User login
American Society of Hematology (ASH): ASH 2014
Long-term side effects of CAR T cells mostly mild
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.

Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The most frequent adverse event was hypogammaglobulinemia in 60% of evaluable patients.
Study details: Prospective observational study of 60 patients with relapsed/refractory CLL or NHL.
Disclosures: Dr. Cordeiro reported having no relevant conflicts of interest.
Source: Cordeiro A et al. ASH 2018, Abstract 223.
Factor XI inhibitor trims DVTs after knee replacement surgery
SAN FRANCISCO – Reducing factor XI levels with the experimental antisense oligonucleotide FXI-ASO lowered venous thromboembolism rates after total knee arthroplasty without increasing bleeding in a phase II study.
Venous thromboembolism (VTE) rates were 30% among controls (21/69) on enoxaparin (Lovenox) 40 mg, compared with 27% for patients (36/134) given FXI-ASO 200 mg and 4% for those (3/71) given FXI-ASO 300 mg. Low-dose FXI-ASO was noninferior to enoxaparin (P = .59), while the high-dose regimen was superior (P < .001).

A 4% VTE rate “has never ever been seen before in patients undergoing knee surgery,” Dr. Harry Büller said during the late-breaking abstract session at the annual meeting of the American Society of Hematology.
The strategy of targeting factor XI is based on the understanding that patients with factor XI deficiency (plasma levels < 20% of normal) have a reduced risk of deep vein thrombosis (DVT). Experimental data in mice and primates also suggest that reducing factor XI attenuates thrombosis without excess bleeding.
Among the 300 patients in the open-label study, major or clinically relevant bleeding occurred in 3% of both FXI-ASO groups and 8% of the enoxaparin group (P = .09).
The findings provide the first evidence in humans that the factor XI intrinsic pathway is one of the drivers of postoperative thrombosis and support the concept that thrombosis and hemostasis can be dissociated, said Dr. Büller of the Academic Medical Center, Amsterdam.
“FXI-ASO is a promising new investigational antithrombotic agent and I believe you are witnessing the birth of a new class of antithrombotic agents,” he concluded.
During a press conference, Dr. Büller confided to reporters that he felt like a boy in a candy store, finally able to reveal the superb study findings.
Dr. Robert Flaumenhaft of Harvard Medical School, Boston, was far less effusive in an editorial that accompanied the simultaneous publication of the study in the New England Journal of Medicine.
“Do these finding prove that reduction in factor XI levels inhibits thrombosis without affecting bleeding? The conservative answer is no,” he wrote.
Dr. Flaumenhaft observed that the incidence of clinically relevant bleeding is relatively low after knee arthroplasty, even when patients receive anticoagulants, and that this safety outcome did not differ significantly between the enoxaparin and 300-mg FXI-ASO groups.
“These results also do not make a compelling case for the clinical use of the factor XI antisense oligonucleotide over anticoagulants that are currently used for prophylaxis in patients undergoing knee arthroplasty,” he wrote.
Central to this argument are issues of convenience and questions regarding reversibility. Treatment began 36 days before surgery and was associated with a high incidence of adverse events at the injection site and factor XI levels remained about 60% lower 70 days after initiation of therapy.
The half-life of FXI-ASO is about 22 days, “which in the classical setting in terms of bleeding could be seen as something of a disadvantage,” Dr. Büller told reporters. “But if we do the next study and it shows to be safe, it turns into an advantage” … because there is the possibility of giving FXI-ASO once every 3 weeks.
Dr. Flaumenhaft closed the editorial by acknowledging that the study challenges “the current paradigm” regarding the primary mechanism responsible for fibrin formation during thrombosis. “The striking observation that reducing factor XI levels prevents thrombosis after knee arthroplasty provides the best clinical evidence to date that the intrinsic pathway is essential for thrombus formation,” he wrote.
The study was conducted at 19 centers in five countries and randomly assigned 300 patients scheduled for elective primary unilateral total-knee arthroplasty to daily enoxaparin 40 mg or three doses of FXI-ASO. The protocol was amended early on to exclude a 100-mg FXI-ASO dose.
FXI-ASO 200 mg or 300 mg was given subcutaneously beginning 36 days before surgery on days 1, 3, 5, 8, 15, 22, and 29, and 6 hours postoperatively, with a final dose on day 39.
Enoxaparin 40 mg was given subcutaneously once daily, beginning the evening before or 6-8 hours after surgery, according to investigator preference, and was continued for at least 8 days postoperatively.
The primary efficacy point was a composite of asymptomatic DVT, detected by venography, and confirmed symptomatic VTE.
At baseline, the average factor XI level was 1.23 units/mL in the enoxaparin group, 1.20 U/mL in the 200-mg group, and 1.16 U/mL in the 300-mg group.
In patients with an average factor XI level of 0.2 U/mL or less, the incidence of the primary efficacy outcome was 5%.
Isis Pharmaceuticals funded the study. Dr. Büller disclosed ties with Isis, Daiichi-Sankyo, Bayer Healthcare, Pfizer, and Bristol-Myers Squibb. Dr. Flaumenhaft reported having no disclosures.
SAN FRANCISCO – Reducing factor XI levels with the experimental antisense oligonucleotide FXI-ASO lowered venous thromboembolism rates after total knee arthroplasty without increasing bleeding in a phase II study.
Venous thromboembolism (VTE) rates were 30% among controls (21/69) on enoxaparin (Lovenox) 40 mg, compared with 27% for patients (36/134) given FXI-ASO 200 mg and 4% for those (3/71) given FXI-ASO 300 mg. Low-dose FXI-ASO was noninferior to enoxaparin (P = .59), while the high-dose regimen was superior (P < .001).
A 4% VTE rate “has never ever been seen before in patients undergoing knee surgery,” Dr. Harry Büller said during the late-breaking abstract session at the annual meeting of the American Society of Hematology.
The strategy of targeting factor XI is based on the understanding that patients with factor XI deficiency (plasma levels < 20% of normal) have a reduced risk of deep vein thrombosis (DVT). Experimental data in mice and primates also suggest that reducing factor XI attenuates thrombosis without excess bleeding.
Among the 300 patients in the open-label study, major or clinically relevant bleeding occurred in 3% of both FXI-ASO groups and 8% of the enoxaparin group (P = .09).
The findings provide the first evidence in humans that the factor XI intrinsic pathway is one of the drivers of postoperative thrombosis and support the concept that thrombosis and hemostasis can be dissociated, said Dr. Büller of the Academic Medical Center, Amsterdam.
“FXI-ASO is a promising new investigational antithrombotic agent and I believe you are witnessing the birth of a new class of antithrombotic agents,” he concluded.
During a press conference, Dr. Büller confided to reporters that he felt like a boy in a candy store, finally able to reveal the superb study findings.
Dr. Robert Flaumenhaft of Harvard Medical School, Boston, was far less effusive in an editorial that accompanied the simultaneous publication of the study in the New England Journal of Medicine.
“Do these finding prove that reduction in factor XI levels inhibits thrombosis without affecting bleeding? The conservative answer is no,” he wrote.
Dr. Flaumenhaft observed that the incidence of clinically relevant bleeding is relatively low after knee arthroplasty, even when patients receive anticoagulants, and that this safety outcome did not differ significantly between the enoxaparin and 300-mg FXI-ASO groups.
“These results also do not make a compelling case for the clinical use of the factor XI antisense oligonucleotide over anticoagulants that are currently used for prophylaxis in patients undergoing knee arthroplasty,” he wrote.
Central to this argument are issues of convenience and questions regarding reversibility. Treatment began 36 days before surgery and was associated with a high incidence of adverse events at the injection site and factor XI levels remained about 60% lower 70 days after initiation of therapy.
The half-life of FXI-ASO is about 22 days, “which in the classical setting in terms of bleeding could be seen as something of a disadvantage,” Dr. Büller told reporters. “But if we do the next study and it shows to be safe, it turns into an advantage” … because there is the possibility of giving FXI-ASO once every 3 weeks.
Dr. Flaumenhaft closed the editorial by acknowledging that the study challenges “the current paradigm” regarding the primary mechanism responsible for fibrin formation during thrombosis. “The striking observation that reducing factor XI levels prevents thrombosis after knee arthroplasty provides the best clinical evidence to date that the intrinsic pathway is essential for thrombus formation,” he wrote.
The study was conducted at 19 centers in five countries and randomly assigned 300 patients scheduled for elective primary unilateral total-knee arthroplasty to daily enoxaparin 40 mg or three doses of FXI-ASO. The protocol was amended early on to exclude a 100-mg FXI-ASO dose.
FXI-ASO 200 mg or 300 mg was given subcutaneously beginning 36 days before surgery on days 1, 3, 5, 8, 15, 22, and 29, and 6 hours postoperatively, with a final dose on day 39.
Enoxaparin 40 mg was given subcutaneously once daily, beginning the evening before or 6-8 hours after surgery, according to investigator preference, and was continued for at least 8 days postoperatively.
The primary efficacy point was a composite of asymptomatic DVT, detected by venography, and confirmed symptomatic VTE.
At baseline, the average factor XI level was 1.23 units/mL in the enoxaparin group, 1.20 U/mL in the 200-mg group, and 1.16 U/mL in the 300-mg group.
In patients with an average factor XI level of 0.2 U/mL or less, the incidence of the primary efficacy outcome was 5%.
Isis Pharmaceuticals funded the study. Dr. Büller disclosed ties with Isis, Daiichi-Sankyo, Bayer Healthcare, Pfizer, and Bristol-Myers Squibb. Dr. Flaumenhaft reported having no disclosures.
SAN FRANCISCO – Reducing factor XI levels with the experimental antisense oligonucleotide FXI-ASO lowered venous thromboembolism rates after total knee arthroplasty without increasing bleeding in a phase II study.
Venous thromboembolism (VTE) rates were 30% among controls (21/69) on enoxaparin (Lovenox) 40 mg, compared with 27% for patients (36/134) given FXI-ASO 200 mg and 4% for those (3/71) given FXI-ASO 300 mg. Low-dose FXI-ASO was noninferior to enoxaparin (P = .59), while the high-dose regimen was superior (P < .001).
A 4% VTE rate “has never ever been seen before in patients undergoing knee surgery,” Dr. Harry Büller said during the late-breaking abstract session at the annual meeting of the American Society of Hematology.
The strategy of targeting factor XI is based on the understanding that patients with factor XI deficiency (plasma levels < 20% of normal) have a reduced risk of deep vein thrombosis (DVT). Experimental data in mice and primates also suggest that reducing factor XI attenuates thrombosis without excess bleeding.
Among the 300 patients in the open-label study, major or clinically relevant bleeding occurred in 3% of both FXI-ASO groups and 8% of the enoxaparin group (P = .09).
The findings provide the first evidence in humans that the factor XI intrinsic pathway is one of the drivers of postoperative thrombosis and support the concept that thrombosis and hemostasis can be dissociated, said Dr. Büller of the Academic Medical Center, Amsterdam.
“FXI-ASO is a promising new investigational antithrombotic agent and I believe you are witnessing the birth of a new class of antithrombotic agents,” he concluded.
During a press conference, Dr. Büller confided to reporters that he felt like a boy in a candy store, finally able to reveal the superb study findings.
Dr. Robert Flaumenhaft of Harvard Medical School, Boston, was far less effusive in an editorial that accompanied the simultaneous publication of the study in the New England Journal of Medicine.
“Do these finding prove that reduction in factor XI levels inhibits thrombosis without affecting bleeding? The conservative answer is no,” he wrote.
Dr. Flaumenhaft observed that the incidence of clinically relevant bleeding is relatively low after knee arthroplasty, even when patients receive anticoagulants, and that this safety outcome did not differ significantly between the enoxaparin and 300-mg FXI-ASO groups.
“These results also do not make a compelling case for the clinical use of the factor XI antisense oligonucleotide over anticoagulants that are currently used for prophylaxis in patients undergoing knee arthroplasty,” he wrote.
Central to this argument are issues of convenience and questions regarding reversibility. Treatment began 36 days before surgery and was associated with a high incidence of adverse events at the injection site and factor XI levels remained about 60% lower 70 days after initiation of therapy.
The half-life of FXI-ASO is about 22 days, “which in the classical setting in terms of bleeding could be seen as something of a disadvantage,” Dr. Büller told reporters. “But if we do the next study and it shows to be safe, it turns into an advantage” … because there is the possibility of giving FXI-ASO once every 3 weeks.
Dr. Flaumenhaft closed the editorial by acknowledging that the study challenges “the current paradigm” regarding the primary mechanism responsible for fibrin formation during thrombosis. “The striking observation that reducing factor XI levels prevents thrombosis after knee arthroplasty provides the best clinical evidence to date that the intrinsic pathway is essential for thrombus formation,” he wrote.
The study was conducted at 19 centers in five countries and randomly assigned 300 patients scheduled for elective primary unilateral total-knee arthroplasty to daily enoxaparin 40 mg or three doses of FXI-ASO. The protocol was amended early on to exclude a 100-mg FXI-ASO dose.
FXI-ASO 200 mg or 300 mg was given subcutaneously beginning 36 days before surgery on days 1, 3, 5, 8, 15, 22, and 29, and 6 hours postoperatively, with a final dose on day 39.
Enoxaparin 40 mg was given subcutaneously once daily, beginning the evening before or 6-8 hours after surgery, according to investigator preference, and was continued for at least 8 days postoperatively.
The primary efficacy point was a composite of asymptomatic DVT, detected by venography, and confirmed symptomatic VTE.
At baseline, the average factor XI level was 1.23 units/mL in the enoxaparin group, 1.20 U/mL in the 200-mg group, and 1.16 U/mL in the 300-mg group.
In patients with an average factor XI level of 0.2 U/mL or less, the incidence of the primary efficacy outcome was 5%.
Isis Pharmaceuticals funded the study. Dr. Büller disclosed ties with Isis, Daiichi-Sankyo, Bayer Healthcare, Pfizer, and Bristol-Myers Squibb. Dr. Flaumenhaft reported having no disclosures.
AT ASH 2014
Key clinical point: Reducing factor XI levels with FXI-ASO was effective in preventing VTE in patients undergoing knee arthroplasty and appeared safe with respect to bleeding risk.
Major finding: The primary VTE endpoint occurred in 4% of patients on FXI-ASO 300 mg, 27% on FXI-ASO 200 mg, and 30% on enoxaparin.
Data source: Open-label, parallel-group phase II study of 300 patients undergoing primary unilateral total-knee arthroplasty.
Disclosures: Isis Pharmaceuticals funded the study. Dr. Büller disclosed ties with Isis, Daiichi-Sankyo, Bayer Healthcare, Pfizer, and Bristol-Myers Squibb. Dr. Flaumenhaft reported having no disclosures.

New test could improve warfarin monitoring

SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
Drug reverses dabigatran’s effects in elderly/impaired

Credit: CDC
SAN FRANCISCO—An investigational, humanized antibody fragment known as idarucizumab can reverse the anticoagulation effects of dabigatran, new research suggests.
In a small study, idarucizumab led to sustained reversal of dabigatran’s effects in healthy subjects, elderly volunteers, and participants with mild to moderate renal impairment.
Furthermore, dabigatran anticoagulation could be re-established 24 hours after the subjects had received idarucizumab.
Joachim Stangier, PhD, of Boehringer Ingelheim Pharma GmbH & Co KG in Biberach, Germany, presented these results at the 2014 ASH Annual Meeting as abstract 344*. Boehringer Ingelheim is the company developing idarucizumab.
Dr Stangier said idarucizumab binds dabigatran with high affinity, off-target binding is not expected, and no procoagulant effects have been observed with the drug.
In a previous study of healthy volunteers, idarucizumab provided immediate, complete, and sustained reversal of dabigatran’s anticoagulant effect. The effect was sustained when idarucizumab was given at 2 g and 4 g doses.
For the current study, Dr Stangier and his colleagues wanted to evaluate whether and to what extent doses of up to 5 g of idarucizumab would reverse the anticoagulant effects of dabigatran in healthy subjects, elderly participants, and renally impaired volunteers. The team also wanted to determine the effects of dabigatran given after subjects had received idarucizumab.
The study included 46 male and female subjects who were 45 to 80 years of age. Healthy subjects received dabigatran etexilate at 220 mg twice daily, and subjects with mild to moderate renal impairment received 150 mg twice daily, both over 4 days.
Subjects then received idarucizumab at 1 g, 2.5 g, 5 g, or 5 g given as 2 x 2.5 g 1 hour apart. They received these doses as 5-minute intravenous infusions 2 hours after their last dose of dabigatran.
The researchers found that dabigatran-prolonged clotting times—thrombin time, diluted thrombin time, ecarin clotting time, and activated partial thromboplastin time—were reversed to baseline immediately after the end of idarucizumab infusion.
And the team observed sustained reversal of dabigatran’s anticoagulant effects in all subjects.
The researchers then readministered dabigatran to healthy subjects 24 hours after idarucizumab treatment and to subjects who received placebo instead of idarucizumab.
Dabigatran-mediated anticoagulation was similar in subjects who received idarucizumab and those who received placebo. And anticoagulation was restored to levels comparable to initial levels.
Dr Stangier said there were no clinically relevant adverse events (AEs) related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. There were no AEs indicative of immunogenic reactions.
AEs and local tolerability reactions were similar for placebo and idarucizumab. And there was no relationship between the frequency of AEs and drug dose, gender, or renal function.
The researchers did observe a dose-dependent, transient increase in urine protein and low-weight proteins, but values returned to the normal range within 4 hours to 24 hours.
Based on these results, Dr Stangier said idarucizumab fulfills the requirements for a fast-acting and specific antidote to dabigatran with a favorable safety profile. Additional clinical testing of the antidote is ongoing. ![]()
*Information in the abstract differs from that presented.
Credit: CDC
SAN FRANCISCO—An investigational, humanized antibody fragment known as idarucizumab can reverse the anticoagulation effects of dabigatran, new research suggests.
In a small study, idarucizumab led to sustained reversal of dabigatran’s effects in healthy subjects, elderly volunteers, and participants with mild to moderate renal impairment.
Furthermore, dabigatran anticoagulation could be re-established 24 hours after the subjects had received idarucizumab.
Joachim Stangier, PhD, of Boehringer Ingelheim Pharma GmbH & Co KG in Biberach, Germany, presented these results at the 2014 ASH Annual Meeting as abstract 344*. Boehringer Ingelheim is the company developing idarucizumab.
Dr Stangier said idarucizumab binds dabigatran with high affinity, off-target binding is not expected, and no procoagulant effects have been observed with the drug.
In a previous study of healthy volunteers, idarucizumab provided immediate, complete, and sustained reversal of dabigatran’s anticoagulant effect. The effect was sustained when idarucizumab was given at 2 g and 4 g doses.
For the current study, Dr Stangier and his colleagues wanted to evaluate whether and to what extent doses of up to 5 g of idarucizumab would reverse the anticoagulant effects of dabigatran in healthy subjects, elderly participants, and renally impaired volunteers. The team also wanted to determine the effects of dabigatran given after subjects had received idarucizumab.
The study included 46 male and female subjects who were 45 to 80 years of age. Healthy subjects received dabigatran etexilate at 220 mg twice daily, and subjects with mild to moderate renal impairment received 150 mg twice daily, both over 4 days.
Subjects then received idarucizumab at 1 g, 2.5 g, 5 g, or 5 g given as 2 x 2.5 g 1 hour apart. They received these doses as 5-minute intravenous infusions 2 hours after their last dose of dabigatran.
The researchers found that dabigatran-prolonged clotting times—thrombin time, diluted thrombin time, ecarin clotting time, and activated partial thromboplastin time—were reversed to baseline immediately after the end of idarucizumab infusion.
And the team observed sustained reversal of dabigatran’s anticoagulant effects in all subjects.
The researchers then readministered dabigatran to healthy subjects 24 hours after idarucizumab treatment and to subjects who received placebo instead of idarucizumab.
Dabigatran-mediated anticoagulation was similar in subjects who received idarucizumab and those who received placebo. And anticoagulation was restored to levels comparable to initial levels.
Dr Stangier said there were no clinically relevant adverse events (AEs) related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. There were no AEs indicative of immunogenic reactions.
AEs and local tolerability reactions were similar for placebo and idarucizumab. And there was no relationship between the frequency of AEs and drug dose, gender, or renal function.
The researchers did observe a dose-dependent, transient increase in urine protein and low-weight proteins, but values returned to the normal range within 4 hours to 24 hours.
Based on these results, Dr Stangier said idarucizumab fulfills the requirements for a fast-acting and specific antidote to dabigatran with a favorable safety profile. Additional clinical testing of the antidote is ongoing. ![]()
*Information in the abstract differs from that presented.
Credit: CDC
SAN FRANCISCO—An investigational, humanized antibody fragment known as idarucizumab can reverse the anticoagulation effects of dabigatran, new research suggests.
In a small study, idarucizumab led to sustained reversal of dabigatran’s effects in healthy subjects, elderly volunteers, and participants with mild to moderate renal impairment.
Furthermore, dabigatran anticoagulation could be re-established 24 hours after the subjects had received idarucizumab.
Joachim Stangier, PhD, of Boehringer Ingelheim Pharma GmbH & Co KG in Biberach, Germany, presented these results at the 2014 ASH Annual Meeting as abstract 344*. Boehringer Ingelheim is the company developing idarucizumab.
Dr Stangier said idarucizumab binds dabigatran with high affinity, off-target binding is not expected, and no procoagulant effects have been observed with the drug.
In a previous study of healthy volunteers, idarucizumab provided immediate, complete, and sustained reversal of dabigatran’s anticoagulant effect. The effect was sustained when idarucizumab was given at 2 g and 4 g doses.
For the current study, Dr Stangier and his colleagues wanted to evaluate whether and to what extent doses of up to 5 g of idarucizumab would reverse the anticoagulant effects of dabigatran in healthy subjects, elderly participants, and renally impaired volunteers. The team also wanted to determine the effects of dabigatran given after subjects had received idarucizumab.
The study included 46 male and female subjects who were 45 to 80 years of age. Healthy subjects received dabigatran etexilate at 220 mg twice daily, and subjects with mild to moderate renal impairment received 150 mg twice daily, both over 4 days.
Subjects then received idarucizumab at 1 g, 2.5 g, 5 g, or 5 g given as 2 x 2.5 g 1 hour apart. They received these doses as 5-minute intravenous infusions 2 hours after their last dose of dabigatran.
The researchers found that dabigatran-prolonged clotting times—thrombin time, diluted thrombin time, ecarin clotting time, and activated partial thromboplastin time—were reversed to baseline immediately after the end of idarucizumab infusion.
And the team observed sustained reversal of dabigatran’s anticoagulant effects in all subjects.
The researchers then readministered dabigatran to healthy subjects 24 hours after idarucizumab treatment and to subjects who received placebo instead of idarucizumab.
Dabigatran-mediated anticoagulation was similar in subjects who received idarucizumab and those who received placebo. And anticoagulation was restored to levels comparable to initial levels.
Dr Stangier said there were no clinically relevant adverse events (AEs) related to idarucizumab, and there were no relevant changes in any of the investigated safety parameters. There were no AEs indicative of immunogenic reactions.
AEs and local tolerability reactions were similar for placebo and idarucizumab. And there was no relationship between the frequency of AEs and drug dose, gender, or renal function.
The researchers did observe a dose-dependent, transient increase in urine protein and low-weight proteins, but values returned to the normal range within 4 hours to 24 hours.
Based on these results, Dr Stangier said idarucizumab fulfills the requirements for a fast-acting and specific antidote to dabigatran with a favorable safety profile. Additional clinical testing of the antidote is ongoing. ![]()
*Information in the abstract differs from that presented.
HIV doesn’t hinder lymphoma patients’ response to ASCT
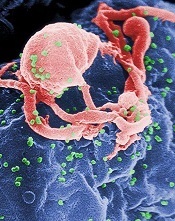
a cultured lymphocyte
Credit: CDC
SAN FRANCISCO—Patients with HIV-related lymphoma (HRL) should not be excluded from clinical trials of autologous stem cell transplant (ASCT) due to their HIV status, new research suggests.
Investigators found no significant difference in rates of treatment failure, disease progression, or survival between transplant-treated historical controls who had lymphoma but not HIV and patients with HRL who received the modified BEAM regimen followed by ASCT on a phase 2 trial.
This suggests patients with chemotherapy-sensitive, relapsed/refractory HRL can be treated successfully with the modified BEAM regimen, said study investigator Joseph Alvarnas, MD, of City of Hope National Medical Center in Duarte, California.
“Patients with treatment-responsive HIV infection and HIV-related lymphoma should be considered candidates for autologous transplant if they meet standard transplant criteria,” he added. “And we would argue that exclusion from clinical trials on the basis of HIV infection alone is no longer justified.”
Dr Alvarnas presented this viewpoint and the research to support it at the 2014 ASH Annual Meeting as abstract 674.
The trial enrolled 43 patients with treatable HIV-1 infection, adequate organ function, and aggressive lymphoma. Three patients were excluded because they could not undergo transplant due to lymphoma progression.
Of the 40 remaining patients, 5 were female, and their median age was 46.9 years (range, 22.5-62.2). They had diffuse large B-cell lymphoma (n=16), plasmablastic lymphoma (n=2), Burkitt/Burkitt-like lymphoma (n=7), and Hodgkin lymphoma (n=15).
The pre-ASCT HIV viral load was undetectable in 31 patients. In the patients with detectable HIV, the median viral load pre-ASCT was 84 copies/μL (range, 50-17,455). The median CD4 count was 250.5/µL (range, 39-797).
Before transplant, 30 patients (75%) were in complete remission, 8 (20%) were in partial remission, and 2 (5%) had relapsed/progressive disease.
The patients underwent ASCT after conditioning with the modified BEAM regimen—carmustine at 300 mg/m2 (day -6), etoposide at 100 mg/m2 twice daily (days -5 to -2), cytarabine at 100 mg/m2 (days -5 to -2), and melphalan at 140 mg/m2 (day -1).
Combination antiretroviral therapy (cART) was held during the preparative regimen and resumed after the resolution of gastrointestinal toxicity. The investigators switched efavirenz to an alternative agent 2 or more weeks prior to the planned interruption of cART, as the drug has a long half-life. And AZT was prohibited due to its myelosuppressive effects.
Treatment results
The median follow-up was 24 months. At 100 days post-transplant, the investigators assessed 39 patients for response. One patient was not evaluable due to early death.
Thirty-six of the patients (92.3%) were in complete remission, 1 (2.6%) was in partial remission, and 2 (5.1%) had relapsed or progressive disease.
Fifteen patients reported grade 3 or higher toxicities within a year of transplant. Of the 13 unexpected grade 3-5 adverse events (reported in 9 patients), 5 were infection/sepsis, 1 was acute appendicitis, 1 was acute coronary syndrome, 2 were deep vein thromboses, 2 were gastrointestinal toxicities, and 2 were metabolic abnormalities.
Seventeen patients reported at least 1 infectious episode, 42 events in total, 9 of which were severe. Fourteen patients required readmission to the hospital after transplant.
Within a year of transplant, 5 patients had died—3 from recurrent/persistent disease, 1 due to a fungal infection, and 1 from cardiac arrest. Two additional patients died after the 1-year mark—1 of recurrent/persistent disease and 1 of heart failure.
At 12 months, the rate of overall survival was 86.6%, progression-free survival was 82.3%, progression was 12.5%, and non-relapse mortality was 5.2%.
“In order to place this within context, we had the opportunity to compare our patient experience with 151 matched controls [without HIV] from CIBMTR,” Dr Alvarnas said. “Ninety-three percent of these patients were actually transplanted within 2 years so that they were the time-concordant group, and they were matched for performance score, disease, and disease stage.”
The investigators found no significant difference between their patient group and the HIV-free controls with regard to overall mortality (P=0.56), treatment failure (P=0.10), progression (P=0.06), and treatment-related mortality (P=0.97).
Likewise, there was no significant difference in overall survival between the HRL patients and controls—86.6% and 87.7%, respectively (P=0.56). And the same was true of progression-free survival—82.3% and 69.5%, respectively (P=0.10). ![]()
a cultured lymphocyte
Credit: CDC
SAN FRANCISCO—Patients with HIV-related lymphoma (HRL) should not be excluded from clinical trials of autologous stem cell transplant (ASCT) due to their HIV status, new research suggests.
Investigators found no significant difference in rates of treatment failure, disease progression, or survival between transplant-treated historical controls who had lymphoma but not HIV and patients with HRL who received the modified BEAM regimen followed by ASCT on a phase 2 trial.
This suggests patients with chemotherapy-sensitive, relapsed/refractory HRL can be treated successfully with the modified BEAM regimen, said study investigator Joseph Alvarnas, MD, of City of Hope National Medical Center in Duarte, California.
“Patients with treatment-responsive HIV infection and HIV-related lymphoma should be considered candidates for autologous transplant if they meet standard transplant criteria,” he added. “And we would argue that exclusion from clinical trials on the basis of HIV infection alone is no longer justified.”
Dr Alvarnas presented this viewpoint and the research to support it at the 2014 ASH Annual Meeting as abstract 674.
The trial enrolled 43 patients with treatable HIV-1 infection, adequate organ function, and aggressive lymphoma. Three patients were excluded because they could not undergo transplant due to lymphoma progression.
Of the 40 remaining patients, 5 were female, and their median age was 46.9 years (range, 22.5-62.2). They had diffuse large B-cell lymphoma (n=16), plasmablastic lymphoma (n=2), Burkitt/Burkitt-like lymphoma (n=7), and Hodgkin lymphoma (n=15).
The pre-ASCT HIV viral load was undetectable in 31 patients. In the patients with detectable HIV, the median viral load pre-ASCT was 84 copies/μL (range, 50-17,455). The median CD4 count was 250.5/µL (range, 39-797).
Before transplant, 30 patients (75%) were in complete remission, 8 (20%) were in partial remission, and 2 (5%) had relapsed/progressive disease.
The patients underwent ASCT after conditioning with the modified BEAM regimen—carmustine at 300 mg/m2 (day -6), etoposide at 100 mg/m2 twice daily (days -5 to -2), cytarabine at 100 mg/m2 (days -5 to -2), and melphalan at 140 mg/m2 (day -1).
Combination antiretroviral therapy (cART) was held during the preparative regimen and resumed after the resolution of gastrointestinal toxicity. The investigators switched efavirenz to an alternative agent 2 or more weeks prior to the planned interruption of cART, as the drug has a long half-life. And AZT was prohibited due to its myelosuppressive effects.
Treatment results
The median follow-up was 24 months. At 100 days post-transplant, the investigators assessed 39 patients for response. One patient was not evaluable due to early death.
Thirty-six of the patients (92.3%) were in complete remission, 1 (2.6%) was in partial remission, and 2 (5.1%) had relapsed or progressive disease.
Fifteen patients reported grade 3 or higher toxicities within a year of transplant. Of the 13 unexpected grade 3-5 adverse events (reported in 9 patients), 5 were infection/sepsis, 1 was acute appendicitis, 1 was acute coronary syndrome, 2 were deep vein thromboses, 2 were gastrointestinal toxicities, and 2 were metabolic abnormalities.
Seventeen patients reported at least 1 infectious episode, 42 events in total, 9 of which were severe. Fourteen patients required readmission to the hospital after transplant.
Within a year of transplant, 5 patients had died—3 from recurrent/persistent disease, 1 due to a fungal infection, and 1 from cardiac arrest. Two additional patients died after the 1-year mark—1 of recurrent/persistent disease and 1 of heart failure.
At 12 months, the rate of overall survival was 86.6%, progression-free survival was 82.3%, progression was 12.5%, and non-relapse mortality was 5.2%.
“In order to place this within context, we had the opportunity to compare our patient experience with 151 matched controls [without HIV] from CIBMTR,” Dr Alvarnas said. “Ninety-three percent of these patients were actually transplanted within 2 years so that they were the time-concordant group, and they were matched for performance score, disease, and disease stage.”
The investigators found no significant difference between their patient group and the HIV-free controls with regard to overall mortality (P=0.56), treatment failure (P=0.10), progression (P=0.06), and treatment-related mortality (P=0.97).
Likewise, there was no significant difference in overall survival between the HRL patients and controls—86.6% and 87.7%, respectively (P=0.56). And the same was true of progression-free survival—82.3% and 69.5%, respectively (P=0.10). ![]()
a cultured lymphocyte
Credit: CDC
SAN FRANCISCO—Patients with HIV-related lymphoma (HRL) should not be excluded from clinical trials of autologous stem cell transplant (ASCT) due to their HIV status, new research suggests.
Investigators found no significant difference in rates of treatment failure, disease progression, or survival between transplant-treated historical controls who had lymphoma but not HIV and patients with HRL who received the modified BEAM regimen followed by ASCT on a phase 2 trial.
This suggests patients with chemotherapy-sensitive, relapsed/refractory HRL can be treated successfully with the modified BEAM regimen, said study investigator Joseph Alvarnas, MD, of City of Hope National Medical Center in Duarte, California.
“Patients with treatment-responsive HIV infection and HIV-related lymphoma should be considered candidates for autologous transplant if they meet standard transplant criteria,” he added. “And we would argue that exclusion from clinical trials on the basis of HIV infection alone is no longer justified.”
Dr Alvarnas presented this viewpoint and the research to support it at the 2014 ASH Annual Meeting as abstract 674.
The trial enrolled 43 patients with treatable HIV-1 infection, adequate organ function, and aggressive lymphoma. Three patients were excluded because they could not undergo transplant due to lymphoma progression.
Of the 40 remaining patients, 5 were female, and their median age was 46.9 years (range, 22.5-62.2). They had diffuse large B-cell lymphoma (n=16), plasmablastic lymphoma (n=2), Burkitt/Burkitt-like lymphoma (n=7), and Hodgkin lymphoma (n=15).
The pre-ASCT HIV viral load was undetectable in 31 patients. In the patients with detectable HIV, the median viral load pre-ASCT was 84 copies/μL (range, 50-17,455). The median CD4 count was 250.5/µL (range, 39-797).
Before transplant, 30 patients (75%) were in complete remission, 8 (20%) were in partial remission, and 2 (5%) had relapsed/progressive disease.
The patients underwent ASCT after conditioning with the modified BEAM regimen—carmustine at 300 mg/m2 (day -6), etoposide at 100 mg/m2 twice daily (days -5 to -2), cytarabine at 100 mg/m2 (days -5 to -2), and melphalan at 140 mg/m2 (day -1).
Combination antiretroviral therapy (cART) was held during the preparative regimen and resumed after the resolution of gastrointestinal toxicity. The investigators switched efavirenz to an alternative agent 2 or more weeks prior to the planned interruption of cART, as the drug has a long half-life. And AZT was prohibited due to its myelosuppressive effects.
Treatment results
The median follow-up was 24 months. At 100 days post-transplant, the investigators assessed 39 patients for response. One patient was not evaluable due to early death.
Thirty-six of the patients (92.3%) were in complete remission, 1 (2.6%) was in partial remission, and 2 (5.1%) had relapsed or progressive disease.
Fifteen patients reported grade 3 or higher toxicities within a year of transplant. Of the 13 unexpected grade 3-5 adverse events (reported in 9 patients), 5 were infection/sepsis, 1 was acute appendicitis, 1 was acute coronary syndrome, 2 were deep vein thromboses, 2 were gastrointestinal toxicities, and 2 were metabolic abnormalities.
Seventeen patients reported at least 1 infectious episode, 42 events in total, 9 of which were severe. Fourteen patients required readmission to the hospital after transplant.
Within a year of transplant, 5 patients had died—3 from recurrent/persistent disease, 1 due to a fungal infection, and 1 from cardiac arrest. Two additional patients died after the 1-year mark—1 of recurrent/persistent disease and 1 of heart failure.
At 12 months, the rate of overall survival was 86.6%, progression-free survival was 82.3%, progression was 12.5%, and non-relapse mortality was 5.2%.
“In order to place this within context, we had the opportunity to compare our patient experience with 151 matched controls [without HIV] from CIBMTR,” Dr Alvarnas said. “Ninety-three percent of these patients were actually transplanted within 2 years so that they were the time-concordant group, and they were matched for performance score, disease, and disease stage.”
The investigators found no significant difference between their patient group and the HIV-free controls with regard to overall mortality (P=0.56), treatment failure (P=0.10), progression (P=0.06), and treatment-related mortality (P=0.97).
Likewise, there was no significant difference in overall survival between the HRL patients and controls—86.6% and 87.7%, respectively (P=0.56). And the same was true of progression-free survival—82.3% and 69.5%, respectively (P=0.10). ![]()
Maintenance prolongs PFS, not OS, in relapsed CLL

Credit: Linda Bartlett
SAN FRANCISCO—Maintenance therapy with the anti-CD20 monoclonal antibody ofatumumab improves progression-free survival (PFS), but not overall survival (OS), in patients with relapsed chronic lymphocytic leukemia (CLL), according to an interim analysis of the PROLONG study.
The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance (P<0.0001).
But there was no significant difference in the median OS, which was not reached in either treatment arm.
Marinus H.J. van Oers, MD, PhD, of the Academisch Medisch Centrum and HOVON in Amsterdam, The Netherlands, reported these results at the 2014 ASH Annual Meeting (abstract 21*). The study was sponsored by GlaxoSmithKline, makers of ofatumumab.
“[A]s of 2014, still we cannot say that we are able to cure CLL,” Dr van Oers noted. “And CLL is characterized by decreasing response duration with subsequent lines of treatment. In this respect, but also a number of other respects, there are similarities in biological behavior between CLL and follicular lymphoma.”
“There is definitely a role—although it’s somewhat debated—for maintenance treatment in follicular lymphoma. Therefore, it is rational to explore safe and effective maintenance treatment in CLL as well.”
To that end, Dr van Oers and his colleagues compared ofatumumab maintenance to observation in patients who were in remission after induction treatment for relapsed CLL. The team enrolled 474 patients who were in complete or partial remission after their 2nd- or 3rd-line treatment for CLL.
Patients were randomized to observation (n=236) or to receive ofatumumab (n=238) at 300 mg, followed 1 week later by 1000 mg every 8 weeks for up to 2 years. Patients on ofatumumab also received premedication with acetaminophen, antihistamine, and glucocorticoid.
The patients were stratified by the number and type of prior therapy, as well as remission status after induction treatment, and baseline characteristics were similar between the two treatment arms.
“The median age was about 65, and about 30% of patients were older than 70 years,” Dr van Oers noted. “[There was] a male preponderance, as you would expect, and the time since diagnosis was somewhere between 5 and 6 years.”
“Most patients were in [partial response], actually 80%, and most patients had received 2 prior regimens, about 70%. As for prior treatments, 80% of patients had received effective immuno-chemotherapy.”
“In both arms, there were only a few patients with unfavorable cytogenetics—11q and 17p deletion. [As for] β2 microglobulin, two-thirds [of patients in both arms] had low levels. And, in both arms, there were almost twice as many IGVH-mutated as unmutated patients.”
Patient outcomes
The median follow-up was 19.1 months. The study’s primary endpoint was PFS, which was defined as the time from randomization to the date of disease progression or death from any cause.
The median PFS was significantly longer in the ofatumumab arm than in the observation arm, at 29.4 months and 15.2 months, respectively (hazard ratio [HR]=0.50; P<0.0001).
Similarly, the time to the start of patients’ next therapy was significantly longer in the ofatumumab arm than in the in observation arm—a median of 38 months and 31.1 months, respectively (HR=0.66, P=0.108).
However, there was no significant difference in OS, which was not reached in either arm (HR=0.85, P=0.4877).
Adverse events (AEs) occurred in 86% of patients in the ofatumumab arm and 72% of patients in the observation arm (P<0.001). Sixty percent of AEs were considered related to ofatumumab. None of the AEs led to study withdrawal.
Grade 3 or higher AEs occurred in 46% of patients in the ofatumumab arm and 28% in the observation arm. They included neutropenia (24% and 10%, respectively; P<0.001), infections (13% and 8%, respectively), thrombocytopenia (2% and 3%, respectively), and infusion-related reactions (1% and 0%, respectively).
There were 5 deaths in the observation arm—1 due to progression and 4 due to causes other than progression, infection, or secondary malignancy. There were 2 deaths in the ofatumumab arm—1 due to infection/sepsis and 1 due to an “other” cause.
“So based on this planned interim analysis, we can conclude that ofatumumab maintenance in relapsed CLL results in a highly significant and clinically meaningful improvement of progression-free survival,” Dr van Oers said in closing.
“It significantly prolongs time to next treatment, it’s well-tolerated, and it’s associated with an adverse event profile which is quite characteristic of anti-CD20 monoclonal antibodies.” ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Linda Bartlett
SAN FRANCISCO—Maintenance therapy with the anti-CD20 monoclonal antibody ofatumumab improves progression-free survival (PFS), but not overall survival (OS), in patients with relapsed chronic lymphocytic leukemia (CLL), according to an interim analysis of the PROLONG study.
The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance (P<0.0001).
But there was no significant difference in the median OS, which was not reached in either treatment arm.
Marinus H.J. van Oers, MD, PhD, of the Academisch Medisch Centrum and HOVON in Amsterdam, The Netherlands, reported these results at the 2014 ASH Annual Meeting (abstract 21*). The study was sponsored by GlaxoSmithKline, makers of ofatumumab.
“[A]s of 2014, still we cannot say that we are able to cure CLL,” Dr van Oers noted. “And CLL is characterized by decreasing response duration with subsequent lines of treatment. In this respect, but also a number of other respects, there are similarities in biological behavior between CLL and follicular lymphoma.”
“There is definitely a role—although it’s somewhat debated—for maintenance treatment in follicular lymphoma. Therefore, it is rational to explore safe and effective maintenance treatment in CLL as well.”
To that end, Dr van Oers and his colleagues compared ofatumumab maintenance to observation in patients who were in remission after induction treatment for relapsed CLL. The team enrolled 474 patients who were in complete or partial remission after their 2nd- or 3rd-line treatment for CLL.
Patients were randomized to observation (n=236) or to receive ofatumumab (n=238) at 300 mg, followed 1 week later by 1000 mg every 8 weeks for up to 2 years. Patients on ofatumumab also received premedication with acetaminophen, antihistamine, and glucocorticoid.
The patients were stratified by the number and type of prior therapy, as well as remission status after induction treatment, and baseline characteristics were similar between the two treatment arms.
“The median age was about 65, and about 30% of patients were older than 70 years,” Dr van Oers noted. “[There was] a male preponderance, as you would expect, and the time since diagnosis was somewhere between 5 and 6 years.”
“Most patients were in [partial response], actually 80%, and most patients had received 2 prior regimens, about 70%. As for prior treatments, 80% of patients had received effective immuno-chemotherapy.”
“In both arms, there were only a few patients with unfavorable cytogenetics—11q and 17p deletion. [As for] β2 microglobulin, two-thirds [of patients in both arms] had low levels. And, in both arms, there were almost twice as many IGVH-mutated as unmutated patients.”
Patient outcomes
The median follow-up was 19.1 months. The study’s primary endpoint was PFS, which was defined as the time from randomization to the date of disease progression or death from any cause.
The median PFS was significantly longer in the ofatumumab arm than in the observation arm, at 29.4 months and 15.2 months, respectively (hazard ratio [HR]=0.50; P<0.0001).
Similarly, the time to the start of patients’ next therapy was significantly longer in the ofatumumab arm than in the in observation arm—a median of 38 months and 31.1 months, respectively (HR=0.66, P=0.108).
However, there was no significant difference in OS, which was not reached in either arm (HR=0.85, P=0.4877).
Adverse events (AEs) occurred in 86% of patients in the ofatumumab arm and 72% of patients in the observation arm (P<0.001). Sixty percent of AEs were considered related to ofatumumab. None of the AEs led to study withdrawal.
Grade 3 or higher AEs occurred in 46% of patients in the ofatumumab arm and 28% in the observation arm. They included neutropenia (24% and 10%, respectively; P<0.001), infections (13% and 8%, respectively), thrombocytopenia (2% and 3%, respectively), and infusion-related reactions (1% and 0%, respectively).
There were 5 deaths in the observation arm—1 due to progression and 4 due to causes other than progression, infection, or secondary malignancy. There were 2 deaths in the ofatumumab arm—1 due to infection/sepsis and 1 due to an “other” cause.
“So based on this planned interim analysis, we can conclude that ofatumumab maintenance in relapsed CLL results in a highly significant and clinically meaningful improvement of progression-free survival,” Dr van Oers said in closing.
“It significantly prolongs time to next treatment, it’s well-tolerated, and it’s associated with an adverse event profile which is quite characteristic of anti-CD20 monoclonal antibodies.” ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Linda Bartlett
SAN FRANCISCO—Maintenance therapy with the anti-CD20 monoclonal antibody ofatumumab improves progression-free survival (PFS), but not overall survival (OS), in patients with relapsed chronic lymphocytic leukemia (CLL), according to an interim analysis of the PROLONG study.
The median PFS was about 29 months in patients who received ofatumumab and about 15 months for patients who did not receive maintenance (P<0.0001).
But there was no significant difference in the median OS, which was not reached in either treatment arm.
Marinus H.J. van Oers, MD, PhD, of the Academisch Medisch Centrum and HOVON in Amsterdam, The Netherlands, reported these results at the 2014 ASH Annual Meeting (abstract 21*). The study was sponsored by GlaxoSmithKline, makers of ofatumumab.
“[A]s of 2014, still we cannot say that we are able to cure CLL,” Dr van Oers noted. “And CLL is characterized by decreasing response duration with subsequent lines of treatment. In this respect, but also a number of other respects, there are similarities in biological behavior between CLL and follicular lymphoma.”
“There is definitely a role—although it’s somewhat debated—for maintenance treatment in follicular lymphoma. Therefore, it is rational to explore safe and effective maintenance treatment in CLL as well.”
To that end, Dr van Oers and his colleagues compared ofatumumab maintenance to observation in patients who were in remission after induction treatment for relapsed CLL. The team enrolled 474 patients who were in complete or partial remission after their 2nd- or 3rd-line treatment for CLL.
Patients were randomized to observation (n=236) or to receive ofatumumab (n=238) at 300 mg, followed 1 week later by 1000 mg every 8 weeks for up to 2 years. Patients on ofatumumab also received premedication with acetaminophen, antihistamine, and glucocorticoid.
The patients were stratified by the number and type of prior therapy, as well as remission status after induction treatment, and baseline characteristics were similar between the two treatment arms.
“The median age was about 65, and about 30% of patients were older than 70 years,” Dr van Oers noted. “[There was] a male preponderance, as you would expect, and the time since diagnosis was somewhere between 5 and 6 years.”
“Most patients were in [partial response], actually 80%, and most patients had received 2 prior regimens, about 70%. As for prior treatments, 80% of patients had received effective immuno-chemotherapy.”
“In both arms, there were only a few patients with unfavorable cytogenetics—11q and 17p deletion. [As for] β2 microglobulin, two-thirds [of patients in both arms] had low levels. And, in both arms, there were almost twice as many IGVH-mutated as unmutated patients.”
Patient outcomes
The median follow-up was 19.1 months. The study’s primary endpoint was PFS, which was defined as the time from randomization to the date of disease progression or death from any cause.
The median PFS was significantly longer in the ofatumumab arm than in the observation arm, at 29.4 months and 15.2 months, respectively (hazard ratio [HR]=0.50; P<0.0001).
Similarly, the time to the start of patients’ next therapy was significantly longer in the ofatumumab arm than in the in observation arm—a median of 38 months and 31.1 months, respectively (HR=0.66, P=0.108).
However, there was no significant difference in OS, which was not reached in either arm (HR=0.85, P=0.4877).
Adverse events (AEs) occurred in 86% of patients in the ofatumumab arm and 72% of patients in the observation arm (P<0.001). Sixty percent of AEs were considered related to ofatumumab. None of the AEs led to study withdrawal.
Grade 3 or higher AEs occurred in 46% of patients in the ofatumumab arm and 28% in the observation arm. They included neutropenia (24% and 10%, respectively; P<0.001), infections (13% and 8%, respectively), thrombocytopenia (2% and 3%, respectively), and infusion-related reactions (1% and 0%, respectively).
There were 5 deaths in the observation arm—1 due to progression and 4 due to causes other than progression, infection, or secondary malignancy. There were 2 deaths in the ofatumumab arm—1 due to infection/sepsis and 1 due to an “other” cause.
“So based on this planned interim analysis, we can conclude that ofatumumab maintenance in relapsed CLL results in a highly significant and clinically meaningful improvement of progression-free survival,” Dr van Oers said in closing.
“It significantly prolongs time to next treatment, it’s well-tolerated, and it’s associated with an adverse event profile which is quite characteristic of anti-CD20 monoclonal antibodies.” ![]()
*Information in the abstract differs from that presented at the meeting.
Older patients benefit from brentuximab treatment

Credit: NIH
SAN FRANCISCO—Younger patients with Hodgkin lymphoma fare well on brentuximab vedotin, experiencing an overall objective response rate (ORR) of 75% and a complete response (CR) rate of 34% in the pivotal phase 2 study of patients with relapsed/refractory disease.
And a retrospective study of patients older than 60 years showed that single-agent therapy was well tolerated, prompting an ORR of 53% and a CR rate of 40% in a relapsed or refractory population.
So investigators decided to explore in a prospective study whether patients 60 years or older could benefit from up-front treatment with brentuximab as a single agent or in combination.
Andres Forero-Torres, MD, of the University of Alabama in Birmingham, presented the results of this trial at the 2014 ASH Annual Meeting (abstract 294).*
Enrolled patients had classic Hodgkin lymphoma, were treatment-naïve, and were ineligible for or declined conventional front-line treatment. The primary endpoint was ORR.
The study is being conducted in 3 parts—brentuximab as a single agent, brentuximab plus dacarbazine, and brentuximab plus bendamustine. At the time of the ASH presentation, data for the brentuximab-bendamustine combination were not available.
Single-agent brentuximab
Twenty-seven patients on the single-agent arm were evaluable for efficacy and safety. They were a median age of 78 (range, 64 to 92). About half (52%) were male, and 78% had an ECOG performance status of grade 0 or 1.
Forty-four percent had moderate renal function impairment with a creatinine clearance between 30 and 60 mL/min. Thirty percent had B symptoms, 22% had bulky disease, and 52% had extra-nodal involvement.
Patients received 1.8 mg/kg of brentuximab intravenously on day 1 of a 21-day cycle. Response was assessed by CT scan during cycles 2, 4, 8, and 16, and by CT plus PET scan during cycles 2 and 8.
The median follow-up was 8.7 months. Dr Forero-Torres pointed out that, initially, “there were no progressions,” and all patients achieved tumor reduction.
The ORR was 93%, the CR rate was 70%, the partial response rate was 22%, and the rate of stable disease was 7%.
The median duration of response was 9.1 months (range, 0.03 to 13.14), and the median progression-free survival was 10.5 months (range, 2.6 to 14.3). For patients who had a CR, the median progression-free survival was about 12 months, Dr Forero-Torres said.
The median number of treatment cycles administered per patient was 8 (range, 3 to 23). Patients discontinued treatment primarily because of progressive disease (41%) or adverse events (AEs, 37%).
AEs occurring in 20% or more of patients were constipation, decreased appetite, diarrhea, peripheral edema, nausea, fatigue, and peripheral sensory neuropathy. All were grade 1 or 2, except for peripheral sensory neuropathy, which also had about 20% grade 3 events.
Grade 3 or higher treatment-related AEs included peripheral sensory neuropathy (n=7), peripheral motor neuropathy (n=2), rash (n=2), and 1 patient each with anemia, increased aspartate aminotransferase, asthenia, neutropenia, orthostatic hypotension, generalized rash, and maculopapular rash.
Serious AEs (SAEs) were minimal, Dr Forero-Torres said, and included 1 patient each with pyrexia, orthostatic hypotension, asthenia and rash, and deep vein thrombosis.
Seven patients discontinued treatment due to peripheral sensory neuropathy, 2 due to peripheral motor neuropathy, and 1 due to orthostatic hypotension.
Dr Forero-Torres emphasized that there were no grade 4 AEs, no AE-related deaths, and no deaths within 30 days of the last dose of medication.
Brentuximab plus dacarbazine
Fourteen of 18 patients in the combination arm were evaluable for efficacy and safety. Their median age was 72.5 (range, 62 to 87), 72% were male, 67% had an ECOG status of grade 0 or 1, and 56% had normal renal function with a creatinine clearance greater than 80 mL/min.
Forty-four percent exhibited B symptoms, 11% had bulky disease, and 50% had extra-nodal involvement.
They received brentuximab at 1.8 mg/kg plus dacarbazine at 375 mg/m2 for cycles 1-12, followed by monotherapy for cycles 13-16.
At the time of the interim analysis, 83% of patients were still on treatment, “so this is very early preliminary data,” Dr Forero-Torres noted.
All of the patients achieved tumor reduction, and 4 patients achieved a CR.
They had a median treatment duration of 16.7 weeks (range, 3 to 36), received a median of 5.5 cycles (range, 1 to 12), and had a median follow-up time of 19.1 weeks (range, 6.1 to 36.1).
The most common grade 1 or 2 AEs were peripheral sensory neuropathy (33%), nausea (33%), diarrhea (28%), constipation (28%), fatigue (22%), alopecia (22%), arthralgia (22%), and headache (22%).
Grade 3 AEs or SAEs, with 1 patient each, were C difficile colitis (SAE), hypotension (SAE), and hyperglycemia.
Dr Forero-Torres noted that investigators observed “robust antitumor activity” among these older patients receiving front-line brentuximab.
The cohort combining brentuximab with bendamustine is currently enrolling patients.
The study is sponsored by Seattle Genetics, Inc., developer of brentuximab vedotin (Adcetris).![]()
*Information in the abstract differs from that presented at the meeting.
Credit: NIH
SAN FRANCISCO—Younger patients with Hodgkin lymphoma fare well on brentuximab vedotin, experiencing an overall objective response rate (ORR) of 75% and a complete response (CR) rate of 34% in the pivotal phase 2 study of patients with relapsed/refractory disease.
And a retrospective study of patients older than 60 years showed that single-agent therapy was well tolerated, prompting an ORR of 53% and a CR rate of 40% in a relapsed or refractory population.
So investigators decided to explore in a prospective study whether patients 60 years or older could benefit from up-front treatment with brentuximab as a single agent or in combination.
Andres Forero-Torres, MD, of the University of Alabama in Birmingham, presented the results of this trial at the 2014 ASH Annual Meeting (abstract 294).*
Enrolled patients had classic Hodgkin lymphoma, were treatment-naïve, and were ineligible for or declined conventional front-line treatment. The primary endpoint was ORR.
The study is being conducted in 3 parts—brentuximab as a single agent, brentuximab plus dacarbazine, and brentuximab plus bendamustine. At the time of the ASH presentation, data for the brentuximab-bendamustine combination were not available.
Single-agent brentuximab
Twenty-seven patients on the single-agent arm were evaluable for efficacy and safety. They were a median age of 78 (range, 64 to 92). About half (52%) were male, and 78% had an ECOG performance status of grade 0 or 1.
Forty-four percent had moderate renal function impairment with a creatinine clearance between 30 and 60 mL/min. Thirty percent had B symptoms, 22% had bulky disease, and 52% had extra-nodal involvement.
Patients received 1.8 mg/kg of brentuximab intravenously on day 1 of a 21-day cycle. Response was assessed by CT scan during cycles 2, 4, 8, and 16, and by CT plus PET scan during cycles 2 and 8.
The median follow-up was 8.7 months. Dr Forero-Torres pointed out that, initially, “there were no progressions,” and all patients achieved tumor reduction.
The ORR was 93%, the CR rate was 70%, the partial response rate was 22%, and the rate of stable disease was 7%.
The median duration of response was 9.1 months (range, 0.03 to 13.14), and the median progression-free survival was 10.5 months (range, 2.6 to 14.3). For patients who had a CR, the median progression-free survival was about 12 months, Dr Forero-Torres said.
The median number of treatment cycles administered per patient was 8 (range, 3 to 23). Patients discontinued treatment primarily because of progressive disease (41%) or adverse events (AEs, 37%).
AEs occurring in 20% or more of patients were constipation, decreased appetite, diarrhea, peripheral edema, nausea, fatigue, and peripheral sensory neuropathy. All were grade 1 or 2, except for peripheral sensory neuropathy, which also had about 20% grade 3 events.
Grade 3 or higher treatment-related AEs included peripheral sensory neuropathy (n=7), peripheral motor neuropathy (n=2), rash (n=2), and 1 patient each with anemia, increased aspartate aminotransferase, asthenia, neutropenia, orthostatic hypotension, generalized rash, and maculopapular rash.
Serious AEs (SAEs) were minimal, Dr Forero-Torres said, and included 1 patient each with pyrexia, orthostatic hypotension, asthenia and rash, and deep vein thrombosis.
Seven patients discontinued treatment due to peripheral sensory neuropathy, 2 due to peripheral motor neuropathy, and 1 due to orthostatic hypotension.
Dr Forero-Torres emphasized that there were no grade 4 AEs, no AE-related deaths, and no deaths within 30 days of the last dose of medication.
Brentuximab plus dacarbazine
Fourteen of 18 patients in the combination arm were evaluable for efficacy and safety. Their median age was 72.5 (range, 62 to 87), 72% were male, 67% had an ECOG status of grade 0 or 1, and 56% had normal renal function with a creatinine clearance greater than 80 mL/min.
Forty-four percent exhibited B symptoms, 11% had bulky disease, and 50% had extra-nodal involvement.
They received brentuximab at 1.8 mg/kg plus dacarbazine at 375 mg/m2 for cycles 1-12, followed by monotherapy for cycles 13-16.
At the time of the interim analysis, 83% of patients were still on treatment, “so this is very early preliminary data,” Dr Forero-Torres noted.
All of the patients achieved tumor reduction, and 4 patients achieved a CR.
They had a median treatment duration of 16.7 weeks (range, 3 to 36), received a median of 5.5 cycles (range, 1 to 12), and had a median follow-up time of 19.1 weeks (range, 6.1 to 36.1).
The most common grade 1 or 2 AEs were peripheral sensory neuropathy (33%), nausea (33%), diarrhea (28%), constipation (28%), fatigue (22%), alopecia (22%), arthralgia (22%), and headache (22%).
Grade 3 AEs or SAEs, with 1 patient each, were C difficile colitis (SAE), hypotension (SAE), and hyperglycemia.
Dr Forero-Torres noted that investigators observed “robust antitumor activity” among these older patients receiving front-line brentuximab.
The cohort combining brentuximab with bendamustine is currently enrolling patients.
The study is sponsored by Seattle Genetics, Inc., developer of brentuximab vedotin (Adcetris).![]()
*Information in the abstract differs from that presented at the meeting.
Credit: NIH
SAN FRANCISCO—Younger patients with Hodgkin lymphoma fare well on brentuximab vedotin, experiencing an overall objective response rate (ORR) of 75% and a complete response (CR) rate of 34% in the pivotal phase 2 study of patients with relapsed/refractory disease.
And a retrospective study of patients older than 60 years showed that single-agent therapy was well tolerated, prompting an ORR of 53% and a CR rate of 40% in a relapsed or refractory population.
So investigators decided to explore in a prospective study whether patients 60 years or older could benefit from up-front treatment with brentuximab as a single agent or in combination.
Andres Forero-Torres, MD, of the University of Alabama in Birmingham, presented the results of this trial at the 2014 ASH Annual Meeting (abstract 294).*
Enrolled patients had classic Hodgkin lymphoma, were treatment-naïve, and were ineligible for or declined conventional front-line treatment. The primary endpoint was ORR.
The study is being conducted in 3 parts—brentuximab as a single agent, brentuximab plus dacarbazine, and brentuximab plus bendamustine. At the time of the ASH presentation, data for the brentuximab-bendamustine combination were not available.
Single-agent brentuximab
Twenty-seven patients on the single-agent arm were evaluable for efficacy and safety. They were a median age of 78 (range, 64 to 92). About half (52%) were male, and 78% had an ECOG performance status of grade 0 or 1.
Forty-four percent had moderate renal function impairment with a creatinine clearance between 30 and 60 mL/min. Thirty percent had B symptoms, 22% had bulky disease, and 52% had extra-nodal involvement.
Patients received 1.8 mg/kg of brentuximab intravenously on day 1 of a 21-day cycle. Response was assessed by CT scan during cycles 2, 4, 8, and 16, and by CT plus PET scan during cycles 2 and 8.
The median follow-up was 8.7 months. Dr Forero-Torres pointed out that, initially, “there were no progressions,” and all patients achieved tumor reduction.
The ORR was 93%, the CR rate was 70%, the partial response rate was 22%, and the rate of stable disease was 7%.
The median duration of response was 9.1 months (range, 0.03 to 13.14), and the median progression-free survival was 10.5 months (range, 2.6 to 14.3). For patients who had a CR, the median progression-free survival was about 12 months, Dr Forero-Torres said.
The median number of treatment cycles administered per patient was 8 (range, 3 to 23). Patients discontinued treatment primarily because of progressive disease (41%) or adverse events (AEs, 37%).
AEs occurring in 20% or more of patients were constipation, decreased appetite, diarrhea, peripheral edema, nausea, fatigue, and peripheral sensory neuropathy. All were grade 1 or 2, except for peripheral sensory neuropathy, which also had about 20% grade 3 events.
Grade 3 or higher treatment-related AEs included peripheral sensory neuropathy (n=7), peripheral motor neuropathy (n=2), rash (n=2), and 1 patient each with anemia, increased aspartate aminotransferase, asthenia, neutropenia, orthostatic hypotension, generalized rash, and maculopapular rash.
Serious AEs (SAEs) were minimal, Dr Forero-Torres said, and included 1 patient each with pyrexia, orthostatic hypotension, asthenia and rash, and deep vein thrombosis.
Seven patients discontinued treatment due to peripheral sensory neuropathy, 2 due to peripheral motor neuropathy, and 1 due to orthostatic hypotension.
Dr Forero-Torres emphasized that there were no grade 4 AEs, no AE-related deaths, and no deaths within 30 days of the last dose of medication.
Brentuximab plus dacarbazine
Fourteen of 18 patients in the combination arm were evaluable for efficacy and safety. Their median age was 72.5 (range, 62 to 87), 72% were male, 67% had an ECOG status of grade 0 or 1, and 56% had normal renal function with a creatinine clearance greater than 80 mL/min.
Forty-four percent exhibited B symptoms, 11% had bulky disease, and 50% had extra-nodal involvement.
They received brentuximab at 1.8 mg/kg plus dacarbazine at 375 mg/m2 for cycles 1-12, followed by monotherapy for cycles 13-16.
At the time of the interim analysis, 83% of patients were still on treatment, “so this is very early preliminary data,” Dr Forero-Torres noted.
All of the patients achieved tumor reduction, and 4 patients achieved a CR.
They had a median treatment duration of 16.7 weeks (range, 3 to 36), received a median of 5.5 cycles (range, 1 to 12), and had a median follow-up time of 19.1 weeks (range, 6.1 to 36.1).
The most common grade 1 or 2 AEs were peripheral sensory neuropathy (33%), nausea (33%), diarrhea (28%), constipation (28%), fatigue (22%), alopecia (22%), arthralgia (22%), and headache (22%).
Grade 3 AEs or SAEs, with 1 patient each, were C difficile colitis (SAE), hypotension (SAE), and hyperglycemia.
Dr Forero-Torres noted that investigators observed “robust antitumor activity” among these older patients receiving front-line brentuximab.
The cohort combining brentuximab with bendamustine is currently enrolling patients.
The study is sponsored by Seattle Genetics, Inc., developer of brentuximab vedotin (Adcetris).![]()
*Information in the abstract differs from that presented at the meeting.
Anti-CD38 antibodies poised to transform myeloma treatment
SAN FRANCISCO – Combining anti-CD38 monoclonal antibodies with standard antimyeloma therapies proved highly active without excessive toxicity in newly diagnosed as well as relapsed or refractory multiple myeloma in a pair of phase Ib studies.
“These anti-CD38 antibodies are the next blockbuster class of agents,” Dr. Thomas Martin III, lead author of one of the studies, said during a press briefing at the annual meeting of the American Society of Hematology. “These are the next agents that are really going to show some benefit for myeloma patients. And the next 5 years are going to be really fun moving them from the refractory setting to the less refractory setting to the frontline setting.”

Three agents are in development that target CD38, a cell surface glycoprotein that is strongly expressed in multiple myeloma. All three – daratumumab, SAR650984, and MOR202 – bind to a different part of the anti-CD39 receptor, but “whether that makes any clinical difference, we certainly don’t know at this point in time,” said Dr. Martin of the University of California, San Francisco.
Immunomodulatory drugs (IMiDs) and proteasome inhibitors (PI) are the current blockbuster agents in myeloma and have advanced overall survival from about 3 years to 7-10 years. But all patients still relapse after IMiD and PI failure, and survival outcomes remain poor at a median of about 9 months for those with advanced relapsed/refractory disease. Further, myeloma has failed to respond like the B-cell lymphomas to anti-CD20 antibodies such as rituximab (Rituxan).
Dr. Martin and his associates launched a phase Ib dose-escalation trial to evaluate SAR650984 in combination with standard doses of lenalidomide (Revlimid) and dexamethasone in adults with relapsed or refractory multiple myeloma failing at least two prior therapies.
SAR650984 was given intravenously on days 1 and 15 of a 28-day cycle at 3 mg/kg to 4 patients (cohort 1), 5 mg/kg to 3 patients (cohort 2), and 10 mg/kg to 6 patients (cohort 3) plus an additional 18 patients in an expansion cohort.
All 31 patients were heavily pretreated, with 94% previously treated with lenalidomide or bortezomib (Velcade), 29% with pomalidomide (Pomalyst), and 48% with carfilzomib (Kyprolis). Many (84%) were considered double refractory, or relapsed and refractory, to their last IMiD therapy, Dr. Martin reported.
Despite this, nearly two-thirds (58%) had a response, including two stringent complete responses, seven very good partial responses, and nine partial responses. The overall response rate (ORR) was 25% in cohort 1, 67% in cohort 2, and 63% at the 10-mg dose level or double what was seen as a single agent, he said. The clinical benefit rates were 50%, 67%, and 67%.
In addition, the ORR was 50% in patients who were IMiD relapsed and refractory and 33% in patients pomalidomide relapsed and refractory, he reported.
At 9 months’ follow-up, median progression-free survival was 6.2 months and had not been reached in patients who received fewer than three lines of prior therapy.
The most common treatment-related adverse events were infusion reactions, fatigue, nausea, upper respiratory tract infection, and diarrhea. Infusion reactions occurred about a third of the time, typically in the first or second cycle, and were mostly mild (grade 1 or 2), Dr. Martin said. Only two patients discontinued treatment because of infusion reactions, one for a serious grade 3 anaphylactic reaction in cycle 1 and the other for a nonserious grade 3 maculopapular rash in cycle 2.
Daratumumab
The second highlighted study looked at the benefit and safety of adding daratumumab to commonly used backbone regimens in patients with newly diagnosed, relapsed, or treatment-resistant myeloma.
Single-agent daratumumab has shown activity in prior studies and received breakthrough therapy designation in May 2013 for patients with multiple myeloma after at least three prior lines of therapy including a proteasome inhibitor and an IMiD or those double refractory to a PI and IMiD.
In the four-arm, open-label study, daratumumab was given at a starting dose of 16 mg/kg in combination with bortezomib-dexamethasone (VD), bortezomib-thalidomide-dexamethasone (VTD), bortezomib-melphalan-prednisone (VMP), and pomalidomide-dexamethasone (POM-D). Treatment was for 18 three-week cycles or until transplantation in the VD and VTD arms, for 9 six-week cycles in the VMP arm, and was given in four-week cycles until disease progression in the POM-D arm.
Newly diagnosed patients were included in the VD and VTD arms irrespective of transplant eligibility, while patients in the VMP arm were newly diagnosed and transplant ineligible. Patients in the POM-D arm were relapsed/refractory to two or more lines of therapy including two consecutive cycles of lenalidomide and bortezomib. In all, 24 patients were evaluable for efficacy.
The ORR in newly diagnosed patients was 100%, including partial responses and very good partial responses, and 50% in the relapsed group, including one complete response, study coauthor Dr. María-Victoria Mateos said at the briefing.

Daratumumab does not appear to have a negative effect on stem cell mobilization, with five patients electively taken off study for autologous stem cell transplantation after cycle 4.
Three of the seven patients in the POM-D arm dropped out (one because of physician decision and two because of disease progression).
The addition of daratumumab to the various backbones was well tolerated in all evaluable patients and did not result in significant additional toxicity, said Dr. Mateos of University Hospital of Salamanca, Spain.
Adverse events possibly or probably related to daratumumab included one grade 3 neutropenia in the VD arm, one grade 3 thrombocytopenia in the VMP arm, and one serious infectious pneumonia in the POM-D arm. There were a few infusion-related reactions, but most were grade 1 or 2 and occurred within the first infusions, she said.
“This is a very new and exciting concept in multiple myeloma, as we are seeing that combining this precision approach with the standard of care is leading to more effective treatment without increased toxicity,” Dr. Philippe Moreau, lead study author, of University Hospital of Nantes, France, said in a statement. “By targeting a simple molecule expressed by the cancer cells, this therapy has the potential to become a potent addition to conventional treatment.”
Dr. Brad S. Kahl, press briefing moderator, of the University of Wisconsin-Madison, was enthusiastic about the potential for anti-CD38 antibodies to transform the treatment of multiple myeloma.
“Obviously it’s very, very early, probably too early to plant a victory flag in the ground,” he said. “Having said that, the early data is very promising and totally justifies all the comments about bringing these drugs forward, aggressively moving them into the front line.”
Phase III studies are ongoing or will be initiated shortly with daratumumab plus VD in relapsed myeloma (MMY3004-CASTOR), with VMP in non–transplant eligible patients (MMY3007-ALCYONE), and with VTD as induction therapy (MMY3006/IFM-HOVON-CASSIOPEIA).
SAN FRANCISCO – Combining anti-CD38 monoclonal antibodies with standard antimyeloma therapies proved highly active without excessive toxicity in newly diagnosed as well as relapsed or refractory multiple myeloma in a pair of phase Ib studies.
“These anti-CD38 antibodies are the next blockbuster class of agents,” Dr. Thomas Martin III, lead author of one of the studies, said during a press briefing at the annual meeting of the American Society of Hematology. “These are the next agents that are really going to show some benefit for myeloma patients. And the next 5 years are going to be really fun moving them from the refractory setting to the less refractory setting to the frontline setting.”
Three agents are in development that target CD38, a cell surface glycoprotein that is strongly expressed in multiple myeloma. All three – daratumumab, SAR650984, and MOR202 – bind to a different part of the anti-CD39 receptor, but “whether that makes any clinical difference, we certainly don’t know at this point in time,” said Dr. Martin of the University of California, San Francisco.
Immunomodulatory drugs (IMiDs) and proteasome inhibitors (PI) are the current blockbuster agents in myeloma and have advanced overall survival from about 3 years to 7-10 years. But all patients still relapse after IMiD and PI failure, and survival outcomes remain poor at a median of about 9 months for those with advanced relapsed/refractory disease. Further, myeloma has failed to respond like the B-cell lymphomas to anti-CD20 antibodies such as rituximab (Rituxan).
Dr. Martin and his associates launched a phase Ib dose-escalation trial to evaluate SAR650984 in combination with standard doses of lenalidomide (Revlimid) and dexamethasone in adults with relapsed or refractory multiple myeloma failing at least two prior therapies.
SAR650984 was given intravenously on days 1 and 15 of a 28-day cycle at 3 mg/kg to 4 patients (cohort 1), 5 mg/kg to 3 patients (cohort 2), and 10 mg/kg to 6 patients (cohort 3) plus an additional 18 patients in an expansion cohort.
All 31 patients were heavily pretreated, with 94% previously treated with lenalidomide or bortezomib (Velcade), 29% with pomalidomide (Pomalyst), and 48% with carfilzomib (Kyprolis). Many (84%) were considered double refractory, or relapsed and refractory, to their last IMiD therapy, Dr. Martin reported.
Despite this, nearly two-thirds (58%) had a response, including two stringent complete responses, seven very good partial responses, and nine partial responses. The overall response rate (ORR) was 25% in cohort 1, 67% in cohort 2, and 63% at the 10-mg dose level or double what was seen as a single agent, he said. The clinical benefit rates were 50%, 67%, and 67%.
In addition, the ORR was 50% in patients who were IMiD relapsed and refractory and 33% in patients pomalidomide relapsed and refractory, he reported.
At 9 months’ follow-up, median progression-free survival was 6.2 months and had not been reached in patients who received fewer than three lines of prior therapy.
The most common treatment-related adverse events were infusion reactions, fatigue, nausea, upper respiratory tract infection, and diarrhea. Infusion reactions occurred about a third of the time, typically in the first or second cycle, and were mostly mild (grade 1 or 2), Dr. Martin said. Only two patients discontinued treatment because of infusion reactions, one for a serious grade 3 anaphylactic reaction in cycle 1 and the other for a nonserious grade 3 maculopapular rash in cycle 2.
Daratumumab
The second highlighted study looked at the benefit and safety of adding daratumumab to commonly used backbone regimens in patients with newly diagnosed, relapsed, or treatment-resistant myeloma.
Single-agent daratumumab has shown activity in prior studies and received breakthrough therapy designation in May 2013 for patients with multiple myeloma after at least three prior lines of therapy including a proteasome inhibitor and an IMiD or those double refractory to a PI and IMiD.
In the four-arm, open-label study, daratumumab was given at a starting dose of 16 mg/kg in combination with bortezomib-dexamethasone (VD), bortezomib-thalidomide-dexamethasone (VTD), bortezomib-melphalan-prednisone (VMP), and pomalidomide-dexamethasone (POM-D). Treatment was for 18 three-week cycles or until transplantation in the VD and VTD arms, for 9 six-week cycles in the VMP arm, and was given in four-week cycles until disease progression in the POM-D arm.
Newly diagnosed patients were included in the VD and VTD arms irrespective of transplant eligibility, while patients in the VMP arm were newly diagnosed and transplant ineligible. Patients in the POM-D arm were relapsed/refractory to two or more lines of therapy including two consecutive cycles of lenalidomide and bortezomib. In all, 24 patients were evaluable for efficacy.
The ORR in newly diagnosed patients was 100%, including partial responses and very good partial responses, and 50% in the relapsed group, including one complete response, study coauthor Dr. María-Victoria Mateos said at the briefing.
Daratumumab does not appear to have a negative effect on stem cell mobilization, with five patients electively taken off study for autologous stem cell transplantation after cycle 4.
Three of the seven patients in the POM-D arm dropped out (one because of physician decision and two because of disease progression).
The addition of daratumumab to the various backbones was well tolerated in all evaluable patients and did not result in significant additional toxicity, said Dr. Mateos of University Hospital of Salamanca, Spain.
Adverse events possibly or probably related to daratumumab included one grade 3 neutropenia in the VD arm, one grade 3 thrombocytopenia in the VMP arm, and one serious infectious pneumonia in the POM-D arm. There were a few infusion-related reactions, but most were grade 1 or 2 and occurred within the first infusions, she said.
“This is a very new and exciting concept in multiple myeloma, as we are seeing that combining this precision approach with the standard of care is leading to more effective treatment without increased toxicity,” Dr. Philippe Moreau, lead study author, of University Hospital of Nantes, France, said in a statement. “By targeting a simple molecule expressed by the cancer cells, this therapy has the potential to become a potent addition to conventional treatment.”
Dr. Brad S. Kahl, press briefing moderator, of the University of Wisconsin-Madison, was enthusiastic about the potential for anti-CD38 antibodies to transform the treatment of multiple myeloma.
“Obviously it’s very, very early, probably too early to plant a victory flag in the ground,” he said. “Having said that, the early data is very promising and totally justifies all the comments about bringing these drugs forward, aggressively moving them into the front line.”
Phase III studies are ongoing or will be initiated shortly with daratumumab plus VD in relapsed myeloma (MMY3004-CASTOR), with VMP in non–transplant eligible patients (MMY3007-ALCYONE), and with VTD as induction therapy (MMY3006/IFM-HOVON-CASSIOPEIA).
SAN FRANCISCO – Combining anti-CD38 monoclonal antibodies with standard antimyeloma therapies proved highly active without excessive toxicity in newly diagnosed as well as relapsed or refractory multiple myeloma in a pair of phase Ib studies.
“These anti-CD38 antibodies are the next blockbuster class of agents,” Dr. Thomas Martin III, lead author of one of the studies, said during a press briefing at the annual meeting of the American Society of Hematology. “These are the next agents that are really going to show some benefit for myeloma patients. And the next 5 years are going to be really fun moving them from the refractory setting to the less refractory setting to the frontline setting.”
Three agents are in development that target CD38, a cell surface glycoprotein that is strongly expressed in multiple myeloma. All three – daratumumab, SAR650984, and MOR202 – bind to a different part of the anti-CD39 receptor, but “whether that makes any clinical difference, we certainly don’t know at this point in time,” said Dr. Martin of the University of California, San Francisco.
Immunomodulatory drugs (IMiDs) and proteasome inhibitors (PI) are the current blockbuster agents in myeloma and have advanced overall survival from about 3 years to 7-10 years. But all patients still relapse after IMiD and PI failure, and survival outcomes remain poor at a median of about 9 months for those with advanced relapsed/refractory disease. Further, myeloma has failed to respond like the B-cell lymphomas to anti-CD20 antibodies such as rituximab (Rituxan).
Dr. Martin and his associates launched a phase Ib dose-escalation trial to evaluate SAR650984 in combination with standard doses of lenalidomide (Revlimid) and dexamethasone in adults with relapsed or refractory multiple myeloma failing at least two prior therapies.
SAR650984 was given intravenously on days 1 and 15 of a 28-day cycle at 3 mg/kg to 4 patients (cohort 1), 5 mg/kg to 3 patients (cohort 2), and 10 mg/kg to 6 patients (cohort 3) plus an additional 18 patients in an expansion cohort.
All 31 patients were heavily pretreated, with 94% previously treated with lenalidomide or bortezomib (Velcade), 29% with pomalidomide (Pomalyst), and 48% with carfilzomib (Kyprolis). Many (84%) were considered double refractory, or relapsed and refractory, to their last IMiD therapy, Dr. Martin reported.
Despite this, nearly two-thirds (58%) had a response, including two stringent complete responses, seven very good partial responses, and nine partial responses. The overall response rate (ORR) was 25% in cohort 1, 67% in cohort 2, and 63% at the 10-mg dose level or double what was seen as a single agent, he said. The clinical benefit rates were 50%, 67%, and 67%.
In addition, the ORR was 50% in patients who were IMiD relapsed and refractory and 33% in patients pomalidomide relapsed and refractory, he reported.
At 9 months’ follow-up, median progression-free survival was 6.2 months and had not been reached in patients who received fewer than three lines of prior therapy.
The most common treatment-related adverse events were infusion reactions, fatigue, nausea, upper respiratory tract infection, and diarrhea. Infusion reactions occurred about a third of the time, typically in the first or second cycle, and were mostly mild (grade 1 or 2), Dr. Martin said. Only two patients discontinued treatment because of infusion reactions, one for a serious grade 3 anaphylactic reaction in cycle 1 and the other for a nonserious grade 3 maculopapular rash in cycle 2.
Daratumumab
The second highlighted study looked at the benefit and safety of adding daratumumab to commonly used backbone regimens in patients with newly diagnosed, relapsed, or treatment-resistant myeloma.
Single-agent daratumumab has shown activity in prior studies and received breakthrough therapy designation in May 2013 for patients with multiple myeloma after at least three prior lines of therapy including a proteasome inhibitor and an IMiD or those double refractory to a PI and IMiD.
In the four-arm, open-label study, daratumumab was given at a starting dose of 16 mg/kg in combination with bortezomib-dexamethasone (VD), bortezomib-thalidomide-dexamethasone (VTD), bortezomib-melphalan-prednisone (VMP), and pomalidomide-dexamethasone (POM-D). Treatment was for 18 three-week cycles or until transplantation in the VD and VTD arms, for 9 six-week cycles in the VMP arm, and was given in four-week cycles until disease progression in the POM-D arm.
Newly diagnosed patients were included in the VD and VTD arms irrespective of transplant eligibility, while patients in the VMP arm were newly diagnosed and transplant ineligible. Patients in the POM-D arm were relapsed/refractory to two or more lines of therapy including two consecutive cycles of lenalidomide and bortezomib. In all, 24 patients were evaluable for efficacy.
The ORR in newly diagnosed patients was 100%, including partial responses and very good partial responses, and 50% in the relapsed group, including one complete response, study coauthor Dr. María-Victoria Mateos said at the briefing.
Daratumumab does not appear to have a negative effect on stem cell mobilization, with five patients electively taken off study for autologous stem cell transplantation after cycle 4.
Three of the seven patients in the POM-D arm dropped out (one because of physician decision and two because of disease progression).
The addition of daratumumab to the various backbones was well tolerated in all evaluable patients and did not result in significant additional toxicity, said Dr. Mateos of University Hospital of Salamanca, Spain.
Adverse events possibly or probably related to daratumumab included one grade 3 neutropenia in the VD arm, one grade 3 thrombocytopenia in the VMP arm, and one serious infectious pneumonia in the POM-D arm. There were a few infusion-related reactions, but most were grade 1 or 2 and occurred within the first infusions, she said.
“This is a very new and exciting concept in multiple myeloma, as we are seeing that combining this precision approach with the standard of care is leading to more effective treatment without increased toxicity,” Dr. Philippe Moreau, lead study author, of University Hospital of Nantes, France, said in a statement. “By targeting a simple molecule expressed by the cancer cells, this therapy has the potential to become a potent addition to conventional treatment.”
Dr. Brad S. Kahl, press briefing moderator, of the University of Wisconsin-Madison, was enthusiastic about the potential for anti-CD38 antibodies to transform the treatment of multiple myeloma.
“Obviously it’s very, very early, probably too early to plant a victory flag in the ground,” he said. “Having said that, the early data is very promising and totally justifies all the comments about bringing these drugs forward, aggressively moving them into the front line.”
Phase III studies are ongoing or will be initiated shortly with daratumumab plus VD in relapsed myeloma (MMY3004-CASTOR), with VMP in non–transplant eligible patients (MMY3007-ALCYONE), and with VTD as induction therapy (MMY3006/IFM-HOVON-CASSIOPEIA).
AT ASH 2014
Key clinical point: The investigational anti-CD38 antibodies SAR650984 and daratumumab in combination with standard regimens were highly active in untreated and relapsed/refractory multiple myeloma.
Major finding: Overall response rates were 58% in patients treated with SAR650984, and 100% and 50%, respectively, in newly diagnosed and relapsed patients given daratumumab.
Data source: Two phase Ib studies in patients with newly diagnosed or relapsed/refractory multiple myeloma.
Disclosures: Sanofi Oncology sponsored the SAR650984 study. Dr. Martin reported research funding from Sanofi and serving as a speaker for Novartis. Genmab sponsored the daratumumab study. Dr. Mateos and Dr. Moreau reported ties with Janssen. Dr. Kahl reported ties with numerous drug companies.

CARs come in different makes and models

Credit: NIAID
SAN FRANCISCO—CTL019, a chimeric antigen receptor (CAR) T cell targeting CD19, is not the only CAR in the production line.
Investigators at the National Cancer Institute in Bethesda, Maryland, and Memorial Sloan Kettering Cancer Center (MSKCC) in New York City are also pursuing CAR T-cell therapy.
These groups are using a retroviral platform to transduce the T cells rather than a lentiviral one, as is the case with CTL019.
Investigators reported progress to date on these makes of CARs at the 2014 ASH Annual Meeting.
Daniel W. Lee III, MD, of the National Cancer Institute, reported on a phase 1 study of CD19 CAR T cells in children and young adults with CD19+ acute lymphoblastic leukemia or non-Hodgkin lymphoma.
And Jae H. Park, MD, of MSKCC, presented data from a trial of JCAR015—autologous T cells genetically modified to express a 19-28z CAR targeting CD19—in patients with B-cell acute lymphoblastic leukemia.
The study is sponsored by MSKCC, but JCAR015 is a product of Juno Therapeutics. ![]()
Credit: NIAID
SAN FRANCISCO—CTL019, a chimeric antigen receptor (CAR) T cell targeting CD19, is not the only CAR in the production line.
Investigators at the National Cancer Institute in Bethesda, Maryland, and Memorial Sloan Kettering Cancer Center (MSKCC) in New York City are also pursuing CAR T-cell therapy.
These groups are using a retroviral platform to transduce the T cells rather than a lentiviral one, as is the case with CTL019.
Investigators reported progress to date on these makes of CARs at the 2014 ASH Annual Meeting.
Daniel W. Lee III, MD, of the National Cancer Institute, reported on a phase 1 study of CD19 CAR T cells in children and young adults with CD19+ acute lymphoblastic leukemia or non-Hodgkin lymphoma.
And Jae H. Park, MD, of MSKCC, presented data from a trial of JCAR015—autologous T cells genetically modified to express a 19-28z CAR targeting CD19—in patients with B-cell acute lymphoblastic leukemia.
The study is sponsored by MSKCC, but JCAR015 is a product of Juno Therapeutics. ![]()
Credit: NIAID
SAN FRANCISCO—CTL019, a chimeric antigen receptor (CAR) T cell targeting CD19, is not the only CAR in the production line.
Investigators at the National Cancer Institute in Bethesda, Maryland, and Memorial Sloan Kettering Cancer Center (MSKCC) in New York City are also pursuing CAR T-cell therapy.
These groups are using a retroviral platform to transduce the T cells rather than a lentiviral one, as is the case with CTL019.
Investigators reported progress to date on these makes of CARs at the 2014 ASH Annual Meeting.
Daniel W. Lee III, MD, of the National Cancer Institute, reported on a phase 1 study of CD19 CAR T cells in children and young adults with CD19+ acute lymphoblastic leukemia or non-Hodgkin lymphoma.
And Jae H. Park, MD, of MSKCC, presented data from a trial of JCAR015—autologous T cells genetically modified to express a 19-28z CAR targeting CD19—in patients with B-cell acute lymphoblastic leukemia.
The study is sponsored by MSKCC, but JCAR015 is a product of Juno Therapeutics. ![]()
CAR is feasible in majority of ALL patients, team says

Credit: Bill Branson
SAN FRANCISCO—A chimeric antigen receptor (CAR) T-cell therapy is feasible in 90% of heavily pretreated or transplanted patients with acute lymphoblastic leukemia (ALL) and can serve as a bridge to transplant, according to investigators.
Daniel W. Lee III, MD, of the National Cancer Institute in Bethesda, Maryland, reported on a phase 1 study of this CD19 CAR T-cell therapy in children and young adults with CD19+ ALL or non-Hodgkin lymphoma at the 2014 ASH Annual Meeting (abstract 381*).
Twenty-one patients were enrolled on the trial. They had a preparative regimen of fludarabine and cyclophosphamide and were infused with CAR T cells 11 days after the peripheral blood mononuclear cells were collected.
Dose levels were 1 x 106 CAR+ T cells/kg, 3 x 106 CAR+ T cells/kg, or the maximum number of cells generated if below either one of these levels. Two patients received less than the dose assigned and were not evaluated for toxicity.
Patients were a median age of 13 years (range, 5 to 27). Fourteen were male, 20 had ALL, and 1 had diffuse large B-cell lymphoma.
All had detectable disease, and 2 were CNS2 at the time of T-cell infusion. Six had primary refractory disease, 8 had at least 1 prior stem cell transplant, and 4 had prior immunotherapy.
The investigators determined that the maximally tolerated dose was 1 x 106 CAR+ T cells/kg. The dose-limiting toxicities were related to cytokine release syndrome (CRS), which was reversible if managed appropriately with tocilizumab, with or without steroids.
Grade 3 adverse events possibly related to therapy included fever (47%), febrile neutropenia (37%), electrolyte disturbance (29%), CRS (16%), hypotension (11%), transaminitis (16%), and 5% each for hypertension, prolonged QTc, dysphasia, LV systolic dysfunction, multiorgan failure, hypoxia, and pulmonary edema.
Grade 4 events possibly related to treatment included electrolyte disturbance (5%), CRS (16%), hypotension (11%), cardiac arrest (5%), and hypoxia (5%). There was no evidence of graft-vs-host disease.
The complete response (CR) rate was 67% in the intent-to-treat population and 70% in patients with ALL.
“Those patients who responded tended to have some degree of cytokine release syndrome, whereas those patients who did not respond or had stable disease did not have any CRS,” Dr Lee said. “But, also, it’s important to note that you don’t have to have severe grade 3 or grade 4 CRS in order to have significant response.”
Dr Lee also pointed out that in vivo CAR T-cell expansion significantly correlated with response (P=0.0028). And CRS severity correlated with IL-6 (P=0.0002), INF-γ (P=0.0002), C-reactive protein (P=0.0015), and CAR (P=0.0011).
At a median follow-up of 10 months, minimal residual disease-negative patients had a 79% leukemia-free survival. Overall survival was 52% for all patients enrolled. Two patients had CD19-negative relapses.
The investigators also found that CAR T cells can eliminate CNS leukemia, with 11 of 17 patients (65%) having CAR T cells detectable in their cerebrospinal fluid.
The team concluded that this therapy is feasible in 90% of heavily pretreated or transplanted ALL patients and can serve as a bridge to transplant. ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Bill Branson
SAN FRANCISCO—A chimeric antigen receptor (CAR) T-cell therapy is feasible in 90% of heavily pretreated or transplanted patients with acute lymphoblastic leukemia (ALL) and can serve as a bridge to transplant, according to investigators.
Daniel W. Lee III, MD, of the National Cancer Institute in Bethesda, Maryland, reported on a phase 1 study of this CD19 CAR T-cell therapy in children and young adults with CD19+ ALL or non-Hodgkin lymphoma at the 2014 ASH Annual Meeting (abstract 381*).
Twenty-one patients were enrolled on the trial. They had a preparative regimen of fludarabine and cyclophosphamide and were infused with CAR T cells 11 days after the peripheral blood mononuclear cells were collected.
Dose levels were 1 x 106 CAR+ T cells/kg, 3 x 106 CAR+ T cells/kg, or the maximum number of cells generated if below either one of these levels. Two patients received less than the dose assigned and were not evaluated for toxicity.
Patients were a median age of 13 years (range, 5 to 27). Fourteen were male, 20 had ALL, and 1 had diffuse large B-cell lymphoma.
All had detectable disease, and 2 were CNS2 at the time of T-cell infusion. Six had primary refractory disease, 8 had at least 1 prior stem cell transplant, and 4 had prior immunotherapy.
The investigators determined that the maximally tolerated dose was 1 x 106 CAR+ T cells/kg. The dose-limiting toxicities were related to cytokine release syndrome (CRS), which was reversible if managed appropriately with tocilizumab, with or without steroids.
Grade 3 adverse events possibly related to therapy included fever (47%), febrile neutropenia (37%), electrolyte disturbance (29%), CRS (16%), hypotension (11%), transaminitis (16%), and 5% each for hypertension, prolonged QTc, dysphasia, LV systolic dysfunction, multiorgan failure, hypoxia, and pulmonary edema.
Grade 4 events possibly related to treatment included electrolyte disturbance (5%), CRS (16%), hypotension (11%), cardiac arrest (5%), and hypoxia (5%). There was no evidence of graft-vs-host disease.
The complete response (CR) rate was 67% in the intent-to-treat population and 70% in patients with ALL.
“Those patients who responded tended to have some degree of cytokine release syndrome, whereas those patients who did not respond or had stable disease did not have any CRS,” Dr Lee said. “But, also, it’s important to note that you don’t have to have severe grade 3 or grade 4 CRS in order to have significant response.”
Dr Lee also pointed out that in vivo CAR T-cell expansion significantly correlated with response (P=0.0028). And CRS severity correlated with IL-6 (P=0.0002), INF-γ (P=0.0002), C-reactive protein (P=0.0015), and CAR (P=0.0011).
At a median follow-up of 10 months, minimal residual disease-negative patients had a 79% leukemia-free survival. Overall survival was 52% for all patients enrolled. Two patients had CD19-negative relapses.
The investigators also found that CAR T cells can eliminate CNS leukemia, with 11 of 17 patients (65%) having CAR T cells detectable in their cerebrospinal fluid.
The team concluded that this therapy is feasible in 90% of heavily pretreated or transplanted ALL patients and can serve as a bridge to transplant. ![]()
*Information in the abstract differs from that presented at the meeting.
Credit: Bill Branson
SAN FRANCISCO—A chimeric antigen receptor (CAR) T-cell therapy is feasible in 90% of heavily pretreated or transplanted patients with acute lymphoblastic leukemia (ALL) and can serve as a bridge to transplant, according to investigators.
Daniel W. Lee III, MD, of the National Cancer Institute in Bethesda, Maryland, reported on a phase 1 study of this CD19 CAR T-cell therapy in children and young adults with CD19+ ALL or non-Hodgkin lymphoma at the 2014 ASH Annual Meeting (abstract 381*).
Twenty-one patients were enrolled on the trial. They had a preparative regimen of fludarabine and cyclophosphamide and were infused with CAR T cells 11 days after the peripheral blood mononuclear cells were collected.
Dose levels were 1 x 106 CAR+ T cells/kg, 3 x 106 CAR+ T cells/kg, or the maximum number of cells generated if below either one of these levels. Two patients received less than the dose assigned and were not evaluated for toxicity.
Patients were a median age of 13 years (range, 5 to 27). Fourteen were male, 20 had ALL, and 1 had diffuse large B-cell lymphoma.
All had detectable disease, and 2 were CNS2 at the time of T-cell infusion. Six had primary refractory disease, 8 had at least 1 prior stem cell transplant, and 4 had prior immunotherapy.
The investigators determined that the maximally tolerated dose was 1 x 106 CAR+ T cells/kg. The dose-limiting toxicities were related to cytokine release syndrome (CRS), which was reversible if managed appropriately with tocilizumab, with or without steroids.
Grade 3 adverse events possibly related to therapy included fever (47%), febrile neutropenia (37%), electrolyte disturbance (29%), CRS (16%), hypotension (11%), transaminitis (16%), and 5% each for hypertension, prolonged QTc, dysphasia, LV systolic dysfunction, multiorgan failure, hypoxia, and pulmonary edema.
Grade 4 events possibly related to treatment included electrolyte disturbance (5%), CRS (16%), hypotension (11%), cardiac arrest (5%), and hypoxia (5%). There was no evidence of graft-vs-host disease.
The complete response (CR) rate was 67% in the intent-to-treat population and 70% in patients with ALL.
“Those patients who responded tended to have some degree of cytokine release syndrome, whereas those patients who did not respond or had stable disease did not have any CRS,” Dr Lee said. “But, also, it’s important to note that you don’t have to have severe grade 3 or grade 4 CRS in order to have significant response.”
Dr Lee also pointed out that in vivo CAR T-cell expansion significantly correlated with response (P=0.0028). And CRS severity correlated with IL-6 (P=0.0002), INF-γ (P=0.0002), C-reactive protein (P=0.0015), and CAR (P=0.0011).
At a median follow-up of 10 months, minimal residual disease-negative patients had a 79% leukemia-free survival. Overall survival was 52% for all patients enrolled. Two patients had CD19-negative relapses.
The investigators also found that CAR T cells can eliminate CNS leukemia, with 11 of 17 patients (65%) having CAR T cells detectable in their cerebrospinal fluid.
The team concluded that this therapy is feasible in 90% of heavily pretreated or transplanted ALL patients and can serve as a bridge to transplant.
*Information in the abstract differs from that presented at the meeting.