User login
How Can Patient History Improve the Diagnosis of Chronic Migraine?
SAN DIEGO—Accurate diagnosis of chronic migraine may improve when patients use a “lumping” strategy versus a “splitting” strategy when sharing their headache history with providers, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society.
Chronic migraine can be challenging to diagnose, especially for less experienced providers, said Priyanka Yadav, MBBS, a neurology resident at the University of Kentucky in Lexington. The International Classification of Headache Disorders, third edition, defines chronic migraine as headaches occurring on at least 15 days per month for three months, at least eight of which have features of migraine. Patients with chronic migraine can experience headaches similar to those of patients with tension-type headache. As a result, headache history is vital to achieving a correct diagnosis, Dr. Yadav added.
Although a provider may have sufficient information about the patient’s headache history, diagnosis may still be confusing if patients describe their headache history using a splitting strategy, in which patients distinguish multiple types of headache. For example, a patient may report that he or she had two types of headache that occurred over the previous three months. “This [history] can be confusing because inexperienced providers may not recognize that patients with chronic migraine experience headache days similar to [those of] tension headache,” said Dr. Yadav.

Dr. Yadav and colleagues hypothesized that a lumping strategy would help neurologists to reach a correct headache diagnosis, as opposed to the splitting strategy. In the lumping strategy, patients describe one type of headache. For example, a woman may report having a continuous, background, mild, pressure-like pain throughout her head, which can evolve into intense, throbbing pain associated with nausea and light and sound sensitivity, said Dr. Yadav.
For the study, researchers sent an unannounced electronic multiple choice quiz to 19 neurology residents at the University of Kentucky. The quiz assessed the residents’ ability to recognize the correct headache diagnoses of various case vignettes of episodic migraine, chronic tension-type headache, and chronic migraine. In addition, residents were asked to recognize the chronic migraine criteria. The main outcome measure was frequency of chronic migraine recognition as a function of history style, presented in either the lumping or splitting format.
The response rate to the quiz was 100%. Results indicated that correct recognition of chronic migraine was more likely when the headache history was presented in a lumping format, as opposed to a splitting format. “When presented with the splitting style, participants often thought that the patient had two headache diagnoses (eg, chronic tension-type headache and episodic migraine), instead of one unifying diagnosis (eg, chronic migraine),” said Jonathan H. Smith, MD, coauthor and Assistant Professor of Neurology at the University of Kentucky College of Medicine.
Participants were poor at recognizing that features of these primary headache disorders can coexist to contribute toward a uniform diagnosis of chronic migraine. “These results have strategic implications for how physicians should organize and teach the headache history,” said Dr. Yadav.
—Erica Robinson
Suggested Reading
Moriarty M, Mallick-Searle T. Diagnosis and treatment for chronic migraine. Nurse Pract. 2016;41(6):18-32.
Eross E, Dodick D, Eross M. The Sinus, Allergy and Migraine Study (SAMS). Headache. 2007;47(2):213-224.
SAN DIEGO—Accurate diagnosis of chronic migraine may improve when patients use a “lumping” strategy versus a “splitting” strategy when sharing their headache history with providers, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society.
Chronic migraine can be challenging to diagnose, especially for less experienced providers, said Priyanka Yadav, MBBS, a neurology resident at the University of Kentucky in Lexington. The International Classification of Headache Disorders, third edition, defines chronic migraine as headaches occurring on at least 15 days per month for three months, at least eight of which have features of migraine. Patients with chronic migraine can experience headaches similar to those of patients with tension-type headache. As a result, headache history is vital to achieving a correct diagnosis, Dr. Yadav added.
Although a provider may have sufficient information about the patient’s headache history, diagnosis may still be confusing if patients describe their headache history using a splitting strategy, in which patients distinguish multiple types of headache. For example, a patient may report that he or she had two types of headache that occurred over the previous three months. “This [history] can be confusing because inexperienced providers may not recognize that patients with chronic migraine experience headache days similar to [those of] tension headache,” said Dr. Yadav.
Dr. Yadav and colleagues hypothesized that a lumping strategy would help neurologists to reach a correct headache diagnosis, as opposed to the splitting strategy. In the lumping strategy, patients describe one type of headache. For example, a woman may report having a continuous, background, mild, pressure-like pain throughout her head, which can evolve into intense, throbbing pain associated with nausea and light and sound sensitivity, said Dr. Yadav.
For the study, researchers sent an unannounced electronic multiple choice quiz to 19 neurology residents at the University of Kentucky. The quiz assessed the residents’ ability to recognize the correct headache diagnoses of various case vignettes of episodic migraine, chronic tension-type headache, and chronic migraine. In addition, residents were asked to recognize the chronic migraine criteria. The main outcome measure was frequency of chronic migraine recognition as a function of history style, presented in either the lumping or splitting format.
The response rate to the quiz was 100%. Results indicated that correct recognition of chronic migraine was more likely when the headache history was presented in a lumping format, as opposed to a splitting format. “When presented with the splitting style, participants often thought that the patient had two headache diagnoses (eg, chronic tension-type headache and episodic migraine), instead of one unifying diagnosis (eg, chronic migraine),” said Jonathan H. Smith, MD, coauthor and Assistant Professor of Neurology at the University of Kentucky College of Medicine.
Participants were poor at recognizing that features of these primary headache disorders can coexist to contribute toward a uniform diagnosis of chronic migraine. “These results have strategic implications for how physicians should organize and teach the headache history,” said Dr. Yadav.
—Erica Robinson
Suggested Reading
Moriarty M, Mallick-Searle T. Diagnosis and treatment for chronic migraine. Nurse Pract. 2016;41(6):18-32.
Eross E, Dodick D, Eross M. The Sinus, Allergy and Migraine Study (SAMS). Headache. 2007;47(2):213-224.
SAN DIEGO—Accurate diagnosis of chronic migraine may improve when patients use a “lumping” strategy versus a “splitting” strategy when sharing their headache history with providers, according to research presented at the 58th Annual Scientific Meeting of the American Headache Society.
Chronic migraine can be challenging to diagnose, especially for less experienced providers, said Priyanka Yadav, MBBS, a neurology resident at the University of Kentucky in Lexington. The International Classification of Headache Disorders, third edition, defines chronic migraine as headaches occurring on at least 15 days per month for three months, at least eight of which have features of migraine. Patients with chronic migraine can experience headaches similar to those of patients with tension-type headache. As a result, headache history is vital to achieving a correct diagnosis, Dr. Yadav added.
Although a provider may have sufficient information about the patient’s headache history, diagnosis may still be confusing if patients describe their headache history using a splitting strategy, in which patients distinguish multiple types of headache. For example, a patient may report that he or she had two types of headache that occurred over the previous three months. “This [history] can be confusing because inexperienced providers may not recognize that patients with chronic migraine experience headache days similar to [those of] tension headache,” said Dr. Yadav.
Dr. Yadav and colleagues hypothesized that a lumping strategy would help neurologists to reach a correct headache diagnosis, as opposed to the splitting strategy. In the lumping strategy, patients describe one type of headache. For example, a woman may report having a continuous, background, mild, pressure-like pain throughout her head, which can evolve into intense, throbbing pain associated with nausea and light and sound sensitivity, said Dr. Yadav.
For the study, researchers sent an unannounced electronic multiple choice quiz to 19 neurology residents at the University of Kentucky. The quiz assessed the residents’ ability to recognize the correct headache diagnoses of various case vignettes of episodic migraine, chronic tension-type headache, and chronic migraine. In addition, residents were asked to recognize the chronic migraine criteria. The main outcome measure was frequency of chronic migraine recognition as a function of history style, presented in either the lumping or splitting format.
The response rate to the quiz was 100%. Results indicated that correct recognition of chronic migraine was more likely when the headache history was presented in a lumping format, as opposed to a splitting format. “When presented with the splitting style, participants often thought that the patient had two headache diagnoses (eg, chronic tension-type headache and episodic migraine), instead of one unifying diagnosis (eg, chronic migraine),” said Jonathan H. Smith, MD, coauthor and Assistant Professor of Neurology at the University of Kentucky College of Medicine.
Participants were poor at recognizing that features of these primary headache disorders can coexist to contribute toward a uniform diagnosis of chronic migraine. “These results have strategic implications for how physicians should organize and teach the headache history,” said Dr. Yadav.
—Erica Robinson
Suggested Reading
Moriarty M, Mallick-Searle T. Diagnosis and treatment for chronic migraine. Nurse Pract. 2016;41(6):18-32.
Eross E, Dodick D, Eross M. The Sinus, Allergy and Migraine Study (SAMS). Headache. 2007;47(2):213-224.
USPSTF considers BP measurements in pregnancy to detect preeclampsia
All pregnant women should be screened with blood pressure measurements for preeclampsia throughout pregnancy, according to a new draft recommendation from the U.S. Preventive Services Task Force.
“Preeclampsia is a complex syndrome. It can quickly evolve into a severe disease that can result in serious, even fatal health outcomes for the mother and infant,” the USPSTF members wrote in their draft. “The ability to screen for preeclampsia using blood pressure measurements is important in order to identify and effectively treat a potentially unpredictable and fatal condition.”
The USPSTF noted that there is “adequate evidence” that supported the superior accuracy of blood pressure measurements over urinalysis as dipstick tests have “low diagnostic accuracy for proteinuria detection in pregnancy.”The USPSTF concluded “with moderate certainty” that screening for preeclampsia in all pregnant women with blood pressure measurements yields a substantial net benefit for mothers and newborns as there are “likely few harms” from screening with blood pressure measurements. The draft recommendation has a “B” grade, meaning that clinicians are encouraged to offer the service.
The American College of Obstetricians and Gynecologists applauded the USPSTF’s draft, noting that the recommendations align with their current guidance, which recommends that physicians use detailed medical histories to evaluate a patient’s risk for developing preeclampsia. Ob.gyns. already take a woman’s blood pressure at each routine visit, along with measuring weight, uterine size, and the presence of fetal heart activity, ACOG noted.
“Importantly, ACOG has found there are no accurate, predictive tests at this time to determine whether a woman will develop preeclampsia and therefore continues to recommend against other methods for predicting preeclampsia,” ACOG president Thomas Gellhaus, MD, said in a statement. “A detailed medical history and routine blood pressure measurements are the best tools available to alert ob.gyns. of a potential risk.”
In July, ACOG recommended an expanded list of risk factors for preeclampsia that include history of the condition, multifetal gestation, chronic hypertension, diabetes, renal disease, and autoimmune disease.
The USPSTF is accepting public comment on the draft recommendation until Oct. 24, 2016.
On Twitter @jessnicolecraig
All pregnant women should be screened with blood pressure measurements for preeclampsia throughout pregnancy, according to a new draft recommendation from the U.S. Preventive Services Task Force.
“Preeclampsia is a complex syndrome. It can quickly evolve into a severe disease that can result in serious, even fatal health outcomes for the mother and infant,” the USPSTF members wrote in their draft. “The ability to screen for preeclampsia using blood pressure measurements is important in order to identify and effectively treat a potentially unpredictable and fatal condition.”
The USPSTF noted that there is “adequate evidence” that supported the superior accuracy of blood pressure measurements over urinalysis as dipstick tests have “low diagnostic accuracy for proteinuria detection in pregnancy.”The USPSTF concluded “with moderate certainty” that screening for preeclampsia in all pregnant women with blood pressure measurements yields a substantial net benefit for mothers and newborns as there are “likely few harms” from screening with blood pressure measurements. The draft recommendation has a “B” grade, meaning that clinicians are encouraged to offer the service.
The American College of Obstetricians and Gynecologists applauded the USPSTF’s draft, noting that the recommendations align with their current guidance, which recommends that physicians use detailed medical histories to evaluate a patient’s risk for developing preeclampsia. Ob.gyns. already take a woman’s blood pressure at each routine visit, along with measuring weight, uterine size, and the presence of fetal heart activity, ACOG noted.
“Importantly, ACOG has found there are no accurate, predictive tests at this time to determine whether a woman will develop preeclampsia and therefore continues to recommend against other methods for predicting preeclampsia,” ACOG president Thomas Gellhaus, MD, said in a statement. “A detailed medical history and routine blood pressure measurements are the best tools available to alert ob.gyns. of a potential risk.”
In July, ACOG recommended an expanded list of risk factors for preeclampsia that include history of the condition, multifetal gestation, chronic hypertension, diabetes, renal disease, and autoimmune disease.
The USPSTF is accepting public comment on the draft recommendation until Oct. 24, 2016.
On Twitter @jessnicolecraig
All pregnant women should be screened with blood pressure measurements for preeclampsia throughout pregnancy, according to a new draft recommendation from the U.S. Preventive Services Task Force.
“Preeclampsia is a complex syndrome. It can quickly evolve into a severe disease that can result in serious, even fatal health outcomes for the mother and infant,” the USPSTF members wrote in their draft. “The ability to screen for preeclampsia using blood pressure measurements is important in order to identify and effectively treat a potentially unpredictable and fatal condition.”
The USPSTF noted that there is “adequate evidence” that supported the superior accuracy of blood pressure measurements over urinalysis as dipstick tests have “low diagnostic accuracy for proteinuria detection in pregnancy.”The USPSTF concluded “with moderate certainty” that screening for preeclampsia in all pregnant women with blood pressure measurements yields a substantial net benefit for mothers and newborns as there are “likely few harms” from screening with blood pressure measurements. The draft recommendation has a “B” grade, meaning that clinicians are encouraged to offer the service.
The American College of Obstetricians and Gynecologists applauded the USPSTF’s draft, noting that the recommendations align with their current guidance, which recommends that physicians use detailed medical histories to evaluate a patient’s risk for developing preeclampsia. Ob.gyns. already take a woman’s blood pressure at each routine visit, along with measuring weight, uterine size, and the presence of fetal heart activity, ACOG noted.
“Importantly, ACOG has found there are no accurate, predictive tests at this time to determine whether a woman will develop preeclampsia and therefore continues to recommend against other methods for predicting preeclampsia,” ACOG president Thomas Gellhaus, MD, said in a statement. “A detailed medical history and routine blood pressure measurements are the best tools available to alert ob.gyns. of a potential risk.”
In July, ACOG recommended an expanded list of risk factors for preeclampsia that include history of the condition, multifetal gestation, chronic hypertension, diabetes, renal disease, and autoimmune disease.
The USPSTF is accepting public comment on the draft recommendation until Oct. 24, 2016.
On Twitter @jessnicolecraig
QUIZ: Treating Infants Hospitalized With Viral Bronchiolitis
[WpProQuiz 14]
[WpProQuiz_toplist 14]
[WpProQuiz 14]
[WpProQuiz_toplist 14]
[WpProQuiz 14]
[WpProQuiz_toplist 14]
Changes in DSM-5 codes in ICD-10 go into effect Oct. 1
Earlier this month, the American Psychiatric Association released a supplement to the DSM-5 that updated codes for 14 diagnoses, including binge-eating disorder, disruptive mood dysregulation disorder, hoarding disorder, and premenstrual dysphoric disorder. On Oct. 1, those DSM-5 coding changes will be reflected within the International Classification of Diseases, Tenth Edition, Clinical Modification (ICD-10-CM).
Some of the changes are minor. For example, the code for binge-eating disorder is now F50.8 and will become F50.81 on Oct. 1. Disruptive mood dysregulation disorder will change from F34.8 to F34.81, and hoarding disorder will change from F42 to F42.3.
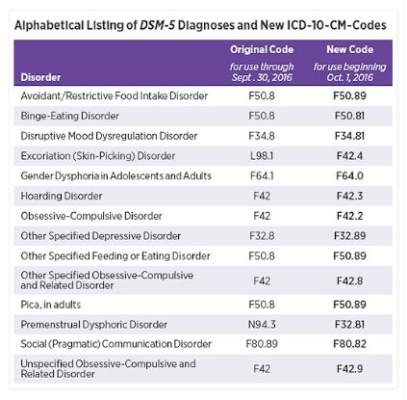
However, some of the changes are significant. The code for premenstrual dysphoric disorder, for example, will change from N94.3 to F32.81. The changes are aimed at improving diagnostic recording, communication among clinicians, and the collection of prevalence data.
To access the DSM-5 supplement, click here. A concise listing of the updated ICD-10-CM changes can be found here.
Earlier this month, the American Psychiatric Association released a supplement to the DSM-5 that updated codes for 14 diagnoses, including binge-eating disorder, disruptive mood dysregulation disorder, hoarding disorder, and premenstrual dysphoric disorder. On Oct. 1, those DSM-5 coding changes will be reflected within the International Classification of Diseases, Tenth Edition, Clinical Modification (ICD-10-CM).
Some of the changes are minor. For example, the code for binge-eating disorder is now F50.8 and will become F50.81 on Oct. 1. Disruptive mood dysregulation disorder will change from F34.8 to F34.81, and hoarding disorder will change from F42 to F42.3.
However, some of the changes are significant. The code for premenstrual dysphoric disorder, for example, will change from N94.3 to F32.81. The changes are aimed at improving diagnostic recording, communication among clinicians, and the collection of prevalence data.
To access the DSM-5 supplement, click here. A concise listing of the updated ICD-10-CM changes can be found here.
Earlier this month, the American Psychiatric Association released a supplement to the DSM-5 that updated codes for 14 diagnoses, including binge-eating disorder, disruptive mood dysregulation disorder, hoarding disorder, and premenstrual dysphoric disorder. On Oct. 1, those DSM-5 coding changes will be reflected within the International Classification of Diseases, Tenth Edition, Clinical Modification (ICD-10-CM).
Some of the changes are minor. For example, the code for binge-eating disorder is now F50.8 and will become F50.81 on Oct. 1. Disruptive mood dysregulation disorder will change from F34.8 to F34.81, and hoarding disorder will change from F42 to F42.3.
However, some of the changes are significant. The code for premenstrual dysphoric disorder, for example, will change from N94.3 to F32.81. The changes are aimed at improving diagnostic recording, communication among clinicians, and the collection of prevalence data.
To access the DSM-5 supplement, click here. A concise listing of the updated ICD-10-CM changes can be found here.
New chikungunya diagnostic assay proves quick, effective
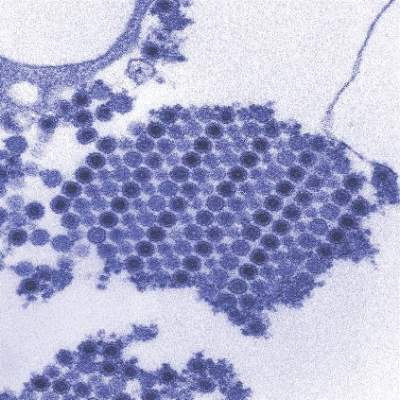
A reverse transcription recombinase polymerase amplification (RT-RPA) assay was able to quickly and effectively identify chikungunya virus (CHIKV), according to a study published in PLOS Neglected Tropical Diseases.
Using chikungunya virus RNA samples, the RT-RPA assay detected down to 80 genome copies per reaction within 15 minutes, a time period four to six times faster than other molecular diagnostic techniques, such as reverse transcription-polymerase chain reaction (RT-PCR). In a sensitivity test involving all chikungunya serotypes and various alphaviruses, flaviviruses, and one phlebovirus, the RT-RPA assay identified all virus genotypes, with the only cross-reaction occurring with O’nyong’nyong virus.
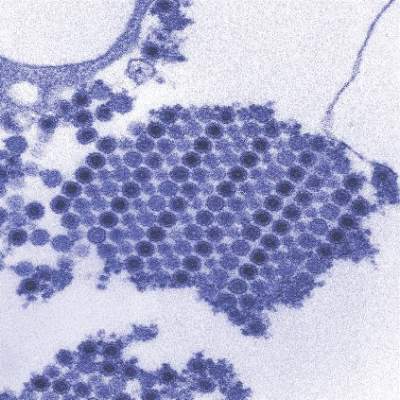
In a test involving 58 plasma samples of suspected chikungunya fever from a trial in Thailand, two real-time RT-PCR tests identified 36 out of 58 samples (62%) as positive for chikungunya. The RT-RPA test successfully detected the virus in all 36 positive samples and did not detect the virus in any of the negative samples, giving a sensitivity and specificity of 100%.
“The CHIKV RPA assay presented here is a promising tool for CHIKV diagnostics at the point of need,” the investigators wrote. “Integration into a multimer or multiplex assay for simultaneous and differential detection of CHIKV, Dengue virus, and Zika virus, as well as an internal positive control would improve outbreak investigations, since the three viruses induce the same clinical picture upon infection and increasingly cocirculate in many parts of the world.”
Find the full study in PLOS Neglected Tropical Diseases (doi: 10.1371/journal.pntd.0004953).
A reverse transcription recombinase polymerase amplification (RT-RPA) assay was able to quickly and effectively identify chikungunya virus (CHIKV), according to a study published in PLOS Neglected Tropical Diseases.
Using chikungunya virus RNA samples, the RT-RPA assay detected down to 80 genome copies per reaction within 15 minutes, a time period four to six times faster than other molecular diagnostic techniques, such as reverse transcription-polymerase chain reaction (RT-PCR). In a sensitivity test involving all chikungunya serotypes and various alphaviruses, flaviviruses, and one phlebovirus, the RT-RPA assay identified all virus genotypes, with the only cross-reaction occurring with O’nyong’nyong virus.
In a test involving 58 plasma samples of suspected chikungunya fever from a trial in Thailand, two real-time RT-PCR tests identified 36 out of 58 samples (62%) as positive for chikungunya. The RT-RPA test successfully detected the virus in all 36 positive samples and did not detect the virus in any of the negative samples, giving a sensitivity and specificity of 100%.
“The CHIKV RPA assay presented here is a promising tool for CHIKV diagnostics at the point of need,” the investigators wrote. “Integration into a multimer or multiplex assay for simultaneous and differential detection of CHIKV, Dengue virus, and Zika virus, as well as an internal positive control would improve outbreak investigations, since the three viruses induce the same clinical picture upon infection and increasingly cocirculate in many parts of the world.”
Find the full study in PLOS Neglected Tropical Diseases (doi: 10.1371/journal.pntd.0004953).
A reverse transcription recombinase polymerase amplification (RT-RPA) assay was able to quickly and effectively identify chikungunya virus (CHIKV), according to a study published in PLOS Neglected Tropical Diseases.
Using chikungunya virus RNA samples, the RT-RPA assay detected down to 80 genome copies per reaction within 15 minutes, a time period four to six times faster than other molecular diagnostic techniques, such as reverse transcription-polymerase chain reaction (RT-PCR). In a sensitivity test involving all chikungunya serotypes and various alphaviruses, flaviviruses, and one phlebovirus, the RT-RPA assay identified all virus genotypes, with the only cross-reaction occurring with O’nyong’nyong virus.
In a test involving 58 plasma samples of suspected chikungunya fever from a trial in Thailand, two real-time RT-PCR tests identified 36 out of 58 samples (62%) as positive for chikungunya. The RT-RPA test successfully detected the virus in all 36 positive samples and did not detect the virus in any of the negative samples, giving a sensitivity and specificity of 100%.
“The CHIKV RPA assay presented here is a promising tool for CHIKV diagnostics at the point of need,” the investigators wrote. “Integration into a multimer or multiplex assay for simultaneous and differential detection of CHIKV, Dengue virus, and Zika virus, as well as an internal positive control would improve outbreak investigations, since the three viruses induce the same clinical picture upon infection and increasingly cocirculate in many parts of the world.”
Find the full study in PLOS Neglected Tropical Diseases (doi: 10.1371/journal.pntd.0004953).
FROM PLOS NEGLECTED TROPICAL DISEASES
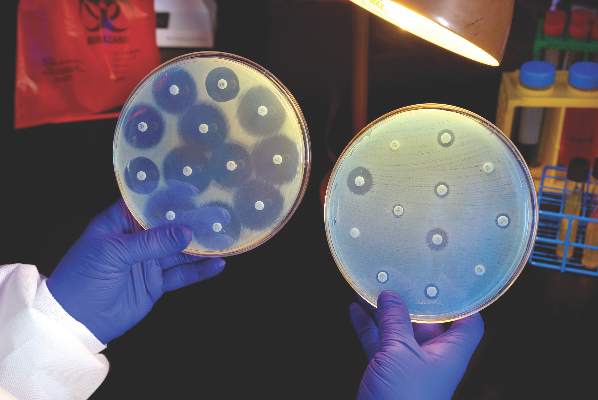
Ceftazidime-avibactam stands up to CRE, but resistance a problem
Intravenous ceftazidime-avibactam successfully treated 59% of carbapenem-resistant Enterobacteriaceae (CRE) infections, and 76% of patients remained alive at 30 days, according to a retrospective cohort study published in Clinical Infectious Diseases.
Those rates resemble previous reports of treatment with in vitro active agents, while the rate of acute kidney injury was about a third lower, said Ryan K. Shields, PharmD, of the University of Pittsburgh, and his associates. But 8% of CRE infections developed ceftazidime-avibactam resistance, which accounted for about a third of microbiological failures, the researchers said. “It is incumbent upon health care providers to share their clinical experiences with ceftazidime-avibactam and other new beta-lactamase inhibitors, so these agents can be used most effectively for the longest period of time,” they added.
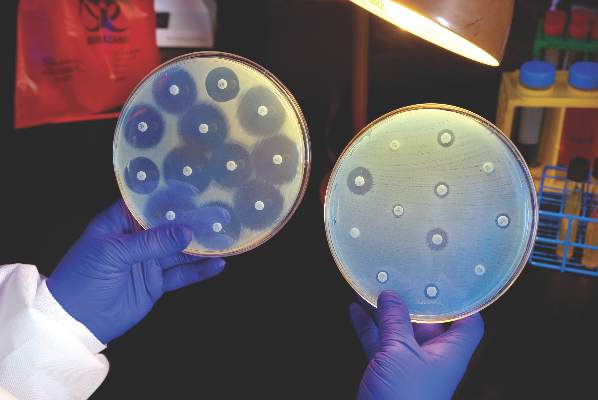
Ceftazidime-avibactam (Avycaz) is a novel beta-lactam/beta-lactamase inhibitor combination approved by the Food and Drug Administration in 2015 for complicated intra-abdominal and urinary tract infections. It was hoped that the newly approved combination would prove safer and more effective than previously developed agents that showed in vitro activity against CRE, such as colistin, gentamicin, and tigecycline, the researchers noted.
They described CRE-infected patients treated with ceftazidime-avibactam (median, 14 days; range, 4-71 days) between April 2015 and February 2016. The average age of the patients was 64 years (range 26-78 years), and 57% were men. Infections included ventilator or health care–associated pneumonia, primary bacteremia, intra-abdominal infection, skin and soft tissue infections, pyelonephritis, mediastinitis, subdural empyema/ventriculitis, and purulent tracheobronchitis. All the CRE isolates were susceptible to ceftazidime-avibactam at baseline. In all, 70% of patients received ceftazidime-avibactam as monotherapy, while 30% received it in combination with intravenous or inhaled gentamicin, intravenous or intrathecal colistin, or tigecycline (Clin Infect Dis. 2016 Sep 13. doi: 10.1093/cid/ciw636).
A total of 28 (76%) patients were alive at 30 days and 62% were alive at 90 days, the investigators said. They calculated a 59% rate of clinical success, defined as absence of recurrence within 30 days of onset, resolution of signs and symptoms, and sterilization of site-specific cultures within 7 days of treatment. Combination therapy did not improve the chances of clinical success, they noted. Among the 15 clinical failures, 9 patients died, 4 developed recurrent CRE infections, and 2 did not clinically improve. Clinical success was less likely when patients needed continuous renal replacement therapy (17% vs. 68% for other patients; P = .03) or had higher Sequential Organ Failure Assessment (SOFA) scores (average, 5.2 in clinical successes vs. 8.8 in clinical failures; P = .047). In addition, 10% of patients developed acute kidney injury within 7 days of starting treatment, which was “considerably lower than the approximately 30% rate we previously reported with carbapenem-colistin or aminoglycoside-based combinations,” the investigators said.
The sample size was too small to definitively answer questions about whether combination regimens can overcome resistance, improve outcomes, or effectively treat specific types of CRE infection, the researchers noted. “Nevertheless, we can conclude that ceftazidime-avibactam offers an important advance in the treatment of CRE infections,” they wrote. “The development of resistance after as few as 10 days of therapy is troubling, and treatment failures and deaths in a significant minority of patients highlight the need for more agents with activity against CRE.”
The University of Pittsburgh Medical Center and the National Institutes of Health provided funding. One coauthor disclosed ties to Meiji, Shionogi, Tetraphase, Achaogen, Merck, and The Medicines Company. The other authors had no disclosures.
In the movie “Jaws,” after confidently setting out with an experienced shark hunter, upon catching his first glimpse of the predator, Chief Brody famously uttered, “We’re gonna need a bigger boat.” Similarly, we rejoiced at our triumph when ceftazidime-avibactam became available to treat our patients infected with [Klebsiella pneumoniae carbapenemase]-producing bacteria, and confidently set out to combat this killer. But like Chief Brody, we appear to have underestimated our foe; we too need a “bigger boat.”
We must not let the past repeat itself; hubris about the sudden availability of effective antibiotics has led to overconfidence and complacency among the medical and microbiological communities on several prior occasions in the last 80 years, with serious societal consequences. Shields and colleagues have provided us with a sobering reminder that there is no endpoint in our struggle against microbes. They will never stop adapting to what we conceive of to combat them, and in turn we must never stop conceiving of new ways to stay one step ahead.
Brad Spellberg, MD, is at Los Angeles County–USC Medical Center in Los Angeles. He disclosed ties to Cempra, The Medicines Company, MedImmune/AstraZeneca, PTC Therapeutics, Entasis, Tetraphase, Merck, Genentech, Dipexium, Motif, BioAIM, and Synthetic Biologics. He has received grants from AstraZeneca, Merck, Melinta, Steris, NIH, and Veterans Affairs Merit Review. These comments are from an editorial (Clin Infect Dis. 2016 Sept 13. doi: 10.1093/cid/ciw639).
In the movie “Jaws,” after confidently setting out with an experienced shark hunter, upon catching his first glimpse of the predator, Chief Brody famously uttered, “We’re gonna need a bigger boat.” Similarly, we rejoiced at our triumph when ceftazidime-avibactam became available to treat our patients infected with [Klebsiella pneumoniae carbapenemase]-producing bacteria, and confidently set out to combat this killer. But like Chief Brody, we appear to have underestimated our foe; we too need a “bigger boat.”
We must not let the past repeat itself; hubris about the sudden availability of effective antibiotics has led to overconfidence and complacency among the medical and microbiological communities on several prior occasions in the last 80 years, with serious societal consequences. Shields and colleagues have provided us with a sobering reminder that there is no endpoint in our struggle against microbes. They will never stop adapting to what we conceive of to combat them, and in turn we must never stop conceiving of new ways to stay one step ahead.
Brad Spellberg, MD, is at Los Angeles County–USC Medical Center in Los Angeles. He disclosed ties to Cempra, The Medicines Company, MedImmune/AstraZeneca, PTC Therapeutics, Entasis, Tetraphase, Merck, Genentech, Dipexium, Motif, BioAIM, and Synthetic Biologics. He has received grants from AstraZeneca, Merck, Melinta, Steris, NIH, and Veterans Affairs Merit Review. These comments are from an editorial (Clin Infect Dis. 2016 Sept 13. doi: 10.1093/cid/ciw639).
In the movie “Jaws,” after confidently setting out with an experienced shark hunter, upon catching his first glimpse of the predator, Chief Brody famously uttered, “We’re gonna need a bigger boat.” Similarly, we rejoiced at our triumph when ceftazidime-avibactam became available to treat our patients infected with [Klebsiella pneumoniae carbapenemase]-producing bacteria, and confidently set out to combat this killer. But like Chief Brody, we appear to have underestimated our foe; we too need a “bigger boat.”
We must not let the past repeat itself; hubris about the sudden availability of effective antibiotics has led to overconfidence and complacency among the medical and microbiological communities on several prior occasions in the last 80 years, with serious societal consequences. Shields and colleagues have provided us with a sobering reminder that there is no endpoint in our struggle against microbes. They will never stop adapting to what we conceive of to combat them, and in turn we must never stop conceiving of new ways to stay one step ahead.
Brad Spellberg, MD, is at Los Angeles County–USC Medical Center in Los Angeles. He disclosed ties to Cempra, The Medicines Company, MedImmune/AstraZeneca, PTC Therapeutics, Entasis, Tetraphase, Merck, Genentech, Dipexium, Motif, BioAIM, and Synthetic Biologics. He has received grants from AstraZeneca, Merck, Melinta, Steris, NIH, and Veterans Affairs Merit Review. These comments are from an editorial (Clin Infect Dis. 2016 Sept 13. doi: 10.1093/cid/ciw639).
Intravenous ceftazidime-avibactam successfully treated 59% of carbapenem-resistant Enterobacteriaceae (CRE) infections, and 76% of patients remained alive at 30 days, according to a retrospective cohort study published in Clinical Infectious Diseases.
Those rates resemble previous reports of treatment with in vitro active agents, while the rate of acute kidney injury was about a third lower, said Ryan K. Shields, PharmD, of the University of Pittsburgh, and his associates. But 8% of CRE infections developed ceftazidime-avibactam resistance, which accounted for about a third of microbiological failures, the researchers said. “It is incumbent upon health care providers to share their clinical experiences with ceftazidime-avibactam and other new beta-lactamase inhibitors, so these agents can be used most effectively for the longest period of time,” they added.
Ceftazidime-avibactam (Avycaz) is a novel beta-lactam/beta-lactamase inhibitor combination approved by the Food and Drug Administration in 2015 for complicated intra-abdominal and urinary tract infections. It was hoped that the newly approved combination would prove safer and more effective than previously developed agents that showed in vitro activity against CRE, such as colistin, gentamicin, and tigecycline, the researchers noted.
They described CRE-infected patients treated with ceftazidime-avibactam (median, 14 days; range, 4-71 days) between April 2015 and February 2016. The average age of the patients was 64 years (range 26-78 years), and 57% were men. Infections included ventilator or health care–associated pneumonia, primary bacteremia, intra-abdominal infection, skin and soft tissue infections, pyelonephritis, mediastinitis, subdural empyema/ventriculitis, and purulent tracheobronchitis. All the CRE isolates were susceptible to ceftazidime-avibactam at baseline. In all, 70% of patients received ceftazidime-avibactam as monotherapy, while 30% received it in combination with intravenous or inhaled gentamicin, intravenous or intrathecal colistin, or tigecycline (Clin Infect Dis. 2016 Sep 13. doi: 10.1093/cid/ciw636).
A total of 28 (76%) patients were alive at 30 days and 62% were alive at 90 days, the investigators said. They calculated a 59% rate of clinical success, defined as absence of recurrence within 30 days of onset, resolution of signs and symptoms, and sterilization of site-specific cultures within 7 days of treatment. Combination therapy did not improve the chances of clinical success, they noted. Among the 15 clinical failures, 9 patients died, 4 developed recurrent CRE infections, and 2 did not clinically improve. Clinical success was less likely when patients needed continuous renal replacement therapy (17% vs. 68% for other patients; P = .03) or had higher Sequential Organ Failure Assessment (SOFA) scores (average, 5.2 in clinical successes vs. 8.8 in clinical failures; P = .047). In addition, 10% of patients developed acute kidney injury within 7 days of starting treatment, which was “considerably lower than the approximately 30% rate we previously reported with carbapenem-colistin or aminoglycoside-based combinations,” the investigators said.
The sample size was too small to definitively answer questions about whether combination regimens can overcome resistance, improve outcomes, or effectively treat specific types of CRE infection, the researchers noted. “Nevertheless, we can conclude that ceftazidime-avibactam offers an important advance in the treatment of CRE infections,” they wrote. “The development of resistance after as few as 10 days of therapy is troubling, and treatment failures and deaths in a significant minority of patients highlight the need for more agents with activity against CRE.”
The University of Pittsburgh Medical Center and the National Institutes of Health provided funding. One coauthor disclosed ties to Meiji, Shionogi, Tetraphase, Achaogen, Merck, and The Medicines Company. The other authors had no disclosures.
Intravenous ceftazidime-avibactam successfully treated 59% of carbapenem-resistant Enterobacteriaceae (CRE) infections, and 76% of patients remained alive at 30 days, according to a retrospective cohort study published in Clinical Infectious Diseases.
Those rates resemble previous reports of treatment with in vitro active agents, while the rate of acute kidney injury was about a third lower, said Ryan K. Shields, PharmD, of the University of Pittsburgh, and his associates. But 8% of CRE infections developed ceftazidime-avibactam resistance, which accounted for about a third of microbiological failures, the researchers said. “It is incumbent upon health care providers to share their clinical experiences with ceftazidime-avibactam and other new beta-lactamase inhibitors, so these agents can be used most effectively for the longest period of time,” they added.
Ceftazidime-avibactam (Avycaz) is a novel beta-lactam/beta-lactamase inhibitor combination approved by the Food and Drug Administration in 2015 for complicated intra-abdominal and urinary tract infections. It was hoped that the newly approved combination would prove safer and more effective than previously developed agents that showed in vitro activity against CRE, such as colistin, gentamicin, and tigecycline, the researchers noted.
They described CRE-infected patients treated with ceftazidime-avibactam (median, 14 days; range, 4-71 days) between April 2015 and February 2016. The average age of the patients was 64 years (range 26-78 years), and 57% were men. Infections included ventilator or health care–associated pneumonia, primary bacteremia, intra-abdominal infection, skin and soft tissue infections, pyelonephritis, mediastinitis, subdural empyema/ventriculitis, and purulent tracheobronchitis. All the CRE isolates were susceptible to ceftazidime-avibactam at baseline. In all, 70% of patients received ceftazidime-avibactam as monotherapy, while 30% received it in combination with intravenous or inhaled gentamicin, intravenous or intrathecal colistin, or tigecycline (Clin Infect Dis. 2016 Sep 13. doi: 10.1093/cid/ciw636).
A total of 28 (76%) patients were alive at 30 days and 62% were alive at 90 days, the investigators said. They calculated a 59% rate of clinical success, defined as absence of recurrence within 30 days of onset, resolution of signs and symptoms, and sterilization of site-specific cultures within 7 days of treatment. Combination therapy did not improve the chances of clinical success, they noted. Among the 15 clinical failures, 9 patients died, 4 developed recurrent CRE infections, and 2 did not clinically improve. Clinical success was less likely when patients needed continuous renal replacement therapy (17% vs. 68% for other patients; P = .03) or had higher Sequential Organ Failure Assessment (SOFA) scores (average, 5.2 in clinical successes vs. 8.8 in clinical failures; P = .047). In addition, 10% of patients developed acute kidney injury within 7 days of starting treatment, which was “considerably lower than the approximately 30% rate we previously reported with carbapenem-colistin or aminoglycoside-based combinations,” the investigators said.
The sample size was too small to definitively answer questions about whether combination regimens can overcome resistance, improve outcomes, or effectively treat specific types of CRE infection, the researchers noted. “Nevertheless, we can conclude that ceftazidime-avibactam offers an important advance in the treatment of CRE infections,” they wrote. “The development of resistance after as few as 10 days of therapy is troubling, and treatment failures and deaths in a significant minority of patients highlight the need for more agents with activity against CRE.”
The University of Pittsburgh Medical Center and the National Institutes of Health provided funding. One coauthor disclosed ties to Meiji, Shionogi, Tetraphase, Achaogen, Merck, and The Medicines Company. The other authors had no disclosures.
FROM CLINICAL INFECTIOUS DISEASES
Key clinical point: Ceftazidime-avibactam effectively treated carbapenem-resistant Enterobacteriaceae (CRE) infections, but resistance emerged rapidly and in some cases led to microbiological failure.
Major finding: The rate of clinical success was 59%; 10% of patients developed acute kidney injury within 7 days of starting treatment; 8% developed resistance.
Data source: A single-center retrospective study of 37 patients with carbapenem-resistant Enterobacteriaceae infections treated with ceftazidime-avibactam.
Disclosures: The University of Pittsburgh Medical Center and the National Institutes of Health provided funding. A coauthor disclosed ties to Meiji, Shionogi, Tetraphase, Achaogen, Merck, and The Medicines Company. The other authors had no disclosures.
Targeted HCV patients improve on sofosbuvir/daclatasvir combination
A combination of sofosbuvir/daclatasvir yielded sustained virological responses at 12 weeks after the last treatment in 95% of hepatitis C virus–infected patients with genotype 1.
“Real-life results of the sofosbuvir + ribavirin or sofosbuvir + simeprevir combination have been extensively reported, but there are few data regarding the sofosbuvir + daclatasvir combination in genotype 1–infected patients,” wrote Stanislas Pol, MD, of Hôpital Cochin, Institut Pasteur, Paris, and his colleagues.
To assess the effectiveness of the combination, researchers reviewed data from 768 patients with HCV genotype 1 who began treatments of 400 mg/day sofosbuvir and 60 mg/day daclatasvir prior to Oct. 1, 2014 (J Hepatol. 2016. doi: 10.1016/j.jhep.2016.08.021). Patients were treated for 12 or 24 weeks, and the primary endpoint was sustained virological response 12 weeks after the last treatment (SVR12).
A total of 92% of patients treated for 12 weeks and 99% of patients treated for 24 weeks with the combination met the primary endpoint of SVR12, for an average of 95% overall. Treatment duration and the presence or absence of ribavirin had no significant impact on the treatment responses in noncirrhotic patients. However, the SVR12 rate was significantly higher among cirrhotic patients in the 24-week treatment group than in the 12-week group (95% vs. 88%).
One patient died from cerebral hemorrhage 6 weeks after beginning treatment, and the death was considered possibly related to the combination treatment; two deaths from septic shock and two deaths from end-stage liver disease were not considered treatment related. Other serious adverse events were reported in 10% of patients independent of treatment duration or use of ribavirin. The six serious adverse events possibly related to treatment included three cardiac disorders. The most common adverse events included insomnia, headache, and asthenia, reported in at least 10% of patients.
Only decompensated cirrhosis and a prothrombin time greater than 70% were independently associated with serious adverse events.
The study was limited by several factors, including its observational nature and relatively low number of patients treated with ribavirin in the 12-week group, the researchers noted. However, the results suggest that “in real life, the sofosbuvir + daclatasvir combination in difficult-to-treat patients with HCV genotype 1 infection was associated with a high rate of SVR12,” they said.
Inserm-ANRS supported the study. The researchers disclosed funding from government organizations and pharmaceutical companies including MSD, Janssen, Gilead, AbbVie, BMS, and Roche.
A combination of sofosbuvir/daclatasvir yielded sustained virological responses at 12 weeks after the last treatment in 95% of hepatitis C virus–infected patients with genotype 1.
“Real-life results of the sofosbuvir + ribavirin or sofosbuvir + simeprevir combination have been extensively reported, but there are few data regarding the sofosbuvir + daclatasvir combination in genotype 1–infected patients,” wrote Stanislas Pol, MD, of Hôpital Cochin, Institut Pasteur, Paris, and his colleagues.
To assess the effectiveness of the combination, researchers reviewed data from 768 patients with HCV genotype 1 who began treatments of 400 mg/day sofosbuvir and 60 mg/day daclatasvir prior to Oct. 1, 2014 (J Hepatol. 2016. doi: 10.1016/j.jhep.2016.08.021). Patients were treated for 12 or 24 weeks, and the primary endpoint was sustained virological response 12 weeks after the last treatment (SVR12).
A total of 92% of patients treated for 12 weeks and 99% of patients treated for 24 weeks with the combination met the primary endpoint of SVR12, for an average of 95% overall. Treatment duration and the presence or absence of ribavirin had no significant impact on the treatment responses in noncirrhotic patients. However, the SVR12 rate was significantly higher among cirrhotic patients in the 24-week treatment group than in the 12-week group (95% vs. 88%).
One patient died from cerebral hemorrhage 6 weeks after beginning treatment, and the death was considered possibly related to the combination treatment; two deaths from septic shock and two deaths from end-stage liver disease were not considered treatment related. Other serious adverse events were reported in 10% of patients independent of treatment duration or use of ribavirin. The six serious adverse events possibly related to treatment included three cardiac disorders. The most common adverse events included insomnia, headache, and asthenia, reported in at least 10% of patients.
Only decompensated cirrhosis and a prothrombin time greater than 70% were independently associated with serious adverse events.
The study was limited by several factors, including its observational nature and relatively low number of patients treated with ribavirin in the 12-week group, the researchers noted. However, the results suggest that “in real life, the sofosbuvir + daclatasvir combination in difficult-to-treat patients with HCV genotype 1 infection was associated with a high rate of SVR12,” they said.
Inserm-ANRS supported the study. The researchers disclosed funding from government organizations and pharmaceutical companies including MSD, Janssen, Gilead, AbbVie, BMS, and Roche.
A combination of sofosbuvir/daclatasvir yielded sustained virological responses at 12 weeks after the last treatment in 95% of hepatitis C virus–infected patients with genotype 1.
“Real-life results of the sofosbuvir + ribavirin or sofosbuvir + simeprevir combination have been extensively reported, but there are few data regarding the sofosbuvir + daclatasvir combination in genotype 1–infected patients,” wrote Stanislas Pol, MD, of Hôpital Cochin, Institut Pasteur, Paris, and his colleagues.
To assess the effectiveness of the combination, researchers reviewed data from 768 patients with HCV genotype 1 who began treatments of 400 mg/day sofosbuvir and 60 mg/day daclatasvir prior to Oct. 1, 2014 (J Hepatol. 2016. doi: 10.1016/j.jhep.2016.08.021). Patients were treated for 12 or 24 weeks, and the primary endpoint was sustained virological response 12 weeks after the last treatment (SVR12).
A total of 92% of patients treated for 12 weeks and 99% of patients treated for 24 weeks with the combination met the primary endpoint of SVR12, for an average of 95% overall. Treatment duration and the presence or absence of ribavirin had no significant impact on the treatment responses in noncirrhotic patients. However, the SVR12 rate was significantly higher among cirrhotic patients in the 24-week treatment group than in the 12-week group (95% vs. 88%).
One patient died from cerebral hemorrhage 6 weeks after beginning treatment, and the death was considered possibly related to the combination treatment; two deaths from septic shock and two deaths from end-stage liver disease were not considered treatment related. Other serious adverse events were reported in 10% of patients independent of treatment duration or use of ribavirin. The six serious adverse events possibly related to treatment included three cardiac disorders. The most common adverse events included insomnia, headache, and asthenia, reported in at least 10% of patients.
Only decompensated cirrhosis and a prothrombin time greater than 70% were independently associated with serious adverse events.
The study was limited by several factors, including its observational nature and relatively low number of patients treated with ribavirin in the 12-week group, the researchers noted. However, the results suggest that “in real life, the sofosbuvir + daclatasvir combination in difficult-to-treat patients with HCV genotype 1 infection was associated with a high rate of SVR12,” they said.
Inserm-ANRS supported the study. The researchers disclosed funding from government organizations and pharmaceutical companies including MSD, Janssen, Gilead, AbbVie, BMS, and Roche.
FROM THE JOURNAL OF HEPATOLOGY
Key clinical point: A sofosbuvir/daclatasvir combination was effective in most patients with HCV genotype 1, independent of an addition of ribavirin.
Major finding: Sustained virological response at 12 weeks after the last treatment occurred in 95% of patients treated with a combination of sofosbuvir and daclatasvir.
Data source: A selection of 768 patients with a HCV genotype 1 who were part of a ongoing multicenter, observational cohort study.
Disclosures: Inserm-ANRS supported the study. The researchers disclosed funding from government organizations and pharmaceutical companies including MSD, Janssen, Gilead, AbbVie, BMS, and Roche.
Hot flashes and sleep disruption contribute independently to depression in menopause
Hot flashes and sleep disruption contribute independently to the development of depression in menopause, judging from the findings of a recent study.
In that study, 29 premenopausal women, aged 18-45 years, received a single dose of the GnRH agonist leuprolide in order to induce hypoestrogenism and ovarian suppression for the study period. The women in the study had no history of primary sleep disturbances, low estrogen levels, or depression, according to Hadine Joffe, MD, director of the Women’s Hormone and Aging Research Program at Harvard Medical School, in Boston, and her associates.
All the study participants underwent baseline mood evaluation using both the Montgomery-Asberg Depression Rating Scale (MADRS) and the Beck Depression Inventory (BDI). Existing sleep disturbances were ruled out at baseline with sleep diaries, questionnaires, and two ambulatory screening polysomnography (PSG) studies.
After 4 weeks of administration of leuprolide, depressive symptoms had developed among most of the women in the study. The mean MADRS score was 4.1, and overall, it was 3.1 points higher than it had been at baseline. One woman had a 15-point increase in her score, suggesting significant depression. The MADRS score increased by at least 5 points in 24% of the women and remained unchanged in 38%, reflecting variability among the women on the impact of leuprolide on depressive symptoms, the investigators reported (J Clin Endocrinol Metab. 2016 Sep 20. doi: 10.1210/jc.2016-2348).
Leuprolide universally suppressed estradiol to postmenopausal levels in the women within 2 weeks. Hot flashes developed in 20 (69%) women, with a median of 3.6 hot flashes during the day and 3.8 at night. The median number of objectively measured nighttime hot flashes per night was 3.
Changes to sleep patterns varied widely for each woman; for example, wake time after sleep onset ranged from an additional 140 minutes to 23 fewer minutes for one woman. There was a correlation between the number of subjectively reported nighttime hot flashes with increased sleep fragmentation as measured by PSG. The number of reported nighttime hot flashes was associated with an increase in depressive symptoms that was disproportionate to the number of nighttime hot flashes reported, according to the findings of a univariate analysis. The number of daytime hot flashes had no such effect.
In light of these findings, Dr. Joffe and her associates urged clinicians to screen women who report nighttime hot flashes and sleep interruption for mood disturbance. “Treatment of those with menopause-related depressive symptoms should encompass therapies that improve sleep interruption as well as nocturnal [hot flashes],” they wrote.
The study was sponsored by the National Institute of Mental Health. Dr. Joffe has received grant support from Merck and has served as a consultant/adviser for Merck, Mitsubishi Tanabe, NeRRe Therapeutics, and Noven.
Hot flashes and sleep disruption contribute independently to the development of depression in menopause, judging from the findings of a recent study.
In that study, 29 premenopausal women, aged 18-45 years, received a single dose of the GnRH agonist leuprolide in order to induce hypoestrogenism and ovarian suppression for the study period. The women in the study had no history of primary sleep disturbances, low estrogen levels, or depression, according to Hadine Joffe, MD, director of the Women’s Hormone and Aging Research Program at Harvard Medical School, in Boston, and her associates.
All the study participants underwent baseline mood evaluation using both the Montgomery-Asberg Depression Rating Scale (MADRS) and the Beck Depression Inventory (BDI). Existing sleep disturbances were ruled out at baseline with sleep diaries, questionnaires, and two ambulatory screening polysomnography (PSG) studies.
After 4 weeks of administration of leuprolide, depressive symptoms had developed among most of the women in the study. The mean MADRS score was 4.1, and overall, it was 3.1 points higher than it had been at baseline. One woman had a 15-point increase in her score, suggesting significant depression. The MADRS score increased by at least 5 points in 24% of the women and remained unchanged in 38%, reflecting variability among the women on the impact of leuprolide on depressive symptoms, the investigators reported (J Clin Endocrinol Metab. 2016 Sep 20. doi: 10.1210/jc.2016-2348).
Leuprolide universally suppressed estradiol to postmenopausal levels in the women within 2 weeks. Hot flashes developed in 20 (69%) women, with a median of 3.6 hot flashes during the day and 3.8 at night. The median number of objectively measured nighttime hot flashes per night was 3.
Changes to sleep patterns varied widely for each woman; for example, wake time after sleep onset ranged from an additional 140 minutes to 23 fewer minutes for one woman. There was a correlation between the number of subjectively reported nighttime hot flashes with increased sleep fragmentation as measured by PSG. The number of reported nighttime hot flashes was associated with an increase in depressive symptoms that was disproportionate to the number of nighttime hot flashes reported, according to the findings of a univariate analysis. The number of daytime hot flashes had no such effect.
In light of these findings, Dr. Joffe and her associates urged clinicians to screen women who report nighttime hot flashes and sleep interruption for mood disturbance. “Treatment of those with menopause-related depressive symptoms should encompass therapies that improve sleep interruption as well as nocturnal [hot flashes],” they wrote.
The study was sponsored by the National Institute of Mental Health. Dr. Joffe has received grant support from Merck and has served as a consultant/adviser for Merck, Mitsubishi Tanabe, NeRRe Therapeutics, and Noven.
Hot flashes and sleep disruption contribute independently to the development of depression in menopause, judging from the findings of a recent study.
In that study, 29 premenopausal women, aged 18-45 years, received a single dose of the GnRH agonist leuprolide in order to induce hypoestrogenism and ovarian suppression for the study period. The women in the study had no history of primary sleep disturbances, low estrogen levels, or depression, according to Hadine Joffe, MD, director of the Women’s Hormone and Aging Research Program at Harvard Medical School, in Boston, and her associates.
All the study participants underwent baseline mood evaluation using both the Montgomery-Asberg Depression Rating Scale (MADRS) and the Beck Depression Inventory (BDI). Existing sleep disturbances were ruled out at baseline with sleep diaries, questionnaires, and two ambulatory screening polysomnography (PSG) studies.
After 4 weeks of administration of leuprolide, depressive symptoms had developed among most of the women in the study. The mean MADRS score was 4.1, and overall, it was 3.1 points higher than it had been at baseline. One woman had a 15-point increase in her score, suggesting significant depression. The MADRS score increased by at least 5 points in 24% of the women and remained unchanged in 38%, reflecting variability among the women on the impact of leuprolide on depressive symptoms, the investigators reported (J Clin Endocrinol Metab. 2016 Sep 20. doi: 10.1210/jc.2016-2348).
Leuprolide universally suppressed estradiol to postmenopausal levels in the women within 2 weeks. Hot flashes developed in 20 (69%) women, with a median of 3.6 hot flashes during the day and 3.8 at night. The median number of objectively measured nighttime hot flashes per night was 3.
Changes to sleep patterns varied widely for each woman; for example, wake time after sleep onset ranged from an additional 140 minutes to 23 fewer minutes for one woman. There was a correlation between the number of subjectively reported nighttime hot flashes with increased sleep fragmentation as measured by PSG. The number of reported nighttime hot flashes was associated with an increase in depressive symptoms that was disproportionate to the number of nighttime hot flashes reported, according to the findings of a univariate analysis. The number of daytime hot flashes had no such effect.
In light of these findings, Dr. Joffe and her associates urged clinicians to screen women who report nighttime hot flashes and sleep interruption for mood disturbance. “Treatment of those with menopause-related depressive symptoms should encompass therapies that improve sleep interruption as well as nocturnal [hot flashes],” they wrote.
The study was sponsored by the National Institute of Mental Health. Dr. Joffe has received grant support from Merck and has served as a consultant/adviser for Merck, Mitsubishi Tanabe, NeRRe Therapeutics, and Noven.
FROM THE JOURNAL OF ENDOCRINOLOGY & METABOLISM
Key clinical point: Hot flashes and sleep disruption contribute independently to the development of depression in menopause.
Major finding: Depression scores increased by 3 points on the MADRS after 4 weeks on GnRH agonist leuprolide. Sleep disruption was also common among the women in the study. Depression developed among many, but not all the women, with univariate analysis showing that hot flashes and sleep disruption contributed independently to their mood changes.
Data source: A prospective study in which 29 young women without depression were subjected to rapid, premature, and reversible menopause with one open-label dose of leuprolide in an experimental model.
Disclosures: The study was sponsored by the National Institute of Mental Health. Dr. Hadine Joffe has received grant support from Merck and has served as a consultant/adviser for Merck, Mitsubishi Tanabe, NeRRe Therapeutics, and Noven.
Congress sends Zika funding bill to President
In a move that narrowly avoids a government shutdown, Congress has passed a long-awaited bill that keeps the government afloat and provides $1.1 billion in funding to combat the Zika virus.
The House cleared H.R. 5325 late Sept. 28 by a 342-85 tally, following a 72-26 vote by the Senate earlier in the day. The final package, which keeps the government operating through Dec. 9, also includes $37 million for opioid addiction and $500 million for flooding in Louisiana. The White House has indicated that President Obama will sign the bill into law.
The American Congress of Obstetricians and Gynecologists praised Congress for passing the long-delayed comprehensive Zika funding package.

“Congress has finally treated Zika like the emergency it is and shown the American people that it is capable of rising above partisanship for the health of its citizens,” Thomas Gellhaus, MD, ACOG president, said in a statement. “ACOG stands with peer organizations and government agencies in the fight to prevent and respond to the Zika virus and support the care and treatment of all people affected by it. ... The fight against the spread of Zika cannot be won without the resources to support responsive and proactive solutions. This comprehensive funding package is essential to our success and the health of women and babies.”
The bill’s passage caps months of fiery debate within Congress over what to include in the measure. The bill stalled earlier this week largely over whether to direct funds to Flint, Mich., to deal with the crisis over lead-tainted water. Leaders agreed to provide aid to Flint residents in a separate water projects bill. Legislators will address final approval of the Flint measure in December.
Of the $1.1 billion included in the final package to fight Zika, $15 million would go to Florida and $60 million to the territory of Puerto Rico to respond to Zika outbreaks in those areas. The remainder of the funding would be used to prevent, prepare for, and respond to Zika; health conditions related to such virus; and other vector-borne diseases, domestically and internationally.

If signed by the President, the money would also go toward developing necessary countermeasures and vaccines, including the development and purchase of vaccines, therapeutics, diagnostics, and necessary medical supplies. Additionally, the funding would aid research on the virology, natural history, and pathogenesis of the Zika virus infection and preclinical and clinical development of vaccines and other medical countermeasures for the Zika virus.
The American Medical Association expressed relief that Congress had finally taken action to provide resources for fighting Zika.
“It has been clear over the past several months that the U.S. has needed additional resources to combat the Zika virus,” AMA president Andrew W. Gurman, MD, said in a statement. “With the threat of the virus continuing to loom, this funding will help protect more people – particularly pregnant women and their children – from the virus’s lasting negative health effects.”
On Twitter @legal_med
In a move that narrowly avoids a government shutdown, Congress has passed a long-awaited bill that keeps the government afloat and provides $1.1 billion in funding to combat the Zika virus.
The House cleared H.R. 5325 late Sept. 28 by a 342-85 tally, following a 72-26 vote by the Senate earlier in the day. The final package, which keeps the government operating through Dec. 9, also includes $37 million for opioid addiction and $500 million for flooding in Louisiana. The White House has indicated that President Obama will sign the bill into law.
The American Congress of Obstetricians and Gynecologists praised Congress for passing the long-delayed comprehensive Zika funding package.
“Congress has finally treated Zika like the emergency it is and shown the American people that it is capable of rising above partisanship for the health of its citizens,” Thomas Gellhaus, MD, ACOG president, said in a statement. “ACOG stands with peer organizations and government agencies in the fight to prevent and respond to the Zika virus and support the care and treatment of all people affected by it. ... The fight against the spread of Zika cannot be won without the resources to support responsive and proactive solutions. This comprehensive funding package is essential to our success and the health of women and babies.”
The bill’s passage caps months of fiery debate within Congress over what to include in the measure. The bill stalled earlier this week largely over whether to direct funds to Flint, Mich., to deal with the crisis over lead-tainted water. Leaders agreed to provide aid to Flint residents in a separate water projects bill. Legislators will address final approval of the Flint measure in December.
Of the $1.1 billion included in the final package to fight Zika, $15 million would go to Florida and $60 million to the territory of Puerto Rico to respond to Zika outbreaks in those areas. The remainder of the funding would be used to prevent, prepare for, and respond to Zika; health conditions related to such virus; and other vector-borne diseases, domestically and internationally.
If signed by the President, the money would also go toward developing necessary countermeasures and vaccines, including the development and purchase of vaccines, therapeutics, diagnostics, and necessary medical supplies. Additionally, the funding would aid research on the virology, natural history, and pathogenesis of the Zika virus infection and preclinical and clinical development of vaccines and other medical countermeasures for the Zika virus.
The American Medical Association expressed relief that Congress had finally taken action to provide resources for fighting Zika.
“It has been clear over the past several months that the U.S. has needed additional resources to combat the Zika virus,” AMA president Andrew W. Gurman, MD, said in a statement. “With the threat of the virus continuing to loom, this funding will help protect more people – particularly pregnant women and their children – from the virus’s lasting negative health effects.”
On Twitter @legal_med
In a move that narrowly avoids a government shutdown, Congress has passed a long-awaited bill that keeps the government afloat and provides $1.1 billion in funding to combat the Zika virus.
The House cleared H.R. 5325 late Sept. 28 by a 342-85 tally, following a 72-26 vote by the Senate earlier in the day. The final package, which keeps the government operating through Dec. 9, also includes $37 million for opioid addiction and $500 million for flooding in Louisiana. The White House has indicated that President Obama will sign the bill into law.
The American Congress of Obstetricians and Gynecologists praised Congress for passing the long-delayed comprehensive Zika funding package.
“Congress has finally treated Zika like the emergency it is and shown the American people that it is capable of rising above partisanship for the health of its citizens,” Thomas Gellhaus, MD, ACOG president, said in a statement. “ACOG stands with peer organizations and government agencies in the fight to prevent and respond to the Zika virus and support the care and treatment of all people affected by it. ... The fight against the spread of Zika cannot be won without the resources to support responsive and proactive solutions. This comprehensive funding package is essential to our success and the health of women and babies.”
The bill’s passage caps months of fiery debate within Congress over what to include in the measure. The bill stalled earlier this week largely over whether to direct funds to Flint, Mich., to deal with the crisis over lead-tainted water. Leaders agreed to provide aid to Flint residents in a separate water projects bill. Legislators will address final approval of the Flint measure in December.
Of the $1.1 billion included in the final package to fight Zika, $15 million would go to Florida and $60 million to the territory of Puerto Rico to respond to Zika outbreaks in those areas. The remainder of the funding would be used to prevent, prepare for, and respond to Zika; health conditions related to such virus; and other vector-borne diseases, domestically and internationally.
If signed by the President, the money would also go toward developing necessary countermeasures and vaccines, including the development and purchase of vaccines, therapeutics, diagnostics, and necessary medical supplies. Additionally, the funding would aid research on the virology, natural history, and pathogenesis of the Zika virus infection and preclinical and clinical development of vaccines and other medical countermeasures for the Zika virus.
The American Medical Association expressed relief that Congress had finally taken action to provide resources for fighting Zika.
“It has been clear over the past several months that the U.S. has needed additional resources to combat the Zika virus,” AMA president Andrew W. Gurman, MD, said in a statement. “With the threat of the virus continuing to loom, this funding will help protect more people – particularly pregnant women and their children – from the virus’s lasting negative health effects.”
On Twitter @legal_med

AATS Mitral Conclave Call for Abstracts & Videos
AATS welcomes you to submit your abstracts and videos to the 2017 Mitral Conclave.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
Submission Deadline:
Sunday, January 8, 2017 @ 11.59 pm EST
Share:
AATS welcomes you to submit your abstracts and videos to the 2017 Mitral Conclave.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
Submission Deadline:
Sunday, January 8, 2017 @ 11.59 pm EST
Share:
AATS welcomes you to submit your abstracts and videos to the 2017 Mitral Conclave.
AATS Mitral Conclave
April 27-28, 2017
New York, NY
Submission Deadline:
Sunday, January 8, 2017 @ 11.59 pm EST
Share: