User login
Simple change increases forced-air warming use in trauma
SAN ANTONIO – A month-long quality improvement project to increase the use of forced-air warming blankets reduced mean hypothermia times in trauma patients at Parkland Memorial Hospital, Dallas, from 229 to 154 minutes.
All it took to get doctors, nurses, and staff to use the forced-air warming blankets more often was a reminder that hypothermia is an independent predictor of death in trauma, and data showing that Parkland, a Level 1 trauma center, used forced-air warming in just 11% of its hypothermic trauma patients. Meetings to get those points across were held in December 2014.
Forced-air warming jumped to 70% of hypothermic patients over the next 4 months in 2015 (P equal to or less than .0001), leading to the 33% drop in rewarming times (P = .009). The improvement came without any shift in the use of the rewarming methods trauma teams were in the habit of using: warm blankets, room air, and IV fluids.
Investigator Dr. Frank Zhao thinks it’s something all trauma centers can and should do. “There’s no reason that we shouldn’t recommend this be part of the rewarming protocol in every trauma center. It took about a month to roll this out so everyone was on the same page and was easily achieved,” said Dr. Zhao, formerly a Parkland surgery resident but now a trauma and surgical critical care fellow at the Oregon Health and Sciences University in Portland.
The blankets are an almost universal presence in operating rooms to keep core temperatures at least 36 degrees Celsius, but “from what I’ve seen at multiple institutions, the Bair Hugger is probably one of the least used warming methods” in trauma. “They’re recommended for trauma rewarming, but we [didn’t] use them very often.” Staff were not in the habit, he said at the Eastern Association for the Surgery of Trauma scientific assembly.
From July to November 2014, before the intervention, 15.2% (114) of Levels 1 and 2 trauma patients arrived at Parkland hypothermic, versus 20.9% (82) during the colder period of January-April 2015. Almost 80% of the trauma patients over that time were male, and the average patient age was about 40 years.
The investigators have no disclosures, and there was no outside funding for the project.
SAN ANTONIO – A month-long quality improvement project to increase the use of forced-air warming blankets reduced mean hypothermia times in trauma patients at Parkland Memorial Hospital, Dallas, from 229 to 154 minutes.
All it took to get doctors, nurses, and staff to use the forced-air warming blankets more often was a reminder that hypothermia is an independent predictor of death in trauma, and data showing that Parkland, a Level 1 trauma center, used forced-air warming in just 11% of its hypothermic trauma patients. Meetings to get those points across were held in December 2014.
Forced-air warming jumped to 70% of hypothermic patients over the next 4 months in 2015 (P equal to or less than .0001), leading to the 33% drop in rewarming times (P = .009). The improvement came without any shift in the use of the rewarming methods trauma teams were in the habit of using: warm blankets, room air, and IV fluids.
Investigator Dr. Frank Zhao thinks it’s something all trauma centers can and should do. “There’s no reason that we shouldn’t recommend this be part of the rewarming protocol in every trauma center. It took about a month to roll this out so everyone was on the same page and was easily achieved,” said Dr. Zhao, formerly a Parkland surgery resident but now a trauma and surgical critical care fellow at the Oregon Health and Sciences University in Portland.
The blankets are an almost universal presence in operating rooms to keep core temperatures at least 36 degrees Celsius, but “from what I’ve seen at multiple institutions, the Bair Hugger is probably one of the least used warming methods” in trauma. “They’re recommended for trauma rewarming, but we [didn’t] use them very often.” Staff were not in the habit, he said at the Eastern Association for the Surgery of Trauma scientific assembly.
From July to November 2014, before the intervention, 15.2% (114) of Levels 1 and 2 trauma patients arrived at Parkland hypothermic, versus 20.9% (82) during the colder period of January-April 2015. Almost 80% of the trauma patients over that time were male, and the average patient age was about 40 years.
The investigators have no disclosures, and there was no outside funding for the project.
SAN ANTONIO – A month-long quality improvement project to increase the use of forced-air warming blankets reduced mean hypothermia times in trauma patients at Parkland Memorial Hospital, Dallas, from 229 to 154 minutes.
All it took to get doctors, nurses, and staff to use the forced-air warming blankets more often was a reminder that hypothermia is an independent predictor of death in trauma, and data showing that Parkland, a Level 1 trauma center, used forced-air warming in just 11% of its hypothermic trauma patients. Meetings to get those points across were held in December 2014.
Forced-air warming jumped to 70% of hypothermic patients over the next 4 months in 2015 (P equal to or less than .0001), leading to the 33% drop in rewarming times (P = .009). The improvement came without any shift in the use of the rewarming methods trauma teams were in the habit of using: warm blankets, room air, and IV fluids.
Investigator Dr. Frank Zhao thinks it’s something all trauma centers can and should do. “There’s no reason that we shouldn’t recommend this be part of the rewarming protocol in every trauma center. It took about a month to roll this out so everyone was on the same page and was easily achieved,” said Dr. Zhao, formerly a Parkland surgery resident but now a trauma and surgical critical care fellow at the Oregon Health and Sciences University in Portland.
The blankets are an almost universal presence in operating rooms to keep core temperatures at least 36 degrees Celsius, but “from what I’ve seen at multiple institutions, the Bair Hugger is probably one of the least used warming methods” in trauma. “They’re recommended for trauma rewarming, but we [didn’t] use them very often.” Staff were not in the habit, he said at the Eastern Association for the Surgery of Trauma scientific assembly.
From July to November 2014, before the intervention, 15.2% (114) of Levels 1 and 2 trauma patients arrived at Parkland hypothermic, versus 20.9% (82) during the colder period of January-April 2015. Almost 80% of the trauma patients over that time were male, and the average patient age was about 40 years.
The investigators have no disclosures, and there was no outside funding for the project.
AT THE EAST SCIENTIFIC ASSEMBLY
Key clinical point: It can take as little as a month to make forced-air warming blankets a routine part of hypothermia care in trauma.
Major finding: A month-long quality improvement project to increase the use of forced-air warming blankets reduced mean hypothermia times at a Level 1 trauma center from 229 to 154 minutes.
Data source: The project involved 196 hypothermic trauma patients
Disclosures: The investigators have no disclosures, and there was no outside funding for the project.
Minimal residual disease a powerful prognostic factor in AML
The presence of minimal residual disease predicts relapse in patients with NMP1-mutated acute myeloid leukemia and is superior to currently used molecular genetic markers in determining whether these patients should be considered for stem cell transplantation, a new study has found.
At 3 years, patients with minimal residual disease (MRD) had a significantly greater risk of relapse than those with no MRD (82% vs. 30%; univariate hazard ratio, 4.80; P less than .001) and a lower rate of survival (24% vs. 75%; univariate HR, 4.38; P less than .001), Adam Ivey of King’s College London reported (N Engl J Med. 2016; doi:10.1056/NEJMoa1507471).
In an editorial that accompanied the study Dr. Michael J. Burke from the Children’s Hospital of Wisconsin in Milwaukee wrote, “Time will tell, but this moment may prove to be a pivotal one in the assessment of minimal residual disease to assign treatment in patients with AML” (N Engl J Med. 2016; doi:10.1056/NEJMe1515525).
In adult AML, assessment of MRD has taken a back seat to analyses of cytogenetic and molecular lesions in determining a patient’s risk and treatment strategy. Typically, allogeneic stem cell transplantation is used for patients with high-risk features such as chromosome 3, 5, or 7 abnormalities or the FLT3-internal tandem duplication (ITD) mutation, while chemotherapy alone is used for low-risk disease.
The role of transplantation is unclear, however, for cytogenetically standard-risk patients, which includes those with a mutation in the gene encoding nucleophosmin (NPM1).
To address this issue, the investigators used a reverse-transcriptase quantitative polymerase chain reaction assay to evaluate 2,569 bone marrow and peripheral-blood samples from 346 patients with NPM1 mutations who had completed two cycles of induction chemotherapy in the U.K. National Cancer Research Institute AML17 trial.
MRD, defined as persistence of NPM1-mutated transcripts in peripheral blood, was present in 15% of patients after the second chemotherapy cycle.
Patients with MRD were significantly more likely than those without MRD to have a high U.K. Medical Research Council clinical risk score and to carry the FLT3-ITD mutation.
On univariate analysis, the risk of relapse was significantly higher with the presence of MRD in peripheral blood, an increased white cell count, and with the DNMT3A and FLT3-ITD mutations.
Only the presence of MRD and an elevated white cell count significantly predicted survival, Mr. Ivey reported.
“We could find no specific molecular subgroup consisting of 10 patients or more that had a rate of survival less than 52%; in contrast, the rate in the group with the presence of minimal residual disease was 24%,” he observed.
In multivariate analysis, the presence of MRD was the only significant prognostic factor for relapse (HR, 5.09; P less than .001) or death (HR, 4.84; P less than .001).
The results were validated in an independent cohort of 91 AML17 study patients. It confirmed that MRD in peripheral blood predicts worse outcome at 2 years than the absence of MRD, with a cumulative incidence of relapse of 70% vs. 31% (P = .001) and overall survival rates of 40% vs. 87% (P = .001), reported the investigators, including senior author Professor David Grimwade, also from King’s College London.
The clinical implications of these results “are substantive” because NPM-1 mutated AML is the most common subtype of AML and because of the uncertainty over the best treatment strategy for patients typically classified as standard risk, editorialist Dr. Burke observed.
“Now with the ability to reclassify standard-risk or low-risk patients as high-risk on the basis of the persistent expression of mutant NPM1 transcripts, it may be possible that stem-cell transplantation is a better approach in patients who otherwise would be treated with chemotherapy alone and that transplantation may be avoidable in high-risk patients who have no evidence of minimal residual disease,” he wrote. “Such predictions will need to be tested prospectively.”
The presence of MRD is also known to be an important independent prognostic factor in acute lymphoblastic leukemia, but since AML has a greater molecular heterogeneity, routine MRD assessment has not been as quickly adopted in AML, Dr. Burke noted.
The Children’s Oncology Group, however, recently adopted MRD assessment by flow cytometry to further stratify children with newly diagnosed AML after first induction therapy into low-risk or high-risk groups.
The study was supported by grants from Bloodwise and the National Institute for Health Research. Mr. Ivey and Dr. Burke reported having no disclosures.
The presence of minimal residual disease predicts relapse in patients with NMP1-mutated acute myeloid leukemia and is superior to currently used molecular genetic markers in determining whether these patients should be considered for stem cell transplantation, a new study has found.
At 3 years, patients with minimal residual disease (MRD) had a significantly greater risk of relapse than those with no MRD (82% vs. 30%; univariate hazard ratio, 4.80; P less than .001) and a lower rate of survival (24% vs. 75%; univariate HR, 4.38; P less than .001), Adam Ivey of King’s College London reported (N Engl J Med. 2016; doi:10.1056/NEJMoa1507471).
In an editorial that accompanied the study Dr. Michael J. Burke from the Children’s Hospital of Wisconsin in Milwaukee wrote, “Time will tell, but this moment may prove to be a pivotal one in the assessment of minimal residual disease to assign treatment in patients with AML” (N Engl J Med. 2016; doi:10.1056/NEJMe1515525).
In adult AML, assessment of MRD has taken a back seat to analyses of cytogenetic and molecular lesions in determining a patient’s risk and treatment strategy. Typically, allogeneic stem cell transplantation is used for patients with high-risk features such as chromosome 3, 5, or 7 abnormalities or the FLT3-internal tandem duplication (ITD) mutation, while chemotherapy alone is used for low-risk disease.
The role of transplantation is unclear, however, for cytogenetically standard-risk patients, which includes those with a mutation in the gene encoding nucleophosmin (NPM1).
To address this issue, the investigators used a reverse-transcriptase quantitative polymerase chain reaction assay to evaluate 2,569 bone marrow and peripheral-blood samples from 346 patients with NPM1 mutations who had completed two cycles of induction chemotherapy in the U.K. National Cancer Research Institute AML17 trial.
MRD, defined as persistence of NPM1-mutated transcripts in peripheral blood, was present in 15% of patients after the second chemotherapy cycle.
Patients with MRD were significantly more likely than those without MRD to have a high U.K. Medical Research Council clinical risk score and to carry the FLT3-ITD mutation.
On univariate analysis, the risk of relapse was significantly higher with the presence of MRD in peripheral blood, an increased white cell count, and with the DNMT3A and FLT3-ITD mutations.
Only the presence of MRD and an elevated white cell count significantly predicted survival, Mr. Ivey reported.
“We could find no specific molecular subgroup consisting of 10 patients or more that had a rate of survival less than 52%; in contrast, the rate in the group with the presence of minimal residual disease was 24%,” he observed.
In multivariate analysis, the presence of MRD was the only significant prognostic factor for relapse (HR, 5.09; P less than .001) or death (HR, 4.84; P less than .001).
The results were validated in an independent cohort of 91 AML17 study patients. It confirmed that MRD in peripheral blood predicts worse outcome at 2 years than the absence of MRD, with a cumulative incidence of relapse of 70% vs. 31% (P = .001) and overall survival rates of 40% vs. 87% (P = .001), reported the investigators, including senior author Professor David Grimwade, also from King’s College London.
The clinical implications of these results “are substantive” because NPM-1 mutated AML is the most common subtype of AML and because of the uncertainty over the best treatment strategy for patients typically classified as standard risk, editorialist Dr. Burke observed.
“Now with the ability to reclassify standard-risk or low-risk patients as high-risk on the basis of the persistent expression of mutant NPM1 transcripts, it may be possible that stem-cell transplantation is a better approach in patients who otherwise would be treated with chemotherapy alone and that transplantation may be avoidable in high-risk patients who have no evidence of minimal residual disease,” he wrote. “Such predictions will need to be tested prospectively.”
The presence of MRD is also known to be an important independent prognostic factor in acute lymphoblastic leukemia, but since AML has a greater molecular heterogeneity, routine MRD assessment has not been as quickly adopted in AML, Dr. Burke noted.
The Children’s Oncology Group, however, recently adopted MRD assessment by flow cytometry to further stratify children with newly diagnosed AML after first induction therapy into low-risk or high-risk groups.
The study was supported by grants from Bloodwise and the National Institute for Health Research. Mr. Ivey and Dr. Burke reported having no disclosures.
The presence of minimal residual disease predicts relapse in patients with NMP1-mutated acute myeloid leukemia and is superior to currently used molecular genetic markers in determining whether these patients should be considered for stem cell transplantation, a new study has found.
At 3 years, patients with minimal residual disease (MRD) had a significantly greater risk of relapse than those with no MRD (82% vs. 30%; univariate hazard ratio, 4.80; P less than .001) and a lower rate of survival (24% vs. 75%; univariate HR, 4.38; P less than .001), Adam Ivey of King’s College London reported (N Engl J Med. 2016; doi:10.1056/NEJMoa1507471).
In an editorial that accompanied the study Dr. Michael J. Burke from the Children’s Hospital of Wisconsin in Milwaukee wrote, “Time will tell, but this moment may prove to be a pivotal one in the assessment of minimal residual disease to assign treatment in patients with AML” (N Engl J Med. 2016; doi:10.1056/NEJMe1515525).
In adult AML, assessment of MRD has taken a back seat to analyses of cytogenetic and molecular lesions in determining a patient’s risk and treatment strategy. Typically, allogeneic stem cell transplantation is used for patients with high-risk features such as chromosome 3, 5, or 7 abnormalities or the FLT3-internal tandem duplication (ITD) mutation, while chemotherapy alone is used for low-risk disease.
The role of transplantation is unclear, however, for cytogenetically standard-risk patients, which includes those with a mutation in the gene encoding nucleophosmin (NPM1).
To address this issue, the investigators used a reverse-transcriptase quantitative polymerase chain reaction assay to evaluate 2,569 bone marrow and peripheral-blood samples from 346 patients with NPM1 mutations who had completed two cycles of induction chemotherapy in the U.K. National Cancer Research Institute AML17 trial.
MRD, defined as persistence of NPM1-mutated transcripts in peripheral blood, was present in 15% of patients after the second chemotherapy cycle.
Patients with MRD were significantly more likely than those without MRD to have a high U.K. Medical Research Council clinical risk score and to carry the FLT3-ITD mutation.
On univariate analysis, the risk of relapse was significantly higher with the presence of MRD in peripheral blood, an increased white cell count, and with the DNMT3A and FLT3-ITD mutations.
Only the presence of MRD and an elevated white cell count significantly predicted survival, Mr. Ivey reported.
“We could find no specific molecular subgroup consisting of 10 patients or more that had a rate of survival less than 52%; in contrast, the rate in the group with the presence of minimal residual disease was 24%,” he observed.
In multivariate analysis, the presence of MRD was the only significant prognostic factor for relapse (HR, 5.09; P less than .001) or death (HR, 4.84; P less than .001).
The results were validated in an independent cohort of 91 AML17 study patients. It confirmed that MRD in peripheral blood predicts worse outcome at 2 years than the absence of MRD, with a cumulative incidence of relapse of 70% vs. 31% (P = .001) and overall survival rates of 40% vs. 87% (P = .001), reported the investigators, including senior author Professor David Grimwade, also from King’s College London.
The clinical implications of these results “are substantive” because NPM-1 mutated AML is the most common subtype of AML and because of the uncertainty over the best treatment strategy for patients typically classified as standard risk, editorialist Dr. Burke observed.
“Now with the ability to reclassify standard-risk or low-risk patients as high-risk on the basis of the persistent expression of mutant NPM1 transcripts, it may be possible that stem-cell transplantation is a better approach in patients who otherwise would be treated with chemotherapy alone and that transplantation may be avoidable in high-risk patients who have no evidence of minimal residual disease,” he wrote. “Such predictions will need to be tested prospectively.”
The presence of MRD is also known to be an important independent prognostic factor in acute lymphoblastic leukemia, but since AML has a greater molecular heterogeneity, routine MRD assessment has not been as quickly adopted in AML, Dr. Burke noted.
The Children’s Oncology Group, however, recently adopted MRD assessment by flow cytometry to further stratify children with newly diagnosed AML after first induction therapy into low-risk or high-risk groups.
The study was supported by grants from Bloodwise and the National Institute for Health Research. Mr. Ivey and Dr. Burke reported having no disclosures.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: The presence of minimal residual disease provides powerful prognostic information independent of other risk factors in patients with NPM1-mutated AML.
Major finding: Minimal residual disease was associated with a significantly greater risk of relapse than absence of MRD (82% vs. 30%; hazard ratio, 4.80; P less than .001) and a lower rate of survival (24% vs. 75%; HR, 4.38, P less than .001).
Data source: Analysis of 346 patients with NPM1-mutated AML.
Disclosures: The study was supported by grants from Bloodwise and the National Institute for Health Research. Mr. Ivey and Dr. Burke reported having no disclosures.
Baux cut-points predict geriatric burn outcomes
SAN ANTONIO – Geriatric burn patients have less than a 50% chance of returning home with a Baux score of about 85, and the risk of death begins to climb steadily after a score 93, approaching 50% at 110 points and almost 100% at 130 points, according to a review of 8,001 elderly patients in the National Burn Repository.
The investigators are developing the findings into a decision-making tool to help counsel families and caregivers about their options when elderly loved ones are seriously burned.

“There’s just not a lot of data out there on prognosis after burn injury in the geriatric population. We thought a simple decision aid for discussion with key stakeholders would provide significant assistance,” said investigator Dr. Erica Hodgman, a surgery research resident at the University of Texas Southwestern Medical Center, Dallas.
The hope is that families and caregivers will be able to better judge if the patient would want to press on with treatment given the odds of returning home, being discharged to a skilled nursing or rehab facility, or dying. “I think it will help people” feel less guilty if they decide to withdraw care or not send patients far away to a burn center, she said at the Eastern Association for the Surgery of Trauma scientific assembly.
The Baux score, a well-known metric in the burn community, adds the patient’s age to the percentage of surface area burned, so a 70 year old patient burned over 23% of their body, for instance, would have a score of 93. A modified Baux score adds points for inhalation injuries, but because the data didn’t include inhalation injury severity, the investigators found it more useful to stick with the original formula.
They queried the repository for patients 65 years or older with second- or third-degree burns from 2002-2011. They excluded patients with a length of stay of a day or less, along with elective admissions, non-burn injuries, and transfers to other burn centers. Next, they calculated Baux scores for each of their 8,001 subjects and noted if the patients were discharged home or to an alternate facility, or if he or she died.
Most patients had moderate scores of 70-100, and almost half were sent home. Of the 1,509 that died in hospital, 264 (17.5%) had care withdraw at a median of 3 days, but a range of 0-231 days. Flames were the most common cause of injury, followed by scalding.
A receiver operating curve analysis found that a Baux score at or below 86.15 predicted discharge home (AUC 0.698, 75.28% sensitivity, 54.64% specificity); a score above 77.12 predicted discharge to an alternate setting (AUC 0.539, 74.91% sensitivity, 34.38% specificity); and a score above 93.3 predicted mortality (AUC 0.779, 57.46% sensitivity, 87.08% specificity).
Dr. Hodgman said she thinks the cut-points will remain useful even as burn care improves with new grafting techniques that require smaller donor sites. Such innovation will apply mostly to moderately injured patients; for the more severely injured, the predictive power of the findings should still hold.
The investigators have no disclosures.
SAN ANTONIO – Geriatric burn patients have less than a 50% chance of returning home with a Baux score of about 85, and the risk of death begins to climb steadily after a score 93, approaching 50% at 110 points and almost 100% at 130 points, according to a review of 8,001 elderly patients in the National Burn Repository.
The investigators are developing the findings into a decision-making tool to help counsel families and caregivers about their options when elderly loved ones are seriously burned.
“There’s just not a lot of data out there on prognosis after burn injury in the geriatric population. We thought a simple decision aid for discussion with key stakeholders would provide significant assistance,” said investigator Dr. Erica Hodgman, a surgery research resident at the University of Texas Southwestern Medical Center, Dallas.
The hope is that families and caregivers will be able to better judge if the patient would want to press on with treatment given the odds of returning home, being discharged to a skilled nursing or rehab facility, or dying. “I think it will help people” feel less guilty if they decide to withdraw care or not send patients far away to a burn center, she said at the Eastern Association for the Surgery of Trauma scientific assembly.
The Baux score, a well-known metric in the burn community, adds the patient’s age to the percentage of surface area burned, so a 70 year old patient burned over 23% of their body, for instance, would have a score of 93. A modified Baux score adds points for inhalation injuries, but because the data didn’t include inhalation injury severity, the investigators found it more useful to stick with the original formula.
They queried the repository for patients 65 years or older with second- or third-degree burns from 2002-2011. They excluded patients with a length of stay of a day or less, along with elective admissions, non-burn injuries, and transfers to other burn centers. Next, they calculated Baux scores for each of their 8,001 subjects and noted if the patients were discharged home or to an alternate facility, or if he or she died.
Most patients had moderate scores of 70-100, and almost half were sent home. Of the 1,509 that died in hospital, 264 (17.5%) had care withdraw at a median of 3 days, but a range of 0-231 days. Flames were the most common cause of injury, followed by scalding.
A receiver operating curve analysis found that a Baux score at or below 86.15 predicted discharge home (AUC 0.698, 75.28% sensitivity, 54.64% specificity); a score above 77.12 predicted discharge to an alternate setting (AUC 0.539, 74.91% sensitivity, 34.38% specificity); and a score above 93.3 predicted mortality (AUC 0.779, 57.46% sensitivity, 87.08% specificity).
Dr. Hodgman said she thinks the cut-points will remain useful even as burn care improves with new grafting techniques that require smaller donor sites. Such innovation will apply mostly to moderately injured patients; for the more severely injured, the predictive power of the findings should still hold.
The investigators have no disclosures.
SAN ANTONIO – Geriatric burn patients have less than a 50% chance of returning home with a Baux score of about 85, and the risk of death begins to climb steadily after a score 93, approaching 50% at 110 points and almost 100% at 130 points, according to a review of 8,001 elderly patients in the National Burn Repository.
The investigators are developing the findings into a decision-making tool to help counsel families and caregivers about their options when elderly loved ones are seriously burned.
“There’s just not a lot of data out there on prognosis after burn injury in the geriatric population. We thought a simple decision aid for discussion with key stakeholders would provide significant assistance,” said investigator Dr. Erica Hodgman, a surgery research resident at the University of Texas Southwestern Medical Center, Dallas.
The hope is that families and caregivers will be able to better judge if the patient would want to press on with treatment given the odds of returning home, being discharged to a skilled nursing or rehab facility, or dying. “I think it will help people” feel less guilty if they decide to withdraw care or not send patients far away to a burn center, she said at the Eastern Association for the Surgery of Trauma scientific assembly.
The Baux score, a well-known metric in the burn community, adds the patient’s age to the percentage of surface area burned, so a 70 year old patient burned over 23% of their body, for instance, would have a score of 93. A modified Baux score adds points for inhalation injuries, but because the data didn’t include inhalation injury severity, the investigators found it more useful to stick with the original formula.
They queried the repository for patients 65 years or older with second- or third-degree burns from 2002-2011. They excluded patients with a length of stay of a day or less, along with elective admissions, non-burn injuries, and transfers to other burn centers. Next, they calculated Baux scores for each of their 8,001 subjects and noted if the patients were discharged home or to an alternate facility, or if he or she died.
Most patients had moderate scores of 70-100, and almost half were sent home. Of the 1,509 that died in hospital, 264 (17.5%) had care withdraw at a median of 3 days, but a range of 0-231 days. Flames were the most common cause of injury, followed by scalding.
A receiver operating curve analysis found that a Baux score at or below 86.15 predicted discharge home (AUC 0.698, 75.28% sensitivity, 54.64% specificity); a score above 77.12 predicted discharge to an alternate setting (AUC 0.539, 74.91% sensitivity, 34.38% specificity); and a score above 93.3 predicted mortality (AUC 0.779, 57.46% sensitivity, 87.08% specificity).
Dr. Hodgman said she thinks the cut-points will remain useful even as burn care improves with new grafting techniques that require smaller donor sites. Such innovation will apply mostly to moderately injured patients; for the more severely injured, the predictive power of the findings should still hold.
The investigators have no disclosures.
AT THE EAST SCIENTIFIC ASSEMBLY
Key clinical point: Baux score cut-points help counsel families about their options when an elderly family member is seriously burned.
Major finding: Geriatric burn patients have less than a 50% chance of returning home with a Baux score of about 85, and the risk of death begins to climb steadily after a score 93.
Data source: Review of 8,001 patients over 65 years old in the National Burn Repository
Disclosures: The investigators have no disclosures.

Health IT Chief, Hospital Medicine ‘Godfather’ Headline SHM Annual Meeting Keynotes
Health information technology (IT) will take center stage early and often at this year’s annual meeting of the Society of Hospital Medicine.
Karen DeSalvo, MD, MPH, MSc, acting assistant secretary for health in the U.S. Department of Health & Human Services (HHS) and the national coordinator for health information technology, will deliver the keynote address. She is scheduled to give her talk an hour into the first day of programming.
Another highly anticipated talk will be delivered by Robert Wachter, MD, MHM, chief of the division of hospital medicine at the University of California at San Francisco, the “godfather” of hospital medicine, and the field’s most well-known practitioner. Dr. Wachter will give his 12th straight meeting-closing talk at noon Wednesday.
Dr. DeSalvo, an internist by training, was the chief of general internal medicine at Tulane University for about 10 years. She also started at Charity Hospital in New Orleans, site of one of the earliest hospital medicine programs.

She says her speech will take a broad look at information technology as a tool for advancing good health, with attention to the role that hospitals and hospitalists play. She also plans to touch on the successes in U.S. healthcare in recent years, including expanded coverage, quality and safety improvements, and the rapid rate of adoption of electronic health records (EHRs), especially in the hospital. She says the hospital setting is “most ripe” for health IT advancement because it is the site of “the most rich data about the patient’s care and care experience and health … and there is the best interoperability right now between hospital systems and the best opportunity to make that more seamless.”
The future, she says, will be about “much more than the electronic health record.”
“I want to talk a bit about what’s happening on the pioneering edge in health IT, ranging anywhere from apps to consumer interface with digital health records to some really on-the-edge things like using telehealth and hologram technology for remote patient care,” she explains.
Dr. DeSalvo also plans to underscore health IT’s key role in HHS’s push for delivery system reform: changing the way care is paid for and delivered and the way information is delivered. HHS’s goal is for 50% of payments to be in the form of alternative or value-based payment models by 2018. Without health IT advancements, that won’t be possible, she says.
Health IT policy at HHS, she notes, has centered largely on “freeing the data” so that information is no longer trapped within a particular EHR system. A rule taking effect in 2018 will require that EHRs be built so that apps can be overlaid onto the data, allowing easier access and the ability to tailor data to an individual’s needs.
“It’s going to get to be more like the way we do our banking or call for transportation with a smartphone or have an interface for our travel arrangements,” she says. “That’s the way that the health IT world is evolving.”
She says hospitalists are “pioneering, early adopters who are by nature very innovative” and are ideal for helping refine health IT. But she also recognizes that the bumps along the way can cause technology to be seen as a hurdle. That’s why HHS policy has focused on making data more readily available, smoothing out clunkiness, making EHR vendors become more transparent about their products, and aligning documentation requirements with real patient outcomes so that unnecessary requirements can be eliminated.
Good systems have been developed, but improvements are needed, she acknowledges.
“We’re working with an intense sense of urgency at HHS because we know that is a source of frustration to doctors on the frontlines,” Dr. DeSalvo says. “We not only hear it all the time when we’re out speaking with folks, but some of us still practice and will shortly be practicing again, so it’s very real to us to know that this has to get better. What we don’t want is for people to be frustrated with the technology. We want it to lift them up and help make their practice better. We also want it to be an enabler for consumers.”
Dr. Wachter has an easy way to remember how many annual meeting lectures he’s given: The 10th was the one where he dressed up as Elton John, sang, and played the piano on stage. That was in Las Vegas, of course.

This year? Don’t expect the piano, or singing for that matter. His HM16 theme will be more sober, one of caution and the importance of perspective.
The early title, he tells The Hospitalist, is “Why Culture Is Key to Improvement … And Why Hospitalists Are the Key to Hospital Culture.” The title might change, and the precise direction and details of his talk are still in flux, he says.
But the thrust will be a concern that, with a blizzard of quality improvement (QI) projects and process analyses being taken on by hospitalists, hospitalists are not immune to the burnout we’re seeing throughout medicine. A bad vibe is creeping in, he fears, and unless there’s more awareness of, and attention to, the culture itself—and not just a grim soldiering on from one initiative to another—the field will suffer.
“There’s a risk that we’ll lose sight of the people and culture within the organization,” Dr. Wachter says. “Even good people are beginning to say, ‘I just can’t do another QI project; I just can’t do another thing.’”
He wants hospitalists to think “more deeply” about the issues of culture, how the workforce is being managed, and “that we’re focusing on the right things in the right way.”
He hopes to call on hospitalists and hospitalist leaders to continue to recognize “the importance of the human spirit in all of this.”
So how worried is he?
“It won’t be a downer,” he says. “I still think we’re in great shape, but I am a bit worried, in part, because of our successes. We grew so fast, and we became so important to our organizations. We have to be sure we’re taking care of ourselves.”
So many hospitalists now have leadership roles. That’s a good thing, he adds, “but it does mean that as people are beginning to be burned out or organizations are struggling with dealing with initiative fatigue, we’re the first ones that are going to feel that because we are disproportionately involved.” TH
Thomas R. Collins is a freelance writer in South Florida.
Health information technology (IT) will take center stage early and often at this year’s annual meeting of the Society of Hospital Medicine.
Karen DeSalvo, MD, MPH, MSc, acting assistant secretary for health in the U.S. Department of Health & Human Services (HHS) and the national coordinator for health information technology, will deliver the keynote address. She is scheduled to give her talk an hour into the first day of programming.
Another highly anticipated talk will be delivered by Robert Wachter, MD, MHM, chief of the division of hospital medicine at the University of California at San Francisco, the “godfather” of hospital medicine, and the field’s most well-known practitioner. Dr. Wachter will give his 12th straight meeting-closing talk at noon Wednesday.
Dr. DeSalvo, an internist by training, was the chief of general internal medicine at Tulane University for about 10 years. She also started at Charity Hospital in New Orleans, site of one of the earliest hospital medicine programs.
She says her speech will take a broad look at information technology as a tool for advancing good health, with attention to the role that hospitals and hospitalists play. She also plans to touch on the successes in U.S. healthcare in recent years, including expanded coverage, quality and safety improvements, and the rapid rate of adoption of electronic health records (EHRs), especially in the hospital. She says the hospital setting is “most ripe” for health IT advancement because it is the site of “the most rich data about the patient’s care and care experience and health … and there is the best interoperability right now between hospital systems and the best opportunity to make that more seamless.”
The future, she says, will be about “much more than the electronic health record.”
“I want to talk a bit about what’s happening on the pioneering edge in health IT, ranging anywhere from apps to consumer interface with digital health records to some really on-the-edge things like using telehealth and hologram technology for remote patient care,” she explains.
Dr. DeSalvo also plans to underscore health IT’s key role in HHS’s push for delivery system reform: changing the way care is paid for and delivered and the way information is delivered. HHS’s goal is for 50% of payments to be in the form of alternative or value-based payment models by 2018. Without health IT advancements, that won’t be possible, she says.
Health IT policy at HHS, she notes, has centered largely on “freeing the data” so that information is no longer trapped within a particular EHR system. A rule taking effect in 2018 will require that EHRs be built so that apps can be overlaid onto the data, allowing easier access and the ability to tailor data to an individual’s needs.
“It’s going to get to be more like the way we do our banking or call for transportation with a smartphone or have an interface for our travel arrangements,” she says. “That’s the way that the health IT world is evolving.”
She says hospitalists are “pioneering, early adopters who are by nature very innovative” and are ideal for helping refine health IT. But she also recognizes that the bumps along the way can cause technology to be seen as a hurdle. That’s why HHS policy has focused on making data more readily available, smoothing out clunkiness, making EHR vendors become more transparent about their products, and aligning documentation requirements with real patient outcomes so that unnecessary requirements can be eliminated.
Good systems have been developed, but improvements are needed, she acknowledges.
“We’re working with an intense sense of urgency at HHS because we know that is a source of frustration to doctors on the frontlines,” Dr. DeSalvo says. “We not only hear it all the time when we’re out speaking with folks, but some of us still practice and will shortly be practicing again, so it’s very real to us to know that this has to get better. What we don’t want is for people to be frustrated with the technology. We want it to lift them up and help make their practice better. We also want it to be an enabler for consumers.”
Dr. Wachter has an easy way to remember how many annual meeting lectures he’s given: The 10th was the one where he dressed up as Elton John, sang, and played the piano on stage. That was in Las Vegas, of course.
This year? Don’t expect the piano, or singing for that matter. His HM16 theme will be more sober, one of caution and the importance of perspective.
The early title, he tells The Hospitalist, is “Why Culture Is Key to Improvement … And Why Hospitalists Are the Key to Hospital Culture.” The title might change, and the precise direction and details of his talk are still in flux, he says.
But the thrust will be a concern that, with a blizzard of quality improvement (QI) projects and process analyses being taken on by hospitalists, hospitalists are not immune to the burnout we’re seeing throughout medicine. A bad vibe is creeping in, he fears, and unless there’s more awareness of, and attention to, the culture itself—and not just a grim soldiering on from one initiative to another—the field will suffer.
“There’s a risk that we’ll lose sight of the people and culture within the organization,” Dr. Wachter says. “Even good people are beginning to say, ‘I just can’t do another QI project; I just can’t do another thing.’”
He wants hospitalists to think “more deeply” about the issues of culture, how the workforce is being managed, and “that we’re focusing on the right things in the right way.”
He hopes to call on hospitalists and hospitalist leaders to continue to recognize “the importance of the human spirit in all of this.”
So how worried is he?
“It won’t be a downer,” he says. “I still think we’re in great shape, but I am a bit worried, in part, because of our successes. We grew so fast, and we became so important to our organizations. We have to be sure we’re taking care of ourselves.”
So many hospitalists now have leadership roles. That’s a good thing, he adds, “but it does mean that as people are beginning to be burned out or organizations are struggling with dealing with initiative fatigue, we’re the first ones that are going to feel that because we are disproportionately involved.” TH
Thomas R. Collins is a freelance writer in South Florida.
Health information technology (IT) will take center stage early and often at this year’s annual meeting of the Society of Hospital Medicine.
Karen DeSalvo, MD, MPH, MSc, acting assistant secretary for health in the U.S. Department of Health & Human Services (HHS) and the national coordinator for health information technology, will deliver the keynote address. She is scheduled to give her talk an hour into the first day of programming.
Another highly anticipated talk will be delivered by Robert Wachter, MD, MHM, chief of the division of hospital medicine at the University of California at San Francisco, the “godfather” of hospital medicine, and the field’s most well-known practitioner. Dr. Wachter will give his 12th straight meeting-closing talk at noon Wednesday.
Dr. DeSalvo, an internist by training, was the chief of general internal medicine at Tulane University for about 10 years. She also started at Charity Hospital in New Orleans, site of one of the earliest hospital medicine programs.
She says her speech will take a broad look at information technology as a tool for advancing good health, with attention to the role that hospitals and hospitalists play. She also plans to touch on the successes in U.S. healthcare in recent years, including expanded coverage, quality and safety improvements, and the rapid rate of adoption of electronic health records (EHRs), especially in the hospital. She says the hospital setting is “most ripe” for health IT advancement because it is the site of “the most rich data about the patient’s care and care experience and health … and there is the best interoperability right now between hospital systems and the best opportunity to make that more seamless.”
The future, she says, will be about “much more than the electronic health record.”
“I want to talk a bit about what’s happening on the pioneering edge in health IT, ranging anywhere from apps to consumer interface with digital health records to some really on-the-edge things like using telehealth and hologram technology for remote patient care,” she explains.
Dr. DeSalvo also plans to underscore health IT’s key role in HHS’s push for delivery system reform: changing the way care is paid for and delivered and the way information is delivered. HHS’s goal is for 50% of payments to be in the form of alternative or value-based payment models by 2018. Without health IT advancements, that won’t be possible, she says.
Health IT policy at HHS, she notes, has centered largely on “freeing the data” so that information is no longer trapped within a particular EHR system. A rule taking effect in 2018 will require that EHRs be built so that apps can be overlaid onto the data, allowing easier access and the ability to tailor data to an individual’s needs.
“It’s going to get to be more like the way we do our banking or call for transportation with a smartphone or have an interface for our travel arrangements,” she says. “That’s the way that the health IT world is evolving.”
She says hospitalists are “pioneering, early adopters who are by nature very innovative” and are ideal for helping refine health IT. But she also recognizes that the bumps along the way can cause technology to be seen as a hurdle. That’s why HHS policy has focused on making data more readily available, smoothing out clunkiness, making EHR vendors become more transparent about their products, and aligning documentation requirements with real patient outcomes so that unnecessary requirements can be eliminated.
Good systems have been developed, but improvements are needed, she acknowledges.
“We’re working with an intense sense of urgency at HHS because we know that is a source of frustration to doctors on the frontlines,” Dr. DeSalvo says. “We not only hear it all the time when we’re out speaking with folks, but some of us still practice and will shortly be practicing again, so it’s very real to us to know that this has to get better. What we don’t want is for people to be frustrated with the technology. We want it to lift them up and help make their practice better. We also want it to be an enabler for consumers.”
Dr. Wachter has an easy way to remember how many annual meeting lectures he’s given: The 10th was the one where he dressed up as Elton John, sang, and played the piano on stage. That was in Las Vegas, of course.
This year? Don’t expect the piano, or singing for that matter. His HM16 theme will be more sober, one of caution and the importance of perspective.
The early title, he tells The Hospitalist, is “Why Culture Is Key to Improvement … And Why Hospitalists Are the Key to Hospital Culture.” The title might change, and the precise direction and details of his talk are still in flux, he says.
But the thrust will be a concern that, with a blizzard of quality improvement (QI) projects and process analyses being taken on by hospitalists, hospitalists are not immune to the burnout we’re seeing throughout medicine. A bad vibe is creeping in, he fears, and unless there’s more awareness of, and attention to, the culture itself—and not just a grim soldiering on from one initiative to another—the field will suffer.
“There’s a risk that we’ll lose sight of the people and culture within the organization,” Dr. Wachter says. “Even good people are beginning to say, ‘I just can’t do another QI project; I just can’t do another thing.’”
He wants hospitalists to think “more deeply” about the issues of culture, how the workforce is being managed, and “that we’re focusing on the right things in the right way.”
He hopes to call on hospitalists and hospitalist leaders to continue to recognize “the importance of the human spirit in all of this.”
So how worried is he?
“It won’t be a downer,” he says. “I still think we’re in great shape, but I am a bit worried, in part, because of our successes. We grew so fast, and we became so important to our organizations. We have to be sure we’re taking care of ourselves.”
So many hospitalists now have leadership roles. That’s a good thing, he adds, “but it does mean that as people are beginning to be burned out or organizations are struggling with dealing with initiative fatigue, we’re the first ones that are going to feel that because we are disproportionately involved.” TH
Thomas R. Collins is a freelance writer in South Florida.
New SHM Members – February 2016
A. Benefield, BSN, FNP, MSN, RN, Alabama
A. Mahajan, MPH, Alabama
B. Meredith, DO, Alabama
L. Ortiz, MD, Arizona
N. Charlton, MD, California
T. Fong, BS, MD, California
B. Gee, MD, California
I. He, California
K. Jones, FAAFP, California
J. Kolev, MD, California
S. Krishnamoorthy, MD, California
A. Kumar, MD, California
D. Pan, California
M. Reaves, MD, California
A. Sones, California
A. Wang, California
S. McCary, MD, Colorado
A. Beg, MD, Connecticut
S. Das, MD, Connecticut
A. Reisner, MD, D.C.
S. Gadalla, MD, Florida
N. Joseph, MBBS, Florida
R. Chowdhury, DO, MPH, Georgia
K. Mann, Georgia
D. Aiken, Idaho
C. Riji Anil, MBBS, Idaho
A. Asare, MACP, MBchB, Illinois
R. Clapp, ACNP, Illinois
A. Kost, MD, Illinois
R. Pavurala, MBBS, Illinois
M. Tsien, Illinois
D. Brewer, Indiana
J. Mast, MD, Indiana
M. Arakane, Iowa
S. Dobler, MD, Kansas
A. Ausef, Louisiana
C. Newby, MD, Louisiana
S. Brown, Massachusetts
A. Gonzalez, MD, Massachusetts
K. Lombardo, Massachusetts
C. Alamelumangapuram, MBBS, Michigan
A. Chakrabarti, Michigan
K. Dazy, MD, FAAP, Michigan
A. John, Michigan
J. Meinke, Michigan
P. Vemula, Michigan
R. Albrecht, PA-C, Minnesota
D. Burgy, Minnesota
T. Ebel, MD, Minnesota
T. Goddard, PA-C, Minnesota
K. Lichter, Minnesota
D. Lorentz, MD, Minnesota
R. Parikh, MD, Minnesota
E. Zygmunt, PA-C, Minnesota
A. Blaes, ACNP, MSN, Missouri
M. Sheikh, MD, Missouri
J. Carrasquillo, APRN-NP, Nebraska
T. Diesing, ACMPE, Nebraska
M. Havekost, MD, FAAFP, Nebraska
E. Guzman, MD, Nevada
S. Gupta, APRN, New Hampshire
M. Scaccia, MD, New Hampshire
M. Ward, New Hampshire
M. Weber, PA-C, New Jersey
Y. Abdou, New Mexico
J. Cruickshank, New Mexico
S. Scott, MD, New Mexico
P. Agrawal, MD, New York
A. Cruz, MD, New York
S. Dutta, New York
B. Fuchs, New York
D. Gill, MD, New York
K. Hernandez, New York
H. Holzer, MD, New York
L. Janson, MD, New York
P. Koukuntla, MBBS, New York
R. Mamun, New York
A. Nair, MD, New York
C. Ramchandani, New York
D. Shah, New York
J. Silver, MD, New York
A. Tusano, New York
J. Virk, MBBS, New York
J. Weisenberger, DO, New York
S. Chow, DO, North Carolina
Z. Hafeez, North Carolina
M. Jagosky, MD, North Carolina
L. Spence, DO, North Carolina
N. Uchendu, MD, MBBS, North Carolina
D. Wefuan, MD, North Carolina
O. Afzal, MD, Ohio
P. Areti, MD, MBBS, Ohio
S. Arobelidze, MD, Ohio
A. Bath, MD, Ohio
D. Bhandari, MD, Ohio
M. Hadley, Ohio
L. Hakam, MD, Ohio
L. Heinemann, MD, Ohio
V. Janamanchi, MD, Ohio
M. Sundhu, MD, Ohio
M. Syed, MD, Ohio
R. Wajahat, MD, Ohio
K. Welch, Ohio
R. Barnhart, MD, Oregon
D. Harmon, Oregon
K. Ordelheide, FACP, Oregon
C. Boyle, BS, Pennsylvania
J. Dong, Pennsylvania
S. Kowsika, MD, Pennsylvania
S. Krauthamer, Pennsylvania
A. Rajaratnam, MD, Pennsylvania
C. Rauscher, MD, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
C. Shoff, MD, Pennsylvania
E. Garcia-Torres, MD, South Carolina
J. Kanter, FAAP, South Carolina
T. Klein, Tennessee
C. Taylor, MBA, Tennessee
J. Tedesco, ACNP, Tennessee
S. Ansari, Texas
A. Corley, MPAS, PA-C, Texas
K. Cunnusamy, Texas
C. Faust, MPH, Texas
B. Gajjar, Texas
S. Gudur, Texas
J. Hinojosa, Texas
R. Imtiaz, BS, Texas
B. Kennedy, MD, Texas
K. Koch, Texas
O. Mirza, Texas
L. Rijos, ACNP, Texas
H. Saikumar, MD, Texas
M. Walker, MD, Texas
M. Zaman, Texas
D. Borg, APRNBC, MSN, NP, Utah
A. Breviu, MD, Utah
R. Cook, Utah
K. Kenealy, MD, Utah
C. Chen, MD, Virginia
K. Malloy, Virginia
L. McDaniel, MD, Virginia
R. Miller, MD, Virginia
T. Schaefer, PA-C, Virginia
D. Vanikar, Virginia
J. Asriel, MD, Washington
T. Corn, Washington
K. Bergquist, DO, Wisconsin
S. Patel, MD, Wisconsin
S. Skogen, APRN-BC, Wisconsin
E. Winkel, Wisconsin
D. Figueroa, MD, Puerto Rico
A. Benefield, BSN, FNP, MSN, RN, Alabama
A. Mahajan, MPH, Alabama
B. Meredith, DO, Alabama
L. Ortiz, MD, Arizona
N. Charlton, MD, California
T. Fong, BS, MD, California
B. Gee, MD, California
I. He, California
K. Jones, FAAFP, California
J. Kolev, MD, California
S. Krishnamoorthy, MD, California
A. Kumar, MD, California
D. Pan, California
M. Reaves, MD, California
A. Sones, California
A. Wang, California
S. McCary, MD, Colorado
A. Beg, MD, Connecticut
S. Das, MD, Connecticut
A. Reisner, MD, D.C.
S. Gadalla, MD, Florida
N. Joseph, MBBS, Florida
R. Chowdhury, DO, MPH, Georgia
K. Mann, Georgia
D. Aiken, Idaho
C. Riji Anil, MBBS, Idaho
A. Asare, MACP, MBchB, Illinois
R. Clapp, ACNP, Illinois
A. Kost, MD, Illinois
R. Pavurala, MBBS, Illinois
M. Tsien, Illinois
D. Brewer, Indiana
J. Mast, MD, Indiana
M. Arakane, Iowa
S. Dobler, MD, Kansas
A. Ausef, Louisiana
C. Newby, MD, Louisiana
S. Brown, Massachusetts
A. Gonzalez, MD, Massachusetts
K. Lombardo, Massachusetts
C. Alamelumangapuram, MBBS, Michigan
A. Chakrabarti, Michigan
K. Dazy, MD, FAAP, Michigan
A. John, Michigan
J. Meinke, Michigan
P. Vemula, Michigan
R. Albrecht, PA-C, Minnesota
D. Burgy, Minnesota
T. Ebel, MD, Minnesota
T. Goddard, PA-C, Minnesota
K. Lichter, Minnesota
D. Lorentz, MD, Minnesota
R. Parikh, MD, Minnesota
E. Zygmunt, PA-C, Minnesota
A. Blaes, ACNP, MSN, Missouri
M. Sheikh, MD, Missouri
J. Carrasquillo, APRN-NP, Nebraska
T. Diesing, ACMPE, Nebraska
M. Havekost, MD, FAAFP, Nebraska
E. Guzman, MD, Nevada
S. Gupta, APRN, New Hampshire
M. Scaccia, MD, New Hampshire
M. Ward, New Hampshire
M. Weber, PA-C, New Jersey
Y. Abdou, New Mexico
J. Cruickshank, New Mexico
S. Scott, MD, New Mexico
P. Agrawal, MD, New York
A. Cruz, MD, New York
S. Dutta, New York
B. Fuchs, New York
D. Gill, MD, New York
K. Hernandez, New York
H. Holzer, MD, New York
L. Janson, MD, New York
P. Koukuntla, MBBS, New York
R. Mamun, New York
A. Nair, MD, New York
C. Ramchandani, New York
D. Shah, New York
J. Silver, MD, New York
A. Tusano, New York
J. Virk, MBBS, New York
J. Weisenberger, DO, New York
S. Chow, DO, North Carolina
Z. Hafeez, North Carolina
M. Jagosky, MD, North Carolina
L. Spence, DO, North Carolina
N. Uchendu, MD, MBBS, North Carolina
D. Wefuan, MD, North Carolina
O. Afzal, MD, Ohio
P. Areti, MD, MBBS, Ohio
S. Arobelidze, MD, Ohio
A. Bath, MD, Ohio
D. Bhandari, MD, Ohio
M. Hadley, Ohio
L. Hakam, MD, Ohio
L. Heinemann, MD, Ohio
V. Janamanchi, MD, Ohio
M. Sundhu, MD, Ohio
M. Syed, MD, Ohio
R. Wajahat, MD, Ohio
K. Welch, Ohio
R. Barnhart, MD, Oregon
D. Harmon, Oregon
K. Ordelheide, FACP, Oregon
C. Boyle, BS, Pennsylvania
J. Dong, Pennsylvania
S. Kowsika, MD, Pennsylvania
S. Krauthamer, Pennsylvania
A. Rajaratnam, MD, Pennsylvania
C. Rauscher, MD, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
C. Shoff, MD, Pennsylvania
E. Garcia-Torres, MD, South Carolina
J. Kanter, FAAP, South Carolina
T. Klein, Tennessee
C. Taylor, MBA, Tennessee
J. Tedesco, ACNP, Tennessee
S. Ansari, Texas
A. Corley, MPAS, PA-C, Texas
K. Cunnusamy, Texas
C. Faust, MPH, Texas
B. Gajjar, Texas
S. Gudur, Texas
J. Hinojosa, Texas
R. Imtiaz, BS, Texas
B. Kennedy, MD, Texas
K. Koch, Texas
O. Mirza, Texas
L. Rijos, ACNP, Texas
H. Saikumar, MD, Texas
M. Walker, MD, Texas
M. Zaman, Texas
D. Borg, APRNBC, MSN, NP, Utah
A. Breviu, MD, Utah
R. Cook, Utah
K. Kenealy, MD, Utah
C. Chen, MD, Virginia
K. Malloy, Virginia
L. McDaniel, MD, Virginia
R. Miller, MD, Virginia
T. Schaefer, PA-C, Virginia
D. Vanikar, Virginia
J. Asriel, MD, Washington
T. Corn, Washington
K. Bergquist, DO, Wisconsin
S. Patel, MD, Wisconsin
S. Skogen, APRN-BC, Wisconsin
E. Winkel, Wisconsin
D. Figueroa, MD, Puerto Rico
A. Benefield, BSN, FNP, MSN, RN, Alabama
A. Mahajan, MPH, Alabama
B. Meredith, DO, Alabama
L. Ortiz, MD, Arizona
N. Charlton, MD, California
T. Fong, BS, MD, California
B. Gee, MD, California
I. He, California
K. Jones, FAAFP, California
J. Kolev, MD, California
S. Krishnamoorthy, MD, California
A. Kumar, MD, California
D. Pan, California
M. Reaves, MD, California
A. Sones, California
A. Wang, California
S. McCary, MD, Colorado
A. Beg, MD, Connecticut
S. Das, MD, Connecticut
A. Reisner, MD, D.C.
S. Gadalla, MD, Florida
N. Joseph, MBBS, Florida
R. Chowdhury, DO, MPH, Georgia
K. Mann, Georgia
D. Aiken, Idaho
C. Riji Anil, MBBS, Idaho
A. Asare, MACP, MBchB, Illinois
R. Clapp, ACNP, Illinois
A. Kost, MD, Illinois
R. Pavurala, MBBS, Illinois
M. Tsien, Illinois
D. Brewer, Indiana
J. Mast, MD, Indiana
M. Arakane, Iowa
S. Dobler, MD, Kansas
A. Ausef, Louisiana
C. Newby, MD, Louisiana
S. Brown, Massachusetts
A. Gonzalez, MD, Massachusetts
K. Lombardo, Massachusetts
C. Alamelumangapuram, MBBS, Michigan
A. Chakrabarti, Michigan
K. Dazy, MD, FAAP, Michigan
A. John, Michigan
J. Meinke, Michigan
P. Vemula, Michigan
R. Albrecht, PA-C, Minnesota
D. Burgy, Minnesota
T. Ebel, MD, Minnesota
T. Goddard, PA-C, Minnesota
K. Lichter, Minnesota
D. Lorentz, MD, Minnesota
R. Parikh, MD, Minnesota
E. Zygmunt, PA-C, Minnesota
A. Blaes, ACNP, MSN, Missouri
M. Sheikh, MD, Missouri
J. Carrasquillo, APRN-NP, Nebraska
T. Diesing, ACMPE, Nebraska
M. Havekost, MD, FAAFP, Nebraska
E. Guzman, MD, Nevada
S. Gupta, APRN, New Hampshire
M. Scaccia, MD, New Hampshire
M. Ward, New Hampshire
M. Weber, PA-C, New Jersey
Y. Abdou, New Mexico
J. Cruickshank, New Mexico
S. Scott, MD, New Mexico
P. Agrawal, MD, New York
A. Cruz, MD, New York
S. Dutta, New York
B. Fuchs, New York
D. Gill, MD, New York
K. Hernandez, New York
H. Holzer, MD, New York
L. Janson, MD, New York
P. Koukuntla, MBBS, New York
R. Mamun, New York
A. Nair, MD, New York
C. Ramchandani, New York
D. Shah, New York
J. Silver, MD, New York
A. Tusano, New York
J. Virk, MBBS, New York
J. Weisenberger, DO, New York
S. Chow, DO, North Carolina
Z. Hafeez, North Carolina
M. Jagosky, MD, North Carolina
L. Spence, DO, North Carolina
N. Uchendu, MD, MBBS, North Carolina
D. Wefuan, MD, North Carolina
O. Afzal, MD, Ohio
P. Areti, MD, MBBS, Ohio
S. Arobelidze, MD, Ohio
A. Bath, MD, Ohio
D. Bhandari, MD, Ohio
M. Hadley, Ohio
L. Hakam, MD, Ohio
L. Heinemann, MD, Ohio
V. Janamanchi, MD, Ohio
M. Sundhu, MD, Ohio
M. Syed, MD, Ohio
R. Wajahat, MD, Ohio
K. Welch, Ohio
R. Barnhart, MD, Oregon
D. Harmon, Oregon
K. Ordelheide, FACP, Oregon
C. Boyle, BS, Pennsylvania
J. Dong, Pennsylvania
S. Kowsika, MD, Pennsylvania
S. Krauthamer, Pennsylvania
A. Rajaratnam, MD, Pennsylvania
C. Rauscher, MD, Pennsylvania
M. Scoulos-Hanson, Pennsylvania
C. Shoff, MD, Pennsylvania
E. Garcia-Torres, MD, South Carolina
J. Kanter, FAAP, South Carolina
T. Klein, Tennessee
C. Taylor, MBA, Tennessee
J. Tedesco, ACNP, Tennessee
S. Ansari, Texas
A. Corley, MPAS, PA-C, Texas
K. Cunnusamy, Texas
C. Faust, MPH, Texas
B. Gajjar, Texas
S. Gudur, Texas
J. Hinojosa, Texas
R. Imtiaz, BS, Texas
B. Kennedy, MD, Texas
K. Koch, Texas
O. Mirza, Texas
L. Rijos, ACNP, Texas
H. Saikumar, MD, Texas
M. Walker, MD, Texas
M. Zaman, Texas
D. Borg, APRNBC, MSN, NP, Utah
A. Breviu, MD, Utah
R. Cook, Utah
K. Kenealy, MD, Utah
C. Chen, MD, Virginia
K. Malloy, Virginia
L. McDaniel, MD, Virginia
R. Miller, MD, Virginia
T. Schaefer, PA-C, Virginia
D. Vanikar, Virginia
J. Asriel, MD, Washington
T. Corn, Washington
K. Bergquist, DO, Wisconsin
S. Patel, MD, Wisconsin
S. Skogen, APRN-BC, Wisconsin
E. Winkel, Wisconsin
D. Figueroa, MD, Puerto Rico
How p53 and telomeres stave off cancer
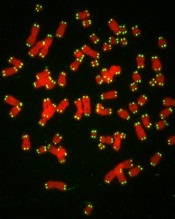
telomeres in green
Image by Claus Azzalin
Research published in The EMBO Journal suggests that p53 has tumor suppressor functions related to telomeres.
The study showed, for the first time, that p53 can suppress accumulated DNA damage at telomeres.
P53 is known to regulate the genome’s integrity. When DNA is damaged, p53 helps activate the transcription of genes that regulate the cell cycle and induce apoptosis.
However, prior studies have shown that p53 can bind at many locations across the genome, including sites that are not responsible for activating these regulatory genes.
Paul Lieberman, PhD, of The Wistar Institute in Philadelphia, Pennsylvania, and his colleagues decided to study these binding sites to see if p53 and telomeres might be more closely related than previous research suggested.
“We believed that p53 may be responsible for a more direct protective effect in telomeres,” Dr Lieberman said.
Using ChIP-sequencing, he and his colleagues identified p53 binding sites in subtelomeres.
They found that when p53 was bound to subtelomeres, the protein was able to suppress the formation of a histone modification called γ-H2AX.
This histone is modified in greater amounts when there is a double-strand break on DNA. If it persists, the break is not repaired, so suppressing its expression means the DNA is being preserved.
Additionally, p53 was able to prevent DNA degradation in telomeres, thereby keeping them intact and allowing them to more properly protect the tips of chromosomes.
“Based on our findings, we propose that the modifications to chromatin made by p53 enhance local DNA repair or protection,” Dr Lieberman said. “This would be yet another tumor suppressor function of p53, thus providing additional framework for just how important this gene is in protecting us from cancer.” ![]()
telomeres in green
Image by Claus Azzalin
Research published in The EMBO Journal suggests that p53 has tumor suppressor functions related to telomeres.
The study showed, for the first time, that p53 can suppress accumulated DNA damage at telomeres.
P53 is known to regulate the genome’s integrity. When DNA is damaged, p53 helps activate the transcription of genes that regulate the cell cycle and induce apoptosis.
However, prior studies have shown that p53 can bind at many locations across the genome, including sites that are not responsible for activating these regulatory genes.
Paul Lieberman, PhD, of The Wistar Institute in Philadelphia, Pennsylvania, and his colleagues decided to study these binding sites to see if p53 and telomeres might be more closely related than previous research suggested.
“We believed that p53 may be responsible for a more direct protective effect in telomeres,” Dr Lieberman said.
Using ChIP-sequencing, he and his colleagues identified p53 binding sites in subtelomeres.
They found that when p53 was bound to subtelomeres, the protein was able to suppress the formation of a histone modification called γ-H2AX.
This histone is modified in greater amounts when there is a double-strand break on DNA. If it persists, the break is not repaired, so suppressing its expression means the DNA is being preserved.
Additionally, p53 was able to prevent DNA degradation in telomeres, thereby keeping them intact and allowing them to more properly protect the tips of chromosomes.
“Based on our findings, we propose that the modifications to chromatin made by p53 enhance local DNA repair or protection,” Dr Lieberman said. “This would be yet another tumor suppressor function of p53, thus providing additional framework for just how important this gene is in protecting us from cancer.” ![]()
telomeres in green
Image by Claus Azzalin
Research published in The EMBO Journal suggests that p53 has tumor suppressor functions related to telomeres.
The study showed, for the first time, that p53 can suppress accumulated DNA damage at telomeres.
P53 is known to regulate the genome’s integrity. When DNA is damaged, p53 helps activate the transcription of genes that regulate the cell cycle and induce apoptosis.
However, prior studies have shown that p53 can bind at many locations across the genome, including sites that are not responsible for activating these regulatory genes.
Paul Lieberman, PhD, of The Wistar Institute in Philadelphia, Pennsylvania, and his colleagues decided to study these binding sites to see if p53 and telomeres might be more closely related than previous research suggested.
“We believed that p53 may be responsible for a more direct protective effect in telomeres,” Dr Lieberman said.
Using ChIP-sequencing, he and his colleagues identified p53 binding sites in subtelomeres.
They found that when p53 was bound to subtelomeres, the protein was able to suppress the formation of a histone modification called γ-H2AX.
This histone is modified in greater amounts when there is a double-strand break on DNA. If it persists, the break is not repaired, so suppressing its expression means the DNA is being preserved.
Additionally, p53 was able to prevent DNA degradation in telomeres, thereby keeping them intact and allowing them to more properly protect the tips of chromosomes.
“Based on our findings, we propose that the modifications to chromatin made by p53 enhance local DNA repair or protection,” Dr Lieberman said. “This would be yet another tumor suppressor function of p53, thus providing additional framework for just how important this gene is in protecting us from cancer.” ![]()
A dermatologist’s bad dream?
My plane doesn’t leave till 2:30. Glad I cut off seeing patients at 11, which should give me plenty of time. I’m getting smarter in my old age.
Smooth morning, paperwork pretty much done. Just one patient left. Look, a nice little old man. He has such a sweet smile.
“How can I help you, Mr. Goldfarb?”
“It’s complicated. This letter explains everything that’s happened the past 3 years,”
Oh-oh, that doesn’t sound good. “OK, let’s have a look.”
“My, you read fast, Doctor.”
When the first line says, ‘The lice all over my body don’t go away even after I apply bug shampoo every day,’ I’m pretty much done.
“Doctor, this bag has everything I’ve used: lice shampoo, insect spray, itch lotion. I forgot to bring in all the little bugs I collected from my combs and sheets.”
No! This can’t be happening! How do I negotiate with a delusion and still make my plane?
“Sometimes it feels like bugs are crawling on my skin.”
“Itching often feels that way ... ”
“I brought pictures. Want to see?”
No! Not an album! Snap after snap: scabs on the scalp, scaling at the corners of the mouth, linear scratches on the extremities.
“You know, Mr. Goldfarb – maybe firm confidence will let me regain control of this interview – what you’re describing does not sound like lice or bugs of any kind ... ”
“But Doctor, how do you explain this?” Another photo, this of a comb filled with brownish epidermal fragments. “I meant to bring some in, but I forgot.”
Enough. Time to look grim and speak briskly. “Mr. Goldfarb, this cannot be lice because ... ”
“I see them coming out of all my pores ... ”
“Mr. Goldfarb!” Now it’s my turn to interrupt. “I would appreciate it if you would let me finish my sentence.”
“Yes, Doctor. If it’s not lice, what do you think it is?”
Must think fast. “Sensitivity. Sensitive skin, especially if you’ve scratched it, can certainly feel as though there are things crawling on you. Patients often say that the skin feels this way. I will therefore treat this sensitivity with anti-inflammatory creams and lotion you will apply to the scalp, face, and the rest of your body respectively.”
Goldfarb is still listening. I’m almost there.
“I want you to use this medication for 2 weeks without stopping, and not use any more of the bug shampoos and creams because they can be irritating and increase itch and sensitivity. Please call me on my private extension at that point with your progress.” Easier to deny a delusion when not standing face to face.
“That’s good news, Doctor. I’ll pick up the medication and let you know.”
At most, he’ll stop scratching for a while. By the time he starts again, I’ll have made my plane and come back to the office. Meantime: Depart exam room briskly!
I can still make it if I leave right away. One last check of my office e-mail. There’s one from Zelda. She has a small scaly patch on one forearm. Claims it’s responded neither to topical antifungals nor steroids.
Here’s the text of her e-mail: “Doctor, I showed my rash to my neighbor Mary. She did some Internet research, and she’s convinced it’s chromoblastomycosis. I’m pretty sure she’s right. What do you think?”
I think I better leave right now.
My reply: “Dear Zelda, pretty unlikely. Try the new cream I’m going to prescribe for 2 weeks, and let me check on how you’re doing.”
How does evil dermatologic karma know that I’m trying to leave town? Parasitosis and chromoblastomycosis! Can this be a bad dream? If so, why don’t I wake up?
Mr. Goldfarb, still looking sweet and mild, sits in the waiting room, awaiting the elder shuttle to take him home.
Walk fast. Do not smile and meet his gaze. This is no time for politeness.
No, sir. I am outta here.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. Write to him at [email protected].
My plane doesn’t leave till 2:30. Glad I cut off seeing patients at 11, which should give me plenty of time. I’m getting smarter in my old age.
Smooth morning, paperwork pretty much done. Just one patient left. Look, a nice little old man. He has such a sweet smile.
“How can I help you, Mr. Goldfarb?”
“It’s complicated. This letter explains everything that’s happened the past 3 years,”
Oh-oh, that doesn’t sound good. “OK, let’s have a look.”
“My, you read fast, Doctor.”
When the first line says, ‘The lice all over my body don’t go away even after I apply bug shampoo every day,’ I’m pretty much done.
“Doctor, this bag has everything I’ve used: lice shampoo, insect spray, itch lotion. I forgot to bring in all the little bugs I collected from my combs and sheets.”
No! This can’t be happening! How do I negotiate with a delusion and still make my plane?
“Sometimes it feels like bugs are crawling on my skin.”
“Itching often feels that way ... ”
“I brought pictures. Want to see?”
No! Not an album! Snap after snap: scabs on the scalp, scaling at the corners of the mouth, linear scratches on the extremities.
“You know, Mr. Goldfarb – maybe firm confidence will let me regain control of this interview – what you’re describing does not sound like lice or bugs of any kind ... ”
“But Doctor, how do you explain this?” Another photo, this of a comb filled with brownish epidermal fragments. “I meant to bring some in, but I forgot.”
Enough. Time to look grim and speak briskly. “Mr. Goldfarb, this cannot be lice because ... ”
“I see them coming out of all my pores ... ”
“Mr. Goldfarb!” Now it’s my turn to interrupt. “I would appreciate it if you would let me finish my sentence.”
“Yes, Doctor. If it’s not lice, what do you think it is?”
Must think fast. “Sensitivity. Sensitive skin, especially if you’ve scratched it, can certainly feel as though there are things crawling on you. Patients often say that the skin feels this way. I will therefore treat this sensitivity with anti-inflammatory creams and lotion you will apply to the scalp, face, and the rest of your body respectively.”
Goldfarb is still listening. I’m almost there.
“I want you to use this medication for 2 weeks without stopping, and not use any more of the bug shampoos and creams because they can be irritating and increase itch and sensitivity. Please call me on my private extension at that point with your progress.” Easier to deny a delusion when not standing face to face.
“That’s good news, Doctor. I’ll pick up the medication and let you know.”
At most, he’ll stop scratching for a while. By the time he starts again, I’ll have made my plane and come back to the office. Meantime: Depart exam room briskly!
I can still make it if I leave right away. One last check of my office e-mail. There’s one from Zelda. She has a small scaly patch on one forearm. Claims it’s responded neither to topical antifungals nor steroids.
Here’s the text of her e-mail: “Doctor, I showed my rash to my neighbor Mary. She did some Internet research, and she’s convinced it’s chromoblastomycosis. I’m pretty sure she’s right. What do you think?”
I think I better leave right now.
My reply: “Dear Zelda, pretty unlikely. Try the new cream I’m going to prescribe for 2 weeks, and let me check on how you’re doing.”
How does evil dermatologic karma know that I’m trying to leave town? Parasitosis and chromoblastomycosis! Can this be a bad dream? If so, why don’t I wake up?
Mr. Goldfarb, still looking sweet and mild, sits in the waiting room, awaiting the elder shuttle to take him home.
Walk fast. Do not smile and meet his gaze. This is no time for politeness.
No, sir. I am outta here.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. Write to him at [email protected].
My plane doesn’t leave till 2:30. Glad I cut off seeing patients at 11, which should give me plenty of time. I’m getting smarter in my old age.
Smooth morning, paperwork pretty much done. Just one patient left. Look, a nice little old man. He has such a sweet smile.
“How can I help you, Mr. Goldfarb?”
“It’s complicated. This letter explains everything that’s happened the past 3 years,”
Oh-oh, that doesn’t sound good. “OK, let’s have a look.”
“My, you read fast, Doctor.”
When the first line says, ‘The lice all over my body don’t go away even after I apply bug shampoo every day,’ I’m pretty much done.
“Doctor, this bag has everything I’ve used: lice shampoo, insect spray, itch lotion. I forgot to bring in all the little bugs I collected from my combs and sheets.”
No! This can’t be happening! How do I negotiate with a delusion and still make my plane?
“Sometimes it feels like bugs are crawling on my skin.”
“Itching often feels that way ... ”
“I brought pictures. Want to see?”
No! Not an album! Snap after snap: scabs on the scalp, scaling at the corners of the mouth, linear scratches on the extremities.
“You know, Mr. Goldfarb – maybe firm confidence will let me regain control of this interview – what you’re describing does not sound like lice or bugs of any kind ... ”
“But Doctor, how do you explain this?” Another photo, this of a comb filled with brownish epidermal fragments. “I meant to bring some in, but I forgot.”
Enough. Time to look grim and speak briskly. “Mr. Goldfarb, this cannot be lice because ... ”
“I see them coming out of all my pores ... ”
“Mr. Goldfarb!” Now it’s my turn to interrupt. “I would appreciate it if you would let me finish my sentence.”
“Yes, Doctor. If it’s not lice, what do you think it is?”
Must think fast. “Sensitivity. Sensitive skin, especially if you’ve scratched it, can certainly feel as though there are things crawling on you. Patients often say that the skin feels this way. I will therefore treat this sensitivity with anti-inflammatory creams and lotion you will apply to the scalp, face, and the rest of your body respectively.”
Goldfarb is still listening. I’m almost there.
“I want you to use this medication for 2 weeks without stopping, and not use any more of the bug shampoos and creams because they can be irritating and increase itch and sensitivity. Please call me on my private extension at that point with your progress.” Easier to deny a delusion when not standing face to face.
“That’s good news, Doctor. I’ll pick up the medication and let you know.”
At most, he’ll stop scratching for a while. By the time he starts again, I’ll have made my plane and come back to the office. Meantime: Depart exam room briskly!
I can still make it if I leave right away. One last check of my office e-mail. There’s one from Zelda. She has a small scaly patch on one forearm. Claims it’s responded neither to topical antifungals nor steroids.
Here’s the text of her e-mail: “Doctor, I showed my rash to my neighbor Mary. She did some Internet research, and she’s convinced it’s chromoblastomycosis. I’m pretty sure she’s right. What do you think?”
I think I better leave right now.
My reply: “Dear Zelda, pretty unlikely. Try the new cream I’m going to prescribe for 2 weeks, and let me check on how you’re doing.”
How does evil dermatologic karma know that I’m trying to leave town? Parasitosis and chromoblastomycosis! Can this be a bad dream? If so, why don’t I wake up?
Mr. Goldfarb, still looking sweet and mild, sits in the waiting room, awaiting the elder shuttle to take him home.
Walk fast. Do not smile and meet his gaze. This is no time for politeness.
No, sir. I am outta here.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. Write to him at [email protected].

Zika virus: What clinicians must know
The recent spike in Zika virus cases in Central and South America brings with it the alarming risk – and even the expectation – of outbreaks occurring in the United States. How should U.S.-based clinicians prepare for the inevitable?
“The current outbreaks of Zika virus are the first of their kind in the Americas, so there isn’t a previous history of Zika virus spreading into the [United States],” explained Dr. Joy St. John, director of surveillance, disease prevention and control at the Caribbean Public Health Agency in Trinidad.
But now that the virus has hit the United States, with a confirmed case in Texas last week and more emerging since then, Dr. St. John said the most important thing is for U.S. health care providers to recognize the signs and symptoms of Zika virus infection. The virus is carried and transmitted by the Aedes aegypti species of mosquito, the same vector that transmits the dengue and chikungunya viruses. Zika virus symptoms are relatively mild, consisting predominantly of maculopapular rash, fever, arthralgia, myalgia, and conjunctivitis. Only one in five individuals with a Zika virus infection develop symptoms, but patients who present as such and who have traveled to Central or South America in the week prior to the onset of symptoms should be considered likely infected.
"At present, there is no rapid test available for diagnosis of Zika,” said Dr. St. John. “Diagnosis is primarily based on detection of viral RNA from clinical serum specimens in acutely ill patients.”
To that end, polymerase chain reaction (PCR) testing can be conducted on serum samples collected within 3-5 days of symptom onset. Beyond that, elevated levels of IgM antibodies can be confirmed by serology, based on the neutralization, seroconversion, or four-fold increase of Zika-specific antibodies in paired samples. However, Dr. St. John warned that “Due to the possibility of cross reactivity with other viruses, for example, dengue, it is strongly recommended samples be collected early enough for PCR testing.”
Zika and pregnancy
Zika virus has now been identified in more than 20 countries and territories worldwide, most of them in the Americas, although outbreaks have occurred in areas of Africa, Southeast Asia, and the Pacific Islands. While most infected patients experience relatively mild symptoms, Zika may be particularly dangerous when it infects a pregnant woman. There have been multiple cases of microcephaly in children whose mothers were infected with Zika virus during pregnancy, although the association of microcephaly with Zika virus infection during pregnancy has not been definitively confirmed. The Centers for Disease Control and Prevention recently issued a warning to Americans – particularly pregnant women – about traveling to high-risk areas.
“Scientifically, we’re not 100% sure if Zika virus is causing microcephaly, [but] what we’re seeing is in certain Brazilian districts, there’s been a 20-fold increase in rates of microcephaly at the same time that there’s been a lot more Zika virus in pregnant women,” explained Dr. Sanjaya Senanayake of Australian National University in Canberra.
According to data from the CDC, 1,248 suspected cases of microcephaly had been reported in Brazil as of Nov. 28, 2015, compared with the annual rate of just 150-200 such cases during 2010-2014. “Examination of the fetus [and] amniotic fluid, in some cases, has shown Zika virus, so there seems to be an association,” Dr. Senanayake clarified, adding that “the [ANVISA – Brazilian Health Surveillance Agency] has told women in certain districts where there’s been a lot of microcephaly not to get pregnant.”
Brazil is set to host millions of guests from around the world as the 2016 Olympics get underway in only a few months’ time. Women who are pregnant or anticipate becoming pregnant should consider the risks if they are planning to travel to Rio de Janeiro. The risk of microcephaly does not apply to infected women who are not pregnant, however, as the CDC states that “Zika virus usually remains in the blood of an infected person for only a few days to a week,” and therefore, “does not pose a risk of birth defects for future pregnancies.”
Dr. St. John also stated that “public health personnel are still cautioning pregnant women to take special care to avoid mosquito bites during their pregnancies,” adding that the “[Pan-American Health Organization] is working on their guidelines for surveillance of congenital abnormalities.”
Clinical insights
With treatment options so sparse – there is no vaccine or drug available specifically meant to combat a Zika virus infection – what can health care providers do for their patients? The CDC advises health care providers to “treat the symptoms,” which means telling patients to stay in bed, stay hydrated, and, most importantly, stay away from aspirin and NSAIDs “until dengue can be ruled out to reduce the risk of hemorrhage.” Acetaminophen is safe to use, in order to mitigate fever symptoms.
Those who are infected are also advised to stay indoors and remain as isolated as possible for at least a week after symptoms first present. While the risk of a domestic outbreak is probably low, Dr. St. John said, the more exposure a Zika virus–infected individual has to the outside world, the more likely he or she is to be bitten by another mosquito, which can then carry and transmit the virus to another person.
“Chikungunya and dengue virus, which are transmitted by the same vectors [as Zika virus], have not managed to establish ongoing transmission in the U.S., despite repeated importations, [so] it is likely that Zika virus’ spread would follow a similar pattern,” Dr. St. John noted.
Though rare, sexual transmission of Zika virus has also been found in at least one case, although it had been previously suspected for some time. In December 2013, a 44-year-old Tahitian man sought treatment for hematospermia. Analysis of his sperm, however, found Zika virus, indicating possible sexual transmission of the virus.
“The observation that [Zika virus] RNA was detectable in urine after viremia clearance in blood suggests that, as found for [dengue] and [West Nile virus] infections, urine samples can yield evidence of [Zika virus] for late diagnosis, but more investigation is needed,” the study concluded.
“The best way to control all this is to control the mosquito,” said Dr. Senanayake. “You get a four-for-one deal; not only do you get rid of Zika virus, but also chikungunya, dengue, and yellow fever.” Dr. Senanayake added that advanced research is currently underway in mosquito-control efforts, including the idea of releasing mosquitoes into the wild which have been genetically modified so they can’t breed.
Now that the Illinois Department of Health has confirmed two new cases of Zika virus infection in that state, with other new cases cropping up in Saint Martin and Guadeloupe and El Salvador, providers should remain vigilant, taking note of patients who have traveled to afflicted regions and show mosquito bites. Person-to-person transmission is “rare as hen’s teeth,” said Dr. Senanayake, which is to say, it is highly unlikely to occur. Nonetheless, he said information and communication is the best way to ensure that Zika virus does not spread widely in the United States.
*This story was updated 1/25/2016.
The recent spike in Zika virus cases in Central and South America brings with it the alarming risk – and even the expectation – of outbreaks occurring in the United States. How should U.S.-based clinicians prepare for the inevitable?
“The current outbreaks of Zika virus are the first of their kind in the Americas, so there isn’t a previous history of Zika virus spreading into the [United States],” explained Dr. Joy St. John, director of surveillance, disease prevention and control at the Caribbean Public Health Agency in Trinidad.
But now that the virus has hit the United States, with a confirmed case in Texas last week and more emerging since then, Dr. St. John said the most important thing is for U.S. health care providers to recognize the signs and symptoms of Zika virus infection. The virus is carried and transmitted by the Aedes aegypti species of mosquito, the same vector that transmits the dengue and chikungunya viruses. Zika virus symptoms are relatively mild, consisting predominantly of maculopapular rash, fever, arthralgia, myalgia, and conjunctivitis. Only one in five individuals with a Zika virus infection develop symptoms, but patients who present as such and who have traveled to Central or South America in the week prior to the onset of symptoms should be considered likely infected.
"At present, there is no rapid test available for diagnosis of Zika,” said Dr. St. John. “Diagnosis is primarily based on detection of viral RNA from clinical serum specimens in acutely ill patients.”
To that end, polymerase chain reaction (PCR) testing can be conducted on serum samples collected within 3-5 days of symptom onset. Beyond that, elevated levels of IgM antibodies can be confirmed by serology, based on the neutralization, seroconversion, or four-fold increase of Zika-specific antibodies in paired samples. However, Dr. St. John warned that “Due to the possibility of cross reactivity with other viruses, for example, dengue, it is strongly recommended samples be collected early enough for PCR testing.”
Zika and pregnancy
Zika virus has now been identified in more than 20 countries and territories worldwide, most of them in the Americas, although outbreaks have occurred in areas of Africa, Southeast Asia, and the Pacific Islands. While most infected patients experience relatively mild symptoms, Zika may be particularly dangerous when it infects a pregnant woman. There have been multiple cases of microcephaly in children whose mothers were infected with Zika virus during pregnancy, although the association of microcephaly with Zika virus infection during pregnancy has not been definitively confirmed. The Centers for Disease Control and Prevention recently issued a warning to Americans – particularly pregnant women – about traveling to high-risk areas.
“Scientifically, we’re not 100% sure if Zika virus is causing microcephaly, [but] what we’re seeing is in certain Brazilian districts, there’s been a 20-fold increase in rates of microcephaly at the same time that there’s been a lot more Zika virus in pregnant women,” explained Dr. Sanjaya Senanayake of Australian National University in Canberra.
According to data from the CDC, 1,248 suspected cases of microcephaly had been reported in Brazil as of Nov. 28, 2015, compared with the annual rate of just 150-200 such cases during 2010-2014. “Examination of the fetus [and] amniotic fluid, in some cases, has shown Zika virus, so there seems to be an association,” Dr. Senanayake clarified, adding that “the [ANVISA – Brazilian Health Surveillance Agency] has told women in certain districts where there’s been a lot of microcephaly not to get pregnant.”
Brazil is set to host millions of guests from around the world as the 2016 Olympics get underway in only a few months’ time. Women who are pregnant or anticipate becoming pregnant should consider the risks if they are planning to travel to Rio de Janeiro. The risk of microcephaly does not apply to infected women who are not pregnant, however, as the CDC states that “Zika virus usually remains in the blood of an infected person for only a few days to a week,” and therefore, “does not pose a risk of birth defects for future pregnancies.”
Dr. St. John also stated that “public health personnel are still cautioning pregnant women to take special care to avoid mosquito bites during their pregnancies,” adding that the “[Pan-American Health Organization] is working on their guidelines for surveillance of congenital abnormalities.”
Clinical insights
With treatment options so sparse – there is no vaccine or drug available specifically meant to combat a Zika virus infection – what can health care providers do for their patients? The CDC advises health care providers to “treat the symptoms,” which means telling patients to stay in bed, stay hydrated, and, most importantly, stay away from aspirin and NSAIDs “until dengue can be ruled out to reduce the risk of hemorrhage.” Acetaminophen is safe to use, in order to mitigate fever symptoms.
Those who are infected are also advised to stay indoors and remain as isolated as possible for at least a week after symptoms first present. While the risk of a domestic outbreak is probably low, Dr. St. John said, the more exposure a Zika virus–infected individual has to the outside world, the more likely he or she is to be bitten by another mosquito, which can then carry and transmit the virus to another person.
“Chikungunya and dengue virus, which are transmitted by the same vectors [as Zika virus], have not managed to establish ongoing transmission in the U.S., despite repeated importations, [so] it is likely that Zika virus’ spread would follow a similar pattern,” Dr. St. John noted.
Though rare, sexual transmission of Zika virus has also been found in at least one case, although it had been previously suspected for some time. In December 2013, a 44-year-old Tahitian man sought treatment for hematospermia. Analysis of his sperm, however, found Zika virus, indicating possible sexual transmission of the virus.
“The observation that [Zika virus] RNA was detectable in urine after viremia clearance in blood suggests that, as found for [dengue] and [West Nile virus] infections, urine samples can yield evidence of [Zika virus] for late diagnosis, but more investigation is needed,” the study concluded.
“The best way to control all this is to control the mosquito,” said Dr. Senanayake. “You get a four-for-one deal; not only do you get rid of Zika virus, but also chikungunya, dengue, and yellow fever.” Dr. Senanayake added that advanced research is currently underway in mosquito-control efforts, including the idea of releasing mosquitoes into the wild which have been genetically modified so they can’t breed.
Now that the Illinois Department of Health has confirmed two new cases of Zika virus infection in that state, with other new cases cropping up in Saint Martin and Guadeloupe and El Salvador, providers should remain vigilant, taking note of patients who have traveled to afflicted regions and show mosquito bites. Person-to-person transmission is “rare as hen’s teeth,” said Dr. Senanayake, which is to say, it is highly unlikely to occur. Nonetheless, he said information and communication is the best way to ensure that Zika virus does not spread widely in the United States.
*This story was updated 1/25/2016.
The recent spike in Zika virus cases in Central and South America brings with it the alarming risk – and even the expectation – of outbreaks occurring in the United States. How should U.S.-based clinicians prepare for the inevitable?
“The current outbreaks of Zika virus are the first of their kind in the Americas, so there isn’t a previous history of Zika virus spreading into the [United States],” explained Dr. Joy St. John, director of surveillance, disease prevention and control at the Caribbean Public Health Agency in Trinidad.
But now that the virus has hit the United States, with a confirmed case in Texas last week and more emerging since then, Dr. St. John said the most important thing is for U.S. health care providers to recognize the signs and symptoms of Zika virus infection. The virus is carried and transmitted by the Aedes aegypti species of mosquito, the same vector that transmits the dengue and chikungunya viruses. Zika virus symptoms are relatively mild, consisting predominantly of maculopapular rash, fever, arthralgia, myalgia, and conjunctivitis. Only one in five individuals with a Zika virus infection develop symptoms, but patients who present as such and who have traveled to Central or South America in the week prior to the onset of symptoms should be considered likely infected.
"At present, there is no rapid test available for diagnosis of Zika,” said Dr. St. John. “Diagnosis is primarily based on detection of viral RNA from clinical serum specimens in acutely ill patients.”
To that end, polymerase chain reaction (PCR) testing can be conducted on serum samples collected within 3-5 days of symptom onset. Beyond that, elevated levels of IgM antibodies can be confirmed by serology, based on the neutralization, seroconversion, or four-fold increase of Zika-specific antibodies in paired samples. However, Dr. St. John warned that “Due to the possibility of cross reactivity with other viruses, for example, dengue, it is strongly recommended samples be collected early enough for PCR testing.”
Zika and pregnancy
Zika virus has now been identified in more than 20 countries and territories worldwide, most of them in the Americas, although outbreaks have occurred in areas of Africa, Southeast Asia, and the Pacific Islands. While most infected patients experience relatively mild symptoms, Zika may be particularly dangerous when it infects a pregnant woman. There have been multiple cases of microcephaly in children whose mothers were infected with Zika virus during pregnancy, although the association of microcephaly with Zika virus infection during pregnancy has not been definitively confirmed. The Centers for Disease Control and Prevention recently issued a warning to Americans – particularly pregnant women – about traveling to high-risk areas.
“Scientifically, we’re not 100% sure if Zika virus is causing microcephaly, [but] what we’re seeing is in certain Brazilian districts, there’s been a 20-fold increase in rates of microcephaly at the same time that there’s been a lot more Zika virus in pregnant women,” explained Dr. Sanjaya Senanayake of Australian National University in Canberra.
According to data from the CDC, 1,248 suspected cases of microcephaly had been reported in Brazil as of Nov. 28, 2015, compared with the annual rate of just 150-200 such cases during 2010-2014. “Examination of the fetus [and] amniotic fluid, in some cases, has shown Zika virus, so there seems to be an association,” Dr. Senanayake clarified, adding that “the [ANVISA – Brazilian Health Surveillance Agency] has told women in certain districts where there’s been a lot of microcephaly not to get pregnant.”
Brazil is set to host millions of guests from around the world as the 2016 Olympics get underway in only a few months’ time. Women who are pregnant or anticipate becoming pregnant should consider the risks if they are planning to travel to Rio de Janeiro. The risk of microcephaly does not apply to infected women who are not pregnant, however, as the CDC states that “Zika virus usually remains in the blood of an infected person for only a few days to a week,” and therefore, “does not pose a risk of birth defects for future pregnancies.”
Dr. St. John also stated that “public health personnel are still cautioning pregnant women to take special care to avoid mosquito bites during their pregnancies,” adding that the “[Pan-American Health Organization] is working on their guidelines for surveillance of congenital abnormalities.”
Clinical insights
With treatment options so sparse – there is no vaccine or drug available specifically meant to combat a Zika virus infection – what can health care providers do for their patients? The CDC advises health care providers to “treat the symptoms,” which means telling patients to stay in bed, stay hydrated, and, most importantly, stay away from aspirin and NSAIDs “until dengue can be ruled out to reduce the risk of hemorrhage.” Acetaminophen is safe to use, in order to mitigate fever symptoms.
Those who are infected are also advised to stay indoors and remain as isolated as possible for at least a week after symptoms first present. While the risk of a domestic outbreak is probably low, Dr. St. John said, the more exposure a Zika virus–infected individual has to the outside world, the more likely he or she is to be bitten by another mosquito, which can then carry and transmit the virus to another person.
“Chikungunya and dengue virus, which are transmitted by the same vectors [as Zika virus], have not managed to establish ongoing transmission in the U.S., despite repeated importations, [so] it is likely that Zika virus’ spread would follow a similar pattern,” Dr. St. John noted.
Though rare, sexual transmission of Zika virus has also been found in at least one case, although it had been previously suspected for some time. In December 2013, a 44-year-old Tahitian man sought treatment for hematospermia. Analysis of his sperm, however, found Zika virus, indicating possible sexual transmission of the virus.
“The observation that [Zika virus] RNA was detectable in urine after viremia clearance in blood suggests that, as found for [dengue] and [West Nile virus] infections, urine samples can yield evidence of [Zika virus] for late diagnosis, but more investigation is needed,” the study concluded.
“The best way to control all this is to control the mosquito,” said Dr. Senanayake. “You get a four-for-one deal; not only do you get rid of Zika virus, but also chikungunya, dengue, and yellow fever.” Dr. Senanayake added that advanced research is currently underway in mosquito-control efforts, including the idea of releasing mosquitoes into the wild which have been genetically modified so they can’t breed.
Now that the Illinois Department of Health has confirmed two new cases of Zika virus infection in that state, with other new cases cropping up in Saint Martin and Guadeloupe and El Salvador, providers should remain vigilant, taking note of patients who have traveled to afflicted regions and show mosquito bites. Person-to-person transmission is “rare as hen’s teeth,” said Dr. Senanayake, which is to say, it is highly unlikely to occur. Nonetheless, he said information and communication is the best way to ensure that Zika virus does not spread widely in the United States.
*This story was updated 1/25/2016.

AACE: New algorithm stresses lifestyle modification in type 2 diabetes
Lifestyle modification must be the cornerstone of any management plan for type 2 diabetes.
Effectively attending to basic problems – obesity, nutrition, and exercise – will dramatically increase the success of any long-term treatment plan for patients with type 2 diabetes (T2DM), and help prevent or delay disease development in those with prediabetes, according to a new treatment algorithm.
The document, published by the American Association of Clinical Endocrinology, is an annual update to its unique strategy of clarifying T2DM management.
The 2016 AACE/ACE Comprehensive Diabetes Management Algorithm presents an easy-to-follow stepwise decision model for diagnosis, blood glucose management, and medical management – including all of the currently approved oral diabetes medications and insulins (Endocr Pract. 2016 Jan;22[1]:84-113). The original algorithm was launched 10 years ago and was last updated in 2013.
This new iteration is the first to place lifestyle intervention as a foundation for the most effective medical management.

“Obviously, this is very important,” said Dr. Paul S. Jellinger, a member of the algorithm writing committee, and an endocrinologist in Fort Lauderdale, Fla. “Weight loss, fitness training, and nutritional management play important roles in the management of blood glucose, lipids, and blood pressure. Appropriate focus in this direction may reduce medication dosage and at times eliminate the need for pharmaceutical intervention. We have all experienced improved therapeutic results in patients who are engaged in effective lifestyle therapy.”
As well as being a text document replete with data-driven details, the algorithm is presented in a colorful poster format that is very helpful for both doctors and patients alike, said Dr. George Grunberger, AACE president. It’s an especially effective tool when considering treatment decisions in the 12 different drug classes used in T2DM management.
“It’s one thing to talk about risk and benefits but to see it graphically displayed is very helpful,” he said in an interview. “I have the poster in every exam room, and every time I am with a patient, I can point out where we are and where we are heading. Rather than me just talking, I can show exactly where we are and where we want to go.”

It is especially helpful for primary care physicians, who care for the vast majority of patients with T2DM, Dr. Grunberger said in an interview.
“We are trying to get better management information into primary care. Most people with diabetes will never see a specialist in their entire life. They need help with glycemic control, obesity, prediabetes, dyslipidemia, and these are the bread and butter of primary care. But there are so many new things going on in this field, and primary care doctors are already in over their heads with the amount of things they deal with. So we have sorted it out and provide practical, practice-oriented guidelines about how to get from A to B.”
Dr. Grunberger noted that most of the recommendations in the document are based on expert opinion. “There are no randomized, controlled trials for most of this stuff. Many of the medications are relatively new, and with 12 drug classes, there’s no way you can ever do a trial with every permutation.”
The new document is similar to the 2013 algorithm with regard to medical management, Dr. Grunberger said. Its focus on lifestyle modification as an integral part of treatment is new, however. “The initial algorithm 10 years ago was solely based on glycemic control. Now we’ve decided to look at more than blood sugar – at obesity, overweight, hypertension and dyslipidemia. You cannot ignore these things in a disease where the major morbidity and mortality are cardiovascular.”
The algorithm stresses that “lifestyle optimization” is essential for all patients with diabetes. “[It] is multifaceted, ongoing, and should engage the entire diabetes team.”
There are several key components to lifestyle modification. All of these should be addressed early.
Medical nutrition therapy
This is a fundamental issue that must be addressed. A primarily plant-based diet high in poly- and monounsaturated fats is recommended, with the goal of a 5%-10% reduction in body weight for overweight or obese patients. In addition to discussing foods that damage and promote metabolic health, patients may need help with carbohydrate and sugar intake. Structured counseling is an excellent way to achieve consistent results.
Physical activity
Regular exercise improves glucose control and lowers lipid and blood pressure levels. It decreases the chance of falls and fractures, promotes functional capacity, and reduces the risk of depression. The goal should be at least 150 minutes of moderate-intense exercise each week. Every patient – and particularly those with complications of diabetes and/or obesity – should have a thorough physical exam before embarking on an exercise program.
Adequate rest
Emerging data continue to confirm the importance of sleep in health and disease. Getting 6-9 hours each night is associated with a reduction in cardiometabolic risk factors. Sleep deprivation aggravates insulin resistance, hypertension, hyperglycemia, and dyslipidemia and increases proinflammatory cytokines. An evaluation for obstructive sleep apnea may be in order, especially for obese patients.
Behavioral support
It’s impossible to overstate the importance of support in a successful lifestyle modification program. Patients should be encouraged to join community groups that facilitate and teach healthy behaviors. Not only will doing so help improve compliance, but being part of a structured group also reaps social and cognitive benefits.
Smoking cessation
The final component of the program, smoking cessation, is critical. All forms of tobacco should be eliminated.
While lifestyle modification is crucial, it should not obviate prompt medical therapy. “Such efforts should not delay needed pharmacotherapy, which can be initiated simultaneously and adjusted based on patient response to lifestyle efforts,” the document notes. “The need for medical therapy should not be interpreted as a failure of lifestyle management but as an adjunct to it.”
Aggressive medical therapy really accelerates effective diabetes treatment, Dr. Jellinger said.
“Clinical inertia has been and remains a huge problem. Some studies demonstrate as much as a 2-year delay in advancing therapy while the patient still remains far from hemoglobin A1c goal. For decades a ‘treat to failure’ concept dominated, i.e., that we should advance therapy only after a prolonged period of failure on existing therapy. One of the major contributions of the earlier AACE algorithms as well as the current version has been the strong therapeutic mandate to re-evaluate the patient and make a therapeutic change in no longer than 3 months. This is a direct attempt to eliminate clinical inertia.”
Dr. Grunberger agreed.
“Why do we wait until people are sick and experiencing complications before we take them seriously? Preventing and dealing with overweight and obesity is complicated, but if you treat obesity, you are treating diabetes. We emphasize starting medical therapy early, going to combination therapy quickly because no one drug usually achieves the target, and trying to be aggressive. Get people on the right treatment as quickly as possible and sustain success – don’t go from one failure to another.”
Dr. Jellinger has received support from Amarin, Boehringer Ingelheim, Bristol-Myers Squibb/AstraZeneca, Janssen Pharmaceuticals, and Novo Nordisk.
Dr. Grunberger has received remuneration and research funding from Eli Lilly, BI-Lilly, Novo Nordisk, Sanofi, Janssen, AstraZeneca, Merck, Medtronic, and GlaxoSmithKline.
Lifestyle modification must be the cornerstone of any management plan for type 2 diabetes.
Effectively attending to basic problems – obesity, nutrition, and exercise – will dramatically increase the success of any long-term treatment plan for patients with type 2 diabetes (T2DM), and help prevent or delay disease development in those with prediabetes, according to a new treatment algorithm.
The document, published by the American Association of Clinical Endocrinology, is an annual update to its unique strategy of clarifying T2DM management.
The 2016 AACE/ACE Comprehensive Diabetes Management Algorithm presents an easy-to-follow stepwise decision model for diagnosis, blood glucose management, and medical management – including all of the currently approved oral diabetes medications and insulins (Endocr Pract. 2016 Jan;22[1]:84-113). The original algorithm was launched 10 years ago and was last updated in 2013.
This new iteration is the first to place lifestyle intervention as a foundation for the most effective medical management.
“Obviously, this is very important,” said Dr. Paul S. Jellinger, a member of the algorithm writing committee, and an endocrinologist in Fort Lauderdale, Fla. “Weight loss, fitness training, and nutritional management play important roles in the management of blood glucose, lipids, and blood pressure. Appropriate focus in this direction may reduce medication dosage and at times eliminate the need for pharmaceutical intervention. We have all experienced improved therapeutic results in patients who are engaged in effective lifestyle therapy.”
As well as being a text document replete with data-driven details, the algorithm is presented in a colorful poster format that is very helpful for both doctors and patients alike, said Dr. George Grunberger, AACE president. It’s an especially effective tool when considering treatment decisions in the 12 different drug classes used in T2DM management.
“It’s one thing to talk about risk and benefits but to see it graphically displayed is very helpful,” he said in an interview. “I have the poster in every exam room, and every time I am with a patient, I can point out where we are and where we are heading. Rather than me just talking, I can show exactly where we are and where we want to go.”
It is especially helpful for primary care physicians, who care for the vast majority of patients with T2DM, Dr. Grunberger said in an interview.
“We are trying to get better management information into primary care. Most people with diabetes will never see a specialist in their entire life. They need help with glycemic control, obesity, prediabetes, dyslipidemia, and these are the bread and butter of primary care. But there are so many new things going on in this field, and primary care doctors are already in over their heads with the amount of things they deal with. So we have sorted it out and provide practical, practice-oriented guidelines about how to get from A to B.”
Dr. Grunberger noted that most of the recommendations in the document are based on expert opinion. “There are no randomized, controlled trials for most of this stuff. Many of the medications are relatively new, and with 12 drug classes, there’s no way you can ever do a trial with every permutation.”
The new document is similar to the 2013 algorithm with regard to medical management, Dr. Grunberger said. Its focus on lifestyle modification as an integral part of treatment is new, however. “The initial algorithm 10 years ago was solely based on glycemic control. Now we’ve decided to look at more than blood sugar – at obesity, overweight, hypertension and dyslipidemia. You cannot ignore these things in a disease where the major morbidity and mortality are cardiovascular.”
The algorithm stresses that “lifestyle optimization” is essential for all patients with diabetes. “[It] is multifaceted, ongoing, and should engage the entire diabetes team.”
There are several key components to lifestyle modification. All of these should be addressed early.
Medical nutrition therapy
This is a fundamental issue that must be addressed. A primarily plant-based diet high in poly- and monounsaturated fats is recommended, with the goal of a 5%-10% reduction in body weight for overweight or obese patients. In addition to discussing foods that damage and promote metabolic health, patients may need help with carbohydrate and sugar intake. Structured counseling is an excellent way to achieve consistent results.
Physical activity
Regular exercise improves glucose control and lowers lipid and blood pressure levels. It decreases the chance of falls and fractures, promotes functional capacity, and reduces the risk of depression. The goal should be at least 150 minutes of moderate-intense exercise each week. Every patient – and particularly those with complications of diabetes and/or obesity – should have a thorough physical exam before embarking on an exercise program.
Adequate rest
Emerging data continue to confirm the importance of sleep in health and disease. Getting 6-9 hours each night is associated with a reduction in cardiometabolic risk factors. Sleep deprivation aggravates insulin resistance, hypertension, hyperglycemia, and dyslipidemia and increases proinflammatory cytokines. An evaluation for obstructive sleep apnea may be in order, especially for obese patients.
Behavioral support
It’s impossible to overstate the importance of support in a successful lifestyle modification program. Patients should be encouraged to join community groups that facilitate and teach healthy behaviors. Not only will doing so help improve compliance, but being part of a structured group also reaps social and cognitive benefits.
Smoking cessation
The final component of the program, smoking cessation, is critical. All forms of tobacco should be eliminated.
While lifestyle modification is crucial, it should not obviate prompt medical therapy. “Such efforts should not delay needed pharmacotherapy, which can be initiated simultaneously and adjusted based on patient response to lifestyle efforts,” the document notes. “The need for medical therapy should not be interpreted as a failure of lifestyle management but as an adjunct to it.”
Aggressive medical therapy really accelerates effective diabetes treatment, Dr. Jellinger said.
“Clinical inertia has been and remains a huge problem. Some studies demonstrate as much as a 2-year delay in advancing therapy while the patient still remains far from hemoglobin A1c goal. For decades a ‘treat to failure’ concept dominated, i.e., that we should advance therapy only after a prolonged period of failure on existing therapy. One of the major contributions of the earlier AACE algorithms as well as the current version has been the strong therapeutic mandate to re-evaluate the patient and make a therapeutic change in no longer than 3 months. This is a direct attempt to eliminate clinical inertia.”
Dr. Grunberger agreed.
“Why do we wait until people are sick and experiencing complications before we take them seriously? Preventing and dealing with overweight and obesity is complicated, but if you treat obesity, you are treating diabetes. We emphasize starting medical therapy early, going to combination therapy quickly because no one drug usually achieves the target, and trying to be aggressive. Get people on the right treatment as quickly as possible and sustain success – don’t go from one failure to another.”
Dr. Jellinger has received support from Amarin, Boehringer Ingelheim, Bristol-Myers Squibb/AstraZeneca, Janssen Pharmaceuticals, and Novo Nordisk.
Dr. Grunberger has received remuneration and research funding from Eli Lilly, BI-Lilly, Novo Nordisk, Sanofi, Janssen, AstraZeneca, Merck, Medtronic, and GlaxoSmithKline.
Lifestyle modification must be the cornerstone of any management plan for type 2 diabetes.
Effectively attending to basic problems – obesity, nutrition, and exercise – will dramatically increase the success of any long-term treatment plan for patients with type 2 diabetes (T2DM), and help prevent or delay disease development in those with prediabetes, according to a new treatment algorithm.
The document, published by the American Association of Clinical Endocrinology, is an annual update to its unique strategy of clarifying T2DM management.
The 2016 AACE/ACE Comprehensive Diabetes Management Algorithm presents an easy-to-follow stepwise decision model for diagnosis, blood glucose management, and medical management – including all of the currently approved oral diabetes medications and insulins (Endocr Pract. 2016 Jan;22[1]:84-113). The original algorithm was launched 10 years ago and was last updated in 2013.
This new iteration is the first to place lifestyle intervention as a foundation for the most effective medical management.
“Obviously, this is very important,” said Dr. Paul S. Jellinger, a member of the algorithm writing committee, and an endocrinologist in Fort Lauderdale, Fla. “Weight loss, fitness training, and nutritional management play important roles in the management of blood glucose, lipids, and blood pressure. Appropriate focus in this direction may reduce medication dosage and at times eliminate the need for pharmaceutical intervention. We have all experienced improved therapeutic results in patients who are engaged in effective lifestyle therapy.”
As well as being a text document replete with data-driven details, the algorithm is presented in a colorful poster format that is very helpful for both doctors and patients alike, said Dr. George Grunberger, AACE president. It’s an especially effective tool when considering treatment decisions in the 12 different drug classes used in T2DM management.
“It’s one thing to talk about risk and benefits but to see it graphically displayed is very helpful,” he said in an interview. “I have the poster in every exam room, and every time I am with a patient, I can point out where we are and where we are heading. Rather than me just talking, I can show exactly where we are and where we want to go.”
It is especially helpful for primary care physicians, who care for the vast majority of patients with T2DM, Dr. Grunberger said in an interview.
“We are trying to get better management information into primary care. Most people with diabetes will never see a specialist in their entire life. They need help with glycemic control, obesity, prediabetes, dyslipidemia, and these are the bread and butter of primary care. But there are so many new things going on in this field, and primary care doctors are already in over their heads with the amount of things they deal with. So we have sorted it out and provide practical, practice-oriented guidelines about how to get from A to B.”
Dr. Grunberger noted that most of the recommendations in the document are based on expert opinion. “There are no randomized, controlled trials for most of this stuff. Many of the medications are relatively new, and with 12 drug classes, there’s no way you can ever do a trial with every permutation.”
The new document is similar to the 2013 algorithm with regard to medical management, Dr. Grunberger said. Its focus on lifestyle modification as an integral part of treatment is new, however. “The initial algorithm 10 years ago was solely based on glycemic control. Now we’ve decided to look at more than blood sugar – at obesity, overweight, hypertension and dyslipidemia. You cannot ignore these things in a disease where the major morbidity and mortality are cardiovascular.”
The algorithm stresses that “lifestyle optimization” is essential for all patients with diabetes. “[It] is multifaceted, ongoing, and should engage the entire diabetes team.”
There are several key components to lifestyle modification. All of these should be addressed early.
Medical nutrition therapy
This is a fundamental issue that must be addressed. A primarily plant-based diet high in poly- and monounsaturated fats is recommended, with the goal of a 5%-10% reduction in body weight for overweight or obese patients. In addition to discussing foods that damage and promote metabolic health, patients may need help with carbohydrate and sugar intake. Structured counseling is an excellent way to achieve consistent results.
Physical activity
Regular exercise improves glucose control and lowers lipid and blood pressure levels. It decreases the chance of falls and fractures, promotes functional capacity, and reduces the risk of depression. The goal should be at least 150 minutes of moderate-intense exercise each week. Every patient – and particularly those with complications of diabetes and/or obesity – should have a thorough physical exam before embarking on an exercise program.
Adequate rest
Emerging data continue to confirm the importance of sleep in health and disease. Getting 6-9 hours each night is associated with a reduction in cardiometabolic risk factors. Sleep deprivation aggravates insulin resistance, hypertension, hyperglycemia, and dyslipidemia and increases proinflammatory cytokines. An evaluation for obstructive sleep apnea may be in order, especially for obese patients.
Behavioral support
It’s impossible to overstate the importance of support in a successful lifestyle modification program. Patients should be encouraged to join community groups that facilitate and teach healthy behaviors. Not only will doing so help improve compliance, but being part of a structured group also reaps social and cognitive benefits.
Smoking cessation
The final component of the program, smoking cessation, is critical. All forms of tobacco should be eliminated.
While lifestyle modification is crucial, it should not obviate prompt medical therapy. “Such efforts should not delay needed pharmacotherapy, which can be initiated simultaneously and adjusted based on patient response to lifestyle efforts,” the document notes. “The need for medical therapy should not be interpreted as a failure of lifestyle management but as an adjunct to it.”
Aggressive medical therapy really accelerates effective diabetes treatment, Dr. Jellinger said.
“Clinical inertia has been and remains a huge problem. Some studies demonstrate as much as a 2-year delay in advancing therapy while the patient still remains far from hemoglobin A1c goal. For decades a ‘treat to failure’ concept dominated, i.e., that we should advance therapy only after a prolonged period of failure on existing therapy. One of the major contributions of the earlier AACE algorithms as well as the current version has been the strong therapeutic mandate to re-evaluate the patient and make a therapeutic change in no longer than 3 months. This is a direct attempt to eliminate clinical inertia.”
Dr. Grunberger agreed.
“Why do we wait until people are sick and experiencing complications before we take them seriously? Preventing and dealing with overweight and obesity is complicated, but if you treat obesity, you are treating diabetes. We emphasize starting medical therapy early, going to combination therapy quickly because no one drug usually achieves the target, and trying to be aggressive. Get people on the right treatment as quickly as possible and sustain success – don’t go from one failure to another.”
Dr. Jellinger has received support from Amarin, Boehringer Ingelheim, Bristol-Myers Squibb/AstraZeneca, Janssen Pharmaceuticals, and Novo Nordisk.
Dr. Grunberger has received remuneration and research funding from Eli Lilly, BI-Lilly, Novo Nordisk, Sanofi, Janssen, AstraZeneca, Merck, Medtronic, and GlaxoSmithKline.

Caring for gender-nonconforming youth in a primary care setting – Part 2
Gender identity typically develops in early childhood, and by age 4 years, most children consistently refer to themselves as a girl or a boy.1 For the majority of children, natal sex or sex assigned at birth, aligns with gender identity (a person’s innate sense of feeling male, female, or somewhere in between). However, this is not always the case. Gender identity can be understood as a spectrum with youth identifying as a gender that aligns with their natal sex (cisgender), is opposite of their natal sex (transgender), no gender (agender), or somewhere in between (genderqueer). The distress that can result from an incongruence between natal sex and gender identity is called gender dysphoria. Youth with gender dysphoria are at increased risk for a number of conditions, including suicide and self-harm. Early identification and appropriate care of these youth can reduce these risks. This month’s column will briefly review assessment of these youth in the pediatric setting.
Many youth who have a gender-nonconforming identity in childhood will not go on to have one in adulthood.2,3 Those who have a consistent, insistent, and persistent nonconforming identity are more likely to have this identity persist into adulthood. Youth who experience increased gender dysphoria with the onset of puberty rarely have this subside.

As it can be difficult to predict the trajectory of gender identity from childhood to adolescence, the approach to the prepubertal and pubertal gender nonconforming patient is different. It is important to note that research suggests that gender identity is innate and cannot be changed with interventions. The goals of care for gender-nonconforming (GN) youth include providing a safe environment where youth can explore their identities, and individualizing treatment to meet the needs of each patient and family.
Care for prepubertal GN youth
For parents:
Have you noticed, or are you concerned about your child’s:
• Preference or rejection of particular toys/games?
• Hair and clothing preferences or rejections?
• Preferred (if any) gender of playmates?
Has your child ever expressed:
• A desire to be or insistence that they are the other gender?
• A dislike of their sexual anatomy?
• A desire for primary (penis, vagina) or secondary (periods, facial hair) sex characteristics of the other gender?
Are you concerned about bullying ?
Do you have any concerns about your child’s mood or concerns for self-harm?
For children:
• Do you feel more like a girl, boy, neither, both?
• How would you like to play, cut your hair, dress?
• What name or pronoun (she for girl, he for boy) fits you?4
The goal for prepubertal youth with nonconforming identities is to ensure that they are safe at home, school, and at play. Some youth may express a desire to “transition” or live as their identified gender by changing their name and dressing as their identified gender. Some youth and families may choose to transition only in certain settings (at home, but not at school). Some youth and families may want a safe space where the child can grow, develop, and continue to explore their identity without transitioning. Mental health providers trained in the care of GN youth can help patients and families decide if transition is appropriate for them and support them with the process and timing of transitioning. For youth who experience depression, anxiety, bullying, or thoughts of self-harm related to their gender identity, care by an experienced mental health provider is essential. It is important to recognize that each patient and family will need an individualized approach based on their needs.
Care for pubertal GN youth

The development of secondary sex characteristics can be particularly distressing for GN youth. Some youth may first experience gender dysphoria at this time. This distress combined with the psychosocial stressors of adolescent development can lead to depression, anxiety, suicidal ideation, self-harm, and other risk taking behaviors. Visits with pubertal GN youth, as with any adolescent, should include confidential time alone with the medical provider to discuss any concerns. Youth should be informed that information will be kept confidential, but parents will need to be notified of any safety concerns (such as suicidality or self-harm). As with prepubertal youth, a history related to hair and clothing preferences; distress related to genital anatomy; and the desire to be the other gender should be obtained. A pubertal history and any related symptoms of distress also should be obtained.
DO
• Ask preferred name and pronoun.
• Perform confidential strength and risk assessment.
• Assess for family and social support.
• Refer to appropriate mental health and transgender providers.
DON’T
• Assume names and pronouns.
• Interview patient only with parent in the room.
• Disclose identity without patient consent.
• Dismiss parents as sources of support.
• Refer for reparative therapy.4
Youth who are suspected to have a diagnosis of gender dysphoria should be referred to mental health and medical providers with experience caring for transgender youth. These specialists can work with patients and families, and determine when and if youth are eligible for puberty blocking therapy with GnRH analogues and/or hormone therapy. GnRH analogues, if appropriate, can be prescribed after patients have reached sexual maturity rating stage 2. The rationale for this treatment is to prevent the development of unwanted secondary sex characteristics while giving the youth a chance to continue with psychotherapy and explore their gender identity.5 Hormone therapy, if appropriate, can be prescribed a few years later under the care of a transgender specialist and mental health provider.
Summary
It is normal to experiment with gender roles and expression in childhood. Providing a safe space to do this is important.
Individuals who have a persistent, consistent, and insistent gender-nonconforming identification and who have increased distress with puberty are unlikely to have this subside.
Pediatricians can assess for gender dysphoria and screen for related mood disorders and behaviors in the primary care setting. Appropriate referral to trained professionals is important.
Care should be individualized and focused on the health and safety of the patient.
Resources
For health care professionals
• World Professional Association for Transgender Health: Standards of care on care of transgender patients and provider directory. www.wpath.org• Physicians for Reproductive Health’s adolescent reproductive and sexual health education program (ARSHEP): Best practices for adolescent and reproductive health: Module on caring for transgender adolescent patients. prh.org/teen-reproductive-health/arshep-downloads/
For patients and families
• Family Acceptance Project: familyproject.sfsu.edu/
• Healthychildren.org: Parenting website supported by the American Academy of Pediatrics. Links to articles on gender nonconforming and transgender children; gender identity development in children. www.healthychildren.org
References
1. Caring for Your School Age Child: Ages 5-12 by the American Academy of Pediatrics (New York: Bantam Books, 1995).
2. Dev Psychol. 2008 Jan;44(1):34-45.
3. J Am Acad Child and Adolesc Psychiatry. 2008;47(12):1413-23
4. Caring for Transgender Adolescent Patients. Physicians for Reproductive Health’s Adolescent Reproductive and Sexual Health Education Program (ARSHEP): Best practices for adolescent and reproductive health: prh.org/teen-reproductive-health/arshep-downloads/
5. World Professional Association of Transgender Health, Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, 7th Edition (International Journal of Transgenderism. 2011;13:165-232)
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at the Ohio State University, both in Columbus.
Gender identity typically develops in early childhood, and by age 4 years, most children consistently refer to themselves as a girl or a boy.1 For the majority of children, natal sex or sex assigned at birth, aligns with gender identity (a person’s innate sense of feeling male, female, or somewhere in between). However, this is not always the case. Gender identity can be understood as a spectrum with youth identifying as a gender that aligns with their natal sex (cisgender), is opposite of their natal sex (transgender), no gender (agender), or somewhere in between (genderqueer). The distress that can result from an incongruence between natal sex and gender identity is called gender dysphoria. Youth with gender dysphoria are at increased risk for a number of conditions, including suicide and self-harm. Early identification and appropriate care of these youth can reduce these risks. This month’s column will briefly review assessment of these youth in the pediatric setting.
Many youth who have a gender-nonconforming identity in childhood will not go on to have one in adulthood.2,3 Those who have a consistent, insistent, and persistent nonconforming identity are more likely to have this identity persist into adulthood. Youth who experience increased gender dysphoria with the onset of puberty rarely have this subside.
As it can be difficult to predict the trajectory of gender identity from childhood to adolescence, the approach to the prepubertal and pubertal gender nonconforming patient is different. It is important to note that research suggests that gender identity is innate and cannot be changed with interventions. The goals of care for gender-nonconforming (GN) youth include providing a safe environment where youth can explore their identities, and individualizing treatment to meet the needs of each patient and family.
Care for prepubertal GN youth
For parents:
Have you noticed, or are you concerned about your child’s:
• Preference or rejection of particular toys/games?
• Hair and clothing preferences or rejections?
• Preferred (if any) gender of playmates?
Has your child ever expressed:
• A desire to be or insistence that they are the other gender?
• A dislike of their sexual anatomy?
• A desire for primary (penis, vagina) or secondary (periods, facial hair) sex characteristics of the other gender?
Are you concerned about bullying ?
Do you have any concerns about your child’s mood or concerns for self-harm?
For children:
• Do you feel more like a girl, boy, neither, both?
• How would you like to play, cut your hair, dress?
• What name or pronoun (she for girl, he for boy) fits you?4
The goal for prepubertal youth with nonconforming identities is to ensure that they are safe at home, school, and at play. Some youth may express a desire to “transition” or live as their identified gender by changing their name and dressing as their identified gender. Some youth and families may choose to transition only in certain settings (at home, but not at school). Some youth and families may want a safe space where the child can grow, develop, and continue to explore their identity without transitioning. Mental health providers trained in the care of GN youth can help patients and families decide if transition is appropriate for them and support them with the process and timing of transitioning. For youth who experience depression, anxiety, bullying, or thoughts of self-harm related to their gender identity, care by an experienced mental health provider is essential. It is important to recognize that each patient and family will need an individualized approach based on their needs.
Care for pubertal GN youth
The development of secondary sex characteristics can be particularly distressing for GN youth. Some youth may first experience gender dysphoria at this time. This distress combined with the psychosocial stressors of adolescent development can lead to depression, anxiety, suicidal ideation, self-harm, and other risk taking behaviors. Visits with pubertal GN youth, as with any adolescent, should include confidential time alone with the medical provider to discuss any concerns. Youth should be informed that information will be kept confidential, but parents will need to be notified of any safety concerns (such as suicidality or self-harm). As with prepubertal youth, a history related to hair and clothing preferences; distress related to genital anatomy; and the desire to be the other gender should be obtained. A pubertal history and any related symptoms of distress also should be obtained.
DO
• Ask preferred name and pronoun.
• Perform confidential strength and risk assessment.
• Assess for family and social support.
• Refer to appropriate mental health and transgender providers.
DON’T
• Assume names and pronouns.
• Interview patient only with parent in the room.
• Disclose identity without patient consent.
• Dismiss parents as sources of support.
• Refer for reparative therapy.4
Youth who are suspected to have a diagnosis of gender dysphoria should be referred to mental health and medical providers with experience caring for transgender youth. These specialists can work with patients and families, and determine when and if youth are eligible for puberty blocking therapy with GnRH analogues and/or hormone therapy. GnRH analogues, if appropriate, can be prescribed after patients have reached sexual maturity rating stage 2. The rationale for this treatment is to prevent the development of unwanted secondary sex characteristics while giving the youth a chance to continue with psychotherapy and explore their gender identity.5 Hormone therapy, if appropriate, can be prescribed a few years later under the care of a transgender specialist and mental health provider.
Summary
It is normal to experiment with gender roles and expression in childhood. Providing a safe space to do this is important.
Individuals who have a persistent, consistent, and insistent gender-nonconforming identification and who have increased distress with puberty are unlikely to have this subside.
Pediatricians can assess for gender dysphoria and screen for related mood disorders and behaviors in the primary care setting. Appropriate referral to trained professionals is important.
Care should be individualized and focused on the health and safety of the patient.
Resources
For health care professionals
• World Professional Association for Transgender Health: Standards of care on care of transgender patients and provider directory. www.wpath.org• Physicians for Reproductive Health’s adolescent reproductive and sexual health education program (ARSHEP): Best practices for adolescent and reproductive health: Module on caring for transgender adolescent patients. prh.org/teen-reproductive-health/arshep-downloads/
For patients and families
• Family Acceptance Project: familyproject.sfsu.edu/
• Healthychildren.org: Parenting website supported by the American Academy of Pediatrics. Links to articles on gender nonconforming and transgender children; gender identity development in children. www.healthychildren.org
References
1. Caring for Your School Age Child: Ages 5-12 by the American Academy of Pediatrics (New York: Bantam Books, 1995).
2. Dev Psychol. 2008 Jan;44(1):34-45.
3. J Am Acad Child and Adolesc Psychiatry. 2008;47(12):1413-23
4. Caring for Transgender Adolescent Patients. Physicians for Reproductive Health’s Adolescent Reproductive and Sexual Health Education Program (ARSHEP): Best practices for adolescent and reproductive health: prh.org/teen-reproductive-health/arshep-downloads/
5. World Professional Association of Transgender Health, Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, 7th Edition (International Journal of Transgenderism. 2011;13:165-232)
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at the Ohio State University, both in Columbus.
Gender identity typically develops in early childhood, and by age 4 years, most children consistently refer to themselves as a girl or a boy.1 For the majority of children, natal sex or sex assigned at birth, aligns with gender identity (a person’s innate sense of feeling male, female, or somewhere in between). However, this is not always the case. Gender identity can be understood as a spectrum with youth identifying as a gender that aligns with their natal sex (cisgender), is opposite of their natal sex (transgender), no gender (agender), or somewhere in between (genderqueer). The distress that can result from an incongruence between natal sex and gender identity is called gender dysphoria. Youth with gender dysphoria are at increased risk for a number of conditions, including suicide and self-harm. Early identification and appropriate care of these youth can reduce these risks. This month’s column will briefly review assessment of these youth in the pediatric setting.
Many youth who have a gender-nonconforming identity in childhood will not go on to have one in adulthood.2,3 Those who have a consistent, insistent, and persistent nonconforming identity are more likely to have this identity persist into adulthood. Youth who experience increased gender dysphoria with the onset of puberty rarely have this subside.
As it can be difficult to predict the trajectory of gender identity from childhood to adolescence, the approach to the prepubertal and pubertal gender nonconforming patient is different. It is important to note that research suggests that gender identity is innate and cannot be changed with interventions. The goals of care for gender-nonconforming (GN) youth include providing a safe environment where youth can explore their identities, and individualizing treatment to meet the needs of each patient and family.
Care for prepubertal GN youth
For parents:
Have you noticed, or are you concerned about your child’s:
• Preference or rejection of particular toys/games?
• Hair and clothing preferences or rejections?
• Preferred (if any) gender of playmates?
Has your child ever expressed:
• A desire to be or insistence that they are the other gender?
• A dislike of their sexual anatomy?
• A desire for primary (penis, vagina) or secondary (periods, facial hair) sex characteristics of the other gender?
Are you concerned about bullying ?
Do you have any concerns about your child’s mood or concerns for self-harm?
For children:
• Do you feel more like a girl, boy, neither, both?
• How would you like to play, cut your hair, dress?
• What name or pronoun (she for girl, he for boy) fits you?4
The goal for prepubertal youth with nonconforming identities is to ensure that they are safe at home, school, and at play. Some youth may express a desire to “transition” or live as their identified gender by changing their name and dressing as their identified gender. Some youth and families may choose to transition only in certain settings (at home, but not at school). Some youth and families may want a safe space where the child can grow, develop, and continue to explore their identity without transitioning. Mental health providers trained in the care of GN youth can help patients and families decide if transition is appropriate for them and support them with the process and timing of transitioning. For youth who experience depression, anxiety, bullying, or thoughts of self-harm related to their gender identity, care by an experienced mental health provider is essential. It is important to recognize that each patient and family will need an individualized approach based on their needs.
Care for pubertal GN youth
The development of secondary sex characteristics can be particularly distressing for GN youth. Some youth may first experience gender dysphoria at this time. This distress combined with the psychosocial stressors of adolescent development can lead to depression, anxiety, suicidal ideation, self-harm, and other risk taking behaviors. Visits with pubertal GN youth, as with any adolescent, should include confidential time alone with the medical provider to discuss any concerns. Youth should be informed that information will be kept confidential, but parents will need to be notified of any safety concerns (such as suicidality or self-harm). As with prepubertal youth, a history related to hair and clothing preferences; distress related to genital anatomy; and the desire to be the other gender should be obtained. A pubertal history and any related symptoms of distress also should be obtained.
DO
• Ask preferred name and pronoun.
• Perform confidential strength and risk assessment.
• Assess for family and social support.
• Refer to appropriate mental health and transgender providers.
DON’T
• Assume names and pronouns.
• Interview patient only with parent in the room.
• Disclose identity without patient consent.
• Dismiss parents as sources of support.
• Refer for reparative therapy.4
Youth who are suspected to have a diagnosis of gender dysphoria should be referred to mental health and medical providers with experience caring for transgender youth. These specialists can work with patients and families, and determine when and if youth are eligible for puberty blocking therapy with GnRH analogues and/or hormone therapy. GnRH analogues, if appropriate, can be prescribed after patients have reached sexual maturity rating stage 2. The rationale for this treatment is to prevent the development of unwanted secondary sex characteristics while giving the youth a chance to continue with psychotherapy and explore their gender identity.5 Hormone therapy, if appropriate, can be prescribed a few years later under the care of a transgender specialist and mental health provider.
Summary
It is normal to experiment with gender roles and expression in childhood. Providing a safe space to do this is important.
Individuals who have a persistent, consistent, and insistent gender-nonconforming identification and who have increased distress with puberty are unlikely to have this subside.
Pediatricians can assess for gender dysphoria and screen for related mood disorders and behaviors in the primary care setting. Appropriate referral to trained professionals is important.
Care should be individualized and focused on the health and safety of the patient.
Resources
For health care professionals
• World Professional Association for Transgender Health: Standards of care on care of transgender patients and provider directory. www.wpath.org• Physicians for Reproductive Health’s adolescent reproductive and sexual health education program (ARSHEP): Best practices for adolescent and reproductive health: Module on caring for transgender adolescent patients. prh.org/teen-reproductive-health/arshep-downloads/
For patients and families
• Family Acceptance Project: familyproject.sfsu.edu/
• Healthychildren.org: Parenting website supported by the American Academy of Pediatrics. Links to articles on gender nonconforming and transgender children; gender identity development in children. www.healthychildren.org
References
1. Caring for Your School Age Child: Ages 5-12 by the American Academy of Pediatrics (New York: Bantam Books, 1995).
2. Dev Psychol. 2008 Jan;44(1):34-45.
3. J Am Acad Child and Adolesc Psychiatry. 2008;47(12):1413-23
4. Caring for Transgender Adolescent Patients. Physicians for Reproductive Health’s Adolescent Reproductive and Sexual Health Education Program (ARSHEP): Best practices for adolescent and reproductive health: prh.org/teen-reproductive-health/arshep-downloads/
5. World Professional Association of Transgender Health, Standards of Care for the Health of Transsexual, Transgender, and Gender-Nonconforming People, 7th Edition (International Journal of Transgenderism. 2011;13:165-232)
Dr. Chelvakumar is an attending physician in the division of adolescent medicine at Nationwide Children’s Hospital and an assistant professor of clinical pediatrics at the Ohio State University, both in Columbus.
