User login
A Multifaceted Case
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
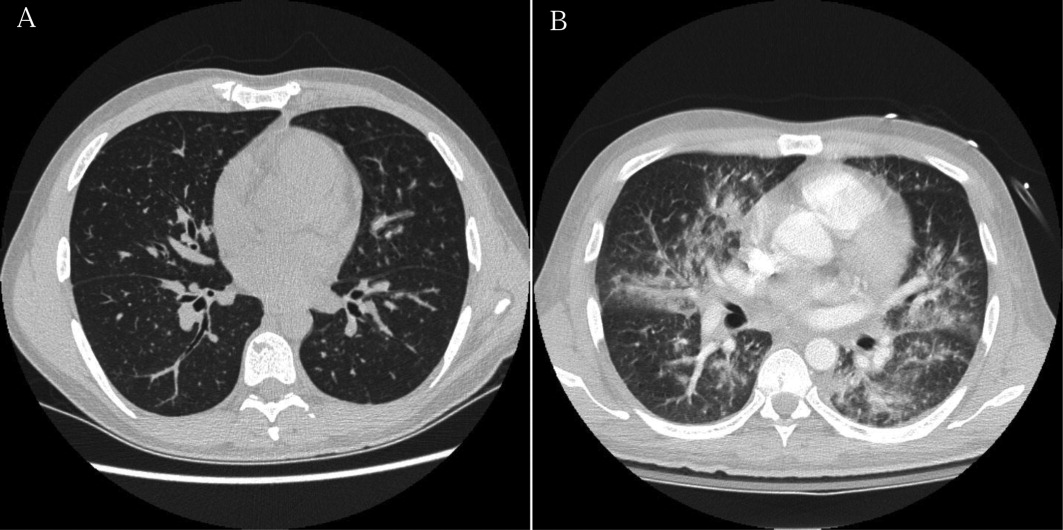
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
Box
1
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Box
2
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
A 67‐year‐old male presented to an outside hospital with a 1‐day history of fevers up to 39.4C, bilateral upper extremity weakness, and confusion. Forty‐eight hours prior to his presentation he had undergone uncomplicated bilateral carpal tunnel release surgery for the complaint of bilateral upper extremity paresthesias.
Bilateral carpal tunnel syndrome should prompt consideration of systemic diseases that infiltrate or impinge both canals (eg, rheumatoid arthritis, acromegaly, hypothyroidism, amyloidosis), although it is most frequently explained by a bilateral repetitive stress (eg, workplace typing). The development of upper extremity weakness suggests that an alternative condition such as cervical myelopathy, bilateral radiculopathy, or a rapidly progressive peripheral neuropathy may be responsible for his paresthesias. It would be unusual for a central nervous system process to selectively cause bilateral upper extremity weakness. Occasionally, patients emerge from surgery with limb weakness caused by peripheral nerve injury sustained from malpositioning of the extremity, but this would have been evident immediately following the operation.
Postoperative fevers are frequently unexplained, but require a search for common healthcare‐associated infections, such as pneumonia, urinary tract infection, intravenous catheter thrombophlebitis, wound infection, or Clostridium difficile colitis. However, such complications are unlikely following an ambulatory procedure. Confusion and fever together point to a central nervous system infection (meningoencephalitis or brain abscess) or a systemic infection that has impaired cognition. Malignancies can cause fever and altered mental status, but these are typically asynchronous events.
His past medical history was notable for hypertension, dyslipidemia, gout, actinic keratosis, and gastroesophageal reflux. His surgical history included bilateral knee replacements, repair of a left rotator cuff injury, and a herniorrhaphy. He was a nonsmoker who consumed 4 to 6 beers daily. His medications included clonidine, colchicine, atorvastatin, extended release metoprolol, triamterene‐hydrochlorothiazide, probenecid, and as‐needed ibuprofen and omeprazole.
Upon presentation he was cooperative and in no distress. Temperature was 38.9C, pulse 119 beats per minute, blood pressure 140/90 mm Hg, and oxygen saturation 94% on room air. He was noted to have logical thinking but impaired concentration. His upper extremity movement was restricted because of postoperative discomfort and swelling rather than true weakness. The rest of the exam was normal.
Metabolic, infectious, structural (intracranial), and toxic disorders can cause altered mental status. His heavy alcohol use puts him at risk for alcohol withdrawal and infections (such as Listeria meningitis), both of which may explain his fever and altered mental status. Signs and symptoms of meningitis are absent at this time. His knee prostheses could have harbored an infection preoperatively and therefore warrant close examination. Patients sometimes have adverse reactions to medications they have been prescribed but are not exposed to until hospitalization, although his surgical procedure was likely done on an outpatient basis. Empiric thiamine should be administered early given his confusion and alcohol habits.
Basic laboratories revealed a hemoglobin of 11.2 g/dL, white blood cell (WBC) count of 6,900/mm3 with 75% neutrophils, platelets of 206,000/mm3. Mean corpuscular volume was 97 mm3. Serum albumin was 2.4 g/dl, sodium 134 mmol/L, potassium 3.9 mmol/L, blood urea nitrogen 12 mg/dL, and creatinine 0.9 mg/dL. The aspartate aminotransferase was 93 U/L, alanine aminotransferase 73 U/L, alkaline phosphatase 254 U/L, and total bilirubin 1.0 mg/dL. Urinalysis was normal. Over the next 16 days fevers and waxing and waning mentation continued. The following studies were normal or negative: blood and urine cultures; transthoracic echocardiogram, antinuclear antibodies, hepatitis B surface antigen, hepatitis C antibody, and human immunodeficiency virus antibody; magnetic resonance imaging of the brain, electroencephalogram, and lower extremity venous ultrasound.
Hypoalbuminemia may signal chronic illness, hypoproduction from liver disease (caused by his heavy alcohol use), or losses from the kidney or gastrointestinal tract. His anemia may reflect chronic disease or point toward a specific underlying disorder. For example, fever and anemia could arise from hemolytic processes such as thrombotic thrombocytopenic purpura or clostridial infections.
An extensive workup has not revealed a cause for his prolonged fever (eg, infection, malignancy, autoimmune condition, or toxin). Likewise, an explanation for confusion is lacking. Because systemic illness and structural brain disease have not been uncovered, a lumbar puncture is indicated.
A lumbar puncture under fluoroscopic guidance revealed a cerebrospinal fluid (CSF) WBC count of 6/mm3, red blood cell count (RBC) 2255/mm3, protein 49 mg/dL, and glucose 54 mg/dL. The WBC differential was not reported. No growth was reported on bacterial cultures. Polymerase chain reactions for enterovirus and herpes simplex viruses 1 and 2 were negative. Cryptococcal antigen and Venereal Disease Research Laboratory serologies were also negative.
A CSF WBC count of 6 is out of the normal range, but could be explained by a traumatic tap given the elevated RBC; the protein and glucose are likewise at the border of normal. Collectively, these are nonspecific findings that could point to an infectious or noninfectious cause of intrathecal or paraspinous inflammation, but are not suggestive of bacterial meningitis.
The patient developed pneumonia, for which he received ertapenem. On hospital day 17 he was intubated for hypoxia and respiratory distress and was extubated after 4 days of mechanical ventilation. Increasing weakness in all extremities prompted magnetic resonance imaging of the spine, which revealed fluid and enhancement involving the soft tissues around C3‐C4 and C5‐C6, raising concerns for discitis and osteomyelitis. Possible septic arthritis at the C3‐C4 and C4‐C5 facets was noted. Ring enhancing fluid collections from T2‐T8 compatible with an epidural abscess with cord compression at T4‐T5 and T6‐T7 were seen. Enhancement and fluid involving the facet joints between T2‐T7 was also consistent with septic arthritis (Figure 1).
His pneumonia appears to have developed many days into his hospitalization, and therefore is unlikely to account for his initial fever and confusion. Blood cultures and echocardiogram have not suggested an endovascular infection that could account for such widespread vertebral and epidural deposition. A wide number of bacteria can cause epidural abscesses and septic arthritis, most commonly Staphylococcus aureus. Less common pathogens with a predilection for osteoarticular involvement, such as Brucella species, warrant consideration when there is appropriate epidemiologic risk.
Systemic bacterial infection remains a concern with his alcoholism rendering him partially immunosuppressed. However, a large number of adjacent spinal joints harboring a bacterial infection is unusual, and a working diagnosis of multilevel spinal infection, therefore, should prompt consideration of noninfectious processes. When a patient develops a swollen peripheral joint and fever in the postoperative setting, gout or pseudogout is a leading consideration. That same thinking should be applied to the vertebrae, where spinal gout can manifest. Surgery itself or associated changes in alcohol consumption patterns or changes in medications (at least 4 of which are relevant to goutcolchicine, hydrochlorothiazide, probenecid, and ibuprofen) could predispose him to a flare.
Aspiration of the epidural collection yielded a negative Gram stain and culture. He developed swelling in the bilateral proximal interphalangeal joints and was treated with steroids and colchicine for suspected gout flare. Vancomycin and piperacillin‐tazobactam were initiated, and on hospital day 22 the patient was transferred to another hospital for further evaluation by neurosurgery.
The negative Gram stain and culture argues against septic arthritis, but these are imperfect tests and will not detect atypical pathogens (eg, spinal tuberculosis). Reexamination of the aspirate for urate and calcium pyrophosphate crystals would be useful. Initiation of steroids in the setting of potentially undiagnosed infection requires a careful risk/benefit analysis. It may be reasonable to treat the patient with colchicine alone while withholding steroids and avoiding nonsteroidal agents in case invasive procedures are planned.
On exam his temperature was 36C, blood pressure 156/92 mm Hg, pulse 100 beats per minute, respirations 21 per minute, and oxygenation 97% on room air. He was not in acute distress and was only oriented to self. Bilateral 2+ lower extremity pitting edema up to the knees was noted. Examination of the heart and lungs was unremarkable. Gouty tophi were noted over both elbows. His joints were normal.
Cranial nerves IIXII were normal. Motor exam revealed normal muscle tone and bulk. Muscle strength was approximately 3/5 in the right upper extremity and 4+/5 in the left upper extremity. Bilateral lower extremity strength was 3/5 in hip flexion, knee flexion, and knee extension. Dorsiflexion and plantar flexion were approximately 2/5 bilaterally. Sensation was intact to light touch and pinprick, and proprioception was normal. Gait was not tested. A Foley catheter was in place.
This examination confirms ongoing encephalopathy and incomplete quadriplegia. The lower extremity weakness is nearly equal proximally and distally, which can be seen with an advanced peripheral neuropathy but is more characteristic of myelopathy. The expected concomitant sensory deficit of myelopathy is not present, although this may be difficult to detect in a confused patient. Reflex testing would help in distinguishing myelopathy (favored because of the imaging findings) from a rapid progressive peripheral motor neuropathy (eg, acute inflammatory demyelinating polyneuropathy or acute intermittent porphyria).
The pitting edema likely represents fluid overload, which can be nonspecific after prolonged immobility during hospitalization; hypoalbuminemia is oftentimes speculated to play a role when this develops. His alcohol use puts him at risk for heart failure (although there is no evidence of this on exam) and liver disease (which his liver function tests suggest). The tophi speak to the extent and chronicity of his hyperuricemia.
On arrival he reported recent onset diarrhea. Medications at transfer included metoprolol, omeprazole, prednisone, piperacillin/tazobactam, vancomycin, and colchicine; acetaminophen, bisacodyl, diphenhydramine, fentanyl, subcutaneous insulin, and labetalol were administered as needed. Laboratory studies included a hemoglobin of 9.5 g/dL, WBC count of 7,300/mm3 with 95% neutrophils, platelets 301,000/mm3, sodium 151 mmol/L, potassium 2.9 mmol/L, blood urea nitrogen 76 mg/dL, creatinine 2.0 mg/dL, aspartate aminotransferase 171 U/L, and alanine aminotransferase 127 U/L. Serum albumin was 1.7 g/dL.
At least 3 of his medicationsdiphenhydramine, fentanyl, and prednisonemay be contributing to his ongoing altered mental status, which may be further compounded by hypernatremia. Although his liver disease remains uncharacterized, hepatic encephalopathy may be contributing to his confusion as well.
Colchicine is likely responsible for his diarrhea, which would be the most readily available explanation for his hypernatremia, hypokalemia, and acute kidney injury (AKI). Acute kidney injury could result from progressive liver disease (hepatorenal syndrome), decreased arterial perfusion (suggested by third spacing or his diarrhea), acute tubular necrosis (from infection or medication), or urinary retention secondary to catheter obstruction. Acute hyperuricemia can also cause AKI (urate nephropathy).
Anemia has progressed and requires evaluation for blood loss as well as hemolysis. Hepatotoxicity from any of his medications (eg, acetaminophen) must be considered. Coagulation studies and review of the previous abdominal computed tomography would help determine the extent of his liver disease.
Neurosurgical consultation was obtained and the patient and his family elected to proceed with a thoracic laminectomy. Cheesy fluid was identified at the facet joints at T6‐T7, which was found to contain rare deposits of monosodium urate crystals. Surgical specimen cultures were sterile. His mental status and strength slowly improved to baseline following the surgery. He was discharged on postoperative day 7 to a rehabilitation facility. On the telephone follow‐up he reported that he has regained his strength completely.
The fluid analysis and clinical course confirms spinal gout. The presenting encephalopathy remains unexplained; I am unaware of gout leading to altered mental status.
COMMENTARY
Gout is an inflammatory condition triggered by the deposition of monosodium urate crystals in tissues in association with hyperuricemia.[1] Based on the 20072008 National Health and Nutrition Examination Survey, the prevalence of gout among US adults was 3.9% (8.3 million individuals).[2] These rates are increasing and are thought to be spurred by the aging population, increasing rates of obesity, and changing dietary habits including increases in the consumption of soft drinks and red meat.[3, 4, 5] The development of gout during hospitalization can prolong length of stay, and the implementation of a management protocol appears to help decrease treatment delays and the inappropriate discontinuation of gout prophylaxis.[6, 7] Surgery, with its associated physiologic stressors, can trigger gout, which is often polyarticular and presents with fever leading to testing and consultations for the febrile episode.[8]
Gout is an ancient disease that is familiar to most clinicians. In 1666, Daniel Sennert, a German physician, described gout as the physician's shame because of its infrequent recognition.[9] Clinical gout spans 3 stages: asymptomatic hyperuricemia, acute and intercritical gout, and chronic gouty arthritis. The typical acute presentation is monoarticular with the abrupt onset of pain, swelling, warmth, and erythema in a peripheral joint. It manifests most characteristically in the first metatarsophalangeal joint (podagra), but also frequently involves the midfoot, ankle, knee, and wrist and sometimes affects multiple joints simultaneously (polyarticular gout).[1, 10] The visualization of monosodium urate crystals either in synovial fluid or from a tophus is diagnostic of gout; however, guidelines recognize that a classic presentation of gout may be diagnosed based on clinical criteria alone.[11] Dual energy computerized tomography and ultrasonography are emerging as techniques for the visualization of monosodium urate crystals; however, they are not currently routinely recommended.[12]
There are many unusual presentations of gout, with an increase in such reports paralleling both the overall increase in the prevalence of gout and improvements in available imaging techniques.[13] Atypical presentations present diagnostic challenges and are often caused by tophaceous deposits in unusual locations. Reports of atypical gout have described entrapment neuropathies (eg, gouty deposits inducing carpal tunnel syndrome), ocular gout manifested as conjunctival deposits and uveitis, pancreatic gout presenting as a mass, and dermatologic manifestations including panniculitis.[13, 14]
Spinal gout (also known as axial gout) manifests when crystal‐induced inflammation, erosive arthritis, and tophaceous deposits occur along the spinal column. A cross‐sectional study of patients with poorly controlled gout reported the prevalence of spinal gout diagnosed by computerized tomography to be 35%. These radiographic findings were not consistently correlated with back pain.[15] Imaging features that are suggestive of spinal gout include intra‐articular and juxta‐articular erosions with sclerotic margins and density greater than the surrounding muscle. Periosteal new bone formation adjacent to bony destruction can form overhanging edges.[16] When retrospectively presented with the final diagnosis, the radiologist at our institution noted that the appearance was typical gout in an atypical location.
Spinal gout can be confused with spinal metastasis, infection, and stenosis. It can remain asymptomatic or present with back pain, radiculopathy, or cord compression. The lumbar spine is the most frequently affected site.[17, 18] Many patients with spinal gout have had chronic tophaceous gout with radiologic evidence of erosions in the peripheral joints.[15] Patients with spinal gout also have elevated urate levels and markers of inflammation.[18] Surgical decompression and stabilization is recommended when there is frank cord compression, progressive neurologic compromise, or lack of improvement with gout therapy alone.[18]
This patient's male gender, history of gout, hypertension, alcohol consumption, and thiazide diuretic use placed him at an increased risk of a gout attack.[19, 20] The possible interruption of urate‐lowering therapy for the surgical procedure and surgery itself further heightened his risk of suffering acute gouty arthritis in the perioperative period.[21] The patient's encephalopathy may have masked back pain and precluded an accurate neurologic exam. There is one case report to our knowledge describing encephalopathy that improved with colchicine and was possibly related to gout.[22] This patient's encephalopathy was deemed multifactorial and attributed to alcohol withdrawal, medications (including opioids and steroids), and infection (pneumonia).
Gout is best known for its peripheral arthritis and is rarely invoked in the consideration of spinal and myelopathic processes where more pressing competing diagnoses, such as infection and malignancy, are typically considered. In addition, when surgical specimens are submitted for examination for pathology in formaldehyde (rather than alcohol), monosodium urate crystals are dissolved and are thus difficult to identify in the specimen.
This case reminds us that gout remains a diagnostic challenge and should be considered in the differential of an inflammatory process. Recognition of the multifaceted nature of gout can allow for the earlier recognition and treatment of the less typical presentations of this ancient malady.
KEY TEACHING POINTS
- Crystalline disease is a common cause of postoperative arthritis.
- Gout (and pseudogout) should be considered in cases of focal inflammation (detected by examination or imaging) when the evidence or predisposition for infection is limited or nonexistent.
- Spinal gout presents with back pain, radiculopathy, or cord compression and may be confused with spinal metastasis, infection, and stenosis.
Acknowledgements
The authors thank Dr. Kari Waddell and Elaine Bammerlin for their assistance in the preparation of this manuscript.
Disclosure: Nothing to report.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.
- , . Clinical features and treatment of gout. In: Firestein GS, Budd RC, Gabriel SE, McInnes IB, O'Dell JR, eds. Kelley's Textbook of Rheumatology. Vol 2. 9th ed. Philadelphia, PA: Elsevier/Saunders; 2013:1544–1575.
- , , . Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–3141.
- , , , . Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31(8):1582–1587.
- , , , , . Purine‐rich foods, dairy and protein intake, and the risk of gout in men. New Engl J Med. 2004;350(11):1093–1103.
- , , . Fructose‐rich beverages and risk of gout in women. JAMA. 2010;304(20):2270–2278.
- , . Healthcare burden of in‐hospital gout. Intern Med J. 2012;42(11):1261–1263.
- , , , , , . Improved management of acute gout during hospitalization following introduction of a protocol. Int J Rheum Dis. 2012;15(6):512–520.
- , , . Postsurgical gout. Am Surg. 1995;61(1):56–59.
- , . Evolution of modern medicine. Arch Intern Med. 1960;105(4):640–644.
- . Clinical practice. Gout. N Engl J Med. 2011;364(5):443–452.
- . Management of gout: a 57‐year‐old man with a history of podagra, hyperuricemia, and mild renal insufficiency. JAMA. 2012;308(20):2133–2141.
- , , , et al. Diagnostic imaging of gout: comparison of high‐resolution US versus conventional X‐ray. Eur Radiol. 2008;18(3):621–630.
- , . The broad spectrum of urate crystal deposition: unusual presentations of gouty tophi. Semin Arthritis Rheum. 2012;42(2):146–154.
- , . Unusual clinical presentations of gout. Curr Opin Rheumatol. 2010;22(2):181–187.
- , , , , , . Correlates of axial gout: a cross‐sectional study. J Rheumatol. 2012;39(7):1445–1449.
- , , , , , . Axial gouty arthropathy. Am J Med Sci. 2009;338(2):140–146.
- , , . Axial (spinal) gout. Curr Rheumatol Rep. 2012;14(2):161–164.
- , , , . Spinal gout in a renal transplant patient: a case report and literature review. Surg Neurol. 2007;67(1):65–73.
- , , , et al. Alcohol consumption as a trigger of recurrent gout attacks. Am J Med. 2006;119(9):800.e11–800.e16.
- , , , , , . Recent diuretic use and the risk of recurrent gout attacks: the online case‐crossover gout study. J Rheumatol. 2006;33(7):1341–1345.
- , , , , . Clinical features and risk factors of postsurgical gout. Ann Rheum Dis. 2008;67(9):1271–1275.
- , , , . Gouty encephalopathy: myth or reality [in French]? Rev Med Interne. 1997;18(6):474–476.
Conservative Management of Pediatric Pleural Empyema Results in Good Long-Term Outcomes

Clinical question: What are the long-term outcomes of pediatric pleural empyema?
Background: Hospitalizations for complicated pneumonia have increased in recent years. In the U.S., early intervention—commonly video-assisted thorascopic surgery (VATS)—has become popular. Although short-term outcomes appear cost-effective with this approach, long-term comparative-effectiveness outcomes are not entirely clear.
Study design: Prospective observational study.
Setting: Tertiary-care children's hospital.
Synopsis: Over a two-year period, 82 patients were enrolled and available for at least one follow-up visit in a 12-month period. Chest drain was used in 62% of children; fibrinolytics were used in 78% of those cases. All patients received antibiotics. Six patients (7%) were readmitted in the first month, with three patients requiring a chest drain. At 12 months, four patients (5%) had mildly abnormal spirometric or radiographic abnormalities but were asymptomatic with normal quality-of-life scores.
This prospective observational study is notable for the relatively conservative approach (antibiotics alone or chest drainage, without VATS) employed in all subjects. The results provide a comprehensive summary of outcomes at 12 months in this population. Unfortunately, comparative-effectiveness data for VATS are not available in a generalizable form. Nevertheless, this single-center snapshot suggests that long-term outcomes are good with a conservative approach.
Given these findings, and the low likelihood that significant advantages of VATS will be demonstrated in the absence of a large multicenter trial, better understanding of parental preferences will become critical to making the right decision for each patient.
Bottom line: Conservative management of pediatric pleural empyema yields good long-term outcomes.
Citation: Cohen E, Mahant S, Dell SD, et al. The long-term outcomes of pediatric pleural empyema: a prospective study. Arch Pediatr Adolesc Med. 2012;166(11):999-1004.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What are the long-term outcomes of pediatric pleural empyema?
Background: Hospitalizations for complicated pneumonia have increased in recent years. In the U.S., early intervention—commonly video-assisted thorascopic surgery (VATS)—has become popular. Although short-term outcomes appear cost-effective with this approach, long-term comparative-effectiveness outcomes are not entirely clear.
Study design: Prospective observational study.
Setting: Tertiary-care children's hospital.
Synopsis: Over a two-year period, 82 patients were enrolled and available for at least one follow-up visit in a 12-month period. Chest drain was used in 62% of children; fibrinolytics were used in 78% of those cases. All patients received antibiotics. Six patients (7%) were readmitted in the first month, with three patients requiring a chest drain. At 12 months, four patients (5%) had mildly abnormal spirometric or radiographic abnormalities but were asymptomatic with normal quality-of-life scores.
This prospective observational study is notable for the relatively conservative approach (antibiotics alone or chest drainage, without VATS) employed in all subjects. The results provide a comprehensive summary of outcomes at 12 months in this population. Unfortunately, comparative-effectiveness data for VATS are not available in a generalizable form. Nevertheless, this single-center snapshot suggests that long-term outcomes are good with a conservative approach.
Given these findings, and the low likelihood that significant advantages of VATS will be demonstrated in the absence of a large multicenter trial, better understanding of parental preferences will become critical to making the right decision for each patient.
Bottom line: Conservative management of pediatric pleural empyema yields good long-term outcomes.
Citation: Cohen E, Mahant S, Dell SD, et al. The long-term outcomes of pediatric pleural empyema: a prospective study. Arch Pediatr Adolesc Med. 2012;166(11):999-1004.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What are the long-term outcomes of pediatric pleural empyema?
Background: Hospitalizations for complicated pneumonia have increased in recent years. In the U.S., early intervention—commonly video-assisted thorascopic surgery (VATS)—has become popular. Although short-term outcomes appear cost-effective with this approach, long-term comparative-effectiveness outcomes are not entirely clear.
Study design: Prospective observational study.
Setting: Tertiary-care children's hospital.
Synopsis: Over a two-year period, 82 patients were enrolled and available for at least one follow-up visit in a 12-month period. Chest drain was used in 62% of children; fibrinolytics were used in 78% of those cases. All patients received antibiotics. Six patients (7%) were readmitted in the first month, with three patients requiring a chest drain. At 12 months, four patients (5%) had mildly abnormal spirometric or radiographic abnormalities but were asymptomatic with normal quality-of-life scores.
This prospective observational study is notable for the relatively conservative approach (antibiotics alone or chest drainage, without VATS) employed in all subjects. The results provide a comprehensive summary of outcomes at 12 months in this population. Unfortunately, comparative-effectiveness data for VATS are not available in a generalizable form. Nevertheless, this single-center snapshot suggests that long-term outcomes are good with a conservative approach.
Given these findings, and the low likelihood that significant advantages of VATS will be demonstrated in the absence of a large multicenter trial, better understanding of parental preferences will become critical to making the right decision for each patient.
Bottom line: Conservative management of pediatric pleural empyema yields good long-term outcomes.
Citation: Cohen E, Mahant S, Dell SD, et al. The long-term outcomes of pediatric pleural empyema: a prospective study. Arch Pediatr Adolesc Med. 2012;166(11):999-1004.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
How Should a Patient with Cocaine-Associated Chest Pain be Treated?

Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
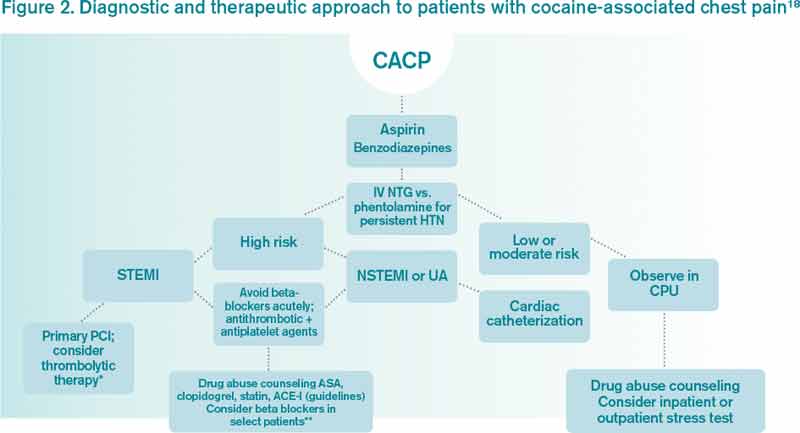
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.

Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
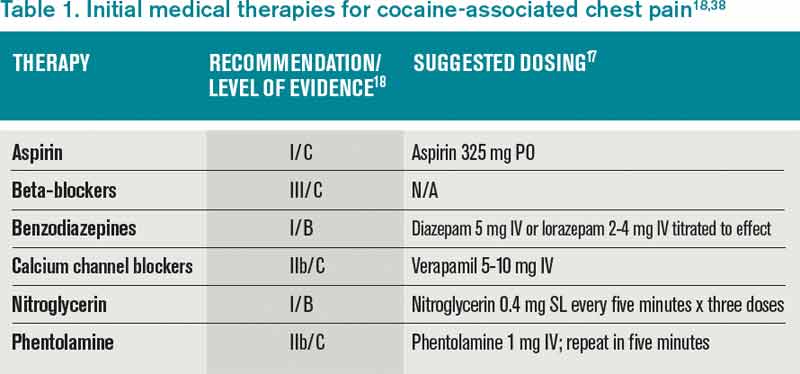
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.
Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
Case
A 38-year-old man with a history of tobacco use presents to the emergency department complaining of constant substernal chest pain for three hours. His temperature is 37.7°C, his heart rate is 110 beats per minute, and his blood pressure is 155/95 mmHg. He appears anxious and diaphoretic but examination is otherwise unremarkable. He admits to cocaine use one hour before the onset of symptoms. What are the appropriate treatments for his condition?
Overview
Cocaine is the second-most-commonly used illicit drug in the U.S. and represents 31% of all ED visits related to substance abuse.1,2 According to recent survey results, 2.1 million people report recent cocaine use, and 1.6 million engage in cocaine abuse or dependence.2 Acute cardiopulmonary complaints are common in individuals who present to the ED after cocaine use, with chest pain being the most frequently reported symptom in 40%.3
Numerous etiologies for cocaine-associated chest pain (CACP) have been discovered, including musculoskeletal pain, pulmonary hypertension, cardiomyopathy, arrhythmias, and endocarditis.4 Only 0.5% of patients with aortic dissection over a four-year period had a recent history of cocaine use, making cocaine a rare cause of a rare condition.5 Cardiac chest pain remains the most frequent underlying etiology, resulting in the most common complication of myocardial infarction (MI) in up to 6% of patients.6,7
The ways in which cocaine use can cause myocardial ischemia and MI are multifactorial. A vigorous central sympathomimetic effect, coronary artery vasoconstriction, stimulation of platelets, and enhanced atherosclerosis all lead to a myocardial oxygen supply-demand imbalance.8 Other key interactions in the cardiovascular system are displayed in Figure 1. Understanding the role of these mechanisms in CACP is crucial to patient care.
Clinician goals in the management of CACP are to rapidly and accurately exclude life-threatening etiologies; assess the need for urgent acute coronary syndrome (ACS) evaluation; risk-stratify patients and ensure appropriate disposition; normalize the toxic effects of cocaine; treat resultant organ damage; and prevent long-term complications. An algorithm detailing this approach is provided in Figure 2.
Review of the Data
Diagnostic evaluation. Given potential differences in treatment regimens, it is imperative to differentiate patients who present with CACP from those whose chest pain is not associated with cocaine either by direct questioning or by screening of urine for cocaine metabolites. Once the presence of cocaine has been confirmed, guideline-based evaluation for potential ACS with serial electrocardiograms (ECG), cardiac biomarkers, and close monitoring of cardiac rhythms and hemodynamics is largely similar to standard management of all patients presenting with chest pain, with a few caveats.
Interpretation of the ECG can be challenging in the setting of cocaine. Studies have shown “abnormal” ECGs in 56% to 84% of patients, with many representing early repolarization or left ventricular hypertrophy.9,10 Likewise, patients with MI are as likely to present with normal or nonspecific ECG findings as with ischemic findings.7,11 ECG interpretation to diagnose ischemia or infarction in patients with CACP yields a sensitivity of 36% and specificity of 90%.7
Creatine kinase (CK), CK-MB fraction, and myoglobin have low specificity for the diagnosis of ischemia, as cocaine can induce skeletal muscle injury and rhabdomyolysis.9,12 Cardiac troponins demonstrate a superior specificity compared to CK and CK-MB and are thus the preferred cardiac biomarkers in diagnosing cocaine-associated MI.12
Initial management and disposition. Patients at high risk for cardiovascular events are generally admitted to a monitored bed.13 Immediate reperfusion therapy with primary percutaneous coronary intervention is recommended in patients with ST-elevation MI (STEMI). Treatment with thrombolytic agents is associated with an increased risk of intracerebral hemorrhage and lacks documented efficacy in patients with CACP. Thrombolysis should therefore only be utilized if the diagnosis of STEMI is unequivocal and an experienced cardiac catheterization laboratory is unavailable.14,15
Patients with unstable angina (UA) or non-ST-elevation MI (NSTEMI) are at higher risk for further cardiac events in a similar manner to those with ACS unrelated to cocaine. These cases might benefit from early cardiac catheterization and revascularization.16 Because of the increased risk of stent thrombosis in cocaine-users, thought to be due to recidivism, a detailed risk-benefit analysis should be undertaken prior to the implantation of cardiac stents.
Other diagnostic tests, such as stress testing and myocardial imaging, have not shown significant accuracy in diagnosing MI in this setting; moreover, these patients are at low overall risk for cardiac events and mortality. Consequently, an extensive diagnostic evaluation might not be cost-effective.7,10,13,17 Patients who have CACP without MI have a very low frequency of delayed complications.3,17 As such, cost-effective evaluation strategies, such as nine- or 12-hour observation periods in a chest pain unit, are appropriate for many of these low- to moderate-risk patients.13 For all CACP patients, the most critical post-discharge interventions are cardiac risk modification and cocaine cessation.13
Normalizing the toxic effects of cocaine with medications.
Aspirin: While no specific study has been performed in patients with CACP and aspirin, CACP guidelines, based on data supporting ACS guidelines for all patients, recommend administration of full-dose aspirin given its associated reduction in morbidity and mortality.18,19 Furthermore, given the platelet-stimulating effects of cocaine, using aspirin in this setting seems very reasonable.
Benzodiazepines: CACP guidelines support the use of benzodiazepines early in management to indirectly combat the agitation, hypertension, and tachycardia resulting from the stimulatory effects of cocaine.18,20 These recommendations are based on several animal and human studies that demonstrate significant reduction in heart rate and systemic arterial pressure with the use of these agents.21,22
Nitroglycerin: Cardiac catheterization studies have shown reversal of vasoconstriction with administration of nitroglycerin. One study demonstrated a benefit of the drug in 49% of participants.23 Additional investigation into the benefit of benzodiazepine and nitroglycerin combination therapy revealed mixed results. In one study, lorazepam plus nitroglycerin was found to be more efficacious than nitroglycerin alone.24 In another, however, use of diazepam in combination with nitroglycerin did not show benefit when evaluating pain relief, cardiac dynamics, and left ventricular function.25
Phentolamine: Phentolamine administration has been studied much less in the literature. This nonselective alpha-adrenergic antagonist exerts a dose-dependent reversal of cocaine’s vasoconstrictive properties in monkeys and humans.26,27 International guidelines for Emergency Cardiovascular Care recommend its use in treatment of cocaine-associated ACS;27 however, the AHA recommends it less strongly.18
Calcium channel blockers: Calcium channel blockers (CCBs) have not shown promise as first-line agents. While catheterization studies demonstrate the vasodilatory properties of verapamil, larger studies looking at all-cause mortality conclude that CCBs might worsen mortality rates,28 and animal studies indicate an increased risk of seizures.29 At this time, CCBs are recommended only if cardiac symptoms continue after both benzodiazepines and nitroglycerin are administered.18
The beta-blocker controversy: The use of beta-blockers in patients with CACP remains controversial given the theoretical risk of unopposed alpha-adrenergic activation. Coronary vasospasm, decreased myocardial oxygen delivery, and increased systemic vascular resistance can result from their use.30
Propranolol, a nonselective beta-blocker, was shown in catheterization studies to potentiate the coronary vasoconstriction of cocaine.31 Labetalol, a combined alpha/beta-blocker, reduced mean arterial pressure after cocaine administration during cardiac catheterization but did not reverse coronary vasoconstriction.32 This was attributed to the predominating beta greater than alpha blockade at doses administered. The selective beta-1 antagonists esmolol and metoprolol have shown no benefit in CACP.33 Carvedilol, a combined alpha/beta-blocker with both peripheral and central nervous system activity, has potential to attenuate both physiologic and behavioral response to cocaine, but it has not been well studied in this patient subset.34
The 2005 ACC/AHA STEMI guidelines recommended against beta-blockers in the setting of STEMI precipitated by cocaine use due to the potential of exacerbating coronary vasoconstriction.35 The 2007 ACC/AHA UA/NSTEMI guidelines stated that the use of a combined alpha/beta-blocker in patients with cocaine-induced ACS may be reasonable for patients with hypertension or tachycardia if pre-treated with a vasodilator.19 The 2008 ACC/AHA guidelines on the management of cocaine-related chest pain and MI recommended against the use of beta-blockers in the acute setting given the low incidence of cocaine-related MI and death.18
In a more recent study, Dattilo et al showed that beta-blockers administered to patients admitted with positive urine toxicology for cocaine significantly reduced MI and in-hospital mortality. Reduction of MI was of borderline significance in those admitted with a chief complaint of chest pain.36 Limitations of this study include unknown time of cocaine ingestion, lack of follow-up on discharge mortality, and a small sample size of 348 patients lacking statistical power.
Another retrospective cohort study examined patients admitted with chest pain and urine toxicology positive for cocaine and found that beta-blocker administration during hospitalization was not associated with increased incident mortality. Further, after a mean follow-up of 2.5 years, there was a statistically significant decrease in cardiovascular death.37 Drawbacks of this study included an older patient population, greater proportion of coronary artery disease, and higher follow-up of cardiovascular mortality rates than in previous studies, suggesting this subset might have received greater benefit from beta-blockers as a result of these characteristics.
The 2008 ACC/AHA guidelines instruct individualized consideration of the risk/benefit ratio for beta-blocker use in patients with CACP given the high rate of recidivism in cocaine abusers. The strongest indication is given to those with documented MI, left ventricular systolic dysfunction, or ventricular arrhythmias.18
It is important to note that these recommendations are based on cardiac catheterization laboratory studies, case reports, retrospective analyses, and animal experiments. No prospective controlled trials evaluating the role of beta-blockers in CACP and MI exist, and no trials regarding therapies to improve outcomes of patients sustaining a cocaine-associated MI have been reported.18
Back to the Case
This patient was experiencing cocaine-associated chest pain, which was confirmed with positive urine toxicology. Initial diagnostic workup with basic laboratory studies and cardiac biomarkers showed mild elevation in CK with troponin levels within normal limits. His ECG showed changes consistent with left ventricular hypertrophy. Chest radiograph was unremarkable.
He received aspirin, benzodiazepines, and nitroglycerin with normalization of vital signs, as well as subjective improvement in chest pain and anxiety. He was deemed to be at low risk for potential cardiac complications; thus, further cardiac testing was not pursued. Rather, he was admitted to an overnight observation unit with telemetry monitoring, where his chest pain did not recur.
He was seen in consultation with social work staff who arranged for drug abuse counseling after discharge. Given the uncertainty of relapse to cocaine use, as well as lack of known cardiac risk factors, he was not discharged on any new medications.
Bottom Line
The treatment of CACP includes normalizing the toxic systemic effects of the drug and minimizing the direct ischemic damage to the myocardium. Management varies slightly from traditional chest pain algorithms and includes benzodiazepines as well as antiplatelet agents and vasodilators to achieve this goal. Initial therapy with beta-blockers remains undefined and is largely discouraged in the acute setting. The role of beta-blockade upon discharge, however, can be beneficial in specific populations, especially those found to have underlying coronary disease.
Dr. Houchens and Dr. Czarnik are clinical instructors and Dr. Mack is a clinical lecturer at the University of Michigan Health System in Ann Arbor.
References
- Hughes A, Sathe N, Spagnola K. State Estimates of Substance Use from the 2005-2006 National Surveys on Drug Use and Health. DHHS Publication No. SMA 08-4311, NSDUH Series H-33. Rockville, MD: Substance Abuse and Mental Health Services Administration, Office of Applied Studies; 2008.
- Volkow ND. Cocaine: Abuse and Addiction. National Institute on Drug Abuse. Washington, DC: U.S. Department of Health and Human Services; 2009.
- Brody SL, Slovis CM, Wrenn KD. Cocaine-related medical problems: consecutive series of 233 patients. Am J Med. 1990;88:325-331.
- Levis JT, Garmel GM. Cocaine-associated chest pain. Emerg Med Clin North Am. 2005;23:1083-1103.
- Eagle KA, Isselbacher EM, DeSanctis RW. Cocaine-related aortic dissection in perspective. Circulation. 2002;105:1529-1530.
- Feldman JA, Fish SS, Beshansky JR, Griffith JL, Woolard RH, Selker HP. Acute cardiac ischemia in patients with cocaine-associated complaints: results of a multicenter trial. Ann Emerg Med. 2000;36:469-476.
- Hollander JE, Hoffman RS, Gennis P, et al. Prospective multicenter evaluation of cocaine associated chest pain. Cocaine Associated Chest Pain (COCHPA) Study Group. Acad Emerg Med. 1994;1:330-339.
- Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation. 2010;122:2558-2569.
- Gitter MJ, Goldsmith SR, Dunbar DN, et al. Cocaine and chest pain: clinical features and outcomes of patients hospitalized to rule out myocardial infarction. Ann Intern Med. 1991;115:277-282.
- Amin M, Gabelman G, Karpel J, et al. Acute myocardial infarction and chest pain syndromes after cocaine use. Am J Cardiol. 1990;66:1434-1437.
- Tokarski GF, Paganussi P, Urbanski R, et al. An evaluation of cocaine-induced chest pain. Ann Emerg Med. 1990;19:1088-1092.
- Hollander JE, Levitt MA, Young GP, Briglia E, Wetli CV, Gawad Y. Effect of recent cocaine use on the specificity of cardiac markers for diagnosis of acute myocardial infarction. Am Heart J. 1998;135(2 Pt 1):245-252.
- Weber JE, Shofer FS, Larkin GL, Kalaria AS, Hollander JE. Validation of a brief observation period for patients with cocaine-associated chest pain. N Engl J Med. 2003;348:510-517.
- Hahn IH, Hoffman RS. Diagnosis and treatment of acute myocardial infarction: cocaine use and acute myocardial infarction. Emerg Med Clin North Am. 2001;19(2):1-18.
- Hoffman RS, Hollander JE. Evaluation of patients with chest pain after cocaine use. Crit Care Clin. 1997;13:809-828. Cannon CP, Weintraub WS, Demopoulos LA, et al.
- Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban. N Engl J Med. 2001;344:1879-1887.
- Hollander JE, Hoffman RS. Cocaine-induced myocardial infarction: an analysis and review of the literature. J Emerg Med. 1992;10:169-177.
- McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897-1907.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 guidelines for the management of patients with unstable angina/non-ST-Elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction) developed in collaboration with the American College of Emergency Physicians, the Society for Cardiovascular Angiography and Interventions, and the Society of Thoracic Surgeons endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation and the Society for Academic Emergency Medicine. J Am Coll Cardiol. 2007;50:E1-E157.
- Hollander JE. Management of cocaine-associated myocardial ischemia. N Engl J Med. 1995;333:1267-1272.
- Brubacher JR, Hoffman RS. Cocaine toxicity. Top Emerg Med. 1997;19(4):1-16.
- Catavas JD, Waters IW. Acute cocaine intoxication in the conscious dog: studies on the mechanism of lethality. J Pharmacol Exp Ther. 1981;217:350-356.
- Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32(3): 243-256.
- Honderick T, Williams D, Seaberg D, Wears R. A prospective, randomized, controlled trial of benzodiazepines and nitroglycerin or nitroglycerin alone in the treatment of cocaine-associated acute coronary syndromes. Am J Emerg Med. 2003;21(1):39-42.
- Baumann BM, Perrone J, Hornig SE, Shofer FS, Hollander JE. Randomized, double-blind, placebo-controlled trial of diazepam, nitroglycerin, or both for treatment of patients with potential cocaine-associated acute coronary syndromes. Acad Emerg Med. 2000;7:878-885.
- Schindler CW, Tella SR, Goldberg SR. Adrenoceptor mechanisms in the cardiovascular effects of cocaine in conscious squirrel monkeys. Life Sci. 1992;51(9):653-660.
- Lange RA, Cigarroa RG, Yancy CW Jr., et al. Cocaine-induced coronary-artery vasoconstriction. N Engl J Med. 1989;321(23):1557-1562.
- Furberg CD, Psaty BM, Meyer JV. Nifedipine. Dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326-1331.
- Derlet RW, Albertson TE. Potentiation of cocaine toxicity with calcium channel blockers. Am J Emerg Med. 1989;7:464-468.
- Lange RA, Hillis LD. Cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351-358.
- Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897-903.
- Boehrer JD, Moliterno DJ, Willard JE, Hillis LD, Lange RA. Influence of labetalol on cocaine-induced coronary vasoconstriction in humans. Am J Med. 1993;94:608-610.
- Sand IC, Brody SL, Wrenn KD, Slovis CM. Experience with esmolol for the treatment of cocaine-associated cardiovascular complications. Am J Emerg Med. 1991;9:161-163.
- Sofuoglo M, Brown S, Babb DA, Pentel PR, Hatsukami DK. Carvedilol affects the physiological and behavioral response to smoked cocaine in humans. Drug Alcohol Depend. 2000;60:69-76.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction: a report of the American College of Cardiology/American Heart Association Task Force of Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1-E211.
- Dattilo PB, Hailpern SM, Fearon K, Sohal D, Nordin C. Beta-blockers are associated with reduced risk of myocardial infarction after cocaine use. Ann Emerg Med. 2008;51:117-125.
- Rangel C, Shu RG, Lazar LD, Vittinghoff E, Hsue PY, Marcus GM. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med. 2010;170:874-879.
A Sheep in Wolf's Clothing
A 51‐year‐old man presented with severe pain and swelling in the lower anterior right thigh. He stated that the symptoms limited his movement, and began 4 days prior to this presentation. He rated the pain severity a 10 on a 10‐point scale. He denied fevers, chills, or history of trauma or weight loss.
Cellulitis of the lower extremity is the most likely possibility, but the presence of severe pain and swelling of an extremity in the absence of trauma should always make the clinician consider deep‐seated infections such as myositis or necrotizing fasciitis. An early clue for necrotizing fasciitis is severe pain that is disproportionate to the physical examination findings. Erythema, bullous lesions, or crepitus can develop later in the course. The absence of fever and chills also raises the possibility of noninfectious causes such as unrecognized trauma, deep vein thrombosis, or tumor.
The patient had a 15‐year history of type 2 diabetes complicated by end‐stage renal disease secondary to diabetic nephropathy for which he had been on hemodialysis for 5 months, proliferative diabetic retinopathy that rendered him legally blind, hypertension, and anemia. He stated that his diabetes had been poorly controlled, especially after he started dialysis.
A history of poorly controlled diabetes mellitus certainly increases the risk of the infectious disorders mentioned above. The patient's long‐standing history of diabetes mellitus with secondary nephropathy and retinopathy puts him at higher risk of atherosclerosis and vascular insufficiency, which consequently increase his risk for ischemic myonecrosis. Diabetic amyotrophy (diabetic lumbosacral plexopathy) is also a possibility, as it usually manifests with acute, unilateral, and focal tenderness followed by weakness involving a proximal leg. However, it typically occurs in patients who have been recently diagnosed with type 2 diabetes mellitus or whose disease has been under fairly good control and usually is associated with significant weight loss.
The patient was on oral medications for his diabetes until 1year before his presentation, at which point he was switched to insulin therapy. His other medications were amlodipine, lisinopril, aspirin, sevelamer, calcitriol, and calcium and iron supplements. He denied using alcohol, tobacco, or illicit drugs. He lives in Chicago and denies a recent travel history. His family history was significant for type 2 diabetes in multiple family members.
The absence of drugs, tobacco, and alcohol lowers the risk of some infectious and ischemic conditions. Patients with alcoholic liver disease who live in the southern United States are predisposed to developing Vibrio vulnificus myositis and fasciitis after ingesting contaminated oysters during the summer months. However, the clinical presentation of Vibrio usually includes septic shock and bullous lesions on the lower extremity. Also, the patient denies any recent travel to the southern United States, which makes Vibrio myositis and fasciitis less likely. Tobacco abuse increases the risk of atherosclerosis, peripheral vascular insufficiency, and ischemic myonecrosis.
The patient had a temperature of 99.1F, blood pressure of 139/85 mm Hg, pulse of 97 beats/minute, and respiratory rate of 18 breaths/minute. His body mass index was 31 kg/m2. Physical examination revealed a firm, warm, severely tender area of swelling in the inferomedial aspect of the right thigh. The knee was also swollen, and effusion could not be ruled out. The range of motion of the knee was markedly limited by pain. The skin overlying the swelling was erythematous but not broken. No crepitus was noted. The strength of the right lower extremity muscles could not be accurately assessed because of the patient's excruciating pain, but the patient was able to move his foot and toes against gravity. Sensation was absent in most of the tested points in the feet but was normal in the legs. The deep tendon reflexes in both ankles were normal. The pedal pulses were mildly decreased in both feet. He also had extremely decreased visual acuity, which has been chronic. The rest of the physical examination was unremarkable.
The absence of fever does not rule out a serious infection in a diabetic patient but does raise the possibility of a noninfectious cause. Also, over‐the‐counter acetaminophen or nonsteroidal anti‐inflammatory drugs could mask a fever. The patient's physical examination was significant for obesity, a risk factor for developing deep‐seated infections, and a firm and severely tender area of swelling near the right knee that limited range of motion. Septic arthritis of the knee is one possibility; arthrocentesis should be performed as soon as possible. The absence of crepitus, because it is a late physical examination finding, does not rule out myositis or necrotizing fasciitis. The presence of unilateral lower extremity swelling also raises the suspicion for a deep vein thrombosis, which warrants compression ultrasonography. The localized tenderness and the lack of dermatological manifestations, such as Gottron's papules, makes an inflammatory myositis such as dermatomyositis much less likely.
Laboratory studies demonstrated a hemoglobin A1C of 13.0% (reference range, 4.36.1%), fasting blood glucose level of 224 mg/dL (reference range, 7099 mg/dL), white blood cell count of 8300 cells/mm3 (reference range, 450011,000 cells/mm3) without band forms, erythrocyte sedimentation rate of 81 mm/hr (reference range, 14 mm/hr), and creatinine kinase level of 582 IU/L (reference range, 30200 IU/L). Routine chemistries were normal otherwise. An x‐ray of the right knee revealed soft tissue edema. The right knee was aspirated, and fluid analysis revealed a white blood cell count of 106 cells/mm3 (reference range, 200 cell/mm3). Compression ultrasonography of the right lower extremity did not reveal thrombosis.
Poor glycemic control, as evidenced by a high hemoglobin A1C level, is associated with a higher probability of infectious complications. An elevated sedimentation rate is compatible with an infection, and an increased creatinine kinase intensifies suspicion of myositis or myonecrosis. A normal white blood cell count decreases, but does not eliminate, the likelihood of a serious bacterial infection. The fluid analysis rules out septic arthritis, and the compression ultrasonography findings make deep vein thrombosis very unlikely. However, the differential diagnosis still includes myositis, clostridial myonecrosis, cellulitis, and necrotizing fasciitis. The patient should undergo magnetic resonance imaging (MRI) of the lower extremity, and a surgical consultation should be obtained to consider the possibility of surgical exploration.
Blood and the aspirated fluids were sent for culturing, and the patient was started on empiric antibiotics. MRI of his right thigh revealed extensive edema involving the vastus medialis and lateralis of the quadriceps as well as subcutaneous edema without fascial enhancement or gas (Figure 1).
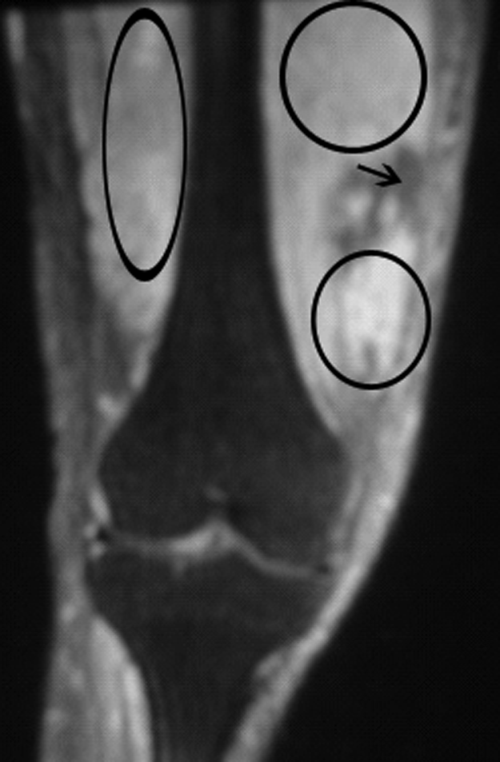
The absence of gas and fascial enhancement makes clostridial myonecrosis or necrotizing fasciitis less likely. The absence of a fluid collection in the muscle makes pyomyositis due to Staphylococci unlikely. Broad‐spectrum antibiotic coverage (usually vancomycin and either piperacillin/tazobactam or a carbapenem) targeting methicillin‐resistant Staphylococcus aureus, anaerobes, Streptococci, and Enterobacteriaceae should be empirically started as soon as cultures are obtained. Clindamycin should be part of the empiric antibiotic regimen to block toxin production in the event that Streptococcus pyogenes is responsible.
Surgical biopsy of the right vastus medialis muscle was performed, and tissue was sent for Gram staining, culture, and routine histopathological analysis. Gram staining was negative, and histopathological analysis revealed ischemic skeletal muscle fibers with areas of necrosis (Figure 2). Cultures from blood, fluid from the right knee, and muscular tissue samples did not grow any bacteria.
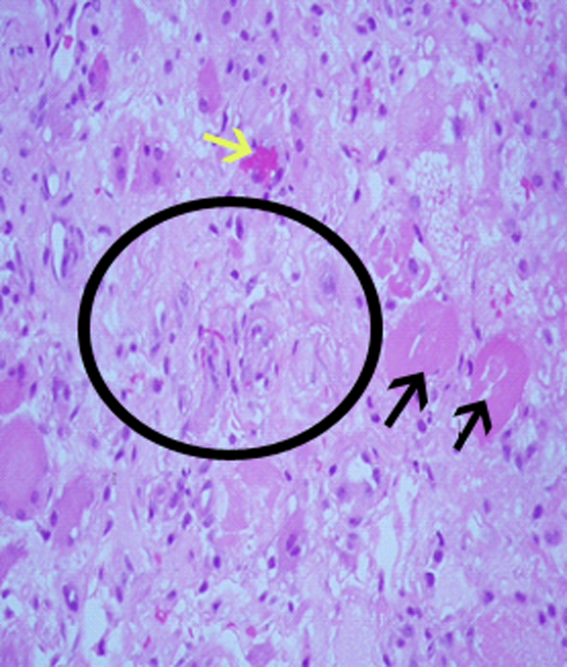
The muscle biopsy results are consistent with myonecrosis. Clostridial myonecrosis is possible but usually is associated with gas in tissues or occurs in the setting of intra‐abdominal pathology or severe trauma, and tissue culture was negative. Ischemic myonecrosis due to severe vascular insufficiency would be unlikely given the presence of pedal pulses and the absence of toes or forefoot cyanosis. A vasculitis syndrome is also unlikely because of the focal nature of the findings and the absence of weight loss, muscle weakness, and chronic joint pain in the patient's history. Calciphylaxis (calcific uremic arteriolopathy) might be considered in a patient with end‐stage renal disease who presents with a thigh pain; however, this condition is usually characterized by areas of ischemic necrosis that develop in the dermis and/or subcutaneous fat and infrequently involve muscles. The absence of the painful subcutaneous nodules typical of calciphylaxis makes it an unlikely diagnosis.
A diagnosis of diabetic myonecrosis was made. Antibiotics were discontinued, and the patient was treated symptomatically. His symptoms improved during the next few days. The patient was discharged from the hospital, and conservative management with bed rest and analgesics for 4 weeks was prescribed. Four months later, however, the patient returned with similar symptoms in the contralateral thigh. The patient was diagnosed with recurrent diabetic myonecrosis by MRI and muscle biopsy findings. Conservative management was advised, and the patient became pain‐free in a few weeks.
DISCUSSION
Diabetic myonecrosis (also known as diabetic muscle infarction) is a rare disorder initially described in 1965[1] that typically presents spontaneously as an acute, localized, severely painful swelling that limits the mobility of the affected extremity, usually without systemic signs of infection. It affects the thighs in 83% of patients and the calves in 17% of patients.[2, 3] Bilateral involvement, which is usually asynchronous, occurs in one‐third of patients.[4] The upper limbs are rarely involved. Diabetic myonecrosis affects patients who have a relatively longstanding history of diabetes. It is commonly associated with the microvascular complications of diabetes, including nephropathy (80% of patients), retinopathy (60% of patents), and/or neuropathy (64% of patients).[3, 5] The pathogenesis of diabetic myonecrosis is unclear, but the disease is likely due to a diffuse microangiopathy and atherosclerosis.[2, 5] Some authors have suggested that abnormalities in the clotting or fibrinolytic pathways play a role in the etiology of the disorder.[6]
Clinical and MRI findings can be used to make the diagnosis with reasonable certainty.[3, 5] Although both ultrasonography and MRI have been used to assess patients with diabetic myonecrosis, MRI with intravenous contrast enhancement appears to be the most useful diagnostic technique. It demonstrates extensive edema within the muscle(s), muscle enlargement, subcutaneous and interfascial edema, a patchwork pattern of involvement, and a high signal intensity on T2‐weighted images.[4, 7] Gadolinium enhancement may reveal an enhanced margin of the infarcted muscle with a central nonenhancing area of necrotic tissue.[4, 5] Muscle biopsy is not typically indicated because it may prolong recovery time and lead to infections.[8, 9, 10, 11] When performed, however, muscle biopsy reveals ischemic muscle fibers in different stages of degeneration and regeneration, with areas of necrosis and edema. Occlusion of arterioles and capillaries by fibrin could also be seen.[1] Although the patient underwent a muscle biopsy because infection could not be excluded definitively on clinical grounds, we believe that repeating the biopsy 4 months later was inappropriate.
Diabetic myonecrosis should be considered in a diabetic patient who presents with severe localized muscle pain and swelling of an extremity, especially if the clinical features favoring infection are absent. The differential diagnosis should include infection (eg, clostridial myonecrosis, myositis, cellulitis, abscess, necrotizing fasciitis, osteomyelitis), trauma (eg, hematoma, muscle rupture, myositis ossificans), peripheral neuropathy (particularly lumbosacral plexopathy), vascular disorders (deep vein thrombosis, and compartment syndrome), tumors, inflammatory muscle diseases, and drug‐related myositis.
No evidence‐based recommendations regarding the management of diabetic myonecrosis are available, although the findings of one retrospective analysis support conservative management with bed rest, leg elevation, and analgesics.[12] Physiotherapy may cause the condition to worsen,[13, 14] but routine daily activity, although often painful, is not harmful.[14] Some authors suggest a cautious use of antiplatelet or anti‐inflammatory medications.[12] We would also recommend achieving good glycemic control during the illness. Owing to the rarity of the disease, however, no studies have definitively shown that this hastens recovery or prevents recurrent diabetic myonecrosis. Surgery may prolong the recovery period; one study found that the recovery period of patients with diabetic myonecrosis who underwent surgery was longer than that of those who were treated conservatively (13 weeks vs 5.5 weeks).[12] Patients with diabetic myonecrosis have a good short‐term prognosis. Longer‐term, however, they have a poor prognosis; their recurrence rate is as high as 40%, and their 2‐year mortality rate is 10%, even after one episode of the disease. Death in these patients is mainly due to macrovascular events.[12]
TEACHING POINTS
- Diabetic myonecrosis is a rare complication of longstanding and poorly controlled diabetes. It usually presents with acute localized muscular pain in the lower extremities.
- Although a definitive diagnosis of diabetic myonecrosis is histopathologic, a clinical diagnosis can be made with reasonable certainty for patients with compatible MRI findings and no clinical or laboratory features suggesting infection.
- Conservative management with bed rest, analgesics, and antiplatelets is recommended. Surgery should be avoided, as it may prolong recovery.
Disclosure
Nothing to report.
- , . Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1:39–42.
- . Diabetic muscle infarction: an underdiagnosed complication of long‐standing diabetes. Diabetes Care. 2003;26:211–215.
- , , . Diabetic muscle infarction: case report and review. J Rheumatol. 2004;31:190–194.
- , , , , , . Muscle infarction in patients with diabetes mellitus: MR imaging findings. Radiology. 1999;211:241–247.
- , , , . Skeletal muscle infarction in diabetes mellitus. J Rheumatol. 2000;27:1063–1068.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 29–1997. A 54‐year‐old diabetic woman with pain and swelling of the leg. N Engl J Med. 1997;337:839–845.
- , , . Clinical and radiological aspects of idiopathic diabetic muscle infarction. Rational approach to diagnosis and treatment. J Bone Joint Surg Br. 1999;81:323–326.
- , , . Diabetic muscular infarction. Preventing morbidity by avoiding excisional biopsy. Arch Intern Med. 1997;157:1611.
- , . Case‐of‐the‐month: painful thigh mass in a young woman: diabetic muscle infarction. Muscle Nerve. 1992;15:850–855.
- . Diabetic muscle infarction: magnetic resonance imaging (MRI) avoids the need for biopsy. Muscle Nerve. 1995;18:129–130.
- , , , . Skeletal muscle infarction in diabetes: MR findings. J Comput Assist Tomogr. 1993;17:986–988.
- , . Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11:8–12.
- , , . Diabetic muscular infarction. Semin Arthritis Rheum. 1993;22:280–287.
- , . Focal infarction of muscle in diabetics. Diabetes Care. 1986;9:623–630.
A 51‐year‐old man presented with severe pain and swelling in the lower anterior right thigh. He stated that the symptoms limited his movement, and began 4 days prior to this presentation. He rated the pain severity a 10 on a 10‐point scale. He denied fevers, chills, or history of trauma or weight loss.
Cellulitis of the lower extremity is the most likely possibility, but the presence of severe pain and swelling of an extremity in the absence of trauma should always make the clinician consider deep‐seated infections such as myositis or necrotizing fasciitis. An early clue for necrotizing fasciitis is severe pain that is disproportionate to the physical examination findings. Erythema, bullous lesions, or crepitus can develop later in the course. The absence of fever and chills also raises the possibility of noninfectious causes such as unrecognized trauma, deep vein thrombosis, or tumor.
The patient had a 15‐year history of type 2 diabetes complicated by end‐stage renal disease secondary to diabetic nephropathy for which he had been on hemodialysis for 5 months, proliferative diabetic retinopathy that rendered him legally blind, hypertension, and anemia. He stated that his diabetes had been poorly controlled, especially after he started dialysis.
A history of poorly controlled diabetes mellitus certainly increases the risk of the infectious disorders mentioned above. The patient's long‐standing history of diabetes mellitus with secondary nephropathy and retinopathy puts him at higher risk of atherosclerosis and vascular insufficiency, which consequently increase his risk for ischemic myonecrosis. Diabetic amyotrophy (diabetic lumbosacral plexopathy) is also a possibility, as it usually manifests with acute, unilateral, and focal tenderness followed by weakness involving a proximal leg. However, it typically occurs in patients who have been recently diagnosed with type 2 diabetes mellitus or whose disease has been under fairly good control and usually is associated with significant weight loss.
The patient was on oral medications for his diabetes until 1year before his presentation, at which point he was switched to insulin therapy. His other medications were amlodipine, lisinopril, aspirin, sevelamer, calcitriol, and calcium and iron supplements. He denied using alcohol, tobacco, or illicit drugs. He lives in Chicago and denies a recent travel history. His family history was significant for type 2 diabetes in multiple family members.
The absence of drugs, tobacco, and alcohol lowers the risk of some infectious and ischemic conditions. Patients with alcoholic liver disease who live in the southern United States are predisposed to developing Vibrio vulnificus myositis and fasciitis after ingesting contaminated oysters during the summer months. However, the clinical presentation of Vibrio usually includes septic shock and bullous lesions on the lower extremity. Also, the patient denies any recent travel to the southern United States, which makes Vibrio myositis and fasciitis less likely. Tobacco abuse increases the risk of atherosclerosis, peripheral vascular insufficiency, and ischemic myonecrosis.
The patient had a temperature of 99.1F, blood pressure of 139/85 mm Hg, pulse of 97 beats/minute, and respiratory rate of 18 breaths/minute. His body mass index was 31 kg/m2. Physical examination revealed a firm, warm, severely tender area of swelling in the inferomedial aspect of the right thigh. The knee was also swollen, and effusion could not be ruled out. The range of motion of the knee was markedly limited by pain. The skin overlying the swelling was erythematous but not broken. No crepitus was noted. The strength of the right lower extremity muscles could not be accurately assessed because of the patient's excruciating pain, but the patient was able to move his foot and toes against gravity. Sensation was absent in most of the tested points in the feet but was normal in the legs. The deep tendon reflexes in both ankles were normal. The pedal pulses were mildly decreased in both feet. He also had extremely decreased visual acuity, which has been chronic. The rest of the physical examination was unremarkable.
The absence of fever does not rule out a serious infection in a diabetic patient but does raise the possibility of a noninfectious cause. Also, over‐the‐counter acetaminophen or nonsteroidal anti‐inflammatory drugs could mask a fever. The patient's physical examination was significant for obesity, a risk factor for developing deep‐seated infections, and a firm and severely tender area of swelling near the right knee that limited range of motion. Septic arthritis of the knee is one possibility; arthrocentesis should be performed as soon as possible. The absence of crepitus, because it is a late physical examination finding, does not rule out myositis or necrotizing fasciitis. The presence of unilateral lower extremity swelling also raises the suspicion for a deep vein thrombosis, which warrants compression ultrasonography. The localized tenderness and the lack of dermatological manifestations, such as Gottron's papules, makes an inflammatory myositis such as dermatomyositis much less likely.
Laboratory studies demonstrated a hemoglobin A1C of 13.0% (reference range, 4.36.1%), fasting blood glucose level of 224 mg/dL (reference range, 7099 mg/dL), white blood cell count of 8300 cells/mm3 (reference range, 450011,000 cells/mm3) without band forms, erythrocyte sedimentation rate of 81 mm/hr (reference range, 14 mm/hr), and creatinine kinase level of 582 IU/L (reference range, 30200 IU/L). Routine chemistries were normal otherwise. An x‐ray of the right knee revealed soft tissue edema. The right knee was aspirated, and fluid analysis revealed a white blood cell count of 106 cells/mm3 (reference range, 200 cell/mm3). Compression ultrasonography of the right lower extremity did not reveal thrombosis.
Poor glycemic control, as evidenced by a high hemoglobin A1C level, is associated with a higher probability of infectious complications. An elevated sedimentation rate is compatible with an infection, and an increased creatinine kinase intensifies suspicion of myositis or myonecrosis. A normal white blood cell count decreases, but does not eliminate, the likelihood of a serious bacterial infection. The fluid analysis rules out septic arthritis, and the compression ultrasonography findings make deep vein thrombosis very unlikely. However, the differential diagnosis still includes myositis, clostridial myonecrosis, cellulitis, and necrotizing fasciitis. The patient should undergo magnetic resonance imaging (MRI) of the lower extremity, and a surgical consultation should be obtained to consider the possibility of surgical exploration.
Blood and the aspirated fluids were sent for culturing, and the patient was started on empiric antibiotics. MRI of his right thigh revealed extensive edema involving the vastus medialis and lateralis of the quadriceps as well as subcutaneous edema without fascial enhancement or gas (Figure 1).
The absence of gas and fascial enhancement makes clostridial myonecrosis or necrotizing fasciitis less likely. The absence of a fluid collection in the muscle makes pyomyositis due to Staphylococci unlikely. Broad‐spectrum antibiotic coverage (usually vancomycin and either piperacillin/tazobactam or a carbapenem) targeting methicillin‐resistant Staphylococcus aureus, anaerobes, Streptococci, and Enterobacteriaceae should be empirically started as soon as cultures are obtained. Clindamycin should be part of the empiric antibiotic regimen to block toxin production in the event that Streptococcus pyogenes is responsible.
Surgical biopsy of the right vastus medialis muscle was performed, and tissue was sent for Gram staining, culture, and routine histopathological analysis. Gram staining was negative, and histopathological analysis revealed ischemic skeletal muscle fibers with areas of necrosis (Figure 2). Cultures from blood, fluid from the right knee, and muscular tissue samples did not grow any bacteria.
The muscle biopsy results are consistent with myonecrosis. Clostridial myonecrosis is possible but usually is associated with gas in tissues or occurs in the setting of intra‐abdominal pathology or severe trauma, and tissue culture was negative. Ischemic myonecrosis due to severe vascular insufficiency would be unlikely given the presence of pedal pulses and the absence of toes or forefoot cyanosis. A vasculitis syndrome is also unlikely because of the focal nature of the findings and the absence of weight loss, muscle weakness, and chronic joint pain in the patient's history. Calciphylaxis (calcific uremic arteriolopathy) might be considered in a patient with end‐stage renal disease who presents with a thigh pain; however, this condition is usually characterized by areas of ischemic necrosis that develop in the dermis and/or subcutaneous fat and infrequently involve muscles. The absence of the painful subcutaneous nodules typical of calciphylaxis makes it an unlikely diagnosis.
A diagnosis of diabetic myonecrosis was made. Antibiotics were discontinued, and the patient was treated symptomatically. His symptoms improved during the next few days. The patient was discharged from the hospital, and conservative management with bed rest and analgesics for 4 weeks was prescribed. Four months later, however, the patient returned with similar symptoms in the contralateral thigh. The patient was diagnosed with recurrent diabetic myonecrosis by MRI and muscle biopsy findings. Conservative management was advised, and the patient became pain‐free in a few weeks.
DISCUSSION
Diabetic myonecrosis (also known as diabetic muscle infarction) is a rare disorder initially described in 1965[1] that typically presents spontaneously as an acute, localized, severely painful swelling that limits the mobility of the affected extremity, usually without systemic signs of infection. It affects the thighs in 83% of patients and the calves in 17% of patients.[2, 3] Bilateral involvement, which is usually asynchronous, occurs in one‐third of patients.[4] The upper limbs are rarely involved. Diabetic myonecrosis affects patients who have a relatively longstanding history of diabetes. It is commonly associated with the microvascular complications of diabetes, including nephropathy (80% of patients), retinopathy (60% of patents), and/or neuropathy (64% of patients).[3, 5] The pathogenesis of diabetic myonecrosis is unclear, but the disease is likely due to a diffuse microangiopathy and atherosclerosis.[2, 5] Some authors have suggested that abnormalities in the clotting or fibrinolytic pathways play a role in the etiology of the disorder.[6]
Clinical and MRI findings can be used to make the diagnosis with reasonable certainty.[3, 5] Although both ultrasonography and MRI have been used to assess patients with diabetic myonecrosis, MRI with intravenous contrast enhancement appears to be the most useful diagnostic technique. It demonstrates extensive edema within the muscle(s), muscle enlargement, subcutaneous and interfascial edema, a patchwork pattern of involvement, and a high signal intensity on T2‐weighted images.[4, 7] Gadolinium enhancement may reveal an enhanced margin of the infarcted muscle with a central nonenhancing area of necrotic tissue.[4, 5] Muscle biopsy is not typically indicated because it may prolong recovery time and lead to infections.[8, 9, 10, 11] When performed, however, muscle biopsy reveals ischemic muscle fibers in different stages of degeneration and regeneration, with areas of necrosis and edema. Occlusion of arterioles and capillaries by fibrin could also be seen.[1] Although the patient underwent a muscle biopsy because infection could not be excluded definitively on clinical grounds, we believe that repeating the biopsy 4 months later was inappropriate.
Diabetic myonecrosis should be considered in a diabetic patient who presents with severe localized muscle pain and swelling of an extremity, especially if the clinical features favoring infection are absent. The differential diagnosis should include infection (eg, clostridial myonecrosis, myositis, cellulitis, abscess, necrotizing fasciitis, osteomyelitis), trauma (eg, hematoma, muscle rupture, myositis ossificans), peripheral neuropathy (particularly lumbosacral plexopathy), vascular disorders (deep vein thrombosis, and compartment syndrome), tumors, inflammatory muscle diseases, and drug‐related myositis.
No evidence‐based recommendations regarding the management of diabetic myonecrosis are available, although the findings of one retrospective analysis support conservative management with bed rest, leg elevation, and analgesics.[12] Physiotherapy may cause the condition to worsen,[13, 14] but routine daily activity, although often painful, is not harmful.[14] Some authors suggest a cautious use of antiplatelet or anti‐inflammatory medications.[12] We would also recommend achieving good glycemic control during the illness. Owing to the rarity of the disease, however, no studies have definitively shown that this hastens recovery or prevents recurrent diabetic myonecrosis. Surgery may prolong the recovery period; one study found that the recovery period of patients with diabetic myonecrosis who underwent surgery was longer than that of those who were treated conservatively (13 weeks vs 5.5 weeks).[12] Patients with diabetic myonecrosis have a good short‐term prognosis. Longer‐term, however, they have a poor prognosis; their recurrence rate is as high as 40%, and their 2‐year mortality rate is 10%, even after one episode of the disease. Death in these patients is mainly due to macrovascular events.[12]
TEACHING POINTS
- Diabetic myonecrosis is a rare complication of longstanding and poorly controlled diabetes. It usually presents with acute localized muscular pain in the lower extremities.
- Although a definitive diagnosis of diabetic myonecrosis is histopathologic, a clinical diagnosis can be made with reasonable certainty for patients with compatible MRI findings and no clinical or laboratory features suggesting infection.
- Conservative management with bed rest, analgesics, and antiplatelets is recommended. Surgery should be avoided, as it may prolong recovery.
Disclosure
Nothing to report.
A 51‐year‐old man presented with severe pain and swelling in the lower anterior right thigh. He stated that the symptoms limited his movement, and began 4 days prior to this presentation. He rated the pain severity a 10 on a 10‐point scale. He denied fevers, chills, or history of trauma or weight loss.
Cellulitis of the lower extremity is the most likely possibility, but the presence of severe pain and swelling of an extremity in the absence of trauma should always make the clinician consider deep‐seated infections such as myositis or necrotizing fasciitis. An early clue for necrotizing fasciitis is severe pain that is disproportionate to the physical examination findings. Erythema, bullous lesions, or crepitus can develop later in the course. The absence of fever and chills also raises the possibility of noninfectious causes such as unrecognized trauma, deep vein thrombosis, or tumor.
The patient had a 15‐year history of type 2 diabetes complicated by end‐stage renal disease secondary to diabetic nephropathy for which he had been on hemodialysis for 5 months, proliferative diabetic retinopathy that rendered him legally blind, hypertension, and anemia. He stated that his diabetes had been poorly controlled, especially after he started dialysis.
A history of poorly controlled diabetes mellitus certainly increases the risk of the infectious disorders mentioned above. The patient's long‐standing history of diabetes mellitus with secondary nephropathy and retinopathy puts him at higher risk of atherosclerosis and vascular insufficiency, which consequently increase his risk for ischemic myonecrosis. Diabetic amyotrophy (diabetic lumbosacral plexopathy) is also a possibility, as it usually manifests with acute, unilateral, and focal tenderness followed by weakness involving a proximal leg. However, it typically occurs in patients who have been recently diagnosed with type 2 diabetes mellitus or whose disease has been under fairly good control and usually is associated with significant weight loss.
The patient was on oral medications for his diabetes until 1year before his presentation, at which point he was switched to insulin therapy. His other medications were amlodipine, lisinopril, aspirin, sevelamer, calcitriol, and calcium and iron supplements. He denied using alcohol, tobacco, or illicit drugs. He lives in Chicago and denies a recent travel history. His family history was significant for type 2 diabetes in multiple family members.
The absence of drugs, tobacco, and alcohol lowers the risk of some infectious and ischemic conditions. Patients with alcoholic liver disease who live in the southern United States are predisposed to developing Vibrio vulnificus myositis and fasciitis after ingesting contaminated oysters during the summer months. However, the clinical presentation of Vibrio usually includes septic shock and bullous lesions on the lower extremity. Also, the patient denies any recent travel to the southern United States, which makes Vibrio myositis and fasciitis less likely. Tobacco abuse increases the risk of atherosclerosis, peripheral vascular insufficiency, and ischemic myonecrosis.
The patient had a temperature of 99.1F, blood pressure of 139/85 mm Hg, pulse of 97 beats/minute, and respiratory rate of 18 breaths/minute. His body mass index was 31 kg/m2. Physical examination revealed a firm, warm, severely tender area of swelling in the inferomedial aspect of the right thigh. The knee was also swollen, and effusion could not be ruled out. The range of motion of the knee was markedly limited by pain. The skin overlying the swelling was erythematous but not broken. No crepitus was noted. The strength of the right lower extremity muscles could not be accurately assessed because of the patient's excruciating pain, but the patient was able to move his foot and toes against gravity. Sensation was absent in most of the tested points in the feet but was normal in the legs. The deep tendon reflexes in both ankles were normal. The pedal pulses were mildly decreased in both feet. He also had extremely decreased visual acuity, which has been chronic. The rest of the physical examination was unremarkable.
The absence of fever does not rule out a serious infection in a diabetic patient but does raise the possibility of a noninfectious cause. Also, over‐the‐counter acetaminophen or nonsteroidal anti‐inflammatory drugs could mask a fever. The patient's physical examination was significant for obesity, a risk factor for developing deep‐seated infections, and a firm and severely tender area of swelling near the right knee that limited range of motion. Septic arthritis of the knee is one possibility; arthrocentesis should be performed as soon as possible. The absence of crepitus, because it is a late physical examination finding, does not rule out myositis or necrotizing fasciitis. The presence of unilateral lower extremity swelling also raises the suspicion for a deep vein thrombosis, which warrants compression ultrasonography. The localized tenderness and the lack of dermatological manifestations, such as Gottron's papules, makes an inflammatory myositis such as dermatomyositis much less likely.
Laboratory studies demonstrated a hemoglobin A1C of 13.0% (reference range, 4.36.1%), fasting blood glucose level of 224 mg/dL (reference range, 7099 mg/dL), white blood cell count of 8300 cells/mm3 (reference range, 450011,000 cells/mm3) without band forms, erythrocyte sedimentation rate of 81 mm/hr (reference range, 14 mm/hr), and creatinine kinase level of 582 IU/L (reference range, 30200 IU/L). Routine chemistries were normal otherwise. An x‐ray of the right knee revealed soft tissue edema. The right knee was aspirated, and fluid analysis revealed a white blood cell count of 106 cells/mm3 (reference range, 200 cell/mm3). Compression ultrasonography of the right lower extremity did not reveal thrombosis.
Poor glycemic control, as evidenced by a high hemoglobin A1C level, is associated with a higher probability of infectious complications. An elevated sedimentation rate is compatible with an infection, and an increased creatinine kinase intensifies suspicion of myositis or myonecrosis. A normal white blood cell count decreases, but does not eliminate, the likelihood of a serious bacterial infection. The fluid analysis rules out septic arthritis, and the compression ultrasonography findings make deep vein thrombosis very unlikely. However, the differential diagnosis still includes myositis, clostridial myonecrosis, cellulitis, and necrotizing fasciitis. The patient should undergo magnetic resonance imaging (MRI) of the lower extremity, and a surgical consultation should be obtained to consider the possibility of surgical exploration.
Blood and the aspirated fluids were sent for culturing, and the patient was started on empiric antibiotics. MRI of his right thigh revealed extensive edema involving the vastus medialis and lateralis of the quadriceps as well as subcutaneous edema without fascial enhancement or gas (Figure 1).
The absence of gas and fascial enhancement makes clostridial myonecrosis or necrotizing fasciitis less likely. The absence of a fluid collection in the muscle makes pyomyositis due to Staphylococci unlikely. Broad‐spectrum antibiotic coverage (usually vancomycin and either piperacillin/tazobactam or a carbapenem) targeting methicillin‐resistant Staphylococcus aureus, anaerobes, Streptococci, and Enterobacteriaceae should be empirically started as soon as cultures are obtained. Clindamycin should be part of the empiric antibiotic regimen to block toxin production in the event that Streptococcus pyogenes is responsible.
Surgical biopsy of the right vastus medialis muscle was performed, and tissue was sent for Gram staining, culture, and routine histopathological analysis. Gram staining was negative, and histopathological analysis revealed ischemic skeletal muscle fibers with areas of necrosis (Figure 2). Cultures from blood, fluid from the right knee, and muscular tissue samples did not grow any bacteria.
The muscle biopsy results are consistent with myonecrosis. Clostridial myonecrosis is possible but usually is associated with gas in tissues or occurs in the setting of intra‐abdominal pathology or severe trauma, and tissue culture was negative. Ischemic myonecrosis due to severe vascular insufficiency would be unlikely given the presence of pedal pulses and the absence of toes or forefoot cyanosis. A vasculitis syndrome is also unlikely because of the focal nature of the findings and the absence of weight loss, muscle weakness, and chronic joint pain in the patient's history. Calciphylaxis (calcific uremic arteriolopathy) might be considered in a patient with end‐stage renal disease who presents with a thigh pain; however, this condition is usually characterized by areas of ischemic necrosis that develop in the dermis and/or subcutaneous fat and infrequently involve muscles. The absence of the painful subcutaneous nodules typical of calciphylaxis makes it an unlikely diagnosis.
A diagnosis of diabetic myonecrosis was made. Antibiotics were discontinued, and the patient was treated symptomatically. His symptoms improved during the next few days. The patient was discharged from the hospital, and conservative management with bed rest and analgesics for 4 weeks was prescribed. Four months later, however, the patient returned with similar symptoms in the contralateral thigh. The patient was diagnosed with recurrent diabetic myonecrosis by MRI and muscle biopsy findings. Conservative management was advised, and the patient became pain‐free in a few weeks.
DISCUSSION
Diabetic myonecrosis (also known as diabetic muscle infarction) is a rare disorder initially described in 1965[1] that typically presents spontaneously as an acute, localized, severely painful swelling that limits the mobility of the affected extremity, usually without systemic signs of infection. It affects the thighs in 83% of patients and the calves in 17% of patients.[2, 3] Bilateral involvement, which is usually asynchronous, occurs in one‐third of patients.[4] The upper limbs are rarely involved. Diabetic myonecrosis affects patients who have a relatively longstanding history of diabetes. It is commonly associated with the microvascular complications of diabetes, including nephropathy (80% of patients), retinopathy (60% of patents), and/or neuropathy (64% of patients).[3, 5] The pathogenesis of diabetic myonecrosis is unclear, but the disease is likely due to a diffuse microangiopathy and atherosclerosis.[2, 5] Some authors have suggested that abnormalities in the clotting or fibrinolytic pathways play a role in the etiology of the disorder.[6]
Clinical and MRI findings can be used to make the diagnosis with reasonable certainty.[3, 5] Although both ultrasonography and MRI have been used to assess patients with diabetic myonecrosis, MRI with intravenous contrast enhancement appears to be the most useful diagnostic technique. It demonstrates extensive edema within the muscle(s), muscle enlargement, subcutaneous and interfascial edema, a patchwork pattern of involvement, and a high signal intensity on T2‐weighted images.[4, 7] Gadolinium enhancement may reveal an enhanced margin of the infarcted muscle with a central nonenhancing area of necrotic tissue.[4, 5] Muscle biopsy is not typically indicated because it may prolong recovery time and lead to infections.[8, 9, 10, 11] When performed, however, muscle biopsy reveals ischemic muscle fibers in different stages of degeneration and regeneration, with areas of necrosis and edema. Occlusion of arterioles and capillaries by fibrin could also be seen.[1] Although the patient underwent a muscle biopsy because infection could not be excluded definitively on clinical grounds, we believe that repeating the biopsy 4 months later was inappropriate.
Diabetic myonecrosis should be considered in a diabetic patient who presents with severe localized muscle pain and swelling of an extremity, especially if the clinical features favoring infection are absent. The differential diagnosis should include infection (eg, clostridial myonecrosis, myositis, cellulitis, abscess, necrotizing fasciitis, osteomyelitis), trauma (eg, hematoma, muscle rupture, myositis ossificans), peripheral neuropathy (particularly lumbosacral plexopathy), vascular disorders (deep vein thrombosis, and compartment syndrome), tumors, inflammatory muscle diseases, and drug‐related myositis.
No evidence‐based recommendations regarding the management of diabetic myonecrosis are available, although the findings of one retrospective analysis support conservative management with bed rest, leg elevation, and analgesics.[12] Physiotherapy may cause the condition to worsen,[13, 14] but routine daily activity, although often painful, is not harmful.[14] Some authors suggest a cautious use of antiplatelet or anti‐inflammatory medications.[12] We would also recommend achieving good glycemic control during the illness. Owing to the rarity of the disease, however, no studies have definitively shown that this hastens recovery or prevents recurrent diabetic myonecrosis. Surgery may prolong the recovery period; one study found that the recovery period of patients with diabetic myonecrosis who underwent surgery was longer than that of those who were treated conservatively (13 weeks vs 5.5 weeks).[12] Patients with diabetic myonecrosis have a good short‐term prognosis. Longer‐term, however, they have a poor prognosis; their recurrence rate is as high as 40%, and their 2‐year mortality rate is 10%, even after one episode of the disease. Death in these patients is mainly due to macrovascular events.[12]
TEACHING POINTS
- Diabetic myonecrosis is a rare complication of longstanding and poorly controlled diabetes. It usually presents with acute localized muscular pain in the lower extremities.
- Although a definitive diagnosis of diabetic myonecrosis is histopathologic, a clinical diagnosis can be made with reasonable certainty for patients with compatible MRI findings and no clinical or laboratory features suggesting infection.
- Conservative management with bed rest, analgesics, and antiplatelets is recommended. Surgery should be avoided, as it may prolong recovery.
Disclosure
Nothing to report.
- , . Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1:39–42.
- . Diabetic muscle infarction: an underdiagnosed complication of long‐standing diabetes. Diabetes Care. 2003;26:211–215.
- , , . Diabetic muscle infarction: case report and review. J Rheumatol. 2004;31:190–194.
- , , , , , . Muscle infarction in patients with diabetes mellitus: MR imaging findings. Radiology. 1999;211:241–247.
- , , , . Skeletal muscle infarction in diabetes mellitus. J Rheumatol. 2000;27:1063–1068.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 29–1997. A 54‐year‐old diabetic woman with pain and swelling of the leg. N Engl J Med. 1997;337:839–845.
- , , . Clinical and radiological aspects of idiopathic diabetic muscle infarction. Rational approach to diagnosis and treatment. J Bone Joint Surg Br. 1999;81:323–326.
- , , . Diabetic muscular infarction. Preventing morbidity by avoiding excisional biopsy. Arch Intern Med. 1997;157:1611.
- , . Case‐of‐the‐month: painful thigh mass in a young woman: diabetic muscle infarction. Muscle Nerve. 1992;15:850–855.
- . Diabetic muscle infarction: magnetic resonance imaging (MRI) avoids the need for biopsy. Muscle Nerve. 1995;18:129–130.
- , , , . Skeletal muscle infarction in diabetes: MR findings. J Comput Assist Tomogr. 1993;17:986–988.
- , . Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11:8–12.
- , , . Diabetic muscular infarction. Semin Arthritis Rheum. 1993;22:280–287.
- , . Focal infarction of muscle in diabetics. Diabetes Care. 1986;9:623–630.
- , . Tumoriform focal muscular degeneration in two diabetic patients. Diabetologia. 1965;1:39–42.
- . Diabetic muscle infarction: an underdiagnosed complication of long‐standing diabetes. Diabetes Care. 2003;26:211–215.
- , , . Diabetic muscle infarction: case report and review. J Rheumatol. 2004;31:190–194.
- , , , , , . Muscle infarction in patients with diabetes mellitus: MR imaging findings. Radiology. 1999;211:241–247.
- , , , . Skeletal muscle infarction in diabetes mellitus. J Rheumatol. 2000;27:1063–1068.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 29–1997. A 54‐year‐old diabetic woman with pain and swelling of the leg. N Engl J Med. 1997;337:839–845.
- , , . Clinical and radiological aspects of idiopathic diabetic muscle infarction. Rational approach to diagnosis and treatment. J Bone Joint Surg Br. 1999;81:323–326.
- , , . Diabetic muscular infarction. Preventing morbidity by avoiding excisional biopsy. Arch Intern Med. 1997;157:1611.
- , . Case‐of‐the‐month: painful thigh mass in a young woman: diabetic muscle infarction. Muscle Nerve. 1992;15:850–855.
- . Diabetic muscle infarction: magnetic resonance imaging (MRI) avoids the need for biopsy. Muscle Nerve. 1995;18:129–130.
- , , , . Skeletal muscle infarction in diabetes: MR findings. J Comput Assist Tomogr. 1993;17:986–988.
- , . Treatment and outcomes of diabetic muscle infarction. J Clin Rheumatol. 2005;11:8–12.
- , , . Diabetic muscular infarction. Semin Arthritis Rheum. 1993;22:280–287.
- , . Focal infarction of muscle in diabetics. Diabetes Care. 1986;9:623–630.
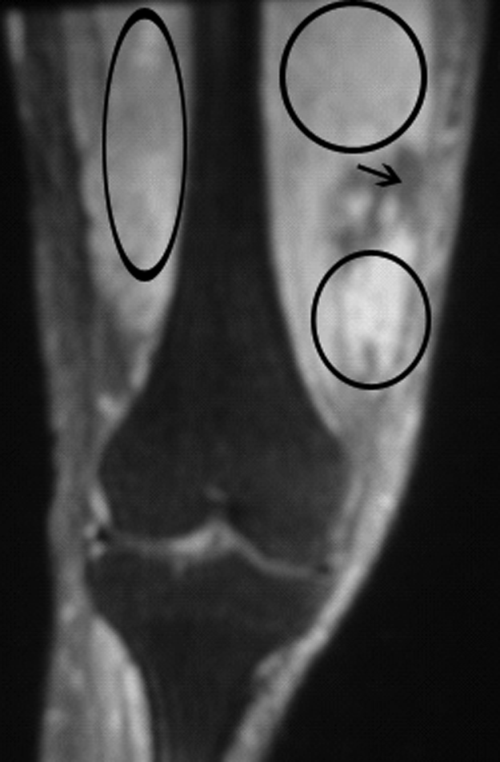
Hospitalwide Reductions in Pediatric Patient Harm are Achievable

Clinical question: Can a broadly constructed improvement initiative significantly reduce serious safety events (SSEs)?
Study design: Single-institution quality-improvement initiative.
Setting: Cincinnati Children’s Hospital Medical Center.
Synopsis: A multidisciplinary team supported by leadership was formed to reduce SSEs across the hospital by 80% within four years. A consulting firm with expertise in the field was also engaged for this process. Multifaceted interventions were clustered according to perceived key drivers of change in the institution: error prevention systems, improved safety governance, cause analysis programs, lessons-learned programs, and specific tactical interventions.
SSEs per 10,000 adjusted patient-days decreased significantly, to a mean of 0.3 from 0.9 (P<0.0001) after implementation, while days between SSEs increased to a mean of 55.2 from 19.4 (P<0.0001).
This work represents one of the most robust single-center approaches to improving patient safety that has been published to date. The authors attribute much of their success to culture change, which required “relentless clarity of vision by the organization.” Although this substantially limits immediate generalizability of any of the specific interventions, the work stands on its own as a prime example of what may be accomplished through focused dedication to reducing patient harm.
Bottom line: Patient harm is preventable through a widespread and multifaceted institutional initiative.
Citation: Muething SE, Goudie A, Schoettker PJ, et al. Quality improvement initiative to reduce serious safety events and improve patient safety culture. Pediatrics. 2012;130:e423-431.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Can a broadly constructed improvement initiative significantly reduce serious safety events (SSEs)?
Study design: Single-institution quality-improvement initiative.
Setting: Cincinnati Children’s Hospital Medical Center.
Synopsis: A multidisciplinary team supported by leadership was formed to reduce SSEs across the hospital by 80% within four years. A consulting firm with expertise in the field was also engaged for this process. Multifaceted interventions were clustered according to perceived key drivers of change in the institution: error prevention systems, improved safety governance, cause analysis programs, lessons-learned programs, and specific tactical interventions.
SSEs per 10,000 adjusted patient-days decreased significantly, to a mean of 0.3 from 0.9 (P<0.0001) after implementation, while days between SSEs increased to a mean of 55.2 from 19.4 (P<0.0001).
This work represents one of the most robust single-center approaches to improving patient safety that has been published to date. The authors attribute much of their success to culture change, which required “relentless clarity of vision by the organization.” Although this substantially limits immediate generalizability of any of the specific interventions, the work stands on its own as a prime example of what may be accomplished through focused dedication to reducing patient harm.
Bottom line: Patient harm is preventable through a widespread and multifaceted institutional initiative.
Citation: Muething SE, Goudie A, Schoettker PJ, et al. Quality improvement initiative to reduce serious safety events and improve patient safety culture. Pediatrics. 2012;130:e423-431.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Can a broadly constructed improvement initiative significantly reduce serious safety events (SSEs)?
Study design: Single-institution quality-improvement initiative.
Setting: Cincinnati Children’s Hospital Medical Center.
Synopsis: A multidisciplinary team supported by leadership was formed to reduce SSEs across the hospital by 80% within four years. A consulting firm with expertise in the field was also engaged for this process. Multifaceted interventions were clustered according to perceived key drivers of change in the institution: error prevention systems, improved safety governance, cause analysis programs, lessons-learned programs, and specific tactical interventions.
SSEs per 10,000 adjusted patient-days decreased significantly, to a mean of 0.3 from 0.9 (P<0.0001) after implementation, while days between SSEs increased to a mean of 55.2 from 19.4 (P<0.0001).
This work represents one of the most robust single-center approaches to improving patient safety that has been published to date. The authors attribute much of their success to culture change, which required “relentless clarity of vision by the organization.” Although this substantially limits immediate generalizability of any of the specific interventions, the work stands on its own as a prime example of what may be accomplished through focused dedication to reducing patient harm.
Bottom line: Patient harm is preventable through a widespread and multifaceted institutional initiative.
Citation: Muething SE, Goudie A, Schoettker PJ, et al. Quality improvement initiative to reduce serious safety events and improve patient safety culture. Pediatrics. 2012;130:e423-431.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
How Should Physicians Assess and Manage Pressure Ulcers in the Hospitalized Patient?
The Case
An 85-year-old woman with stroke, functional quadriplegia, and diabetes mellitus presents with altered mental status. She is febrile (38.5°C) with leukocytosis (14,400 cells/mm3) and has a 5 cm x 4 cm x 2 cm Stage III malodorous sacral ulcer without surrounding erythema, tunneling, or pain. The ulcer base is partially covered by green slough. How should this pressure ulcer be evaluated and treated?
Overview
Pressure ulcers in vulnerable populations, such as the elderly and those with limited mobility, are exceedingly common. In the acute-care setting, the incidence of pressure ulcers ranges from 0.4% to 38%, with 2.5 million cases treated annually at an estimated cost of $11 billion per year.1,2 Moreover, as of Oct. 1, 2008, the Centers for Medicare & Medicaid Services (CMS) guideline states that hospitals will no longer receive additional payment when a hospitalized patient develops Stage III or IV pressure ulcers that are not present on admission.
A pressure ulcer is a localized injury to skin and underlying soft tissue over a bony prominence due to sustained external pressure.3 Prolonged pressure on these weight-bearing areas leads to reduced blood flow, ischemia, cell death, and necrosis of local tissues.4 Risk factors for developing pressure ulcers include increased external pressure, shear, friction, moisture, poor perfusion, immobility, incontinence, malnutrition, and impaired mental status.4 Inadequately treated pressure ulcers can lead to pain, tunneling, fistula formation, disfigurement, infection, prolonged hospitalization, lower quality of life, and increased mortality.4
Because of the significant morbidities and high costs associated with the care of pressure ulcers in acute care, hospitalists must be familiar with the assessment and treatment of pressure ulcers in vulnerable patients.
Review of the Data
The management of pressure ulcers in the hospitalized patient starts with a comprehensive assessment of the patient’s medical comorbidities, risk factors, and wound-staging. Considerations must be given to differentiate an infected pressure ulcer from a noninfected ulcer. These evaluations then guide the appropriate treatments of pressure ulcers, including the prevention of progression or formation of new ulcers, debridement, application of wound dressing, and antibiotic use.
Assessing pressure ulcer stage. The National Pressure Ulcer Advisory Panel (NPUAP) Classification System is the most commonly used staging tool. It describes four stages of pressure ulcers (see Table 1).3 A Stage 1 pressure ulcer is characterized by intact skin with nonblanchable erythema and may be discolored, painful, soft, firm, and warmer or cooler compared to adjacent area. A Stage II pressure ulcer presents with partial thickness skin loss with a shallow red-pink wound bed without slough, or as an intact or ruptured serum-filled blister. Stage II pressure ulcers do not include skin tears, tape burns, macerations, or excoriations. A Stage III pressure ulcer has full thickness skin loss with or without visible subcutaneous fat. Bone, tendon, or muscle are not exposed or directly palpable. Slough may be present but it does not obscure the depth of ulcer. Deep ulcers can develop in anatomical regions with high adiposity, such as the pelvic girdle. A Stage IV pressure ulcer has full thickness tissue loss with exposed and palpable bone, tendon, or muscle. Slough, eschar, undermining, and tunneling may be present. The depth of a Stage IV ulcer varies depending on anatomical location and adiposity. Stage IV ulcers also create a nidus for osteomyelitis.
NPUAP describes two additional categories of pressure ulcers: unstageable and deep tissue injury.3 An unstageable ulcer has full thickness skin or tissue loss of unknown depth because the wound base is completely obscured by slough or eschar. The ulcer can only be accurately categorized as Stage III or IV after sufficient slough or eschar is removed to identify wound depth. Lastly, suspected deep tissue injury describes a localized area of discolored intact skin (purple or maroon) or blood-filled blister due to damage of underlying tissue from pressure or shear.
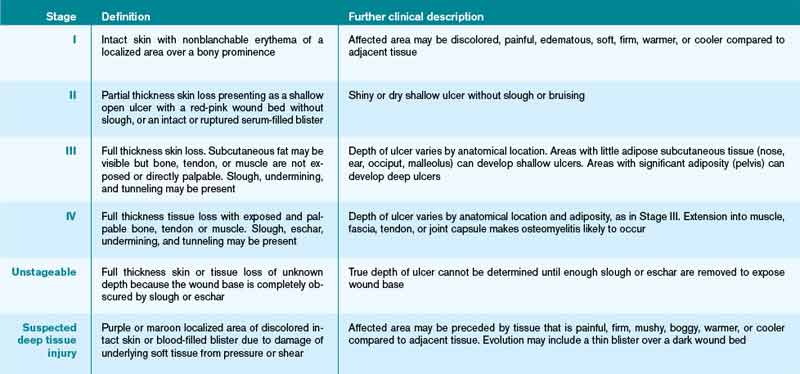
Diagnosing infected pressure ulcers. Pressure ulcer infection delays wound healing and increases risks for sepsis, cellulitis, osteomyelitis, and death.5,6 Clinical evidence of soft tissue involvement, such as erythema, warmth, tenderness, foul odor, or purulent discharge, and systemic inflammatory response (fever, tachycardia, or leukocytosis) are suggestive of a wound infection.3,5 However, these clinical signs may be absent and thus make the distinction between chronic wound and infected pressure ulcer difficult.7 Delayed healing with friable granulation tissue and increased pain in a treated wound may be the only signs of a pressure ulcer infection.3,5,7
Routine laboratory tests (i.e. white blood cell count, C-reactive protein, and erythrocyte sedimentation rate) are neither sensitive nor specific in diagnosing wound infection. Moreover, because pressure ulcers are typically colonized with ≥105 organisms/mL of normal skin flora and bacteria from adjacent gastrointestinal or urogenital environments, swab cultures identify colonizing organisms and are not recommended as a diagnostic test for pressure ulcer microbiologic evaluation.5,6 If microbiological data are needed to guide antibiotic use, cultures of blood, bone, or deep tissue biopsied from a surgically debrided wound should be used.5 Importantly, a higher index of suspicion should be maintained for infection of Stage III or IV pressure ulcers because they are more commonly infected than Stage I or II ulcers.3
Prevention. The prevention of wound progression is essential in treating acute, chronic, or infected pressure ulcers. Although management guidelines are limited by few high-quality, randomized controlled trials, NPUAP recommends a number of prevention strategies targeting risk factors that contribute to pressure ulcer development.2,3,8
For all bed-bound and chair-bound persons with impaired ability to self-reposition, risk assessment for pressure ulcer should be done on admission and repeated every 24 hours. The presence of such risk factors as immobility, shear, friction, moisture, incontinence, and malnutrition should be used to guide preventive treatments. Pressure relief on an ulcer can be achieved by repositioning the immobile patient at one- to two-hour intervals. Pressure-redistributing support surfaces (static, overlays, or dynamic) reduce tissue pressure and decrease overall incidence of pressure ulcers. Due to a lack of relative efficacy data, the selection of a support surface should be determined by the patient’s individual needs in order to reduce pressure and shear.3 For instance, dynamic support is an appropriate surface for an immobile patient with multiple or nonhealing ulcers. Shearing force and friction can be reduced by limiting head-of-bed elevation to <30° and using such transfer aids as bed linens while repositioning patients. The use of pillows, foam wedges, or other devices should be used to eliminate direct contact of bony prominences or reduce pressure on heels.8
Skin care should be optimized to limit excessive dryness or moisture. This includes using moisturizers for dry skin, particularly for the sacrum, and implementing bowel and bladder programs and absorbent underpads in patients with bowel or bladder incontinence.2 Given that patients with pressure ulcers are in a catabolic state, those who are nutritionally compromised may benefit from nutritional supplementation.3 Lastly, appropriate use of local and systemic pain regimen for painful pressure ulcers can improve patient cooperation in repositioning, dressing change, and quality of life.
Debridement. Wound debridement removes necrotic tissue often present in infected or chronic pressure ulcers, reduces risk for further infection, and promotes granulation tissue formation and wound healing. Debridement, however, is not indicated for ulcers of an ischemic limb or dry eschar of the heel, due to propensity for complications.3,4 The five common debridement methods are sharp, mechanical, autolytic, enzymatic, and biosurgical. The debridement method of choice is determined by clinician preference and availability.4
Sharp debridement results in rapid removal of large amounts of nonviable necrotic tissues and eschar using sharp instruments and, therefore, is indicated if wound infection or sepsis is present. Mechanical debridement by wet-to-dry dressing or whirlpool nonselectively removes granulation tissue and, thus, should be used cautiously. Autolytic debridement uses occlusive dressings (i.e. hydrocolloid or hydrogel) to maintain a moist wound environment in order to optimize the body’s inherent ability to selectively self-digest necrotic tissues. Enzymatic debridement with concentrated topical proteolytic enzymes (i.e. collagenase) digests necrotic tissues and achieves faster debridement than autolysis while being less invasive than surgical intervention. Biosurgery utilizes maggots (i.e. Lucilia sericata) that produce enzymes to effectively debride necrotic tissues.
Wound care and dressing. Pressure ulcers should be cleansed with each dressing change using such physiologic solutions as normal saline. Cleansing with antimicrobial solutions for ulcers with large necrotic debris or infection needs to be thoughtfully administered due to the potential impairment on wound healing.4 Wound dressing should maintain a moist wound environment to allow epithelialization and limit excessive exudates in order to prevent maceration. Although there are many categories of moisture retentive dressings, their comparative effectiveness remain unclear.4 Table 2 summarizes characteristics of common wound dressings and their applications.
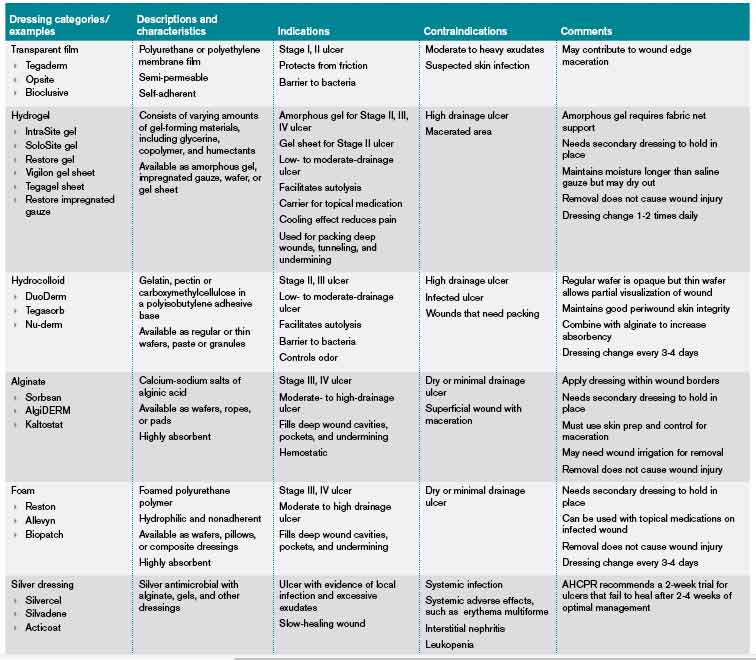
Antibiotic use. Topical antibiotics are appropriate for Stage III or IV ulcers with signs of local infection, including periwound erythema and friable granulation tissue.4 The Agency for Health Care Policy and Research recommends a two-week trial of a topical antibiotic, such as silver sulfadiazine, for pressure ulcers that fail to heal after two to four weeks of optimal care.6 Systemic antibiotics should be used for patients who demonstrate evidence of systemic infection including sepsis, osteomyelitis, or cellulitis with associated fever and leukocytosis. The choice of systemic antibiotics should be based on cultures from blood, bone, or deep tissue biopsied from a surgically debrided wound.4,6
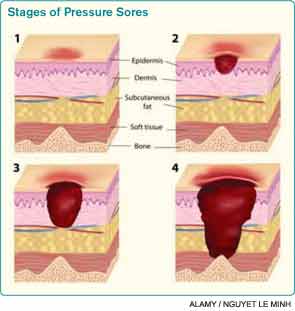
Back to the Case
The patient was hospitalized for altered mental status. She was at high risk for the progression of her sacral ulcer and the development of new pressure ulcers due to immobility, incontinence, malnutrition, and impaired mental status. The sacral wound was a chronic, Stage III pressure ulcer without evidence of local tissue infection. However, the presence of leukocytosis and fever were suggestive of an underlying infection. Her urine analysis was consistent with a urinary tract infection.
Trimethoprim/sulfamethoxazole was administered with subsequent resolution of leukocytosis, fever, and delirium. The sacral ulcer was cleansed with normal saline and covered with hydrocolloid dressing every 72 hours in order to maintain a moist wound environment and facilitate autolysis. Preventive interventions guided by her risk factors for pressure ulcer were implemented. Interventions included:
- Daily skin and wound assessment;
- Pressure relief with repositioning every two hours;
- Use of a dynamic support surface;
- Head-of-bed elevation of no more than <30° to reduce shear and friction;
- Use of transfer aids;
- Use of devices to eliminate direct contact of bony prominences;
- Optimizing skin care with moisturizers for dry skin and frequent changing of absorbent under pads for incontinence; and
- Consulting nutrition service to optimize nutritional intake.
Bottom Line
Assessments of pressure ulcer stage, wound infection, and risk factors guide targeted therapeutic interventions that facilitate wound healing and prevent new pressure ulcer formation.
Dr. Prager is a fellow in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai School of Medicine in New York City. Dr. Ko is a hospitalist and an assistant professor in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai.
References
- Pressure ulcers in America: prevalence, incidence, and implications for the future. An executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care. 2001;14(4):208-215.
- Reddy M, Gill SS, Rochon PA. Preventing pressure ulcers: a systematic review. JAMA. 2006;296(8):974-984.
- European Pressure Ulcer Advisory Panel and National Pressure Ulcer Advisory Panel. Treatment of Pressure Ulcers: Quick Reference Guide. Washington, D.C.: National Pressure Ulcer Advisory Panel; 2009.
- Bates-Jensen BM. Chapter 58. Pressure Ulcers. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York: McGraw-Hill; 2009.
- Livesley NJ, Chow AW. Infected pressure ulcers in elderly individuals. Clin Infect Dis. 2002;35(11):1390-1396.
- Agency for Health Care Policy and Research (AHCPR). Treatment of Pressure Ulcers. Clinical Practice Guideline Number 15. U.S. Department of Health and Human Services. 1994.
- Reddy M, Gill SS, Wu W, Kalkar SR, Rochon PA. Does this patient have an infection of a chronic wound? JAMA. 2012;307(6):605-611.
- National Pressure Ulcer Advisory Panel. Pressure Ulcer Prevention Points. National Pressure Ulcer Advisory Panel website. Available at: http://www.npuap.org/resources/educational-and-clinical-resources/pressure-ulcer-prevention-points/. Accessed Aug. 1, 2012.
- Reuben DB, Herr KA, Pacala JT, et al. Skin Ulcers. In: Geriatrics At Your Fingertips. 12th ed. New York: The American Geriatrics Society; 2010.
The Case
An 85-year-old woman with stroke, functional quadriplegia, and diabetes mellitus presents with altered mental status. She is febrile (38.5°C) with leukocytosis (14,400 cells/mm3) and has a 5 cm x 4 cm x 2 cm Stage III malodorous sacral ulcer without surrounding erythema, tunneling, or pain. The ulcer base is partially covered by green slough. How should this pressure ulcer be evaluated and treated?
Overview
Pressure ulcers in vulnerable populations, such as the elderly and those with limited mobility, are exceedingly common. In the acute-care setting, the incidence of pressure ulcers ranges from 0.4% to 38%, with 2.5 million cases treated annually at an estimated cost of $11 billion per year.1,2 Moreover, as of Oct. 1, 2008, the Centers for Medicare & Medicaid Services (CMS) guideline states that hospitals will no longer receive additional payment when a hospitalized patient develops Stage III or IV pressure ulcers that are not present on admission.
A pressure ulcer is a localized injury to skin and underlying soft tissue over a bony prominence due to sustained external pressure.3 Prolonged pressure on these weight-bearing areas leads to reduced blood flow, ischemia, cell death, and necrosis of local tissues.4 Risk factors for developing pressure ulcers include increased external pressure, shear, friction, moisture, poor perfusion, immobility, incontinence, malnutrition, and impaired mental status.4 Inadequately treated pressure ulcers can lead to pain, tunneling, fistula formation, disfigurement, infection, prolonged hospitalization, lower quality of life, and increased mortality.4
Because of the significant morbidities and high costs associated with the care of pressure ulcers in acute care, hospitalists must be familiar with the assessment and treatment of pressure ulcers in vulnerable patients.
Review of the Data
The management of pressure ulcers in the hospitalized patient starts with a comprehensive assessment of the patient’s medical comorbidities, risk factors, and wound-staging. Considerations must be given to differentiate an infected pressure ulcer from a noninfected ulcer. These evaluations then guide the appropriate treatments of pressure ulcers, including the prevention of progression or formation of new ulcers, debridement, application of wound dressing, and antibiotic use.
Assessing pressure ulcer stage. The National Pressure Ulcer Advisory Panel (NPUAP) Classification System is the most commonly used staging tool. It describes four stages of pressure ulcers (see Table 1).3 A Stage 1 pressure ulcer is characterized by intact skin with nonblanchable erythema and may be discolored, painful, soft, firm, and warmer or cooler compared to adjacent area. A Stage II pressure ulcer presents with partial thickness skin loss with a shallow red-pink wound bed without slough, or as an intact or ruptured serum-filled blister. Stage II pressure ulcers do not include skin tears, tape burns, macerations, or excoriations. A Stage III pressure ulcer has full thickness skin loss with or without visible subcutaneous fat. Bone, tendon, or muscle are not exposed or directly palpable. Slough may be present but it does not obscure the depth of ulcer. Deep ulcers can develop in anatomical regions with high adiposity, such as the pelvic girdle. A Stage IV pressure ulcer has full thickness tissue loss with exposed and palpable bone, tendon, or muscle. Slough, eschar, undermining, and tunneling may be present. The depth of a Stage IV ulcer varies depending on anatomical location and adiposity. Stage IV ulcers also create a nidus for osteomyelitis.
NPUAP describes two additional categories of pressure ulcers: unstageable and deep tissue injury.3 An unstageable ulcer has full thickness skin or tissue loss of unknown depth because the wound base is completely obscured by slough or eschar. The ulcer can only be accurately categorized as Stage III or IV after sufficient slough or eschar is removed to identify wound depth. Lastly, suspected deep tissue injury describes a localized area of discolored intact skin (purple or maroon) or blood-filled blister due to damage of underlying tissue from pressure or shear.
Diagnosing infected pressure ulcers. Pressure ulcer infection delays wound healing and increases risks for sepsis, cellulitis, osteomyelitis, and death.5,6 Clinical evidence of soft tissue involvement, such as erythema, warmth, tenderness, foul odor, or purulent discharge, and systemic inflammatory response (fever, tachycardia, or leukocytosis) are suggestive of a wound infection.3,5 However, these clinical signs may be absent and thus make the distinction between chronic wound and infected pressure ulcer difficult.7 Delayed healing with friable granulation tissue and increased pain in a treated wound may be the only signs of a pressure ulcer infection.3,5,7
Routine laboratory tests (i.e. white blood cell count, C-reactive protein, and erythrocyte sedimentation rate) are neither sensitive nor specific in diagnosing wound infection. Moreover, because pressure ulcers are typically colonized with ≥105 organisms/mL of normal skin flora and bacteria from adjacent gastrointestinal or urogenital environments, swab cultures identify colonizing organisms and are not recommended as a diagnostic test for pressure ulcer microbiologic evaluation.5,6 If microbiological data are needed to guide antibiotic use, cultures of blood, bone, or deep tissue biopsied from a surgically debrided wound should be used.5 Importantly, a higher index of suspicion should be maintained for infection of Stage III or IV pressure ulcers because they are more commonly infected than Stage I or II ulcers.3
Prevention. The prevention of wound progression is essential in treating acute, chronic, or infected pressure ulcers. Although management guidelines are limited by few high-quality, randomized controlled trials, NPUAP recommends a number of prevention strategies targeting risk factors that contribute to pressure ulcer development.2,3,8
For all bed-bound and chair-bound persons with impaired ability to self-reposition, risk assessment for pressure ulcer should be done on admission and repeated every 24 hours. The presence of such risk factors as immobility, shear, friction, moisture, incontinence, and malnutrition should be used to guide preventive treatments. Pressure relief on an ulcer can be achieved by repositioning the immobile patient at one- to two-hour intervals. Pressure-redistributing support surfaces (static, overlays, or dynamic) reduce tissue pressure and decrease overall incidence of pressure ulcers. Due to a lack of relative efficacy data, the selection of a support surface should be determined by the patient’s individual needs in order to reduce pressure and shear.3 For instance, dynamic support is an appropriate surface for an immobile patient with multiple or nonhealing ulcers. Shearing force and friction can be reduced by limiting head-of-bed elevation to <30° and using such transfer aids as bed linens while repositioning patients. The use of pillows, foam wedges, or other devices should be used to eliminate direct contact of bony prominences or reduce pressure on heels.8
Skin care should be optimized to limit excessive dryness or moisture. This includes using moisturizers for dry skin, particularly for the sacrum, and implementing bowel and bladder programs and absorbent underpads in patients with bowel or bladder incontinence.2 Given that patients with pressure ulcers are in a catabolic state, those who are nutritionally compromised may benefit from nutritional supplementation.3 Lastly, appropriate use of local and systemic pain regimen for painful pressure ulcers can improve patient cooperation in repositioning, dressing change, and quality of life.
Debridement. Wound debridement removes necrotic tissue often present in infected or chronic pressure ulcers, reduces risk for further infection, and promotes granulation tissue formation and wound healing. Debridement, however, is not indicated for ulcers of an ischemic limb or dry eschar of the heel, due to propensity for complications.3,4 The five common debridement methods are sharp, mechanical, autolytic, enzymatic, and biosurgical. The debridement method of choice is determined by clinician preference and availability.4
Sharp debridement results in rapid removal of large amounts of nonviable necrotic tissues and eschar using sharp instruments and, therefore, is indicated if wound infection or sepsis is present. Mechanical debridement by wet-to-dry dressing or whirlpool nonselectively removes granulation tissue and, thus, should be used cautiously. Autolytic debridement uses occlusive dressings (i.e. hydrocolloid or hydrogel) to maintain a moist wound environment in order to optimize the body’s inherent ability to selectively self-digest necrotic tissues. Enzymatic debridement with concentrated topical proteolytic enzymes (i.e. collagenase) digests necrotic tissues and achieves faster debridement than autolysis while being less invasive than surgical intervention. Biosurgery utilizes maggots (i.e. Lucilia sericata) that produce enzymes to effectively debride necrotic tissues.
Wound care and dressing. Pressure ulcers should be cleansed with each dressing change using such physiologic solutions as normal saline. Cleansing with antimicrobial solutions for ulcers with large necrotic debris or infection needs to be thoughtfully administered due to the potential impairment on wound healing.4 Wound dressing should maintain a moist wound environment to allow epithelialization and limit excessive exudates in order to prevent maceration. Although there are many categories of moisture retentive dressings, their comparative effectiveness remain unclear.4 Table 2 summarizes characteristics of common wound dressings and their applications.
Antibiotic use. Topical antibiotics are appropriate for Stage III or IV ulcers with signs of local infection, including periwound erythema and friable granulation tissue.4 The Agency for Health Care Policy and Research recommends a two-week trial of a topical antibiotic, such as silver sulfadiazine, for pressure ulcers that fail to heal after two to four weeks of optimal care.6 Systemic antibiotics should be used for patients who demonstrate evidence of systemic infection including sepsis, osteomyelitis, or cellulitis with associated fever and leukocytosis. The choice of systemic antibiotics should be based on cultures from blood, bone, or deep tissue biopsied from a surgically debrided wound.4,6
Back to the Case
The patient was hospitalized for altered mental status. She was at high risk for the progression of her sacral ulcer and the development of new pressure ulcers due to immobility, incontinence, malnutrition, and impaired mental status. The sacral wound was a chronic, Stage III pressure ulcer without evidence of local tissue infection. However, the presence of leukocytosis and fever were suggestive of an underlying infection. Her urine analysis was consistent with a urinary tract infection.
Trimethoprim/sulfamethoxazole was administered with subsequent resolution of leukocytosis, fever, and delirium. The sacral ulcer was cleansed with normal saline and covered with hydrocolloid dressing every 72 hours in order to maintain a moist wound environment and facilitate autolysis. Preventive interventions guided by her risk factors for pressure ulcer were implemented. Interventions included:
- Daily skin and wound assessment;
- Pressure relief with repositioning every two hours;
- Use of a dynamic support surface;
- Head-of-bed elevation of no more than <30° to reduce shear and friction;
- Use of transfer aids;
- Use of devices to eliminate direct contact of bony prominences;
- Optimizing skin care with moisturizers for dry skin and frequent changing of absorbent under pads for incontinence; and
- Consulting nutrition service to optimize nutritional intake.
Bottom Line
Assessments of pressure ulcer stage, wound infection, and risk factors guide targeted therapeutic interventions that facilitate wound healing and prevent new pressure ulcer formation.
Dr. Prager is a fellow in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai School of Medicine in New York City. Dr. Ko is a hospitalist and an assistant professor in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai.
References
- Pressure ulcers in America: prevalence, incidence, and implications for the future. An executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care. 2001;14(4):208-215.
- Reddy M, Gill SS, Rochon PA. Preventing pressure ulcers: a systematic review. JAMA. 2006;296(8):974-984.
- European Pressure Ulcer Advisory Panel and National Pressure Ulcer Advisory Panel. Treatment of Pressure Ulcers: Quick Reference Guide. Washington, D.C.: National Pressure Ulcer Advisory Panel; 2009.
- Bates-Jensen BM. Chapter 58. Pressure Ulcers. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York: McGraw-Hill; 2009.
- Livesley NJ, Chow AW. Infected pressure ulcers in elderly individuals. Clin Infect Dis. 2002;35(11):1390-1396.
- Agency for Health Care Policy and Research (AHCPR). Treatment of Pressure Ulcers. Clinical Practice Guideline Number 15. U.S. Department of Health and Human Services. 1994.
- Reddy M, Gill SS, Wu W, Kalkar SR, Rochon PA. Does this patient have an infection of a chronic wound? JAMA. 2012;307(6):605-611.
- National Pressure Ulcer Advisory Panel. Pressure Ulcer Prevention Points. National Pressure Ulcer Advisory Panel website. Available at: http://www.npuap.org/resources/educational-and-clinical-resources/pressure-ulcer-prevention-points/. Accessed Aug. 1, 2012.
- Reuben DB, Herr KA, Pacala JT, et al. Skin Ulcers. In: Geriatrics At Your Fingertips. 12th ed. New York: The American Geriatrics Society; 2010.
The Case
An 85-year-old woman with stroke, functional quadriplegia, and diabetes mellitus presents with altered mental status. She is febrile (38.5°C) with leukocytosis (14,400 cells/mm3) and has a 5 cm x 4 cm x 2 cm Stage III malodorous sacral ulcer without surrounding erythema, tunneling, or pain. The ulcer base is partially covered by green slough. How should this pressure ulcer be evaluated and treated?
Overview
Pressure ulcers in vulnerable populations, such as the elderly and those with limited mobility, are exceedingly common. In the acute-care setting, the incidence of pressure ulcers ranges from 0.4% to 38%, with 2.5 million cases treated annually at an estimated cost of $11 billion per year.1,2 Moreover, as of Oct. 1, 2008, the Centers for Medicare & Medicaid Services (CMS) guideline states that hospitals will no longer receive additional payment when a hospitalized patient develops Stage III or IV pressure ulcers that are not present on admission.
A pressure ulcer is a localized injury to skin and underlying soft tissue over a bony prominence due to sustained external pressure.3 Prolonged pressure on these weight-bearing areas leads to reduced blood flow, ischemia, cell death, and necrosis of local tissues.4 Risk factors for developing pressure ulcers include increased external pressure, shear, friction, moisture, poor perfusion, immobility, incontinence, malnutrition, and impaired mental status.4 Inadequately treated pressure ulcers can lead to pain, tunneling, fistula formation, disfigurement, infection, prolonged hospitalization, lower quality of life, and increased mortality.4
Because of the significant morbidities and high costs associated with the care of pressure ulcers in acute care, hospitalists must be familiar with the assessment and treatment of pressure ulcers in vulnerable patients.
Review of the Data
The management of pressure ulcers in the hospitalized patient starts with a comprehensive assessment of the patient’s medical comorbidities, risk factors, and wound-staging. Considerations must be given to differentiate an infected pressure ulcer from a noninfected ulcer. These evaluations then guide the appropriate treatments of pressure ulcers, including the prevention of progression or formation of new ulcers, debridement, application of wound dressing, and antibiotic use.
Assessing pressure ulcer stage. The National Pressure Ulcer Advisory Panel (NPUAP) Classification System is the most commonly used staging tool. It describes four stages of pressure ulcers (see Table 1).3 A Stage 1 pressure ulcer is characterized by intact skin with nonblanchable erythema and may be discolored, painful, soft, firm, and warmer or cooler compared to adjacent area. A Stage II pressure ulcer presents with partial thickness skin loss with a shallow red-pink wound bed without slough, or as an intact or ruptured serum-filled blister. Stage II pressure ulcers do not include skin tears, tape burns, macerations, or excoriations. A Stage III pressure ulcer has full thickness skin loss with or without visible subcutaneous fat. Bone, tendon, or muscle are not exposed or directly palpable. Slough may be present but it does not obscure the depth of ulcer. Deep ulcers can develop in anatomical regions with high adiposity, such as the pelvic girdle. A Stage IV pressure ulcer has full thickness tissue loss with exposed and palpable bone, tendon, or muscle. Slough, eschar, undermining, and tunneling may be present. The depth of a Stage IV ulcer varies depending on anatomical location and adiposity. Stage IV ulcers also create a nidus for osteomyelitis.
NPUAP describes two additional categories of pressure ulcers: unstageable and deep tissue injury.3 An unstageable ulcer has full thickness skin or tissue loss of unknown depth because the wound base is completely obscured by slough or eschar. The ulcer can only be accurately categorized as Stage III or IV after sufficient slough or eschar is removed to identify wound depth. Lastly, suspected deep tissue injury describes a localized area of discolored intact skin (purple or maroon) or blood-filled blister due to damage of underlying tissue from pressure or shear.
Diagnosing infected pressure ulcers. Pressure ulcer infection delays wound healing and increases risks for sepsis, cellulitis, osteomyelitis, and death.5,6 Clinical evidence of soft tissue involvement, such as erythema, warmth, tenderness, foul odor, or purulent discharge, and systemic inflammatory response (fever, tachycardia, or leukocytosis) are suggestive of a wound infection.3,5 However, these clinical signs may be absent and thus make the distinction between chronic wound and infected pressure ulcer difficult.7 Delayed healing with friable granulation tissue and increased pain in a treated wound may be the only signs of a pressure ulcer infection.3,5,7
Routine laboratory tests (i.e. white blood cell count, C-reactive protein, and erythrocyte sedimentation rate) are neither sensitive nor specific in diagnosing wound infection. Moreover, because pressure ulcers are typically colonized with ≥105 organisms/mL of normal skin flora and bacteria from adjacent gastrointestinal or urogenital environments, swab cultures identify colonizing organisms and are not recommended as a diagnostic test for pressure ulcer microbiologic evaluation.5,6 If microbiological data are needed to guide antibiotic use, cultures of blood, bone, or deep tissue biopsied from a surgically debrided wound should be used.5 Importantly, a higher index of suspicion should be maintained for infection of Stage III or IV pressure ulcers because they are more commonly infected than Stage I or II ulcers.3
Prevention. The prevention of wound progression is essential in treating acute, chronic, or infected pressure ulcers. Although management guidelines are limited by few high-quality, randomized controlled trials, NPUAP recommends a number of prevention strategies targeting risk factors that contribute to pressure ulcer development.2,3,8
For all bed-bound and chair-bound persons with impaired ability to self-reposition, risk assessment for pressure ulcer should be done on admission and repeated every 24 hours. The presence of such risk factors as immobility, shear, friction, moisture, incontinence, and malnutrition should be used to guide preventive treatments. Pressure relief on an ulcer can be achieved by repositioning the immobile patient at one- to two-hour intervals. Pressure-redistributing support surfaces (static, overlays, or dynamic) reduce tissue pressure and decrease overall incidence of pressure ulcers. Due to a lack of relative efficacy data, the selection of a support surface should be determined by the patient’s individual needs in order to reduce pressure and shear.3 For instance, dynamic support is an appropriate surface for an immobile patient with multiple or nonhealing ulcers. Shearing force and friction can be reduced by limiting head-of-bed elevation to <30° and using such transfer aids as bed linens while repositioning patients. The use of pillows, foam wedges, or other devices should be used to eliminate direct contact of bony prominences or reduce pressure on heels.8
Skin care should be optimized to limit excessive dryness or moisture. This includes using moisturizers for dry skin, particularly for the sacrum, and implementing bowel and bladder programs and absorbent underpads in patients with bowel or bladder incontinence.2 Given that patients with pressure ulcers are in a catabolic state, those who are nutritionally compromised may benefit from nutritional supplementation.3 Lastly, appropriate use of local and systemic pain regimen for painful pressure ulcers can improve patient cooperation in repositioning, dressing change, and quality of life.
Debridement. Wound debridement removes necrotic tissue often present in infected or chronic pressure ulcers, reduces risk for further infection, and promotes granulation tissue formation and wound healing. Debridement, however, is not indicated for ulcers of an ischemic limb or dry eschar of the heel, due to propensity for complications.3,4 The five common debridement methods are sharp, mechanical, autolytic, enzymatic, and biosurgical. The debridement method of choice is determined by clinician preference and availability.4
Sharp debridement results in rapid removal of large amounts of nonviable necrotic tissues and eschar using sharp instruments and, therefore, is indicated if wound infection or sepsis is present. Mechanical debridement by wet-to-dry dressing or whirlpool nonselectively removes granulation tissue and, thus, should be used cautiously. Autolytic debridement uses occlusive dressings (i.e. hydrocolloid or hydrogel) to maintain a moist wound environment in order to optimize the body’s inherent ability to selectively self-digest necrotic tissues. Enzymatic debridement with concentrated topical proteolytic enzymes (i.e. collagenase) digests necrotic tissues and achieves faster debridement than autolysis while being less invasive than surgical intervention. Biosurgery utilizes maggots (i.e. Lucilia sericata) that produce enzymes to effectively debride necrotic tissues.
Wound care and dressing. Pressure ulcers should be cleansed with each dressing change using such physiologic solutions as normal saline. Cleansing with antimicrobial solutions for ulcers with large necrotic debris or infection needs to be thoughtfully administered due to the potential impairment on wound healing.4 Wound dressing should maintain a moist wound environment to allow epithelialization and limit excessive exudates in order to prevent maceration. Although there are many categories of moisture retentive dressings, their comparative effectiveness remain unclear.4 Table 2 summarizes characteristics of common wound dressings and their applications.
Antibiotic use. Topical antibiotics are appropriate for Stage III or IV ulcers with signs of local infection, including periwound erythema and friable granulation tissue.4 The Agency for Health Care Policy and Research recommends a two-week trial of a topical antibiotic, such as silver sulfadiazine, for pressure ulcers that fail to heal after two to four weeks of optimal care.6 Systemic antibiotics should be used for patients who demonstrate evidence of systemic infection including sepsis, osteomyelitis, or cellulitis with associated fever and leukocytosis. The choice of systemic antibiotics should be based on cultures from blood, bone, or deep tissue biopsied from a surgically debrided wound.4,6
Back to the Case
The patient was hospitalized for altered mental status. She was at high risk for the progression of her sacral ulcer and the development of new pressure ulcers due to immobility, incontinence, malnutrition, and impaired mental status. The sacral wound was a chronic, Stage III pressure ulcer without evidence of local tissue infection. However, the presence of leukocytosis and fever were suggestive of an underlying infection. Her urine analysis was consistent with a urinary tract infection.
Trimethoprim/sulfamethoxazole was administered with subsequent resolution of leukocytosis, fever, and delirium. The sacral ulcer was cleansed with normal saline and covered with hydrocolloid dressing every 72 hours in order to maintain a moist wound environment and facilitate autolysis. Preventive interventions guided by her risk factors for pressure ulcer were implemented. Interventions included:
- Daily skin and wound assessment;
- Pressure relief with repositioning every two hours;
- Use of a dynamic support surface;
- Head-of-bed elevation of no more than <30° to reduce shear and friction;
- Use of transfer aids;
- Use of devices to eliminate direct contact of bony prominences;
- Optimizing skin care with moisturizers for dry skin and frequent changing of absorbent under pads for incontinence; and
- Consulting nutrition service to optimize nutritional intake.
Bottom Line
Assessments of pressure ulcer stage, wound infection, and risk factors guide targeted therapeutic interventions that facilitate wound healing and prevent new pressure ulcer formation.
Dr. Prager is a fellow in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai School of Medicine in New York City. Dr. Ko is a hospitalist and an assistant professor in the Brookdale Department of Geriatrics and Palliative Medicine at Mount Sinai.
References
- Pressure ulcers in America: prevalence, incidence, and implications for the future. An executive summary of the National Pressure Ulcer Advisory Panel monograph. Adv Skin Wound Care. 2001;14(4):208-215.
- Reddy M, Gill SS, Rochon PA. Preventing pressure ulcers: a systematic review. JAMA. 2006;296(8):974-984.
- European Pressure Ulcer Advisory Panel and National Pressure Ulcer Advisory Panel. Treatment of Pressure Ulcers: Quick Reference Guide. Washington, D.C.: National Pressure Ulcer Advisory Panel; 2009.
- Bates-Jensen BM. Chapter 58. Pressure Ulcers. In: Halter JB, Ouslander JG, Tinetti ME, Studenski S, High KP, Asthana S, eds. Hazzard’s Geriatric Medicine and Gerontology. 6th ed. New York: McGraw-Hill; 2009.
- Livesley NJ, Chow AW. Infected pressure ulcers in elderly individuals. Clin Infect Dis. 2002;35(11):1390-1396.
- Agency for Health Care Policy and Research (AHCPR). Treatment of Pressure Ulcers. Clinical Practice Guideline Number 15. U.S. Department of Health and Human Services. 1994.
- Reddy M, Gill SS, Wu W, Kalkar SR, Rochon PA. Does this patient have an infection of a chronic wound? JAMA. 2012;307(6):605-611.
- National Pressure Ulcer Advisory Panel. Pressure Ulcer Prevention Points. National Pressure Ulcer Advisory Panel website. Available at: http://www.npuap.org/resources/educational-and-clinical-resources/pressure-ulcer-prevention-points/. Accessed Aug. 1, 2012.
- Reuben DB, Herr KA, Pacala JT, et al. Skin Ulcers. In: Geriatrics At Your Fingertips. 12th ed. New York: The American Geriatrics Society; 2010.
New Anticoagulants Offer Promise, but Obstacles Remain

I see more and more people taking one of the newer anticoagulants. I’ve also seen a few disasters with these drugs. What’s the story?
Stacy M. Harper, Green Bay, Wis.
Dr. Hospitalist responds:
Although warfarin (Coumadin) has been a mainstay anticoagulant for decades, it can often be a frustrating medicine to manage due to its myriad drug interactions and the constant need for therapeutic testing. Recently, we have seen new medications hit the market (with one more likely to be approved soon), each with its pros and cons. Here’s an overview:
- Dabigatran (Pradaxa): It’s a direct thrombin inhibitor, taken twice daily. It has been approved for use in stroke prevention for atrial fibrillation (afib) (RELY trial) at 150 mg bid. It’s also been extensively studied for VTE prevention after orthopedic surgery, but it has not yet been approved in the U.S. for this indication.
As with all of these drugs, there is no reversal agent and there are no levels to measure. A recent report noted an increased risk of bleeding in patients who are older, have a low BMI, or have renal dysfunction. The manufacturer recommends a dose of 75 mg bid for patients with renal dysfunction, defined as a GFR of 15 to 30 mL/min; however, that dosing regimen was never explicitly studied.
Overall, it’s become quite a popular drug with the cardiologists in my neck of the woods. GERD can be a bothersome side effect. I avoid using it in patients older than 80, or in a patient with any renal dysfunction. Also, remember that it is not approved for VTE prevention or treatment.
- Rivaroxaban (Xarelto): An oral factor Xa inhibitor. Usually taken once daily at 10 mg for VTE prevention (RECORD trials). It is dosed at 20 mg/day for stroke prevention in afib (ROCKET-AF trial). Just recently, it was approved by the FDA for use in the acute treatment of DVT and PE (EINSTEIN trial), dosed at 15 mg BID for the first 21 days, and then continued at 20 mg daily after the initial period (see “Game-Changer,” p. 41). It is more hepatically metabolized than dabigatran, but it still has a significant renal clearance component. When compared to lovenox in orthopedic patients, it’s as effective but with a slightly higher risk of bleeding. I would avoid using it in any patients with significant renal or hepatic dysfunction.
- Apixaban (Eliquis): Another oral factor Xa inhibitor. Studied at 2.5 mg BID for VTE prevention in orthopedic patients (ADVANCE trials). Studied at 5 mg BID for stroke prevention in afib (ARISTOTLE trial). It is not yet approved in the U.S for any indication, but a final decision is expected from the FDA by March. Overall, the data are fairly compelling, and it looks like a strong candidate. The data show a drug that is potentially more effective than lovenox, with less risk of bleeding for orthopedic patients. It is mainly hepatically metabolized.
So, with no drug company relationships to disclose, here are my general observations: For starters, I think dabigatran is being overused in older patients with renal dysfunction. I seem to stop it more than I recommend it, and it is far from my favorite drug. With rivaroxaban, it looks appropriate for VTE prevention, and now having the option of being able to transition patients who develop a clot onto a treatment dose of the drug is appealing. Apixaban’s data look the best out of all three agents in terms of both efficacy and bleeding, and although it is yet to be approved here, I imagine that will change in the near future. For all of these drugs, remember that we have no long-term safety data, and no reversal agents. It will be interesting to see how this plays out and which of these drugs have staying power. For all of warfarin’s faults, at least we know how to measure it and how to stop it.
I see more and more people taking one of the newer anticoagulants. I’ve also seen a few disasters with these drugs. What’s the story?
Stacy M. Harper, Green Bay, Wis.
Dr. Hospitalist responds:
Although warfarin (Coumadin) has been a mainstay anticoagulant for decades, it can often be a frustrating medicine to manage due to its myriad drug interactions and the constant need for therapeutic testing. Recently, we have seen new medications hit the market (with one more likely to be approved soon), each with its pros and cons. Here’s an overview:
- Dabigatran (Pradaxa): It’s a direct thrombin inhibitor, taken twice daily. It has been approved for use in stroke prevention for atrial fibrillation (afib) (RELY trial) at 150 mg bid. It’s also been extensively studied for VTE prevention after orthopedic surgery, but it has not yet been approved in the U.S. for this indication.
As with all of these drugs, there is no reversal agent and there are no levels to measure. A recent report noted an increased risk of bleeding in patients who are older, have a low BMI, or have renal dysfunction. The manufacturer recommends a dose of 75 mg bid for patients with renal dysfunction, defined as a GFR of 15 to 30 mL/min; however, that dosing regimen was never explicitly studied.
Overall, it’s become quite a popular drug with the cardiologists in my neck of the woods. GERD can be a bothersome side effect. I avoid using it in patients older than 80, or in a patient with any renal dysfunction. Also, remember that it is not approved for VTE prevention or treatment.
- Rivaroxaban (Xarelto): An oral factor Xa inhibitor. Usually taken once daily at 10 mg for VTE prevention (RECORD trials). It is dosed at 20 mg/day for stroke prevention in afib (ROCKET-AF trial). Just recently, it was approved by the FDA for use in the acute treatment of DVT and PE (EINSTEIN trial), dosed at 15 mg BID for the first 21 days, and then continued at 20 mg daily after the initial period (see “Game-Changer,” p. 41). It is more hepatically metabolized than dabigatran, but it still has a significant renal clearance component. When compared to lovenox in orthopedic patients, it’s as effective but with a slightly higher risk of bleeding. I would avoid using it in any patients with significant renal or hepatic dysfunction.
- Apixaban (Eliquis): Another oral factor Xa inhibitor. Studied at 2.5 mg BID for VTE prevention in orthopedic patients (ADVANCE trials). Studied at 5 mg BID for stroke prevention in afib (ARISTOTLE trial). It is not yet approved in the U.S for any indication, but a final decision is expected from the FDA by March. Overall, the data are fairly compelling, and it looks like a strong candidate. The data show a drug that is potentially more effective than lovenox, with less risk of bleeding for orthopedic patients. It is mainly hepatically metabolized.
So, with no drug company relationships to disclose, here are my general observations: For starters, I think dabigatran is being overused in older patients with renal dysfunction. I seem to stop it more than I recommend it, and it is far from my favorite drug. With rivaroxaban, it looks appropriate for VTE prevention, and now having the option of being able to transition patients who develop a clot onto a treatment dose of the drug is appealing. Apixaban’s data look the best out of all three agents in terms of both efficacy and bleeding, and although it is yet to be approved here, I imagine that will change in the near future. For all of these drugs, remember that we have no long-term safety data, and no reversal agents. It will be interesting to see how this plays out and which of these drugs have staying power. For all of warfarin’s faults, at least we know how to measure it and how to stop it.
I see more and more people taking one of the newer anticoagulants. I’ve also seen a few disasters with these drugs. What’s the story?
Stacy M. Harper, Green Bay, Wis.
Dr. Hospitalist responds:
Although warfarin (Coumadin) has been a mainstay anticoagulant for decades, it can often be a frustrating medicine to manage due to its myriad drug interactions and the constant need for therapeutic testing. Recently, we have seen new medications hit the market (with one more likely to be approved soon), each with its pros and cons. Here’s an overview:
- Dabigatran (Pradaxa): It’s a direct thrombin inhibitor, taken twice daily. It has been approved for use in stroke prevention for atrial fibrillation (afib) (RELY trial) at 150 mg bid. It’s also been extensively studied for VTE prevention after orthopedic surgery, but it has not yet been approved in the U.S. for this indication.
As with all of these drugs, there is no reversal agent and there are no levels to measure. A recent report noted an increased risk of bleeding in patients who are older, have a low BMI, or have renal dysfunction. The manufacturer recommends a dose of 75 mg bid for patients with renal dysfunction, defined as a GFR of 15 to 30 mL/min; however, that dosing regimen was never explicitly studied.
Overall, it’s become quite a popular drug with the cardiologists in my neck of the woods. GERD can be a bothersome side effect. I avoid using it in patients older than 80, or in a patient with any renal dysfunction. Also, remember that it is not approved for VTE prevention or treatment.
- Rivaroxaban (Xarelto): An oral factor Xa inhibitor. Usually taken once daily at 10 mg for VTE prevention (RECORD trials). It is dosed at 20 mg/day for stroke prevention in afib (ROCKET-AF trial). Just recently, it was approved by the FDA for use in the acute treatment of DVT and PE (EINSTEIN trial), dosed at 15 mg BID for the first 21 days, and then continued at 20 mg daily after the initial period (see “Game-Changer,” p. 41). It is more hepatically metabolized than dabigatran, but it still has a significant renal clearance component. When compared to lovenox in orthopedic patients, it’s as effective but with a slightly higher risk of bleeding. I would avoid using it in any patients with significant renal or hepatic dysfunction.
- Apixaban (Eliquis): Another oral factor Xa inhibitor. Studied at 2.5 mg BID for VTE prevention in orthopedic patients (ADVANCE trials). Studied at 5 mg BID for stroke prevention in afib (ARISTOTLE trial). It is not yet approved in the U.S for any indication, but a final decision is expected from the FDA by March. Overall, the data are fairly compelling, and it looks like a strong candidate. The data show a drug that is potentially more effective than lovenox, with less risk of bleeding for orthopedic patients. It is mainly hepatically metabolized.
So, with no drug company relationships to disclose, here are my general observations: For starters, I think dabigatran is being overused in older patients with renal dysfunction. I seem to stop it more than I recommend it, and it is far from my favorite drug. With rivaroxaban, it looks appropriate for VTE prevention, and now having the option of being able to transition patients who develop a clot onto a treatment dose of the drug is appealing. Apixaban’s data look the best out of all three agents in terms of both efficacy and bleeding, and although it is yet to be approved here, I imagine that will change in the near future. For all of these drugs, remember that we have no long-term safety data, and no reversal agents. It will be interesting to see how this plays out and which of these drugs have staying power. For all of warfarin’s faults, at least we know how to measure it and how to stop it.
Increased Ordering of Diagnostic Tests Associated with Longer Lengths of Stay in Pediatric Pneumonia

Clinical question: What is the relationship between variation in resource utilization and outcomes in children with community-acquired pneumonia (CAP)?
Background: Variation in clinical care, particularly when driven by provider preferences, often highlights opportunities for improvement in the quality of our care. CAP is one of the most common reasons for hospitalization in children. The relationship between variation in care processes, utilization, and outcomes in pediatric CAP is not well defined.
Study design: Retrospective database review.
Setting: Twenty-nine freestanding children's hospitals.
Synopsis: The Pediatric Health Information System (PHIS) database was used to review utilization and outcomes data on 43,819 children admitted with nonsevere CAP during a five-year period. Substantial degrees of variation in test ordering, empiric antibiotic selection, length of stay (LOS), and 14-day readmissions were found. An association was noted between increased resource utilization—specifically, ordering of diagnostic tests—and longer LOS.
The association between increased resource utilization and LOS has been suggested in other work in respiratory illness in children. Although the retrospective nature of this work precludes detailed resolution of whether confounding by severity was an issue, this appears unlikely based on the relatively homogeneous patient populations and hospital types. Additional limitations of this work exist, and include an inability to further assess the appropriateness of the testing that was ordered—as well as relatively crude rankings of hospitals based on resource utilization. Nevertheless, in an era where a premium is placed on finding value in clinical medicine, these results should prompt further exploration of the link between testing and LOS in children hospitalized with CAP.
Bottom line: Unnecessary testing in children hospitalized with pneumonia may lead to longer LOS.
Citation: Brogan TV, Hall M, Williams DJ, et al. Variability in processes of care and outcomes among children hospitalized with community-acquired pneumonia. Ped Infect Dis J. 2012;31:1036-1041.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What is the relationship between variation in resource utilization and outcomes in children with community-acquired pneumonia (CAP)?
Background: Variation in clinical care, particularly when driven by provider preferences, often highlights opportunities for improvement in the quality of our care. CAP is one of the most common reasons for hospitalization in children. The relationship between variation in care processes, utilization, and outcomes in pediatric CAP is not well defined.
Study design: Retrospective database review.
Setting: Twenty-nine freestanding children's hospitals.
Synopsis: The Pediatric Health Information System (PHIS) database was used to review utilization and outcomes data on 43,819 children admitted with nonsevere CAP during a five-year period. Substantial degrees of variation in test ordering, empiric antibiotic selection, length of stay (LOS), and 14-day readmissions were found. An association was noted between increased resource utilization—specifically, ordering of diagnostic tests—and longer LOS.
The association between increased resource utilization and LOS has been suggested in other work in respiratory illness in children. Although the retrospective nature of this work precludes detailed resolution of whether confounding by severity was an issue, this appears unlikely based on the relatively homogeneous patient populations and hospital types. Additional limitations of this work exist, and include an inability to further assess the appropriateness of the testing that was ordered—as well as relatively crude rankings of hospitals based on resource utilization. Nevertheless, in an era where a premium is placed on finding value in clinical medicine, these results should prompt further exploration of the link between testing and LOS in children hospitalized with CAP.
Bottom line: Unnecessary testing in children hospitalized with pneumonia may lead to longer LOS.
Citation: Brogan TV, Hall M, Williams DJ, et al. Variability in processes of care and outcomes among children hospitalized with community-acquired pneumonia. Ped Infect Dis J. 2012;31:1036-1041.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What is the relationship between variation in resource utilization and outcomes in children with community-acquired pneumonia (CAP)?
Background: Variation in clinical care, particularly when driven by provider preferences, often highlights opportunities for improvement in the quality of our care. CAP is one of the most common reasons for hospitalization in children. The relationship between variation in care processes, utilization, and outcomes in pediatric CAP is not well defined.
Study design: Retrospective database review.
Setting: Twenty-nine freestanding children's hospitals.
Synopsis: The Pediatric Health Information System (PHIS) database was used to review utilization and outcomes data on 43,819 children admitted with nonsevere CAP during a five-year period. Substantial degrees of variation in test ordering, empiric antibiotic selection, length of stay (LOS), and 14-day readmissions were found. An association was noted between increased resource utilization—specifically, ordering of diagnostic tests—and longer LOS.
The association between increased resource utilization and LOS has been suggested in other work in respiratory illness in children. Although the retrospective nature of this work precludes detailed resolution of whether confounding by severity was an issue, this appears unlikely based on the relatively homogeneous patient populations and hospital types. Additional limitations of this work exist, and include an inability to further assess the appropriateness of the testing that was ordered—as well as relatively crude rankings of hospitals based on resource utilization. Nevertheless, in an era where a premium is placed on finding value in clinical medicine, these results should prompt further exploration of the link between testing and LOS in children hospitalized with CAP.
Bottom line: Unnecessary testing in children hospitalized with pneumonia may lead to longer LOS.
Citation: Brogan TV, Hall M, Williams DJ, et al. Variability in processes of care and outcomes among children hospitalized with community-acquired pneumonia. Ped Infect Dis J. 2012;31:1036-1041.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
What Is the Best Treatment for an Adult Patient with Staphylococcus aureus Bacteremia?
Case
An 82-year-old man with non-Hodgkin’s lymphoma in remission and a history of congestive heart failure and hypertension presents with one week of generalized malaise and intermittent fevers. Vitals show a temperature of 101oF, blood pressure of 130/60 mmHg, and heart rate of 100. His exam is notable for an erythematous and tender chest port site, with no murmurs. Blood cultures drawn upon presentation show gram-positive cocci speciated to Staphylococcus aureus. What are the next steps in management of this patient?
Overview
S. aureus bacteremia (SAB) is a common infectious cause of morbidity and mortality worldwide, causing both community-acquired and hospital-acquired bacteremia. In the U.S. alone, it accounts for 23% of all bloodstream infections and is the bacterial pathogen most strongly associated with death.1 Mortality rates are approximately 42% in those with methicillin-resistant S. aureus (MRSA) bacteremia and 28% in those with methicillin-sensitive S. aureus (MSSA) bacteremia.2
Recognizing the severity of SAB, the Infectious Disease Society of America (IDSA) published treatment guidelines in 2011 to help direct the clinical care of this disease process.3 However, the majority of the recommendations are based on observational studies and expert opinion, as less than 1,500 patients have been enrolled in randomized controlled trials specifically targeted to investigate the treatment of SAB.4
Review of the Data
A clinically significant SAB usually is defined as the isolation of S. aureus from a venous blood culture with associated symptoms and signs of systemic infection.5 As SAB contamination is rare and can be associated with multiple complications, including metastatic infections, embolic stroke, recurrent infection, and death, any finding of a positive blood culture must be taken seriously.4
SAB treatment is multifaceted and should focus on the removal of any nidus of infection, such as a catheter or a prosthetic device, the use of prolonged antimicrobial therapy, and the evaluation of potential complications. In a retrospective study, Johnson et al showed that failure to remove the source is one of the strongest independent predictors of relapse in patients with SAB.6 However, 10% to 40% of patients have no identifiable focus, which increases the impetus to evaluate for complications.7-8 Overall, approximately one-third of patients with SAB develop metastatic complications, either from hematogenous seeding or local extension of infection.9
In addition to advanced age and such comorbid conditions as cirrhosis, the strongest predictor of complications is a positive blood culture at 48 to 96 hours after an initial positive blood culture, as shown in a large prospective cohort study by Fowler et al.7,10-11 Additional independent risk factors (see Table 1) include community acquisition (likely due to prolonged duration of bacteremia), skin examination suggesting the presence of acute systemic infection, and persistent fever at 72 hours after the first positive blood culture. Patients with even one of these risk factors are at high risk for a complicated course (which occurs in about 35%). In a case-control study, Chihara et al showed that S. aureus bacteruria in the absence of urinary tract pathology or recent urinary tract instrumentation might be associated with threefold increased mortality compared with those without bacteriuria, even after adjustment for comorbid conditions.12
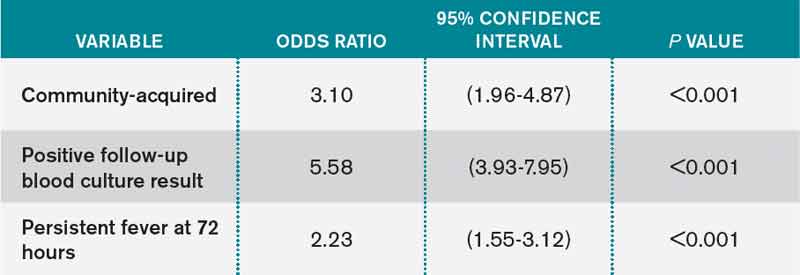
Antimicrobial Treatment
The initial choice of antibiotic therapy for SAB must take into account the MRSA prevalence in the community and hospital. If suspicion is high enough for MRSA, the IDSA’s 2011 guidelines suggest treatment with vancomycin or daptomycin.3 Although there are no published RCTs to support a particular antibiotic regimen, there are trials to suggest that a delay in treatment could be harmful. One study, by Lordis et al, showed that a delay in treatment, as defined by treatment after 44.75 hours, was associated with a longer hospital stay, with the delayed treatment group being hospitalized for 20.2 days and the early treatment group being hospitalized for 14.3 days.13 A delay in treatment was also found to be an independent predictor of mortality.13
Once susceptibilities are known, it is important to appropriately tailor antibiotics, as studies have shown lower treatment failure rates with the use of beta-lactam antibiotics when compared with empiric MRSA coverage.14-15 In one prospective study of 123 hemodialysis patients with MSSA bacteremia, Stryjewski et al showed that those treated with vancomycin were at higher risk of experiencing treatment failure than those treated with cefazolin.15 In another prospective observational study of 505 patients with SAB, Chang et al found that treatment with nafcillin was superior to vancomycin in preventing persistent bacteremia or relapse for MSSA bacteremia.14 These studies highlight the benefits of adjusting the empirically selected antibiotics, as narrowing the spectrum can result in less treatment failure.
If susceptibilities confirm MRSA, the IDSA recommends continued treatment with vancomycin or daptomycin.3 Although vancomycin is most commonly used, partly because of low cost and familiarity, Fowler et al published a study of 246 patients with SAB with or without endocarditis, assigning them to treatment with daptomycin, initial low-dose gentamicin plus vancomycin or an antistaphylococcal penicillin.16 The study found that daptomycin was not inferior to the other therapies, confirming that daptomycin is a reasonable choice in the treatment of MRSA infections.
Oral antibiotics are an option to treat SAB when necessary. A RCT by Heldman et al of 85 intravenous drug users with SAB (and suspected right-sided endocarditis, 65% of which had HIV) showed similar efficacy of ciprofloxacin plus rifampin versus standard intravenous therapy.17 A subsequent randomized trial of 104 patients with SAB comparing oral fleroxacin plus rifampin against conventional intravenous therapy also showed similar cure rates, with the added benefit of earlier discharge.18 Furthermore, in a meta-analysis of five randomized studies by Shorr et al (see Table 2), linezolid was found to have outcomes that were not inferior to vancomycin (clinical cure/microbiological success of 56%/69% in the linezolid group and 46%/73% in the vancomycin group).19
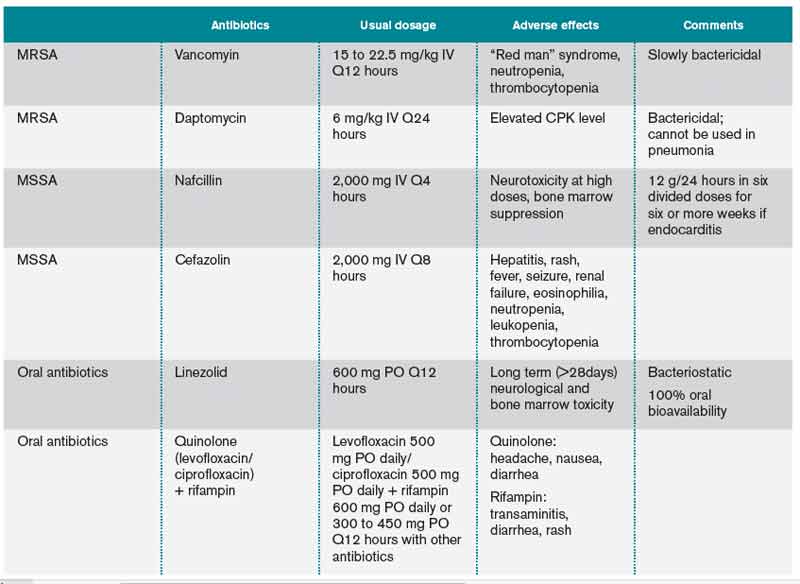
Treatment Duration
Recommendations for the duration of antibiotic treatment for SAB are mainly based on observational studies, which show mixed results. In one study done in the 1950s, about two-thirds of cases of SAB were associated with endocarditis, and longer courses of intravenous therapy (greater than four weeks) were recommended.20
More recently, with the increasing rates of catheter-related SAB and its relatively high rate of expeditious blood culture clearance, a shorter duration has been evaluated in several studies. In 1992, an analysis of published data and a retrospective case series concluded that fewer than 10 days of intravenous antibiotics might be associated with an increased risk of recurrence, but 10 to 14 days of intravenous therapy was effective for most cases of catheter-associated SAB.5 In another prospective study, Fowler et al found that a seven-day course of intravenous antibiotic therapy may be sufficient for simple, catheter-related infections.21 A subsequent prospective study by Jensen et al reported that a course of antibiotic therapy of less than 14 days might be associated with higher mortality compared to a longer course.9 A prospective study of 276 patients by Thomas et al found there was no relationship between relapse and duration of treatment (seven to 15 days) in catheter-related SAB, concluding that more than 14 days of antibiotic therapy was unnecessary.22
Per IDSA guidelines, uncomplicated SAB (no implanted prosthesis, negative blood cultures within two to four days, defervescence within 72 hours of initiating therapy, and lack of metastatic complication) can be treated with a two-week course of antibiotics, while complicated bacteremia (any of above criteria) should be treated within four to six weeks.3
Monitoring for Complications: Echos
Based on the IDSA guidelines, echocardiography is recommended in all patients with bacteremia, with a preference of transesophageal echocardiography (TEE) over transthoracic echocardiography (TTE).3 More recently, Kaasch et al developed simple criteria to identify patients with nosocomial SAB at low risk for infective endocarditis based on two prospective cohort studies.23 Lack of any of these criteria, which include prolonged bacteremia of more than four days’ duration, presence of a permanent intracardiac device, hemodialysis dependency, spinal infection and nonvertebral osteomyelitis, along with a negative TTE indicates that a TEE is not necessary (see Table 3). However, these patients need close follow-up to ensure that bacteremia clears and no new signs or symptoms concerning for metastatic infection develop.
ID Consultation
Several studies have shown that ID consultation not only improves adherence to evidence-based management of SAB, but it also reduces mortality.24-27 In a recent prospective cohort study in a tertiary-care center, even after adjusting for pre-existing comorbidities and severity of disease, an ID consult was associated with a 56% reduction in 28-day mortality.24 The patients who were followed by an ID consult service were more likely to receive appropriate duration of antibiotics (81% vs. 29%, respectively) and undergo appropriate workup for the evaluation of metastatic infections (34% and 8%, respectively). This study concluded that routine ID consult should be considered in patients with SAB, especially those with severe illness and multiple comorbid conditions.
Back to the Case
The patient was started on empiric therapy with vancomycin and serial blood cultures were obtained. He remained hemodynamically stable but febrile, with persistently positive blood and urine cultures. Given concern for the port being the source of his infection, his chest port was removed. A high-quality TTE was performed and was unremarkable.
ID was consulted. Blood cultures subsequently grew MSSA and vancomycin was switched to cefazolin 2g every eight hours. On hospital Day 5, his fever resolved and blood cultures turned negative. There were no clinical signs or symptoms for metastatic infections. A PICC line was placed after blood cultures remained negative for 48 hours. The decision was made to treat him with four weeks of antibiotics from his last positive blood culture, with follow-up in ID clinic.
Bottom Line
SAB is a common worldwide cause of morbidity and mortality. Treatment should include removing the nidus if present, finding and administering the appropriate antimicrobial therapy, evaluating for possible complications, and consulting with ID.
Dr. Ward is an assistant professor, Dr. Kim a clinical instructor, and Dr. Stojan a clinical lecturer at the University of Michigan Health System in Ann Arbor.
Acknowledgement
The authors would like to thank Dr. Jeffrey Rohde for reviewing the manuscript.
References
- Shorr AF, Tabak YP, Killian AD, et al. Healthcare-associated bloodstream infection: a distinct entity? Insights from a large U.S. database. Crit Care Med. 2006;34:3588-3595.
- Mylotte JM, McDermott C, Spooner JA. Prospective study of 114 consecutive episodes of Staphylococcus aureus bacteremia. Rev Infect Dis. 1987;9:891-907.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3).
- Naber CK, Baddour LM, Giamarellos-Bourboulis EJ, et al. Clinical consensus conference: survey on gram-positive bloodstream infections with a focus on Staphylococcus aureus. Clin Infect Dis. 2011;52(3):285-292.
- Thwaites GE, Edgeworth JD, Gkrania-Klotsas E, et al. Clinical management of Staphylococcus bacteremia. Lancet Infect Dis. 2011;11(3):208-222.
- Johnson LB, Almoujahed MO, Ilg K, et al. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand J Infec Dis. 2003;35(11-12):782-789.
- Fowler VG Jr., Olsen MK, Corey GR, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med. 2003;163:2066-2072.
- Mylotte JM, Tayara A. Staphylococcus aureus bacteremia: predictors of 30-day mortality in a large cohort. Clin Infect Dis. 2000;31:1170-1174.
- Jensen AG, Wachmann CH, Espersen F, et al. Treatment and outcome of Staphylococcus aureus bacteremia: a prospective study of 278 cases. Arch Intern Med. 2002;162(1):25-32.
- Malani PN, Rana MM, Banerjee M, et al. Staphylococcus aureus bloodstream infections: the association between age and mortality and functional status. J Am Geriatr Soc. 2008;56(8):1485-1489.
- Kim SH, Park WB, Lee KD, et al. Outcome of Staphylococcus aureus bacteremia in patients with eradicable foci versus noneradicable foci. Clin Infect Dis. 2003;37(6):794-799.
- Chihara S. Popovich KJ, Weinstein RA, et al. Staphylococcus aureus bacteriuria as a prognosticator for outcome of Staphylococcus aureus bacteremia: a case control study. BMC Inf Dis. 2010;10:225.
- Lodise TP, McKinnon PS, Swiderski L, Rybak MJ. Outcomes analysis of delayed antibiotic treatment for hospital-acquired Staphylococcus aureus bacteremia. Clin Infect Dis. 2003;36:1418-1423.
- Chang FY, Peacock JE Jr., Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore). 2003;82:333.
- Stryjewski ME, Szczech LA, Benjamin DK Jr., et al. Use of vancomycin or first-generation cephalosporins for the treatment of hemodialysisdependent patients with methicillin-susceptible Staphylococcus aureus bacteremia. Clin Infect Dis. 2007;44:190-196.
- Fowler VG Jr., Boucher HW, Corey GR, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med. 2006;355:653.
- Heldman AW, Hartert TV, Ray SC, et al. Oral antibiotic treatment of right-sided staphylococcal endocarditis in injection drug users: prospective randomized comparison with parenteral therapy. Am J Med. 1996;101:68-76.
- Schrenzel J, Harbarth S, Schockmel G, et al. A randomized clinical trial to compare fleroxacin-rifampicin with flucloxacillin or vancomycin for the treatment of Staphylococcal infection. Clin Infect Dis. 2004;39:1285-1292.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother. 2005;56:923-929.
- Wilson R, Hamburger M. Fifteen years’ experience with Staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med. 1957;22:437-457.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendation of infectious disease specialists: experience with 244 patients. Clin Infect Dis. 1998;27:478-486.
- Thomas MG, Morris AJ. Cannula-associated Staphylococcus aureus bacteremia: outcome in relation to treatment. Intern Med J. 2005;35:319-330.
- Kaasch AJ, Fowler VG Jr, Rieg S, et al. Use of a simple criteria set for guiding echocardiography in nosocomidal Staphylococcus aureus bacteremia. Clin Infect Dis. 2011;53:1-9.
- Honda H, Krauss MJ, Jones JC, et al. The value of infectious disease consultation in Staphylococcus aureus bacteremia. Am J Med. 2010;123:631-637.
- Nagao M, Iinuma Y, Saito T, et al. Close cooperation between infectious disease physicians and attending physicians can result in better management and outcome for patients with Staphylococcus aureus bacteremia. Euro Soc Clin Microbiology Infect Dis. 2010;16:1783-1788.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendations of infectious diseases specialist: experience with 244 patients. Clin Infect Dis. 1998;27(3):478-486.
- Jenkins TC, Price CS, Sabel AL et al. Impact of routine infectious diseases service consultation on the evaluation, management, and outcome of Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(7):1000-1008.
Case
An 82-year-old man with non-Hodgkin’s lymphoma in remission and a history of congestive heart failure and hypertension presents with one week of generalized malaise and intermittent fevers. Vitals show a temperature of 101oF, blood pressure of 130/60 mmHg, and heart rate of 100. His exam is notable for an erythematous and tender chest port site, with no murmurs. Blood cultures drawn upon presentation show gram-positive cocci speciated to Staphylococcus aureus. What are the next steps in management of this patient?
Overview
S. aureus bacteremia (SAB) is a common infectious cause of morbidity and mortality worldwide, causing both community-acquired and hospital-acquired bacteremia. In the U.S. alone, it accounts for 23% of all bloodstream infections and is the bacterial pathogen most strongly associated with death.1 Mortality rates are approximately 42% in those with methicillin-resistant S. aureus (MRSA) bacteremia and 28% in those with methicillin-sensitive S. aureus (MSSA) bacteremia.2
Recognizing the severity of SAB, the Infectious Disease Society of America (IDSA) published treatment guidelines in 2011 to help direct the clinical care of this disease process.3 However, the majority of the recommendations are based on observational studies and expert opinion, as less than 1,500 patients have been enrolled in randomized controlled trials specifically targeted to investigate the treatment of SAB.4
Review of the Data
A clinically significant SAB usually is defined as the isolation of S. aureus from a venous blood culture with associated symptoms and signs of systemic infection.5 As SAB contamination is rare and can be associated with multiple complications, including metastatic infections, embolic stroke, recurrent infection, and death, any finding of a positive blood culture must be taken seriously.4
SAB treatment is multifaceted and should focus on the removal of any nidus of infection, such as a catheter or a prosthetic device, the use of prolonged antimicrobial therapy, and the evaluation of potential complications. In a retrospective study, Johnson et al showed that failure to remove the source is one of the strongest independent predictors of relapse in patients with SAB.6 However, 10% to 40% of patients have no identifiable focus, which increases the impetus to evaluate for complications.7-8 Overall, approximately one-third of patients with SAB develop metastatic complications, either from hematogenous seeding or local extension of infection.9
In addition to advanced age and such comorbid conditions as cirrhosis, the strongest predictor of complications is a positive blood culture at 48 to 96 hours after an initial positive blood culture, as shown in a large prospective cohort study by Fowler et al.7,10-11 Additional independent risk factors (see Table 1) include community acquisition (likely due to prolonged duration of bacteremia), skin examination suggesting the presence of acute systemic infection, and persistent fever at 72 hours after the first positive blood culture. Patients with even one of these risk factors are at high risk for a complicated course (which occurs in about 35%). In a case-control study, Chihara et al showed that S. aureus bacteruria in the absence of urinary tract pathology or recent urinary tract instrumentation might be associated with threefold increased mortality compared with those without bacteriuria, even after adjustment for comorbid conditions.12
Antimicrobial Treatment
The initial choice of antibiotic therapy for SAB must take into account the MRSA prevalence in the community and hospital. If suspicion is high enough for MRSA, the IDSA’s 2011 guidelines suggest treatment with vancomycin or daptomycin.3 Although there are no published RCTs to support a particular antibiotic regimen, there are trials to suggest that a delay in treatment could be harmful. One study, by Lordis et al, showed that a delay in treatment, as defined by treatment after 44.75 hours, was associated with a longer hospital stay, with the delayed treatment group being hospitalized for 20.2 days and the early treatment group being hospitalized for 14.3 days.13 A delay in treatment was also found to be an independent predictor of mortality.13
Once susceptibilities are known, it is important to appropriately tailor antibiotics, as studies have shown lower treatment failure rates with the use of beta-lactam antibiotics when compared with empiric MRSA coverage.14-15 In one prospective study of 123 hemodialysis patients with MSSA bacteremia, Stryjewski et al showed that those treated with vancomycin were at higher risk of experiencing treatment failure than those treated with cefazolin.15 In another prospective observational study of 505 patients with SAB, Chang et al found that treatment with nafcillin was superior to vancomycin in preventing persistent bacteremia or relapse for MSSA bacteremia.14 These studies highlight the benefits of adjusting the empirically selected antibiotics, as narrowing the spectrum can result in less treatment failure.
If susceptibilities confirm MRSA, the IDSA recommends continued treatment with vancomycin or daptomycin.3 Although vancomycin is most commonly used, partly because of low cost and familiarity, Fowler et al published a study of 246 patients with SAB with or without endocarditis, assigning them to treatment with daptomycin, initial low-dose gentamicin plus vancomycin or an antistaphylococcal penicillin.16 The study found that daptomycin was not inferior to the other therapies, confirming that daptomycin is a reasonable choice in the treatment of MRSA infections.
Oral antibiotics are an option to treat SAB when necessary. A RCT by Heldman et al of 85 intravenous drug users with SAB (and suspected right-sided endocarditis, 65% of which had HIV) showed similar efficacy of ciprofloxacin plus rifampin versus standard intravenous therapy.17 A subsequent randomized trial of 104 patients with SAB comparing oral fleroxacin plus rifampin against conventional intravenous therapy also showed similar cure rates, with the added benefit of earlier discharge.18 Furthermore, in a meta-analysis of five randomized studies by Shorr et al (see Table 2), linezolid was found to have outcomes that were not inferior to vancomycin (clinical cure/microbiological success of 56%/69% in the linezolid group and 46%/73% in the vancomycin group).19
Treatment Duration
Recommendations for the duration of antibiotic treatment for SAB are mainly based on observational studies, which show mixed results. In one study done in the 1950s, about two-thirds of cases of SAB were associated with endocarditis, and longer courses of intravenous therapy (greater than four weeks) were recommended.20
More recently, with the increasing rates of catheter-related SAB and its relatively high rate of expeditious blood culture clearance, a shorter duration has been evaluated in several studies. In 1992, an analysis of published data and a retrospective case series concluded that fewer than 10 days of intravenous antibiotics might be associated with an increased risk of recurrence, but 10 to 14 days of intravenous therapy was effective for most cases of catheter-associated SAB.5 In another prospective study, Fowler et al found that a seven-day course of intravenous antibiotic therapy may be sufficient for simple, catheter-related infections.21 A subsequent prospective study by Jensen et al reported that a course of antibiotic therapy of less than 14 days might be associated with higher mortality compared to a longer course.9 A prospective study of 276 patients by Thomas et al found there was no relationship between relapse and duration of treatment (seven to 15 days) in catheter-related SAB, concluding that more than 14 days of antibiotic therapy was unnecessary.22
Per IDSA guidelines, uncomplicated SAB (no implanted prosthesis, negative blood cultures within two to four days, defervescence within 72 hours of initiating therapy, and lack of metastatic complication) can be treated with a two-week course of antibiotics, while complicated bacteremia (any of above criteria) should be treated within four to six weeks.3
Monitoring for Complications: Echos
Based on the IDSA guidelines, echocardiography is recommended in all patients with bacteremia, with a preference of transesophageal echocardiography (TEE) over transthoracic echocardiography (TTE).3 More recently, Kaasch et al developed simple criteria to identify patients with nosocomial SAB at low risk for infective endocarditis based on two prospective cohort studies.23 Lack of any of these criteria, which include prolonged bacteremia of more than four days’ duration, presence of a permanent intracardiac device, hemodialysis dependency, spinal infection and nonvertebral osteomyelitis, along with a negative TTE indicates that a TEE is not necessary (see Table 3). However, these patients need close follow-up to ensure that bacteremia clears and no new signs or symptoms concerning for metastatic infection develop.
ID Consultation
Several studies have shown that ID consultation not only improves adherence to evidence-based management of SAB, but it also reduces mortality.24-27 In a recent prospective cohort study in a tertiary-care center, even after adjusting for pre-existing comorbidities and severity of disease, an ID consult was associated with a 56% reduction in 28-day mortality.24 The patients who were followed by an ID consult service were more likely to receive appropriate duration of antibiotics (81% vs. 29%, respectively) and undergo appropriate workup for the evaluation of metastatic infections (34% and 8%, respectively). This study concluded that routine ID consult should be considered in patients with SAB, especially those with severe illness and multiple comorbid conditions.
Back to the Case
The patient was started on empiric therapy with vancomycin and serial blood cultures were obtained. He remained hemodynamically stable but febrile, with persistently positive blood and urine cultures. Given concern for the port being the source of his infection, his chest port was removed. A high-quality TTE was performed and was unremarkable.
ID was consulted. Blood cultures subsequently grew MSSA and vancomycin was switched to cefazolin 2g every eight hours. On hospital Day 5, his fever resolved and blood cultures turned negative. There were no clinical signs or symptoms for metastatic infections. A PICC line was placed after blood cultures remained negative for 48 hours. The decision was made to treat him with four weeks of antibiotics from his last positive blood culture, with follow-up in ID clinic.
Bottom Line
SAB is a common worldwide cause of morbidity and mortality. Treatment should include removing the nidus if present, finding and administering the appropriate antimicrobial therapy, evaluating for possible complications, and consulting with ID.
Dr. Ward is an assistant professor, Dr. Kim a clinical instructor, and Dr. Stojan a clinical lecturer at the University of Michigan Health System in Ann Arbor.
Acknowledgement
The authors would like to thank Dr. Jeffrey Rohde for reviewing the manuscript.
References
- Shorr AF, Tabak YP, Killian AD, et al. Healthcare-associated bloodstream infection: a distinct entity? Insights from a large U.S. database. Crit Care Med. 2006;34:3588-3595.
- Mylotte JM, McDermott C, Spooner JA. Prospective study of 114 consecutive episodes of Staphylococcus aureus bacteremia. Rev Infect Dis. 1987;9:891-907.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3).
- Naber CK, Baddour LM, Giamarellos-Bourboulis EJ, et al. Clinical consensus conference: survey on gram-positive bloodstream infections with a focus on Staphylococcus aureus. Clin Infect Dis. 2011;52(3):285-292.
- Thwaites GE, Edgeworth JD, Gkrania-Klotsas E, et al. Clinical management of Staphylococcus bacteremia. Lancet Infect Dis. 2011;11(3):208-222.
- Johnson LB, Almoujahed MO, Ilg K, et al. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand J Infec Dis. 2003;35(11-12):782-789.
- Fowler VG Jr., Olsen MK, Corey GR, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med. 2003;163:2066-2072.
- Mylotte JM, Tayara A. Staphylococcus aureus bacteremia: predictors of 30-day mortality in a large cohort. Clin Infect Dis. 2000;31:1170-1174.
- Jensen AG, Wachmann CH, Espersen F, et al. Treatment and outcome of Staphylococcus aureus bacteremia: a prospective study of 278 cases. Arch Intern Med. 2002;162(1):25-32.
- Malani PN, Rana MM, Banerjee M, et al. Staphylococcus aureus bloodstream infections: the association between age and mortality and functional status. J Am Geriatr Soc. 2008;56(8):1485-1489.
- Kim SH, Park WB, Lee KD, et al. Outcome of Staphylococcus aureus bacteremia in patients with eradicable foci versus noneradicable foci. Clin Infect Dis. 2003;37(6):794-799.
- Chihara S. Popovich KJ, Weinstein RA, et al. Staphylococcus aureus bacteriuria as a prognosticator for outcome of Staphylococcus aureus bacteremia: a case control study. BMC Inf Dis. 2010;10:225.
- Lodise TP, McKinnon PS, Swiderski L, Rybak MJ. Outcomes analysis of delayed antibiotic treatment for hospital-acquired Staphylococcus aureus bacteremia. Clin Infect Dis. 2003;36:1418-1423.
- Chang FY, Peacock JE Jr., Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore). 2003;82:333.
- Stryjewski ME, Szczech LA, Benjamin DK Jr., et al. Use of vancomycin or first-generation cephalosporins for the treatment of hemodialysisdependent patients with methicillin-susceptible Staphylococcus aureus bacteremia. Clin Infect Dis. 2007;44:190-196.
- Fowler VG Jr., Boucher HW, Corey GR, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med. 2006;355:653.
- Heldman AW, Hartert TV, Ray SC, et al. Oral antibiotic treatment of right-sided staphylococcal endocarditis in injection drug users: prospective randomized comparison with parenteral therapy. Am J Med. 1996;101:68-76.
- Schrenzel J, Harbarth S, Schockmel G, et al. A randomized clinical trial to compare fleroxacin-rifampicin with flucloxacillin or vancomycin for the treatment of Staphylococcal infection. Clin Infect Dis. 2004;39:1285-1292.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother. 2005;56:923-929.
- Wilson R, Hamburger M. Fifteen years’ experience with Staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med. 1957;22:437-457.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendation of infectious disease specialists: experience with 244 patients. Clin Infect Dis. 1998;27:478-486.
- Thomas MG, Morris AJ. Cannula-associated Staphylococcus aureus bacteremia: outcome in relation to treatment. Intern Med J. 2005;35:319-330.
- Kaasch AJ, Fowler VG Jr, Rieg S, et al. Use of a simple criteria set for guiding echocardiography in nosocomidal Staphylococcus aureus bacteremia. Clin Infect Dis. 2011;53:1-9.
- Honda H, Krauss MJ, Jones JC, et al. The value of infectious disease consultation in Staphylococcus aureus bacteremia. Am J Med. 2010;123:631-637.
- Nagao M, Iinuma Y, Saito T, et al. Close cooperation between infectious disease physicians and attending physicians can result in better management and outcome for patients with Staphylococcus aureus bacteremia. Euro Soc Clin Microbiology Infect Dis. 2010;16:1783-1788.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendations of infectious diseases specialist: experience with 244 patients. Clin Infect Dis. 1998;27(3):478-486.
- Jenkins TC, Price CS, Sabel AL et al. Impact of routine infectious diseases service consultation on the evaluation, management, and outcome of Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(7):1000-1008.
Case
An 82-year-old man with non-Hodgkin’s lymphoma in remission and a history of congestive heart failure and hypertension presents with one week of generalized malaise and intermittent fevers. Vitals show a temperature of 101oF, blood pressure of 130/60 mmHg, and heart rate of 100. His exam is notable for an erythematous and tender chest port site, with no murmurs. Blood cultures drawn upon presentation show gram-positive cocci speciated to Staphylococcus aureus. What are the next steps in management of this patient?
Overview
S. aureus bacteremia (SAB) is a common infectious cause of morbidity and mortality worldwide, causing both community-acquired and hospital-acquired bacteremia. In the U.S. alone, it accounts for 23% of all bloodstream infections and is the bacterial pathogen most strongly associated with death.1 Mortality rates are approximately 42% in those with methicillin-resistant S. aureus (MRSA) bacteremia and 28% in those with methicillin-sensitive S. aureus (MSSA) bacteremia.2
Recognizing the severity of SAB, the Infectious Disease Society of America (IDSA) published treatment guidelines in 2011 to help direct the clinical care of this disease process.3 However, the majority of the recommendations are based on observational studies and expert opinion, as less than 1,500 patients have been enrolled in randomized controlled trials specifically targeted to investigate the treatment of SAB.4
Review of the Data
A clinically significant SAB usually is defined as the isolation of S. aureus from a venous blood culture with associated symptoms and signs of systemic infection.5 As SAB contamination is rare and can be associated with multiple complications, including metastatic infections, embolic stroke, recurrent infection, and death, any finding of a positive blood culture must be taken seriously.4
SAB treatment is multifaceted and should focus on the removal of any nidus of infection, such as a catheter or a prosthetic device, the use of prolonged antimicrobial therapy, and the evaluation of potential complications. In a retrospective study, Johnson et al showed that failure to remove the source is one of the strongest independent predictors of relapse in patients with SAB.6 However, 10% to 40% of patients have no identifiable focus, which increases the impetus to evaluate for complications.7-8 Overall, approximately one-third of patients with SAB develop metastatic complications, either from hematogenous seeding or local extension of infection.9
In addition to advanced age and such comorbid conditions as cirrhosis, the strongest predictor of complications is a positive blood culture at 48 to 96 hours after an initial positive blood culture, as shown in a large prospective cohort study by Fowler et al.7,10-11 Additional independent risk factors (see Table 1) include community acquisition (likely due to prolonged duration of bacteremia), skin examination suggesting the presence of acute systemic infection, and persistent fever at 72 hours after the first positive blood culture. Patients with even one of these risk factors are at high risk for a complicated course (which occurs in about 35%). In a case-control study, Chihara et al showed that S. aureus bacteruria in the absence of urinary tract pathology or recent urinary tract instrumentation might be associated with threefold increased mortality compared with those without bacteriuria, even after adjustment for comorbid conditions.12
Antimicrobial Treatment
The initial choice of antibiotic therapy for SAB must take into account the MRSA prevalence in the community and hospital. If suspicion is high enough for MRSA, the IDSA’s 2011 guidelines suggest treatment with vancomycin or daptomycin.3 Although there are no published RCTs to support a particular antibiotic regimen, there are trials to suggest that a delay in treatment could be harmful. One study, by Lordis et al, showed that a delay in treatment, as defined by treatment after 44.75 hours, was associated with a longer hospital stay, with the delayed treatment group being hospitalized for 20.2 days and the early treatment group being hospitalized for 14.3 days.13 A delay in treatment was also found to be an independent predictor of mortality.13
Once susceptibilities are known, it is important to appropriately tailor antibiotics, as studies have shown lower treatment failure rates with the use of beta-lactam antibiotics when compared with empiric MRSA coverage.14-15 In one prospective study of 123 hemodialysis patients with MSSA bacteremia, Stryjewski et al showed that those treated with vancomycin were at higher risk of experiencing treatment failure than those treated with cefazolin.15 In another prospective observational study of 505 patients with SAB, Chang et al found that treatment with nafcillin was superior to vancomycin in preventing persistent bacteremia or relapse for MSSA bacteremia.14 These studies highlight the benefits of adjusting the empirically selected antibiotics, as narrowing the spectrum can result in less treatment failure.
If susceptibilities confirm MRSA, the IDSA recommends continued treatment with vancomycin or daptomycin.3 Although vancomycin is most commonly used, partly because of low cost and familiarity, Fowler et al published a study of 246 patients with SAB with or without endocarditis, assigning them to treatment with daptomycin, initial low-dose gentamicin plus vancomycin or an antistaphylococcal penicillin.16 The study found that daptomycin was not inferior to the other therapies, confirming that daptomycin is a reasonable choice in the treatment of MRSA infections.
Oral antibiotics are an option to treat SAB when necessary. A RCT by Heldman et al of 85 intravenous drug users with SAB (and suspected right-sided endocarditis, 65% of which had HIV) showed similar efficacy of ciprofloxacin plus rifampin versus standard intravenous therapy.17 A subsequent randomized trial of 104 patients with SAB comparing oral fleroxacin plus rifampin against conventional intravenous therapy also showed similar cure rates, with the added benefit of earlier discharge.18 Furthermore, in a meta-analysis of five randomized studies by Shorr et al (see Table 2), linezolid was found to have outcomes that were not inferior to vancomycin (clinical cure/microbiological success of 56%/69% in the linezolid group and 46%/73% in the vancomycin group).19
Treatment Duration
Recommendations for the duration of antibiotic treatment for SAB are mainly based on observational studies, which show mixed results. In one study done in the 1950s, about two-thirds of cases of SAB were associated with endocarditis, and longer courses of intravenous therapy (greater than four weeks) were recommended.20
More recently, with the increasing rates of catheter-related SAB and its relatively high rate of expeditious blood culture clearance, a shorter duration has been evaluated in several studies. In 1992, an analysis of published data and a retrospective case series concluded that fewer than 10 days of intravenous antibiotics might be associated with an increased risk of recurrence, but 10 to 14 days of intravenous therapy was effective for most cases of catheter-associated SAB.5 In another prospective study, Fowler et al found that a seven-day course of intravenous antibiotic therapy may be sufficient for simple, catheter-related infections.21 A subsequent prospective study by Jensen et al reported that a course of antibiotic therapy of less than 14 days might be associated with higher mortality compared to a longer course.9 A prospective study of 276 patients by Thomas et al found there was no relationship between relapse and duration of treatment (seven to 15 days) in catheter-related SAB, concluding that more than 14 days of antibiotic therapy was unnecessary.22
Per IDSA guidelines, uncomplicated SAB (no implanted prosthesis, negative blood cultures within two to four days, defervescence within 72 hours of initiating therapy, and lack of metastatic complication) can be treated with a two-week course of antibiotics, while complicated bacteremia (any of above criteria) should be treated within four to six weeks.3
Monitoring for Complications: Echos
Based on the IDSA guidelines, echocardiography is recommended in all patients with bacteremia, with a preference of transesophageal echocardiography (TEE) over transthoracic echocardiography (TTE).3 More recently, Kaasch et al developed simple criteria to identify patients with nosocomial SAB at low risk for infective endocarditis based on two prospective cohort studies.23 Lack of any of these criteria, which include prolonged bacteremia of more than four days’ duration, presence of a permanent intracardiac device, hemodialysis dependency, spinal infection and nonvertebral osteomyelitis, along with a negative TTE indicates that a TEE is not necessary (see Table 3). However, these patients need close follow-up to ensure that bacteremia clears and no new signs or symptoms concerning for metastatic infection develop.
ID Consultation
Several studies have shown that ID consultation not only improves adherence to evidence-based management of SAB, but it also reduces mortality.24-27 In a recent prospective cohort study in a tertiary-care center, even after adjusting for pre-existing comorbidities and severity of disease, an ID consult was associated with a 56% reduction in 28-day mortality.24 The patients who were followed by an ID consult service were more likely to receive appropriate duration of antibiotics (81% vs. 29%, respectively) and undergo appropriate workup for the evaluation of metastatic infections (34% and 8%, respectively). This study concluded that routine ID consult should be considered in patients with SAB, especially those with severe illness and multiple comorbid conditions.
Back to the Case
The patient was started on empiric therapy with vancomycin and serial blood cultures were obtained. He remained hemodynamically stable but febrile, with persistently positive blood and urine cultures. Given concern for the port being the source of his infection, his chest port was removed. A high-quality TTE was performed and was unremarkable.
ID was consulted. Blood cultures subsequently grew MSSA and vancomycin was switched to cefazolin 2g every eight hours. On hospital Day 5, his fever resolved and blood cultures turned negative. There were no clinical signs or symptoms for metastatic infections. A PICC line was placed after blood cultures remained negative for 48 hours. The decision was made to treat him with four weeks of antibiotics from his last positive blood culture, with follow-up in ID clinic.
Bottom Line
SAB is a common worldwide cause of morbidity and mortality. Treatment should include removing the nidus if present, finding and administering the appropriate antimicrobial therapy, evaluating for possible complications, and consulting with ID.
Dr. Ward is an assistant professor, Dr. Kim a clinical instructor, and Dr. Stojan a clinical lecturer at the University of Michigan Health System in Ann Arbor.
Acknowledgement
The authors would like to thank Dr. Jeffrey Rohde for reviewing the manuscript.
References
- Shorr AF, Tabak YP, Killian AD, et al. Healthcare-associated bloodstream infection: a distinct entity? Insights from a large U.S. database. Crit Care Med. 2006;34:3588-3595.
- Mylotte JM, McDermott C, Spooner JA. Prospective study of 114 consecutive episodes of Staphylococcus aureus bacteremia. Rev Infect Dis. 1987;9:891-907.
- Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3).
- Naber CK, Baddour LM, Giamarellos-Bourboulis EJ, et al. Clinical consensus conference: survey on gram-positive bloodstream infections with a focus on Staphylococcus aureus. Clin Infect Dis. 2011;52(3):285-292.
- Thwaites GE, Edgeworth JD, Gkrania-Klotsas E, et al. Clinical management of Staphylococcus bacteremia. Lancet Infect Dis. 2011;11(3):208-222.
- Johnson LB, Almoujahed MO, Ilg K, et al. Staphylococcus aureus bacteremia: compliance with standard treatment, long-term outcome and predictors of relapse. Scand J Infec Dis. 2003;35(11-12):782-789.
- Fowler VG Jr., Olsen MK, Corey GR, et al. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med. 2003;163:2066-2072.
- Mylotte JM, Tayara A. Staphylococcus aureus bacteremia: predictors of 30-day mortality in a large cohort. Clin Infect Dis. 2000;31:1170-1174.
- Jensen AG, Wachmann CH, Espersen F, et al. Treatment and outcome of Staphylococcus aureus bacteremia: a prospective study of 278 cases. Arch Intern Med. 2002;162(1):25-32.
- Malani PN, Rana MM, Banerjee M, et al. Staphylococcus aureus bloodstream infections: the association between age and mortality and functional status. J Am Geriatr Soc. 2008;56(8):1485-1489.
- Kim SH, Park WB, Lee KD, et al. Outcome of Staphylococcus aureus bacteremia in patients with eradicable foci versus noneradicable foci. Clin Infect Dis. 2003;37(6):794-799.
- Chihara S. Popovich KJ, Weinstein RA, et al. Staphylococcus aureus bacteriuria as a prognosticator for outcome of Staphylococcus aureus bacteremia: a case control study. BMC Inf Dis. 2010;10:225.
- Lodise TP, McKinnon PS, Swiderski L, Rybak MJ. Outcomes analysis of delayed antibiotic treatment for hospital-acquired Staphylococcus aureus bacteremia. Clin Infect Dis. 2003;36:1418-1423.
- Chang FY, Peacock JE Jr., Musher DM, et al. Staphylococcus aureus bacteremia: recurrence and the impact of antibiotic treatment in a prospective multicenter study. Medicine (Baltimore). 2003;82:333.
- Stryjewski ME, Szczech LA, Benjamin DK Jr., et al. Use of vancomycin or first-generation cephalosporins for the treatment of hemodialysisdependent patients with methicillin-susceptible Staphylococcus aureus bacteremia. Clin Infect Dis. 2007;44:190-196.
- Fowler VG Jr., Boucher HW, Corey GR, et al. Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureus. N Engl J Med. 2006;355:653.
- Heldman AW, Hartert TV, Ray SC, et al. Oral antibiotic treatment of right-sided staphylococcal endocarditis in injection drug users: prospective randomized comparison with parenteral therapy. Am J Med. 1996;101:68-76.
- Schrenzel J, Harbarth S, Schockmel G, et al. A randomized clinical trial to compare fleroxacin-rifampicin with flucloxacillin or vancomycin for the treatment of Staphylococcal infection. Clin Infect Dis. 2004;39:1285-1292.
- Shorr AF, Kunkel MJ, Kollef M. Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studies. J Antimicrob Chemother. 2005;56:923-929.
- Wilson R, Hamburger M. Fifteen years’ experience with Staphylococcus septicemia in a large city hospital; analysis of fifty-five cases in the Cincinnati General Hospital 1940 to 1954. Am J Med. 1957;22:437-457.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendation of infectious disease specialists: experience with 244 patients. Clin Infect Dis. 1998;27:478-486.
- Thomas MG, Morris AJ. Cannula-associated Staphylococcus aureus bacteremia: outcome in relation to treatment. Intern Med J. 2005;35:319-330.
- Kaasch AJ, Fowler VG Jr, Rieg S, et al. Use of a simple criteria set for guiding echocardiography in nosocomidal Staphylococcus aureus bacteremia. Clin Infect Dis. 2011;53:1-9.
- Honda H, Krauss MJ, Jones JC, et al. The value of infectious disease consultation in Staphylococcus aureus bacteremia. Am J Med. 2010;123:631-637.
- Nagao M, Iinuma Y, Saito T, et al. Close cooperation between infectious disease physicians and attending physicians can result in better management and outcome for patients with Staphylococcus aureus bacteremia. Euro Soc Clin Microbiology Infect Dis. 2010;16:1783-1788.
- Fowler VG Jr., Sanders LL, Sexton DJ, et al. Outcome of Staphylococcus aureus bacteremia according to compliance with recommendations of infectious diseases specialist: experience with 244 patients. Clin Infect Dis. 1998;27(3):478-486.
- Jenkins TC, Price CS, Sabel AL et al. Impact of routine infectious diseases service consultation on the evaluation, management, and outcome of Staphylococcus aureus bacteremia. Clin Infect Dis. 2008;46(7):1000-1008.
A Double‐Edged Sword
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.

Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
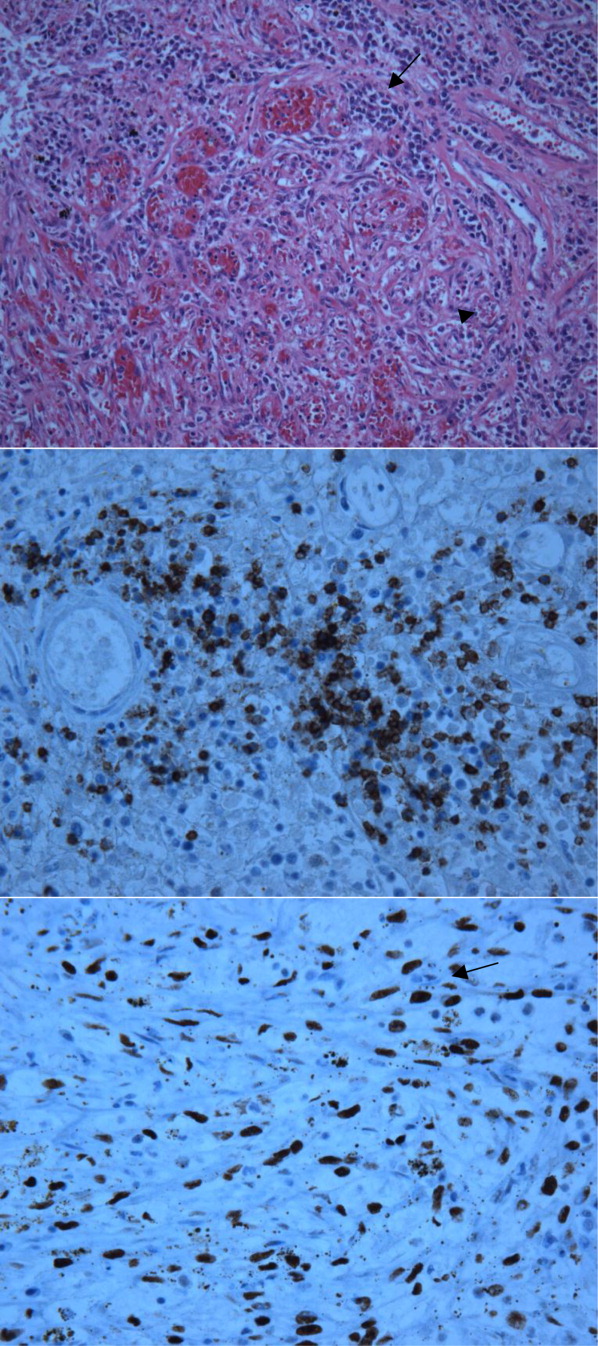
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 40‐year‐old man with human immunodeficiency virus (HIV) infection and a CD4 count of 58 cells/L was admitted to the hospital with 1 month of fevers, night sweats, a 5‐kg weight loss, several weeks of progressive dyspnea on exertion, and a nonproductive cough. He denied headaches, vision changes, odynophagia, diarrhea, or rash. He had no history of opportunistic infections, HIV‐associated neoplasms, or other relevant past medical history. He was diagnosed with HIV 3 years ago and had been off antiretroviral therapy (ART) for the last 10 months. Two weeks prior to this presentation, he was seen in clinic but did not report his symptoms. He was prescribed trimethoprim/sulfamethoxazole (TMP/SMX) for prophylaxis against Pneumocystis jirovecii pneumonia (PCP). He had recently moved from New York City to San Francisco, had quit smoking within the last month, and denied alcohol or illicit drug use.
At a CD4 cell count of 58 cells/L, the patient is at risk for the entire spectrum of HIV‐associated opportunistic infections and neoplasms. The presence of fevers, night sweats, and weight loss suggests the possibility of a disseminated infection, although a neoplastic process with accompanying B symptoms should also be considered. Dyspnea and nonproductive cough indicate cardiopulmonary involvement. The duration of these complaints is more suggestive of a nonbacterial infectious etiology (e.g., PCP, mycobacterial or fungal disease) than a bacterial etiology (e.g., Streptococcus pneumoniae). Irrespective of CD4 count, patients with HIV are at increased risk for cardiovascular events and pulmonary arterial hypertension, although the time course and presence of constitutional symptoms makes these diagnoses less likely. Similarly, patients with HIV are at increased risk for chronic obstructive pulmonary disease (COPD), and the patient does have a history of cigarette smoking, but the clinical history and systemic involvement make COPD unlikely.
On physical examination, the patient was in no acute distress. The temperature was 36C, the blood pressure 117/68 mm Hg, the heart rate 106 beats per minute, the respiratory rate 18 breaths per minute, and the oxygen saturation 100% on ambient air. No oral lesions were noted, and his neck was supple with nontender bilateral cervical lymphadenopathy measuring up to 1.5 cm. There was no jugular venous distension or peripheral edema. The cardiovascular exam revealed tachycardia with a regular rhythm and no murmurs or gallops. His lungs were clear to auscultation. The spleen tipwas palpable. No rashes were identified. The neurological examination, including mental status, was normal.
The white blood cell count was 2400/mm3, the hemoglobin 7 g/dL with mean corpuscular volume of 86 fL, and the platelet count 162,000/mm3. Basic chemistry, liver, and glucose‐6‐phosphate dehydrogenase (G6PD) tests were within the laboratory's normal range. The HIV viral load was 150,000 copies/mL. Chest radiography revealed bibasilar hazy opacities, and computerized tomography (CT) of the chest revealed a focal nodular consolidation in the right middle lobe along with subcentimeter bilateral axillary and mediastinal lymphadenopathy. There were no ground‐glass opacities.
The patient's physical examination does not support a cardiac disorder. Lymphadenopathy is nonspecific, but it is consistent with a potential infectious or neoplastic process. Leukopenia and anemia suggest potential bone‐marrow infiltration or suppression by TMP/SMX. Although the pulmonary exam was nonfocal, chest imaging is the cornerstone of the evaluation of suspected pulmonary disease in persons with HIV. The focal nodular consolidation on chest CT is nonspecific but is more characteristic of typical or atypical bacterial pneumonia, mycobacterial disease such as tuberculosis, or fungal pneumonia than PCP or viral pneumonia. A lack of ground‐glass opacities also makes PCP and interstitial lung diseases less likely.
The patient was treated for community‐acquired pneumonia with ceftriaxone and doxycycline with improvement in dyspnea. Antiretroviral therapy with darunavir, ritonavir, tenofovir, and emtricitabine was initiated. Azithromycin was started for prophylaxis against Mycobacterium avium complex (MAC). The TMP/SMX was changed to dapsone, given concern for bone‐marrow suppression. Blood cultures for bacteria, fungi, and mycobacteria were negative. Polymerase chain reaction from pharyngeal swab for influenza A and B, parainfluenza types 13, rhinovirus, and respiratory syncytial virus were negative. Several attempts to obtain sputum for acid‐fast bacillus staining and culture were unsuccessful because the patient was unable to expectorate sputum. Serum interferon‐gamma release assay for M. tuberculosis and thefollowing serologic studies were also negative: cytomegalovirus, Epstein‐Barr virus, parvovirus, Bartonella species, Coccidioides immitis, and Cryptococcus neoformans antigen. Given his improvement, the patient was discharged from the hospital on ART, doxycycline for community‐acquired pneumonia, and prophylactic azithromycin and dapsone with scheduled outpatient follow‐up.
Ten days later, he was seen in clinic. Though his dyspnea had improved after completing the doxycycline, he noted a persistent dry cough and daily fevers to 40C. The physical exam was unchanged, including persistent cervical lymphadenopathy. Laboratories revealed a white blood cell count of 2400/mm3, hemoglobin of 4.8 g/dL, and a platelet count of 122,000/mm3. The absolute reticulocyte count was 21,000/L (normal value, 20,000100,000/L). A peripheral blood smear was unremarkable, and serum lactate dehydrogenase (LDH) was within normal limits. The direct antiglobulin test (DAT) was negative. The patient was readmitted to the hospital.
The initial improvement in dyspnea but persistent fevers and cough and worsening pancytopenia are suggestive of multiple processes occurring simultaneously. Dapsone can cause both hemolytic anemia and aplastic anemia, although the peripheral smear, normal LDH and G6PD, and negative DAT are not consistent with the former. Bone‐marrow suppression from a combination of ART medications and dapsone cannot be ruled out. An infiltrative process involving the bone marrow, including tuberculosis, MAC, disseminated fungal infection, or malignancy, remains a possibility. Repeat chest imaging is warranted to assess the prior right middle lobe consolidation and to further evaluate the persistent respiratory complaints.
Prophylaxis of PCP with dapsone was switched to atovaquone due to persistent anemia. A repeat CT of the chest and a concurrent abdominal CT revealed interval enlargement of mediastinal lymph nodes with multiple periportal, retroperitoneal, and hilar nodes not present on prior chest imaging, in addition to new bilateral centrilobular nodules and interval development of small bilateral pleural effusions. The abdominal CT also showed hepatosplenomegaly with splenic‐vein engorgement. Empiric treatment for disseminated MAC infection with clarithromycin and ethambutol was initiated in addition to vancomycin and cefepime for possible healthcare‐associated pneumonia. Over the next several days, the patient continued to have daily fevers up to 39.8C. A repeat CD4 count 3 weeks after starting ART was 121 cells/L. The HIV RNA level had decreased to 854 copies/mL.
The patient has developed progressive, generalized lymphadenopathy, worsening pancytopenia, and persistent fevers in the setting of negative cultures and serologic studies and despite treatment for MAC. This constellation, along with the radiographic findings of hilar lymphadenopathy and pleural effusions, is suggestive of non‐Hodgkin lymphoma (NHL). Alternatively, Kaposi sarcoma (KS) or tuberculosis can have a similar radiographic and clinical presentation, although pancytopenia from KS seems unusual. The lymphadenopathy could be consistent with multicentric Castleman disease or bacillary angiomatosis (BA), although the latter diagnosis would be unlikely given recent antibiotic therapy. At this time, a careful search for other manifestations and reasonable targets for biopsy is warranted. An appropriate suppression of the HIV viral load after initiation of ART, with improvement in CD4 count, is the proper context for the immune reconstitution inflammatory syndrome (IRIS), which is characterized by paradoxical worsening or unmasking of a disseminated process.
A bone‐marrow biopsy revealed marked dysmegakaryopoiesis and mild dyserythropoiesis, but no other abnormalities. Flow cytometry and histoimmunochemical staining did not show evidence of lymphoproliferative disorder in the marrow. Smears and cultures of the bone marrow for bacteria, acid‐fast bacilli, and fungi were negative. A right cervical lymph node biopsy was performed, with multiple fine‐needle aspiration and core samples taken. Bacterial, fungal, and acid‐fast bacilli tissue cultures were without growth, and initial pathology results were concerning for high‐grade lymphoma. A monoclonal proliferation of lymphocytes was noted on flow cytometry of the tissue sample. The patient developed progressive dyspnea, tachypnea, and hypoxemia. A chest x‐ray revealed worsening perihilar and basilar opacities.
The possibility of bone‐marrow sampling error must be considered in a patient that has such a high pretest probability for lymphoma or infection, but staining, immunological assays, cultures, and direct assessment by pathologists generally give some suggestion of an alternative diagnosis. The bone‐marrow findings are compatible with HIV‐related changes, but continued vigilance for infection and malignancy is warranted. Although the diagnosis of NHL based on the cervical biopsy result is only preliminary, the patient's rapidly deteriorating clinical status warrants initiation of treatment with steroids while awaiting definitive results, particularly given his poor response to aggressive management of potential infectious causes. A bronchoscopy should be considered given the predominance of pulmonary symptoms and his rapid respiratory decline.
Approximately 1 week after admission, high‐dose systemic corticosteroids were administered for presumed aggressive lymphoma. Over the next 48 hours, the patient's hypoxemia worsened, and he was intubated for hypoxemic respiratory failure. A repeat chest CT (see Fig. 1) showed bilateral peribronchovascular patchy consolidations and pleural effusions without evidence of pulmonary embolism. The patient was also noted to have a single, discrete violaceous nodule on the hard palate as well as a nodule with similar appearance on his upper chest (neither lesion was present on admission). A skin biopsy was obtained. Despite steroids, antibiotic therapy, and aggressive critical‐care management, severe acidosis, progressive acute kidney injury, and anuria ensued. Continuous venovenous hemodialysis was initiated.
Discrete violaceous nodules with mucocutaneous localization in the context of AIDS are virtually pathognomonic for KS. Rarely, BA may be misdiagnosed as KS, or they may occur concurrently. The patient's current clinical deterioration, radiographic findings, and development of new skin lesions in the setting of response to ART are concerning for KS‐related IRIS with visceral involvement. It is likely that systemic corticosteroids are potentiating KS‐related IRIS. At this point, there is compelling evidence of 2 distinct systemic disease processes: lymphoma and KS‐related IRIS, both of which may be contributing to respiratory failure. Steroids can be highly effective in the treatment of high‐grade lymphoma but can be harmful in patients with KS, where they have been shown to potentially exacerbate underlying disease. Given the patient's worsening respiratory status, discontinuation of corticosteroids and initiation of chemotherapy against both opportunistic malignancies should be considered.
The patient's condition deteriorated with progressive acidosis and hypoxemia, and he died shortly after being transitioned to comfort‐care measures. Review of the skin biopsy revealed KS. Autopsy revealed disseminated KS involving the skin, lymph nodes, and lungs, and high‐grade anaplastic plasmablastic lymphoma infiltrating multiple lymph nodes and organs, including the lungs (see Fig. 2). There was no evidence of infection.
COMMENTARY
This case demonstrates the simultaneous fatal progression of 2 treatable HIV‐associated malignancies in an era in which the end‐stage manifestations of untreated HIV are becoming less common, particularly in developed countries. Modern ARTthe centerpiece of progress with HIVhas yielded dramatic improvements in prognosis, but in this case, by precipitating KS‐IRIS, ART paradoxically contributed to this patient's demise. Similarly, high‐dose systemic corticosteroids, which were deemed necessary to stabilize the progression of his high‐grade lymphoma, likely accelerated his KS. This corticosteroid‐mediated worsening appears to be unique to KS given that corticosteroids are often recommended to treat severe presentations of IRIS in other diseases (eg, tuberculosis, MAC, PCP).
Immune reconstitution inflammatory syndrome is the paradoxical worsening of well‐controlled disease or progression of previously occult disease after initiation of ART.1
Although infectious diseasesincluding mycobacteria, cytomegalovirus, cryptococcosis, or PCPare best known for their ability to recrudesce or manifest with a recovering immune system, opportunistic malignancies such as KS can do the same. Risk factors for development of IRIS are low pre‐ART CD4 count, high pre‐ART viral load, and rapid response to ART.2 In 1 large series, the median time to diagnosis of IRIS was 33 days.2 Immune reconstitution inflammatory syndrome is a clinical diagnosis without specific pathologic findings. Because IRIS is a diagnosis of exclusion, other explanations for worsening disease, including drug resistance, drug reactions (eg, abacavir hypersensitivity syndrome), and poor adherence to medications, should be ruled out before making the diagnosis.
Kaposi sarcoma is a vascular tumor associated with infection by human herpesvirus 8 (HHV‐8). The incidence of AIDS‐related KS has declined substantially in the post‐ART era.2, 4 The classic radiographic presentation of pulmonary KS includes central bilateral opacities with a peribronchovascular distribution as well as pulmonary nodules, intraseptal thickening, mediastinal lymphadenopathy, and associated pleural effusions.5, 6 Kaposi sarcomarelated IRIS has been described as developing within weeks of ART initiation and is associated with substantial morbidity and mortality, particularly in the context of pulmonary involvement, with 1 recent series showing 100% mortality in patients who did not receive chemotherapy.79
Human immunodeficiency virusassociated KS can respond well to ART alone. Indications for systemic chemotherapy for KS include extensive mucocutaneous disease, symptomatic visceral disease, or KS‐related IRIS.10 The main chemotherapeutic agents used systemically for KS are liposomal anthracyclines such as doxorubicin or daunorubicin, or taxanes such as paclitaxel.11 An association between corticosteroids and progression of KS has been previously described, even as early as several days after steroid administration.1214 Recently, revised diagnostic criteria for corticosteroid‐associated KS‐IRIS have been proposed; this patient met those criteria.15
Plasmablastic lymphoma is a highly aggressive systemic NHL seen predominantly in HIV‐positive patients. There is a strong association with Epstein‐Barr virus; HHV‐8 is more variably associated and is of unclear significance.16 Most HIV‐infected patients have extranodal involvement at diagnosis; in a series of 53 HIV‐positive patients, the oral cavity was the most frequent site, and lung involvement was seen in 12%. The prognosis is poor, with a mean survival of approximately 1 year.17
Treatment for systemic NHL in HIV‐positive patients generally consists of a chemotherapy regimen while ART is continued or initiated.18 The most commonly used chemotherapy combination is cyclophosphamide, doxorubicin, vincristine, and prednisone, often supplemented with the anti‐CD20 monoclonal antibody rituximab. In the case of aggressive systemic NHL, more intensive treatment regimens are often utilized, though it remains unclear if they are associated with improved outcomes.17, 19 Antiretroviral therapy is continued, as it has been shown to reduce the rate of opportunistic infections and decrease mortality.20
Despite the remarkable progress that has been made in the past 30 years, HIV/AIDS remains a devastating and remarkably complex disease. As the landscape of HIV/AIDS evolves, clinicians will continue to be faced with new challenging and vexing decisions. Perhaps no greater challenge exists than the presence of 2 simultaneous, rapidly fatal malignancies with directly competing therapeutic strategies, as in this case, where the ART and steroids employed to address NHL fostered widespread KS‐IRIS. This case reminds us that a single unifying diagnosis can often be the exception rather than the rule in the care of patients with advanced HIV. It also illustrates how the mainstay of HIV treatment, ART, can be a double‐edged sword.
KEY TEACHING POINTS
-
In HIV/AIDS patients receiving ART who become paradoxically more ill despite improvements in their CD4 counts, consider IRIS.
-
Though corticosteroids are a hallmark of treatment for most types of IRIS‐and for aggressive lymphomas‐they can worsen KS.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.
- , , , et al. Defining immune reconstitution inflammatory syndrome: evaluation of expert opinion versus 2 case definitions in a South African cohort. Clin Infect Dis. 2009;49:1424–1432.
- , , , et al. Risk factor analyses for immune reconstitution inflammatory syndrome in a randomized study of early vs. deferred ART during an opportunistic infection. PLoS One. 2010;5:e11416.
- , . Kaposi's sarcoma. N Engl J Med. 2000;342:1027–1038.
- , , , et al. The changing pattern of Kaposi sarcoma in patients with HIV, 1994–2003: the EuroSIDA Study. Cancer. 2004;100:2644–2654.
- , , , , , . Imaging features of pulmonary Kaposi sarcoma–associated immune reconstitution syndrome. AJR Am J Roentgenol. 2007;189:956–965.
- , , , et al. Pulmonary involvement in Kaposi sarcoma: correlation between imaging and pathology. Orphanet J Rare Dis. 2009;4:18.
- , , , et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228.
- , . Recrudescent Kaposi's sarcoma after initiation of HAART: a manifestation of immune reconstitution syndrome. AIDS Patient Care STDS. 2005;19:635–644.
- , , , , , . Paradoxical immune reconstitution inflammatory syndrome in HIV‐infected patients treated with combination antiretroviral therapy after AIDS‐defining opportunistic infection. Clin Infect Dis. 2012;54:424–433.
- , , , et al. British HIV Association guidelines for HIV‐associated malignancies 2008. HIV Med. 2008;9:336–388.
- , , , , . HIV/AIDS: epidemiology, pathophysiology, and treatment of Kaposi sarcoma–associated herpesvirus disease: Kaposi sarcoma, primary effusion lymphoma, and multicentric Castleman disease. Clin Infect Dis. 2008;47(9):1209–1215.
- , , . A 36‐year‐old man with AIDS and relapsing, nonproductive cough. Chest. 2007;131:1929–1931.
- , , , , . Life‐threatening exacerbation of Kaposi's sarcoma after prednisone treatment for immune reconstitution inflammatory syndrome. AIDS. 2008;22:663–665.
- , , , , , . Clinical effect of glucocorticoids on Kaposi sarcoma related to the acquired immunodeficiency syndrome (AIDS). Ann Intern Med. 1989;110:937–940.
- , , , . Kaposi sarcoma–associated immune reconstitution inflammatory syndrome: in need of a specific case definition. Clin Infect Dis. 2012;55(1):157–158.
- , , , , , . Plasmablastic lymphoma in HIV‐positive patients: an aggressive Epstein‐Barr virus–associated extramedullary plasmacytic neoplasm. Am J Surg Pathol. 2005;29:1633–1641.
- , , , et al. Human immunodeficiency virus–associated plasmablastic lymphoma: poor prognosis in the era of highly active antiretroviral therapy. Cancer. 2012;118:5270–5277.
- , , . Modern management of non‐Hodgkin lymphoma in HIV‐infected patients. Br J Haematol. 2007;136(5):685–698.
- , , , et al. CD20‐negative large‐cell lymphoma with plasmablastic features: a clinically heterogeneous spectrum in both HIV‐positive and ‐negative patients. Ann Oncol. 2004;15(11):1673–1679.
- , , , et al. Concomitant cyclophosphamide, doxorubicin, vincristine, and prednisone chemotherapy plus highly active antiretroviral therapy in patients with human immunodeficiency virus–related, non‐Hodgkin lymphoma. Cancer. 2001;91(1):155–163.