User login
Gastroesophageal Reflux Disease
- Heartburn on 2 or more days a week warrants medical attention, as patients are likely to suffer from gastroesophageal reflux disease (GERD).Chronic GERD can lead to the development of complications including erosive esophagitis, stricture formation, and Barrett’s esophagus, which increases the risk of esophageal adenocarcinoma.
- A trial with a proton pump inhibitor (PPI) is the quickest and most cost-effective way to diagnose GERD, and is at least as sensitive as 24-hour intraesophageal pH monitoring.
- As PPIs only bind to actively secreting proton pumps, they should be dosed 30 to 60 minutes before a meal.Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take a PPI with or after a meal or did not specify the timing of dosing.
- The patients who will have the best response to surgical therapy for GERD are those who had clearly documented acid reflux with typical symptoms, and who have responded to PPI treatment. Unfortunately, the same survey found that most physicians recommend antireflux surgery for patients in whom medical therapy has failed.
Gastroesophageal reflux disease (GERD) is a common, multifactorial condition that often results in decreased quality of life with interruptions of sleep, work, and social activities. Patients have reported that GERD affects emotional well-being to a greater degree than diabetes or hypertension.1,2 GERD is also associated with well-established complications, including Barrett’s esophagus. The role of reflux in carcinogenesis is controversial; the possibility of an association, however, implies that GERD should be treated aggressively and early.3
Symptoms of gerd
The typical symptoms of GERD are heartburn and regurgitation. Heartburn is best defined as a burning retrosternal discomfort starting in the epigastrium or lower chest and moving upwards towards the neck. Regurgitation is the effortless movement of gastric contents up into the esophagus or pharynx.
Most patients with GERD do not have endoscopically visible lesions; a careful analysis of symptoms generally forms the basis of a preliminary diagnosis.
The occurrence of heartburn on 2 or more days a week has been suggested as a basis for further investigation for GERD.4 However, symptoms vary greatly. Patients may be asymptomatic or experience symptoms that more closely resemble gastric disorders, infectious and motor disorders of the esophagus, biliary tract disease, or even coronary artery disease.
Extraesophageal manifestations
Adding to the complexity of diagnosis, GERD has been shown to have extraesophageal manifestations, including chronic cough, asthma, recurrent aspiration, chronic sore throat, reflux laryngitis, and paroxysmal laryngospasm or voice changes.
Although the relationship between asthma and GERD remains unclear, it has been estimated that 24% to 98% of patients with asthma also have GERD.5 Some patients with asthma have been shown to have excess acid reflux into the esophagus. Reflux-like symptoms may precede episodes of asthma that occur after meals or when lying down.6 8
Additionally, GERD has been noted in 10% to 50% of patients with non-cardiac chest pain.9,10
Diagnostic strategies
Trial of treatment
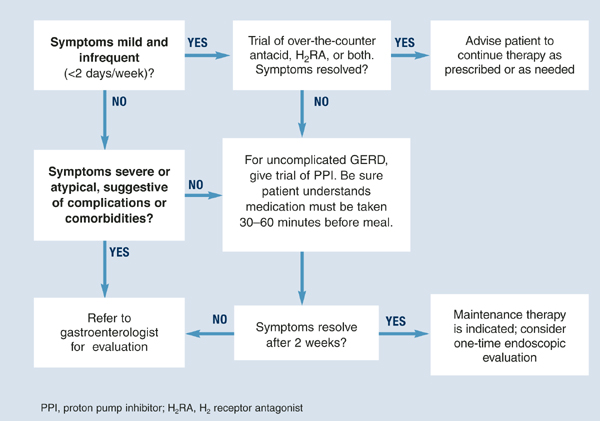
Diagnosis is usually based on typical symptoms—heartburn or regurgitation—in the clinical history. (The Figure shows a treatment algorithm for both severe and mild symptoms.)
A 2-week trial of treatment with a proton pump inhibitor (PPI) provides the quickest and most cost-effective confirmation of diagnosis and is recommended for the patient whose history suggests uncomplicated GERD. A positive response to PPI treatment in a patient with symptoms suggestive of GERD is at least as sensitive and specific as 24-hour intraesophageal pH monitoring, which is still often considered the “gold standard” for the diagnosis of GERD. Furthermore, complete lack of improvement in response to PPI treatment is highly predictive that the patient does not have GERD and indicates the need for further evaluation and a possible revision of diagnosis.11,12
H2 receptor antagonists (H2RAs) have also been investigated in empirical trials for usefulness in diagnosing GERD. H2RAs are less effective than PPIs.13,14
FIGURE Medical management of suspected GERD
Endoscopy
No data support routine endoscopy for patients with the recent onset of uncomplicated heartburn who respond to medical therapy. Endoscopy is recommended, however, for patients with severe or atypical GERD symptoms, when other diseases may be present, or when a treatment trial with a PPI is ineffective.15 Endoscopy is useful for diagnosing complications of GERD, such as Barrett’s esophagus, esophagitis, and strictures. Fewer than 50% of patients with GERD symptoms have evidence of esophagitis on endoscopy.16
The American Society for Gastrointestinal Endoscopy recommends endoscopy when there are clinical suggestions of severe reflux or other disease.17 The American College of Gastroenterology recommends further testing
- when empiric therapy has failed
- when symptoms of complicated disease exist
- when there is dysphagia, bleeding, weight loss, choking, chest pain, or long-standing symptoms
- when continuous therapy is required
- to screen for Barrett’s esophagus.18
The Canadian Consensus Conference recommends endoscopy in the presence of
- dysphagia
- odynophagia
- bleeding
- weight loss
- noncardiac chest pain
- failure to respond to 4 to 8 weeks of pharmacologic therapy.19
It also recommends a single test if maintenance therapy is required.
Other diagnostic tests
Other diagnostic tools may be of use in some settings.
A barium esophagram can document reflux, and Bernstein testing (esophageal acid infusion test) can identify esophageal hypersensitivity to acid, although neither establishes a diagnosis of GERD. Ambulatory 24-hour intraesophageal pH monitoring can help to establish the presence of GERD by documenting the proportion of time during which the intraesophageal pH is acidic (<4) and can also establish the degree of association between patients’ symptoms and episodes of esophageal acidification.
Esophageal manometry is not recommended as a routine diagnostic test for GERD. It is important in selected patients to exclude an esophageal motility disorder and may be necessary as part of the pre-operative evaluation for patients in whom a surgical operation for GERD is being considered.
Management of gerd
GERD commonly requires long-term management that includes dietary, lifestyle, and pharmacological interventions. Surgery may be considered for the long-term management of the condition in carefully selected patients.
Diet and lifestyle
Dietary modifications. Patients should not consume large meals and should avoid lying down for 3 to 4 hours after eating. Caffeinated products, peppermint, fatty foods, chocolate, spicy foods, citrus fruits and juices, tomato-based products, and alcohol may contribute to episodes of GERD.18,21 Lozenges of any kind are able to stimulate salivary secretion, help clear refluxed acid, and hence, help relieve symptoms.
Lifestyle modifications. Changes in lifestyle may include such seemingly sensible interventions as sleeping with the head elevated, stopping smoking, and losing weight. There is little or no established evidence for the efficacy of these and other lifestyle modifications in the management of GERD. However, in1 trial of 63 patients, elevating the head of the bed with 6-inch blocks resulted in 1 less episode of heartburn or acid regurgitation per night when compared with lying flat.22 In another trial of 71 patients with esophagitis, elevating the bed was nearly as effective as ranitidine for reducing symptoms and producing endoscopically verifiable healing.23
Arecent survey20 of 1046 primary care physicians found that:
- 36% instructed patients to take PPIs during or after a meal or did not specify a time of dosing
- 75% referred patients for surgical antireflux therapy and 20% referred patients directly to a surgeon without gastrointestinal consultation
- 15% reported that a trial with a H2 receptor antagonist was required by their healthsystem or insurance company prior to using a PPI.
Drug interventions
Pharmacological interventions include over-the-counter remedies such as antacids and H2RAs (Table 1), as well as prescription-only doses of H2RAs and PPIs. At the time of writing, no PPI was available in an over-the-counter preparation in the United States, although over-the-counter omeprazole may soon be approved. Many authorities believe an incremental approach to the management of GERD is appropriate, beginning with lifestyle modifications and over-the-counter preparations, continuing with H2 blockers, and reserving PPIs for nonresponders. While this approach may have appeal from a cost perspective, we believe another approach (as illustrated in the Figure) is clinically superior.
Antacids. Over-the-counter antacids rapidly increase the pH of the intraesophageal contents and also neutralize acidic gastric contents that might be refluxed. They are frequently used to treat heartburn. However, few clinical trials have evaluated the efficacy of antacids. Published trials24-26 are limited by small sample sizes and a lack of intention-to-treat analysis. Only 1 showed positive evidence for antacid efficacy.25
The utility of antacids is limited by the need for frequent dosing and possible interactions with such drugs as fluoroquinolones, tetracycline, and ferrous sulfate.27 Alginate/antacids have shown statistically significant benefit compared with placebo for relief of mild-to-moderate GERD symptoms and healing of esophagitis.24,28-34
H 2 receptor antagonists. H2RAs have shown positive effects on symptoms in some studies, although symptomatic response rates observed were only around 60% to 70%. Additionally, most of the trials to date have been for 2 to 6 weeks in duration.35 43 An issue worthy of consideration with the H2RAs is the development of tolerance with continuous use.44
An H2RA-antacid combination was recently evaluated in a trial that compared it with monotherapy using either agent. Of the patients receiving combination therapy, 81% reported an excellent or good symptom response. Those receiving famotidine or atacid alone reported a 72% excellent or good symptom response.3
Proton pump inhibitors. PPIs potently reduce gastric acid secretion by inhibiting the H+-K+adenosine triphosphatase pump of the parietal cell. As a result, they suppress gastric acid secretion for a longer period than H2RAs.45 Evidence from randomized, controlled trials has demonstrated the superiority of PPIs over any other class of drugs for the relief of GERD symptoms, for healing esophagitis, and for maintaining patients in remission. Standard doses of omeprazole, lansoprazole, panto-prazole, esomeprazole, and rabeprazole have, for the most part, shown comparable rates of healing and remission in erosive esophagitis.46-52
PPIs are best absorbed in the absence of food. Ingestion of food after a PPI stimulates parietal cell activity when blood levels of the PPI are increasing; this promotes uptake of the PPI by the parietal cells. Therefore, patients should be advised to take their PPI between 30 and 60 minutes before eating. For patients on a once-daily PPI, the best time to take it is about 30 to 60 minutes before breakfast. Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take their PPI with or after a meal or did not specify the timing of dosing.53
Clinical evidence indicates that a trial with a PPI provides the quickest and most cost-effective method for diagnosing GERD. Despite this, many physicians use a trial of H2 receptor antagonists prior to initiation of PPI therapy.
- Clinicians should clearly instruct their patients regarding optimal timing of the dose, since this can have a significant effect on the success of therapy.
- Patients for whom antireflux surgery is being considered should first be referred for consultation with a gastroenterologist to assist in patient selection, to ensure that appropriate preoperative evaluation has been performed and to help exclude other possible causes of their symptoms.21,54
PPI therapy can be tailored to control GERD symptoms. Treatment can start with the most effective dosage and then be stepped down, or start with a minimum dosage and then be stepped up (Table 2). Patients with predominantly daytime symptoms should take PPIs before breakfast. Concerns that were once expressed about the long-term use of PPIs, such as predisposing patients to stomach cancer, have been refuted by extensive clinical experience and intensive monitoring (Table 3).3
TABLE 1
Over-the-counter therapy for GERD
|
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. |
| Bethesda, Md.:American Gastroenterological Association;2002. |
TABLE 2
Step-down and step-up treatments: advantages and disadvantages
| Regimen | Advantages | Disadvantages |
|---|---|---|
| Step-down therapy (high-dose initial therapy) | Rapid symptom relief | Potential overtreatment |
| Efficient for physician | Higher initial drug cost | |
| Avoids overinvestigation and associated costs | ||
| Step-up therapy (minimum-dose initial therapy) | Avoids overtreatment | Patient may continue with symptoms unnecessarily |
| Lower initial drug cost | Inefficient for physician | |
| May lead to overinvestigation | ||
| Uncertain end point (partial symptom relief) | ||
| Adapted from Dent J, et al. Management of gastro-oesophageal reflux disease in general practice.BMJ 2001;322:344-347. | ||
TABLE 3
Potential concerns associated with the use of proton pump inhibitors
| Potential concern | Level of Evidence* | Grade † | Comments |
|---|---|---|---|
| Long-term PPI treatment may lead to reduced serum cobalamin levels | 2b | B | This is most likely to occur in individuals with atrophic gastritis |
| Increased acid output has been seen after stopping a PPI | 2b | B | Effects of PPI treatment on corpus glandular atrophy in H pylori-infected individuals are difficult to interpret due to possible sampling error and short study duration |
| PPI treatment may predispose to bacterial enteric infection | 3 | B | Only shown in a single case control study |
| *Level of evidence:1, Evidence for and/or general agreement that treatment is useful and effective;1a, systematic review with homogeneity of randomized controlled trials (RCTs);1b, individual RCTs (with narrow confidence interval);2, conflicting evidence and/or divergent opinion about efficacy and use;2a, evidence or opinion is in favor of treatment;2b, use and efficacy is less well established by evidence or opinion;3, evidence and/or general agreement that treatment is not useful or effective and may be harmful in some cases. | |||
| †Quality grading:A, well-designed, clinical trials;B, well-designed cohort or case-control studies;C, case reports, flawed trials;D, personal clinical experience;E, insufficient evidence to form opinion. | |||
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. Bethesda, Md:American Gastroenterological Association, 2002. | |||
Surgery
Surgical antireflux therapy is an option in carefully selected patients. Those who respond best to surgical therapy will have had clearly documented acid reflux, typical symptoms, and symptomatic improvement while on PPI treatment.54
Unfortunately, a recent survey suggests that physicians tend to recommend surgery for patients in whom medical therapy has failed.53 However, patients who failed to respond to PPI therapy are unlikely to have GERD and, therefore, are highly unlikely to have a good outcome from antireflux surgery. Recent studies suggest that up to 62% of patients who have had open surgery for GERD continue to require medical treatment afterward. Although some studies demonstrate that surgery has greater efficacy over medical therapy initially, long-term follow-up has shown that surgically treated patients often need further medical therapy for persistent GERD symptoms.55 Community-based studies of antireflux surgery indicate that many patients develop new symptoms that they did not have before surgery and that these substantially diminish quality of life.
New endoscopic therapies, including radiofrequency energy delivery to the region of the lower esophageal sphincter and endoscopic suturing, have recently been approved for use by the FDA. This approval was based largely on safety rather than efficacy data. Clinical evidence is limited to uncontrolled studies in patients with no or mild esophagitis.3 These techniques should not be used in preference to established medical treatment unless and until data from randomized, controlled trials become available that demonstrate safety and efficacy.56
1. Dimenas E. Methodological aspects of evaluation of quality of life in upper gastrointestinal diseases. Scand J Gastroenterol Suppl 1993;199:18-21.
2. Revicki DA, Wood M, Maton PN, Sorensen S. The impact of gastroesophageal reflux disease on health-related quality of life. Am J Med 1998;104:252-8.
3. Peterson WL. GERD: Evidence-based therapeutic strategies. Bethesda, Md: American Gastroenterological Association; 2002.
4. Dent J, Jones R, Kahrilas P, Talley NJ. Management of gastro-oesophageal reflux disease in general practice. BMJ 2001;322:344-7.
5. Harding SM. Nocturnal asthma: role of gastroesophageal reflux. Chronobiol Int 1999;16:641-2.
6. Kjellen G, Wranne B. The prevalence of asymptomatic gastro-oesophageal relfux in adult patients with asthma. Eur J Respir Dis 1984;65:233.-
7. Schnatz PF, Castell JA, Castell DO. Pulmonary symptoms associated with gastroesophageal reflux: use of ambulatory pH monitoring to diagnose and to direct therapy. Am J Gastroenterol 1996;91:1715-8.
8. Sontag SJ, Schnell TG, Miller TQ, Khandelwal S, O’Connell S, Chejfec G, et al. Prevalence of oesophagitis in asthmatics. Gut 1992;33:872-6.
9. Schofield PM, Bennett DH, Whorwell PJ, Brooks NH, Bray CL, Ward C, et al. Exertional gastro-oesophageal reflux: a mechanism for symptoms in patients with angina pectoris and normal coronary angiograms. Br Med J (Clin Res Ed) 1987;294:1459-61.
10. Hewson EG, Sinclair JW, Dalton CB, Richter JE. Twenty-four-hour esophageal pH monitoring: the most useful test for evaluating noncardiac chest pain. Am J Med 1991;90:576-83.
11. Goyal RK. Diseases of the esophagus. In: Braunwald E, ed. Harrison’s principles of internal medicine. 15th ed. New York: McGraw-Hill; 2001;1642-9.
12. Schenk BE, Kuipers EJ, Klinkenberg-Knol EC, Festen HP, Jansen EH, Tuynman HA, et al. Omeprazole as a diagnostic tool in gastroesophageal reflux disease. Am J Gastroenterol 1997;92:1997-2000.
13. van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastrooesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2001;(4):CD002095.-
14. Brun J, Sorngard H. High dose proton pump inhibitor response as an initial strategy for a clinical diagnosis of gastro-oesophageal reflux disease (GERD).Swedish multi-centre group in primary health care. Fam Pract 2000;17:401-4.
15. Lundell L. Anti-reflux surgery in the laparoscopic era. Baillieres Best Pract Res Clin Gastroenterol 2000;14:793-810.
16. Chen MY, Ott DJ, Sinclair JW, Wu WC, Gelfand DW. Gastroesophageal reflux disease: correlation of esophageal pH testing and radiographic findings. Radiology 1992;185:483-6.
17. The role of endoscopy in the management of GERD: guidelines for clinical application. From the ASGE. American Society for Gastrointestinal Endoscopy. Gastrointest Endosc 1999;49:834-5.
18. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434-42.
19. Beck IT, Champion MC, Lemire S, Thomson AB, Anvari M, Armstrong D, et al. The Second Canadian Consensus Conference on the Management of Patients with Gastroesophageal Reflux Disease. Can J Gastroenterol 1997;11(suppl B):7B-20B.
20. Chey WD, Inadomi JM, Booher AK, Fendrick AM. Primary care physicians’ perceptions and practices of the management of GERD: results of a national survey. Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
21. Fennerty MB, Castell D, Fendrick AM, Halpern M, Johnson D, Kahrilas PJ, et al. The diagnosis and treatment of gastroesophageal reflux disease in a managed care environment, Suggested disease management guidelines. Arch Intern Med 1996;156:477-84.
22. Stanciu C, Bennett JR. Effects of posture on gastro-oesophageal reflux. Digestion 1977;15:104-9.
23. Harvey RF, Gordon PC, Hadley N, Long DE, Gill TR, Macpherson RI, et al. Effects of sleeping with the bed-head raised and of ranitidine in patients with severe peptic oesophagitis. Lancet 1987;2:1200-3.
24. Graham DY, Patterson DJ. Double-blind comparison of liquid antacid and placebo in the treatment of symptomatic reflux esophagitis. Dig Dis Sci 1983;28:559-63.
25. Grove O, Bekker C, Jeppe-Hansen MG, Karstoft E, Sanchez G, Axelsson CK, et al. Ranitidine and high-dose antacid in reflux oesophagitis. A randomized, placebo-controlled trial. Scand J Gastroenterol 1985;20:457-61.
26. Weberg R, Berstad A. Symptomatic effect of a low-dose antacid regimen in reflux oesophagitis. Scand J Gastroenterol 1989;24:401-6.
27. Welage LS, Berardi RR. Evaluation of omeprazole, lansoprazole, pantopra-zole, and rabeprazole in the treatment of acid-related diseases. J Am Pharm Assoc (Wash) 2000;40:52-62.
28. Barnardo DE, Lancaster-Smith M, Strickland ID, Wright JT. A double-blind controlled trial of ‘Gaviscon’ in patients with symptomatic gastro-oesophageal reflux. Curr Med Res Opin 1975;3:388-91.
29. Beeley M, Warner JO. Medical treatment of symptomatic hiatus hernia with low-density compounds. Curr Med Res Opin 1972;1:63-9.
30. Chevrel B. A comparative crossover study on the treatment of heartburn and epigastric pain: liquid Gaviscon and a magnesium-aluminium antacid gel. J Int Med Res. 1980;8:300-2.
31. Laitinen S, Stahlberg M, Kairaluoma MI, Kiviniemi H, Paakkonen M, Lahtinen J, et al. Sucralfate and algi-nate/antacid in reflux esophagitis. Scand J Gastroenterol 1985;20:229-32.
32. Lanza FL, Smith V, Page-Castell JA, Castell DO. Effectiveness of foaming antacid in relieving induced heartburn. South Med J 1986;79:327-30.
33. McHardy G. A multicentric, randomized clinical trial of Gaviscon in reflux esophagitis. South Med J 1978;71(suppl 1):16-21.
34. Stanciu C, Bennett JR. Alginate-antacid in the reduction of gastro-oesophageal reflux. Lancet 1974;1:109-11.
35. Paul K, Redman CM, Chen M. Effectiveness and safety of nizatidine, 75 mg, for the relief of episodic heartburn. Aliment Pharmacol Ther 2001;15:1571-7.
36. Spiegel JE, Thoden WR, Pappas K, Fratarcangelo P, Furey SA. A double-blind, placebo-controlled study of the effectiveness and safety of nizatidine in the prevention of postprandial heartburn. Arch Intern Med 1997;157:1594-9.
37. Pappa KA, Buaron K, Payne JE, Sirgo MA, Giefer EE. An evaluation of increasing doses of ranitidine for treatment of heartburn. Aliment Pharmacol Ther 1999;13:475-81.
38. Pappa KA, Gooch WM, Buaron K, Payne JE, Giefer EE, Sirgo MA, et al. Low-dose ranitidine for the relief of heartburn. Aliment Pharmacol Ther 1999;13:459-65.
39. Pappa KA, Williams BO, Payne JE, Buaron KS, Mussari KL, Ciociola AA. A double-blind, placebo-controlled study of the efficacy and safety of non-pre-scription ranitidine 75 mg in the prevention of meal-induced heartburn. Aliment Pharmacol Ther 1999;13:467-73.
40. Ciociola AA, Pappa KA, Sirgo MA. Nonprescription doses of ranitidine are effective in the relief of episodic heartburn. Am J Ther 2001;8:399-408.
41. Gottlieb S, Decktor DL, Eckert JM, Simon TJ, Stauffer L, Ciccone PE. Efficacy and tolerability of famotidine in preventing heartburn and related symptoms of upper gastrointestinal discomfort. Am J Ther 1995;2:314-9.
42. Simon TJ, Berlin RG, Gardner AH, Stauffer LA, Gould AL, Getson AJ. Self-directed treatment of intermittent heartburn: a randomized, multicenter, double-blind, placebo-controlled evaluation of antacid and low doses of an H(2)-receptor antagonist (famotidine). Am J Ther 1995;2:304-13.
43. Galmiche JP, Shi G, Simon B, Casset-Semanza F, Slama A. On-demand treatment of gastro-oesophageal reflux symptoms: a comparison of ranitidine 75 mg with cimetidine 200 mg or place-bo. Aliment Pharmacol Ther 1998;12:909-17.
44. Qvigstad G, Arnestad JS, Brenna E, Waldum HL. Treatment with proton pump inhibitors induces tolerance to histamine-2 receptor antagonists in Helicobacter pylori-negative patients. Scand J Gastroenterol 1998;33:1244-8.
45. Howden CW. Optimizing the pharmacology of acid control in acid-related disorders. Am J Gastroenterol 1997;92(suppl):17S-21S.
46. Sharma VK, Leontiadis GI, Howden CW. Meta-analysis of randomized controlled trials comparing standard clinical doses of omeprazole and lansopra-zole in erosive oesophagitis. Aliment Pharmacol Ther 2001;15:227-31.
47. Edwards SJ, Lind T, Lundell L. Systematic review of proton pump inhibitors for the acute treatment of reflux oesophagitis. Aliment Pharmacol Ther 2001;15:1729-36.
48. Dupas JL, Houcke P, Samoyeau R. Pantoprazole versus lansoprazole in French patients with reflux esophagi-tis. Gastroenterol Clin Biol 2001;25:245-50.
49. Castell DO, Kahrilas PJ, Richter JE, Vakil NB, Johnson DA, Zuckerman S, et al. Esomeprazole (40 mg) compared with lansoprazole (30 mg) in the treatment of erosive esophagitis. Am J Gastroenterol 2002;97:575-83.
50. Howden CW, Ballard ED, Robison W. Evidence for therapeutic equivalence of lansoprazole 30 mg and esomeprazole 40 mg in the treatment of erosive oesophagitis. Clin Drug Invest 2002;22:99-109.
51. Thjodleifsson B, Beker JA, Dekkers C, Bjaaland T, Finnegan V, Humphries TJ. Rabeprazole versus omeprazole in preventing relapse of erosive or ulcerative gastroesophageal reflux disease: a dou-ble-blind, multicenter, European trial. The European Rabeprazole Study Group. Dig Dis Sci 2000;45:845-53.
52. Carling L, Axelsson CK, Forssell H, Stubberod A, Kraglund K, Bonnevie O, et al. Lansoprazole and omeprazole in the prevention of relapse of reflux oesophagitis: a long-term comparative study. Aliment Pharmacol Ther 1998;12:985-90.
53. Chey WD, Inadmoni JM, Boojer AK, Fendrick AM. What do primary care physicians think about Barrett’s esoph-agus, the relationship between GERD and H. pylori, and treatment of nocturnal heartburn? Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
54. Sampliner RE and The Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett’s esophagus. Am J Gastroenterol 2002;97:1888-95.
55. Spechler SJ, Lee E, Ahnen D, Goyal RK, Hirano I, Ramirez F, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow-up of a randomized controlled trial. JAMA 2001;285:2331-8.
56. Katz PO. Gastroesophageal reflux disease: new treatments. Rev Gastroenterol Disord 2002;2:66-74.
- Heartburn on 2 or more days a week warrants medical attention, as patients are likely to suffer from gastroesophageal reflux disease (GERD).Chronic GERD can lead to the development of complications including erosive esophagitis, stricture formation, and Barrett’s esophagus, which increases the risk of esophageal adenocarcinoma.
- A trial with a proton pump inhibitor (PPI) is the quickest and most cost-effective way to diagnose GERD, and is at least as sensitive as 24-hour intraesophageal pH monitoring.
- As PPIs only bind to actively secreting proton pumps, they should be dosed 30 to 60 minutes before a meal.Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take a PPI with or after a meal or did not specify the timing of dosing.
- The patients who will have the best response to surgical therapy for GERD are those who had clearly documented acid reflux with typical symptoms, and who have responded to PPI treatment. Unfortunately, the same survey found that most physicians recommend antireflux surgery for patients in whom medical therapy has failed.
Gastroesophageal reflux disease (GERD) is a common, multifactorial condition that often results in decreased quality of life with interruptions of sleep, work, and social activities. Patients have reported that GERD affects emotional well-being to a greater degree than diabetes or hypertension.1,2 GERD is also associated with well-established complications, including Barrett’s esophagus. The role of reflux in carcinogenesis is controversial; the possibility of an association, however, implies that GERD should be treated aggressively and early.3
Symptoms of gerd
The typical symptoms of GERD are heartburn and regurgitation. Heartburn is best defined as a burning retrosternal discomfort starting in the epigastrium or lower chest and moving upwards towards the neck. Regurgitation is the effortless movement of gastric contents up into the esophagus or pharynx.
Most patients with GERD do not have endoscopically visible lesions; a careful analysis of symptoms generally forms the basis of a preliminary diagnosis.
The occurrence of heartburn on 2 or more days a week has been suggested as a basis for further investigation for GERD.4 However, symptoms vary greatly. Patients may be asymptomatic or experience symptoms that more closely resemble gastric disorders, infectious and motor disorders of the esophagus, biliary tract disease, or even coronary artery disease.
Extraesophageal manifestations
Adding to the complexity of diagnosis, GERD has been shown to have extraesophageal manifestations, including chronic cough, asthma, recurrent aspiration, chronic sore throat, reflux laryngitis, and paroxysmal laryngospasm or voice changes.
Although the relationship between asthma and GERD remains unclear, it has been estimated that 24% to 98% of patients with asthma also have GERD.5 Some patients with asthma have been shown to have excess acid reflux into the esophagus. Reflux-like symptoms may precede episodes of asthma that occur after meals or when lying down.6 8
Additionally, GERD has been noted in 10% to 50% of patients with non-cardiac chest pain.9,10
Diagnostic strategies
Trial of treatment
Diagnosis is usually based on typical symptoms—heartburn or regurgitation—in the clinical history. (The Figure shows a treatment algorithm for both severe and mild symptoms.)
A 2-week trial of treatment with a proton pump inhibitor (PPI) provides the quickest and most cost-effective confirmation of diagnosis and is recommended for the patient whose history suggests uncomplicated GERD. A positive response to PPI treatment in a patient with symptoms suggestive of GERD is at least as sensitive and specific as 24-hour intraesophageal pH monitoring, which is still often considered the “gold standard” for the diagnosis of GERD. Furthermore, complete lack of improvement in response to PPI treatment is highly predictive that the patient does not have GERD and indicates the need for further evaluation and a possible revision of diagnosis.11,12
H2 receptor antagonists (H2RAs) have also been investigated in empirical trials for usefulness in diagnosing GERD. H2RAs are less effective than PPIs.13,14
FIGURE Medical management of suspected GERD
Endoscopy
No data support routine endoscopy for patients with the recent onset of uncomplicated heartburn who respond to medical therapy. Endoscopy is recommended, however, for patients with severe or atypical GERD symptoms, when other diseases may be present, or when a treatment trial with a PPI is ineffective.15 Endoscopy is useful for diagnosing complications of GERD, such as Barrett’s esophagus, esophagitis, and strictures. Fewer than 50% of patients with GERD symptoms have evidence of esophagitis on endoscopy.16
The American Society for Gastrointestinal Endoscopy recommends endoscopy when there are clinical suggestions of severe reflux or other disease.17 The American College of Gastroenterology recommends further testing
- when empiric therapy has failed
- when symptoms of complicated disease exist
- when there is dysphagia, bleeding, weight loss, choking, chest pain, or long-standing symptoms
- when continuous therapy is required
- to screen for Barrett’s esophagus.18
The Canadian Consensus Conference recommends endoscopy in the presence of
- dysphagia
- odynophagia
- bleeding
- weight loss
- noncardiac chest pain
- failure to respond to 4 to 8 weeks of pharmacologic therapy.19
It also recommends a single test if maintenance therapy is required.
Other diagnostic tests
Other diagnostic tools may be of use in some settings.
A barium esophagram can document reflux, and Bernstein testing (esophageal acid infusion test) can identify esophageal hypersensitivity to acid, although neither establishes a diagnosis of GERD. Ambulatory 24-hour intraesophageal pH monitoring can help to establish the presence of GERD by documenting the proportion of time during which the intraesophageal pH is acidic (<4) and can also establish the degree of association between patients’ symptoms and episodes of esophageal acidification.
Esophageal manometry is not recommended as a routine diagnostic test for GERD. It is important in selected patients to exclude an esophageal motility disorder and may be necessary as part of the pre-operative evaluation for patients in whom a surgical operation for GERD is being considered.
Management of gerd
GERD commonly requires long-term management that includes dietary, lifestyle, and pharmacological interventions. Surgery may be considered for the long-term management of the condition in carefully selected patients.
Diet and lifestyle
Dietary modifications. Patients should not consume large meals and should avoid lying down for 3 to 4 hours after eating. Caffeinated products, peppermint, fatty foods, chocolate, spicy foods, citrus fruits and juices, tomato-based products, and alcohol may contribute to episodes of GERD.18,21 Lozenges of any kind are able to stimulate salivary secretion, help clear refluxed acid, and hence, help relieve symptoms.
Lifestyle modifications. Changes in lifestyle may include such seemingly sensible interventions as sleeping with the head elevated, stopping smoking, and losing weight. There is little or no established evidence for the efficacy of these and other lifestyle modifications in the management of GERD. However, in1 trial of 63 patients, elevating the head of the bed with 6-inch blocks resulted in 1 less episode of heartburn or acid regurgitation per night when compared with lying flat.22 In another trial of 71 patients with esophagitis, elevating the bed was nearly as effective as ranitidine for reducing symptoms and producing endoscopically verifiable healing.23
Arecent survey20 of 1046 primary care physicians found that:
- 36% instructed patients to take PPIs during or after a meal or did not specify a time of dosing
- 75% referred patients for surgical antireflux therapy and 20% referred patients directly to a surgeon without gastrointestinal consultation
- 15% reported that a trial with a H2 receptor antagonist was required by their healthsystem or insurance company prior to using a PPI.
Drug interventions
Pharmacological interventions include over-the-counter remedies such as antacids and H2RAs (Table 1), as well as prescription-only doses of H2RAs and PPIs. At the time of writing, no PPI was available in an over-the-counter preparation in the United States, although over-the-counter omeprazole may soon be approved. Many authorities believe an incremental approach to the management of GERD is appropriate, beginning with lifestyle modifications and over-the-counter preparations, continuing with H2 blockers, and reserving PPIs for nonresponders. While this approach may have appeal from a cost perspective, we believe another approach (as illustrated in the Figure) is clinically superior.
Antacids. Over-the-counter antacids rapidly increase the pH of the intraesophageal contents and also neutralize acidic gastric contents that might be refluxed. They are frequently used to treat heartburn. However, few clinical trials have evaluated the efficacy of antacids. Published trials24-26 are limited by small sample sizes and a lack of intention-to-treat analysis. Only 1 showed positive evidence for antacid efficacy.25
The utility of antacids is limited by the need for frequent dosing and possible interactions with such drugs as fluoroquinolones, tetracycline, and ferrous sulfate.27 Alginate/antacids have shown statistically significant benefit compared with placebo for relief of mild-to-moderate GERD symptoms and healing of esophagitis.24,28-34
H 2 receptor antagonists. H2RAs have shown positive effects on symptoms in some studies, although symptomatic response rates observed were only around 60% to 70%. Additionally, most of the trials to date have been for 2 to 6 weeks in duration.35 43 An issue worthy of consideration with the H2RAs is the development of tolerance with continuous use.44
An H2RA-antacid combination was recently evaluated in a trial that compared it with monotherapy using either agent. Of the patients receiving combination therapy, 81% reported an excellent or good symptom response. Those receiving famotidine or atacid alone reported a 72% excellent or good symptom response.3
Proton pump inhibitors. PPIs potently reduce gastric acid secretion by inhibiting the H+-K+adenosine triphosphatase pump of the parietal cell. As a result, they suppress gastric acid secretion for a longer period than H2RAs.45 Evidence from randomized, controlled trials has demonstrated the superiority of PPIs over any other class of drugs for the relief of GERD symptoms, for healing esophagitis, and for maintaining patients in remission. Standard doses of omeprazole, lansoprazole, panto-prazole, esomeprazole, and rabeprazole have, for the most part, shown comparable rates of healing and remission in erosive esophagitis.46-52
PPIs are best absorbed in the absence of food. Ingestion of food after a PPI stimulates parietal cell activity when blood levels of the PPI are increasing; this promotes uptake of the PPI by the parietal cells. Therefore, patients should be advised to take their PPI between 30 and 60 minutes before eating. For patients on a once-daily PPI, the best time to take it is about 30 to 60 minutes before breakfast. Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take their PPI with or after a meal or did not specify the timing of dosing.53
Clinical evidence indicates that a trial with a PPI provides the quickest and most cost-effective method for diagnosing GERD. Despite this, many physicians use a trial of H2 receptor antagonists prior to initiation of PPI therapy.
- Clinicians should clearly instruct their patients regarding optimal timing of the dose, since this can have a significant effect on the success of therapy.
- Patients for whom antireflux surgery is being considered should first be referred for consultation with a gastroenterologist to assist in patient selection, to ensure that appropriate preoperative evaluation has been performed and to help exclude other possible causes of their symptoms.21,54
PPI therapy can be tailored to control GERD symptoms. Treatment can start with the most effective dosage and then be stepped down, or start with a minimum dosage and then be stepped up (Table 2). Patients with predominantly daytime symptoms should take PPIs before breakfast. Concerns that were once expressed about the long-term use of PPIs, such as predisposing patients to stomach cancer, have been refuted by extensive clinical experience and intensive monitoring (Table 3).3
TABLE 1
Over-the-counter therapy for GERD
|
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. |
| Bethesda, Md.:American Gastroenterological Association;2002. |
TABLE 2
Step-down and step-up treatments: advantages and disadvantages
| Regimen | Advantages | Disadvantages |
|---|---|---|
| Step-down therapy (high-dose initial therapy) | Rapid symptom relief | Potential overtreatment |
| Efficient for physician | Higher initial drug cost | |
| Avoids overinvestigation and associated costs | ||
| Step-up therapy (minimum-dose initial therapy) | Avoids overtreatment | Patient may continue with symptoms unnecessarily |
| Lower initial drug cost | Inefficient for physician | |
| May lead to overinvestigation | ||
| Uncertain end point (partial symptom relief) | ||
| Adapted from Dent J, et al. Management of gastro-oesophageal reflux disease in general practice.BMJ 2001;322:344-347. | ||
TABLE 3
Potential concerns associated with the use of proton pump inhibitors
| Potential concern | Level of Evidence* | Grade † | Comments |
|---|---|---|---|
| Long-term PPI treatment may lead to reduced serum cobalamin levels | 2b | B | This is most likely to occur in individuals with atrophic gastritis |
| Increased acid output has been seen after stopping a PPI | 2b | B | Effects of PPI treatment on corpus glandular atrophy in H pylori-infected individuals are difficult to interpret due to possible sampling error and short study duration |
| PPI treatment may predispose to bacterial enteric infection | 3 | B | Only shown in a single case control study |
| *Level of evidence:1, Evidence for and/or general agreement that treatment is useful and effective;1a, systematic review with homogeneity of randomized controlled trials (RCTs);1b, individual RCTs (with narrow confidence interval);2, conflicting evidence and/or divergent opinion about efficacy and use;2a, evidence or opinion is in favor of treatment;2b, use and efficacy is less well established by evidence or opinion;3, evidence and/or general agreement that treatment is not useful or effective and may be harmful in some cases. | |||
| †Quality grading:A, well-designed, clinical trials;B, well-designed cohort or case-control studies;C, case reports, flawed trials;D, personal clinical experience;E, insufficient evidence to form opinion. | |||
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. Bethesda, Md:American Gastroenterological Association, 2002. | |||
Surgery
Surgical antireflux therapy is an option in carefully selected patients. Those who respond best to surgical therapy will have had clearly documented acid reflux, typical symptoms, and symptomatic improvement while on PPI treatment.54
Unfortunately, a recent survey suggests that physicians tend to recommend surgery for patients in whom medical therapy has failed.53 However, patients who failed to respond to PPI therapy are unlikely to have GERD and, therefore, are highly unlikely to have a good outcome from antireflux surgery. Recent studies suggest that up to 62% of patients who have had open surgery for GERD continue to require medical treatment afterward. Although some studies demonstrate that surgery has greater efficacy over medical therapy initially, long-term follow-up has shown that surgically treated patients often need further medical therapy for persistent GERD symptoms.55 Community-based studies of antireflux surgery indicate that many patients develop new symptoms that they did not have before surgery and that these substantially diminish quality of life.
New endoscopic therapies, including radiofrequency energy delivery to the region of the lower esophageal sphincter and endoscopic suturing, have recently been approved for use by the FDA. This approval was based largely on safety rather than efficacy data. Clinical evidence is limited to uncontrolled studies in patients with no or mild esophagitis.3 These techniques should not be used in preference to established medical treatment unless and until data from randomized, controlled trials become available that demonstrate safety and efficacy.56
- Heartburn on 2 or more days a week warrants medical attention, as patients are likely to suffer from gastroesophageal reflux disease (GERD).Chronic GERD can lead to the development of complications including erosive esophagitis, stricture formation, and Barrett’s esophagus, which increases the risk of esophageal adenocarcinoma.
- A trial with a proton pump inhibitor (PPI) is the quickest and most cost-effective way to diagnose GERD, and is at least as sensitive as 24-hour intraesophageal pH monitoring.
- As PPIs only bind to actively secreting proton pumps, they should be dosed 30 to 60 minutes before a meal.Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take a PPI with or after a meal or did not specify the timing of dosing.
- The patients who will have the best response to surgical therapy for GERD are those who had clearly documented acid reflux with typical symptoms, and who have responded to PPI treatment. Unfortunately, the same survey found that most physicians recommend antireflux surgery for patients in whom medical therapy has failed.
Gastroesophageal reflux disease (GERD) is a common, multifactorial condition that often results in decreased quality of life with interruptions of sleep, work, and social activities. Patients have reported that GERD affects emotional well-being to a greater degree than diabetes or hypertension.1,2 GERD is also associated with well-established complications, including Barrett’s esophagus. The role of reflux in carcinogenesis is controversial; the possibility of an association, however, implies that GERD should be treated aggressively and early.3
Symptoms of gerd
The typical symptoms of GERD are heartburn and regurgitation. Heartburn is best defined as a burning retrosternal discomfort starting in the epigastrium or lower chest and moving upwards towards the neck. Regurgitation is the effortless movement of gastric contents up into the esophagus or pharynx.
Most patients with GERD do not have endoscopically visible lesions; a careful analysis of symptoms generally forms the basis of a preliminary diagnosis.
The occurrence of heartburn on 2 or more days a week has been suggested as a basis for further investigation for GERD.4 However, symptoms vary greatly. Patients may be asymptomatic or experience symptoms that more closely resemble gastric disorders, infectious and motor disorders of the esophagus, biliary tract disease, or even coronary artery disease.
Extraesophageal manifestations
Adding to the complexity of diagnosis, GERD has been shown to have extraesophageal manifestations, including chronic cough, asthma, recurrent aspiration, chronic sore throat, reflux laryngitis, and paroxysmal laryngospasm or voice changes.
Although the relationship between asthma and GERD remains unclear, it has been estimated that 24% to 98% of patients with asthma also have GERD.5 Some patients with asthma have been shown to have excess acid reflux into the esophagus. Reflux-like symptoms may precede episodes of asthma that occur after meals or when lying down.6 8
Additionally, GERD has been noted in 10% to 50% of patients with non-cardiac chest pain.9,10
Diagnostic strategies
Trial of treatment
Diagnosis is usually based on typical symptoms—heartburn or regurgitation—in the clinical history. (The Figure shows a treatment algorithm for both severe and mild symptoms.)
A 2-week trial of treatment with a proton pump inhibitor (PPI) provides the quickest and most cost-effective confirmation of diagnosis and is recommended for the patient whose history suggests uncomplicated GERD. A positive response to PPI treatment in a patient with symptoms suggestive of GERD is at least as sensitive and specific as 24-hour intraesophageal pH monitoring, which is still often considered the “gold standard” for the diagnosis of GERD. Furthermore, complete lack of improvement in response to PPI treatment is highly predictive that the patient does not have GERD and indicates the need for further evaluation and a possible revision of diagnosis.11,12
H2 receptor antagonists (H2RAs) have also been investigated in empirical trials for usefulness in diagnosing GERD. H2RAs are less effective than PPIs.13,14
FIGURE Medical management of suspected GERD
Endoscopy
No data support routine endoscopy for patients with the recent onset of uncomplicated heartburn who respond to medical therapy. Endoscopy is recommended, however, for patients with severe or atypical GERD symptoms, when other diseases may be present, or when a treatment trial with a PPI is ineffective.15 Endoscopy is useful for diagnosing complications of GERD, such as Barrett’s esophagus, esophagitis, and strictures. Fewer than 50% of patients with GERD symptoms have evidence of esophagitis on endoscopy.16
The American Society for Gastrointestinal Endoscopy recommends endoscopy when there are clinical suggestions of severe reflux or other disease.17 The American College of Gastroenterology recommends further testing
- when empiric therapy has failed
- when symptoms of complicated disease exist
- when there is dysphagia, bleeding, weight loss, choking, chest pain, or long-standing symptoms
- when continuous therapy is required
- to screen for Barrett’s esophagus.18
The Canadian Consensus Conference recommends endoscopy in the presence of
- dysphagia
- odynophagia
- bleeding
- weight loss
- noncardiac chest pain
- failure to respond to 4 to 8 weeks of pharmacologic therapy.19
It also recommends a single test if maintenance therapy is required.
Other diagnostic tests
Other diagnostic tools may be of use in some settings.
A barium esophagram can document reflux, and Bernstein testing (esophageal acid infusion test) can identify esophageal hypersensitivity to acid, although neither establishes a diagnosis of GERD. Ambulatory 24-hour intraesophageal pH monitoring can help to establish the presence of GERD by documenting the proportion of time during which the intraesophageal pH is acidic (<4) and can also establish the degree of association between patients’ symptoms and episodes of esophageal acidification.
Esophageal manometry is not recommended as a routine diagnostic test for GERD. It is important in selected patients to exclude an esophageal motility disorder and may be necessary as part of the pre-operative evaluation for patients in whom a surgical operation for GERD is being considered.
Management of gerd
GERD commonly requires long-term management that includes dietary, lifestyle, and pharmacological interventions. Surgery may be considered for the long-term management of the condition in carefully selected patients.
Diet and lifestyle
Dietary modifications. Patients should not consume large meals and should avoid lying down for 3 to 4 hours after eating. Caffeinated products, peppermint, fatty foods, chocolate, spicy foods, citrus fruits and juices, tomato-based products, and alcohol may contribute to episodes of GERD.18,21 Lozenges of any kind are able to stimulate salivary secretion, help clear refluxed acid, and hence, help relieve symptoms.
Lifestyle modifications. Changes in lifestyle may include such seemingly sensible interventions as sleeping with the head elevated, stopping smoking, and losing weight. There is little or no established evidence for the efficacy of these and other lifestyle modifications in the management of GERD. However, in1 trial of 63 patients, elevating the head of the bed with 6-inch blocks resulted in 1 less episode of heartburn or acid regurgitation per night when compared with lying flat.22 In another trial of 71 patients with esophagitis, elevating the bed was nearly as effective as ranitidine for reducing symptoms and producing endoscopically verifiable healing.23
Arecent survey20 of 1046 primary care physicians found that:
- 36% instructed patients to take PPIs during or after a meal or did not specify a time of dosing
- 75% referred patients for surgical antireflux therapy and 20% referred patients directly to a surgeon without gastrointestinal consultation
- 15% reported that a trial with a H2 receptor antagonist was required by their healthsystem or insurance company prior to using a PPI.
Drug interventions
Pharmacological interventions include over-the-counter remedies such as antacids and H2RAs (Table 1), as well as prescription-only doses of H2RAs and PPIs. At the time of writing, no PPI was available in an over-the-counter preparation in the United States, although over-the-counter omeprazole may soon be approved. Many authorities believe an incremental approach to the management of GERD is appropriate, beginning with lifestyle modifications and over-the-counter preparations, continuing with H2 blockers, and reserving PPIs for nonresponders. While this approach may have appeal from a cost perspective, we believe another approach (as illustrated in the Figure) is clinically superior.
Antacids. Over-the-counter antacids rapidly increase the pH of the intraesophageal contents and also neutralize acidic gastric contents that might be refluxed. They are frequently used to treat heartburn. However, few clinical trials have evaluated the efficacy of antacids. Published trials24-26 are limited by small sample sizes and a lack of intention-to-treat analysis. Only 1 showed positive evidence for antacid efficacy.25
The utility of antacids is limited by the need for frequent dosing and possible interactions with such drugs as fluoroquinolones, tetracycline, and ferrous sulfate.27 Alginate/antacids have shown statistically significant benefit compared with placebo for relief of mild-to-moderate GERD symptoms and healing of esophagitis.24,28-34
H 2 receptor antagonists. H2RAs have shown positive effects on symptoms in some studies, although symptomatic response rates observed were only around 60% to 70%. Additionally, most of the trials to date have been for 2 to 6 weeks in duration.35 43 An issue worthy of consideration with the H2RAs is the development of tolerance with continuous use.44
An H2RA-antacid combination was recently evaluated in a trial that compared it with monotherapy using either agent. Of the patients receiving combination therapy, 81% reported an excellent or good symptom response. Those receiving famotidine or atacid alone reported a 72% excellent or good symptom response.3
Proton pump inhibitors. PPIs potently reduce gastric acid secretion by inhibiting the H+-K+adenosine triphosphatase pump of the parietal cell. As a result, they suppress gastric acid secretion for a longer period than H2RAs.45 Evidence from randomized, controlled trials has demonstrated the superiority of PPIs over any other class of drugs for the relief of GERD symptoms, for healing esophagitis, and for maintaining patients in remission. Standard doses of omeprazole, lansoprazole, panto-prazole, esomeprazole, and rabeprazole have, for the most part, shown comparable rates of healing and remission in erosive esophagitis.46-52
PPIs are best absorbed in the absence of food. Ingestion of food after a PPI stimulates parietal cell activity when blood levels of the PPI are increasing; this promotes uptake of the PPI by the parietal cells. Therefore, patients should be advised to take their PPI between 30 and 60 minutes before eating. For patients on a once-daily PPI, the best time to take it is about 30 to 60 minutes before breakfast. Despite these recommendations, a recent survey of over 1000 US primary care physicians found that 36% instructed their patients to take their PPI with or after a meal or did not specify the timing of dosing.53
Clinical evidence indicates that a trial with a PPI provides the quickest and most cost-effective method for diagnosing GERD. Despite this, many physicians use a trial of H2 receptor antagonists prior to initiation of PPI therapy.
- Clinicians should clearly instruct their patients regarding optimal timing of the dose, since this can have a significant effect on the success of therapy.
- Patients for whom antireflux surgery is being considered should first be referred for consultation with a gastroenterologist to assist in patient selection, to ensure that appropriate preoperative evaluation has been performed and to help exclude other possible causes of their symptoms.21,54
PPI therapy can be tailored to control GERD symptoms. Treatment can start with the most effective dosage and then be stepped down, or start with a minimum dosage and then be stepped up (Table 2). Patients with predominantly daytime symptoms should take PPIs before breakfast. Concerns that were once expressed about the long-term use of PPIs, such as predisposing patients to stomach cancer, have been refuted by extensive clinical experience and intensive monitoring (Table 3).3
TABLE 1
Over-the-counter therapy for GERD
|
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. |
| Bethesda, Md.:American Gastroenterological Association;2002. |
TABLE 2
Step-down and step-up treatments: advantages and disadvantages
| Regimen | Advantages | Disadvantages |
|---|---|---|
| Step-down therapy (high-dose initial therapy) | Rapid symptom relief | Potential overtreatment |
| Efficient for physician | Higher initial drug cost | |
| Avoids overinvestigation and associated costs | ||
| Step-up therapy (minimum-dose initial therapy) | Avoids overtreatment | Patient may continue with symptoms unnecessarily |
| Lower initial drug cost | Inefficient for physician | |
| May lead to overinvestigation | ||
| Uncertain end point (partial symptom relief) | ||
| Adapted from Dent J, et al. Management of gastro-oesophageal reflux disease in general practice.BMJ 2001;322:344-347. | ||
TABLE 3
Potential concerns associated with the use of proton pump inhibitors
| Potential concern | Level of Evidence* | Grade † | Comments |
|---|---|---|---|
| Long-term PPI treatment may lead to reduced serum cobalamin levels | 2b | B | This is most likely to occur in individuals with atrophic gastritis |
| Increased acid output has been seen after stopping a PPI | 2b | B | Effects of PPI treatment on corpus glandular atrophy in H pylori-infected individuals are difficult to interpret due to possible sampling error and short study duration |
| PPI treatment may predispose to bacterial enteric infection | 3 | B | Only shown in a single case control study |
| *Level of evidence:1, Evidence for and/or general agreement that treatment is useful and effective;1a, systematic review with homogeneity of randomized controlled trials (RCTs);1b, individual RCTs (with narrow confidence interval);2, conflicting evidence and/or divergent opinion about efficacy and use;2a, evidence or opinion is in favor of treatment;2b, use and efficacy is less well established by evidence or opinion;3, evidence and/or general agreement that treatment is not useful or effective and may be harmful in some cases. | |||
| †Quality grading:A, well-designed, clinical trials;B, well-designed cohort or case-control studies;C, case reports, flawed trials;D, personal clinical experience;E, insufficient evidence to form opinion. | |||
| Adapted from Peterson, WL.GERD:Evidence-based therapeutic strategies. Bethesda, Md:American Gastroenterological Association, 2002. | |||
Surgery
Surgical antireflux therapy is an option in carefully selected patients. Those who respond best to surgical therapy will have had clearly documented acid reflux, typical symptoms, and symptomatic improvement while on PPI treatment.54
Unfortunately, a recent survey suggests that physicians tend to recommend surgery for patients in whom medical therapy has failed.53 However, patients who failed to respond to PPI therapy are unlikely to have GERD and, therefore, are highly unlikely to have a good outcome from antireflux surgery. Recent studies suggest that up to 62% of patients who have had open surgery for GERD continue to require medical treatment afterward. Although some studies demonstrate that surgery has greater efficacy over medical therapy initially, long-term follow-up has shown that surgically treated patients often need further medical therapy for persistent GERD symptoms.55 Community-based studies of antireflux surgery indicate that many patients develop new symptoms that they did not have before surgery and that these substantially diminish quality of life.
New endoscopic therapies, including radiofrequency energy delivery to the region of the lower esophageal sphincter and endoscopic suturing, have recently been approved for use by the FDA. This approval was based largely on safety rather than efficacy data. Clinical evidence is limited to uncontrolled studies in patients with no or mild esophagitis.3 These techniques should not be used in preference to established medical treatment unless and until data from randomized, controlled trials become available that demonstrate safety and efficacy.56
1. Dimenas E. Methodological aspects of evaluation of quality of life in upper gastrointestinal diseases. Scand J Gastroenterol Suppl 1993;199:18-21.
2. Revicki DA, Wood M, Maton PN, Sorensen S. The impact of gastroesophageal reflux disease on health-related quality of life. Am J Med 1998;104:252-8.
3. Peterson WL. GERD: Evidence-based therapeutic strategies. Bethesda, Md: American Gastroenterological Association; 2002.
4. Dent J, Jones R, Kahrilas P, Talley NJ. Management of gastro-oesophageal reflux disease in general practice. BMJ 2001;322:344-7.
5. Harding SM. Nocturnal asthma: role of gastroesophageal reflux. Chronobiol Int 1999;16:641-2.
6. Kjellen G, Wranne B. The prevalence of asymptomatic gastro-oesophageal relfux in adult patients with asthma. Eur J Respir Dis 1984;65:233.-
7. Schnatz PF, Castell JA, Castell DO. Pulmonary symptoms associated with gastroesophageal reflux: use of ambulatory pH monitoring to diagnose and to direct therapy. Am J Gastroenterol 1996;91:1715-8.
8. Sontag SJ, Schnell TG, Miller TQ, Khandelwal S, O’Connell S, Chejfec G, et al. Prevalence of oesophagitis in asthmatics. Gut 1992;33:872-6.
9. Schofield PM, Bennett DH, Whorwell PJ, Brooks NH, Bray CL, Ward C, et al. Exertional gastro-oesophageal reflux: a mechanism for symptoms in patients with angina pectoris and normal coronary angiograms. Br Med J (Clin Res Ed) 1987;294:1459-61.
10. Hewson EG, Sinclair JW, Dalton CB, Richter JE. Twenty-four-hour esophageal pH monitoring: the most useful test for evaluating noncardiac chest pain. Am J Med 1991;90:576-83.
11. Goyal RK. Diseases of the esophagus. In: Braunwald E, ed. Harrison’s principles of internal medicine. 15th ed. New York: McGraw-Hill; 2001;1642-9.
12. Schenk BE, Kuipers EJ, Klinkenberg-Knol EC, Festen HP, Jansen EH, Tuynman HA, et al. Omeprazole as a diagnostic tool in gastroesophageal reflux disease. Am J Gastroenterol 1997;92:1997-2000.
13. van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastrooesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2001;(4):CD002095.-
14. Brun J, Sorngard H. High dose proton pump inhibitor response as an initial strategy for a clinical diagnosis of gastro-oesophageal reflux disease (GERD).Swedish multi-centre group in primary health care. Fam Pract 2000;17:401-4.
15. Lundell L. Anti-reflux surgery in the laparoscopic era. Baillieres Best Pract Res Clin Gastroenterol 2000;14:793-810.
16. Chen MY, Ott DJ, Sinclair JW, Wu WC, Gelfand DW. Gastroesophageal reflux disease: correlation of esophageal pH testing and radiographic findings. Radiology 1992;185:483-6.
17. The role of endoscopy in the management of GERD: guidelines for clinical application. From the ASGE. American Society for Gastrointestinal Endoscopy. Gastrointest Endosc 1999;49:834-5.
18. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434-42.
19. Beck IT, Champion MC, Lemire S, Thomson AB, Anvari M, Armstrong D, et al. The Second Canadian Consensus Conference on the Management of Patients with Gastroesophageal Reflux Disease. Can J Gastroenterol 1997;11(suppl B):7B-20B.
20. Chey WD, Inadomi JM, Booher AK, Fendrick AM. Primary care physicians’ perceptions and practices of the management of GERD: results of a national survey. Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
21. Fennerty MB, Castell D, Fendrick AM, Halpern M, Johnson D, Kahrilas PJ, et al. The diagnosis and treatment of gastroesophageal reflux disease in a managed care environment, Suggested disease management guidelines. Arch Intern Med 1996;156:477-84.
22. Stanciu C, Bennett JR. Effects of posture on gastro-oesophageal reflux. Digestion 1977;15:104-9.
23. Harvey RF, Gordon PC, Hadley N, Long DE, Gill TR, Macpherson RI, et al. Effects of sleeping with the bed-head raised and of ranitidine in patients with severe peptic oesophagitis. Lancet 1987;2:1200-3.
24. Graham DY, Patterson DJ. Double-blind comparison of liquid antacid and placebo in the treatment of symptomatic reflux esophagitis. Dig Dis Sci 1983;28:559-63.
25. Grove O, Bekker C, Jeppe-Hansen MG, Karstoft E, Sanchez G, Axelsson CK, et al. Ranitidine and high-dose antacid in reflux oesophagitis. A randomized, placebo-controlled trial. Scand J Gastroenterol 1985;20:457-61.
26. Weberg R, Berstad A. Symptomatic effect of a low-dose antacid regimen in reflux oesophagitis. Scand J Gastroenterol 1989;24:401-6.
27. Welage LS, Berardi RR. Evaluation of omeprazole, lansoprazole, pantopra-zole, and rabeprazole in the treatment of acid-related diseases. J Am Pharm Assoc (Wash) 2000;40:52-62.
28. Barnardo DE, Lancaster-Smith M, Strickland ID, Wright JT. A double-blind controlled trial of ‘Gaviscon’ in patients with symptomatic gastro-oesophageal reflux. Curr Med Res Opin 1975;3:388-91.
29. Beeley M, Warner JO. Medical treatment of symptomatic hiatus hernia with low-density compounds. Curr Med Res Opin 1972;1:63-9.
30. Chevrel B. A comparative crossover study on the treatment of heartburn and epigastric pain: liquid Gaviscon and a magnesium-aluminium antacid gel. J Int Med Res. 1980;8:300-2.
31. Laitinen S, Stahlberg M, Kairaluoma MI, Kiviniemi H, Paakkonen M, Lahtinen J, et al. Sucralfate and algi-nate/antacid in reflux esophagitis. Scand J Gastroenterol 1985;20:229-32.
32. Lanza FL, Smith V, Page-Castell JA, Castell DO. Effectiveness of foaming antacid in relieving induced heartburn. South Med J 1986;79:327-30.
33. McHardy G. A multicentric, randomized clinical trial of Gaviscon in reflux esophagitis. South Med J 1978;71(suppl 1):16-21.
34. Stanciu C, Bennett JR. Alginate-antacid in the reduction of gastro-oesophageal reflux. Lancet 1974;1:109-11.
35. Paul K, Redman CM, Chen M. Effectiveness and safety of nizatidine, 75 mg, for the relief of episodic heartburn. Aliment Pharmacol Ther 2001;15:1571-7.
36. Spiegel JE, Thoden WR, Pappas K, Fratarcangelo P, Furey SA. A double-blind, placebo-controlled study of the effectiveness and safety of nizatidine in the prevention of postprandial heartburn. Arch Intern Med 1997;157:1594-9.
37. Pappa KA, Buaron K, Payne JE, Sirgo MA, Giefer EE. An evaluation of increasing doses of ranitidine for treatment of heartburn. Aliment Pharmacol Ther 1999;13:475-81.
38. Pappa KA, Gooch WM, Buaron K, Payne JE, Giefer EE, Sirgo MA, et al. Low-dose ranitidine for the relief of heartburn. Aliment Pharmacol Ther 1999;13:459-65.
39. Pappa KA, Williams BO, Payne JE, Buaron KS, Mussari KL, Ciociola AA. A double-blind, placebo-controlled study of the efficacy and safety of non-pre-scription ranitidine 75 mg in the prevention of meal-induced heartburn. Aliment Pharmacol Ther 1999;13:467-73.
40. Ciociola AA, Pappa KA, Sirgo MA. Nonprescription doses of ranitidine are effective in the relief of episodic heartburn. Am J Ther 2001;8:399-408.
41. Gottlieb S, Decktor DL, Eckert JM, Simon TJ, Stauffer L, Ciccone PE. Efficacy and tolerability of famotidine in preventing heartburn and related symptoms of upper gastrointestinal discomfort. Am J Ther 1995;2:314-9.
42. Simon TJ, Berlin RG, Gardner AH, Stauffer LA, Gould AL, Getson AJ. Self-directed treatment of intermittent heartburn: a randomized, multicenter, double-blind, placebo-controlled evaluation of antacid and low doses of an H(2)-receptor antagonist (famotidine). Am J Ther 1995;2:304-13.
43. Galmiche JP, Shi G, Simon B, Casset-Semanza F, Slama A. On-demand treatment of gastro-oesophageal reflux symptoms: a comparison of ranitidine 75 mg with cimetidine 200 mg or place-bo. Aliment Pharmacol Ther 1998;12:909-17.
44. Qvigstad G, Arnestad JS, Brenna E, Waldum HL. Treatment with proton pump inhibitors induces tolerance to histamine-2 receptor antagonists in Helicobacter pylori-negative patients. Scand J Gastroenterol 1998;33:1244-8.
45. Howden CW. Optimizing the pharmacology of acid control in acid-related disorders. Am J Gastroenterol 1997;92(suppl):17S-21S.
46. Sharma VK, Leontiadis GI, Howden CW. Meta-analysis of randomized controlled trials comparing standard clinical doses of omeprazole and lansopra-zole in erosive oesophagitis. Aliment Pharmacol Ther 2001;15:227-31.
47. Edwards SJ, Lind T, Lundell L. Systematic review of proton pump inhibitors for the acute treatment of reflux oesophagitis. Aliment Pharmacol Ther 2001;15:1729-36.
48. Dupas JL, Houcke P, Samoyeau R. Pantoprazole versus lansoprazole in French patients with reflux esophagi-tis. Gastroenterol Clin Biol 2001;25:245-50.
49. Castell DO, Kahrilas PJ, Richter JE, Vakil NB, Johnson DA, Zuckerman S, et al. Esomeprazole (40 mg) compared with lansoprazole (30 mg) in the treatment of erosive esophagitis. Am J Gastroenterol 2002;97:575-83.
50. Howden CW, Ballard ED, Robison W. Evidence for therapeutic equivalence of lansoprazole 30 mg and esomeprazole 40 mg in the treatment of erosive oesophagitis. Clin Drug Invest 2002;22:99-109.
51. Thjodleifsson B, Beker JA, Dekkers C, Bjaaland T, Finnegan V, Humphries TJ. Rabeprazole versus omeprazole in preventing relapse of erosive or ulcerative gastroesophageal reflux disease: a dou-ble-blind, multicenter, European trial. The European Rabeprazole Study Group. Dig Dis Sci 2000;45:845-53.
52. Carling L, Axelsson CK, Forssell H, Stubberod A, Kraglund K, Bonnevie O, et al. Lansoprazole and omeprazole in the prevention of relapse of reflux oesophagitis: a long-term comparative study. Aliment Pharmacol Ther 1998;12:985-90.
53. Chey WD, Inadmoni JM, Boojer AK, Fendrick AM. What do primary care physicians think about Barrett’s esoph-agus, the relationship between GERD and H. pylori, and treatment of nocturnal heartburn? Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
54. Sampliner RE and The Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett’s esophagus. Am J Gastroenterol 2002;97:1888-95.
55. Spechler SJ, Lee E, Ahnen D, Goyal RK, Hirano I, Ramirez F, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow-up of a randomized controlled trial. JAMA 2001;285:2331-8.
56. Katz PO. Gastroesophageal reflux disease: new treatments. Rev Gastroenterol Disord 2002;2:66-74.
1. Dimenas E. Methodological aspects of evaluation of quality of life in upper gastrointestinal diseases. Scand J Gastroenterol Suppl 1993;199:18-21.
2. Revicki DA, Wood M, Maton PN, Sorensen S. The impact of gastroesophageal reflux disease on health-related quality of life. Am J Med 1998;104:252-8.
3. Peterson WL. GERD: Evidence-based therapeutic strategies. Bethesda, Md: American Gastroenterological Association; 2002.
4. Dent J, Jones R, Kahrilas P, Talley NJ. Management of gastro-oesophageal reflux disease in general practice. BMJ 2001;322:344-7.
5. Harding SM. Nocturnal asthma: role of gastroesophageal reflux. Chronobiol Int 1999;16:641-2.
6. Kjellen G, Wranne B. The prevalence of asymptomatic gastro-oesophageal relfux in adult patients with asthma. Eur J Respir Dis 1984;65:233.-
7. Schnatz PF, Castell JA, Castell DO. Pulmonary symptoms associated with gastroesophageal reflux: use of ambulatory pH monitoring to diagnose and to direct therapy. Am J Gastroenterol 1996;91:1715-8.
8. Sontag SJ, Schnell TG, Miller TQ, Khandelwal S, O’Connell S, Chejfec G, et al. Prevalence of oesophagitis in asthmatics. Gut 1992;33:872-6.
9. Schofield PM, Bennett DH, Whorwell PJ, Brooks NH, Bray CL, Ward C, et al. Exertional gastro-oesophageal reflux: a mechanism for symptoms in patients with angina pectoris and normal coronary angiograms. Br Med J (Clin Res Ed) 1987;294:1459-61.
10. Hewson EG, Sinclair JW, Dalton CB, Richter JE. Twenty-four-hour esophageal pH monitoring: the most useful test for evaluating noncardiac chest pain. Am J Med 1991;90:576-83.
11. Goyal RK. Diseases of the esophagus. In: Braunwald E, ed. Harrison’s principles of internal medicine. 15th ed. New York: McGraw-Hill; 2001;1642-9.
12. Schenk BE, Kuipers EJ, Klinkenberg-Knol EC, Festen HP, Jansen EH, Tuynman HA, et al. Omeprazole as a diagnostic tool in gastroesophageal reflux disease. Am J Gastroenterol 1997;92:1997-2000.
13. van Pinxteren B, Numans ME, Bonis PA, Lau J. Short-term treatment with proton pump inhibitors, H2-receptor antagonists and prokinetics for gastrooesophageal reflux disease-like symptoms and endoscopy negative reflux disease. Cochrane Database Syst Rev 2001;(4):CD002095.-
14. Brun J, Sorngard H. High dose proton pump inhibitor response as an initial strategy for a clinical diagnosis of gastro-oesophageal reflux disease (GERD).Swedish multi-centre group in primary health care. Fam Pract 2000;17:401-4.
15. Lundell L. Anti-reflux surgery in the laparoscopic era. Baillieres Best Pract Res Clin Gastroenterol 2000;14:793-810.
16. Chen MY, Ott DJ, Sinclair JW, Wu WC, Gelfand DW. Gastroesophageal reflux disease: correlation of esophageal pH testing and radiographic findings. Radiology 1992;185:483-6.
17. The role of endoscopy in the management of GERD: guidelines for clinical application. From the ASGE. American Society for Gastrointestinal Endoscopy. Gastrointest Endosc 1999;49:834-5.
18. DeVault KR, Castell DO. Updated guidelines for the diagnosis and treatment of gastroesophageal reflux disease. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol 1999;94:1434-42.
19. Beck IT, Champion MC, Lemire S, Thomson AB, Anvari M, Armstrong D, et al. The Second Canadian Consensus Conference on the Management of Patients with Gastroesophageal Reflux Disease. Can J Gastroenterol 1997;11(suppl B):7B-20B.
20. Chey WD, Inadomi JM, Booher AK, Fendrick AM. Primary care physicians’ perceptions and practices of the management of GERD: results of a national survey. Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
21. Fennerty MB, Castell D, Fendrick AM, Halpern M, Johnson D, Kahrilas PJ, et al. The diagnosis and treatment of gastroesophageal reflux disease in a managed care environment, Suggested disease management guidelines. Arch Intern Med 1996;156:477-84.
22. Stanciu C, Bennett JR. Effects of posture on gastro-oesophageal reflux. Digestion 1977;15:104-9.
23. Harvey RF, Gordon PC, Hadley N, Long DE, Gill TR, Macpherson RI, et al. Effects of sleeping with the bed-head raised and of ranitidine in patients with severe peptic oesophagitis. Lancet 1987;2:1200-3.
24. Graham DY, Patterson DJ. Double-blind comparison of liquid antacid and placebo in the treatment of symptomatic reflux esophagitis. Dig Dis Sci 1983;28:559-63.
25. Grove O, Bekker C, Jeppe-Hansen MG, Karstoft E, Sanchez G, Axelsson CK, et al. Ranitidine and high-dose antacid in reflux oesophagitis. A randomized, placebo-controlled trial. Scand J Gastroenterol 1985;20:457-61.
26. Weberg R, Berstad A. Symptomatic effect of a low-dose antacid regimen in reflux oesophagitis. Scand J Gastroenterol 1989;24:401-6.
27. Welage LS, Berardi RR. Evaluation of omeprazole, lansoprazole, pantopra-zole, and rabeprazole in the treatment of acid-related diseases. J Am Pharm Assoc (Wash) 2000;40:52-62.
28. Barnardo DE, Lancaster-Smith M, Strickland ID, Wright JT. A double-blind controlled trial of ‘Gaviscon’ in patients with symptomatic gastro-oesophageal reflux. Curr Med Res Opin 1975;3:388-91.
29. Beeley M, Warner JO. Medical treatment of symptomatic hiatus hernia with low-density compounds. Curr Med Res Opin 1972;1:63-9.
30. Chevrel B. A comparative crossover study on the treatment of heartburn and epigastric pain: liquid Gaviscon and a magnesium-aluminium antacid gel. J Int Med Res. 1980;8:300-2.
31. Laitinen S, Stahlberg M, Kairaluoma MI, Kiviniemi H, Paakkonen M, Lahtinen J, et al. Sucralfate and algi-nate/antacid in reflux esophagitis. Scand J Gastroenterol 1985;20:229-32.
32. Lanza FL, Smith V, Page-Castell JA, Castell DO. Effectiveness of foaming antacid in relieving induced heartburn. South Med J 1986;79:327-30.
33. McHardy G. A multicentric, randomized clinical trial of Gaviscon in reflux esophagitis. South Med J 1978;71(suppl 1):16-21.
34. Stanciu C, Bennett JR. Alginate-antacid in the reduction of gastro-oesophageal reflux. Lancet 1974;1:109-11.
35. Paul K, Redman CM, Chen M. Effectiveness and safety of nizatidine, 75 mg, for the relief of episodic heartburn. Aliment Pharmacol Ther 2001;15:1571-7.
36. Spiegel JE, Thoden WR, Pappas K, Fratarcangelo P, Furey SA. A double-blind, placebo-controlled study of the effectiveness and safety of nizatidine in the prevention of postprandial heartburn. Arch Intern Med 1997;157:1594-9.
37. Pappa KA, Buaron K, Payne JE, Sirgo MA, Giefer EE. An evaluation of increasing doses of ranitidine for treatment of heartburn. Aliment Pharmacol Ther 1999;13:475-81.
38. Pappa KA, Gooch WM, Buaron K, Payne JE, Giefer EE, Sirgo MA, et al. Low-dose ranitidine for the relief of heartburn. Aliment Pharmacol Ther 1999;13:459-65.
39. Pappa KA, Williams BO, Payne JE, Buaron KS, Mussari KL, Ciociola AA. A double-blind, placebo-controlled study of the efficacy and safety of non-pre-scription ranitidine 75 mg in the prevention of meal-induced heartburn. Aliment Pharmacol Ther 1999;13:467-73.
40. Ciociola AA, Pappa KA, Sirgo MA. Nonprescription doses of ranitidine are effective in the relief of episodic heartburn. Am J Ther 2001;8:399-408.
41. Gottlieb S, Decktor DL, Eckert JM, Simon TJ, Stauffer L, Ciccone PE. Efficacy and tolerability of famotidine in preventing heartburn and related symptoms of upper gastrointestinal discomfort. Am J Ther 1995;2:314-9.
42. Simon TJ, Berlin RG, Gardner AH, Stauffer LA, Gould AL, Getson AJ. Self-directed treatment of intermittent heartburn: a randomized, multicenter, double-blind, placebo-controlled evaluation of antacid and low doses of an H(2)-receptor antagonist (famotidine). Am J Ther 1995;2:304-13.
43. Galmiche JP, Shi G, Simon B, Casset-Semanza F, Slama A. On-demand treatment of gastro-oesophageal reflux symptoms: a comparison of ranitidine 75 mg with cimetidine 200 mg or place-bo. Aliment Pharmacol Ther 1998;12:909-17.
44. Qvigstad G, Arnestad JS, Brenna E, Waldum HL. Treatment with proton pump inhibitors induces tolerance to histamine-2 receptor antagonists in Helicobacter pylori-negative patients. Scand J Gastroenterol 1998;33:1244-8.
45. Howden CW. Optimizing the pharmacology of acid control in acid-related disorders. Am J Gastroenterol 1997;92(suppl):17S-21S.
46. Sharma VK, Leontiadis GI, Howden CW. Meta-analysis of randomized controlled trials comparing standard clinical doses of omeprazole and lansopra-zole in erosive oesophagitis. Aliment Pharmacol Ther 2001;15:227-31.
47. Edwards SJ, Lind T, Lundell L. Systematic review of proton pump inhibitors for the acute treatment of reflux oesophagitis. Aliment Pharmacol Ther 2001;15:1729-36.
48. Dupas JL, Houcke P, Samoyeau R. Pantoprazole versus lansoprazole in French patients with reflux esophagi-tis. Gastroenterol Clin Biol 2001;25:245-50.
49. Castell DO, Kahrilas PJ, Richter JE, Vakil NB, Johnson DA, Zuckerman S, et al. Esomeprazole (40 mg) compared with lansoprazole (30 mg) in the treatment of erosive esophagitis. Am J Gastroenterol 2002;97:575-83.
50. Howden CW, Ballard ED, Robison W. Evidence for therapeutic equivalence of lansoprazole 30 mg and esomeprazole 40 mg in the treatment of erosive oesophagitis. Clin Drug Invest 2002;22:99-109.
51. Thjodleifsson B, Beker JA, Dekkers C, Bjaaland T, Finnegan V, Humphries TJ. Rabeprazole versus omeprazole in preventing relapse of erosive or ulcerative gastroesophageal reflux disease: a dou-ble-blind, multicenter, European trial. The European Rabeprazole Study Group. Dig Dis Sci 2000;45:845-53.
52. Carling L, Axelsson CK, Forssell H, Stubberod A, Kraglund K, Bonnevie O, et al. Lansoprazole and omeprazole in the prevention of relapse of reflux oesophagitis: a long-term comparative study. Aliment Pharmacol Ther 1998;12:985-90.
53. Chey WD, Inadmoni JM, Boojer AK, Fendrick AM. What do primary care physicians think about Barrett’s esoph-agus, the relationship between GERD and H. pylori, and treatment of nocturnal heartburn? Abstract presented at: DDW 2003; May 17-23, 2003; Orlando, Fla.
54. Sampliner RE and The Practice Parameters Committee of the American College of Gastroenterology. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett’s esophagus. Am J Gastroenterol 2002;97:1888-95.
55. Spechler SJ, Lee E, Ahnen D, Goyal RK, Hirano I, Ramirez F, et al. Long-term outcome of medical and surgical therapies for gastroesophageal reflux disease: follow-up of a randomized controlled trial. JAMA 2001;285:2331-8.
56. Katz PO. Gastroesophageal reflux disease: new treatments. Rev Gastroenterol Disord 2002;2:66-74.
Diagnosing skin malignancy: Assessment of predictive clinical criteria and risk factors
- Expect to encounter 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma ever y year.
- There is good evidence for using the American Cancer Society’s ABCDE criteria as a clinical diagnostic test to rule out malignant melanoma (A).
- The revised 7-point checklist has high sensitivity and is therefore useful for ruling out a diagnosis of malignant melanoma. However, its low specificity yields many false-positive results (B).
- The gold standard for diagnosis of skin malignancies is a tissue biopsy. If any doubt exists about the diagnosis, a biopsy should be performed (A).
The American Cancer Society’s ABCDE criteria and the revised 7-point checklist are the most reliable means of detecting or ruling out malignant melanoma. Each has its strengths and weaknesses, a knowledge of which will increase the accuracy of assessment and minimize chances of misdiagnosis.
In addition to these 2 clinical prediction rules, we examine the evidence on physician’s global assessment of nonmelanoma skin cancers and review the risk factors for the major types of skin cancer. As a result of a comprehensive evidence-based review on the incidence, risk factors, and diagnosis of skin malignancies, we present an algorithm for evaluating skin lesions.
Impact of skin cancer
The incidence of malignant melanoma has increased from 1 in 1500 in 1930 to 1 in 75 for the year 2000.1 Although it is the rarest skin cancer (1% of skin malignancies), it is also the deadliest, accounting for 60% of skin cancer deaths.2
Nonmelanoma skin cancers, which include squamous cell cancers and basal cell cancers, account for one third of all cancers in the United States. Approximately 1 million cases were diagnosed in 1999.3 Deaths from nonmelanoma skin cancers are in steady decline, and the overall 5-year survival rate is high (over 95%).4 Recurrent nonmelanoma skin cancer, however, carries a very poor prognosis, with only a 50% cure rate.5
Treatment of nonmelanoma skin cancer costs over $500 million yearly in the US.4
Primary care physicians help improve prognosis
More persons visit primary care physicians (38.2%) than dermatologists (29.9%) for evaluation of suspicious skin lesions.6 Such lesions are usually benign, but a malignancy must be excluded. A primary care physician can expect to diagnose 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma every year, according to population-based studies.4
Primary care practitioners contribute to a more favorable prognosis. For each additional family physician per 10,000 population, the chances of diagnosing malignant melanoma earlier increase significantly (odds ratio= 1.21, 95% confidence interval, 1.09–1.33, P<.001).7
Primary care physicians who diagnose non-melanoma skin cancers can select therapies that offer maximum efficacy and cost-effectiveness.
Differential diagnosis
According to a study of 1215 biopsies conducted in a primary care population, over 80% of biopsied lesions were benign and included nevi, seborrheic keratoses, cysts, dermatofibromas, fibrous histiocytomas, and polyps or skin tags. Pre-malignant lesions (including actinic keratoses and lentigo maligna) represented 7% of the total. Thirteen percent were malignancies: basal cell carcinomas (73%), followed by squamous cell carcinomas (14%), and malignant melanomas (12%). One metastatic adenomacarcinoma was included in the series (1%) (level of evidence [LOE]: 4).8
The differential diagnosis for basal cell carcinoma includes superficial basal cell carcinoma, pigmented basal cell carcinoma, infiltrating basal cell carcinoma, tricoepithelioma, keloid, molluscum contagiosum, and dermatofibromas.
For squamous cell cancer, the differential includes squamous cell carcinoma, keratoacanthoma, eczema and atopic dermatitis, contact dermatitis, psoriasis, and seborrheic dermatitis.
The differential diagnosis for malignant melanoma includes seborrheic keratosis, traumatized or irritated nevus, pigmented basal cell carcinoma, lentigo, blue nevus, angiokeratoma, traumatic hematoma, venous lake, hemangioma, dermatofibroma, and pigmented actinic keratosis.
Using the history and physical examination
Nearly 70% of melanomas are discovered by patients or their family (LOE: 4).9 Patients may express concern about changed size or appearance of a lesion; associated pain, pruritis, ulceration, or bleeding; location in a cosmetically sensitive area; or worry voiced by a family member. Additionally, a patient may have a family or personal history of skin malignancy, history of skin biopsy, or predisposition to sunburns.
Nurses and physicians identify lesions before a patient does approximately one quarter of the time while examining a patient for an unrelated condition or as part of a comprehensive work-up (LOE: 4).9
Types of skin malignancies
See Photo Rounds, page 219, for images of many types of skin cancer.
Basal cell carcinoma
The patterns of basal cell carcinoma are nodular, superficial, micronodular, infiltrative, morpheaform, and mixed.10 They may be pigmented and are sometimes misdiagnosed as melanoma.11 However, most basal cell carcinomas are typical in appearance and easily diagnosed by visual and tactile inspection.
The most common nodular type is a smooth, skin-colored, indurated, dome-shaped papule with a rolled edge. Other attributes include a pearly appearance, overlying telangiectatic vessels, and a history of bleeding with minor trauma.7,11
Superficial basal cell carcinoma is similar to dermatitis but more often has distinct borders and a bright pink appearance.11 If in doubt about the diagnosis, obtain a tissue sample for pathology.
Squamous cell carcinoma
Squamous cell carcinoma most often is a small, firm, hyperkaratotic nodule sitting atop an inflamed base. It may also be skin-colored and smooth. The history can include itching, pain, and nonhealing after minor trauma.7,11,12 As with basal cell carcinoma, diagnosis is made by tissue pathology.
Malignant melanoma
Malignant melanoma usually appears as a changing or unusual mole with haphazard color variegation, including combinations of brown, black, blue, gray, white, and (rarely) pink. Most melanomas are larger than 5 mm in diameter at time of diagnosis.13
There are 4 main types of malignant melanoma:
- Superficial spreading melanoma accounts for 50% of cases and occurs more frequently in younger adults.
- Nodular melanoma also occurs in younger adults, representing 20% to 25% of cases.
- Lentigo maligna melanoma occurs in older adults and accounts for only 15% of cases.
- Acral or acral-lentiginous melanomas are the least common form (10% of cases). They appear on the palms, soles, and around the first toenail.14
Risk factors for skin malignancies
Factors conferring the highest relative risk for malignant melanoma include:13
- atypical nevus syndrome with a personal and family history of melanoma
- history of a changing mole
- atypical nevus syndrome with just a family history of melanoma
- age greater than or equal to 15 years
- history of dysplastic moles.
Table 1 provides a list of risk factors that should prompt an annual skin survey (LOE: 5).
For nonmelanoma skin cancers, the strongest risk factors ( Table 2) include Caucasian race; age 55 to 75 years; and male sex.2 There is good evidence that a history of nonmelanoma skin cancer confers a 10-fold risk for recurrence (LOE: 2a).15 A distinct risk factor for squamous cell carcinoma is immunosuppression.2 Table 2 also provides a complete list of risk factors for nonmelanoma skin cancer.
Precursor lesions for nonmelanoma skin cancers include Bowen’s disease and erythroplasia of Queyrat (forms of squamous cell carcinoma in situ that will progress if left untreated). Actinic keratoses are common precursor lesions, but their overall annual rate of malignant transformation is only 1 in 400. In the case of SCC, up to 60% of cancers develop from an existing actinic keratosis.2
TABLE 1
Risk factors for malignant melanoma 13
| Risk factors that should prompt an annual skin survey | RR (LOE)* |
|---|---|
| Atypical nevus syndrome with personal and family history of melanoma | 500 (1b) |
| Changing mole | >400 (4) |
| Atypical nevus syndrome with family history of melanoma | 148 (1b)† |
| Age ≥ 15 | 88 (2c) |
| Dysplastic moles | 7–70 (3b) |
| History of melanoma before age 40 | 23 (2b) |
| Large congenital nevus (≥15 cm) | 17 (2b) |
| Caucasian race | 12 (2b) |
| Lentigo maligna | 10 (2c) |
| Atypical nevi | 7–27 (3b) |
| Regular use of tanning bed before age 30 | 7.7‡ (3b) |
| Multiple nevi | 5–12 (3b) |
| Personal history of melanoma | 5–9 (2b) |
| Immunosupression | 4–8 (2b) |
| Family history (first degree) of melanoma | 3–8 (3b) |
| Nonmelanoma skin cancer | 3–5 (3b) |
| Sun sensitivity or tendency to burn | 2–3 (3b) |
| *See page 239 for a description of levels of evidence | |
| †(95% CI, 40–379) | |
| ‡(95% CI, 1–63.6) | |
| RR, relative risk (compared with person without risk factors); | |
| LOE, level of evidence; | |
| CI, confidence interval | |
TABLE 2
Risk factors for nonmelanoma skin cancer
| Significant risk factors | RR | LOE* |
|---|---|---|
| Caucasian race | 70 | 2c |
| Immunosuppression | 5–20 | 2c |
| Previous nonmelanoma skin cancer | 10 | 2a |
| Age 55–75 | 4–8 | 2c |
| Male sex | 2 | 2c |
| Genetic risk factors associated with nonmelonoma skin cancer 3 | ||
| ||
| Chemical exposure risk factors associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| Environmental factors and medical conditions associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| *See page 239 for a description of levels of evidence | ||
| RR, relative risk (compared with person without risk factors); LOE, level of evidence | ||
Clinical prediction rules for skin malignancies
Malignant melanoma
ABCDE criteria. A useful clinical prediction rule for malignant melanoma is the American Cancer Society’s “ABCDE criteria” (Table 3). This rule was validated in 4 dermatology clinics, studying a total of 1118 lesions, although the studies were not homogenous (strength of recommendation [SOR]: A).16-19 Results of the study are summarized in Table 4. The test is normally considered positive if one or more of the criteria are met; however, as more criteria are met, specificity increases while sensitivity decreases.17-19
For lesions lacking any of the ABCDE criteria, 99.8% are something other than melanoma (using a prevalence of 1% found in the US population) (SOR: A). Use caution, however, as this rule will miss amelanotic melanomas, as well as smaller melanomas that are changing in size or have other features suggestive of malignant melanoma.
Conversely, if one of the criteria is met, there is nearly a 1.5% (positive predictive value) probability it is melanoma. Excisional biopsy of the lesion is indicated if good clinical judgment is used and it cannot be identified with certainty as a typical benign lesion (SOR: A). This test thus guides clinicians when making a decision to biopsy, as well as in choosing a biopsy technique.
The ABCDE criteria establish a risk of malignancy if the lesion is 6 mm in diameter or greater. Some evidence, however, suggests that this value should not be used as an absolute cutoff for diagnosing malignant melanoma. A large retrospective study performed in Australia found that 31% of biopsy-confirmed melanomas were less than 6 mm in diameter (LOE: 2b).20
Revised 7-point checklist. Another potentially useful diagnostic test is the revised 7-point checklist developed in the United Kingdom ( Table 5). This test was found to have a high sensitivity, but low specificity ( Table 4) . Therefore, it has a low false-negative rate, and is useful for ruling out the diagnosis of melanoma when negative. However, the test yields a significant number of false positive results, leading to possibly unnecessary biopsies and increased patient anxiety (SOR: B).16,21,22
Note: the described sensitivities and specificities for both tests apply only to malignant melanomas, and their accuracy decreases when including basal cell and squamous cell carcinomas. Also, the 2 tests were scored differently in some of the validation studies, making attempts to generalize problematic.
Studies of physicians’ global assessments to detect melanomas ( Table 4) vary widely for sensitivity (50% to 97%) but are consistent for specificity (96% to 99%) (SOR: B).23-28 Additionally, some studies have shown higher percentages of correct diagnosis of malignant melanoma among dermatologists compared with nondermatologists, but all these studies (except for a small subset of patients in one) used lesion images rather than patient examinations (SOR: B).23,29-33
More importantly, when the choice of correct treatment was evaluated, no statistically significant difference was found between the two groups. Further prospective cohort trials using patient examinations are needed to evaluate dermatologist performance versus nondermatologist performance.
TABLE 3
American Cancer Society's ABCDE criteria
| The test is considered positive if a lesion exhibits 1 or more of the 5 criteria |
| Assymetry—one half of the lesion not identical to the other |
| Border irregularity—lesion has an uneven or ragged border |
| Color variegation—lesion has more than one color (ie, black, blue, pink, red, or white) |
| Diameter—lesion has a diameter greater than 6 mm |
| Elevation or Enlargement—elevation of lesion above skin surface or enlargement by patient report |
 |
TABLE 4
Clinical prediction tests for skin malignancies
| Diagnostic test | Study quality (SOR)* | Sensitivity % (average) | Specificity % (average) | LR+ (1% pretest) | LR– | PV+ | PV– |
|---|---|---|---|---|---|---|---|
| ABCDE criteria (1 criterion positive)16-19 | A | 92–97 (93) | 13–63 (37) | 1.5 | 0.2 | 1.5% | 99.8% |
| Revised 7-point checklist16,21,22 | B | 79–100 (90) | 30–37 (34) | 1.4 | 0.3 | 1.4% | 99.7% |
| Physician global assessments23-28 | B | 50–97 (74) | 96–99 (98) | 37 | 0.3 | 27.2% | 99.7% |
| *See page 239 for a description of strength of recommendation | |||||||
| Note: These calculations are based on simple averages. Statistical homogeneity could not be fully evaluated due to study data limitations. | |||||||
| SOR, strength of recommendation; LR+, positive likelihood ratio; LR–, negative likelihood ratio; PV+, positive predictive value; PV–, negative predictive value | |||||||
TABLE 5
Revised 7-point checklist for assessing risk of melanoma
Suspect melanoma if there are 1 or more major signs:
|
3 or 4 minor signs without a major sign can also indicate a need to biopsy suspicious moles:
|
No validated tool for diagnosis of nonmelanoma skin cancers
A useful diagnostic tool has not yet been validated for nonmelanoma skin cancers. Over 60% of non-melanoma skin cancers occur on the face and neck, and these areas bear careful inspection. Lesions behind the ear, at the medial canthus, and within the nasolabial folds are most easily missed.
How to proceed in assessing lesions
When evaluating skin lesions, remember the gold standard for diagnosis of skin malignancies is a tissue biopsy. If you or your patient has any doubt about the diagnosis, a biopsy should be performed.
To review: Good evidence supports the use the ABCDE criteria or the revised 7-point checklist in determining whether lesions are likely to be malignant melanomas. No similar diagnostic rules exist for basal cell and squamous cell carcinomas. The decision to biopsy these lesions must be based on global assessment and typical characteristics.
Based on this information, we developed an algorithm for evaluating patients at risk for skin malignancies ( Figure) . The first step is to apply the ABCDE criteria and the revised 7-point checklist to identify or rule out possible malignant melanomas. An excisional biopsy should be performed if either test is positive (and the lesion is not clinically benign), or if you or your patient has any doubt.
If neither of these diagnostic tests yields a positive result, the lesion should be classified as typically benign or as having characteristics suggestive of a squamous cell or basal cell carcinoma.
Lesions that have characteristics of squamous cell or basal cell cancer should be biopsied, and benign lesions can be observed and the patient reassured.
FIGURE
Approach to the patient with a skin lesion
Acknowledgments
The authors wish to thank Barbara Zuckerman and Michael Campese, PhD for their assistance in preparation of this manuscript. We also gratefully acknowledge Dr. Richard P. Usatine for preparing the accompanying Photo Rounds.
1. Rigel DS, Friedman RJ, Kopf AW. The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Dermatol 1996;34:839-847.
2. Skin Tumors. In: Sauer GC, Hall JC, eds. A manual of skin diseases Philadelphia, Pa: Lippincott-Raven, 1996;342.-
3. Landis SH, Murray T, Bolden S, Wingo PA. Cancer Statistics, 1999. CA Cancer J Clin 1999;49:8-31.
4. Gloster HM, , Jr. Brodland DG. The epidemiology of skin cancer. Dermatol Surg 1996;22:217-226.
5. Garner KL, Rodney WM. Basal and squamous cell carcinoma. Prim Care 2000;27:447-458.
6. Schappert SM, Nelson C. National Ambulatory Medical Care Survey, 1995-96 Summary. Vital Health Stat 1999;13:1-122.
7. Roetzheim RG, Naazneen P, Van Durme DJ. Increasing supplies of dermatologists and family physicians are associated with earlier stage of melanoma detection. J Am Acad Dermatol 2000;43:211-218.
8. Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. JABFP 1996;9:397-404.
9. Koh HK, Miller DR, Geller AC, Clapp RW, Mercer MB, Lew RA. Who discovers melanoma: patterns from a population-based survey. J Am Acad Dermatol 1992;26:914-919.
10. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol 1995;13:617-620.
11. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc 2000;75:491-500.
12. Alam M, Ratner D. Primary care: cutaneous squamous-cell carcinoma. N Engl J Med 2001;344:975-983.
13. Rhodes AR, Weinstock MA, , Jr, Fitzpatrick B, Mihm MC, Jr, Sober AJ. Risk factors for cutaneous melanoma: a practical method of recognizing predisposed individuals. JAMA 1987;258:3146-3154.
14. Austoker J. Melanoma: prevention and early diagnosis. BMJ 1994;308:1682-1686.
15. Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol 2000;136:1524-1530.
16. Healsmith MF, Bourke JF, Osborne JE, Graham-Brown RAC. An evaluation of the revised seven-point checklist for the early diagnosis of cutaneous melanoma. Br J Dermatol 1994;130:48-50.
17. McGovern TW, Litaker MS. Clinical predictors of malignant pigmented lesions: a comparson of the Glasgow seven-point checklist and the American Cancer Society’s ABCDs of pigmented lesions. J Dermatol Surg Oncol 1992;18:22-26.
18. Benelli C, Roscetti E, Dal Pozzo V, Gasparini G, Cavicchini S. The dermoscopic versus the clinical diagnosis of melanoma. Eur J Dermatol 1999;9:470-476.
19. Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998;197:11-17.
20. Shaw HM, McCarthy WH. Small-diameter malignant melanoma: a common diagnosis in New South Wales, Australia. J Am Acad Dermatol 1992;27:679-682.
21. Du Vivier AWP, Williams HC, Brett JV, Higgins EM. How do malignant melanomas present and does this correlate with the seven-point checklist? Clin Exp Dermatol 1991;16:344-347.
22. Higgins EM, Hall P, Todd P, Murthi R, Du Vivier AWP. The application of the seven-point check-list in the assessment of benign pigmented lesions. Clin Exp Dermatol 1992;17:313-315.
23. Whited JD, Grichnik JM. Does this patient have a more or a melanoma? JAMA 2002;279:696-701.
24. Curley RK, Cook MG, Fallowfield ME, Marsden RA. Accuracy in clinically evaluating pigmented lesions. Br Med J 1989;299:16-18.
25. DeCoste SD, Stern RS. Diagnosis and treatment of nevomelanocytic lesion of the skin: a community-based study. Arch Dermatol 1993;129:57-62.
26. Grin CM, Kopf AW, Welkovich B, Bart RS, Levenstein MJ. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol 1990;126:763-766.
27. Koh HK, Caruso A, Gage I, Geller AC, Prout MN, White H, et al. Evaluation of melanoma/skin cancer screening in Massachusetts: preliminary results. Cancer 1990;65:375-379.
28. McMullan FH, Hubener LF. Malignant melanoma: a statistical review of clinical and histological diagnoses. Arch Dermatol 1956;74:618-619.
29. Cassileth BR, Clark WHJ, Lusk EJ, Frederick BE, Thompson CJ, Walsh WP. How well do physicians recognize melanoma and other problem lesions? J Am Acad Dermatol 1986;14:555-560.
30. Gerbert G, Maurer T, Berger T, Pantilat S, McPhee SJ, Wolff M, et al. Primary care physicians as gatekeepers in managed care. Arch Dermatol 1996;132:1030-1038.
31. McGee R, Elwood M, Sneyd MJ, Williams S, Tilyard M. The recognition and management of melanoma and other skin lesions by general practitioners in New Zealand. N Z Med J 1994;107:287-290.
32. Paine SL, Cockburn J, Noy SM, Marks R. Early detection of skin cancer: knowledge, perceptions, and practices of general practitioners in Victoria. Med J Aust 1994;161:188-195.
33. Ramsay DL, Fox AB. The ability of primary care physicians to recognize the common dermatoses. Arch Dermatol 1981;117:620-622.
- Expect to encounter 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma ever y year.
- There is good evidence for using the American Cancer Society’s ABCDE criteria as a clinical diagnostic test to rule out malignant melanoma (A).
- The revised 7-point checklist has high sensitivity and is therefore useful for ruling out a diagnosis of malignant melanoma. However, its low specificity yields many false-positive results (B).
- The gold standard for diagnosis of skin malignancies is a tissue biopsy. If any doubt exists about the diagnosis, a biopsy should be performed (A).
The American Cancer Society’s ABCDE criteria and the revised 7-point checklist are the most reliable means of detecting or ruling out malignant melanoma. Each has its strengths and weaknesses, a knowledge of which will increase the accuracy of assessment and minimize chances of misdiagnosis.
In addition to these 2 clinical prediction rules, we examine the evidence on physician’s global assessment of nonmelanoma skin cancers and review the risk factors for the major types of skin cancer. As a result of a comprehensive evidence-based review on the incidence, risk factors, and diagnosis of skin malignancies, we present an algorithm for evaluating skin lesions.
Impact of skin cancer
The incidence of malignant melanoma has increased from 1 in 1500 in 1930 to 1 in 75 for the year 2000.1 Although it is the rarest skin cancer (1% of skin malignancies), it is also the deadliest, accounting for 60% of skin cancer deaths.2
Nonmelanoma skin cancers, which include squamous cell cancers and basal cell cancers, account for one third of all cancers in the United States. Approximately 1 million cases were diagnosed in 1999.3 Deaths from nonmelanoma skin cancers are in steady decline, and the overall 5-year survival rate is high (over 95%).4 Recurrent nonmelanoma skin cancer, however, carries a very poor prognosis, with only a 50% cure rate.5
Treatment of nonmelanoma skin cancer costs over $500 million yearly in the US.4
Primary care physicians help improve prognosis
More persons visit primary care physicians (38.2%) than dermatologists (29.9%) for evaluation of suspicious skin lesions.6 Such lesions are usually benign, but a malignancy must be excluded. A primary care physician can expect to diagnose 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma every year, according to population-based studies.4
Primary care practitioners contribute to a more favorable prognosis. For each additional family physician per 10,000 population, the chances of diagnosing malignant melanoma earlier increase significantly (odds ratio= 1.21, 95% confidence interval, 1.09–1.33, P<.001).7
Primary care physicians who diagnose non-melanoma skin cancers can select therapies that offer maximum efficacy and cost-effectiveness.
Differential diagnosis
According to a study of 1215 biopsies conducted in a primary care population, over 80% of biopsied lesions were benign and included nevi, seborrheic keratoses, cysts, dermatofibromas, fibrous histiocytomas, and polyps or skin tags. Pre-malignant lesions (including actinic keratoses and lentigo maligna) represented 7% of the total. Thirteen percent were malignancies: basal cell carcinomas (73%), followed by squamous cell carcinomas (14%), and malignant melanomas (12%). One metastatic adenomacarcinoma was included in the series (1%) (level of evidence [LOE]: 4).8
The differential diagnosis for basal cell carcinoma includes superficial basal cell carcinoma, pigmented basal cell carcinoma, infiltrating basal cell carcinoma, tricoepithelioma, keloid, molluscum contagiosum, and dermatofibromas.
For squamous cell cancer, the differential includes squamous cell carcinoma, keratoacanthoma, eczema and atopic dermatitis, contact dermatitis, psoriasis, and seborrheic dermatitis.
The differential diagnosis for malignant melanoma includes seborrheic keratosis, traumatized or irritated nevus, pigmented basal cell carcinoma, lentigo, blue nevus, angiokeratoma, traumatic hematoma, venous lake, hemangioma, dermatofibroma, and pigmented actinic keratosis.
Using the history and physical examination
Nearly 70% of melanomas are discovered by patients or their family (LOE: 4).9 Patients may express concern about changed size or appearance of a lesion; associated pain, pruritis, ulceration, or bleeding; location in a cosmetically sensitive area; or worry voiced by a family member. Additionally, a patient may have a family or personal history of skin malignancy, history of skin biopsy, or predisposition to sunburns.
Nurses and physicians identify lesions before a patient does approximately one quarter of the time while examining a patient for an unrelated condition or as part of a comprehensive work-up (LOE: 4).9
Types of skin malignancies
See Photo Rounds, page 219, for images of many types of skin cancer.
Basal cell carcinoma
The patterns of basal cell carcinoma are nodular, superficial, micronodular, infiltrative, morpheaform, and mixed.10 They may be pigmented and are sometimes misdiagnosed as melanoma.11 However, most basal cell carcinomas are typical in appearance and easily diagnosed by visual and tactile inspection.
The most common nodular type is a smooth, skin-colored, indurated, dome-shaped papule with a rolled edge. Other attributes include a pearly appearance, overlying telangiectatic vessels, and a history of bleeding with minor trauma.7,11
Superficial basal cell carcinoma is similar to dermatitis but more often has distinct borders and a bright pink appearance.11 If in doubt about the diagnosis, obtain a tissue sample for pathology.
Squamous cell carcinoma
Squamous cell carcinoma most often is a small, firm, hyperkaratotic nodule sitting atop an inflamed base. It may also be skin-colored and smooth. The history can include itching, pain, and nonhealing after minor trauma.7,11,12 As with basal cell carcinoma, diagnosis is made by tissue pathology.
Malignant melanoma
Malignant melanoma usually appears as a changing or unusual mole with haphazard color variegation, including combinations of brown, black, blue, gray, white, and (rarely) pink. Most melanomas are larger than 5 mm in diameter at time of diagnosis.13
There are 4 main types of malignant melanoma:
- Superficial spreading melanoma accounts for 50% of cases and occurs more frequently in younger adults.
- Nodular melanoma also occurs in younger adults, representing 20% to 25% of cases.
- Lentigo maligna melanoma occurs in older adults and accounts for only 15% of cases.
- Acral or acral-lentiginous melanomas are the least common form (10% of cases). They appear on the palms, soles, and around the first toenail.14
Risk factors for skin malignancies
Factors conferring the highest relative risk for malignant melanoma include:13
- atypical nevus syndrome with a personal and family history of melanoma
- history of a changing mole
- atypical nevus syndrome with just a family history of melanoma
- age greater than or equal to 15 years
- history of dysplastic moles.
Table 1 provides a list of risk factors that should prompt an annual skin survey (LOE: 5).
For nonmelanoma skin cancers, the strongest risk factors ( Table 2) include Caucasian race; age 55 to 75 years; and male sex.2 There is good evidence that a history of nonmelanoma skin cancer confers a 10-fold risk for recurrence (LOE: 2a).15 A distinct risk factor for squamous cell carcinoma is immunosuppression.2 Table 2 also provides a complete list of risk factors for nonmelanoma skin cancer.
Precursor lesions for nonmelanoma skin cancers include Bowen’s disease and erythroplasia of Queyrat (forms of squamous cell carcinoma in situ that will progress if left untreated). Actinic keratoses are common precursor lesions, but their overall annual rate of malignant transformation is only 1 in 400. In the case of SCC, up to 60% of cancers develop from an existing actinic keratosis.2
TABLE 1
Risk factors for malignant melanoma 13
| Risk factors that should prompt an annual skin survey | RR (LOE)* |
|---|---|
| Atypical nevus syndrome with personal and family history of melanoma | 500 (1b) |
| Changing mole | >400 (4) |
| Atypical nevus syndrome with family history of melanoma | 148 (1b)† |
| Age ≥ 15 | 88 (2c) |
| Dysplastic moles | 7–70 (3b) |
| History of melanoma before age 40 | 23 (2b) |
| Large congenital nevus (≥15 cm) | 17 (2b) |
| Caucasian race | 12 (2b) |
| Lentigo maligna | 10 (2c) |
| Atypical nevi | 7–27 (3b) |
| Regular use of tanning bed before age 30 | 7.7‡ (3b) |
| Multiple nevi | 5–12 (3b) |
| Personal history of melanoma | 5–9 (2b) |
| Immunosupression | 4–8 (2b) |
| Family history (first degree) of melanoma | 3–8 (3b) |
| Nonmelanoma skin cancer | 3–5 (3b) |
| Sun sensitivity or tendency to burn | 2–3 (3b) |
| *See page 239 for a description of levels of evidence | |
| †(95% CI, 40–379) | |
| ‡(95% CI, 1–63.6) | |
| RR, relative risk (compared with person without risk factors); | |
| LOE, level of evidence; | |
| CI, confidence interval | |
TABLE 2
Risk factors for nonmelanoma skin cancer
| Significant risk factors | RR | LOE* |
|---|---|---|
| Caucasian race | 70 | 2c |
| Immunosuppression | 5–20 | 2c |
| Previous nonmelanoma skin cancer | 10 | 2a |
| Age 55–75 | 4–8 | 2c |
| Male sex | 2 | 2c |
| Genetic risk factors associated with nonmelonoma skin cancer 3 | ||
| ||
| Chemical exposure risk factors associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| Environmental factors and medical conditions associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| *See page 239 for a description of levels of evidence | ||
| RR, relative risk (compared with person without risk factors); LOE, level of evidence | ||
Clinical prediction rules for skin malignancies
Malignant melanoma
ABCDE criteria. A useful clinical prediction rule for malignant melanoma is the American Cancer Society’s “ABCDE criteria” (Table 3). This rule was validated in 4 dermatology clinics, studying a total of 1118 lesions, although the studies were not homogenous (strength of recommendation [SOR]: A).16-19 Results of the study are summarized in Table 4. The test is normally considered positive if one or more of the criteria are met; however, as more criteria are met, specificity increases while sensitivity decreases.17-19
For lesions lacking any of the ABCDE criteria, 99.8% are something other than melanoma (using a prevalence of 1% found in the US population) (SOR: A). Use caution, however, as this rule will miss amelanotic melanomas, as well as smaller melanomas that are changing in size or have other features suggestive of malignant melanoma.
Conversely, if one of the criteria is met, there is nearly a 1.5% (positive predictive value) probability it is melanoma. Excisional biopsy of the lesion is indicated if good clinical judgment is used and it cannot be identified with certainty as a typical benign lesion (SOR: A). This test thus guides clinicians when making a decision to biopsy, as well as in choosing a biopsy technique.
The ABCDE criteria establish a risk of malignancy if the lesion is 6 mm in diameter or greater. Some evidence, however, suggests that this value should not be used as an absolute cutoff for diagnosing malignant melanoma. A large retrospective study performed in Australia found that 31% of biopsy-confirmed melanomas were less than 6 mm in diameter (LOE: 2b).20
Revised 7-point checklist. Another potentially useful diagnostic test is the revised 7-point checklist developed in the United Kingdom ( Table 5). This test was found to have a high sensitivity, but low specificity ( Table 4) . Therefore, it has a low false-negative rate, and is useful for ruling out the diagnosis of melanoma when negative. However, the test yields a significant number of false positive results, leading to possibly unnecessary biopsies and increased patient anxiety (SOR: B).16,21,22
Note: the described sensitivities and specificities for both tests apply only to malignant melanomas, and their accuracy decreases when including basal cell and squamous cell carcinomas. Also, the 2 tests were scored differently in some of the validation studies, making attempts to generalize problematic.
Studies of physicians’ global assessments to detect melanomas ( Table 4) vary widely for sensitivity (50% to 97%) but are consistent for specificity (96% to 99%) (SOR: B).23-28 Additionally, some studies have shown higher percentages of correct diagnosis of malignant melanoma among dermatologists compared with nondermatologists, but all these studies (except for a small subset of patients in one) used lesion images rather than patient examinations (SOR: B).23,29-33
More importantly, when the choice of correct treatment was evaluated, no statistically significant difference was found between the two groups. Further prospective cohort trials using patient examinations are needed to evaluate dermatologist performance versus nondermatologist performance.
TABLE 3
American Cancer Society's ABCDE criteria
| The test is considered positive if a lesion exhibits 1 or more of the 5 criteria |
| Assymetry—one half of the lesion not identical to the other |
| Border irregularity—lesion has an uneven or ragged border |
| Color variegation—lesion has more than one color (ie, black, blue, pink, red, or white) |
| Diameter—lesion has a diameter greater than 6 mm |
| Elevation or Enlargement—elevation of lesion above skin surface or enlargement by patient report |
|
TABLE 4
Clinical prediction tests for skin malignancies
| Diagnostic test | Study quality (SOR)* | Sensitivity % (average) | Specificity % (average) | LR+ (1% pretest) | LR– | PV+ | PV– |
|---|---|---|---|---|---|---|---|
| ABCDE criteria (1 criterion positive)16-19 | A | 92–97 (93) | 13–63 (37) | 1.5 | 0.2 | 1.5% | 99.8% |
| Revised 7-point checklist16,21,22 | B | 79–100 (90) | 30–37 (34) | 1.4 | 0.3 | 1.4% | 99.7% |
| Physician global assessments23-28 | B | 50–97 (74) | 96–99 (98) | 37 | 0.3 | 27.2% | 99.7% |
| *See page 239 for a description of strength of recommendation | |||||||
| Note: These calculations are based on simple averages. Statistical homogeneity could not be fully evaluated due to study data limitations. | |||||||
| SOR, strength of recommendation; LR+, positive likelihood ratio; LR–, negative likelihood ratio; PV+, positive predictive value; PV–, negative predictive value | |||||||
TABLE 5
Revised 7-point checklist for assessing risk of melanoma
Suspect melanoma if there are 1 or more major signs:
|
3 or 4 minor signs without a major sign can also indicate a need to biopsy suspicious moles:
|
No validated tool for diagnosis of nonmelanoma skin cancers
A useful diagnostic tool has not yet been validated for nonmelanoma skin cancers. Over 60% of non-melanoma skin cancers occur on the face and neck, and these areas bear careful inspection. Lesions behind the ear, at the medial canthus, and within the nasolabial folds are most easily missed.
How to proceed in assessing lesions
When evaluating skin lesions, remember the gold standard for diagnosis of skin malignancies is a tissue biopsy. If you or your patient has any doubt about the diagnosis, a biopsy should be performed.
To review: Good evidence supports the use the ABCDE criteria or the revised 7-point checklist in determining whether lesions are likely to be malignant melanomas. No similar diagnostic rules exist for basal cell and squamous cell carcinomas. The decision to biopsy these lesions must be based on global assessment and typical characteristics.
Based on this information, we developed an algorithm for evaluating patients at risk for skin malignancies ( Figure) . The first step is to apply the ABCDE criteria and the revised 7-point checklist to identify or rule out possible malignant melanomas. An excisional biopsy should be performed if either test is positive (and the lesion is not clinically benign), or if you or your patient has any doubt.
If neither of these diagnostic tests yields a positive result, the lesion should be classified as typically benign or as having characteristics suggestive of a squamous cell or basal cell carcinoma.
Lesions that have characteristics of squamous cell or basal cell cancer should be biopsied, and benign lesions can be observed and the patient reassured.
FIGURE
Approach to the patient with a skin lesion
Acknowledgments
The authors wish to thank Barbara Zuckerman and Michael Campese, PhD for their assistance in preparation of this manuscript. We also gratefully acknowledge Dr. Richard P. Usatine for preparing the accompanying Photo Rounds.
- Expect to encounter 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma ever y year.
- There is good evidence for using the American Cancer Society’s ABCDE criteria as a clinical diagnostic test to rule out malignant melanoma (A).
- The revised 7-point checklist has high sensitivity and is therefore useful for ruling out a diagnosis of malignant melanoma. However, its low specificity yields many false-positive results (B).
- The gold standard for diagnosis of skin malignancies is a tissue biopsy. If any doubt exists about the diagnosis, a biopsy should be performed (A).
The American Cancer Society’s ABCDE criteria and the revised 7-point checklist are the most reliable means of detecting or ruling out malignant melanoma. Each has its strengths and weaknesses, a knowledge of which will increase the accuracy of assessment and minimize chances of misdiagnosis.
In addition to these 2 clinical prediction rules, we examine the evidence on physician’s global assessment of nonmelanoma skin cancers and review the risk factors for the major types of skin cancer. As a result of a comprehensive evidence-based review on the incidence, risk factors, and diagnosis of skin malignancies, we present an algorithm for evaluating skin lesions.
Impact of skin cancer
The incidence of malignant melanoma has increased from 1 in 1500 in 1930 to 1 in 75 for the year 2000.1 Although it is the rarest skin cancer (1% of skin malignancies), it is also the deadliest, accounting for 60% of skin cancer deaths.2
Nonmelanoma skin cancers, which include squamous cell cancers and basal cell cancers, account for one third of all cancers in the United States. Approximately 1 million cases were diagnosed in 1999.3 Deaths from nonmelanoma skin cancers are in steady decline, and the overall 5-year survival rate is high (over 95%).4 Recurrent nonmelanoma skin cancer, however, carries a very poor prognosis, with only a 50% cure rate.5
Treatment of nonmelanoma skin cancer costs over $500 million yearly in the US.4
Primary care physicians help improve prognosis
More persons visit primary care physicians (38.2%) than dermatologists (29.9%) for evaluation of suspicious skin lesions.6 Such lesions are usually benign, but a malignancy must be excluded. A primary care physician can expect to diagnose 6 to 7 cases of basal cell cancer, 1 to 2 cases of squamous cell cancer, and approximately 1 case of melanoma every year, according to population-based studies.4
Primary care practitioners contribute to a more favorable prognosis. For each additional family physician per 10,000 population, the chances of diagnosing malignant melanoma earlier increase significantly (odds ratio= 1.21, 95% confidence interval, 1.09–1.33, P<.001).7
Primary care physicians who diagnose non-melanoma skin cancers can select therapies that offer maximum efficacy and cost-effectiveness.
Differential diagnosis
According to a study of 1215 biopsies conducted in a primary care population, over 80% of biopsied lesions were benign and included nevi, seborrheic keratoses, cysts, dermatofibromas, fibrous histiocytomas, and polyps or skin tags. Pre-malignant lesions (including actinic keratoses and lentigo maligna) represented 7% of the total. Thirteen percent were malignancies: basal cell carcinomas (73%), followed by squamous cell carcinomas (14%), and malignant melanomas (12%). One metastatic adenomacarcinoma was included in the series (1%) (level of evidence [LOE]: 4).8
The differential diagnosis for basal cell carcinoma includes superficial basal cell carcinoma, pigmented basal cell carcinoma, infiltrating basal cell carcinoma, tricoepithelioma, keloid, molluscum contagiosum, and dermatofibromas.
For squamous cell cancer, the differential includes squamous cell carcinoma, keratoacanthoma, eczema and atopic dermatitis, contact dermatitis, psoriasis, and seborrheic dermatitis.
The differential diagnosis for malignant melanoma includes seborrheic keratosis, traumatized or irritated nevus, pigmented basal cell carcinoma, lentigo, blue nevus, angiokeratoma, traumatic hematoma, venous lake, hemangioma, dermatofibroma, and pigmented actinic keratosis.
Using the history and physical examination
Nearly 70% of melanomas are discovered by patients or their family (LOE: 4).9 Patients may express concern about changed size or appearance of a lesion; associated pain, pruritis, ulceration, or bleeding; location in a cosmetically sensitive area; or worry voiced by a family member. Additionally, a patient may have a family or personal history of skin malignancy, history of skin biopsy, or predisposition to sunburns.
Nurses and physicians identify lesions before a patient does approximately one quarter of the time while examining a patient for an unrelated condition or as part of a comprehensive work-up (LOE: 4).9
Types of skin malignancies
See Photo Rounds, page 219, for images of many types of skin cancer.
Basal cell carcinoma
The patterns of basal cell carcinoma are nodular, superficial, micronodular, infiltrative, morpheaform, and mixed.10 They may be pigmented and are sometimes misdiagnosed as melanoma.11 However, most basal cell carcinomas are typical in appearance and easily diagnosed by visual and tactile inspection.
The most common nodular type is a smooth, skin-colored, indurated, dome-shaped papule with a rolled edge. Other attributes include a pearly appearance, overlying telangiectatic vessels, and a history of bleeding with minor trauma.7,11
Superficial basal cell carcinoma is similar to dermatitis but more often has distinct borders and a bright pink appearance.11 If in doubt about the diagnosis, obtain a tissue sample for pathology.
Squamous cell carcinoma
Squamous cell carcinoma most often is a small, firm, hyperkaratotic nodule sitting atop an inflamed base. It may also be skin-colored and smooth. The history can include itching, pain, and nonhealing after minor trauma.7,11,12 As with basal cell carcinoma, diagnosis is made by tissue pathology.
Malignant melanoma
Malignant melanoma usually appears as a changing or unusual mole with haphazard color variegation, including combinations of brown, black, blue, gray, white, and (rarely) pink. Most melanomas are larger than 5 mm in diameter at time of diagnosis.13
There are 4 main types of malignant melanoma:
- Superficial spreading melanoma accounts for 50% of cases and occurs more frequently in younger adults.
- Nodular melanoma also occurs in younger adults, representing 20% to 25% of cases.
- Lentigo maligna melanoma occurs in older adults and accounts for only 15% of cases.
- Acral or acral-lentiginous melanomas are the least common form (10% of cases). They appear on the palms, soles, and around the first toenail.14
Risk factors for skin malignancies
Factors conferring the highest relative risk for malignant melanoma include:13
- atypical nevus syndrome with a personal and family history of melanoma
- history of a changing mole
- atypical nevus syndrome with just a family history of melanoma
- age greater than or equal to 15 years
- history of dysplastic moles.
Table 1 provides a list of risk factors that should prompt an annual skin survey (LOE: 5).
For nonmelanoma skin cancers, the strongest risk factors ( Table 2) include Caucasian race; age 55 to 75 years; and male sex.2 There is good evidence that a history of nonmelanoma skin cancer confers a 10-fold risk for recurrence (LOE: 2a).15 A distinct risk factor for squamous cell carcinoma is immunosuppression.2 Table 2 also provides a complete list of risk factors for nonmelanoma skin cancer.
Precursor lesions for nonmelanoma skin cancers include Bowen’s disease and erythroplasia of Queyrat (forms of squamous cell carcinoma in situ that will progress if left untreated). Actinic keratoses are common precursor lesions, but their overall annual rate of malignant transformation is only 1 in 400. In the case of SCC, up to 60% of cancers develop from an existing actinic keratosis.2
TABLE 1
Risk factors for malignant melanoma 13
| Risk factors that should prompt an annual skin survey | RR (LOE)* |
|---|---|
| Atypical nevus syndrome with personal and family history of melanoma | 500 (1b) |
| Changing mole | >400 (4) |
| Atypical nevus syndrome with family history of melanoma | 148 (1b)† |
| Age ≥ 15 | 88 (2c) |
| Dysplastic moles | 7–70 (3b) |
| History of melanoma before age 40 | 23 (2b) |
| Large congenital nevus (≥15 cm) | 17 (2b) |
| Caucasian race | 12 (2b) |
| Lentigo maligna | 10 (2c) |
| Atypical nevi | 7–27 (3b) |
| Regular use of tanning bed before age 30 | 7.7‡ (3b) |
| Multiple nevi | 5–12 (3b) |
| Personal history of melanoma | 5–9 (2b) |
| Immunosupression | 4–8 (2b) |
| Family history (first degree) of melanoma | 3–8 (3b) |
| Nonmelanoma skin cancer | 3–5 (3b) |
| Sun sensitivity or tendency to burn | 2–3 (3b) |
| *See page 239 for a description of levels of evidence | |
| †(95% CI, 40–379) | |
| ‡(95% CI, 1–63.6) | |
| RR, relative risk (compared with person without risk factors); | |
| LOE, level of evidence; | |
| CI, confidence interval | |
TABLE 2
Risk factors for nonmelanoma skin cancer
| Significant risk factors | RR | LOE* |
|---|---|---|
| Caucasian race | 70 | 2c |
| Immunosuppression | 5–20 | 2c |
| Previous nonmelanoma skin cancer | 10 | 2a |
| Age 55–75 | 4–8 | 2c |
| Male sex | 2 | 2c |
| Genetic risk factors associated with nonmelonoma skin cancer 3 | ||
| ||
| Chemical exposure risk factors associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| Environmental factors and medical conditions associated with nonmelonoma skin cancer (particularly squamous cell carcinoma) 3 | ||
| ||
| *See page 239 for a description of levels of evidence | ||
| RR, relative risk (compared with person without risk factors); LOE, level of evidence | ||
Clinical prediction rules for skin malignancies
Malignant melanoma
ABCDE criteria. A useful clinical prediction rule for malignant melanoma is the American Cancer Society’s “ABCDE criteria” (Table 3). This rule was validated in 4 dermatology clinics, studying a total of 1118 lesions, although the studies were not homogenous (strength of recommendation [SOR]: A).16-19 Results of the study are summarized in Table 4. The test is normally considered positive if one or more of the criteria are met; however, as more criteria are met, specificity increases while sensitivity decreases.17-19
For lesions lacking any of the ABCDE criteria, 99.8% are something other than melanoma (using a prevalence of 1% found in the US population) (SOR: A). Use caution, however, as this rule will miss amelanotic melanomas, as well as smaller melanomas that are changing in size or have other features suggestive of malignant melanoma.
Conversely, if one of the criteria is met, there is nearly a 1.5% (positive predictive value) probability it is melanoma. Excisional biopsy of the lesion is indicated if good clinical judgment is used and it cannot be identified with certainty as a typical benign lesion (SOR: A). This test thus guides clinicians when making a decision to biopsy, as well as in choosing a biopsy technique.
The ABCDE criteria establish a risk of malignancy if the lesion is 6 mm in diameter or greater. Some evidence, however, suggests that this value should not be used as an absolute cutoff for diagnosing malignant melanoma. A large retrospective study performed in Australia found that 31% of biopsy-confirmed melanomas were less than 6 mm in diameter (LOE: 2b).20
Revised 7-point checklist. Another potentially useful diagnostic test is the revised 7-point checklist developed in the United Kingdom ( Table 5). This test was found to have a high sensitivity, but low specificity ( Table 4) . Therefore, it has a low false-negative rate, and is useful for ruling out the diagnosis of melanoma when negative. However, the test yields a significant number of false positive results, leading to possibly unnecessary biopsies and increased patient anxiety (SOR: B).16,21,22
Note: the described sensitivities and specificities for both tests apply only to malignant melanomas, and their accuracy decreases when including basal cell and squamous cell carcinomas. Also, the 2 tests were scored differently in some of the validation studies, making attempts to generalize problematic.
Studies of physicians’ global assessments to detect melanomas ( Table 4) vary widely for sensitivity (50% to 97%) but are consistent for specificity (96% to 99%) (SOR: B).23-28 Additionally, some studies have shown higher percentages of correct diagnosis of malignant melanoma among dermatologists compared with nondermatologists, but all these studies (except for a small subset of patients in one) used lesion images rather than patient examinations (SOR: B).23,29-33
More importantly, when the choice of correct treatment was evaluated, no statistically significant difference was found between the two groups. Further prospective cohort trials using patient examinations are needed to evaluate dermatologist performance versus nondermatologist performance.
TABLE 3
American Cancer Society's ABCDE criteria
| The test is considered positive if a lesion exhibits 1 or more of the 5 criteria |
| Assymetry—one half of the lesion not identical to the other |
| Border irregularity—lesion has an uneven or ragged border |
| Color variegation—lesion has more than one color (ie, black, blue, pink, red, or white) |
| Diameter—lesion has a diameter greater than 6 mm |
| Elevation or Enlargement—elevation of lesion above skin surface or enlargement by patient report |
|
TABLE 4
Clinical prediction tests for skin malignancies
| Diagnostic test | Study quality (SOR)* | Sensitivity % (average) | Specificity % (average) | LR+ (1% pretest) | LR– | PV+ | PV– |
|---|---|---|---|---|---|---|---|
| ABCDE criteria (1 criterion positive)16-19 | A | 92–97 (93) | 13–63 (37) | 1.5 | 0.2 | 1.5% | 99.8% |
| Revised 7-point checklist16,21,22 | B | 79–100 (90) | 30–37 (34) | 1.4 | 0.3 | 1.4% | 99.7% |
| Physician global assessments23-28 | B | 50–97 (74) | 96–99 (98) | 37 | 0.3 | 27.2% | 99.7% |
| *See page 239 for a description of strength of recommendation | |||||||
| Note: These calculations are based on simple averages. Statistical homogeneity could not be fully evaluated due to study data limitations. | |||||||
| SOR, strength of recommendation; LR+, positive likelihood ratio; LR–, negative likelihood ratio; PV+, positive predictive value; PV–, negative predictive value | |||||||
TABLE 5
Revised 7-point checklist for assessing risk of melanoma
Suspect melanoma if there are 1 or more major signs:
|
3 or 4 minor signs without a major sign can also indicate a need to biopsy suspicious moles:
|
No validated tool for diagnosis of nonmelanoma skin cancers
A useful diagnostic tool has not yet been validated for nonmelanoma skin cancers. Over 60% of non-melanoma skin cancers occur on the face and neck, and these areas bear careful inspection. Lesions behind the ear, at the medial canthus, and within the nasolabial folds are most easily missed.
How to proceed in assessing lesions
When evaluating skin lesions, remember the gold standard for diagnosis of skin malignancies is a tissue biopsy. If you or your patient has any doubt about the diagnosis, a biopsy should be performed.
To review: Good evidence supports the use the ABCDE criteria or the revised 7-point checklist in determining whether lesions are likely to be malignant melanomas. No similar diagnostic rules exist for basal cell and squamous cell carcinomas. The decision to biopsy these lesions must be based on global assessment and typical characteristics.
Based on this information, we developed an algorithm for evaluating patients at risk for skin malignancies ( Figure) . The first step is to apply the ABCDE criteria and the revised 7-point checklist to identify or rule out possible malignant melanomas. An excisional biopsy should be performed if either test is positive (and the lesion is not clinically benign), or if you or your patient has any doubt.
If neither of these diagnostic tests yields a positive result, the lesion should be classified as typically benign or as having characteristics suggestive of a squamous cell or basal cell carcinoma.
Lesions that have characteristics of squamous cell or basal cell cancer should be biopsied, and benign lesions can be observed and the patient reassured.
FIGURE
Approach to the patient with a skin lesion
Acknowledgments
The authors wish to thank Barbara Zuckerman and Michael Campese, PhD for their assistance in preparation of this manuscript. We also gratefully acknowledge Dr. Richard P. Usatine for preparing the accompanying Photo Rounds.
1. Rigel DS, Friedman RJ, Kopf AW. The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Dermatol 1996;34:839-847.
2. Skin Tumors. In: Sauer GC, Hall JC, eds. A manual of skin diseases Philadelphia, Pa: Lippincott-Raven, 1996;342.-
3. Landis SH, Murray T, Bolden S, Wingo PA. Cancer Statistics, 1999. CA Cancer J Clin 1999;49:8-31.
4. Gloster HM, , Jr. Brodland DG. The epidemiology of skin cancer. Dermatol Surg 1996;22:217-226.
5. Garner KL, Rodney WM. Basal and squamous cell carcinoma. Prim Care 2000;27:447-458.
6. Schappert SM, Nelson C. National Ambulatory Medical Care Survey, 1995-96 Summary. Vital Health Stat 1999;13:1-122.
7. Roetzheim RG, Naazneen P, Van Durme DJ. Increasing supplies of dermatologists and family physicians are associated with earlier stage of melanoma detection. J Am Acad Dermatol 2000;43:211-218.
8. Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. JABFP 1996;9:397-404.
9. Koh HK, Miller DR, Geller AC, Clapp RW, Mercer MB, Lew RA. Who discovers melanoma: patterns from a population-based survey. J Am Acad Dermatol 1992;26:914-919.
10. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol 1995;13:617-620.
11. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc 2000;75:491-500.
12. Alam M, Ratner D. Primary care: cutaneous squamous-cell carcinoma. N Engl J Med 2001;344:975-983.
13. Rhodes AR, Weinstock MA, , Jr, Fitzpatrick B, Mihm MC, Jr, Sober AJ. Risk factors for cutaneous melanoma: a practical method of recognizing predisposed individuals. JAMA 1987;258:3146-3154.
14. Austoker J. Melanoma: prevention and early diagnosis. BMJ 1994;308:1682-1686.
15. Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol 2000;136:1524-1530.
16. Healsmith MF, Bourke JF, Osborne JE, Graham-Brown RAC. An evaluation of the revised seven-point checklist for the early diagnosis of cutaneous melanoma. Br J Dermatol 1994;130:48-50.
17. McGovern TW, Litaker MS. Clinical predictors of malignant pigmented lesions: a comparson of the Glasgow seven-point checklist and the American Cancer Society’s ABCDs of pigmented lesions. J Dermatol Surg Oncol 1992;18:22-26.
18. Benelli C, Roscetti E, Dal Pozzo V, Gasparini G, Cavicchini S. The dermoscopic versus the clinical diagnosis of melanoma. Eur J Dermatol 1999;9:470-476.
19. Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998;197:11-17.
20. Shaw HM, McCarthy WH. Small-diameter malignant melanoma: a common diagnosis in New South Wales, Australia. J Am Acad Dermatol 1992;27:679-682.
21. Du Vivier AWP, Williams HC, Brett JV, Higgins EM. How do malignant melanomas present and does this correlate with the seven-point checklist? Clin Exp Dermatol 1991;16:344-347.
22. Higgins EM, Hall P, Todd P, Murthi R, Du Vivier AWP. The application of the seven-point check-list in the assessment of benign pigmented lesions. Clin Exp Dermatol 1992;17:313-315.
23. Whited JD, Grichnik JM. Does this patient have a more or a melanoma? JAMA 2002;279:696-701.
24. Curley RK, Cook MG, Fallowfield ME, Marsden RA. Accuracy in clinically evaluating pigmented lesions. Br Med J 1989;299:16-18.
25. DeCoste SD, Stern RS. Diagnosis and treatment of nevomelanocytic lesion of the skin: a community-based study. Arch Dermatol 1993;129:57-62.
26. Grin CM, Kopf AW, Welkovich B, Bart RS, Levenstein MJ. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol 1990;126:763-766.
27. Koh HK, Caruso A, Gage I, Geller AC, Prout MN, White H, et al. Evaluation of melanoma/skin cancer screening in Massachusetts: preliminary results. Cancer 1990;65:375-379.
28. McMullan FH, Hubener LF. Malignant melanoma: a statistical review of clinical and histological diagnoses. Arch Dermatol 1956;74:618-619.
29. Cassileth BR, Clark WHJ, Lusk EJ, Frederick BE, Thompson CJ, Walsh WP. How well do physicians recognize melanoma and other problem lesions? J Am Acad Dermatol 1986;14:555-560.
30. Gerbert G, Maurer T, Berger T, Pantilat S, McPhee SJ, Wolff M, et al. Primary care physicians as gatekeepers in managed care. Arch Dermatol 1996;132:1030-1038.
31. McGee R, Elwood M, Sneyd MJ, Williams S, Tilyard M. The recognition and management of melanoma and other skin lesions by general practitioners in New Zealand. N Z Med J 1994;107:287-290.
32. Paine SL, Cockburn J, Noy SM, Marks R. Early detection of skin cancer: knowledge, perceptions, and practices of general practitioners in Victoria. Med J Aust 1994;161:188-195.
33. Ramsay DL, Fox AB. The ability of primary care physicians to recognize the common dermatoses. Arch Dermatol 1981;117:620-622.
1. Rigel DS, Friedman RJ, Kopf AW. The incidence of malignant melanoma in the United States: issues as we approach the 21st century. J Am Acad Dermatol 1996;34:839-847.
2. Skin Tumors. In: Sauer GC, Hall JC, eds. A manual of skin diseases Philadelphia, Pa: Lippincott-Raven, 1996;342.-
3. Landis SH, Murray T, Bolden S, Wingo PA. Cancer Statistics, 1999. CA Cancer J Clin 1999;49:8-31.
4. Gloster HM, , Jr. Brodland DG. The epidemiology of skin cancer. Dermatol Surg 1996;22:217-226.
5. Garner KL, Rodney WM. Basal and squamous cell carcinoma. Prim Care 2000;27:447-458.
6. Schappert SM, Nelson C. National Ambulatory Medical Care Survey, 1995-96 Summary. Vital Health Stat 1999;13:1-122.
7. Roetzheim RG, Naazneen P, Van Durme DJ. Increasing supplies of dermatologists and family physicians are associated with earlier stage of melanoma detection. J Am Acad Dermatol 2000;43:211-218.
8. Jones TP, Boiko PE, Piepkorn MW. Skin biopsy indications in primary care practice: a population-based study. JABFP 1996;9:397-404.
9. Koh HK, Miller DR, Geller AC, Clapp RW, Mercer MB, Lew RA. Who discovers melanoma: patterns from a population-based survey. J Am Acad Dermatol 1992;26:914-919.
10. Rowe DE. Comparison of treatment modalities for basal cell carcinoma. Clin Dermatol 1995;13:617-620.
11. Bruce AJ, Brodland DG. Overview of skin cancer detection and prevention for the primary care physician. Mayo Clin Proc 2000;75:491-500.
12. Alam M, Ratner D. Primary care: cutaneous squamous-cell carcinoma. N Engl J Med 2001;344:975-983.
13. Rhodes AR, Weinstock MA, , Jr, Fitzpatrick B, Mihm MC, Jr, Sober AJ. Risk factors for cutaneous melanoma: a practical method of recognizing predisposed individuals. JAMA 1987;258:3146-3154.
14. Austoker J. Melanoma: prevention and early diagnosis. BMJ 1994;308:1682-1686.
15. Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Arch Dermatol 2000;136:1524-1530.
16. Healsmith MF, Bourke JF, Osborne JE, Graham-Brown RAC. An evaluation of the revised seven-point checklist for the early diagnosis of cutaneous melanoma. Br J Dermatol 1994;130:48-50.
17. McGovern TW, Litaker MS. Clinical predictors of malignant pigmented lesions: a comparson of the Glasgow seven-point checklist and the American Cancer Society’s ABCDs of pigmented lesions. J Dermatol Surg Oncol 1992;18:22-26.
18. Benelli C, Roscetti E, Dal Pozzo V, Gasparini G, Cavicchini S. The dermoscopic versus the clinical diagnosis of melanoma. Eur J Dermatol 1999;9:470-476.
19. Thomas L, Tranchand P, Berard F, Secchi T, Colin C, Moulin G. Semiological value of ABCDE criteria in the diagnosis of cutaneous pigmented tumors. Dermatology 1998;197:11-17.
20. Shaw HM, McCarthy WH. Small-diameter malignant melanoma: a common diagnosis in New South Wales, Australia. J Am Acad Dermatol 1992;27:679-682.
21. Du Vivier AWP, Williams HC, Brett JV, Higgins EM. How do malignant melanomas present and does this correlate with the seven-point checklist? Clin Exp Dermatol 1991;16:344-347.
22. Higgins EM, Hall P, Todd P, Murthi R, Du Vivier AWP. The application of the seven-point check-list in the assessment of benign pigmented lesions. Clin Exp Dermatol 1992;17:313-315.
23. Whited JD, Grichnik JM. Does this patient have a more or a melanoma? JAMA 2002;279:696-701.
24. Curley RK, Cook MG, Fallowfield ME, Marsden RA. Accuracy in clinically evaluating pigmented lesions. Br Med J 1989;299:16-18.
25. DeCoste SD, Stern RS. Diagnosis and treatment of nevomelanocytic lesion of the skin: a community-based study. Arch Dermatol 1993;129:57-62.
26. Grin CM, Kopf AW, Welkovich B, Bart RS, Levenstein MJ. Accuracy in the clinical diagnosis of malignant melanoma. Arch Dermatol 1990;126:763-766.
27. Koh HK, Caruso A, Gage I, Geller AC, Prout MN, White H, et al. Evaluation of melanoma/skin cancer screening in Massachusetts: preliminary results. Cancer 1990;65:375-379.
28. McMullan FH, Hubener LF. Malignant melanoma: a statistical review of clinical and histological diagnoses. Arch Dermatol 1956;74:618-619.
29. Cassileth BR, Clark WHJ, Lusk EJ, Frederick BE, Thompson CJ, Walsh WP. How well do physicians recognize melanoma and other problem lesions? J Am Acad Dermatol 1986;14:555-560.
30. Gerbert G, Maurer T, Berger T, Pantilat S, McPhee SJ, Wolff M, et al. Primary care physicians as gatekeepers in managed care. Arch Dermatol 1996;132:1030-1038.
31. McGee R, Elwood M, Sneyd MJ, Williams S, Tilyard M. The recognition and management of melanoma and other skin lesions by general practitioners in New Zealand. N Z Med J 1994;107:287-290.
32. Paine SL, Cockburn J, Noy SM, Marks R. Early detection of skin cancer: knowledge, perceptions, and practices of general practitioners in Victoria. Med J Aust 1994;161:188-195.
33. Ramsay DL, Fox AB. The ability of primary care physicians to recognize the common dermatoses. Arch Dermatol 1981;117:620-622.
Achieving the best outcome in treatment of depression
- Combined treatment with psychotherapy or psychiatric consult and drug therapy has shown better response in several studies than either therapy alone (A).
- Although not proven by clinical trials, selecting a medication by matching its side-effect profile to patient characteristics is supported by case reports and likely enhances compliance.
- Patients who do not improve with initial therapy often benefit from being switched to another class of antidepressants (A), or having a drug from another class added to their therapy (B).
You are more likely to see depression in your practice than any other disorder except hypertension.1 Given the prevalence of depression* and the variability of its clinical symptoms and comorbidities, how do you determine the optimal therapy for a given patient?
A sobering thought: nearly half of all patients stop taking their antidepressant prescription medication within the first month of treatment.1 We discuss the critical factors you can address to help patients stick with treatment and achieve the best outcome.
Therapeutic Options
Pharmacotherapy
Antidepressants are thought to exert their therapeutic and adverse effects through 3 chemical monamine neurotransmission systems; by increasing levels of norepinephrine, serotonin, or dopamine in the synapse; and by resultant secondary changes in presynaptic and postsynaptic receptor physiology.3,8,9 Newer medications—such as selective serotonin reuptake inhibitors (SSRIs)—have simpler dose schedules, different (and for some patients more favorable) adverse effect profiles, and less likelihood of causing death from overdose compared with older tricyclic antidepressants (TCAs) and monamine oxidase inhibitors (MAOIs).
Patients are less likely to discontinue treatment with SSRIs than with TCAs (odds ratio=1.21; 95% confidence interval [CI], 1.12–1.30).10
However, there are no clinically significant differences in effectiveness between SSRIs and TCAs (strength of recommendation [SOR]: A).11 Importantly, although practice patterns in the use of antidepressants have changed, some reasons for the preference of newer effective agents have not been substantiated. For instance, we do not know whether the patient population taking newer agents has a lower rate of suicide, despite the difference in fatality risk mentioned earlier.
Combined pharmacotherapy and psychiatric consultation
Combining pharmacotherapy and psychotherapy can be more effective than either modality alone. In one study, 73% of patients with chronic depression treated with combination therapy showed a reduction of 50% or more on the Hamilton Rating Scale for Depression (HRSD), compared with just 48% in the nefazodone-only and psychotherapy-only groups (SOR: A). Among those who completed the study, the rates of response were 85%, 55%, and 52%, respectively (although the results considered compliant patients only, which biases the results in favor of treatment).2
Among elderly depressed patients who received home care, 58% of those who underwent intervention by a psychogeriatric team recovered, compared with just 25% in the control group (SOR: A).12 The intervention group received a multidisciplinary team evaluation and an individualized management plan, which could include any combination of physical, psychological, or social interventions. The control group received usual care from their general practitioner.
Studies of combination therapy have yielded mixed results, but guidelines from the psychiatric literature based on clinical experience advocate concomitant psychotherapy and medication (SOR: A).13 For patients with persistent symptoms after 6 to 8 weeks of taking antidepressant medication, concomitant psychotherapy improved compliance, satisfaction, and outcomes when compared with usual care.14
At any one time, at least 3% of the US population suffers from chronic depression.2 More than 17% of the population have had a major depressive episode in their lifetime, and more than 10% have experienced an episode within the past 12 months.3 The incidence and prevalence of depression in women are approximately twice that seen in men.4
Major depression is the fourth leading cause of worldwide disease burden.5
Natural history and prognosis
An untreated episode of depression usually lasts 6 months or longer. About half of persons experiencing major depression will have a second episode; a second episode increases the risk for a third episode to 80%.1,3 Patients diagnosed with depression average 5 depressive episodes in their life and may have recurrences every 4 to 6 years. Episodes usually become longer and more frequent with advancing age. In about 20% to 35% of cases, only partial remission occurs and functioning remains impaired.1
Fifteen percent of severely depressed patients commit suicide. The 2 most powerful predictors of suicide are a history of major depression or schizophrenia and a history of addictive disorders.6
Outpatient treatment of depression has increased markedly in the United States, with greater involvement on the part of physicians, greater use of psychotropic medications, expanding availability of third-party payment, and less use of psychotherapy.7
The concomitant therapy group participated in a multifaceted program including education, psychiatric referral, pharmacy utilization records, and primary physician feedback. The usual care group received standard antidepressants and follow-up visits from their family physician, with optional referral to a mental health provider.
Psychotherapy has also been shown to decrease the risk of relapse once symptoms have remitted.15 Primary care physicians can also incorporate counseling as adjunctive therapy.
Herbal and nutritional products
St. John’s wort. St. John’s wort (Hypericum perforatum L.) has been used as an herbal medication for more than 2000 years. Its efficacy in the treatment of depression has been studied extensively. Some studies demonstrated that these extracts are more effective than placebo for the short-term treatment of mild and moderate depression.16,17,18 Two randomized controlled trials demonstrated minimal efficacy of St. John’s wort in moderately severe major depression.19,20 The National Institutes of Health is sponsoring a placebo-controlled, double-blinded trial comparing St. John’s wort with SSRIs.21
Omega-3 fatty acids. Chronic deficiencies of essential fatty acids may adversely affect central nervous system function. In a small, 4-week double-blind study, outpatients receiving antidepressant therapy who were also given eicosapentaenoic acid exhibited improvement in core depressive symptoms (eg, worthlessness, guilt, insomnia) compared with the antidepressantplus-placebo group. Larger, long-term prospective trials are needed to confirm an antidepressant effect with omega-3 fatty acids.22
S-adenosyl-L-methionine. S-adenosyl-L-methionine is possibly effective for short-term treatment of major depression. Data for other herbal or nutritional remedies are negligible.23
Exercise
Physical activity may play an important role in relieving depression. One randomized controlled trial showed that an aerobic exercise program, sertraline therapy, or a combination of both were equally effective in the treatment of depression, although there was a more rapid initial response with sertraline.24
A systematic review and meta-analysis concluded that exercise may reduce depression symptoms short term, but much of the evidence is of poor quality.25 Well-controlled studies are needed to clarify the role of exercise in the treatment of depression. However, exercise is promising enough to consider implementation in clinical practice at this time.
Treatment strategy
Guidelines for medicating patients and setting expectations
Start antidepressant therapy promptly when depression is diagnosed. Maintain the initial dosage for at least 3 to 4 weeks before increasing it. A trial of 6 to 8 weeks at maximum dosage (or the maximum tolerated dosage) is necessary to confirm treatment success or failure.26,27,28
An improvement in symptoms will usually not be noted until after 2 to 6 weeks of therapy. Depending on depression severity, schedule weekly or monthly visits for patients during the initial treatment phase. The response rate to initial treatment is only 50% to 60%, but more than 80% of depressed patients will respond to at least 1 medication.1
Response to placebo is highly variable. It is often substantial and has increased in recent years. In an analysis of 75 trials between 1981 and 2001, the mean proportion of patients in the placebo group who responded (50% improvement on the HRSD) was 29.7%, compared with 50.1% in the active medication group.29 The placebo effect may reflect some combination of patient expectations, the natural history of depression with possible spontaneous remission, and limitations of study methods.
Antidepressant therapy is effective compared with placebo for depression secondary to medical illness (number needed to treat [NNT], 4.2; 95% CI, 3.2–6.4), with minimal treatment dropout (number needed to harm, 9.8; 95% CI, 5.4–42.9).30 Although many patients settle for partial improvement of their symptoms, the treatment goal should be complete remission.
Factors in drug selection
Selecting an antidepressant can be challenging: more than 24 drugs are on the market, each working through 1 or more of 7 pharmacologic mechanisms. Theoretically, choosing a drug is made easier by matching patient symptoms to likely medication side effects or by knowing that the patient or a family member responded favorably to a particular antidepressant in the past.
This intuitive model has not been proven superior to any other model of selecting antidepres-sants, but it is clinically sound, pharmacodynamically appealing, and supported by case reports. Its strength may lie in enhancing patient adherence during the critical initial phase of treatment.
A recent randomized, prospective comparison of the SSRIs paroxetine, fluoxetine, and sertraline showed similar effectiveness and tolerability (SOR: A).31 This suggests that efforts to individualize therapy based on comorbidities or likely side effects may not be as useful when choosing from among analogous SSRIs.
Nevertheless, choosing a drug that is effective, convenient, and well tolerated will improve the likelihood of achieving and maintaining a full remission. The data on adverse effects of antidepressants are widely available and well understood. Also consider cost (Table).
Preferences based on characteristics. For a patient whose depression is not complicated by other clinical conditions, the initial choice of antidepressant would usually be an SSRI. But nefazodone, mirtazapine, bupropion, or low-dose venlafaxine may be equally appropriate.
For a patient whose depression has other specific components, use your knowledge of drugs’ common side effects to fit the patient’s clinical profile.
- If there is generalized anxiety, agitation, and insomnia, both nefazodone8 and mirtazapine32 are excellent choices. Trazodone at low doses is often used as a sedative with nonsedating antidepressants.8
- If weight gain is desired, mirtazapine is indicated.32
- If tobacco cessation is a secondary goal, bupropion is preferred.31
- Those suffering from hypersomnia, retarded depression, cognitive slowing, and pseudodementia would benefit from bupropion or venlafaxine.9
- For more severely depressed patients, venlafaxine may be advantageous due to its dual serotonergic and noradrenergic activity at moderate to high doses.34,35,36 Mirtazapine and TCAs are also useful in severe depression, as well as for coexisting chronic pain syndromes.8 For refractory or atypical depression in motivated and compliant patients, MAOIs my be useful.8
When to avoid specific drugs.
- Patients with hypersomnia and motor retardation should avoid nefazodone and mirtazapine.8,32
- With obesity, mirtazapine and TCAs are least preferred.8,32
- If sexual dysfunction preceded depression, avoid giving SSRIs and venlafaxine.3
- Those experiencing agitation and insomnia should avoid bupropion and venlafaxine.3
- Seizure disorder is a contraindication to bupropion.3
- Hypertension is a relative contraindication to venlafaxine.3
- Liver disease is a contraindication to nefazodone.37
- Preexisting heart disease and increased suicide risk are both relative contraindications to TCAs.8
TABLE
Comparative dosages and costs of antidepressant drugs
| Agents | Initial target dose | Maximum effective dose | Monthly cost of initial target dose* |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Citalopram (Celexa) | 20 mg qd | 60 mg qd | $61.58 |
| Escitalopram (Lexapro) | 10 mg qd | 20 mg qd | $65.28 |
| Fluoxetine (Prozac) | 20 mg qd | 80 mg qd | $81.78 |
| Fluoxetine (generic) | 20 mg qd | 80 mg qd | $61.80 |
| Fluoxetine (Prozac Weekly) | 90 mg qwk | $71.04 | |
| Fluvoxamine (Luvox) | 50 mg qd | 150 mg bid | $59.70 |
| Paroxetine (Paxil) | 20 mg qd | 60 mg qd | $70.98 |
| Paroxetine (Paxil CR) | 25 mg qd | 75 mg qd | $75.86 |
| Sertraline (Zoloft) | 50 mg qd | 200 mg qd | $65.24 |
| Tricyclic antidepressants | |||
| Amitriptyline (Elavil, Endep, Vanatrip) | 100 mg qhs | 300 mg qhs | $ 7.99 |
| Desipramine (Norpramin) | 100 mg qhs | 200 mg qhs | $18.54 |
| Doxepin (Adapin, Sinequan) | 100 mg qhs | 300 mg qhs | $8.12 |
| Imipramine (Tofranil) | 100 mg qhs | 300 mg qhs | $31.96 |
| Nortriptyline (Aventil, Pamelor) | 75 mg qhs | 150 mg qhs | $ 8.71 |
| Others | |||
| Bupropion (Wellbutrin) | 100 mg tid | 150 mg tid | $92.33 |
| Bupropion (generic) | 100 mg tid | 150 mg tid | $64.62 |
| Bupropion (Wellbutrin SR) | 150 mg bid | 200 mg bid | $87.09 |
| Mirtazapine (Remeron) | 30 mg qhs | 45 mg qhs | $80.79 |
| Nefazodone (Serzone) | 100 mg bid | 300 mg bid | $74.94 |
| Trazodone (Desyrel) | 100 mg bid | 300 mg bid | $15.98 |
| Venlafaxine (Effexor) | 37.5 mg bid | 150 mg tid | $74.39 |
| Venlafaxine (Effexor XL) | 75 mg qd | 225 mg qd | $66.25 |
| Lithium (Eskalith, Lithobid, Lithonate, Lithotabs) | 300 mg bid | 600 mg bid | $13.70 |
| *Costs from www.drugstore.com, November 2002. | |||
Helping nonresponders
Patients whose symptoms do not improve with therapy could be switched to a different monotherapy or to multiple drugs. Drug choices for treatment-refractory and nonresponding patients have evolved more by anecdote than by systematic study.9
Switch drugs. The benefit of switching patients to another category of antidepressant was recently demonstrated in a study where nearly half of patients who did not respond to an initial antidepressant, whether SSRI or TCA, responded when switched to the alternate agent (SOR: A).38 It is also beneficial to switch medications within a category (SOR: B).27,39,40
Add a drug. Adding a second antidepressant from a category with a different mechanism of action often enhances clinical efficacy. This has been demonstrated in combining an SSRI with a TCA (SOR: B).41 Though response rates are very similar for various antidepressants, complete remission and rates of response in severely depressed patients may be higher in dual-action antidepressants (SOR: A).34,35,36
Add lithium. A great deal of evidence supports the use of lithium augmentation (SOR: A).42,43 This agent should be used more in primary care and not only by psychiatrists. A recent meta-analysis of double-blind, placebo-controlled studies of lithium (given at a dosage of at least 800 mg/d or at a level high enough to achieve a serum drug concentration of 0.5 mEq/L for at least 2 weeks) found a summary pooled odds ratio of response to lithium of 3.31 (95% CI, 1.46–7.53) with a NNT of 3.7.44 Other studies have been less clear on the optimal dose or blood level, so a starting dose of 300 mg twice daily with a serum drug concentration of 0.4 mEq/L has been recommended.
If renal function is normal, the concentration of lithium can be checked 5 days after a patient has received a stable dosage, at least 8 hours fol-lowing the last dose. Lithium may cause thyroid abnormalities; monitoring should include a measurement of thyroid-stimulating hormone, repeated at 6 months and 1 year.
Other augmentation options. Augmentation of antidepressants with buspirone has been proven useful in major depression (SOR: B).45 Thyroid supplementation may also increase the effectiveness of antidepressant therapy using triiodothyronine (T3), at doses not to exceed 50 mcg per day (SOR: B).46,47 Electroconvulsive therapy (ECT) has a high rate of therapeutic success, including speed and safety, but it is not administered as first-line treatment by psychiatrists except in severe cases (SOR: A).48,49 Augmentation with antipsychotic or anticonvulsants is another strategy that shows some benefit for select patients.50
Texas medication algorithm project
The process of drug selection just described can avoid treatment-threatening side effects, enable patient adherence to treatment, and maximize the potential for therapeutic response. However, the model can become disorienting for the clinician and the patient if 1 or 2 initial selections for treatment do not succeed. A useful synergy may be achieved by adapting the intuitive model to an algorithmic model—the Texas Medication Algorithm Project (TMAP). TMAP is an evolving model that reflects ongoing clinical research in the treatment of depression.27
Developed in 1995 from a review of existing antidepressant research and several consensus conferences, the TMAP (continually updated with new research findings) has developed algorithms for treatment of schizophrenia and bipolar disorder in addition to major depression. At each stage in the depression algorithm, treatment plans similar in efficacy and safety are grouped together, and the clinician is given a limited number of options. The later stages in the algorithm are more complex, admittedly with a greater potential for medical complications (Figure).51
The algorithm represents a tentative foundation for a sequenced medication plan. Research pertaining to the selection of antidepressant medicationis underway, sponsored by the National Institute of Mental Health. Unlike most antide-pressant trials, this study includes subjects with significant concomitant medical illnesses.
FIGURE
Treatment of chronic major depression*
When to refer
Patients requiring referral to a psychiatrist include those with suicidal ideation or severe depression, aggressive ideation, bipolar disorder, atypical depression, psychotic depression, substance abuse, or treatment resistance.52 Referral to a licensed counselor should be offered to most patients with depression, with or without psychiatric involvement, though many factors (eg, patient motivation, capacity for insight, patient perceptions of therapist) will affect follow-through and outcome.
Maintenance therapy
Once full remission has been achieved, 6 to 12 months of continued pharmacotherapy at the same dose is recommended, as it decreases the risk of relapse by 70%.5,21,26 More than half of patients will have a recurrence of depression in their lifetime, and they should be advised about this risk.1
A second episode of major depression confers an 80% chance of additional recurrences, and patients should therefore be maintained on medication for 1 to 2 years.
A third episode requires indefinite maintenance treatment because of a 90% recurrence rate.3,26
Follow-up visits after remission can be tapered gradually to once every 3 months. Discontinuation of therapy should be done gradually to minimize withdrawal reactions; it also necessitates follow-up visits or phone calls.
* For a review of screening for depression, see Nease DE, Malouin JM. Depression screening: A practical strategy. J Fam Pract 2003; 52(2):118–126.
1. Whooley MA, Simon GE. Managing depression in medical outpatients. N Engl J Med 2000;343:1942-1950.
2. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system for psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-1470.
3. Cohen L. Rational drug use in the treatment of depression. Pharmacotherapy 1997;17:45-61.
4. Bhatia SC, Bhatia SK. Depression in women: diagnostic and treatment considerations. Am Fam Physician 1999;60:225-240.
5. Glass RM. Treating depression as a recurrent or chronic disease. JAMA 1999;281:83.-
6. Harwitz D, Ravizza L. Psychiatric emergencies; suicide and depression. Emergency Medical Clinics of North America 2000;18:263-271.
7. Olfson M, Marcus S, Druss B, et al. National trends in the out-patient treatment of depression. JAMA 2002;287:203-209.
8. Stahl SM. Selecting an antidepressant by using mechanism of action to enhance efficacy and avoid side effects. J Clin Psychiatry 1998;59(supp1 18):23-29.
9. Stahl SM. Depression and bipolar disorders. In: Stahl SM. Essential Psychopharmacology, Neuroscientific Basis and Clinical Applications. Cambridge: Cambridge University Press, 1996;135-295.
10. Barbui C, Hotopf M, Freemantle N, et al. Treatment discontinuation with selective serotonin reuptake inhibitors (SSRIs) versus tricyclic antidepressants (TCAs) (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
11. Geddes JR, Freemantle N, Mason J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford: Update Software.
12. Banerjee S, Shamash K, Macdonald AJD, Mann AH. Randomized controlled trial of effect of intervention by psychogeriatric team on depression in frail elderly people at home. BMJ 1996;313:1058-1061.
13. Reynolds III CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression. JAMA 1999;281:39-45.
14. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry 1999;56:1109-1115.
15. Fava GA, Rafanelli C, Grandi S, Grandi S, Conti S, Belluardo P. Prevention of recurrent depression with cognitive behavioral therapy: preliminary findings. Arch Gen Psychiatry 1998;55:816-820.
16. Linde K, Mulrow CD. St John’s wort for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
17. Williams Jr JW, Mulrow CD, Chiquette E, et al. A systematic review of newer pharmacotherapies for depression in adults: evidence report summary. Ann Intern Med 2000;132:743-756.
18. Lecrubier Y, Clerc G, Didi R, Kieser M. Efficacy of St. John’s wort extract WS 5570 in major depression: a double-blind, placebo-controlled trial. Am J Psychiatry 2002;159:1361-1366.
19. Shelton RC, Keller MB, Gelenberg A, et al. Effectiveness of St. John’s wort in major depression, a randomized controlled trial. JAMA 2001;285:1978-1986.
20. Hypericum Depression Trial Study Group. Effect of Hypericum perforatum (St. John’s wort) in major depressive disorder, a randomized controlled trial. JAMA 2002;287:1807-1814.
21. Evidence Report/Technology Assessment. Number 7, Treatment of depression-newer pharmacotherapies (AHCPR Publication No. 99-E014). Rockville, Md: US Department of Health and Human Services; 1999.
22. Nemets B, Stahl Z, Belmaker RH. Addition of Omega-3 fatty acid to maintenance medication treatment for recurrent unipolar depressive disorder. Am J Psychiatry 2002;159:477-479.
23. Jellin JM, Gregory P, Balz F, et al. Pharmacist’s Letter/Prescriber’s Letter. Natural Medicines Comprehensive Database. 3rd ed. Stockton, Calif: Therapeutic Research Faculty; 2000;925-928.
24. Blumenthal JA, Babyak MA, Moore KA, Craighead WE, et al. Effects of exercise training on older patients with major depression. Arch Intern Med 1999;159:2349-2356.
25. Lawler DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomized controlled trials. Br Med J 2001;322:763-767.
26. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder in adults. Am J Psychiatry 2000;157(4 suppl):1-45.
27. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference. Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-156.
28. Quitkin FM, Rabkin JG, Ross D, McGrath PJ. Duration of antidepressant drug treatment: what is an adequate trial? Arch Gen Psychiatry 1984;41:238-245.
29. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression, variable, substantial, and growing. JAMA 2002;287:1840-1847.
30. Gill D, Hatcher S. Antidepressants for depression in medical illness (Cochrane Review). In: The Cochrane Library,. 1, 2002. Oxford; Update Software.
31. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: A randomized trial. JAMA 2001;286:2947-2955.
32. Hartmann PM. Mirtazapine: a newer antidepressant. Am Fam Physician 1999;59:159-161.
33. Hurt RD, Sachs DPL, Glover ED, Offord KP, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
34. Clerc GE, Ruimy P, Verdeau-Pailles J, et al. A double-blind comparison of venlafaxine and fluoxetine in patients hospitalized for major depression and melancholia. Int Clin Psychopharmacology 1994;9:139-143.
35. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J of Psychiatry 2001;178:234-241.
36. Schweitzer E, Feighner J, Mandos L, Rickles K. Comparison of venlafaxine and imipramine in the acute treatment of major depression in outpatients. J Clin Psychiatry 1994;55:104-108.
37. Product information. Serzone(c) (nefazodone). Princeton, NJ Bristol Myers Squibb Co., 2002 (January).
38. Thase ME, Rush AJ, Howland RH, Kornstein SG, Kocsis JH, Gelenberg AJ, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59:233-239.
39. Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ. A proposed algorithm for improved recognition and treatment of depression/anxiety spectrum in primary care. J Clin Psychiatry 2001;3:44-52.
40. Thase ME, Feighner JP, Lydiard RB. Citalopram treatment of fluoxetine nonresponders. J Clin Psychiatry 2001;62:683-687.
41. Nelson JC, Mazure CM, Bowers MB, Jatlow PI. A preliminary, open study of the combination of fluoxetine and desipramine for rapid treatment of major depression. Arch Gen Psychiatry 1991;48:303-307.
42. Stein G, Bernardt M. Lithium augmentation therapy in tri-cyclic-resistant depression: a controlled trial using lithium in low and normal doses. Br J Psychiatry 1993;162:634-640.
43. Joffe RT, Singer W, Levitt AJ, et al. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-393.
44. Bauer M, Dopfmer S. Lithium augmentation in treatment-resistant depression: meta-analysis of placebo-controlled studies. J Clin Psychopharmacol 1999;19:427-434.
45. Harvey KV, Balon R. Augmentation with buspirone: a review. Ann Clin Psychiatry 1995;2:143-147.
46. Thase ME, Rush AJ. Treatment-resistant depression. In Bloom FE, Kupfer DJ. Psychopharmacology: The Fourth Generation of Progress. New York, NY: Karen Press; 1995;1081-1097.
47. Prange AJ Jr, Loosen PT, Wilson IC, Lipton MA. The therapeutic use of hormone of the thyroid axis in depression. In: Post R, Ballenger J. Neurobiology of mood disorders.. Vol 1. Baltimore, Md: Williams & Wilkins; 1980;311-322.
48. Gagne GG, Furman MJ, Carpenter LL, Price LH. Efficacy of continuation ECT and antidepressant drugs compared to long-term antidepressants alone in depressed patients. Am J Psychiatry 2000;157:1960-1965.
49. American Psychiatric Association.The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. Washington, DC: American Psychiatric Association, 1990.
50. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-134.
51. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6):22-29.
52. Montana CB. Recognition and treatment of depression in a primary care setting. J Clin Psychiatry 1994;55(suppl 1):18-34.
- Combined treatment with psychotherapy or psychiatric consult and drug therapy has shown better response in several studies than either therapy alone (A).
- Although not proven by clinical trials, selecting a medication by matching its side-effect profile to patient characteristics is supported by case reports and likely enhances compliance.
- Patients who do not improve with initial therapy often benefit from being switched to another class of antidepressants (A), or having a drug from another class added to their therapy (B).
You are more likely to see depression in your practice than any other disorder except hypertension.1 Given the prevalence of depression* and the variability of its clinical symptoms and comorbidities, how do you determine the optimal therapy for a given patient?
A sobering thought: nearly half of all patients stop taking their antidepressant prescription medication within the first month of treatment.1 We discuss the critical factors you can address to help patients stick with treatment and achieve the best outcome.
Therapeutic Options
Pharmacotherapy
Antidepressants are thought to exert their therapeutic and adverse effects through 3 chemical monamine neurotransmission systems; by increasing levels of norepinephrine, serotonin, or dopamine in the synapse; and by resultant secondary changes in presynaptic and postsynaptic receptor physiology.3,8,9 Newer medications—such as selective serotonin reuptake inhibitors (SSRIs)—have simpler dose schedules, different (and for some patients more favorable) adverse effect profiles, and less likelihood of causing death from overdose compared with older tricyclic antidepressants (TCAs) and monamine oxidase inhibitors (MAOIs).
Patients are less likely to discontinue treatment with SSRIs than with TCAs (odds ratio=1.21; 95% confidence interval [CI], 1.12–1.30).10
However, there are no clinically significant differences in effectiveness between SSRIs and TCAs (strength of recommendation [SOR]: A).11 Importantly, although practice patterns in the use of antidepressants have changed, some reasons for the preference of newer effective agents have not been substantiated. For instance, we do not know whether the patient population taking newer agents has a lower rate of suicide, despite the difference in fatality risk mentioned earlier.
Combined pharmacotherapy and psychiatric consultation
Combining pharmacotherapy and psychotherapy can be more effective than either modality alone. In one study, 73% of patients with chronic depression treated with combination therapy showed a reduction of 50% or more on the Hamilton Rating Scale for Depression (HRSD), compared with just 48% in the nefazodone-only and psychotherapy-only groups (SOR: A). Among those who completed the study, the rates of response were 85%, 55%, and 52%, respectively (although the results considered compliant patients only, which biases the results in favor of treatment).2
Among elderly depressed patients who received home care, 58% of those who underwent intervention by a psychogeriatric team recovered, compared with just 25% in the control group (SOR: A).12 The intervention group received a multidisciplinary team evaluation and an individualized management plan, which could include any combination of physical, psychological, or social interventions. The control group received usual care from their general practitioner.
Studies of combination therapy have yielded mixed results, but guidelines from the psychiatric literature based on clinical experience advocate concomitant psychotherapy and medication (SOR: A).13 For patients with persistent symptoms after 6 to 8 weeks of taking antidepressant medication, concomitant psychotherapy improved compliance, satisfaction, and outcomes when compared with usual care.14
At any one time, at least 3% of the US population suffers from chronic depression.2 More than 17% of the population have had a major depressive episode in their lifetime, and more than 10% have experienced an episode within the past 12 months.3 The incidence and prevalence of depression in women are approximately twice that seen in men.4
Major depression is the fourth leading cause of worldwide disease burden.5
Natural history and prognosis
An untreated episode of depression usually lasts 6 months or longer. About half of persons experiencing major depression will have a second episode; a second episode increases the risk for a third episode to 80%.1,3 Patients diagnosed with depression average 5 depressive episodes in their life and may have recurrences every 4 to 6 years. Episodes usually become longer and more frequent with advancing age. In about 20% to 35% of cases, only partial remission occurs and functioning remains impaired.1
Fifteen percent of severely depressed patients commit suicide. The 2 most powerful predictors of suicide are a history of major depression or schizophrenia and a history of addictive disorders.6
Outpatient treatment of depression has increased markedly in the United States, with greater involvement on the part of physicians, greater use of psychotropic medications, expanding availability of third-party payment, and less use of psychotherapy.7
The concomitant therapy group participated in a multifaceted program including education, psychiatric referral, pharmacy utilization records, and primary physician feedback. The usual care group received standard antidepressants and follow-up visits from their family physician, with optional referral to a mental health provider.
Psychotherapy has also been shown to decrease the risk of relapse once symptoms have remitted.15 Primary care physicians can also incorporate counseling as adjunctive therapy.
Herbal and nutritional products
St. John’s wort. St. John’s wort (Hypericum perforatum L.) has been used as an herbal medication for more than 2000 years. Its efficacy in the treatment of depression has been studied extensively. Some studies demonstrated that these extracts are more effective than placebo for the short-term treatment of mild and moderate depression.16,17,18 Two randomized controlled trials demonstrated minimal efficacy of St. John’s wort in moderately severe major depression.19,20 The National Institutes of Health is sponsoring a placebo-controlled, double-blinded trial comparing St. John’s wort with SSRIs.21
Omega-3 fatty acids. Chronic deficiencies of essential fatty acids may adversely affect central nervous system function. In a small, 4-week double-blind study, outpatients receiving antidepressant therapy who were also given eicosapentaenoic acid exhibited improvement in core depressive symptoms (eg, worthlessness, guilt, insomnia) compared with the antidepressantplus-placebo group. Larger, long-term prospective trials are needed to confirm an antidepressant effect with omega-3 fatty acids.22
S-adenosyl-L-methionine. S-adenosyl-L-methionine is possibly effective for short-term treatment of major depression. Data for other herbal or nutritional remedies are negligible.23
Exercise
Physical activity may play an important role in relieving depression. One randomized controlled trial showed that an aerobic exercise program, sertraline therapy, or a combination of both were equally effective in the treatment of depression, although there was a more rapid initial response with sertraline.24
A systematic review and meta-analysis concluded that exercise may reduce depression symptoms short term, but much of the evidence is of poor quality.25 Well-controlled studies are needed to clarify the role of exercise in the treatment of depression. However, exercise is promising enough to consider implementation in clinical practice at this time.
Treatment strategy
Guidelines for medicating patients and setting expectations
Start antidepressant therapy promptly when depression is diagnosed. Maintain the initial dosage for at least 3 to 4 weeks before increasing it. A trial of 6 to 8 weeks at maximum dosage (or the maximum tolerated dosage) is necessary to confirm treatment success or failure.26,27,28
An improvement in symptoms will usually not be noted until after 2 to 6 weeks of therapy. Depending on depression severity, schedule weekly or monthly visits for patients during the initial treatment phase. The response rate to initial treatment is only 50% to 60%, but more than 80% of depressed patients will respond to at least 1 medication.1
Response to placebo is highly variable. It is often substantial and has increased in recent years. In an analysis of 75 trials between 1981 and 2001, the mean proportion of patients in the placebo group who responded (50% improvement on the HRSD) was 29.7%, compared with 50.1% in the active medication group.29 The placebo effect may reflect some combination of patient expectations, the natural history of depression with possible spontaneous remission, and limitations of study methods.
Antidepressant therapy is effective compared with placebo for depression secondary to medical illness (number needed to treat [NNT], 4.2; 95% CI, 3.2–6.4), with minimal treatment dropout (number needed to harm, 9.8; 95% CI, 5.4–42.9).30 Although many patients settle for partial improvement of their symptoms, the treatment goal should be complete remission.
Factors in drug selection
Selecting an antidepressant can be challenging: more than 24 drugs are on the market, each working through 1 or more of 7 pharmacologic mechanisms. Theoretically, choosing a drug is made easier by matching patient symptoms to likely medication side effects or by knowing that the patient or a family member responded favorably to a particular antidepressant in the past.
This intuitive model has not been proven superior to any other model of selecting antidepres-sants, but it is clinically sound, pharmacodynamically appealing, and supported by case reports. Its strength may lie in enhancing patient adherence during the critical initial phase of treatment.
A recent randomized, prospective comparison of the SSRIs paroxetine, fluoxetine, and sertraline showed similar effectiveness and tolerability (SOR: A).31 This suggests that efforts to individualize therapy based on comorbidities or likely side effects may not be as useful when choosing from among analogous SSRIs.
Nevertheless, choosing a drug that is effective, convenient, and well tolerated will improve the likelihood of achieving and maintaining a full remission. The data on adverse effects of antidepressants are widely available and well understood. Also consider cost (Table).
Preferences based on characteristics. For a patient whose depression is not complicated by other clinical conditions, the initial choice of antidepressant would usually be an SSRI. But nefazodone, mirtazapine, bupropion, or low-dose venlafaxine may be equally appropriate.
For a patient whose depression has other specific components, use your knowledge of drugs’ common side effects to fit the patient’s clinical profile.
- If there is generalized anxiety, agitation, and insomnia, both nefazodone8 and mirtazapine32 are excellent choices. Trazodone at low doses is often used as a sedative with nonsedating antidepressants.8
- If weight gain is desired, mirtazapine is indicated.32
- If tobacco cessation is a secondary goal, bupropion is preferred.31
- Those suffering from hypersomnia, retarded depression, cognitive slowing, and pseudodementia would benefit from bupropion or venlafaxine.9
- For more severely depressed patients, venlafaxine may be advantageous due to its dual serotonergic and noradrenergic activity at moderate to high doses.34,35,36 Mirtazapine and TCAs are also useful in severe depression, as well as for coexisting chronic pain syndromes.8 For refractory or atypical depression in motivated and compliant patients, MAOIs my be useful.8
When to avoid specific drugs.
- Patients with hypersomnia and motor retardation should avoid nefazodone and mirtazapine.8,32
- With obesity, mirtazapine and TCAs are least preferred.8,32
- If sexual dysfunction preceded depression, avoid giving SSRIs and venlafaxine.3
- Those experiencing agitation and insomnia should avoid bupropion and venlafaxine.3
- Seizure disorder is a contraindication to bupropion.3
- Hypertension is a relative contraindication to venlafaxine.3
- Liver disease is a contraindication to nefazodone.37
- Preexisting heart disease and increased suicide risk are both relative contraindications to TCAs.8
TABLE
Comparative dosages and costs of antidepressant drugs
| Agents | Initial target dose | Maximum effective dose | Monthly cost of initial target dose* |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Citalopram (Celexa) | 20 mg qd | 60 mg qd | $61.58 |
| Escitalopram (Lexapro) | 10 mg qd | 20 mg qd | $65.28 |
| Fluoxetine (Prozac) | 20 mg qd | 80 mg qd | $81.78 |
| Fluoxetine (generic) | 20 mg qd | 80 mg qd | $61.80 |
| Fluoxetine (Prozac Weekly) | 90 mg qwk | $71.04 | |
| Fluvoxamine (Luvox) | 50 mg qd | 150 mg bid | $59.70 |
| Paroxetine (Paxil) | 20 mg qd | 60 mg qd | $70.98 |
| Paroxetine (Paxil CR) | 25 mg qd | 75 mg qd | $75.86 |
| Sertraline (Zoloft) | 50 mg qd | 200 mg qd | $65.24 |
| Tricyclic antidepressants | |||
| Amitriptyline (Elavil, Endep, Vanatrip) | 100 mg qhs | 300 mg qhs | $ 7.99 |
| Desipramine (Norpramin) | 100 mg qhs | 200 mg qhs | $18.54 |
| Doxepin (Adapin, Sinequan) | 100 mg qhs | 300 mg qhs | $8.12 |
| Imipramine (Tofranil) | 100 mg qhs | 300 mg qhs | $31.96 |
| Nortriptyline (Aventil, Pamelor) | 75 mg qhs | 150 mg qhs | $ 8.71 |
| Others | |||
| Bupropion (Wellbutrin) | 100 mg tid | 150 mg tid | $92.33 |
| Bupropion (generic) | 100 mg tid | 150 mg tid | $64.62 |
| Bupropion (Wellbutrin SR) | 150 mg bid | 200 mg bid | $87.09 |
| Mirtazapine (Remeron) | 30 mg qhs | 45 mg qhs | $80.79 |
| Nefazodone (Serzone) | 100 mg bid | 300 mg bid | $74.94 |
| Trazodone (Desyrel) | 100 mg bid | 300 mg bid | $15.98 |
| Venlafaxine (Effexor) | 37.5 mg bid | 150 mg tid | $74.39 |
| Venlafaxine (Effexor XL) | 75 mg qd | 225 mg qd | $66.25 |
| Lithium (Eskalith, Lithobid, Lithonate, Lithotabs) | 300 mg bid | 600 mg bid | $13.70 |
| *Costs from www.drugstore.com, November 2002. | |||
Helping nonresponders
Patients whose symptoms do not improve with therapy could be switched to a different monotherapy or to multiple drugs. Drug choices for treatment-refractory and nonresponding patients have evolved more by anecdote than by systematic study.9
Switch drugs. The benefit of switching patients to another category of antidepressant was recently demonstrated in a study where nearly half of patients who did not respond to an initial antidepressant, whether SSRI or TCA, responded when switched to the alternate agent (SOR: A).38 It is also beneficial to switch medications within a category (SOR: B).27,39,40
Add a drug. Adding a second antidepressant from a category with a different mechanism of action often enhances clinical efficacy. This has been demonstrated in combining an SSRI with a TCA (SOR: B).41 Though response rates are very similar for various antidepressants, complete remission and rates of response in severely depressed patients may be higher in dual-action antidepressants (SOR: A).34,35,36
Add lithium. A great deal of evidence supports the use of lithium augmentation (SOR: A).42,43 This agent should be used more in primary care and not only by psychiatrists. A recent meta-analysis of double-blind, placebo-controlled studies of lithium (given at a dosage of at least 800 mg/d or at a level high enough to achieve a serum drug concentration of 0.5 mEq/L for at least 2 weeks) found a summary pooled odds ratio of response to lithium of 3.31 (95% CI, 1.46–7.53) with a NNT of 3.7.44 Other studies have been less clear on the optimal dose or blood level, so a starting dose of 300 mg twice daily with a serum drug concentration of 0.4 mEq/L has been recommended.
If renal function is normal, the concentration of lithium can be checked 5 days after a patient has received a stable dosage, at least 8 hours fol-lowing the last dose. Lithium may cause thyroid abnormalities; monitoring should include a measurement of thyroid-stimulating hormone, repeated at 6 months and 1 year.
Other augmentation options. Augmentation of antidepressants with buspirone has been proven useful in major depression (SOR: B).45 Thyroid supplementation may also increase the effectiveness of antidepressant therapy using triiodothyronine (T3), at doses not to exceed 50 mcg per day (SOR: B).46,47 Electroconvulsive therapy (ECT) has a high rate of therapeutic success, including speed and safety, but it is not administered as first-line treatment by psychiatrists except in severe cases (SOR: A).48,49 Augmentation with antipsychotic or anticonvulsants is another strategy that shows some benefit for select patients.50
Texas medication algorithm project
The process of drug selection just described can avoid treatment-threatening side effects, enable patient adherence to treatment, and maximize the potential for therapeutic response. However, the model can become disorienting for the clinician and the patient if 1 or 2 initial selections for treatment do not succeed. A useful synergy may be achieved by adapting the intuitive model to an algorithmic model—the Texas Medication Algorithm Project (TMAP). TMAP is an evolving model that reflects ongoing clinical research in the treatment of depression.27
Developed in 1995 from a review of existing antidepressant research and several consensus conferences, the TMAP (continually updated with new research findings) has developed algorithms for treatment of schizophrenia and bipolar disorder in addition to major depression. At each stage in the depression algorithm, treatment plans similar in efficacy and safety are grouped together, and the clinician is given a limited number of options. The later stages in the algorithm are more complex, admittedly with a greater potential for medical complications (Figure).51
The algorithm represents a tentative foundation for a sequenced medication plan. Research pertaining to the selection of antidepressant medicationis underway, sponsored by the National Institute of Mental Health. Unlike most antide-pressant trials, this study includes subjects with significant concomitant medical illnesses.
FIGURE
Treatment of chronic major depression*
When to refer
Patients requiring referral to a psychiatrist include those with suicidal ideation or severe depression, aggressive ideation, bipolar disorder, atypical depression, psychotic depression, substance abuse, or treatment resistance.52 Referral to a licensed counselor should be offered to most patients with depression, with or without psychiatric involvement, though many factors (eg, patient motivation, capacity for insight, patient perceptions of therapist) will affect follow-through and outcome.
Maintenance therapy
Once full remission has been achieved, 6 to 12 months of continued pharmacotherapy at the same dose is recommended, as it decreases the risk of relapse by 70%.5,21,26 More than half of patients will have a recurrence of depression in their lifetime, and they should be advised about this risk.1
A second episode of major depression confers an 80% chance of additional recurrences, and patients should therefore be maintained on medication for 1 to 2 years.
A third episode requires indefinite maintenance treatment because of a 90% recurrence rate.3,26
Follow-up visits after remission can be tapered gradually to once every 3 months. Discontinuation of therapy should be done gradually to minimize withdrawal reactions; it also necessitates follow-up visits or phone calls.
* For a review of screening for depression, see Nease DE, Malouin JM. Depression screening: A practical strategy. J Fam Pract 2003; 52(2):118–126.
- Combined treatment with psychotherapy or psychiatric consult and drug therapy has shown better response in several studies than either therapy alone (A).
- Although not proven by clinical trials, selecting a medication by matching its side-effect profile to patient characteristics is supported by case reports and likely enhances compliance.
- Patients who do not improve with initial therapy often benefit from being switched to another class of antidepressants (A), or having a drug from another class added to their therapy (B).
You are more likely to see depression in your practice than any other disorder except hypertension.1 Given the prevalence of depression* and the variability of its clinical symptoms and comorbidities, how do you determine the optimal therapy for a given patient?
A sobering thought: nearly half of all patients stop taking their antidepressant prescription medication within the first month of treatment.1 We discuss the critical factors you can address to help patients stick with treatment and achieve the best outcome.
Therapeutic Options
Pharmacotherapy
Antidepressants are thought to exert their therapeutic and adverse effects through 3 chemical monamine neurotransmission systems; by increasing levels of norepinephrine, serotonin, or dopamine in the synapse; and by resultant secondary changes in presynaptic and postsynaptic receptor physiology.3,8,9 Newer medications—such as selective serotonin reuptake inhibitors (SSRIs)—have simpler dose schedules, different (and for some patients more favorable) adverse effect profiles, and less likelihood of causing death from overdose compared with older tricyclic antidepressants (TCAs) and monamine oxidase inhibitors (MAOIs).
Patients are less likely to discontinue treatment with SSRIs than with TCAs (odds ratio=1.21; 95% confidence interval [CI], 1.12–1.30).10
However, there are no clinically significant differences in effectiveness between SSRIs and TCAs (strength of recommendation [SOR]: A).11 Importantly, although practice patterns in the use of antidepressants have changed, some reasons for the preference of newer effective agents have not been substantiated. For instance, we do not know whether the patient population taking newer agents has a lower rate of suicide, despite the difference in fatality risk mentioned earlier.
Combined pharmacotherapy and psychiatric consultation
Combining pharmacotherapy and psychotherapy can be more effective than either modality alone. In one study, 73% of patients with chronic depression treated with combination therapy showed a reduction of 50% or more on the Hamilton Rating Scale for Depression (HRSD), compared with just 48% in the nefazodone-only and psychotherapy-only groups (SOR: A). Among those who completed the study, the rates of response were 85%, 55%, and 52%, respectively (although the results considered compliant patients only, which biases the results in favor of treatment).2
Among elderly depressed patients who received home care, 58% of those who underwent intervention by a psychogeriatric team recovered, compared with just 25% in the control group (SOR: A).12 The intervention group received a multidisciplinary team evaluation and an individualized management plan, which could include any combination of physical, psychological, or social interventions. The control group received usual care from their general practitioner.
Studies of combination therapy have yielded mixed results, but guidelines from the psychiatric literature based on clinical experience advocate concomitant psychotherapy and medication (SOR: A).13 For patients with persistent symptoms after 6 to 8 weeks of taking antidepressant medication, concomitant psychotherapy improved compliance, satisfaction, and outcomes when compared with usual care.14
At any one time, at least 3% of the US population suffers from chronic depression.2 More than 17% of the population have had a major depressive episode in their lifetime, and more than 10% have experienced an episode within the past 12 months.3 The incidence and prevalence of depression in women are approximately twice that seen in men.4
Major depression is the fourth leading cause of worldwide disease burden.5
Natural history and prognosis
An untreated episode of depression usually lasts 6 months or longer. About half of persons experiencing major depression will have a second episode; a second episode increases the risk for a third episode to 80%.1,3 Patients diagnosed with depression average 5 depressive episodes in their life and may have recurrences every 4 to 6 years. Episodes usually become longer and more frequent with advancing age. In about 20% to 35% of cases, only partial remission occurs and functioning remains impaired.1
Fifteen percent of severely depressed patients commit suicide. The 2 most powerful predictors of suicide are a history of major depression or schizophrenia and a history of addictive disorders.6
Outpatient treatment of depression has increased markedly in the United States, with greater involvement on the part of physicians, greater use of psychotropic medications, expanding availability of third-party payment, and less use of psychotherapy.7
The concomitant therapy group participated in a multifaceted program including education, psychiatric referral, pharmacy utilization records, and primary physician feedback. The usual care group received standard antidepressants and follow-up visits from their family physician, with optional referral to a mental health provider.
Psychotherapy has also been shown to decrease the risk of relapse once symptoms have remitted.15 Primary care physicians can also incorporate counseling as adjunctive therapy.
Herbal and nutritional products
St. John’s wort. St. John’s wort (Hypericum perforatum L.) has been used as an herbal medication for more than 2000 years. Its efficacy in the treatment of depression has been studied extensively. Some studies demonstrated that these extracts are more effective than placebo for the short-term treatment of mild and moderate depression.16,17,18 Two randomized controlled trials demonstrated minimal efficacy of St. John’s wort in moderately severe major depression.19,20 The National Institutes of Health is sponsoring a placebo-controlled, double-blinded trial comparing St. John’s wort with SSRIs.21
Omega-3 fatty acids. Chronic deficiencies of essential fatty acids may adversely affect central nervous system function. In a small, 4-week double-blind study, outpatients receiving antidepressant therapy who were also given eicosapentaenoic acid exhibited improvement in core depressive symptoms (eg, worthlessness, guilt, insomnia) compared with the antidepressantplus-placebo group. Larger, long-term prospective trials are needed to confirm an antidepressant effect with omega-3 fatty acids.22
S-adenosyl-L-methionine. S-adenosyl-L-methionine is possibly effective for short-term treatment of major depression. Data for other herbal or nutritional remedies are negligible.23
Exercise
Physical activity may play an important role in relieving depression. One randomized controlled trial showed that an aerobic exercise program, sertraline therapy, or a combination of both were equally effective in the treatment of depression, although there was a more rapid initial response with sertraline.24
A systematic review and meta-analysis concluded that exercise may reduce depression symptoms short term, but much of the evidence is of poor quality.25 Well-controlled studies are needed to clarify the role of exercise in the treatment of depression. However, exercise is promising enough to consider implementation in clinical practice at this time.
Treatment strategy
Guidelines for medicating patients and setting expectations
Start antidepressant therapy promptly when depression is diagnosed. Maintain the initial dosage for at least 3 to 4 weeks before increasing it. A trial of 6 to 8 weeks at maximum dosage (or the maximum tolerated dosage) is necessary to confirm treatment success or failure.26,27,28
An improvement in symptoms will usually not be noted until after 2 to 6 weeks of therapy. Depending on depression severity, schedule weekly or monthly visits for patients during the initial treatment phase. The response rate to initial treatment is only 50% to 60%, but more than 80% of depressed patients will respond to at least 1 medication.1
Response to placebo is highly variable. It is often substantial and has increased in recent years. In an analysis of 75 trials between 1981 and 2001, the mean proportion of patients in the placebo group who responded (50% improvement on the HRSD) was 29.7%, compared with 50.1% in the active medication group.29 The placebo effect may reflect some combination of patient expectations, the natural history of depression with possible spontaneous remission, and limitations of study methods.
Antidepressant therapy is effective compared with placebo for depression secondary to medical illness (number needed to treat [NNT], 4.2; 95% CI, 3.2–6.4), with minimal treatment dropout (number needed to harm, 9.8; 95% CI, 5.4–42.9).30 Although many patients settle for partial improvement of their symptoms, the treatment goal should be complete remission.
Factors in drug selection
Selecting an antidepressant can be challenging: more than 24 drugs are on the market, each working through 1 or more of 7 pharmacologic mechanisms. Theoretically, choosing a drug is made easier by matching patient symptoms to likely medication side effects or by knowing that the patient or a family member responded favorably to a particular antidepressant in the past.
This intuitive model has not been proven superior to any other model of selecting antidepres-sants, but it is clinically sound, pharmacodynamically appealing, and supported by case reports. Its strength may lie in enhancing patient adherence during the critical initial phase of treatment.
A recent randomized, prospective comparison of the SSRIs paroxetine, fluoxetine, and sertraline showed similar effectiveness and tolerability (SOR: A).31 This suggests that efforts to individualize therapy based on comorbidities or likely side effects may not be as useful when choosing from among analogous SSRIs.
Nevertheless, choosing a drug that is effective, convenient, and well tolerated will improve the likelihood of achieving and maintaining a full remission. The data on adverse effects of antidepressants are widely available and well understood. Also consider cost (Table).
Preferences based on characteristics. For a patient whose depression is not complicated by other clinical conditions, the initial choice of antidepressant would usually be an SSRI. But nefazodone, mirtazapine, bupropion, or low-dose venlafaxine may be equally appropriate.
For a patient whose depression has other specific components, use your knowledge of drugs’ common side effects to fit the patient’s clinical profile.
- If there is generalized anxiety, agitation, and insomnia, both nefazodone8 and mirtazapine32 are excellent choices. Trazodone at low doses is often used as a sedative with nonsedating antidepressants.8
- If weight gain is desired, mirtazapine is indicated.32
- If tobacco cessation is a secondary goal, bupropion is preferred.31
- Those suffering from hypersomnia, retarded depression, cognitive slowing, and pseudodementia would benefit from bupropion or venlafaxine.9
- For more severely depressed patients, venlafaxine may be advantageous due to its dual serotonergic and noradrenergic activity at moderate to high doses.34,35,36 Mirtazapine and TCAs are also useful in severe depression, as well as for coexisting chronic pain syndromes.8 For refractory or atypical depression in motivated and compliant patients, MAOIs my be useful.8
When to avoid specific drugs.
- Patients with hypersomnia and motor retardation should avoid nefazodone and mirtazapine.8,32
- With obesity, mirtazapine and TCAs are least preferred.8,32
- If sexual dysfunction preceded depression, avoid giving SSRIs and venlafaxine.3
- Those experiencing agitation and insomnia should avoid bupropion and venlafaxine.3
- Seizure disorder is a contraindication to bupropion.3
- Hypertension is a relative contraindication to venlafaxine.3
- Liver disease is a contraindication to nefazodone.37
- Preexisting heart disease and increased suicide risk are both relative contraindications to TCAs.8
TABLE
Comparative dosages and costs of antidepressant drugs
| Agents | Initial target dose | Maximum effective dose | Monthly cost of initial target dose* |
|---|---|---|---|
| Selective serotonin reuptake inhibitors | |||
| Citalopram (Celexa) | 20 mg qd | 60 mg qd | $61.58 |
| Escitalopram (Lexapro) | 10 mg qd | 20 mg qd | $65.28 |
| Fluoxetine (Prozac) | 20 mg qd | 80 mg qd | $81.78 |
| Fluoxetine (generic) | 20 mg qd | 80 mg qd | $61.80 |
| Fluoxetine (Prozac Weekly) | 90 mg qwk | $71.04 | |
| Fluvoxamine (Luvox) | 50 mg qd | 150 mg bid | $59.70 |
| Paroxetine (Paxil) | 20 mg qd | 60 mg qd | $70.98 |
| Paroxetine (Paxil CR) | 25 mg qd | 75 mg qd | $75.86 |
| Sertraline (Zoloft) | 50 mg qd | 200 mg qd | $65.24 |
| Tricyclic antidepressants | |||
| Amitriptyline (Elavil, Endep, Vanatrip) | 100 mg qhs | 300 mg qhs | $ 7.99 |
| Desipramine (Norpramin) | 100 mg qhs | 200 mg qhs | $18.54 |
| Doxepin (Adapin, Sinequan) | 100 mg qhs | 300 mg qhs | $8.12 |
| Imipramine (Tofranil) | 100 mg qhs | 300 mg qhs | $31.96 |
| Nortriptyline (Aventil, Pamelor) | 75 mg qhs | 150 mg qhs | $ 8.71 |
| Others | |||
| Bupropion (Wellbutrin) | 100 mg tid | 150 mg tid | $92.33 |
| Bupropion (generic) | 100 mg tid | 150 mg tid | $64.62 |
| Bupropion (Wellbutrin SR) | 150 mg bid | 200 mg bid | $87.09 |
| Mirtazapine (Remeron) | 30 mg qhs | 45 mg qhs | $80.79 |
| Nefazodone (Serzone) | 100 mg bid | 300 mg bid | $74.94 |
| Trazodone (Desyrel) | 100 mg bid | 300 mg bid | $15.98 |
| Venlafaxine (Effexor) | 37.5 mg bid | 150 mg tid | $74.39 |
| Venlafaxine (Effexor XL) | 75 mg qd | 225 mg qd | $66.25 |
| Lithium (Eskalith, Lithobid, Lithonate, Lithotabs) | 300 mg bid | 600 mg bid | $13.70 |
| *Costs from www.drugstore.com, November 2002. | |||
Helping nonresponders
Patients whose symptoms do not improve with therapy could be switched to a different monotherapy or to multiple drugs. Drug choices for treatment-refractory and nonresponding patients have evolved more by anecdote than by systematic study.9
Switch drugs. The benefit of switching patients to another category of antidepressant was recently demonstrated in a study where nearly half of patients who did not respond to an initial antidepressant, whether SSRI or TCA, responded when switched to the alternate agent (SOR: A).38 It is also beneficial to switch medications within a category (SOR: B).27,39,40
Add a drug. Adding a second antidepressant from a category with a different mechanism of action often enhances clinical efficacy. This has been demonstrated in combining an SSRI with a TCA (SOR: B).41 Though response rates are very similar for various antidepressants, complete remission and rates of response in severely depressed patients may be higher in dual-action antidepressants (SOR: A).34,35,36
Add lithium. A great deal of evidence supports the use of lithium augmentation (SOR: A).42,43 This agent should be used more in primary care and not only by psychiatrists. A recent meta-analysis of double-blind, placebo-controlled studies of lithium (given at a dosage of at least 800 mg/d or at a level high enough to achieve a serum drug concentration of 0.5 mEq/L for at least 2 weeks) found a summary pooled odds ratio of response to lithium of 3.31 (95% CI, 1.46–7.53) with a NNT of 3.7.44 Other studies have been less clear on the optimal dose or blood level, so a starting dose of 300 mg twice daily with a serum drug concentration of 0.4 mEq/L has been recommended.
If renal function is normal, the concentration of lithium can be checked 5 days after a patient has received a stable dosage, at least 8 hours fol-lowing the last dose. Lithium may cause thyroid abnormalities; monitoring should include a measurement of thyroid-stimulating hormone, repeated at 6 months and 1 year.
Other augmentation options. Augmentation of antidepressants with buspirone has been proven useful in major depression (SOR: B).45 Thyroid supplementation may also increase the effectiveness of antidepressant therapy using triiodothyronine (T3), at doses not to exceed 50 mcg per day (SOR: B).46,47 Electroconvulsive therapy (ECT) has a high rate of therapeutic success, including speed and safety, but it is not administered as first-line treatment by psychiatrists except in severe cases (SOR: A).48,49 Augmentation with antipsychotic or anticonvulsants is another strategy that shows some benefit for select patients.50
Texas medication algorithm project
The process of drug selection just described can avoid treatment-threatening side effects, enable patient adherence to treatment, and maximize the potential for therapeutic response. However, the model can become disorienting for the clinician and the patient if 1 or 2 initial selections for treatment do not succeed. A useful synergy may be achieved by adapting the intuitive model to an algorithmic model—the Texas Medication Algorithm Project (TMAP). TMAP is an evolving model that reflects ongoing clinical research in the treatment of depression.27
Developed in 1995 from a review of existing antidepressant research and several consensus conferences, the TMAP (continually updated with new research findings) has developed algorithms for treatment of schizophrenia and bipolar disorder in addition to major depression. At each stage in the depression algorithm, treatment plans similar in efficacy and safety are grouped together, and the clinician is given a limited number of options. The later stages in the algorithm are more complex, admittedly with a greater potential for medical complications (Figure).51
The algorithm represents a tentative foundation for a sequenced medication plan. Research pertaining to the selection of antidepressant medicationis underway, sponsored by the National Institute of Mental Health. Unlike most antide-pressant trials, this study includes subjects with significant concomitant medical illnesses.
FIGURE
Treatment of chronic major depression*
When to refer
Patients requiring referral to a psychiatrist include those with suicidal ideation or severe depression, aggressive ideation, bipolar disorder, atypical depression, psychotic depression, substance abuse, or treatment resistance.52 Referral to a licensed counselor should be offered to most patients with depression, with or without psychiatric involvement, though many factors (eg, patient motivation, capacity for insight, patient perceptions of therapist) will affect follow-through and outcome.
Maintenance therapy
Once full remission has been achieved, 6 to 12 months of continued pharmacotherapy at the same dose is recommended, as it decreases the risk of relapse by 70%.5,21,26 More than half of patients will have a recurrence of depression in their lifetime, and they should be advised about this risk.1
A second episode of major depression confers an 80% chance of additional recurrences, and patients should therefore be maintained on medication for 1 to 2 years.
A third episode requires indefinite maintenance treatment because of a 90% recurrence rate.3,26
Follow-up visits after remission can be tapered gradually to once every 3 months. Discontinuation of therapy should be done gradually to minimize withdrawal reactions; it also necessitates follow-up visits or phone calls.
* For a review of screening for depression, see Nease DE, Malouin JM. Depression screening: A practical strategy. J Fam Pract 2003; 52(2):118–126.
1. Whooley MA, Simon GE. Managing depression in medical outpatients. N Engl J Med 2000;343:1942-1950.
2. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system for psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-1470.
3. Cohen L. Rational drug use in the treatment of depression. Pharmacotherapy 1997;17:45-61.
4. Bhatia SC, Bhatia SK. Depression in women: diagnostic and treatment considerations. Am Fam Physician 1999;60:225-240.
5. Glass RM. Treating depression as a recurrent or chronic disease. JAMA 1999;281:83.-
6. Harwitz D, Ravizza L. Psychiatric emergencies; suicide and depression. Emergency Medical Clinics of North America 2000;18:263-271.
7. Olfson M, Marcus S, Druss B, et al. National trends in the out-patient treatment of depression. JAMA 2002;287:203-209.
8. Stahl SM. Selecting an antidepressant by using mechanism of action to enhance efficacy and avoid side effects. J Clin Psychiatry 1998;59(supp1 18):23-29.
9. Stahl SM. Depression and bipolar disorders. In: Stahl SM. Essential Psychopharmacology, Neuroscientific Basis and Clinical Applications. Cambridge: Cambridge University Press, 1996;135-295.
10. Barbui C, Hotopf M, Freemantle N, et al. Treatment discontinuation with selective serotonin reuptake inhibitors (SSRIs) versus tricyclic antidepressants (TCAs) (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
11. Geddes JR, Freemantle N, Mason J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford: Update Software.
12. Banerjee S, Shamash K, Macdonald AJD, Mann AH. Randomized controlled trial of effect of intervention by psychogeriatric team on depression in frail elderly people at home. BMJ 1996;313:1058-1061.
13. Reynolds III CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression. JAMA 1999;281:39-45.
14. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry 1999;56:1109-1115.
15. Fava GA, Rafanelli C, Grandi S, Grandi S, Conti S, Belluardo P. Prevention of recurrent depression with cognitive behavioral therapy: preliminary findings. Arch Gen Psychiatry 1998;55:816-820.
16. Linde K, Mulrow CD. St John’s wort for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
17. Williams Jr JW, Mulrow CD, Chiquette E, et al. A systematic review of newer pharmacotherapies for depression in adults: evidence report summary. Ann Intern Med 2000;132:743-756.
18. Lecrubier Y, Clerc G, Didi R, Kieser M. Efficacy of St. John’s wort extract WS 5570 in major depression: a double-blind, placebo-controlled trial. Am J Psychiatry 2002;159:1361-1366.
19. Shelton RC, Keller MB, Gelenberg A, et al. Effectiveness of St. John’s wort in major depression, a randomized controlled trial. JAMA 2001;285:1978-1986.
20. Hypericum Depression Trial Study Group. Effect of Hypericum perforatum (St. John’s wort) in major depressive disorder, a randomized controlled trial. JAMA 2002;287:1807-1814.
21. Evidence Report/Technology Assessment. Number 7, Treatment of depression-newer pharmacotherapies (AHCPR Publication No. 99-E014). Rockville, Md: US Department of Health and Human Services; 1999.
22. Nemets B, Stahl Z, Belmaker RH. Addition of Omega-3 fatty acid to maintenance medication treatment for recurrent unipolar depressive disorder. Am J Psychiatry 2002;159:477-479.
23. Jellin JM, Gregory P, Balz F, et al. Pharmacist’s Letter/Prescriber’s Letter. Natural Medicines Comprehensive Database. 3rd ed. Stockton, Calif: Therapeutic Research Faculty; 2000;925-928.
24. Blumenthal JA, Babyak MA, Moore KA, Craighead WE, et al. Effects of exercise training on older patients with major depression. Arch Intern Med 1999;159:2349-2356.
25. Lawler DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomized controlled trials. Br Med J 2001;322:763-767.
26. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder in adults. Am J Psychiatry 2000;157(4 suppl):1-45.
27. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference. Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-156.
28. Quitkin FM, Rabkin JG, Ross D, McGrath PJ. Duration of antidepressant drug treatment: what is an adequate trial? Arch Gen Psychiatry 1984;41:238-245.
29. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression, variable, substantial, and growing. JAMA 2002;287:1840-1847.
30. Gill D, Hatcher S. Antidepressants for depression in medical illness (Cochrane Review). In: The Cochrane Library,. 1, 2002. Oxford; Update Software.
31. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: A randomized trial. JAMA 2001;286:2947-2955.
32. Hartmann PM. Mirtazapine: a newer antidepressant. Am Fam Physician 1999;59:159-161.
33. Hurt RD, Sachs DPL, Glover ED, Offord KP, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
34. Clerc GE, Ruimy P, Verdeau-Pailles J, et al. A double-blind comparison of venlafaxine and fluoxetine in patients hospitalized for major depression and melancholia. Int Clin Psychopharmacology 1994;9:139-143.
35. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J of Psychiatry 2001;178:234-241.
36. Schweitzer E, Feighner J, Mandos L, Rickles K. Comparison of venlafaxine and imipramine in the acute treatment of major depression in outpatients. J Clin Psychiatry 1994;55:104-108.
37. Product information. Serzone(c) (nefazodone). Princeton, NJ Bristol Myers Squibb Co., 2002 (January).
38. Thase ME, Rush AJ, Howland RH, Kornstein SG, Kocsis JH, Gelenberg AJ, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59:233-239.
39. Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ. A proposed algorithm for improved recognition and treatment of depression/anxiety spectrum in primary care. J Clin Psychiatry 2001;3:44-52.
40. Thase ME, Feighner JP, Lydiard RB. Citalopram treatment of fluoxetine nonresponders. J Clin Psychiatry 2001;62:683-687.
41. Nelson JC, Mazure CM, Bowers MB, Jatlow PI. A preliminary, open study of the combination of fluoxetine and desipramine for rapid treatment of major depression. Arch Gen Psychiatry 1991;48:303-307.
42. Stein G, Bernardt M. Lithium augmentation therapy in tri-cyclic-resistant depression: a controlled trial using lithium in low and normal doses. Br J Psychiatry 1993;162:634-640.
43. Joffe RT, Singer W, Levitt AJ, et al. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-393.
44. Bauer M, Dopfmer S. Lithium augmentation in treatment-resistant depression: meta-analysis of placebo-controlled studies. J Clin Psychopharmacol 1999;19:427-434.
45. Harvey KV, Balon R. Augmentation with buspirone: a review. Ann Clin Psychiatry 1995;2:143-147.
46. Thase ME, Rush AJ. Treatment-resistant depression. In Bloom FE, Kupfer DJ. Psychopharmacology: The Fourth Generation of Progress. New York, NY: Karen Press; 1995;1081-1097.
47. Prange AJ Jr, Loosen PT, Wilson IC, Lipton MA. The therapeutic use of hormone of the thyroid axis in depression. In: Post R, Ballenger J. Neurobiology of mood disorders.. Vol 1. Baltimore, Md: Williams & Wilkins; 1980;311-322.
48. Gagne GG, Furman MJ, Carpenter LL, Price LH. Efficacy of continuation ECT and antidepressant drugs compared to long-term antidepressants alone in depressed patients. Am J Psychiatry 2000;157:1960-1965.
49. American Psychiatric Association.The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. Washington, DC: American Psychiatric Association, 1990.
50. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-134.
51. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6):22-29.
52. Montana CB. Recognition and treatment of depression in a primary care setting. J Clin Psychiatry 1994;55(suppl 1):18-34.
1. Whooley MA, Simon GE. Managing depression in medical outpatients. N Engl J Med 2000;343:1942-1950.
2. Keller MB, McCullough JP, Klein DN, et al. A comparison of nefazodone, the cognitive behavioral-analysis system for psychotherapy, and their combination for the treatment of chronic depression. N Engl J Med 2000;342:1462-1470.
3. Cohen L. Rational drug use in the treatment of depression. Pharmacotherapy 1997;17:45-61.
4. Bhatia SC, Bhatia SK. Depression in women: diagnostic and treatment considerations. Am Fam Physician 1999;60:225-240.
5. Glass RM. Treating depression as a recurrent or chronic disease. JAMA 1999;281:83.-
6. Harwitz D, Ravizza L. Psychiatric emergencies; suicide and depression. Emergency Medical Clinics of North America 2000;18:263-271.
7. Olfson M, Marcus S, Druss B, et al. National trends in the out-patient treatment of depression. JAMA 2002;287:203-209.
8. Stahl SM. Selecting an antidepressant by using mechanism of action to enhance efficacy and avoid side effects. J Clin Psychiatry 1998;59(supp1 18):23-29.
9. Stahl SM. Depression and bipolar disorders. In: Stahl SM. Essential Psychopharmacology, Neuroscientific Basis and Clinical Applications. Cambridge: Cambridge University Press, 1996;135-295.
10. Barbui C, Hotopf M, Freemantle N, et al. Treatment discontinuation with selective serotonin reuptake inhibitors (SSRIs) versus tricyclic antidepressants (TCAs) (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
11. Geddes JR, Freemantle N, Mason J, et al. Selective serotonin reuptake inhibitors (SSRIs) for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford: Update Software.
12. Banerjee S, Shamash K, Macdonald AJD, Mann AH. Randomized controlled trial of effect of intervention by psychogeriatric team on depression in frail elderly people at home. BMJ 1996;313:1058-1061.
13. Reynolds III CF, Frank E, Perel JM, et al. Nortriptyline and interpersonal psychotherapy as maintenance therapies for recurrent major depression. JAMA 1999;281:39-45.
14. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry 1999;56:1109-1115.
15. Fava GA, Rafanelli C, Grandi S, Grandi S, Conti S, Belluardo P. Prevention of recurrent depression with cognitive behavioral therapy: preliminary findings. Arch Gen Psychiatry 1998;55:816-820.
16. Linde K, Mulrow CD. St John’s wort for depression (Cochrane Review). In: The Cochrane Library, 1, 2002. Oxford; Update Software.
17. Williams Jr JW, Mulrow CD, Chiquette E, et al. A systematic review of newer pharmacotherapies for depression in adults: evidence report summary. Ann Intern Med 2000;132:743-756.
18. Lecrubier Y, Clerc G, Didi R, Kieser M. Efficacy of St. John’s wort extract WS 5570 in major depression: a double-blind, placebo-controlled trial. Am J Psychiatry 2002;159:1361-1366.
19. Shelton RC, Keller MB, Gelenberg A, et al. Effectiveness of St. John’s wort in major depression, a randomized controlled trial. JAMA 2001;285:1978-1986.
20. Hypericum Depression Trial Study Group. Effect of Hypericum perforatum (St. John’s wort) in major depressive disorder, a randomized controlled trial. JAMA 2002;287:1807-1814.
21. Evidence Report/Technology Assessment. Number 7, Treatment of depression-newer pharmacotherapies (AHCPR Publication No. 99-E014). Rockville, Md: US Department of Health and Human Services; 1999.
22. Nemets B, Stahl Z, Belmaker RH. Addition of Omega-3 fatty acid to maintenance medication treatment for recurrent unipolar depressive disorder. Am J Psychiatry 2002;159:477-479.
23. Jellin JM, Gregory P, Balz F, et al. Pharmacist’s Letter/Prescriber’s Letter. Natural Medicines Comprehensive Database. 3rd ed. Stockton, Calif: Therapeutic Research Faculty; 2000;925-928.
24. Blumenthal JA, Babyak MA, Moore KA, Craighead WE, et al. Effects of exercise training on older patients with major depression. Arch Intern Med 1999;159:2349-2356.
25. Lawler DA, Hopker SW. The effectiveness of exercise as an intervention in the management of depression: systematic review and meta-regression analysis of randomized controlled trials. Br Med J 2001;322:763-767.
26. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder in adults. Am J Psychiatry 2000;157(4 suppl):1-45.
27. Crismon ML, Trivedi M, Pigott TA, et al. The Texas Medication Algorithm Project: Report of the Texas Consensus Conference. Panel on medication treatment of major depressive disorder. J Clin Psychiatry 1999;60:142-156.
28. Quitkin FM, Rabkin JG, Ross D, McGrath PJ. Duration of antidepressant drug treatment: what is an adequate trial? Arch Gen Psychiatry 1984;41:238-245.
29. Walsh BT, Seidman SN, Sysko R, Gould M. Placebo response in studies of major depression, variable, substantial, and growing. JAMA 2002;287:1840-1847.
30. Gill D, Hatcher S. Antidepressants for depression in medical illness (Cochrane Review). In: The Cochrane Library,. 1, 2002. Oxford; Update Software.
31. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: A randomized trial. JAMA 2001;286:2947-2955.
32. Hartmann PM. Mirtazapine: a newer antidepressant. Am Fam Physician 1999;59:159-161.
33. Hurt RD, Sachs DPL, Glover ED, Offord KP, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med 1997;337:1195-1202.
34. Clerc GE, Ruimy P, Verdeau-Pailles J, et al. A double-blind comparison of venlafaxine and fluoxetine in patients hospitalized for major depression and melancholia. Int Clin Psychopharmacology 1994;9:139-143.
35. Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J of Psychiatry 2001;178:234-241.
36. Schweitzer E, Feighner J, Mandos L, Rickles K. Comparison of venlafaxine and imipramine in the acute treatment of major depression in outpatients. J Clin Psychiatry 1994;55:104-108.
37. Product information. Serzone(c) (nefazodone). Princeton, NJ Bristol Myers Squibb Co., 2002 (January).
38. Thase ME, Rush AJ, Howland RH, Kornstein SG, Kocsis JH, Gelenberg AJ, et al. Double-blind switch study of imipramine or sertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry 2002;59:233-239.
39. Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ. A proposed algorithm for improved recognition and treatment of depression/anxiety spectrum in primary care. J Clin Psychiatry 2001;3:44-52.
40. Thase ME, Feighner JP, Lydiard RB. Citalopram treatment of fluoxetine nonresponders. J Clin Psychiatry 2001;62:683-687.
41. Nelson JC, Mazure CM, Bowers MB, Jatlow PI. A preliminary, open study of the combination of fluoxetine and desipramine for rapid treatment of major depression. Arch Gen Psychiatry 1991;48:303-307.
42. Stein G, Bernardt M. Lithium augmentation therapy in tri-cyclic-resistant depression: a controlled trial using lithium in low and normal doses. Br J Psychiatry 1993;162:634-640.
43. Joffe RT, Singer W, Levitt AJ, et al. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-393.
44. Bauer M, Dopfmer S. Lithium augmentation in treatment-resistant depression: meta-analysis of placebo-controlled studies. J Clin Psychopharmacol 1999;19:427-434.
45. Harvey KV, Balon R. Augmentation with buspirone: a review. Ann Clin Psychiatry 1995;2:143-147.
46. Thase ME, Rush AJ. Treatment-resistant depression. In Bloom FE, Kupfer DJ. Psychopharmacology: The Fourth Generation of Progress. New York, NY: Karen Press; 1995;1081-1097.
47. Prange AJ Jr, Loosen PT, Wilson IC, Lipton MA. The therapeutic use of hormone of the thyroid axis in depression. In: Post R, Ballenger J. Neurobiology of mood disorders.. Vol 1. Baltimore, Md: Williams & Wilkins; 1980;311-322.
48. Gagne GG, Furman MJ, Carpenter LL, Price LH. Efficacy of continuation ECT and antidepressant drugs compared to long-term antidepressants alone in depressed patients. Am J Psychiatry 2000;157:1960-1965.
49. American Psychiatric Association.The practice of electroconvulsive therapy: recommendations for treatment, training, and privileging. Washington, DC: American Psychiatric Association, 1990.
50. Shelton RC, Tollefson GD, Tohen M, et al. A novel augmentation strategy for treating resistant major depression. Am J Psychiatry 2001;158:131-134.
51. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry 2001;62(suppl 6):22-29.
52. Montana CB. Recognition and treatment of depression in a primary care setting. J Clin Psychiatry 1994;55(suppl 1):18-34.
A thorough yet efficient exam identifies most problems in school athletes
- A complete medical history, preferably from the student and a parent, will reveal approximately 75% of problems affecting initial athletic participation (D).
- For asymptomatic athletes with no previous injuries, a 90-second screening musculoskeletal test will detect 90% of significant musculoskeletal injuries (A).
- A routine screening need not include noninvasive cardiac testing or laboratory tests such as ur inalysis, blood count, chemistry profile, lipid profile, ferritin level, or spirometry (B).
Is the preparticipation physical examination the best way to determine whether a student athlete can participate fully in his or her chosen sport? This examination has become the standard of care for the over 6 million high school and college students. While most athletes pass the exam without significant medical or orthopedic abnormalities being noted, it often detects conditions that may predispose an athlete to injury or limit full participation in certain activities. We describe an efficient approach to the preparticipation examination.
Although many organizations have adopted the preparticipation exam there has been considerable debate on its content and usefulness.1-4 Nevertheless, sponsoring institutions continue to require the medical evaluation prior to competition in organized athletics, so family physicians should be knowledgeable about the objectives and limitations of the exam.
The American Academy of Family Physicians, the American Academy of Pediatrics, the American Medical Society for Sports Medicine, the American Orthopedic Society for Sports Medicine, and the American Osteopathic Academy of Sports Medicine established the Preparticipation Physical Examination Task Force. The recommendations of this task force serve as a guide for the physician conducting these examinations for high school and collegiate athletes.5,6
Assessing risks of mortality and morbidity
The mortality associated with athletic participation is most often the result of sudden cardiac death, which occurs in about 0.5 per 100,000 high school athletes per academic year and is most commonly due to hypertrophic cardiomyopathy.7,8 Screening for predisposing conditions is limited by the low prevalence of relevant cardiovascular lesions in the general youth population, the low risk of sudden death even among persons with an unsuspected abnormality, and the large number of school athletes.7-9
An estimated 200,000 children and adolescents would have to be screened to detect the 500 athletes who are at risk for sudden cardiac death and the 1 person who would actually experience it.10 Even when cardiac abnormalities are detected, the findings leading to disqualification are most often rhythm and conduction abnormalities, valvular abnormalities, and systemic hypertension, which are not the cardiac abnormalities usually associated with sudden cardiac death in athletes.11,12
The majority of sudden deaths are associated with 4 sports: football, basketball, track, and soccer. Approximately 90% of athletic-field deaths have occurred in males, mostly high school athletes.7,13
More frequently than mortality, athletic participation places the individual at risk for acute injury or worsening of an underlying medical condition. These conditions are most commonly musculoskeletal, cardiovascular, or ophthalmologic (Table 1).5,9,21
Nine studies of the preparticipation exam done between 1980 and 1999 show general agreement on the rates at which it qualifies (84.8% to 96.6%), qualifies with conditions (3.1% to 13.9%), and disqualifies students for sports participation (0.2% to 2.6%).14-22
TABLE 1
Medical and orthopedic conditions resulting in additional evaluations
| Rifat, 1995* | Lively, 1999† | ||
|---|---|---|---|
| n=2,574 | n=596 | ||
| Pass with follow-up and/or restriction (12.6%) | Fail with follow-up (2.6%) | Follow-up or restriction (14.1%) | |
| Medical (% of overall total) | 76.6 | 74.1 | 55.4 |
| Cardiovascular | 18.3 | 35.0 | 63.0 |
| Dermatologic | 6.8 | ||
| Endocrinologic | 0.4 | ||
| Ear, nose, and throat | 9.6 | 2.5 | |
| Gastrointestinal | 0.9 | 2.2 | |
| Genitourinary | 9.6 | 12.5 | 8.7 |
| Gynecologic | 4.4 | ||
| Infectious | 0.4 | 6.5 | |
| Neurologic | 6.5 | ||
| Ophthalmologic | 26.0 | 25.0 | 6.5 |
| Psychological | 2.2 | ||
| Pulmonary | 14.2 | 2.5 | |
| Other‡ | 13.7 | 22.5 | |
| Total medical (%) | 100.0 | 100.0 | 100.0 |
| Orthopedic (% of overall total) | 23.4 | 25.9 | 44.6 |
| Ankle/Foot | 14.9 | 7.7 | 2.7 |
| Back/Neck | 22.4 | 14.3 | 5.4 |
| Elbow | 5.4 | ||
| Hand/Wrist | 1.5 | 10.9 | |
| Knee | 41.8 | 7.1 | 43.2 |
| Leg | 5.4 | ||
| Shoulder | 27.0 | ||
| Nonspecific pain/injury | 19.4 | 71.4 | |
| Total orthopedic (%) | 100.0 | 100.0 | 100.0 |
| ∗Studied junior high and high school students. Two individual s failed (nonspecific pain/injury). | |||
| †Studied college-aged students. One individual failed (complicated pregnancy). | |||
| ‡“Other ” includes abdominal pain, allergy, bruising, chest pain, chronic/recurrent illness, dizziness/syncope with exercise, surgery (recent). | |||
What should the medical history include?
The examining physician should obtain a medical history from each participant (strength of recommendation [SOR]: D). A complete medical history will identify approximately 75% of problems that will affect initial athletic participation and serves as the cornerstone of the exam.14,19 Most conditions requiring further evaluation or restriction will be identified from the medical history. Rifat and colleagues21 noted that a complete medical history accounted for 88% of the abnormal findings and 57% of the reasons cited for activity restriction. The Preparticipation Physical Evaluation Task Force has developed a history form that emphasizes the areas of greatest concern.5
In particular, examining physicians should ask regarding risk factors and symptoms of cardiovascular disease ( Table 2 ). You should confirm a positive response to any of these questions, and conduct further evaluation if necessary. Unfortunately, most athletes with hypertrophic cardiomyopathy do not report a history of syncope with exercise or a family history of premature sudden cardiac death due to the disease.
Musculoskeletal injury is a common cause for disqualification of an athlete.14,19,21 The most common injury to restrict participation is a knee injury, with an ankle injury the next most common.23 The strongest independent predictor of sports injuries is a previous injury (odds ratio [OR]=9.4) and exposure time (OR=6.9).24 DuRant and colleagues23 found that a previous knee injury or knee surgery was significantly associated with further knee injuries during the subsequent sports season when compared with individuals who did not report previous knee injury or surgery (30.6% vs. 7.2%, P=.0001).
Additional historical information has been recommended for inclusion (SOR: D). For example, the examining physician should question the athlete about wheezing during exercise. Due to the high rate of recurrence and potential for long-term adverse effects, he or she should also obtain a history of previous concussions. Other issues to be addressed include presence of a single bilateral organ and use of performance-enhancing medication. Finally, physicians should question female athletes regarding their menstrual history and other symptoms or signs of the female athletic triad (eating disorder, amenorrhea, and osteoporosis).
Always carefully review the information provided by the athlete and his or her parents. In 2 separate studies, minimal agreement was found between histories obtained from athletes and parents independently.19,25 We do not know which source provides the most accurate history; therefore, both the parents and student athlete should be questioned.
TABLE 2
Questions to help discern cardiovascular risk
| Have you ever passed out during or after exercise? |
| Have you ever been dizzy during or after exercise? |
| Have you ever had chest pain during or after exercise? |
| Do you get tired more quickly than your friends during exercise? |
| Have you ever had racing of your heart or skipped heartbeats? |
| Have you ever had high blood pressure or high cholesterol? |
| Have you been told you have a heart murmur? |
| Has any family member or relative died of heart problems or of sudden death before age 50? |
| Have you had a severe viral infection (for example, myocarditis or mononucleosis) within the last month? |
| Has a physician ever denied or restricted your participation in sports for any heart problem? |
What should the physical examination include ?
A complete physical examination is not necessary (SOR: D).5 The screening physical examination should include vital signs (ie, height, weight, and blood pressure) and visual acuity testing as well as a cardiovascular, pulmonary, abdominal, skin, genital (for males), and musculoskeletal examination. Further examination should be based on issues elicited during the history.
Cardiovascular examination
The cardiovascular examination requires an additional level of detail. Perform auscultation of the heart initially with the patient in both standing and supine position, and during various maneuvers (squat-to-stand, deep inspiration, or Valsalva’s maneuver), as these maneuvers can clarify the type of murmur.
Any systolic murmur grade III/VI or louder, any murmur that disrupts normal heart sounds, any diastolic murmur, or any murmur that intensifies with the previously described maneuvers should be evaluated further through diagnostic studies (echocardiography) or consultation prior to participation. Sinus bradycardia and systolic murmurs are commonly found, occurring in over 50% and between 30% and 50% of athletes, respectively; they do not warrant further evaluation in the asymptomatic athlete.26 Third and fourth heart sounds are also commonly found in asymptomatic athletes without underlying heart disease.26,27
Noninvasive cardiac testing (eg, electrocardiography, echocardiography, or exercise stress testing) should not be a routine part of the screening preparticipation exam (SOR: B ).7 These tests are not cost-effective in a population at relatively low risk for cardiac abnormalities and cannot consistently identify athletes at actual risk.28-32 For example, a substantial minority of subjects (11%) were found to have a clinically significant increased ventricular wall thickness, which made clinical interpretation of the echocardiographic findings difficult in individual athletes.28 Furthermore, some patients with hypertrophic cardiomyopathy are able to tolerate particularly intense athletic training and competition for many years, and even maintain high levels of achievement without incurring symptoms, disease progression, or sudden death.29
Echocardiography and stress testing are the most commonly recommended diagnostic tests for patients with an abnormal cardiovascular history or examination. With the assistance of clinical information, echocardiography is able to distinguish the nonobstructive hypertrophic cardiomyopathy from the athletic heart syndrome.33
Musculos keletal examination
A screening musculoskeletal history and examination in combination can be used for asymptomatic athletes with no previous injuries (Table 3) (SOR: A).34 An accurate history is able to detect over 90% of significant musculoskeletal injuries. The screening physical examination is 51% sensitive and 97% specific.34 If the athlete has either a previous injury or other signs or symptoms (ie, pain; tenderness; asymmetries in muscle bulk, strength, or range of motion; any obvious deformity) detected by the general screening examination or history, the general screening should be supplemented with relevant elements of a site-specific examination.
Additional forms of musculoskeletal evaluation are often performed for athletes to determine their general state of flexibility and muscular strength. While various degrees of hyperlaxity, muscular tightness, weakness, asymmetry of strength or flexibility, poor endurance, and abnormal foot configuration may predispose an athlete to increased risk of injury during sports competition, studies have failed to demonstrate conclusively that injuries are prevented by interventions aimed at correcting such abnormalities.35-37
TABLE 3
The “90-second” musculoskeletal screening examination
| Instruction | Observations |
|---|---|
| Stand facing examiner | Acromiclavicular joints: general habitus |
| Look at ceiling, floor, over both shoulders, touch ears to shoulder | Cervical spine motion |
| Shrug shoulders (resistance) | Trapezius strength |
| Abduct shoulders to 90° (resistance at 90°) | Deltoid strength |
| Full external rotation of arms | Shoulder motion |
| Flex and extend elbows | Elbow motion |
| Arms at sides, elbows at 90° flexed; pronate and supinate wrists | Elbow and wrist motion |
| Spread fingers; make fist | Hand and finger motion, strength, and deformities |
| Tighten (contract) quadriceps; quadriceps | Symmetry and knee effusions, ankle effusion relax |
| “Duck walk” away and towards examiner | Hip, knee, and ankle motions |
| Back to examiner | Shoulder symmetry; scoliosis |
| Knees straight, touch toes | Scoliosis, hip motion, hamstring tightness |
| Raise upon toes, heels | Calf symmetry, leg strength |
Role for lab tests?
Studies do not support the use of routine laboratory or other screening tests such as urinalysis, complete blood count, chemistry profile, lipid profile, ferritin level, or spirometry as part of the exam (SOR: B).38-41
Determining clearance
Occasionally, an abnormality or condition is found that may limit an athlete’s participation or predispose him or her to further injury. In these cases, the examining physician should review the following questions:5
- Does the problem place the athlete at increased risk for injury?
- Is another participant at risk for injury because of the problem?
- Can the athlete safely participate with treatment (ie, medication, rehabilitation, bracing, or padding)?
- Can limited participation be allowed while treatment is being completed?
- If clearance is denied only for certain sports or sport categories, in what activities can the athlete safely participate?
Physicians should base clearance to participate in a particular sport on previously published guidelines, such as the recommendations by the American Academy of Pediatrics, the 26th Bethesda Conference, and the American Heart Association.7,43,44 Participation recommendations are based on the specific diagnosis, though multiple factors such as the classification of the sport and the specific health status of the athlete affect the decision.44
Approach to the patient
While current research demonstrates that the preparticipation physical examination has no effect on the overall morbidity and mortality rates in athletes, these exams may fulfill other objectives. Furthermore, no harmful effects of these examinations have been reported, and the exam has become institutionalized in the athletic and sports medicine community. As such, physicians should base their evaluation on the best available evidence using the standard form shown in “Preparticipation physical evaluation for athletics.”6 (A copy of the Preparticipation Physical Evaluation form can be found at www.jfponline.com.) This may require that the physician work with local school systems to assure that they understand what constitutes an appropriate examination.
To assist future patient care decisions and research efforts, a standardized preparticipation physical examination with an associated form similar to the evaluation recommended by the Preparticipation Physical Evaluation Task Force should be uniformly implemented throughout the country. The use of consistent clearance criteria as recommended by the Preparticipation Physical Evaluation Task Force or the American Academy of Pediatrics (“Medical conditions and sports participation,” also available at www.jfponline.com) should be used, studied, and revised as needed.5,44
In addition to the exam, physicians should consider exploring other aspects of sports participation to assist athletes in reducing the risk of injury. Rules, equipment, or other factors may have a greater effect on decreasing the mortality and morbidity associated with athletic participation. A marked decrease in cervical spine injuries occurred following the rule change in football banning deliberate “spearing”—the use of the top of the helmet as the initial point of contact in making a tackle.41
1. MacAuley D. Does the preseason screening for cardiac disease really work?: the British perspective. Med Sci Sports Exerc 1998;30(Suppl):S345-S350.
2. Glover DW, Maron BJ. Profile of preparticipation cardiovascular screening for high school athletes. JAMA 1998;279:1817-9.
3. Pfister GC, Puffer JC, Maron BJ. Preparticipation cardiovascular screening for US collegiate student-athletes. JAMA 2000;283:1597-9.
4. Reich JD. It won’t be me next time: an opinion on preparticipation sports physicals. Am Fam Physician 2000;61:2618, 2620, 2625, 2629.-
5. Smith DM, Kovan JR, Rich BSE, Tanner SM. Preparticipation Physical Evaluation. 2nd ed. Minneapolis, Minn: McGraw-Hill Co; 1997;1-46.
6. Lombardo JA, Robinson JB, Smith DM, et al. Preparticipation physical examination. 1st ed. Kansas City, Mo: American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Orthopedic Society for Sports Medicine, American Osteopathic Academy of Sports Medicine; 1992.
7. Maron BJ, Thompson PD, Puffer JC, et al. Cardiovascular preparticipation screening of competitive athletes. A statement for health professionals from the Sudden Death Committee (clinical cardiology) and Congenital Cardiac Defects Committee (cardiovascular disease in the young), American Heart Association. Circulation 1996;94:850-6.
8. Maron BJ, Gohman TE, Aeppli D. Prevalence of sudden cardiac death during competitive sports activities in Minnesota high school athletes. J Am Coll Cardiol 1998;32:1881-4.
9. American Medical Association Board of Trustees, Group on Science and Technology. Athletic participation examinations for adolescents. Arch Pediatr Adolesc Med 1994;148:93-8.
10. Epstein SE, Maron BJ. Sudden death and the competitive athlete: perspectives on preparticipation screening studies. J Am Coll Cardiol 1986;7:220-30.
11. Pelliccia A, Maron BJ. Preparticipation cardiovascular evaluation of the competitive athlete: Perspectives from the 30-year Italian experience. Am J Cardiol 1995;75:827-9.
12. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364-9.
13. Cantu RC, Mueller FO. Fatalities and catastrophic injuries in high school and college sports, 1982-1997. Phys Sportsmed 1999;27:35-48.
14. Goldberg B, Saraniti A, Witman P, et al. Preparticipation sports assessment: an objective evaluation. Pediatrics 1980;66:736-45.
15. Linder CW, DuRant RH, Seklecki RM, Strong WB. Preparticipation health screening of young athletes: results of 1268 examinations. Am J Sports Med 1981;9:187-93.
16. Tennant FS, Jr, Sorenson K, Day CM. Benefits of preparticipation sports examinations. J Fam Pract 1981;13:287-8.
17. Thompson TR, Andrish JT, Bergfeld JA. A prospective study of preparticipation sports examinations of 2670 young athletes: method and results. Cleve Clin Q 1982;49:225-33.
18. DuRant R, Seymore C, Linder CW, Jay S. The preparticipation examination of athletes. Comparison of single and multiple examiners. Am J Dis Child. 1985;139:657-61.
19. Risser WL, Hoffman HM, Bellah GG, Jr. Frequency of preparticipation sports examinations in secondary school athletes: are the University Interscholastic League guidelines appropriate? Tex Med 1985;81:35-9.
20. Magnes SA, Henderson JM, Hunter SC. What limits sports participation: experience with 10,540 athletes. Phys Sportsmed 1992;20:143-60.
21. Rifat SF, Ruffin MT, Gorenflo DW. Disqualifying criteria in preparticipation sports evaluation. J Fam Pract 1995;41:42-50.
22. Lively MW. Preparticipation physical examinations: a collegiate experience. Clin J Sports Med 1999;9:38.-
23. DuRant RH, Pendergrast RA, Seymore C, Gaillard G, Donner J. Findings from the preparticipation athletic examination and athletic injuries. Am J Dis Child 1992;146:85-91.
24. Van Mechelen W, Twisk J, Molendijk A, Blom B, Snel J, Kemper HC. Subject-related risk factors for sports injuries: a 1-yr prospective study in young adults. Med Sci Sports Exerc 1996;28:1171-9.
25. Carek PJ, Futrell MA. Athlete’s view of the preparticipation physical examination: Attitudes toward certain health screening questions. Arch Fam Med 1999;8:307-12.
26. Huston TP, Puffer JC, Rodney WM. The athletic heart syndrome. N Engl J Med 1985;313:24-32.
27. Crawford MH, O’Rourke RA. The athlete’s heart. Adv Intern Med 1979;24:311-29.
28. Lewis JF, Maron BJ, Diggs JA, Spencer JE, Mehrotra PP, Curry CL. Preparticipation echocardiographic screening for cardiovascular disease in a large, predominately black population of collegiate athletes. Am J Cardiol 1989;64:1029-33.
29. Maron BJ, Klues HG. Surviving competitive athletes with hypertrophic cardiomyopathy. Am J Cardiol 1994;73:1098-104.
30. Fuller CM, McNulty CM, Spring DA, et al. Prospective screening of 5,615 high school athletes for risk of sudden death. Med Sci Sports Exer 1997;29:1131-8.
31. Fuller CM. Cost effectiveness of analysis of high school athletes for risks of sudden cardiac death. Med Sci Sports Exer 2000;32:887-90.
32. Pelliccia A, Maron BJ, Culasso F, et al. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000;102:278-84.
33. Maron BJ, Pelliccia A, Spirito P. Cardiac disease in young trained athletes: insights into methods for distinguishing athlete’s heart from structural heart disease with particular emphasis on hypertrophic cardiomyopathy. Circulation 1995;91:1596-1601.
34. Gomez JE, Landry GL, Bernhardt DT. Critical evaluation of the 2-minute orthopedic screening examination. Am J Dis Child 1993;147:1109-13.
35. Abbott HG, Kress JB. Preconditioning in the prevention of knee injuries. Arch Phys Med Rehabil 1969;50:326-33.
36. Jackson DW, Jarrett H, Bailey D, Kausek J, Swanson J, Powell JW. Injury prediction in the young athlete: a preliminary report. Am J Sports Med 1978;6:6-14.
37. Nicholas JA. Injuries in knee ligaments: Relationship to looseness and tightness in football players. JAMA 1970;212:2236-9.
38. Dodge WF, West EF, Smith EH, Harvey B 3rd. Proteinuria and hematuria in schoolchildren: epidemiology and early natural history. J Pediatr 1976;88:327-47.
39. Peggs JF, Reinhardt RW, O’Brien JM. Proteinuria in adolescent sports physical examinations. J Fam Pract 1986;22:80-1.
40. Rupp NT, Brudno DS, Guill MF. The value of screening for risk of exercise-induced asthma in high school athletes. Ann Allergy 1993;70:339-42.
41. Feinstein RA, LaRussa J, Wang-Dohlman A, Bartolucci AA. Screening adolescent athletes for exercise-induced asthma. Clin J Sports Med 1996;6:119-23.
42. Torg JS, Vegso JJ, Sennett B, Das M. The National Football Head and Neck Injury Registry. 14-year report on cervical quadriplegia, 1971 through 1984. JAMA 1985;254:3439-43.
43. 26th Bethesda Conference: Recommendations for determining eligibility for competition in athletes with cardiovascular abnormalities Med Sci Sports Exerc 1994;26(Suppl):S223-S283.
44. American Academy of Pediatrics. Medical conditions affecting sports participation. Pediatrics 2001;107:1206-7.
- A complete medical history, preferably from the student and a parent, will reveal approximately 75% of problems affecting initial athletic participation (D).
- For asymptomatic athletes with no previous injuries, a 90-second screening musculoskeletal test will detect 90% of significant musculoskeletal injuries (A).
- A routine screening need not include noninvasive cardiac testing or laboratory tests such as ur inalysis, blood count, chemistry profile, lipid profile, ferritin level, or spirometry (B).
Is the preparticipation physical examination the best way to determine whether a student athlete can participate fully in his or her chosen sport? This examination has become the standard of care for the over 6 million high school and college students. While most athletes pass the exam without significant medical or orthopedic abnormalities being noted, it often detects conditions that may predispose an athlete to injury or limit full participation in certain activities. We describe an efficient approach to the preparticipation examination.
Although many organizations have adopted the preparticipation exam there has been considerable debate on its content and usefulness.1-4 Nevertheless, sponsoring institutions continue to require the medical evaluation prior to competition in organized athletics, so family physicians should be knowledgeable about the objectives and limitations of the exam.
The American Academy of Family Physicians, the American Academy of Pediatrics, the American Medical Society for Sports Medicine, the American Orthopedic Society for Sports Medicine, and the American Osteopathic Academy of Sports Medicine established the Preparticipation Physical Examination Task Force. The recommendations of this task force serve as a guide for the physician conducting these examinations for high school and collegiate athletes.5,6
Assessing risks of mortality and morbidity
The mortality associated with athletic participation is most often the result of sudden cardiac death, which occurs in about 0.5 per 100,000 high school athletes per academic year and is most commonly due to hypertrophic cardiomyopathy.7,8 Screening for predisposing conditions is limited by the low prevalence of relevant cardiovascular lesions in the general youth population, the low risk of sudden death even among persons with an unsuspected abnormality, and the large number of school athletes.7-9
An estimated 200,000 children and adolescents would have to be screened to detect the 500 athletes who are at risk for sudden cardiac death and the 1 person who would actually experience it.10 Even when cardiac abnormalities are detected, the findings leading to disqualification are most often rhythm and conduction abnormalities, valvular abnormalities, and systemic hypertension, which are not the cardiac abnormalities usually associated with sudden cardiac death in athletes.11,12
The majority of sudden deaths are associated with 4 sports: football, basketball, track, and soccer. Approximately 90% of athletic-field deaths have occurred in males, mostly high school athletes.7,13
More frequently than mortality, athletic participation places the individual at risk for acute injury or worsening of an underlying medical condition. These conditions are most commonly musculoskeletal, cardiovascular, or ophthalmologic (Table 1).5,9,21
Nine studies of the preparticipation exam done between 1980 and 1999 show general agreement on the rates at which it qualifies (84.8% to 96.6%), qualifies with conditions (3.1% to 13.9%), and disqualifies students for sports participation (0.2% to 2.6%).14-22
TABLE 1
Medical and orthopedic conditions resulting in additional evaluations
| Rifat, 1995* | Lively, 1999† | ||
|---|---|---|---|
| n=2,574 | n=596 | ||
| Pass with follow-up and/or restriction (12.6%) | Fail with follow-up (2.6%) | Follow-up or restriction (14.1%) | |
| Medical (% of overall total) | 76.6 | 74.1 | 55.4 |
| Cardiovascular | 18.3 | 35.0 | 63.0 |
| Dermatologic | 6.8 | ||
| Endocrinologic | 0.4 | ||
| Ear, nose, and throat | 9.6 | 2.5 | |
| Gastrointestinal | 0.9 | 2.2 | |
| Genitourinary | 9.6 | 12.5 | 8.7 |
| Gynecologic | 4.4 | ||
| Infectious | 0.4 | 6.5 | |
| Neurologic | 6.5 | ||
| Ophthalmologic | 26.0 | 25.0 | 6.5 |
| Psychological | 2.2 | ||
| Pulmonary | 14.2 | 2.5 | |
| Other‡ | 13.7 | 22.5 | |
| Total medical (%) | 100.0 | 100.0 | 100.0 |
| Orthopedic (% of overall total) | 23.4 | 25.9 | 44.6 |
| Ankle/Foot | 14.9 | 7.7 | 2.7 |
| Back/Neck | 22.4 | 14.3 | 5.4 |
| Elbow | 5.4 | ||
| Hand/Wrist | 1.5 | 10.9 | |
| Knee | 41.8 | 7.1 | 43.2 |
| Leg | 5.4 | ||
| Shoulder | 27.0 | ||
| Nonspecific pain/injury | 19.4 | 71.4 | |
| Total orthopedic (%) | 100.0 | 100.0 | 100.0 |
| ∗Studied junior high and high school students. Two individual s failed (nonspecific pain/injury). | |||
| †Studied college-aged students. One individual failed (complicated pregnancy). | |||
| ‡“Other ” includes abdominal pain, allergy, bruising, chest pain, chronic/recurrent illness, dizziness/syncope with exercise, surgery (recent). | |||
What should the medical history include?
The examining physician should obtain a medical history from each participant (strength of recommendation [SOR]: D). A complete medical history will identify approximately 75% of problems that will affect initial athletic participation and serves as the cornerstone of the exam.14,19 Most conditions requiring further evaluation or restriction will be identified from the medical history. Rifat and colleagues21 noted that a complete medical history accounted for 88% of the abnormal findings and 57% of the reasons cited for activity restriction. The Preparticipation Physical Evaluation Task Force has developed a history form that emphasizes the areas of greatest concern.5
In particular, examining physicians should ask regarding risk factors and symptoms of cardiovascular disease ( Table 2 ). You should confirm a positive response to any of these questions, and conduct further evaluation if necessary. Unfortunately, most athletes with hypertrophic cardiomyopathy do not report a history of syncope with exercise or a family history of premature sudden cardiac death due to the disease.
Musculoskeletal injury is a common cause for disqualification of an athlete.14,19,21 The most common injury to restrict participation is a knee injury, with an ankle injury the next most common.23 The strongest independent predictor of sports injuries is a previous injury (odds ratio [OR]=9.4) and exposure time (OR=6.9).24 DuRant and colleagues23 found that a previous knee injury or knee surgery was significantly associated with further knee injuries during the subsequent sports season when compared with individuals who did not report previous knee injury or surgery (30.6% vs. 7.2%, P=.0001).
Additional historical information has been recommended for inclusion (SOR: D). For example, the examining physician should question the athlete about wheezing during exercise. Due to the high rate of recurrence and potential for long-term adverse effects, he or she should also obtain a history of previous concussions. Other issues to be addressed include presence of a single bilateral organ and use of performance-enhancing medication. Finally, physicians should question female athletes regarding their menstrual history and other symptoms or signs of the female athletic triad (eating disorder, amenorrhea, and osteoporosis).
Always carefully review the information provided by the athlete and his or her parents. In 2 separate studies, minimal agreement was found between histories obtained from athletes and parents independently.19,25 We do not know which source provides the most accurate history; therefore, both the parents and student athlete should be questioned.
TABLE 2
Questions to help discern cardiovascular risk
| Have you ever passed out during or after exercise? |
| Have you ever been dizzy during or after exercise? |
| Have you ever had chest pain during or after exercise? |
| Do you get tired more quickly than your friends during exercise? |
| Have you ever had racing of your heart or skipped heartbeats? |
| Have you ever had high blood pressure or high cholesterol? |
| Have you been told you have a heart murmur? |
| Has any family member or relative died of heart problems or of sudden death before age 50? |
| Have you had a severe viral infection (for example, myocarditis or mononucleosis) within the last month? |
| Has a physician ever denied or restricted your participation in sports for any heart problem? |
What should the physical examination include ?
A complete physical examination is not necessary (SOR: D).5 The screening physical examination should include vital signs (ie, height, weight, and blood pressure) and visual acuity testing as well as a cardiovascular, pulmonary, abdominal, skin, genital (for males), and musculoskeletal examination. Further examination should be based on issues elicited during the history.
Cardiovascular examination
The cardiovascular examination requires an additional level of detail. Perform auscultation of the heart initially with the patient in both standing and supine position, and during various maneuvers (squat-to-stand, deep inspiration, or Valsalva’s maneuver), as these maneuvers can clarify the type of murmur.
Any systolic murmur grade III/VI or louder, any murmur that disrupts normal heart sounds, any diastolic murmur, or any murmur that intensifies with the previously described maneuvers should be evaluated further through diagnostic studies (echocardiography) or consultation prior to participation. Sinus bradycardia and systolic murmurs are commonly found, occurring in over 50% and between 30% and 50% of athletes, respectively; they do not warrant further evaluation in the asymptomatic athlete.26 Third and fourth heart sounds are also commonly found in asymptomatic athletes without underlying heart disease.26,27
Noninvasive cardiac testing (eg, electrocardiography, echocardiography, or exercise stress testing) should not be a routine part of the screening preparticipation exam (SOR: B ).7 These tests are not cost-effective in a population at relatively low risk for cardiac abnormalities and cannot consistently identify athletes at actual risk.28-32 For example, a substantial minority of subjects (11%) were found to have a clinically significant increased ventricular wall thickness, which made clinical interpretation of the echocardiographic findings difficult in individual athletes.28 Furthermore, some patients with hypertrophic cardiomyopathy are able to tolerate particularly intense athletic training and competition for many years, and even maintain high levels of achievement without incurring symptoms, disease progression, or sudden death.29
Echocardiography and stress testing are the most commonly recommended diagnostic tests for patients with an abnormal cardiovascular history or examination. With the assistance of clinical information, echocardiography is able to distinguish the nonobstructive hypertrophic cardiomyopathy from the athletic heart syndrome.33
Musculos keletal examination
A screening musculoskeletal history and examination in combination can be used for asymptomatic athletes with no previous injuries (Table 3) (SOR: A).34 An accurate history is able to detect over 90% of significant musculoskeletal injuries. The screening physical examination is 51% sensitive and 97% specific.34 If the athlete has either a previous injury or other signs or symptoms (ie, pain; tenderness; asymmetries in muscle bulk, strength, or range of motion; any obvious deformity) detected by the general screening examination or history, the general screening should be supplemented with relevant elements of a site-specific examination.
Additional forms of musculoskeletal evaluation are often performed for athletes to determine their general state of flexibility and muscular strength. While various degrees of hyperlaxity, muscular tightness, weakness, asymmetry of strength or flexibility, poor endurance, and abnormal foot configuration may predispose an athlete to increased risk of injury during sports competition, studies have failed to demonstrate conclusively that injuries are prevented by interventions aimed at correcting such abnormalities.35-37
TABLE 3
The “90-second” musculoskeletal screening examination
| Instruction | Observations |
|---|---|
| Stand facing examiner | Acromiclavicular joints: general habitus |
| Look at ceiling, floor, over both shoulders, touch ears to shoulder | Cervical spine motion |
| Shrug shoulders (resistance) | Trapezius strength |
| Abduct shoulders to 90° (resistance at 90°) | Deltoid strength |
| Full external rotation of arms | Shoulder motion |
| Flex and extend elbows | Elbow motion |
| Arms at sides, elbows at 90° flexed; pronate and supinate wrists | Elbow and wrist motion |
| Spread fingers; make fist | Hand and finger motion, strength, and deformities |
| Tighten (contract) quadriceps; quadriceps | Symmetry and knee effusions, ankle effusion relax |
| “Duck walk” away and towards examiner | Hip, knee, and ankle motions |
| Back to examiner | Shoulder symmetry; scoliosis |
| Knees straight, touch toes | Scoliosis, hip motion, hamstring tightness |
| Raise upon toes, heels | Calf symmetry, leg strength |
Role for lab tests?
Studies do not support the use of routine laboratory or other screening tests such as urinalysis, complete blood count, chemistry profile, lipid profile, ferritin level, or spirometry as part of the exam (SOR: B).38-41
Determining clearance
Occasionally, an abnormality or condition is found that may limit an athlete’s participation or predispose him or her to further injury. In these cases, the examining physician should review the following questions:5
- Does the problem place the athlete at increased risk for injury?
- Is another participant at risk for injury because of the problem?
- Can the athlete safely participate with treatment (ie, medication, rehabilitation, bracing, or padding)?
- Can limited participation be allowed while treatment is being completed?
- If clearance is denied only for certain sports or sport categories, in what activities can the athlete safely participate?
Physicians should base clearance to participate in a particular sport on previously published guidelines, such as the recommendations by the American Academy of Pediatrics, the 26th Bethesda Conference, and the American Heart Association.7,43,44 Participation recommendations are based on the specific diagnosis, though multiple factors such as the classification of the sport and the specific health status of the athlete affect the decision.44
Approach to the patient
While current research demonstrates that the preparticipation physical examination has no effect on the overall morbidity and mortality rates in athletes, these exams may fulfill other objectives. Furthermore, no harmful effects of these examinations have been reported, and the exam has become institutionalized in the athletic and sports medicine community. As such, physicians should base their evaluation on the best available evidence using the standard form shown in “Preparticipation physical evaluation for athletics.”6 (A copy of the Preparticipation Physical Evaluation form can be found at www.jfponline.com.) This may require that the physician work with local school systems to assure that they understand what constitutes an appropriate examination.
To assist future patient care decisions and research efforts, a standardized preparticipation physical examination with an associated form similar to the evaluation recommended by the Preparticipation Physical Evaluation Task Force should be uniformly implemented throughout the country. The use of consistent clearance criteria as recommended by the Preparticipation Physical Evaluation Task Force or the American Academy of Pediatrics (“Medical conditions and sports participation,” also available at www.jfponline.com) should be used, studied, and revised as needed.5,44
In addition to the exam, physicians should consider exploring other aspects of sports participation to assist athletes in reducing the risk of injury. Rules, equipment, or other factors may have a greater effect on decreasing the mortality and morbidity associated with athletic participation. A marked decrease in cervical spine injuries occurred following the rule change in football banning deliberate “spearing”—the use of the top of the helmet as the initial point of contact in making a tackle.41
- A complete medical history, preferably from the student and a parent, will reveal approximately 75% of problems affecting initial athletic participation (D).
- For asymptomatic athletes with no previous injuries, a 90-second screening musculoskeletal test will detect 90% of significant musculoskeletal injuries (A).
- A routine screening need not include noninvasive cardiac testing or laboratory tests such as ur inalysis, blood count, chemistry profile, lipid profile, ferritin level, or spirometry (B).
Is the preparticipation physical examination the best way to determine whether a student athlete can participate fully in his or her chosen sport? This examination has become the standard of care for the over 6 million high school and college students. While most athletes pass the exam without significant medical or orthopedic abnormalities being noted, it often detects conditions that may predispose an athlete to injury or limit full participation in certain activities. We describe an efficient approach to the preparticipation examination.
Although many organizations have adopted the preparticipation exam there has been considerable debate on its content and usefulness.1-4 Nevertheless, sponsoring institutions continue to require the medical evaluation prior to competition in organized athletics, so family physicians should be knowledgeable about the objectives and limitations of the exam.
The American Academy of Family Physicians, the American Academy of Pediatrics, the American Medical Society for Sports Medicine, the American Orthopedic Society for Sports Medicine, and the American Osteopathic Academy of Sports Medicine established the Preparticipation Physical Examination Task Force. The recommendations of this task force serve as a guide for the physician conducting these examinations for high school and collegiate athletes.5,6
Assessing risks of mortality and morbidity
The mortality associated with athletic participation is most often the result of sudden cardiac death, which occurs in about 0.5 per 100,000 high school athletes per academic year and is most commonly due to hypertrophic cardiomyopathy.7,8 Screening for predisposing conditions is limited by the low prevalence of relevant cardiovascular lesions in the general youth population, the low risk of sudden death even among persons with an unsuspected abnormality, and the large number of school athletes.7-9
An estimated 200,000 children and adolescents would have to be screened to detect the 500 athletes who are at risk for sudden cardiac death and the 1 person who would actually experience it.10 Even when cardiac abnormalities are detected, the findings leading to disqualification are most often rhythm and conduction abnormalities, valvular abnormalities, and systemic hypertension, which are not the cardiac abnormalities usually associated with sudden cardiac death in athletes.11,12
The majority of sudden deaths are associated with 4 sports: football, basketball, track, and soccer. Approximately 90% of athletic-field deaths have occurred in males, mostly high school athletes.7,13
More frequently than mortality, athletic participation places the individual at risk for acute injury or worsening of an underlying medical condition. These conditions are most commonly musculoskeletal, cardiovascular, or ophthalmologic (Table 1).5,9,21
Nine studies of the preparticipation exam done between 1980 and 1999 show general agreement on the rates at which it qualifies (84.8% to 96.6%), qualifies with conditions (3.1% to 13.9%), and disqualifies students for sports participation (0.2% to 2.6%).14-22
TABLE 1
Medical and orthopedic conditions resulting in additional evaluations
| Rifat, 1995* | Lively, 1999† | ||
|---|---|---|---|
| n=2,574 | n=596 | ||
| Pass with follow-up and/or restriction (12.6%) | Fail with follow-up (2.6%) | Follow-up or restriction (14.1%) | |
| Medical (% of overall total) | 76.6 | 74.1 | 55.4 |
| Cardiovascular | 18.3 | 35.0 | 63.0 |
| Dermatologic | 6.8 | ||
| Endocrinologic | 0.4 | ||
| Ear, nose, and throat | 9.6 | 2.5 | |
| Gastrointestinal | 0.9 | 2.2 | |
| Genitourinary | 9.6 | 12.5 | 8.7 |
| Gynecologic | 4.4 | ||
| Infectious | 0.4 | 6.5 | |
| Neurologic | 6.5 | ||
| Ophthalmologic | 26.0 | 25.0 | 6.5 |
| Psychological | 2.2 | ||
| Pulmonary | 14.2 | 2.5 | |
| Other‡ | 13.7 | 22.5 | |
| Total medical (%) | 100.0 | 100.0 | 100.0 |
| Orthopedic (% of overall total) | 23.4 | 25.9 | 44.6 |
| Ankle/Foot | 14.9 | 7.7 | 2.7 |
| Back/Neck | 22.4 | 14.3 | 5.4 |
| Elbow | 5.4 | ||
| Hand/Wrist | 1.5 | 10.9 | |
| Knee | 41.8 | 7.1 | 43.2 |
| Leg | 5.4 | ||
| Shoulder | 27.0 | ||
| Nonspecific pain/injury | 19.4 | 71.4 | |
| Total orthopedic (%) | 100.0 | 100.0 | 100.0 |
| ∗Studied junior high and high school students. Two individual s failed (nonspecific pain/injury). | |||
| †Studied college-aged students. One individual failed (complicated pregnancy). | |||
| ‡“Other ” includes abdominal pain, allergy, bruising, chest pain, chronic/recurrent illness, dizziness/syncope with exercise, surgery (recent). | |||
What should the medical history include?
The examining physician should obtain a medical history from each participant (strength of recommendation [SOR]: D). A complete medical history will identify approximately 75% of problems that will affect initial athletic participation and serves as the cornerstone of the exam.14,19 Most conditions requiring further evaluation or restriction will be identified from the medical history. Rifat and colleagues21 noted that a complete medical history accounted for 88% of the abnormal findings and 57% of the reasons cited for activity restriction. The Preparticipation Physical Evaluation Task Force has developed a history form that emphasizes the areas of greatest concern.5
In particular, examining physicians should ask regarding risk factors and symptoms of cardiovascular disease ( Table 2 ). You should confirm a positive response to any of these questions, and conduct further evaluation if necessary. Unfortunately, most athletes with hypertrophic cardiomyopathy do not report a history of syncope with exercise or a family history of premature sudden cardiac death due to the disease.
Musculoskeletal injury is a common cause for disqualification of an athlete.14,19,21 The most common injury to restrict participation is a knee injury, with an ankle injury the next most common.23 The strongest independent predictor of sports injuries is a previous injury (odds ratio [OR]=9.4) and exposure time (OR=6.9).24 DuRant and colleagues23 found that a previous knee injury or knee surgery was significantly associated with further knee injuries during the subsequent sports season when compared with individuals who did not report previous knee injury or surgery (30.6% vs. 7.2%, P=.0001).
Additional historical information has been recommended for inclusion (SOR: D). For example, the examining physician should question the athlete about wheezing during exercise. Due to the high rate of recurrence and potential for long-term adverse effects, he or she should also obtain a history of previous concussions. Other issues to be addressed include presence of a single bilateral organ and use of performance-enhancing medication. Finally, physicians should question female athletes regarding their menstrual history and other symptoms or signs of the female athletic triad (eating disorder, amenorrhea, and osteoporosis).
Always carefully review the information provided by the athlete and his or her parents. In 2 separate studies, minimal agreement was found between histories obtained from athletes and parents independently.19,25 We do not know which source provides the most accurate history; therefore, both the parents and student athlete should be questioned.
TABLE 2
Questions to help discern cardiovascular risk
| Have you ever passed out during or after exercise? |
| Have you ever been dizzy during or after exercise? |
| Have you ever had chest pain during or after exercise? |
| Do you get tired more quickly than your friends during exercise? |
| Have you ever had racing of your heart or skipped heartbeats? |
| Have you ever had high blood pressure or high cholesterol? |
| Have you been told you have a heart murmur? |
| Has any family member or relative died of heart problems or of sudden death before age 50? |
| Have you had a severe viral infection (for example, myocarditis or mononucleosis) within the last month? |
| Has a physician ever denied or restricted your participation in sports for any heart problem? |
What should the physical examination include ?
A complete physical examination is not necessary (SOR: D).5 The screening physical examination should include vital signs (ie, height, weight, and blood pressure) and visual acuity testing as well as a cardiovascular, pulmonary, abdominal, skin, genital (for males), and musculoskeletal examination. Further examination should be based on issues elicited during the history.
Cardiovascular examination
The cardiovascular examination requires an additional level of detail. Perform auscultation of the heart initially with the patient in both standing and supine position, and during various maneuvers (squat-to-stand, deep inspiration, or Valsalva’s maneuver), as these maneuvers can clarify the type of murmur.
Any systolic murmur grade III/VI or louder, any murmur that disrupts normal heart sounds, any diastolic murmur, or any murmur that intensifies with the previously described maneuvers should be evaluated further through diagnostic studies (echocardiography) or consultation prior to participation. Sinus bradycardia and systolic murmurs are commonly found, occurring in over 50% and between 30% and 50% of athletes, respectively; they do not warrant further evaluation in the asymptomatic athlete.26 Third and fourth heart sounds are also commonly found in asymptomatic athletes without underlying heart disease.26,27
Noninvasive cardiac testing (eg, electrocardiography, echocardiography, or exercise stress testing) should not be a routine part of the screening preparticipation exam (SOR: B ).7 These tests are not cost-effective in a population at relatively low risk for cardiac abnormalities and cannot consistently identify athletes at actual risk.28-32 For example, a substantial minority of subjects (11%) were found to have a clinically significant increased ventricular wall thickness, which made clinical interpretation of the echocardiographic findings difficult in individual athletes.28 Furthermore, some patients with hypertrophic cardiomyopathy are able to tolerate particularly intense athletic training and competition for many years, and even maintain high levels of achievement without incurring symptoms, disease progression, or sudden death.29
Echocardiography and stress testing are the most commonly recommended diagnostic tests for patients with an abnormal cardiovascular history or examination. With the assistance of clinical information, echocardiography is able to distinguish the nonobstructive hypertrophic cardiomyopathy from the athletic heart syndrome.33
Musculos keletal examination
A screening musculoskeletal history and examination in combination can be used for asymptomatic athletes with no previous injuries (Table 3) (SOR: A).34 An accurate history is able to detect over 90% of significant musculoskeletal injuries. The screening physical examination is 51% sensitive and 97% specific.34 If the athlete has either a previous injury or other signs or symptoms (ie, pain; tenderness; asymmetries in muscle bulk, strength, or range of motion; any obvious deformity) detected by the general screening examination or history, the general screening should be supplemented with relevant elements of a site-specific examination.
Additional forms of musculoskeletal evaluation are often performed for athletes to determine their general state of flexibility and muscular strength. While various degrees of hyperlaxity, muscular tightness, weakness, asymmetry of strength or flexibility, poor endurance, and abnormal foot configuration may predispose an athlete to increased risk of injury during sports competition, studies have failed to demonstrate conclusively that injuries are prevented by interventions aimed at correcting such abnormalities.35-37
TABLE 3
The “90-second” musculoskeletal screening examination
| Instruction | Observations |
|---|---|
| Stand facing examiner | Acromiclavicular joints: general habitus |
| Look at ceiling, floor, over both shoulders, touch ears to shoulder | Cervical spine motion |
| Shrug shoulders (resistance) | Trapezius strength |
| Abduct shoulders to 90° (resistance at 90°) | Deltoid strength |
| Full external rotation of arms | Shoulder motion |
| Flex and extend elbows | Elbow motion |
| Arms at sides, elbows at 90° flexed; pronate and supinate wrists | Elbow and wrist motion |
| Spread fingers; make fist | Hand and finger motion, strength, and deformities |
| Tighten (contract) quadriceps; quadriceps | Symmetry and knee effusions, ankle effusion relax |
| “Duck walk” away and towards examiner | Hip, knee, and ankle motions |
| Back to examiner | Shoulder symmetry; scoliosis |
| Knees straight, touch toes | Scoliosis, hip motion, hamstring tightness |
| Raise upon toes, heels | Calf symmetry, leg strength |
Role for lab tests?
Studies do not support the use of routine laboratory or other screening tests such as urinalysis, complete blood count, chemistry profile, lipid profile, ferritin level, or spirometry as part of the exam (SOR: B).38-41
Determining clearance
Occasionally, an abnormality or condition is found that may limit an athlete’s participation or predispose him or her to further injury. In these cases, the examining physician should review the following questions:5
- Does the problem place the athlete at increased risk for injury?
- Is another participant at risk for injury because of the problem?
- Can the athlete safely participate with treatment (ie, medication, rehabilitation, bracing, or padding)?
- Can limited participation be allowed while treatment is being completed?
- If clearance is denied only for certain sports or sport categories, in what activities can the athlete safely participate?
Physicians should base clearance to participate in a particular sport on previously published guidelines, such as the recommendations by the American Academy of Pediatrics, the 26th Bethesda Conference, and the American Heart Association.7,43,44 Participation recommendations are based on the specific diagnosis, though multiple factors such as the classification of the sport and the specific health status of the athlete affect the decision.44
Approach to the patient
While current research demonstrates that the preparticipation physical examination has no effect on the overall morbidity and mortality rates in athletes, these exams may fulfill other objectives. Furthermore, no harmful effects of these examinations have been reported, and the exam has become institutionalized in the athletic and sports medicine community. As such, physicians should base their evaluation on the best available evidence using the standard form shown in “Preparticipation physical evaluation for athletics.”6 (A copy of the Preparticipation Physical Evaluation form can be found at www.jfponline.com.) This may require that the physician work with local school systems to assure that they understand what constitutes an appropriate examination.
To assist future patient care decisions and research efforts, a standardized preparticipation physical examination with an associated form similar to the evaluation recommended by the Preparticipation Physical Evaluation Task Force should be uniformly implemented throughout the country. The use of consistent clearance criteria as recommended by the Preparticipation Physical Evaluation Task Force or the American Academy of Pediatrics (“Medical conditions and sports participation,” also available at www.jfponline.com) should be used, studied, and revised as needed.5,44
In addition to the exam, physicians should consider exploring other aspects of sports participation to assist athletes in reducing the risk of injury. Rules, equipment, or other factors may have a greater effect on decreasing the mortality and morbidity associated with athletic participation. A marked decrease in cervical spine injuries occurred following the rule change in football banning deliberate “spearing”—the use of the top of the helmet as the initial point of contact in making a tackle.41
1. MacAuley D. Does the preseason screening for cardiac disease really work?: the British perspective. Med Sci Sports Exerc 1998;30(Suppl):S345-S350.
2. Glover DW, Maron BJ. Profile of preparticipation cardiovascular screening for high school athletes. JAMA 1998;279:1817-9.
3. Pfister GC, Puffer JC, Maron BJ. Preparticipation cardiovascular screening for US collegiate student-athletes. JAMA 2000;283:1597-9.
4. Reich JD. It won’t be me next time: an opinion on preparticipation sports physicals. Am Fam Physician 2000;61:2618, 2620, 2625, 2629.-
5. Smith DM, Kovan JR, Rich BSE, Tanner SM. Preparticipation Physical Evaluation. 2nd ed. Minneapolis, Minn: McGraw-Hill Co; 1997;1-46.
6. Lombardo JA, Robinson JB, Smith DM, et al. Preparticipation physical examination. 1st ed. Kansas City, Mo: American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Orthopedic Society for Sports Medicine, American Osteopathic Academy of Sports Medicine; 1992.
7. Maron BJ, Thompson PD, Puffer JC, et al. Cardiovascular preparticipation screening of competitive athletes. A statement for health professionals from the Sudden Death Committee (clinical cardiology) and Congenital Cardiac Defects Committee (cardiovascular disease in the young), American Heart Association. Circulation 1996;94:850-6.
8. Maron BJ, Gohman TE, Aeppli D. Prevalence of sudden cardiac death during competitive sports activities in Minnesota high school athletes. J Am Coll Cardiol 1998;32:1881-4.
9. American Medical Association Board of Trustees, Group on Science and Technology. Athletic participation examinations for adolescents. Arch Pediatr Adolesc Med 1994;148:93-8.
10. Epstein SE, Maron BJ. Sudden death and the competitive athlete: perspectives on preparticipation screening studies. J Am Coll Cardiol 1986;7:220-30.
11. Pelliccia A, Maron BJ. Preparticipation cardiovascular evaluation of the competitive athlete: Perspectives from the 30-year Italian experience. Am J Cardiol 1995;75:827-9.
12. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364-9.
13. Cantu RC, Mueller FO. Fatalities and catastrophic injuries in high school and college sports, 1982-1997. Phys Sportsmed 1999;27:35-48.
14. Goldberg B, Saraniti A, Witman P, et al. Preparticipation sports assessment: an objective evaluation. Pediatrics 1980;66:736-45.
15. Linder CW, DuRant RH, Seklecki RM, Strong WB. Preparticipation health screening of young athletes: results of 1268 examinations. Am J Sports Med 1981;9:187-93.
16. Tennant FS, Jr, Sorenson K, Day CM. Benefits of preparticipation sports examinations. J Fam Pract 1981;13:287-8.
17. Thompson TR, Andrish JT, Bergfeld JA. A prospective study of preparticipation sports examinations of 2670 young athletes: method and results. Cleve Clin Q 1982;49:225-33.
18. DuRant R, Seymore C, Linder CW, Jay S. The preparticipation examination of athletes. Comparison of single and multiple examiners. Am J Dis Child. 1985;139:657-61.
19. Risser WL, Hoffman HM, Bellah GG, Jr. Frequency of preparticipation sports examinations in secondary school athletes: are the University Interscholastic League guidelines appropriate? Tex Med 1985;81:35-9.
20. Magnes SA, Henderson JM, Hunter SC. What limits sports participation: experience with 10,540 athletes. Phys Sportsmed 1992;20:143-60.
21. Rifat SF, Ruffin MT, Gorenflo DW. Disqualifying criteria in preparticipation sports evaluation. J Fam Pract 1995;41:42-50.
22. Lively MW. Preparticipation physical examinations: a collegiate experience. Clin J Sports Med 1999;9:38.-
23. DuRant RH, Pendergrast RA, Seymore C, Gaillard G, Donner J. Findings from the preparticipation athletic examination and athletic injuries. Am J Dis Child 1992;146:85-91.
24. Van Mechelen W, Twisk J, Molendijk A, Blom B, Snel J, Kemper HC. Subject-related risk factors for sports injuries: a 1-yr prospective study in young adults. Med Sci Sports Exerc 1996;28:1171-9.
25. Carek PJ, Futrell MA. Athlete’s view of the preparticipation physical examination: Attitudes toward certain health screening questions. Arch Fam Med 1999;8:307-12.
26. Huston TP, Puffer JC, Rodney WM. The athletic heart syndrome. N Engl J Med 1985;313:24-32.
27. Crawford MH, O’Rourke RA. The athlete’s heart. Adv Intern Med 1979;24:311-29.
28. Lewis JF, Maron BJ, Diggs JA, Spencer JE, Mehrotra PP, Curry CL. Preparticipation echocardiographic screening for cardiovascular disease in a large, predominately black population of collegiate athletes. Am J Cardiol 1989;64:1029-33.
29. Maron BJ, Klues HG. Surviving competitive athletes with hypertrophic cardiomyopathy. Am J Cardiol 1994;73:1098-104.
30. Fuller CM, McNulty CM, Spring DA, et al. Prospective screening of 5,615 high school athletes for risk of sudden death. Med Sci Sports Exer 1997;29:1131-8.
31. Fuller CM. Cost effectiveness of analysis of high school athletes for risks of sudden cardiac death. Med Sci Sports Exer 2000;32:887-90.
32. Pelliccia A, Maron BJ, Culasso F, et al. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000;102:278-84.
33. Maron BJ, Pelliccia A, Spirito P. Cardiac disease in young trained athletes: insights into methods for distinguishing athlete’s heart from structural heart disease with particular emphasis on hypertrophic cardiomyopathy. Circulation 1995;91:1596-1601.
34. Gomez JE, Landry GL, Bernhardt DT. Critical evaluation of the 2-minute orthopedic screening examination. Am J Dis Child 1993;147:1109-13.
35. Abbott HG, Kress JB. Preconditioning in the prevention of knee injuries. Arch Phys Med Rehabil 1969;50:326-33.
36. Jackson DW, Jarrett H, Bailey D, Kausek J, Swanson J, Powell JW. Injury prediction in the young athlete: a preliminary report. Am J Sports Med 1978;6:6-14.
37. Nicholas JA. Injuries in knee ligaments: Relationship to looseness and tightness in football players. JAMA 1970;212:2236-9.
38. Dodge WF, West EF, Smith EH, Harvey B 3rd. Proteinuria and hematuria in schoolchildren: epidemiology and early natural history. J Pediatr 1976;88:327-47.
39. Peggs JF, Reinhardt RW, O’Brien JM. Proteinuria in adolescent sports physical examinations. J Fam Pract 1986;22:80-1.
40. Rupp NT, Brudno DS, Guill MF. The value of screening for risk of exercise-induced asthma in high school athletes. Ann Allergy 1993;70:339-42.
41. Feinstein RA, LaRussa J, Wang-Dohlman A, Bartolucci AA. Screening adolescent athletes for exercise-induced asthma. Clin J Sports Med 1996;6:119-23.
42. Torg JS, Vegso JJ, Sennett B, Das M. The National Football Head and Neck Injury Registry. 14-year report on cervical quadriplegia, 1971 through 1984. JAMA 1985;254:3439-43.
43. 26th Bethesda Conference: Recommendations for determining eligibility for competition in athletes with cardiovascular abnormalities Med Sci Sports Exerc 1994;26(Suppl):S223-S283.
44. American Academy of Pediatrics. Medical conditions affecting sports participation. Pediatrics 2001;107:1206-7.
1. MacAuley D. Does the preseason screening for cardiac disease really work?: the British perspective. Med Sci Sports Exerc 1998;30(Suppl):S345-S350.
2. Glover DW, Maron BJ. Profile of preparticipation cardiovascular screening for high school athletes. JAMA 1998;279:1817-9.
3. Pfister GC, Puffer JC, Maron BJ. Preparticipation cardiovascular screening for US collegiate student-athletes. JAMA 2000;283:1597-9.
4. Reich JD. It won’t be me next time: an opinion on preparticipation sports physicals. Am Fam Physician 2000;61:2618, 2620, 2625, 2629.-
5. Smith DM, Kovan JR, Rich BSE, Tanner SM. Preparticipation Physical Evaluation. 2nd ed. Minneapolis, Minn: McGraw-Hill Co; 1997;1-46.
6. Lombardo JA, Robinson JB, Smith DM, et al. Preparticipation physical examination. 1st ed. Kansas City, Mo: American Academy of Family Physicians, American Academy of Pediatrics, American Medical Society for Sports Medicine, American Orthopedic Society for Sports Medicine, American Osteopathic Academy of Sports Medicine; 1992.
7. Maron BJ, Thompson PD, Puffer JC, et al. Cardiovascular preparticipation screening of competitive athletes. A statement for health professionals from the Sudden Death Committee (clinical cardiology) and Congenital Cardiac Defects Committee (cardiovascular disease in the young), American Heart Association. Circulation 1996;94:850-6.
8. Maron BJ, Gohman TE, Aeppli D. Prevalence of sudden cardiac death during competitive sports activities in Minnesota high school athletes. J Am Coll Cardiol 1998;32:1881-4.
9. American Medical Association Board of Trustees, Group on Science and Technology. Athletic participation examinations for adolescents. Arch Pediatr Adolesc Med 1994;148:93-8.
10. Epstein SE, Maron BJ. Sudden death and the competitive athlete: perspectives on preparticipation screening studies. J Am Coll Cardiol 1986;7:220-30.
11. Pelliccia A, Maron BJ. Preparticipation cardiovascular evaluation of the competitive athlete: Perspectives from the 30-year Italian experience. Am J Cardiol 1995;75:827-9.
12. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364-9.
13. Cantu RC, Mueller FO. Fatalities and catastrophic injuries in high school and college sports, 1982-1997. Phys Sportsmed 1999;27:35-48.
14. Goldberg B, Saraniti A, Witman P, et al. Preparticipation sports assessment: an objective evaluation. Pediatrics 1980;66:736-45.
15. Linder CW, DuRant RH, Seklecki RM, Strong WB. Preparticipation health screening of young athletes: results of 1268 examinations. Am J Sports Med 1981;9:187-93.
16. Tennant FS, Jr, Sorenson K, Day CM. Benefits of preparticipation sports examinations. J Fam Pract 1981;13:287-8.
17. Thompson TR, Andrish JT, Bergfeld JA. A prospective study of preparticipation sports examinations of 2670 young athletes: method and results. Cleve Clin Q 1982;49:225-33.
18. DuRant R, Seymore C, Linder CW, Jay S. The preparticipation examination of athletes. Comparison of single and multiple examiners. Am J Dis Child. 1985;139:657-61.
19. Risser WL, Hoffman HM, Bellah GG, Jr. Frequency of preparticipation sports examinations in secondary school athletes: are the University Interscholastic League guidelines appropriate? Tex Med 1985;81:35-9.
20. Magnes SA, Henderson JM, Hunter SC. What limits sports participation: experience with 10,540 athletes. Phys Sportsmed 1992;20:143-60.
21. Rifat SF, Ruffin MT, Gorenflo DW. Disqualifying criteria in preparticipation sports evaluation. J Fam Pract 1995;41:42-50.
22. Lively MW. Preparticipation physical examinations: a collegiate experience. Clin J Sports Med 1999;9:38.-
23. DuRant RH, Pendergrast RA, Seymore C, Gaillard G, Donner J. Findings from the preparticipation athletic examination and athletic injuries. Am J Dis Child 1992;146:85-91.
24. Van Mechelen W, Twisk J, Molendijk A, Blom B, Snel J, Kemper HC. Subject-related risk factors for sports injuries: a 1-yr prospective study in young adults. Med Sci Sports Exerc 1996;28:1171-9.
25. Carek PJ, Futrell MA. Athlete’s view of the preparticipation physical examination: Attitudes toward certain health screening questions. Arch Fam Med 1999;8:307-12.
26. Huston TP, Puffer JC, Rodney WM. The athletic heart syndrome. N Engl J Med 1985;313:24-32.
27. Crawford MH, O’Rourke RA. The athlete’s heart. Adv Intern Med 1979;24:311-29.
28. Lewis JF, Maron BJ, Diggs JA, Spencer JE, Mehrotra PP, Curry CL. Preparticipation echocardiographic screening for cardiovascular disease in a large, predominately black population of collegiate athletes. Am J Cardiol 1989;64:1029-33.
29. Maron BJ, Klues HG. Surviving competitive athletes with hypertrophic cardiomyopathy. Am J Cardiol 1994;73:1098-104.
30. Fuller CM, McNulty CM, Spring DA, et al. Prospective screening of 5,615 high school athletes for risk of sudden death. Med Sci Sports Exer 1997;29:1131-8.
31. Fuller CM. Cost effectiveness of analysis of high school athletes for risks of sudden cardiac death. Med Sci Sports Exer 2000;32:887-90.
32. Pelliccia A, Maron BJ, Culasso F, et al. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 2000;102:278-84.
33. Maron BJ, Pelliccia A, Spirito P. Cardiac disease in young trained athletes: insights into methods for distinguishing athlete’s heart from structural heart disease with particular emphasis on hypertrophic cardiomyopathy. Circulation 1995;91:1596-1601.
34. Gomez JE, Landry GL, Bernhardt DT. Critical evaluation of the 2-minute orthopedic screening examination. Am J Dis Child 1993;147:1109-13.
35. Abbott HG, Kress JB. Preconditioning in the prevention of knee injuries. Arch Phys Med Rehabil 1969;50:326-33.
36. Jackson DW, Jarrett H, Bailey D, Kausek J, Swanson J, Powell JW. Injury prediction in the young athlete: a preliminary report. Am J Sports Med 1978;6:6-14.
37. Nicholas JA. Injuries in knee ligaments: Relationship to looseness and tightness in football players. JAMA 1970;212:2236-9.
38. Dodge WF, West EF, Smith EH, Harvey B 3rd. Proteinuria and hematuria in schoolchildren: epidemiology and early natural history. J Pediatr 1976;88:327-47.
39. Peggs JF, Reinhardt RW, O’Brien JM. Proteinuria in adolescent sports physical examinations. J Fam Pract 1986;22:80-1.
40. Rupp NT, Brudno DS, Guill MF. The value of screening for risk of exercise-induced asthma in high school athletes. Ann Allergy 1993;70:339-42.
41. Feinstein RA, LaRussa J, Wang-Dohlman A, Bartolucci AA. Screening adolescent athletes for exercise-induced asthma. Clin J Sports Med 1996;6:119-23.
42. Torg JS, Vegso JJ, Sennett B, Das M. The National Football Head and Neck Injury Registry. 14-year report on cervical quadriplegia, 1971 through 1984. JAMA 1985;254:3439-43.
43. 26th Bethesda Conference: Recommendations for determining eligibility for competition in athletes with cardiovascular abnormalities Med Sci Sports Exerc 1994;26(Suppl):S223-S283.
44. American Academy of Pediatrics. Medical conditions affecting sports participation. Pediatrics 2001;107:1206-7.
Depression screening: a practical strategy
- A 2-stage strategy, combining an assessment of severity with depression criteria, can help a physician focus on the most severe cases without missing less severe ones that still need treatment (B).
- Because of its brevity, relatively high positive predictive value, and ability to inform the clinician on both depression severity and diagnostic criteria, the PRIME-MD Patient Health Questionnaire (PHQ-9) is the best available depression screening tool for primary care (B).
- One-time screening is cost-effective; physicians may elect to screen more often based on risk factors (A).
What is the most efficient and accurate way for a busy primary care physician to screen patients for depression? Many screening tools exist, but they are not equally effective.
A careful review of the literature strongly favors a 2-stage strategy assessing both depression severity and criteria. In this article, we describe this optimal approach against the background of other available resources.
Health and economic impact of depression
In the average family practice, around 6 cases of depression go unrecognized each week. This real-world estimate derives from studies that consistently report a 10% prevalence of depression in primary care patients1 but a rate of recognition by primary care clinicians of only 29% to 35%.2-4 Depression is a common condition with a large impact on quality of life and productivity, one that indirectly affects other health states, including cardiovascular disease.5-9 It is responsible for an estimated economic cost in the US of over $40 billion annually. As a result, depression screening has been an active area of research, and a variety of organizations have issued guidelines recommending routine screening for depression in primary care.
The need for an efficient, reliable screening tool
Based on a recent review of the evidence on depression screening outcomes in primary care settings,10 the US Preventive Services Task Force (USPSTF) updated its screening recommendation in 2002 to include an endorsement of depression screening in adults “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up” (strength of recommendation [SOR]=A).11 This endorsement leaves the primary care clinician with no guidance about how or when to screen for depression.
Despite lack of guidance in the USPTF guidelines, we believe depression screening can be done efficiently and reliably in primary care. However, one must begin by understanding that depression screening is different from screening for cancer or cardiovascular risk factors (Table 1). The burdens of interpretation of depression screening results are especially noteworthy. For example, the PRIME-MD Patient Health Questionnaire (PHQ) is reported to have a sensitivity of 61% and specificity of 94% for any mood or depressive disorder.12 This results in a positive predictive value (PPV) of 50% using a reasonable estimate of 10% prevalence for depression in primary care settings.13
Put simply, following administration and scoring of the PHQ, the clinician is left with little better odds than a coin toss of identifying a patient that has an active major depressive disorder requiring treatment. If there was no objective help, clinicians would have only their clinical judgment to resolve this, all during an office visit that contains many other competing agendas and demands.14,15
We have reviewed the evidence on depression screening instruments with the intent to highlight an instrument that clinicians can efficiently and reliably use to find depressed and impaired patients in their practice whom they might otherwise miss.
TABLE 1
Burdens of screening for cancer, hyperlipidemia, and depression
| Cancer | Hyperlipidemia | Depression | ||||
|---|---|---|---|---|---|---|
| Burden of performance | Low | Simple test or performance of billable procedure | Low | Blood test | High | Time-intensive administration & scoring |
| Burden of interpretation | Low | Confirmatory testing often referred to specialists | Low | No confirmatory reference standard testing | High | High false positive rate w/burdensome reference standard |
| Burden of treatment | Low | Treatment done by specialists | High | Requires activation of patient & frequent monitoring | High | Requires activation of patient & frequent monitoring |
Two types of screening instruments
Depression screening instruments can be grouped into 2 categories:
- depression assessment scales, which ask patients to rate the severity or frequency of various symptoms
- symptom count instruments, which are based on depression criteria.
Depression assessment scales preceded symptom count instruments, and many were developed prior to the establishment of formal diagnostic criteria within the Diagnostic and Statistical Manual ofMental Disorders (DSM) system.16 Table 2 lists available examples of depression assessment scales and symptom count instruments, along with websites where you may access further information and the instruments themselves.
TABLE 2
Accuracy and ease of administration of commonly available screening instruments
| Instrument | Time and scoring | LR+ (95% CI) | LR– (95% CI) | PPV (95% CI) | Web source |
|---|---|---|---|---|---|
| Assessment scale | |||||
| Beck Depression Inventory (BDI)32 | 2–5 min; simple | 4.2 (1.2–13.6) | 0.17 (0.1–0.3) | 29.6% (10.7–57.6) | www.psychcorpcenter.com/content/bdi-II.htm |
| Center for Epidemiologic Studies Depression Scale (CES-D)34 | 2–5 min; simple | 3.3 (2.5–4.4) | 0.24 (0.2–0.3) | 24.8% (20–30.6) | http://www.mhhe.com/hper/health/personal health/labs/Stress/activ2-2.html |
| Geriatric Depression Scale (GDS)35 | 2–5 min; simple> | 3.3 (2.4–4.7) | 0.16 (0.1–0.3) | 24.8% (19.4–32) | http://www.stanford.edu/~yesavage/GDS.html |
| Hospital Anxiety and Depression Scale* (HADS)20 | 2–5 min; simple | 7.0 (2.9–11.2) | 0.3 (0.3–0.4) | 41.3% (22.6–52.8) | www.clinical-supervision.com/hads.htm |
| Zung Self Assessment Depression Scale (Zung SDS)33 | 2–5 min; simple | 3.3 (1.3–8.1) | 0.35 (0.2–0.8) | 24.8% (11.5–44.8) | http://fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom count | |||||
| Primary Care Evaluation of Mental Disorders †(PRIME-MD)27 | 2 min; complex | 2.7 (2.0–3.7) | 0.14 (0.1–0.3) | 21.3% (16.7–27) | Available upon request to Robert Spitzer, MD: [email protected] |
| PRIME-MD Patient Health Questionnaire (PHQ) | 5–7 min; simple | 10.2‡ (6.5–17.5) | 0.4‡ (0.3–0.5) | 50.4% (39.4–63.6) | fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom-Driven Diagnostic System for Primary Care†(SDDS-PC) | 2 min; simple | 3.5 (2.4–5.1) | 0.2 (0.1–0.4) | 25.9% (19.4–33.8) | No website available |
| PRIME-MD Patient Health Questionnaire (PHQ-9) | 2 –5 min; simple | 12.2 (8.4–18) | 0.28 (0.2–0.5) | 55% (45.7–64.3) | www.depression-primarycare.org/ap1.html |
| * Unless noted by (*), adapted from Williams et al.18 | |||||
| † Values reflect the initial brief screening portion of these instruments. | |||||
| ‡ PHQ vaues obtained from original position and reflect diagnosis of “any mood disorders.” | |||||
| LR+, positive likelihood ratio; LR–, negative likelihood ratio; PPV, positive predictive value; CI, confidence interval | |||||
Pros and cons of assessment scales
The advantages of using a scale are due to the manner in which patients experience depressive symptoms, along a continuum of mild to severe. A scale is able to represent these gradations in severity and may be helpful in guiding the need for treatment and treatment adjustments.
Unfortunately, this ability to measure the dimensional nature of depression is also a weakness, as a threshold must be identified above which the patient is classified as warranting further investigation. Ideally, these thresholds should be established in a representative primary care sample and predict functional status as well as likelihood of meeting DSM-IV diagnostic criteria. The ability of a scale to accurately identify patients in need of attention depends directly on the threshold.
Pros and cons of symptom counts
Instruments based on depression criteria are a relatively new innovation, appearing since the establishment of DSM-IV criteria that define reference symptoms, a minimum number of which must be present to diagnose depression. Depression criteria–based instruments have the advantage of not being dependent on a threshold of symptom severity.
However, in primary care settings this can also be a weakness because the presence of depression criteria alone may not be a reliable indicator of depression-related impairment.17 Instruments that can be used in both a diagnostic criteria and scale modes have a particular advantage in that the weaknesses of each are offset.
Characteristics of selected screening instruments
We searched MEDLINE and the Cochrane databases for reviews of depression screening, with particular attention to reviews of primary care-based trials. Forty-one papers emerged, 3 of which were systematic reviews. For this paper, we focused on the review published by Williams and colleagues,18 which summarizes primary care data on the depression screening instruments most widely used. They examined 379 studies that compared the primary care performance of these instruments with a reference standard diagnostic interview, such as the Structured Clinical Interview for DSM-IV (SCID).19 Twenty-eight studies met their criteria and were included in the systematic review.
In Table 2 we have adapted the information from Williams’s review and added a calculation of PPV based on a 10% prevalence estimate for depression in primary care populations. We chose to exclude information on the Single Question (SQ) screen because of its very low PPV and the Hopkins Symptom Checklist (HSCL) because of its length (25 questions). In addition, we chose to add the Hospital Anxiety and Depression Scale (HADS), using operating characteristic information from 2 studies,20,21 because of its purported advantages in medically ill populations.
Beyond the SQ, it is useful to comment on “2-question screening” as suggested by the USPSTF. We are unable to find justification for this in the paper by Pingone and colleagues, which served as background for the recommendations.10 Although Pingone et al did cite the report of Wells and colleagues as using a 2-item screener, their study used not only 2 questions on mood and anhedonia but also other criteria in screening their population.22 Therefore, it is not appropriate as a source for 2-item screening performance characteristics.
Comparison of the operating characteristics of the selected instruments reveals that most yield PPV values in the 20% to 30% range, with the exception of the HADS, the PHQ, and the PHQ-9, which yield PPV values of 41.3%, 50%, and 55%, respectively.
The PHQ-9 (included in the (Appendix) offers a further advantage over the HADS and other instruments listed in that within a 9-item instrument both the presence of diagnostic criteria and severity may be assessed. Kroenke and colleagues have examined the use of the PHQ-9 as a severity instrument and found it to be a reliable and valid measure of depression severity when compared with the Medical Outcomes Study Short Form (SF-20).23
We purposely have not examined negative predictive values (NPV) for the listed instruments. NPV is useful when screening using biomedical markers where a negative result allows extrapolation into the future due to a known, predictable time course for development of the screened-for condition. For example, a negative screening colonoscopy has value not just because of its current predictive value, but because we know something about how long it may take to develop precancerous polyps in a negative screened patient. However, this is not the case with depression. A patient that fails to meet criteria for depression today could fully meet criteria in 2 weeks and be quite depressed. Therefore we have chosen to focus on PPV in comparing depression screening instruments.
Selection and use of a screening instrument
How should a busy clinician select a depression screening instrument? Ease of administration and interpretation are key. Ideally, a depression screen should function similarly to a vital sign, providing an easy-to-assess yet reliable marker of the need to address a patient’s depression. It is not enough to know that formal depression criteria are met; it is also important to know whether a patient’s functioning is impaired. Research indicates that it is difficult in primary care to “clinically” assess functioning in the face of numerous competing demands,15 even when clinicians know from a screening test that a patient meets criteria for depression.24 For this reason, even watchful waiting for the “positive screening/low impairment” patients25 may be difficult to put into practice.
Two-stage strategy to assess impairment
Use of a 2-stage strategy, combining an assessment of severity with an assessment of depression criteria, appears to answer this dilemma. One study26 has attempted to assess whether this strategy could identify the appropriate patients for clinician attention, using an existing data set that included the PRIME-MD27 and 6 items identified from the original data via factor analyses that assess depression severity.
The results suggest that a combined assessment of depression severity and criteria could help clinicians focus on the most severely depressed patients without missing less severely impaired patients that need treatment (SOR=B).
We suggest the PHQ-9 as the instrument of choice for primary care depression screening because it measures both depression criteria and severity. The PHQ-9 provides a simple way to assess both diagnostic criteria and severity with a single, well-validated instrument. While its PPV is not appreciably greater than 50%, this reflects use in a purely “diagnostic mode,” ie, a cut-point of 10.
A well done, primary care evaluation of the PHQ-9 suggests that a score of 15 or greater reliably indicates both satisfaction of DSM-IV depression criteria and a moderate to severe level of impairment (SOR=A).28 Patients screening positive at this level should be targeted by their physician for a discussion of their symptoms and a recommendation for treatment (SOR=B). Patients with a score of 10–14 meet diagnostic criteria for depression but at a lower level of severity; these patients could be candidates for a strategy of repeat testing or watchful waiting (SOR=B).
Before leaving the topic, a comment is warranted regarding 2-stage screening using an initial 1-or 2-question screen followed by a more lengthy instrument. This type of strategy was embodied in the original PRIME-MD with its 2-question Patient Questionnaire (PQ).27 The intent is to reduce the burden of applying a full diagnostic instrument to an entire practice population. By giving the full instrument only to patients that are positive on the initial 2-question screen, the screening performance burden (as identified in Table 1) is reduced. Use of a brief instrument such as the PHQ-9, which requires only 2 to 5 minutes to fully complete, makes it possible to accurately assess both diagnostic criteria and depression severity in an entire patient population, with little administration burden.
When to screen
Once a decision is made to screen, and an instrument is selected, an interval for screening must be determined. Suggested ranges vary greatly from one-time to annual screening. The recent USPSTF recommendations provide little guidance, stating simply, “the optimal interval for screening is unknown.”11
Regular intervals. One-time screening was found to be cost-effective by Valenstein and colleagues,13 suggesting that, at a minimum, screening should occur when a new patient enters a practice (SOR=A). If a more frequent schedule of screening is desired, depression screening should be linked to other periodic preventive services provided in a practice, such as routine Pap smears or health maintenance exams, to ensure that screening occurs in a systematic fashion (SOR=C).
Risk factors. A practice may also elect to screen based on risk factors (SOR=D). Important risk factors to consider include prior history of treated depression, family history of depression, postpartum status, and any history of substance abuse.
Patients with chronic diseases known to have a high rate of comorbidity with depression—ie, diabetes, congestive heart failure, myocardial infarction—should also be considered as having risk factors for depression.
Ease of implementation
The depression screening instruments reviewed in this paper may all be completed by a patient with a sixth- to ninth-grade reading level, and can therefore be given to patients to complete in an exam room while they wait for their physician. Scoring may be then quickly completed either by the patient or by the physician.
Positive screens should prompt the physician to engage the patient in a discussion of their symptoms, the need for treatment, and a quick assessment for the presence of any suicidal ideation.
Finally, when depression is identified by screening, the potential presence of other psychiatric disorders should be noted. Anxiety disorders are frequently diagnosable in depressed patients, although it is unclear whether comorbid anxiety necessitates a change in treatment plans.29 In contrast, a comorbid substance abuse should be recognized and addressed. Similarly, coexisting dysthymia may contribute to depressed patients’ functional impairment.30
Phq-9 reasonable for monitoring treatment
It is important to note that the USPSTF recommendation specifies screening “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and followup.” Routine, periodic monitoring is an important aspect of a systems approach to depression care. The PHQ-9, when scored as an assessment scale, and the depression assessment scales listed in Table 2 should be considered for periodic monitoring of patients being treated for depression (SOR=B). Active monitoring may alert the clinician to improvement in symptoms or to a need for treatment adjustment when symptoms do not improve.
The Hamilton Rating Scale for Depression (HAM-D) is often used as a reference standard for monitoring of outcomes in clinical trials, but it is administered by trained interviewers and is therefore impractical to administer in a routine patient care setting. The Beck Depression Inventory (BDI) and Zung Self-rating Depression Scale (SDS) have been used as outcome measures as well, but they are not as sensitive to change over time as the HAM-D.31
The sensitivity to change over time of the PHQ-9 has not yet been formally compared to the HAM-D, but it still represents a reasonable option until the results of such a comparison are available.
1. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
2. Magruder-Habib K, Zung WW, Feussner JR. Improving physicians’ recognition and treatment of depression in general medical care. Results from a randomized clinical trial. Med Care 1990;28:239-50.
3. Coyne JC, Schwenk TL, Fechner-Bates S. Nondetection of depression by primary care physicians reconsidered. Gen Hosp Psychiatry 1995;17:3-12.
4. Williams JW, Mulrow CD, Kroenke K, et al. Case-finding for depression in primary care: a randomized trial. Am J Med 1999;106:36-43.
5. Greenberg PE, Stiglin LE, Finkelstein SN, Berndt ER. The economic burden of depression in 1990. J Clin Psychiatry 1993;54:405-18.
6. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
7. Von Korff M, Ormel J, Katon W, Lin EH. Disability and depression among high utilizers of health care. A longitudinal analysis. Arch Gen Psychiatry 1992;49:91-100.
8. Wells KB, Stewart A, Hays RD, et al. The functioning and well-being of depressed patients. Results from the Medical Outcomes Study. JAMA 1989;262:914-9.
9. Ford DE, Mead LA, Chang PP, Cooper-Patrick L, Wang NY, Klag MJ. Depression is a risk factor for coronary artery disease in men: the precursors study. Arch Intern Med 1998;158:1422-6.
10. Pignone MP, Gaynes BN, Rushton JL, et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002;136:765-76.
11. US. Preventive Services Task Force. Screening for depression: recommendations and rationale. Ann Intern Med 2002;136:760-4.
12. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999;282:1737-44.
13. Valenstein M, Vijan S, Zeber JE, Boehm K, Buttar A. The cost-utility of screening for depression in primary care. Ann Intern Med 2001;134:345-60.
14. Jaen CR, Stange KC, Nutting PA. Competing demands of primary care: a model for the delivery of clinical preventive services. J Fam Pract 1994;38:166-71.
15. Klinkman MS. Competing demands in psychosocial care. A model for the identification and treatment of depressive disorders in primary care. Gen Hosp Psychiatry 1997;19:98-111.
16. American Psychiatric Association, American Psychiatric Association, Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. 4th ed. Washington, DC: American Psychiatric Association; 2000.
17. Schwenk TL, Coyne JC, Fechner-Bates S. Differences between detected and undetected patients in primary care and depressed psychiatric patients. Gen Hosp Psychiatry 1996;18:407-15.
18. Williams JW, Jr, Noel PH, Cordes JA, Ramirez G, Pignone M. Is this patient clinically depressed? JAMA 2002;287:1160-70.
19. Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry 1992;49:624-9.
20. Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70.
21. Silverstone PH. Poor efficacy of the Hospital Anxiety and Depression Scale in the diagnosis of major depressive disorder in both medical and psychiatric patients. J Psychosom Res 1994;38:441-50.
22. Wells KB, Sherbourne C, Schoenbaum M, et al. Impact of disseminating quality improvement programs for depression in managed primary care: a randomized controlled trial. JAMA 2000;283:212-20.
23. Stewart AL, Hays RD, Ware JE, Jr. The MOS short-form general health survey. Reliability and validity in a patient population. Med Care 1988;26:724-35.
24. Rost K, Nutting P, Smith J, Coyne JC, Cooper-Patrick L, Rubenstein L. The role of competing demands in the treatment provided primary care patients with major depression. Arch Fam Med 2000;9:150-4.
25. Leon AC, Portera L, Olfson M, et al. False positive results: a challenge for psychiatric screening in primary care. Am J Psychiatry 1997;154:1462-4.
26. Nease DE, Jr, Klinkman MA, Volk RJ. Improved detection of depression in primary care through severity detection. J Fam Pract 2002;51:1065-70.
27. Spitzer RL, Williams J, Kroenke K, Linzer M, deGruy FV, Hann SR, et al. Utility of a new procedure for diagnosing mental disorders in primary care: the PRIME-MD 1000 study. JAMA 1994;272:1749-56.
28. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
29. Coyne JC, Fechner-Bates S, Schwenk TL. Prevalence, nature, and comorbidity of depressive disorders in primary care. Gen Hosp Psychiatry 1994;16:267-76.
30. Wells K, Burnam M, Rogers W, Hays R, Camp P. The course of depression in adult outpatients: results from the Medical Outcomes Study. Arch Gen Psychiatry 1992;49:788-94.
31. Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta-analytic comparison. J Consult Clin Psychol 1986;54:54-9.
32. Beck A, Ward C, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
33. Zung WW, Richards CB, Short MJ. Self-rating depression scale in an outpatient clinic. Further validation of the SDS. Arch Gen Psychiatry 1965;13:508-15.
34. Radloff LS. The CES-D Scale: A self-report depression scale for research in the general population. Applied Psychological Measurement 1977;1:385-401.
35. Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version. In: Clinical Gerontology: A Guide to Assessment and Intervention. New York: Haworth Press; 1986;165-73.
- A 2-stage strategy, combining an assessment of severity with depression criteria, can help a physician focus on the most severe cases without missing less severe ones that still need treatment (B).
- Because of its brevity, relatively high positive predictive value, and ability to inform the clinician on both depression severity and diagnostic criteria, the PRIME-MD Patient Health Questionnaire (PHQ-9) is the best available depression screening tool for primary care (B).
- One-time screening is cost-effective; physicians may elect to screen more often based on risk factors (A).
What is the most efficient and accurate way for a busy primary care physician to screen patients for depression? Many screening tools exist, but they are not equally effective.
A careful review of the literature strongly favors a 2-stage strategy assessing both depression severity and criteria. In this article, we describe this optimal approach against the background of other available resources.
Health and economic impact of depression
In the average family practice, around 6 cases of depression go unrecognized each week. This real-world estimate derives from studies that consistently report a 10% prevalence of depression in primary care patients1 but a rate of recognition by primary care clinicians of only 29% to 35%.2-4 Depression is a common condition with a large impact on quality of life and productivity, one that indirectly affects other health states, including cardiovascular disease.5-9 It is responsible for an estimated economic cost in the US of over $40 billion annually. As a result, depression screening has been an active area of research, and a variety of organizations have issued guidelines recommending routine screening for depression in primary care.
The need for an efficient, reliable screening tool
Based on a recent review of the evidence on depression screening outcomes in primary care settings,10 the US Preventive Services Task Force (USPSTF) updated its screening recommendation in 2002 to include an endorsement of depression screening in adults “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up” (strength of recommendation [SOR]=A).11 This endorsement leaves the primary care clinician with no guidance about how or when to screen for depression.
Despite lack of guidance in the USPTF guidelines, we believe depression screening can be done efficiently and reliably in primary care. However, one must begin by understanding that depression screening is different from screening for cancer or cardiovascular risk factors (Table 1). The burdens of interpretation of depression screening results are especially noteworthy. For example, the PRIME-MD Patient Health Questionnaire (PHQ) is reported to have a sensitivity of 61% and specificity of 94% for any mood or depressive disorder.12 This results in a positive predictive value (PPV) of 50% using a reasonable estimate of 10% prevalence for depression in primary care settings.13
Put simply, following administration and scoring of the PHQ, the clinician is left with little better odds than a coin toss of identifying a patient that has an active major depressive disorder requiring treatment. If there was no objective help, clinicians would have only their clinical judgment to resolve this, all during an office visit that contains many other competing agendas and demands.14,15
We have reviewed the evidence on depression screening instruments with the intent to highlight an instrument that clinicians can efficiently and reliably use to find depressed and impaired patients in their practice whom they might otherwise miss.
TABLE 1
Burdens of screening for cancer, hyperlipidemia, and depression
| Cancer | Hyperlipidemia | Depression | ||||
|---|---|---|---|---|---|---|
| Burden of performance | Low | Simple test or performance of billable procedure | Low | Blood test | High | Time-intensive administration & scoring |
| Burden of interpretation | Low | Confirmatory testing often referred to specialists | Low | No confirmatory reference standard testing | High | High false positive rate w/burdensome reference standard |
| Burden of treatment | Low | Treatment done by specialists | High | Requires activation of patient & frequent monitoring | High | Requires activation of patient & frequent monitoring |
Two types of screening instruments
Depression screening instruments can be grouped into 2 categories:
- depression assessment scales, which ask patients to rate the severity or frequency of various symptoms
- symptom count instruments, which are based on depression criteria.
Depression assessment scales preceded symptom count instruments, and many were developed prior to the establishment of formal diagnostic criteria within the Diagnostic and Statistical Manual ofMental Disorders (DSM) system.16 Table 2 lists available examples of depression assessment scales and symptom count instruments, along with websites where you may access further information and the instruments themselves.
TABLE 2
Accuracy and ease of administration of commonly available screening instruments
| Instrument | Time and scoring | LR+ (95% CI) | LR– (95% CI) | PPV (95% CI) | Web source |
|---|---|---|---|---|---|
| Assessment scale | |||||
| Beck Depression Inventory (BDI)32 | 2–5 min; simple | 4.2 (1.2–13.6) | 0.17 (0.1–0.3) | 29.6% (10.7–57.6) | www.psychcorpcenter.com/content/bdi-II.htm |
| Center for Epidemiologic Studies Depression Scale (CES-D)34 | 2–5 min; simple | 3.3 (2.5–4.4) | 0.24 (0.2–0.3) | 24.8% (20–30.6) | http://www.mhhe.com/hper/health/personal health/labs/Stress/activ2-2.html |
| Geriatric Depression Scale (GDS)35 | 2–5 min; simple> | 3.3 (2.4–4.7) | 0.16 (0.1–0.3) | 24.8% (19.4–32) | http://www.stanford.edu/~yesavage/GDS.html |
| Hospital Anxiety and Depression Scale* (HADS)20 | 2–5 min; simple | 7.0 (2.9–11.2) | 0.3 (0.3–0.4) | 41.3% (22.6–52.8) | www.clinical-supervision.com/hads.htm |
| Zung Self Assessment Depression Scale (Zung SDS)33 | 2–5 min; simple | 3.3 (1.3–8.1) | 0.35 (0.2–0.8) | 24.8% (11.5–44.8) | http://fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom count | |||||
| Primary Care Evaluation of Mental Disorders †(PRIME-MD)27 | 2 min; complex | 2.7 (2.0–3.7) | 0.14 (0.1–0.3) | 21.3% (16.7–27) | Available upon request to Robert Spitzer, MD: [email protected] |
| PRIME-MD Patient Health Questionnaire (PHQ) | 5–7 min; simple | 10.2‡ (6.5–17.5) | 0.4‡ (0.3–0.5) | 50.4% (39.4–63.6) | fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom-Driven Diagnostic System for Primary Care†(SDDS-PC) | 2 min; simple | 3.5 (2.4–5.1) | 0.2 (0.1–0.4) | 25.9% (19.4–33.8) | No website available |
| PRIME-MD Patient Health Questionnaire (PHQ-9) | 2 –5 min; simple | 12.2 (8.4–18) | 0.28 (0.2–0.5) | 55% (45.7–64.3) | www.depression-primarycare.org/ap1.html |
| * Unless noted by (*), adapted from Williams et al.18 | |||||
| † Values reflect the initial brief screening portion of these instruments. | |||||
| ‡ PHQ vaues obtained from original position and reflect diagnosis of “any mood disorders.” | |||||
| LR+, positive likelihood ratio; LR–, negative likelihood ratio; PPV, positive predictive value; CI, confidence interval | |||||
Pros and cons of assessment scales
The advantages of using a scale are due to the manner in which patients experience depressive symptoms, along a continuum of mild to severe. A scale is able to represent these gradations in severity and may be helpful in guiding the need for treatment and treatment adjustments.
Unfortunately, this ability to measure the dimensional nature of depression is also a weakness, as a threshold must be identified above which the patient is classified as warranting further investigation. Ideally, these thresholds should be established in a representative primary care sample and predict functional status as well as likelihood of meeting DSM-IV diagnostic criteria. The ability of a scale to accurately identify patients in need of attention depends directly on the threshold.
Pros and cons of symptom counts
Instruments based on depression criteria are a relatively new innovation, appearing since the establishment of DSM-IV criteria that define reference symptoms, a minimum number of which must be present to diagnose depression. Depression criteria–based instruments have the advantage of not being dependent on a threshold of symptom severity.
However, in primary care settings this can also be a weakness because the presence of depression criteria alone may not be a reliable indicator of depression-related impairment.17 Instruments that can be used in both a diagnostic criteria and scale modes have a particular advantage in that the weaknesses of each are offset.
Characteristics of selected screening instruments
We searched MEDLINE and the Cochrane databases for reviews of depression screening, with particular attention to reviews of primary care-based trials. Forty-one papers emerged, 3 of which were systematic reviews. For this paper, we focused on the review published by Williams and colleagues,18 which summarizes primary care data on the depression screening instruments most widely used. They examined 379 studies that compared the primary care performance of these instruments with a reference standard diagnostic interview, such as the Structured Clinical Interview for DSM-IV (SCID).19 Twenty-eight studies met their criteria and were included in the systematic review.
In Table 2 we have adapted the information from Williams’s review and added a calculation of PPV based on a 10% prevalence estimate for depression in primary care populations. We chose to exclude information on the Single Question (SQ) screen because of its very low PPV and the Hopkins Symptom Checklist (HSCL) because of its length (25 questions). In addition, we chose to add the Hospital Anxiety and Depression Scale (HADS), using operating characteristic information from 2 studies,20,21 because of its purported advantages in medically ill populations.
Beyond the SQ, it is useful to comment on “2-question screening” as suggested by the USPSTF. We are unable to find justification for this in the paper by Pingone and colleagues, which served as background for the recommendations.10 Although Pingone et al did cite the report of Wells and colleagues as using a 2-item screener, their study used not only 2 questions on mood and anhedonia but also other criteria in screening their population.22 Therefore, it is not appropriate as a source for 2-item screening performance characteristics.
Comparison of the operating characteristics of the selected instruments reveals that most yield PPV values in the 20% to 30% range, with the exception of the HADS, the PHQ, and the PHQ-9, which yield PPV values of 41.3%, 50%, and 55%, respectively.
The PHQ-9 (included in the (Appendix) offers a further advantage over the HADS and other instruments listed in that within a 9-item instrument both the presence of diagnostic criteria and severity may be assessed. Kroenke and colleagues have examined the use of the PHQ-9 as a severity instrument and found it to be a reliable and valid measure of depression severity when compared with the Medical Outcomes Study Short Form (SF-20).23
We purposely have not examined negative predictive values (NPV) for the listed instruments. NPV is useful when screening using biomedical markers where a negative result allows extrapolation into the future due to a known, predictable time course for development of the screened-for condition. For example, a negative screening colonoscopy has value not just because of its current predictive value, but because we know something about how long it may take to develop precancerous polyps in a negative screened patient. However, this is not the case with depression. A patient that fails to meet criteria for depression today could fully meet criteria in 2 weeks and be quite depressed. Therefore we have chosen to focus on PPV in comparing depression screening instruments.
Selection and use of a screening instrument
How should a busy clinician select a depression screening instrument? Ease of administration and interpretation are key. Ideally, a depression screen should function similarly to a vital sign, providing an easy-to-assess yet reliable marker of the need to address a patient’s depression. It is not enough to know that formal depression criteria are met; it is also important to know whether a patient’s functioning is impaired. Research indicates that it is difficult in primary care to “clinically” assess functioning in the face of numerous competing demands,15 even when clinicians know from a screening test that a patient meets criteria for depression.24 For this reason, even watchful waiting for the “positive screening/low impairment” patients25 may be difficult to put into practice.
Two-stage strategy to assess impairment
Use of a 2-stage strategy, combining an assessment of severity with an assessment of depression criteria, appears to answer this dilemma. One study26 has attempted to assess whether this strategy could identify the appropriate patients for clinician attention, using an existing data set that included the PRIME-MD27 and 6 items identified from the original data via factor analyses that assess depression severity.
The results suggest that a combined assessment of depression severity and criteria could help clinicians focus on the most severely depressed patients without missing less severely impaired patients that need treatment (SOR=B).
We suggest the PHQ-9 as the instrument of choice for primary care depression screening because it measures both depression criteria and severity. The PHQ-9 provides a simple way to assess both diagnostic criteria and severity with a single, well-validated instrument. While its PPV is not appreciably greater than 50%, this reflects use in a purely “diagnostic mode,” ie, a cut-point of 10.
A well done, primary care evaluation of the PHQ-9 suggests that a score of 15 or greater reliably indicates both satisfaction of DSM-IV depression criteria and a moderate to severe level of impairment (SOR=A).28 Patients screening positive at this level should be targeted by their physician for a discussion of their symptoms and a recommendation for treatment (SOR=B). Patients with a score of 10–14 meet diagnostic criteria for depression but at a lower level of severity; these patients could be candidates for a strategy of repeat testing or watchful waiting (SOR=B).
Before leaving the topic, a comment is warranted regarding 2-stage screening using an initial 1-or 2-question screen followed by a more lengthy instrument. This type of strategy was embodied in the original PRIME-MD with its 2-question Patient Questionnaire (PQ).27 The intent is to reduce the burden of applying a full diagnostic instrument to an entire practice population. By giving the full instrument only to patients that are positive on the initial 2-question screen, the screening performance burden (as identified in Table 1) is reduced. Use of a brief instrument such as the PHQ-9, which requires only 2 to 5 minutes to fully complete, makes it possible to accurately assess both diagnostic criteria and depression severity in an entire patient population, with little administration burden.
When to screen
Once a decision is made to screen, and an instrument is selected, an interval for screening must be determined. Suggested ranges vary greatly from one-time to annual screening. The recent USPSTF recommendations provide little guidance, stating simply, “the optimal interval for screening is unknown.”11
Regular intervals. One-time screening was found to be cost-effective by Valenstein and colleagues,13 suggesting that, at a minimum, screening should occur when a new patient enters a practice (SOR=A). If a more frequent schedule of screening is desired, depression screening should be linked to other periodic preventive services provided in a practice, such as routine Pap smears or health maintenance exams, to ensure that screening occurs in a systematic fashion (SOR=C).
Risk factors. A practice may also elect to screen based on risk factors (SOR=D). Important risk factors to consider include prior history of treated depression, family history of depression, postpartum status, and any history of substance abuse.
Patients with chronic diseases known to have a high rate of comorbidity with depression—ie, diabetes, congestive heart failure, myocardial infarction—should also be considered as having risk factors for depression.
Ease of implementation
The depression screening instruments reviewed in this paper may all be completed by a patient with a sixth- to ninth-grade reading level, and can therefore be given to patients to complete in an exam room while they wait for their physician. Scoring may be then quickly completed either by the patient or by the physician.
Positive screens should prompt the physician to engage the patient in a discussion of their symptoms, the need for treatment, and a quick assessment for the presence of any suicidal ideation.
Finally, when depression is identified by screening, the potential presence of other psychiatric disorders should be noted. Anxiety disorders are frequently diagnosable in depressed patients, although it is unclear whether comorbid anxiety necessitates a change in treatment plans.29 In contrast, a comorbid substance abuse should be recognized and addressed. Similarly, coexisting dysthymia may contribute to depressed patients’ functional impairment.30
Phq-9 reasonable for monitoring treatment
It is important to note that the USPSTF recommendation specifies screening “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and followup.” Routine, periodic monitoring is an important aspect of a systems approach to depression care. The PHQ-9, when scored as an assessment scale, and the depression assessment scales listed in Table 2 should be considered for periodic monitoring of patients being treated for depression (SOR=B). Active monitoring may alert the clinician to improvement in symptoms or to a need for treatment adjustment when symptoms do not improve.
The Hamilton Rating Scale for Depression (HAM-D) is often used as a reference standard for monitoring of outcomes in clinical trials, but it is administered by trained interviewers and is therefore impractical to administer in a routine patient care setting. The Beck Depression Inventory (BDI) and Zung Self-rating Depression Scale (SDS) have been used as outcome measures as well, but they are not as sensitive to change over time as the HAM-D.31
The sensitivity to change over time of the PHQ-9 has not yet been formally compared to the HAM-D, but it still represents a reasonable option until the results of such a comparison are available.
- A 2-stage strategy, combining an assessment of severity with depression criteria, can help a physician focus on the most severe cases without missing less severe ones that still need treatment (B).
- Because of its brevity, relatively high positive predictive value, and ability to inform the clinician on both depression severity and diagnostic criteria, the PRIME-MD Patient Health Questionnaire (PHQ-9) is the best available depression screening tool for primary care (B).
- One-time screening is cost-effective; physicians may elect to screen more often based on risk factors (A).
What is the most efficient and accurate way for a busy primary care physician to screen patients for depression? Many screening tools exist, but they are not equally effective.
A careful review of the literature strongly favors a 2-stage strategy assessing both depression severity and criteria. In this article, we describe this optimal approach against the background of other available resources.
Health and economic impact of depression
In the average family practice, around 6 cases of depression go unrecognized each week. This real-world estimate derives from studies that consistently report a 10% prevalence of depression in primary care patients1 but a rate of recognition by primary care clinicians of only 29% to 35%.2-4 Depression is a common condition with a large impact on quality of life and productivity, one that indirectly affects other health states, including cardiovascular disease.5-9 It is responsible for an estimated economic cost in the US of over $40 billion annually. As a result, depression screening has been an active area of research, and a variety of organizations have issued guidelines recommending routine screening for depression in primary care.
The need for an efficient, reliable screening tool
Based on a recent review of the evidence on depression screening outcomes in primary care settings,10 the US Preventive Services Task Force (USPSTF) updated its screening recommendation in 2002 to include an endorsement of depression screening in adults “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and follow-up” (strength of recommendation [SOR]=A).11 This endorsement leaves the primary care clinician with no guidance about how or when to screen for depression.
Despite lack of guidance in the USPTF guidelines, we believe depression screening can be done efficiently and reliably in primary care. However, one must begin by understanding that depression screening is different from screening for cancer or cardiovascular risk factors (Table 1). The burdens of interpretation of depression screening results are especially noteworthy. For example, the PRIME-MD Patient Health Questionnaire (PHQ) is reported to have a sensitivity of 61% and specificity of 94% for any mood or depressive disorder.12 This results in a positive predictive value (PPV) of 50% using a reasonable estimate of 10% prevalence for depression in primary care settings.13
Put simply, following administration and scoring of the PHQ, the clinician is left with little better odds than a coin toss of identifying a patient that has an active major depressive disorder requiring treatment. If there was no objective help, clinicians would have only their clinical judgment to resolve this, all during an office visit that contains many other competing agendas and demands.14,15
We have reviewed the evidence on depression screening instruments with the intent to highlight an instrument that clinicians can efficiently and reliably use to find depressed and impaired patients in their practice whom they might otherwise miss.
TABLE 1
Burdens of screening for cancer, hyperlipidemia, and depression
| Cancer | Hyperlipidemia | Depression | ||||
|---|---|---|---|---|---|---|
| Burden of performance | Low | Simple test or performance of billable procedure | Low | Blood test | High | Time-intensive administration & scoring |
| Burden of interpretation | Low | Confirmatory testing often referred to specialists | Low | No confirmatory reference standard testing | High | High false positive rate w/burdensome reference standard |
| Burden of treatment | Low | Treatment done by specialists | High | Requires activation of patient & frequent monitoring | High | Requires activation of patient & frequent monitoring |
Two types of screening instruments
Depression screening instruments can be grouped into 2 categories:
- depression assessment scales, which ask patients to rate the severity or frequency of various symptoms
- symptom count instruments, which are based on depression criteria.
Depression assessment scales preceded symptom count instruments, and many were developed prior to the establishment of formal diagnostic criteria within the Diagnostic and Statistical Manual ofMental Disorders (DSM) system.16 Table 2 lists available examples of depression assessment scales and symptom count instruments, along with websites where you may access further information and the instruments themselves.
TABLE 2
Accuracy and ease of administration of commonly available screening instruments
| Instrument | Time and scoring | LR+ (95% CI) | LR– (95% CI) | PPV (95% CI) | Web source |
|---|---|---|---|---|---|
| Assessment scale | |||||
| Beck Depression Inventory (BDI)32 | 2–5 min; simple | 4.2 (1.2–13.6) | 0.17 (0.1–0.3) | 29.6% (10.7–57.6) | www.psychcorpcenter.com/content/bdi-II.htm |
| Center for Epidemiologic Studies Depression Scale (CES-D)34 | 2–5 min; simple | 3.3 (2.5–4.4) | 0.24 (0.2–0.3) | 24.8% (20–30.6) | http://www.mhhe.com/hper/health/personal health/labs/Stress/activ2-2.html |
| Geriatric Depression Scale (GDS)35 | 2–5 min; simple> | 3.3 (2.4–4.7) | 0.16 (0.1–0.3) | 24.8% (19.4–32) | http://www.stanford.edu/~yesavage/GDS.html |
| Hospital Anxiety and Depression Scale* (HADS)20 | 2–5 min; simple | 7.0 (2.9–11.2) | 0.3 (0.3–0.4) | 41.3% (22.6–52.8) | www.clinical-supervision.com/hads.htm |
| Zung Self Assessment Depression Scale (Zung SDS)33 | 2–5 min; simple | 3.3 (1.3–8.1) | 0.35 (0.2–0.8) | 24.8% (11.5–44.8) | http://fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom count | |||||
| Primary Care Evaluation of Mental Disorders †(PRIME-MD)27 | 2 min; complex | 2.7 (2.0–3.7) | 0.14 (0.1–0.3) | 21.3% (16.7–27) | Available upon request to Robert Spitzer, MD: [email protected] |
| PRIME-MD Patient Health Questionnaire (PHQ) | 5–7 min; simple | 10.2‡ (6.5–17.5) | 0.4‡ (0.3–0.5) | 50.4% (39.4–63.6) | fpinfo.medicine.uiowa.edu/calculat.htm |
| Symptom-Driven Diagnostic System for Primary Care†(SDDS-PC) | 2 min; simple | 3.5 (2.4–5.1) | 0.2 (0.1–0.4) | 25.9% (19.4–33.8) | No website available |
| PRIME-MD Patient Health Questionnaire (PHQ-9) | 2 –5 min; simple | 12.2 (8.4–18) | 0.28 (0.2–0.5) | 55% (45.7–64.3) | www.depression-primarycare.org/ap1.html |
| * Unless noted by (*), adapted from Williams et al.18 | |||||
| † Values reflect the initial brief screening portion of these instruments. | |||||
| ‡ PHQ vaues obtained from original position and reflect diagnosis of “any mood disorders.” | |||||
| LR+, positive likelihood ratio; LR–, negative likelihood ratio; PPV, positive predictive value; CI, confidence interval | |||||
Pros and cons of assessment scales
The advantages of using a scale are due to the manner in which patients experience depressive symptoms, along a continuum of mild to severe. A scale is able to represent these gradations in severity and may be helpful in guiding the need for treatment and treatment adjustments.
Unfortunately, this ability to measure the dimensional nature of depression is also a weakness, as a threshold must be identified above which the patient is classified as warranting further investigation. Ideally, these thresholds should be established in a representative primary care sample and predict functional status as well as likelihood of meeting DSM-IV diagnostic criteria. The ability of a scale to accurately identify patients in need of attention depends directly on the threshold.
Pros and cons of symptom counts
Instruments based on depression criteria are a relatively new innovation, appearing since the establishment of DSM-IV criteria that define reference symptoms, a minimum number of which must be present to diagnose depression. Depression criteria–based instruments have the advantage of not being dependent on a threshold of symptom severity.
However, in primary care settings this can also be a weakness because the presence of depression criteria alone may not be a reliable indicator of depression-related impairment.17 Instruments that can be used in both a diagnostic criteria and scale modes have a particular advantage in that the weaknesses of each are offset.
Characteristics of selected screening instruments
We searched MEDLINE and the Cochrane databases for reviews of depression screening, with particular attention to reviews of primary care-based trials. Forty-one papers emerged, 3 of which were systematic reviews. For this paper, we focused on the review published by Williams and colleagues,18 which summarizes primary care data on the depression screening instruments most widely used. They examined 379 studies that compared the primary care performance of these instruments with a reference standard diagnostic interview, such as the Structured Clinical Interview for DSM-IV (SCID).19 Twenty-eight studies met their criteria and were included in the systematic review.
In Table 2 we have adapted the information from Williams’s review and added a calculation of PPV based on a 10% prevalence estimate for depression in primary care populations. We chose to exclude information on the Single Question (SQ) screen because of its very low PPV and the Hopkins Symptom Checklist (HSCL) because of its length (25 questions). In addition, we chose to add the Hospital Anxiety and Depression Scale (HADS), using operating characteristic information from 2 studies,20,21 because of its purported advantages in medically ill populations.
Beyond the SQ, it is useful to comment on “2-question screening” as suggested by the USPSTF. We are unable to find justification for this in the paper by Pingone and colleagues, which served as background for the recommendations.10 Although Pingone et al did cite the report of Wells and colleagues as using a 2-item screener, their study used not only 2 questions on mood and anhedonia but also other criteria in screening their population.22 Therefore, it is not appropriate as a source for 2-item screening performance characteristics.
Comparison of the operating characteristics of the selected instruments reveals that most yield PPV values in the 20% to 30% range, with the exception of the HADS, the PHQ, and the PHQ-9, which yield PPV values of 41.3%, 50%, and 55%, respectively.
The PHQ-9 (included in the (Appendix) offers a further advantage over the HADS and other instruments listed in that within a 9-item instrument both the presence of diagnostic criteria and severity may be assessed. Kroenke and colleagues have examined the use of the PHQ-9 as a severity instrument and found it to be a reliable and valid measure of depression severity when compared with the Medical Outcomes Study Short Form (SF-20).23
We purposely have not examined negative predictive values (NPV) for the listed instruments. NPV is useful when screening using biomedical markers where a negative result allows extrapolation into the future due to a known, predictable time course for development of the screened-for condition. For example, a negative screening colonoscopy has value not just because of its current predictive value, but because we know something about how long it may take to develop precancerous polyps in a negative screened patient. However, this is not the case with depression. A patient that fails to meet criteria for depression today could fully meet criteria in 2 weeks and be quite depressed. Therefore we have chosen to focus on PPV in comparing depression screening instruments.
Selection and use of a screening instrument
How should a busy clinician select a depression screening instrument? Ease of administration and interpretation are key. Ideally, a depression screen should function similarly to a vital sign, providing an easy-to-assess yet reliable marker of the need to address a patient’s depression. It is not enough to know that formal depression criteria are met; it is also important to know whether a patient’s functioning is impaired. Research indicates that it is difficult in primary care to “clinically” assess functioning in the face of numerous competing demands,15 even when clinicians know from a screening test that a patient meets criteria for depression.24 For this reason, even watchful waiting for the “positive screening/low impairment” patients25 may be difficult to put into practice.
Two-stage strategy to assess impairment
Use of a 2-stage strategy, combining an assessment of severity with an assessment of depression criteria, appears to answer this dilemma. One study26 has attempted to assess whether this strategy could identify the appropriate patients for clinician attention, using an existing data set that included the PRIME-MD27 and 6 items identified from the original data via factor analyses that assess depression severity.
The results suggest that a combined assessment of depression severity and criteria could help clinicians focus on the most severely depressed patients without missing less severely impaired patients that need treatment (SOR=B).
We suggest the PHQ-9 as the instrument of choice for primary care depression screening because it measures both depression criteria and severity. The PHQ-9 provides a simple way to assess both diagnostic criteria and severity with a single, well-validated instrument. While its PPV is not appreciably greater than 50%, this reflects use in a purely “diagnostic mode,” ie, a cut-point of 10.
A well done, primary care evaluation of the PHQ-9 suggests that a score of 15 or greater reliably indicates both satisfaction of DSM-IV depression criteria and a moderate to severe level of impairment (SOR=A).28 Patients screening positive at this level should be targeted by their physician for a discussion of their symptoms and a recommendation for treatment (SOR=B). Patients with a score of 10–14 meet diagnostic criteria for depression but at a lower level of severity; these patients could be candidates for a strategy of repeat testing or watchful waiting (SOR=B).
Before leaving the topic, a comment is warranted regarding 2-stage screening using an initial 1-or 2-question screen followed by a more lengthy instrument. This type of strategy was embodied in the original PRIME-MD with its 2-question Patient Questionnaire (PQ).27 The intent is to reduce the burden of applying a full diagnostic instrument to an entire practice population. By giving the full instrument only to patients that are positive on the initial 2-question screen, the screening performance burden (as identified in Table 1) is reduced. Use of a brief instrument such as the PHQ-9, which requires only 2 to 5 minutes to fully complete, makes it possible to accurately assess both diagnostic criteria and depression severity in an entire patient population, with little administration burden.
When to screen
Once a decision is made to screen, and an instrument is selected, an interval for screening must be determined. Suggested ranges vary greatly from one-time to annual screening. The recent USPSTF recommendations provide little guidance, stating simply, “the optimal interval for screening is unknown.”11
Regular intervals. One-time screening was found to be cost-effective by Valenstein and colleagues,13 suggesting that, at a minimum, screening should occur when a new patient enters a practice (SOR=A). If a more frequent schedule of screening is desired, depression screening should be linked to other periodic preventive services provided in a practice, such as routine Pap smears or health maintenance exams, to ensure that screening occurs in a systematic fashion (SOR=C).
Risk factors. A practice may also elect to screen based on risk factors (SOR=D). Important risk factors to consider include prior history of treated depression, family history of depression, postpartum status, and any history of substance abuse.
Patients with chronic diseases known to have a high rate of comorbidity with depression—ie, diabetes, congestive heart failure, myocardial infarction—should also be considered as having risk factors for depression.
Ease of implementation
The depression screening instruments reviewed in this paper may all be completed by a patient with a sixth- to ninth-grade reading level, and can therefore be given to patients to complete in an exam room while they wait for their physician. Scoring may be then quickly completed either by the patient or by the physician.
Positive screens should prompt the physician to engage the patient in a discussion of their symptoms, the need for treatment, and a quick assessment for the presence of any suicidal ideation.
Finally, when depression is identified by screening, the potential presence of other psychiatric disorders should be noted. Anxiety disorders are frequently diagnosable in depressed patients, although it is unclear whether comorbid anxiety necessitates a change in treatment plans.29 In contrast, a comorbid substance abuse should be recognized and addressed. Similarly, coexisting dysthymia may contribute to depressed patients’ functional impairment.30
Phq-9 reasonable for monitoring treatment
It is important to note that the USPSTF recommendation specifies screening “in clinical practices that have systems in place to assure accurate diagnosis, effective treatment, and followup.” Routine, periodic monitoring is an important aspect of a systems approach to depression care. The PHQ-9, when scored as an assessment scale, and the depression assessment scales listed in Table 2 should be considered for periodic monitoring of patients being treated for depression (SOR=B). Active monitoring may alert the clinician to improvement in symptoms or to a need for treatment adjustment when symptoms do not improve.
The Hamilton Rating Scale for Depression (HAM-D) is often used as a reference standard for monitoring of outcomes in clinical trials, but it is administered by trained interviewers and is therefore impractical to administer in a routine patient care setting. The Beck Depression Inventory (BDI) and Zung Self-rating Depression Scale (SDS) have been used as outcome measures as well, but they are not as sensitive to change over time as the HAM-D.31
The sensitivity to change over time of the PHQ-9 has not yet been formally compared to the HAM-D, but it still represents a reasonable option until the results of such a comparison are available.
1. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
2. Magruder-Habib K, Zung WW, Feussner JR. Improving physicians’ recognition and treatment of depression in general medical care. Results from a randomized clinical trial. Med Care 1990;28:239-50.
3. Coyne JC, Schwenk TL, Fechner-Bates S. Nondetection of depression by primary care physicians reconsidered. Gen Hosp Psychiatry 1995;17:3-12.
4. Williams JW, Mulrow CD, Kroenke K, et al. Case-finding for depression in primary care: a randomized trial. Am J Med 1999;106:36-43.
5. Greenberg PE, Stiglin LE, Finkelstein SN, Berndt ER. The economic burden of depression in 1990. J Clin Psychiatry 1993;54:405-18.
6. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
7. Von Korff M, Ormel J, Katon W, Lin EH. Disability and depression among high utilizers of health care. A longitudinal analysis. Arch Gen Psychiatry 1992;49:91-100.
8. Wells KB, Stewart A, Hays RD, et al. The functioning and well-being of depressed patients. Results from the Medical Outcomes Study. JAMA 1989;262:914-9.
9. Ford DE, Mead LA, Chang PP, Cooper-Patrick L, Wang NY, Klag MJ. Depression is a risk factor for coronary artery disease in men: the precursors study. Arch Intern Med 1998;158:1422-6.
10. Pignone MP, Gaynes BN, Rushton JL, et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002;136:765-76.
11. US. Preventive Services Task Force. Screening for depression: recommendations and rationale. Ann Intern Med 2002;136:760-4.
12. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999;282:1737-44.
13. Valenstein M, Vijan S, Zeber JE, Boehm K, Buttar A. The cost-utility of screening for depression in primary care. Ann Intern Med 2001;134:345-60.
14. Jaen CR, Stange KC, Nutting PA. Competing demands of primary care: a model for the delivery of clinical preventive services. J Fam Pract 1994;38:166-71.
15. Klinkman MS. Competing demands in psychosocial care. A model for the identification and treatment of depressive disorders in primary care. Gen Hosp Psychiatry 1997;19:98-111.
16. American Psychiatric Association, American Psychiatric Association, Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. 4th ed. Washington, DC: American Psychiatric Association; 2000.
17. Schwenk TL, Coyne JC, Fechner-Bates S. Differences between detected and undetected patients in primary care and depressed psychiatric patients. Gen Hosp Psychiatry 1996;18:407-15.
18. Williams JW, Jr, Noel PH, Cordes JA, Ramirez G, Pignone M. Is this patient clinically depressed? JAMA 2002;287:1160-70.
19. Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry 1992;49:624-9.
20. Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70.
21. Silverstone PH. Poor efficacy of the Hospital Anxiety and Depression Scale in the diagnosis of major depressive disorder in both medical and psychiatric patients. J Psychosom Res 1994;38:441-50.
22. Wells KB, Sherbourne C, Schoenbaum M, et al. Impact of disseminating quality improvement programs for depression in managed primary care: a randomized controlled trial. JAMA 2000;283:212-20.
23. Stewart AL, Hays RD, Ware JE, Jr. The MOS short-form general health survey. Reliability and validity in a patient population. Med Care 1988;26:724-35.
24. Rost K, Nutting P, Smith J, Coyne JC, Cooper-Patrick L, Rubenstein L. The role of competing demands in the treatment provided primary care patients with major depression. Arch Fam Med 2000;9:150-4.
25. Leon AC, Portera L, Olfson M, et al. False positive results: a challenge for psychiatric screening in primary care. Am J Psychiatry 1997;154:1462-4.
26. Nease DE, Jr, Klinkman MA, Volk RJ. Improved detection of depression in primary care through severity detection. J Fam Pract 2002;51:1065-70.
27. Spitzer RL, Williams J, Kroenke K, Linzer M, deGruy FV, Hann SR, et al. Utility of a new procedure for diagnosing mental disorders in primary care: the PRIME-MD 1000 study. JAMA 1994;272:1749-56.
28. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
29. Coyne JC, Fechner-Bates S, Schwenk TL. Prevalence, nature, and comorbidity of depressive disorders in primary care. Gen Hosp Psychiatry 1994;16:267-76.
30. Wells K, Burnam M, Rogers W, Hays R, Camp P. The course of depression in adult outpatients: results from the Medical Outcomes Study. Arch Gen Psychiatry 1992;49:788-94.
31. Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta-analytic comparison. J Consult Clin Psychol 1986;54:54-9.
32. Beck A, Ward C, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
33. Zung WW, Richards CB, Short MJ. Self-rating depression scale in an outpatient clinic. Further validation of the SDS. Arch Gen Psychiatry 1965;13:508-15.
34. Radloff LS. The CES-D Scale: A self-report depression scale for research in the general population. Applied Psychological Measurement 1977;1:385-401.
35. Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version. In: Clinical Gerontology: A Guide to Assessment and Intervention. New York: Haworth Press; 1986;165-73.
1. Katon W, Schulberg H. Epidemiology of depression in primary care. Gen Hosp Psychiatry 1992;14:237-47.
2. Magruder-Habib K, Zung WW, Feussner JR. Improving physicians’ recognition and treatment of depression in general medical care. Results from a randomized clinical trial. Med Care 1990;28:239-50.
3. Coyne JC, Schwenk TL, Fechner-Bates S. Nondetection of depression by primary care physicians reconsidered. Gen Hosp Psychiatry 1995;17:3-12.
4. Williams JW, Mulrow CD, Kroenke K, et al. Case-finding for depression in primary care: a randomized trial. Am J Med 1999;106:36-43.
5. Greenberg PE, Stiglin LE, Finkelstein SN, Berndt ER. The economic burden of depression in 1990. J Clin Psychiatry 1993;54:405-18.
6. Katon W, Von Korff M, Lin E, et al. Distressed high utilizers of medical care. DSM-III-R diagnoses and treatment needs. Gen Hosp Psychiatry 1990;12:355-62.
7. Von Korff M, Ormel J, Katon W, Lin EH. Disability and depression among high utilizers of health care. A longitudinal analysis. Arch Gen Psychiatry 1992;49:91-100.
8. Wells KB, Stewart A, Hays RD, et al. The functioning and well-being of depressed patients. Results from the Medical Outcomes Study. JAMA 1989;262:914-9.
9. Ford DE, Mead LA, Chang PP, Cooper-Patrick L, Wang NY, Klag MJ. Depression is a risk factor for coronary artery disease in men: the precursors study. Arch Intern Med 1998;158:1422-6.
10. Pignone MP, Gaynes BN, Rushton JL, et al. Screening for depression in adults: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med 2002;136:765-76.
11. US. Preventive Services Task Force. Screening for depression: recommendations and rationale. Ann Intern Med 2002;136:760-4.
12. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999;282:1737-44.
13. Valenstein M, Vijan S, Zeber JE, Boehm K, Buttar A. The cost-utility of screening for depression in primary care. Ann Intern Med 2001;134:345-60.
14. Jaen CR, Stange KC, Nutting PA. Competing demands of primary care: a model for the delivery of clinical preventive services. J Fam Pract 1994;38:166-71.
15. Klinkman MS. Competing demands in psychosocial care. A model for the identification and treatment of depressive disorders in primary care. Gen Hosp Psychiatry 1997;19:98-111.
16. American Psychiatric Association, American Psychiatric Association, Task Force on DSM-IV. Diagnostic and statistical manual of mental disorders: DSM-IV-TR. 4th ed. Washington, DC: American Psychiatric Association; 2000.
17. Schwenk TL, Coyne JC, Fechner-Bates S. Differences between detected and undetected patients in primary care and depressed psychiatric patients. Gen Hosp Psychiatry 1996;18:407-15.
18. Williams JW, Jr, Noel PH, Cordes JA, Ramirez G, Pignone M. Is this patient clinically depressed? JAMA 2002;287:1160-70.
19. Spitzer RL, Williams JB, Gibbon M, First MB. The Structured Clinical Interview for DSM-III-R (SCID). I: History, rationale, and description. Arch Gen Psychiatry 1992;49:624-9.
20. Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70.
21. Silverstone PH. Poor efficacy of the Hospital Anxiety and Depression Scale in the diagnosis of major depressive disorder in both medical and psychiatric patients. J Psychosom Res 1994;38:441-50.
22. Wells KB, Sherbourne C, Schoenbaum M, et al. Impact of disseminating quality improvement programs for depression in managed primary care: a randomized controlled trial. JAMA 2000;283:212-20.
23. Stewart AL, Hays RD, Ware JE, Jr. The MOS short-form general health survey. Reliability and validity in a patient population. Med Care 1988;26:724-35.
24. Rost K, Nutting P, Smith J, Coyne JC, Cooper-Patrick L, Rubenstein L. The role of competing demands in the treatment provided primary care patients with major depression. Arch Fam Med 2000;9:150-4.
25. Leon AC, Portera L, Olfson M, et al. False positive results: a challenge for psychiatric screening in primary care. Am J Psychiatry 1997;154:1462-4.
26. Nease DE, Jr, Klinkman MA, Volk RJ. Improved detection of depression in primary care through severity detection. J Fam Pract 2002;51:1065-70.
27. Spitzer RL, Williams J, Kroenke K, Linzer M, deGruy FV, Hann SR, et al. Utility of a new procedure for diagnosing mental disorders in primary care: the PRIME-MD 1000 study. JAMA 1994;272:1749-56.
28. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med 2001;16:606-13.
29. Coyne JC, Fechner-Bates S, Schwenk TL. Prevalence, nature, and comorbidity of depressive disorders in primary care. Gen Hosp Psychiatry 1994;16:267-76.
30. Wells K, Burnam M, Rogers W, Hays R, Camp P. The course of depression in adult outpatients: results from the Medical Outcomes Study. Arch Gen Psychiatry 1992;49:788-94.
31. Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta-analytic comparison. J Consult Clin Psychol 1986;54:54-9.
32. Beck A, Ward C, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561-71.
33. Zung WW, Richards CB, Short MJ. Self-rating depression scale in an outpatient clinic. Further validation of the SDS. Arch Gen Psychiatry 1965;13:508-15.
34. Radloff LS. The CES-D Scale: A self-report depression scale for research in the general population. Applied Psychological Measurement 1977;1:385-401.
35. Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): Recent evidence and development of a shorter version. In: Clinical Gerontology: A Guide to Assessment and Intervention. New York: Haworth Press; 1986;165-73.
Management of acne
- Select medication based on the type and severity of a patient’s acne, as well as the patient’s skin type: creams, lotions, or ointments for dry skin; solutions or gels for oily skin.
- Choose topical therapy whenever possible to minimize side effects.
- Allow 6 to 8 weeks for most treatments to work before deciding to try another regimen or add other agents.
The surest route to success in treating acne vulgaris follows 3 steps. First, establish the type and severity of acne. Second, select medication appropriate for the patient’s condition and skin type. In general, patients with oily skin benefit from solutions or gels, while those with dry skin do better with creams, lotions, or ointments.1 Third, educate the patient about the disease, the different types of medications and their side effects, and expectations for improvement that are realistic. Realistic expectations should enhance compliance and lead to the successful resolution of a debilitating disease.
Types And Severity Of Acne
There are 3 types of acne: comedonal, papulopustular, and nodular (Table 1), all of which result from a multifactorial pathophysiologic process in the pilosebaceous unit: (1) sebum production, (2) follicular hyperkeratinization, (3) proliferation and colonization by Propionibacterium acnes, and (4) the release of inflammatory mediators.2 The resulting lesions include noninflammatory open (blackheads) and closed (whiteheads) comedones, as well as inflammatory papules, pustules, and nodules.
Acne severity is rated according to the Combined Acne Severity Classification that classifies acne into mild, moderate, and severe, based on the number and type of lesions (Table 2).3 Determining acne type and severity serves as a guide to treatment (Table 3).
Table 1
Types of acne
TABLE 2
Combined acne severity classification
| Severity | Definition |
|---|---|
| Mild acne | Fewer than 20 comedones, or Fewer than 15 inflammatory lesions, or Total lesion count fewer than 30 |
| Moderate acne | 20–100 comedones, or 15–50 inflammatory lesions, or total lesion count 30–125 |
| Severe acne | More than 5 nodules, or Total inflammatory count greater than 50, or Total lesion count greater than 125 |
TABLE 3
Treatment options based on type of acne
| Non-inflammatory acne | Inflammatory acne | ||
|---|---|---|---|
| Treatment | Comedonal | Papulo-pustular | Nodulocystic |
| Topical | |||
| Tretinoin (Renova et al) | X | X | |
| Benzoyl Peroxide | X | X | |
| Adapalene (Differin) | X | X | |
| Antibiotics | X | X | |
| Azelaic acid (Azelex) | X | X | |
| Tazarotene (Tazorac) | X | X | |
| Systemic | |||
| Oral contraceptives | X | X | X |
| Erythromycin | X | X | |
| Tetracycline | X | X | |
| Doxycycline | X | X | |
| Minocycline | X | X | |
| Isotretinoin (Accutane) | X | ||
| Adapted from Use of Systemic Agents in the Treatment of Acne Vulgaris, Am Fam Physician 2000;62. | |||
Treatment options
A variety of medications are available for the treatment of acne vulgaris. Note that most treatment regimens should be used for at least 6 to 8 weeks to judge their effectiveness before considering alternative treatments or adding other agents. The Figure 2, an algorithmic guide to the treatment of acne, represents the author’s assessment of the current literature. Alternative approaches may be appropriate after discussing options with individual patients. Table 4 summarizes the strength of evidence for acne interventions and how each compares with other treatments for the same type of acne.
Topical agents
Salicylic acid, found in a number over-the-counter cleansers, has both anti-inflammatory and mild comedolytic effects. It can be used as initial therapy for mild acne or as an adjunctive agent in a broader therapeutic regimen. In a placebo-controlled study of 114 patients, 2% salicylic acid lotion demonstrated a statistically significant improvement from baseline at 12 weeks (SOR: B).4 No side effect data were provided.
Tea tree oil comes from the Australian tree Melaleuca alternifolia and has had success anecdotally in treating various skin conditions. In a single-blind trial of 124 patients, 5% tea tree oil gel was compared with 5% benzoyl peroxide lotion in the treatment of mild-to-moderate acne. Both agents significantly reduced the number of inflammatory lesions and comedones (SOR: B).5 However, benzoyl peroxide was statistically superior to tea tree oil in reducing inflammatory lesions and had a faster onset of action. Encouragingly, patients treated with tea tree oil experienced fewer side effects.
Benzoyl peroxide (BPO) is a potent bactericidal agent with mild keratolytic properties. Several trials have shown the 5% concentration to be consistently superior to placebo at a statistically significant level in the treatment of mild-to-moderate acne, with a 30% decrease in lesion counts (SOR: A).6,7 In addition, two small trials involving 153 patients with mild-to-moderate acne compared the efficacy of different concentrations of topical benzoyl peroxide (2.5% vs 5% and 2.5% vs 10%) used twice daily.8 These trials demonstrated no differences in efficacy among the various preparations based on lesion counts, and there was no dose-response effect (SOR: A). Erythema and scaling occurred with almost identical frequency with the 2.5% and 5% concentrations but more often with the 10% concentration. Thus, the 2.5% and 5% concentrations appear to be preferable based on the balance of risks and benefits.
Topical tretinoin (Renova, Retin-A, Avita) is a comedolytic agent effective as monotherapy for noninflammatory acne. In two randomized controlled trials involving 292 patients of comparable age, use of 0.02% and 0.05% tretinoin strengths showed a statistically significant reduction in comedones and papules with a dose-response effect after at least 4 to 8 weeks of treatment when compared with placebo (SOR: A).9,10 However, there was also a statistically significant increase in erythema and peeling that was maximal after 1 to 3 weeks and decreasing thereafter. In addition, an exacerbation of inflammatory lesions (pustular flare) may occur within 2 to 4 weeks of onset of therapy.11
Adapalene (Differin) is a synthetic naphthoic acid derivative with retinoid activity. Several large, randomized studies have shown that adapalene gel 0.1% and tretinoin concentrations ranging from 0.025% to 0.1% were comparable in reducing total lesion counts by 50% in 4 to 12 weeks (SOR: A).12-14 One trial found that adapalene 0.1% produced a statistically significant reduction greater than that with tretinoin 0.025% in both noninflammatory and inflammatory lesions at 12 weeks (SOR: A).12 Adapalene was also significantly better tolerated than tretinoin, as evidenced by less erythema, scaling, and dryness.12,15 Thus far, there have been no significant studies comparing adapalene to other topical agents such as benzoyl peroxide.
Azelaic acid (Azelex) is a dicarboxylic acid that possesses bacteriostatic properties and is structurally unrelated to any of the conventional acne therapies. In a single-blind trial of 309 patients comparing 20% AZA with 5% benzoyl peroxide and placebo, AZA yielded a significant decrease in papulo-pustular lesion counts by 35% compared with placebo. There was equivalent efficacy between AZA and benzoyl peroxide (SOR: B).16 Patients tolerated AZA better than benzoyl peroxide, with 9% of AZA recipients reporting a burning sensation that subsided after 2 weeks, and 15% of the benzoyl peroxide group reporting local side effects. In another controlled comparison, 20% AZA cream used twice daily for 5 to 6 months was comparable in efficacy to 0.05% tretinoin cream for patients with comedonal acne but statistically more effective in reducing the number of papules (SOR: B).17 Tretinoin caused significantly more erythema and scaling than did AZA.
Tazarotene (Tazorac) is the first of a family of receptor-selective acetylenic retinoids. In a multicenter, randomized controlled trial including 334 patients, tazarotene 0.1% and 0.05%, applied once daily for mild-to-moderate acne significantly reduced noninflammatory acne and total lesion counts when compared with placebo at 4 to 8 weeks (SOR: B).18 The 0.1% gel also significantly reduced inflammatory lesions at 12 weeks. Adverse effects were dose related, ranging from 5% to 13% and included erythema and burning. There are no published trials comparing tazarotene with other retinoids or benzoyl peroxide. Tazarotene is 30% to 70% more expensive than comparable topical agents such as tretinoin, benzoyl peroxide, and antibiotics.
FIGURE
Treatment of acne according to type and severity
TABLE 4
Medication options for acne vulgaris
| Evidence Strength* | Medication | Cost per month** | Relative efficacy | Comparator | Comment |
|---|---|---|---|---|---|
| Comedonal, papulopustular, or nodulocystic | |||||
| A | Norgestimate/ethinyl estradiol | $31.08 | > | Placebo | Decreases comedone and inflammatory lesion counts |
| Comedonal or papulopustular | |||||
| A | Adapalene | $34.47 (gel) | = | Tretinoin | Adapalene has better side-effect profile |
| A | Benzoyl peroxide | $7.99–$16.19 | > | Placebo | Price depends on generic vs brand, not concentration |
| A | Clindamycin | $34.73 (gel) | > | Placebo | Topical |
| A | Erythromycin | $18.31 (gel) | > | Placebo | Topical |
| A or B | Tretinoin | $23.91 | > | Placebo | Evidence strength A for noninflammatory and B for inflammatory |
| B | Azelaic acid | $44.40 | > | Placebo | Topical |
| B | Azelaic acid | $44.40 | = | Benzoyl peroxide | Azelaic acid has better side-effect profile |
| B | Azelaic acid | $44.40 | = | Tretinoin | Azelaic acid has better side-effect profile |
| B | Clindamycin | $34.73 | = | Erythromycin | Topical |
| B | Clindamycin | $34.73 | = | Benzoyl peroxide | Topical |
| B | Salicylic Acid | > | Placebo | Topical | |
| B | Tazarotene | $64.75 (.05%) $68.74 (0.1%) | > | Placebo | Side effects similar to those of topical retinoids |
| B | Tretinoin | $23.91 | > | Benzoyl peroxide | Tretinoin: stronger effect on comedones; BPO: stronger effect on papules |
| Papulopustular or nodulocystic | |||||
| A | Tetracycline | $8.38 | > | Placebo | Oral |
| B | Doxycycline | $24.82 | > | Placebo | Oral |
| B | Erythromycin | $27.15 | = | Tetracycline | Oral. Higher resistance levels of P acnes to erythromycin |
| B | Minocycline | $21.90 | > | Placebo | Oral |
| B | Minocycline | $21.90 | = | Tetracycline | Oral |
| KEY: > is more effective than; < is less effective than; = is equivalent to. | |||||
| *Evidence Strength: | |||||
| A = At least two trials of acceptable quality showing moderate to strong statistical evidence for clinically meaningful endpoint and effect. | |||||
| B = Evidence is of modest strength, such as when only one trial addresses a comparison, there is significant heterogeneity, large differences are not statistically significant, or poor trial quality prevents accepting strong statistical evidence at face value. | |||||
| **Cost: Referenced from a major on-line retail pharmacy. | |||||
Topical antibiotics
Topical antibiotics are effective in the treatment of mild-to-moderate inflammatory acne by reducing the population of P acnes in sebaceous follicles and by suppressing chemotaxis.11 Several large randomized controlled trials demonstrated that topical clindamycin 1% and topical erythromycin 2% applied twice daily were consistently superior to placebo in reducing the number of papules and pustules in patients with moderate-to-severe acne (SOR: A).19-23 Erythema and peeling were rare, comparable to that seen with placebo. Moreover, a randomized trial of 102 patients comparing 1% clindamycin with 2% erythromycin demonstrated that both medications significantly reduced the number of papules and comedones with no significant differences between the two (SOR: B).24,25 Furthermore, in two double-blind, randomized trials involving 334 patients, a combination gel containing clindamycin 1% and benzoyl peroxide 5% proved superior to each component alone in reducing inflammatory lesions, and superior to the clindamycin-only gel in reducing noninflammatory lesions (SOR: B).26 Trial data on the combination gel containing erythromycin 3% and benzoyl peroxide 5% are of poor quality; thus the same conclusion cannot be made.
Oral antibiotics
Oral antibiotics are most often used for moderate-to-severe inflammatory acne. They work by suppressing P acnes growth, thereby reducing the production of inflammatory mediators.27 However, as systemicagents, they cause more significant and diverse side effects than do topical agents. Unfortunately there are no head-to-head trials comparing different oral antibiotics, or comparing oral and topical antibiotics.
Erythromycin. In a randomized study of 200 patients with moderate-to-severe acne, erythromycin, 1 g in 2 divided doses daily, significantly reduced the comedone, papule, and pustule count after 8 weeks (SOR: B).28 The side-effect rate was 8%, usually associated with gastrointestinal irritation. Studies have shown that P acnes exhibits greater resistance to erythromycin than to tetracycline.29
Tetracycline/doxycycline/minocycline.Tetracycline and its lipophilic derivatives, doxycycline and minocycline, are the most commonly prescribed oral agents for acne vulgaris. As a class, tetracyclines should not be prescribed for pregnant women or for those younger than 9 years of age, to avoid the risks of tooth discoloration and bone growth retardation in the fetus or child.30 Various double-blinded, randomized controlled trials involving patients with mild-to-moderate acne have shown that tetracycline confers a statistically significant improvement over placebo as early as 6 weeks (SOR: A).31-33 Adverse effects include gastrointestinal upset, vaginal yeast infection, and a theoretical decrease in the efficacy of oral contraceptives.
Doxycycline, 100mg/d, has been shown to significantly reduce inflammatory lesions in a crossover trial of 62 patients (SOR: B).34 Its adverse effect profile is similar to that of tetracycline, though it tends to cause more photosensitivity (4% vs 1%).35
In a recent Cochrane review of 27 studies, minocycline was shown to be an effective treatment for acne, but no randomized controlled trial evidence was found to support the benefits of minocycline in acne resistant to other therapies (SOR: A).36 A recent study demonstrated that minocycline has a greater tendency than tetracycline or doxycycline to cause rare adverse side effects, including serumsickness-like reactions, drug-induced lupus, and hypersensitivity reactions.35 These factors, in addition to the higher cost, suggest that minocycline should not be a first line antibiotic choice for treating acne.
Oral contraceptives
Oral contraceptives (OCs) reduce the severity of acne vulgaris by decreasing the amount of circulating androgens.37 In 1997, a triphasic combination OC containing ethinyl estradiol 0.035 mg and increasing doses of norgestimate (0.180 mg, 0.215 mg, and 0.250 mg) was approved by the FDA for the treatment of acne in women. This decision was based on the results of a randomized, double-blind trial involving 257 patients in which the triphasic contraceptive was compared with placebo over 6 months (SOR: A).38 The OC group showed statistically significant improvement greater than that of the placebo group in all types of acne lesions. It also reduced total lesion counts by more than 53% in female subjects at 26 weeks, compared with lesion reductions of about 27% in controls. The principal adverse effect noted in this study was nausea.
Isotretinoin
Isotretinoin (Accutane) is an oral retinoid labeled for use in patients with severe, refractory, nodulocystic acne. In a randomized, crossover trial that included 33 patients, isotretinoin significantly decreased the number of nodulocystic lesions by 95% when compared with placebo, with only rare side effects of cheilitis and dermatitis (SOR: B).39 However, other studies suggest that cheilitis is fairly common and its absence may imply noncompliance or malabsorption of the drug.40 In addition, the FDA issued a warning in 1998 regarding possible increased risks in depression, psychosis, suicidal thoughts, and suicide attempts, though no conclusive evidence has been found.40 The typical dosage of isotretinoin is 0.5 to 1 mg/kg daily in two divided doses, with a standard cumulative maximum dose of 120 to 150 mg/kg per treatment course.41
In April 2002, Roche Laboratories released the System to Manage Accutane Related Teratogenicity (S.M.A.R.T) program, aimed at preventing pregnant women from receiving isotretinoin.42 Major malformations may occur in 25% to 30% of fetuses exposed to isotretinoin.43 Under this program, female patients must have both a screening and confirmation pregnancy test (urine or serum) prior to receiving a prescription for isotretinoin. In addition, patients must commit to using 2 forms of birth control for at least 1 month prior to initiation of therapy, during therapy, and 1 month after discontinuing isotretinoin. During monthly visits, a pregnancy test must be obtained and no more than a 30-day supply of isotretinoin may be prescribed. Finally, pharmacists will fill prescriptions only if an isotretinoin qualification sticker is affixed, obtained after signing and returning the S.M.A.R.T Letter of Understanding.
In addition to procedural safeguards, it is necessary to monitor lipid levels (hypertriglyceridemia and hypercholesterolemia) and liver enzymes at the start of therapy and at each monthly visit. If elevations occur in these measurements, dosage reduction or drug discontinuation should be considered. Given the intensity of monitoring that is required, some family physicians may wish to refer patients who require Accutane to a dermatologist.
1. Thiboutot DM. An overview of acne and its treatment. Cutis 1996;57:8-12.
2. Leyden JJ. Therapy for acne vulgaris. N Engl J Med 1997;336:1156-62.
3. Issued by funding/sponsoring agency: Management of Acne Volume 1: Evidence Report and Appendixes. Rockville, Md: Dept. of Health and Human Services (US), Public Health Service; 2001 Sep. Report No.: 01-E019. Issued by performing agency: Lehmann HP, Andrews JS, Robinson KA, Holloway VL, Goodman SN. Johns Hopkins Evidence-based Practice Center. Contract No.: 290-97-006. Sponsored by the Agency for Healthcare Research and Quality.
4. Eady EA, Burke BM, Pulling K, Cunliffe WJ. The benefit of 2% salicylic acid lotion in acne—A placebo-controlled study. J Dermatol Treat 1996;7:93-6.
5. Basset OB, Pannowitz DL, Barnetson RS. A comparative study of tea-tree oil versus benzoyl peroxide in the treatment of acne. Med J Aust 1990;153:455-8.
6. Hughes BR, Norris JF, Cunliffe WJ. A double-blind evaluation of topical isotretinoin 0.05%, benzoyl peroxide gel 5% and placebo in patients with acne. Clin Exp Dermatol 1992;17:165-8.
7. Ede M. A double-blind, comparative study of benzoyl peroxide, benzoyl peroxide-chlorhydroxyquinoline, benzoyl peroxide-chlorhydroxyquinolone-hydrocortisone, and placebo lotions in acne. Curr Ther Res Clin Exp 1973;15:624-9.
8. Mills OH, Jr, Kligman AM, Pochi P, Comite H. Comparing 2.5%, 5%, and 10% benzoyl peroxide on inflammatory acne vulgaris. Int J Dermatol 1986;25:664-7.
9. Pedace FJ, Stoughton R. Topical retinoic acid in acne vulgaris. Br J Dermatol 1971;84:465-9.
10. Christiansen JV, Gadborg E, Ludvigsen K, et al. Topical tretinoin, vitamin A acid (Airol) in acne vulgaris. A controlled clinical trial. Dermatologica 1974;148:82-9.
11. Berson DS, Shalita AR. The treatment of acne: the role of combination therapies. J Am Acad Dermatol 1995;32:531-41.
12. Shalita A, Weiss JS, Chalker DK, et al. A comparison of the efficacy and safety of adapalene gel 0.1% and tretinoin gel 0.025% in the treatment of acne vulgaris: a multicenter trial. J Am Acad Dermatol 1996;34:482-5.
13. Cunliffe W, Caputo R, Dreno B, et al. Clinical efficacy and safety comparison of adapalene gel and tretinoin gel in the treatment of acne vulgaris: Europe and U.S.multicenter trials. J Am Acad Dermatol 1997;36:S126-34.
14. Galvin SA, Gilbert R, Baker M, et al. Comparative tolerance of adapalene 0.1% gel and six different tretinoin formulations. Br J Dermatol 1998;139:S34-40.
15. Clucas A, Verschoore M, Sorba V, et al. Adapalene 0.1% gel is better tolerated than tretinoin 0.025% gel in acne patients. J Am Acad Dermatol 1997;36:S116-8.
16. Cavicchini S, Caputo R. Long-term treatment of acne with 20% azelaic acid cream. Acta Derm Venereol Suppl. 1989;143:40-4.
17. Katsambas A, Graupe K, Stratigos J. Clinical studies of 20% azelaic cream in the treatment of acne vulgaris. Comparison with vehicle and topical tretinoin. Acta Derm Venereol Suppl 1989;143:35-9.
18. Shalita AR, Chalker DK, Griffith RF, et al. Tazarotene gel is safe and effective in the treatment of acne vulgaris: a multicenter, double blind, vehicle-controlled study. Cutis 1999;63:349-53.
19. Becker LE, Bergstresser PR, Whiting DA, et al. Topical clindamycin therapy for acne vulgaris. A cooperative clinical study Arch Dermatol 1981;117:482-5.
20. Ellis CN, Gammon WR, Stone DZ, Heezen-Wehner JL. A comparison of Cleocin T Solution, Cleocin T Gel, and placebo in the treatment of acne vulgaris. Cutis 1988;42:245-7.
21. Pochi PE, Bagatell FK, Ellis CN, et al. Erythromycin 2 percent gel in the treatment of acne vulgaris. Cutis 1988;41:132-6.
22. Lesher JL, Jr, Chalker DK, Smith JG, Jr, et al. An evaluation of a 2% erythromycin ointment in the topical therapy of acne vulgaris. J Am Acad Dermatol 1985;12:526-31.
23. Dobson RL, Belknap BS. Topical erythromycin solution in acne. Results of a multiclinic trial. J Am Acad Dermatol 1980;3:478-82.
24. Leyden JJ, Shalita AR, Saatjian GD, Sefton J. Erythromycin 2% gel in comparison with clindamycin phosphate 1% solution in acne vulgaris. J Am Acad Dermatol 1987;16:822-7.
25. Shalita AR, Smith EB, Bauer E. Topical erythromycin v clindamycin therapy for acne. A multicenter, double-blind comparison. Arch Dermatol 1984;120:351-5.
26. Lookingbill DP, Chalker DK, Lindholm JS, et al. Treatment of acne with a combination clindamycin/benzoyl peroxide gel compared with clindamycin gel, benzoyl peroxide gel and vehicle gel: combined results of two double-blind investigations. J Am Acad Dermatol 1997;37:590-5.
27. Sykes NL, Webster GF. Acne: a review of optimum treatment. Drugs 1994;48:59-70.
28. Gammon WR, Meyer C, Lantis S, et al. Comparative efficacy of oral erythromycin versus oral tetracycline in the treatment of acne vulgaris. A double-blind study. J Am Acad Dermatol 1986;14:183-6.
29. Eady EA, Jones CE, Tipper JL, et al. Antibiotic resistant Propionibacteria in acne: need for policies to modify antibiotic usage. Br Med J 1993;306:555-6.
30. McEvoy GK, editor. AHFS Drug Information. Bethesda, Md: American Society of Health System Pharmacists; 1996.
31. Blaney DJ, Cook CH. Topical use of tetracycline in the treatment of acne: a double-blind study comparing topical and oral tetracycline therapy and placebo. Arch Dermatol 1976;112:971-3.
32. Lane P, Williamson DM. Treatment of acne vulgaris with tetracycline hydrochloride: a double-blind trial with 51 patients. Br Med J 1969;2:76-9.
33. Wong RC, Kang S, Heezen JL, et al. Oral ibuprofen and tetracycline for the treatment of acne vulgaris. J Am Acad Dermatol 1984;11:1076-81.
34. Plewig G, Petrozzi JW, Berendes U. Double-blind study of doxycycline in acne vulgaris. Arch Dermatol 1970;101:435-8.
35. Shapiro LE, Knowles SR, Shear NH. Comparative safety of tetracycline, minocycline, and doxycycline. Arch Dermatol 1997;133:1224-30.
36. Garner SE, Eady EA, Popescu C, Newton J, Li Wan Po A. Minocycline for acne vulgaris: efficacy and safety (Cochrane Review). In: The Cochrane Library., Issue 4, 2001.
37. Lucky AW. Hormonal correlates of acne and hirsutism. Am J Med 1995;98:89S-94S.
38. Lucky AW, Henderson TA, Olson WH, et al. Effectiveness of norgestimate and ethinyl estradiol in treating moderate acne vulgaris. J Am Acad Dermatol 1997;37:746-54.
39. Peck GL, Olsen TG, Butkus D, et al. Isotretinoin versus placebo in the treatment of cystic acne. A randomized double-blind study. J Am Acad Dermatol 1982;6:735-45.
40. Hanson N, Leachman S. Safety Issues in Isotretinoin Therapy. Semin Cutan Med Surg 2001;20:166-83.
41. Layton AM, Knaggs H, Taylor J, Cunliffe WJ. Isotretinoin for acne vulgaris—10 years later: a safe and successful treatment. Br J Dermatol 1993;129:292-6.
42. Accutane prescribing information. Nutley, N.J.: Roche Pharmaceuticals, 1998.
43. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, et al. Retinoic acid embryopathy. N Engl J Med 1985;313:837-41.
44. Helms SE, Bredle DL, Zajic J, et al. Oral contraceptive failure rates and oral antibiotics. J Am Acad Dermatol 1997;36:705-10.
- Select medication based on the type and severity of a patient’s acne, as well as the patient’s skin type: creams, lotions, or ointments for dry skin; solutions or gels for oily skin.
- Choose topical therapy whenever possible to minimize side effects.
- Allow 6 to 8 weeks for most treatments to work before deciding to try another regimen or add other agents.
The surest route to success in treating acne vulgaris follows 3 steps. First, establish the type and severity of acne. Second, select medication appropriate for the patient’s condition and skin type. In general, patients with oily skin benefit from solutions or gels, while those with dry skin do better with creams, lotions, or ointments.1 Third, educate the patient about the disease, the different types of medications and their side effects, and expectations for improvement that are realistic. Realistic expectations should enhance compliance and lead to the successful resolution of a debilitating disease.
Types And Severity Of Acne
There are 3 types of acne: comedonal, papulopustular, and nodular (Table 1), all of which result from a multifactorial pathophysiologic process in the pilosebaceous unit: (1) sebum production, (2) follicular hyperkeratinization, (3) proliferation and colonization by Propionibacterium acnes, and (4) the release of inflammatory mediators.2 The resulting lesions include noninflammatory open (blackheads) and closed (whiteheads) comedones, as well as inflammatory papules, pustules, and nodules.
Acne severity is rated according to the Combined Acne Severity Classification that classifies acne into mild, moderate, and severe, based on the number and type of lesions (Table 2).3 Determining acne type and severity serves as a guide to treatment (Table 3).
Table 1
Types of acne
TABLE 2
Combined acne severity classification
| Severity | Definition |
|---|---|
| Mild acne | Fewer than 20 comedones, or Fewer than 15 inflammatory lesions, or Total lesion count fewer than 30 |
| Moderate acne | 20–100 comedones, or 15–50 inflammatory lesions, or total lesion count 30–125 |
| Severe acne | More than 5 nodules, or Total inflammatory count greater than 50, or Total lesion count greater than 125 |
TABLE 3
Treatment options based on type of acne
| Non-inflammatory acne | Inflammatory acne | ||
|---|---|---|---|
| Treatment | Comedonal | Papulo-pustular | Nodulocystic |
| Topical | |||
| Tretinoin (Renova et al) | X | X | |
| Benzoyl Peroxide | X | X | |
| Adapalene (Differin) | X | X | |
| Antibiotics | X | X | |
| Azelaic acid (Azelex) | X | X | |
| Tazarotene (Tazorac) | X | X | |
| Systemic | |||
| Oral contraceptives | X | X | X |
| Erythromycin | X | X | |
| Tetracycline | X | X | |
| Doxycycline | X | X | |
| Minocycline | X | X | |
| Isotretinoin (Accutane) | X | ||
| Adapted from Use of Systemic Agents in the Treatment of Acne Vulgaris, Am Fam Physician 2000;62. | |||
Treatment options
A variety of medications are available for the treatment of acne vulgaris. Note that most treatment regimens should be used for at least 6 to 8 weeks to judge their effectiveness before considering alternative treatments or adding other agents. The Figure 2, an algorithmic guide to the treatment of acne, represents the author’s assessment of the current literature. Alternative approaches may be appropriate after discussing options with individual patients. Table 4 summarizes the strength of evidence for acne interventions and how each compares with other treatments for the same type of acne.
Topical agents
Salicylic acid, found in a number over-the-counter cleansers, has both anti-inflammatory and mild comedolytic effects. It can be used as initial therapy for mild acne or as an adjunctive agent in a broader therapeutic regimen. In a placebo-controlled study of 114 patients, 2% salicylic acid lotion demonstrated a statistically significant improvement from baseline at 12 weeks (SOR: B).4 No side effect data were provided.
Tea tree oil comes from the Australian tree Melaleuca alternifolia and has had success anecdotally in treating various skin conditions. In a single-blind trial of 124 patients, 5% tea tree oil gel was compared with 5% benzoyl peroxide lotion in the treatment of mild-to-moderate acne. Both agents significantly reduced the number of inflammatory lesions and comedones (SOR: B).5 However, benzoyl peroxide was statistically superior to tea tree oil in reducing inflammatory lesions and had a faster onset of action. Encouragingly, patients treated with tea tree oil experienced fewer side effects.
Benzoyl peroxide (BPO) is a potent bactericidal agent with mild keratolytic properties. Several trials have shown the 5% concentration to be consistently superior to placebo at a statistically significant level in the treatment of mild-to-moderate acne, with a 30% decrease in lesion counts (SOR: A).6,7 In addition, two small trials involving 153 patients with mild-to-moderate acne compared the efficacy of different concentrations of topical benzoyl peroxide (2.5% vs 5% and 2.5% vs 10%) used twice daily.8 These trials demonstrated no differences in efficacy among the various preparations based on lesion counts, and there was no dose-response effect (SOR: A). Erythema and scaling occurred with almost identical frequency with the 2.5% and 5% concentrations but more often with the 10% concentration. Thus, the 2.5% and 5% concentrations appear to be preferable based on the balance of risks and benefits.
Topical tretinoin (Renova, Retin-A, Avita) is a comedolytic agent effective as monotherapy for noninflammatory acne. In two randomized controlled trials involving 292 patients of comparable age, use of 0.02% and 0.05% tretinoin strengths showed a statistically significant reduction in comedones and papules with a dose-response effect after at least 4 to 8 weeks of treatment when compared with placebo (SOR: A).9,10 However, there was also a statistically significant increase in erythema and peeling that was maximal after 1 to 3 weeks and decreasing thereafter. In addition, an exacerbation of inflammatory lesions (pustular flare) may occur within 2 to 4 weeks of onset of therapy.11
Adapalene (Differin) is a synthetic naphthoic acid derivative with retinoid activity. Several large, randomized studies have shown that adapalene gel 0.1% and tretinoin concentrations ranging from 0.025% to 0.1% were comparable in reducing total lesion counts by 50% in 4 to 12 weeks (SOR: A).12-14 One trial found that adapalene 0.1% produced a statistically significant reduction greater than that with tretinoin 0.025% in both noninflammatory and inflammatory lesions at 12 weeks (SOR: A).12 Adapalene was also significantly better tolerated than tretinoin, as evidenced by less erythema, scaling, and dryness.12,15 Thus far, there have been no significant studies comparing adapalene to other topical agents such as benzoyl peroxide.
Azelaic acid (Azelex) is a dicarboxylic acid that possesses bacteriostatic properties and is structurally unrelated to any of the conventional acne therapies. In a single-blind trial of 309 patients comparing 20% AZA with 5% benzoyl peroxide and placebo, AZA yielded a significant decrease in papulo-pustular lesion counts by 35% compared with placebo. There was equivalent efficacy between AZA and benzoyl peroxide (SOR: B).16 Patients tolerated AZA better than benzoyl peroxide, with 9% of AZA recipients reporting a burning sensation that subsided after 2 weeks, and 15% of the benzoyl peroxide group reporting local side effects. In another controlled comparison, 20% AZA cream used twice daily for 5 to 6 months was comparable in efficacy to 0.05% tretinoin cream for patients with comedonal acne but statistically more effective in reducing the number of papules (SOR: B).17 Tretinoin caused significantly more erythema and scaling than did AZA.
Tazarotene (Tazorac) is the first of a family of receptor-selective acetylenic retinoids. In a multicenter, randomized controlled trial including 334 patients, tazarotene 0.1% and 0.05%, applied once daily for mild-to-moderate acne significantly reduced noninflammatory acne and total lesion counts when compared with placebo at 4 to 8 weeks (SOR: B).18 The 0.1% gel also significantly reduced inflammatory lesions at 12 weeks. Adverse effects were dose related, ranging from 5% to 13% and included erythema and burning. There are no published trials comparing tazarotene with other retinoids or benzoyl peroxide. Tazarotene is 30% to 70% more expensive than comparable topical agents such as tretinoin, benzoyl peroxide, and antibiotics.
FIGURE
Treatment of acne according to type and severity
TABLE 4
Medication options for acne vulgaris
| Evidence Strength* | Medication | Cost per month** | Relative efficacy | Comparator | Comment |
|---|---|---|---|---|---|
| Comedonal, papulopustular, or nodulocystic | |||||
| A | Norgestimate/ethinyl estradiol | $31.08 | > | Placebo | Decreases comedone and inflammatory lesion counts |
| Comedonal or papulopustular | |||||
| A | Adapalene | $34.47 (gel) | = | Tretinoin | Adapalene has better side-effect profile |
| A | Benzoyl peroxide | $7.99–$16.19 | > | Placebo | Price depends on generic vs brand, not concentration |
| A | Clindamycin | $34.73 (gel) | > | Placebo | Topical |
| A | Erythromycin | $18.31 (gel) | > | Placebo | Topical |
| A or B | Tretinoin | $23.91 | > | Placebo | Evidence strength A for noninflammatory and B for inflammatory |
| B | Azelaic acid | $44.40 | > | Placebo | Topical |
| B | Azelaic acid | $44.40 | = | Benzoyl peroxide | Azelaic acid has better side-effect profile |
| B | Azelaic acid | $44.40 | = | Tretinoin | Azelaic acid has better side-effect profile |
| B | Clindamycin | $34.73 | = | Erythromycin | Topical |
| B | Clindamycin | $34.73 | = | Benzoyl peroxide | Topical |
| B | Salicylic Acid | > | Placebo | Topical | |
| B | Tazarotene | $64.75 (.05%) $68.74 (0.1%) | > | Placebo | Side effects similar to those of topical retinoids |
| B | Tretinoin | $23.91 | > | Benzoyl peroxide | Tretinoin: stronger effect on comedones; BPO: stronger effect on papules |
| Papulopustular or nodulocystic | |||||
| A | Tetracycline | $8.38 | > | Placebo | Oral |
| B | Doxycycline | $24.82 | > | Placebo | Oral |
| B | Erythromycin | $27.15 | = | Tetracycline | Oral. Higher resistance levels of P acnes to erythromycin |
| B | Minocycline | $21.90 | > | Placebo | Oral |
| B | Minocycline | $21.90 | = | Tetracycline | Oral |
| KEY: > is more effective than; < is less effective than; = is equivalent to. | |||||
| *Evidence Strength: | |||||
| A = At least two trials of acceptable quality showing moderate to strong statistical evidence for clinically meaningful endpoint and effect. | |||||
| B = Evidence is of modest strength, such as when only one trial addresses a comparison, there is significant heterogeneity, large differences are not statistically significant, or poor trial quality prevents accepting strong statistical evidence at face value. | |||||
| **Cost: Referenced from a major on-line retail pharmacy. | |||||
Topical antibiotics
Topical antibiotics are effective in the treatment of mild-to-moderate inflammatory acne by reducing the population of P acnes in sebaceous follicles and by suppressing chemotaxis.11 Several large randomized controlled trials demonstrated that topical clindamycin 1% and topical erythromycin 2% applied twice daily were consistently superior to placebo in reducing the number of papules and pustules in patients with moderate-to-severe acne (SOR: A).19-23 Erythema and peeling were rare, comparable to that seen with placebo. Moreover, a randomized trial of 102 patients comparing 1% clindamycin with 2% erythromycin demonstrated that both medications significantly reduced the number of papules and comedones with no significant differences between the two (SOR: B).24,25 Furthermore, in two double-blind, randomized trials involving 334 patients, a combination gel containing clindamycin 1% and benzoyl peroxide 5% proved superior to each component alone in reducing inflammatory lesions, and superior to the clindamycin-only gel in reducing noninflammatory lesions (SOR: B).26 Trial data on the combination gel containing erythromycin 3% and benzoyl peroxide 5% are of poor quality; thus the same conclusion cannot be made.
Oral antibiotics
Oral antibiotics are most often used for moderate-to-severe inflammatory acne. They work by suppressing P acnes growth, thereby reducing the production of inflammatory mediators.27 However, as systemicagents, they cause more significant and diverse side effects than do topical agents. Unfortunately there are no head-to-head trials comparing different oral antibiotics, or comparing oral and topical antibiotics.
Erythromycin. In a randomized study of 200 patients with moderate-to-severe acne, erythromycin, 1 g in 2 divided doses daily, significantly reduced the comedone, papule, and pustule count after 8 weeks (SOR: B).28 The side-effect rate was 8%, usually associated with gastrointestinal irritation. Studies have shown that P acnes exhibits greater resistance to erythromycin than to tetracycline.29
Tetracycline/doxycycline/minocycline.Tetracycline and its lipophilic derivatives, doxycycline and minocycline, are the most commonly prescribed oral agents for acne vulgaris. As a class, tetracyclines should not be prescribed for pregnant women or for those younger than 9 years of age, to avoid the risks of tooth discoloration and bone growth retardation in the fetus or child.30 Various double-blinded, randomized controlled trials involving patients with mild-to-moderate acne have shown that tetracycline confers a statistically significant improvement over placebo as early as 6 weeks (SOR: A).31-33 Adverse effects include gastrointestinal upset, vaginal yeast infection, and a theoretical decrease in the efficacy of oral contraceptives.
Doxycycline, 100mg/d, has been shown to significantly reduce inflammatory lesions in a crossover trial of 62 patients (SOR: B).34 Its adverse effect profile is similar to that of tetracycline, though it tends to cause more photosensitivity (4% vs 1%).35
In a recent Cochrane review of 27 studies, minocycline was shown to be an effective treatment for acne, but no randomized controlled trial evidence was found to support the benefits of minocycline in acne resistant to other therapies (SOR: A).36 A recent study demonstrated that minocycline has a greater tendency than tetracycline or doxycycline to cause rare adverse side effects, including serumsickness-like reactions, drug-induced lupus, and hypersensitivity reactions.35 These factors, in addition to the higher cost, suggest that minocycline should not be a first line antibiotic choice for treating acne.
Oral contraceptives
Oral contraceptives (OCs) reduce the severity of acne vulgaris by decreasing the amount of circulating androgens.37 In 1997, a triphasic combination OC containing ethinyl estradiol 0.035 mg and increasing doses of norgestimate (0.180 mg, 0.215 mg, and 0.250 mg) was approved by the FDA for the treatment of acne in women. This decision was based on the results of a randomized, double-blind trial involving 257 patients in which the triphasic contraceptive was compared with placebo over 6 months (SOR: A).38 The OC group showed statistically significant improvement greater than that of the placebo group in all types of acne lesions. It also reduced total lesion counts by more than 53% in female subjects at 26 weeks, compared with lesion reductions of about 27% in controls. The principal adverse effect noted in this study was nausea.
Isotretinoin
Isotretinoin (Accutane) is an oral retinoid labeled for use in patients with severe, refractory, nodulocystic acne. In a randomized, crossover trial that included 33 patients, isotretinoin significantly decreased the number of nodulocystic lesions by 95% when compared with placebo, with only rare side effects of cheilitis and dermatitis (SOR: B).39 However, other studies suggest that cheilitis is fairly common and its absence may imply noncompliance or malabsorption of the drug.40 In addition, the FDA issued a warning in 1998 regarding possible increased risks in depression, psychosis, suicidal thoughts, and suicide attempts, though no conclusive evidence has been found.40 The typical dosage of isotretinoin is 0.5 to 1 mg/kg daily in two divided doses, with a standard cumulative maximum dose of 120 to 150 mg/kg per treatment course.41
In April 2002, Roche Laboratories released the System to Manage Accutane Related Teratogenicity (S.M.A.R.T) program, aimed at preventing pregnant women from receiving isotretinoin.42 Major malformations may occur in 25% to 30% of fetuses exposed to isotretinoin.43 Under this program, female patients must have both a screening and confirmation pregnancy test (urine or serum) prior to receiving a prescription for isotretinoin. In addition, patients must commit to using 2 forms of birth control for at least 1 month prior to initiation of therapy, during therapy, and 1 month after discontinuing isotretinoin. During monthly visits, a pregnancy test must be obtained and no more than a 30-day supply of isotretinoin may be prescribed. Finally, pharmacists will fill prescriptions only if an isotretinoin qualification sticker is affixed, obtained after signing and returning the S.M.A.R.T Letter of Understanding.
In addition to procedural safeguards, it is necessary to monitor lipid levels (hypertriglyceridemia and hypercholesterolemia) and liver enzymes at the start of therapy and at each monthly visit. If elevations occur in these measurements, dosage reduction or drug discontinuation should be considered. Given the intensity of monitoring that is required, some family physicians may wish to refer patients who require Accutane to a dermatologist.
- Select medication based on the type and severity of a patient’s acne, as well as the patient’s skin type: creams, lotions, or ointments for dry skin; solutions or gels for oily skin.
- Choose topical therapy whenever possible to minimize side effects.
- Allow 6 to 8 weeks for most treatments to work before deciding to try another regimen or add other agents.
The surest route to success in treating acne vulgaris follows 3 steps. First, establish the type and severity of acne. Second, select medication appropriate for the patient’s condition and skin type. In general, patients with oily skin benefit from solutions or gels, while those with dry skin do better with creams, lotions, or ointments.1 Third, educate the patient about the disease, the different types of medications and their side effects, and expectations for improvement that are realistic. Realistic expectations should enhance compliance and lead to the successful resolution of a debilitating disease.
Types And Severity Of Acne
There are 3 types of acne: comedonal, papulopustular, and nodular (Table 1), all of which result from a multifactorial pathophysiologic process in the pilosebaceous unit: (1) sebum production, (2) follicular hyperkeratinization, (3) proliferation and colonization by Propionibacterium acnes, and (4) the release of inflammatory mediators.2 The resulting lesions include noninflammatory open (blackheads) and closed (whiteheads) comedones, as well as inflammatory papules, pustules, and nodules.
Acne severity is rated according to the Combined Acne Severity Classification that classifies acne into mild, moderate, and severe, based on the number and type of lesions (Table 2).3 Determining acne type and severity serves as a guide to treatment (Table 3).
Table 1
Types of acne
TABLE 2
Combined acne severity classification
| Severity | Definition |
|---|---|
| Mild acne | Fewer than 20 comedones, or Fewer than 15 inflammatory lesions, or Total lesion count fewer than 30 |
| Moderate acne | 20–100 comedones, or 15–50 inflammatory lesions, or total lesion count 30–125 |
| Severe acne | More than 5 nodules, or Total inflammatory count greater than 50, or Total lesion count greater than 125 |
TABLE 3
Treatment options based on type of acne
| Non-inflammatory acne | Inflammatory acne | ||
|---|---|---|---|
| Treatment | Comedonal | Papulo-pustular | Nodulocystic |
| Topical | |||
| Tretinoin (Renova et al) | X | X | |
| Benzoyl Peroxide | X | X | |
| Adapalene (Differin) | X | X | |
| Antibiotics | X | X | |
| Azelaic acid (Azelex) | X | X | |
| Tazarotene (Tazorac) | X | X | |
| Systemic | |||
| Oral contraceptives | X | X | X |
| Erythromycin | X | X | |
| Tetracycline | X | X | |
| Doxycycline | X | X | |
| Minocycline | X | X | |
| Isotretinoin (Accutane) | X | ||
| Adapted from Use of Systemic Agents in the Treatment of Acne Vulgaris, Am Fam Physician 2000;62. | |||
Treatment options
A variety of medications are available for the treatment of acne vulgaris. Note that most treatment regimens should be used for at least 6 to 8 weeks to judge their effectiveness before considering alternative treatments or adding other agents. The Figure 2, an algorithmic guide to the treatment of acne, represents the author’s assessment of the current literature. Alternative approaches may be appropriate after discussing options with individual patients. Table 4 summarizes the strength of evidence for acne interventions and how each compares with other treatments for the same type of acne.
Topical agents
Salicylic acid, found in a number over-the-counter cleansers, has both anti-inflammatory and mild comedolytic effects. It can be used as initial therapy for mild acne or as an adjunctive agent in a broader therapeutic regimen. In a placebo-controlled study of 114 patients, 2% salicylic acid lotion demonstrated a statistically significant improvement from baseline at 12 weeks (SOR: B).4 No side effect data were provided.
Tea tree oil comes from the Australian tree Melaleuca alternifolia and has had success anecdotally in treating various skin conditions. In a single-blind trial of 124 patients, 5% tea tree oil gel was compared with 5% benzoyl peroxide lotion in the treatment of mild-to-moderate acne. Both agents significantly reduced the number of inflammatory lesions and comedones (SOR: B).5 However, benzoyl peroxide was statistically superior to tea tree oil in reducing inflammatory lesions and had a faster onset of action. Encouragingly, patients treated with tea tree oil experienced fewer side effects.
Benzoyl peroxide (BPO) is a potent bactericidal agent with mild keratolytic properties. Several trials have shown the 5% concentration to be consistently superior to placebo at a statistically significant level in the treatment of mild-to-moderate acne, with a 30% decrease in lesion counts (SOR: A).6,7 In addition, two small trials involving 153 patients with mild-to-moderate acne compared the efficacy of different concentrations of topical benzoyl peroxide (2.5% vs 5% and 2.5% vs 10%) used twice daily.8 These trials demonstrated no differences in efficacy among the various preparations based on lesion counts, and there was no dose-response effect (SOR: A). Erythema and scaling occurred with almost identical frequency with the 2.5% and 5% concentrations but more often with the 10% concentration. Thus, the 2.5% and 5% concentrations appear to be preferable based on the balance of risks and benefits.
Topical tretinoin (Renova, Retin-A, Avita) is a comedolytic agent effective as monotherapy for noninflammatory acne. In two randomized controlled trials involving 292 patients of comparable age, use of 0.02% and 0.05% tretinoin strengths showed a statistically significant reduction in comedones and papules with a dose-response effect after at least 4 to 8 weeks of treatment when compared with placebo (SOR: A).9,10 However, there was also a statistically significant increase in erythema and peeling that was maximal after 1 to 3 weeks and decreasing thereafter. In addition, an exacerbation of inflammatory lesions (pustular flare) may occur within 2 to 4 weeks of onset of therapy.11
Adapalene (Differin) is a synthetic naphthoic acid derivative with retinoid activity. Several large, randomized studies have shown that adapalene gel 0.1% and tretinoin concentrations ranging from 0.025% to 0.1% were comparable in reducing total lesion counts by 50% in 4 to 12 weeks (SOR: A).12-14 One trial found that adapalene 0.1% produced a statistically significant reduction greater than that with tretinoin 0.025% in both noninflammatory and inflammatory lesions at 12 weeks (SOR: A).12 Adapalene was also significantly better tolerated than tretinoin, as evidenced by less erythema, scaling, and dryness.12,15 Thus far, there have been no significant studies comparing adapalene to other topical agents such as benzoyl peroxide.
Azelaic acid (Azelex) is a dicarboxylic acid that possesses bacteriostatic properties and is structurally unrelated to any of the conventional acne therapies. In a single-blind trial of 309 patients comparing 20% AZA with 5% benzoyl peroxide and placebo, AZA yielded a significant decrease in papulo-pustular lesion counts by 35% compared with placebo. There was equivalent efficacy between AZA and benzoyl peroxide (SOR: B).16 Patients tolerated AZA better than benzoyl peroxide, with 9% of AZA recipients reporting a burning sensation that subsided after 2 weeks, and 15% of the benzoyl peroxide group reporting local side effects. In another controlled comparison, 20% AZA cream used twice daily for 5 to 6 months was comparable in efficacy to 0.05% tretinoin cream for patients with comedonal acne but statistically more effective in reducing the number of papules (SOR: B).17 Tretinoin caused significantly more erythema and scaling than did AZA.
Tazarotene (Tazorac) is the first of a family of receptor-selective acetylenic retinoids. In a multicenter, randomized controlled trial including 334 patients, tazarotene 0.1% and 0.05%, applied once daily for mild-to-moderate acne significantly reduced noninflammatory acne and total lesion counts when compared with placebo at 4 to 8 weeks (SOR: B).18 The 0.1% gel also significantly reduced inflammatory lesions at 12 weeks. Adverse effects were dose related, ranging from 5% to 13% and included erythema and burning. There are no published trials comparing tazarotene with other retinoids or benzoyl peroxide. Tazarotene is 30% to 70% more expensive than comparable topical agents such as tretinoin, benzoyl peroxide, and antibiotics.
FIGURE
Treatment of acne according to type and severity
TABLE 4
Medication options for acne vulgaris
| Evidence Strength* | Medication | Cost per month** | Relative efficacy | Comparator | Comment |
|---|---|---|---|---|---|
| Comedonal, papulopustular, or nodulocystic | |||||
| A | Norgestimate/ethinyl estradiol | $31.08 | > | Placebo | Decreases comedone and inflammatory lesion counts |
| Comedonal or papulopustular | |||||
| A | Adapalene | $34.47 (gel) | = | Tretinoin | Adapalene has better side-effect profile |
| A | Benzoyl peroxide | $7.99–$16.19 | > | Placebo | Price depends on generic vs brand, not concentration |
| A | Clindamycin | $34.73 (gel) | > | Placebo | Topical |
| A | Erythromycin | $18.31 (gel) | > | Placebo | Topical |
| A or B | Tretinoin | $23.91 | > | Placebo | Evidence strength A for noninflammatory and B for inflammatory |
| B | Azelaic acid | $44.40 | > | Placebo | Topical |
| B | Azelaic acid | $44.40 | = | Benzoyl peroxide | Azelaic acid has better side-effect profile |
| B | Azelaic acid | $44.40 | = | Tretinoin | Azelaic acid has better side-effect profile |
| B | Clindamycin | $34.73 | = | Erythromycin | Topical |
| B | Clindamycin | $34.73 | = | Benzoyl peroxide | Topical |
| B | Salicylic Acid | > | Placebo | Topical | |
| B | Tazarotene | $64.75 (.05%) $68.74 (0.1%) | > | Placebo | Side effects similar to those of topical retinoids |
| B | Tretinoin | $23.91 | > | Benzoyl peroxide | Tretinoin: stronger effect on comedones; BPO: stronger effect on papules |
| Papulopustular or nodulocystic | |||||
| A | Tetracycline | $8.38 | > | Placebo | Oral |
| B | Doxycycline | $24.82 | > | Placebo | Oral |
| B | Erythromycin | $27.15 | = | Tetracycline | Oral. Higher resistance levels of P acnes to erythromycin |
| B | Minocycline | $21.90 | > | Placebo | Oral |
| B | Minocycline | $21.90 | = | Tetracycline | Oral |
| KEY: > is more effective than; < is less effective than; = is equivalent to. | |||||
| *Evidence Strength: | |||||
| A = At least two trials of acceptable quality showing moderate to strong statistical evidence for clinically meaningful endpoint and effect. | |||||
| B = Evidence is of modest strength, such as when only one trial addresses a comparison, there is significant heterogeneity, large differences are not statistically significant, or poor trial quality prevents accepting strong statistical evidence at face value. | |||||
| **Cost: Referenced from a major on-line retail pharmacy. | |||||
Topical antibiotics
Topical antibiotics are effective in the treatment of mild-to-moderate inflammatory acne by reducing the population of P acnes in sebaceous follicles and by suppressing chemotaxis.11 Several large randomized controlled trials demonstrated that topical clindamycin 1% and topical erythromycin 2% applied twice daily were consistently superior to placebo in reducing the number of papules and pustules in patients with moderate-to-severe acne (SOR: A).19-23 Erythema and peeling were rare, comparable to that seen with placebo. Moreover, a randomized trial of 102 patients comparing 1% clindamycin with 2% erythromycin demonstrated that both medications significantly reduced the number of papules and comedones with no significant differences between the two (SOR: B).24,25 Furthermore, in two double-blind, randomized trials involving 334 patients, a combination gel containing clindamycin 1% and benzoyl peroxide 5% proved superior to each component alone in reducing inflammatory lesions, and superior to the clindamycin-only gel in reducing noninflammatory lesions (SOR: B).26 Trial data on the combination gel containing erythromycin 3% and benzoyl peroxide 5% are of poor quality; thus the same conclusion cannot be made.
Oral antibiotics
Oral antibiotics are most often used for moderate-to-severe inflammatory acne. They work by suppressing P acnes growth, thereby reducing the production of inflammatory mediators.27 However, as systemicagents, they cause more significant and diverse side effects than do topical agents. Unfortunately there are no head-to-head trials comparing different oral antibiotics, or comparing oral and topical antibiotics.
Erythromycin. In a randomized study of 200 patients with moderate-to-severe acne, erythromycin, 1 g in 2 divided doses daily, significantly reduced the comedone, papule, and pustule count after 8 weeks (SOR: B).28 The side-effect rate was 8%, usually associated with gastrointestinal irritation. Studies have shown that P acnes exhibits greater resistance to erythromycin than to tetracycline.29
Tetracycline/doxycycline/minocycline.Tetracycline and its lipophilic derivatives, doxycycline and minocycline, are the most commonly prescribed oral agents for acne vulgaris. As a class, tetracyclines should not be prescribed for pregnant women or for those younger than 9 years of age, to avoid the risks of tooth discoloration and bone growth retardation in the fetus or child.30 Various double-blinded, randomized controlled trials involving patients with mild-to-moderate acne have shown that tetracycline confers a statistically significant improvement over placebo as early as 6 weeks (SOR: A).31-33 Adverse effects include gastrointestinal upset, vaginal yeast infection, and a theoretical decrease in the efficacy of oral contraceptives.
Doxycycline, 100mg/d, has been shown to significantly reduce inflammatory lesions in a crossover trial of 62 patients (SOR: B).34 Its adverse effect profile is similar to that of tetracycline, though it tends to cause more photosensitivity (4% vs 1%).35
In a recent Cochrane review of 27 studies, minocycline was shown to be an effective treatment for acne, but no randomized controlled trial evidence was found to support the benefits of minocycline in acne resistant to other therapies (SOR: A).36 A recent study demonstrated that minocycline has a greater tendency than tetracycline or doxycycline to cause rare adverse side effects, including serumsickness-like reactions, drug-induced lupus, and hypersensitivity reactions.35 These factors, in addition to the higher cost, suggest that minocycline should not be a first line antibiotic choice for treating acne.
Oral contraceptives
Oral contraceptives (OCs) reduce the severity of acne vulgaris by decreasing the amount of circulating androgens.37 In 1997, a triphasic combination OC containing ethinyl estradiol 0.035 mg and increasing doses of norgestimate (0.180 mg, 0.215 mg, and 0.250 mg) was approved by the FDA for the treatment of acne in women. This decision was based on the results of a randomized, double-blind trial involving 257 patients in which the triphasic contraceptive was compared with placebo over 6 months (SOR: A).38 The OC group showed statistically significant improvement greater than that of the placebo group in all types of acne lesions. It also reduced total lesion counts by more than 53% in female subjects at 26 weeks, compared with lesion reductions of about 27% in controls. The principal adverse effect noted in this study was nausea.
Isotretinoin
Isotretinoin (Accutane) is an oral retinoid labeled for use in patients with severe, refractory, nodulocystic acne. In a randomized, crossover trial that included 33 patients, isotretinoin significantly decreased the number of nodulocystic lesions by 95% when compared with placebo, with only rare side effects of cheilitis and dermatitis (SOR: B).39 However, other studies suggest that cheilitis is fairly common and its absence may imply noncompliance or malabsorption of the drug.40 In addition, the FDA issued a warning in 1998 regarding possible increased risks in depression, psychosis, suicidal thoughts, and suicide attempts, though no conclusive evidence has been found.40 The typical dosage of isotretinoin is 0.5 to 1 mg/kg daily in two divided doses, with a standard cumulative maximum dose of 120 to 150 mg/kg per treatment course.41
In April 2002, Roche Laboratories released the System to Manage Accutane Related Teratogenicity (S.M.A.R.T) program, aimed at preventing pregnant women from receiving isotretinoin.42 Major malformations may occur in 25% to 30% of fetuses exposed to isotretinoin.43 Under this program, female patients must have both a screening and confirmation pregnancy test (urine or serum) prior to receiving a prescription for isotretinoin. In addition, patients must commit to using 2 forms of birth control for at least 1 month prior to initiation of therapy, during therapy, and 1 month after discontinuing isotretinoin. During monthly visits, a pregnancy test must be obtained and no more than a 30-day supply of isotretinoin may be prescribed. Finally, pharmacists will fill prescriptions only if an isotretinoin qualification sticker is affixed, obtained after signing and returning the S.M.A.R.T Letter of Understanding.
In addition to procedural safeguards, it is necessary to monitor lipid levels (hypertriglyceridemia and hypercholesterolemia) and liver enzymes at the start of therapy and at each monthly visit. If elevations occur in these measurements, dosage reduction or drug discontinuation should be considered. Given the intensity of monitoring that is required, some family physicians may wish to refer patients who require Accutane to a dermatologist.
1. Thiboutot DM. An overview of acne and its treatment. Cutis 1996;57:8-12.
2. Leyden JJ. Therapy for acne vulgaris. N Engl J Med 1997;336:1156-62.
3. Issued by funding/sponsoring agency: Management of Acne Volume 1: Evidence Report and Appendixes. Rockville, Md: Dept. of Health and Human Services (US), Public Health Service; 2001 Sep. Report No.: 01-E019. Issued by performing agency: Lehmann HP, Andrews JS, Robinson KA, Holloway VL, Goodman SN. Johns Hopkins Evidence-based Practice Center. Contract No.: 290-97-006. Sponsored by the Agency for Healthcare Research and Quality.
4. Eady EA, Burke BM, Pulling K, Cunliffe WJ. The benefit of 2% salicylic acid lotion in acne—A placebo-controlled study. J Dermatol Treat 1996;7:93-6.
5. Basset OB, Pannowitz DL, Barnetson RS. A comparative study of tea-tree oil versus benzoyl peroxide in the treatment of acne. Med J Aust 1990;153:455-8.
6. Hughes BR, Norris JF, Cunliffe WJ. A double-blind evaluation of topical isotretinoin 0.05%, benzoyl peroxide gel 5% and placebo in patients with acne. Clin Exp Dermatol 1992;17:165-8.
7. Ede M. A double-blind, comparative study of benzoyl peroxide, benzoyl peroxide-chlorhydroxyquinoline, benzoyl peroxide-chlorhydroxyquinolone-hydrocortisone, and placebo lotions in acne. Curr Ther Res Clin Exp 1973;15:624-9.
8. Mills OH, Jr, Kligman AM, Pochi P, Comite H. Comparing 2.5%, 5%, and 10% benzoyl peroxide on inflammatory acne vulgaris. Int J Dermatol 1986;25:664-7.
9. Pedace FJ, Stoughton R. Topical retinoic acid in acne vulgaris. Br J Dermatol 1971;84:465-9.
10. Christiansen JV, Gadborg E, Ludvigsen K, et al. Topical tretinoin, vitamin A acid (Airol) in acne vulgaris. A controlled clinical trial. Dermatologica 1974;148:82-9.
11. Berson DS, Shalita AR. The treatment of acne: the role of combination therapies. J Am Acad Dermatol 1995;32:531-41.
12. Shalita A, Weiss JS, Chalker DK, et al. A comparison of the efficacy and safety of adapalene gel 0.1% and tretinoin gel 0.025% in the treatment of acne vulgaris: a multicenter trial. J Am Acad Dermatol 1996;34:482-5.
13. Cunliffe W, Caputo R, Dreno B, et al. Clinical efficacy and safety comparison of adapalene gel and tretinoin gel in the treatment of acne vulgaris: Europe and U.S.multicenter trials. J Am Acad Dermatol 1997;36:S126-34.
14. Galvin SA, Gilbert R, Baker M, et al. Comparative tolerance of adapalene 0.1% gel and six different tretinoin formulations. Br J Dermatol 1998;139:S34-40.
15. Clucas A, Verschoore M, Sorba V, et al. Adapalene 0.1% gel is better tolerated than tretinoin 0.025% gel in acne patients. J Am Acad Dermatol 1997;36:S116-8.
16. Cavicchini S, Caputo R. Long-term treatment of acne with 20% azelaic acid cream. Acta Derm Venereol Suppl. 1989;143:40-4.
17. Katsambas A, Graupe K, Stratigos J. Clinical studies of 20% azelaic cream in the treatment of acne vulgaris. Comparison with vehicle and topical tretinoin. Acta Derm Venereol Suppl 1989;143:35-9.
18. Shalita AR, Chalker DK, Griffith RF, et al. Tazarotene gel is safe and effective in the treatment of acne vulgaris: a multicenter, double blind, vehicle-controlled study. Cutis 1999;63:349-53.
19. Becker LE, Bergstresser PR, Whiting DA, et al. Topical clindamycin therapy for acne vulgaris. A cooperative clinical study Arch Dermatol 1981;117:482-5.
20. Ellis CN, Gammon WR, Stone DZ, Heezen-Wehner JL. A comparison of Cleocin T Solution, Cleocin T Gel, and placebo in the treatment of acne vulgaris. Cutis 1988;42:245-7.
21. Pochi PE, Bagatell FK, Ellis CN, et al. Erythromycin 2 percent gel in the treatment of acne vulgaris. Cutis 1988;41:132-6.
22. Lesher JL, Jr, Chalker DK, Smith JG, Jr, et al. An evaluation of a 2% erythromycin ointment in the topical therapy of acne vulgaris. J Am Acad Dermatol 1985;12:526-31.
23. Dobson RL, Belknap BS. Topical erythromycin solution in acne. Results of a multiclinic trial. J Am Acad Dermatol 1980;3:478-82.
24. Leyden JJ, Shalita AR, Saatjian GD, Sefton J. Erythromycin 2% gel in comparison with clindamycin phosphate 1% solution in acne vulgaris. J Am Acad Dermatol 1987;16:822-7.
25. Shalita AR, Smith EB, Bauer E. Topical erythromycin v clindamycin therapy for acne. A multicenter, double-blind comparison. Arch Dermatol 1984;120:351-5.
26. Lookingbill DP, Chalker DK, Lindholm JS, et al. Treatment of acne with a combination clindamycin/benzoyl peroxide gel compared with clindamycin gel, benzoyl peroxide gel and vehicle gel: combined results of two double-blind investigations. J Am Acad Dermatol 1997;37:590-5.
27. Sykes NL, Webster GF. Acne: a review of optimum treatment. Drugs 1994;48:59-70.
28. Gammon WR, Meyer C, Lantis S, et al. Comparative efficacy of oral erythromycin versus oral tetracycline in the treatment of acne vulgaris. A double-blind study. J Am Acad Dermatol 1986;14:183-6.
29. Eady EA, Jones CE, Tipper JL, et al. Antibiotic resistant Propionibacteria in acne: need for policies to modify antibiotic usage. Br Med J 1993;306:555-6.
30. McEvoy GK, editor. AHFS Drug Information. Bethesda, Md: American Society of Health System Pharmacists; 1996.
31. Blaney DJ, Cook CH. Topical use of tetracycline in the treatment of acne: a double-blind study comparing topical and oral tetracycline therapy and placebo. Arch Dermatol 1976;112:971-3.
32. Lane P, Williamson DM. Treatment of acne vulgaris with tetracycline hydrochloride: a double-blind trial with 51 patients. Br Med J 1969;2:76-9.
33. Wong RC, Kang S, Heezen JL, et al. Oral ibuprofen and tetracycline for the treatment of acne vulgaris. J Am Acad Dermatol 1984;11:1076-81.
34. Plewig G, Petrozzi JW, Berendes U. Double-blind study of doxycycline in acne vulgaris. Arch Dermatol 1970;101:435-8.
35. Shapiro LE, Knowles SR, Shear NH. Comparative safety of tetracycline, minocycline, and doxycycline. Arch Dermatol 1997;133:1224-30.
36. Garner SE, Eady EA, Popescu C, Newton J, Li Wan Po A. Minocycline for acne vulgaris: efficacy and safety (Cochrane Review). In: The Cochrane Library., Issue 4, 2001.
37. Lucky AW. Hormonal correlates of acne and hirsutism. Am J Med 1995;98:89S-94S.
38. Lucky AW, Henderson TA, Olson WH, et al. Effectiveness of norgestimate and ethinyl estradiol in treating moderate acne vulgaris. J Am Acad Dermatol 1997;37:746-54.
39. Peck GL, Olsen TG, Butkus D, et al. Isotretinoin versus placebo in the treatment of cystic acne. A randomized double-blind study. J Am Acad Dermatol 1982;6:735-45.
40. Hanson N, Leachman S. Safety Issues in Isotretinoin Therapy. Semin Cutan Med Surg 2001;20:166-83.
41. Layton AM, Knaggs H, Taylor J, Cunliffe WJ. Isotretinoin for acne vulgaris—10 years later: a safe and successful treatment. Br J Dermatol 1993;129:292-6.
42. Accutane prescribing information. Nutley, N.J.: Roche Pharmaceuticals, 1998.
43. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, et al. Retinoic acid embryopathy. N Engl J Med 1985;313:837-41.
44. Helms SE, Bredle DL, Zajic J, et al. Oral contraceptive failure rates and oral antibiotics. J Am Acad Dermatol 1997;36:705-10.
1. Thiboutot DM. An overview of acne and its treatment. Cutis 1996;57:8-12.
2. Leyden JJ. Therapy for acne vulgaris. N Engl J Med 1997;336:1156-62.
3. Issued by funding/sponsoring agency: Management of Acne Volume 1: Evidence Report and Appendixes. Rockville, Md: Dept. of Health and Human Services (US), Public Health Service; 2001 Sep. Report No.: 01-E019. Issued by performing agency: Lehmann HP, Andrews JS, Robinson KA, Holloway VL, Goodman SN. Johns Hopkins Evidence-based Practice Center. Contract No.: 290-97-006. Sponsored by the Agency for Healthcare Research and Quality.
4. Eady EA, Burke BM, Pulling K, Cunliffe WJ. The benefit of 2% salicylic acid lotion in acne—A placebo-controlled study. J Dermatol Treat 1996;7:93-6.
5. Basset OB, Pannowitz DL, Barnetson RS. A comparative study of tea-tree oil versus benzoyl peroxide in the treatment of acne. Med J Aust 1990;153:455-8.
6. Hughes BR, Norris JF, Cunliffe WJ. A double-blind evaluation of topical isotretinoin 0.05%, benzoyl peroxide gel 5% and placebo in patients with acne. Clin Exp Dermatol 1992;17:165-8.
7. Ede M. A double-blind, comparative study of benzoyl peroxide, benzoyl peroxide-chlorhydroxyquinoline, benzoyl peroxide-chlorhydroxyquinolone-hydrocortisone, and placebo lotions in acne. Curr Ther Res Clin Exp 1973;15:624-9.
8. Mills OH, Jr, Kligman AM, Pochi P, Comite H. Comparing 2.5%, 5%, and 10% benzoyl peroxide on inflammatory acne vulgaris. Int J Dermatol 1986;25:664-7.
9. Pedace FJ, Stoughton R. Topical retinoic acid in acne vulgaris. Br J Dermatol 1971;84:465-9.
10. Christiansen JV, Gadborg E, Ludvigsen K, et al. Topical tretinoin, vitamin A acid (Airol) in acne vulgaris. A controlled clinical trial. Dermatologica 1974;148:82-9.
11. Berson DS, Shalita AR. The treatment of acne: the role of combination therapies. J Am Acad Dermatol 1995;32:531-41.
12. Shalita A, Weiss JS, Chalker DK, et al. A comparison of the efficacy and safety of adapalene gel 0.1% and tretinoin gel 0.025% in the treatment of acne vulgaris: a multicenter trial. J Am Acad Dermatol 1996;34:482-5.
13. Cunliffe W, Caputo R, Dreno B, et al. Clinical efficacy and safety comparison of adapalene gel and tretinoin gel in the treatment of acne vulgaris: Europe and U.S.multicenter trials. J Am Acad Dermatol 1997;36:S126-34.
14. Galvin SA, Gilbert R, Baker M, et al. Comparative tolerance of adapalene 0.1% gel and six different tretinoin formulations. Br J Dermatol 1998;139:S34-40.
15. Clucas A, Verschoore M, Sorba V, et al. Adapalene 0.1% gel is better tolerated than tretinoin 0.025% gel in acne patients. J Am Acad Dermatol 1997;36:S116-8.
16. Cavicchini S, Caputo R. Long-term treatment of acne with 20% azelaic acid cream. Acta Derm Venereol Suppl. 1989;143:40-4.
17. Katsambas A, Graupe K, Stratigos J. Clinical studies of 20% azelaic cream in the treatment of acne vulgaris. Comparison with vehicle and topical tretinoin. Acta Derm Venereol Suppl 1989;143:35-9.
18. Shalita AR, Chalker DK, Griffith RF, et al. Tazarotene gel is safe and effective in the treatment of acne vulgaris: a multicenter, double blind, vehicle-controlled study. Cutis 1999;63:349-53.
19. Becker LE, Bergstresser PR, Whiting DA, et al. Topical clindamycin therapy for acne vulgaris. A cooperative clinical study Arch Dermatol 1981;117:482-5.
20. Ellis CN, Gammon WR, Stone DZ, Heezen-Wehner JL. A comparison of Cleocin T Solution, Cleocin T Gel, and placebo in the treatment of acne vulgaris. Cutis 1988;42:245-7.
21. Pochi PE, Bagatell FK, Ellis CN, et al. Erythromycin 2 percent gel in the treatment of acne vulgaris. Cutis 1988;41:132-6.
22. Lesher JL, Jr, Chalker DK, Smith JG, Jr, et al. An evaluation of a 2% erythromycin ointment in the topical therapy of acne vulgaris. J Am Acad Dermatol 1985;12:526-31.
23. Dobson RL, Belknap BS. Topical erythromycin solution in acne. Results of a multiclinic trial. J Am Acad Dermatol 1980;3:478-82.
24. Leyden JJ, Shalita AR, Saatjian GD, Sefton J. Erythromycin 2% gel in comparison with clindamycin phosphate 1% solution in acne vulgaris. J Am Acad Dermatol 1987;16:822-7.
25. Shalita AR, Smith EB, Bauer E. Topical erythromycin v clindamycin therapy for acne. A multicenter, double-blind comparison. Arch Dermatol 1984;120:351-5.
26. Lookingbill DP, Chalker DK, Lindholm JS, et al. Treatment of acne with a combination clindamycin/benzoyl peroxide gel compared with clindamycin gel, benzoyl peroxide gel and vehicle gel: combined results of two double-blind investigations. J Am Acad Dermatol 1997;37:590-5.
27. Sykes NL, Webster GF. Acne: a review of optimum treatment. Drugs 1994;48:59-70.
28. Gammon WR, Meyer C, Lantis S, et al. Comparative efficacy of oral erythromycin versus oral tetracycline in the treatment of acne vulgaris. A double-blind study. J Am Acad Dermatol 1986;14:183-6.
29. Eady EA, Jones CE, Tipper JL, et al. Antibiotic resistant Propionibacteria in acne: need for policies to modify antibiotic usage. Br Med J 1993;306:555-6.
30. McEvoy GK, editor. AHFS Drug Information. Bethesda, Md: American Society of Health System Pharmacists; 1996.
31. Blaney DJ, Cook CH. Topical use of tetracycline in the treatment of acne: a double-blind study comparing topical and oral tetracycline therapy and placebo. Arch Dermatol 1976;112:971-3.
32. Lane P, Williamson DM. Treatment of acne vulgaris with tetracycline hydrochloride: a double-blind trial with 51 patients. Br Med J 1969;2:76-9.
33. Wong RC, Kang S, Heezen JL, et al. Oral ibuprofen and tetracycline for the treatment of acne vulgaris. J Am Acad Dermatol 1984;11:1076-81.
34. Plewig G, Petrozzi JW, Berendes U. Double-blind study of doxycycline in acne vulgaris. Arch Dermatol 1970;101:435-8.
35. Shapiro LE, Knowles SR, Shear NH. Comparative safety of tetracycline, minocycline, and doxycycline. Arch Dermatol 1997;133:1224-30.
36. Garner SE, Eady EA, Popescu C, Newton J, Li Wan Po A. Minocycline for acne vulgaris: efficacy and safety (Cochrane Review). In: The Cochrane Library., Issue 4, 2001.
37. Lucky AW. Hormonal correlates of acne and hirsutism. Am J Med 1995;98:89S-94S.
38. Lucky AW, Henderson TA, Olson WH, et al. Effectiveness of norgestimate and ethinyl estradiol in treating moderate acne vulgaris. J Am Acad Dermatol 1997;37:746-54.
39. Peck GL, Olsen TG, Butkus D, et al. Isotretinoin versus placebo in the treatment of cystic acne. A randomized double-blind study. J Am Acad Dermatol 1982;6:735-45.
40. Hanson N, Leachman S. Safety Issues in Isotretinoin Therapy. Semin Cutan Med Surg 2001;20:166-83.
41. Layton AM, Knaggs H, Taylor J, Cunliffe WJ. Isotretinoin for acne vulgaris—10 years later: a safe and successful treatment. Br J Dermatol 1993;129:292-6.
42. Accutane prescribing information. Nutley, N.J.: Roche Pharmaceuticals, 1998.
43. Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, et al. Retinoic acid embryopathy. N Engl J Med 1985;313:837-41.
44. Helms SE, Bredle DL, Zajic J, et al. Oral contraceptive failure rates and oral antibiotics. J Am Acad Dermatol 1997;36:705-10.
Effective management of obesity
- Diet and exercise coupled with behavioral modification can improve both patient-related and disease-related outcomes in the short term; however, long-term efficacy is lacking (A).
- Drugs such as sibutramine or orlistat may achieve modest, short-term weight loss, but their long-term effectiveness is unproven (A).
- For patients with a BMI >40, gastric bypass procedures can lead to long-term weight loss (B).
Physician intervention to encourage and assist obese patients to lose weight is warranted, for these reasons:
- Prevention of adverse outcomes. Left untreated, obesity is clearly related to the development of many adverse health outcomes, including diabetes mellitus, hypertension, stroke, hyperlipidemia, coronary artery disease, gallstones, osteoarthritis, obstructive sleep apnea, vascular disease, depression, and certain cancers (breast, endometrial, prostate, colon).1-4
- Reduction of morbidity and mortality. A causal effect between intentional weight loss and mortality has been difficult to prove, but even modest weight loss can reduce the morbidity of obesity-related disease, such as arthritis and obstructive sleep apnea (Strength of recommendation: A).5-7 For those at increased risk of death from cardiovascular disease, such as persons with obesity and diabetes mellitus, intentional weight loss coupled with lifestyle change can significantly reduce mortality (SOR: C).8 Nearly 280,000 deaths per year are attributable to obesity.9
- Cost reduction. Nearly 25% of American adults are obese, and more than half are overweight.1,2 Obesity burdens society with significant costs, including more than $50 billion annually for direct care. With an additional $30 billion spent each year on weight-loss products and services, this disease accounts for over 5% of annual health care expenditures in the United States.5
Nature or nurture?
Excess fat is created when energy intake exceeds cellular energy consumption. The complex relationship between the human body’s environment and the development of obesity is poorly understood, but recent genetic investigations have elucidated new mechanisms in the regulation of both satiety and energy expenditure. Using data from heritability studies, some researchers have estimated that up to 70% of the variability in weight among humans can be explained by genetic influences, but it is unlikely that changes in human genes account for the recent change in obesity prevalence.10
Risk factors
Identifiable risks factors for obesity in adulthood include parental history of obesity, low socio-economic background, and a history of high birth weight.11
Prevention
Recently, factors including consumption of sugar-sweetened beverages, lack of breast-feeding, and television viewing have been identified as risk factors for childhood and adolescent obesity.12-14 Because obesity at a young age can lead to adult obesity, these factors may be targeted to prevent adult obesity.
School-based programs for diet and exercise appear to be ineffective for preventing obesity (SOR: B).15,16 However, most research has been of limited quality. In one recent randomized study, reduction of television viewing (including videotape and videogame use) through school intervention was associated with significant reduction in BMI (SOR: B).14
Screening recommendations
The United States Preventive Service Task Force recommends periodic measurement of both height and weight in adults (SOR: B). The waist-to-hip ratio is thought to have insufficient evidence for recommendation as a routine screening tool because studies identifying a benefit to screening using only the waist-to-hip ratio have not been completed (SOR: C).17
Initial determination of obesity
Although the standard for body fat measurement is densitometry, which determines the density of a body submersed in water, the cost and technical requirements prohibit routine use in the clinical setting.18-20 The waist-to-hip ratio and waist circumference are used to identify central (or android) obesity in which adipose tissue in the abdomen is associated with atherosclerosis.21,22 The waist circumference is found by measuring the circumference around the waist at the level of the iliac crest. Values above 40 inches for men and 35 inches for women are indicative of increased risk of adverse health outcomes.5,19 The waist-to-hip ratio is calculated by dividing the circumference of the waist at the level of the L3 by the hip circumference measured at the largest area of the gluteal region.19 For men, waist-to-hip ratios greater than 1.0 are associated with significantly increased risk of cardiovascular events. For women, a ratio greater than 0.85 indicates increased risk.19
Body-mass index calculation
The body-mass index (BMI), also known as the Quetelet index, is the most commonly used measure of obesity.20 BMI is a patient’s weight in kilograms divided by his or her height in meters squared (kg/m2 ).5,19 A free online BMI calculator is available through the National Heart, Lung and Blood Institute at www.nhlbisupport.com/bmi/bmicalc.htm.
Although the BMI estimates total body fat and compares well with densitometry, it may be less accurate in selected populations (eg, the elderly, certain ethnic groups, and persons with large muscle mass).19 Generally, when BMI exceeds 25, the greater the BMI, the greater the obesity-related morbidity and mortality.5,19,20,23 Table 1 shows the classification of obesity based on BMI, and Table 2 shows the BMI for combinations of height and weight in inches and pounds.5
TABLE 1
Body mass index (BMI) and degrees of obesity
| BMI | Category | Therapeutic options |
|---|---|---|
| <18.5 | Underweight | |
| 18.5–25.9 | Normal weight | Reinforce positive lifestyle |
| 25.0–29.9 | Overweight | Diet, exercise, behavior modification |
| 30.0–34.9 | Obese (Class I) | Diet, exercise, behavior modification; consider pharmacologic therapy* |
| 35.0–39.9 | Obese (Class II) | Diet, exercise, behavior modification; consider pharmacologic therapy |
| ≥40.0 | Obese (Class III or “Morbid Obesity”) | Consider surgical management |
| *For patients with multiple cardiovascular risk factors (eg, diabetes, hyperlipidemia), BMI >27 may be an indication for pharmacologic intervention at; at BMI >37, patients may be considered candidates for surgical therapy. | ||
| From the National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: the Evidence Report. Bethesda, Md: US Department of Health and Human Services, 1998. | ||
Evaluation of documented obesity
In history taking and physical examination, look for reversible causes of obesity (including medications and endocrine disorders), consider the degree of obesity, and determine whether comorbid conditions are present, to help estimate prognosis. Evaluate the patient’s dietary and exercise habits, as well as willingness to modify these habits if necessary.1,5 Finally, review the patient’s weight history and any attempts at weight loss.
Medications associated with weight gain include psychotropic drugs, anticonvulsant agents, steroid hormones, insulin, and many oral hypoglycemic agents.1
Endocrine disorders such as Cushing’s syndrome and hypothyroidism may also contribute to obesity, but only rarely. Physical findings that increase the likelihood of Cushing’s syndrome, and their respective positive likelihood ratios (LR+)— the higher the value, the greater the likelihood of disease—include: hypertension (2.3), moon facies (1.6), thin skinfold (115.6), ecchymoses (4.5), and acne (2.2).19 Findings, and their likelihood ratios, associated with hypothyroidism include coarse skin (5.6), cool/dry skin (4.7), bradycardia (4.1), enlarged thyroid (2.8), and hoarse voice (5.4).19 The very high LR for skinfold thickness was determined for women of childbearing age who had elevated risk of having Cushing’s syndrome because of a history of both menstrual irregularities and hirsutism. Skinfold thickness is determined by using calipers on an area of minimal subcutaneous fat (eg, back of the hand). For women of reproductive age, skinfold thickness is normally greater than 1.8 mm.19
Laboratory assessment of the obese patient will rarely find a cause of weight gain (eg, hypothyroidism), but the addition of selected diagnostic tests will aid in the determination of prognosis. An abnormal fasting glucose level or impaired glucose tolerance is a major risk factor for cardiovascular disease. Abnormal lipid profiles heighten that risk for obese patients. All patients with documented obesity should undergo assessment for abnormal lipids and impaired glucose tolerance5 (SOR: D).
Treatment
The most important step in treating obesity is to establish a calorie deficit. The deficit can be achieved by increasing energy expenditure or by reducing energy intake or absorption. On average, a caloric deficit of 500 kilocalories per day will result in a weight loss of 1 pound per week.5 Reasonable expectations of therapy include weight loss of 1–2 pounds a week and a loss of 10% of total body weight in 5 months.5
Interventions for weight loss fall into 4 categories: lifestyle modifications (diet, exercise, and behavioral modification), drug therapy, complementary or alternative measures, and surgery. Table 3 summarizes the levels of evidence to support each intervention.
TABLE 3
Efficacy of weight-loss interventions
| Weight loss | ||||
|---|---|---|---|---|
| SOR* | Intervention | Short-term | Long-term | Comments |
| Diet, exercise, and behavioral modification | ||||
| A | Low/very-lowcalorie diet | 8% average weight loss from 3–12 months | Weight nears baseline in studies >24 months | Very-low-calorie diets may require laboratory assessment of metabolic function |
| High rate of noncompliance | ||||
| A | Low/very low fat with reduced calories | Similar to lowcalorie with moderate fat | Weight nears baseline in studies >24 months | No known side effects |
| A | Exercise | Less weight loss than diet therapy | Likely no significant weight loss | Improved cardiovascular fitness |
| May be effective in preventing weight gain | ||||
| A | Low-calorie diet + exercise | Increased weight loss vs. diet or exercise alone | Weight nears baseline in long-term studies | Improved cardiovascular fitness |
| Compliance a major problem | ||||
| B | Behavior modification | Increases effectiveness of diet, exercise | No significant effect at 5 years | No reported harms |
| Only studied when used with other methods | ||||
| C | Lowcarbohydrate diet | Not significant if calories are not reduced | No long-term data available | No known side effects, but creates nutritional imbalance and ketosis |
| Needs additional study | ||||
| Medication | ||||
| A | Sibutramine | ˜ 4 kg for trials less than 1 year | Modest weight loss when used for >1 year | Can elevate blood pressure |
| Number needed to treat (NNT) for 5% weight loss at 1 year = 3 | ||||
| NNT for 10% weight loss at 1 year = 5 | ||||
| A | Orlistat | ˜ 2–3 kg for trials less than 1 year | ˜ 3 kg at 2 years | GI side effects common, possible vitamin deficiencies |
| NNT for 5% weight loss at 1 year = 5 | ||||
| NNT for 10% weight loss at 1 year = 7 | ||||
| Surgery | ||||
| B | Roux-en-Y Gastric bypass | ˜ 50 kg (110 lbs) at 1 year | ˜ 50 kg (110 lbs) at up to 4 years | Significant operative risk and post-operative GI side effects |
| Nadir for weight loss occurs at 12–24 months | ||||
| B | Gastric banding | ˜ 30 Kg (66 lbs) at one year | 10–15% of initial weight lost may be regained long-term | Significant operative risk and post-operative GI side effects |
| Generally considered less effective that gastric bypass | ||||
| Complementary/alternative medicine | ||||
| B | Hypnosis | Minimal reduction | No statistically significant difference | Studied in combination with cognitive behavioral therapy |
| Systematic reviews reveal significant heterogeneity of low-quality randomized controlled trials | ||||
| B | Acupuncture | No significant difference | No significant difference | Systematic review reveals poor quality |
| RCTs, which limits ability to determine effect | ||||
| * Strength of recommendation | ||||
| A = Systematic review of randomized controlled trials (RCT) (with homogeneity) or individual RCT with narrow confidence interval | ||||
| B = Systematic review of cohort studies (with homogeneity), individual cohort studies or low-quality RCT, individual case-control study or SR of case-control studies (with homogeneity) | ||||
| C = Case series and poor quality cohort and case control studies | ||||
Lifestyle modifications
Management of obesity in every case should include dietary changes, exercise, and behavioral modification.
Dietary changes. Diets create a caloric deficit by reducing the intake of calories. Average weight loss with low-calorie diets is approximately 8% at 3–12 months (SOR: A),5 with most of the loss occurring in the first 3–5 months.24 There are many kinds of diets for weight loss, including low calorie, very-low calorie, low fat, very low fat, and low carbohydrate, but long-term compliance with all types of dietary interventions is a significant problem. When diet alone is used as therapy, between one third and one half of weight loss will not be maintained.24 Emerging strategies to help improve dietary compliance include behavioral modification (see below) and meal substitutes. Recent reports of interventions such as meal-replacement shakes indicate that long-term weight loss can be significantly improved (SOR: B).25-27
Examples of low-calorie, nutritionally balanced diets are Weight Watchers, Jenny Craig, Nutrisystem, the National Cholesterol Education Program Step I and Step II diets, and the Dietary Approaches to Stop Hypertension (DASH) diet. Low-calorie diets provide 800–1500 kilocalories per day.5,28,29 Very-low-calorie diets (400–500 kilocalories per day) may increase rates of weight loss initially, but at 1 year, results are similar to those of low-calorie diets (SOR: A).5,30
A low-fat diet (fat content 10%–19%) without a decrease in total calorie intake does not promote weight loss (SOR: A).5,31 Very-low-fat diets containing less than 10% fat have been described by such authors as Ornish and Pritikin.25 Obese patients using either the low-fat or very-low-fat diet can lose body weight and body fat, but only if calories are also decreased (SOR: A).5,28
Low-carbohydrate diets, such as Dr Atkin’s diet, are associated with modest (approximately 5 kg, or 11 lbs) weight loss (SOR: C).28,32 Improved study design is required to further evaluate the effectiveness and safety of low-carbohydrate diets in the clinical setting.
Exercise. Most studies of exercise are based on 30–50 minutes of moderately intense aerobic exercise, repeated 3–7 times per week.5 When it is the only prescribed therapy, exercise can be expected to produce modest weight loss only (SOR: A).5,33 Exercise combined with dietary intervention, however, increases weight lost (SOR: A), and exercise by itself may prevent weight gain (SOR: C).5
Behavioral Modification. Behavioral modification has been evaluated in combination with diet or exercise, and has been shown to increase compliance and weight loss for durations of 1 year or less (SOR: A).5,34 Weight gain is common when therapy is discontinued, and at 5 years, there is no difference between those who received behavioral therapy and those who were in control groups (SOR: A).5
Medications
Medications for treatment of obesity act through 1 or more of 3 mechanisms:
- Appetite suppression (eg, sibutramine, antidepressants such as fluoxetine)
- Increased metabolic activity (eg, stimulants such as ephedra with caffeine, Β-3 agonists)
- Decreased absorption of caloric load (orlistat)
For mild-to-moderate obesity (BMI >30 and <40), medications can be beneficial (SOR: A), but long-term weight loss beyond 2 years has not been studied.35 Pharmacologic intervention without lifestyle intervention actually decreases a person’s ability to lose weight (SOR: B).36
Two medications are approved by the United States Food and Drug Administration for long-term obesity management: sibutramine and orlistat. Both drugs reduce weight modestly (SOR: A).37-39 Both medications have similar indications for use: BMI >30, or BMI >27 with the presence of other cardiovascular risk factors (ie, diabetes or hyperlipidemia). Both should be used in conjunction with reduced-calorie diet and exercise (SOR: B).36
Sibutramine is usually started at 10 mg once a day, given with or without food. The dose may be titrated to a maximum of 15 mg/d after 4 weeks if weight loss has been inadequate.1 Sibutramine is known to increase pulse rate and blood pressure in a significant number of patients; because of this, regular evaluation of vital signs is required. At present, long-term use of sibutramine cannot be recommended, and safety data are unavailable beyond 1 year of use. Sibutramine should be avoided if these conditions are present: hypertension, coronary heart disease, congestive heart failure, an arrhythmic condition, pregnancy, renal impairment, concomitant use of MAOI, or a history of stroke.
Orlistat is started at 120 mg three times a day, and is taken with meals that contain fat. It may still be effective if taken up to one hour after eating. Orlistat may be avoided if the meal contains no fat. This drug may interfere with the absorption of some fat-soluble vitamins, and it is therefore recommended that patients take a multivitamin that has fat-soluble vitamins at least 2 hours before or after ingesting orlistat. Orlistat is not absorbed into the body and, at this time, no laboratory follow-up is needed. Regular evaluation of weight is needed to assess the efficacy of treatment. Orlistat should be avoided by those who have cholestasis or malabsorptive disorders or by those taking cyclosporine.40
Other medications that have been used include phentermine, ephedra, dexfenfluramine, phenyl-propanolamine (PPA), and mazindol.1,2 All of these medications produce significant weight loss in the short term (SOR: A), but they are not indicated for long-term use.2 In fact, phenylpropanolamine and dexfenfluramine are no longer available because of their severe side effects.2 Antidepressants do not yield a consistent benefit in well-designed studies of obesity management.2
Table 3 summarizes the effects of medications on weight loss.
Surgery
Surgical management of obesity is reserved for extremely obese persons because of the significant morbidity and mortality associated with the interventions. Currently, gastric bypass procedures result in less than 1% perioperative mortality and about 10% perioperative morbidity.41 Patients with a BMI >40 (or >37 with weight-related comorbidities) are candidates for surgery.42
It has been estimated that, for these patients, the cost per pound lost is less with surgery than with medications.43 In most series, the average morbidly obese patient can expect to lose 50% of excess body weight at 5 years after bypass surgery, and 50% of excess weight will be lost even 10 years post-operatively (SOR: B).44
Several options are available for surgical management of obesity. While the technical aspects of surgery are beyond the scope of this article, some generalizations can be made. Procedures may reduce the size of the stomach to decrease the volume of intake (gastroplasty), or may create a malabsorption condition (intestinal bypass) to decrease absorption of calories. The combination of a restrictive procedure with malabsorption (Rouxen-Y gastic bypass) is superior to a restrictive procedure alone (SOR: B).44
The surgical management of morbid obesity improves quality of life for patients,43 but no published studies to date have been able to evaluate the effect of the surgical management on mortality in the morbidly obese patient.
Complementary and alternative therapies
In addition to the traditional methods of weight loss, acupuncture and hypnosis have been studied in the treatment of obesity. Acupuncture does not appear to have any benefit greater than placebo (SOR: B).45 Hypnosis has also been reviewed and likely adds little, if any, benefit beyond that of placebo (SOR: B).45 Most studies of both acupuncture and hypnosis suffer from the difficulties of performing adequate control groups, and meta-analyses have demonstrated mixed results.45,46
Maintenance programs
There is significant evidence that when patients discontinue effective weight loss interventions (eg, diet or behavioral modification) they will return to their baseline weight. Because of this, it is important to consider maintenance programs as part of overall treatment and to imbue in patients the expectation that treatment will be lifelong. Examples of an approach to maintenance therapy include attendance at regular exercise or therapy sessions even after achieving weight-loss goals, or continued participation at commercial weight-loss program meetings or support groups.
ACKNOWLEDGEMENTS
The author was supported by grant 1 D45 PE 50175 -01, “Faculty Development in Family Medicine” funded by Health Resources and Services Administration (HRSA). The author wishes to thank Bill Hueston, MD, Peter Carek, MD, Arch Mainous III, PhD, and Lori Dickerson, PharmD, for their help with manuscript review. The author wishes to thank Tara Hogue for her for help with the preparation of this manuscript.
1. Dickerson L, Carek PJ. Drug therapy for obesity. Am Fam Physician 2000;51:2131-8.
2. Arterburn D, Hitchcock-Noel P. Obesity. Clinical Evidence 2001;5:512-9.
3. Mun EC, Blackburn GL, Matthews JB. Current status of medical and surgical therapy for obesity. Gastroenterology 2001;120:559-81.
4. Field AE, Coakley EH, Must A, Spandano JL, Laird N, Dietz WH, et al. Impact of overweight on the risk of developing common chronic diseases during a 10 year period. Arch Intern Med 2001;151:1581-5.
5. National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: The evidence report. Bethesda, Md: US Department of Health and Human Services; 1998.
6. Messier SP, Loeser RF, Mitchell MN, Valle G, Morgan TP, Rejeski WJ, et al. Exercise and weight loss in obese older adults with knee osteoarthritis: a preliminary study. J Am Geriatr Soc 2000;48:1052-72.
7. Shneerson J, Wright J. Lifestyle modification for obstructive sleep apnoea (Cochrane Review). In: The Cochrane Library, Issue 4, 2001. Oxford: Update Software.
8. Williamson DF, Thompson TS, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000;10:1499-1504.
9. Allison DB, Fontaine KR, Manson JE, Stevens J, Van Itallie TB. Annual deaths attributable to obesity in the United States. JAMA 1999;282:1530-8.
10. Yanovski J, Yanovski S. Recent advances in basic obesity research. JAMA 1999;282:1504-6.
11. Parsons TJ, Power C, Logan S, Summerbell CD. Childhood predictors of adult obesity: a systematic review. Int J Obes Relat Metab Disord 1999;8 (suppl):S1-107.
12. Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet 2001;357:505-8.
13. Gillman MW, Rifas-Shiman SL, Camargo CA, Berkey CS, Frazier AL, Rocket HRH, et al. Risk of overweight among adolescents who were breastfed as infants. JAMA 2001;285:2451-7.
14. Robinson T. Reducing children’s television viewing to prevent obesity. JAMA 1999;282:1551-7.
15. Campbell K, Waters E, O’Meara S, Summerbell C. Interventions for preventing obesity in children (Cochrane Review). In: The Cochrane Library, Issue 5, 2001. Oxford: Update Software.
16. Sahota P, Rudolf MC, Dixey R, Hill AJ, Barth JH, Cade J. Randomized controlled trial of primary school based intervention to reduce risk factors for obesity. BMJ 2001;323:1029-32.
17. US Preventive Services Task Force. Screening for obesity. In: US Preventive Services Task Force: Guide to Clinical Preventative Services: Report of the US Preventive Services Task Force. 2nd ed. Baltimore, Md: Williams and Wilkins; 1995;219-29.
18. Flier JS, Foster DW. Eating Disorders: Obesity, anorexia nervosa, and bullemia nervosa. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia, Pa: WB Saunders; 1998;1051-97.
19. McGee S. Evidence-based physical diagnosis. Philadelphia, Pa: WB Saunders; 2001.
20. Willett WC, Dietz WH, Colditz GA. Guidelines for healthy weight. N Engl J Med 1999;351:527-35.
21. Rexrode KM, Carey VJ, Hennekens CH, Walters EE, Colditz GA, Stampfer MJ, et al. Abdominal adiposity and coronary heart disease in women. JAMA 1998;280:1843-8.
22. Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Does waist circumference add to the predictive power of the body mass index for coronary risk? Obesity Research. 2001;9:585-95.
23. Toriano RP, Frongillo EA, Sobal J, Levitsky DA. The relationship between body weight and mortality: a quantitative analysis of combined information from existing studies. Int J Obes Relat Metab Disord 1996;20:63-75.
24. Hensrud DD. Dietary treatment and long-term weight loss and maintenance in type 2 diabetes. Obesity Research 2001;9:348S-353S.
25. Ditschuneit HH, Flechtner-Mors M. Value of structured meals for weight management: risk factors and long-term weight maintenance. Obesity Research 2001;9(suppl 4):284S-289S.
26. Ditschuneit HH, Flechtner-Mors M, Johnson TD, Adler G. Metabolic and weight-loss effects of a long-term dietary intervention in obese patients. Am J Clin Nutr 1999;69:198-204.
27. Flechtner-Mors M, Ditschuneit HH, Johnson TD, Suchard MA, Adler G. Metabolic and weight loss effects of long-term dietary interventions in obese patients: four-year results. Obesity Research 2000;8:399-402.
28. Freedman MR, King J, Kennedy E. Popular diets: a scientific review. Obesity Research 2001;9(suppl 1):1S-50S.
29. Finer N. Low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):290S-292S.
30. Saris W. Very-low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):292S-301S.
31. Pirozzo S, Summerbell C, Cameron C, Glasziou P. Advice on low-fat diets or obesity (Cochrane Review). In: The Cochrane Library, Issue 2, 2002. Oxford: Update Software.
32. Skov AR, Toubro S, Ronn B, Holm L, Astrup A. Randomized trial on protein vs.carbohydrate in ad libi-tum reduced diet for the treatment of obesity. Int J Obes 1999;23:528-36.
33. Bray GA. Role of physical activity and exercise in obesity. Up To Date (online 9.3). Retrieval date 11/5/2001. Up To Date, Inc. Wellesley, Mass, USA.
34. Thorogood M, Hillsdon M, Summerbell C. Changing behaviour. Clinical Evidence 2000;5:25-52.
35. Weintraub M, Sundaresan PR, Schuster B, Averbuch M, Stein EC, Byrne L. Long-term weight control study V. Clin Pharmacol Ther 1992;51:615-8.
36. Wadden T. Benefits of lifestyle modification in the phar-macologic treatment of obesity: a randomized trial. Arch Intern Med 2001;151:218-27.
37. Smith MB. Randomized placebo-controlled trial of long-term treatment with sibutramine in mild to moderate obesity. J Fam Pract 2001;50:505-12.
38. Sjostrom L, Rissanen A, Anderson T, Boldrin M, Golay A, Koppeschaan HPF, et al. Randomized placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet 1998;352:157-72.
39. O’meara S, Riemsma R, Shirran L, Mather L, ter Riet G. A rapid and systematic review of the clinical effectiveness and cost-effectiveness of orlistat in the management of obesity. Health Technology Assess 2001;5:18.
40. Physician’s Desk Reference. PGS. Montvale, NJ: Medical Economics; 2001;2809-13.
41. Balsiger BM, Kennedy FP, Abu-Lebdeh HS, Collazo-Clavell M, Jensen MD, O’Brien T, et al. Prospective evaluation of Roux-en Y gastric bypass as primary operation for medically complicated obesity. Mayo Clin Proc 2000;75:573-80.
42. Gastrointestinal Surgery for Severe Obesity. NIH Consensus Statement Online 1991; 25-27. Cited August 1, 2001;9(1):1-20.
43. Van Gemert WG, Van Dielen F, Soeters PB, Greve JW. Quality of life before and after weight-reducing surgery and cost-effectiveness analysis. In: Deitel M,†Cowan SM, eds. Update: Surgery for the Morbidly Obese Patient. Toronto: FD Communications; 2000;595-602.
44. Sugerman HJ. The epidemic of severe obesity: value of surgical treatment. Mayo Clin Proc 2000;75:559-572.
45. Ernst E. Acupuncture/acupressure for weight reduction? A systematic review. Wien Klin Wochenschr 1997;109:50-2.
46. Allison DB, Faith MS. Hypnosis as an adjunct to cogni-tive-behavioral psychotherapy for obesity: a meta-analyt-ic reappraisal. J Consult Clin Psychol 1995;54:513-5.
- Diet and exercise coupled with behavioral modification can improve both patient-related and disease-related outcomes in the short term; however, long-term efficacy is lacking (A).
- Drugs such as sibutramine or orlistat may achieve modest, short-term weight loss, but their long-term effectiveness is unproven (A).
- For patients with a BMI >40, gastric bypass procedures can lead to long-term weight loss (B).
Physician intervention to encourage and assist obese patients to lose weight is warranted, for these reasons:
- Prevention of adverse outcomes. Left untreated, obesity is clearly related to the development of many adverse health outcomes, including diabetes mellitus, hypertension, stroke, hyperlipidemia, coronary artery disease, gallstones, osteoarthritis, obstructive sleep apnea, vascular disease, depression, and certain cancers (breast, endometrial, prostate, colon).1-4
- Reduction of morbidity and mortality. A causal effect between intentional weight loss and mortality has been difficult to prove, but even modest weight loss can reduce the morbidity of obesity-related disease, such as arthritis and obstructive sleep apnea (Strength of recommendation: A).5-7 For those at increased risk of death from cardiovascular disease, such as persons with obesity and diabetes mellitus, intentional weight loss coupled with lifestyle change can significantly reduce mortality (SOR: C).8 Nearly 280,000 deaths per year are attributable to obesity.9
- Cost reduction. Nearly 25% of American adults are obese, and more than half are overweight.1,2 Obesity burdens society with significant costs, including more than $50 billion annually for direct care. With an additional $30 billion spent each year on weight-loss products and services, this disease accounts for over 5% of annual health care expenditures in the United States.5
Nature or nurture?
Excess fat is created when energy intake exceeds cellular energy consumption. The complex relationship between the human body’s environment and the development of obesity is poorly understood, but recent genetic investigations have elucidated new mechanisms in the regulation of both satiety and energy expenditure. Using data from heritability studies, some researchers have estimated that up to 70% of the variability in weight among humans can be explained by genetic influences, but it is unlikely that changes in human genes account for the recent change in obesity prevalence.10
Risk factors
Identifiable risks factors for obesity in adulthood include parental history of obesity, low socio-economic background, and a history of high birth weight.11
Prevention
Recently, factors including consumption of sugar-sweetened beverages, lack of breast-feeding, and television viewing have been identified as risk factors for childhood and adolescent obesity.12-14 Because obesity at a young age can lead to adult obesity, these factors may be targeted to prevent adult obesity.
School-based programs for diet and exercise appear to be ineffective for preventing obesity (SOR: B).15,16 However, most research has been of limited quality. In one recent randomized study, reduction of television viewing (including videotape and videogame use) through school intervention was associated with significant reduction in BMI (SOR: B).14
Screening recommendations
The United States Preventive Service Task Force recommends periodic measurement of both height and weight in adults (SOR: B). The waist-to-hip ratio is thought to have insufficient evidence for recommendation as a routine screening tool because studies identifying a benefit to screening using only the waist-to-hip ratio have not been completed (SOR: C).17
Initial determination of obesity
Although the standard for body fat measurement is densitometry, which determines the density of a body submersed in water, the cost and technical requirements prohibit routine use in the clinical setting.18-20 The waist-to-hip ratio and waist circumference are used to identify central (or android) obesity in which adipose tissue in the abdomen is associated with atherosclerosis.21,22 The waist circumference is found by measuring the circumference around the waist at the level of the iliac crest. Values above 40 inches for men and 35 inches for women are indicative of increased risk of adverse health outcomes.5,19 The waist-to-hip ratio is calculated by dividing the circumference of the waist at the level of the L3 by the hip circumference measured at the largest area of the gluteal region.19 For men, waist-to-hip ratios greater than 1.0 are associated with significantly increased risk of cardiovascular events. For women, a ratio greater than 0.85 indicates increased risk.19
Body-mass index calculation
The body-mass index (BMI), also known as the Quetelet index, is the most commonly used measure of obesity.20 BMI is a patient’s weight in kilograms divided by his or her height in meters squared (kg/m2 ).5,19 A free online BMI calculator is available through the National Heart, Lung and Blood Institute at www.nhlbisupport.com/bmi/bmicalc.htm.
Although the BMI estimates total body fat and compares well with densitometry, it may be less accurate in selected populations (eg, the elderly, certain ethnic groups, and persons with large muscle mass).19 Generally, when BMI exceeds 25, the greater the BMI, the greater the obesity-related morbidity and mortality.5,19,20,23 Table 1 shows the classification of obesity based on BMI, and Table 2 shows the BMI for combinations of height and weight in inches and pounds.5
TABLE 1
Body mass index (BMI) and degrees of obesity
| BMI | Category | Therapeutic options |
|---|---|---|
| <18.5 | Underweight | |
| 18.5–25.9 | Normal weight | Reinforce positive lifestyle |
| 25.0–29.9 | Overweight | Diet, exercise, behavior modification |
| 30.0–34.9 | Obese (Class I) | Diet, exercise, behavior modification; consider pharmacologic therapy* |
| 35.0–39.9 | Obese (Class II) | Diet, exercise, behavior modification; consider pharmacologic therapy |
| ≥40.0 | Obese (Class III or “Morbid Obesity”) | Consider surgical management |
| *For patients with multiple cardiovascular risk factors (eg, diabetes, hyperlipidemia), BMI >27 may be an indication for pharmacologic intervention at; at BMI >37, patients may be considered candidates for surgical therapy. | ||
| From the National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: the Evidence Report. Bethesda, Md: US Department of Health and Human Services, 1998. | ||
Evaluation of documented obesity
In history taking and physical examination, look for reversible causes of obesity (including medications and endocrine disorders), consider the degree of obesity, and determine whether comorbid conditions are present, to help estimate prognosis. Evaluate the patient’s dietary and exercise habits, as well as willingness to modify these habits if necessary.1,5 Finally, review the patient’s weight history and any attempts at weight loss.
Medications associated with weight gain include psychotropic drugs, anticonvulsant agents, steroid hormones, insulin, and many oral hypoglycemic agents.1
Endocrine disorders such as Cushing’s syndrome and hypothyroidism may also contribute to obesity, but only rarely. Physical findings that increase the likelihood of Cushing’s syndrome, and their respective positive likelihood ratios (LR+)— the higher the value, the greater the likelihood of disease—include: hypertension (2.3), moon facies (1.6), thin skinfold (115.6), ecchymoses (4.5), and acne (2.2).19 Findings, and their likelihood ratios, associated with hypothyroidism include coarse skin (5.6), cool/dry skin (4.7), bradycardia (4.1), enlarged thyroid (2.8), and hoarse voice (5.4).19 The very high LR for skinfold thickness was determined for women of childbearing age who had elevated risk of having Cushing’s syndrome because of a history of both menstrual irregularities and hirsutism. Skinfold thickness is determined by using calipers on an area of minimal subcutaneous fat (eg, back of the hand). For women of reproductive age, skinfold thickness is normally greater than 1.8 mm.19
Laboratory assessment of the obese patient will rarely find a cause of weight gain (eg, hypothyroidism), but the addition of selected diagnostic tests will aid in the determination of prognosis. An abnormal fasting glucose level or impaired glucose tolerance is a major risk factor for cardiovascular disease. Abnormal lipid profiles heighten that risk for obese patients. All patients with documented obesity should undergo assessment for abnormal lipids and impaired glucose tolerance5 (SOR: D).
Treatment
The most important step in treating obesity is to establish a calorie deficit. The deficit can be achieved by increasing energy expenditure or by reducing energy intake or absorption. On average, a caloric deficit of 500 kilocalories per day will result in a weight loss of 1 pound per week.5 Reasonable expectations of therapy include weight loss of 1–2 pounds a week and a loss of 10% of total body weight in 5 months.5
Interventions for weight loss fall into 4 categories: lifestyle modifications (diet, exercise, and behavioral modification), drug therapy, complementary or alternative measures, and surgery. Table 3 summarizes the levels of evidence to support each intervention.
TABLE 3
Efficacy of weight-loss interventions
| Weight loss | ||||
|---|---|---|---|---|
| SOR* | Intervention | Short-term | Long-term | Comments |
| Diet, exercise, and behavioral modification | ||||
| A | Low/very-lowcalorie diet | 8% average weight loss from 3–12 months | Weight nears baseline in studies >24 months | Very-low-calorie diets may require laboratory assessment of metabolic function |
| High rate of noncompliance | ||||
| A | Low/very low fat with reduced calories | Similar to lowcalorie with moderate fat | Weight nears baseline in studies >24 months | No known side effects |
| A | Exercise | Less weight loss than diet therapy | Likely no significant weight loss | Improved cardiovascular fitness |
| May be effective in preventing weight gain | ||||
| A | Low-calorie diet + exercise | Increased weight loss vs. diet or exercise alone | Weight nears baseline in long-term studies | Improved cardiovascular fitness |
| Compliance a major problem | ||||
| B | Behavior modification | Increases effectiveness of diet, exercise | No significant effect at 5 years | No reported harms |
| Only studied when used with other methods | ||||
| C | Lowcarbohydrate diet | Not significant if calories are not reduced | No long-term data available | No known side effects, but creates nutritional imbalance and ketosis |
| Needs additional study | ||||
| Medication | ||||
| A | Sibutramine | ˜ 4 kg for trials less than 1 year | Modest weight loss when used for >1 year | Can elevate blood pressure |
| Number needed to treat (NNT) for 5% weight loss at 1 year = 3 | ||||
| NNT for 10% weight loss at 1 year = 5 | ||||
| A | Orlistat | ˜ 2–3 kg for trials less than 1 year | ˜ 3 kg at 2 years | GI side effects common, possible vitamin deficiencies |
| NNT for 5% weight loss at 1 year = 5 | ||||
| NNT for 10% weight loss at 1 year = 7 | ||||
| Surgery | ||||
| B | Roux-en-Y Gastric bypass | ˜ 50 kg (110 lbs) at 1 year | ˜ 50 kg (110 lbs) at up to 4 years | Significant operative risk and post-operative GI side effects |
| Nadir for weight loss occurs at 12–24 months | ||||
| B | Gastric banding | ˜ 30 Kg (66 lbs) at one year | 10–15% of initial weight lost may be regained long-term | Significant operative risk and post-operative GI side effects |
| Generally considered less effective that gastric bypass | ||||
| Complementary/alternative medicine | ||||
| B | Hypnosis | Minimal reduction | No statistically significant difference | Studied in combination with cognitive behavioral therapy |
| Systematic reviews reveal significant heterogeneity of low-quality randomized controlled trials | ||||
| B | Acupuncture | No significant difference | No significant difference | Systematic review reveals poor quality |
| RCTs, which limits ability to determine effect | ||||
| * Strength of recommendation | ||||
| A = Systematic review of randomized controlled trials (RCT) (with homogeneity) or individual RCT with narrow confidence interval | ||||
| B = Systematic review of cohort studies (with homogeneity), individual cohort studies or low-quality RCT, individual case-control study or SR of case-control studies (with homogeneity) | ||||
| C = Case series and poor quality cohort and case control studies | ||||
Lifestyle modifications
Management of obesity in every case should include dietary changes, exercise, and behavioral modification.
Dietary changes. Diets create a caloric deficit by reducing the intake of calories. Average weight loss with low-calorie diets is approximately 8% at 3–12 months (SOR: A),5 with most of the loss occurring in the first 3–5 months.24 There are many kinds of diets for weight loss, including low calorie, very-low calorie, low fat, very low fat, and low carbohydrate, but long-term compliance with all types of dietary interventions is a significant problem. When diet alone is used as therapy, between one third and one half of weight loss will not be maintained.24 Emerging strategies to help improve dietary compliance include behavioral modification (see below) and meal substitutes. Recent reports of interventions such as meal-replacement shakes indicate that long-term weight loss can be significantly improved (SOR: B).25-27
Examples of low-calorie, nutritionally balanced diets are Weight Watchers, Jenny Craig, Nutrisystem, the National Cholesterol Education Program Step I and Step II diets, and the Dietary Approaches to Stop Hypertension (DASH) diet. Low-calorie diets provide 800–1500 kilocalories per day.5,28,29 Very-low-calorie diets (400–500 kilocalories per day) may increase rates of weight loss initially, but at 1 year, results are similar to those of low-calorie diets (SOR: A).5,30
A low-fat diet (fat content 10%–19%) without a decrease in total calorie intake does not promote weight loss (SOR: A).5,31 Very-low-fat diets containing less than 10% fat have been described by such authors as Ornish and Pritikin.25 Obese patients using either the low-fat or very-low-fat diet can lose body weight and body fat, but only if calories are also decreased (SOR: A).5,28
Low-carbohydrate diets, such as Dr Atkin’s diet, are associated with modest (approximately 5 kg, or 11 lbs) weight loss (SOR: C).28,32 Improved study design is required to further evaluate the effectiveness and safety of low-carbohydrate diets in the clinical setting.
Exercise. Most studies of exercise are based on 30–50 minutes of moderately intense aerobic exercise, repeated 3–7 times per week.5 When it is the only prescribed therapy, exercise can be expected to produce modest weight loss only (SOR: A).5,33 Exercise combined with dietary intervention, however, increases weight lost (SOR: A), and exercise by itself may prevent weight gain (SOR: C).5
Behavioral Modification. Behavioral modification has been evaluated in combination with diet or exercise, and has been shown to increase compliance and weight loss for durations of 1 year or less (SOR: A).5,34 Weight gain is common when therapy is discontinued, and at 5 years, there is no difference between those who received behavioral therapy and those who were in control groups (SOR: A).5
Medications
Medications for treatment of obesity act through 1 or more of 3 mechanisms:
- Appetite suppression (eg, sibutramine, antidepressants such as fluoxetine)
- Increased metabolic activity (eg, stimulants such as ephedra with caffeine, Β-3 agonists)
- Decreased absorption of caloric load (orlistat)
For mild-to-moderate obesity (BMI >30 and <40), medications can be beneficial (SOR: A), but long-term weight loss beyond 2 years has not been studied.35 Pharmacologic intervention without lifestyle intervention actually decreases a person’s ability to lose weight (SOR: B).36
Two medications are approved by the United States Food and Drug Administration for long-term obesity management: sibutramine and orlistat. Both drugs reduce weight modestly (SOR: A).37-39 Both medications have similar indications for use: BMI >30, or BMI >27 with the presence of other cardiovascular risk factors (ie, diabetes or hyperlipidemia). Both should be used in conjunction with reduced-calorie diet and exercise (SOR: B).36
Sibutramine is usually started at 10 mg once a day, given with or without food. The dose may be titrated to a maximum of 15 mg/d after 4 weeks if weight loss has been inadequate.1 Sibutramine is known to increase pulse rate and blood pressure in a significant number of patients; because of this, regular evaluation of vital signs is required. At present, long-term use of sibutramine cannot be recommended, and safety data are unavailable beyond 1 year of use. Sibutramine should be avoided if these conditions are present: hypertension, coronary heart disease, congestive heart failure, an arrhythmic condition, pregnancy, renal impairment, concomitant use of MAOI, or a history of stroke.
Orlistat is started at 120 mg three times a day, and is taken with meals that contain fat. It may still be effective if taken up to one hour after eating. Orlistat may be avoided if the meal contains no fat. This drug may interfere with the absorption of some fat-soluble vitamins, and it is therefore recommended that patients take a multivitamin that has fat-soluble vitamins at least 2 hours before or after ingesting orlistat. Orlistat is not absorbed into the body and, at this time, no laboratory follow-up is needed. Regular evaluation of weight is needed to assess the efficacy of treatment. Orlistat should be avoided by those who have cholestasis or malabsorptive disorders or by those taking cyclosporine.40
Other medications that have been used include phentermine, ephedra, dexfenfluramine, phenyl-propanolamine (PPA), and mazindol.1,2 All of these medications produce significant weight loss in the short term (SOR: A), but they are not indicated for long-term use.2 In fact, phenylpropanolamine and dexfenfluramine are no longer available because of their severe side effects.2 Antidepressants do not yield a consistent benefit in well-designed studies of obesity management.2
Table 3 summarizes the effects of medications on weight loss.
Surgery
Surgical management of obesity is reserved for extremely obese persons because of the significant morbidity and mortality associated with the interventions. Currently, gastric bypass procedures result in less than 1% perioperative mortality and about 10% perioperative morbidity.41 Patients with a BMI >40 (or >37 with weight-related comorbidities) are candidates for surgery.42
It has been estimated that, for these patients, the cost per pound lost is less with surgery than with medications.43 In most series, the average morbidly obese patient can expect to lose 50% of excess body weight at 5 years after bypass surgery, and 50% of excess weight will be lost even 10 years post-operatively (SOR: B).44
Several options are available for surgical management of obesity. While the technical aspects of surgery are beyond the scope of this article, some generalizations can be made. Procedures may reduce the size of the stomach to decrease the volume of intake (gastroplasty), or may create a malabsorption condition (intestinal bypass) to decrease absorption of calories. The combination of a restrictive procedure with malabsorption (Rouxen-Y gastic bypass) is superior to a restrictive procedure alone (SOR: B).44
The surgical management of morbid obesity improves quality of life for patients,43 but no published studies to date have been able to evaluate the effect of the surgical management on mortality in the morbidly obese patient.
Complementary and alternative therapies
In addition to the traditional methods of weight loss, acupuncture and hypnosis have been studied in the treatment of obesity. Acupuncture does not appear to have any benefit greater than placebo (SOR: B).45 Hypnosis has also been reviewed and likely adds little, if any, benefit beyond that of placebo (SOR: B).45 Most studies of both acupuncture and hypnosis suffer from the difficulties of performing adequate control groups, and meta-analyses have demonstrated mixed results.45,46
Maintenance programs
There is significant evidence that when patients discontinue effective weight loss interventions (eg, diet or behavioral modification) they will return to their baseline weight. Because of this, it is important to consider maintenance programs as part of overall treatment and to imbue in patients the expectation that treatment will be lifelong. Examples of an approach to maintenance therapy include attendance at regular exercise or therapy sessions even after achieving weight-loss goals, or continued participation at commercial weight-loss program meetings or support groups.
ACKNOWLEDGEMENTS
The author was supported by grant 1 D45 PE 50175 -01, “Faculty Development in Family Medicine” funded by Health Resources and Services Administration (HRSA). The author wishes to thank Bill Hueston, MD, Peter Carek, MD, Arch Mainous III, PhD, and Lori Dickerson, PharmD, for their help with manuscript review. The author wishes to thank Tara Hogue for her for help with the preparation of this manuscript.
- Diet and exercise coupled with behavioral modification can improve both patient-related and disease-related outcomes in the short term; however, long-term efficacy is lacking (A).
- Drugs such as sibutramine or orlistat may achieve modest, short-term weight loss, but their long-term effectiveness is unproven (A).
- For patients with a BMI >40, gastric bypass procedures can lead to long-term weight loss (B).
Physician intervention to encourage and assist obese patients to lose weight is warranted, for these reasons:
- Prevention of adverse outcomes. Left untreated, obesity is clearly related to the development of many adverse health outcomes, including diabetes mellitus, hypertension, stroke, hyperlipidemia, coronary artery disease, gallstones, osteoarthritis, obstructive sleep apnea, vascular disease, depression, and certain cancers (breast, endometrial, prostate, colon).1-4
- Reduction of morbidity and mortality. A causal effect between intentional weight loss and mortality has been difficult to prove, but even modest weight loss can reduce the morbidity of obesity-related disease, such as arthritis and obstructive sleep apnea (Strength of recommendation: A).5-7 For those at increased risk of death from cardiovascular disease, such as persons with obesity and diabetes mellitus, intentional weight loss coupled with lifestyle change can significantly reduce mortality (SOR: C).8 Nearly 280,000 deaths per year are attributable to obesity.9
- Cost reduction. Nearly 25% of American adults are obese, and more than half are overweight.1,2 Obesity burdens society with significant costs, including more than $50 billion annually for direct care. With an additional $30 billion spent each year on weight-loss products and services, this disease accounts for over 5% of annual health care expenditures in the United States.5
Nature or nurture?
Excess fat is created when energy intake exceeds cellular energy consumption. The complex relationship between the human body’s environment and the development of obesity is poorly understood, but recent genetic investigations have elucidated new mechanisms in the regulation of both satiety and energy expenditure. Using data from heritability studies, some researchers have estimated that up to 70% of the variability in weight among humans can be explained by genetic influences, but it is unlikely that changes in human genes account for the recent change in obesity prevalence.10
Risk factors
Identifiable risks factors for obesity in adulthood include parental history of obesity, low socio-economic background, and a history of high birth weight.11
Prevention
Recently, factors including consumption of sugar-sweetened beverages, lack of breast-feeding, and television viewing have been identified as risk factors for childhood and adolescent obesity.12-14 Because obesity at a young age can lead to adult obesity, these factors may be targeted to prevent adult obesity.
School-based programs for diet and exercise appear to be ineffective for preventing obesity (SOR: B).15,16 However, most research has been of limited quality. In one recent randomized study, reduction of television viewing (including videotape and videogame use) through school intervention was associated with significant reduction in BMI (SOR: B).14
Screening recommendations
The United States Preventive Service Task Force recommends periodic measurement of both height and weight in adults (SOR: B). The waist-to-hip ratio is thought to have insufficient evidence for recommendation as a routine screening tool because studies identifying a benefit to screening using only the waist-to-hip ratio have not been completed (SOR: C).17
Initial determination of obesity
Although the standard for body fat measurement is densitometry, which determines the density of a body submersed in water, the cost and technical requirements prohibit routine use in the clinical setting.18-20 The waist-to-hip ratio and waist circumference are used to identify central (or android) obesity in which adipose tissue in the abdomen is associated with atherosclerosis.21,22 The waist circumference is found by measuring the circumference around the waist at the level of the iliac crest. Values above 40 inches for men and 35 inches for women are indicative of increased risk of adverse health outcomes.5,19 The waist-to-hip ratio is calculated by dividing the circumference of the waist at the level of the L3 by the hip circumference measured at the largest area of the gluteal region.19 For men, waist-to-hip ratios greater than 1.0 are associated with significantly increased risk of cardiovascular events. For women, a ratio greater than 0.85 indicates increased risk.19
Body-mass index calculation
The body-mass index (BMI), also known as the Quetelet index, is the most commonly used measure of obesity.20 BMI is a patient’s weight in kilograms divided by his or her height in meters squared (kg/m2 ).5,19 A free online BMI calculator is available through the National Heart, Lung and Blood Institute at www.nhlbisupport.com/bmi/bmicalc.htm.
Although the BMI estimates total body fat and compares well with densitometry, it may be less accurate in selected populations (eg, the elderly, certain ethnic groups, and persons with large muscle mass).19 Generally, when BMI exceeds 25, the greater the BMI, the greater the obesity-related morbidity and mortality.5,19,20,23 Table 1 shows the classification of obesity based on BMI, and Table 2 shows the BMI for combinations of height and weight in inches and pounds.5
TABLE 1
Body mass index (BMI) and degrees of obesity
| BMI | Category | Therapeutic options |
|---|---|---|
| <18.5 | Underweight | |
| 18.5–25.9 | Normal weight | Reinforce positive lifestyle |
| 25.0–29.9 | Overweight | Diet, exercise, behavior modification |
| 30.0–34.9 | Obese (Class I) | Diet, exercise, behavior modification; consider pharmacologic therapy* |
| 35.0–39.9 | Obese (Class II) | Diet, exercise, behavior modification; consider pharmacologic therapy |
| ≥40.0 | Obese (Class III or “Morbid Obesity”) | Consider surgical management |
| *For patients with multiple cardiovascular risk factors (eg, diabetes, hyperlipidemia), BMI >27 may be an indication for pharmacologic intervention at; at BMI >37, patients may be considered candidates for surgical therapy. | ||
| From the National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: the Evidence Report. Bethesda, Md: US Department of Health and Human Services, 1998. | ||
Evaluation of documented obesity
In history taking and physical examination, look for reversible causes of obesity (including medications and endocrine disorders), consider the degree of obesity, and determine whether comorbid conditions are present, to help estimate prognosis. Evaluate the patient’s dietary and exercise habits, as well as willingness to modify these habits if necessary.1,5 Finally, review the patient’s weight history and any attempts at weight loss.
Medications associated with weight gain include psychotropic drugs, anticonvulsant agents, steroid hormones, insulin, and many oral hypoglycemic agents.1
Endocrine disorders such as Cushing’s syndrome and hypothyroidism may also contribute to obesity, but only rarely. Physical findings that increase the likelihood of Cushing’s syndrome, and their respective positive likelihood ratios (LR+)— the higher the value, the greater the likelihood of disease—include: hypertension (2.3), moon facies (1.6), thin skinfold (115.6), ecchymoses (4.5), and acne (2.2).19 Findings, and their likelihood ratios, associated with hypothyroidism include coarse skin (5.6), cool/dry skin (4.7), bradycardia (4.1), enlarged thyroid (2.8), and hoarse voice (5.4).19 The very high LR for skinfold thickness was determined for women of childbearing age who had elevated risk of having Cushing’s syndrome because of a history of both menstrual irregularities and hirsutism. Skinfold thickness is determined by using calipers on an area of minimal subcutaneous fat (eg, back of the hand). For women of reproductive age, skinfold thickness is normally greater than 1.8 mm.19
Laboratory assessment of the obese patient will rarely find a cause of weight gain (eg, hypothyroidism), but the addition of selected diagnostic tests will aid in the determination of prognosis. An abnormal fasting glucose level or impaired glucose tolerance is a major risk factor for cardiovascular disease. Abnormal lipid profiles heighten that risk for obese patients. All patients with documented obesity should undergo assessment for abnormal lipids and impaired glucose tolerance5 (SOR: D).
Treatment
The most important step in treating obesity is to establish a calorie deficit. The deficit can be achieved by increasing energy expenditure or by reducing energy intake or absorption. On average, a caloric deficit of 500 kilocalories per day will result in a weight loss of 1 pound per week.5 Reasonable expectations of therapy include weight loss of 1–2 pounds a week and a loss of 10% of total body weight in 5 months.5
Interventions for weight loss fall into 4 categories: lifestyle modifications (diet, exercise, and behavioral modification), drug therapy, complementary or alternative measures, and surgery. Table 3 summarizes the levels of evidence to support each intervention.
TABLE 3
Efficacy of weight-loss interventions
| Weight loss | ||||
|---|---|---|---|---|
| SOR* | Intervention | Short-term | Long-term | Comments |
| Diet, exercise, and behavioral modification | ||||
| A | Low/very-lowcalorie diet | 8% average weight loss from 3–12 months | Weight nears baseline in studies >24 months | Very-low-calorie diets may require laboratory assessment of metabolic function |
| High rate of noncompliance | ||||
| A | Low/very low fat with reduced calories | Similar to lowcalorie with moderate fat | Weight nears baseline in studies >24 months | No known side effects |
| A | Exercise | Less weight loss than diet therapy | Likely no significant weight loss | Improved cardiovascular fitness |
| May be effective in preventing weight gain | ||||
| A | Low-calorie diet + exercise | Increased weight loss vs. diet or exercise alone | Weight nears baseline in long-term studies | Improved cardiovascular fitness |
| Compliance a major problem | ||||
| B | Behavior modification | Increases effectiveness of diet, exercise | No significant effect at 5 years | No reported harms |
| Only studied when used with other methods | ||||
| C | Lowcarbohydrate diet | Not significant if calories are not reduced | No long-term data available | No known side effects, but creates nutritional imbalance and ketosis |
| Needs additional study | ||||
| Medication | ||||
| A | Sibutramine | ˜ 4 kg for trials less than 1 year | Modest weight loss when used for >1 year | Can elevate blood pressure |
| Number needed to treat (NNT) for 5% weight loss at 1 year = 3 | ||||
| NNT for 10% weight loss at 1 year = 5 | ||||
| A | Orlistat | ˜ 2–3 kg for trials less than 1 year | ˜ 3 kg at 2 years | GI side effects common, possible vitamin deficiencies |
| NNT for 5% weight loss at 1 year = 5 | ||||
| NNT for 10% weight loss at 1 year = 7 | ||||
| Surgery | ||||
| B | Roux-en-Y Gastric bypass | ˜ 50 kg (110 lbs) at 1 year | ˜ 50 kg (110 lbs) at up to 4 years | Significant operative risk and post-operative GI side effects |
| Nadir for weight loss occurs at 12–24 months | ||||
| B | Gastric banding | ˜ 30 Kg (66 lbs) at one year | 10–15% of initial weight lost may be regained long-term | Significant operative risk and post-operative GI side effects |
| Generally considered less effective that gastric bypass | ||||
| Complementary/alternative medicine | ||||
| B | Hypnosis | Minimal reduction | No statistically significant difference | Studied in combination with cognitive behavioral therapy |
| Systematic reviews reveal significant heterogeneity of low-quality randomized controlled trials | ||||
| B | Acupuncture | No significant difference | No significant difference | Systematic review reveals poor quality |
| RCTs, which limits ability to determine effect | ||||
| * Strength of recommendation | ||||
| A = Systematic review of randomized controlled trials (RCT) (with homogeneity) or individual RCT with narrow confidence interval | ||||
| B = Systematic review of cohort studies (with homogeneity), individual cohort studies or low-quality RCT, individual case-control study or SR of case-control studies (with homogeneity) | ||||
| C = Case series and poor quality cohort and case control studies | ||||
Lifestyle modifications
Management of obesity in every case should include dietary changes, exercise, and behavioral modification.
Dietary changes. Diets create a caloric deficit by reducing the intake of calories. Average weight loss with low-calorie diets is approximately 8% at 3–12 months (SOR: A),5 with most of the loss occurring in the first 3–5 months.24 There are many kinds of diets for weight loss, including low calorie, very-low calorie, low fat, very low fat, and low carbohydrate, but long-term compliance with all types of dietary interventions is a significant problem. When diet alone is used as therapy, between one third and one half of weight loss will not be maintained.24 Emerging strategies to help improve dietary compliance include behavioral modification (see below) and meal substitutes. Recent reports of interventions such as meal-replacement shakes indicate that long-term weight loss can be significantly improved (SOR: B).25-27
Examples of low-calorie, nutritionally balanced diets are Weight Watchers, Jenny Craig, Nutrisystem, the National Cholesterol Education Program Step I and Step II diets, and the Dietary Approaches to Stop Hypertension (DASH) diet. Low-calorie diets provide 800–1500 kilocalories per day.5,28,29 Very-low-calorie diets (400–500 kilocalories per day) may increase rates of weight loss initially, but at 1 year, results are similar to those of low-calorie diets (SOR: A).5,30
A low-fat diet (fat content 10%–19%) without a decrease in total calorie intake does not promote weight loss (SOR: A).5,31 Very-low-fat diets containing less than 10% fat have been described by such authors as Ornish and Pritikin.25 Obese patients using either the low-fat or very-low-fat diet can lose body weight and body fat, but only if calories are also decreased (SOR: A).5,28
Low-carbohydrate diets, such as Dr Atkin’s diet, are associated with modest (approximately 5 kg, or 11 lbs) weight loss (SOR: C).28,32 Improved study design is required to further evaluate the effectiveness and safety of low-carbohydrate diets in the clinical setting.
Exercise. Most studies of exercise are based on 30–50 minutes of moderately intense aerobic exercise, repeated 3–7 times per week.5 When it is the only prescribed therapy, exercise can be expected to produce modest weight loss only (SOR: A).5,33 Exercise combined with dietary intervention, however, increases weight lost (SOR: A), and exercise by itself may prevent weight gain (SOR: C).5
Behavioral Modification. Behavioral modification has been evaluated in combination with diet or exercise, and has been shown to increase compliance and weight loss for durations of 1 year or less (SOR: A).5,34 Weight gain is common when therapy is discontinued, and at 5 years, there is no difference between those who received behavioral therapy and those who were in control groups (SOR: A).5
Medications
Medications for treatment of obesity act through 1 or more of 3 mechanisms:
- Appetite suppression (eg, sibutramine, antidepressants such as fluoxetine)
- Increased metabolic activity (eg, stimulants such as ephedra with caffeine, Β-3 agonists)
- Decreased absorption of caloric load (orlistat)
For mild-to-moderate obesity (BMI >30 and <40), medications can be beneficial (SOR: A), but long-term weight loss beyond 2 years has not been studied.35 Pharmacologic intervention without lifestyle intervention actually decreases a person’s ability to lose weight (SOR: B).36
Two medications are approved by the United States Food and Drug Administration for long-term obesity management: sibutramine and orlistat. Both drugs reduce weight modestly (SOR: A).37-39 Both medications have similar indications for use: BMI >30, or BMI >27 with the presence of other cardiovascular risk factors (ie, diabetes or hyperlipidemia). Both should be used in conjunction with reduced-calorie diet and exercise (SOR: B).36
Sibutramine is usually started at 10 mg once a day, given with or without food. The dose may be titrated to a maximum of 15 mg/d after 4 weeks if weight loss has been inadequate.1 Sibutramine is known to increase pulse rate and blood pressure in a significant number of patients; because of this, regular evaluation of vital signs is required. At present, long-term use of sibutramine cannot be recommended, and safety data are unavailable beyond 1 year of use. Sibutramine should be avoided if these conditions are present: hypertension, coronary heart disease, congestive heart failure, an arrhythmic condition, pregnancy, renal impairment, concomitant use of MAOI, or a history of stroke.
Orlistat is started at 120 mg three times a day, and is taken with meals that contain fat. It may still be effective if taken up to one hour after eating. Orlistat may be avoided if the meal contains no fat. This drug may interfere with the absorption of some fat-soluble vitamins, and it is therefore recommended that patients take a multivitamin that has fat-soluble vitamins at least 2 hours before or after ingesting orlistat. Orlistat is not absorbed into the body and, at this time, no laboratory follow-up is needed. Regular evaluation of weight is needed to assess the efficacy of treatment. Orlistat should be avoided by those who have cholestasis or malabsorptive disorders or by those taking cyclosporine.40
Other medications that have been used include phentermine, ephedra, dexfenfluramine, phenyl-propanolamine (PPA), and mazindol.1,2 All of these medications produce significant weight loss in the short term (SOR: A), but they are not indicated for long-term use.2 In fact, phenylpropanolamine and dexfenfluramine are no longer available because of their severe side effects.2 Antidepressants do not yield a consistent benefit in well-designed studies of obesity management.2
Table 3 summarizes the effects of medications on weight loss.
Surgery
Surgical management of obesity is reserved for extremely obese persons because of the significant morbidity and mortality associated with the interventions. Currently, gastric bypass procedures result in less than 1% perioperative mortality and about 10% perioperative morbidity.41 Patients with a BMI >40 (or >37 with weight-related comorbidities) are candidates for surgery.42
It has been estimated that, for these patients, the cost per pound lost is less with surgery than with medications.43 In most series, the average morbidly obese patient can expect to lose 50% of excess body weight at 5 years after bypass surgery, and 50% of excess weight will be lost even 10 years post-operatively (SOR: B).44
Several options are available for surgical management of obesity. While the technical aspects of surgery are beyond the scope of this article, some generalizations can be made. Procedures may reduce the size of the stomach to decrease the volume of intake (gastroplasty), or may create a malabsorption condition (intestinal bypass) to decrease absorption of calories. The combination of a restrictive procedure with malabsorption (Rouxen-Y gastic bypass) is superior to a restrictive procedure alone (SOR: B).44
The surgical management of morbid obesity improves quality of life for patients,43 but no published studies to date have been able to evaluate the effect of the surgical management on mortality in the morbidly obese patient.
Complementary and alternative therapies
In addition to the traditional methods of weight loss, acupuncture and hypnosis have been studied in the treatment of obesity. Acupuncture does not appear to have any benefit greater than placebo (SOR: B).45 Hypnosis has also been reviewed and likely adds little, if any, benefit beyond that of placebo (SOR: B).45 Most studies of both acupuncture and hypnosis suffer from the difficulties of performing adequate control groups, and meta-analyses have demonstrated mixed results.45,46
Maintenance programs
There is significant evidence that when patients discontinue effective weight loss interventions (eg, diet or behavioral modification) they will return to their baseline weight. Because of this, it is important to consider maintenance programs as part of overall treatment and to imbue in patients the expectation that treatment will be lifelong. Examples of an approach to maintenance therapy include attendance at regular exercise or therapy sessions even after achieving weight-loss goals, or continued participation at commercial weight-loss program meetings or support groups.
ACKNOWLEDGEMENTS
The author was supported by grant 1 D45 PE 50175 -01, “Faculty Development in Family Medicine” funded by Health Resources and Services Administration (HRSA). The author wishes to thank Bill Hueston, MD, Peter Carek, MD, Arch Mainous III, PhD, and Lori Dickerson, PharmD, for their help with manuscript review. The author wishes to thank Tara Hogue for her for help with the preparation of this manuscript.
1. Dickerson L, Carek PJ. Drug therapy for obesity. Am Fam Physician 2000;51:2131-8.
2. Arterburn D, Hitchcock-Noel P. Obesity. Clinical Evidence 2001;5:512-9.
3. Mun EC, Blackburn GL, Matthews JB. Current status of medical and surgical therapy for obesity. Gastroenterology 2001;120:559-81.
4. Field AE, Coakley EH, Must A, Spandano JL, Laird N, Dietz WH, et al. Impact of overweight on the risk of developing common chronic diseases during a 10 year period. Arch Intern Med 2001;151:1581-5.
5. National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: The evidence report. Bethesda, Md: US Department of Health and Human Services; 1998.
6. Messier SP, Loeser RF, Mitchell MN, Valle G, Morgan TP, Rejeski WJ, et al. Exercise and weight loss in obese older adults with knee osteoarthritis: a preliminary study. J Am Geriatr Soc 2000;48:1052-72.
7. Shneerson J, Wright J. Lifestyle modification for obstructive sleep apnoea (Cochrane Review). In: The Cochrane Library, Issue 4, 2001. Oxford: Update Software.
8. Williamson DF, Thompson TS, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000;10:1499-1504.
9. Allison DB, Fontaine KR, Manson JE, Stevens J, Van Itallie TB. Annual deaths attributable to obesity in the United States. JAMA 1999;282:1530-8.
10. Yanovski J, Yanovski S. Recent advances in basic obesity research. JAMA 1999;282:1504-6.
11. Parsons TJ, Power C, Logan S, Summerbell CD. Childhood predictors of adult obesity: a systematic review. Int J Obes Relat Metab Disord 1999;8 (suppl):S1-107.
12. Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet 2001;357:505-8.
13. Gillman MW, Rifas-Shiman SL, Camargo CA, Berkey CS, Frazier AL, Rocket HRH, et al. Risk of overweight among adolescents who were breastfed as infants. JAMA 2001;285:2451-7.
14. Robinson T. Reducing children’s television viewing to prevent obesity. JAMA 1999;282:1551-7.
15. Campbell K, Waters E, O’Meara S, Summerbell C. Interventions for preventing obesity in children (Cochrane Review). In: The Cochrane Library, Issue 5, 2001. Oxford: Update Software.
16. Sahota P, Rudolf MC, Dixey R, Hill AJ, Barth JH, Cade J. Randomized controlled trial of primary school based intervention to reduce risk factors for obesity. BMJ 2001;323:1029-32.
17. US Preventive Services Task Force. Screening for obesity. In: US Preventive Services Task Force: Guide to Clinical Preventative Services: Report of the US Preventive Services Task Force. 2nd ed. Baltimore, Md: Williams and Wilkins; 1995;219-29.
18. Flier JS, Foster DW. Eating Disorders: Obesity, anorexia nervosa, and bullemia nervosa. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia, Pa: WB Saunders; 1998;1051-97.
19. McGee S. Evidence-based physical diagnosis. Philadelphia, Pa: WB Saunders; 2001.
20. Willett WC, Dietz WH, Colditz GA. Guidelines for healthy weight. N Engl J Med 1999;351:527-35.
21. Rexrode KM, Carey VJ, Hennekens CH, Walters EE, Colditz GA, Stampfer MJ, et al. Abdominal adiposity and coronary heart disease in women. JAMA 1998;280:1843-8.
22. Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Does waist circumference add to the predictive power of the body mass index for coronary risk? Obesity Research. 2001;9:585-95.
23. Toriano RP, Frongillo EA, Sobal J, Levitsky DA. The relationship between body weight and mortality: a quantitative analysis of combined information from existing studies. Int J Obes Relat Metab Disord 1996;20:63-75.
24. Hensrud DD. Dietary treatment and long-term weight loss and maintenance in type 2 diabetes. Obesity Research 2001;9:348S-353S.
25. Ditschuneit HH, Flechtner-Mors M. Value of structured meals for weight management: risk factors and long-term weight maintenance. Obesity Research 2001;9(suppl 4):284S-289S.
26. Ditschuneit HH, Flechtner-Mors M, Johnson TD, Adler G. Metabolic and weight-loss effects of a long-term dietary intervention in obese patients. Am J Clin Nutr 1999;69:198-204.
27. Flechtner-Mors M, Ditschuneit HH, Johnson TD, Suchard MA, Adler G. Metabolic and weight loss effects of long-term dietary interventions in obese patients: four-year results. Obesity Research 2000;8:399-402.
28. Freedman MR, King J, Kennedy E. Popular diets: a scientific review. Obesity Research 2001;9(suppl 1):1S-50S.
29. Finer N. Low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):290S-292S.
30. Saris W. Very-low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):292S-301S.
31. Pirozzo S, Summerbell C, Cameron C, Glasziou P. Advice on low-fat diets or obesity (Cochrane Review). In: The Cochrane Library, Issue 2, 2002. Oxford: Update Software.
32. Skov AR, Toubro S, Ronn B, Holm L, Astrup A. Randomized trial on protein vs.carbohydrate in ad libi-tum reduced diet for the treatment of obesity. Int J Obes 1999;23:528-36.
33. Bray GA. Role of physical activity and exercise in obesity. Up To Date (online 9.3). Retrieval date 11/5/2001. Up To Date, Inc. Wellesley, Mass, USA.
34. Thorogood M, Hillsdon M, Summerbell C. Changing behaviour. Clinical Evidence 2000;5:25-52.
35. Weintraub M, Sundaresan PR, Schuster B, Averbuch M, Stein EC, Byrne L. Long-term weight control study V. Clin Pharmacol Ther 1992;51:615-8.
36. Wadden T. Benefits of lifestyle modification in the phar-macologic treatment of obesity: a randomized trial. Arch Intern Med 2001;151:218-27.
37. Smith MB. Randomized placebo-controlled trial of long-term treatment with sibutramine in mild to moderate obesity. J Fam Pract 2001;50:505-12.
38. Sjostrom L, Rissanen A, Anderson T, Boldrin M, Golay A, Koppeschaan HPF, et al. Randomized placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet 1998;352:157-72.
39. O’meara S, Riemsma R, Shirran L, Mather L, ter Riet G. A rapid and systematic review of the clinical effectiveness and cost-effectiveness of orlistat in the management of obesity. Health Technology Assess 2001;5:18.
40. Physician’s Desk Reference. PGS. Montvale, NJ: Medical Economics; 2001;2809-13.
41. Balsiger BM, Kennedy FP, Abu-Lebdeh HS, Collazo-Clavell M, Jensen MD, O’Brien T, et al. Prospective evaluation of Roux-en Y gastric bypass as primary operation for medically complicated obesity. Mayo Clin Proc 2000;75:573-80.
42. Gastrointestinal Surgery for Severe Obesity. NIH Consensus Statement Online 1991; 25-27. Cited August 1, 2001;9(1):1-20.
43. Van Gemert WG, Van Dielen F, Soeters PB, Greve JW. Quality of life before and after weight-reducing surgery and cost-effectiveness analysis. In: Deitel M,†Cowan SM, eds. Update: Surgery for the Morbidly Obese Patient. Toronto: FD Communications; 2000;595-602.
44. Sugerman HJ. The epidemic of severe obesity: value of surgical treatment. Mayo Clin Proc 2000;75:559-572.
45. Ernst E. Acupuncture/acupressure for weight reduction? A systematic review. Wien Klin Wochenschr 1997;109:50-2.
46. Allison DB, Faith MS. Hypnosis as an adjunct to cogni-tive-behavioral psychotherapy for obesity: a meta-analyt-ic reappraisal. J Consult Clin Psychol 1995;54:513-5.
1. Dickerson L, Carek PJ. Drug therapy for obesity. Am Fam Physician 2000;51:2131-8.
2. Arterburn D, Hitchcock-Noel P. Obesity. Clinical Evidence 2001;5:512-9.
3. Mun EC, Blackburn GL, Matthews JB. Current status of medical and surgical therapy for obesity. Gastroenterology 2001;120:559-81.
4. Field AE, Coakley EH, Must A, Spandano JL, Laird N, Dietz WH, et al. Impact of overweight on the risk of developing common chronic diseases during a 10 year period. Arch Intern Med 2001;151:1581-5.
5. National Institutes of Health. Clinical guidelines on the identification, evaluation, and treatment of obesity in adults: The evidence report. Bethesda, Md: US Department of Health and Human Services; 1998.
6. Messier SP, Loeser RF, Mitchell MN, Valle G, Morgan TP, Rejeski WJ, et al. Exercise and weight loss in obese older adults with knee osteoarthritis: a preliminary study. J Am Geriatr Soc 2000;48:1052-72.
7. Shneerson J, Wright J. Lifestyle modification for obstructive sleep apnoea (Cochrane Review). In: The Cochrane Library, Issue 4, 2001. Oxford: Update Software.
8. Williamson DF, Thompson TS, Thun M, Flanders D, Pamuk E, Byers T. Intentional weight loss and mortality among overweight individuals with diabetes. Diabetes Care 2000;10:1499-1504.
9. Allison DB, Fontaine KR, Manson JE, Stevens J, Van Itallie TB. Annual deaths attributable to obesity in the United States. JAMA 1999;282:1530-8.
10. Yanovski J, Yanovski S. Recent advances in basic obesity research. JAMA 1999;282:1504-6.
11. Parsons TJ, Power C, Logan S, Summerbell CD. Childhood predictors of adult obesity: a systematic review. Int J Obes Relat Metab Disord 1999;8 (suppl):S1-107.
12. Ludwig DS, Peterson KE, Gortmaker SL. Relation between consumption of sugar-sweetened drinks and childhood obesity: a prospective, observational analysis. Lancet 2001;357:505-8.
13. Gillman MW, Rifas-Shiman SL, Camargo CA, Berkey CS, Frazier AL, Rocket HRH, et al. Risk of overweight among adolescents who were breastfed as infants. JAMA 2001;285:2451-7.
14. Robinson T. Reducing children’s television viewing to prevent obesity. JAMA 1999;282:1551-7.
15. Campbell K, Waters E, O’Meara S, Summerbell C. Interventions for preventing obesity in children (Cochrane Review). In: The Cochrane Library, Issue 5, 2001. Oxford: Update Software.
16. Sahota P, Rudolf MC, Dixey R, Hill AJ, Barth JH, Cade J. Randomized controlled trial of primary school based intervention to reduce risk factors for obesity. BMJ 2001;323:1029-32.
17. US Preventive Services Task Force. Screening for obesity. In: US Preventive Services Task Force: Guide to Clinical Preventative Services: Report of the US Preventive Services Task Force. 2nd ed. Baltimore, Md: Williams and Wilkins; 1995;219-29.
18. Flier JS, Foster DW. Eating Disorders: Obesity, anorexia nervosa, and bullemia nervosa. In: Wilson JD, Foster DW, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia, Pa: WB Saunders; 1998;1051-97.
19. McGee S. Evidence-based physical diagnosis. Philadelphia, Pa: WB Saunders; 2001.
20. Willett WC, Dietz WH, Colditz GA. Guidelines for healthy weight. N Engl J Med 1999;351:527-35.
21. Rexrode KM, Carey VJ, Hennekens CH, Walters EE, Colditz GA, Stampfer MJ, et al. Abdominal adiposity and coronary heart disease in women. JAMA 1998;280:1843-8.
22. Iwao S, Iwao N, Muller DC, Elahi D, Shimokata H, Andres R. Does waist circumference add to the predictive power of the body mass index for coronary risk? Obesity Research. 2001;9:585-95.
23. Toriano RP, Frongillo EA, Sobal J, Levitsky DA. The relationship between body weight and mortality: a quantitative analysis of combined information from existing studies. Int J Obes Relat Metab Disord 1996;20:63-75.
24. Hensrud DD. Dietary treatment and long-term weight loss and maintenance in type 2 diabetes. Obesity Research 2001;9:348S-353S.
25. Ditschuneit HH, Flechtner-Mors M. Value of structured meals for weight management: risk factors and long-term weight maintenance. Obesity Research 2001;9(suppl 4):284S-289S.
26. Ditschuneit HH, Flechtner-Mors M, Johnson TD, Adler G. Metabolic and weight-loss effects of a long-term dietary intervention in obese patients. Am J Clin Nutr 1999;69:198-204.
27. Flechtner-Mors M, Ditschuneit HH, Johnson TD, Suchard MA, Adler G. Metabolic and weight loss effects of long-term dietary interventions in obese patients: four-year results. Obesity Research 2000;8:399-402.
28. Freedman MR, King J, Kennedy E. Popular diets: a scientific review. Obesity Research 2001;9(suppl 1):1S-50S.
29. Finer N. Low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):290S-292S.
30. Saris W. Very-low-calorie diets and sustained weight loss. Obesity Research 2001;9(suppl 4):292S-301S.
31. Pirozzo S, Summerbell C, Cameron C, Glasziou P. Advice on low-fat diets or obesity (Cochrane Review). In: The Cochrane Library, Issue 2, 2002. Oxford: Update Software.
32. Skov AR, Toubro S, Ronn B, Holm L, Astrup A. Randomized trial on protein vs.carbohydrate in ad libi-tum reduced diet for the treatment of obesity. Int J Obes 1999;23:528-36.
33. Bray GA. Role of physical activity and exercise in obesity. Up To Date (online 9.3). Retrieval date 11/5/2001. Up To Date, Inc. Wellesley, Mass, USA.
34. Thorogood M, Hillsdon M, Summerbell C. Changing behaviour. Clinical Evidence 2000;5:25-52.
35. Weintraub M, Sundaresan PR, Schuster B, Averbuch M, Stein EC, Byrne L. Long-term weight control study V. Clin Pharmacol Ther 1992;51:615-8.
36. Wadden T. Benefits of lifestyle modification in the phar-macologic treatment of obesity: a randomized trial. Arch Intern Med 2001;151:218-27.
37. Smith MB. Randomized placebo-controlled trial of long-term treatment with sibutramine in mild to moderate obesity. J Fam Pract 2001;50:505-12.
38. Sjostrom L, Rissanen A, Anderson T, Boldrin M, Golay A, Koppeschaan HPF, et al. Randomized placebo-controlled trial of orlistat for weight loss and prevention of weight regain in obese patients. European Multicentre Orlistat Study Group. Lancet 1998;352:157-72.
39. O’meara S, Riemsma R, Shirran L, Mather L, ter Riet G. A rapid and systematic review of the clinical effectiveness and cost-effectiveness of orlistat in the management of obesity. Health Technology Assess 2001;5:18.
40. Physician’s Desk Reference. PGS. Montvale, NJ: Medical Economics; 2001;2809-13.
41. Balsiger BM, Kennedy FP, Abu-Lebdeh HS, Collazo-Clavell M, Jensen MD, O’Brien T, et al. Prospective evaluation of Roux-en Y gastric bypass as primary operation for medically complicated obesity. Mayo Clin Proc 2000;75:573-80.
42. Gastrointestinal Surgery for Severe Obesity. NIH Consensus Statement Online 1991; 25-27. Cited August 1, 2001;9(1):1-20.
43. Van Gemert WG, Van Dielen F, Soeters PB, Greve JW. Quality of life before and after weight-reducing surgery and cost-effectiveness analysis. In: Deitel M,†Cowan SM, eds. Update: Surgery for the Morbidly Obese Patient. Toronto: FD Communications; 2000;595-602.
44. Sugerman HJ. The epidemic of severe obesity: value of surgical treatment. Mayo Clin Proc 2000;75:559-572.
45. Ernst E. Acupuncture/acupressure for weight reduction? A systematic review. Wien Klin Wochenschr 1997;109:50-2.
46. Allison DB, Faith MS. Hypnosis as an adjunct to cogni-tive-behavioral psychotherapy for obesity: a meta-analyt-ic reappraisal. J Consult Clin Psychol 1995;54:513-5.
Evaluation and treatment of the patient with allergic rhinitis
- Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”).
- Skin prick testing can detect IgE antibodies in patients with reliable histories of exposure to allergens.
- Intranasal corticosteroids are superior to other medications in achieving desired clinical outcomes, including quality of life.
- For some cases of allergic rhinitis, subcutaneous immunotherapy can achieve clinical remission for up to 3 years after cessation of therapy.
While allergic rhinitis is merely a nuisance to most people afflicted by it, the condition can lead to complications if it is severe or exists undetected for too long. In this article, I review the most reliable means of diagnosing allergic rhinitis, and outline a recommended approach to treatment.
Prevalence and pathophysiology
An estimated 20 to 40 million Americans are affected by allergic rhinitis. The actual prevalence of the condition is difficult to discern as many sufferers self-medicate without seeking medical care. One survey stated that up to 92% of patients had self-medicated prior to seeking medical care.1 Even when accounting for self-treatment, allergic rhinitis is the most commonly encountered form of chronic rhinitis, representing about 3% of all primary care office visits.2,3 Direct and indirect clinical costs run between $1.2 and $5.3 billion per year.4-6 Although the disease can develop in persons of any age, in 80% of cases symptoms will develop before the patient is 20 years old.5 Symptoms often wane as a patient grows older, and it is uncommon for persons older than 65 to experience new onset of allergic rhinitis.3,7
Allergic rhinitis stems from a type I hypersensitivity reaction.4 During an initial sensitization phase, the immune system identifies an allergen as foreign and generates specific antibodies to act against that allergen. Atopic patients exhibit an exaggerated response, generating high levels of Type 2 T-helper (Th2) cells and, subsequently, IgE antibodies.8 On reexposure to the allergen, specific IgE antibodies bound to mast cells form cross-links resulting in mast cell degranulation and the release of histamine and other chemical mediators. The patient then immediately develops such allergy symptoms as itching, sneezing, and rhinorrhea. A cellular inflammatory response, chiefly involving eosinophils, monocytes, and basophils, characterizes the secondary phase of the allergic reaction. Nasal congestion tends to dominate this later response phase.
Seasonal allergic rhinitis is usually triggered by pollens or molds. Perennial allergic rhinitis, triggered by dust mites, molds, cockroach or animal allergens, is defined as occurring 9 months out of the year.9
Clinical evaluation
The diagnosis of allergic rhinitis is usually made on the basis of the patient’s history and the results of your physical examination. In addition to classic symptoms of nasal congestion, itchy nose, sneezing, rhinorrhea, or itchy, watery eyes, patients may also complain of chronic cough, dry scratchy throat, otalgia, or recurrent sinusitis.4 Other important historical considerations include a family history of allergic rhinitis, a history of other atopic disease, previous treatment experiences, and suspected triggers.5
Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”). Chronic nasal congestion may also precipitate darkening of the skin under the eyes or “allergic shiners.”2,6 Concurrent conjunctivitis is common. Polyps, seen on direct nasal examination, may occur both in allergic and non-allergic patients.
No studies have evaluated the accuracy of the history or physical examination in confirming the diagnosis of allergic rhinitis. The differential diagnosis is extensive and includes infectious rhinitis, non-allergic rhinitis with eosinophilia syndrome (NARES), occupational rhinitis, mechanical obstruction, vasomotor rhinitis, drug-induced rhinitis, and nasal polyps.5
Diagnostic tests
Published guidelines from the American Academy of Asthma, Allergy and Immunology, as well as other expert panels, recommend confirmatory testing when allergic rhinitis is clinically suspected.2,5,10 There is no evidence to support the superiority of this recommendation over an empiric trial of medication, and most primary care physicians choose to treat empirically based upon the history and physical examination.
Although further testing should be done when the diagnosis is unclear, be aware that there is uncertainty associated with allergy testing. Because an individual may become sensitized to an allergen without exhibiting symptoms of allergic rhinitis, there is no clearly defined reference standard for the confirmation of allergic rhinitis.11 Likewise, a history of sensitivity is not always followed by expected IgE test results. Challenge methods developed for studies of airborne allergens are used as reference standards in the evaluation of clinical tests.12
Diagnostic tests include skin prick testing, intradermal testing, and in vitro blood tests. Nasal challenge testing, nasal smears, sinus transillumination, and nasopharyngoscopy are nonspecific tests. They are not recommended for routine evaluation but may be useful in selected cases when allergen-specific tests have failed to clarify the cause of the rhinitis. An expert panel has stated that no studies address the cost-effectiveness of any of these methods.2
Skin prick testing (SPT) is considered the most convenient and least expensive screening test. SPT can detect IgE antibodies in patients with reliable exposure histories.13 Sensitivity and specificity are difficult to determine, for a number of reasons. First, as previously mentioned, there is no clearly defined reference standard.11 Second, only 5 allergen extracts have been standardized for defined quantities known to induce biologic activity. Standardized extracts in the United States include ragweed pollen, cat dander, house dust mites, Hymenoptera venoms, and some grasses. All other extracts are local or regional preparations, and skin tests with nonstandard extracts are not necessarily reproducible.11 Third, even with a single individual, there can be wide variation in skin reaction to the same reagent, depending on the device used.14 As a result, correlation between SPT and inhalation challenges vary from 60% to 90%.13
Intradermal skin tests (IDST) are usually done when SPT yields a negative result despite a history compatible with allergic rhinitis.13 The primary advantage of IDST is sensitivity afforded by a fixed concentration of allergen. Because of this sensitivity, not all reactions are clinically relevant.13 In fact, IDST is often used as a reference standard in studies of the accuracy of SPT and in vitro tests.
Several in vitro assays of specific IgE antibodies are available. They are all modeled after the original radioallergosorbent tests (RAST); the term “RAST” is often used interchangeably with any type of in vitro blood test.13 IgE antibody tests have a high false-positive rate, meaning the test is positive in patients without allergy symptoms. RAST tests are less sensitive than SPT, with a mean sensitivity of 75% and a range of approximately 50% to 95%.13
The 3 primary diagnostic tests for allergic rhinitis are usually compared with each other and not to a recognized standard. Table 1 summarizes data from a study that compared all 3 tests with subjects who were placed in a small room with 2 cats and their bed.12 While this is one of very few studies that contrasts all 3 tests to a reasonable reference standard, the findings cannot necessarily be extrapolated to other airborne allergens.
In the hope of limiting referrals to allergists for testing, and reducing the uncertainty in making a diagnosis, one study looked at the RAST response to 19 allergens. The authors found that of all the patients who responded to any allergen, 95% exhibited responses specifically to grass pollen, dust mites, or cat dander. They went on to conclude that 96.3% of patients with allergic disease could be correctly identified with a combination of a standardized history (available in the study text), a total serum IgE of greater than 40 U/mL, and in vitro tests for cat dander, dust mites, and grass pollen.15
TABLE 1
Accuracy of diagnostic tests for diagnosis of cat allergy13
| Test | Sensitivity | Specificity | PV+ | PV- | LR+ | LR- |
|---|---|---|---|---|---|---|
| Skin prick test | 79.2 | 90.6 | 92.6 | 74.3 | 8.4 | 0.2 |
| Intradermal test | 60.0 | 31.0 | 23.1 | 69.2 | 0.9 | 1.3 |
| RAST | 69.2 | 100 | 100 | 72.7 | 69.2 | 0.3 |
| Note: Results are based upon any upper or lower symptoms when exposed to cat challenge. Intradermal test done if negative skin prick test. LR+ = positive likelihood ratio, LR- = negative likelihood ratio, PV+ = positive predictive value, PV- = negative predictive value. | ||||||
Treatment
Untreated allergic rhinitis can have a significant impact on quality of life. Patients are bothered by nose blowing, disrupted sleep, fatigue, and decreased concentration.1 In one 1996 survey, 32 % of patients said that allergy attacks embarrassed them or interfered with their quality of life.16 As a result, most patient-oriented studies on treatment evaluate the impact on health-related quality of life.17
The initial form of treatment is usually avoidance of the allergen, although this can be difficult. For animal allergens, washing pets and using high-efficiency particulate air (HEPA) filters have been shown to temporarily reduce the volume of airborne allergens but not to improve patient-oriented outcomes.18,19 Removing the pet from the home is the only sure remedy.18 More studies are needed to evaluate the benefit of multiple home treatments to reduce exposure to cockroach and fungal allergens.18 A systematic review of several studies showed that maternal antigen avoidance during lactation reduced the incidence of atopic dermatitis in at-risk infants.20 A meta-analysis of measures to avoid house-dust mites showed no clear benefit for patients with asthma10 It is unclear if these findings can be extrapolated to other atopic conditions such as allergic rhinitis.
Intranasal corticosteroids. Intranasal corticosteroids are the most effective medication in the treatment of allergic rhinitis. Available preparations in the US include beclomethasone diproprionate, budesonide, funisolide, fluticasone propionate, mometa-sone furoate, and triamcinalone acetonide. A meta-analysis identified 16 randomized controlled trials (RCTs) that compared antihistamines with intranasal corticosteroids in a total of 2767 patients. Intranasal corticosteroids provided significantly greater relief from nasal discharge, sneezing, pruritis, and postnasal drip. There was no statistically significant difference between the 2 in reduction of eye symptoms.21
Although this review did not address quality of life, other studies have shown that both triamcinolone acetonide and fluticasone propionate are superior to loratadine in improving quality of life.22,23 Few studies provide any guidance in choosing one intranasal steroid over another. Generally, they are of equal efficacy in patient-oriented outcomes.24,25 Although intranasal corticosteroids are considered daily or “maintenance” medications, a single small RCT of 26 patients showed that fluticasone propionate improved quality of life and reduced symptoms compared with placebo when used on an as-needed basis over a 4-week period.26 More studies are needed to confirm this preliminary finding, though.
Antihistamines. Although not as effective as intranasal steroids, antihistamines do reduce symptoms of rhinorrhea, sneezing, and itching.27 First-gen-eration antihistamines (diphenhydramine, chlorpheniramine, etc.) are lipophilic and cross the blood-brain barrier, resulting in varying degrees of anticholinergic side effects. Placebo-controlled studies have confirmed that these agents cause psychomotor retardation, sleepiness, and decreased work production.5,28 Specifically they seem to affect attention, memory, and vigilance. These symptoms may persist even after an overnight period of sleep.28,29 Second-generation antihistamines (fexofenadine, loratadine, etc.) do not penetrate the brain as well and are less likely to cause central nervous system effects.
However, a recent RCT involving 63 elementary school students challenges findings from previous studies. Children who received diphenhydramine, 25 mg twice daily, performed no differently on computerized reaction-time tests or multiple-choice learning tests than did children who received placebo or loratadine, 10 mg daily.30 Another RCT involving 845 patients from ages 12 to 65 years evaluated quality of life as well as work and school performance of patients who received fexofenadine or placebo. While quality-of-life scores and work performance improved significantly with fexofenadine, there was no significant difference between the groups in school performance.31 Direct comparisons of antihistamines are rare and the results are conflicting. There are no data to show that one of the first-generation antihistamines is superior to the others. Similarly, second-generation drugs are no more effective than the older medications; they only have fewer side effects. Among the second-generation antihistamines, fexofenadine and cetirizine appear to be more effective then loratadine.29
Decongestants. Systemic and topical decongestants relieve the congestion that accompanies the secondary phase of an allergic reaction.4 They have limited effects on other allergic symptoms and, as a result, are often used in combination with antihistamines.32 When used for more than 10 days, topical decongestants (oxymetazoline, xylometazoline) are associated with rebound congestion (rhinitis medicamentosa).33
Leukotriene receptor antagonists. Although not approved by the FDA for treatment of allergic rhinitis, the leukotriene receptor antagonist montelukast was shown in a randomized double-blinded trial to be as effective as loratadine in relieving symptoms. There was minimal additional benefit in using the medications concomitantly.34
Cromolyn sodium. Cromolyn sodium has been shown to prevent the onset of allergic rhinitis symptoms in multiple placebo-controlled trials.35 It is extremely safe but requires regular use and is not as effective as other medications for acute symptoms. Direct comparison studies have shown that cromolyn is not as effective as intranasal corticosteroids.35,36
Immunotherapy. Subcutaneous immunotherapy (SIT) is recommended by all guidelines for patients who fail to respond to pharmacotherapy and allergen avoidance.2,5,10,27 It is recommended in particular for allergic rhinitis secondary to ragweed, grasses, molds, and dust mites. Immunotherapy induces the creation of protective IgG and inhibits the inflammatory response to allergens.27 SIT requires specific allergen confirmation with either a skin test or in vitro assay. Preparation of SIT doses should be done by a practitioner well trained in mixing and diluting extracts.5,37 Forty-three placebo-controlled, double-blind studies have evaluated the efficacy of SIT for 12 different allergens since 1980.10 Thirty-two trials showed clinical efficacy, which can be long lasting. A study of patients treated for 3 to 4 years with immunotherapy for grass pollen allergy showed continued clinical remission for at least 3 years after treatment was stopped.38
Herbal therapies. Alternative approaches to the treatment of allergic rhinitis warrant further investigation. Herbal medications, such as licorice, gingko, and ginseng, are currently used to treat allergic rhinitis, although there are no large studies to confirm their effectiveness.39
Probiotics. Epidemiologic studies suggest that the increase in atopic disease may be related to a clean environment and widespread use of antibiotics in Western countries. The environment may deprive fetal and infant immune systems of bacterial antigens that stimulate type 1 T-helper (Th1) cells.8 In light of this theory, Finnish researchers randomly assigned 159 pregnant women with a family history of atopy to receive capsules of Lactobacillus GG (a potentially beneficial bacteria or “probiotic”) or placebo, beginning 2 to 4 weeks prior to delivery and continuing 6 months postpartum. Infants were followed for 2 years. Frequency of atopic dermatitis was reduced by 50% among those infants whose mothers received Lactobacillus.40 Further study of this association in allergic rhinitis would be beneficial.
Treatment recommendations.Table 2 summarizes treatment-related evidence in the management of allergic rhinitis, and the Figure illustrates a proposed treatment algorithm. Another algorithm by the European Academy of Allergy and Clinical Immunology recommends initial therapy with oral or nasal antihistamines for mild disease, nasal corticosteroids for moderate disease, and both for severe disease.10 This was a consensus opinion. Of the 2 most commonly used medications for allergy, nasal steroids are favored over antihistamines for overall safety, tolerability, effectiveness, and simplicity in all cases. In one study of 61 adults, patients were randomized to receive either a nasal steroid or an antihistamine as initial therapy, with the other agent reserved as “back-up.” After 6 weeks, 86% of patients started on an antihistamine had added their steroid back-up, while 51% of the group started on a steroid remained on that agent alone.41 Starting all patients on both an antihistamine and a nasal steroid is inappropriate.
TABLE 2
Evidence to support treatment recommendations
| Strength of recommendation | Treatment | Comment |
|---|---|---|
| A | Immunotherapy | Can have long lasting clinical benefit. |
| A | Intranasal Consistently | superior to antihistamines corticosteroids in head to head trials. Not clear if all steroids are equally effective. |
| A | Antihistamines | Effective, but inferior to intranasal steroids in most clinical outcomes. |
| A | Cromolyn sodium | Intranasal steroids superior in all clinical outcomes. |
| B | Decongestants | Less effective than antihistamines in direct comparisons, many trials involve combination products. |
| D | Probiotics | Larger trials needed, limited evidence. |
| D | Herbal medications (licorice, gingko, ginseng) | Limited evidence. |
FIGUREA guide to evaluation and treatment of allergic rhinitis
Prognosis
The long-term prognosis for allergic rhinitis is excellent. For most patients, the illness is primarily a nuisance with no significant morbidity. However, for patients whose rhinitis is moderate to severe and poorly controlled, there can be significant complications. These complications include asthma, sinusitis, otitis media, nasal polyposis, respiratory infections, and orthodontic malocclusions.42 In one study of 605 children with allergic rhinitis, 21% had chronic otitis media with effusion (OME). Conversely, in another study of 259 children with OME, 50% had allergic rhinitis.43 Even among patients without asthma, 20% to 30% will have bronchial hyper-responsiveness. Additionally, poorly controlled allergic rhinitis can contribute to sleep loss, daytime fatigue, and learning impairment.44
Potential complications related to long-term treatment in children remain controversial. In a 1998 study of intranasal beclomethasone, children receiving the study medication grew an average of only 5 centimeters (cm) in 1 year, compared with an average of 5.9 cm in the placebo group.45 However, a similar study done 2 years later with intranasal mometa-sone showed no evidence of growth suppression.45 Further studies are needed before the true impact of intranasal steroids on children can be determined.
· Acknowledgments ·
The opinions and assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the U.S. Army or the U.S. Department of Defense. The author wishes to thank Kathleen Conner, JD, for her assistance in the preparation of the manuscript.
1. Meltzer EO. The prevalence and medical and economic impact of allergic rhinitis in the United States. J Allergy Clin Immunol 1997;99:S805-28.
2. Fornadley JA, et al. Allergic rhinitis: Clinical practice guideline. Otolaryngol Head Neck Surg 1996;115(1):115-122.
3. Wagner W, Kavuru M. Diagnosis and Management of Rhinitis. 1st Ed. Professional Communications, Inc. 1996. Available at www.medscape.com. Accessed Sept. 6, 2001.
4. Hadley JA. Evaluation and management of allergic rhinitis. Med Clin North Am 1999;83(1):13-25.
5. Dykewicz MS, Fineman S, et al. Diagnosis and management of rhinitis: complete guidelines of the joint task force on practice parameters in allergy, asthma and immunology. Ann Allergy Asthma Immunol 1998;81:478-518.
6. Weiss KB. The health economics of asthma and rhinitis. I. Assessing the economic impact. J Allergy Clin Immunol 2001;107:3-8.
7. Gentile DA, Friday GA, Skoner DP. Management of allergic rhinitis: antihistamines and decongestants. Immunol Allergy Clin North Am 2000;20(2):355-368.
8. Kay AB. Allergy and allergic diseases. N Engl J Med 2001;344:30-36.
9. Skoner DP. Allergic rhinitis: definition, epidemiology, pathophysiology, detection and diagnosis. J Allergy Clin Immunol 2001;108:S2-8.
10. van Cauwenberge P, et al. Consensus statement on the treatment of allergic rhinitis. Allergy 2000;55:116-134.
11. Ownby DR. Skin tests in comparison to other diagnostic methods. Immunol Allergy Clin North Am 2001;21(2):355-367.
12. Wood RA, Phipatanakul W, Hamilton RG, Eggleston PA. A comparison of skin prick tests, intradermal skin tests, and RASTs in the diagnosis of cat allergy. J Allergy Clin Immunol 1999;103:773-779.
13. Bernstein IL, et al. Practice parameters for allergy diagnostic testing. Ann Allergy Asthma Immunol 1995;75:543-625.
14. Nelson HS, Lahr J, Buchmeier A, McCormick D. Evaluation of devices for skin prick testing. J Allergy Clin Immunol 1998;101:153-156.
15. Merrett TG, Pantin CF, Dimond AH, Merrett J. Screening for IgE-mediated allergy. Allergy 1980 Sep;35(6):491-501.
16. Ferguson BJ. Cost effective pharmacotherapy for allergic rhinitis. Otolaryngol Clin North Am 1998;31(1):91-110.
17. Thompson AK, Juniper E, Meltzer EO. Quality of life in patients with allergic rhinitis. Ann Allergy Asthma Immunol 2000;85:338-348.
18. Eggleston PA, Bush RK. Environmental allergen avoidance: an overview. J Allergy Clin Immunol 2001;107:S403-405.
19. Wood RA, Johnson EF, van Atta ML, et al. A placebo-controlled trial of a HEPA air filter in the treatment of cat allergy. Am J Resp CC Med 1998;158:115-120.
20. Kramer MS. Maternal antigen avoidance during lactation for preventing atopic disease in infants of women at high risk (Cochrane Review). In: The Cochrane Library, Issue 3, 2001. Oxford: Update software.
21. Weiner JM, Abramson MJ, Puy RM. Intranasal corticosteroids versus H1 receptor antagonists in allergic rhinitis: systematic review of randomised controlled trials. BMJ 1998;317:1624-1629.
22. Condemi J, Schulz R, Lim J. Triamcinolone acetonide aqueous nasal spray versus loratadine in seasonal allergic rhinitis: efficacy and quality of life. Ann Allergy Asthma Immunol 2000;84:533-538.
23. Ratner PH, van Bavel JH, Martin BG, et al. A comparison of the efficacy of fluticasone propionate aqueous nasal spray and loratadine, alone and in combination, for the treatment of seasonal allergic rhinitis. J Fam Pract 1998;47:118-125.
24. Small P, et al. Comparison of triamcinalone acetonide nasal aerosol spray and fluticasone propionate aqueous solution spray in treatment of spring allergic rhinitis. J Allergy Clin Immunol 1997;100(5):592-5.
25. Day J, Carrillo T. Comparison of the efficacy of budesonide and fluticasone propionate aqueous nasal spray for once daily treatment of perennial allergic rhinitis. J Allergy Clin Immunol 1998;102:902-908.
26. Jen A, Baroody F, de Tineo M, et al. As-needed use of fluticasone propionate nasal spray reduces symptoms of seasonal allergic rhinitis. J Allergy Clin Immunol 2000;105:732-738
27. Naclerio RM. Allergic rhinitis. N Engl J Med 1991;325:860-869.
28. Kay GG. The effects of antihistamines on cognition and performance. J Allergy Clin Immunol 2000;105:S622-627.
29. Abramowicz M. (Ed.) Newer antihistamines. Med Letter 2001;43:35.-
30. Bender BG, McCormick DR, Milgrom H. Children’s school performance is not impaired by short-term administration of diphenhydramine or loratadine. J Peds 2001;138:656-660.
31. Meltzer EO. Once-daily fexofenadine HCl improves quality of life and reduces work and activity impairment in patients with seasonal allergic rhinitis. Ann Allergy Asthma Immunol 1999;83:311-317.
32. Sussman GL, Mason J, Compton D, Stewart J, Ricard N. The efficacy and safety of fexofenadine HCL and pseudophedrine, alone and in combination, in seasonal allergic rhinitis. J Allergy Clin Immunol 1999;104:100-106.
33. Graf P, Enerdal J, Hallen H. Ten days’use of oxymetazoline nasal spray with benzalkonioum chloride in patients with vasomotor rhinitis. Arch Otolaryngol Head and Neck Surg 1999;125:1128-1132.
34. Meltzer EO, Malmstrom K, Lu S, et al. Concomitant montelukast and loratadine as treatment for seasonal allergic rhinitis: A randomized placebo-controlled clinical trial. J Allergy Clin Immunol 2000;105:917-922.
35. LaForce C. Use of nasal steroids in managing allergic rhinitis. J Allergy Clin Immunol 1999;103:S388-394.
36. Williams PV. Treatment of rhinitis: corticosteroids and cromolyn sodium. Immunol Allergy Clin North Am 2000;20:369-381.
37. Craig T, Sawyer AM, Fornadley JA. Use of immunotherapy in a primary care office. Am Fam Phys 1998;57:1888-1894.
38. Durham SR, Walker SM, Varga EM, et al. Long-term efficacy of grass-pollen immunotherapy. N Engl J Med 1999;341:468-475.
39. Bielory L, Lupoli K. Herbal interventions in asthma and allergy. J Asthma 1999;36(1):1-65.
40. Kalliomaki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 2001;357:1076-1079.
41. Juniper EF, Guyatt GJ, Ferrie PJ, Griffith LE. First-line treatment of seasonal (ragweed) rhinoconjunctivitis. Can Med Assoc J 1997;156:1123-1131.
42. Spector SL. Overview of comorbid associations of allergic rhinitis. J Allergy Clin Immunol 1997;99:S773-780.
43. Skoner DP. Complications of allergic rhinitis. J Allergy Clin Immunol 2000;105:S605-609.
44. Settipane RA. Complications of allergic rhinitis. Allergy Asthma Proc 1999;20(4):209-213.
45. Buck ML. Intranasal steroids for Children with Allergic Rhinitis. Pediatric Pharmacotherapy 2000;(7)5:1-12.
- Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”).
- Skin prick testing can detect IgE antibodies in patients with reliable histories of exposure to allergens.
- Intranasal corticosteroids are superior to other medications in achieving desired clinical outcomes, including quality of life.
- For some cases of allergic rhinitis, subcutaneous immunotherapy can achieve clinical remission for up to 3 years after cessation of therapy.
While allergic rhinitis is merely a nuisance to most people afflicted by it, the condition can lead to complications if it is severe or exists undetected for too long. In this article, I review the most reliable means of diagnosing allergic rhinitis, and outline a recommended approach to treatment.
Prevalence and pathophysiology
An estimated 20 to 40 million Americans are affected by allergic rhinitis. The actual prevalence of the condition is difficult to discern as many sufferers self-medicate without seeking medical care. One survey stated that up to 92% of patients had self-medicated prior to seeking medical care.1 Even when accounting for self-treatment, allergic rhinitis is the most commonly encountered form of chronic rhinitis, representing about 3% of all primary care office visits.2,3 Direct and indirect clinical costs run between $1.2 and $5.3 billion per year.4-6 Although the disease can develop in persons of any age, in 80% of cases symptoms will develop before the patient is 20 years old.5 Symptoms often wane as a patient grows older, and it is uncommon for persons older than 65 to experience new onset of allergic rhinitis.3,7
Allergic rhinitis stems from a type I hypersensitivity reaction.4 During an initial sensitization phase, the immune system identifies an allergen as foreign and generates specific antibodies to act against that allergen. Atopic patients exhibit an exaggerated response, generating high levels of Type 2 T-helper (Th2) cells and, subsequently, IgE antibodies.8 On reexposure to the allergen, specific IgE antibodies bound to mast cells form cross-links resulting in mast cell degranulation and the release of histamine and other chemical mediators. The patient then immediately develops such allergy symptoms as itching, sneezing, and rhinorrhea. A cellular inflammatory response, chiefly involving eosinophils, monocytes, and basophils, characterizes the secondary phase of the allergic reaction. Nasal congestion tends to dominate this later response phase.
Seasonal allergic rhinitis is usually triggered by pollens or molds. Perennial allergic rhinitis, triggered by dust mites, molds, cockroach or animal allergens, is defined as occurring 9 months out of the year.9
Clinical evaluation
The diagnosis of allergic rhinitis is usually made on the basis of the patient’s history and the results of your physical examination. In addition to classic symptoms of nasal congestion, itchy nose, sneezing, rhinorrhea, or itchy, watery eyes, patients may also complain of chronic cough, dry scratchy throat, otalgia, or recurrent sinusitis.4 Other important historical considerations include a family history of allergic rhinitis, a history of other atopic disease, previous treatment experiences, and suspected triggers.5
Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”). Chronic nasal congestion may also precipitate darkening of the skin under the eyes or “allergic shiners.”2,6 Concurrent conjunctivitis is common. Polyps, seen on direct nasal examination, may occur both in allergic and non-allergic patients.
No studies have evaluated the accuracy of the history or physical examination in confirming the diagnosis of allergic rhinitis. The differential diagnosis is extensive and includes infectious rhinitis, non-allergic rhinitis with eosinophilia syndrome (NARES), occupational rhinitis, mechanical obstruction, vasomotor rhinitis, drug-induced rhinitis, and nasal polyps.5
Diagnostic tests
Published guidelines from the American Academy of Asthma, Allergy and Immunology, as well as other expert panels, recommend confirmatory testing when allergic rhinitis is clinically suspected.2,5,10 There is no evidence to support the superiority of this recommendation over an empiric trial of medication, and most primary care physicians choose to treat empirically based upon the history and physical examination.
Although further testing should be done when the diagnosis is unclear, be aware that there is uncertainty associated with allergy testing. Because an individual may become sensitized to an allergen without exhibiting symptoms of allergic rhinitis, there is no clearly defined reference standard for the confirmation of allergic rhinitis.11 Likewise, a history of sensitivity is not always followed by expected IgE test results. Challenge methods developed for studies of airborne allergens are used as reference standards in the evaluation of clinical tests.12
Diagnostic tests include skin prick testing, intradermal testing, and in vitro blood tests. Nasal challenge testing, nasal smears, sinus transillumination, and nasopharyngoscopy are nonspecific tests. They are not recommended for routine evaluation but may be useful in selected cases when allergen-specific tests have failed to clarify the cause of the rhinitis. An expert panel has stated that no studies address the cost-effectiveness of any of these methods.2
Skin prick testing (SPT) is considered the most convenient and least expensive screening test. SPT can detect IgE antibodies in patients with reliable exposure histories.13 Sensitivity and specificity are difficult to determine, for a number of reasons. First, as previously mentioned, there is no clearly defined reference standard.11 Second, only 5 allergen extracts have been standardized for defined quantities known to induce biologic activity. Standardized extracts in the United States include ragweed pollen, cat dander, house dust mites, Hymenoptera venoms, and some grasses. All other extracts are local or regional preparations, and skin tests with nonstandard extracts are not necessarily reproducible.11 Third, even with a single individual, there can be wide variation in skin reaction to the same reagent, depending on the device used.14 As a result, correlation between SPT and inhalation challenges vary from 60% to 90%.13
Intradermal skin tests (IDST) are usually done when SPT yields a negative result despite a history compatible with allergic rhinitis.13 The primary advantage of IDST is sensitivity afforded by a fixed concentration of allergen. Because of this sensitivity, not all reactions are clinically relevant.13 In fact, IDST is often used as a reference standard in studies of the accuracy of SPT and in vitro tests.
Several in vitro assays of specific IgE antibodies are available. They are all modeled after the original radioallergosorbent tests (RAST); the term “RAST” is often used interchangeably with any type of in vitro blood test.13 IgE antibody tests have a high false-positive rate, meaning the test is positive in patients without allergy symptoms. RAST tests are less sensitive than SPT, with a mean sensitivity of 75% and a range of approximately 50% to 95%.13
The 3 primary diagnostic tests for allergic rhinitis are usually compared with each other and not to a recognized standard. Table 1 summarizes data from a study that compared all 3 tests with subjects who were placed in a small room with 2 cats and their bed.12 While this is one of very few studies that contrasts all 3 tests to a reasonable reference standard, the findings cannot necessarily be extrapolated to other airborne allergens.
In the hope of limiting referrals to allergists for testing, and reducing the uncertainty in making a diagnosis, one study looked at the RAST response to 19 allergens. The authors found that of all the patients who responded to any allergen, 95% exhibited responses specifically to grass pollen, dust mites, or cat dander. They went on to conclude that 96.3% of patients with allergic disease could be correctly identified with a combination of a standardized history (available in the study text), a total serum IgE of greater than 40 U/mL, and in vitro tests for cat dander, dust mites, and grass pollen.15
TABLE 1
Accuracy of diagnostic tests for diagnosis of cat allergy13
| Test | Sensitivity | Specificity | PV+ | PV- | LR+ | LR- |
|---|---|---|---|---|---|---|
| Skin prick test | 79.2 | 90.6 | 92.6 | 74.3 | 8.4 | 0.2 |
| Intradermal test | 60.0 | 31.0 | 23.1 | 69.2 | 0.9 | 1.3 |
| RAST | 69.2 | 100 | 100 | 72.7 | 69.2 | 0.3 |
| Note: Results are based upon any upper or lower symptoms when exposed to cat challenge. Intradermal test done if negative skin prick test. LR+ = positive likelihood ratio, LR- = negative likelihood ratio, PV+ = positive predictive value, PV- = negative predictive value. | ||||||
Treatment
Untreated allergic rhinitis can have a significant impact on quality of life. Patients are bothered by nose blowing, disrupted sleep, fatigue, and decreased concentration.1 In one 1996 survey, 32 % of patients said that allergy attacks embarrassed them or interfered with their quality of life.16 As a result, most patient-oriented studies on treatment evaluate the impact on health-related quality of life.17
The initial form of treatment is usually avoidance of the allergen, although this can be difficult. For animal allergens, washing pets and using high-efficiency particulate air (HEPA) filters have been shown to temporarily reduce the volume of airborne allergens but not to improve patient-oriented outcomes.18,19 Removing the pet from the home is the only sure remedy.18 More studies are needed to evaluate the benefit of multiple home treatments to reduce exposure to cockroach and fungal allergens.18 A systematic review of several studies showed that maternal antigen avoidance during lactation reduced the incidence of atopic dermatitis in at-risk infants.20 A meta-analysis of measures to avoid house-dust mites showed no clear benefit for patients with asthma10 It is unclear if these findings can be extrapolated to other atopic conditions such as allergic rhinitis.
Intranasal corticosteroids. Intranasal corticosteroids are the most effective medication in the treatment of allergic rhinitis. Available preparations in the US include beclomethasone diproprionate, budesonide, funisolide, fluticasone propionate, mometa-sone furoate, and triamcinalone acetonide. A meta-analysis identified 16 randomized controlled trials (RCTs) that compared antihistamines with intranasal corticosteroids in a total of 2767 patients. Intranasal corticosteroids provided significantly greater relief from nasal discharge, sneezing, pruritis, and postnasal drip. There was no statistically significant difference between the 2 in reduction of eye symptoms.21
Although this review did not address quality of life, other studies have shown that both triamcinolone acetonide and fluticasone propionate are superior to loratadine in improving quality of life.22,23 Few studies provide any guidance in choosing one intranasal steroid over another. Generally, they are of equal efficacy in patient-oriented outcomes.24,25 Although intranasal corticosteroids are considered daily or “maintenance” medications, a single small RCT of 26 patients showed that fluticasone propionate improved quality of life and reduced symptoms compared with placebo when used on an as-needed basis over a 4-week period.26 More studies are needed to confirm this preliminary finding, though.
Antihistamines. Although not as effective as intranasal steroids, antihistamines do reduce symptoms of rhinorrhea, sneezing, and itching.27 First-gen-eration antihistamines (diphenhydramine, chlorpheniramine, etc.) are lipophilic and cross the blood-brain barrier, resulting in varying degrees of anticholinergic side effects. Placebo-controlled studies have confirmed that these agents cause psychomotor retardation, sleepiness, and decreased work production.5,28 Specifically they seem to affect attention, memory, and vigilance. These symptoms may persist even after an overnight period of sleep.28,29 Second-generation antihistamines (fexofenadine, loratadine, etc.) do not penetrate the brain as well and are less likely to cause central nervous system effects.
However, a recent RCT involving 63 elementary school students challenges findings from previous studies. Children who received diphenhydramine, 25 mg twice daily, performed no differently on computerized reaction-time tests or multiple-choice learning tests than did children who received placebo or loratadine, 10 mg daily.30 Another RCT involving 845 patients from ages 12 to 65 years evaluated quality of life as well as work and school performance of patients who received fexofenadine or placebo. While quality-of-life scores and work performance improved significantly with fexofenadine, there was no significant difference between the groups in school performance.31 Direct comparisons of antihistamines are rare and the results are conflicting. There are no data to show that one of the first-generation antihistamines is superior to the others. Similarly, second-generation drugs are no more effective than the older medications; they only have fewer side effects. Among the second-generation antihistamines, fexofenadine and cetirizine appear to be more effective then loratadine.29
Decongestants. Systemic and topical decongestants relieve the congestion that accompanies the secondary phase of an allergic reaction.4 They have limited effects on other allergic symptoms and, as a result, are often used in combination with antihistamines.32 When used for more than 10 days, topical decongestants (oxymetazoline, xylometazoline) are associated with rebound congestion (rhinitis medicamentosa).33
Leukotriene receptor antagonists. Although not approved by the FDA for treatment of allergic rhinitis, the leukotriene receptor antagonist montelukast was shown in a randomized double-blinded trial to be as effective as loratadine in relieving symptoms. There was minimal additional benefit in using the medications concomitantly.34
Cromolyn sodium. Cromolyn sodium has been shown to prevent the onset of allergic rhinitis symptoms in multiple placebo-controlled trials.35 It is extremely safe but requires regular use and is not as effective as other medications for acute symptoms. Direct comparison studies have shown that cromolyn is not as effective as intranasal corticosteroids.35,36
Immunotherapy. Subcutaneous immunotherapy (SIT) is recommended by all guidelines for patients who fail to respond to pharmacotherapy and allergen avoidance.2,5,10,27 It is recommended in particular for allergic rhinitis secondary to ragweed, grasses, molds, and dust mites. Immunotherapy induces the creation of protective IgG and inhibits the inflammatory response to allergens.27 SIT requires specific allergen confirmation with either a skin test or in vitro assay. Preparation of SIT doses should be done by a practitioner well trained in mixing and diluting extracts.5,37 Forty-three placebo-controlled, double-blind studies have evaluated the efficacy of SIT for 12 different allergens since 1980.10 Thirty-two trials showed clinical efficacy, which can be long lasting. A study of patients treated for 3 to 4 years with immunotherapy for grass pollen allergy showed continued clinical remission for at least 3 years after treatment was stopped.38
Herbal therapies. Alternative approaches to the treatment of allergic rhinitis warrant further investigation. Herbal medications, such as licorice, gingko, and ginseng, are currently used to treat allergic rhinitis, although there are no large studies to confirm their effectiveness.39
Probiotics. Epidemiologic studies suggest that the increase in atopic disease may be related to a clean environment and widespread use of antibiotics in Western countries. The environment may deprive fetal and infant immune systems of bacterial antigens that stimulate type 1 T-helper (Th1) cells.8 In light of this theory, Finnish researchers randomly assigned 159 pregnant women with a family history of atopy to receive capsules of Lactobacillus GG (a potentially beneficial bacteria or “probiotic”) or placebo, beginning 2 to 4 weeks prior to delivery and continuing 6 months postpartum. Infants were followed for 2 years. Frequency of atopic dermatitis was reduced by 50% among those infants whose mothers received Lactobacillus.40 Further study of this association in allergic rhinitis would be beneficial.
Treatment recommendations.Table 2 summarizes treatment-related evidence in the management of allergic rhinitis, and the Figure illustrates a proposed treatment algorithm. Another algorithm by the European Academy of Allergy and Clinical Immunology recommends initial therapy with oral or nasal antihistamines for mild disease, nasal corticosteroids for moderate disease, and both for severe disease.10 This was a consensus opinion. Of the 2 most commonly used medications for allergy, nasal steroids are favored over antihistamines for overall safety, tolerability, effectiveness, and simplicity in all cases. In one study of 61 adults, patients were randomized to receive either a nasal steroid or an antihistamine as initial therapy, with the other agent reserved as “back-up.” After 6 weeks, 86% of patients started on an antihistamine had added their steroid back-up, while 51% of the group started on a steroid remained on that agent alone.41 Starting all patients on both an antihistamine and a nasal steroid is inappropriate.
TABLE 2
Evidence to support treatment recommendations
| Strength of recommendation | Treatment | Comment |
|---|---|---|
| A | Immunotherapy | Can have long lasting clinical benefit. |
| A | Intranasal Consistently | superior to antihistamines corticosteroids in head to head trials. Not clear if all steroids are equally effective. |
| A | Antihistamines | Effective, but inferior to intranasal steroids in most clinical outcomes. |
| A | Cromolyn sodium | Intranasal steroids superior in all clinical outcomes. |
| B | Decongestants | Less effective than antihistamines in direct comparisons, many trials involve combination products. |
| D | Probiotics | Larger trials needed, limited evidence. |
| D | Herbal medications (licorice, gingko, ginseng) | Limited evidence. |
FIGUREA guide to evaluation and treatment of allergic rhinitis
Prognosis
The long-term prognosis for allergic rhinitis is excellent. For most patients, the illness is primarily a nuisance with no significant morbidity. However, for patients whose rhinitis is moderate to severe and poorly controlled, there can be significant complications. These complications include asthma, sinusitis, otitis media, nasal polyposis, respiratory infections, and orthodontic malocclusions.42 In one study of 605 children with allergic rhinitis, 21% had chronic otitis media with effusion (OME). Conversely, in another study of 259 children with OME, 50% had allergic rhinitis.43 Even among patients without asthma, 20% to 30% will have bronchial hyper-responsiveness. Additionally, poorly controlled allergic rhinitis can contribute to sleep loss, daytime fatigue, and learning impairment.44
Potential complications related to long-term treatment in children remain controversial. In a 1998 study of intranasal beclomethasone, children receiving the study medication grew an average of only 5 centimeters (cm) in 1 year, compared with an average of 5.9 cm in the placebo group.45 However, a similar study done 2 years later with intranasal mometa-sone showed no evidence of growth suppression.45 Further studies are needed before the true impact of intranasal steroids on children can be determined.
· Acknowledgments ·
The opinions and assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the U.S. Army or the U.S. Department of Defense. The author wishes to thank Kathleen Conner, JD, for her assistance in the preparation of the manuscript.
- Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”).
- Skin prick testing can detect IgE antibodies in patients with reliable histories of exposure to allergens.
- Intranasal corticosteroids are superior to other medications in achieving desired clinical outcomes, including quality of life.
- For some cases of allergic rhinitis, subcutaneous immunotherapy can achieve clinical remission for up to 3 years after cessation of therapy.
While allergic rhinitis is merely a nuisance to most people afflicted by it, the condition can lead to complications if it is severe or exists undetected for too long. In this article, I review the most reliable means of diagnosing allergic rhinitis, and outline a recommended approach to treatment.
Prevalence and pathophysiology
An estimated 20 to 40 million Americans are affected by allergic rhinitis. The actual prevalence of the condition is difficult to discern as many sufferers self-medicate without seeking medical care. One survey stated that up to 92% of patients had self-medicated prior to seeking medical care.1 Even when accounting for self-treatment, allergic rhinitis is the most commonly encountered form of chronic rhinitis, representing about 3% of all primary care office visits.2,3 Direct and indirect clinical costs run between $1.2 and $5.3 billion per year.4-6 Although the disease can develop in persons of any age, in 80% of cases symptoms will develop before the patient is 20 years old.5 Symptoms often wane as a patient grows older, and it is uncommon for persons older than 65 to experience new onset of allergic rhinitis.3,7
Allergic rhinitis stems from a type I hypersensitivity reaction.4 During an initial sensitization phase, the immune system identifies an allergen as foreign and generates specific antibodies to act against that allergen. Atopic patients exhibit an exaggerated response, generating high levels of Type 2 T-helper (Th2) cells and, subsequently, IgE antibodies.8 On reexposure to the allergen, specific IgE antibodies bound to mast cells form cross-links resulting in mast cell degranulation and the release of histamine and other chemical mediators. The patient then immediately develops such allergy symptoms as itching, sneezing, and rhinorrhea. A cellular inflammatory response, chiefly involving eosinophils, monocytes, and basophils, characterizes the secondary phase of the allergic reaction. Nasal congestion tends to dominate this later response phase.
Seasonal allergic rhinitis is usually triggered by pollens or molds. Perennial allergic rhinitis, triggered by dust mites, molds, cockroach or animal allergens, is defined as occurring 9 months out of the year.9
Clinical evaluation
The diagnosis of allergic rhinitis is usually made on the basis of the patient’s history and the results of your physical examination. In addition to classic symptoms of nasal congestion, itchy nose, sneezing, rhinorrhea, or itchy, watery eyes, patients may also complain of chronic cough, dry scratchy throat, otalgia, or recurrent sinusitis.4 Other important historical considerations include a family history of allergic rhinitis, a history of other atopic disease, previous treatment experiences, and suspected triggers.5
Physical clues to allergic rhinitis include boggy, pale, or “bluish” nasal turbinates, with watery discharge on nasal speculum exam. Patients may also have a nasal crease on the external nose caused by repeated rubbing or itching (the so-called “allergic salute”). Chronic nasal congestion may also precipitate darkening of the skin under the eyes or “allergic shiners.”2,6 Concurrent conjunctivitis is common. Polyps, seen on direct nasal examination, may occur both in allergic and non-allergic patients.
No studies have evaluated the accuracy of the history or physical examination in confirming the diagnosis of allergic rhinitis. The differential diagnosis is extensive and includes infectious rhinitis, non-allergic rhinitis with eosinophilia syndrome (NARES), occupational rhinitis, mechanical obstruction, vasomotor rhinitis, drug-induced rhinitis, and nasal polyps.5
Diagnostic tests
Published guidelines from the American Academy of Asthma, Allergy and Immunology, as well as other expert panels, recommend confirmatory testing when allergic rhinitis is clinically suspected.2,5,10 There is no evidence to support the superiority of this recommendation over an empiric trial of medication, and most primary care physicians choose to treat empirically based upon the history and physical examination.
Although further testing should be done when the diagnosis is unclear, be aware that there is uncertainty associated with allergy testing. Because an individual may become sensitized to an allergen without exhibiting symptoms of allergic rhinitis, there is no clearly defined reference standard for the confirmation of allergic rhinitis.11 Likewise, a history of sensitivity is not always followed by expected IgE test results. Challenge methods developed for studies of airborne allergens are used as reference standards in the evaluation of clinical tests.12
Diagnostic tests include skin prick testing, intradermal testing, and in vitro blood tests. Nasal challenge testing, nasal smears, sinus transillumination, and nasopharyngoscopy are nonspecific tests. They are not recommended for routine evaluation but may be useful in selected cases when allergen-specific tests have failed to clarify the cause of the rhinitis. An expert panel has stated that no studies address the cost-effectiveness of any of these methods.2
Skin prick testing (SPT) is considered the most convenient and least expensive screening test. SPT can detect IgE antibodies in patients with reliable exposure histories.13 Sensitivity and specificity are difficult to determine, for a number of reasons. First, as previously mentioned, there is no clearly defined reference standard.11 Second, only 5 allergen extracts have been standardized for defined quantities known to induce biologic activity. Standardized extracts in the United States include ragweed pollen, cat dander, house dust mites, Hymenoptera venoms, and some grasses. All other extracts are local or regional preparations, and skin tests with nonstandard extracts are not necessarily reproducible.11 Third, even with a single individual, there can be wide variation in skin reaction to the same reagent, depending on the device used.14 As a result, correlation between SPT and inhalation challenges vary from 60% to 90%.13
Intradermal skin tests (IDST) are usually done when SPT yields a negative result despite a history compatible with allergic rhinitis.13 The primary advantage of IDST is sensitivity afforded by a fixed concentration of allergen. Because of this sensitivity, not all reactions are clinically relevant.13 In fact, IDST is often used as a reference standard in studies of the accuracy of SPT and in vitro tests.
Several in vitro assays of specific IgE antibodies are available. They are all modeled after the original radioallergosorbent tests (RAST); the term “RAST” is often used interchangeably with any type of in vitro blood test.13 IgE antibody tests have a high false-positive rate, meaning the test is positive in patients without allergy symptoms. RAST tests are less sensitive than SPT, with a mean sensitivity of 75% and a range of approximately 50% to 95%.13
The 3 primary diagnostic tests for allergic rhinitis are usually compared with each other and not to a recognized standard. Table 1 summarizes data from a study that compared all 3 tests with subjects who were placed in a small room with 2 cats and their bed.12 While this is one of very few studies that contrasts all 3 tests to a reasonable reference standard, the findings cannot necessarily be extrapolated to other airborne allergens.
In the hope of limiting referrals to allergists for testing, and reducing the uncertainty in making a diagnosis, one study looked at the RAST response to 19 allergens. The authors found that of all the patients who responded to any allergen, 95% exhibited responses specifically to grass pollen, dust mites, or cat dander. They went on to conclude that 96.3% of patients with allergic disease could be correctly identified with a combination of a standardized history (available in the study text), a total serum IgE of greater than 40 U/mL, and in vitro tests for cat dander, dust mites, and grass pollen.15
TABLE 1
Accuracy of diagnostic tests for diagnosis of cat allergy13
| Test | Sensitivity | Specificity | PV+ | PV- | LR+ | LR- |
|---|---|---|---|---|---|---|
| Skin prick test | 79.2 | 90.6 | 92.6 | 74.3 | 8.4 | 0.2 |
| Intradermal test | 60.0 | 31.0 | 23.1 | 69.2 | 0.9 | 1.3 |
| RAST | 69.2 | 100 | 100 | 72.7 | 69.2 | 0.3 |
| Note: Results are based upon any upper or lower symptoms when exposed to cat challenge. Intradermal test done if negative skin prick test. LR+ = positive likelihood ratio, LR- = negative likelihood ratio, PV+ = positive predictive value, PV- = negative predictive value. | ||||||
Treatment
Untreated allergic rhinitis can have a significant impact on quality of life. Patients are bothered by nose blowing, disrupted sleep, fatigue, and decreased concentration.1 In one 1996 survey, 32 % of patients said that allergy attacks embarrassed them or interfered with their quality of life.16 As a result, most patient-oriented studies on treatment evaluate the impact on health-related quality of life.17
The initial form of treatment is usually avoidance of the allergen, although this can be difficult. For animal allergens, washing pets and using high-efficiency particulate air (HEPA) filters have been shown to temporarily reduce the volume of airborne allergens but not to improve patient-oriented outcomes.18,19 Removing the pet from the home is the only sure remedy.18 More studies are needed to evaluate the benefit of multiple home treatments to reduce exposure to cockroach and fungal allergens.18 A systematic review of several studies showed that maternal antigen avoidance during lactation reduced the incidence of atopic dermatitis in at-risk infants.20 A meta-analysis of measures to avoid house-dust mites showed no clear benefit for patients with asthma10 It is unclear if these findings can be extrapolated to other atopic conditions such as allergic rhinitis.
Intranasal corticosteroids. Intranasal corticosteroids are the most effective medication in the treatment of allergic rhinitis. Available preparations in the US include beclomethasone diproprionate, budesonide, funisolide, fluticasone propionate, mometa-sone furoate, and triamcinalone acetonide. A meta-analysis identified 16 randomized controlled trials (RCTs) that compared antihistamines with intranasal corticosteroids in a total of 2767 patients. Intranasal corticosteroids provided significantly greater relief from nasal discharge, sneezing, pruritis, and postnasal drip. There was no statistically significant difference between the 2 in reduction of eye symptoms.21
Although this review did not address quality of life, other studies have shown that both triamcinolone acetonide and fluticasone propionate are superior to loratadine in improving quality of life.22,23 Few studies provide any guidance in choosing one intranasal steroid over another. Generally, they are of equal efficacy in patient-oriented outcomes.24,25 Although intranasal corticosteroids are considered daily or “maintenance” medications, a single small RCT of 26 patients showed that fluticasone propionate improved quality of life and reduced symptoms compared with placebo when used on an as-needed basis over a 4-week period.26 More studies are needed to confirm this preliminary finding, though.
Antihistamines. Although not as effective as intranasal steroids, antihistamines do reduce symptoms of rhinorrhea, sneezing, and itching.27 First-gen-eration antihistamines (diphenhydramine, chlorpheniramine, etc.) are lipophilic and cross the blood-brain barrier, resulting in varying degrees of anticholinergic side effects. Placebo-controlled studies have confirmed that these agents cause psychomotor retardation, sleepiness, and decreased work production.5,28 Specifically they seem to affect attention, memory, and vigilance. These symptoms may persist even after an overnight period of sleep.28,29 Second-generation antihistamines (fexofenadine, loratadine, etc.) do not penetrate the brain as well and are less likely to cause central nervous system effects.
However, a recent RCT involving 63 elementary school students challenges findings from previous studies. Children who received diphenhydramine, 25 mg twice daily, performed no differently on computerized reaction-time tests or multiple-choice learning tests than did children who received placebo or loratadine, 10 mg daily.30 Another RCT involving 845 patients from ages 12 to 65 years evaluated quality of life as well as work and school performance of patients who received fexofenadine or placebo. While quality-of-life scores and work performance improved significantly with fexofenadine, there was no significant difference between the groups in school performance.31 Direct comparisons of antihistamines are rare and the results are conflicting. There are no data to show that one of the first-generation antihistamines is superior to the others. Similarly, second-generation drugs are no more effective than the older medications; they only have fewer side effects. Among the second-generation antihistamines, fexofenadine and cetirizine appear to be more effective then loratadine.29
Decongestants. Systemic and topical decongestants relieve the congestion that accompanies the secondary phase of an allergic reaction.4 They have limited effects on other allergic symptoms and, as a result, are often used in combination with antihistamines.32 When used for more than 10 days, topical decongestants (oxymetazoline, xylometazoline) are associated with rebound congestion (rhinitis medicamentosa).33
Leukotriene receptor antagonists. Although not approved by the FDA for treatment of allergic rhinitis, the leukotriene receptor antagonist montelukast was shown in a randomized double-blinded trial to be as effective as loratadine in relieving symptoms. There was minimal additional benefit in using the medications concomitantly.34
Cromolyn sodium. Cromolyn sodium has been shown to prevent the onset of allergic rhinitis symptoms in multiple placebo-controlled trials.35 It is extremely safe but requires regular use and is not as effective as other medications for acute symptoms. Direct comparison studies have shown that cromolyn is not as effective as intranasal corticosteroids.35,36
Immunotherapy. Subcutaneous immunotherapy (SIT) is recommended by all guidelines for patients who fail to respond to pharmacotherapy and allergen avoidance.2,5,10,27 It is recommended in particular for allergic rhinitis secondary to ragweed, grasses, molds, and dust mites. Immunotherapy induces the creation of protective IgG and inhibits the inflammatory response to allergens.27 SIT requires specific allergen confirmation with either a skin test or in vitro assay. Preparation of SIT doses should be done by a practitioner well trained in mixing and diluting extracts.5,37 Forty-three placebo-controlled, double-blind studies have evaluated the efficacy of SIT for 12 different allergens since 1980.10 Thirty-two trials showed clinical efficacy, which can be long lasting. A study of patients treated for 3 to 4 years with immunotherapy for grass pollen allergy showed continued clinical remission for at least 3 years after treatment was stopped.38
Herbal therapies. Alternative approaches to the treatment of allergic rhinitis warrant further investigation. Herbal medications, such as licorice, gingko, and ginseng, are currently used to treat allergic rhinitis, although there are no large studies to confirm their effectiveness.39
Probiotics. Epidemiologic studies suggest that the increase in atopic disease may be related to a clean environment and widespread use of antibiotics in Western countries. The environment may deprive fetal and infant immune systems of bacterial antigens that stimulate type 1 T-helper (Th1) cells.8 In light of this theory, Finnish researchers randomly assigned 159 pregnant women with a family history of atopy to receive capsules of Lactobacillus GG (a potentially beneficial bacteria or “probiotic”) or placebo, beginning 2 to 4 weeks prior to delivery and continuing 6 months postpartum. Infants were followed for 2 years. Frequency of atopic dermatitis was reduced by 50% among those infants whose mothers received Lactobacillus.40 Further study of this association in allergic rhinitis would be beneficial.
Treatment recommendations.Table 2 summarizes treatment-related evidence in the management of allergic rhinitis, and the Figure illustrates a proposed treatment algorithm. Another algorithm by the European Academy of Allergy and Clinical Immunology recommends initial therapy with oral or nasal antihistamines for mild disease, nasal corticosteroids for moderate disease, and both for severe disease.10 This was a consensus opinion. Of the 2 most commonly used medications for allergy, nasal steroids are favored over antihistamines for overall safety, tolerability, effectiveness, and simplicity in all cases. In one study of 61 adults, patients were randomized to receive either a nasal steroid or an antihistamine as initial therapy, with the other agent reserved as “back-up.” After 6 weeks, 86% of patients started on an antihistamine had added their steroid back-up, while 51% of the group started on a steroid remained on that agent alone.41 Starting all patients on both an antihistamine and a nasal steroid is inappropriate.
TABLE 2
Evidence to support treatment recommendations
| Strength of recommendation | Treatment | Comment |
|---|---|---|
| A | Immunotherapy | Can have long lasting clinical benefit. |
| A | Intranasal Consistently | superior to antihistamines corticosteroids in head to head trials. Not clear if all steroids are equally effective. |
| A | Antihistamines | Effective, but inferior to intranasal steroids in most clinical outcomes. |
| A | Cromolyn sodium | Intranasal steroids superior in all clinical outcomes. |
| B | Decongestants | Less effective than antihistamines in direct comparisons, many trials involve combination products. |
| D | Probiotics | Larger trials needed, limited evidence. |
| D | Herbal medications (licorice, gingko, ginseng) | Limited evidence. |
FIGUREA guide to evaluation and treatment of allergic rhinitis
Prognosis
The long-term prognosis for allergic rhinitis is excellent. For most patients, the illness is primarily a nuisance with no significant morbidity. However, for patients whose rhinitis is moderate to severe and poorly controlled, there can be significant complications. These complications include asthma, sinusitis, otitis media, nasal polyposis, respiratory infections, and orthodontic malocclusions.42 In one study of 605 children with allergic rhinitis, 21% had chronic otitis media with effusion (OME). Conversely, in another study of 259 children with OME, 50% had allergic rhinitis.43 Even among patients without asthma, 20% to 30% will have bronchial hyper-responsiveness. Additionally, poorly controlled allergic rhinitis can contribute to sleep loss, daytime fatigue, and learning impairment.44
Potential complications related to long-term treatment in children remain controversial. In a 1998 study of intranasal beclomethasone, children receiving the study medication grew an average of only 5 centimeters (cm) in 1 year, compared with an average of 5.9 cm in the placebo group.45 However, a similar study done 2 years later with intranasal mometa-sone showed no evidence of growth suppression.45 Further studies are needed before the true impact of intranasal steroids on children can be determined.
· Acknowledgments ·
The opinions and assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the U.S. Army or the U.S. Department of Defense. The author wishes to thank Kathleen Conner, JD, for her assistance in the preparation of the manuscript.
1. Meltzer EO. The prevalence and medical and economic impact of allergic rhinitis in the United States. J Allergy Clin Immunol 1997;99:S805-28.
2. Fornadley JA, et al. Allergic rhinitis: Clinical practice guideline. Otolaryngol Head Neck Surg 1996;115(1):115-122.
3. Wagner W, Kavuru M. Diagnosis and Management of Rhinitis. 1st Ed. Professional Communications, Inc. 1996. Available at www.medscape.com. Accessed Sept. 6, 2001.
4. Hadley JA. Evaluation and management of allergic rhinitis. Med Clin North Am 1999;83(1):13-25.
5. Dykewicz MS, Fineman S, et al. Diagnosis and management of rhinitis: complete guidelines of the joint task force on practice parameters in allergy, asthma and immunology. Ann Allergy Asthma Immunol 1998;81:478-518.
6. Weiss KB. The health economics of asthma and rhinitis. I. Assessing the economic impact. J Allergy Clin Immunol 2001;107:3-8.
7. Gentile DA, Friday GA, Skoner DP. Management of allergic rhinitis: antihistamines and decongestants. Immunol Allergy Clin North Am 2000;20(2):355-368.
8. Kay AB. Allergy and allergic diseases. N Engl J Med 2001;344:30-36.
9. Skoner DP. Allergic rhinitis: definition, epidemiology, pathophysiology, detection and diagnosis. J Allergy Clin Immunol 2001;108:S2-8.
10. van Cauwenberge P, et al. Consensus statement on the treatment of allergic rhinitis. Allergy 2000;55:116-134.
11. Ownby DR. Skin tests in comparison to other diagnostic methods. Immunol Allergy Clin North Am 2001;21(2):355-367.
12. Wood RA, Phipatanakul W, Hamilton RG, Eggleston PA. A comparison of skin prick tests, intradermal skin tests, and RASTs in the diagnosis of cat allergy. J Allergy Clin Immunol 1999;103:773-779.
13. Bernstein IL, et al. Practice parameters for allergy diagnostic testing. Ann Allergy Asthma Immunol 1995;75:543-625.
14. Nelson HS, Lahr J, Buchmeier A, McCormick D. Evaluation of devices for skin prick testing. J Allergy Clin Immunol 1998;101:153-156.
15. Merrett TG, Pantin CF, Dimond AH, Merrett J. Screening for IgE-mediated allergy. Allergy 1980 Sep;35(6):491-501.
16. Ferguson BJ. Cost effective pharmacotherapy for allergic rhinitis. Otolaryngol Clin North Am 1998;31(1):91-110.
17. Thompson AK, Juniper E, Meltzer EO. Quality of life in patients with allergic rhinitis. Ann Allergy Asthma Immunol 2000;85:338-348.
18. Eggleston PA, Bush RK. Environmental allergen avoidance: an overview. J Allergy Clin Immunol 2001;107:S403-405.
19. Wood RA, Johnson EF, van Atta ML, et al. A placebo-controlled trial of a HEPA air filter in the treatment of cat allergy. Am J Resp CC Med 1998;158:115-120.
20. Kramer MS. Maternal antigen avoidance during lactation for preventing atopic disease in infants of women at high risk (Cochrane Review). In: The Cochrane Library, Issue 3, 2001. Oxford: Update software.
21. Weiner JM, Abramson MJ, Puy RM. Intranasal corticosteroids versus H1 receptor antagonists in allergic rhinitis: systematic review of randomised controlled trials. BMJ 1998;317:1624-1629.
22. Condemi J, Schulz R, Lim J. Triamcinolone acetonide aqueous nasal spray versus loratadine in seasonal allergic rhinitis: efficacy and quality of life. Ann Allergy Asthma Immunol 2000;84:533-538.
23. Ratner PH, van Bavel JH, Martin BG, et al. A comparison of the efficacy of fluticasone propionate aqueous nasal spray and loratadine, alone and in combination, for the treatment of seasonal allergic rhinitis. J Fam Pract 1998;47:118-125.
24. Small P, et al. Comparison of triamcinalone acetonide nasal aerosol spray and fluticasone propionate aqueous solution spray in treatment of spring allergic rhinitis. J Allergy Clin Immunol 1997;100(5):592-5.
25. Day J, Carrillo T. Comparison of the efficacy of budesonide and fluticasone propionate aqueous nasal spray for once daily treatment of perennial allergic rhinitis. J Allergy Clin Immunol 1998;102:902-908.
26. Jen A, Baroody F, de Tineo M, et al. As-needed use of fluticasone propionate nasal spray reduces symptoms of seasonal allergic rhinitis. J Allergy Clin Immunol 2000;105:732-738
27. Naclerio RM. Allergic rhinitis. N Engl J Med 1991;325:860-869.
28. Kay GG. The effects of antihistamines on cognition and performance. J Allergy Clin Immunol 2000;105:S622-627.
29. Abramowicz M. (Ed.) Newer antihistamines. Med Letter 2001;43:35.-
30. Bender BG, McCormick DR, Milgrom H. Children’s school performance is not impaired by short-term administration of diphenhydramine or loratadine. J Peds 2001;138:656-660.
31. Meltzer EO. Once-daily fexofenadine HCl improves quality of life and reduces work and activity impairment in patients with seasonal allergic rhinitis. Ann Allergy Asthma Immunol 1999;83:311-317.
32. Sussman GL, Mason J, Compton D, Stewart J, Ricard N. The efficacy and safety of fexofenadine HCL and pseudophedrine, alone and in combination, in seasonal allergic rhinitis. J Allergy Clin Immunol 1999;104:100-106.
33. Graf P, Enerdal J, Hallen H. Ten days’use of oxymetazoline nasal spray with benzalkonioum chloride in patients with vasomotor rhinitis. Arch Otolaryngol Head and Neck Surg 1999;125:1128-1132.
34. Meltzer EO, Malmstrom K, Lu S, et al. Concomitant montelukast and loratadine as treatment for seasonal allergic rhinitis: A randomized placebo-controlled clinical trial. J Allergy Clin Immunol 2000;105:917-922.
35. LaForce C. Use of nasal steroids in managing allergic rhinitis. J Allergy Clin Immunol 1999;103:S388-394.
36. Williams PV. Treatment of rhinitis: corticosteroids and cromolyn sodium. Immunol Allergy Clin North Am 2000;20:369-381.
37. Craig T, Sawyer AM, Fornadley JA. Use of immunotherapy in a primary care office. Am Fam Phys 1998;57:1888-1894.
38. Durham SR, Walker SM, Varga EM, et al. Long-term efficacy of grass-pollen immunotherapy. N Engl J Med 1999;341:468-475.
39. Bielory L, Lupoli K. Herbal interventions in asthma and allergy. J Asthma 1999;36(1):1-65.
40. Kalliomaki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 2001;357:1076-1079.
41. Juniper EF, Guyatt GJ, Ferrie PJ, Griffith LE. First-line treatment of seasonal (ragweed) rhinoconjunctivitis. Can Med Assoc J 1997;156:1123-1131.
42. Spector SL. Overview of comorbid associations of allergic rhinitis. J Allergy Clin Immunol 1997;99:S773-780.
43. Skoner DP. Complications of allergic rhinitis. J Allergy Clin Immunol 2000;105:S605-609.
44. Settipane RA. Complications of allergic rhinitis. Allergy Asthma Proc 1999;20(4):209-213.
45. Buck ML. Intranasal steroids for Children with Allergic Rhinitis. Pediatric Pharmacotherapy 2000;(7)5:1-12.
1. Meltzer EO. The prevalence and medical and economic impact of allergic rhinitis in the United States. J Allergy Clin Immunol 1997;99:S805-28.
2. Fornadley JA, et al. Allergic rhinitis: Clinical practice guideline. Otolaryngol Head Neck Surg 1996;115(1):115-122.
3. Wagner W, Kavuru M. Diagnosis and Management of Rhinitis. 1st Ed. Professional Communications, Inc. 1996. Available at www.medscape.com. Accessed Sept. 6, 2001.
4. Hadley JA. Evaluation and management of allergic rhinitis. Med Clin North Am 1999;83(1):13-25.
5. Dykewicz MS, Fineman S, et al. Diagnosis and management of rhinitis: complete guidelines of the joint task force on practice parameters in allergy, asthma and immunology. Ann Allergy Asthma Immunol 1998;81:478-518.
6. Weiss KB. The health economics of asthma and rhinitis. I. Assessing the economic impact. J Allergy Clin Immunol 2001;107:3-8.
7. Gentile DA, Friday GA, Skoner DP. Management of allergic rhinitis: antihistamines and decongestants. Immunol Allergy Clin North Am 2000;20(2):355-368.
8. Kay AB. Allergy and allergic diseases. N Engl J Med 2001;344:30-36.
9. Skoner DP. Allergic rhinitis: definition, epidemiology, pathophysiology, detection and diagnosis. J Allergy Clin Immunol 2001;108:S2-8.
10. van Cauwenberge P, et al. Consensus statement on the treatment of allergic rhinitis. Allergy 2000;55:116-134.
11. Ownby DR. Skin tests in comparison to other diagnostic methods. Immunol Allergy Clin North Am 2001;21(2):355-367.
12. Wood RA, Phipatanakul W, Hamilton RG, Eggleston PA. A comparison of skin prick tests, intradermal skin tests, and RASTs in the diagnosis of cat allergy. J Allergy Clin Immunol 1999;103:773-779.
13. Bernstein IL, et al. Practice parameters for allergy diagnostic testing. Ann Allergy Asthma Immunol 1995;75:543-625.
14. Nelson HS, Lahr J, Buchmeier A, McCormick D. Evaluation of devices for skin prick testing. J Allergy Clin Immunol 1998;101:153-156.
15. Merrett TG, Pantin CF, Dimond AH, Merrett J. Screening for IgE-mediated allergy. Allergy 1980 Sep;35(6):491-501.
16. Ferguson BJ. Cost effective pharmacotherapy for allergic rhinitis. Otolaryngol Clin North Am 1998;31(1):91-110.
17. Thompson AK, Juniper E, Meltzer EO. Quality of life in patients with allergic rhinitis. Ann Allergy Asthma Immunol 2000;85:338-348.
18. Eggleston PA, Bush RK. Environmental allergen avoidance: an overview. J Allergy Clin Immunol 2001;107:S403-405.
19. Wood RA, Johnson EF, van Atta ML, et al. A placebo-controlled trial of a HEPA air filter in the treatment of cat allergy. Am J Resp CC Med 1998;158:115-120.
20. Kramer MS. Maternal antigen avoidance during lactation for preventing atopic disease in infants of women at high risk (Cochrane Review). In: The Cochrane Library, Issue 3, 2001. Oxford: Update software.
21. Weiner JM, Abramson MJ, Puy RM. Intranasal corticosteroids versus H1 receptor antagonists in allergic rhinitis: systematic review of randomised controlled trials. BMJ 1998;317:1624-1629.
22. Condemi J, Schulz R, Lim J. Triamcinolone acetonide aqueous nasal spray versus loratadine in seasonal allergic rhinitis: efficacy and quality of life. Ann Allergy Asthma Immunol 2000;84:533-538.
23. Ratner PH, van Bavel JH, Martin BG, et al. A comparison of the efficacy of fluticasone propionate aqueous nasal spray and loratadine, alone and in combination, for the treatment of seasonal allergic rhinitis. J Fam Pract 1998;47:118-125.
24. Small P, et al. Comparison of triamcinalone acetonide nasal aerosol spray and fluticasone propionate aqueous solution spray in treatment of spring allergic rhinitis. J Allergy Clin Immunol 1997;100(5):592-5.
25. Day J, Carrillo T. Comparison of the efficacy of budesonide and fluticasone propionate aqueous nasal spray for once daily treatment of perennial allergic rhinitis. J Allergy Clin Immunol 1998;102:902-908.
26. Jen A, Baroody F, de Tineo M, et al. As-needed use of fluticasone propionate nasal spray reduces symptoms of seasonal allergic rhinitis. J Allergy Clin Immunol 2000;105:732-738
27. Naclerio RM. Allergic rhinitis. N Engl J Med 1991;325:860-869.
28. Kay GG. The effects of antihistamines on cognition and performance. J Allergy Clin Immunol 2000;105:S622-627.
29. Abramowicz M. (Ed.) Newer antihistamines. Med Letter 2001;43:35.-
30. Bender BG, McCormick DR, Milgrom H. Children’s school performance is not impaired by short-term administration of diphenhydramine or loratadine. J Peds 2001;138:656-660.
31. Meltzer EO. Once-daily fexofenadine HCl improves quality of life and reduces work and activity impairment in patients with seasonal allergic rhinitis. Ann Allergy Asthma Immunol 1999;83:311-317.
32. Sussman GL, Mason J, Compton D, Stewart J, Ricard N. The efficacy and safety of fexofenadine HCL and pseudophedrine, alone and in combination, in seasonal allergic rhinitis. J Allergy Clin Immunol 1999;104:100-106.
33. Graf P, Enerdal J, Hallen H. Ten days’use of oxymetazoline nasal spray with benzalkonioum chloride in patients with vasomotor rhinitis. Arch Otolaryngol Head and Neck Surg 1999;125:1128-1132.
34. Meltzer EO, Malmstrom K, Lu S, et al. Concomitant montelukast and loratadine as treatment for seasonal allergic rhinitis: A randomized placebo-controlled clinical trial. J Allergy Clin Immunol 2000;105:917-922.
35. LaForce C. Use of nasal steroids in managing allergic rhinitis. J Allergy Clin Immunol 1999;103:S388-394.
36. Williams PV. Treatment of rhinitis: corticosteroids and cromolyn sodium. Immunol Allergy Clin North Am 2000;20:369-381.
37. Craig T, Sawyer AM, Fornadley JA. Use of immunotherapy in a primary care office. Am Fam Phys 1998;57:1888-1894.
38. Durham SR, Walker SM, Varga EM, et al. Long-term efficacy of grass-pollen immunotherapy. N Engl J Med 1999;341:468-475.
39. Bielory L, Lupoli K. Herbal interventions in asthma and allergy. J Asthma 1999;36(1):1-65.
40. Kalliomaki M, Salminen S, Arvilommi H, et al. Probiotics in primary prevention of atopic disease: a randomised placebo-controlled trial. Lancet 2001;357:1076-1079.
41. Juniper EF, Guyatt GJ, Ferrie PJ, Griffith LE. First-line treatment of seasonal (ragweed) rhinoconjunctivitis. Can Med Assoc J 1997;156:1123-1131.
42. Spector SL. Overview of comorbid associations of allergic rhinitis. J Allergy Clin Immunol 1997;99:S773-780.
43. Skoner DP. Complications of allergic rhinitis. J Allergy Clin Immunol 2000;105:S605-609.
44. Settipane RA. Complications of allergic rhinitis. Allergy Asthma Proc 1999;20(4):209-213.
45. Buck ML. Intranasal steroids for Children with Allergic Rhinitis. Pediatric Pharmacotherapy 2000;(7)5:1-12.
Prevention and Treatment of Osteoporosis in Postmenopausal Women
The last decade has witnessed important technological advances in the diagnosis of osteoporosis and an increase in therapeutic options. However, there is still considerable uncertainty about optimal strategies for screening and primary preventive treatment.
In 1994, a World Health Organization working group proposed that the diagnosis of osteoporosis be made when BMD, assessed by a dual-energy x-ray absorptiometry (DXA), is at least 2.5 standard deviations below the mean for young adult women (T-score) at the spine, hip, or wrist, or when a history of a traumatic fracture is present.2 A T-score between −1 and −2.5 is designated as osteopenia.
Osteoporosis is defined as “a skeletal disorder characterized by compromised bone strength predisposing to an increased risk of fracture.”1 While no accurate overall measurement of bone strength exists, bone mineral density (BMD) is frequently used as a proxy.
These facts underscore the importance of osteoporotic fractures:
- Only one third of patients regain their prior level of functioning after hip fracture, and one third are discharged to nursing homes.3
- About 1 in 5 patients dies within a year after a hip fracture.
- Vertebral fracture may result in chronic back pain and disability.4
- Existence of fracture greatly increases risk of subsequent fracture.5
- Direct medical costs for osteoporotic fractures are estimated at $13.8 billion in 1995 dollars.6
Prevalence of osteoporosis and fractures
Of American women over age 50 of all races, an estimated 15%, or 5 million, have osteoporosis (based on DXA T-score at the femoral neck) and an additional 40%, or 14 million, have osteopenia.7 In African Americans, the prevalence is about half that of whites.8 The prevalence of osteoporosis assessed by BMD testing increases with age—from 4% of white women aged 50 to 59 to 48% of women aged 80 to 89.9
At least 1 vertebral fracture, as indicated by radiographic criteria, has occurred in 5% of white women aged 50 to 59, and in 25% at age 80.3 The lifetime risk of hip fracture for 50-year-old white women and men is 14% and 5%, respectively; for African American women and men, 6% and 3%, respectively.3 Hip and symptomatic vertebral fractures occur mainly in women over 75,3,10 and the risk for wrist fractures increases starting in the late 50s.11
Age is a particularly important risk factor for hip fractures, reflecting deterioration in bone strength beyond that detectable with BMD testing. The National Osteoporosis Foundation12 observed that the 5-year risk of hip fracture for women with the same T-score (−3) increases dramatically with advancing age (Figure): from 2.4% at age 50 to 9.7% at age 90, with the steepest increase occurring during the 10 years between ages 70 (5.5%) and 80 (9%).
FIGURE
Five-year risk for hip fracture for women with T-score of −3 by age12
Bone mineral density testing
Screening recommendations
The clinical value of different screening strategies is not established, although recommendations have been made within guidelines and consensus statements that discuss prevention and treatment of osteoporosis. Guidelines are consistent in recommending that BMD screening be done only if results will influence treatment decisions. The US Preventive Services Task Force,13 The National Osteoporosis Foundation,14 and American Association of Clinical Endocrinologists15 recommend screening all women over 65, as well as younger women with risk factors for osteoporosis. The National Institutes of Health3 and the North American Menopause Society16 recommend an individualized decision-making approach to screening. The National Osteoporosis Foundation developed nomograms that integrate risk factors into decision-making for testing and treatment,12 which seem promising and merit testing in prospective studies.
Diagnostic testing
DXA. Although several technologies are available, DXA of the hip is considered the best predictor of hip fracture and an equivalent predictor of other fractures.10 The likelihood of making a diagnosis of osteoporosis based on BMD, however, varies and is related to type of test, equipment, anatomic site tested, number of sites tested, technique, and relevance of the reference range to the local population. For example, when the same group of people is tested with DXA equipment from different manufacturers, the proportion diagnosed with osteoporosis varies by as much as 15%.11
Quantitative ultrasound (QUS) and radiographic absorptiometry (RA). Testing by QUS of the heel and RA of the hand are less expensive than DXA and have become popular. While QUS of the heel has been shown to predict hip fracture and all nonvertebral fractures nearly as well as DXA,3,10 it does not highly correlate with DXA and appears to reflect other aspects of bone quality.10 Since QUS and DXA results frequently disagree and can cause confusion, DXA is the most appropriate test of BMD at present. If QUS and RA are used for screening, confirmation with DXA is recommended before therapy is initiated.
Calculations based on risk factors. In a comparison of strategies using risk factors to predict low BMD in postmenopausal women, 2 decision rules performed well: the Osteoporosis Risk Assessment Instrument, which is based on age and weight (Table 1),17 and the Simple Calculated Osteoporosis Risk Estimation (SCORE).17 Research to test these instruments with fracture rather than BMD as outcome is needed.18
Biochemical markers. Levels of markers in serum and/or urine reflect bone turnover and have potential use in diagnosing and monitoring therapy of osteoporosis. They are not yet widely available and have not been consistently associated with identifying patients at risk for fracture.10 They are not recommended at this time.
TABLE 1
Osteoporosis risk assessment instrument17
| Patient characteristic | Points |
|---|---|
| Age (years) | |
| 75 or older | 15 |
| 65 to 74 | 9 |
| 55 to 64 | 5 |
| 54 or younger | 0 |
| Weight | |
| <132 lb (60 kg) | 9 |
| 132 to 153.9 lb (60 to 70 kg) | 3 |
| >154 lb (>70 kg) | 0 |
| No current estrogen use | 2 |
| Total: | |
| Patients with a score of 9 or higher are at risk for diagnosis of osteoporosis by bone mineral density measurement. Sensitivity 97.5%, specificity 28%, positive predictive value 28%, negative predictive value 99.6%, given a 10% baseline risk of a bone mineral density 2.5 SD less than the mean. | |
Importance of primary prevention
At least half of bone strength is attributable to genetic factors12; modifiable factors may contribute almost equally as a group, and therefore warrant attention. Genetic risk factors include age, family history, female sex, low weight, small frame, and white or Asian race. Primary prevention efforts should begin in childhood and continue throughout the life span to maximize bone mass.3
Prevention efforts that target the modifiable factors described below should be a routine part of the health-maintenance visit.
Fall reduction
Falls are the direct cause of more than 90% of osteoporotic hip fractures,19 and the tendency to fall increases with age. Some studies have shown that, for women over age 70, the most important predictors of hip fractures are fall-related factors20,21 such as poor cognitive function, slow gait and otherwise impaired mobility, poor vision, drugs that impair alertness or balance, and history of falls. In women over 75, age and slow gait are equal to low BMD of the femoral neck as predictors of hip fracture.22 Unfortunately, labeling women as osteopenic or osteoporotic can cause fear of falling and lack of activity, leading to further acceleration of bone loss.10
Medications that interfere with balance or alertness should be avoided if possible. Environmental hazards such as loose rugs and uneven or slippery surfaces are also well-recognized modifiable risks for falls23,24 that should be eliminated. Hip protectors effectively reduce fractures in the frail elderly25 and can boost confidence for beneficial increases in physical activity levels,26 but they are often poorly accepted by patients.25,27 Other options include referral for gait training, home visits by a physician or nurse to identify problems in the home that increase the risk of falls, or providing information on home modification (such as installing bathtub rails, removing throw rugs, etc.).
Improvement of nutritional intake
Adequate consumption of calcium is essential for bone health. Calcium balance also can be adversely affected by dietary habits, including high intake of protein, phosphorus, and sodium, although these effects appear to be less important when dietary calcium is sufficient.3 The recommended calcium intake for postmenopausal women (1200–1500 mg/day)28 can be met with food sources, but supplements should be added if needed. Most postmenopausal women in the United States consume only about 600 mg/day.28 High-calcium foods include milk (290–300 mg/cup), sardines in oil, with bones (370 mg/3 oz), yogurt (300–500 mg depending on container size), cheese (165–270 mg/slice), canned salmon, with bones (170–210 mg/3 oz), broccoli (160–180 mg/cup), and tofu (144–155 mg/4 oz).15
Vitamin D is essential for intestinal absorption of calcium. The recommended intake for women is 400 IU/day for ages 51 to 70, 600 IU/day over age 70, and 800 IU/day for all high-risk women, including those who are homebound, institutionalized, on chronic glucocorticoids, or who live in northern latitudes and therefore have limited exposure to sunlight.29 Sources of vitamin D include sunlight, vitamin D–fortified foods, fish oils, and supplements. Multivitamins typically contain 400 IU of vitamin D.
Phytoestrogens, particularly in the form of soy products, have received attention for bone health. Overall, studies do not support the use of soy foods to prevent osteoporosis.3 A well-designed trial in postmenopausal women found that ipriflavone, a synthetic phytoestrogen, did not decrease bone loss.30 Furthermore, use was associated with subclinical lymphocytopenia.
Regular exercise
Weight-bearing physical activity such as walking or running in early life contributes to higher peak bone mass. Limited data suggest weight-bearing exercise in postmenopausal women produces small increases in bone density at the hip31 and improvement in balance and strength.32 For women with established osteoporosis, activities that place an anterior load on the vertebral bodies, such as forward flexion exercises, are associated with an increased incidence of new vertebral deformities, and patients should be advised to avoid them.33
Avoidance of adverse health habits
Current smoking, compared with never smoking, doubles the risk of hip fracture.34 Consumption of more than 1 alcoholic drink/day or more than 7/week is associated with osteoporosis and fracture, while moderate consumption of 1 drink/day or less is associated with decreased risk.35 Excessive caffeine intake is also associated with increased osteoporosis risk and should be avoided. This effect appears to result from substitution of calcium-containing beverages such as milk or fortified orange juice with caffeinated, non–calcium-containing beverages such as colas.
Treatment for fracture prevention and pain relief
The goals of therapy for osteoporosis are fracture prevention and pain relief to maximize physical function.15 Prior fracture is associated with a fivefold risk of future fractures.5 About 20% of women who experience a vertebral fracture have another fracture within 1 year.36 Currently available therapies (Table 2) are antiresorptive: they slow bone turnover and allow bone formation to exceed resorption. Trials of antiresorptive agents in elderly women with osteoporosis and baseline vertebral fracture demonstrate that 1 new vertebral fracture is prevented for each 12 to 35 women treated for 2 to 3 years.14,37Table 3 summarizes the results of key treatment studies and provides information on the number of women that need to be treated (NNT) for the study period to prevent 1 fracture.
TABLE 2
Drug therapy for prevention and treatment of postmenopausal osteoporosis
| Drug (trade name) | Indication and dosage | Possible side effects (% of patients) | Cost per month* |
|---|---|---|---|
| Calcium and vitamin D (generic, Tums, Citracal, and others) | Prevention and treatment: 1200–1500 mg/day calcium and 800 IU/day vitamin D | Nausea, dyspepsia (uncommon), constipation (10%) | $5 (both) |
| Estrogen† (Premarin, Ogen, Estrace, Estraderm, and others) | Prevention: 0.625 mg/day conjugated equine estrogen or the equivalent; 0.3 mg/day may be effective | Nausea, breast tenderness, vaginal bleeding, mood alterations, headache, bloating | $14–$28 |
| Alendronate (Fosamax) | Prevention and treatment: 5 mg/day or 35 mg/week | Nausea, dyspepsia esophageal irritation | $67 |
| Risedronate (Actonel) | Prevention and treatment: 5 mg/day or 35 mg/week | Abdominal pain, nausea, diarrhea | $67 |
| Raloxifene (Evista) | Treatment: 60 mg/day | Hot flashes (6%), leg cramps (3%) | $70 |
| Calcitonin nasal spray (Miacalcin) | Treatment: 200 IU/day (1 spray in 1 nostril per day) | Rhinitis (5%), epistaxis, sinusitis | $66 |
| *Average wholesale cost to the pharmacy for 30 days of therapy; (Drug Topics Red Book. Montvale, NJ; Medical Economics Co., Inc, 2002.) | |||
| †Women with a uterus need to take a progestin such as medroxyprogesterone acetate (Provera $30/month, generic $9/month) or a combination estrogen/progestin product (Prempro $33/month, FemHRT $26/month). | |||
TABLE 3
Clinical trials of drug therapy for the prevention of fracture in postmenopausal women with osteoporosis
| Trial, year | Therapy | Outcome prevented | Number needed to treat for n years |
|---|---|---|---|
| Elderly, postmenopausal women | |||
| Chapuy, 199267 | Calcium/vitamin D | Hip fracture | 48 women for 1.5 years |
| Postmenopausal women | |||
| WHI, 200242 | Hormone replacement therapy | Hip fracture | 2000 women for 5 years |
| Postmenopausal women with osteoporosis | |||
| Ettinger, 199956 | Raloxifene | Vertebral fracture | 29 women for 3 years |
| Liberman, 199554 | Alendronate | Vertebral fracture | 34 women for 3 years |
| Heaney, 200253 | Risendronate | Vertebral fracture | 15 women for 3 years |
| McClung, 200170 | Risedronate | Hip fracture | 91 women for 3 years |
| Postmenopausal women with osteoporosis and previous vertebral racture | |||
| Harris, 199951 | Risedronate | Vertebral fracture | 20 women for 3 years |
| Black, 199652 | Alendronate | Vertebral fracture | 35 women for 3 years |
| Black, 199652 | Alendronate | Hip fracture | 86 women for 3 years |
Calcium and vitamin D
Calcium with or without vitamin D has been reported to positively affect fracture incidence.14 Vitamin D alone does not decrease the incidence of hip fractures.38 Calcium, 1200 to 1500 mg/day, and vitamin D, 800 IU/day, should be used concurrently with other forms of pharmacologic treatment. Calcium supplements are best absorbed with meals; for maximum absorption, calcium should be taken in doses of 500 mg or less.29,39
Minor gastrointestinal adverse effects may occur (most often constipation, 10%),14 which is often resolved by switching to a different preparation.40 Calcium in doses up to 1500 mg/day does not increase the risk for renal calculi and may, in fact, decrease risk.40,41 Calcium interferes with the absorption of certain medications, including tetracycline and quinolone antibiotics, which should be taken several hours apart from calcium. Calcium carbonate requires an acidic environment to dissolve. Patients taking stomach acid–suppressant therapy should use calcium citrate because it does not require an acidic environment for dissolution or should take their calcium supplement with meals. Traces of lead may be present in natural sources of calcium (bone meal, oyster shell, limestone, and dolomite), but can be avoided by use of over-the-counter calcium carbonate tablets (Tums).
Estrogen
Data from the Women’s Health Initiative (WHI) demonstrated that hormone replacement therapy (HRT) combining an estrogen and a progestin reduced hip and vertebral fractures by 1 in 2000 women per year and reduced all fractures by 1 in 333 women per year.42 Estrogen has a positive effect on BMD whether given in early or late postmenopause.43 Rapid bone loss as assessed by BMD does not occur after stopping HRT.44 However, in elderly women who have never used HRT, BMD is similar to those who have used it for 10 years and then stopped for at least 10 years.43 The effect of short-term (< 5 years) HRT during perimenopause on lifetime risk of osteoporotic fracture is unknown.
Common side effects (Table 2) can often be addressed by altering dosages, specific products, or regimens. The risk for breast cancer increases with duration of treatment and with combination HRT, compared with estrogen-only preparations.42,45,46 Combination HRT products are, however, essential for endometrial protection in women who have a uterus. The WHI reported an increase in breast cancer cases of 1 in 1250 women per year during an average 5-year follow-up.42 An increased risk for venous thromboembolism of 1 in 555 women per year was also observed with HRT use. Both the Heart and Estrogen/Progestin Study47 and the WHI studies found that venous thromboembolism occurred more frequently in the first 2 years of HRT use. An increased risk of myocardial infarction in the first 2 years of use was also noted among women with coronary heart disease47 and those without heart disease (1 in 1429 women per year).42 A small increase in stroke risk has also been documented.42,48-50 Contraindications to estrogen use include active thromboembolism, estrogen-related cancers, and liver disease.
Bisphosphonates
Two bisphosphonates, alendronate and risedronate, are approved in the United States for both prevention and treatment of postmenopausal osteoporosis. Clinical trials have demonstrated that both rapidly reduce the risk for symptomatic fractures in women with previous fracture and osteoporosis.37,51,52 The extent of fracture reduction is significant: Recent studies have shown that, over 3 years, the number of patients who would need to be treated with risedronate to prevent a vertebral fracture is 15, with alendronate, 34.53,54 Prevention of fractures among women without a prior vertebral fracture is less well established. No published data demonstrate that one bisphosphonate is more effective than another at preventing clinical fractures. A bisphosphonate is, therefore, the drug of choice for severe osteoporosis.
Bisphosphonates are generally well tolerated.55,56 However, postmarketing surveillance has demonstrated esophagitis and esophageal ulcer associated with alendronate.55 A pooled analysis of trials of risedronate found no increase in upper gastrointestinal (GI) adverse events even in patients with history of peptic ulcers, heartburn, and esophagitis, or among those taking nonsteroidal anti-inflammatory drugs, including aspirin.57
Oral bisphosphonates are not well absorbed (less than 1% of each dose).55,56 Therefore, to maximize absorption and to decrease the likelihood of adverse GI effects, the manufacturers of both bisphosphonates recommend that patients take the medication with a full glass of water, remain upright (sitting or standing) for at least 30 minutes following the dose, and not recline until food is consumed. Both bisphosphonates should be used with caution in patients with active GI disorders. Bisphosphonates are eliminated via the kidney and are not recommended for patients with a creatinine clearance below 30 mL/min.
Selective estrogen receptor modulators (SERMs)
Raloxifene is currently the only selective estrogen receptor modulator approved in the United States for prevention and treatment of osteoporosis. It has been shown to significantly decrease new vertebral fractures in women with a previous history of fracture and osteoporosis.58 The magnitude of fracture reduction is similar to that of bisphosphonates, although improvement in BMD is less marked.59 Raloxifene may confer other benefits. It has been shown to reduce the risk of breast cancer60,61 except in women with low estradiol levels,60 and may reduce the risk of myocardial infarction in women at high risk.60 Raloxifene does not increase the risk for endometrial cancer.60
Hot flashes and leg cramps are relatively common side effects of raloxifene.60 The observed risk of venous thromboembolism, 1 in 465 women per year during 3 years of treatment, is similar to that observed with HRT.60 Raloxifene is teratogenic and should not be used in premenopausal women.
Salmon calcitonin
Salmon calcitonin has demonstrated an analgesic effect for osteoporotic fracture.63,64 A large trial of salmon calcitonin at dosages of 100, 200, and 400 IU/day versus placebo found that salmon calcitonin at 200 IU/day decreased new vertebral fractures among women with a previous osteoporotic vertebral fracture based on radiographic assessment.65 No benefit was observed at the 100 and 400 IU/day dosages. The effect of calcitonin on clinical (symptomatic) fractures has not been reported. Calcitonin is approved for use in treatment, but not prevention, of osteoporosis.
Nasal calcitonin can cause minor rhinitis symptoms.30 Saline nasal solution may be useful to prevent or resolve irritation and dryness. Administration using alternate nostrils helps minimize local side effects. Unopened bottles (14 doses) must be stored in the refrigerator. Open bottles are stable at room temperature for up to 30 days.
Monitoring therapy
The value of serial densitometry to monitor the therapy of individual patients has not been established by randomized trials comparing different monitoring intervals or monitoring versus no monitoring.10,66 One important limitation is the relative imprecision of BMD testing: it takes almost a year to detect a 3% change in BMD.10 Disconcerting decreases in BMD scores are seen in yearly testing and may be offset by larger increases later, without a change in therapy. In studies of alendronate and raloxifene, disproportionately large fracture reductions cannot be explained by improvement in BMD alone.66,68 Bone densitometry is therefore not recommended until the patient has been treated for 2 years, and is of uncertain value beyond that point.
1. Osteoporosis Prevention, Diagnosis, and Therapy. NIH Consensus Statement Online 2000 March 27-29; [cited 9/2/2]; 17(1):1-36.
2. World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. WHO Technical Report Series 843. Geneva: World Health Organization; 1994.
3. NIH Consensus Development Panel on Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285:785-795.
4. Gold DT. The clinical impact of vertebral fractures: quality of life in women with osteoporosis. Bone. 1996;18(suppl 3):185S-189S.
5. Black DM, Arden NK, Palermo I, Pearson J, Cummings SR. Prevalent vertebral deformities predict hip fractures and new vertebral deformities but not wrist fractures. Study of Osteoporotic Fractures Research Group. J Bone Miner Res. 1999;14:821-828.
6. Ray NF, Chan JK, Thamer M, Melton LJ, 3rd. Medical expenditures for the treatment of osteoporotic fractures in the United States in 1995: report from the National Osteoporosis Foundation. J Bone Miner Res. 1997;12:24-35.
7. Looker AC, Wahner HW, Dunn WL, et al. Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int. 1998;8:468-489.
8. Aloia JF, Vaswani A, Yeh JK, Flaster E. Risk for osteoporosis in black women. Calcif Tissue Int. 1996;59:415-423.
9. Melton LJ. How many women have osteoporosis now? J Bone Miner Res. 1995;10:175-177.
10. Nelson HD, Morris CD, Mahon S, Carney N, Nygren PM, Helfand M. Osteoporosis in postmenopausal women: diagnosis and monitoring. Evidence Report/Technology Assessment Number 28. Rockville, MD: Agency for Healthcare Research and Quality; November 2001. 01-E032.
11. Melton LJ, 3rd, Amadio PC, Crowson CS, O’Fallon WM. Long-term trends in the incidence of distal forearm fractures. Osteoporos Int. 1998;8:341-348.
12. National Osteoporosis Foundation. Osteoporosis: review of the evidence for prevention, diagnosis, and treatment and cost-effectiveness analysis. Osteoporos Int. 1998;8(suppl):S7-S80.
13. U.S. Preventive Services Task Force. Screening for Osteoporosis in Postmenopausal Women. September 2002. Originally in Annals of Internal Medicine 2002;137:526-8.Agency for Healthcare Research and Quality, Rockville, MD. http://www.ahrq.gov/clinic/3rduspstf/osteoporosis/osteorr.htm.
14. Physician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 1998.
15. American Association of Clinical Endocrinologists 2001 medical guidelines for clinical practice for the prevention and management of osteoporosis. Endocr Pract. 2001;7:293-312.
16. North American Menopause Society. Management of postmenopausal osteoporosis: position statement of The North American Menopause Society. Menopause. 2002;9:84-101.
17. Cadarette SM, Jaglal SB, Murray TM, McIsaac WJ, Joseph L, Brown JP. Evaluation of decision rules for referring women for bone densitometry by dual energy x-ray absorptiometry. JAMA. 2001;286:57-63.
18. Wasnich RD. Consensus and the T-score fallacy. Clin Rheumatol. 1997;16:337-339.
19. Grisso JA, Kelsey JL, Strom BL, et al. Risk factors for falls as a cause of hip fracture in women.The Northeast Hip Fracture Study Group. N Engl J Med. 1991;324:1326-1331.
20. Cooper C, Barker DJ, Morris J, Briggs RS. Osteoporosis, falls, and age in fracture of the proximal femur. Br Med J (Clin Res Ed). 1987;295:13-15.
21. Gardsell P, Johnell O, Nilsson BE, Nilsson JA. The predictive value of fracture, disease, and falling tendency for fragility fractures in women. Calcif Tissue Int. 1989;45:327-330.
22. Dargent-Molina P, Schott AM, Hans D, et al. Separate and combined value of bone mass and gait speed measurements in screening for hip fracture risk: results from the EPIDOS study. Epidemiologie de l’Osteoporose. Osteoporos Int. 1999;9:188-192.
23. Clemson L, Cumming RG, Roland M. Case-control study of hazards in the home and risk of falls and hip fractures. Age Ageing. 1996;25:97-101.
24. Norton R, Campbell AJ, Lee-Joe T, Robinson E, Butler M. Circumstances of falls resulting in hip fractures among older people. J Am Geriatr Soc 1997;45:1108-1112.
25. Kannus P, Parkkari J, Niemi S, et al. Prevention of hip fracture in elderly people with use of a hip protector. N Engl J Med. 2000;343:1506-1513.
26. Cameron ID, Stafford B, Cumming RG, et al. Hip protectors improve falls self-efficacy. Age Ageing. 2000;29:57-62.
27. Hubacher M, Wettstein A. Acceptance of hip protectors for hip fracture prevention in nursing homes. Osteoporos Int. 2001;12:794-799.
28. NIH Consensus Development Panel on Optimal Calcium Intake. Optimal Calcium Intake. JAMA. 1994;272:1942-1948.
29. Morgan SL. Calcium and vitamin D in osteoporosis. Rheum Dis Clin North Am. 2001;27:101-130.
30. Tsourounis C. Clinical effects of phytoestrogens. Clin Obstet Gynecol. 2001;44:836-842.
31. Kelley GA. Aerobic exercise and bone density at the hip in postmenopausal women: a meta-analysis. Prev Med. 1998;27:798-807.
32. Marcus R. Role of exercise in preventing and treating osteoporosis. Rheum Dis Clin North Am. 2001;27:131-141,vi.-
33. Sinaki M, Mikkelsen BA. Postmenopausal spinal osteoporosis: flexion versus extension exercises. Arch Phys Med Rehabil. 1984;65:593-596.
34. Law M, Hackshaw AK. A meta-analysis of cigarette smoking, bone mineral density and risk of hip fracture: recognition of major effect. BMJ. 1997;315:841-846.
35. Felson DT, Zhang Y, Hannan MT, Kannel WB, Kiel DP. Alcohol intake and bone mineral density in elderly men and women. The Framingham Study. Am J Epidemiol. 1995;142:485-492.
36. Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA. 2001;285:320-323.
37. Marcus R, Wong M, Heath H, 3rd, Stock JL. Antiresorptive treatment of postmenopausal osteoporosis: comparison of study designs and outcomes in large clinical trials with fracture as an endpoint. Endocr Rev. 2002;23:16-37.
38. Lips P, Graafmans WC, Ooms ME, Bezemer PD, Bouter LM. Vitamin D supplementation and fracture incidence in elderly persons. A randomized, placebo-controlled clinical trial. Ann Intern Med. 1996;124:400-406.
39. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346:77-84.
40. North American Menopause Society. The role of calcium in peri- and postmenopausal women; consensus opinion of The North American Menopause Society. Menopause. 2001;8:84-95.
41. Williams CP, Child DF, Hudson PR, et al. Why oral calcium supplements may reduce renal stone disease: report of a clinical pilot study. J Clin Pathol. 2001;54:54-62.
42. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results for the Women’s Health Initiative Randomized Controlled Trial. JAMA. 2002;288:321-333.
43. Barrett-Connor E, Hendrix S, Ettinger B. International position paper on women’s health and menopause: a comprehensive approach. National Heart, Lung, and Blood Institute; 2002.
44. Greendale GA, Espeland M, Slone S, Marcus R, Barrett-Connor E. Bone mass response to discontinuation of long-term hormone replacement therapy: results from the Postmenopausal Estrogen/Progestin Interventions (PEPI) Safety Follow-up Study. Arch Intern Med. 2002;162:665-672.
45. Schairer C, Lubin J, Triosi R, Sturgeon S, Brinton L, Hoover R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000;283:485-491.
46. Ross R, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/Progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
48. Oger E, Scarabin PY. Risk of stroke among users of hormone replacement therapy. Annals d’Endocrinologie. 1999;60:232-241.
49. Simon J, Hsia J, Cauley JA, et al. Postmenopausal hormone therapy and risk of stroke: the Heart and Estrogen/Progestin Replacement Study (HERS). Circulation. 2001;103:638-642.
50. Viscoli C, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345:1243-1249.
51. Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344-1352.
52. Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect alendronate on risk of fracture in women with existing vertebral fractures. Lancet. 1996;348:1535-1541.
53. Heaney RP, Zizic TM, Fogelman I, et al. Risedronate reduces the risk of first vertebral fracture in osteoporotic women. Osteoporos Int. 2002;13:501-505.
54. Liberman UA, Weiss SR, Broll J, et al. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437-1443.
55. Sharpe M, Noble S, Spencer CM. Alendronate: an update of its use in osteoporosis. Drugs. 2001;61:999-1039.
56. Dunn CJ, Goa KL. Risedronate: a review of its pharmacological properties and clinical use in resorptive bone disease. Drugs. 2001;61:685-712.
57. Taggart H, Bolognese MA, Lindsay R, et al. Upper gastrointestinal tract safety of risedronate: a pooled analysis of 9 clinical trials. Mayo Clin Proc. 2002;77:262-270.
58. Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. JAMA. 1999;282:637-645.
59. Sarkar S, Mitlak B, Wong M, Stock J, Black D, Harper K. Raloxifene-induced fracture reductions not directly associated with BMD changes. J Bone Miner Res. 2002;1:1-10.
60. Barrett-Connor E. Raloxifene: risks and benefits. Ann N Y Acad Sci. 2001;949:295-303.
61. Cummings S, Duong T, Kenyon E, Cauley J, Whitehead M, Krueger K. Serum estradiol level and risk of breast cancer during treatment with raloxifene. JAMA. 2002;287:216-220.
62. Barrett-Connor E, Grady D, Sashegyi A, et al. Raloxifene and cardiovascular events in osteoporotic postmenopausal women: four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA. 2002;287:847-857.
63. Lyritis GP, Paspati I, Karachalios T, Ioakimidis D, Skarantavos G, Lyritis PG. Pain relief from nasal salmon calcitonin in osteoporotic vertebral crush fractures. A double blind, placebo-controlled clinical study. Acta Orthop Scand Suppl. 1997;275:112-114.
64. Pun KK, Chan LW. Analgesic effect of intranasal salmon calcitonin in the treatment of osteoporotic vertebral fractures. Clin Ther. 1989;11:205-209.
65. Chesnut CH, 3rd. Calcitonin in the prevention and treatment of osteoporosis. Osteoporos Int. 1993;3(suppl 1):206-207.
66. Crandall C. The role of serial bone mineral density testing for osteoporosis. J Womens Health Gend Based Med. 2001;10:887-895.
67. Cummings SR. The paradox of small changes in bone density and reductions in risk of fracture with raloxifene. Ann N Y Acad Sci. 2001;949:198-201.
68. Cummings SR, Karpf DB, Harris F, et al. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med. 2002;112:281-289.
69. Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in the elderly women. N Engl J Med. 1992;327:1637-1642.
70. McClung M, Geusens P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. N Engl J Med. 2001;344:333-340.
The last decade has witnessed important technological advances in the diagnosis of osteoporosis and an increase in therapeutic options. However, there is still considerable uncertainty about optimal strategies for screening and primary preventive treatment.
In 1994, a World Health Organization working group proposed that the diagnosis of osteoporosis be made when BMD, assessed by a dual-energy x-ray absorptiometry (DXA), is at least 2.5 standard deviations below the mean for young adult women (T-score) at the spine, hip, or wrist, or when a history of a traumatic fracture is present.2 A T-score between −1 and −2.5 is designated as osteopenia.
Osteoporosis is defined as “a skeletal disorder characterized by compromised bone strength predisposing to an increased risk of fracture.”1 While no accurate overall measurement of bone strength exists, bone mineral density (BMD) is frequently used as a proxy.
These facts underscore the importance of osteoporotic fractures:
- Only one third of patients regain their prior level of functioning after hip fracture, and one third are discharged to nursing homes.3
- About 1 in 5 patients dies within a year after a hip fracture.
- Vertebral fracture may result in chronic back pain and disability.4
- Existence of fracture greatly increases risk of subsequent fracture.5
- Direct medical costs for osteoporotic fractures are estimated at $13.8 billion in 1995 dollars.6
Prevalence of osteoporosis and fractures
Of American women over age 50 of all races, an estimated 15%, or 5 million, have osteoporosis (based on DXA T-score at the femoral neck) and an additional 40%, or 14 million, have osteopenia.7 In African Americans, the prevalence is about half that of whites.8 The prevalence of osteoporosis assessed by BMD testing increases with age—from 4% of white women aged 50 to 59 to 48% of women aged 80 to 89.9
At least 1 vertebral fracture, as indicated by radiographic criteria, has occurred in 5% of white women aged 50 to 59, and in 25% at age 80.3 The lifetime risk of hip fracture for 50-year-old white women and men is 14% and 5%, respectively; for African American women and men, 6% and 3%, respectively.3 Hip and symptomatic vertebral fractures occur mainly in women over 75,3,10 and the risk for wrist fractures increases starting in the late 50s.11
Age is a particularly important risk factor for hip fractures, reflecting deterioration in bone strength beyond that detectable with BMD testing. The National Osteoporosis Foundation12 observed that the 5-year risk of hip fracture for women with the same T-score (−3) increases dramatically with advancing age (Figure): from 2.4% at age 50 to 9.7% at age 90, with the steepest increase occurring during the 10 years between ages 70 (5.5%) and 80 (9%).
FIGURE
Five-year risk for hip fracture for women with T-score of −3 by age12
Bone mineral density testing
Screening recommendations
The clinical value of different screening strategies is not established, although recommendations have been made within guidelines and consensus statements that discuss prevention and treatment of osteoporosis. Guidelines are consistent in recommending that BMD screening be done only if results will influence treatment decisions. The US Preventive Services Task Force,13 The National Osteoporosis Foundation,14 and American Association of Clinical Endocrinologists15 recommend screening all women over 65, as well as younger women with risk factors for osteoporosis. The National Institutes of Health3 and the North American Menopause Society16 recommend an individualized decision-making approach to screening. The National Osteoporosis Foundation developed nomograms that integrate risk factors into decision-making for testing and treatment,12 which seem promising and merit testing in prospective studies.
Diagnostic testing
DXA. Although several technologies are available, DXA of the hip is considered the best predictor of hip fracture and an equivalent predictor of other fractures.10 The likelihood of making a diagnosis of osteoporosis based on BMD, however, varies and is related to type of test, equipment, anatomic site tested, number of sites tested, technique, and relevance of the reference range to the local population. For example, when the same group of people is tested with DXA equipment from different manufacturers, the proportion diagnosed with osteoporosis varies by as much as 15%.11
Quantitative ultrasound (QUS) and radiographic absorptiometry (RA). Testing by QUS of the heel and RA of the hand are less expensive than DXA and have become popular. While QUS of the heel has been shown to predict hip fracture and all nonvertebral fractures nearly as well as DXA,3,10 it does not highly correlate with DXA and appears to reflect other aspects of bone quality.10 Since QUS and DXA results frequently disagree and can cause confusion, DXA is the most appropriate test of BMD at present. If QUS and RA are used for screening, confirmation with DXA is recommended before therapy is initiated.
Calculations based on risk factors. In a comparison of strategies using risk factors to predict low BMD in postmenopausal women, 2 decision rules performed well: the Osteoporosis Risk Assessment Instrument, which is based on age and weight (Table 1),17 and the Simple Calculated Osteoporosis Risk Estimation (SCORE).17 Research to test these instruments with fracture rather than BMD as outcome is needed.18
Biochemical markers. Levels of markers in serum and/or urine reflect bone turnover and have potential use in diagnosing and monitoring therapy of osteoporosis. They are not yet widely available and have not been consistently associated with identifying patients at risk for fracture.10 They are not recommended at this time.
TABLE 1
Osteoporosis risk assessment instrument17
| Patient characteristic | Points |
|---|---|
| Age (years) | |
| 75 or older | 15 |
| 65 to 74 | 9 |
| 55 to 64 | 5 |
| 54 or younger | 0 |
| Weight | |
| <132 lb (60 kg) | 9 |
| 132 to 153.9 lb (60 to 70 kg) | 3 |
| >154 lb (>70 kg) | 0 |
| No current estrogen use | 2 |
| Total: | |
| Patients with a score of 9 or higher are at risk for diagnosis of osteoporosis by bone mineral density measurement. Sensitivity 97.5%, specificity 28%, positive predictive value 28%, negative predictive value 99.6%, given a 10% baseline risk of a bone mineral density 2.5 SD less than the mean. | |
Importance of primary prevention
At least half of bone strength is attributable to genetic factors12; modifiable factors may contribute almost equally as a group, and therefore warrant attention. Genetic risk factors include age, family history, female sex, low weight, small frame, and white or Asian race. Primary prevention efforts should begin in childhood and continue throughout the life span to maximize bone mass.3
Prevention efforts that target the modifiable factors described below should be a routine part of the health-maintenance visit.
Fall reduction
Falls are the direct cause of more than 90% of osteoporotic hip fractures,19 and the tendency to fall increases with age. Some studies have shown that, for women over age 70, the most important predictors of hip fractures are fall-related factors20,21 such as poor cognitive function, slow gait and otherwise impaired mobility, poor vision, drugs that impair alertness or balance, and history of falls. In women over 75, age and slow gait are equal to low BMD of the femoral neck as predictors of hip fracture.22 Unfortunately, labeling women as osteopenic or osteoporotic can cause fear of falling and lack of activity, leading to further acceleration of bone loss.10
Medications that interfere with balance or alertness should be avoided if possible. Environmental hazards such as loose rugs and uneven or slippery surfaces are also well-recognized modifiable risks for falls23,24 that should be eliminated. Hip protectors effectively reduce fractures in the frail elderly25 and can boost confidence for beneficial increases in physical activity levels,26 but they are often poorly accepted by patients.25,27 Other options include referral for gait training, home visits by a physician or nurse to identify problems in the home that increase the risk of falls, or providing information on home modification (such as installing bathtub rails, removing throw rugs, etc.).
Improvement of nutritional intake
Adequate consumption of calcium is essential for bone health. Calcium balance also can be adversely affected by dietary habits, including high intake of protein, phosphorus, and sodium, although these effects appear to be less important when dietary calcium is sufficient.3 The recommended calcium intake for postmenopausal women (1200–1500 mg/day)28 can be met with food sources, but supplements should be added if needed. Most postmenopausal women in the United States consume only about 600 mg/day.28 High-calcium foods include milk (290–300 mg/cup), sardines in oil, with bones (370 mg/3 oz), yogurt (300–500 mg depending on container size), cheese (165–270 mg/slice), canned salmon, with bones (170–210 mg/3 oz), broccoli (160–180 mg/cup), and tofu (144–155 mg/4 oz).15
Vitamin D is essential for intestinal absorption of calcium. The recommended intake for women is 400 IU/day for ages 51 to 70, 600 IU/day over age 70, and 800 IU/day for all high-risk women, including those who are homebound, institutionalized, on chronic glucocorticoids, or who live in northern latitudes and therefore have limited exposure to sunlight.29 Sources of vitamin D include sunlight, vitamin D–fortified foods, fish oils, and supplements. Multivitamins typically contain 400 IU of vitamin D.
Phytoestrogens, particularly in the form of soy products, have received attention for bone health. Overall, studies do not support the use of soy foods to prevent osteoporosis.3 A well-designed trial in postmenopausal women found that ipriflavone, a synthetic phytoestrogen, did not decrease bone loss.30 Furthermore, use was associated with subclinical lymphocytopenia.
Regular exercise
Weight-bearing physical activity such as walking or running in early life contributes to higher peak bone mass. Limited data suggest weight-bearing exercise in postmenopausal women produces small increases in bone density at the hip31 and improvement in balance and strength.32 For women with established osteoporosis, activities that place an anterior load on the vertebral bodies, such as forward flexion exercises, are associated with an increased incidence of new vertebral deformities, and patients should be advised to avoid them.33
Avoidance of adverse health habits
Current smoking, compared with never smoking, doubles the risk of hip fracture.34 Consumption of more than 1 alcoholic drink/day or more than 7/week is associated with osteoporosis and fracture, while moderate consumption of 1 drink/day or less is associated with decreased risk.35 Excessive caffeine intake is also associated with increased osteoporosis risk and should be avoided. This effect appears to result from substitution of calcium-containing beverages such as milk or fortified orange juice with caffeinated, non–calcium-containing beverages such as colas.
Treatment for fracture prevention and pain relief
The goals of therapy for osteoporosis are fracture prevention and pain relief to maximize physical function.15 Prior fracture is associated with a fivefold risk of future fractures.5 About 20% of women who experience a vertebral fracture have another fracture within 1 year.36 Currently available therapies (Table 2) are antiresorptive: they slow bone turnover and allow bone formation to exceed resorption. Trials of antiresorptive agents in elderly women with osteoporosis and baseline vertebral fracture demonstrate that 1 new vertebral fracture is prevented for each 12 to 35 women treated for 2 to 3 years.14,37Table 3 summarizes the results of key treatment studies and provides information on the number of women that need to be treated (NNT) for the study period to prevent 1 fracture.
TABLE 2
Drug therapy for prevention and treatment of postmenopausal osteoporosis
| Drug (trade name) | Indication and dosage | Possible side effects (% of patients) | Cost per month* |
|---|---|---|---|
| Calcium and vitamin D (generic, Tums, Citracal, and others) | Prevention and treatment: 1200–1500 mg/day calcium and 800 IU/day vitamin D | Nausea, dyspepsia (uncommon), constipation (10%) | $5 (both) |
| Estrogen† (Premarin, Ogen, Estrace, Estraderm, and others) | Prevention: 0.625 mg/day conjugated equine estrogen or the equivalent; 0.3 mg/day may be effective | Nausea, breast tenderness, vaginal bleeding, mood alterations, headache, bloating | $14–$28 |
| Alendronate (Fosamax) | Prevention and treatment: 5 mg/day or 35 mg/week | Nausea, dyspepsia esophageal irritation | $67 |
| Risedronate (Actonel) | Prevention and treatment: 5 mg/day or 35 mg/week | Abdominal pain, nausea, diarrhea | $67 |
| Raloxifene (Evista) | Treatment: 60 mg/day | Hot flashes (6%), leg cramps (3%) | $70 |
| Calcitonin nasal spray (Miacalcin) | Treatment: 200 IU/day (1 spray in 1 nostril per day) | Rhinitis (5%), epistaxis, sinusitis | $66 |
| *Average wholesale cost to the pharmacy for 30 days of therapy; (Drug Topics Red Book. Montvale, NJ; Medical Economics Co., Inc, 2002.) | |||
| †Women with a uterus need to take a progestin such as medroxyprogesterone acetate (Provera $30/month, generic $9/month) or a combination estrogen/progestin product (Prempro $33/month, FemHRT $26/month). | |||
TABLE 3
Clinical trials of drug therapy for the prevention of fracture in postmenopausal women with osteoporosis
| Trial, year | Therapy | Outcome prevented | Number needed to treat for n years |
|---|---|---|---|
| Elderly, postmenopausal women | |||
| Chapuy, 199267 | Calcium/vitamin D | Hip fracture | 48 women for 1.5 years |
| Postmenopausal women | |||
| WHI, 200242 | Hormone replacement therapy | Hip fracture | 2000 women for 5 years |
| Postmenopausal women with osteoporosis | |||
| Ettinger, 199956 | Raloxifene | Vertebral fracture | 29 women for 3 years |
| Liberman, 199554 | Alendronate | Vertebral fracture | 34 women for 3 years |
| Heaney, 200253 | Risendronate | Vertebral fracture | 15 women for 3 years |
| McClung, 200170 | Risedronate | Hip fracture | 91 women for 3 years |
| Postmenopausal women with osteoporosis and previous vertebral racture | |||
| Harris, 199951 | Risedronate | Vertebral fracture | 20 women for 3 years |
| Black, 199652 | Alendronate | Vertebral fracture | 35 women for 3 years |
| Black, 199652 | Alendronate | Hip fracture | 86 women for 3 years |
Calcium and vitamin D
Calcium with or without vitamin D has been reported to positively affect fracture incidence.14 Vitamin D alone does not decrease the incidence of hip fractures.38 Calcium, 1200 to 1500 mg/day, and vitamin D, 800 IU/day, should be used concurrently with other forms of pharmacologic treatment. Calcium supplements are best absorbed with meals; for maximum absorption, calcium should be taken in doses of 500 mg or less.29,39
Minor gastrointestinal adverse effects may occur (most often constipation, 10%),14 which is often resolved by switching to a different preparation.40 Calcium in doses up to 1500 mg/day does not increase the risk for renal calculi and may, in fact, decrease risk.40,41 Calcium interferes with the absorption of certain medications, including tetracycline and quinolone antibiotics, which should be taken several hours apart from calcium. Calcium carbonate requires an acidic environment to dissolve. Patients taking stomach acid–suppressant therapy should use calcium citrate because it does not require an acidic environment for dissolution or should take their calcium supplement with meals. Traces of lead may be present in natural sources of calcium (bone meal, oyster shell, limestone, and dolomite), but can be avoided by use of over-the-counter calcium carbonate tablets (Tums).
Estrogen
Data from the Women’s Health Initiative (WHI) demonstrated that hormone replacement therapy (HRT) combining an estrogen and a progestin reduced hip and vertebral fractures by 1 in 2000 women per year and reduced all fractures by 1 in 333 women per year.42 Estrogen has a positive effect on BMD whether given in early or late postmenopause.43 Rapid bone loss as assessed by BMD does not occur after stopping HRT.44 However, in elderly women who have never used HRT, BMD is similar to those who have used it for 10 years and then stopped for at least 10 years.43 The effect of short-term (< 5 years) HRT during perimenopause on lifetime risk of osteoporotic fracture is unknown.
Common side effects (Table 2) can often be addressed by altering dosages, specific products, or regimens. The risk for breast cancer increases with duration of treatment and with combination HRT, compared with estrogen-only preparations.42,45,46 Combination HRT products are, however, essential for endometrial protection in women who have a uterus. The WHI reported an increase in breast cancer cases of 1 in 1250 women per year during an average 5-year follow-up.42 An increased risk for venous thromboembolism of 1 in 555 women per year was also observed with HRT use. Both the Heart and Estrogen/Progestin Study47 and the WHI studies found that venous thromboembolism occurred more frequently in the first 2 years of HRT use. An increased risk of myocardial infarction in the first 2 years of use was also noted among women with coronary heart disease47 and those without heart disease (1 in 1429 women per year).42 A small increase in stroke risk has also been documented.42,48-50 Contraindications to estrogen use include active thromboembolism, estrogen-related cancers, and liver disease.
Bisphosphonates
Two bisphosphonates, alendronate and risedronate, are approved in the United States for both prevention and treatment of postmenopausal osteoporosis. Clinical trials have demonstrated that both rapidly reduce the risk for symptomatic fractures in women with previous fracture and osteoporosis.37,51,52 The extent of fracture reduction is significant: Recent studies have shown that, over 3 years, the number of patients who would need to be treated with risedronate to prevent a vertebral fracture is 15, with alendronate, 34.53,54 Prevention of fractures among women without a prior vertebral fracture is less well established. No published data demonstrate that one bisphosphonate is more effective than another at preventing clinical fractures. A bisphosphonate is, therefore, the drug of choice for severe osteoporosis.
Bisphosphonates are generally well tolerated.55,56 However, postmarketing surveillance has demonstrated esophagitis and esophageal ulcer associated with alendronate.55 A pooled analysis of trials of risedronate found no increase in upper gastrointestinal (GI) adverse events even in patients with history of peptic ulcers, heartburn, and esophagitis, or among those taking nonsteroidal anti-inflammatory drugs, including aspirin.57
Oral bisphosphonates are not well absorbed (less than 1% of each dose).55,56 Therefore, to maximize absorption and to decrease the likelihood of adverse GI effects, the manufacturers of both bisphosphonates recommend that patients take the medication with a full glass of water, remain upright (sitting or standing) for at least 30 minutes following the dose, and not recline until food is consumed. Both bisphosphonates should be used with caution in patients with active GI disorders. Bisphosphonates are eliminated via the kidney and are not recommended for patients with a creatinine clearance below 30 mL/min.
Selective estrogen receptor modulators (SERMs)
Raloxifene is currently the only selective estrogen receptor modulator approved in the United States for prevention and treatment of osteoporosis. It has been shown to significantly decrease new vertebral fractures in women with a previous history of fracture and osteoporosis.58 The magnitude of fracture reduction is similar to that of bisphosphonates, although improvement in BMD is less marked.59 Raloxifene may confer other benefits. It has been shown to reduce the risk of breast cancer60,61 except in women with low estradiol levels,60 and may reduce the risk of myocardial infarction in women at high risk.60 Raloxifene does not increase the risk for endometrial cancer.60
Hot flashes and leg cramps are relatively common side effects of raloxifene.60 The observed risk of venous thromboembolism, 1 in 465 women per year during 3 years of treatment, is similar to that observed with HRT.60 Raloxifene is teratogenic and should not be used in premenopausal women.
Salmon calcitonin
Salmon calcitonin has demonstrated an analgesic effect for osteoporotic fracture.63,64 A large trial of salmon calcitonin at dosages of 100, 200, and 400 IU/day versus placebo found that salmon calcitonin at 200 IU/day decreased new vertebral fractures among women with a previous osteoporotic vertebral fracture based on radiographic assessment.65 No benefit was observed at the 100 and 400 IU/day dosages. The effect of calcitonin on clinical (symptomatic) fractures has not been reported. Calcitonin is approved for use in treatment, but not prevention, of osteoporosis.
Nasal calcitonin can cause minor rhinitis symptoms.30 Saline nasal solution may be useful to prevent or resolve irritation and dryness. Administration using alternate nostrils helps minimize local side effects. Unopened bottles (14 doses) must be stored in the refrigerator. Open bottles are stable at room temperature for up to 30 days.
Monitoring therapy
The value of serial densitometry to monitor the therapy of individual patients has not been established by randomized trials comparing different monitoring intervals or monitoring versus no monitoring.10,66 One important limitation is the relative imprecision of BMD testing: it takes almost a year to detect a 3% change in BMD.10 Disconcerting decreases in BMD scores are seen in yearly testing and may be offset by larger increases later, without a change in therapy. In studies of alendronate and raloxifene, disproportionately large fracture reductions cannot be explained by improvement in BMD alone.66,68 Bone densitometry is therefore not recommended until the patient has been treated for 2 years, and is of uncertain value beyond that point.
The last decade has witnessed important technological advances in the diagnosis of osteoporosis and an increase in therapeutic options. However, there is still considerable uncertainty about optimal strategies for screening and primary preventive treatment.
In 1994, a World Health Organization working group proposed that the diagnosis of osteoporosis be made when BMD, assessed by a dual-energy x-ray absorptiometry (DXA), is at least 2.5 standard deviations below the mean for young adult women (T-score) at the spine, hip, or wrist, or when a history of a traumatic fracture is present.2 A T-score between −1 and −2.5 is designated as osteopenia.
Osteoporosis is defined as “a skeletal disorder characterized by compromised bone strength predisposing to an increased risk of fracture.”1 While no accurate overall measurement of bone strength exists, bone mineral density (BMD) is frequently used as a proxy.
These facts underscore the importance of osteoporotic fractures:
- Only one third of patients regain their prior level of functioning after hip fracture, and one third are discharged to nursing homes.3
- About 1 in 5 patients dies within a year after a hip fracture.
- Vertebral fracture may result in chronic back pain and disability.4
- Existence of fracture greatly increases risk of subsequent fracture.5
- Direct medical costs for osteoporotic fractures are estimated at $13.8 billion in 1995 dollars.6
Prevalence of osteoporosis and fractures
Of American women over age 50 of all races, an estimated 15%, or 5 million, have osteoporosis (based on DXA T-score at the femoral neck) and an additional 40%, or 14 million, have osteopenia.7 In African Americans, the prevalence is about half that of whites.8 The prevalence of osteoporosis assessed by BMD testing increases with age—from 4% of white women aged 50 to 59 to 48% of women aged 80 to 89.9
At least 1 vertebral fracture, as indicated by radiographic criteria, has occurred in 5% of white women aged 50 to 59, and in 25% at age 80.3 The lifetime risk of hip fracture for 50-year-old white women and men is 14% and 5%, respectively; for African American women and men, 6% and 3%, respectively.3 Hip and symptomatic vertebral fractures occur mainly in women over 75,3,10 and the risk for wrist fractures increases starting in the late 50s.11
Age is a particularly important risk factor for hip fractures, reflecting deterioration in bone strength beyond that detectable with BMD testing. The National Osteoporosis Foundation12 observed that the 5-year risk of hip fracture for women with the same T-score (−3) increases dramatically with advancing age (Figure): from 2.4% at age 50 to 9.7% at age 90, with the steepest increase occurring during the 10 years between ages 70 (5.5%) and 80 (9%).
FIGURE
Five-year risk for hip fracture for women with T-score of −3 by age12
Bone mineral density testing
Screening recommendations
The clinical value of different screening strategies is not established, although recommendations have been made within guidelines and consensus statements that discuss prevention and treatment of osteoporosis. Guidelines are consistent in recommending that BMD screening be done only if results will influence treatment decisions. The US Preventive Services Task Force,13 The National Osteoporosis Foundation,14 and American Association of Clinical Endocrinologists15 recommend screening all women over 65, as well as younger women with risk factors for osteoporosis. The National Institutes of Health3 and the North American Menopause Society16 recommend an individualized decision-making approach to screening. The National Osteoporosis Foundation developed nomograms that integrate risk factors into decision-making for testing and treatment,12 which seem promising and merit testing in prospective studies.
Diagnostic testing
DXA. Although several technologies are available, DXA of the hip is considered the best predictor of hip fracture and an equivalent predictor of other fractures.10 The likelihood of making a diagnosis of osteoporosis based on BMD, however, varies and is related to type of test, equipment, anatomic site tested, number of sites tested, technique, and relevance of the reference range to the local population. For example, when the same group of people is tested with DXA equipment from different manufacturers, the proportion diagnosed with osteoporosis varies by as much as 15%.11
Quantitative ultrasound (QUS) and radiographic absorptiometry (RA). Testing by QUS of the heel and RA of the hand are less expensive than DXA and have become popular. While QUS of the heel has been shown to predict hip fracture and all nonvertebral fractures nearly as well as DXA,3,10 it does not highly correlate with DXA and appears to reflect other aspects of bone quality.10 Since QUS and DXA results frequently disagree and can cause confusion, DXA is the most appropriate test of BMD at present. If QUS and RA are used for screening, confirmation with DXA is recommended before therapy is initiated.
Calculations based on risk factors. In a comparison of strategies using risk factors to predict low BMD in postmenopausal women, 2 decision rules performed well: the Osteoporosis Risk Assessment Instrument, which is based on age and weight (Table 1),17 and the Simple Calculated Osteoporosis Risk Estimation (SCORE).17 Research to test these instruments with fracture rather than BMD as outcome is needed.18
Biochemical markers. Levels of markers in serum and/or urine reflect bone turnover and have potential use in diagnosing and monitoring therapy of osteoporosis. They are not yet widely available and have not been consistently associated with identifying patients at risk for fracture.10 They are not recommended at this time.
TABLE 1
Osteoporosis risk assessment instrument17
| Patient characteristic | Points |
|---|---|
| Age (years) | |
| 75 or older | 15 |
| 65 to 74 | 9 |
| 55 to 64 | 5 |
| 54 or younger | 0 |
| Weight | |
| <132 lb (60 kg) | 9 |
| 132 to 153.9 lb (60 to 70 kg) | 3 |
| >154 lb (>70 kg) | 0 |
| No current estrogen use | 2 |
| Total: | |
| Patients with a score of 9 or higher are at risk for diagnosis of osteoporosis by bone mineral density measurement. Sensitivity 97.5%, specificity 28%, positive predictive value 28%, negative predictive value 99.6%, given a 10% baseline risk of a bone mineral density 2.5 SD less than the mean. | |
Importance of primary prevention
At least half of bone strength is attributable to genetic factors12; modifiable factors may contribute almost equally as a group, and therefore warrant attention. Genetic risk factors include age, family history, female sex, low weight, small frame, and white or Asian race. Primary prevention efforts should begin in childhood and continue throughout the life span to maximize bone mass.3
Prevention efforts that target the modifiable factors described below should be a routine part of the health-maintenance visit.
Fall reduction
Falls are the direct cause of more than 90% of osteoporotic hip fractures,19 and the tendency to fall increases with age. Some studies have shown that, for women over age 70, the most important predictors of hip fractures are fall-related factors20,21 such as poor cognitive function, slow gait and otherwise impaired mobility, poor vision, drugs that impair alertness or balance, and history of falls. In women over 75, age and slow gait are equal to low BMD of the femoral neck as predictors of hip fracture.22 Unfortunately, labeling women as osteopenic or osteoporotic can cause fear of falling and lack of activity, leading to further acceleration of bone loss.10
Medications that interfere with balance or alertness should be avoided if possible. Environmental hazards such as loose rugs and uneven or slippery surfaces are also well-recognized modifiable risks for falls23,24 that should be eliminated. Hip protectors effectively reduce fractures in the frail elderly25 and can boost confidence for beneficial increases in physical activity levels,26 but they are often poorly accepted by patients.25,27 Other options include referral for gait training, home visits by a physician or nurse to identify problems in the home that increase the risk of falls, or providing information on home modification (such as installing bathtub rails, removing throw rugs, etc.).
Improvement of nutritional intake
Adequate consumption of calcium is essential for bone health. Calcium balance also can be adversely affected by dietary habits, including high intake of protein, phosphorus, and sodium, although these effects appear to be less important when dietary calcium is sufficient.3 The recommended calcium intake for postmenopausal women (1200–1500 mg/day)28 can be met with food sources, but supplements should be added if needed. Most postmenopausal women in the United States consume only about 600 mg/day.28 High-calcium foods include milk (290–300 mg/cup), sardines in oil, with bones (370 mg/3 oz), yogurt (300–500 mg depending on container size), cheese (165–270 mg/slice), canned salmon, with bones (170–210 mg/3 oz), broccoli (160–180 mg/cup), and tofu (144–155 mg/4 oz).15
Vitamin D is essential for intestinal absorption of calcium. The recommended intake for women is 400 IU/day for ages 51 to 70, 600 IU/day over age 70, and 800 IU/day for all high-risk women, including those who are homebound, institutionalized, on chronic glucocorticoids, or who live in northern latitudes and therefore have limited exposure to sunlight.29 Sources of vitamin D include sunlight, vitamin D–fortified foods, fish oils, and supplements. Multivitamins typically contain 400 IU of vitamin D.
Phytoestrogens, particularly in the form of soy products, have received attention for bone health. Overall, studies do not support the use of soy foods to prevent osteoporosis.3 A well-designed trial in postmenopausal women found that ipriflavone, a synthetic phytoestrogen, did not decrease bone loss.30 Furthermore, use was associated with subclinical lymphocytopenia.
Regular exercise
Weight-bearing physical activity such as walking or running in early life contributes to higher peak bone mass. Limited data suggest weight-bearing exercise in postmenopausal women produces small increases in bone density at the hip31 and improvement in balance and strength.32 For women with established osteoporosis, activities that place an anterior load on the vertebral bodies, such as forward flexion exercises, are associated with an increased incidence of new vertebral deformities, and patients should be advised to avoid them.33
Avoidance of adverse health habits
Current smoking, compared with never smoking, doubles the risk of hip fracture.34 Consumption of more than 1 alcoholic drink/day or more than 7/week is associated with osteoporosis and fracture, while moderate consumption of 1 drink/day or less is associated with decreased risk.35 Excessive caffeine intake is also associated with increased osteoporosis risk and should be avoided. This effect appears to result from substitution of calcium-containing beverages such as milk or fortified orange juice with caffeinated, non–calcium-containing beverages such as colas.
Treatment for fracture prevention and pain relief
The goals of therapy for osteoporosis are fracture prevention and pain relief to maximize physical function.15 Prior fracture is associated with a fivefold risk of future fractures.5 About 20% of women who experience a vertebral fracture have another fracture within 1 year.36 Currently available therapies (Table 2) are antiresorptive: they slow bone turnover and allow bone formation to exceed resorption. Trials of antiresorptive agents in elderly women with osteoporosis and baseline vertebral fracture demonstrate that 1 new vertebral fracture is prevented for each 12 to 35 women treated for 2 to 3 years.14,37Table 3 summarizes the results of key treatment studies and provides information on the number of women that need to be treated (NNT) for the study period to prevent 1 fracture.
TABLE 2
Drug therapy for prevention and treatment of postmenopausal osteoporosis
| Drug (trade name) | Indication and dosage | Possible side effects (% of patients) | Cost per month* |
|---|---|---|---|
| Calcium and vitamin D (generic, Tums, Citracal, and others) | Prevention and treatment: 1200–1500 mg/day calcium and 800 IU/day vitamin D | Nausea, dyspepsia (uncommon), constipation (10%) | $5 (both) |
| Estrogen† (Premarin, Ogen, Estrace, Estraderm, and others) | Prevention: 0.625 mg/day conjugated equine estrogen or the equivalent; 0.3 mg/day may be effective | Nausea, breast tenderness, vaginal bleeding, mood alterations, headache, bloating | $14–$28 |
| Alendronate (Fosamax) | Prevention and treatment: 5 mg/day or 35 mg/week | Nausea, dyspepsia esophageal irritation | $67 |
| Risedronate (Actonel) | Prevention and treatment: 5 mg/day or 35 mg/week | Abdominal pain, nausea, diarrhea | $67 |
| Raloxifene (Evista) | Treatment: 60 mg/day | Hot flashes (6%), leg cramps (3%) | $70 |
| Calcitonin nasal spray (Miacalcin) | Treatment: 200 IU/day (1 spray in 1 nostril per day) | Rhinitis (5%), epistaxis, sinusitis | $66 |
| *Average wholesale cost to the pharmacy for 30 days of therapy; (Drug Topics Red Book. Montvale, NJ; Medical Economics Co., Inc, 2002.) | |||
| †Women with a uterus need to take a progestin such as medroxyprogesterone acetate (Provera $30/month, generic $9/month) or a combination estrogen/progestin product (Prempro $33/month, FemHRT $26/month). | |||
TABLE 3
Clinical trials of drug therapy for the prevention of fracture in postmenopausal women with osteoporosis
| Trial, year | Therapy | Outcome prevented | Number needed to treat for n years |
|---|---|---|---|
| Elderly, postmenopausal women | |||
| Chapuy, 199267 | Calcium/vitamin D | Hip fracture | 48 women for 1.5 years |
| Postmenopausal women | |||
| WHI, 200242 | Hormone replacement therapy | Hip fracture | 2000 women for 5 years |
| Postmenopausal women with osteoporosis | |||
| Ettinger, 199956 | Raloxifene | Vertebral fracture | 29 women for 3 years |
| Liberman, 199554 | Alendronate | Vertebral fracture | 34 women for 3 years |
| Heaney, 200253 | Risendronate | Vertebral fracture | 15 women for 3 years |
| McClung, 200170 | Risedronate | Hip fracture | 91 women for 3 years |
| Postmenopausal women with osteoporosis and previous vertebral racture | |||
| Harris, 199951 | Risedronate | Vertebral fracture | 20 women for 3 years |
| Black, 199652 | Alendronate | Vertebral fracture | 35 women for 3 years |
| Black, 199652 | Alendronate | Hip fracture | 86 women for 3 years |
Calcium and vitamin D
Calcium with or without vitamin D has been reported to positively affect fracture incidence.14 Vitamin D alone does not decrease the incidence of hip fractures.38 Calcium, 1200 to 1500 mg/day, and vitamin D, 800 IU/day, should be used concurrently with other forms of pharmacologic treatment. Calcium supplements are best absorbed with meals; for maximum absorption, calcium should be taken in doses of 500 mg or less.29,39
Minor gastrointestinal adverse effects may occur (most often constipation, 10%),14 which is often resolved by switching to a different preparation.40 Calcium in doses up to 1500 mg/day does not increase the risk for renal calculi and may, in fact, decrease risk.40,41 Calcium interferes with the absorption of certain medications, including tetracycline and quinolone antibiotics, which should be taken several hours apart from calcium. Calcium carbonate requires an acidic environment to dissolve. Patients taking stomach acid–suppressant therapy should use calcium citrate because it does not require an acidic environment for dissolution or should take their calcium supplement with meals. Traces of lead may be present in natural sources of calcium (bone meal, oyster shell, limestone, and dolomite), but can be avoided by use of over-the-counter calcium carbonate tablets (Tums).
Estrogen
Data from the Women’s Health Initiative (WHI) demonstrated that hormone replacement therapy (HRT) combining an estrogen and a progestin reduced hip and vertebral fractures by 1 in 2000 women per year and reduced all fractures by 1 in 333 women per year.42 Estrogen has a positive effect on BMD whether given in early or late postmenopause.43 Rapid bone loss as assessed by BMD does not occur after stopping HRT.44 However, in elderly women who have never used HRT, BMD is similar to those who have used it for 10 years and then stopped for at least 10 years.43 The effect of short-term (< 5 years) HRT during perimenopause on lifetime risk of osteoporotic fracture is unknown.
Common side effects (Table 2) can often be addressed by altering dosages, specific products, or regimens. The risk for breast cancer increases with duration of treatment and with combination HRT, compared with estrogen-only preparations.42,45,46 Combination HRT products are, however, essential for endometrial protection in women who have a uterus. The WHI reported an increase in breast cancer cases of 1 in 1250 women per year during an average 5-year follow-up.42 An increased risk for venous thromboembolism of 1 in 555 women per year was also observed with HRT use. Both the Heart and Estrogen/Progestin Study47 and the WHI studies found that venous thromboembolism occurred more frequently in the first 2 years of HRT use. An increased risk of myocardial infarction in the first 2 years of use was also noted among women with coronary heart disease47 and those without heart disease (1 in 1429 women per year).42 A small increase in stroke risk has also been documented.42,48-50 Contraindications to estrogen use include active thromboembolism, estrogen-related cancers, and liver disease.
Bisphosphonates
Two bisphosphonates, alendronate and risedronate, are approved in the United States for both prevention and treatment of postmenopausal osteoporosis. Clinical trials have demonstrated that both rapidly reduce the risk for symptomatic fractures in women with previous fracture and osteoporosis.37,51,52 The extent of fracture reduction is significant: Recent studies have shown that, over 3 years, the number of patients who would need to be treated with risedronate to prevent a vertebral fracture is 15, with alendronate, 34.53,54 Prevention of fractures among women without a prior vertebral fracture is less well established. No published data demonstrate that one bisphosphonate is more effective than another at preventing clinical fractures. A bisphosphonate is, therefore, the drug of choice for severe osteoporosis.
Bisphosphonates are generally well tolerated.55,56 However, postmarketing surveillance has demonstrated esophagitis and esophageal ulcer associated with alendronate.55 A pooled analysis of trials of risedronate found no increase in upper gastrointestinal (GI) adverse events even in patients with history of peptic ulcers, heartburn, and esophagitis, or among those taking nonsteroidal anti-inflammatory drugs, including aspirin.57
Oral bisphosphonates are not well absorbed (less than 1% of each dose).55,56 Therefore, to maximize absorption and to decrease the likelihood of adverse GI effects, the manufacturers of both bisphosphonates recommend that patients take the medication with a full glass of water, remain upright (sitting or standing) for at least 30 minutes following the dose, and not recline until food is consumed. Both bisphosphonates should be used with caution in patients with active GI disorders. Bisphosphonates are eliminated via the kidney and are not recommended for patients with a creatinine clearance below 30 mL/min.
Selective estrogen receptor modulators (SERMs)
Raloxifene is currently the only selective estrogen receptor modulator approved in the United States for prevention and treatment of osteoporosis. It has been shown to significantly decrease new vertebral fractures in women with a previous history of fracture and osteoporosis.58 The magnitude of fracture reduction is similar to that of bisphosphonates, although improvement in BMD is less marked.59 Raloxifene may confer other benefits. It has been shown to reduce the risk of breast cancer60,61 except in women with low estradiol levels,60 and may reduce the risk of myocardial infarction in women at high risk.60 Raloxifene does not increase the risk for endometrial cancer.60
Hot flashes and leg cramps are relatively common side effects of raloxifene.60 The observed risk of venous thromboembolism, 1 in 465 women per year during 3 years of treatment, is similar to that observed with HRT.60 Raloxifene is teratogenic and should not be used in premenopausal women.
Salmon calcitonin
Salmon calcitonin has demonstrated an analgesic effect for osteoporotic fracture.63,64 A large trial of salmon calcitonin at dosages of 100, 200, and 400 IU/day versus placebo found that salmon calcitonin at 200 IU/day decreased new vertebral fractures among women with a previous osteoporotic vertebral fracture based on radiographic assessment.65 No benefit was observed at the 100 and 400 IU/day dosages. The effect of calcitonin on clinical (symptomatic) fractures has not been reported. Calcitonin is approved for use in treatment, but not prevention, of osteoporosis.
Nasal calcitonin can cause minor rhinitis symptoms.30 Saline nasal solution may be useful to prevent or resolve irritation and dryness. Administration using alternate nostrils helps minimize local side effects. Unopened bottles (14 doses) must be stored in the refrigerator. Open bottles are stable at room temperature for up to 30 days.
Monitoring therapy
The value of serial densitometry to monitor the therapy of individual patients has not been established by randomized trials comparing different monitoring intervals or monitoring versus no monitoring.10,66 One important limitation is the relative imprecision of BMD testing: it takes almost a year to detect a 3% change in BMD.10 Disconcerting decreases in BMD scores are seen in yearly testing and may be offset by larger increases later, without a change in therapy. In studies of alendronate and raloxifene, disproportionately large fracture reductions cannot be explained by improvement in BMD alone.66,68 Bone densitometry is therefore not recommended until the patient has been treated for 2 years, and is of uncertain value beyond that point.
1. Osteoporosis Prevention, Diagnosis, and Therapy. NIH Consensus Statement Online 2000 March 27-29; [cited 9/2/2]; 17(1):1-36.
2. World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. WHO Technical Report Series 843. Geneva: World Health Organization; 1994.
3. NIH Consensus Development Panel on Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285:785-795.
4. Gold DT. The clinical impact of vertebral fractures: quality of life in women with osteoporosis. Bone. 1996;18(suppl 3):185S-189S.
5. Black DM, Arden NK, Palermo I, Pearson J, Cummings SR. Prevalent vertebral deformities predict hip fractures and new vertebral deformities but not wrist fractures. Study of Osteoporotic Fractures Research Group. J Bone Miner Res. 1999;14:821-828.
6. Ray NF, Chan JK, Thamer M, Melton LJ, 3rd. Medical expenditures for the treatment of osteoporotic fractures in the United States in 1995: report from the National Osteoporosis Foundation. J Bone Miner Res. 1997;12:24-35.
7. Looker AC, Wahner HW, Dunn WL, et al. Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int. 1998;8:468-489.
8. Aloia JF, Vaswani A, Yeh JK, Flaster E. Risk for osteoporosis in black women. Calcif Tissue Int. 1996;59:415-423.
9. Melton LJ. How many women have osteoporosis now? J Bone Miner Res. 1995;10:175-177.
10. Nelson HD, Morris CD, Mahon S, Carney N, Nygren PM, Helfand M. Osteoporosis in postmenopausal women: diagnosis and monitoring. Evidence Report/Technology Assessment Number 28. Rockville, MD: Agency for Healthcare Research and Quality; November 2001. 01-E032.
11. Melton LJ, 3rd, Amadio PC, Crowson CS, O’Fallon WM. Long-term trends in the incidence of distal forearm fractures. Osteoporos Int. 1998;8:341-348.
12. National Osteoporosis Foundation. Osteoporosis: review of the evidence for prevention, diagnosis, and treatment and cost-effectiveness analysis. Osteoporos Int. 1998;8(suppl):S7-S80.
13. U.S. Preventive Services Task Force. Screening for Osteoporosis in Postmenopausal Women. September 2002. Originally in Annals of Internal Medicine 2002;137:526-8.Agency for Healthcare Research and Quality, Rockville, MD. http://www.ahrq.gov/clinic/3rduspstf/osteoporosis/osteorr.htm.
14. Physician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 1998.
15. American Association of Clinical Endocrinologists 2001 medical guidelines for clinical practice for the prevention and management of osteoporosis. Endocr Pract. 2001;7:293-312.
16. North American Menopause Society. Management of postmenopausal osteoporosis: position statement of The North American Menopause Society. Menopause. 2002;9:84-101.
17. Cadarette SM, Jaglal SB, Murray TM, McIsaac WJ, Joseph L, Brown JP. Evaluation of decision rules for referring women for bone densitometry by dual energy x-ray absorptiometry. JAMA. 2001;286:57-63.
18. Wasnich RD. Consensus and the T-score fallacy. Clin Rheumatol. 1997;16:337-339.
19. Grisso JA, Kelsey JL, Strom BL, et al. Risk factors for falls as a cause of hip fracture in women.The Northeast Hip Fracture Study Group. N Engl J Med. 1991;324:1326-1331.
20. Cooper C, Barker DJ, Morris J, Briggs RS. Osteoporosis, falls, and age in fracture of the proximal femur. Br Med J (Clin Res Ed). 1987;295:13-15.
21. Gardsell P, Johnell O, Nilsson BE, Nilsson JA. The predictive value of fracture, disease, and falling tendency for fragility fractures in women. Calcif Tissue Int. 1989;45:327-330.
22. Dargent-Molina P, Schott AM, Hans D, et al. Separate and combined value of bone mass and gait speed measurements in screening for hip fracture risk: results from the EPIDOS study. Epidemiologie de l’Osteoporose. Osteoporos Int. 1999;9:188-192.
23. Clemson L, Cumming RG, Roland M. Case-control study of hazards in the home and risk of falls and hip fractures. Age Ageing. 1996;25:97-101.
24. Norton R, Campbell AJ, Lee-Joe T, Robinson E, Butler M. Circumstances of falls resulting in hip fractures among older people. J Am Geriatr Soc 1997;45:1108-1112.
25. Kannus P, Parkkari J, Niemi S, et al. Prevention of hip fracture in elderly people with use of a hip protector. N Engl J Med. 2000;343:1506-1513.
26. Cameron ID, Stafford B, Cumming RG, et al. Hip protectors improve falls self-efficacy. Age Ageing. 2000;29:57-62.
27. Hubacher M, Wettstein A. Acceptance of hip protectors for hip fracture prevention in nursing homes. Osteoporos Int. 2001;12:794-799.
28. NIH Consensus Development Panel on Optimal Calcium Intake. Optimal Calcium Intake. JAMA. 1994;272:1942-1948.
29. Morgan SL. Calcium and vitamin D in osteoporosis. Rheum Dis Clin North Am. 2001;27:101-130.
30. Tsourounis C. Clinical effects of phytoestrogens. Clin Obstet Gynecol. 2001;44:836-842.
31. Kelley GA. Aerobic exercise and bone density at the hip in postmenopausal women: a meta-analysis. Prev Med. 1998;27:798-807.
32. Marcus R. Role of exercise in preventing and treating osteoporosis. Rheum Dis Clin North Am. 2001;27:131-141,vi.-
33. Sinaki M, Mikkelsen BA. Postmenopausal spinal osteoporosis: flexion versus extension exercises. Arch Phys Med Rehabil. 1984;65:593-596.
34. Law M, Hackshaw AK. A meta-analysis of cigarette smoking, bone mineral density and risk of hip fracture: recognition of major effect. BMJ. 1997;315:841-846.
35. Felson DT, Zhang Y, Hannan MT, Kannel WB, Kiel DP. Alcohol intake and bone mineral density in elderly men and women. The Framingham Study. Am J Epidemiol. 1995;142:485-492.
36. Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA. 2001;285:320-323.
37. Marcus R, Wong M, Heath H, 3rd, Stock JL. Antiresorptive treatment of postmenopausal osteoporosis: comparison of study designs and outcomes in large clinical trials with fracture as an endpoint. Endocr Rev. 2002;23:16-37.
38. Lips P, Graafmans WC, Ooms ME, Bezemer PD, Bouter LM. Vitamin D supplementation and fracture incidence in elderly persons. A randomized, placebo-controlled clinical trial. Ann Intern Med. 1996;124:400-406.
39. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346:77-84.
40. North American Menopause Society. The role of calcium in peri- and postmenopausal women; consensus opinion of The North American Menopause Society. Menopause. 2001;8:84-95.
41. Williams CP, Child DF, Hudson PR, et al. Why oral calcium supplements may reduce renal stone disease: report of a clinical pilot study. J Clin Pathol. 2001;54:54-62.
42. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results for the Women’s Health Initiative Randomized Controlled Trial. JAMA. 2002;288:321-333.
43. Barrett-Connor E, Hendrix S, Ettinger B. International position paper on women’s health and menopause: a comprehensive approach. National Heart, Lung, and Blood Institute; 2002.
44. Greendale GA, Espeland M, Slone S, Marcus R, Barrett-Connor E. Bone mass response to discontinuation of long-term hormone replacement therapy: results from the Postmenopausal Estrogen/Progestin Interventions (PEPI) Safety Follow-up Study. Arch Intern Med. 2002;162:665-672.
45. Schairer C, Lubin J, Triosi R, Sturgeon S, Brinton L, Hoover R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000;283:485-491.
46. Ross R, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/Progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
48. Oger E, Scarabin PY. Risk of stroke among users of hormone replacement therapy. Annals d’Endocrinologie. 1999;60:232-241.
49. Simon J, Hsia J, Cauley JA, et al. Postmenopausal hormone therapy and risk of stroke: the Heart and Estrogen/Progestin Replacement Study (HERS). Circulation. 2001;103:638-642.
50. Viscoli C, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345:1243-1249.
51. Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344-1352.
52. Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect alendronate on risk of fracture in women with existing vertebral fractures. Lancet. 1996;348:1535-1541.
53. Heaney RP, Zizic TM, Fogelman I, et al. Risedronate reduces the risk of first vertebral fracture in osteoporotic women. Osteoporos Int. 2002;13:501-505.
54. Liberman UA, Weiss SR, Broll J, et al. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437-1443.
55. Sharpe M, Noble S, Spencer CM. Alendronate: an update of its use in osteoporosis. Drugs. 2001;61:999-1039.
56. Dunn CJ, Goa KL. Risedronate: a review of its pharmacological properties and clinical use in resorptive bone disease. Drugs. 2001;61:685-712.
57. Taggart H, Bolognese MA, Lindsay R, et al. Upper gastrointestinal tract safety of risedronate: a pooled analysis of 9 clinical trials. Mayo Clin Proc. 2002;77:262-270.
58. Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. JAMA. 1999;282:637-645.
59. Sarkar S, Mitlak B, Wong M, Stock J, Black D, Harper K. Raloxifene-induced fracture reductions not directly associated with BMD changes. J Bone Miner Res. 2002;1:1-10.
60. Barrett-Connor E. Raloxifene: risks and benefits. Ann N Y Acad Sci. 2001;949:295-303.
61. Cummings S, Duong T, Kenyon E, Cauley J, Whitehead M, Krueger K. Serum estradiol level and risk of breast cancer during treatment with raloxifene. JAMA. 2002;287:216-220.
62. Barrett-Connor E, Grady D, Sashegyi A, et al. Raloxifene and cardiovascular events in osteoporotic postmenopausal women: four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA. 2002;287:847-857.
63. Lyritis GP, Paspati I, Karachalios T, Ioakimidis D, Skarantavos G, Lyritis PG. Pain relief from nasal salmon calcitonin in osteoporotic vertebral crush fractures. A double blind, placebo-controlled clinical study. Acta Orthop Scand Suppl. 1997;275:112-114.
64. Pun KK, Chan LW. Analgesic effect of intranasal salmon calcitonin in the treatment of osteoporotic vertebral fractures. Clin Ther. 1989;11:205-209.
65. Chesnut CH, 3rd. Calcitonin in the prevention and treatment of osteoporosis. Osteoporos Int. 1993;3(suppl 1):206-207.
66. Crandall C. The role of serial bone mineral density testing for osteoporosis. J Womens Health Gend Based Med. 2001;10:887-895.
67. Cummings SR. The paradox of small changes in bone density and reductions in risk of fracture with raloxifene. Ann N Y Acad Sci. 2001;949:198-201.
68. Cummings SR, Karpf DB, Harris F, et al. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med. 2002;112:281-289.
69. Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in the elderly women. N Engl J Med. 1992;327:1637-1642.
70. McClung M, Geusens P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. N Engl J Med. 2001;344:333-340.
1. Osteoporosis Prevention, Diagnosis, and Therapy. NIH Consensus Statement Online 2000 March 27-29; [cited 9/2/2]; 17(1):1-36.
2. World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. WHO Technical Report Series 843. Geneva: World Health Organization; 1994.
3. NIH Consensus Development Panel on Osteoporosis Prevention Diagnosis and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285:785-795.
4. Gold DT. The clinical impact of vertebral fractures: quality of life in women with osteoporosis. Bone. 1996;18(suppl 3):185S-189S.
5. Black DM, Arden NK, Palermo I, Pearson J, Cummings SR. Prevalent vertebral deformities predict hip fractures and new vertebral deformities but not wrist fractures. Study of Osteoporotic Fractures Research Group. J Bone Miner Res. 1999;14:821-828.
6. Ray NF, Chan JK, Thamer M, Melton LJ, 3rd. Medical expenditures for the treatment of osteoporotic fractures in the United States in 1995: report from the National Osteoporosis Foundation. J Bone Miner Res. 1997;12:24-35.
7. Looker AC, Wahner HW, Dunn WL, et al. Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int. 1998;8:468-489.
8. Aloia JF, Vaswani A, Yeh JK, Flaster E. Risk for osteoporosis in black women. Calcif Tissue Int. 1996;59:415-423.
9. Melton LJ. How many women have osteoporosis now? J Bone Miner Res. 1995;10:175-177.
10. Nelson HD, Morris CD, Mahon S, Carney N, Nygren PM, Helfand M. Osteoporosis in postmenopausal women: diagnosis and monitoring. Evidence Report/Technology Assessment Number 28. Rockville, MD: Agency for Healthcare Research and Quality; November 2001. 01-E032.
11. Melton LJ, 3rd, Amadio PC, Crowson CS, O’Fallon WM. Long-term trends in the incidence of distal forearm fractures. Osteoporos Int. 1998;8:341-348.
12. National Osteoporosis Foundation. Osteoporosis: review of the evidence for prevention, diagnosis, and treatment and cost-effectiveness analysis. Osteoporos Int. 1998;8(suppl):S7-S80.
13. U.S. Preventive Services Task Force. Screening for Osteoporosis in Postmenopausal Women. September 2002. Originally in Annals of Internal Medicine 2002;137:526-8.Agency for Healthcare Research and Quality, Rockville, MD. http://www.ahrq.gov/clinic/3rduspstf/osteoporosis/osteorr.htm.
14. Physician’s guide to prevention and treatment of osteoporosis. Washington, DC: National Osteoporosis Foundation; 1998.
15. American Association of Clinical Endocrinologists 2001 medical guidelines for clinical practice for the prevention and management of osteoporosis. Endocr Pract. 2001;7:293-312.
16. North American Menopause Society. Management of postmenopausal osteoporosis: position statement of The North American Menopause Society. Menopause. 2002;9:84-101.
17. Cadarette SM, Jaglal SB, Murray TM, McIsaac WJ, Joseph L, Brown JP. Evaluation of decision rules for referring women for bone densitometry by dual energy x-ray absorptiometry. JAMA. 2001;286:57-63.
18. Wasnich RD. Consensus and the T-score fallacy. Clin Rheumatol. 1997;16:337-339.
19. Grisso JA, Kelsey JL, Strom BL, et al. Risk factors for falls as a cause of hip fracture in women.The Northeast Hip Fracture Study Group. N Engl J Med. 1991;324:1326-1331.
20. Cooper C, Barker DJ, Morris J, Briggs RS. Osteoporosis, falls, and age in fracture of the proximal femur. Br Med J (Clin Res Ed). 1987;295:13-15.
21. Gardsell P, Johnell O, Nilsson BE, Nilsson JA. The predictive value of fracture, disease, and falling tendency for fragility fractures in women. Calcif Tissue Int. 1989;45:327-330.
22. Dargent-Molina P, Schott AM, Hans D, et al. Separate and combined value of bone mass and gait speed measurements in screening for hip fracture risk: results from the EPIDOS study. Epidemiologie de l’Osteoporose. Osteoporos Int. 1999;9:188-192.
23. Clemson L, Cumming RG, Roland M. Case-control study of hazards in the home and risk of falls and hip fractures. Age Ageing. 1996;25:97-101.
24. Norton R, Campbell AJ, Lee-Joe T, Robinson E, Butler M. Circumstances of falls resulting in hip fractures among older people. J Am Geriatr Soc 1997;45:1108-1112.
25. Kannus P, Parkkari J, Niemi S, et al. Prevention of hip fracture in elderly people with use of a hip protector. N Engl J Med. 2000;343:1506-1513.
26. Cameron ID, Stafford B, Cumming RG, et al. Hip protectors improve falls self-efficacy. Age Ageing. 2000;29:57-62.
27. Hubacher M, Wettstein A. Acceptance of hip protectors for hip fracture prevention in nursing homes. Osteoporos Int. 2001;12:794-799.
28. NIH Consensus Development Panel on Optimal Calcium Intake. Optimal Calcium Intake. JAMA. 1994;272:1942-1948.
29. Morgan SL. Calcium and vitamin D in osteoporosis. Rheum Dis Clin North Am. 2001;27:101-130.
30. Tsourounis C. Clinical effects of phytoestrogens. Clin Obstet Gynecol. 2001;44:836-842.
31. Kelley GA. Aerobic exercise and bone density at the hip in postmenopausal women: a meta-analysis. Prev Med. 1998;27:798-807.
32. Marcus R. Role of exercise in preventing and treating osteoporosis. Rheum Dis Clin North Am. 2001;27:131-141,vi.-
33. Sinaki M, Mikkelsen BA. Postmenopausal spinal osteoporosis: flexion versus extension exercises. Arch Phys Med Rehabil. 1984;65:593-596.
34. Law M, Hackshaw AK. A meta-analysis of cigarette smoking, bone mineral density and risk of hip fracture: recognition of major effect. BMJ. 1997;315:841-846.
35. Felson DT, Zhang Y, Hannan MT, Kannel WB, Kiel DP. Alcohol intake and bone mineral density in elderly men and women. The Framingham Study. Am J Epidemiol. 1995;142:485-492.
36. Lindsay R, Silverman SL, Cooper C, et al. Risk of new vertebral fracture in the year following a fracture. JAMA. 2001;285:320-323.
37. Marcus R, Wong M, Heath H, 3rd, Stock JL. Antiresorptive treatment of postmenopausal osteoporosis: comparison of study designs and outcomes in large clinical trials with fracture as an endpoint. Endocr Rev. 2002;23:16-37.
38. Lips P, Graafmans WC, Ooms ME, Bezemer PD, Bouter LM. Vitamin D supplementation and fracture incidence in elderly persons. A randomized, placebo-controlled clinical trial. Ann Intern Med. 1996;124:400-406.
39. Borghi L, Schianchi T, Meschi T, et al. Comparison of two diets for the prevention of recurrent stones in idiopathic hypercalciuria. N Engl J Med. 2002;346:77-84.
40. North American Menopause Society. The role of calcium in peri- and postmenopausal women; consensus opinion of The North American Menopause Society. Menopause. 2001;8:84-95.
41. Williams CP, Child DF, Hudson PR, et al. Why oral calcium supplements may reduce renal stone disease: report of a clinical pilot study. J Clin Pathol. 2001;54:54-62.
42. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results for the Women’s Health Initiative Randomized Controlled Trial. JAMA. 2002;288:321-333.
43. Barrett-Connor E, Hendrix S, Ettinger B. International position paper on women’s health and menopause: a comprehensive approach. National Heart, Lung, and Blood Institute; 2002.
44. Greendale GA, Espeland M, Slone S, Marcus R, Barrett-Connor E. Bone mass response to discontinuation of long-term hormone replacement therapy: results from the Postmenopausal Estrogen/Progestin Interventions (PEPI) Safety Follow-up Study. Arch Intern Med. 2002;162:665-672.
45. Schairer C, Lubin J, Triosi R, Sturgeon S, Brinton L, Hoover R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA. 2000;283:485-491.
46. Ross R, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92:328-332.
47. Hulley S, Grady D, Bush T, et al. Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. Heart and Estrogen/Progestin Replacement Study (HERS) Research Group. JAMA. 1998;280:605-613.
48. Oger E, Scarabin PY. Risk of stroke among users of hormone replacement therapy. Annals d’Endocrinologie. 1999;60:232-241.
49. Simon J, Hsia J, Cauley JA, et al. Postmenopausal hormone therapy and risk of stroke: the Heart and Estrogen/Progestin Replacement Study (HERS). Circulation. 2001;103:638-642.
50. Viscoli C, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI. A clinical trial of estrogen-replacement therapy after ischemic stroke. N Engl J Med. 2001;345:1243-1249.
51. Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: a randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344-1352.
52. Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect alendronate on risk of fracture in women with existing vertebral fractures. Lancet. 1996;348:1535-1541.
53. Heaney RP, Zizic TM, Fogelman I, et al. Risedronate reduces the risk of first vertebral fracture in osteoporotic women. Osteoporos Int. 2002;13:501-505.
54. Liberman UA, Weiss SR, Broll J, et al. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437-1443.
55. Sharpe M, Noble S, Spencer CM. Alendronate: an update of its use in osteoporosis. Drugs. 2001;61:999-1039.
56. Dunn CJ, Goa KL. Risedronate: a review of its pharmacological properties and clinical use in resorptive bone disease. Drugs. 2001;61:685-712.
57. Taggart H, Bolognese MA, Lindsay R, et al. Upper gastrointestinal tract safety of risedronate: a pooled analysis of 9 clinical trials. Mayo Clin Proc. 2002;77:262-270.
58. Ettinger B, Black DM, Mitlak BH, et al. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial. JAMA. 1999;282:637-645.
59. Sarkar S, Mitlak B, Wong M, Stock J, Black D, Harper K. Raloxifene-induced fracture reductions not directly associated with BMD changes. J Bone Miner Res. 2002;1:1-10.
60. Barrett-Connor E. Raloxifene: risks and benefits. Ann N Y Acad Sci. 2001;949:295-303.
61. Cummings S, Duong T, Kenyon E, Cauley J, Whitehead M, Krueger K. Serum estradiol level and risk of breast cancer during treatment with raloxifene. JAMA. 2002;287:216-220.
62. Barrett-Connor E, Grady D, Sashegyi A, et al. Raloxifene and cardiovascular events in osteoporotic postmenopausal women: four-year results from the MORE (Multiple Outcomes of Raloxifene Evaluation) randomized trial. JAMA. 2002;287:847-857.
63. Lyritis GP, Paspati I, Karachalios T, Ioakimidis D, Skarantavos G, Lyritis PG. Pain relief from nasal salmon calcitonin in osteoporotic vertebral crush fractures. A double blind, placebo-controlled clinical study. Acta Orthop Scand Suppl. 1997;275:112-114.
64. Pun KK, Chan LW. Analgesic effect of intranasal salmon calcitonin in the treatment of osteoporotic vertebral fractures. Clin Ther. 1989;11:205-209.
65. Chesnut CH, 3rd. Calcitonin in the prevention and treatment of osteoporosis. Osteoporos Int. 1993;3(suppl 1):206-207.
66. Crandall C. The role of serial bone mineral density testing for osteoporosis. J Womens Health Gend Based Med. 2001;10:887-895.
67. Cummings SR. The paradox of small changes in bone density and reductions in risk of fracture with raloxifene. Ann N Y Acad Sci. 2001;949:198-201.
68. Cummings SR, Karpf DB, Harris F, et al. Improvement in spine bone density and reduction in risk of vertebral fractures during treatment with antiresorptive drugs. Am J Med. 2002;112:281-289.
69. Chapuy MC, Arlot ME, Duboeuf F, et al. Vitamin D3 and calcium to prevent hip fractures in the elderly women. N Engl J Med. 1992;327:1637-1642.
70. McClung M, Geusens P, Miller PD, et al. Effect of risedronate on the risk of hip fracture in elderly women. N Engl J Med. 2001;344:333-340.
The Active Management of Depression
While family physicians play a leading role in caring for patients with major depression, the quality of that care that could be greatly improved. A 1997 to 1998 survey of a national sample of adults with depressive or anxiety disorders revealed that 83% of these patients visited a health care provider.1 Of this total, 84% were treated by primary care clinicians, compared with 16% who were treated by mental health professionals. However, about 90% of those cared for by mental health professionals received treatment that met criteria for adequacy outlined in treatment guidelines, compared with 19% of those cared for by primary care professionals.
A critical role for family physicians is to integrate treatment of depression with that of other conditions, especially in light of the association of depression with a variety of chronic diseases. The Institute of Medicine has concluded that depression is strongly associated with the occurrence of, and death following, myocardial infarctions.2 In diabetes, depression is associated with a 2% increase in glycosylated hemoglobin levels3 and can predict occurrence of diabetic complications. Additionally, chronic illnesses may, in themselves, exacerbate depression several fold.
Primary care clinicians are ideally positioned to serve as the central health care providers for patients with major depression. These physicians have many attributes that support this role, including their longitudinal relationship with patients, response to undifferentiated problems, frequent use of the biopsychosocial model, and ability to integrate care of mental and medical conditions. However, challenges in fulfilling this role also exist, including difficulties in recognizing patients with major depression, developing an adequate diagnostic initial assessment, implementing effective short- and long-term treatment and management strategies, and integrating care of depression with that of other conditions affecting patients.4 This article will review each of these challenges.
Recognition of major depression
DeGruy has eloquently described the barriers to recognition and management of mental disorders in primary care, including infrequent use of diagnostic criteria, concern regarding treatment effectiveness, availability of time and resources, the presence of other pressing clinical problems, and issues of third-party reimbursement and other organizational concerns.4
Family physicians and their patients often do not recognize somatic symptoms as originating from depression. In one study, primary care physicians correctly identified 94% of depressed patients presenting with psychological complaints, but they failed to recognize the psychiatric nature of somatic complaints in about half of the patients. This finding is of concern because 83% of depressed patients presented with somatic complaints.5
The attribution patients assign to their problems can also contribute to lack of recognition. In one general practice study, patients’ attributions were classified as somatizing (5%), psychologizing (23%), normalizing (48%), or no predominate attribution (24%).6 For example, patients in this study might attribute fatigue to anemia (somatizing), emotional exhaustion (psychologizing), or being over-extended (normalizing). The likelihood of a missed diagnosis in patients who met criteria for depression or anxiety was strongly associated with attribution: Physicians diagnosed 72% of psychologizing patients accurately, but they reported a correct diagnosis in only 17% of somatizing patients, 15% of normalizing patients, and 31% of patients with no predominate attribution.
Initial diagnostic assessment
The United States Preventive Services Task Force suggests that primary care physicians screen for major depression. The Task Force recommends using 2 simple questions about mood and anhedonia (Table 1) that are generally as effective as longer instruments.7 The Patient Health Questionnaire-9 (PHQ-9) or the longer Prime-MD can be used for further evaluation of patients who respond positively to either question, thus helping to both confirm the diagnosis of depression and measure severity.8,9 Other instruments include the Beck Depression Inventory,10 the Zung scale,11 and the General Health Questionnaire.12 These tools take longer to administer, are not specific in measuring the criteria for major depression, and do not measure severity well.
In family practices, pregnant and postpartum women represent a special population at increased risk for depression.13 About 5% of middle class women and up to one quarter of low income women experience postpartum depression.14 In about half, onset of the depressive disorder occurs before delivery.15 Women who have previously suffered postpartum depression are at high risk, as are those with histories of depression or premenstrual dysphoric disorder. The Edinburgh Postnatal Depression Scale is a useful 10-item self-report instrument available in Spanish and English (Table 1).16,17 Similar instruments have not been developed for pregnant women.
A patient who responds positively to the 2 screening questions in Table 1 or to another screening approach should be further evaluated to confirm the diagnosis of major depression. Many primary care clinicians do this through unstructured history taking. Others use an instrument such as the previously discussed PHQ-9. This tool offers an advantage because it provides a reliable symptom assessment, measures severity, and can be repeated over time to evaluate therapeutic response.8
The physician should consider bereavement and substance abuse as possible causes of depression; bereaved patients who continue to meet criteria for major depression at 2 months often benefit from treatment. By that time, the sadness, poor concentration, and other symptoms associated with normal grief are no longer constant and occur in waves brought on by memories. Conversely, persons also suffering from depression report these symptoms as enduring and autonomous.18
The primary care physician also should inquire about agitation and symptoms of anxiety disorders. These are experienced by 85% of depressed patients; 50% have comorbid anxiety disorders.19-21 Identification of such comorbidity is helpful in determining treatment, evaluating response, and managing patients over the long term. The Prime-MD, available in multiple languages, is also useful for screening for both anxiety and substance abuse, which can complicate both the recognition and treatment of comorbid depression.9
Sexual function is often affected by depression. The physician should inquire about sexual arousal, erection or lubrication, and orgasm during the initial assessment.22 Approximately 50% of women and 40% of men with major depression report sexual-arousal problems, and 15% to 20% report orgasm problems during the month prior to diagnosis.23 Further questioning can assess whether this dysfunction is caused by another disorder (eg, diabetes) or whether it is part of the depressive syndrome. This provides a baseline for later assessment of side effects and treatment effectiveness, and it communicates to the patient that the physician will be attentive to this area. In discussing sexual function with depressed patients, it may be helpful to tell patients that a study of the effectiveness of treatment of depression with selective serotonin reuptake inhibitors (SSRIs) found that patients reported modestly improved sexual function with treatment.24
TABLE 1
Screening for depression
| Outpatient adults | |
| |
| Postpartum women (Edinburgh Postnatal Depression Scale) | |
1. I have been able to laugh and see the funny side of things
| 6. Things have been getting on top of me
|
2. I have looked forward with enjoyment to things
| 7. I have been so unhappy that I have had difficulty sleeping
|
3. I have blamed myself unnecessarily when things went wrong
| 8. I have felt sad or miserable
|
4. I have been anxious or worried for no good reason
| 9. I have been so unhappy that I have been crying
|
5. I have felt scared or panicky for no very good reason
| 10. The thought of harming myself has occurred to me
|
| Reprinted with permission, from Cox JL et al. British Journal of Psychiatry. 1987; 150:782-786. | |
Management of major depression
The acute management of the patient with major depression includes patient education, shared decision-making regarding a treatment modality, supportive counseling, and treatment-specific counseling.25 Education and counseling should extend over the initial weeks of treatment and be combined with monitoring response, identifying and managing any treatment-emergent side effects, and adjusting medications. Long-term management goals include attaining full remission of symptoms, assisting the patient to return to full functional status, integrating depression care with the treatment of other chronic illnesses, maintaining or tapering pharmacologic treatment, and monitoring for and preventing relapse or recurrence.
Education
Education should help patients understand and accept the diagnosis, reduce any stigma they or their families might attach to major depression, and build increased adherence to subsequent treatment.26 It might be helpful to provide a brief explanation of the biologic basis of depression (including biochemical changes in brain function and “chemical imbalances” of serotonin and other neurotransmitters). Explaining pharmacotherapeutic effects (if medication is desired) as mechanisms to help rebalance brain chemistry further emphasizes the biologic basis of depression and decreases any perceptions that depression is a result of moral or character weakness. This educational message should also stress that antidepressants are not habit-forming or addictive, are not “uppers” or “downers,” and are not tranquilizers. The physician also should convey a positive prognosis but note that several weeks and, possibly, adjustments in treatments, may be required. For patients choosing antidepressants, the McArthur Foundation Initiative has identified 7 key educational messages (Table 2).27
TABLE 2
Key messages for patient education about depression
|
Counseling
Patients often benefit from counseling regarding sleep, exercise, and substance use. Many patients with depression experience early morning awakening. Those with agitated depression also often experience delayed sleep onset associated with worry. Providing the patient with information on basic sleep hygiene, exercise, and encouraging abstinence from or moderation in consumption of alcohol might all help.28-30 Additionally, sleep disturbances can indicate the possibility of comorbid disorders. A report that a patient fears going to sleep because of nightmares suggests posttraumatic stress disorder.
For some patients, counseling by the family physician or through referral may be a helpful treatment adjunct. Often depressed patients have deficient coping mechanisms and need assistance in developing strategies to resolve issues in their life. Principles used in cognitive behavioral therapy might be helpful in patient education and counseling.31 These include problem-solving strategies to resolve stressful concerns and cognitive techniques to identify and correct distorted or maladaptive thought patterns.29
As patients respond to depression treatment, an additional component of primary-care-based counseling should target reinvolvement with pleasurable social and physical activities. This may simply involve identifying activities the patient enjoyed prior to the onset of depression but has since stopped, and focusing on the steps required to reactivate these interests.
Shared decision-making with regard to treatment will improve subsequent patient adherence.27 Treatment options include psychotherapy, particularly cognitive behavioral therapy, pharmacotherapy, and electroconvulsive therapy. The latter should be considered for severely depressed patients, particularly persons with few social supports who are at significant risk of suicide.25
Cognitive behavioral therapy and other psychotherapies can show effectiveness equal to that of pharmacotherapy, although response usually lags by a month to 6 weeks compared with that attained by pharmacotherapy.32 For moderately to severely depressed patients, pharmacotherapy is the treatment of choice in part because of its more rapid onset of action.25
Pharmacotherapy
Pharmacotherapy, most often in the form of an SSRI, is the treatment of choice for depression as a result of patient preference, insurance coverage limitations, or time constraints. In choosing an anti-depressant, the family physician should be guided by effectiveness and potential for drug–drug interactions and for both short-and long-term side effects.33
Tricyclics, the SSRIs, and other newer antidepressants offer similar efficacy.34 While efficacy assesses outcome under ideal treatment conditions, the primary care physician is more concerned with effectiveness, defined as the proportion of patients started on an antidepressant during routine clinical practice who attain lasting benefit. Effectiveness includes consideration of patients who discontinue treatment because of side effects or drug–drug interactions, as well as those who do not obtain adequate therapeutic response. Since about 25% of patients discontinue SSRIs because of side effects, this is an important concern.24 Few studies have been conducted comparing the effectiveness of antidepressants.
Drug–drug interactions are mediated predominately by the cytochrome P450 isoenzymes responsible for drug metabolism in the liver.35-37 The 2D6 isoenzyme is responsible for 50% of drug metabolism in the liver; the 3A4 isoenzyme is responsible for another 30%.38 As a clinical example of the importance of such inhibition, codeine requires 2D6-mediated metabolism to become morphine and is ineffective for pain in many patients who are prescribed a 2D6 inhibitor. Patients receiving such agents also can have a 300% to 400% increase in blood levels of previously stable ß-blockers. Paroxetine and fluoxetine, the two SSRIs that strongly inhibit the 2D6 isoenzyme, cause clinically significant interactions; fluoxetine is also a moderate inhibitor of the 3A4 isoenzyme.35 Because of the number of potential drug–drug interactions through these isoenzymes, physicians must check for interactions before prescribing these medications or adding other new medications in patients already receiving these agents. This also is a consideration for patients who might require additional medications acutely, for instance in response to a cardiac or other emergency.
Side effects of concern include gastrointestinal effects, particularly nausea, and central nervous system (CNS) effects, including anxiety and agitation, sleep disturbance, and tremor. When these occur, they often decrease rapidly over the first 1 to 3 weeks. If severe, they can be managed by a temporary dosage decrease. For patients with significant CNS side effects, altering the timing of the daily dose might provide relief from daytime somnolence or agitation or from nighttime insomnia.
Long-term side effects of concern include weight gain and sexual dysfunction. While other SSRIs have low rates for weight gain, paroxetine causes a weight gain of more than 7% (about 10 lbs for a patient of average weight) in 20% to 25% of patients.39 Some element of sexual dysfunction, most often delayed orgasm, is estimated to occur in 30% to 40% of individuals receiving SSRIs.40,41 Management options include delaying dosage of agents with a half-life of about 24 hours (escitalopram, citalopram, sertraline).42 For instance, an individual who usually takes one of these agents in the morning may delay a day’s dose until after engaging in sexual intercourse in the evening. While open-label studies support augmentation, particularly with bupropion or buspirone, the few small randomized double-blind trials available suggest that positive results should be interpreted with caution.43 Alternatively, patients may benefit from sildenafil44 or a switch to a non-SSRI antidepressant.
While management of side effects presents one option, the best clinical approach may be to select an agent with minimal side-effect potential. In double-blind randomized trials, escitalopram, a new SSRI treatment option, was demonstrated to require treatment termination in less than 5% of recipients at its usual dose of 10 mg, a rate no different from that of placebo.45 In contrast, rates of 15% to 30% have been reported for other SSRIs and newer antidepressants at the time of their initial release.
Adjusting treatment
One recent primary care trial examined the effectiveness of 3 SSRIs: fluoxetine, sertraline, and paroxetine. At the time this study was designed, citalopram was not in common use. While about 75% of patients attained remission, only 40% to 50% of patients were maintained on the first prescribed agent.24 Additionally, about 20% of depression “treatment resistance” resulted because patients did not fill their prescriptions or adhere to treatment.46 For patients who do not respond within the first month, increasing the dosage is appropriate.47 About 25% of patients respond to this adjustment.48 For patients who do not respond, reassessment of the diagnosis, as well as assessment of potential psychiatric comorbidities and suicidal ideation, is indicated. For nonresponders, and for those with intolerable side effects, switching to a second SSRI is a reasonable next step.49 About 50% of patients switched to a second agent respond.50 For those who do not respond, the primary care physician might consider a second medication switch or psychiatric consultation.
Further treatment adjustment is indicated for patients who experience partial response. This might take the form of augmentation with psychotherapy51 or with another agent.52 Lithium and thyroid hormone (often as 25 to 50 mg T3 daily) are the most frequently used options, although stimulants, other antidepressants, and atypical antipsychotics are all of value in some patients.48,49,53
When indicated, treatment should be discontinued by tapering the dose over several weeks to months, depending on the duration and severity of past episodes. Patients should be educated to be alert for recurrence. They should also be monitored for recurrence and restarted on full-dose therapy if this occurs. If patients stop therapy abruptly, the likelihood of withdrawal symptoms (agitation, irritability, dizziness, ataxia, nausea, paresthesias, sleep disturbances) is highly related to the half-life of the SSRI.39 For paroxetine, which has the shortest half-life, withdrawal is frequent; the extended release preparation does not decrease the likelihood of withdrawal. Withdrawal symptoms are infrequent (< 2%) for sertraline, citalopram, and escitalopram, and they do not occur with fluoxetine.
Duration of treatment
A major challenge in family practice is maintaining patient adherence to treatment for the recommended interval to prevent relapse and to avoid recurrence in those with a history of prior episodes. In one study, 25% to 33% of primary care patients stopped depression therapy within 1 month and over 40% within 3 months. Additionally, 62% failed to inform their physicians.54 Depression also adversely affects compliance with treatment of comorbid medical conditions; in one meta-analysis, depression increased noncompliance 3-fold.54
For the first lifetime episode, the recommended duration of treatment is 6 to 9 months (4 to 6 months after recovery).55 Longer therapy is appropriate for those with comorbid anxiety disorders, severe initial symptoms, difficulty in attaining therapeutic response, deficient social support, or a history of substance abuse, as well as for older adults. For patients with 3 or more previous episodes, long-term maintenance therapy is recommended.55 For those with even one past episode, extended maintenance therapy might be beneficial. Maintenance therapy should be at the full dose required to attain initial response. In one study, only about 20% to 30% (depending on the treatment) experienced recurrence over 3 years if maintained at full dose, compared with 70% maintained at half the initial treatment dose, and 78% of those receiving placebo.56 For women who have previously suffered from postpartum depression, postpartum prophylaxis can be very effective. In one randomized trial, 62.5% of women on place-bo experienced recurrence compared with only 6.7% of those receiving prophylaxis.57
Practice strategies to improve care
A number of primary care investigators have demonstrated the value of practice management and quality improvement techniques to increase the portion of patients who achieve and maintain response to depression therapy. These studies share an approach of “active management” to promote adherence to treatment guidelines.58-63 For instance, Simon and colleagues demonstrated the value of initial and monthly phone contact.64
Active management techniques include the following:
- Initial and ongoing patient education and counseling, as discussed above
- Patient involvement and agreement in treatment choice
- Initial phone contact to assure the prescription has been filled and initial dose taken
- Periodic contact to inquire about adherence, treatment response, side effects, and to answer patient questions
- Adjustment of therapy for those not responding adequately by 4 to 6 weeks
- Establishment of a collaborative relationship with a psychiatrist for consultation and telephone advice
Additionally, primary care clinicians may find it helpful to add depression to their medical record preventive health maintenance flow chart, especially for patients with any past history of depression. Using the PHQ-9 can be beneficial in providing both the patient and physician with an objective measure of monitoring response and remission.
Conclusions
Effective and available treatments can have a major beneficial impact on patients with depression. To be maximally effective, primary care clinicians must actively manage the care of their depressed patients, using screening strategies to recognize depression in addition to targeted educational messages and active follow-up to improve treatment adherence. Long-term maintenance treatment prevents further recurrences in those who have already experienced multiple episodes. Choice of treatment should be guided by patient preference. For pharmacologic agents, selection should be based on effectiveness, likelihood of side effects and resultant premature discontinuation, and potential for drug–drug interaction. The majority of individuals with depression are managed solely in primary-care settings. With adequate treatment, remission of symptoms, significant improvement in quality of life, and return to full function at home and at work can be attained.
1. Young AS, Klap R, Sherbourne CD, Wells KB. The quality of care for depressive and anxiety disorders in the United States. Arch Gen Psychiatry. 2001;58:55-61.
2. Institute of Medicine (U.S.). Committee on Health and Behavior: Research Practice and Policy. Health and Behavior: The Interplay of Biological, Behavioral, and Societal Influences. Washington, DC: National Academy Press. 2001.
3. Lustman PJ, Griffith LS, Freedland KE, Clouse RE. The course of major depression in diabetes. Gen Hosp Psychiatry. 1997;19:138-143.
4. deGruy F, III. Mental health care in the primary care setting. In: Donaldson MS, ed. Primary Care: America’s Health in a New Era. Washington, DC: National Academy Press; 1996;285-311.
5. Bridges KW, Goldberg DP. Somatic presentation of DSM III psychiatric disorders in primary care. J Psychosom Res. 1985;29:563-569.
6. Kessler D, Lloyd K, Lewis G, Gray DP. Cross sectional study of symptom attribution and recognition of depression and anxiety in primary care. BMJ. 1999;318:436-439.
7. Whooley MA, Avins AL, Miranda J, Browner WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med. 1997;12:439-445.
8. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
9. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA. 1999;282:1737-1744.
10. Steer RA, Cavalieri TA, Leonard DM, Beck AT. Use of the Beck Depression Inventory for Primary Care to screen for major depression disorders. Gen Hosp Psychiatry. 1999;21:106-111.
11. Biggs JT, Wylie LT, Ziegler VE. Validity of the Zung Self-rating Depression Scale. Br J Psychiatry. 1978;132:381-385.
12. Goldberg DP, Gater R, Sartorius N, et al. The validity of two versions of the GHQ in the WHO study of mental illness in general health care. Psychol Med. 1997;27:191-197.
13. Susman JL. Postpartum depressive disorders. J Fam Pract. 1996;43(6 suppl):S17-24.
14. O’Hara MW, Schlechte JA, Lewis DA, Varner MW. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100:63-73.
15. Yonkers KA, Ramin SM, Rush AJ, et al. Onset and persistence of postpartum depression in an inner-city maternal health clinic system. Am J Psychiatry. 2001;158:1856-1863.
16. Georgiopoulos AM, Bryan TL, Wollan P, Yawn BP. Routine screening for postpartum depression. J Fam Pract. 2001;50:117-122.
17. Eberhard-Gran M, Eskild A, Tambs K, Opjordsmoen S, Samuelsen SO. Review of validation studies of the Edinburgh Postnatal Depression Scale. Acta Psychiatr Scand. 2001;104:243-249.
18. Osterweis M, Solomon F, Green M. Institute of Medicine (U.S.). Committee for the Study of Health Consequences of the Stress of Bereavement. Bereavement: Reactions, Consequences, and Care. Washington, DC: National Academy Press; 1984.
19. Keller MB, Hanks DL. The natural history and heterogeneity of depressive disorders: implications for rational antidepressant therapy. J Clin Psychiatry. 1994;55 (suppl A):25-31;discussion32-23,98-100.
20. Keller MB, Hanks DL. Anxiety symptom relief in depression treatment outcomes. J Clin Psychiatry. 1995;56(suppl 6):22-29.
21. Kravitz HM, Fogg L, Fawcett J, Edwards J. Antidepressant or antianxiety? A study of the efficacy of antidepressant medication. Psychiatry Res. 1990;32:141-149.
22. Clayton AH. Recognition and assessment of sexual dysfunction associated with depression. J Clin Psychiatry. 2001;62(suppl 3):5-9.
23. Kennedy SH, Dickens SE, Eisfeld BS, et al. Sexual dysfunction before antidepressant therapy in major depression. J Affect Disord. 1999;201-208.
24. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: a randomized trial. JAMA. 2001;286:2947-2955.
25. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical Practice Guideline, Number 5. Rockville, MD: U.S. Dept. of Health and Human Services, Agency for Health Care Policy and Research; April 1993. AHCPR publication 93-0551.
26. Hegner RE. Dispelling the myths and stigma of mental illness: the Surgeon General’s report on mental health. Issue Brief Natl Health Policy Forum. 2000;(754):1-7.
27. Lin EH, Von Korff M, Katon W, et al. The role of the primary care physician in patients’ adherence to antidepressant therapy. Med Care. 1995;33:67-74.
28. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. In: Hauri P, ed. Case Studies in Insomnia. New York: Plenum Medical Book; 1991:xiv, 254.
29. Culpepper L. Worries and anxiety. In: Staton EW, ed. 20 Common Problems in Behavioral Health. New York: McGraw-Hill; 2002;385-404.
30. Miser WF. Exercise as an effective treatment option for major depression in older adults. J Fam Pract. 2000;49:109-110.
31. Robinson P, Bush T, Von Korff M, et al. Primary care physician use of cognitive behavioral techniques with depressed patients. J Fam Pract. 1995;40:352-357.
32. Rush AJ, Thase ME. Psychotherapies for depressive disorders: a review. In: Sartorius N, ed. Depressive Disorders. New York: John Wiley and Sons; 1999.
33. Preskorn SH. Selection of an antidepressant: mirtazapine. J Clin Psychiatry. 1997;58(suppl 6):3-8.
34. Geddes JR, Freemantle N, Mason J, Eccles MP, Boynton J. SSRIs versus other antidepressants for depressive disorder. Cochrane Database Syst Rev. 2000;CD001851.-
35. Preskorn SH. Debate resolved: there are differential effects of serotonin selective reuptake inhibitors on cytochrome P450 enzymes. J Psychopharmacol. 1998;12(3 suppl B):S89-97.
36. Preskorn SH. Antidepressant options in primary care. Clin Cornerstone. 1999;1:31-55.
37. Greenblatt DJ, von Moltke LL, Harmatz JS, Shader RI. Drug interactions with newer antidepressants: role of human cytochromes P450. J Clin Psychiatry. 1998;59 (suppl. 15):19-27.
38. Preskorn SH. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin Pharmacokinet. 1997;32(suppl 1):1-21.
39. Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863-867.
40. Montejo-Gonzalez AL, Llorca G, Izquierdo JA, et al. SSRI-induced sexual dysfunction: fluoxetine, paroxetine, sertraline, and fluvoxamine in a prospective, multicenter, and descriptive clinical study of 344 patients. J Sex Marital Ther. 1997;23:176-194.
41. Fava M, Rankin M. Sexual functioning and SSRIs. J Clin Psychiatry. 2002;63(suppl 5):13-16;discussion 23-15.
42. Zajecka J. Strategies for the treatment of antidepressant-related sexual dysfunction. J Clin Psychiatry. 2001;62 (suppl 3):35-43.
43. Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Practice. 2002;51:681.-
44. Nurnberg HG, Hensley PL, Lauriello J, Parker LM, Keith SJ. Sildenafil for women patients with antidepressant-induced sexual dysfunction. Psychiatr Serv. 1999;50:1076-1078.
45. Wade A, Michael Lemming O, Bang Hedegaard K. Escitalopram 10mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2002;95-102.
46. Souery D, Mendlewicz J. Compliance and therapeutic issues in resistant depression. Int Clin Psychopharmacol. 1998;13 (suppl 2):S13-18.
47. Thase ME, Rush AJ. Treatment-resistant depression. In: Kupfer DJ, ed. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995;1081-1097.
48. Thase ME. What role do atypical antipsychotic drugs have in treatment-resistant depression. J Clin Psychiatry. 2002;63:95-103.
49. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(4 suppl):1-45.
50. Howland RH, Thase ME. What to do with SSRI non-responders? J Pract Psychiatry Behav Health. 1999;5:216-233.
51. Thase ME, Friedman ES, Howland RH. Management of treatment-resistant depression: psychotherapeutic perspectives. J Clin Psychiatry. 2001;62(suppl 18):18-24.
52. Fava M. Augmentation and combination strategies in treatment-resistant depression. J Clin Psychiatry. 2001;62 (suppl 18):4-11.
53. Thase ME, Howland RH, Friedman ES. Treating antidepressant nonresponders with augmentation strategies: an overview. J Clin Psychiatry. 1998;59(suppl 5):5-12;discussion 13-15.
54. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160:2101-2107.
55. Keller MB. The long-term treatment of depression. J Clin Psychiatry. 1999;60(suppl 17):41-45;discussion 46-48.
56. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49:782-787.
57. Wisner KL, Wheeler SB. Prevention of recurrent postpartum major depression. Hosp Community Psychiatry. 1994;45:1191-1196.
58. Schulberg HC, Katon W, Simon GE, Rush AJ. Treating major depression in primary care practice: an update of the Agency for Health Care Policy and Research Practice Guidelines. Arch Gen Psychiatry. 1998;55:1121-1127.
59. Schulberg HC. Treating depression in primary care practice: applications of research findings. J Fam Pract. 2001;50:535-537.
60. Katon W, Von Korff M, Lin E, et al. Collaborative management to achieve treatment guidelines. Impact on depression in primary care. JAMA. 1995;273:1026-1031.
61. Katon W, Robinson P, Von Korff M, et al. A multifaceted intervention to improve treatment of depression in primary care. Arch Gen Psychiatry. 1996;53:924-932.
62. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry. 1999;56:1109-1115.
63. Von Korff M, Katon W, Unutzer J, Wells K, Wagner EH. Improving depression care: barriers, solutions, and research needs. J Fam Pract. 2001;50:E1.-
64. Simon GE, VonKorff M, Rutter C, Wagner E. Randomised trial of monitoring, feedback, and management of care by telephone to improve treatment of depression in primary care. BMJ. 2000;320:550-554.
While family physicians play a leading role in caring for patients with major depression, the quality of that care that could be greatly improved. A 1997 to 1998 survey of a national sample of adults with depressive or anxiety disorders revealed that 83% of these patients visited a health care provider.1 Of this total, 84% were treated by primary care clinicians, compared with 16% who were treated by mental health professionals. However, about 90% of those cared for by mental health professionals received treatment that met criteria for adequacy outlined in treatment guidelines, compared with 19% of those cared for by primary care professionals.
A critical role for family physicians is to integrate treatment of depression with that of other conditions, especially in light of the association of depression with a variety of chronic diseases. The Institute of Medicine has concluded that depression is strongly associated with the occurrence of, and death following, myocardial infarctions.2 In diabetes, depression is associated with a 2% increase in glycosylated hemoglobin levels3 and can predict occurrence of diabetic complications. Additionally, chronic illnesses may, in themselves, exacerbate depression several fold.
Primary care clinicians are ideally positioned to serve as the central health care providers for patients with major depression. These physicians have many attributes that support this role, including their longitudinal relationship with patients, response to undifferentiated problems, frequent use of the biopsychosocial model, and ability to integrate care of mental and medical conditions. However, challenges in fulfilling this role also exist, including difficulties in recognizing patients with major depression, developing an adequate diagnostic initial assessment, implementing effective short- and long-term treatment and management strategies, and integrating care of depression with that of other conditions affecting patients.4 This article will review each of these challenges.
Recognition of major depression
DeGruy has eloquently described the barriers to recognition and management of mental disorders in primary care, including infrequent use of diagnostic criteria, concern regarding treatment effectiveness, availability of time and resources, the presence of other pressing clinical problems, and issues of third-party reimbursement and other organizational concerns.4
Family physicians and their patients often do not recognize somatic symptoms as originating from depression. In one study, primary care physicians correctly identified 94% of depressed patients presenting with psychological complaints, but they failed to recognize the psychiatric nature of somatic complaints in about half of the patients. This finding is of concern because 83% of depressed patients presented with somatic complaints.5
The attribution patients assign to their problems can also contribute to lack of recognition. In one general practice study, patients’ attributions were classified as somatizing (5%), psychologizing (23%), normalizing (48%), or no predominate attribution (24%).6 For example, patients in this study might attribute fatigue to anemia (somatizing), emotional exhaustion (psychologizing), or being over-extended (normalizing). The likelihood of a missed diagnosis in patients who met criteria for depression or anxiety was strongly associated with attribution: Physicians diagnosed 72% of psychologizing patients accurately, but they reported a correct diagnosis in only 17% of somatizing patients, 15% of normalizing patients, and 31% of patients with no predominate attribution.
Initial diagnostic assessment
The United States Preventive Services Task Force suggests that primary care physicians screen for major depression. The Task Force recommends using 2 simple questions about mood and anhedonia (Table 1) that are generally as effective as longer instruments.7 The Patient Health Questionnaire-9 (PHQ-9) or the longer Prime-MD can be used for further evaluation of patients who respond positively to either question, thus helping to both confirm the diagnosis of depression and measure severity.8,9 Other instruments include the Beck Depression Inventory,10 the Zung scale,11 and the General Health Questionnaire.12 These tools take longer to administer, are not specific in measuring the criteria for major depression, and do not measure severity well.
In family practices, pregnant and postpartum women represent a special population at increased risk for depression.13 About 5% of middle class women and up to one quarter of low income women experience postpartum depression.14 In about half, onset of the depressive disorder occurs before delivery.15 Women who have previously suffered postpartum depression are at high risk, as are those with histories of depression or premenstrual dysphoric disorder. The Edinburgh Postnatal Depression Scale is a useful 10-item self-report instrument available in Spanish and English (Table 1).16,17 Similar instruments have not been developed for pregnant women.
A patient who responds positively to the 2 screening questions in Table 1 or to another screening approach should be further evaluated to confirm the diagnosis of major depression. Many primary care clinicians do this through unstructured history taking. Others use an instrument such as the previously discussed PHQ-9. This tool offers an advantage because it provides a reliable symptom assessment, measures severity, and can be repeated over time to evaluate therapeutic response.8
The physician should consider bereavement and substance abuse as possible causes of depression; bereaved patients who continue to meet criteria for major depression at 2 months often benefit from treatment. By that time, the sadness, poor concentration, and other symptoms associated with normal grief are no longer constant and occur in waves brought on by memories. Conversely, persons also suffering from depression report these symptoms as enduring and autonomous.18
The primary care physician also should inquire about agitation and symptoms of anxiety disorders. These are experienced by 85% of depressed patients; 50% have comorbid anxiety disorders.19-21 Identification of such comorbidity is helpful in determining treatment, evaluating response, and managing patients over the long term. The Prime-MD, available in multiple languages, is also useful for screening for both anxiety and substance abuse, which can complicate both the recognition and treatment of comorbid depression.9
Sexual function is often affected by depression. The physician should inquire about sexual arousal, erection or lubrication, and orgasm during the initial assessment.22 Approximately 50% of women and 40% of men with major depression report sexual-arousal problems, and 15% to 20% report orgasm problems during the month prior to diagnosis.23 Further questioning can assess whether this dysfunction is caused by another disorder (eg, diabetes) or whether it is part of the depressive syndrome. This provides a baseline for later assessment of side effects and treatment effectiveness, and it communicates to the patient that the physician will be attentive to this area. In discussing sexual function with depressed patients, it may be helpful to tell patients that a study of the effectiveness of treatment of depression with selective serotonin reuptake inhibitors (SSRIs) found that patients reported modestly improved sexual function with treatment.24
TABLE 1
Screening for depression
| Outpatient adults | |
| |
| Postpartum women (Edinburgh Postnatal Depression Scale) | |
1. I have been able to laugh and see the funny side of things
| 6. Things have been getting on top of me
|
2. I have looked forward with enjoyment to things
| 7. I have been so unhappy that I have had difficulty sleeping
|
3. I have blamed myself unnecessarily when things went wrong
| 8. I have felt sad or miserable
|
4. I have been anxious or worried for no good reason
| 9. I have been so unhappy that I have been crying
|
5. I have felt scared or panicky for no very good reason
| 10. The thought of harming myself has occurred to me
|
| Reprinted with permission, from Cox JL et al. British Journal of Psychiatry. 1987; 150:782-786. | |
Management of major depression
The acute management of the patient with major depression includes patient education, shared decision-making regarding a treatment modality, supportive counseling, and treatment-specific counseling.25 Education and counseling should extend over the initial weeks of treatment and be combined with monitoring response, identifying and managing any treatment-emergent side effects, and adjusting medications. Long-term management goals include attaining full remission of symptoms, assisting the patient to return to full functional status, integrating depression care with the treatment of other chronic illnesses, maintaining or tapering pharmacologic treatment, and monitoring for and preventing relapse or recurrence.
Education
Education should help patients understand and accept the diagnosis, reduce any stigma they or their families might attach to major depression, and build increased adherence to subsequent treatment.26 It might be helpful to provide a brief explanation of the biologic basis of depression (including biochemical changes in brain function and “chemical imbalances” of serotonin and other neurotransmitters). Explaining pharmacotherapeutic effects (if medication is desired) as mechanisms to help rebalance brain chemistry further emphasizes the biologic basis of depression and decreases any perceptions that depression is a result of moral or character weakness. This educational message should also stress that antidepressants are not habit-forming or addictive, are not “uppers” or “downers,” and are not tranquilizers. The physician also should convey a positive prognosis but note that several weeks and, possibly, adjustments in treatments, may be required. For patients choosing antidepressants, the McArthur Foundation Initiative has identified 7 key educational messages (Table 2).27
TABLE 2
Key messages for patient education about depression
|
Counseling
Patients often benefit from counseling regarding sleep, exercise, and substance use. Many patients with depression experience early morning awakening. Those with agitated depression also often experience delayed sleep onset associated with worry. Providing the patient with information on basic sleep hygiene, exercise, and encouraging abstinence from or moderation in consumption of alcohol might all help.28-30 Additionally, sleep disturbances can indicate the possibility of comorbid disorders. A report that a patient fears going to sleep because of nightmares suggests posttraumatic stress disorder.
For some patients, counseling by the family physician or through referral may be a helpful treatment adjunct. Often depressed patients have deficient coping mechanisms and need assistance in developing strategies to resolve issues in their life. Principles used in cognitive behavioral therapy might be helpful in patient education and counseling.31 These include problem-solving strategies to resolve stressful concerns and cognitive techniques to identify and correct distorted or maladaptive thought patterns.29
As patients respond to depression treatment, an additional component of primary-care-based counseling should target reinvolvement with pleasurable social and physical activities. This may simply involve identifying activities the patient enjoyed prior to the onset of depression but has since stopped, and focusing on the steps required to reactivate these interests.
Shared decision-making with regard to treatment will improve subsequent patient adherence.27 Treatment options include psychotherapy, particularly cognitive behavioral therapy, pharmacotherapy, and electroconvulsive therapy. The latter should be considered for severely depressed patients, particularly persons with few social supports who are at significant risk of suicide.25
Cognitive behavioral therapy and other psychotherapies can show effectiveness equal to that of pharmacotherapy, although response usually lags by a month to 6 weeks compared with that attained by pharmacotherapy.32 For moderately to severely depressed patients, pharmacotherapy is the treatment of choice in part because of its more rapid onset of action.25
Pharmacotherapy
Pharmacotherapy, most often in the form of an SSRI, is the treatment of choice for depression as a result of patient preference, insurance coverage limitations, or time constraints. In choosing an anti-depressant, the family physician should be guided by effectiveness and potential for drug–drug interactions and for both short-and long-term side effects.33
Tricyclics, the SSRIs, and other newer antidepressants offer similar efficacy.34 While efficacy assesses outcome under ideal treatment conditions, the primary care physician is more concerned with effectiveness, defined as the proportion of patients started on an antidepressant during routine clinical practice who attain lasting benefit. Effectiveness includes consideration of patients who discontinue treatment because of side effects or drug–drug interactions, as well as those who do not obtain adequate therapeutic response. Since about 25% of patients discontinue SSRIs because of side effects, this is an important concern.24 Few studies have been conducted comparing the effectiveness of antidepressants.
Drug–drug interactions are mediated predominately by the cytochrome P450 isoenzymes responsible for drug metabolism in the liver.35-37 The 2D6 isoenzyme is responsible for 50% of drug metabolism in the liver; the 3A4 isoenzyme is responsible for another 30%.38 As a clinical example of the importance of such inhibition, codeine requires 2D6-mediated metabolism to become morphine and is ineffective for pain in many patients who are prescribed a 2D6 inhibitor. Patients receiving such agents also can have a 300% to 400% increase in blood levels of previously stable ß-blockers. Paroxetine and fluoxetine, the two SSRIs that strongly inhibit the 2D6 isoenzyme, cause clinically significant interactions; fluoxetine is also a moderate inhibitor of the 3A4 isoenzyme.35 Because of the number of potential drug–drug interactions through these isoenzymes, physicians must check for interactions before prescribing these medications or adding other new medications in patients already receiving these agents. This also is a consideration for patients who might require additional medications acutely, for instance in response to a cardiac or other emergency.
Side effects of concern include gastrointestinal effects, particularly nausea, and central nervous system (CNS) effects, including anxiety and agitation, sleep disturbance, and tremor. When these occur, they often decrease rapidly over the first 1 to 3 weeks. If severe, they can be managed by a temporary dosage decrease. For patients with significant CNS side effects, altering the timing of the daily dose might provide relief from daytime somnolence or agitation or from nighttime insomnia.
Long-term side effects of concern include weight gain and sexual dysfunction. While other SSRIs have low rates for weight gain, paroxetine causes a weight gain of more than 7% (about 10 lbs for a patient of average weight) in 20% to 25% of patients.39 Some element of sexual dysfunction, most often delayed orgasm, is estimated to occur in 30% to 40% of individuals receiving SSRIs.40,41 Management options include delaying dosage of agents with a half-life of about 24 hours (escitalopram, citalopram, sertraline).42 For instance, an individual who usually takes one of these agents in the morning may delay a day’s dose until after engaging in sexual intercourse in the evening. While open-label studies support augmentation, particularly with bupropion or buspirone, the few small randomized double-blind trials available suggest that positive results should be interpreted with caution.43 Alternatively, patients may benefit from sildenafil44 or a switch to a non-SSRI antidepressant.
While management of side effects presents one option, the best clinical approach may be to select an agent with minimal side-effect potential. In double-blind randomized trials, escitalopram, a new SSRI treatment option, was demonstrated to require treatment termination in less than 5% of recipients at its usual dose of 10 mg, a rate no different from that of placebo.45 In contrast, rates of 15% to 30% have been reported for other SSRIs and newer antidepressants at the time of their initial release.
Adjusting treatment
One recent primary care trial examined the effectiveness of 3 SSRIs: fluoxetine, sertraline, and paroxetine. At the time this study was designed, citalopram was not in common use. While about 75% of patients attained remission, only 40% to 50% of patients were maintained on the first prescribed agent.24 Additionally, about 20% of depression “treatment resistance” resulted because patients did not fill their prescriptions or adhere to treatment.46 For patients who do not respond within the first month, increasing the dosage is appropriate.47 About 25% of patients respond to this adjustment.48 For patients who do not respond, reassessment of the diagnosis, as well as assessment of potential psychiatric comorbidities and suicidal ideation, is indicated. For nonresponders, and for those with intolerable side effects, switching to a second SSRI is a reasonable next step.49 About 50% of patients switched to a second agent respond.50 For those who do not respond, the primary care physician might consider a second medication switch or psychiatric consultation.
Further treatment adjustment is indicated for patients who experience partial response. This might take the form of augmentation with psychotherapy51 or with another agent.52 Lithium and thyroid hormone (often as 25 to 50 mg T3 daily) are the most frequently used options, although stimulants, other antidepressants, and atypical antipsychotics are all of value in some patients.48,49,53
When indicated, treatment should be discontinued by tapering the dose over several weeks to months, depending on the duration and severity of past episodes. Patients should be educated to be alert for recurrence. They should also be monitored for recurrence and restarted on full-dose therapy if this occurs. If patients stop therapy abruptly, the likelihood of withdrawal symptoms (agitation, irritability, dizziness, ataxia, nausea, paresthesias, sleep disturbances) is highly related to the half-life of the SSRI.39 For paroxetine, which has the shortest half-life, withdrawal is frequent; the extended release preparation does not decrease the likelihood of withdrawal. Withdrawal symptoms are infrequent (< 2%) for sertraline, citalopram, and escitalopram, and they do not occur with fluoxetine.
Duration of treatment
A major challenge in family practice is maintaining patient adherence to treatment for the recommended interval to prevent relapse and to avoid recurrence in those with a history of prior episodes. In one study, 25% to 33% of primary care patients stopped depression therapy within 1 month and over 40% within 3 months. Additionally, 62% failed to inform their physicians.54 Depression also adversely affects compliance with treatment of comorbid medical conditions; in one meta-analysis, depression increased noncompliance 3-fold.54
For the first lifetime episode, the recommended duration of treatment is 6 to 9 months (4 to 6 months after recovery).55 Longer therapy is appropriate for those with comorbid anxiety disorders, severe initial symptoms, difficulty in attaining therapeutic response, deficient social support, or a history of substance abuse, as well as for older adults. For patients with 3 or more previous episodes, long-term maintenance therapy is recommended.55 For those with even one past episode, extended maintenance therapy might be beneficial. Maintenance therapy should be at the full dose required to attain initial response. In one study, only about 20% to 30% (depending on the treatment) experienced recurrence over 3 years if maintained at full dose, compared with 70% maintained at half the initial treatment dose, and 78% of those receiving placebo.56 For women who have previously suffered from postpartum depression, postpartum prophylaxis can be very effective. In one randomized trial, 62.5% of women on place-bo experienced recurrence compared with only 6.7% of those receiving prophylaxis.57
Practice strategies to improve care
A number of primary care investigators have demonstrated the value of practice management and quality improvement techniques to increase the portion of patients who achieve and maintain response to depression therapy. These studies share an approach of “active management” to promote adherence to treatment guidelines.58-63 For instance, Simon and colleagues demonstrated the value of initial and monthly phone contact.64
Active management techniques include the following:
- Initial and ongoing patient education and counseling, as discussed above
- Patient involvement and agreement in treatment choice
- Initial phone contact to assure the prescription has been filled and initial dose taken
- Periodic contact to inquire about adherence, treatment response, side effects, and to answer patient questions
- Adjustment of therapy for those not responding adequately by 4 to 6 weeks
- Establishment of a collaborative relationship with a psychiatrist for consultation and telephone advice
Additionally, primary care clinicians may find it helpful to add depression to their medical record preventive health maintenance flow chart, especially for patients with any past history of depression. Using the PHQ-9 can be beneficial in providing both the patient and physician with an objective measure of monitoring response and remission.
Conclusions
Effective and available treatments can have a major beneficial impact on patients with depression. To be maximally effective, primary care clinicians must actively manage the care of their depressed patients, using screening strategies to recognize depression in addition to targeted educational messages and active follow-up to improve treatment adherence. Long-term maintenance treatment prevents further recurrences in those who have already experienced multiple episodes. Choice of treatment should be guided by patient preference. For pharmacologic agents, selection should be based on effectiveness, likelihood of side effects and resultant premature discontinuation, and potential for drug–drug interaction. The majority of individuals with depression are managed solely in primary-care settings. With adequate treatment, remission of symptoms, significant improvement in quality of life, and return to full function at home and at work can be attained.
While family physicians play a leading role in caring for patients with major depression, the quality of that care that could be greatly improved. A 1997 to 1998 survey of a national sample of adults with depressive or anxiety disorders revealed that 83% of these patients visited a health care provider.1 Of this total, 84% were treated by primary care clinicians, compared with 16% who were treated by mental health professionals. However, about 90% of those cared for by mental health professionals received treatment that met criteria for adequacy outlined in treatment guidelines, compared with 19% of those cared for by primary care professionals.
A critical role for family physicians is to integrate treatment of depression with that of other conditions, especially in light of the association of depression with a variety of chronic diseases. The Institute of Medicine has concluded that depression is strongly associated with the occurrence of, and death following, myocardial infarctions.2 In diabetes, depression is associated with a 2% increase in glycosylated hemoglobin levels3 and can predict occurrence of diabetic complications. Additionally, chronic illnesses may, in themselves, exacerbate depression several fold.
Primary care clinicians are ideally positioned to serve as the central health care providers for patients with major depression. These physicians have many attributes that support this role, including their longitudinal relationship with patients, response to undifferentiated problems, frequent use of the biopsychosocial model, and ability to integrate care of mental and medical conditions. However, challenges in fulfilling this role also exist, including difficulties in recognizing patients with major depression, developing an adequate diagnostic initial assessment, implementing effective short- and long-term treatment and management strategies, and integrating care of depression with that of other conditions affecting patients.4 This article will review each of these challenges.
Recognition of major depression
DeGruy has eloquently described the barriers to recognition and management of mental disorders in primary care, including infrequent use of diagnostic criteria, concern regarding treatment effectiveness, availability of time and resources, the presence of other pressing clinical problems, and issues of third-party reimbursement and other organizational concerns.4
Family physicians and their patients often do not recognize somatic symptoms as originating from depression. In one study, primary care physicians correctly identified 94% of depressed patients presenting with psychological complaints, but they failed to recognize the psychiatric nature of somatic complaints in about half of the patients. This finding is of concern because 83% of depressed patients presented with somatic complaints.5
The attribution patients assign to their problems can also contribute to lack of recognition. In one general practice study, patients’ attributions were classified as somatizing (5%), psychologizing (23%), normalizing (48%), or no predominate attribution (24%).6 For example, patients in this study might attribute fatigue to anemia (somatizing), emotional exhaustion (psychologizing), or being over-extended (normalizing). The likelihood of a missed diagnosis in patients who met criteria for depression or anxiety was strongly associated with attribution: Physicians diagnosed 72% of psychologizing patients accurately, but they reported a correct diagnosis in only 17% of somatizing patients, 15% of normalizing patients, and 31% of patients with no predominate attribution.
Initial diagnostic assessment
The United States Preventive Services Task Force suggests that primary care physicians screen for major depression. The Task Force recommends using 2 simple questions about mood and anhedonia (Table 1) that are generally as effective as longer instruments.7 The Patient Health Questionnaire-9 (PHQ-9) or the longer Prime-MD can be used for further evaluation of patients who respond positively to either question, thus helping to both confirm the diagnosis of depression and measure severity.8,9 Other instruments include the Beck Depression Inventory,10 the Zung scale,11 and the General Health Questionnaire.12 These tools take longer to administer, are not specific in measuring the criteria for major depression, and do not measure severity well.
In family practices, pregnant and postpartum women represent a special population at increased risk for depression.13 About 5% of middle class women and up to one quarter of low income women experience postpartum depression.14 In about half, onset of the depressive disorder occurs before delivery.15 Women who have previously suffered postpartum depression are at high risk, as are those with histories of depression or premenstrual dysphoric disorder. The Edinburgh Postnatal Depression Scale is a useful 10-item self-report instrument available in Spanish and English (Table 1).16,17 Similar instruments have not been developed for pregnant women.
A patient who responds positively to the 2 screening questions in Table 1 or to another screening approach should be further evaluated to confirm the diagnosis of major depression. Many primary care clinicians do this through unstructured history taking. Others use an instrument such as the previously discussed PHQ-9. This tool offers an advantage because it provides a reliable symptom assessment, measures severity, and can be repeated over time to evaluate therapeutic response.8
The physician should consider bereavement and substance abuse as possible causes of depression; bereaved patients who continue to meet criteria for major depression at 2 months often benefit from treatment. By that time, the sadness, poor concentration, and other symptoms associated with normal grief are no longer constant and occur in waves brought on by memories. Conversely, persons also suffering from depression report these symptoms as enduring and autonomous.18
The primary care physician also should inquire about agitation and symptoms of anxiety disorders. These are experienced by 85% of depressed patients; 50% have comorbid anxiety disorders.19-21 Identification of such comorbidity is helpful in determining treatment, evaluating response, and managing patients over the long term. The Prime-MD, available in multiple languages, is also useful for screening for both anxiety and substance abuse, which can complicate both the recognition and treatment of comorbid depression.9
Sexual function is often affected by depression. The physician should inquire about sexual arousal, erection or lubrication, and orgasm during the initial assessment.22 Approximately 50% of women and 40% of men with major depression report sexual-arousal problems, and 15% to 20% report orgasm problems during the month prior to diagnosis.23 Further questioning can assess whether this dysfunction is caused by another disorder (eg, diabetes) or whether it is part of the depressive syndrome. This provides a baseline for later assessment of side effects and treatment effectiveness, and it communicates to the patient that the physician will be attentive to this area. In discussing sexual function with depressed patients, it may be helpful to tell patients that a study of the effectiveness of treatment of depression with selective serotonin reuptake inhibitors (SSRIs) found that patients reported modestly improved sexual function with treatment.24
TABLE 1
Screening for depression
| Outpatient adults | |
| |
| Postpartum women (Edinburgh Postnatal Depression Scale) | |
1. I have been able to laugh and see the funny side of things
| 6. Things have been getting on top of me
|
2. I have looked forward with enjoyment to things
| 7. I have been so unhappy that I have had difficulty sleeping
|
3. I have blamed myself unnecessarily when things went wrong
| 8. I have felt sad or miserable
|
4. I have been anxious or worried for no good reason
| 9. I have been so unhappy that I have been crying
|
5. I have felt scared or panicky for no very good reason
| 10. The thought of harming myself has occurred to me
|
| Reprinted with permission, from Cox JL et al. British Journal of Psychiatry. 1987; 150:782-786. | |
Management of major depression
The acute management of the patient with major depression includes patient education, shared decision-making regarding a treatment modality, supportive counseling, and treatment-specific counseling.25 Education and counseling should extend over the initial weeks of treatment and be combined with monitoring response, identifying and managing any treatment-emergent side effects, and adjusting medications. Long-term management goals include attaining full remission of symptoms, assisting the patient to return to full functional status, integrating depression care with the treatment of other chronic illnesses, maintaining or tapering pharmacologic treatment, and monitoring for and preventing relapse or recurrence.
Education
Education should help patients understand and accept the diagnosis, reduce any stigma they or their families might attach to major depression, and build increased adherence to subsequent treatment.26 It might be helpful to provide a brief explanation of the biologic basis of depression (including biochemical changes in brain function and “chemical imbalances” of serotonin and other neurotransmitters). Explaining pharmacotherapeutic effects (if medication is desired) as mechanisms to help rebalance brain chemistry further emphasizes the biologic basis of depression and decreases any perceptions that depression is a result of moral or character weakness. This educational message should also stress that antidepressants are not habit-forming or addictive, are not “uppers” or “downers,” and are not tranquilizers. The physician also should convey a positive prognosis but note that several weeks and, possibly, adjustments in treatments, may be required. For patients choosing antidepressants, the McArthur Foundation Initiative has identified 7 key educational messages (Table 2).27
TABLE 2
Key messages for patient education about depression
|
Counseling
Patients often benefit from counseling regarding sleep, exercise, and substance use. Many patients with depression experience early morning awakening. Those with agitated depression also often experience delayed sleep onset associated with worry. Providing the patient with information on basic sleep hygiene, exercise, and encouraging abstinence from or moderation in consumption of alcohol might all help.28-30 Additionally, sleep disturbances can indicate the possibility of comorbid disorders. A report that a patient fears going to sleep because of nightmares suggests posttraumatic stress disorder.
For some patients, counseling by the family physician or through referral may be a helpful treatment adjunct. Often depressed patients have deficient coping mechanisms and need assistance in developing strategies to resolve issues in their life. Principles used in cognitive behavioral therapy might be helpful in patient education and counseling.31 These include problem-solving strategies to resolve stressful concerns and cognitive techniques to identify and correct distorted or maladaptive thought patterns.29
As patients respond to depression treatment, an additional component of primary-care-based counseling should target reinvolvement with pleasurable social and physical activities. This may simply involve identifying activities the patient enjoyed prior to the onset of depression but has since stopped, and focusing on the steps required to reactivate these interests.
Shared decision-making with regard to treatment will improve subsequent patient adherence.27 Treatment options include psychotherapy, particularly cognitive behavioral therapy, pharmacotherapy, and electroconvulsive therapy. The latter should be considered for severely depressed patients, particularly persons with few social supports who are at significant risk of suicide.25
Cognitive behavioral therapy and other psychotherapies can show effectiveness equal to that of pharmacotherapy, although response usually lags by a month to 6 weeks compared with that attained by pharmacotherapy.32 For moderately to severely depressed patients, pharmacotherapy is the treatment of choice in part because of its more rapid onset of action.25
Pharmacotherapy
Pharmacotherapy, most often in the form of an SSRI, is the treatment of choice for depression as a result of patient preference, insurance coverage limitations, or time constraints. In choosing an anti-depressant, the family physician should be guided by effectiveness and potential for drug–drug interactions and for both short-and long-term side effects.33
Tricyclics, the SSRIs, and other newer antidepressants offer similar efficacy.34 While efficacy assesses outcome under ideal treatment conditions, the primary care physician is more concerned with effectiveness, defined as the proportion of patients started on an antidepressant during routine clinical practice who attain lasting benefit. Effectiveness includes consideration of patients who discontinue treatment because of side effects or drug–drug interactions, as well as those who do not obtain adequate therapeutic response. Since about 25% of patients discontinue SSRIs because of side effects, this is an important concern.24 Few studies have been conducted comparing the effectiveness of antidepressants.
Drug–drug interactions are mediated predominately by the cytochrome P450 isoenzymes responsible for drug metabolism in the liver.35-37 The 2D6 isoenzyme is responsible for 50% of drug metabolism in the liver; the 3A4 isoenzyme is responsible for another 30%.38 As a clinical example of the importance of such inhibition, codeine requires 2D6-mediated metabolism to become morphine and is ineffective for pain in many patients who are prescribed a 2D6 inhibitor. Patients receiving such agents also can have a 300% to 400% increase in blood levels of previously stable ß-blockers. Paroxetine and fluoxetine, the two SSRIs that strongly inhibit the 2D6 isoenzyme, cause clinically significant interactions; fluoxetine is also a moderate inhibitor of the 3A4 isoenzyme.35 Because of the number of potential drug–drug interactions through these isoenzymes, physicians must check for interactions before prescribing these medications or adding other new medications in patients already receiving these agents. This also is a consideration for patients who might require additional medications acutely, for instance in response to a cardiac or other emergency.
Side effects of concern include gastrointestinal effects, particularly nausea, and central nervous system (CNS) effects, including anxiety and agitation, sleep disturbance, and tremor. When these occur, they often decrease rapidly over the first 1 to 3 weeks. If severe, they can be managed by a temporary dosage decrease. For patients with significant CNS side effects, altering the timing of the daily dose might provide relief from daytime somnolence or agitation or from nighttime insomnia.
Long-term side effects of concern include weight gain and sexual dysfunction. While other SSRIs have low rates for weight gain, paroxetine causes a weight gain of more than 7% (about 10 lbs for a patient of average weight) in 20% to 25% of patients.39 Some element of sexual dysfunction, most often delayed orgasm, is estimated to occur in 30% to 40% of individuals receiving SSRIs.40,41 Management options include delaying dosage of agents with a half-life of about 24 hours (escitalopram, citalopram, sertraline).42 For instance, an individual who usually takes one of these agents in the morning may delay a day’s dose until after engaging in sexual intercourse in the evening. While open-label studies support augmentation, particularly with bupropion or buspirone, the few small randomized double-blind trials available suggest that positive results should be interpreted with caution.43 Alternatively, patients may benefit from sildenafil44 or a switch to a non-SSRI antidepressant.
While management of side effects presents one option, the best clinical approach may be to select an agent with minimal side-effect potential. In double-blind randomized trials, escitalopram, a new SSRI treatment option, was demonstrated to require treatment termination in less than 5% of recipients at its usual dose of 10 mg, a rate no different from that of placebo.45 In contrast, rates of 15% to 30% have been reported for other SSRIs and newer antidepressants at the time of their initial release.
Adjusting treatment
One recent primary care trial examined the effectiveness of 3 SSRIs: fluoxetine, sertraline, and paroxetine. At the time this study was designed, citalopram was not in common use. While about 75% of patients attained remission, only 40% to 50% of patients were maintained on the first prescribed agent.24 Additionally, about 20% of depression “treatment resistance” resulted because patients did not fill their prescriptions or adhere to treatment.46 For patients who do not respond within the first month, increasing the dosage is appropriate.47 About 25% of patients respond to this adjustment.48 For patients who do not respond, reassessment of the diagnosis, as well as assessment of potential psychiatric comorbidities and suicidal ideation, is indicated. For nonresponders, and for those with intolerable side effects, switching to a second SSRI is a reasonable next step.49 About 50% of patients switched to a second agent respond.50 For those who do not respond, the primary care physician might consider a second medication switch or psychiatric consultation.
Further treatment adjustment is indicated for patients who experience partial response. This might take the form of augmentation with psychotherapy51 or with another agent.52 Lithium and thyroid hormone (often as 25 to 50 mg T3 daily) are the most frequently used options, although stimulants, other antidepressants, and atypical antipsychotics are all of value in some patients.48,49,53
When indicated, treatment should be discontinued by tapering the dose over several weeks to months, depending on the duration and severity of past episodes. Patients should be educated to be alert for recurrence. They should also be monitored for recurrence and restarted on full-dose therapy if this occurs. If patients stop therapy abruptly, the likelihood of withdrawal symptoms (agitation, irritability, dizziness, ataxia, nausea, paresthesias, sleep disturbances) is highly related to the half-life of the SSRI.39 For paroxetine, which has the shortest half-life, withdrawal is frequent; the extended release preparation does not decrease the likelihood of withdrawal. Withdrawal symptoms are infrequent (< 2%) for sertraline, citalopram, and escitalopram, and they do not occur with fluoxetine.
Duration of treatment
A major challenge in family practice is maintaining patient adherence to treatment for the recommended interval to prevent relapse and to avoid recurrence in those with a history of prior episodes. In one study, 25% to 33% of primary care patients stopped depression therapy within 1 month and over 40% within 3 months. Additionally, 62% failed to inform their physicians.54 Depression also adversely affects compliance with treatment of comorbid medical conditions; in one meta-analysis, depression increased noncompliance 3-fold.54
For the first lifetime episode, the recommended duration of treatment is 6 to 9 months (4 to 6 months after recovery).55 Longer therapy is appropriate for those with comorbid anxiety disorders, severe initial symptoms, difficulty in attaining therapeutic response, deficient social support, or a history of substance abuse, as well as for older adults. For patients with 3 or more previous episodes, long-term maintenance therapy is recommended.55 For those with even one past episode, extended maintenance therapy might be beneficial. Maintenance therapy should be at the full dose required to attain initial response. In one study, only about 20% to 30% (depending on the treatment) experienced recurrence over 3 years if maintained at full dose, compared with 70% maintained at half the initial treatment dose, and 78% of those receiving placebo.56 For women who have previously suffered from postpartum depression, postpartum prophylaxis can be very effective. In one randomized trial, 62.5% of women on place-bo experienced recurrence compared with only 6.7% of those receiving prophylaxis.57
Practice strategies to improve care
A number of primary care investigators have demonstrated the value of practice management and quality improvement techniques to increase the portion of patients who achieve and maintain response to depression therapy. These studies share an approach of “active management” to promote adherence to treatment guidelines.58-63 For instance, Simon and colleagues demonstrated the value of initial and monthly phone contact.64
Active management techniques include the following:
- Initial and ongoing patient education and counseling, as discussed above
- Patient involvement and agreement in treatment choice
- Initial phone contact to assure the prescription has been filled and initial dose taken
- Periodic contact to inquire about adherence, treatment response, side effects, and to answer patient questions
- Adjustment of therapy for those not responding adequately by 4 to 6 weeks
- Establishment of a collaborative relationship with a psychiatrist for consultation and telephone advice
Additionally, primary care clinicians may find it helpful to add depression to their medical record preventive health maintenance flow chart, especially for patients with any past history of depression. Using the PHQ-9 can be beneficial in providing both the patient and physician with an objective measure of monitoring response and remission.
Conclusions
Effective and available treatments can have a major beneficial impact on patients with depression. To be maximally effective, primary care clinicians must actively manage the care of their depressed patients, using screening strategies to recognize depression in addition to targeted educational messages and active follow-up to improve treatment adherence. Long-term maintenance treatment prevents further recurrences in those who have already experienced multiple episodes. Choice of treatment should be guided by patient preference. For pharmacologic agents, selection should be based on effectiveness, likelihood of side effects and resultant premature discontinuation, and potential for drug–drug interaction. The majority of individuals with depression are managed solely in primary-care settings. With adequate treatment, remission of symptoms, significant improvement in quality of life, and return to full function at home and at work can be attained.
1. Young AS, Klap R, Sherbourne CD, Wells KB. The quality of care for depressive and anxiety disorders in the United States. Arch Gen Psychiatry. 2001;58:55-61.
2. Institute of Medicine (U.S.). Committee on Health and Behavior: Research Practice and Policy. Health and Behavior: The Interplay of Biological, Behavioral, and Societal Influences. Washington, DC: National Academy Press. 2001.
3. Lustman PJ, Griffith LS, Freedland KE, Clouse RE. The course of major depression in diabetes. Gen Hosp Psychiatry. 1997;19:138-143.
4. deGruy F, III. Mental health care in the primary care setting. In: Donaldson MS, ed. Primary Care: America’s Health in a New Era. Washington, DC: National Academy Press; 1996;285-311.
5. Bridges KW, Goldberg DP. Somatic presentation of DSM III psychiatric disorders in primary care. J Psychosom Res. 1985;29:563-569.
6. Kessler D, Lloyd K, Lewis G, Gray DP. Cross sectional study of symptom attribution and recognition of depression and anxiety in primary care. BMJ. 1999;318:436-439.
7. Whooley MA, Avins AL, Miranda J, Browner WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med. 1997;12:439-445.
8. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
9. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA. 1999;282:1737-1744.
10. Steer RA, Cavalieri TA, Leonard DM, Beck AT. Use of the Beck Depression Inventory for Primary Care to screen for major depression disorders. Gen Hosp Psychiatry. 1999;21:106-111.
11. Biggs JT, Wylie LT, Ziegler VE. Validity of the Zung Self-rating Depression Scale. Br J Psychiatry. 1978;132:381-385.
12. Goldberg DP, Gater R, Sartorius N, et al. The validity of two versions of the GHQ in the WHO study of mental illness in general health care. Psychol Med. 1997;27:191-197.
13. Susman JL. Postpartum depressive disorders. J Fam Pract. 1996;43(6 suppl):S17-24.
14. O’Hara MW, Schlechte JA, Lewis DA, Varner MW. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100:63-73.
15. Yonkers KA, Ramin SM, Rush AJ, et al. Onset and persistence of postpartum depression in an inner-city maternal health clinic system. Am J Psychiatry. 2001;158:1856-1863.
16. Georgiopoulos AM, Bryan TL, Wollan P, Yawn BP. Routine screening for postpartum depression. J Fam Pract. 2001;50:117-122.
17. Eberhard-Gran M, Eskild A, Tambs K, Opjordsmoen S, Samuelsen SO. Review of validation studies of the Edinburgh Postnatal Depression Scale. Acta Psychiatr Scand. 2001;104:243-249.
18. Osterweis M, Solomon F, Green M. Institute of Medicine (U.S.). Committee for the Study of Health Consequences of the Stress of Bereavement. Bereavement: Reactions, Consequences, and Care. Washington, DC: National Academy Press; 1984.
19. Keller MB, Hanks DL. The natural history and heterogeneity of depressive disorders: implications for rational antidepressant therapy. J Clin Psychiatry. 1994;55 (suppl A):25-31;discussion32-23,98-100.
20. Keller MB, Hanks DL. Anxiety symptom relief in depression treatment outcomes. J Clin Psychiatry. 1995;56(suppl 6):22-29.
21. Kravitz HM, Fogg L, Fawcett J, Edwards J. Antidepressant or antianxiety? A study of the efficacy of antidepressant medication. Psychiatry Res. 1990;32:141-149.
22. Clayton AH. Recognition and assessment of sexual dysfunction associated with depression. J Clin Psychiatry. 2001;62(suppl 3):5-9.
23. Kennedy SH, Dickens SE, Eisfeld BS, et al. Sexual dysfunction before antidepressant therapy in major depression. J Affect Disord. 1999;201-208.
24. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: a randomized trial. JAMA. 2001;286:2947-2955.
25. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical Practice Guideline, Number 5. Rockville, MD: U.S. Dept. of Health and Human Services, Agency for Health Care Policy and Research; April 1993. AHCPR publication 93-0551.
26. Hegner RE. Dispelling the myths and stigma of mental illness: the Surgeon General’s report on mental health. Issue Brief Natl Health Policy Forum. 2000;(754):1-7.
27. Lin EH, Von Korff M, Katon W, et al. The role of the primary care physician in patients’ adherence to antidepressant therapy. Med Care. 1995;33:67-74.
28. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. In: Hauri P, ed. Case Studies in Insomnia. New York: Plenum Medical Book; 1991:xiv, 254.
29. Culpepper L. Worries and anxiety. In: Staton EW, ed. 20 Common Problems in Behavioral Health. New York: McGraw-Hill; 2002;385-404.
30. Miser WF. Exercise as an effective treatment option for major depression in older adults. J Fam Pract. 2000;49:109-110.
31. Robinson P, Bush T, Von Korff M, et al. Primary care physician use of cognitive behavioral techniques with depressed patients. J Fam Pract. 1995;40:352-357.
32. Rush AJ, Thase ME. Psychotherapies for depressive disorders: a review. In: Sartorius N, ed. Depressive Disorders. New York: John Wiley and Sons; 1999.
33. Preskorn SH. Selection of an antidepressant: mirtazapine. J Clin Psychiatry. 1997;58(suppl 6):3-8.
34. Geddes JR, Freemantle N, Mason J, Eccles MP, Boynton J. SSRIs versus other antidepressants for depressive disorder. Cochrane Database Syst Rev. 2000;CD001851.-
35. Preskorn SH. Debate resolved: there are differential effects of serotonin selective reuptake inhibitors on cytochrome P450 enzymes. J Psychopharmacol. 1998;12(3 suppl B):S89-97.
36. Preskorn SH. Antidepressant options in primary care. Clin Cornerstone. 1999;1:31-55.
37. Greenblatt DJ, von Moltke LL, Harmatz JS, Shader RI. Drug interactions with newer antidepressants: role of human cytochromes P450. J Clin Psychiatry. 1998;59 (suppl. 15):19-27.
38. Preskorn SH. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin Pharmacokinet. 1997;32(suppl 1):1-21.
39. Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863-867.
40. Montejo-Gonzalez AL, Llorca G, Izquierdo JA, et al. SSRI-induced sexual dysfunction: fluoxetine, paroxetine, sertraline, and fluvoxamine in a prospective, multicenter, and descriptive clinical study of 344 patients. J Sex Marital Ther. 1997;23:176-194.
41. Fava M, Rankin M. Sexual functioning and SSRIs. J Clin Psychiatry. 2002;63(suppl 5):13-16;discussion 23-15.
42. Zajecka J. Strategies for the treatment of antidepressant-related sexual dysfunction. J Clin Psychiatry. 2001;62 (suppl 3):35-43.
43. Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Practice. 2002;51:681.-
44. Nurnberg HG, Hensley PL, Lauriello J, Parker LM, Keith SJ. Sildenafil for women patients with antidepressant-induced sexual dysfunction. Psychiatr Serv. 1999;50:1076-1078.
45. Wade A, Michael Lemming O, Bang Hedegaard K. Escitalopram 10mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2002;95-102.
46. Souery D, Mendlewicz J. Compliance and therapeutic issues in resistant depression. Int Clin Psychopharmacol. 1998;13 (suppl 2):S13-18.
47. Thase ME, Rush AJ. Treatment-resistant depression. In: Kupfer DJ, ed. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995;1081-1097.
48. Thase ME. What role do atypical antipsychotic drugs have in treatment-resistant depression. J Clin Psychiatry. 2002;63:95-103.
49. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(4 suppl):1-45.
50. Howland RH, Thase ME. What to do with SSRI non-responders? J Pract Psychiatry Behav Health. 1999;5:216-233.
51. Thase ME, Friedman ES, Howland RH. Management of treatment-resistant depression: psychotherapeutic perspectives. J Clin Psychiatry. 2001;62(suppl 18):18-24.
52. Fava M. Augmentation and combination strategies in treatment-resistant depression. J Clin Psychiatry. 2001;62 (suppl 18):4-11.
53. Thase ME, Howland RH, Friedman ES. Treating antidepressant nonresponders with augmentation strategies: an overview. J Clin Psychiatry. 1998;59(suppl 5):5-12;discussion 13-15.
54. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160:2101-2107.
55. Keller MB. The long-term treatment of depression. J Clin Psychiatry. 1999;60(suppl 17):41-45;discussion 46-48.
56. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49:782-787.
57. Wisner KL, Wheeler SB. Prevention of recurrent postpartum major depression. Hosp Community Psychiatry. 1994;45:1191-1196.
58. Schulberg HC, Katon W, Simon GE, Rush AJ. Treating major depression in primary care practice: an update of the Agency for Health Care Policy and Research Practice Guidelines. Arch Gen Psychiatry. 1998;55:1121-1127.
59. Schulberg HC. Treating depression in primary care practice: applications of research findings. J Fam Pract. 2001;50:535-537.
60. Katon W, Von Korff M, Lin E, et al. Collaborative management to achieve treatment guidelines. Impact on depression in primary care. JAMA. 1995;273:1026-1031.
61. Katon W, Robinson P, Von Korff M, et al. A multifaceted intervention to improve treatment of depression in primary care. Arch Gen Psychiatry. 1996;53:924-932.
62. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry. 1999;56:1109-1115.
63. Von Korff M, Katon W, Unutzer J, Wells K, Wagner EH. Improving depression care: barriers, solutions, and research needs. J Fam Pract. 2001;50:E1.-
64. Simon GE, VonKorff M, Rutter C, Wagner E. Randomised trial of monitoring, feedback, and management of care by telephone to improve treatment of depression in primary care. BMJ. 2000;320:550-554.
1. Young AS, Klap R, Sherbourne CD, Wells KB. The quality of care for depressive and anxiety disorders in the United States. Arch Gen Psychiatry. 2001;58:55-61.
2. Institute of Medicine (U.S.). Committee on Health and Behavior: Research Practice and Policy. Health and Behavior: The Interplay of Biological, Behavioral, and Societal Influences. Washington, DC: National Academy Press. 2001.
3. Lustman PJ, Griffith LS, Freedland KE, Clouse RE. The course of major depression in diabetes. Gen Hosp Psychiatry. 1997;19:138-143.
4. deGruy F, III. Mental health care in the primary care setting. In: Donaldson MS, ed. Primary Care: America’s Health in a New Era. Washington, DC: National Academy Press; 1996;285-311.
5. Bridges KW, Goldberg DP. Somatic presentation of DSM III psychiatric disorders in primary care. J Psychosom Res. 1985;29:563-569.
6. Kessler D, Lloyd K, Lewis G, Gray DP. Cross sectional study of symptom attribution and recognition of depression and anxiety in primary care. BMJ. 1999;318:436-439.
7. Whooley MA, Avins AL, Miranda J, Browner WS. Case-finding instruments for depression: two questions are as good as many. J Gen Intern Med. 1997;12:439-445.
8. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16:606-613.
9. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self-report version of PRIME-MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA. 1999;282:1737-1744.
10. Steer RA, Cavalieri TA, Leonard DM, Beck AT. Use of the Beck Depression Inventory for Primary Care to screen for major depression disorders. Gen Hosp Psychiatry. 1999;21:106-111.
11. Biggs JT, Wylie LT, Ziegler VE. Validity of the Zung Self-rating Depression Scale. Br J Psychiatry. 1978;132:381-385.
12. Goldberg DP, Gater R, Sartorius N, et al. The validity of two versions of the GHQ in the WHO study of mental illness in general health care. Psychol Med. 1997;27:191-197.
13. Susman JL. Postpartum depressive disorders. J Fam Pract. 1996;43(6 suppl):S17-24.
14. O’Hara MW, Schlechte JA, Lewis DA, Varner MW. Controlled prospective study of postpartum mood disorders: psychological, environmental, and hormonal variables. J Abnorm Psychol. 1991;100:63-73.
15. Yonkers KA, Ramin SM, Rush AJ, et al. Onset and persistence of postpartum depression in an inner-city maternal health clinic system. Am J Psychiatry. 2001;158:1856-1863.
16. Georgiopoulos AM, Bryan TL, Wollan P, Yawn BP. Routine screening for postpartum depression. J Fam Pract. 2001;50:117-122.
17. Eberhard-Gran M, Eskild A, Tambs K, Opjordsmoen S, Samuelsen SO. Review of validation studies of the Edinburgh Postnatal Depression Scale. Acta Psychiatr Scand. 2001;104:243-249.
18. Osterweis M, Solomon F, Green M. Institute of Medicine (U.S.). Committee for the Study of Health Consequences of the Stress of Bereavement. Bereavement: Reactions, Consequences, and Care. Washington, DC: National Academy Press; 1984.
19. Keller MB, Hanks DL. The natural history and heterogeneity of depressive disorders: implications for rational antidepressant therapy. J Clin Psychiatry. 1994;55 (suppl A):25-31;discussion32-23,98-100.
20. Keller MB, Hanks DL. Anxiety symptom relief in depression treatment outcomes. J Clin Psychiatry. 1995;56(suppl 6):22-29.
21. Kravitz HM, Fogg L, Fawcett J, Edwards J. Antidepressant or antianxiety? A study of the efficacy of antidepressant medication. Psychiatry Res. 1990;32:141-149.
22. Clayton AH. Recognition and assessment of sexual dysfunction associated with depression. J Clin Psychiatry. 2001;62(suppl 3):5-9.
23. Kennedy SH, Dickens SE, Eisfeld BS, et al. Sexual dysfunction before antidepressant therapy in major depression. J Affect Disord. 1999;201-208.
24. Kroenke K, West SL, Swindle R, et al. Similar effectiveness of paroxetine, fluoxetine, and sertraline in primary care: a randomized trial. JAMA. 2001;286:2947-2955.
25. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical Practice Guideline, Number 5. Rockville, MD: U.S. Dept. of Health and Human Services, Agency for Health Care Policy and Research; April 1993. AHCPR publication 93-0551.
26. Hegner RE. Dispelling the myths and stigma of mental illness: the Surgeon General’s report on mental health. Issue Brief Natl Health Policy Forum. 2000;(754):1-7.
27. Lin EH, Von Korff M, Katon W, et al. The role of the primary care physician in patients’ adherence to antidepressant therapy. Med Care. 1995;33:67-74.
28. Bootzin RR, Epstein D, Wood JM. Stimulus control instructions. In: Hauri P, ed. Case Studies in Insomnia. New York: Plenum Medical Book; 1991:xiv, 254.
29. Culpepper L. Worries and anxiety. In: Staton EW, ed. 20 Common Problems in Behavioral Health. New York: McGraw-Hill; 2002;385-404.
30. Miser WF. Exercise as an effective treatment option for major depression in older adults. J Fam Pract. 2000;49:109-110.
31. Robinson P, Bush T, Von Korff M, et al. Primary care physician use of cognitive behavioral techniques with depressed patients. J Fam Pract. 1995;40:352-357.
32. Rush AJ, Thase ME. Psychotherapies for depressive disorders: a review. In: Sartorius N, ed. Depressive Disorders. New York: John Wiley and Sons; 1999.
33. Preskorn SH. Selection of an antidepressant: mirtazapine. J Clin Psychiatry. 1997;58(suppl 6):3-8.
34. Geddes JR, Freemantle N, Mason J, Eccles MP, Boynton J. SSRIs versus other antidepressants for depressive disorder. Cochrane Database Syst Rev. 2000;CD001851.-
35. Preskorn SH. Debate resolved: there are differential effects of serotonin selective reuptake inhibitors on cytochrome P450 enzymes. J Psychopharmacol. 1998;12(3 suppl B):S89-97.
36. Preskorn SH. Antidepressant options in primary care. Clin Cornerstone. 1999;1:31-55.
37. Greenblatt DJ, von Moltke LL, Harmatz JS, Shader RI. Drug interactions with newer antidepressants: role of human cytochromes P450. J Clin Psychiatry. 1998;59 (suppl. 15):19-27.
38. Preskorn SH. Clinically relevant pharmacology of selective serotonin reuptake inhibitors. An overview with emphasis on pharmacokinetics and effects on oxidative drug metabolism. Clin Pharmacokinet. 1997;32(suppl 1):1-21.
39. Fava M, Judge R, Hoog SL, Nilsson ME, Koke SC. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000;61:863-867.
40. Montejo-Gonzalez AL, Llorca G, Izquierdo JA, et al. SSRI-induced sexual dysfunction: fluoxetine, paroxetine, sertraline, and fluvoxamine in a prospective, multicenter, and descriptive clinical study of 344 patients. J Sex Marital Ther. 1997;23:176-194.
41. Fava M, Rankin M. Sexual functioning and SSRIs. J Clin Psychiatry. 2002;63(suppl 5):13-16;discussion 23-15.
42. Zajecka J. Strategies for the treatment of antidepressant-related sexual dysfunction. J Clin Psychiatry. 2001;62 (suppl 3):35-43.
43. Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Practice. 2002;51:681.-
44. Nurnberg HG, Hensley PL, Lauriello J, Parker LM, Keith SJ. Sildenafil for women patients with antidepressant-induced sexual dysfunction. Psychiatr Serv. 1999;50:1076-1078.
45. Wade A, Michael Lemming O, Bang Hedegaard K. Escitalopram 10mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2002;95-102.
46. Souery D, Mendlewicz J. Compliance and therapeutic issues in resistant depression. Int Clin Psychopharmacol. 1998;13 (suppl 2):S13-18.
47. Thase ME, Rush AJ. Treatment-resistant depression. In: Kupfer DJ, ed. Psychopharmacology: The Fourth Generation of Progress. New York: Raven Press; 1995;1081-1097.
48. Thase ME. What role do atypical antipsychotic drugs have in treatment-resistant depression. J Clin Psychiatry. 2002;63:95-103.
49. Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(4 suppl):1-45.
50. Howland RH, Thase ME. What to do with SSRI non-responders? J Pract Psychiatry Behav Health. 1999;5:216-233.
51. Thase ME, Friedman ES, Howland RH. Management of treatment-resistant depression: psychotherapeutic perspectives. J Clin Psychiatry. 2001;62(suppl 18):18-24.
52. Fava M. Augmentation and combination strategies in treatment-resistant depression. J Clin Psychiatry. 2001;62 (suppl 18):4-11.
53. Thase ME, Howland RH, Friedman ES. Treating antidepressant nonresponders with augmentation strategies: an overview. J Clin Psychiatry. 1998;59(suppl 5):5-12;discussion 13-15.
54. DiMatteo MR, Lepper HS, Croghan TW. Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med. 2000;160:2101-2107.
55. Keller MB. The long-term treatment of depression. J Clin Psychiatry. 1999;60(suppl 17):41-45;discussion 46-48.
56. Shea MT, Elkin I, Imber SD, et al. Course of depressive symptoms over follow-up. Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch Gen Psychiatry. 1992;49:782-787.
57. Wisner KL, Wheeler SB. Prevention of recurrent postpartum major depression. Hosp Community Psychiatry. 1994;45:1191-1196.
58. Schulberg HC, Katon W, Simon GE, Rush AJ. Treating major depression in primary care practice: an update of the Agency for Health Care Policy and Research Practice Guidelines. Arch Gen Psychiatry. 1998;55:1121-1127.
59. Schulberg HC. Treating depression in primary care practice: applications of research findings. J Fam Pract. 2001;50:535-537.
60. Katon W, Von Korff M, Lin E, et al. Collaborative management to achieve treatment guidelines. Impact on depression in primary care. JAMA. 1995;273:1026-1031.
61. Katon W, Robinson P, Von Korff M, et al. A multifaceted intervention to improve treatment of depression in primary care. Arch Gen Psychiatry. 1996;53:924-932.
62. Katon W, Von Korff M, Lin E, et al. Stepped collaborative care for primary care patients with persistent symptoms of depression: a randomized trial. Arch Gen Psychiatry. 1999;56:1109-1115.
63. Von Korff M, Katon W, Unutzer J, Wells K, Wagner EH. Improving depression care: barriers, solutions, and research needs. J Fam Pract. 2001;50:E1.-
64. Simon GE, VonKorff M, Rutter C, Wagner E. Randomised trial of monitoring, feedback, and management of care by telephone to improve treatment of depression in primary care. BMJ. 2000;320:550-554.