User login
Bringing you the latest case reports, original research, clinical trial reviews, perspectives, patient resources, and more.
More Reports from the Connective Tissue Oncology Society 2018 annual meeting in Rome, November 14-17
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Early Results Find Olaratumab Combo With Doxorubicin Plus Ifosfamide Safe
Initial results of the phase 1b study of olaratumab plus doxorubicin and ifosfamide have shown the combination to be safe, reported Sebastian Bauer, MD, of the West German Cancer Center, University of Duisburg-Essen, Essen, Germany, and his colleagues at CTOS 2018.
The phase 1 trial (NCT03283696) enrolled 16 patients with advanced or metastatic soft tissue sarcomas. Patients had received no prior lines of systemic therapy and had an ECOG performance status of 0-1. Adequate follow-up data were available for 10 patients.
Olaratumab (Lartruvo), which binds platelet-derived growth factor receptor alpha (PDGFRα), was given at 15 mg/kg in combination with doxorubicin (75 mg/m2 on days 1-3) and ifosfamide (10 g/m2 on days 1-4). This was followed by mandatory granulocyte-colony-stimulating factor therapy in cycles 1-6 on a 21-day cycle. Doxorubicin could be administered by continuous infusion or bolus administration and with cardiac protection. Mesna dosing was at least 60% of the ifosfamide dose.
Two of the 10 patients had dose-limiting toxicities; one had grade 4 febrile neutropenia and the other had grade 3 febrile neutropenia and grade 3 mucositis. Common related adverse events occurring in over 30% of patients included fatigue, anemia, neutropenia, thrombocytopenia, constipation, and nausea. One patient discontinued study treatment due to progressive disease, and all others were on study treatment as of the data cutoff. Among 7 patients evaluated for tumor response, 3 patients had a partial response according to RECIST and 3 other patients had stabilized disease as best overall response, for a disease control rate of 86%.
Given that 8 of 10 evaluable patients have completed the dose-limiting toxicity period without dose-limiting toxicities at the 15 mg/kg dose level of olaratumab, the study has proceeded to the next cohort. In those patients, an olaratumab loading dose of 20 mg/kg will be evaluated in cycle 1, followed by 15 mg/kg of olaratumab in subsequent cycles with the same doses of doxorubicin plus ifosfamide, the researchers wrote in their abstract.
NOTE: Since CTOS 2018, olaratumab plus doxorubicin did not meet its phase 3 endpoint of overall survival (OS) advantage in the full study population or in the leiomyosarcoma subpopulation compared to doxorubicin alone.
Anthracycline-Based Regimen Excels in FIGO-1 Uterine Leiomyosarcoma
Patients with uterine leiomyosarcomas treated with anthracycline-based regimens experienced longer disease-free survival compared to patients treated with gemcitabine and docetaxel, according to a retrospective analysis reported at CTOS 2018.
Roberta Sanfilippo, MD, of Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues reviewed all patients with FIGO stage I uterine leiomyosarcomas at two Italian centers who underwent hysterectomy with or without oophorectomy and were then treated with adjuvant chemotherapy with anthracycline-based or gemcitabine-based regimens.
Of 145 patients, 97 were treated with an anthracycline-based regimen and 48 with gemcitabine and docetaxel. The median number of cycles of anthracycline-based therapy patients received was 4 (range 2-6) and the median number of cycles with gemcitabine and docetaxel was 5 (range 3-7). Disease-free survival was 31 months in patients treated with anthracycline-based chemotherapy and 19 months in patients treated with gemcitabine and docetaxel.
These results suggest that future trials to assess the efficacy of adjuvant chemotherapy in uterine leiomyosarcoma should incorporate anthracyclines, the investigators maintain.
Trabectedin and Concurrent Low-Dose Radiotherapy Feasible
Trabectedin concurrent with lowdose radiotherapy is being examined as an option for patients with pulmonary metastatic soft tissue sarcoma (NCT02275286).
In a phase 1 study, long-lasting dimensional responses were seen in 71% of the irradiated lesions. Based on those results, trabectedin (Yondelis) at 1.5 mg/m2 will be the recommended dose for phase 2, according to Javier Martín-Broto, MD, of the Institute of Biomedicine Research (IBIS)-University Hospital Virgen del Rocio/CSIC/University of Seville, Spain, and his colleagues, reporting at CTOS 2018.
For the study, trabectedin was given along with radiotherapy (30 Gy) in 10 fractions (3 Gy/fraction). Three dose levels of trabectedin were administered: -1 (1.1 mg/m2), 1 (1.3 mg/m2), and 2 (1.5 mg/m2). Dose-limiting toxicity was defined as grade 3 or greater events excluding grade 3/4 neutropenia lasting less than 5 days, grade 3 transaminitis if it did not lead to trabectedin delay, and grade 3/4 nausea/vomiting due to inadequate prophylaxis.
Ten of the 18 patients enrolled had synovial sarcoma; 3 had undifferentiated pleomorphic sarcomas, and the other patients had either myxoid liposarcoma, dedifferentiated liposarcoma, G3 not otherwise specified sarcoma, leiomyosarcoma, or malignant peripheral nerve sheath tumor.
Patients received a median of 1 prior line of chemotherapy (range: 0-3). Twelve patients received trabectedin at dose level 1 and 6 patients at dose level 2. Grade 3/4 adverse events were neutropenia, seen in 8 patients; alanine aminotransferase (ALT) elevation, seen in 2 patients; gamma-glutamyl transferase (GGT) elevation, seen in 2 patients; anemia, seen in 2 patients; febrile neutropenia, seen in 1 patient; and pneumonitis, seen in 1 patient.
There were two dose-limiting toxicities: transient grade 4 ALT elevation at the level 1 dose and grade 4 neutropenia for more than 5 days at the level 2 dose.
Based on central radiological review of 17 evaluable patients, 2 patients achieved complete response, 3 had partial responses, 6 had stable disease, and 6 had progressive disease. The local review reported complete responses in 2 patients, partial responses in 5, stable disease in 4, and progressive disease in 6.
Of the irradiated lesions, 71% had long-lasting dimensional responses: 4 completely responded, 8 responded partially, 4 were stable, and 1 progressed.
With a median follow-up of 18 months, median progression-free survival was 2.83 months (95%CI: 2.3-3.3 months). Thirteen patients have died, with a median overall survival of 8.77 months (95%CI: 3.6-13.9) and a 12-month overall survival rate of 48%.
The investigators concluded trabectedin with concurrent radiotherapy was feasible in patients with pulmonary metastatic soft tissue sarcoma regardless of their histologic subtype.
Health care resource utilization leading to a diagnosis of soft tissue sarcoma
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1. 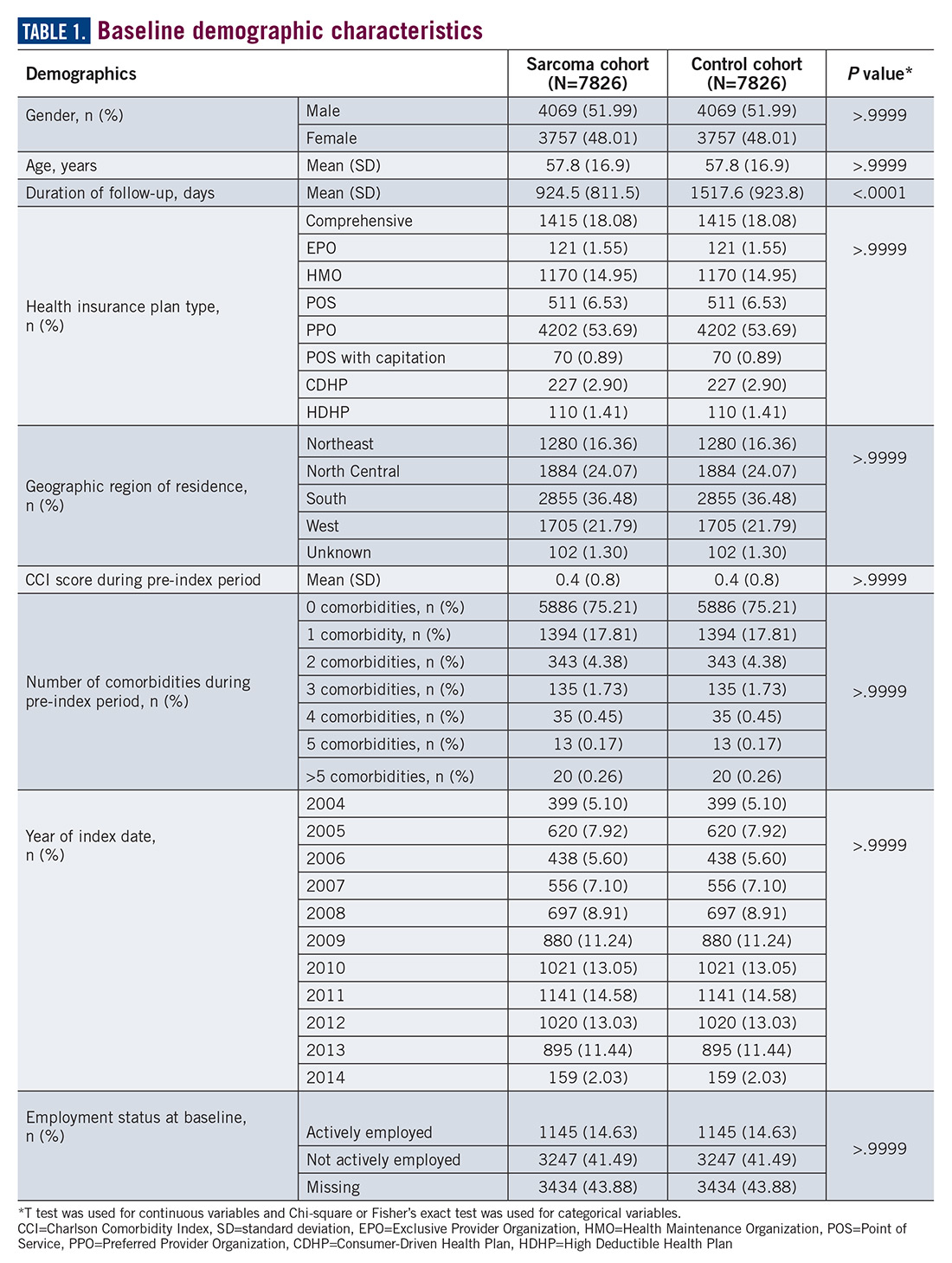
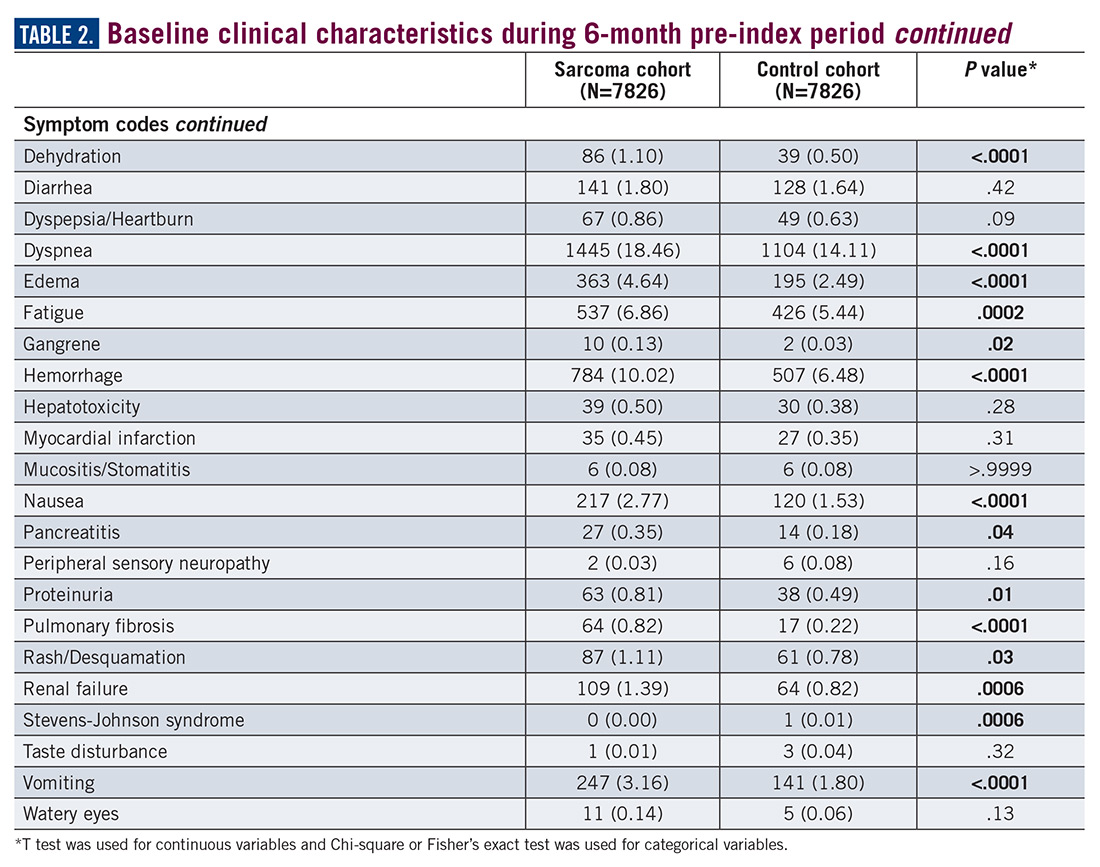
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2). 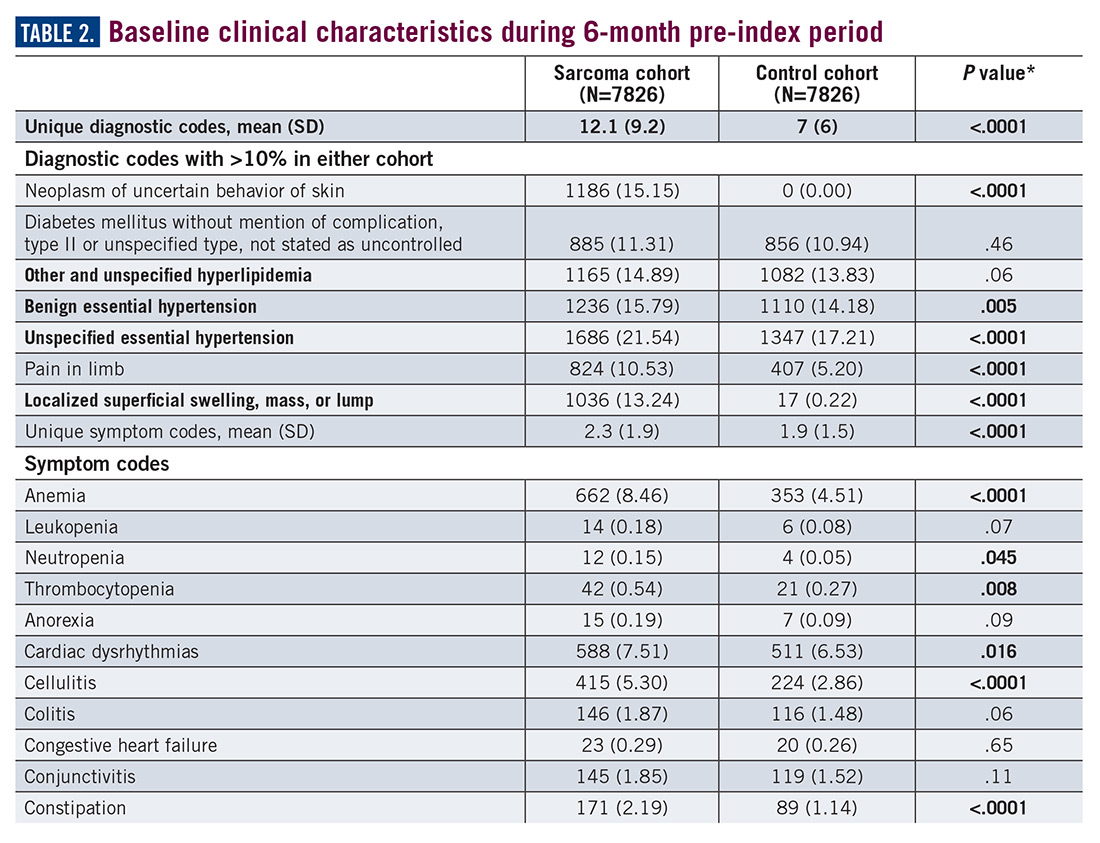
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3). 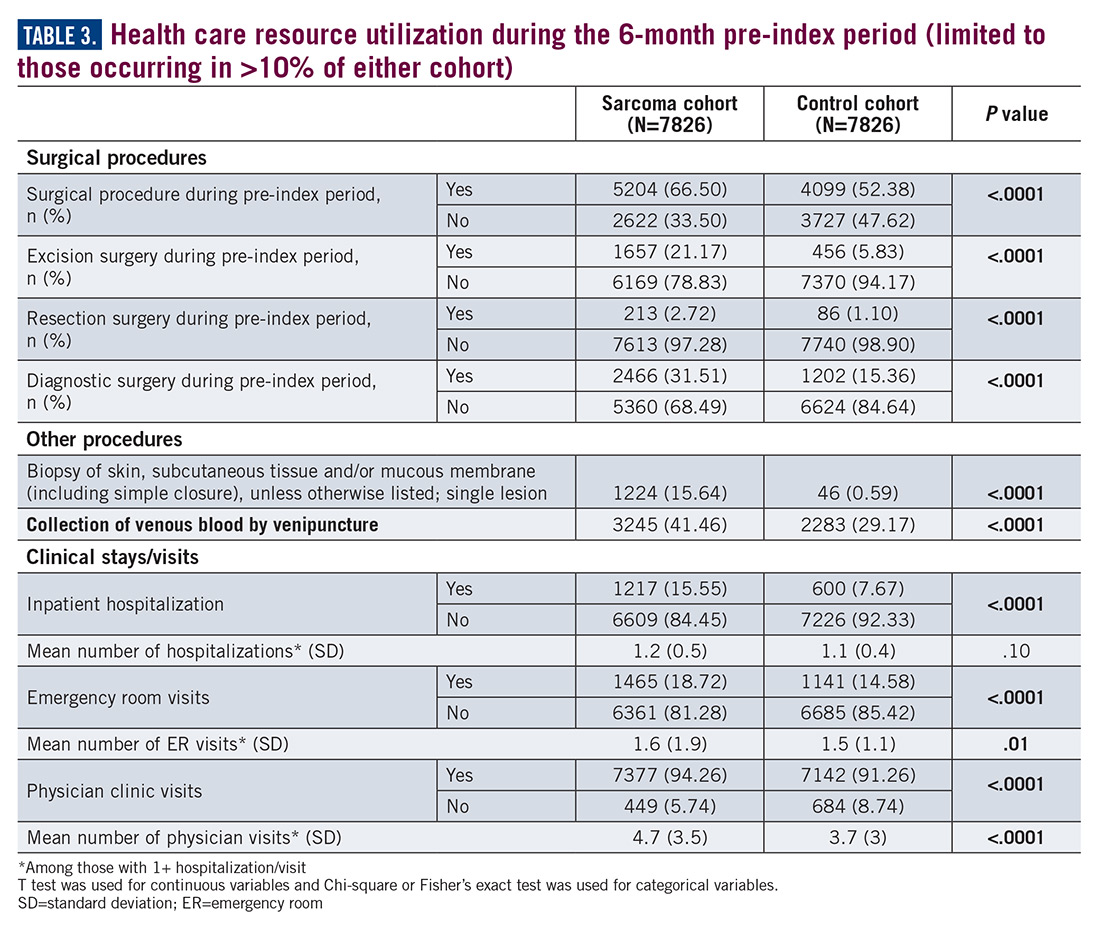
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1.
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2).
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3).
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
Introduction
Soft tissue sarcomas (STS) are a heterogeneous group of cancerous tumors, comprised of more than 50 histological subtypes that develop from soft tissues of the body (eg, fat, muscles, nerve tissue, deep skin tissue, visceral nonepithelial tissue). Due to many factors, not limited to the heterogeneity of this set of diseases and lack of screening tests, reaching a diagnosis of STS is challenging for the general practitioner as well as for the oncologist. Sarcomas may present with nonspecific and often indolent symptomology, depending on the specific histological subtype. According to the American Cancer Society, the signs and symptoms of a sarcoma include a new or growing lump, worsening abdominal pain, blood in stool or vomit, and black stools (due to abdominal bleeding).1 Unfortunately, these symptoms could be indicative of any number of other health conditions and are nonspecific to sarcoma.
As with many cancers, the early detection of disease when it may be completely resected could lead to a cure, whereas diagnosis when the disease is no longer amenable to surgery will impact patient survival. Among all forms of STS, early diagnosis when the patient has only localized disease is associated with an 80.8% five-year survival rate, which decreases to 16.4% for patients whose disease has already metastasized to other parts of the body at the time of diagnosis.2
Previous work has evaluated the relationship between duration of symptoms that may lead to a diagnosis of sarcoma and cancer outcomes. A retrospective analysis of a cohort of adults with bone or STS found no correlation between patient recall of duration of prediagnosis symptoms and survival or metastatic disease at diagnosis.3,4 Little other research was identified that examined the challenges of identifying a potential sarcoma. Despite the gap in knowledge, advocacy and patient-centered organizations emphasize the risk of delayed diagnosis and report high levels of stress and frustration among patients by the time an accurate diagnosis is obtained.5 The objective of this study was to quantify the health care experience and misdiagnoses that occurred prior to a sarcoma diagnosis compared to a cohort of matched controls.
Methods
A retrospective observational database study was conducted using detailed resource utilization and cost data from the Truven MarketScan claims database. Truven MarketScan® is a HIPAA-compliant, fully integrated patient-level database containing inpatient, outpatient, drug, lab, health risk assessment, and benefit design information from commercial and Medicare supplemental insurance plans. Additionally, the Health and Productivity Management (HPM) database, containing workplace absence, short-term disability, long-term disability, and worker’s compensation data, is linked at the individual patient level. The linkage of the claims and HPM database was used for this study.
Patients were eligible for inclusion in the cohort of a sarcoma if they had at least two ICD-9 codes of 171.x on two different days between July 1, 2004, and March 30, 2014. The date of the first eligible code was considered the index date. Patients were required to have at least 6 months of health care plan enrollment prior to the first eligible ICD-9 code to allow for prediagnosis activity to be identified in the database. Patients were also required to be 18 years of age or older on the first eligible ICD-9 code date. Patients were excluded who had evidence suggesting a diagnosis of osteosarcoma, Kaposi’s sarcoma, or gastrointestinal stromal tumors (treatment with methotrexate, ICD-9 codes of 176.x, 171.x, or 238.1), a history of any cancer before the eligible sarcoma ICD-9 code, or history of systemic anticancer therapy during the 6-month pre-index period. All patients meeting eligibility criteria were included in the matching algorithm to identify the control cohort.
The matched control cohort was required to have at least the same duration of follow-up at the case level as the matched sarcoma patient, could not have any evidence of any malignancy at any time in the database, nor could have received any systemic anticancer therapy at any time. Controls were randomly selected from the more than 100 million individual patient cases in the MarketScan database to be matched to the eligible sarcoma patient cohort exactly on age, geographic region of residence, health insurance plan type, gender, noncancer comorbid conditions (measured by Charlson Comorbidity Index items), and employment status. All factors were exact matched at the sarcoma cohort index diagnosis date. In the case of missing variables, patients were matched on missingness (eg, a case with missing employment status would be matched to a control with missing employment status).
The eligible time period for the index date of the possible sarcoma cohort and matched controls was between July 1, 2004, and March 30, 2014, which allowed for a minimum of 1-year follow-up through the end of the database available at the time of analysis.
All ICD-9 diagnostic and procedure codes present in the matched 6-month time period pre-index diagnosis were compared to explore factors that may be more likely to be present in the sarcoma cohort compared to matched controls. Univariate analysis was conducted for each prediagnosis variable. Analyses were conducted using T test for continuous variables, and Chi-square or Fisher’s exact test was used for categorical variables.
Number of physician visits, inpatient hospital stays, surgical procedures, and emergency room visits were compared between those in the sarcoma cohort and matched controls during the matched 6-month pre-index period. The post-index diagnosis employment status was also compared between groups using the HPM database. Comparisons between the sarcoma cohort and control cohort were made among the actively employed patients at baseline related to the proportion of patients who continued active employment, the proportion who permanently discontinued work, and the proportion who initially discontinued work and then returned to work at a later time. No adjustments were made for multiple comparisons.
Results
A total of 7826 controls were each matched to patients in the sarcoma cohort. The baseline characteristics of the study cohorts are provided in Table 1.
During the 6-month period before the sarcoma diagnosis (prediagnosis period), patients had significantly greater frequency of diagnoses identified than controls for uncertain neoplasms, limb pain, and hypertension (all P<.001, Table 2).
Similarly, the majority of health care resource utilization factors evaluated showed statistically higher health care use among patients later suspected of having sarcoma than matched controls (Table 3).
Employment status was missing for 44% of the cohort at baseline and approximately half the cohort during follow-up (Table 4).
Discussion
The symptoms experienced by patients that were recorded in claims were significantly higher across multiple categories than matched controls. However, the rates were relatively low, demonstrating the wide variability in the presentation of sarcoma. Patients had a variety of recorded problems, not limited to a lump or pain, but including hematologic, gastric, and cardiac concerns, that differed from those who had no suspected sarcoma. These factors highlight the challenges that may be facing patients who have an undetected sarcoma.
An expected finding was the difference in duration of follow-up between cohorts. This could be due to longer survival of those without a sarcoma diagnosis or due to insurance changes among those who had a sarcoma diagnosis. The absence of death data did not allow for further exploration of this finding within this study. Future research may wish to identify more comprehensive datasets to allow for the objective evaluation of the differences in time to diagnosis and stage of disease and survival, which would be the ultimate goal in order to develop potential strategies to improve patient outcomes.
This study was limited in that the sarcoma diagnosis could not be verified in a clinical record due to the de-identified nature of the claims data used for this study. Prior work has shown that the ICD coding for sarcoma is incomplete6,7; therefore it is likely there are many other patients in the claims dataset who had a suspected sarcoma but who did not have a 171.x code recorded. Hence, this study is limited to a comparison of a cohort for whom the provider specified a sarcoma code in their billing records. While there are gaps in the ability to identify the entire population of sarcoma patients, the patients with ICD codes used in this study are likely true sarcoma cases. Prior work has demonstrated that the presence of these codes accurately reflects a true sarcoma diagnosis.7 However, given the concerns with ICD coding, two sarcoma codes were required on unique days to reduce the risk of single rule-out codes or data entry error. Patients diagnosed with sarcoma demonstrate significantly greater health care resource use across variables as matched controls during the 6-month period leading to diagnosis, supporting the observations within advocacy and patient reports of the challenges faced during the process to reach an accurate diagnosis. This work may provide the initial basis for the development of strategies to more rapidly identify a potential sarcoma. Future research could also evaluate more than 6 months prior to diagnosis, to quantify the duration of time during which these differences versus controls may exist. Additionally, the cost of care may be of interest to future research to better quantify the burden of misdiagnosis on the health care system.
Acknowledgement
The authors would like to acknowledge Yun Fang, MS, for her support in the SAS coding for the analysis of this study.
Corresponding Author
Lisa M. Hess, PhD, Eli Lilly and Company. [email protected]
Disclosures
No funding was received or exchanged in the conceptualization, conduct, data collection, analysis, interpretation, or writing related to this study. This unfunded study was conducted by employees of Eli Lilly and Company.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
1. ACS. Signs and Symptoms of Soft Tissue Sarcomas. 2018. https://www.cancer.org/cancer/soft-tissue-sarcoma/detection-diagnosis-staging/signs-symptoms.html. Accessed September 27, 2018.
2. SEER. Cancer Stat Facts: Soft Tissue including Heart Cancer. National Cancer Institute Surveillance, Epidemiology, and End Results Program; 2018. https://seer.cancer.gov/statfacts/html/soft.html. Accessed February 20, 2019.
3. Rougraff BT, Davis K, Lawrence J. Does length of symptoms before diagnosis of sarcoma affect patient survival? Clin Orthop Relat Res. 2007;462:181-189.
4. Rougraff BT, Lawrence J, Davis K. Length of symptoms before referral: prognostic variable for high-grade soft tissue sarcoma? Clin Orthop Relat Res. 2012;470(3):706-711.
5. LSSI. Liddy Shriver Sarcoma Initiative. Sarcoma: A diagnosis of patience. http://sarcomahelp.org/articles/patience.html. Accessed September 20, 2018.
6. Hess LM, Zhu EY, Sugihara T, Fang Y, Collins N, Nicol S. Challenges with use of the International Classification of Disease Coding (ICD-9-CM/ICD-10-CM) for soft tissue sarcoma. Perspect Health Inf Manage. 2019;16 (Spring). eCollection 2019.
7. Lyu HG, Stein LA, Saadat LV, Phicil SN, Haider A, Raut CP. Assessment of the accuracy of disease coding among patients diagnosed with sarcoma. JAMA Oncol. 2018;4(9):1293-1295.
Abstract
Introduction: The challenges of diagnosing soft tissue sarcoma are not well studied; however, the heterogeneity of its presentation would suggest that patients may experience a complex journey in the health care system prior to reaching an accurate diagnosis. This study was designed to evaluate the diagnoses, procedures, and health care resource utilization of patients with soft tissue sarcoma compared to a matched healthy control cohort.
Methods: Patients in the sarcoma cohort were identified in claims data by the presence of diagnosis codes for soft tissue sarcoma. Controls were matched using exact methods on demographic, employment, and insurance variables at the date of the index sarcoma diagnosis. Health care resource utilization and diagnosis and procedure codes were compared between the cohorts during the prediagnosis period (6 months prior to the index and matched date). T test was used for continuous variables and Chi-square or Fisher’s exact test was used for categorical variables.
Results: A total of 7826 sarcoma patients were matched to 7826 controls on demographic, employment, and insurance variables. Diagnoses of uncertain neoplasms, limb pain, and hypertension, as well as anemia, neutropenia, thrombocytopenia, cardiac dysrhythmia, cellulitis, constipation, dehydration, diarrhea, dyspnea, edema, fatigue, gangrene, hemorrhage, nausea, pancreatitis, proteinuria, pulmonary fibrosis, rash, renal failure, vomiting, and watery eyes were significantly greater in the sarcoma cohort versus controls (all P <.05). The majority of health care resource utilization evaluated showed statistically higher utilization in the sarcoma cohort versus matched controls.
Conclusions: Sarcoma patients had many health conditions and diagnoses that significantly differed from controls during the 6-month period prior to diagnosis. These data provide initial evidence regarding the quantity and frequency of additional health care resources used and symptoms experienced leading to the diagnosis of sarcoma.
Key words: sarcoma, diagnosis, health care resource utilization, health care economics
The Sarcoma Journal enters its second full year with renewed commitment and energy
We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.

We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.
We begin this second full year publishing The Sarcoma Journal—Official Journal of the Sarcoma Foundation of AmericaTM—with renewed energy and dedication to its founding principle of communicating authoritative and comprehensive scientific information on the diagnosis and treatment of sarcomas and sarcoma subtypes.
Despite advances in treatment and management options for our patients, the need still exists for more effective treatment strategies, new treatment paradigms, and improved understanding of disease biology. This journal, along with its online platform, plays an important role in the dissemination of reliable, peer-reviewed content to the sarcoma community and aims to bridge the gap between bench and bedside.
To this end, we have again recruited an outstanding advisory board, many of whom have long-standing affiliations with the Sarcoma Foundation of America. With their help, the editorial staff and I will continue to build the journal and make it the number one academic and practice resource for the sarcoma community.
We invite you and your colleagues to submit original research, review articles, case reports, opinion pieces, and meeting summaries for publication. In this way we will create the robust forum we all desire.

Recurrence of a small gastric gastrointestinal stromal tumor with high mitotic index
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
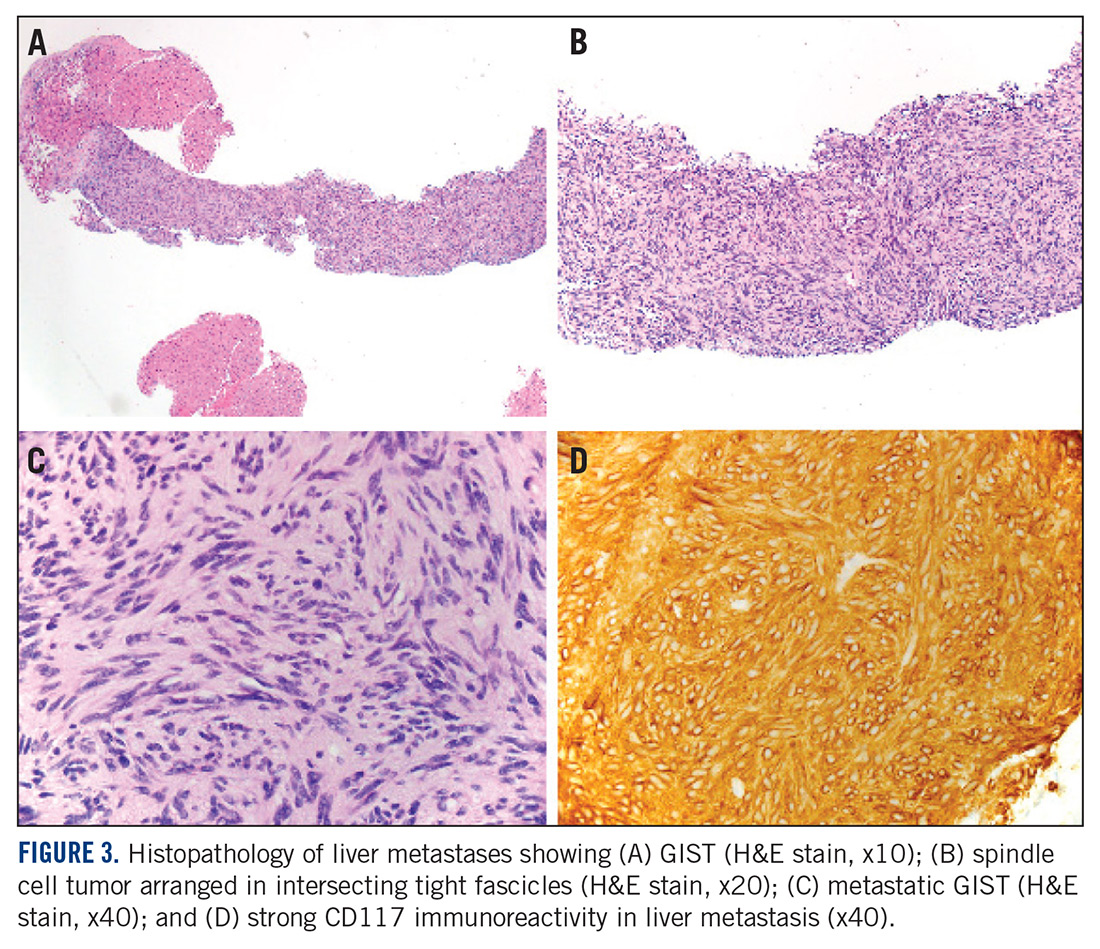
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
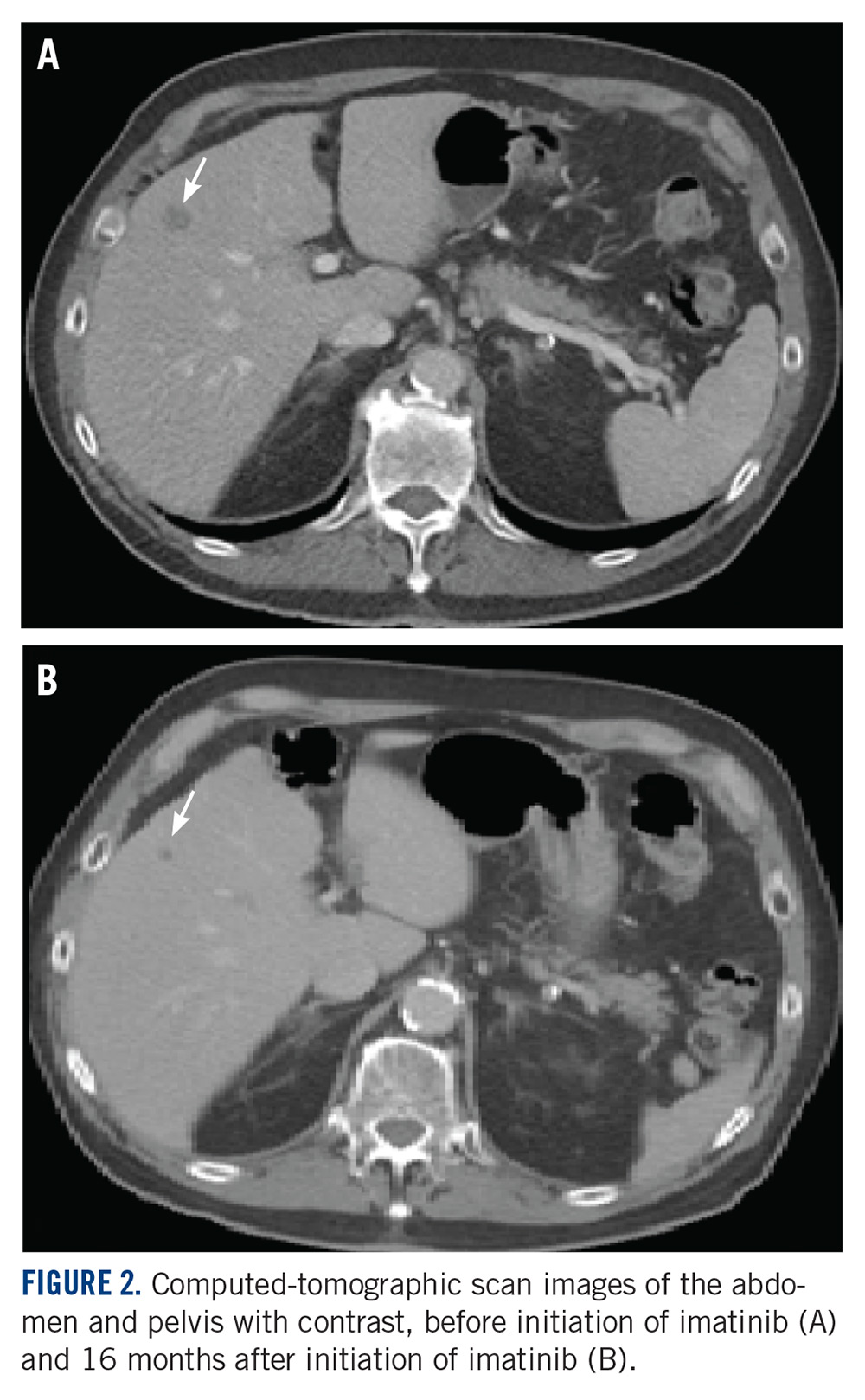
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A). 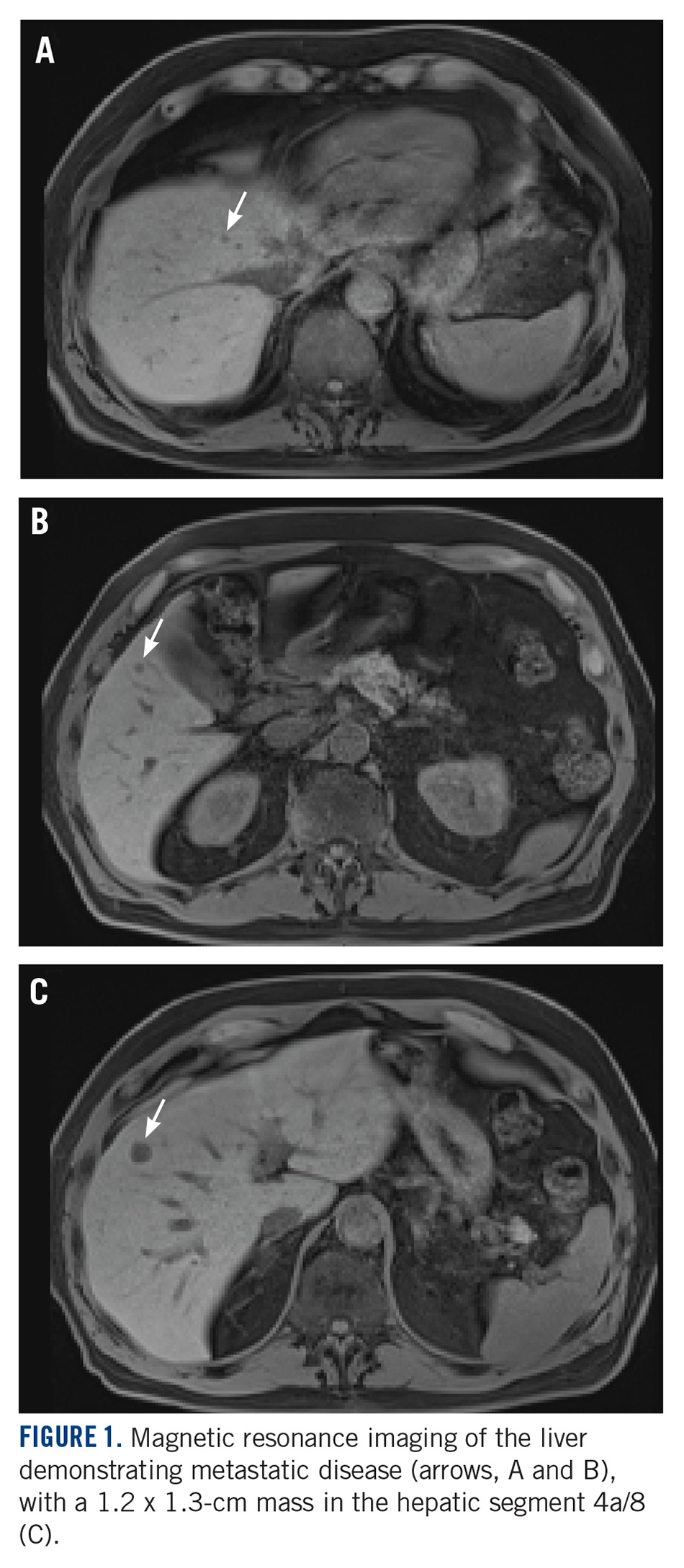
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRα). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRα.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8-cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case Presentation and Summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computed tomographic (CT) scan showed no abnormalities. An esophagogastroduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 x 1.5-cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRα D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size. TSJ
Acknowlegement
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
aDepartment of Medicine, University of Minnesota Medical School; bDepartment of Laboratory Medicine and Pathology, University of Minnesota Medical School; and cMasonic Cancer Center, University of Minnesota Medical School, Minneapolis, Minnesota.
Disclosures
The authors report no disclosures or conflicts of interest. This article was originally published in The Journal of Community and Supportive Oncology JCSO. 2018;16(3):e163-e166. ©Frontline Medical Communications. doi:10.12788/jcso.0402. It is reproduced with permission from the copyright owner. Further reproduction prohibited without permission.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, Kolesnikova V, Maki RG, Pisters PW, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, Bearzi I, Mazzoleni G, Capella C, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org/professionals/physician_gls/default.aspx#age. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, Hu TH, Lin CN, Uen YH, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, Lee HJ, Sohn TS, Hyung WJ, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study from Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, Hartmann JT, Pink D, Schütte J, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, Steigen SE, Brabec P, Plank L, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, Tschernyavsky SJ, Gonen M, Segal NH, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, Kindblom LG, Ahlman H, Nilsson B, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, Kim WK, Kim H, Pantaleo MA, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, Lasota J, Smolkin M, Petroni G, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, Brulard C, Dapremont V, Hostein I, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, Ichikawa H, Haseqawa T, Shitashige M, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, Hishima T, Iwasaki Y, Saito K, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
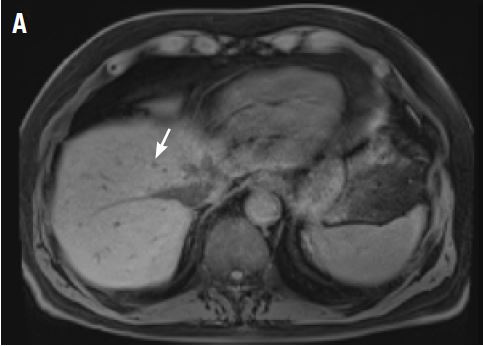
Novel immunostimulant combo shows early efficacy
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.

Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.
Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
SAN FRANCISCO – A combination of two novel immune-stimulating agents has shown early evidence of efficacy against malignant melanoma, leiomyosarcoma, and triple-negative breast cancer in a phase 1b, dose-escalating study.
Among 11 evaluable patients enrolled in a trial of NKTR-262, a small molecule agonist of toll-like receptors (TLR) 7/8, and bempegaldesleukin, an interleukin-2 pathway agonist, 2 had a partial response and 3 had stable disease, reported Adi Diab, MD, from the University of Texas MD Anderson Cancer Center, Houston, and his colleagues.
Patients tolerated the combination well, and there have been no serious adverse events or dose-limiting toxicities.
“Pharmacodynamic data demonstrate both activation of the systemic adaptive and the local innate immune system, and we have seen early evidence of clinical activity in patients who are refractory to checkpoint inhibitors with immunotherapy regimens,” Dr. Diab said at the American Society of Clinical Oncology (ASCO) – Society for Immunotherapy of Cancer (SITC): Clinical Immuno-Oncology Symposium.
NKTR-262 is injected into tumors and is designed to be retained in the tumor microenvironment where it helps to activate antigen-presenting cells, such as dendritic cells, and primes development of new, antigen-specific cytotoxic T cells. Bempegaldesleukin is a cytokine that works within the IL-2 pathway to increase CD8-positive T cells and natural killer (NK) cells in the tumor microenvironment.
The rationale for the combination is that NKTR-262 can activate innate immunity in cells surrounding the tumor microenvironment and activate the machinery of antigen-presenting cells, and bempegaldesleukin can prime and boost a systemic tumor immune response that can ultimately mediate antitumor activity in distant lesions, Dr. Adib said.
In preclinical models, the combination of these agents led to a robust antitumor effect that also involved distant lesions through mediation of the abscopal effect, in which treatment of a tumor activates an immune response against distant tumor cells as well, Dr. Diab said.
The REVEAL study is an ongoing, phase 1b/2 trial looking at the combination in melanoma, Merkel cell carcinoma, triple-negative breast cancer (TNBC), ovarian cancer, renal cell carcinoma, colorectal cancer, urothelial carcinoma, and sarcoma.
The primary goal of the study is to evaluate safety and determine the optimal phase 2 dose of the combination, evaluate biomarkers of response, and assess antitumor activity. As of Jan. 23, 2019, 13 patients were enrolled and evaluable for safety, and 11 were evaluable for the preliminary efficacy analysis.
The most common treatment-related adverse events (TRAEs) with the combination were transient grade 1 or 2 flu-like symptoms, rash, fatigue, pruritus, and nausea. One patients developed grade 3 maculopapular rash and leukocytosis.
Most of the TRAEs are attributable to bempegaldesleukin. There were no immune-mediated AEs and no TRAEs resulted in study discontinuation.
Tumor biopsies obtained 24 hours after injection of NKTR-262 confirmed the activation of TLR 7/8 and robust induction of type 1 interferon, interferon-alpha, and interferon-beta gene-related signatures necessary for optimal antigen presentation.
Dr. Diab noted that in a different trial of bempegaldesleukin monotherapy there was no significant increase in the type 1 interferon gene signature, but the agent did promote activation of the adaptive immune system.
The complementary nature of the two novel agents could also be demonstrated in evaluation of peripheral blood samples, which showed that, although there was no proliferation of T or NK cells following NKTR-262 injection, the addition of bempegaldesleukin resulted in the proliferation of both effector T cells and NK cells to enhance the systemic immune response.
The preliminary efficacy analysis showed that two of five patients with stage IV melanoma who experienced disease progression on prior immune checkpoint inhibitors had partial responses, including one who had a 100% reduction in target lesions and the other with a 50% reduction. In addition, two patients with heavily pretreated leiomyosarcoma had stable disease as the best response, as did the single patient with TNBC.
The maximum tolerated dose of the combination has not been identified, and the investigators are continuing to enroll patients.
The REVEAL study is supported by Nektar Therapeutics. Dr. Diab reported institutional research funding, consulting fees, and advisory board participation from Nektar, Bristol-Myers Squib, Idera Pharmaceuticals, Jounce Therapeutics, and Array BioPharma.
SOURCE: Diab A et al. ASCO-SITC, Abstract 26.
REPORTING FROM ASCO-SITC
Checkpoint inhibitors ‘viable treatment option’ in HIV-infected individuals
Immune checkpoint inhibitors are safe and effective in HIV-infected patients with advanced cancers, according to authors of a recently published systematic review.
The treatment was well tolerated and associated with a 9% rate of grade 3 or higher immune-related adverse events, according to results of the review of 73 patient cases.
There were no adverse impacts on HIV load or CD4 cell count detected in the patients, according to researchers Michael R. Cook, MD, and Chul Kim, MD, MPH, of Georgetown University, Washington.
Antitumor activity of the checkpoint inhibitors in lung cancer patients was comparable to what has been seen in previous randomized clinical trials that excluded HIV-infected individuals, Dr. Cook and Dr. Kim reported in JAMA Oncology.
“Based on the results of the present systematic review, and in the absence of definitive prospective data suggesting an unfavorable risk-to-benefit ratio, immune checkpoint inhibitor therapy may be considered as a viable treatment option for HIV-infected patients with advanced cancer,” they said.
There are preclinical data suggesting that immune checkpoint modulation could improve function of HIV-specific T cells, the investigators added.
“Prospective trials of immune checkpoint inhibitors are necessary to elucidate the antiviral efficacy of immune checkpoint inhibitor therapy in patients with HIV infection and cancer,” they said.
Several such trials are underway to evaluate the role of the pembrolizumab, nivolumab, nivolumab plus ipilimumab, and durvalumab in HIV-infected patients with advanced-stage cancers, according to the review authors.
In the present systematic review, Dr. Cook and Dr. Kim conducted a literature search and reviewed presentations from major annual medical conferences.
Of the 73 HIV-infected patients they identified, most had non–small cell lung cancer (34.2%), melanoma (21.9%), or Kaposi sarcoma (12.3%), while the rest had anal cancer, head and neck cancer, or other malignancies. Most patients had received either nivolumab (39.7%) or pembrolizumab (35.6%).
There were “no concerning findings” among these patients with regard to immune-mediated toxicities or changes in HIV-related parameters.
Six of 70 patients had immune-related adverse events of grade 3 or greater.
Thirty-four patients had documented HIV loads before and after receiving an immune checkpoint inhibitor. Of those, 28 had undetectable HIV loads at baseline, and all but 2 (7%) maintained undetectable loads in the posttreatment evaluation.
Of the remaining six with detectable HIV loads before treatment, five had a decrease in viral load, to the point that four had undetectable HIV viral load in the posttreatment evaluation, the investigators reported.
The overall response rate was 30% for the lung cancer patients, 27% for melanoma, and 63% for Kaposi sarcoma.
In the non–small cell lung cancer subset, response rates were 26% for those who had received previous systemic treatment, and 50% for those who had not, which was similar to findings from major checkpoint inhibitor trials that excluded HIV-infected individuals, the investigators said.
The American Society of Clinical Oncology Conquer Cancer Foundation and Georgetown University supported the study. Dr. Kim reported disclosures related to CARIS Life Science and AstraZeneca.
SOURCE: Cook MR and Kim C. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6737.
Immune checkpoint inhibitors are safe and effective in HIV-infected patients with advanced cancers, according to authors of a recently published systematic review.
The treatment was well tolerated and associated with a 9% rate of grade 3 or higher immune-related adverse events, according to results of the review of 73 patient cases.
There were no adverse impacts on HIV load or CD4 cell count detected in the patients, according to researchers Michael R. Cook, MD, and Chul Kim, MD, MPH, of Georgetown University, Washington.
Antitumor activity of the checkpoint inhibitors in lung cancer patients was comparable to what has been seen in previous randomized clinical trials that excluded HIV-infected individuals, Dr. Cook and Dr. Kim reported in JAMA Oncology.
“Based on the results of the present systematic review, and in the absence of definitive prospective data suggesting an unfavorable risk-to-benefit ratio, immune checkpoint inhibitor therapy may be considered as a viable treatment option for HIV-infected patients with advanced cancer,” they said.
There are preclinical data suggesting that immune checkpoint modulation could improve function of HIV-specific T cells, the investigators added.
“Prospective trials of immune checkpoint inhibitors are necessary to elucidate the antiviral efficacy of immune checkpoint inhibitor therapy in patients with HIV infection and cancer,” they said.
Several such trials are underway to evaluate the role of the pembrolizumab, nivolumab, nivolumab plus ipilimumab, and durvalumab in HIV-infected patients with advanced-stage cancers, according to the review authors.
In the present systematic review, Dr. Cook and Dr. Kim conducted a literature search and reviewed presentations from major annual medical conferences.
Of the 73 HIV-infected patients they identified, most had non–small cell lung cancer (34.2%), melanoma (21.9%), or Kaposi sarcoma (12.3%), while the rest had anal cancer, head and neck cancer, or other malignancies. Most patients had received either nivolumab (39.7%) or pembrolizumab (35.6%).
There were “no concerning findings” among these patients with regard to immune-mediated toxicities or changes in HIV-related parameters.
Six of 70 patients had immune-related adverse events of grade 3 or greater.
Thirty-four patients had documented HIV loads before and after receiving an immune checkpoint inhibitor. Of those, 28 had undetectable HIV loads at baseline, and all but 2 (7%) maintained undetectable loads in the posttreatment evaluation.
Of the remaining six with detectable HIV loads before treatment, five had a decrease in viral load, to the point that four had undetectable HIV viral load in the posttreatment evaluation, the investigators reported.
The overall response rate was 30% for the lung cancer patients, 27% for melanoma, and 63% for Kaposi sarcoma.
In the non–small cell lung cancer subset, response rates were 26% for those who had received previous systemic treatment, and 50% for those who had not, which was similar to findings from major checkpoint inhibitor trials that excluded HIV-infected individuals, the investigators said.
The American Society of Clinical Oncology Conquer Cancer Foundation and Georgetown University supported the study. Dr. Kim reported disclosures related to CARIS Life Science and AstraZeneca.
SOURCE: Cook MR and Kim C. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6737.
Immune checkpoint inhibitors are safe and effective in HIV-infected patients with advanced cancers, according to authors of a recently published systematic review.
The treatment was well tolerated and associated with a 9% rate of grade 3 or higher immune-related adverse events, according to results of the review of 73 patient cases.
There were no adverse impacts on HIV load or CD4 cell count detected in the patients, according to researchers Michael R. Cook, MD, and Chul Kim, MD, MPH, of Georgetown University, Washington.
Antitumor activity of the checkpoint inhibitors in lung cancer patients was comparable to what has been seen in previous randomized clinical trials that excluded HIV-infected individuals, Dr. Cook and Dr. Kim reported in JAMA Oncology.
“Based on the results of the present systematic review, and in the absence of definitive prospective data suggesting an unfavorable risk-to-benefit ratio, immune checkpoint inhibitor therapy may be considered as a viable treatment option for HIV-infected patients with advanced cancer,” they said.
There are preclinical data suggesting that immune checkpoint modulation could improve function of HIV-specific T cells, the investigators added.
“Prospective trials of immune checkpoint inhibitors are necessary to elucidate the antiviral efficacy of immune checkpoint inhibitor therapy in patients with HIV infection and cancer,” they said.
Several such trials are underway to evaluate the role of the pembrolizumab, nivolumab, nivolumab plus ipilimumab, and durvalumab in HIV-infected patients with advanced-stage cancers, according to the review authors.
In the present systematic review, Dr. Cook and Dr. Kim conducted a literature search and reviewed presentations from major annual medical conferences.
Of the 73 HIV-infected patients they identified, most had non–small cell lung cancer (34.2%), melanoma (21.9%), or Kaposi sarcoma (12.3%), while the rest had anal cancer, head and neck cancer, or other malignancies. Most patients had received either nivolumab (39.7%) or pembrolizumab (35.6%).
There were “no concerning findings” among these patients with regard to immune-mediated toxicities or changes in HIV-related parameters.
Six of 70 patients had immune-related adverse events of grade 3 or greater.
Thirty-four patients had documented HIV loads before and after receiving an immune checkpoint inhibitor. Of those, 28 had undetectable HIV loads at baseline, and all but 2 (7%) maintained undetectable loads in the posttreatment evaluation.
Of the remaining six with detectable HIV loads before treatment, five had a decrease in viral load, to the point that four had undetectable HIV viral load in the posttreatment evaluation, the investigators reported.
The overall response rate was 30% for the lung cancer patients, 27% for melanoma, and 63% for Kaposi sarcoma.
In the non–small cell lung cancer subset, response rates were 26% for those who had received previous systemic treatment, and 50% for those who had not, which was similar to findings from major checkpoint inhibitor trials that excluded HIV-infected individuals, the investigators said.
The American Society of Clinical Oncology Conquer Cancer Foundation and Georgetown University supported the study. Dr. Kim reported disclosures related to CARIS Life Science and AstraZeneca.
SOURCE: Cook MR and Kim C. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6737.
FROM JAMA ONCOLOGY
Key clinical point: Immune checkpoint inhibitors are a viable treatment option for HIV-infected patients, according to data supporting their safety and efficacy in this patient population.
Major finding: The treatment was well tolerated, with an 8.6% rate of grade 3 or greater immune-related adverse events, and no impact on HIV-related parameters.
Study details: A systematic review of 73 patients with HIV infection who had received treatment with a checkpoint inhibitor.
Disclosures: The American Society of Clinical Oncology Conquer Cancer Foundation and Georgetown University supported the study. One study author reported disclosures related to CARIS Life Science and AstraZeneca.
Source: Cook MR and Kim C. JAMA Oncol. 2019 Feb 7. doi: 10.1001/jamaoncol.2018.6737.