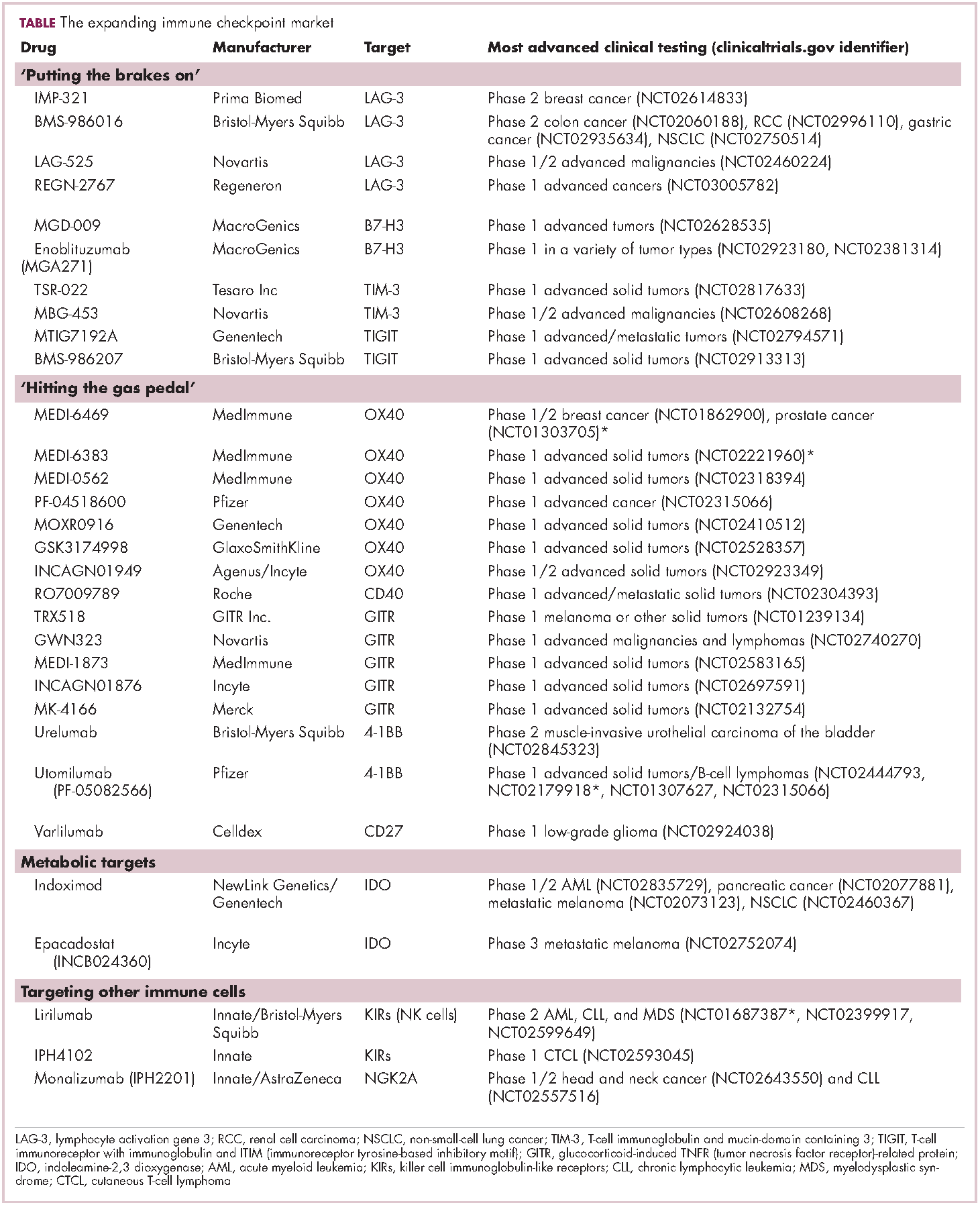
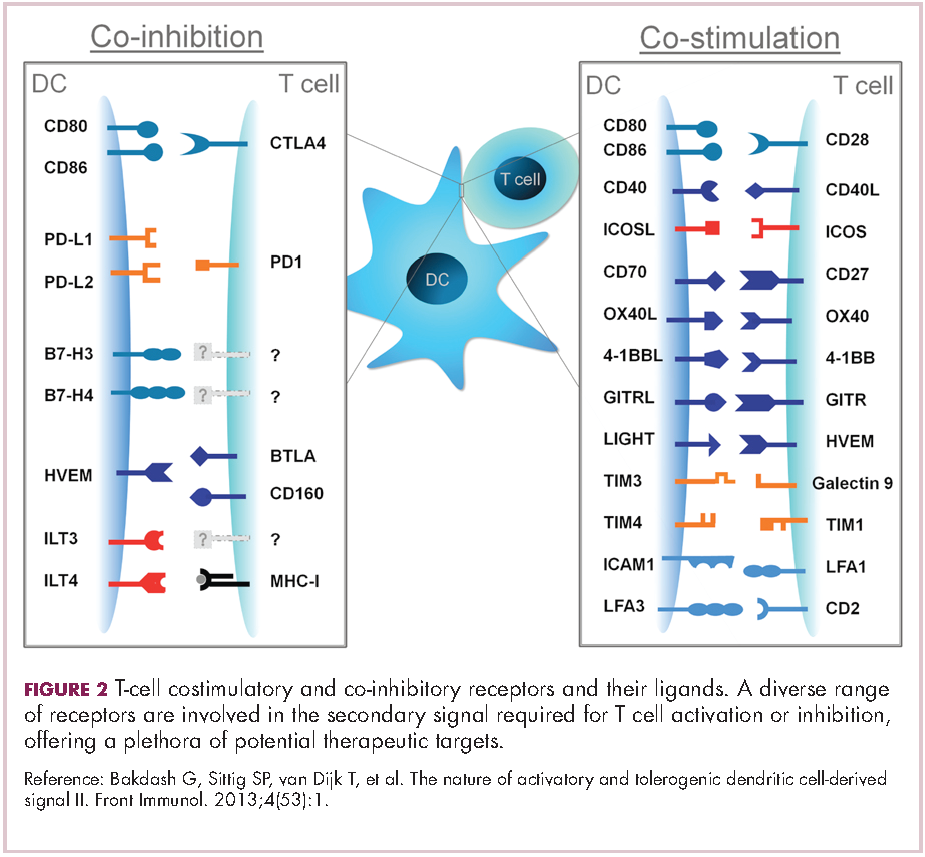
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).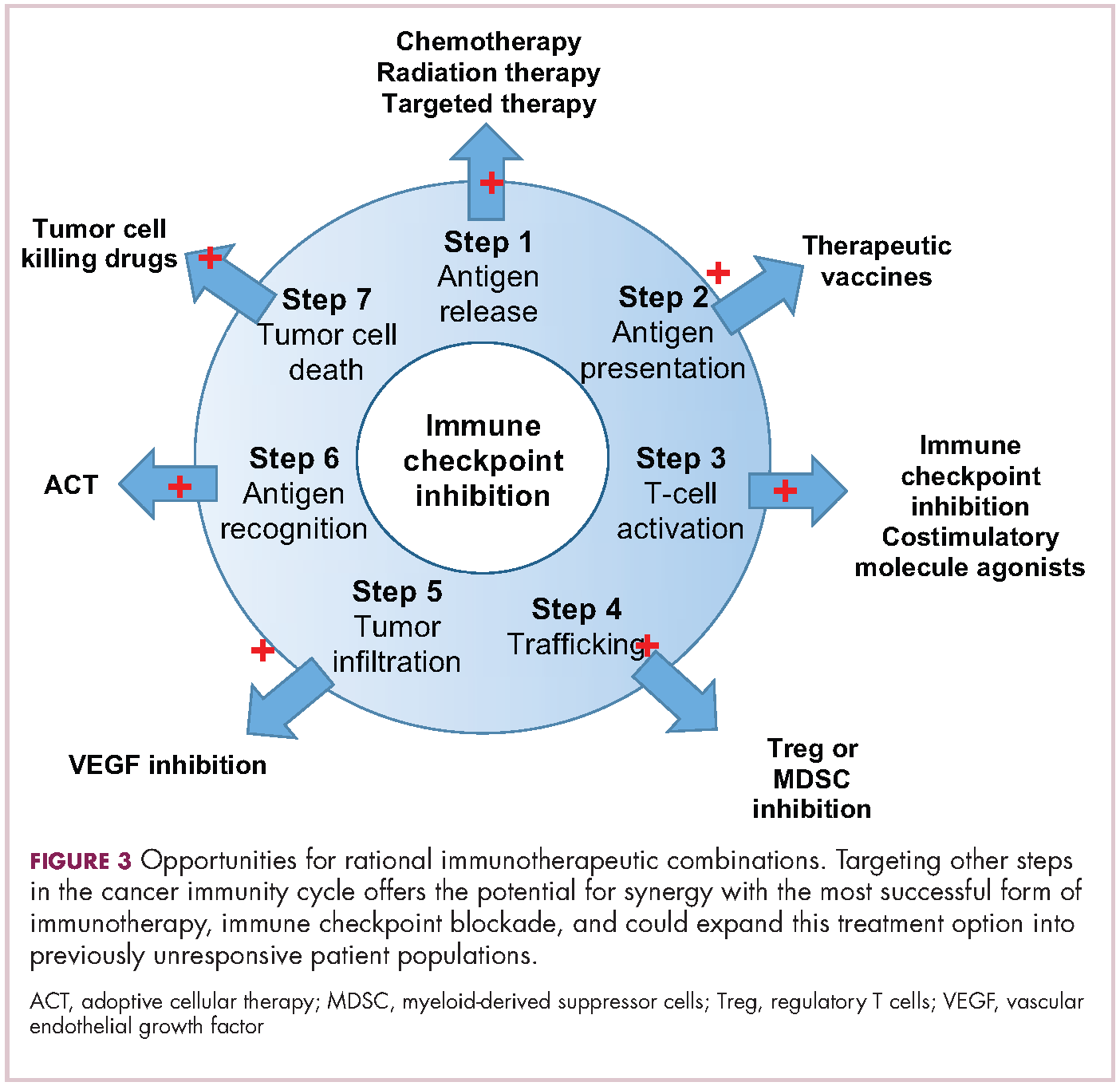
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Durable response to pralatrexate for aggressive PTCL subtypes
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.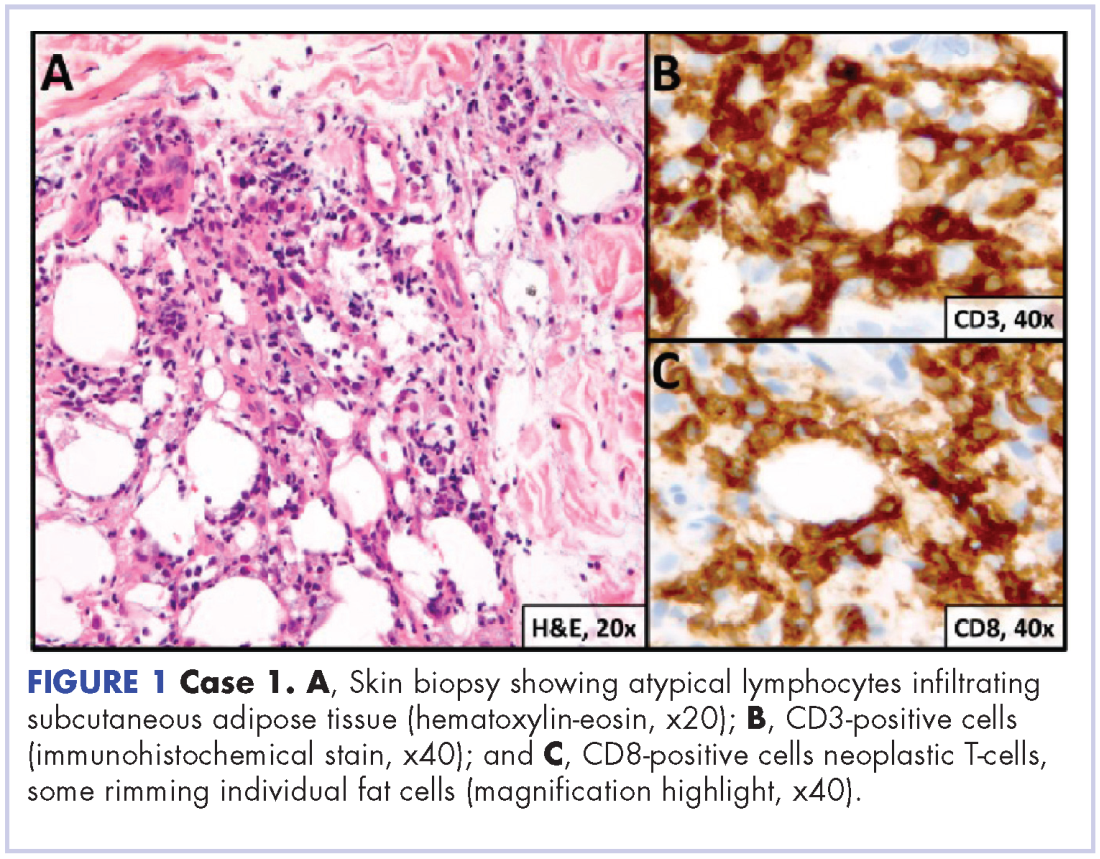
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).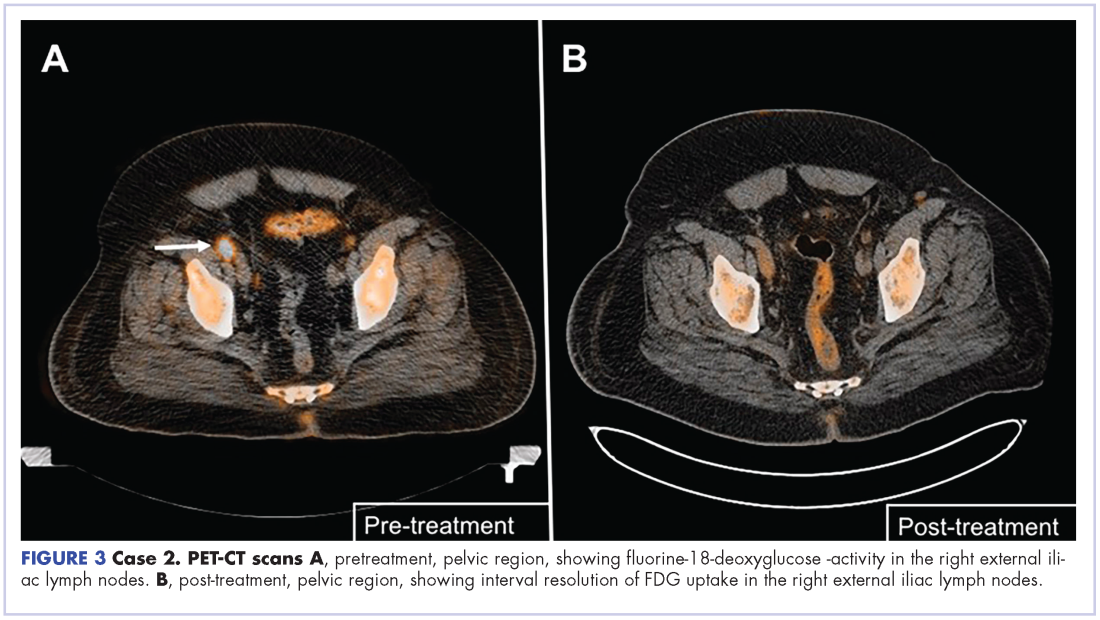
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
Peripheral T-cell lymphoma (PTCL) is a heterogeneous group of mature T- and natural killer-cell neoplasms that comprise about 10%-15% of all non-Hodgkin lymphomas in the United States.1,2 The development of effective therapies for PTCL has been challenging because of the rare nature and heterogeneity of these lymphomas. Most therapies are a derivative of aggressive B-cell lymphoma therapies, including CHOP (cyclophosphamide, hydroxydaunorubicin, vinicristine, prednisone) and CHOEP (cyclophosphamide, hydroxydaunorubicin, vinicristine, etoposide, prednisone).1 Many centers use autologous or allogeneic stem cell transplant in this setting,1 but outcomes remain poor and progress in developing effective treatments has been slow.
Pralatrexate is the first drug to have been approved by the US Food and Drug Administration specifically for treating patients with relapsed or refractory PTCL.3 As a folate analog metabolic inhibitor, pralatrexate competitively inhibits dihydrofolate reductase and reduces cellular levels of thymidine monophosphate, which prevents the cell from synthesizing genetic material and triggers it to undergo apoptosis.4 The agency’s approval of pralatrexate was based on results from the PROPEL study, which is possibly the largest prospective study conducted in patients with relapsed or refractory PTCL (109 evaluable patients).2 Findings from the study showed an overall response rate (ORR) of 29%, and a median duration of response (DoR) of 10 months.2
Pralatrexate is administered intravenously at 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. It is generally continued until disease progression or an unacceptable level of toxicity.2 Alternative dosing schedules have been described, including 15 mg/m2 once weekly for 3 weeks of a 4-week treatment cycle for cutaneous T-cell lymphomas.5
In this case series, we examine the outcomes of 2 patients with particularly aggressive subtypes of PTCL who were treated with pralatrexate. The significance of this report is in describing the long duration of response and reporting on a PTCL subtype – subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type – that was underrepresented in the PROPEL study and is underreported in the literature.
Case presentations and summaries
Case 1
A 23-year-old Asian American man with a medical history of osteogenesis imperfecta presented to Emergency Department at the Hospital of University of Pennsylvania with bilateral lower extremity edema, low-grade fevers, a weight loss of 25 lb, and flat hyperpigmented scaly skin patches across his torso. Symptoms had started manifesting around five months prior to the visit. A punch biopsy of a skin lesion revealed skin tissue with focal infiltrate of small- to medium-sized, atypical lymphocytes infiltrating subcutaneous adipose tissue (panniculitis-like) and adnexa. Immunohistochemical stains showed that the abnormal lymphocytes were positive for CD3, CD8, perforin, granzyme B, TIA-1 (minor subset), and TCR beta; and negative for CD4, CD56, and CD30. Proliferation index (Ki67) was 70%. The findings were consistent with primary subcutaneous panniculitis-like T-cell lymphoma, alpha/beta type (Figure 1). A staging positron-emission tomography–computed tomography (PET–CT) scan demonstrated stage IVB lymphoma with subcutaneous involvement without nodal disease.
He was initially treated with aggressive combination regimens including EPOCH (etoposide, prednisolone, vincristine, cyclophosphamide, hydroxydaunorubicin) and ICE (ifosfamide, carboplatin, etoposide), but he had no response and his disease was primary refractory. Because of his osteogenesis imperfecta, he was not a candidate for allogenic stem cell transplant.
He responded to hyperCVAD B combination therapy (methotrexate and cytarabine), but the course was complicated by cytarabine-induced ataxia and dysarthia. He was then treated with 3 months of intravenous alemtuzumab without response. Intravenous methotrexate (2,000 mg/m2) was then used for 3 cycles, but this exacerbated his previous cytarabine-induced neurological symptoms and resulted in only partial response with persistent fluorine-18-deoxyglucose (FDG) avid lesions on a subsequent PET–CT scan.
At that point, the patient was started on pralatrexate at 15 mg/m2 weekly for 3 weeks on a 4-week cycle schedule. This was his fifth line of therapy and at 16 months from his initial diagnosis. This dosage was continued for 6 months, and he tolerated the therapy well. He reported no exacerbations of his dysarthia, and by the second month, he had achieved clinical and radiographic remission with complete resolution of B symptoms (fevers, night sweats, and weight loss). The dosing was modified to 15 mg/m2 every 2 weeks for 3 months. A whole body PET–CT scan showed resolution of previously FDG avid lesions.
The patient was then continued on 15 mg/m2 pralatrexate every 3 weeks for 1 year and he has been maintained on once-a-month dosing for a second and now third year of therapy. He continues to tolerate the therapy and remains disease free at nearly 2 years since starting pralatrexate.
Case 2
A 64-year-old white man with a medical history of myasthenia gravis (in remission) and invasive thymoma (after thymectomy) presented with diffuse bulky lymphadenopathy and lung lesions to outpatient clinic at the Abramson Cancer Center at the University of Pennsylvania. His LDH was elevated (278 U/L, reference range 98-192 U/L). Excisional biopsy of a left inguinal lymph node revealed sheets of mitotically active large cells with oval to irregular nuclei, clumped chromatin, conspicuous and sometimes multiple nucleoli, and ample eosinophilic cytoplasm. Immunohistochemical staining showed that the neoplastic cells were positive for CD3, CD4, CD30, BCL2 (variable), and MUM1; and negative for ALK 1, CD5, CD8, CD15, CD43, and CD56. Proliferation index (Ki67) was 90% (Figure 2). PET-CT scan showed widespread hypermetabolic lymphoma in the chest, neck, abdomen, and pelvis with pulmonary metastases. Imaging also demonstrated FDG-avid lesions in the gastric and sinus area. The findings were consistent with ALK-negative, anaplastic large cell lymphoma. He was stage IVA; had gastric, lung, and sinus involvement; and disease above and below the diaphragm.
The patient was initially treated with 6 cycles of CHOP and intrathecal methotrexate injections. His post-treatment PET–CT scan showed persistent FDG-avid disease and his LDH level remained elevated. He underwent 1 cycle of ICE and then BCV (busulfan, cyclophosphamide, etoposide) autologous stem cell transplant. Post-transplant PET–CT scan showed improvement from previous 2 scans but still showed several hypermetabolic lymph nodes consistent with persistent disease.
The patient was started on a pralatrexate regimen of 30 mg/m2 once weekly for 6 weeks of a 7-week treatment cycle. After 5 doses, he developed thrombocytopenia and mucositis, which were deemed pralatrexate related. The dosage was reduced to 20 mg/m2 once weekly with variable frequency depending on tolerability. His response assessment with PET–CT scan demonstrated radiographic complete response with resolution of hypermetabolic lesions (Figure 3B).
He then proceeded with pralatrexate for 4 more doses. PET-CT imaging 2 months after the last dose of pralatrexate was consistent with metabolic complete response, and he opted to hold further therapy. His last imaging at 4 years after completion of therapy showed continued remission. At press time, he had been clinically disease free for more than 6 years after his last dose of pralatrexate.
Discussion
PTCL is a rare and heterogeneous lymphoma with poor prognosis. Only 3 agents – pralatrexate, belinostat, and romidespin – have been approved specifically for the treatment of PTCL and all of them have an ORR of less than 30%, based on findings from phase 2 studies.2,6,7 In the PROPEL study, pralatrexate showed an ORR of 29% and a median DoR of 10 months.2 Those results could be considered discouraging, but some PTCL patients may have durable response to pralatrexate monotherapy.
In this case series, each of the patients presented with a particularly aggressive subtype of PTCL, and 1 suffered from a notably rare subtype for which there was scant clinical data to guide treatment. Both patients went through several lines of aggressive treatment that were ineffective and resulted in minimal response. However, both were able to achieve complete resolution of their disease and maintained remission for a significant duration of time after treatment with pralatrexate. In addition, each patient has maintained his remission – one for 6 years after the last dose. These are noteworthy results, and give both patients and clinicians hope that this therapy can be highly effective in some settings.
A better understanding at the molecular level of the oncogenic mechanisms in PTCL patients will be necessary to guide our therapy choices. In these 2 cases, it is likely that the tumor demonstrated superior sensitivity to dihydrofolate reductase inhibition by pralatrexate. In the future, we hope that analysis of the tumor tissue from PTCL patients will allow us to better categorize the tumor sensitivities to particular therapeutic agents. We believe that individualized treatment will lead to better overall outcomes in this challenging group of lymphomas.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
1. d'Amore F, Relander T, Lauritzsen GF, et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012;30(25):3093-3099.
2. O'Connor OA, Pro B, Pinter-Brown L, et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: results from the pivotal PROPEL study. J Clin Oncol. 2011;29(9):1182-1189.
3. Dondi A, Bari A, Pozzi S, Ferri P, Sacchi S. The potential of pralatrexate as a treatment of peripheral T-cell lymphoma. Expert Opin Investig Drugs. 2014;23(5):711-718.
4. Hui J, Przespo E, Elefante A. Pralatrexate: a novel synthetic antifolate for relapsed or refractory peripheral T-cell lymphoma and other potential uses. J Oncol Pharm Pract. 2012;18(2):275-283.
5. Horwitz SM, Kim YH, Foss F, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012;119(18):4115-4122.
6. O'Connor OA, Horwitz S, Masszi T, et al. Belinostat in patients with relapsed or refractory peripheral T-cell lymphoma: Results of the pivotal phase II BELIEF (CLN-19) study. J Clin Oncol. 2015;33(23):2492-2499.
7. Coiffier B, Pro B, Prince HM, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012;30(6):631-636.
New myeloma drugs improve response and extend survival
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.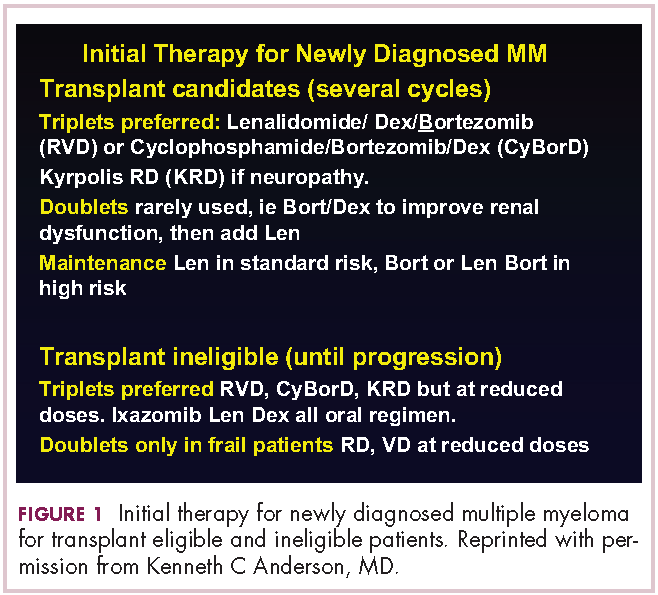
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.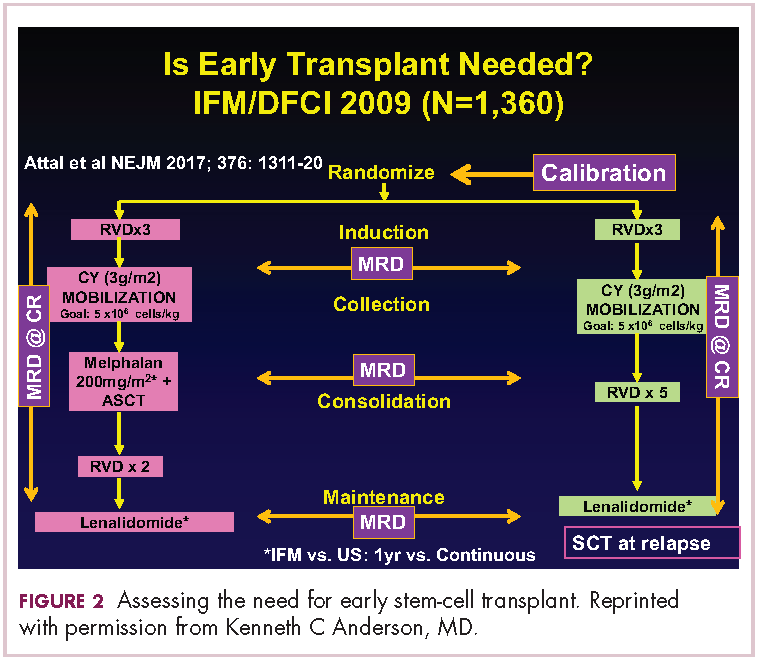
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.

We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.
We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
DR HENRY I thought we might discuss some cases of patients with myeloma, starting with a relatively simple case and ending with one that is a little more complicated. For the first case, we have a 56-year-old healthy man with IgG kappa myeloma whose work-up shows he has multiple lytic bone lesions. He has normal renal function, normal calcium, and he’s transplant-eligible by other health issues. I’ll leave the cytogenetics up to you if that changes your approach. How would you develop or pose some options for this man’s treatment to begin with?
DR ANDERSON It’s important to start out by saying that we, in myeloma, have many new classes of drugs and many new opportunities to choose from to treat this patient.1 As you know, we have proteasome inhibitors, the first-generation bortezomib, then carfilzomib and ixazomib. We have the immunomodulatory drugs (IMiDs), thalidomide, and now lenalidomide and pomalidomide. We have a histone deacetylase (HDAC) inhibitor approved called panobinostat, and we have 2 monoclonal antibodies approved, elotuzumab and daratumumab. These classes of medicine have made it possible for 20 different Food and Drug Administration (FDA) approvals in the last 10-15 years. These agents, having been tested in advanced myeloma, have moved toward initial management.
This person is 50 years old. He has adequate liver, heart, lung, and kidney function, so he would be eligible for high-dose therapy and stem-cell transplantation. In terms of initial management, there are many options (Figure 1). We strongly recommend that triplet therapy be used initially. The most common triplets would be lenalidomide, bortezomib, and dexamethasone (RVD)2,3 or cyclophosphamide, bortezomib, and dexamethasone (CyBorD).4 If this man had neuropathy, perhaps carfilzomib, the second-generation proteasome inhibitor, with lenalidomide and dexamethasone could have been used. Why do we use these? The extent and frequency of response with these triplets is nearly universal overall response rate, with three-quarters very good partial and half-complete responses, including minimal residual disease negative responses. In this patient, we would therefore recommend treatment with either RVD or CyBorD for several cycles to maximal response.
He would then have autologous stem cells collected, and it is still the standard of care to proceed to high-dose melphalan and a single high-dose therapy and stem-cell transplantation. The cytogenetics are important: if this patient has standard-risk multiple myeloma, then lenalidomide maintenance would be given after transplant. It is now FDA-approved for this purpose because it can prolong both progression-free and – most importantly – overall survival.5 Standard-risk cytogenetics might, for example, include hyperdiploidy or translocation 11;14. On the other hand, if his myeloma were high-risk and characterized, for example, by 17p deletion, we would carry out the same induction and transplantation, but we would alter the maintenance to incorporate a proteasome inhibitor. Lenalidomide and bortezomib, for example, could be combined. Early data show that using combined maintenance therapy with lenalidomide and bortezomib, can overcome the early relapses that are characteristic of high-risk disease.6
Because of the extent and frequency of response to combination novel therapies, we have undertaken with our French colleagues a clinical trial of RVD in newly diagnosed patients – such as this patient – followed by stem-cell collection in all patients (Figure 2). Then there is a randomization to either early high-dose therapy, melphalan, and autologous stem-cell transplantation, followed by lenalidomide maintenance; or in the other cohort, harvesting of stem cells, additional RVD, and then maintenance with lenalidomide, saving the stem-cell transplant for later.
The French portion of this trial was reported in the New England Journal of Medicine earlier in 2017.7 It showed that patients who received RVD, high-dose melphalan, stem-cell transplant, and had 1 year of lenalidomide maintenance, had a progression-free survival advantage of about 1 year, without an overall survival advantage; compared with those patients who received RVD and lenalidomide maintenance, saving the transplant for later. I would hasten to add that lenalidomide maintenance was given for only 1 year in this trial, and patients in the RVD-only or RVD-and-transplant arms of this trial relapsed after the lenalidomide maintenance was discontinued.
The American portion of this trial is identical. That is, RVD induction is being given and all patients have a stem-cell collection. Half of the patients then go to high-dose melphalan and stem-cell transplant early, and half of them have the transplant only later at the time of relapse. A major difference, however, is that in both the RVD-only and RVD-and-transplant cohorts, patients receive lenalidomide maintenance until progression. This trial has been ongoing since 2009 and is still ongoing, which tells us that patients in both arms – the RVD-only as well as the RVD-and-transplant arms – are doing well.
In the recent STAMINA trial, all patients underwent a single high-dose therapy and transplant. Then there was a randomization to lenalidomide maintenance only in 1 cohort; a randomization to consolidation with RVD posttransplant followed by lenalidomide maintenance in the second cohort; or a randomization to a second high-dose melphalan and stem-cell transplant followed by lenalidomide maintenance in the third cohort.8 I mention this because the outcomes in all 3 cohorts was similar.
I believe this tells us strongly that high-dose therapy and stem-cell transplantation twice – so-called tandem transplant – is no longer a major option in multiple myeloma. For now, however, in this patient, the standard of care would be to undergo induction therapy with triplet, novel combination treatment. Then, stem cells would be collected and high-dose therapy stem-cell transplant would be done, followed either by lenalidomide maintenance for standard disease or lenalidomide and bortezomib maintenance for high-risk disease. We won’t really know if we can delay transplant until the trials I’ve mentioned totally read out. In my clinical practice, if patients have had a major response to their induction therapy and have stem cells harvested, we can then offer them the opportunity to use maintenance therapy and save the transplant as a potential option for later, when myeloma relapses.
DR HENRY In summary then, this would be, in 2017, off-protocol while the data is pending: it’s reasonable to get a deep induction response, collect stem cells, have a discussion with the patient, and then consider high-dose therapy or not.
DR ANDERSON Yes. I think it’s reasonable to discuss it. We need to be open and honest with patients that the standard of care remains transplant, that you incorporate novel treatments before the transplant and novel treatments as maintenance after the transplant. The happy news is that the outcome, especially for patients who have standard-risk myeloma, is at least a decade or longer progression-free survival. It’s an optimistic picture. The data in terms of transplant being needed or not, will come within the next several years.
For now, it is a standard of care to use 1 high-dose melphalan and stem-cell transplant in this setting. I will add into our discussion with patients – besides the opportunity to harvest stem cells and think about whether one needs to do a transplant early on or not – is the issue of toxicity. High-dose melphalan by itself has a small but real secondary incidence of cancer, myelodysplasia, or leukemia. If one uses lenalidomide maintenance after melphalan transplantation treatment, that risk of secondary cancer is slightly increased.
In my experience, if patients have achieved a complete response with induction therapy only, it’s not unreasonable to offer early transplant and be clear that’s the standard of care. The alternative is maintenance with lenalidomide, knowing once the stem cells have been harvested, that transplantation can be an option to treat relapsed myeloma. We have many other options available as well. Time will tell in terms of the ongoing randomized trials as to whether transplant remains central to our treatment paradigm.
DR HENRY This leads us to our second patient. Here we have an older man of 74 years. He’s a professional piano player, so we want to try to avoid peripheral neuropathy in him. He has some mild renal insufficiency and some coronary artery disease, so he’s deemed transplant-ineligible. He has IgG kappa myeloma, and he’s brand new. What would you consider to be options for him for treatment?
DR ANDERSON This brings up the issue of a transplant-ineligible patient. He has significant comorbidity that would make transplantation an increased risk. What we would recommend in such a patient is still triplet induction therapy incorporating novel agents (Figure 1). Lenalidomide, the immunomodulatory drug, can safely be given in the context of neuropathy because it does not cause significant neuropathy. It would need to be dose modified, depending on the degree of renal insufficiency. We would recommend also including proteasome inhibitors. Bortezomib, the first-generation proteasome inhibitor, would be contraindicated because it does have a small but real attendant neuropathy. If, however, it is given weekly and subcutaneously, the risk of attendant neuropathy is quite low. In this patient, therefore, one could start with lenalidomide and bortezomib weekly and subcutaneously,1,2 with a very early and vigilant follow-up for the earliest signs of neuropathy, so as not to allow it to develop and compromise his career.
Alternatively, one could use a proteasome inhibitor that does not have attendant neuropathy. Carfilzomib, the second-generation proteasome inhibitor, does not have neuropathy.9 But we would need to have caution here, because this patient has a history of coronary artery disease, and carfilzomib has a very small, but real, incidence of cardiac toxicity so would need to be used judiciously in this setting. The third proteasome inhibitor, ixazomib, is the next-generation bortezomib-class proteasome inhibitor, and it’s oral.10 It has less neuropathy than does bortezomib, so in my view is a very realistic option for him together with lenalidomide. It does have a small incidence of neuropathy, so close monitoring for neuropathy would be indicated. We could use lenalidomide–dexamethasone as a doublet and avoid neuropathy,11 but usually doublets are reserved only for frail patients.
My recommendation, therefore, would be RVD with the bortezomib weekly or subcutaneously, or alternatively, lenalidomide, ixazomib, dexamethasone as an all-oral regimen as induction therapy. In my view, this 74-year-old patient with comorbidity is not a transplant candidate. However, one can be very optimistic with this patient. The likelihood that he could have myeloma as a chronic illness and die from something else is quite high. Initial induction triplet therapy would achieve a very high response extent and frequency. The durability would be long, especially with lenalidomide maintenance if it’s standard-risk myeloma or lenalidomide and a proteasome inhibitor, probably ixazomib in this setting, if he were to have high-risk myeloma.
If myeloma relapses, then there are many options that could be used in this patient and achieve years of progression-free and overall survival. Indeed, he is 74 years old and will respond very well to induction triplet therapy, with many years’ duration of response due to continuous lenalidomide or lenalidomide and proteasome inhibitor maintenance. Then there are many effective options to treat relapsed therapy using triplet novel agents. Therefore, his lifespan is unlikely to be shortened by multiple myeloma.
DR HENRY It’s so incredible compared with what it was when I trained. The next patient, a 45-year-old woman with IgG lambda myeloma, has had RVD induction and responded. She had lenalidomide maintenance, but then she progressed, and she got her stem-cell transplant, and she’s progressing after that. I guess we’re looking here to fold in some of the newer agents. How you would you do that in this patient?DR ANDERSON Yes. I think one of the most remarkable and exciting developments with myeloma is the rapid approval of the novel classes of agents that I mentioned earlier – the proteasome inhibitors, the immunomodulatory drugs, the HDAC inhibitor, and the monoclonal antibodies.1 They’re particularly relevant in a patient such as this one, whose myeloma has relapsed after what would be considered standard therapy for a young person with standard-risk myeloma. This patient had RVD and maintenance therapy, and then progressed. The transplant was given for relapsed myeloma. The opportunity to use stem-cell transplant in patients when myeloma becomes active after maintenance should not be forgotten as it can be very effective. In all the trials done to date in which early versus late transplant are compared, there have been similar outcomes. Therefore, if the transplant isn’t done early, don’t forget that it’s an option at the time the myeloma progresses. I do want to mention, that there are lots of options for relapsed myeloma (Figures 3 and 4). I mentioned RVD or CyBorD as initial triplet therapies.2-4 In North America, those are the 2 most common regimens. If myeloma then relapses and is resistant to RVD or to CyBorD, then we need to identify alternatives.
We also need to think about the comorbidities in the patient – issues such as age, neuropathy, presence of renal dysfunction, and other clinical factors. And we need to think about what treatment they’ve had in the past. This patient has had RVD, maintenance with lenalidomide, and a stem-cell transplant. We can offer patients a variety of therapies, but in the context of resistance to the first-generation proteasome inhibitor bortezomib and the first-generation immunomodulatory drug lenalidomide, we would strongly recommend the second-generation immunomodulatory drug pomalidomide12 together with a second-generation proteasome inhibitor, be that carfilzomib13 or ixazomib.14 When one uses the second-generation IMiDs and proteasome inhibitors together, there’s a very high frequency of response in the order of 70%-80%, which lasts years.
Besides carfilzomib and ixazomib proteasome inhibitors, we also have elotuzumab and daratumumab, the monoclonal antibodies.15-17 These agents have been FDA approved to treat patients such as this one who has had 1-3 previous therapies for their myeloma. All of them have been approved in randomized phase 3 trials compared with lenalidomide-dexamethasone in the control arm.13-15,17 They’ve all been found to be superior. Although lenalidomide-dexamethasone combined with daratumumab, ixazomib, elotuzumab, or carfilzomib is superior to lenalidomide in relapsed myeloma, the situation in North America, as in this patient, is usually that patients have had lenalidomide-dexamethasone as part of their initial treatment and their myeloma is refractory to lenalidomide.
Hence, we recommend, that we go to the second-generation pomalidomide and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Having said that, the treatment paradigm is evolving. For example, the monoclonal antibody daratumumab was initially approved by the FDA in multiply relapsed disease as a single agent because it achieves a 30% response rate.16 It now has been moved earlier into the first relapse of multiple myeloma, where it achieves much higher response rates when combined with lenalidomide–dexamethasone or combined with bortezomib–dexamethasone.17,18 Response rates of 70%-80% can be achieved, including minimal residual disease negative complete responses.
Today, in a patient who has had RVD transplant and myeloma has returned, we would recommend second-generation IMiDs, pomalidomide, and second-generation proteasome inhibitors, either carfilzomib or ixazomib. Data for daratumumab combined with lenalidomide-dexamethasone or with bortezomib-dexamethasone, look very promising. We need, however, to see more experience of daratumumab together with lenalidomide-dexamethasone or daratumumab together with bortezomib-dexamethasone in patients whose myeloma is refractory to RVD, that is, patients whose myeloma has returned after RVD induction treatment. Of note, pomalidomide, dexamethasone, and daratumumab have just been approved by the FDA and may also be active even in myeloma recurring after RVD treatment.19
Daratumumab in combination will be moving earlier and earlier and may be appropriate to treat the first relapse. I do want to stress, however, that at present I save daratumumab for the second or greater relapse. Daratumumab is active even when relapse occurs after treatment with second-generation IMiDs and proteasome inhibitors.
DR HENRY Before we close, I have a couple practical questions with these antibodies. Daratumumab has the track record of first-treatment severe reactions and long infusion times. How long are you anticipating the first daratumumab treatment takes? There has been some talk that maybe splitting it in half and going over 2 days is easier on the patient and the infusion center. Have you done that?
DR ANDERSON Yes, I think that’s a very important point. We need to be thinking – first and foremost – about efficacy of our therapy. Equally important, however, are the safety profile and the user-friendliness for the patient. Daratumumab infusions are quite long – on the order of 8 hours or longer on day 1 of infusion. And to date, all the clinical trials have used daratumumab infusions weekly for 8 treatments, followed by 8 treatments given every 2 weeks. Then monthly daratumumab is given as a maintenance therapy. Thus, there is a requirement for multiple outpatient clinic visits that can be prolonged.
One of the opportunities that’s being tested is to give daratumumab subcutaneously. While this is being evaluated in protocols now, the results that have been reported at our national meetings look to be quite promising in terms of efficacy, similar to results with the intravenous administration. Obviously, this would allow for a much more convenient clinic visit and shorter time for the patients being treated.
I should mention that the other antibody, elotuzumab, has been approved in combination with lenalidomide and dexamethasone.15 The infusions with lenalidomide, dexamethasone, and the antibody elotuzumab are much shorter, on the order of 2- or 3-hour visits. The place for elotuzumab in the management of relapsed myeloma is yet to be totally defined. We tend to use it now in the setting of more indolent relapses, where patients might have a slowly rising monoclonal protein. Elotuzumab-lenalidomide-dexamethasone has maintained an overall survival advantage at 4 years compared with lenalidomide-dexamethasone when used in relapsed myeloma.
We are quite excited about both antibodies. Daratumumab tends to get most of the activity, as it achieves responses as a single agent,16 and the depth of the responses are markedly increased when it’s combined with lenalidomide-dexamethasone or bortezomib-dexamethasone.17,18 However, one shouldn’t forget elotuzumab15 based on its tolerability and the survival advantage I mentioned at 4 years.
The final point is that we think about myeloma genetically at the time of diagnosis and relapse in terms of standard or high-risk disease. One of the hallmarks of high-risk disease has been 17P deletion or P53 dysfunction. One of the most exciting outcomes of the development of monoclonal antibodies has been the responses observed even in the context of P53 deletion. Clearly, antibody-mediated cellular cytotoxicity, complement-mediated cytotoxicity, and other mechanisms of action of these antibodies do not require normal P53 function. The important point, therefore, is that what has previously been thought of as high-risk disease can nowadays be effectively treated with these new immune treatments, correlating with the marked improvement in survival and overall outcome.
DR HENRY We have outlined 3 kinds of myeloma patients we see, and especially interesting is the last patient, who has relapsed and then progressed, and in whom newer drugs have a role. Thank you for such a complete and thorough discussion, Dr Anderson.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
1. Kumar SK, Callander NS, Alsina M, et al. Multiple Myeloma, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15:230-269.
2. Richardson PG, Weller E, Lonial S, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly-diagnosed multiple myeloma. Blood. 2010;116:679-686.
3. Durie BG, Hoering A, Abidi MH, et al. Bortezomib with lenalidomide and dexamethasone versus lenalidomide and dexamethasone alone in patients with newly diagnosed myeloma without intent for immediate autologous stem-cell transplant (SWOG S0777: a randomized, open-label, phase 3 trial. Lancet. 2017;389(10068):519-527.
4. Moreau P, Hulin C, Macro M, et al. VTD is superior to VCD prior to intensive therapy in multiple myeloma: results of the prospective IFM2013-04 trial. Blood. 2016;127:2569-2574.
5. McCarthy PL, Owzar K, Hofmeister CC, et al. A phase III study of lenalidomide after transplant for multiple myeloma. N Engl J Med. 2012;366:1770-1781.
6. Nooka AK, Kaufman JL, Muppidi S, et al. Consolidation and maintenance therapy with lenalidomide, bortezomib, and dexamethasone (RVD) in high risk myeloma patients. Leukemia. 2014;28:690-693.
7. Attal M, Lauwers-Cances V, Hulin C, et al. Lenalidomide, bortezomib and dexamethasone with transplantation in myeloma. N Engl J Med. 2017;376:1311-1320.
8. Stadtmauer EA, Pasquini MC, Blackwell B, et al. Comparison of autologous hematopoietic cell transplant (autoHCT), bortezomib, lenalidomide (len) and dexamethasone (RVD) consolidation with len maintenance (ACM), tandem autohct with len maintenance (TAM) and autohct with len maintenance (AM) for up-front treatment of patients with multiple myeloma (MM): primary results from the randomized phase III trial of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN 0702 – StaMINA Trial). Abstract LBA-1. Presented at the 2016 ASH Annual Meeting, December 6, 2016; San Diego, CA.
9. Dytfeld D, Jasielec J, Griffith KA, et al: Carfilzomib, lenalidomide, and low-dose dexamethasone in elderly patients with newly diagnosed multiple myeloma. Haematologica. 2014;99:e162-164.
10. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15:1503-1512
11. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. for the FIRST Trial Team. Lenalidomide and dexamethasone in transplant-ineligible patients with myeloma. N Engl J Med. 2014;371:906-917.
12. Richardson P, Siegel D, Vij R, et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood. 2014;123:1826-1832.
13. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. for the ASPIRE Investigators. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372:142-152
14. Moreau P, Masszi T, Grzasko N, et al. for the TOURMALINE-MM1 Study Group. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;28;374:1621-1634.
15. Lonial S, Dimopoulos M, Palumbo A, et al. for the ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621-631.
16. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD 38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373:1207-1219.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
18. Palumbo A, Chanan-Khan A, Weisel K, et al. for the CASTOR Investigators. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
19. Chari A, Suvannasankha A, Fay JW, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood. 2017;130(8):974-981.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.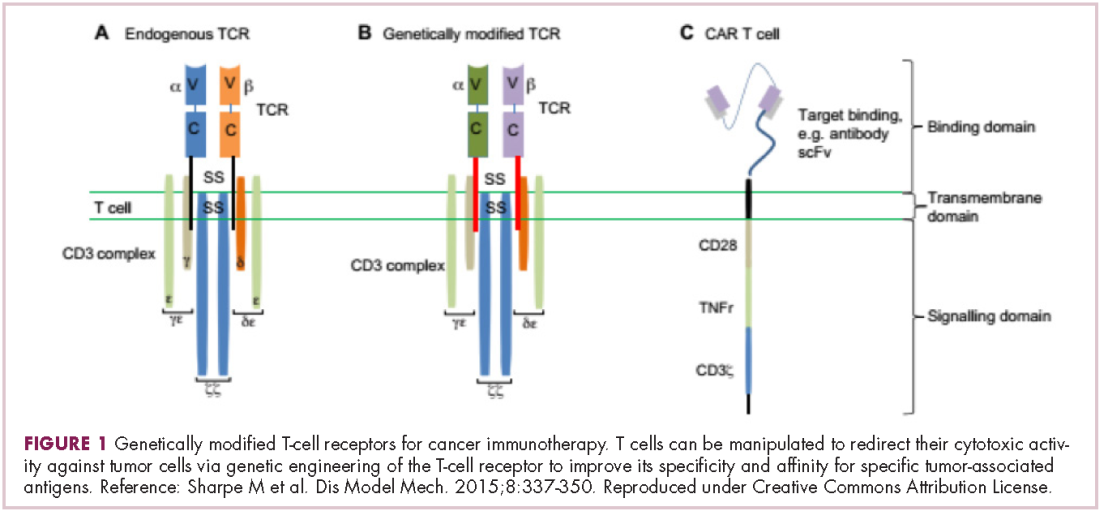
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5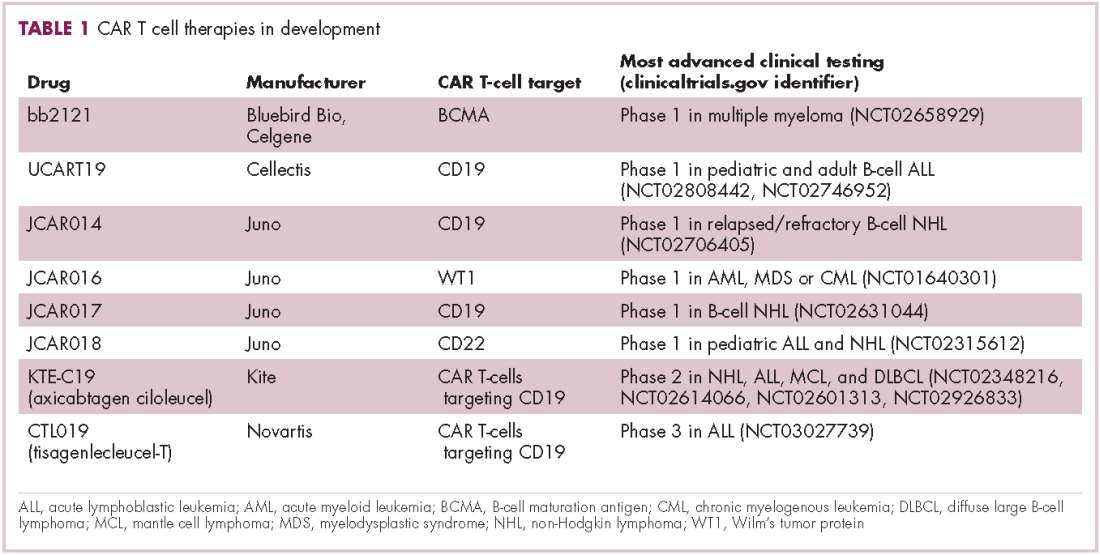
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12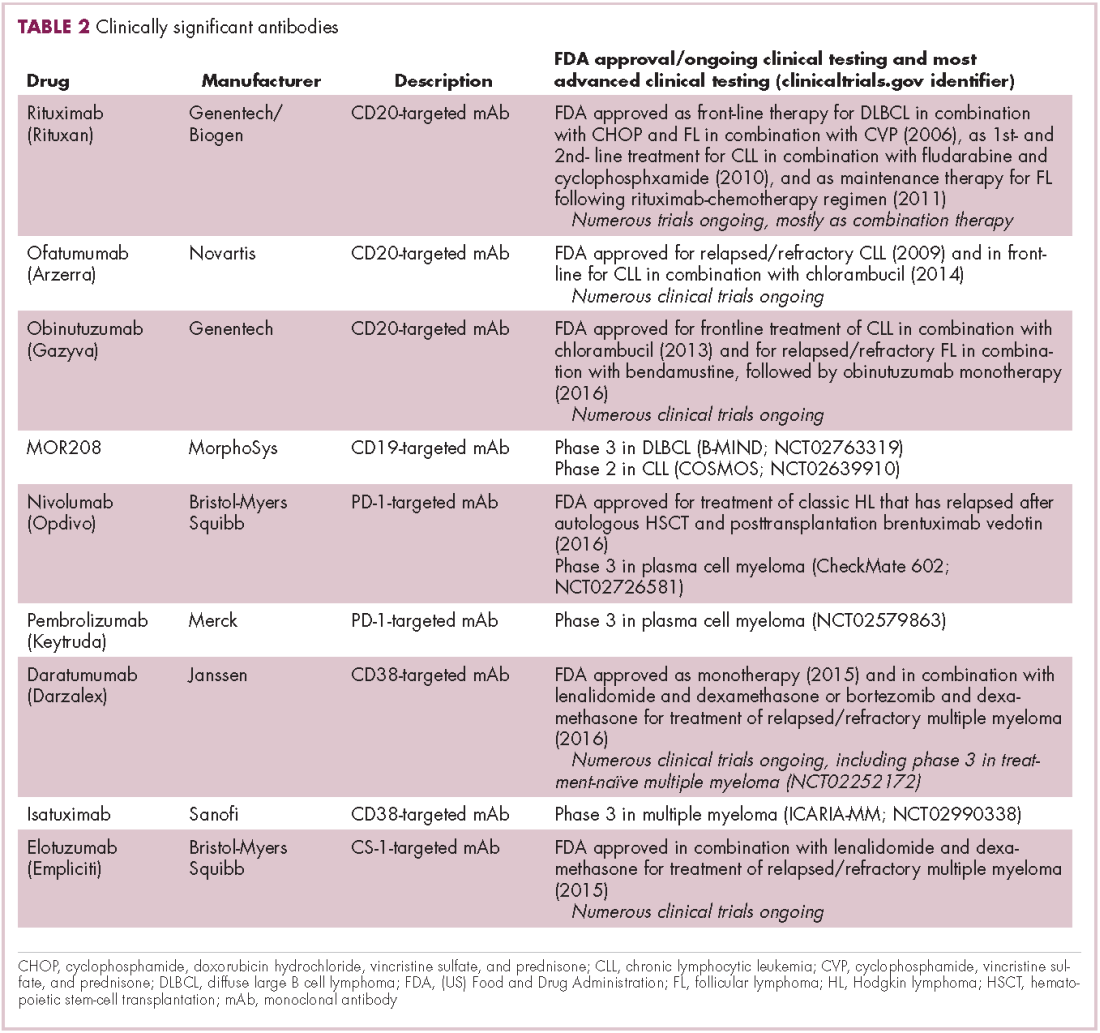
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20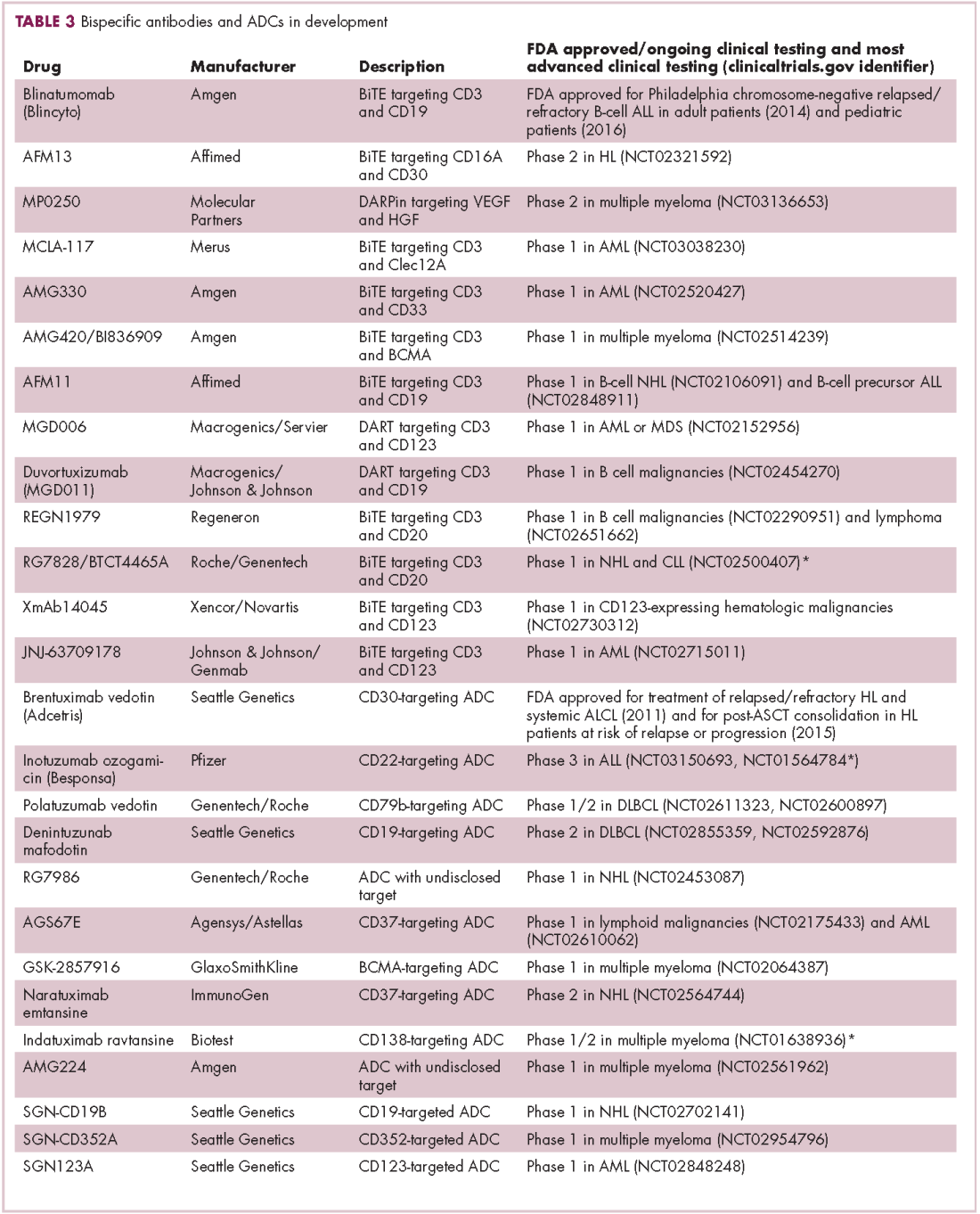
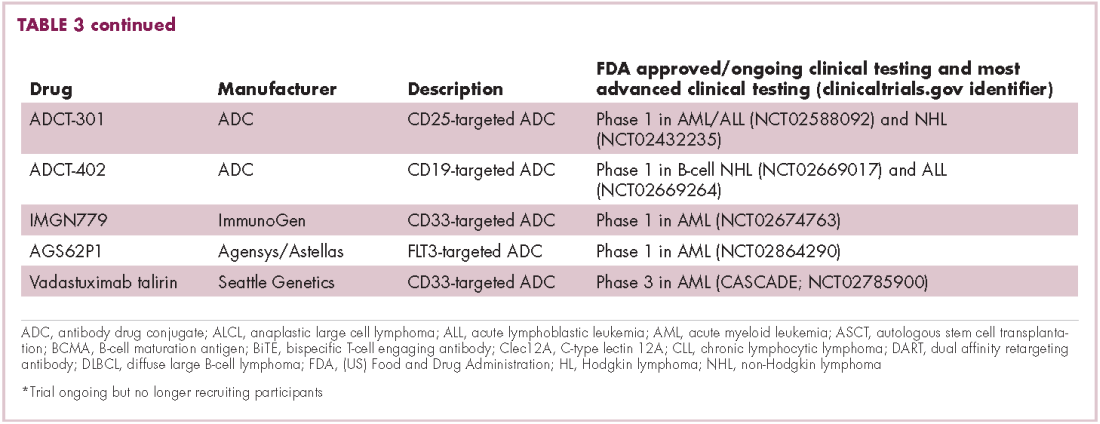
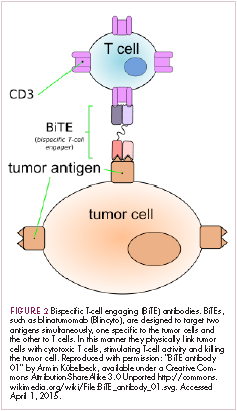
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.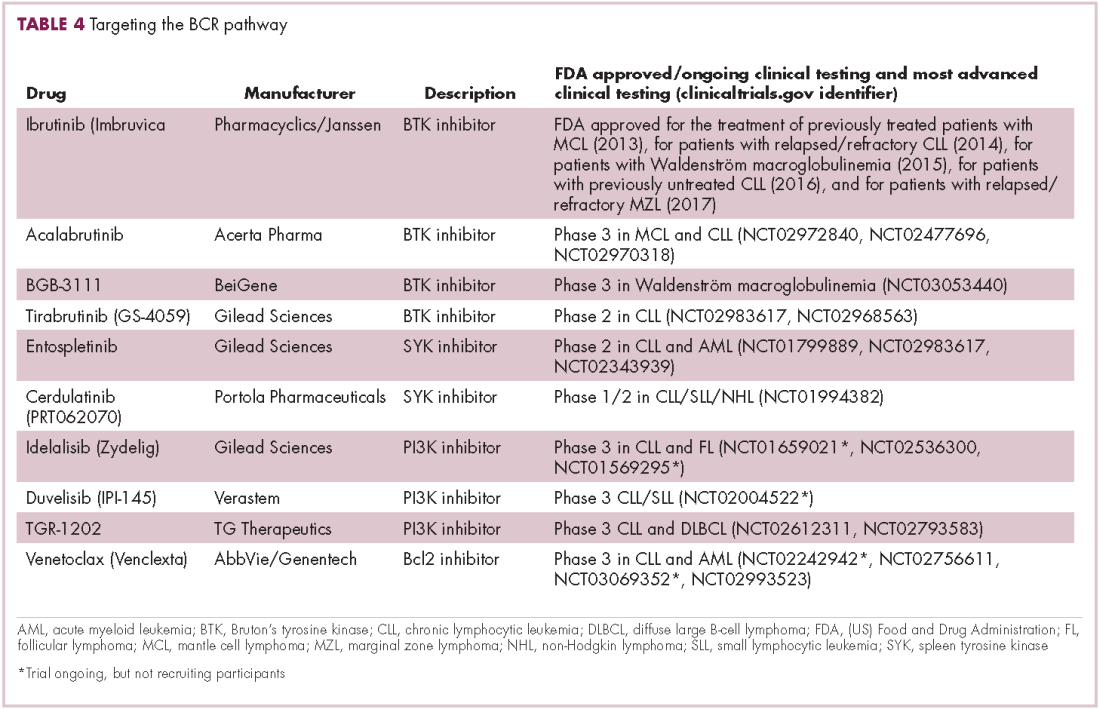
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
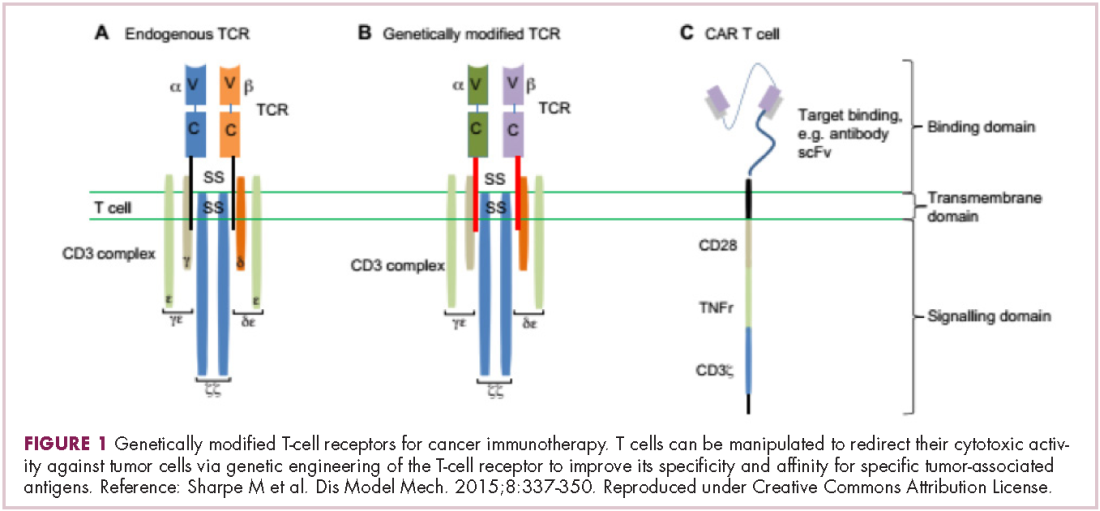
Treating immunotherapy-related AEs in the emergency department
In this interview, Dr David Henry and Dr Maura Sammon discuss some of the most common immunotherapy-related side effects – lung, gastrointestinal, rash, and endocrine-related problems – and Dr Sammon describes in detail how physicians in the ED would triage and treat the patient. However, the overarching takeaway is the importance of communication: first, between the oncologist and patient, so that the patient is aware of these nuances in advance of an emergency, and second, between the ED physician and the treating oncologist soon after the patient has presented and undergone an initial assessment.
Dr Henry is Editor-in-Chief of the JCSO, and Dr Sammon is at the Lewis Katz School of Medicine, Temple University, in Philadelphia, Pennsylvania
Listen here:
In this interview, Dr David Henry and Dr Maura Sammon discuss some of the most common immunotherapy-related side effects – lung, gastrointestinal, rash, and endocrine-related problems – and Dr Sammon describes in detail how physicians in the ED would triage and treat the patient. However, the overarching takeaway is the importance of communication: first, between the oncologist and patient, so that the patient is aware of these nuances in advance of an emergency, and second, between the ED physician and the treating oncologist soon after the patient has presented and undergone an initial assessment.
Dr Henry is Editor-in-Chief of the JCSO, and Dr Sammon is at the Lewis Katz School of Medicine, Temple University, in Philadelphia, Pennsylvania
Listen here:
In this interview, Dr David Henry and Dr Maura Sammon discuss some of the most common immunotherapy-related side effects – lung, gastrointestinal, rash, and endocrine-related problems – and Dr Sammon describes in detail how physicians in the ED would triage and treat the patient. However, the overarching takeaway is the importance of communication: first, between the oncologist and patient, so that the patient is aware of these nuances in advance of an emergency, and second, between the ED physician and the treating oncologist soon after the patient has presented and undergone an initial assessment.
Dr Henry is Editor-in-Chief of the JCSO, and Dr Sammon is at the Lewis Katz School of Medicine, Temple University, in Philadelphia, Pennsylvania
Listen here:
Lenalidomide becomes standard of care for multiple myeloma in the maintenance setting
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
The treatment of multiple myeloma has been revolutionized in the past few decades, with the introduction of numerous novel drug classes that have more than doubled median survival times. The immunomodulatory drug (IMiD), lenalidomide, forms the backbone of the majority of treatment paradigms, first receiving US Food and Drug Administration approval in 2006 for use in combination with dexamethasone in previously treated patients with multiple myeloma. Since then, approved indications for lenalidomide in multiple myeloma have continued to expand.
Most recently, on February 22, 2017, lenalidomide was approved for use as maintenance therapy following autologous stem cell transplant (ASCT), making it the first and only treatment available in this setting. This approval was based on 2 randomized, controlled trials that evaluated the efficacy and safety of lenalidomide in more than 1,000 patients in this setting and demonstrated a significant advantage in progression-free survival (PFS) compared with patients receiving placebo.
CALGB 1001041 and IFM 2005-022 were randomized, double-blind phase 3 trials conducted at 47 locations across the United States and 78 centers in France, Belgium, and Switzerland, respectively. In the CALGB trial, eligible patients were 18-70 years of age, with a European Cooperative Oncology Group (ECOG) performance status of 0 or 1, symptomatic disease requiring treatment (Durie-Salmon stage ≥1), and who received any induction therapy of 2-12 months duration. In the IFM trial, eligible patients were younger than 65 years, with multiple myeloma that had not progressed in the interval between first-line ASCT, performed within the previous 6 months, and randomization, and who had normal liver function tests and blood cell counts.
In CALGB 100104, after undergoing ASCT, 460 patients were randomly assigned to lenalidomide (starting at a dose of 10 mg/day) or placebo between day 100 and day 110 after transplantation. In IFM 2005-02, after undergoing ASCT, 614 patients were randomized 1:1 to receive either consolidation treatment with lenalidomide (at a dose of 25 mg/day on days 1-21 of each 28-day cycle for 2 cycles) followed by maintenance with lenalidomide (10 mg/day for the first 3 months, increasing to 15 mg if tolerated), or the same consolidation treatment followed by maintenance therapy with placebo.
The primary endpoint of CALGB 100104 was time to progression (TTP) and lenalidomide was associated with a significantly longer TTP. Median PFS was also improved by around 15 months (hazard ratio [HR], 0.38; P < .001). In a more recent long-term PFS analysis, median PFS was 5.7 years in the lenalidomide arm compared with 1.9 years with placebo, a difference of 3.8 years (HR, 0.38).3
The primary endpoint for IFM 2005-02 was PFS and lenalidomide maintenance therapy resulted in a significant improvement in PFS in both the originally published study (18-month PFS advantage) and long-term follow-up. The most recent PFS analysis demonstrated a PFS of 3.9 years for lenalidomide, compared with 2 years for no maintenance, a difference of 1.9 years (HR, 0.53). Although the studies were not powered for an overall survival (OS) endpoint, a descriptive analysis showed a median OS of 9.3 years, compared with 7 years in CALGB 100104, and 8.8 years compared with 7.3 years in IFM 2005-02.
In a meta-analysis of data pooled from these 2 studies and a third randomized trial (GIMEMA-RVMM-PI-209),4 which was presented at the 2016 annual meeting of the American Society of Clinical Oncology, maintenance therapy with lenalidomide following frontline treatment with high-dose melphalan and ASCT reduced the risk of death by 26% compared with placebo or no maintenance therapy, prompting suggestions that lenalidomide become standard of care in this setting.
The safety profile of lenalidomide in this setting was similar to that previously described in other studies. The most frequently reported adverse events (AEs), across both studies, were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, asthenia, muscle spasm, and pyrexia. The most common grade 3/4 AEs included neutropenia, thrombocytopenia, and leukopenia. AEs were generally most common in the first 6 months of treatment and subsequently declined in frequency over time or remained stable.
The prescribing information carries warnings and precautions about embryo-fetal toxicity, hematologic toxicity, venous/arterial thromboembolic events, secondary primary malignancies, hepatotoxicity, allergic reactions, tumor lysis syndrome, and thyroid disorders.5 Given its teratogenic effects, lenalidomide is only available through a restricted program under a risk evaluation mitigation strategy.
Patients with neutropenia should be monitored for signs of infections, patients advised to look for signs of bleeding or bruising, and weekly complete blood count performed for the first 2 cycles, on days 1 and 15 of cycle 3 and every 4 weeks thereafter.
Action should be taken to try to reduce the risk of venous and arterial thromboembolic events where possible and thrombophylaxis is recommended, based on the assessment of the underlying risk. Since lenalidomide can increase the risk of secondary primary malignancies, each case should be evaluated for risk-to-benefit ratio.
Liver enzymes should be monitored periodically and treatment interrupted upon their elevation, resuming at a lower dose if levels return to baseline values. Patients who have a history of grade 4 rash following thalidomide treatment should not receive lenalidomide. If grade 2-3 skin rash occurs, treatment interruption or discontinuation should be considered and lenalidomide should be discontinued in the event of angioedema, grade 4 rash, exfoliative or bullous rash, or if Stevens-Johnson s
Patients with high tumor burden prior to treatment are at highest risk of tumor lysis syndrome and should be monitored closely and appropriate precautions taken, and thyroid function should be measured before and during lenalidomide treatment to address potential thyroid disorders. Lenalidomide is marketed as Revlimid by Celgene Corporation.
Expanded approval for daratumumab in multiple myeloma
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.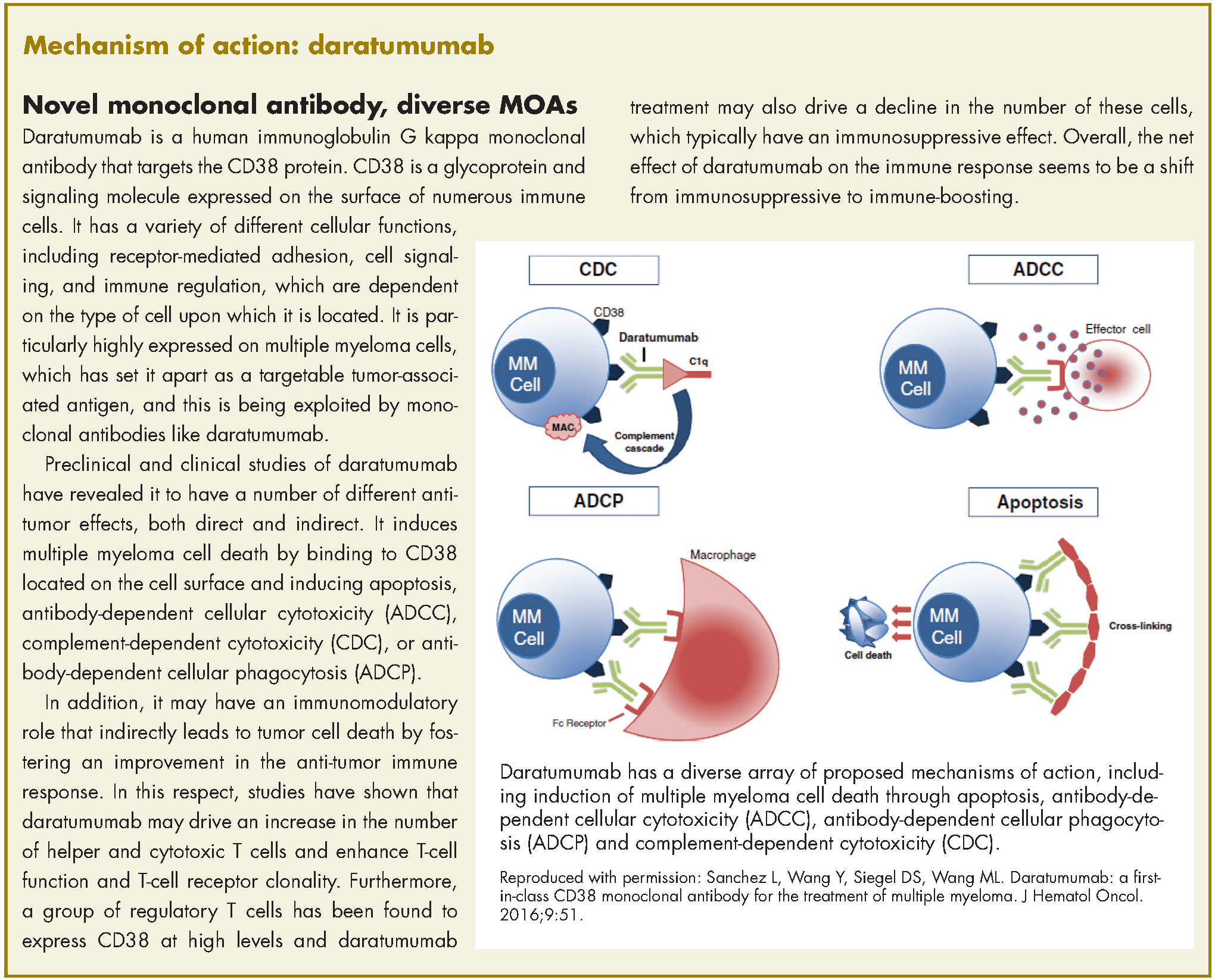
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
In November 2016, the US Food and Drug Administration expanded the approval of daratumumab for patients with multiple myeloma. The monoclonal antibody, which targets CD38, a protein that is highly expressed on the surface of multiple myeloma cells, was previously granted approval by the agency as a single agent for the treatment of patients who had received at least three previous therapies.
The current approval was for the use of daratumumab in two different combination regimens for the treatment of patients who have received one previous line of treatment. On the basis of improved progression-free survival (PFS), demonstrated in two randomized, open-label, phase 3 trials, daratumumab can now be used in combination with the immunomodulatory agent lenalidomide and dexamethasone, or the proteasome inhibitor bortezomib and dexamethasone, both standard therapies for the treatment of multiple myeloma.
In the POLLUX trial, 569 patients with relapsed/refractory multiple myeloma were randomized 1:1 to receive daratumumab in combination with lenalidomide-dexamethasone or lenalidomide-dexamethasone alone. The CASTOR trial randomized 498 patients with relapsed/refractory multiple myeloma 1:1 to daratumumab in combination with bortezomib-dexamethasone, or bortezomib-dexamethasone alone.
The eligibility and exclusion criteria for both trials were similar; patients had received at least one previous line of therapy, had documented progressive disease according to International Myeloma Working Group criteria, and had measurable disease on the basis of urine and/or serum assessments or serum-free, light-chain assay.
Patients with a neutrophil count of ≤1,000 cells/mm3, hemoglobin level of ≤7.5 g/dL, platelet count of <75,000 cells/mm3, creatinine clearance of ≤20 mL/min per 1.73m2 body surface area (or <30 mL/min in the POLLUX trial), alanine aminotransferase or aspartate aminotransferase level ≥2.5 times the upper limit of normal (ULN) range, bilirubin level of ≥1.5 or more times the ULN range, disease refractory to bortezomib or lenalidomide, and unacceptable side effects from bortezomib or lenalidomide, were ineligible for these studies. In addition, patients with grade 2 or higher peripheral neuropathy or neuropathic pain, were excluded from the CASTOR study.
Randomization was stratified according to International Staging System disease stage at the time of screening (stage I, II or III, with higher stage indicating more severe disease), number of previous lines of therapy (1 vs 2, or 3 vs >3), and previous receipt of lenalidomide or bortezomib.
In the CASTOR trial, patients received up to eight 21-day cycles of bortezomib, administered subcutaneously at a dose of 1.3 mg/m2 on days 1, 4, 8, and 11 of cycles 1-8, and dexamethasone, administered orally or intravenously at a dose of 20 mg on days 1, 2, 4, 5, 8, 9, 11, and 12 for a total dose of 160 mg per cycle. Daratumumab was administered at a dose of 16 mg/kg intravenously once weekly on days 1, 8, and 15 during cycles 1 to 3, once every 3 weeks on day 1 of cycles 4-8, and once every 4 weeks thereafter.
In the POLLUX trial, patients were treated in 28-day cycles. Daratumumab was administered at the same dose as in the CASTOR trial, but on days 1, 8, 15 and 22 for 8 weeks during cycles 1 and 2, every 2 weeks on days 1 and 15 for 16 weeks during cycles 3 through 7, and every 4 weeks from then onwards. Lenalidomide was administered at a dose of 25 mg orally on days 1-21 of each cycle, and dexamethasone at a dose of 20 mg before infusion and 20 mg the following day.
The combination of daratumumab with lenalidomide-dexamethasone demonstrated a substantial improvement in PFS, compared with lenalidomide-dexamethasone alone (estimated PFS not yet reached vs 18.4 months, respectively; HR, 0.37; P < .0001), representing a 63% reduction in the risk of disease progression or death. Meanwhile, there was a 61% reduction in the risk of disease progression or death for the combination of daratumumab with bortezomib-dexamethasone in the CASTOR trial (estimated PFS not yet reached vs 7.2 months; HR: 0.39; P < .0001). The PFS benefit was observed across all prespecified subgroups in both studies.
In the CASTOR trial, over a median follow-up of 7.4 months, the overall response rate (ORR) was 82.9% for the combination arm, compared with 63.2% for the bortezomib-dexamethasone arm (P < .001), with a very good partial response (VGPR) or better rate of 59.2% compared with 29.1%, and a complete response (CR) rate of 19.2% compared with 9%. In the POLLUX trial, over a median follow-up of 13.5 months, ORR was 92.9% for the combination arm, compared with 76.4% for lenalidomide-dexamethasone, with a VGPR or better rate of 75.8% versus 44% and a CR rate of 43.1% versus 19.2%.
Overall, the safety profile for both combinations was consistent with what is usually observed with daratumumab monotherapy and lenalidomide-dexamethasone or bortezomib-dexamethasone combinations. The most frequently reported adverse events (AEs) were similar in both studies and included infusion reactions, diarrhea, and upper respiratory tract infection. In the POLLUX trial they also included nausea, fatigue, pyrexia, muscle spasm, cough, and dyspnea, whereas in the CASTOR trial patients also frequently experienced peripheral edema.
The most common grade 3/4 AEs in both trials were neutropenia (51.9% vs 37% in the POLLUX trial and 12.8 vs 4.2% in the CASTOR trial), thrombocytopenia (12.7% vs 13.5% and 45.3% vs 32.9%, respectively), and anemia (12.4% vs 19.6% and 14.4% vs 16%, respectively). The percentage of patients who discontinued treatment due to AEs was similar in both groups across the two studies; in the CASTOR trial discontinuations resulted most commonly from peripheral sensory neuropathy and pneumonia, while in the POLLUX trial, from pneumonia, pulmonary embolism and deterioration in general physical health.
The recommended dose for daratumumab in both combination regimens is 16 mg/kg intravenously, calculated on actual body weight. The dosing schedules begin with weekly administration during weeks 1-8 (when used in combination with lenalidomide-dexamethasone) and weeks 1-9 (for use with the bortezomib-dexamethasone combination), decreasing to every 2 weeks between weeks 9 and 24 or 10 and 24, respectively, and progressing to every 4 weeks from week 25 onward until disease progression and unacceptable toxicity.
Daratumumab is marketed as Darzalex by Janssen Biotech Inc. Neutropenia and thrombocytopenia have been added to the list of warnings and precautions for the prescribing information for these new indications. Complete blood cell count should be monitored periodically during treatment and daratumumab administration delayed to allow recovery of neutrophils or platelets. Supportive care with growth factors or transfusion should be considered in the event of neutropenia or thrombocytopenia, respectively.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.
1. Darzalex (daratumumab) injection, for intravenous use. Prescribing information. Janssen Biotech Inc. https://www.darzalexhcp.com/shared/product/darzalex/darzalex-prescribing-information.pdf. Released November 2016. Accessed January 8, 2017.
2. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:754-766.
3. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375:1319-1331.