User login
45-year-old woman • fever and chills • diffuse abdominal pain • shortness of breath • Dx?
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
THE CASE
A 45-year-old white woman presented to our emergency department (ED) with a 3-day history of fever, chills, diffuse abdominal pain, severe headache, and shortness of breath.
The patient’s medical and surgical history was notable for acromegaly secondary to pituitary microadenoma, pituitary resection, and complete thyroidectomy 4 years earlier. Her medications included lanreotide, levothyroxine, gabapentin, alprazolam, and zolpidem. She had no history of cardiac disease, diabetes mellitus, immunodeficiency, or injection drug use. Three months prior to presenting to the ED, she underwent an outpatient gynecologic procedure for insertion of a levonorgestrel-releasing intrauterine device (IUD) for menorrhagia.
In the ED, the patient had a fever (101.5°F) and an elevated white blood cell count of 13,600/mm3 (reference range, 4,000–10,000/mm3). Cardiac auscultation revealed a regular heart rate and rhythm, with normal S1 and S2 sounds without murmur. Electrocardiogram documented normal sinus rhythm with no abnormalities. The physical examination revealed a diffusely tender lower abdomen without rebound or guarding. A pelvic examination was not conducted, and there was no collection of a vaginal swab sample to test for gonorrhea, chlamydia, or group B Streptococcus (GBS). Further workups for infection, including urinalysis, lumbar puncture, and chest x-ray, all yielded normal results.
Shortly after she was discharged from the ED, the patient was called to return to the hospital after blood cultures grew GBS; she was admitted for treatment.
THE DIAGNOSIS
A diagnosis of sepsis secondary to GBS bacteremia was made. However, the source of the GBS bacteremia and the patient’s abdominal symptoms remained unclear. Further workup included computed tomography (CT) of the abdomen, pelvis, and head, and magnetic resonance imaging of the brain; all imaging revealed no acute findings. Blood work (chem-7 panel, complete blood count, human immunodeficiency virus testing) was unremarkable except for an elevated level of C-reactive protein of 90 mg/L (reference range, 0–10 mg/L).
Radiography confirmed that the IUD was in the correct intrauterine position. However, transesophageal echocardiography (TEE) showed vegetations on the mitral and aortic valves, with preserved cardiac function. A diagnosis of GBS endocarditis was made, and infectious disease specialists were consulted. Because the patient had an anaphylactic allergy to penicillin, she was treated with intravenous vancomycin for 4 weeks. One month later, she had the IUD removed because of persistent abdominal pain.
DISCUSSION
Although the source of GBS bacteremia and endocarditis in our patient remained nondefinitive, the recent insertion of the IUD continued to be the suspected source and leading diagnosis.
Continue to: Other sources of GBS bacteremia...
Other sources of GBS bacteremia were unlikely based on the examination and imaging results. The patient’s abdominal exam was benign, and no intra-abdominal abscess was detected on CT. Although Streptococcus viridans, S bovis, and enterococcus are far more common pathogens for infective endocarditis,1 there was no evidence of dental caries, gastrointestinal pathology, or urinary tract infection to suggest misidentification of bacteria.
Theoretically, GBS bacteremia after a gynecologic procedure is possible since GBS frequently colonizes the vagina.2 However, most reports document transient rather than persistent bacteremia and/or endocarditis.3,4
IUD insertion as a cause of bacteremia. The medical literature offers scant evidence of endocarditis or severe GBS bacteremia related to IUD insertion. Of 124 gynecology-related reports of infective endocarditis between 1946 and 1986, only 3 were associated with IUDs.5 All 3 women had underlying cardiac disease, and 2 of the 3 had identifiable pelvic infections.5
Among 12 case reports of endocarditis related to gynecologic procedures from 1985 to 2003, therapeutic abortion was the most common antecedent event, and no cases were related to IUD insertion.2 Compared with cases reported before 1985, in these cases most patients (64%) did not have underlying valvular disease, and most had a subacute course with low mortality but high morbidity (8 of 11 patients had clinically significant emboli).2 The study authors also mentioned a case of endocarditis following a Pap smear test, suggesting that minimally invasive procedures may result in infective endocarditis.2
THE TAKEAWAY
Our patient presented with fever, fatigue, and abdominal pain in the setting of recent IUD insertion. She was found to have GBS bacteremia with endocarditis based on TEE and positive blood culture growth. Her clinical situation was suspicious for a gynecologic source of bacteremia.
Continue to: There is no definitive way...
There is no definitive way to confirm that IUD insertion 3 months prior caused the GBS bacteremia. However, this case illustrates that it is important to consider a usually benign gynecologic procedure as the source of clinically significant persistent bacteremia.
Evidence is insufficient to recommend prophylactic antibiotic use prior to a gynecologic procedure, and it is not recommended by current practice guidelines of the American College of Obstetricians and Gynecologists or the European Society of Cardiology.6,7
This patient case raises our suspicion for IUD-related bacteremia as an adverse reaction in healthy women with recent IUD insertion who present with fever and diffuse abdominal pain without apparent signs of a pelvic infection. Prompt antibiotic treatment is necessary to prevent significant morbidity and mortality.
CORRESPONDENCE
Lauren Cowen, MD, 777 South Clinton Avenue, Rochester, NY 14620; [email protected]
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.
1. Baddour LM, Wilson WR, Bayer AS, et al; American Heart Association Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease of the Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and Stroke Council. Infective endocarditis in adults: diagnosis, antimicrobial therapy, and management of complications: a scientific statement for healthcare professionals from the American Heart Association. Circulation. 2015;132:1435-1486.
2. Crespo A, Retter AS, Lorber B. Group B streptococcal endocarditis in obstetric and gynecologic practice. Infect Dis Obstet Gynecol. 2003;11:109-115.
3. Murray S, Hickey JB, Houang E. Significant bacteremia associated with replacement of intrauterine contraceptive device. Am J Obstet Gynecol. 1987;156:698-700.
4. Everett ED, Reller LB, Droegemueller W, et al. Absence of bacteremia after insertion or removal of intrauterine devices. Obstet Gynecol. 1976;47:207-209.
5. Seaworth BJ, Durack DT. Infective endocarditis in obstetric and gynecologic practice. Am J Obstet Gynecol. 1986;154:180-188.
6. ACOG Committee on Practice Bulletins–Gynecology. Practice bulletin no. 186: Long-acting reversible contraception: implants and intrauterine devices. Obstet Gynecol. 2017;130:e251-e269.
7. Habib G, Lancellotti P, Antunes MJ, et al; ESC Scientific Document Group. 2015 ESC guidelines for the management of infective endocarditis: the Task Force for the Management of Infective Endocarditis of the European Society of Cardiology (ESC). Eur Heart J. 2015;36:3075-3128.

Worsening nausea, vomiting, and dizziness • 20-pound weight loss in 2 months • mild hearing loss • reoccurring episodes of falls • Dx?
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.

Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.
Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
THE CASE
A 26-year-old Hispanic/African American woman presented to our clinic with a 2-month history of nausea and vomiting, along with dizziness. The nausea and vomiting persistently worsened, and she was only able to tolerate apples and berries. During this 2-month period, she lost 20 pounds and her symptoms progressed to include pruritus, ataxia, and mild hearing loss, with reoccurring episodes of falls.
THE DIAGNOSIS
On examination, she was found to be bradycardic with a heart rate of 47 beats/min, right- axis deviation, and inverted T waves in leads I, II, and augmented vector left. Her family history included the death of an aunt who was in her early 30s due to an unknown heart condition.
Echocardiogram identified mild mitral valve regurgitation with an ejection fraction of 55% to 60% (reference range: 55%-70%). Cardiology determined that her bradycardia was not the source of her symptoms. A neurologic exam identified 3+ hyperreflexia (indicating the reflex was increased), tandem gait instability, and left oculomotor dysfunction.
Brain magnetic resonance imaging (MRI) identified bilateral parietal white matter lesions where a demyelinating process could not be excluded (FIGURE 1A). The patient’s symptoms of nausea and vomiting continued, and she only tolerated peanuts and liquids. An MRI of the spine was negative.
Laboratory testing revealed that the patient was negative for human immunodeficiency virus (HIV), syphilis, Lyme disease, and lupus. Her thyroid-stimulating hormone level was 1.7 mIU/L (reference range: 0.4-4.2 mIU/L), and her vitamin B12 level was 504 pg/mL (reference range: 160-950 pg/mL).
The patient’s lumbar puncture was negative for oligoclonal bands. The IgG synthesis rate/index cerebrospinal fluid (CSF) was –3.9, ruling out multiple sclerosis. Her CSF culture was negative, with a glucose level of 42 mg/dL (reference range: 70-110 mg/dL), colorless appearance, 1 white blood cell, and spinal albumin of 12.2 mg/dL (reference range: 8-42 mg/dL). The visual evoked potential was negative. The aquaporin-4 (AQP4) antibody was positive at 3.4 U/mL, and the myelin oligodendrocyte glycoprotein (MOG) antibody was positive.
Gastroenterology concluded a normal gastric accommodation and unremarkable computed tomography (CT) enterography. Moderate erosions were identified in the stomach with an erythematous gastropathy. The patient was placed on a proton pump inhibitor.
Continue to: Following the examination...
Following the examination and laboratory testing, the patient was admitted under our family medicine service for neuromyelitis optica (NMO) affecting the area postrema. NMO, also known as Devic’s disease, is an autoimmune disorder that affects the spinal cord and optic nerves. Autoantibodies against AQP4 are created in the periphery and are directed against astrocytes in the central nervous system. These antibodies bind to the foot processes of astrocytes, inducing complement-mediated cell damage and granulocyte infiltration.1-5
Intravenous methylprednisolone was initiated at 250 mg every 6 hours for 3 days. A repeat brain MRI demonstrated nonspecific multiple scattered foci of hyperintense signal involving the subcortical supratentorial white matter without abnormal enhancement, most likely representing nonactive demyelinating plaques (FIGURES 1B and 1C).
Dx is revisited. Our patient was referred to an NMO clinic for evaluation. After further testing (including a repeat MRI based on the neurologist’s specifications, anti-aquaporin antibody testing, and MOG-antibody testing) and case discussion, it was determined that the patient had MOG-antibody disease. This disease, along with NMO, comprise a spectrum of disorders referred to as neuromyelitis optica spectrum disorder (NMOSD).
The patient was subsequently prescribed a rituximab infusion, 500 mg/50 mL, to treat the current attack. One infusion was to be completed weekly for 2 weeks with plans to repeat treatment every 6 months to prevent flares of NMO. During the first dose, the patient had a reaction to the treatment, which caused pruritus and chest tightness. She was able to complete the infusion after being treated with diphenhydramine.
Tx continued. In order to complete the second of 2 infusions of rituximab, the patient was pretreated with oral methylprednisolone the night before the infusion, along with diphenhydramine and acetaminophen on the day of treatment. Fortunately, the patient tolerated the infusion well with no adverse effects or reactions.
Continue to: DISCUSSION
DISCUSSION
Within the NMO spectrum, the MOG antibody is positive in up to 42% of AQP4-seronegative cases.6 MOG is a minor myelin component that is expressed exclusively in the central nervous system on the surface of myelin and oligodendrocyte processes. The role of this glycoprotein is not well understood but is hypothesized to function as a cell surface receptor or cell adhesion molecule.7
Among a cohort of 252 patients from the United Kingdom who tested positive for the MOG-IgG1 antibody, optic neuritis was seen in 55%, while 18% experienced transverse myelitis, and 15% had a history of area postrema syndrome. A brain MRI identified lesions in all areas of the brain including the brain stem, cerebellum, and cerebral hemispheres.8
Risk factors for NMOSD include female gender, Asian and African ethnicities, Epstein Barr virus seropositivity, and tobacco abuse.
Differential diagnosis. Many diseases or conditions that are inflammatory, autoimmune, infectious, or neoplastic can involve the central nervous system and mimic the clinical and radiologic phenotypes of NMOSD-AQP4. They include lupus, SjÖgren’s syndrome, multiple sclerosis, sarcoidosis, acute disseminated encephalomyelitis, HIV, and vitamin B12 deficiency.
Treatment. The standard treatment is intravenous methylprednisolone, 1 g/d for 3 to 5 days followed by a steroid taper. Therapeutic plasma exchange is recommended for refractory cases and in patients with spinal cord demyelination.9-11 Rituximab is the first-line therapy for attack prevention12-15 in NMOSD broadly and may be effective in MOG antibody disease, as well. In an open-label study of patients with NMOSD treated with rituximab, 64% were relapse free at follow-up, which ranged from 12 to 67 months.13 In a long-term study of patients treated with rituximab, 87% maintained a reduced relapse rate and 93% had improvement or stability over a 5-year follow-up.14
Continue to: Our patient
Our patient. After her diagnosis of NMOSD/MOG-antibody disease, our patient’s symptoms progressed to include vertigo, vestibular ataxia, pruritus, left foot drop, lower extremity numbness, and decreased hearing. After the second rituximab infusion her symptoms continued, but over time stabilized and have not worsened. She currently receives gabapentin 300 mg every 8 hours, as needed, for extremity numbness (which has been working well) along with sertraline 100 mg/d for depression.
Subsequent office visits have showed no further weight loss. Based on the current response to the rituximab, her prognosis is undetermined by Neurology as they continue to monitor for progression.
THE TAKEAWAY
Vestibular ataxia, foot drop, pruritus, vertigo, decreased hearing, numbness, and oculomotor dysfunction in the presence of nausea and vomiting should raise suspicion for NMOSD. The presence of AQP4 antibodies along with demyelinating central nervous system lesions, is highly indicative of NMO. The presence of MOG antibodies may indicate NMOSD/MOG-antibody disease. The initial treatment of NMOSD is intravenous methylprednisolone, which can be followed by treatment with rituximab to achieve remission.
CORRESPONDENCE
Daniel Murphy, MD, FAAFP, Department of Family and Community Medicine, Texas Tech University Health Science Center El Paso, 9849 Kenworthy Street, El Paso, Texas 79924; [email protected]
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.
1. Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221-2231.
2. Ratelade J, Zhang H, Saadoun S, et al. Neuromyelitis optica IgG and natural killer cells Produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123:861-872.
3. Saadoun S, Waters P, Bell BA, et al. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349-361.
4. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titer. Brain. 2007;130:1235-1243.
5. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain. 2008;131:3072-3080.
6. Hamid SHM, Whittam D, Mutch K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are Mog-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017; 264:2088-2094.
7. Peschl P, Bradi M, Hoftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529.
8. Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128-3138.
9. Sellner J, Boggild M, Clanet M, et al. EFNS Guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019-1032.
10. Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206-216.
11. Watanabe S, Nakashima I, Misu T, et al. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13:128-132.
12. Collongues N, Brassat D, Maillart E, et al. Efficacy of rituximab in refractory neuromyelitis optica. Mult Scler. 2016;22:955-959.
13. Collongues N, de Seze J. An update on the evidence for the efficacy and safety of rituximab in the management of neuromyelitis optica. Ther Adv Neurol Disord. 2016;9:180-188.
14. Kim SH, Huh SY, Lee SJ, et al. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110-1117.
15. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68:1412-1420.

Adiposis Dolorosa Pain Management
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
Adiposis dolorosa (AD), or Dercum disease, is a rare disorder that was first described in 1888 and characterized by the National Organization of Rare Disorders (NORD) as a chronic pain condition of the adipose tissue generally found in patients who are overweight or obese.1,2 AD is more common in females aged 35 to 50 years and proposed to be a disease of postmenopausal women, though no prevalence studies exist.2 The etiology remains unclear.2 Several theories have been proposed, including endocrine and nervous system dysfunction, adipose tissue dysregulation, or pressure on peripheral nerves and chronic inflammation.2-4 Genetic, autoimmune, and trauma also have been proposed as a mechanism for developing the disease. Treatment modalities focusing on narcotic analgesics have been ineffective in long-term management.3
The objective of the case presentation is to report a variety of approaches for AD and their relative successes at pain control in order to assist other medical professionals who may come across patients with this rare condition.
Case Presentation
A 53-year-old male with a history of blast exposure-related traumatic brain injury, subsequent stroke with residual left hemiparesis, and seizure disorder presented with a 10-year history of nodule formation in his lower extremities causing restriction of motion and pain. The patient had previously undergone lower extremity fasciotomies for compartment syndrome with minimal pain relief. In addition, nodules over his abdomen and chest wall had been increasing over the past 5 years. He also experienced worsening fatigue, cramping, tightness, and paresthesias of the affected areas during this time. Erythema and temperature allodynia were noted in addition to an 80-pound weight gain. From the above symptoms and nodule excision showing histologic signs of lipomatous growth, a diagnosis of AD was made.
The following constitutes the approximate timetable of his treatments for 9 years. He was first diagnosed incidentally at the beginning of this period with AD during an electrodiagnostic examination. He had noticed the lipomas when he was in his 30s, but initially they were not painful. He was referred for treatment of pain to the physical medicine and rehabilitation department.
For the next 3 years, he was treated with prolotherapy. Five percent dextrose in water was injected around many of the painful lipomas in the upper extremities. He noted after the second round of neural prolotherapy that he had reduced swelling of his upper extremities and the lipomas decreased in size. He experienced mild improvement in pain and functional usage of his arms.
He continued to receive neural prolotherapy into the nodules in the arms, legs, abdomen, and chest wall. The number of painful nodules continued to increase, and the patient was started on hydrocodone 10 mg/acetaminophen 325 mg (1 tablet every 6 hours as needed) and methadone for pain relief. He was initially started on 5 mg per day of methadone and then was increased in a stepwise, gradual fashion to 10 mg in the morning and 15 mg in the evening. He transitioned to morphine sulfate, which was increased to a maximum dose of 45 mg twice daily. This medication was slowly tapered due to adverse effects (AEs), including sedation.
After weaning off morphine sulfate, the patient was started on lidocaine infusions every 3 months. Each infusion provided at least 50% pain reduction for 6 to 8 weeks. He was approved by the US Department of Veterans Affairs (VA) to have Vaser (Bausch Health, Laval, Canada) minimally invasive ultrasound liposuction treatment, performed at an outside facility. The patient was satisfied with the pain relief that he received and noted that the number of lipomas greatly diminished. However, due to funding issues, this treatment was discontinued after several months.
The patient had moderately good pain relief with methadone 5 mg in the morning, and 15 mg in the evening. However, the patient reported significant somnolence during the daytime with the regimen. Attempts to wean the patient off methadone was met with uncontrollable daytime pain. With suboptimal oral pain regimen, difficulty obtaining Vaser treatments, and limitation in frequency of neural prolotherapy, the decision was made to initiate 12 treatments of Calmare (Fairfield, CT) cutaneous electrostimulation.
During his first treatment, he had the electrodes placed on his lower extremities. The pre- and posttreatment 10-point visual analog scale (VAS) scores were 9 and 0, respectively, after the first visit. The position of the electrodes varied, depending on the location of his pain, including upper extremities and abdominal wall. During the treatment course, the patient experienced an improvement in subjective functional status. He was able to sleep in the same bed as his wife, shake hands without severe pain, and walk .25 mile, all of which he was unable to do before the electrostimulative treatment. He also reported overall improvement in emotional well-being, resumption of his hobbies (eg, playing the guitar), and social engagement. Methadone was successfully weaned off during this trial without breakthrough pain. This improvement in pain and functional status continued for several weeks; however, he had an exacerbation of his pain following a long plane flight. Due to uncertain reliability of pain relief with the procedure, the pain management service initiated a regimen of methadone 10 mg twice daily to be initiated when a procedure does not provide the desired duration of pain relief and gradually discontinued following the next interventional procedure.
The patient continued a regimen that included lidocaine infusions, neural prolotherapy, Calmare electrostimulative therapy, as well as lymphedema massage. Additionally, he began receiving weekly acupuncture treatments. He started with traditional full body acupuncture and then transitioned to battlefield acupuncture (BFA). Each acupuncture treatment provided about 50% improvement in pain on the VAS, and improved sleep for 3 days posttreatment.
However, after 18 months of the above treatment protocol, the patient experienced a general tonic-clonic seizure at home. Due to concern for the lowered seizure threshold, lidocaine infusions and methadone were discontinued. Long-acting oral morphine was initiated. The patient continued Calmare treatments and neural prolotherapy after a seizure-free interval. This regimen provided the patient with temporary pain relief but for a shorter duration than prior interventions.
Ketamine infusions were eventually initiated about 5 years after the diagnosis of AD was made, with postprocedure pain as 0/10 on the VAS. Pain relief was sustained for 3 months, with the notable AEs of hallucinations in the immediate postinfusion period. Administration consisted of the following: 500 mg of ketamine in a 500 mL bag of 0.9% NaCl. A 60-mg slow IV push was given followed by 60 mg/h increased every 15 min by 10 mg/h for a maximum dose of 150 mg/h. In a single visit the maximum total dose of ketamine administered was 500 mg. The protocol, which usually delivered 200 mg in a visit but was increased to 500 mg because the 200-mg dose was ineffective, was based on protocols at other institutions to accommodate the level of monitoring available in the Interventional Pain Clinic. The clinic also developed an infusion protocol with at least 1 month between treatments. The patient continues to undergo scheduled ketamine infusions every 14 weeks in addition to monthly BFA. The patient reported near total pain relief for about a month following ketamine infusion, with about 3 months of sustained pain relief. Each BFA session continues to provide 3 days of relief from insomnia. Calmare treatments and the neural prolotherapy regimen continue to provide effective but temporary relief from pain.
Discussion
Currently there is no curative treatment for AD. The majority of the literature is composed of case reports without summaries of potential interventions and their efficacies. AD therapies focus on symptom relief and mainly include pharmacologic and surgical intervention. In this case report several novel treatment modalities have been shown to be partially effective.
Surgical Intervention
Liposuction and lipoma resection have been described as effective only in the short term for AD.2,4-6 Hansson and colleagues suggested liposuction avulsion for sensory nerves and a portion of the proposed abnormal nerve connections between the peripheral nervous system and sensory nerves as a potential therapy for pain improvement.5 But the clinical significance of pain relief from liposuction is unclear and is contraindicated in recurrent lipomas.5
Pharmaceutical Approach
Although relief with nonsteroidal anti-inflammatory drugs and narcotic analgesics have been unpredictable, Herbst and Asare-Bediako described significant pain relief in a subset of patients with AD with a variety of oral analgesics.7,8 However, the duration of this relief was not clearly stated, and the types or medications or combinations were not discussed. Other pharmacologic agents trialed in the treatment of AD include methotrexate, infliximab, Interferon α-2b, and calcium channel modulators (pregabalin and oxcarbazepine).2,9-11 However, the mechanism and significance of pain relief from these medications remain unclear.
Subanesthesia Therapy
Lidocaine has been used as both a topical agent and an IV infusion in the treatment of chronic pain due to AD for decades. Desai and colleagues described 60% sustained pain reduction in a patient using lidocaine 5% transdermal patches.4 IV infusion of lidocaine has been described in various dosages, though the mechanism of pain relief is ambiguous, and the duration of effect is longer than the biologic half-life.2-4,9 Kosseifi and colleagues describe a patient treated with local injections of lidocaine 1% and obtained symptomatic relief for 3 weeks.9 Animal studies suggest the action of lidocaine involves the sodium channels in peripheral nerves, while another study suggested there may be an increase in sympathetic nervous system activity after the infusion of lidocaine.2,9
Ketamine infusions not previously described in the treatment of AD have long been used to treat other chronic pain syndromes (chronic cancer pain, complex regional pain syndrome [CRPS], fibromyalgia, migraine, ischemic pain, and neuropathic pain).9,12,13 Ketamine has been shown to decrease pain intensity and reduce the amount of opioid analgesic necessary to achieve pain relief, likely through the antagonism of N-methyl-D-aspartate receptors.12 A retrospective review by Patil and Anitescu described subanesthetic ketamine infusions used as a last-line therapy in refractory pain syndromes. They found ketamine reduced VAS scores from mean 8.5 prior to infusion to 0.8 after infusion in patients with CRPS and from 7.0 prior to infusion to 1.0 in patient with non-CRPS refractory pain syndromes.13 Hypertension and sedation were the most frequent AEs of ketamine infusion, though a higher incidence of hallucination and confusion were noted in non-CRPS patients. Hocking and Cousins suggest that psychotomimetic AEs of ketamine infusion may be more likely in patients with anxiety.14 However, it is important to note that ketamine infusion studies have been heterogeneous in their protocol, and only recently have standardization guidelines been proposed.
Physical Modalities
Manual lymphatic massage has been described in multiple reports for symptom relief in patients with cancer with malignant growth causing outflow lymphatic obstruction. This technique also has been used to treat the obstructive symptoms seen with the lipomatous growths of AD. Lange and colleagues described a case as providing reduction in pain and the diameter of extremities with twice weekly massage.14 Herbst and colleagues noted that patients had an equivocal response to massage, with some patients finding that it worsened the progression of lipomatous growths.7
Electrocutaneous Stimulation
In a case study by Martinenghi and colleagues, a patient with AD improved following transcutaneous frequency rhythmic electrical modulation system (FREMS) treatment.16 The treatment involved 4 cycles of 30 minutes each for 6 months. The patient had an improvement of pain on the VAS from 6.4 to 1.7 and an increase from 12 to 18 on the 100-point Barthel index scale for performance in activities of daily living, suggesting an improvement of functional independence as well.16
The MC5-A Calmare is another cutaneous electrostimulation modality that previously has been used for chronic cancer pain management. This FDA-cleared device is indicated for the treatment of various chronic pain syndromes. The device is proposed to stimulate 5 separate pain areas via cutaneous electrodes applied beyond and above the painful areas in order to “scramble” pain information and reduce perception of chronic pain intensity. Ricci and colleagues included cancer and noncancer subjects in their study and observed reduction in pain intensity by 74% (on numeric rating scale) in the entire subject group after 10 days of treatments. Further, no AEs were reported in either group, and most of the subjects were willing to continue treatment.17 However, this modality was limited by concerns with insurance coverage, access to a Calmare machine, operator training, and reproducibility of electrode placement to achieve “zero pain” as is the determinant of device treatment cycle output by the manufacturer.
Perineural Injection/Prolotherapy
Perineural injection therapy (PIT) involves the injection of dextrose solution into tissues surrounding an inflamed nerve to reduce neuropathic inflammation. The proposed source of this inflammation is the stimulation of the superficial branches of peptidergic peripheral nerves. Injections are SC and target the affected superficial nerve pathway. Pain relief is usually immediate but requires several treatments to ensure a lasting benefit. There have been no research studies or case reports on the use of PIT or prolotherapy and AD. Although there is a paucity of published literature on the efficacy of PIT, it remains an alternative modality for treatment of chronic pain syndromes. In a systematic review of prolotherapy for chronic musculoskeletal pain, Hauser and colleagues supported the use of dextrose prolotherapy to treat chronic tendinopathies, osteoarthritis of finger and knee joints, spinal and pelvic pain if conservative measures had failed. However, the efficacy on acute musculoskeletal pain was uncertain.18 In addition to the paucity of published literature, prolotherapy is not available to many patients due to lack of insurance coverage or lack of providers able to perform the procedure.
Hypobaric Pressure Therapy
Hypobaric pressure therapy has been offered as an alternative “touch-free” method for treatment of pain associated with edema. Herbst and Rutledge describe a pilot study focusing on hypobaric pressure therapy in patients with AD using a cyclic altitude conditioning system, which significantly decreased the Pain Catastrophizing Scale (tendency to catastrophize pain symptoms) in patients with AD after 5 days of therapy. VAS scores also demonstrated a linear decrease over 5 days.8
Acupuncture
There have been no research studies or case reports regarding the use of either traditional full body acupuncture or BFA in management of AD. However, prior studies have been performed that suggest that acupuncture can be beneficial in chronic pain relief. For examples, a Cochrane review by Manheimer and colleagues showed that acupuncture had a significant benefit in pain relief in subjects with peripheral joint arthritis.19 In another Cochrane review there was low-to-moderate level evidence compared with no treatment in pain relief, but moderate-level evidence that the effect of acupuncture does not differ from sham (placebo) acupuncture.20,21
Conclusion
Current therapeutic approaches to AD focus on invasive surgical intervention, chronic opiate and oral medication management. However, we have detailed several additional approaches to AD treatment. Ketamine infusions, which have long been a treatment in other chronic pain syndromes may present a viable alternative to lidocaine infusions in patients with AD. Electrocutaneous stimulation is a validated treatment of chronic pain syndromes, including chronic neuropathic pain and offers an alternative to surgical or pharmacologic management. Further, PIT offers another approach to neuropathic pain management, which has yet to be fully explored. As no standard treatment approach exists for patients with AD, multimodal therapies should be considered to optimize pain management and reduce dependency on opiate mediations.
Acknowledgments
Hunter Holmes McGuire Research Institute and the Physical Medicine and Rehabilitation Department provided the resources and facilities to make this work possible.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
1. Dercum FX. A subcutaneous dystrophy. In: University of Pennsylvania. University of Pennsylvania Medical Bulletin. Vol 1. Philadelphia, PA; University of Pennsylvania Press; 1888:140-150. Accessed October 4, 2019.
2. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnositc criteria, diagnositic methods, classification and management. Orphanet J Rare Dis. 2012;7:1-15.
3. Amine B, Leguilchard F, Benhamou CL. Dercum’s disease (adiposis dolorosa): a new case-report. Joint Bone Spine. 2004;71(2):147-149.
4. Desai MJ, Siriki R, Wang D. Treatment of pain in Dercum’s disease with lidoderm (lidocaine 5% patch): a case report. Pain Med. 2008;9(8):1224-1226.
5. Hansson E, Svensson H, Brorson H. Liposuction may reduce pain in Dercum’s disease (adiposis dolorosa). Pain Med. 2011;12:942-952.
6. Kosseifi S, Anaya E, Dronovalli G, Leicht S. Dercum’s disease: an unusual presentation. Pain Med. 2010;11(9):1430-1434.
7. Herbst KL, Asare-Bediako S. Adiposis dolorasa is more than painful fat. Endocrinologist. 2007;17(6):326-334.
8. Herbst KL, Rutledge T. Pilot study: rapidly cycling hypobaric pressure improves pain after 5 days in adiposis dolorosa. J Pain Res. 2010;3:147-153.
9. Lange U, Oelzner P, Uhlemann C. Dercum’s disease (lipomatosis dolorosa): successful therapy with pregabalin and manual lymphatic drainage and a current overview. Rheumatol Int. 2008;29(1):17-22
10. Schaffer PR, Hale CS, Meehan SA, Shupack JL, Ramachandran S. Adoposis dolorosa. Dermatol Online J. 2014;20(12):1-3.
11. Singal A, Janiga JJ, Bossenbroek NM, Lim HW. Dercum’s disease (adiposis dolorosa): a report of improvement with infliximab and methotrexate. J Eur Acad Dermatol Venerol. 2007;21(5):717.
12. Loftus RW, Yeager MP, Clark JA, et al. Intraoperative ketamine reduces perioperative opiate consumption in opiate-dependent patients with chronic back pain undergoing back surgery. Anesthesiology. 2010;113(3):639-646.
13. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis. Pain Med. 2012;13(2):263-269.
14. Hocking G, Cousins MJ. Ketamine in chronic pain management: an evidence-based review. Anesth Analg. 2003;97(6):1730-1739.
15. Cohen SP, Bhatia A, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for chronic pain from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):521-546.
16. Martinenghi S, Caretto A, Losio C, Scavini M, Bosi E. Successful treatment of Dercum’s disease by transcutaneous electrical stimulation: a case report. Medicine (Baltimore). 2015;94(24):e950.
17. Ricci M, Pirotti S, Scarpi E, et al. Managing chronic pain: results from an open-label study using MC5-A Calmare device. Support Care Cancer. 2012;20(2):405-412.
18. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. 2016;9:139-159.
19. Manheimer E, Cheng K, Linde K, et al. Acupuncture for peripheral joint osteoarthritis. Cochrane Database Syst Rev. 2010;(1):CD001977.
20. Deare JC, Zheng Z, Xue CC, et al. Acupuncture for treating fibromyalgia. Cochrane Database Syst Rev. 2013;(5):CD007070.
21. Chan MWC, Wu XY, Wu JCY, Wong SYS, Chung VCH. Safety of acupuncture: overview of systematic reviews. Sci Rep. 2017;7(1):3369.
Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
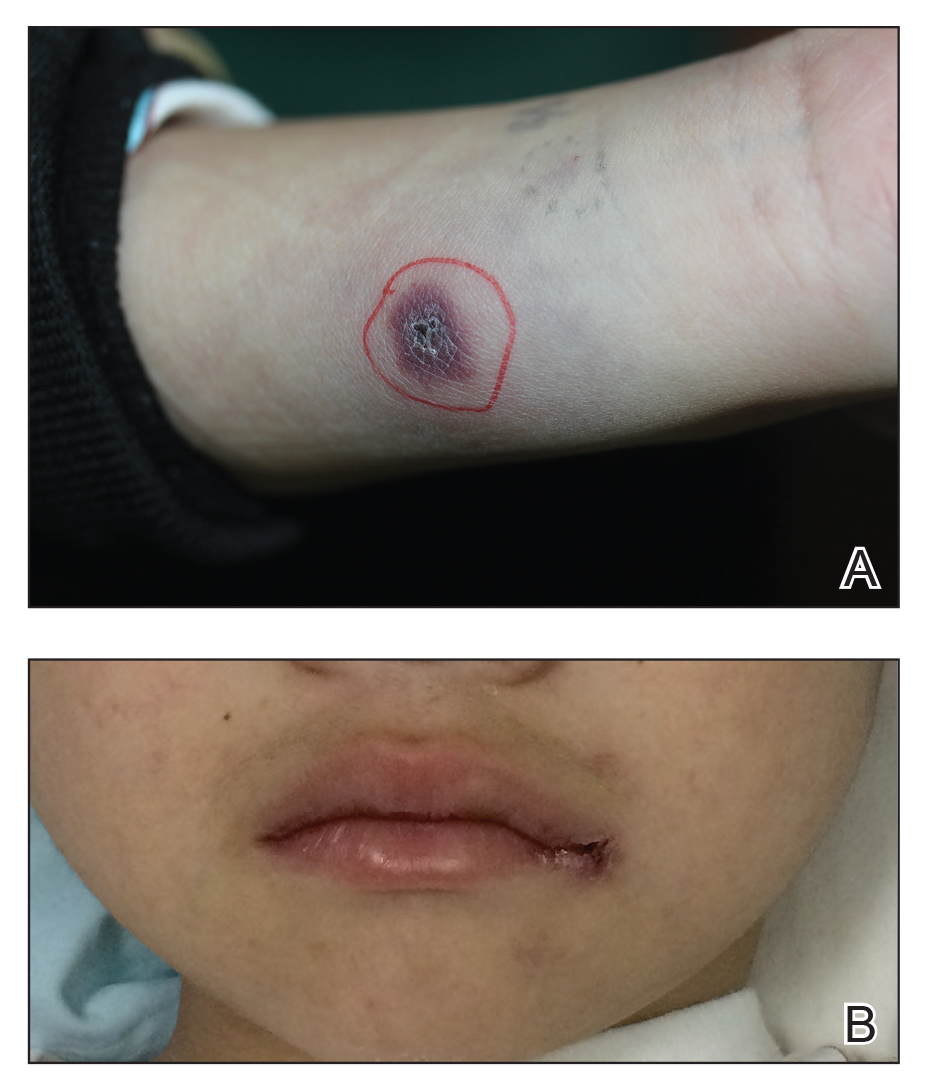
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
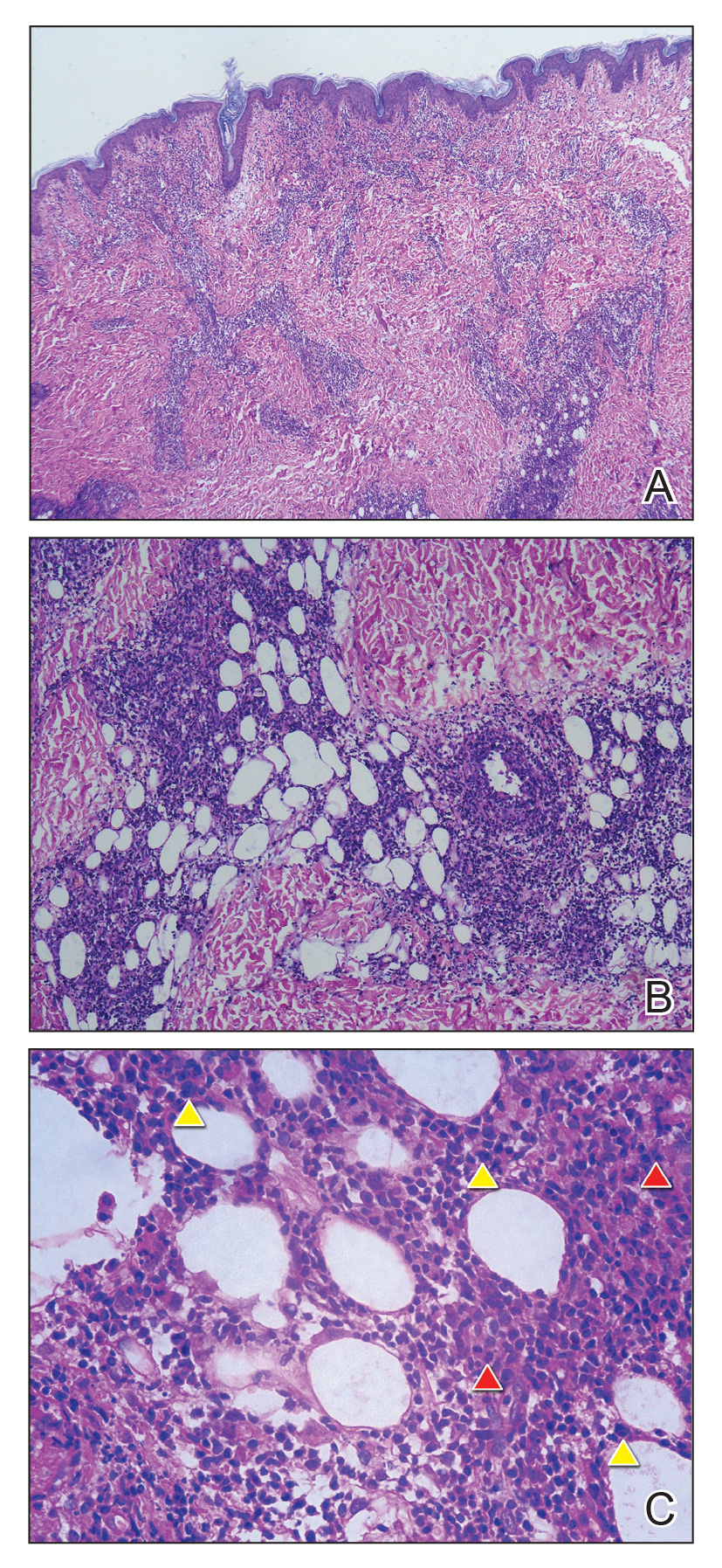
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.
Seborrhea Herpeticum: Cutaneous Herpes Simplex Virus Infection Within Infantile Seborrheic Dermatitis
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
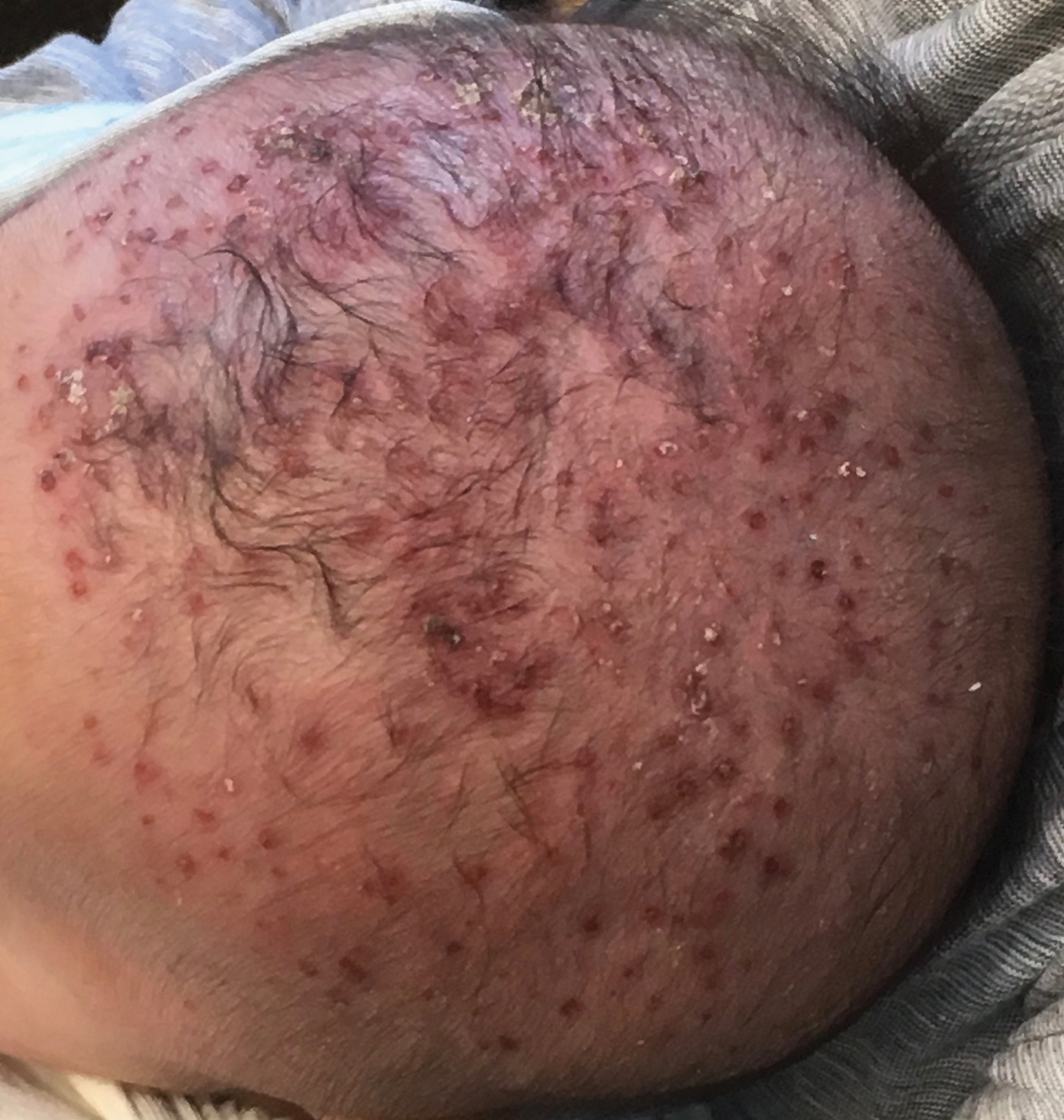
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
Practice Points
- Cutaneous herpes simplex virus may present in a seborrheic distribution within infantile seborrheic dermatitis, suggesting underlying dysfunction secondary to seborrheic dermatitis.
- Treatment of seborrhea herpeticum involves antiviral therapy to treat the secondary viral infection and gentle skin care precautions for the primary condition.
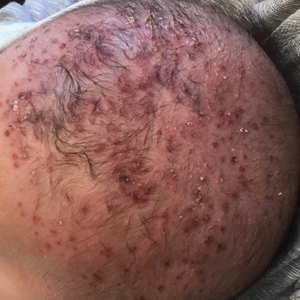
Secondary Syphilis Mimicking Molluscum Contagiosum in the Beard Area of an AIDS Patient
To the Editor:
A 46-year-old man with a history of AIDS (viral load, 28,186 copies/mL; CD4 count, 22 cells/μL) presented with a 40-lb weight loss over the last 6 months as well as dysphagia and a new-onset pruritic facial eruption of 1 week’s duration. The facial lesions quickly spread to involve the beard area and the upper neck. His medical history was notable for nicotine dependence, seborrheic dermatitis, molluscum contagiosum (MC), treated neurosyphilis and latent tuberculosis, hypertension, a liver mass suspected to be a hemangioma, and erythrocytosis. He was diagnosed with human immunodeficiency virus infection 19 years prior to presentation and was not compliant with the prescribed highly active antiretroviral therapy.
Skin examination revealed multiple discrete and coalescing, 2- to 12-mm, nonumbilicated, hyperkeratotic papules and nodules localized to the left and right beard areas (Figure 1A). A few discrete, 2- to 5-mm, umbilicated papules were noted in the right beard area (Figure 1B), as well as on the right side of the neck (Figure 1C), buttocks, and legs. Mild erythema with yellow-white scale was present in the alar creases. Examination of the oropharyngeal mucosa revealed multiple thick white plaques that were easily scraped off with a tongue depressor. Examination of the palms, soles, and anogenital areas was normal.
A punch biopsy of a nonumbilicated hyperkeratotic papule from the left beard area demonstrated spongiosis; neutrophilic microabscess formation; plasma cells; and a superficial and deep perivascular, predominantly lymphohistiocytic infiltrate (Figure 2A). Spirochete immunohistochemical staining of tissue highlighted abundant organisms in the dermoepidermal junction and vascular endothelial cells (Figure 2B). Other tissue stains for bacteria, including acid-fast bacilli, and fungi were negative. Bacterial culture of tissue from the lesion in the left beard area grew Staphylococcus aureus. Results of acid-fast and fungal cultures of tissue were negative. Shave biopsy of the umbilicated papule on the right side of the neck demonstrated classic invagination of the epidermis into the dermis and intracytoplasmic viral inclusions consistent with MC (Figure 2C). Spirochete immunohistochemical staining of the same tissue sample was negative (Figure 2D).
Serum rapid plasma reagin was reactive with a titer of 1:128 compared to the last known reactive rapid plasma reagin titer of 1:1 five years prior to presentation. A fluorescent treponemal antibody absorption test and VDRL test of cerebrospinal fluid was nonreactive. Fungal, bacterial, and acid-fast cultures of cerebral spinal fluid and a cryptococcal antigen test were negative. Serum cryptococcal antigen and coccidioides complement fixation tests were negative. Cytomegalovirus plasma polymerase chain reaction and urine histoplasma antigen testing were negative. Computed tomography of the chest revealed a new 1.9×1.6×2.1-cm3 cavitary lesion with distal tree-in-bud opacities in the lingula of the left lung. Acid-fast blood culture was negative, and acid-fast sputum culture was positive for Mycobacterium kansasii.
The cutaneous pathology findings and serologic findings confirmed the diagnoses of cutaneous secondary syphilis (SS) in the beard area and MC on the right side of the neck. Clinical diagnoses of seborrheic dermatitis of the alar creases and esophageal candidiasis also were made. The patient was treated with intramuscular penicillin G 2.4 million U once weekly for 3 weeks. The lesions confined to the beard area rapidly resolved within 7 days after the first dose of antibiotics, which further supported the diagnosis of localized cutaneous SS. Fluconazole 100 mg once daily was prescribed for the esophageal candidiasis, and he also was started on a regimen of rifampin 600 mg once daily, isoniazid 300 mg once daily, ethambutol 1200 mg once daily, and pyrazinamide 1500 mg once daily.
Syphilis is well known as the great masquerader due to its many possible manifestations. Many patients present with typical palmar and plantar dermatoses.1 Other documented SS presentations include eruptions ranging from a few to diffusely disseminated maculopapular lesions with or without scale on the trunk and upper extremities; pustular and nodular lesions of the face; alopecia; grayish white patches on the oral mucosa; and ulcerative, psoriasiform, follicular, and lichenoid lesions.2 Cutaneous SS has not been commonly reported in a localized distribution to the beard area with a clinical appearance mimicking hyperkeratotic MC lesions.3 Secondary syphilis is not known to spread through autoinoculation, presumably from shaving (as in our case), as might occur with other cutaneous infectious processes such as MC, verruca vulgaris, S aureus, and dermatophytosis in the beard area.
The differential diagnosis for hyperkeratotic papules and nodules localized to the beard area in human immunodeficiency virus–infected males includes MC, verruca vulgaris, chronic verrucous varicella-zoster virus, crusted scabies, tuberculosis verrucosa cutis, hypertrophic lichen planus, and disseminated deep fungal infections including cryptococcosis and coccidioidomycosis. In the setting of immunosuppression, the diagnosis of hyperkeratotic MC was favored in our patient given the co-location of classic umbilicated MC lesions with the hyperkeratotic papules and nodules. It is common to see MC autoinoculated in the beard area in men from shaving, as well as for MC to present in an atypical manner, particularly as hyperkeratotic lesions, in patients with AIDS.4 The predominant localized beard distribution and lack of other mucocutaneous manifestations of SS at presentation supported a clinical diagnosis of hyperkeratotic MC in our patient.
Unique presentations of SS have been documented, including nodular lesions of the face, but they typically have been accompanied by other stigmata of SS such as the classic palmoplantar or truncal maculopapular rash.3 One notable difference in our case was the localized beard distribution of the syphilitic cutaneous lesions in a man with AIDS. Our case reinforces the importance of cutaneous biopsies in immunocompromised patients. It is known that SS spreads hematogenously; however, in our case it was suspected that the new lesions may have spread locally through autoinoculation via beard hair removal, as the hyperkeratotic lesions were limited to the beard area. Koebnerization secondary to trauma induced by beard hair removal was considered in this case; however, koebnerization is known to occur in noninfectious dermatologic conditions, such as psoriasis, lichen planus, lichen nitidus, and vitiligo, but not in infections such as syphilis. Our case is pivotal in raising the question of whether SS can be autoinoculated in the beard area.
- Baughn RE, Musher DM. Secondary syphilitic lesions. Clin Microbiol Rev. 2005;18:205-216.
- Dourmishev LA, Dourmishev AL. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Cohen SE, Klausner JD, Engelman J, et al. Syphilis in the modern era: an update for physicians. Infect Dis Clin North Am. 2013;27:705-722.
- Filo-Rogulska M, Pindycka-Plaszcznska M, Januszewski K, et al. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol. 2013;30:56-58.
To the Editor:
A 46-year-old man with a history of AIDS (viral load, 28,186 copies/mL; CD4 count, 22 cells/μL) presented with a 40-lb weight loss over the last 6 months as well as dysphagia and a new-onset pruritic facial eruption of 1 week’s duration. The facial lesions quickly spread to involve the beard area and the upper neck. His medical history was notable for nicotine dependence, seborrheic dermatitis, molluscum contagiosum (MC), treated neurosyphilis and latent tuberculosis, hypertension, a liver mass suspected to be a hemangioma, and erythrocytosis. He was diagnosed with human immunodeficiency virus infection 19 years prior to presentation and was not compliant with the prescribed highly active antiretroviral therapy.
Skin examination revealed multiple discrete and coalescing, 2- to 12-mm, nonumbilicated, hyperkeratotic papules and nodules localized to the left and right beard areas (Figure 1A). A few discrete, 2- to 5-mm, umbilicated papules were noted in the right beard area (Figure 1B), as well as on the right side of the neck (Figure 1C), buttocks, and legs. Mild erythema with yellow-white scale was present in the alar creases. Examination of the oropharyngeal mucosa revealed multiple thick white plaques that were easily scraped off with a tongue depressor. Examination of the palms, soles, and anogenital areas was normal.
A punch biopsy of a nonumbilicated hyperkeratotic papule from the left beard area demonstrated spongiosis; neutrophilic microabscess formation; plasma cells; and a superficial and deep perivascular, predominantly lymphohistiocytic infiltrate (Figure 2A). Spirochete immunohistochemical staining of tissue highlighted abundant organisms in the dermoepidermal junction and vascular endothelial cells (Figure 2B). Other tissue stains for bacteria, including acid-fast bacilli, and fungi were negative. Bacterial culture of tissue from the lesion in the left beard area grew Staphylococcus aureus. Results of acid-fast and fungal cultures of tissue were negative. Shave biopsy of the umbilicated papule on the right side of the neck demonstrated classic invagination of the epidermis into the dermis and intracytoplasmic viral inclusions consistent with MC (Figure 2C). Spirochete immunohistochemical staining of the same tissue sample was negative (Figure 2D).
Serum rapid plasma reagin was reactive with a titer of 1:128 compared to the last known reactive rapid plasma reagin titer of 1:1 five years prior to presentation. A fluorescent treponemal antibody absorption test and VDRL test of cerebrospinal fluid was nonreactive. Fungal, bacterial, and acid-fast cultures of cerebral spinal fluid and a cryptococcal antigen test were negative. Serum cryptococcal antigen and coccidioides complement fixation tests were negative. Cytomegalovirus plasma polymerase chain reaction and urine histoplasma antigen testing were negative. Computed tomography of the chest revealed a new 1.9×1.6×2.1-cm3 cavitary lesion with distal tree-in-bud opacities in the lingula of the left lung. Acid-fast blood culture was negative, and acid-fast sputum culture was positive for Mycobacterium kansasii.
The cutaneous pathology findings and serologic findings confirmed the diagnoses of cutaneous secondary syphilis (SS) in the beard area and MC on the right side of the neck. Clinical diagnoses of seborrheic dermatitis of the alar creases and esophageal candidiasis also were made. The patient was treated with intramuscular penicillin G 2.4 million U once weekly for 3 weeks. The lesions confined to the beard area rapidly resolved within 7 days after the first dose of antibiotics, which further supported the diagnosis of localized cutaneous SS. Fluconazole 100 mg once daily was prescribed for the esophageal candidiasis, and he also was started on a regimen of rifampin 600 mg once daily, isoniazid 300 mg once daily, ethambutol 1200 mg once daily, and pyrazinamide 1500 mg once daily.
Syphilis is well known as the great masquerader due to its many possible manifestations. Many patients present with typical palmar and plantar dermatoses.1 Other documented SS presentations include eruptions ranging from a few to diffusely disseminated maculopapular lesions with or without scale on the trunk and upper extremities; pustular and nodular lesions of the face; alopecia; grayish white patches on the oral mucosa; and ulcerative, psoriasiform, follicular, and lichenoid lesions.2 Cutaneous SS has not been commonly reported in a localized distribution to the beard area with a clinical appearance mimicking hyperkeratotic MC lesions.3 Secondary syphilis is not known to spread through autoinoculation, presumably from shaving (as in our case), as might occur with other cutaneous infectious processes such as MC, verruca vulgaris, S aureus, and dermatophytosis in the beard area.
The differential diagnosis for hyperkeratotic papules and nodules localized to the beard area in human immunodeficiency virus–infected males includes MC, verruca vulgaris, chronic verrucous varicella-zoster virus, crusted scabies, tuberculosis verrucosa cutis, hypertrophic lichen planus, and disseminated deep fungal infections including cryptococcosis and coccidioidomycosis. In the setting of immunosuppression, the diagnosis of hyperkeratotic MC was favored in our patient given the co-location of classic umbilicated MC lesions with the hyperkeratotic papules and nodules. It is common to see MC autoinoculated in the beard area in men from shaving, as well as for MC to present in an atypical manner, particularly as hyperkeratotic lesions, in patients with AIDS.4 The predominant localized beard distribution and lack of other mucocutaneous manifestations of SS at presentation supported a clinical diagnosis of hyperkeratotic MC in our patient.
Unique presentations of SS have been documented, including nodular lesions of the face, but they typically have been accompanied by other stigmata of SS such as the classic palmoplantar or truncal maculopapular rash.3 One notable difference in our case was the localized beard distribution of the syphilitic cutaneous lesions in a man with AIDS. Our case reinforces the importance of cutaneous biopsies in immunocompromised patients. It is known that SS spreads hematogenously; however, in our case it was suspected that the new lesions may have spread locally through autoinoculation via beard hair removal, as the hyperkeratotic lesions were limited to the beard area. Koebnerization secondary to trauma induced by beard hair removal was considered in this case; however, koebnerization is known to occur in noninfectious dermatologic conditions, such as psoriasis, lichen planus, lichen nitidus, and vitiligo, but not in infections such as syphilis. Our case is pivotal in raising the question of whether SS can be autoinoculated in the beard area.
To the Editor:
A 46-year-old man with a history of AIDS (viral load, 28,186 copies/mL; CD4 count, 22 cells/μL) presented with a 40-lb weight loss over the last 6 months as well as dysphagia and a new-onset pruritic facial eruption of 1 week’s duration. The facial lesions quickly spread to involve the beard area and the upper neck. His medical history was notable for nicotine dependence, seborrheic dermatitis, molluscum contagiosum (MC), treated neurosyphilis and latent tuberculosis, hypertension, a liver mass suspected to be a hemangioma, and erythrocytosis. He was diagnosed with human immunodeficiency virus infection 19 years prior to presentation and was not compliant with the prescribed highly active antiretroviral therapy.
Skin examination revealed multiple discrete and coalescing, 2- to 12-mm, nonumbilicated, hyperkeratotic papules and nodules localized to the left and right beard areas (Figure 1A). A few discrete, 2- to 5-mm, umbilicated papules were noted in the right beard area (Figure 1B), as well as on the right side of the neck (Figure 1C), buttocks, and legs. Mild erythema with yellow-white scale was present in the alar creases. Examination of the oropharyngeal mucosa revealed multiple thick white plaques that were easily scraped off with a tongue depressor. Examination of the palms, soles, and anogenital areas was normal.
A punch biopsy of a nonumbilicated hyperkeratotic papule from the left beard area demonstrated spongiosis; neutrophilic microabscess formation; plasma cells; and a superficial and deep perivascular, predominantly lymphohistiocytic infiltrate (Figure 2A). Spirochete immunohistochemical staining of tissue highlighted abundant organisms in the dermoepidermal junction and vascular endothelial cells (Figure 2B). Other tissue stains for bacteria, including acid-fast bacilli, and fungi were negative. Bacterial culture of tissue from the lesion in the left beard area grew Staphylococcus aureus. Results of acid-fast and fungal cultures of tissue were negative. Shave biopsy of the umbilicated papule on the right side of the neck demonstrated classic invagination of the epidermis into the dermis and intracytoplasmic viral inclusions consistent with MC (Figure 2C). Spirochete immunohistochemical staining of the same tissue sample was negative (Figure 2D).
Serum rapid plasma reagin was reactive with a titer of 1:128 compared to the last known reactive rapid plasma reagin titer of 1:1 five years prior to presentation. A fluorescent treponemal antibody absorption test and VDRL test of cerebrospinal fluid was nonreactive. Fungal, bacterial, and acid-fast cultures of cerebral spinal fluid and a cryptococcal antigen test were negative. Serum cryptococcal antigen and coccidioides complement fixation tests were negative. Cytomegalovirus plasma polymerase chain reaction and urine histoplasma antigen testing were negative. Computed tomography of the chest revealed a new 1.9×1.6×2.1-cm3 cavitary lesion with distal tree-in-bud opacities in the lingula of the left lung. Acid-fast blood culture was negative, and acid-fast sputum culture was positive for Mycobacterium kansasii.
The cutaneous pathology findings and serologic findings confirmed the diagnoses of cutaneous secondary syphilis (SS) in the beard area and MC on the right side of the neck. Clinical diagnoses of seborrheic dermatitis of the alar creases and esophageal candidiasis also were made. The patient was treated with intramuscular penicillin G 2.4 million U once weekly for 3 weeks. The lesions confined to the beard area rapidly resolved within 7 days after the first dose of antibiotics, which further supported the diagnosis of localized cutaneous SS. Fluconazole 100 mg once daily was prescribed for the esophageal candidiasis, and he also was started on a regimen of rifampin 600 mg once daily, isoniazid 300 mg once daily, ethambutol 1200 mg once daily, and pyrazinamide 1500 mg once daily.
Syphilis is well known as the great masquerader due to its many possible manifestations. Many patients present with typical palmar and plantar dermatoses.1 Other documented SS presentations include eruptions ranging from a few to diffusely disseminated maculopapular lesions with or without scale on the trunk and upper extremities; pustular and nodular lesions of the face; alopecia; grayish white patches on the oral mucosa; and ulcerative, psoriasiform, follicular, and lichenoid lesions.2 Cutaneous SS has not been commonly reported in a localized distribution to the beard area with a clinical appearance mimicking hyperkeratotic MC lesions.3 Secondary syphilis is not known to spread through autoinoculation, presumably from shaving (as in our case), as might occur with other cutaneous infectious processes such as MC, verruca vulgaris, S aureus, and dermatophytosis in the beard area.
The differential diagnosis for hyperkeratotic papules and nodules localized to the beard area in human immunodeficiency virus–infected males includes MC, verruca vulgaris, chronic verrucous varicella-zoster virus, crusted scabies, tuberculosis verrucosa cutis, hypertrophic lichen planus, and disseminated deep fungal infections including cryptococcosis and coccidioidomycosis. In the setting of immunosuppression, the diagnosis of hyperkeratotic MC was favored in our patient given the co-location of classic umbilicated MC lesions with the hyperkeratotic papules and nodules. It is common to see MC autoinoculated in the beard area in men from shaving, as well as for MC to present in an atypical manner, particularly as hyperkeratotic lesions, in patients with AIDS.4 The predominant localized beard distribution and lack of other mucocutaneous manifestations of SS at presentation supported a clinical diagnosis of hyperkeratotic MC in our patient.
Unique presentations of SS have been documented, including nodular lesions of the face, but they typically have been accompanied by other stigmata of SS such as the classic palmoplantar or truncal maculopapular rash.3 One notable difference in our case was the localized beard distribution of the syphilitic cutaneous lesions in a man with AIDS. Our case reinforces the importance of cutaneous biopsies in immunocompromised patients. It is known that SS spreads hematogenously; however, in our case it was suspected that the new lesions may have spread locally through autoinoculation via beard hair removal, as the hyperkeratotic lesions were limited to the beard area. Koebnerization secondary to trauma induced by beard hair removal was considered in this case; however, koebnerization is known to occur in noninfectious dermatologic conditions, such as psoriasis, lichen planus, lichen nitidus, and vitiligo, but not in infections such as syphilis. Our case is pivotal in raising the question of whether SS can be autoinoculated in the beard area.
- Baughn RE, Musher DM. Secondary syphilitic lesions. Clin Microbiol Rev. 2005;18:205-216.
- Dourmishev LA, Dourmishev AL. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Cohen SE, Klausner JD, Engelman J, et al. Syphilis in the modern era: an update for physicians. Infect Dis Clin North Am. 2013;27:705-722.
- Filo-Rogulska M, Pindycka-Plaszcznska M, Januszewski K, et al. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol. 2013;30:56-58.
- Baughn RE, Musher DM. Secondary syphilitic lesions. Clin Microbiol Rev. 2005;18:205-216.
- Dourmishev LA, Dourmishev AL. Syphilis: uncommon presentations in adults. Clin Dermatol. 2005;23:555-564.
- Cohen SE, Klausner JD, Engelman J, et al. Syphilis in the modern era: an update for physicians. Infect Dis Clin North Am. 2013;27:705-722.
- Filo-Rogulska M, Pindycka-Plaszcznska M, Januszewski K, et al. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol. 2013;30:56-58.
Practice Points
- Recognize typical and atypical presentations of secondary syphilis (SS).
- This case reinforces the importance of cutaneous biopsies in immunocompromised patients.
- Consider the possibility of autoinoculation in SS.
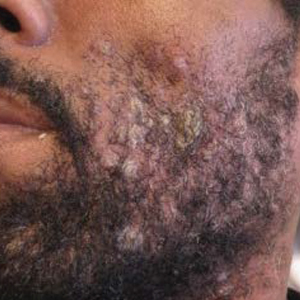
Larval Tick Infestation Causing an Eruption of Pruritic Papules and Pustules
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.


Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.
Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
Case Reports
Patient 1
A 65-year-old woman presented to the dermatology clinic in July with a pruritic rash of 2 days’ duration that started on the back and spread diffusely. The patient gardened regularly. Physical examination showed inflammatory papules and pustules on the back (Figure 1), as well as the groin, breasts, and ears. There was a punctate black dot in the center of some papules, and dermoscopy revealed ticks (Figure 2). Removal and microscopic examination confirmed larval-stage lone star ticks (Figure 3). The patient was prescribed topical steroids for pruritus as well as oral doxycycline for prophylaxis against tick-borne illnesses.
Patient 2
A 54-year-old man presented to the same clinic in July with pruritic lesions on the back, legs, ankles, and scrotum of 3 days’ duration that first appeared 24 hours after performing yardwork. Physical examination revealed diffusely distributed papules, pustules, and vesicles on the back (Figure 4). Some papules featured a punctate black dot in the center (similar to patient 1), and dermoscopy again revealed ticks. Removal and microscopic examination confirmed larval-stage ticks. The patient was treated with topical steroids and oral antihistamines for pruritus as well as prophylactic oral doxycycline.
Comment
Ticks are well-known human parasites, representing the second most common vector of human infectious disease.1 Ticks have 3 motile stages: larva (or “seed”), nymph, and adult. They can bite humans during all stages. Larval ticks, distinguished by having 6 legs rather than 8 legs in nymphs and adults, can attack in droves and cause an infestation that presents as diffuse, pruritic, erythematous papules and pustules.2-4 The first report of larval tick infestation in humans may have been in 1728 by William Byrd who described finding ticks on the skin that were too small to see without a microscope.5
Identification
The ticks in both of our cases were lone star ticks (Amblyomma americanum). The larval stage of A americanum is a proven cause of cutaneous reaction.6,7 A PubMed search of articles indexed for MEDLINE as well as a Google Scholar search using the terms tick, seed tick, or tick bite in combination with rash, eruption, infestation, papule, pustule, or pruritic revealed 6 reported cases of larval tick infestation in the literature (including our case); 5 were caused by A americanum and 1 by Ixodes dammini (now known as Ixodes scapularis); all occurred in July or August.3,7-10 This time frame is consistent with the general tick life cycle across species: Adults feed from April to June, then lay eggs that hatch into larval ticks within 4 to 6 weeks. After hatching, larval ticks climb grass and weeds awaiting a passing host.4
Diagnosis
Larval tick infestation remains a frequently misdiagnosed etiology of diffuse pruritic papules and pustules, especially in urban settings where physicians are less likely to be familiar with this type of manifestation.3,9-11 Larval ticks are submillimeter in size and difficult to appreciate with the naked eye, contributing to misdiagnosis. A punctate black dot may sometimes be seen in papules; however, dermoscopy is critical for accurate diagnosis, as hemorrhagic crust is a frequent misdiagnosis.
Management
In addition to symptomatic therapy, both of our patients received doxycycline as antibiotic prophylaxis for tick-borne illnesses given that a high number of ticks had been attached for more than 2 days.12,13 Antibiotic prophylaxis for tick-borne illness is controversial. The exception is Lyme disease transmitted by nymphal or adult I scapularis when specific conditions are met: the bite must have occurred in an endemic area, doxycycline cannot be contraindicated, estimated duration of attachment is at least 36 hours, and prophylaxis must be started within 72 hours of tick removal.13 There are no official recommendations for the A americanum species or for larval-stage ticks of any species. Larval-stage ticks acting as vectors for disease transmission is not well documented in recent literature, and there currently is limited evidence supporting prophylactic antibiotics for larval tick bites. The presence of spotted fever rickettsioses has been reported (with the exception of Rickettsia rickettsii and Ehrlichia chaffeensis) in larval A americanum ticks, suggesting a theoretical possibility that they could act as disease vectors.3,8,11,14-17 At a minimum, both prompt tick removal and close patient follow-up is warranted.
Conclusion
Human infestation with larval ticks is a common occurrence but can present a diagnostic challenge to an unfamiliar physician. We encourage consideration of larval tick infestation as the etiology of multiple or diffuse pruritic papules with a history of outdoor exposure.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
- Sonenshine DE. Biology of Ticks. New York, NY: Oxford University; 1991.
- Alexander JOD. The effects of tick bites. In: Alexander JOD. Arthropods and Human Skin. London, England: Springer London; 1984:363-382.
- Duckworth PF Jr, Hayden GF, Reed CN. Human infestation by Amblyomma americanum larvae (“seed ticks”). South Med J. 1985;78:751-753.
- Parola P, Raoult D. Ticks and tickborne bacterial diseases in humans: an emerging infectious threat. Clin Infect Dis. 2001;32:897-928.
- Cropley TG. William Byrd on ticks, 1728. Arch Dermatol. 2009;145:187.
- Goddard J. A ten-year study of tick biting in Mississippi: implications for human disease transmission. J Agromedicine. 2002;8:25-32.
- Goddard J, Portugal JS. Cutaneous lesions due to bites by larval Amblyomma americanum ticks. JAMA Dermatol. 2015;151:1373-1375.
- Fibeger EA, Erickson QL, Weintraub BD, et al. Larval tick infestation: a case report and review of tick-borne disease. Cutis. 2008;82:38-46.
- Jones BE. Human ‘seed tick’ infestation: Amblyomma americanum larvae. Arch Dermatol. 1981;117:812-814.
- Fisher EJ, Mo J, Lucky AW. Multiple pruritic papules from lone star tick larvae bites. Arch Dermatol. 2006;142:491-494.
- Culp JS. Seed ticks. Am Fam Physician. 1987;36:121-123.
- Perea AE, Hinckley AF, Mead PS. Tick bite prophylaxis: results from a 2012 survey of healthcare providers. Zoonoses Public Health. 2015;62:388-392.
- Tick bites/prevention. Centers for Disease Control and Prevention website. https://www.cdc.gov/ticks/tickbornediseases/tick-bites-prevention.html. Revised January 10, 2019. Accessed September 17, 2019.
- Moncayo AC, Cohen SB, Fritzen CM, et al. Absence of Rickettsia rickettsii and occurrence of other spotted fever group rickettsiae in ticks from Tennessee. Am J Trop Med Hyg. 2010;83:653-657.
- Castellaw AH, Showers J, Goddard J, et al. Detection of vector-borne agents in lone star ticks, Amblyomma americanum (Acari: Ixodidae), from Mississippi. J Med Entomol. 2010;47:473-476.
- Stromdahl EY, Vince MA, Billingsley PM, et al. Rickettsia amblyommii infecting Amblyomma americanum larvae. Vector Borne Zoonotic Dis. 2008;8:15-24.
- Long SW, Zhang X, Zhang J, et al. Evaluation of transovarial transmission and transmissibility of Ehrlichia chaffeensis (Rickettsiales: Anaplasmataceae) in Amblyomma americanum (Acari: Ixodidae). J Med Entomol. 2003;40:1000-1004.
Practice Points
- Larval (“seed”) ticks can attack in droves, causing a widespread rash consisting of pruritic erythematous papules and pustules.
- Tiny black dots can be seen in some papules, which are the seed ticks themselves. Careful dermoscopic examination is critical to avoid easy misdiagnosis as hemorrhagic crust.
- We encourage providers to include larval tick infestation in the differential for eruptive pruritic papules and pustules with a history of outdoor exposure, especially during the summer months.

20-year-old male college basketball prospect • wrist pain after falling on wrist • normal ROM • pain with active/passive wrist extension • Dx?
THE CASE
A 20-year-old man presented to our family medicine clinic with right wrist pain 4 days after falling on his wrist and hand while playing basketball. He denied any other previous injury or trauma. The pain was unchanged since the injury occurred.
Examination demonstrated mild edema over the palmar and ulnar aspect of the patient’s right wrist with no apparent ecchymosis. He had normal range of motion of his right wrist and hand. However, he experienced pain with active and passive wrist extension and ulnar deviation. There was significant tenderness in the palmar and ulnar aspects of his right wrist just distal to the ulnar styloid process.
THE DIAGNOSIS
Standard plain x-rays of the right wrist revealed an isolated fracture of the body of the triquetrum (FIGURE 1). Since the patient refused to have a cast placed, his wrist was immobilized with a wrist brace. By Day 16 post injury, the pain and edema had improved significantly. After talking with the patient about the potential risks and benefits of continuing to play basketball—and despite our recommendation that he not play—he decided to continue playing since he was a college basketball prospect.

At 4 weeks post injury, x-rays demonstrated mild interval healing (FIGURE 2). At the 8-week visit, the patient had only very mild pain and tenderness, and x-ray images showed improvement (FIGURE 3). Within a few months, his symptoms resolved completely. No further imaging was performed.

DISCUSSION
In general, carpal fractures are uncommon.1 The triquetrum is the second most commonly injured carpal bone, involved in up to 18% of all carpal fractures.2,3 Triquetrum fractures most commonly occur as isolated injuries and are typically classified in 2 general categories: avulsion fractures (dorsal cortex or volar cortex) and fractures of the triquetrum body.4-8 Isolated avulsion fractures of the triquetral dorsal cortex are relatively common, occurring in about 95% of triquetrum injuries.4-9 Isolated fractures of the triquetrum body are less common, occurring in about 4% of triquetrum injuries, and can go unnoticed on conventional x-rays.4-9

Basketball presents a unique risk for hand or wrist fracture due to its high-impact nature, hard playing surfaces, and frequent use of the hands for dribbling, shooting, rebounding, and passing the ball.
In a retrospective study of sports-related fractures conducted at the Royal Infirmary of Edinburgh, basketball had the highest incidence of carpal injuries compared with other sports, including football, rugby, skiing, snowboarding, and ice-skating.4 Similarly, a retrospective study conducted at the University of California, Los Angeles, found that of all Division 1 collegiate athletes at the school, basketball players had the highest incidence of primary fractures, and the most common fracture location was the hand.10
Continue to: An injury that's easy to miss
An injury that’s easy to miss
Because the incidence of hand and wrist injuries is high among basketball players, it is imperative that triquetrum body fractures are not missed or misdiagnosed as more common hand and wrist injuries, such as triquetral dorsal avulsion fractures.
Our patient, who had an isolated triquetrum body fracture, presented with focal tenderness on the palmar and ulnar aspects of his wrist and pain with ulnar deviation. Since triquetral body fractures often have a clinical presentation quite similar to that of triquetral dorsal avulsion fractures, patients presenting with symptoms of wrist tenderness and pain should be treated with a high degree of clinical suspicion.
With our patient, anteroposterior and lateral x-rays were sufficient to demonstrate an isolated triquetrum body fracture; however, triquetral fractures can be missed in up to 20% of x-rays.4 Both magnetic resonance imaging and computerized tomography are useful in diagnosing occult triquetrum fractures and should be used to confirm clinical suspicion when traditional x-rays are inconclusive.11,12
Management varies
Management of isolated triquetrum body fractures varies depending on the fracture pattern and the status of bone consolidation. Triquetral body fractures typically heal well; it’s very rare that there is a nonunion. As our patient’s fracture was nondisplaced and stable, brace immobilization for 4 weeks was sufficient to facilitate healing and restore long-term hand and wrist functionality. This course of treatment is consistent with other cases of nondisplaced triquetrum body fractures reported in the literature.13
Long-term outcomes. The literature is sparse regarding the long-term functional outcome of nonsurgical treatment for nondisplaced triquetrum body fractures. Multiple carpal fractures, displaced triquetrum body fractures, and persistent pain for multiple months after nonsurgical management all indicate the need for referral to orthopedic surgery. In instances of fracture displacement or nonunion, management tends to be surgical, with open reduction and internal fixation (ORIF) used in multiple cases of nonunion for isolated triquetrum body fractures.3,14 Any diagnostic imaging that reveals displacement, malunion, or nonunion of the fracture is an indication for referral to an orthopedic surgeon.
Continue to: Return to play
Return to play. There is no evidence-based return-to-play recommendation for patients with a triquetrum fracture. However, our patient continued to play basketball through the early stages of injury management because he was a collegiate prospect. While medical, social, and economic factors should be considered when discussing treatment options with athletes, injuries should be managed so that there is no long-term loss of function or risk of injury exacerbation. When discussing early return from injury with athletes who have outside pressure to return to play, it’s important to make them aware of the associated long- and short-term risks.15
THE TAKEAWAY
Management of an isolated triquetrum body fracture is typically straightforward; however, if the fracture is displaced, refer the patient to an orthopedic surgeon as ORIF may be required. For this reason, it’s important to be able to promptly identify isolated triquetrum body fractures and to avoid confusing them with triquetrum dorsal avulsion fractures.
Depending on the sport played and the severity of the injury, athletes with conservatively managed nondisplaced triquetral body fractures may be candidates for early return to play. Nonetheless, athletes should understand both the short- and the long-term risks of playing with an injury, and they should never be advised to continue playing with an injury if it jeopardizes their well-being or the long-term functionality of the affected body part.
CORRESPONDENCE
Morteza Khodaee, MD, MPH, University of Colorado School of Medicine, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
1. Suh N, Ek ET, Wolfe SW. Carpal fractures. J Hand Surg Am. 2014;39:785-791.
2. Hey HW, Chong AK, Murphy D. Prevalence of carpal fracture in Singapore. J Hand Surg Am. 2011;36:278-283.
3. Al Rashid M, Rasoli S, Khan WS. Non-union of isolated displaced triquetral body fracture—a case report. Ortop Traumatol Rehabil. 2012;14:71-74.
4. Becce F, Theumann N, Bollmann C, et al. Dorsal fractures of the triquetrum: MRI findings with an emphasis on dorsal carpal ligament injuries. AJR Am J Roentgenol. 2013;200:608-617.
5. Court-Brown CM, Wood AM, Aitken S. The epidemiology of acute sports-related fractures in adults. Injury. 2008;39:1365-1372.
6. Urch EY, Lee SK. Carpal fractures other than scaphoid. Clin Sports Med. 2015;34:51-67.
7. deWeber K. Triquetrum fractures. UpToDate. 2016. www.uptodate.com/contents/triquetrum-fractures. Accessed September 3, 2019.
8. Höcker K, Menschik A. Chip fractures of the triquetrum. Mechanism, classification and results. J Hand Surg Br. 1994;19:584-588.
9. Jarraya M, Hayashi D, Roemer FW, et al. Radiographically occult and subtle fractures: a pictorial review. Radiol Res Pract. 2013;2013:370169.
10. Hame SL, LaFemina JM, McAllister DR, et al. Fractures in the collegiate athlete. Am J Sports Med. 2004;32:446-451.
11. Hindman BW, Kulik WJ, Lee G, et al. Occult fractures of the carpals and metacarpals: demonstration by CT. AJR Am J Roentgenol. 1989;153:529-532.
12. Pierre-Jerome C, Moncayo V, Albastaki U, et al. Multiple occult wrist bone injuries and joint effusions: prevalence and distribution on MRI. Emerg Radiol. 2010;17:179-184.
13. Yildirim C, Akmaz I, Keklikçi K, et al. An unusual combined fracture pattern of the triquetrum. J Hand Surg Eur Vol. 2008;33:385-386.
14. Rasoli S, Ricks M, Packer G. Isolated displaced non-union of a triquetral body fracture: a case report. J Med Case Rep. 2012;6:54.
15. Strickland JW. Considerations for the treatment of the injured athlete. Clin Sports Med. 1998;17:397-400.
THE CASE
A 20-year-old man presented to our family medicine clinic with right wrist pain 4 days after falling on his wrist and hand while playing basketball. He denied any other previous injury or trauma. The pain was unchanged since the injury occurred.
Examination demonstrated mild edema over the palmar and ulnar aspect of the patient’s right wrist with no apparent ecchymosis. He had normal range of motion of his right wrist and hand. However, he experienced pain with active and passive wrist extension and ulnar deviation. There was significant tenderness in the palmar and ulnar aspects of his right wrist just distal to the ulnar styloid process.
THE DIAGNOSIS
Standard plain x-rays of the right wrist revealed an isolated fracture of the body of the triquetrum (FIGURE 1). Since the patient refused to have a cast placed, his wrist was immobilized with a wrist brace. By Day 16 post injury, the pain and edema had improved significantly. After talking with the patient about the potential risks and benefits of continuing to play basketball—and despite our recommendation that he not play—he decided to continue playing since he was a college basketball prospect.
At 4 weeks post injury, x-rays demonstrated mild interval healing (FIGURE 2). At the 8-week visit, the patient had only very mild pain and tenderness, and x-ray images showed improvement (FIGURE 3). Within a few months, his symptoms resolved completely. No further imaging was performed.
DISCUSSION
In general, carpal fractures are uncommon.1 The triquetrum is the second most commonly injured carpal bone, involved in up to 18% of all carpal fractures.2,3 Triquetrum fractures most commonly occur as isolated injuries and are typically classified in 2 general categories: avulsion fractures (dorsal cortex or volar cortex) and fractures of the triquetrum body.4-8 Isolated avulsion fractures of the triquetral dorsal cortex are relatively common, occurring in about 95% of triquetrum injuries.4-9 Isolated fractures of the triquetrum body are less common, occurring in about 4% of triquetrum injuries, and can go unnoticed on conventional x-rays.4-9
Basketball presents a unique risk for hand or wrist fracture due to its high-impact nature, hard playing surfaces, and frequent use of the hands for dribbling, shooting, rebounding, and passing the ball.
In a retrospective study of sports-related fractures conducted at the Royal Infirmary of Edinburgh, basketball had the highest incidence of carpal injuries compared with other sports, including football, rugby, skiing, snowboarding, and ice-skating.4 Similarly, a retrospective study conducted at the University of California, Los Angeles, found that of all Division 1 collegiate athletes at the school, basketball players had the highest incidence of primary fractures, and the most common fracture location was the hand.10
Continue to: An injury that's easy to miss
An injury that’s easy to miss
Because the incidence of hand and wrist injuries is high among basketball players, it is imperative that triquetrum body fractures are not missed or misdiagnosed as more common hand and wrist injuries, such as triquetral dorsal avulsion fractures.
Our patient, who had an isolated triquetrum body fracture, presented with focal tenderness on the palmar and ulnar aspects of his wrist and pain with ulnar deviation. Since triquetral body fractures often have a clinical presentation quite similar to that of triquetral dorsal avulsion fractures, patients presenting with symptoms of wrist tenderness and pain should be treated with a high degree of clinical suspicion.
With our patient, anteroposterior and lateral x-rays were sufficient to demonstrate an isolated triquetrum body fracture; however, triquetral fractures can be missed in up to 20% of x-rays.4 Both magnetic resonance imaging and computerized tomography are useful in diagnosing occult triquetrum fractures and should be used to confirm clinical suspicion when traditional x-rays are inconclusive.11,12
Management varies
Management of isolated triquetrum body fractures varies depending on the fracture pattern and the status of bone consolidation. Triquetral body fractures typically heal well; it’s very rare that there is a nonunion. As our patient’s fracture was nondisplaced and stable, brace immobilization for 4 weeks was sufficient to facilitate healing and restore long-term hand and wrist functionality. This course of treatment is consistent with other cases of nondisplaced triquetrum body fractures reported in the literature.13
Long-term outcomes. The literature is sparse regarding the long-term functional outcome of nonsurgical treatment for nondisplaced triquetrum body fractures. Multiple carpal fractures, displaced triquetrum body fractures, and persistent pain for multiple months after nonsurgical management all indicate the need for referral to orthopedic surgery. In instances of fracture displacement or nonunion, management tends to be surgical, with open reduction and internal fixation (ORIF) used in multiple cases of nonunion for isolated triquetrum body fractures.3,14 Any diagnostic imaging that reveals displacement, malunion, or nonunion of the fracture is an indication for referral to an orthopedic surgeon.
Continue to: Return to play
Return to play. There is no evidence-based return-to-play recommendation for patients with a triquetrum fracture. However, our patient continued to play basketball through the early stages of injury management because he was a collegiate prospect. While medical, social, and economic factors should be considered when discussing treatment options with athletes, injuries should be managed so that there is no long-term loss of function or risk of injury exacerbation. When discussing early return from injury with athletes who have outside pressure to return to play, it’s important to make them aware of the associated long- and short-term risks.15
THE TAKEAWAY
Management of an isolated triquetrum body fracture is typically straightforward; however, if the fracture is displaced, refer the patient to an orthopedic surgeon as ORIF may be required. For this reason, it’s important to be able to promptly identify isolated triquetrum body fractures and to avoid confusing them with triquetrum dorsal avulsion fractures.
Depending on the sport played and the severity of the injury, athletes with conservatively managed nondisplaced triquetral body fractures may be candidates for early return to play. Nonetheless, athletes should understand both the short- and the long-term risks of playing with an injury, and they should never be advised to continue playing with an injury if it jeopardizes their well-being or the long-term functionality of the affected body part.
CORRESPONDENCE
Morteza Khodaee, MD, MPH, University of Colorado School of Medicine, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
THE CASE
A 20-year-old man presented to our family medicine clinic with right wrist pain 4 days after falling on his wrist and hand while playing basketball. He denied any other previous injury or trauma. The pain was unchanged since the injury occurred.
Examination demonstrated mild edema over the palmar and ulnar aspect of the patient’s right wrist with no apparent ecchymosis. He had normal range of motion of his right wrist and hand. However, he experienced pain with active and passive wrist extension and ulnar deviation. There was significant tenderness in the palmar and ulnar aspects of his right wrist just distal to the ulnar styloid process.
THE DIAGNOSIS
Standard plain x-rays of the right wrist revealed an isolated fracture of the body of the triquetrum (FIGURE 1). Since the patient refused to have a cast placed, his wrist was immobilized with a wrist brace. By Day 16 post injury, the pain and edema had improved significantly. After talking with the patient about the potential risks and benefits of continuing to play basketball—and despite our recommendation that he not play—he decided to continue playing since he was a college basketball prospect.
At 4 weeks post injury, x-rays demonstrated mild interval healing (FIGURE 2). At the 8-week visit, the patient had only very mild pain and tenderness, and x-ray images showed improvement (FIGURE 3). Within a few months, his symptoms resolved completely. No further imaging was performed.
DISCUSSION
In general, carpal fractures are uncommon.1 The triquetrum is the second most commonly injured carpal bone, involved in up to 18% of all carpal fractures.2,3 Triquetrum fractures most commonly occur as isolated injuries and are typically classified in 2 general categories: avulsion fractures (dorsal cortex or volar cortex) and fractures of the triquetrum body.4-8 Isolated avulsion fractures of the triquetral dorsal cortex are relatively common, occurring in about 95% of triquetrum injuries.4-9 Isolated fractures of the triquetrum body are less common, occurring in about 4% of triquetrum injuries, and can go unnoticed on conventional x-rays.4-9
Basketball presents a unique risk for hand or wrist fracture due to its high-impact nature, hard playing surfaces, and frequent use of the hands for dribbling, shooting, rebounding, and passing the ball.
In a retrospective study of sports-related fractures conducted at the Royal Infirmary of Edinburgh, basketball had the highest incidence of carpal injuries compared with other sports, including football, rugby, skiing, snowboarding, and ice-skating.4 Similarly, a retrospective study conducted at the University of California, Los Angeles, found that of all Division 1 collegiate athletes at the school, basketball players had the highest incidence of primary fractures, and the most common fracture location was the hand.10
Continue to: An injury that's easy to miss
An injury that’s easy to miss
Because the incidence of hand and wrist injuries is high among basketball players, it is imperative that triquetrum body fractures are not missed or misdiagnosed as more common hand and wrist injuries, such as triquetral dorsal avulsion fractures.
Our patient, who had an isolated triquetrum body fracture, presented with focal tenderness on the palmar and ulnar aspects of his wrist and pain with ulnar deviation. Since triquetral body fractures often have a clinical presentation quite similar to that of triquetral dorsal avulsion fractures, patients presenting with symptoms of wrist tenderness and pain should be treated with a high degree of clinical suspicion.
With our patient, anteroposterior and lateral x-rays were sufficient to demonstrate an isolated triquetrum body fracture; however, triquetral fractures can be missed in up to 20% of x-rays.4 Both magnetic resonance imaging and computerized tomography are useful in diagnosing occult triquetrum fractures and should be used to confirm clinical suspicion when traditional x-rays are inconclusive.11,12
Management varies
Management of isolated triquetrum body fractures varies depending on the fracture pattern and the status of bone consolidation. Triquetral body fractures typically heal well; it’s very rare that there is a nonunion. As our patient’s fracture was nondisplaced and stable, brace immobilization for 4 weeks was sufficient to facilitate healing and restore long-term hand and wrist functionality. This course of treatment is consistent with other cases of nondisplaced triquetrum body fractures reported in the literature.13
Long-term outcomes. The literature is sparse regarding the long-term functional outcome of nonsurgical treatment for nondisplaced triquetrum body fractures. Multiple carpal fractures, displaced triquetrum body fractures, and persistent pain for multiple months after nonsurgical management all indicate the need for referral to orthopedic surgery. In instances of fracture displacement or nonunion, management tends to be surgical, with open reduction and internal fixation (ORIF) used in multiple cases of nonunion for isolated triquetrum body fractures.3,14 Any diagnostic imaging that reveals displacement, malunion, or nonunion of the fracture is an indication for referral to an orthopedic surgeon.
Continue to: Return to play
Return to play. There is no evidence-based return-to-play recommendation for patients with a triquetrum fracture. However, our patient continued to play basketball through the early stages of injury management because he was a collegiate prospect. While medical, social, and economic factors should be considered when discussing treatment options with athletes, injuries should be managed so that there is no long-term loss of function or risk of injury exacerbation. When discussing early return from injury with athletes who have outside pressure to return to play, it’s important to make them aware of the associated long- and short-term risks.15
THE TAKEAWAY
Management of an isolated triquetrum body fracture is typically straightforward; however, if the fracture is displaced, refer the patient to an orthopedic surgeon as ORIF may be required. For this reason, it’s important to be able to promptly identify isolated triquetrum body fractures and to avoid confusing them with triquetrum dorsal avulsion fractures.
Depending on the sport played and the severity of the injury, athletes with conservatively managed nondisplaced triquetral body fractures may be candidates for early return to play. Nonetheless, athletes should understand both the short- and the long-term risks of playing with an injury, and they should never be advised to continue playing with an injury if it jeopardizes their well-being or the long-term functionality of the affected body part.
CORRESPONDENCE
Morteza Khodaee, MD, MPH, University of Colorado School of Medicine, AFW Clinic, 3055 Roslyn Street, Denver, CO 80238; [email protected]
1. Suh N, Ek ET, Wolfe SW. Carpal fractures. J Hand Surg Am. 2014;39:785-791.
2. Hey HW, Chong AK, Murphy D. Prevalence of carpal fracture in Singapore. J Hand Surg Am. 2011;36:278-283.
3. Al Rashid M, Rasoli S, Khan WS. Non-union of isolated displaced triquetral body fracture—a case report. Ortop Traumatol Rehabil. 2012;14:71-74.
4. Becce F, Theumann N, Bollmann C, et al. Dorsal fractures of the triquetrum: MRI findings with an emphasis on dorsal carpal ligament injuries. AJR Am J Roentgenol. 2013;200:608-617.
5. Court-Brown CM, Wood AM, Aitken S. The epidemiology of acute sports-related fractures in adults. Injury. 2008;39:1365-1372.
6. Urch EY, Lee SK. Carpal fractures other than scaphoid. Clin Sports Med. 2015;34:51-67.
7. deWeber K. Triquetrum fractures. UpToDate. 2016. www.uptodate.com/contents/triquetrum-fractures. Accessed September 3, 2019.
8. Höcker K, Menschik A. Chip fractures of the triquetrum. Mechanism, classification and results. J Hand Surg Br. 1994;19:584-588.
9. Jarraya M, Hayashi D, Roemer FW, et al. Radiographically occult and subtle fractures: a pictorial review. Radiol Res Pract. 2013;2013:370169.
10. Hame SL, LaFemina JM, McAllister DR, et al. Fractures in the collegiate athlete. Am J Sports Med. 2004;32:446-451.
11. Hindman BW, Kulik WJ, Lee G, et al. Occult fractures of the carpals and metacarpals: demonstration by CT. AJR Am J Roentgenol. 1989;153:529-532.
12. Pierre-Jerome C, Moncayo V, Albastaki U, et al. Multiple occult wrist bone injuries and joint effusions: prevalence and distribution on MRI. Emerg Radiol. 2010;17:179-184.
13. Yildirim C, Akmaz I, Keklikçi K, et al. An unusual combined fracture pattern of the triquetrum. J Hand Surg Eur Vol. 2008;33:385-386.
14. Rasoli S, Ricks M, Packer G. Isolated displaced non-union of a triquetral body fracture: a case report. J Med Case Rep. 2012;6:54.
15. Strickland JW. Considerations for the treatment of the injured athlete. Clin Sports Med. 1998;17:397-400.
1. Suh N, Ek ET, Wolfe SW. Carpal fractures. J Hand Surg Am. 2014;39:785-791.
2. Hey HW, Chong AK, Murphy D. Prevalence of carpal fracture in Singapore. J Hand Surg Am. 2011;36:278-283.
3. Al Rashid M, Rasoli S, Khan WS. Non-union of isolated displaced triquetral body fracture—a case report. Ortop Traumatol Rehabil. 2012;14:71-74.
4. Becce F, Theumann N, Bollmann C, et al. Dorsal fractures of the triquetrum: MRI findings with an emphasis on dorsal carpal ligament injuries. AJR Am J Roentgenol. 2013;200:608-617.
5. Court-Brown CM, Wood AM, Aitken S. The epidemiology of acute sports-related fractures in adults. Injury. 2008;39:1365-1372.
6. Urch EY, Lee SK. Carpal fractures other than scaphoid. Clin Sports Med. 2015;34:51-67.
7. deWeber K. Triquetrum fractures. UpToDate. 2016. www.uptodate.com/contents/triquetrum-fractures. Accessed September 3, 2019.
8. Höcker K, Menschik A. Chip fractures of the triquetrum. Mechanism, classification and results. J Hand Surg Br. 1994;19:584-588.
9. Jarraya M, Hayashi D, Roemer FW, et al. Radiographically occult and subtle fractures: a pictorial review. Radiol Res Pract. 2013;2013:370169.
10. Hame SL, LaFemina JM, McAllister DR, et al. Fractures in the collegiate athlete. Am J Sports Med. 2004;32:446-451.
11. Hindman BW, Kulik WJ, Lee G, et al. Occult fractures of the carpals and metacarpals: demonstration by CT. AJR Am J Roentgenol. 1989;153:529-532.
12. Pierre-Jerome C, Moncayo V, Albastaki U, et al. Multiple occult wrist bone injuries and joint effusions: prevalence and distribution on MRI. Emerg Radiol. 2010;17:179-184.
13. Yildirim C, Akmaz I, Keklikçi K, et al. An unusual combined fracture pattern of the triquetrum. J Hand Surg Eur Vol. 2008;33:385-386.
14. Rasoli S, Ricks M, Packer G. Isolated displaced non-union of a triquetral body fracture: a case report. J Med Case Rep. 2012;6:54.
15. Strickland JW. Considerations for the treatment of the injured athlete. Clin Sports Med. 1998;17:397-400.
