User login
Hereditary Hemochromatosis Linked to Increased Arthropathies and Joint Surgery
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.

Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
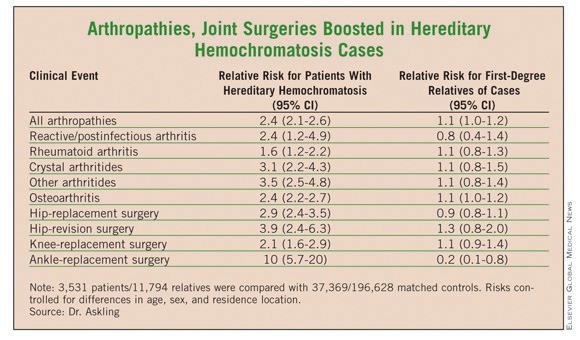
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.
Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.
Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Patients with hereditary hemochromatosis had a 2.4-fold increased prevalence of arthropathies and a 2.9-fold increased rate of hip surgery, compared with matched controls. In contrast, first-degree relatives of hereditary hemochromatosis patients had no significantly different rates for arthropathy or need for joint-replacement surgery, compared with controls.
Data Source: Case-control study of records from Swedish national registries during 1999-2006, with 3,531 patients diagnosed with hereditary hemochromatosis and 37,369 matched controls, and 11,794 first-degree relatives of the patients and 196,628 matched controls.
Disclosures: Dr. Askling said that he had no disclosures.
Hereditary Hemochromatosis Linked to Increased Arthropathies and Joint Surgery
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.
Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.
Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
LONDON – Patients with hereditary hemochromatosis have a significantly increased prevalence of various arthropathies and an elevated need for joint-replacement surgery, compared with the general population, according to findings from a study of Swedish national registry data.
The analysis also showed that first-degree relatives of people with hereditary hemochromatosis do not have an increased rate of arthropathies or need for joint replacement, even though genetic models predict that a majority of these relatives carry one copy of an autosomal recessive mutation that causes hereditary hemochromatosis.
"This dissociation between the genotype and the phenotype" relative to the risk for arthropathy and need for joint replacement "suggests to me that the gene itself is not involved. It suggests to me that you need more than just the gene" to boost the risk for arthropathy and joint failure, noted Dr. Johan Askling.
Arthropathy is a classic phenotypic feature of patients with hereditary hemochromatosis, a genetic disease in people who carry two mutated copies of the hemochromatosis gene (HFE) associated with iron overload. But the nature of the relationship between the disease and arthropathies remains poorly understood. The new finding that increased arthropathies occur only in homozygous, affected individuals suggests that the risk is linked to iron overload itself, rather than to the causative mutated genes.
Dr. Askling and his associates identified 3,531 patients with a diagnosis of hereditary hemochromatosis from Swedish national records for the period 1999-2006. The investigators also identified another 11,794 first-degree relatives of these patients. They then identified 37,369 people as matched controls for the patients from the general Swedish population and 196,628 people as matched controls for the first-degree relatives.
The researchers then tallied the incidence of consultations or hospitalizations for rheumatoid arthritis, osteoarthritis, and other arthritides in the cases, their first-degree relatives, and the controls during the study period. They calculated a relative risk for these complications in affected people and in their relatives, compared with the controls that adjusted for differences in age, sex, and residence location. The researchers also ran similar analyses for the incidence of various joint-replacement surgeries (see table).
The results showed that the patients with hereditary hemochromatosis had consistent, statistically significant increased rates of arthropathies. For example, for all arthropathies the rate was 2.4-fold higher in the patients than in the controls. But this increased rate did not exist among the first-degree relatives. For all arthropathies, their rate was just 10% higher than among the matched controls, a difference that was not significant, reported Dr. Askling, an epidemiologist at the Karolinska Institute in Stockholm.
A similar pattern existed for joint-replacement surgeries. Hip surgery, for example, was 2.9-fold more frequent among the hereditary hemochromatosis patients than among their matched controls, while among the first-degree relatives the incidence of hip-replacement surgery was 10% less common than among the matched controls, a difference that was not statistically significant.
Dr. Askling said that he had no disclosures.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Patients with hereditary hemochromatosis had a 2.4-fold increased prevalence of arthropathies and a 2.9-fold increased rate of hip surgery, compared with matched controls. In contrast, first-degree relatives of hereditary hemochromatosis patients had no significantly different rates for arthropathy or need for joint-replacement surgery, compared with controls.
Data Source: Case-control study of records from Swedish national registries during 1999-2006, with 3,531 patients diagnosed with hereditary hemochromatosis and 37,369 matched controls, and 11,794 first-degree relatives of the patients and 196,628 matched controls.
Disclosures: Dr. Askling said that he had no disclosures.
Tocilizumab Continues to Benefit sJIA Patients Over Time
LONDON – Children with systemic juvenile idiopathic arthritis continued to improve on treatment with the interleukin-6 inhibitor tocilizumab as their time on the drug extended out to 1 year, in follow-up of the open-label phase of the drug’s pivotal trial for this disease.
During continued tocilizumab treatment, the percent of systemic juvenile idiopathic arthritis (sJIA) patients who had an American College of Rheumatology (ACR) 90 response and no fever rose from 37% of the treated group after the end of the 12-week randomized trial, to about 55% after an additional 40 weeks of open-label treatment. There was no active joint disease in 49 of the 99 patients (49%) treated for 52 weeks; the children appeared to be in full remission, Dr. Fabrizio De Benedetti said at the meeting. In addition, 52 of the patients treated for 52 weeks fully withdrew from treatment with oral corticosteroids. At their entry onto tocilizumab treatment, their average corticosteroid dosage was 0.30 mg/kg per day.

Over 52 weeks, tocilizumab treatment also had an "acceptable" safety profile. Thirteen patients had a serious adverse event, and 6 had an adverse event leading to withdrawal from treatment. "There was no increase in the rate of serious adverse events between weeks 12 and 52. It is reassuring that there was no accumulation of safety issues with time. But it’s still only 1 year, so we still need more time" to fully assess safety, said Dr. De Benedetti, who is director of the division of rheumatology at Ospedale Pediatrico Bambino Gesù in Rome and lead investigator for the TENDER trial and extension phase.
Based on the results from the 12-week randomized phase III trial, Roche – the company that developed and markets tocilizumab (Actemra) – in April received approval from the Food and Drug Administration for an expanded indication for tocilizumab to treat sJIA. Tocilizumab received FDA approval last year for treatment of rheumatoid arthritis.
Tocilizumab "is very effective; it’s the best drug we have for sJIA. But the experience we have using it is still short" Dr. Patricia Woo, a collaborator on the TENDER trial, said in an interview. "We have 1-year experience, compared with [potentially] using it for many years" to treat children with sJIA. The next phase of the TENDER protocol is to attempt gradually to wean off the drug those children who achieve complete remission on tocilizumab for 6 months to see if they can remain in remission without ongoing treatment.
"We hope that patients will stay in remission. If we inhibit a mediator of inflammation, interleukin-6, you may give the system a chance to reset and end up with complete and durable remission," Dr. De Benedetti said in an interview.
The continued improvement that the sJIA patients showed as their duration on tocilizumab extended from 12 to 52 weeks didn’t surprise either Dr. Woo or Dr. De Benedetti.
"It takes time for these patients to get better," said Dr. Woo, emeritus professor of rheumatology at University College, London.
But the two differed in their current approach to starting tocilizumab treatment in children with sJIA, now that the drug is on the market and has a sJIA indication in both the United States and in Europe. Dr. Woo recommended a trial of methotrexate treatment first, for 2-3 months, and then a move to tocilizumab if patients don’t respond. In contrast, Dr. De Benedetti sees tocilizumab as a reasonable first-line agent.
Methotrexate has not shown efficacy in sJIA. Some patients may respond, but there is no reason to force people to use methotrexate before anything else," he said. However, another new treatment approach that warrants investigation for treating sJIA is the IL-1 inhibitors, such as anakinra (Kineret) and canakinumab (Ilaris), he added.
The original TENDER cohort included 112 children with an average age of 10 years who were randomized to treatment with tocilizumab or placebo for 12 weeks. Children weighing 30 kg or more received an 8 mg/kg tocilizumab infusion every 2 weeks; those weighing less than 30 kg received a 12-mg/kg dosage.
At the end of 12 weeks, 110 of the participants remained on or crossed over to tocilizumab, and 99 children completed either 52 weeks or 40 weeks (if they started in the placebo group) on the active drug. After 1 year, about 80%-90% of patients had ACR30 and ACR50 responses and no fever, roughly similar to the levels seen after 12 weeks. But the percent with ACR70 responses and no fever rose from about 65% after 12 weeks to about 80% after 1 year. The overall average oral corticosteroid dosage needed by the tocilizumab-treated patients after 1 year was 0.06 mg/kg per day, a significant drop from the average 0.30 mg/kg per day at baseline. The average number of active joints in patients at 1 year was about 3, compared with an average of about 6 active joints after 12 weeks and an average of 20 average joints in each patient at baseline.
The TENDER trial was sponsored by Roche, the company that markets tocilizumab. Dr. De Benedetti said he has been a consultant to Bristol-Myers Squibb, Hoffmann-La Roche, and Pfizer, and he has received research support from Hoffmann-La Roche. Dr. Woo said she had no disclosures.
LONDON – Children with systemic juvenile idiopathic arthritis continued to improve on treatment with the interleukin-6 inhibitor tocilizumab as their time on the drug extended out to 1 year, in follow-up of the open-label phase of the drug’s pivotal trial for this disease.
During continued tocilizumab treatment, the percent of systemic juvenile idiopathic arthritis (sJIA) patients who had an American College of Rheumatology (ACR) 90 response and no fever rose from 37% of the treated group after the end of the 12-week randomized trial, to about 55% after an additional 40 weeks of open-label treatment. There was no active joint disease in 49 of the 99 patients (49%) treated for 52 weeks; the children appeared to be in full remission, Dr. Fabrizio De Benedetti said at the meeting. In addition, 52 of the patients treated for 52 weeks fully withdrew from treatment with oral corticosteroids. At their entry onto tocilizumab treatment, their average corticosteroid dosage was 0.30 mg/kg per day.
Over 52 weeks, tocilizumab treatment also had an "acceptable" safety profile. Thirteen patients had a serious adverse event, and 6 had an adverse event leading to withdrawal from treatment. "There was no increase in the rate of serious adverse events between weeks 12 and 52. It is reassuring that there was no accumulation of safety issues with time. But it’s still only 1 year, so we still need more time" to fully assess safety, said Dr. De Benedetti, who is director of the division of rheumatology at Ospedale Pediatrico Bambino Gesù in Rome and lead investigator for the TENDER trial and extension phase.
Based on the results from the 12-week randomized phase III trial, Roche – the company that developed and markets tocilizumab (Actemra) – in April received approval from the Food and Drug Administration for an expanded indication for tocilizumab to treat sJIA. Tocilizumab received FDA approval last year for treatment of rheumatoid arthritis.
Tocilizumab "is very effective; it’s the best drug we have for sJIA. But the experience we have using it is still short" Dr. Patricia Woo, a collaborator on the TENDER trial, said in an interview. "We have 1-year experience, compared with [potentially] using it for many years" to treat children with sJIA. The next phase of the TENDER protocol is to attempt gradually to wean off the drug those children who achieve complete remission on tocilizumab for 6 months to see if they can remain in remission without ongoing treatment.
"We hope that patients will stay in remission. If we inhibit a mediator of inflammation, interleukin-6, you may give the system a chance to reset and end up with complete and durable remission," Dr. De Benedetti said in an interview.
The continued improvement that the sJIA patients showed as their duration on tocilizumab extended from 12 to 52 weeks didn’t surprise either Dr. Woo or Dr. De Benedetti.
"It takes time for these patients to get better," said Dr. Woo, emeritus professor of rheumatology at University College, London.
But the two differed in their current approach to starting tocilizumab treatment in children with sJIA, now that the drug is on the market and has a sJIA indication in both the United States and in Europe. Dr. Woo recommended a trial of methotrexate treatment first, for 2-3 months, and then a move to tocilizumab if patients don’t respond. In contrast, Dr. De Benedetti sees tocilizumab as a reasonable first-line agent.
Methotrexate has not shown efficacy in sJIA. Some patients may respond, but there is no reason to force people to use methotrexate before anything else," he said. However, another new treatment approach that warrants investigation for treating sJIA is the IL-1 inhibitors, such as anakinra (Kineret) and canakinumab (Ilaris), he added.
The original TENDER cohort included 112 children with an average age of 10 years who were randomized to treatment with tocilizumab or placebo for 12 weeks. Children weighing 30 kg or more received an 8 mg/kg tocilizumab infusion every 2 weeks; those weighing less than 30 kg received a 12-mg/kg dosage.
At the end of 12 weeks, 110 of the participants remained on or crossed over to tocilizumab, and 99 children completed either 52 weeks or 40 weeks (if they started in the placebo group) on the active drug. After 1 year, about 80%-90% of patients had ACR30 and ACR50 responses and no fever, roughly similar to the levels seen after 12 weeks. But the percent with ACR70 responses and no fever rose from about 65% after 12 weeks to about 80% after 1 year. The overall average oral corticosteroid dosage needed by the tocilizumab-treated patients after 1 year was 0.06 mg/kg per day, a significant drop from the average 0.30 mg/kg per day at baseline. The average number of active joints in patients at 1 year was about 3, compared with an average of about 6 active joints after 12 weeks and an average of 20 average joints in each patient at baseline.
The TENDER trial was sponsored by Roche, the company that markets tocilizumab. Dr. De Benedetti said he has been a consultant to Bristol-Myers Squibb, Hoffmann-La Roche, and Pfizer, and he has received research support from Hoffmann-La Roche. Dr. Woo said she had no disclosures.
LONDON – Children with systemic juvenile idiopathic arthritis continued to improve on treatment with the interleukin-6 inhibitor tocilizumab as their time on the drug extended out to 1 year, in follow-up of the open-label phase of the drug’s pivotal trial for this disease.
During continued tocilizumab treatment, the percent of systemic juvenile idiopathic arthritis (sJIA) patients who had an American College of Rheumatology (ACR) 90 response and no fever rose from 37% of the treated group after the end of the 12-week randomized trial, to about 55% after an additional 40 weeks of open-label treatment. There was no active joint disease in 49 of the 99 patients (49%) treated for 52 weeks; the children appeared to be in full remission, Dr. Fabrizio De Benedetti said at the meeting. In addition, 52 of the patients treated for 52 weeks fully withdrew from treatment with oral corticosteroids. At their entry onto tocilizumab treatment, their average corticosteroid dosage was 0.30 mg/kg per day.
Over 52 weeks, tocilizumab treatment also had an "acceptable" safety profile. Thirteen patients had a serious adverse event, and 6 had an adverse event leading to withdrawal from treatment. "There was no increase in the rate of serious adverse events between weeks 12 and 52. It is reassuring that there was no accumulation of safety issues with time. But it’s still only 1 year, so we still need more time" to fully assess safety, said Dr. De Benedetti, who is director of the division of rheumatology at Ospedale Pediatrico Bambino Gesù in Rome and lead investigator for the TENDER trial and extension phase.
Based on the results from the 12-week randomized phase III trial, Roche – the company that developed and markets tocilizumab (Actemra) – in April received approval from the Food and Drug Administration for an expanded indication for tocilizumab to treat sJIA. Tocilizumab received FDA approval last year for treatment of rheumatoid arthritis.
Tocilizumab "is very effective; it’s the best drug we have for sJIA. But the experience we have using it is still short" Dr. Patricia Woo, a collaborator on the TENDER trial, said in an interview. "We have 1-year experience, compared with [potentially] using it for many years" to treat children with sJIA. The next phase of the TENDER protocol is to attempt gradually to wean off the drug those children who achieve complete remission on tocilizumab for 6 months to see if they can remain in remission without ongoing treatment.
"We hope that patients will stay in remission. If we inhibit a mediator of inflammation, interleukin-6, you may give the system a chance to reset and end up with complete and durable remission," Dr. De Benedetti said in an interview.
The continued improvement that the sJIA patients showed as their duration on tocilizumab extended from 12 to 52 weeks didn’t surprise either Dr. Woo or Dr. De Benedetti.
"It takes time for these patients to get better," said Dr. Woo, emeritus professor of rheumatology at University College, London.
But the two differed in their current approach to starting tocilizumab treatment in children with sJIA, now that the drug is on the market and has a sJIA indication in both the United States and in Europe. Dr. Woo recommended a trial of methotrexate treatment first, for 2-3 months, and then a move to tocilizumab if patients don’t respond. In contrast, Dr. De Benedetti sees tocilizumab as a reasonable first-line agent.
Methotrexate has not shown efficacy in sJIA. Some patients may respond, but there is no reason to force people to use methotrexate before anything else," he said. However, another new treatment approach that warrants investigation for treating sJIA is the IL-1 inhibitors, such as anakinra (Kineret) and canakinumab (Ilaris), he added.
The original TENDER cohort included 112 children with an average age of 10 years who were randomized to treatment with tocilizumab or placebo for 12 weeks. Children weighing 30 kg or more received an 8 mg/kg tocilizumab infusion every 2 weeks; those weighing less than 30 kg received a 12-mg/kg dosage.
At the end of 12 weeks, 110 of the participants remained on or crossed over to tocilizumab, and 99 children completed either 52 weeks or 40 weeks (if they started in the placebo group) on the active drug. After 1 year, about 80%-90% of patients had ACR30 and ACR50 responses and no fever, roughly similar to the levels seen after 12 weeks. But the percent with ACR70 responses and no fever rose from about 65% after 12 weeks to about 80% after 1 year. The overall average oral corticosteroid dosage needed by the tocilizumab-treated patients after 1 year was 0.06 mg/kg per day, a significant drop from the average 0.30 mg/kg per day at baseline. The average number of active joints in patients at 1 year was about 3, compared with an average of about 6 active joints after 12 weeks and an average of 20 average joints in each patient at baseline.
The TENDER trial was sponsored by Roche, the company that markets tocilizumab. Dr. De Benedetti said he has been a consultant to Bristol-Myers Squibb, Hoffmann-La Roche, and Pfizer, and he has received research support from Hoffmann-La Roche. Dr. Woo said she had no disclosures.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Treatment with the interleukin-6 blocker tocilizumab led to ongoing improvements in 99 children with systemic juvenile idiopathic arthritis; 49% had no active joints, 53% required no treatment with an oral corticosteroid, and about 55% had no fever and an ACR90 response.
Data Source: Forty-week, open-label extension of the original 12-week TENDER trial.
Disclosures: The TENDER trial was sponsored by Roche, the company that markets tocilizumab. Dr. De Benedetti said he has been a consultant to Bristol-Myers Squibb, Hoffmann-La Roche, and Pfizer, and he has received research support from Hoffmann-La Roche. Dr. Woo said she had no disclosures.
HPV Vaccine Does Not Induce Lupus Flares
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: The quadrivalent human papillomavirus vaccine does not exacerbate disease activity in women with SLE.
Data Source: A prospective study of 50 female SLE patients and an unvaccinated cohort of SLE patients who were observed over a 5-year period at the same institution.
Disclosures: Dr. Mok disclosed having no financial conflicts of interest.
HPV Vaccine Does Not Induce Lupus Flares
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: The quadrivalent human papillomavirus vaccine does not exacerbate disease activity in women with SLE.
Data Source: A prospective study of 50 female SLE patients and an unvaccinated cohort of SLE patients who were observed over a 5-year period at the same institution.
Disclosures: Dr. Mok disclosed having no financial conflicts of interest.
HPV Vaccine Does Not Induce Lupus Flares
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
LONDON – The quadrivalent human papillomavirus vaccine is safe for patients with systemic lupus erythematosus and should be considered for women with inactive disease who receive stable doses of standard immunomodulatory therapy, according to data presented by Dr. Chi Chiu Mok at the annual European Congress of Rheumatology.
Multiple studies have demonstrated higher rates of persistent HPV infections and precancerous lesions in women with SLE, compared with women in the general population, said Dr. Mok. Although the increased risk of HPV infection suggests that SLE patients would be good candidates for immunization against the virus, it has been hypothesized that immune dysfunction related to SLE or to treatment-induced immune suppression may prevent patients with the condition from being able to produce an effective immune response to the vaccine, and could possibly lead to disease flares or the production of new autoantibodies, he explained.
The recombinant quadrivalent HPV vaccine (Gardisil) provides protection against infection of the HPV serotypes 6, 11, 16, and 18, and it has been demonstrated to be safe and efficacious in female patients in the general population aged 9-26 years, according to Dr. Mok. Along with colleagues at Tuen Mun Hospital in Hong Kong, Dr. Mok sought to evaluate the safety and immunogenicity of the vaccine in a cohort of SLE patients.
Toward this end, the investigators recruited 50 female patients aged 18-35 years who fulfilled at least four American College of Rheumatology criteria for SLE, and who had received a stable dose of prednisolone or other immunosuppressive agent within the previous 3 months, to participate in the prospective investigation. The mean age of the study participants was 25.8 years, and their mean disease duration was 6.6 years, he reported.
All of the study subjects received intramuscular injections of the vaccine and were evaluated at baseline and at 2 and 6 months post vaccination via the SLEDAI (SLE Disease Activity Index), PGA (Physicians’ Global Assessment), and the SELENA (Safety of Estrogens in Lupus Erythematosus – National Assessment) disease flare index. Additionally, complement levels (C3 and C4) and anti-dsDNA (anti–double-stranded DNA) titers were assessed and patient-reported adverse events were recorded at the same time points, said Dr. Mok. With respect to baseline disease characteristics, the median SLEDAI score was 4; the mean anti-dsDNA titers, C3 levels, and C4 levels were 139 IU/mL, 0.81 g/dL, and 0.15 g/dL, respectively; and none of the patients had SELENA flares at baseline compared to preceding status, he said.
There were no significant changes in anti-dsDNA titers, C3 or C4 levels, or SLEDAI and PGA scores at any of the time points, Dr. Mok reported. "There were three mild to moderate mucocutaneous flares during the study period (one at month 2 and two at month 6), all of which were controlled with usual treatment," he said. "It’s unclear whether a causal relationship exists between the vaccination and the three lupus flares, but the rate of flares [0.08 per patient per year] was lower than the rate observed in our lupus cohort during the previous 5 years [0.10 per patient per year], and no other adverse events associated with the vaccination were reported."
The study findings indicate that the vaccine is safe in SLE patients, and the lack of significant alterations in the various SLE antibody measures suggests it does not induce an increased incidence of lupus flares, Dr. Mok stated. Considering the increased susceptibility to HPV infection in these patients and the link between HPV infection and cervical cancer, "vaccination is an important consideration in protecting them," he said.
Dr. Mok disclosed having no financial conflicts of interest.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: The quadrivalent human papillomavirus vaccine does not exacerbate disease activity in women with SLE.
Data Source: A prospective study of 50 female SLE patients and an unvaccinated cohort of SLE patients who were observed over a 5-year period at the same institution.
Disclosures: Dr. Mok disclosed having no financial conflicts of interest.
Fulranumab Shows Efficacy for Osteoarthritis Pain
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Treatment with several different dosages of fulranumab led to statistically significant improvements in a number of efficacy measures and was well tolerated. The primary efficacy end point of change in average pain intensity at 12 weeks from the start of treatment showed significant drops, compared with the placebo group for the three largest dosages of fulranumab tested.
Data Source: Phase II randomized, placebo-controlled trial that assessed the efficacy and safety of five dosages of fulranumab after 12 weeks of treatment in patients with moderate to severely painful osteoarthritis of the hip or knee.
Disclosures: Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
Fulranumab Shows Efficacy for Osteoarthritis Pain
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
LONDON – An investigational nerve growth factor inhibitor, fulranumab, showed promising efficacy and safety as a pain reliever for patients with hip or knee osteoarthritis in 12-week results from a phase II study of 466 patients.
Further study of fulranumab in osteoarthritis had been on hold. The Food and Drug Administration issued a moratorium last December that halted clinical testing of fulranumab and most other investigational agents in the anti–nerve growth factor class, following reports that some of these drugs appeared to trigger episodes of rapid progression of hip or knee osteoarthritis (OA) that led to joint replacement and possible osteonecrosis. The FDA lifted that moratorium on research with fulranumab in cancer pain this month. The moratorium on research involving OA pain remains in place, according to investigators.
Whether or not osteoarthritis progressed rapidly in any patient treated with fulranumab remains unknown. "Cases of joint replacement reported during the entire trial are under investigation, and will be reported in a future publication," Dr. John Thipphawong said at the annual European Congress of Rheumatology.
The safety data Dr. Thipphawong presented for 12 weeks of treatment showed a well-tolerated profile, compared with placebo. Specifically, serious adverse events occurred in 1% of patients treated with fulranumab, compared with 2% of those on placebo. Adverse events led to discontinuation of the assigned drug in 2% of fulranumab recipients and 1% of those on placebo. Adverse events that occurred more often in fulranumab-treated patients were paresthesia, with a 6%-10% rate in the higher fulranumab dosage subgroups, compared with a 3% rate for patients on placebo, and a hypoesthesia rate of 5%-6% in the higher dosage fulranumab subgroups, compared with a 1% rate with placebo. The fulranumab-treated patients also had no significant changes in laboratory values, ECG, or vital signs at 12 weeks after treatment began.
The study enrolled patients with documented hip or knee osteoarthritis who met the diagnostic criteria of the American College of Rheumatology and showed radiographic evidence of the disease, with a Kellgren-Lawrence grade of 2 or greater. All patients also reported moderate to severe pain, with a painscore of at least 5 on a 0-10 numerical rating scale despite treatment with an opioid, a nonsteroidal anti-inflammatory drug, or both.
The study randomized patients to receive fulranumab or placebo once every 4 or 8 weeks as a subcutaneous injection in addition to standard pain medications. The protocol tested five different fulranumab dosages: 1 mg every 4 weeks, 3 mg every 4 weeks, 3 mg every 8 weeks, 6 mg every 8 weeks, or 10 mg every 8 weeks. Fulranumab is a fully human, recombinant monoclonal antibody that neutralizes the biological actions of human nerve growth factor. About 78 patients entered each of the five active-treatment arms as well as a placebo arm. The study’s primary efficacy end point was the change in average pain score from baseline to the end of week 12 of the study.
The patients’ average age was 61 years, 58% were women, and two-thirds were white. Their average body mass index was 32 kg/m2, and 60% weighed at least 85 kg. Knee OA predominated as the affected joint, in 77% of patients.
At 12 weeks after the start of treatment, average pain reduction with fulranumab significantly surpassed the placebo group in the 3 mg every 4 weeks, 6 mg every 8 weeks, and 10 mg every 8 weeks subgroups. In these three groups, pain scores fell by an average of 3.05, 2.64, and 2.65 points, respectively, compared with an average drop of 1.91 points in the placebo group, reported Dr. Thipphawong, who is senior director of clinical development, Johnson & Johnson Pharmaceutical Research & Development.
The study also included several secondary efficacy measures. The three highest-dosage subgroups, as well as the 3 mg every 8 weeks subgroup, showed statistically significant declines, compared with placebo after 12 weeks in the average levels of the Western Ontario and McMaster University Osteoarthritis Index (WOMAC) subscales for pain and global function. For the WOMAC subscales of physical function and stiffness, all five fulranumab dosage subgroups showed significant reductions, compared with placebo.
On the Brief Pain Inventory-Short Form, patients in the 3 mg every 4 weeks and 10 mg every 8 weeks subgroups had significant average reductions, compared with the placebo group for the subscales of pain intensity and pain interference with activities. The three highest-dosage subgroups also produced average drops in patient global assessment of disease status that were statistically significant, compared with the placebo group’s average.
In a separate poster at the meeting, Dr. Thipphawong and his associates also reported that several of the fulranumab subgroups showed statistically significant average improvements, with placebo in several subscale measures on the Short From-36, specifically bodily pain, vitality, and physical component. The four highest-dosage subgroups also had significant average improvements in pain interference with sleep, compared with placebo, and all five fulranumab dosage subgroups had significant average improvements in sleep adequacy, compared with the placebo group.
Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Treatment with several different dosages of fulranumab led to statistically significant improvements in a number of efficacy measures and was well tolerated. The primary efficacy end point of change in average pain intensity at 12 weeks from the start of treatment showed significant drops, compared with the placebo group for the three largest dosages of fulranumab tested.
Data Source: Phase II randomized, placebo-controlled trial that assessed the efficacy and safety of five dosages of fulranumab after 12 weeks of treatment in patients with moderate to severely painful osteoarthritis of the hip or knee.
Disclosures: Dr. Thipphawong and several of his associates are employees of Johnson & Johnson, the company developing fulranumab.
RA and SLE Risk Factors Remain for Mothers and Babies
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Adjusted odds ratios for preeclampsia or eclampsia were 2.8 (95% CI 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
Data Source: Inpatient administrative database of obstetric admissions and discharges for 38 women with RA, 95 with SLE, and 525 women with neither condition as controls, between 1998-1999 and 2008-2009.
Disclosures: Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
RA and SLE Risk Factors Remain for Mothers and Babies
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
LONDON – Women with both rheumatoid arthritis and systemic lupus erythematosus were more than twice as likely as women without these diseases to have pregnancy-related hypertension, and the risk has not lessened in recent years despite improved treatment, according to 10-years’ worth of admission and discharge data from several Canadian hospitals.
Such women were more likely to undergo a cesarean section, and their newborns were more likely to be born prematurely, be small for gestational age (SGA), and require intensive care, compared with children born to women without RA or SLE.
"We wanted to look at what obstetrical and neonatal outcomes have been like over the past 10 years in both RA and in lupus," study author and rheumatologist Dr. Cheryl Barnabe said in an interview during a poster session at the annual European Congress of Rheumatology.
To do so, Dr. Barnabe and colleagues used an administrative database maintained by the Canadian government to examine the outcomes of 38 women with RA and 95 with SLE who were admitted and discharged from hospitals in Calgary for obstetric reasons. Outcomes were compared with expectant women who did not have either condition, with four times as many controls as cases.
"We found that the RA and lupus patients stayed in hospital longer [after delivery], approximately double the percentage of patients [in both groups] had C-sections done, and there were more postpartum infections in the lupus patients compared to the lupus controls [6.3% vs. 1.3%, P less than .004]," observed Dr. Barnabe, a clinical scholar in the department of medicine at the University of Calgary in Alberta.
Adjusted odds ratios (ORs) for preeclampsia or eclampsia were 2.8 (95% confidence interval 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
The mean length of hospital stay was 0.9 days longer for women with RA than for those without (P less than .003) and 1.8 days longer for those with SLE than for those without (P less than .001).
One-third (34.2%) of women with RA and 43.2% of women with SLE underwent cesarean section compared with 21.2% of RA controls and 23.7% of SLE controls. The adjusted ORs for cesarean section in RA and SLE were 2.3 (95% CI 0.9-4.7) and 2.8 (95% CI 1.5-4.0), respectively.
Babies were more frequently premature, with 2.6% vs. 0.7% born between 28 and 34 weeks’ gestation in women with and without RA, and 18.4% vs. 8% born at 34-37 weeks’ gestation. Corresponding values for the children of women with SLE were 8.4% vs. 1.9% at 28-34 weeks and 23.2% vs. 5.1% at 34-37 weeks. Three (3.2%) women with SLE but none without gave birth at less than 28 weeks’ gestation.
The adjusted ORs for premature delivery were 2.7 (95% CI 1.0-7.0) for RA and 6.6 (95% CI 3.5-12.3) for SLE.
Not surprisingly, the need for intensive care was higher among children born to women with RA or SLE than among the babies of women in the control groups. Adjusted ORs for SGA were 3.0 and 2.8 for RA and SLE, respectively.
These findings suggest the need for improved multidisciplinary management of women with arthritic conditions who are pregnant, Dr. Barnabe suggested.
"I think this shows that we can’t just assume that if we treat disease activity well enough we will affect obstetrical outcomes," she said.
"We should liaise with the obstetricians and the maternal-fetal medicine specialists and others to ensure that blood pressures are well controlled during pregnancy and make sure the disease activity is well controlled, and that will hopefully optimize these outcomes," Dr. Barnabe added. "I think a combined clinic is probably the best way to approach this."
Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.
FROM THE ANNUAL EUROPEAN CONGRESS OF RHEUMATOLOGY
Major Finding: Adjusted odds ratios for preeclampsia or eclampsia were 2.8 (95% CI 1.0-7.8) for RA and 2.0 (95% CI 1.0-3.7) for SLE.
Data Source: Inpatient administrative database of obstetric admissions and discharges for 38 women with RA, 95 with SLE, and 525 women with neither condition as controls, between 1998-1999 and 2008-2009.
Disclosures: Dr. Barnabe disclosed receiving a scholarship from the Arthritis Society, UCB, and the Canadian Rheumatologists Association for her master’s degree. A coauthor has received financial support from Alberta Innovative Health Solutions.