User login
Cut to the Quick
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
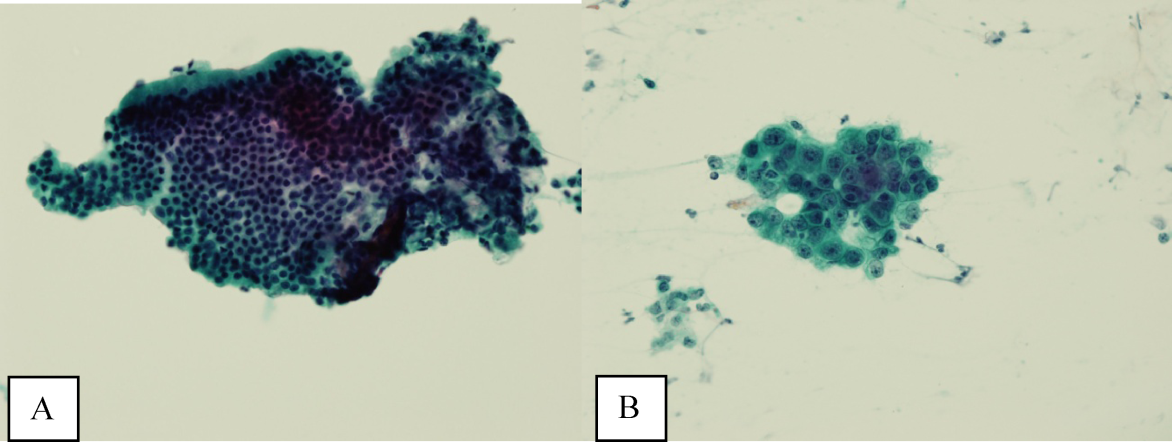
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
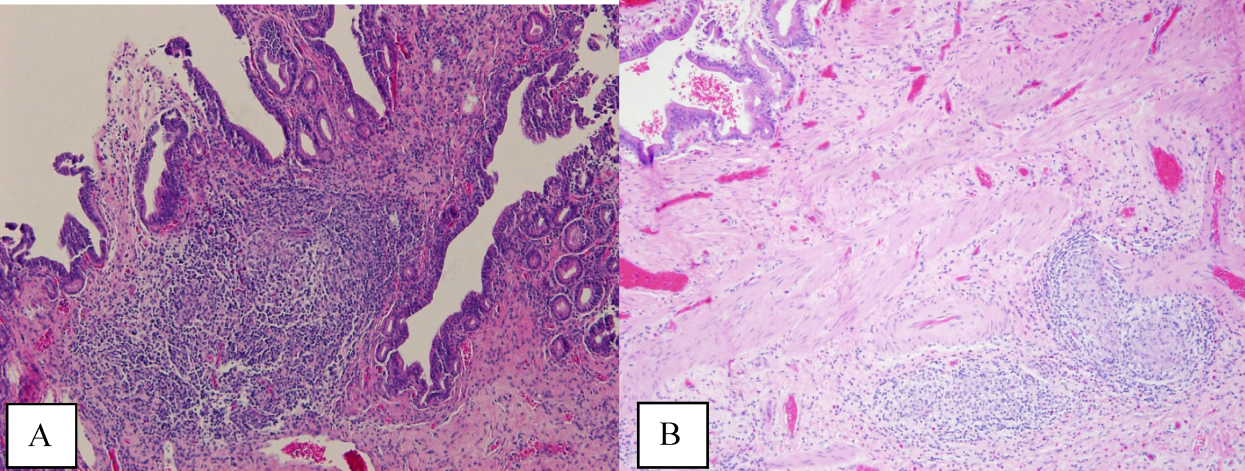
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
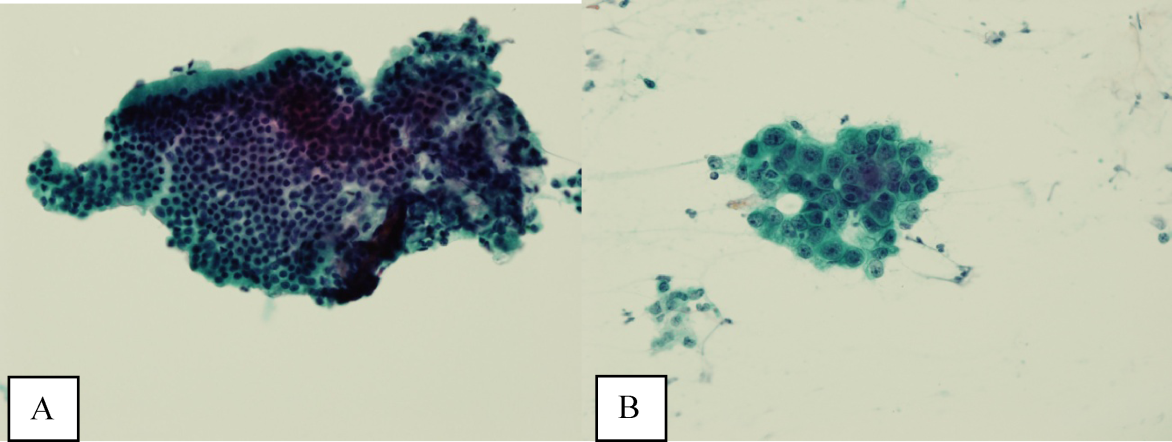
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
A 41‐year‐old woman with dwarfism was referred for evaluation of an isolated elevated alkaline phosphatase (ALP) of 792 U/L (normal value, 3195 U/L) and a gamma‐glutamyl transferase (GGT) of 729 U/L (normal value, 737 U/L), found incidentally on routine laboratory screening. She denied any fevers, chills, weight loss, abdominal pain, nausea, or vomiting.
The presence of an isolated ALP elevation, presumably of hepatobiliary origin given the increase in GGT, in a relatively young woman immediately calls to mind the diagnosis of primary biliary cirrhosis, and I would specifically inquire about pruritus, which occurs commonly in this setting. The absence of abdominal pain argues against the diagnosis of extrahepatic biliary obstruction. Other processes that could result in this asymptomatic presentation include infiltrative diseases such as amyloidosis, sarcoidosis, and other causes of granulomatous hepatitis. The absence of systemic symptoms makes disseminated infection or malignancy with hepatic involvement less likely. I would query whether underlying dwarfism can be associated with metabolic abnormalities that cause infiltrative liver disease, functional or anatomical hepatobiliary abnormalities, or malignancy.
The patient's medical history was notable for chronic constipation, allergic rhinitis, and basal‐cell carcinoma. She had reconstructive surgeries of the left hip and knee 28 years ago without complications. She underwent a right total hip replacement for hip dysplasia 6 months prior, which was complicated by a postoperative joint infection with Enterobacter cloacae. The hardware was retained, and she was treated with incision and drainage and a prolonged fluoroquinolone course. Furthermore, she had a history of immune thrombocytopenic purpura (ITP), which manifested at the age of 20 years. A bone‐marrow biopsy at that time showed no evidence of hematologic malignancy. For her ITP, she had initially received intravenous immunoglobulin (Ig) and cyclosporine without sustained benefit. She underwent a splenectomy at the age of 26 years and was treated intermittently with rituximab over 11 years prior to admission. Her medications included cetirizine. Her parents were nonconsanguineous, of European and Southeast Asian ancestry, and healthy. She was in a long‐term monogamous relationship. The patient had been employed as an educator.
The history of immune‐mediated thrombocytopenia raises the possibility that the present illness may be part of a broader autoimmune diathesis. Other causes of secondary ITP, such as drug‐induced reactions, hematologic malignancies, and viral infections, are unlikely, as her ITP has been persistent for more than 20 years. She has not evolved into a common phenotypic pattern of autoimmune disease such as systemic lupus erythematosus after the appearance of ITP, nor does she endorse a history of thromboembolic complications that would suggest antiphospholipid syndrome.
Ultrasound of the abdomen demonstrated narrowing of the extrahepatic biliary duct in the region of the pancreas without evidence of a mass lesion. Computerized tomography (CT) of the abdomen and pelvis similarly showed mild intrahepatic biliary ductal dilatation with narrowing of the extrahepatic duct in the region of the pancreas without apparent pancreatic mass. Endoscopic retrograde cholangiopancreatography (ERCP) confirmed a stricture in the distal common bile duct and dilatation of the common bile duct. Cytology brushings obtained during ERCP showed groups of overlapping, enlarged cells with pleomorphic irregular nuclei, one or more prominent nucleoli, and focal nuclear molding, leading to a diagnosis of adenocarcinoma (Figure 1).
The absence of jaundice and pruritus indicates incomplete biliary obstruction. Commonbile duct strictures are most commonly seen after manipulation of the biliary tree. Neoplasms including pancreatic cancer, adenocarcinoma of the ampulla of Vater, and cholangiocarcinoma may cause compression and obstruction of the common bile duct, as well as stricture formation mediated by a desmoplastic reaction to the tumor. Occasionally, metastatic malignancy or lymphoma may involve the porta hepatis and cause extrinsic compression of the common bile duct. Other etiologies of strictures include sclerosing cholangitis and opportunistic infections such as Cryptosporidium, cytomegalovirus, and microsporidiosis, which are not supported by this patient's history.
The atypical cells seen on ERCP brushings were interpreted as evidence of cholangiocarcinoma. The patient underwent a pylorus‐sparing Whipple procedure. Examination of the surgical pathology specimens revealed diffuse non‐necrotizing granulomatous inflammation involving the bile duct and gallbladder (Figure 2). There was focal atypia of the bile‐duct epithelial cells, but no evidence of malignancy. There were non‐necrotizing granulomas in numerous lymph nodes, some with significant sclerosis; stains and cultures for acid‐fast bacilli and fungi were negative, and stains for IgG4 and CD1a for Langerhans‐cell histiocytosis were negative.
Granulomatous inflammation may be caused by a variety of intracellular infections, environmental and occupational exposures, and drug hypersensitivity, or may be associated with malignancy such as lymphoma. In the absence of an alternative explanation, the presence of non‐necrotizing granulomas in multiple organs suggests the diagnosis of sarcoidosis, even if classic intrathoracic involvement is not present. Hepatic involvement with sarcoidosis is common but rarely symptomatic, whereas biliary disease is distinctly uncommon. Interestingly, there is an association between both primary biliary cirrhosis and sclerosing cholangitis with sarcoidosis. The pathologic findings could indicate an autoimmune process that has led to widespread granulomas with this unusual distribution. Disseminated infections such as mycobacterial or fungal diseases seem much less plausible in this woman, who had no prior systemic complaints. The atypical cells seen on the ERCP brushings were almost certainly caused by inflammation and a fibroproliferative response rather than malignancy.
On further questioning, the patient endorsed a history of multiple childhood ear infections that required bilateral myringotomy tubes, and multiple episodes of sinusitis, but both problems improved in adulthood. She had experienced 2 episodes of dermatomal zoster in her lifetime. She also noted frequent vaginal yeast infections. She denied any history of pneumonias or thrush. In her second decade of life, she developed allergic rhinitis and eczema. She denied any chemical or environmental exposures. She had had negative tuberculin skin tests as part of her occupational screening and denied any recent travel.
The additional history of recurrent upper‐respiratory infections early in life and subsequent episodes of dermatomal zoster and candidal infections increases the likelihood that this patient has a primary immunodeficiency. A combined cellular and humoral immunodeficiency would predispose to both bacterial sinopulmonary infections, generally a result of Ig isotype or IgG subclass deficiencies, and recurrent zoster and candidal infection. Any evaluation of her Igs at this time may be confounded by her receipt of anti‐CD20 monoclonal antibody therapy, which may decrease serum Ig levels.
The relatively benign course in terms of infection is consistent with the heterogeneous immunodeficiencies classified as combined immunodeficiency (CID), a less‐penetrant phenotype of severe combined immunodeficiency (SCID), or common variable immunodeficiency (CVID). Autoimmunity is a frequent manifestation of CID and CVID, and affected patients have an increased risk of lymphoma and other malignancies. Granulomatous disease may also be a manifestation of both CID and CVID.
Postoperatively, she developed progressive abdominal distension and pain. A CT of the abdomen and pelvis showed colonic dilatation consistent with Ogilvie pseudo‐obstruction. On postoperative day 9, she developed fevers. On physical examination, her temperature was 38.5C, the blood pressure was 104/56 mm Hg, and the heart rate was 131 beats per minute. Her oxygen saturation was 95% on room air. Her height was 105 cm. She had diffuse alopecia without scarring. She did not have a malar rash or oral ulcerations. Both lungs were clear to auscultation. A cardiac examination showed tachycardia with a regular rhythm, normal heart sounds, and no murmurs. Her musculoskeletal exam was notable for short limbs and phalanges, without synovitis. Bilateral hip exam demonstrated internal and external range of motion without abnormalities. No rashes were present. Her abdominal exam revealed diffuse tenderness with postoperative drains in place. She had nonbloody loose stools.
Although autoimmune diseases such as sarcoidosis can rarely manifest with fevers, evaluation of postoperative fever in this patient should focus first on common processes that also occur in immunocompetent patients. Since she has had a splenectomy and we are now suspicious of an underlying immunodeficiency, appropriate cultures should be obtained and broad‐spectrum intravenous antibiotics should be initiated without delay. The presence of nonscarring alopecia could either represent autoimmune alopecia, if the onset was recent, or it could be part of this patient's underlying skeletal dysplasia syndrome.
Piperacillin/tazobactam and oral metronidazole were started for presumed intra‐abdominal infection. The white cell count was 20,500/mm3 with 96% neutrophils, 1.4% lymphocytes with an absolute lymphocyte count 0.33 109/L (normal value, >1.0 109/L), and 2.6% monocytes. The hematocrit was 27.8% with a mean corpuscular volume of 95 fL. The platelet count was 323,000/mm3. Serum aminotransferase and total bilirubin levels were normal, and ALP was 904 U/L. The serum albumin was 1.2 g/dL (normal value, 3.54.8 g/dL) and prealbumin was 6 mg/dL (normal value, 2037 mg/dL).
Blood cultures returned positive for E. cloacae. Clostridium difficile toxin assay was negative. Piperacillin/tazobactam was switched to meroperem, and metronidazole was discontinued. She continued to have fevers, and on postoperative day 16, repeat blood cultures and urine cultures grew Candida albicans; caspofungin was initiated.
In addition to the neutrophilic leukocytosis in response to gram‐negative bacteremia, there is marked lymphopenia. Although sepsis may cause transient declines in the total lymphocyte count, I do not believe that this entirely accounts for such severe lymphopenia. The albumin is also profoundly low. Her catabolic postsurgical state might explain part of this abnormality, but taken together with her prior gastrointestinal symptoms, these findings could be consistent with intestinal malabsorption or a protein‐losing enteropathy, which can also be associated with primary immunodeficiency.
Serum angiotensin‐converting enzyme was 32 U/L (normal value, 967 U/L). A CT of the chest was performed and did not reveal mediastinal lymphadenopathy, nodules, or consolidations. Antinuclear, antismooth muscle, and antimitochondrial antibodies were negative. Human immunodeficiency virus antibody was negative. Serum quantitative Igs, including IgG, IgM, IgA, and IgE, were undetectable.
Serum lymphocyte subset analysis revealed a CD3 T‐cell count of 101 106/L (normal value, >690 106/L), CD4 T cells 46 106/L (normal value, >410 106/L), CD8 T cells 55 106/L (normal value, >190 106/L), CD19 B cells undetectable at 2 106/L (normal value, >90 106/L), CD16 CD56 NK cells 134 106/L (normal value, >90 106/L). T‐cell lymphocyte proliferation assay showed a completely absent response to candida and tetanus antigens, and a very low response to mitogens.
The immunologic evaluation is confounded by her critical illness and by the prior administration of anti‐CD20 monoclonal antibody. Despite these caveats, the results of these studies are profoundly abnormal and suggest a combined B‐cell and T‐cell immunodeficiency that is more severe from a laboratory standpoint than her history prior to surgery has suggested. Low T lymphocyte numbers, with or without functional abnormalities, are a hallmark of CID and can be also be seen in CVID. The extremely low Ig levels in the presence of severe infections warrant replacement with intravenous Ig.
Combined immunodeficiency and CVID may be associated with a number of mutations; elucidating the genetics and molecular mechanism of immunodeficiency may be important in identifying patients whose immunodeficiency may be cured by stem‐cell transplantation.
Intravenous Ig was administered. Her serum was sent for sequencing of the RMRP gene, mutations of which are found in patients who have cartilage‐hair hypoplasia (CHH), a rare autosomal recessive skeletal dysplasia characterized by short‐limbed dwarfism; fine, sparse hair; and variable degrees of immunodeficiency. She was found to have 2 RMRP mutations, a 126 CT transition and a 218 AC transversion.
The patient developed multiple abdominal abscesses, which were drained and grew vancomycin‐resistant enterococcus (VRE) and C. albicans. Blood cultures also turned positive for VRE. A colonoscopy was performed because of radiographic evidence suggestive of colitis. Biopsies taken from the colonoscopy were negative for cytomegalovirus or other infections, but did reveal rare non‐necrotizing granulomas. The patient developed progressive multiorgan failure requiring mechanical ventilation and continuous venovenous hemofiltration. On postoperative day 36, the patient was transitioned to comfort care, and she expired the next day. A unifying diagnosis of CHH‐related immunodeficiency and disseminated granulomatous disease, complicated by postoperative sepsis, was made. An autopsy was declined.
COMMENTARY
Evaluation of abnormal liver tests is a frequent diagnostic challenge faced by clinicians in both ambulatory and inpatient settings. Identifying the pattern of liver injuryhepatocellular, cholestatic, or infiltrativemay guide the initial workup. This patient's presentation of a normal bilirubin and transaminases with elevations in ALP was consistent with infiltrative hepatic disease. The radiographic finding of extrahepatic biliary strictures, on the other hand, raised concern for an obstructive etiology and prompted an ERCP. Brush cytology has high specificity for malignancy, but interpretation of atypical cells can rarely be inconclusive or be associated with false positives.[1]
The suspicion for infiltrative hepatitis was supported postoperatively by the discovery of diffuse hepatobiliary granulomatous disease, which can be associated with a spectrum of disease states including sarcoidosis, autoimmune disorders, intracellular infections, immunodeficiency, malignancy, environmental or occupational exposures, and drug reactions.[2, 3] During the patient's hospital course and case presentation to the discussant, the possibility of sarcoidosis was raised based on the operative findings. Additional history‐taking was essential to evaluate other etiologies of granulomatous inflammation, and this clinical correlation prevented a second erroneous pathologic diagnosis.
Multiple elements of this patient's presentation led to recognition of an underlying primary immunodeficiency. Her prior history of recurrent childhood infections, dermatomal zoster, and vaginal infections suggested a congenital immunodeficiency. The additional features of refractory autoimmune cytopenias (ie, ITP), granulomatous inflammation, undetectable serum Igs, and low T‐cell and B‐cell counts, were consistent with CID or CVID. By definition, CID involves defects in both B and T cells; CVID represents a predominantly B‐cell disorder characterized by abnormalities in Ig production, though concomitant T‐cell dysfunction may also be found.[4] It is worth noting that although this patient had previously received anti‐CD20 monoclonal antibody, which depletes CD20‐positive B lymphocytes, Ig levels are not typically depleted by anti‐CD20 unless there is preexisting antibody deficiency.[5]
We were able to make the unifying diagnosis of CHH to explain her constellation of physical findings, laboratory abnormalities, and histopathology. Also known as McKusick type metaphyseal chondrodysplasia, CHH has a relatively high carrier frequency in the Amish (1:19) and Finnish (1:76) populations.[6, 7] Additional clinical features can include gastrointestinal disorders, poorly pigmented skin and hair, and joint disorders. Dysregulation of immunity is a particular challenge and can be manifested by malignancy, lymphoproliferative disease, cytopenias, or primary immunodeficiencies. Combined immunodeficiency and T cellmediated defects are most common, although there are case reports of CHH associated with severe humoral defects.[8, 9] Primary immunodeficiency, if severe and recognized early, can be treated with bone‐marrow transplantation.[10, 11] Granulomatous inflammation also has been described in CHH.[12]
Although tissue biopsy is often viewed as the gold standard for establishing a definitive diagnosis, this case highlights the significance of applying clinical context to pathologic interpretation and medical decision‐making. Prior to any diagnostic procedure, the patient's history of dwarfism, recurrent infections, and refractory ITP provided clues to an immunodeficiency syndrome, CHH. Knowledge of this immunodeficiency might have better informed the initial pathologic interpretation of atypical cells, which were misread as adenocarcinoma. Furthermore, awareness of the patient's profound immunodeficiency would have given pause to proceeding with invasive surgery without prior Ig and antibiotic support and may have averted a fatal outcome.
KEY TEACHING POINTS
- Infiltrative hepatobiliary diseases may manifest with isolated elevations in ALP.
- Granulomas and autoimmune cytopenias may be features of primary immunodeficiency states.
- A history of recurrent childhood infections should raise suspicion for congenital immunodeficiencies.
- Unique medical complications, including immunodeficiency, can be associated with dwarfism subtypes.
Acknowledgements
The authors thank Jennifer M. Puck, MD, from the University of California San Francisco, Departments of Immunology and Pediatrics, for her invaluable contribution to the discussion on immunodeficiencies.
Disclosure
Nothing to report.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
- , , , . Brush cytology of ductal strictures during ERCP. Acta Gastroenterol Belg. 2000;63:254–259.
- , . Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134;667–690.
- James DG, Zumla A, eds. The Granulomatous Disorders. Cambridge, UK: Cambridge University Press; 1999:17–27.
- , , , et al. Unraveling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–3943.
- , , , et al. Does rituximab aggravate pre‐existing hypogammaglobulinaemia? J Clin Pathol. 2010;63:275–277.
- . Cartilage‐hair hypoplasia in Finland: epidemiological and genetic aspects of 107 patients. J Med Genet. 1992;29:652–655.
- , , , et al. High‐resolution genetic mapping of the cartilage‐hair hypoplasia (CHH) gene in Amish and Finnish families. Genomics. 1994;20:347–353.
- , , , et al. Combined immunodeficiency and vaccine‐related poliomyelitis in a child with cartilage‐hair hypoplasia. J Pediatr. 1975;86:868–872.
- , , . Deficiency of humoral immunity in cartilage‐hair hypoplasia. J Pediatr. 2000;137:487–492.
- , , , , . Bone marrow transplantation for cartilage‐hair hypoplasia. Bone Marrow Transplant. 2006;38:751–756.
- , , , et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation [published corrections appear in Blood. 2010;116:2402 and Blood. 2011;117:2077]. Blood. 2010;116:27–35.
- , , , et al. Granulomatous inflammation in cartilage‐hair hypoplasia: risks and benefits of anti‐TNF‐α mAbs. J Allergy Clin Immunol. 2011;128:847–853.
Pediatric Hospitalists Trust Their Gut with Serious Infections

Clinical question: How helpful is the “gut feeling” that clinicians might have that a child is more ill than they look?
Background: Timely recognition of serious infections in children is difficult but critical to improved outcomes. Numerous studies have examined clinical criteria and laboratory tests to distinguish viral infections from more serious bacterial infections but have demonstrated mixed results. Clinicians’ subjective impressions of patients continue to drive many care patterns, and the relevance of a gut feeling that something is wrong remains unclear.
Study design: Prospective observational study.
Setting: Primary-care clinics in Flanders, Belgium.
Synopsis: Nearly 4,000 children 0-16 years of age were evaluated after presentation for acute illness in primary-care settings. Clinical features, overall “clinical impression” (serious illness present or absent), and “gut feelings” (present, absent or unsure) that something was wrong were prospectively recorded. Serious infections were defined as hospital admissions for potential bacterial infections.
The presence of a gut feeling significantly increased the risk of serious illness (likelihood ratio 25.5, 95% confidence interval 7.9 to 82.0) and had a consistently higher specificity than clinical impression alone. The overall sensitivity of the gut feeling in this cohort was 61.9% with a specificity of 97.2%, while the positive predictive value was 10.8% and negative predictive value 99.8%. A history of convulsions and parental concerns were independently strongly associated with a positive gut feeling.
Similar to other clinical and laboratory evaluations designed to detect serious illness, the absence of a gut feeling might be the more useful finding from this study. Limitations of the data include an inability to further delve into what gives rise to a gut feeling in clinical practice as well as a moderate level of variance in the multivariate models. Additionally, only 21 children were ultimately admitted to the hospital, which, in conjunction with the subsequent power limitations, highlights the proverbial difficulty of finding that “needle in the haystack.”
Bottom line: The presence or absence of a gut feeling that something is wrong might be an important component of the history in acute childhood illness.
Citation: Van den Bruel A, Thompson M, Buntinx F, Mant D. Clinicians’ gut feeling about serious infections in children: observational study. BMJ. 2012;345:e6144.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: How helpful is the “gut feeling” that clinicians might have that a child is more ill than they look?
Background: Timely recognition of serious infections in children is difficult but critical to improved outcomes. Numerous studies have examined clinical criteria and laboratory tests to distinguish viral infections from more serious bacterial infections but have demonstrated mixed results. Clinicians’ subjective impressions of patients continue to drive many care patterns, and the relevance of a gut feeling that something is wrong remains unclear.
Study design: Prospective observational study.
Setting: Primary-care clinics in Flanders, Belgium.
Synopsis: Nearly 4,000 children 0-16 years of age were evaluated after presentation for acute illness in primary-care settings. Clinical features, overall “clinical impression” (serious illness present or absent), and “gut feelings” (present, absent or unsure) that something was wrong were prospectively recorded. Serious infections were defined as hospital admissions for potential bacterial infections.
The presence of a gut feeling significantly increased the risk of serious illness (likelihood ratio 25.5, 95% confidence interval 7.9 to 82.0) and had a consistently higher specificity than clinical impression alone. The overall sensitivity of the gut feeling in this cohort was 61.9% with a specificity of 97.2%, while the positive predictive value was 10.8% and negative predictive value 99.8%. A history of convulsions and parental concerns were independently strongly associated with a positive gut feeling.
Similar to other clinical and laboratory evaluations designed to detect serious illness, the absence of a gut feeling might be the more useful finding from this study. Limitations of the data include an inability to further delve into what gives rise to a gut feeling in clinical practice as well as a moderate level of variance in the multivariate models. Additionally, only 21 children were ultimately admitted to the hospital, which, in conjunction with the subsequent power limitations, highlights the proverbial difficulty of finding that “needle in the haystack.”
Bottom line: The presence or absence of a gut feeling that something is wrong might be an important component of the history in acute childhood illness.
Citation: Van den Bruel A, Thompson M, Buntinx F, Mant D. Clinicians’ gut feeling about serious infections in children: observational study. BMJ. 2012;345:e6144.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: How helpful is the “gut feeling” that clinicians might have that a child is more ill than they look?
Background: Timely recognition of serious infections in children is difficult but critical to improved outcomes. Numerous studies have examined clinical criteria and laboratory tests to distinguish viral infections from more serious bacterial infections but have demonstrated mixed results. Clinicians’ subjective impressions of patients continue to drive many care patterns, and the relevance of a gut feeling that something is wrong remains unclear.
Study design: Prospective observational study.
Setting: Primary-care clinics in Flanders, Belgium.
Synopsis: Nearly 4,000 children 0-16 years of age were evaluated after presentation for acute illness in primary-care settings. Clinical features, overall “clinical impression” (serious illness present or absent), and “gut feelings” (present, absent or unsure) that something was wrong were prospectively recorded. Serious infections were defined as hospital admissions for potential bacterial infections.
The presence of a gut feeling significantly increased the risk of serious illness (likelihood ratio 25.5, 95% confidence interval 7.9 to 82.0) and had a consistently higher specificity than clinical impression alone. The overall sensitivity of the gut feeling in this cohort was 61.9% with a specificity of 97.2%, while the positive predictive value was 10.8% and negative predictive value 99.8%. A history of convulsions and parental concerns were independently strongly associated with a positive gut feeling.
Similar to other clinical and laboratory evaluations designed to detect serious illness, the absence of a gut feeling might be the more useful finding from this study. Limitations of the data include an inability to further delve into what gives rise to a gut feeling in clinical practice as well as a moderate level of variance in the multivariate models. Additionally, only 21 children were ultimately admitted to the hospital, which, in conjunction with the subsequent power limitations, highlights the proverbial difficulty of finding that “needle in the haystack.”
Bottom line: The presence or absence of a gut feeling that something is wrong might be an important component of the history in acute childhood illness.
Citation: Van den Bruel A, Thompson M, Buntinx F, Mant D. Clinicians’ gut feeling about serious infections in children: observational study. BMJ. 2012;345:e6144.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
When Should an Abdominal Aortic Aneurysm Be Treated?
Case
A generally healthy, 74-year-old man presents with sudden-onset abdominal pain due to acute pancreatitis. Computed tomography (CT) of his abdomen shows pancreatic inflammation and an incidental finding of a 4.5-cm abdominal aortic aneurysm. He had never had any imaging of his abdomen prior to this study and described no prior episodes of abdominal pain.
When should his abdominal aortic aneurysm be treated?
Overview
An abdominal aortic aneurysm (AAA) is an abnormal dilation of the abdominal aorta between the diaphragm and the aortic bifurcation of the iliac arteries. An AAA is usually defined as a dilatation with a diameter of >3 cm or 50% greater than the typical diameter. Most AAAs are located in the infrarenal aorta, proximal to the iliac bifurcation.
Population screening programs show a prevalence of AAA of 4% to 8% in men aged 65 to 80 years.1 AAA prevalence is approximately six times greater in men than women, though the prevalence in women might be increasing.1 AAA is most common in white men, with black men and those of Asian heritage having lower risk. A combination of genetic predisposition and environmental and physiologic factors lead to initiation and progression of AAAs; family history, male sex, advanced age, and history of smoking are major risk factors.
Mortality after AAA rupture is high. Approximately 62% of patients die prior to hospital arrival.2 Of those who undergo emergent AAA surgery, 50% will die.1
Aortic repair with a prosthetic vascular graft reduces morbidity and mortality from rupture, but the risks of repair are not trivial.2
Review of the Data
Risk of rupture. An AAA should be repaired when the risk of rupture outweighs the risks of surgical repair. Symptomatic aneurysms—such as those causing back or abdominal pain—have a higher risk of rupture than asymptomatic aneurysms. Most AAAs are asymptomatic and, in the absence of imaging, not identified until the time of rupture. Given the significant mortality associated with rupture, there is benefit to intervening on asymptomatic aneurysms before rupture.
The risk of AAA rupture has been studied in patients who either have been unfit for surgical repair or uninterested in intervention. Risk of rupture increases substantially with aneurysm size. Lederle et al estimated a two-year aneurysm rupture risk of 22.1% for AAA with a diameter of 5.0 to 5.9 cm, 18.9% for 6.0 to 6.9 cm, and 43.4% for a diameter ≥7.0 cm.3 In another study of 476 patients, the average risks of rupture in male and female patients with an AAA of 5.0 to 5.9 cm were 1.0% and 3.9% per year, respectively. For male and female patients with ≥6.0 cm AAAs, risks of rupture were 14.1% and 22.3% per year.4 Women with AAA have been found to have a higher risk of rupture in all studies in which female patients were included.
Because rupture risk increases with size, predicting the rate of growth is clinically important. Powell et al conducted a systematic review of growth rates of small AAAs.5 In 15 studies that examined 7,630 patients, the growth rate for a 3.5-cm aneurysm was estimated at 1.9 mm/year and for a 4.5-cm aneurysm was 3.5 mm/year. Given an exponentially increasing aneurysm diameter, this suggests an elapsed time of 6.2 years for a 3.5-cm aneurysm to grow to 5.5 cm, and 2.3 years for a 4.5-cm AAA to grow to 5.5 cm. This prediction does not account for individual variability in growth rate. Some AAAs grow quickly, others erratically, and others not at all. This growth variability is influenced by individual characteristics including cigarette smoking, sex, age, and other factors.
Medical prevention of progression and rupture. Studies have assessed whether modification of risk factors can delay progression of growth of AAAs. In a small aneurysm trial in the United Kingdom, self-reported smoking status was associated with an incrementally increased growth rate of 0.4 mm per year.5 Each year of smoking increases the relative risk of AAA by 4%, and continued smoking leads to more rapid AAA expansion.6 There is no clear relationship between cholesterol levels and AAA expansion rate. Observational studies suggest that aneurysm expansion decreases with statin use, but there is not sufficient evidence to recommend statin therapy for AAA alone.6
Many patients with AAA, however, are candidates for statins because of concomitant coronary artery or peripheral vascular disease. Small, randomized controlled trials have shown that macrolides and tetracycline antibiotics might inhibit AAA growth, but prescribing them for this purpose is not currently the standard of care.7 Elevated mean blood pressure has been associated with rupture, but there is not good evidence showing delay of progression with treatment of hypertension.6 Early observational studies suggested that beta-blocker use would decrease AAA progression, but further evidence has not supported their benefit in slowing progression of size.8 Likewise, use of angiotensin-converting enzyme inhibitors has also shown no growth inhibition.7 An ongoing Cochrane review is evaluating evidence for these medical treatments of AAA.9
Surgical prevention of rupture. There are two surgical methods of AAA repair: open repair and endovascular aneurysm repair (EVAR). Both involve use of a prosthetic graft to prevent the aneurysm from enlarging. The EVAR procedure typically involves entry at the femoral artery, with use of catheters and guide wires to advance a graft to the desired location and anchor it in place. Because this utilizes an endovascular approach, regional rather than general anesthesia can be used.
Multiple investigators have evaluated for differences in outcomes between the two methods of surgical AAA repair. Studies have shown increased 30-day postoperative mortality with open repair, as well as significantly higher rates of postoperative cardiac, pulmonary, and renal complications. One randomized controlled study found 30-day operative mortality of 1.8% in the EVAR group and 4.3% in the open repair group.10 However, after a median six-year follow-up of patients after EVAR or open repair, there is no difference in total mortality or aneurysm-related mortality.10 Compared with open repair, the need for long-term surveillance and re-intervention post-EVAR is higher, with endoleak and graft migration the most common complications. This accounts for the loss of early survival advantage in post-EVAR patients. By two years post-operation, complication after repair with either technique is not statistically different. De Bruin et al found that six years after randomization for repair type, cumulative survival rates were 69.9% for patients after open repair and 68.9% with EVAR.11
Studies also have focused on subgroups of patients with a higher operative risk and shorter life expectancy, such as the elderly.12 A pooled analysis of 13,419 patients aged ≥80 years from six observational studies showed 8.6% immediate mortality after open repair and 2.3% after EVAR (risk difference 6.2%, 95% CI 5.4-7.0%).13 Pooled analysis of three longer-term studies showed similar overall survival at three years after EVAR and open repair.13 When EVAR is not available, open repair has acceptable short- and long-term survival in patients aged ≥80 years with an AAA at high risk of rupture.14
Screening. A Cochrane review evaluated the effect of ultrasound screening of asymptomatic AAA on mortality. In 127,891 men and 9,342 women aged 65 to 79, researchers found a significant decrease in mortality in men aged 65 to 79 who were screened (odds ratio 0.6, 95% CI 0.47-0.78) but no benefit to screening of women.15 The current U.S. Preventive Services Task Force (USPSTF) guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 who have any history of tobacco use. For men in this age group who have never smoked, the balance between benefits and harms of screening is too close for the USPSTF to make recommendations. Because of the lower prevalence in women, the USPSTF recommends against screening women for AAA.18
Timing of repair. Early repair of small AAAs (4 cm to 5.5 cm) has no long-term survival benefit compared to ultrasound surveillance without repair.16,17 Therefore, AAAs <5.5 cm should be followed with regular ultrasound surveillance every six months, with referral to surgery if the diameter reaches 5.5 cm, or grows >1 cm a year. The size at which surgery should be performed might be lower in women, given that their risk of rupture is higher than men. A thoughtful discussion of individual risks should take place in every case, but in many patients, even the elderly, repair of a large asymptomatic AAA is indicated. 5
Back to the Case
Our patient should have repeat imaging of his AAA in six months and regular surveillance afterward to monitor for growth every six months. When the AAA is >5.5 cm or if it grows >1 cm a year, he should be evaluated for EVAR or open repair.
Bottom Line
The current USPSTF guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 with a history of smoking. If an AAA >3 cm is found, the patient should undergo regular USG screening every six months. The AAA should be repaired if >5.5 cm or symptomatic, via either an endoscopic or open approach.
Dr. Best is a hospitalist at University of Washington Medicine at Harborview and associate program director of the internal-medicine residency program at the University of Washington, Seattle. Dr. Carpenter is a fellow in the division of geriatrics at the University of California at San Franscisco.
References
- Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8, 92-102.
- Ernst CB. Abdominal aortic aneurysm. N Engl J Med. 1993;328(16):1167-1172.
- Lederle FA, Johnson GR, Wilson SE, et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA. 2002;287:2968-2972
- Brown PM, Zelt DT, Sobolev B. The risk of rupture in untreated aneurysms: the impact of size, gender, and expansion rate. J Vasc Surg. 2003;37:280-284.
- Powell JT, Sweeting MJ, Brown LC, Gotensparre SM, Fowkes FG, Thompson SG. Systematic review and meta-analysis of growth rates of small abdominal aortic aneurysms. British Journal of Surgery. 2011;98:609-618.
- Brown LC, Powell JT. Risk factors for aneurysm rupture in patients kept under ultrasound surveillance. UK Small Aneurysm Trial Participants. Ann Surg. 1999;230(3):289-297.
- Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883-1889.
- The Propranolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg. 2002:35:72-79.
- Ruhani G, Robertson L, Clarke M. Medical treatment for small abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012. Sep 12;9:CD009536. doi: 10.1002/14651858.
- The United Kingdom EVAR Trial Investigators. Endovascular versus open repair of abdominal aortic aneurysm. N Engl J Med. 2010; 362:1863-1871.
- De Bruin JL, Baas AF, Buth J, et al. Long-term outcome of open or endovascular repair of abdominal aortic aneurysm. N Engl J Med. 2010;362:1881-1889.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs. endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA. 2012;307(15):1621-1628.
- Biancari F, Catania A, D’Andrea V. Elective endovascular vs. open repair of abdominal aortic aneurysm in patients aged 80 years and older: systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2011;42:571-576.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs. open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med. 2008;358:464-474.
- Cosford PA, Leng GC, Tomas J. Screening for abdominal aortic aneurysm. Cochrane Database for Systematic Reviews 2007, Issue 2. Art. No.: CD002945. DOI: 10.1002/14651858.CD002945.pub2.
- Filardo G, Powell JT, Martinez MA, Ballard DJ. Surgery for small asymptomatic abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012, Issue 3. Art. No.:CD001835. DOI: 10.1002/14651858.CD001835.pub3.
- The UK Small Aneurysm Trial Participants. Final 12-year follow-up of surgery versus surveillance in the UK small aneurysm trial. Brit J Surg. 2007;94:702-708.
- U.S. Preventive Services Task Force. Screening for Abdominal Aortic Aneurysm: Recommendation Statement. AHRQ Publication No. 05-0569-A, February 2005.
Case
A generally healthy, 74-year-old man presents with sudden-onset abdominal pain due to acute pancreatitis. Computed tomography (CT) of his abdomen shows pancreatic inflammation and an incidental finding of a 4.5-cm abdominal aortic aneurysm. He had never had any imaging of his abdomen prior to this study and described no prior episodes of abdominal pain.
When should his abdominal aortic aneurysm be treated?
Overview
An abdominal aortic aneurysm (AAA) is an abnormal dilation of the abdominal aorta between the diaphragm and the aortic bifurcation of the iliac arteries. An AAA is usually defined as a dilatation with a diameter of >3 cm or 50% greater than the typical diameter. Most AAAs are located in the infrarenal aorta, proximal to the iliac bifurcation.
Population screening programs show a prevalence of AAA of 4% to 8% in men aged 65 to 80 years.1 AAA prevalence is approximately six times greater in men than women, though the prevalence in women might be increasing.1 AAA is most common in white men, with black men and those of Asian heritage having lower risk. A combination of genetic predisposition and environmental and physiologic factors lead to initiation and progression of AAAs; family history, male sex, advanced age, and history of smoking are major risk factors.
Mortality after AAA rupture is high. Approximately 62% of patients die prior to hospital arrival.2 Of those who undergo emergent AAA surgery, 50% will die.1
Aortic repair with a prosthetic vascular graft reduces morbidity and mortality from rupture, but the risks of repair are not trivial.2
Review of the Data
Risk of rupture. An AAA should be repaired when the risk of rupture outweighs the risks of surgical repair. Symptomatic aneurysms—such as those causing back or abdominal pain—have a higher risk of rupture than asymptomatic aneurysms. Most AAAs are asymptomatic and, in the absence of imaging, not identified until the time of rupture. Given the significant mortality associated with rupture, there is benefit to intervening on asymptomatic aneurysms before rupture.
The risk of AAA rupture has been studied in patients who either have been unfit for surgical repair or uninterested in intervention. Risk of rupture increases substantially with aneurysm size. Lederle et al estimated a two-year aneurysm rupture risk of 22.1% for AAA with a diameter of 5.0 to 5.9 cm, 18.9% for 6.0 to 6.9 cm, and 43.4% for a diameter ≥7.0 cm.3 In another study of 476 patients, the average risks of rupture in male and female patients with an AAA of 5.0 to 5.9 cm were 1.0% and 3.9% per year, respectively. For male and female patients with ≥6.0 cm AAAs, risks of rupture were 14.1% and 22.3% per year.4 Women with AAA have been found to have a higher risk of rupture in all studies in which female patients were included.
Because rupture risk increases with size, predicting the rate of growth is clinically important. Powell et al conducted a systematic review of growth rates of small AAAs.5 In 15 studies that examined 7,630 patients, the growth rate for a 3.5-cm aneurysm was estimated at 1.9 mm/year and for a 4.5-cm aneurysm was 3.5 mm/year. Given an exponentially increasing aneurysm diameter, this suggests an elapsed time of 6.2 years for a 3.5-cm aneurysm to grow to 5.5 cm, and 2.3 years for a 4.5-cm AAA to grow to 5.5 cm. This prediction does not account for individual variability in growth rate. Some AAAs grow quickly, others erratically, and others not at all. This growth variability is influenced by individual characteristics including cigarette smoking, sex, age, and other factors.
Medical prevention of progression and rupture. Studies have assessed whether modification of risk factors can delay progression of growth of AAAs. In a small aneurysm trial in the United Kingdom, self-reported smoking status was associated with an incrementally increased growth rate of 0.4 mm per year.5 Each year of smoking increases the relative risk of AAA by 4%, and continued smoking leads to more rapid AAA expansion.6 There is no clear relationship between cholesterol levels and AAA expansion rate. Observational studies suggest that aneurysm expansion decreases with statin use, but there is not sufficient evidence to recommend statin therapy for AAA alone.6
Many patients with AAA, however, are candidates for statins because of concomitant coronary artery or peripheral vascular disease. Small, randomized controlled trials have shown that macrolides and tetracycline antibiotics might inhibit AAA growth, but prescribing them for this purpose is not currently the standard of care.7 Elevated mean blood pressure has been associated with rupture, but there is not good evidence showing delay of progression with treatment of hypertension.6 Early observational studies suggested that beta-blocker use would decrease AAA progression, but further evidence has not supported their benefit in slowing progression of size.8 Likewise, use of angiotensin-converting enzyme inhibitors has also shown no growth inhibition.7 An ongoing Cochrane review is evaluating evidence for these medical treatments of AAA.9
Surgical prevention of rupture. There are two surgical methods of AAA repair: open repair and endovascular aneurysm repair (EVAR). Both involve use of a prosthetic graft to prevent the aneurysm from enlarging. The EVAR procedure typically involves entry at the femoral artery, with use of catheters and guide wires to advance a graft to the desired location and anchor it in place. Because this utilizes an endovascular approach, regional rather than general anesthesia can be used.
Multiple investigators have evaluated for differences in outcomes between the two methods of surgical AAA repair. Studies have shown increased 30-day postoperative mortality with open repair, as well as significantly higher rates of postoperative cardiac, pulmonary, and renal complications. One randomized controlled study found 30-day operative mortality of 1.8% in the EVAR group and 4.3% in the open repair group.10 However, after a median six-year follow-up of patients after EVAR or open repair, there is no difference in total mortality or aneurysm-related mortality.10 Compared with open repair, the need for long-term surveillance and re-intervention post-EVAR is higher, with endoleak and graft migration the most common complications. This accounts for the loss of early survival advantage in post-EVAR patients. By two years post-operation, complication after repair with either technique is not statistically different. De Bruin et al found that six years after randomization for repair type, cumulative survival rates were 69.9% for patients after open repair and 68.9% with EVAR.11
Studies also have focused on subgroups of patients with a higher operative risk and shorter life expectancy, such as the elderly.12 A pooled analysis of 13,419 patients aged ≥80 years from six observational studies showed 8.6% immediate mortality after open repair and 2.3% after EVAR (risk difference 6.2%, 95% CI 5.4-7.0%).13 Pooled analysis of three longer-term studies showed similar overall survival at three years after EVAR and open repair.13 When EVAR is not available, open repair has acceptable short- and long-term survival in patients aged ≥80 years with an AAA at high risk of rupture.14
Screening. A Cochrane review evaluated the effect of ultrasound screening of asymptomatic AAA on mortality. In 127,891 men and 9,342 women aged 65 to 79, researchers found a significant decrease in mortality in men aged 65 to 79 who were screened (odds ratio 0.6, 95% CI 0.47-0.78) but no benefit to screening of women.15 The current U.S. Preventive Services Task Force (USPSTF) guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 who have any history of tobacco use. For men in this age group who have never smoked, the balance between benefits and harms of screening is too close for the USPSTF to make recommendations. Because of the lower prevalence in women, the USPSTF recommends against screening women for AAA.18
Timing of repair. Early repair of small AAAs (4 cm to 5.5 cm) has no long-term survival benefit compared to ultrasound surveillance without repair.16,17 Therefore, AAAs <5.5 cm should be followed with regular ultrasound surveillance every six months, with referral to surgery if the diameter reaches 5.5 cm, or grows >1 cm a year. The size at which surgery should be performed might be lower in women, given that their risk of rupture is higher than men. A thoughtful discussion of individual risks should take place in every case, but in many patients, even the elderly, repair of a large asymptomatic AAA is indicated. 5
Back to the Case
Our patient should have repeat imaging of his AAA in six months and regular surveillance afterward to monitor for growth every six months. When the AAA is >5.5 cm or if it grows >1 cm a year, he should be evaluated for EVAR or open repair.
Bottom Line
The current USPSTF guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 with a history of smoking. If an AAA >3 cm is found, the patient should undergo regular USG screening every six months. The AAA should be repaired if >5.5 cm or symptomatic, via either an endoscopic or open approach.
Dr. Best is a hospitalist at University of Washington Medicine at Harborview and associate program director of the internal-medicine residency program at the University of Washington, Seattle. Dr. Carpenter is a fellow in the division of geriatrics at the University of California at San Franscisco.
References
- Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8, 92-102.
- Ernst CB. Abdominal aortic aneurysm. N Engl J Med. 1993;328(16):1167-1172.
- Lederle FA, Johnson GR, Wilson SE, et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA. 2002;287:2968-2972
- Brown PM, Zelt DT, Sobolev B. The risk of rupture in untreated aneurysms: the impact of size, gender, and expansion rate. J Vasc Surg. 2003;37:280-284.
- Powell JT, Sweeting MJ, Brown LC, Gotensparre SM, Fowkes FG, Thompson SG. Systematic review and meta-analysis of growth rates of small abdominal aortic aneurysms. British Journal of Surgery. 2011;98:609-618.
- Brown LC, Powell JT. Risk factors for aneurysm rupture in patients kept under ultrasound surveillance. UK Small Aneurysm Trial Participants. Ann Surg. 1999;230(3):289-297.
- Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883-1889.
- The Propranolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg. 2002:35:72-79.
- Ruhani G, Robertson L, Clarke M. Medical treatment for small abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012. Sep 12;9:CD009536. doi: 10.1002/14651858.
- The United Kingdom EVAR Trial Investigators. Endovascular versus open repair of abdominal aortic aneurysm. N Engl J Med. 2010; 362:1863-1871.
- De Bruin JL, Baas AF, Buth J, et al. Long-term outcome of open or endovascular repair of abdominal aortic aneurysm. N Engl J Med. 2010;362:1881-1889.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs. endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA. 2012;307(15):1621-1628.
- Biancari F, Catania A, D’Andrea V. Elective endovascular vs. open repair of abdominal aortic aneurysm in patients aged 80 years and older: systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2011;42:571-576.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs. open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med. 2008;358:464-474.
- Cosford PA, Leng GC, Tomas J. Screening for abdominal aortic aneurysm. Cochrane Database for Systematic Reviews 2007, Issue 2. Art. No.: CD002945. DOI: 10.1002/14651858.CD002945.pub2.
- Filardo G, Powell JT, Martinez MA, Ballard DJ. Surgery for small asymptomatic abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012, Issue 3. Art. No.:CD001835. DOI: 10.1002/14651858.CD001835.pub3.
- The UK Small Aneurysm Trial Participants. Final 12-year follow-up of surgery versus surveillance in the UK small aneurysm trial. Brit J Surg. 2007;94:702-708.
- U.S. Preventive Services Task Force. Screening for Abdominal Aortic Aneurysm: Recommendation Statement. AHRQ Publication No. 05-0569-A, February 2005.
Case
A generally healthy, 74-year-old man presents with sudden-onset abdominal pain due to acute pancreatitis. Computed tomography (CT) of his abdomen shows pancreatic inflammation and an incidental finding of a 4.5-cm abdominal aortic aneurysm. He had never had any imaging of his abdomen prior to this study and described no prior episodes of abdominal pain.
When should his abdominal aortic aneurysm be treated?
Overview
An abdominal aortic aneurysm (AAA) is an abnormal dilation of the abdominal aorta between the diaphragm and the aortic bifurcation of the iliac arteries. An AAA is usually defined as a dilatation with a diameter of >3 cm or 50% greater than the typical diameter. Most AAAs are located in the infrarenal aorta, proximal to the iliac bifurcation.
Population screening programs show a prevalence of AAA of 4% to 8% in men aged 65 to 80 years.1 AAA prevalence is approximately six times greater in men than women, though the prevalence in women might be increasing.1 AAA is most common in white men, with black men and those of Asian heritage having lower risk. A combination of genetic predisposition and environmental and physiologic factors lead to initiation and progression of AAAs; family history, male sex, advanced age, and history of smoking are major risk factors.
Mortality after AAA rupture is high. Approximately 62% of patients die prior to hospital arrival.2 Of those who undergo emergent AAA surgery, 50% will die.1
Aortic repair with a prosthetic vascular graft reduces morbidity and mortality from rupture, but the risks of repair are not trivial.2
Review of the Data
Risk of rupture. An AAA should be repaired when the risk of rupture outweighs the risks of surgical repair. Symptomatic aneurysms—such as those causing back or abdominal pain—have a higher risk of rupture than asymptomatic aneurysms. Most AAAs are asymptomatic and, in the absence of imaging, not identified until the time of rupture. Given the significant mortality associated with rupture, there is benefit to intervening on asymptomatic aneurysms before rupture.
The risk of AAA rupture has been studied in patients who either have been unfit for surgical repair or uninterested in intervention. Risk of rupture increases substantially with aneurysm size. Lederle et al estimated a two-year aneurysm rupture risk of 22.1% for AAA with a diameter of 5.0 to 5.9 cm, 18.9% for 6.0 to 6.9 cm, and 43.4% for a diameter ≥7.0 cm.3 In another study of 476 patients, the average risks of rupture in male and female patients with an AAA of 5.0 to 5.9 cm were 1.0% and 3.9% per year, respectively. For male and female patients with ≥6.0 cm AAAs, risks of rupture were 14.1% and 22.3% per year.4 Women with AAA have been found to have a higher risk of rupture in all studies in which female patients were included.
Because rupture risk increases with size, predicting the rate of growth is clinically important. Powell et al conducted a systematic review of growth rates of small AAAs.5 In 15 studies that examined 7,630 patients, the growth rate for a 3.5-cm aneurysm was estimated at 1.9 mm/year and for a 4.5-cm aneurysm was 3.5 mm/year. Given an exponentially increasing aneurysm diameter, this suggests an elapsed time of 6.2 years for a 3.5-cm aneurysm to grow to 5.5 cm, and 2.3 years for a 4.5-cm AAA to grow to 5.5 cm. This prediction does not account for individual variability in growth rate. Some AAAs grow quickly, others erratically, and others not at all. This growth variability is influenced by individual characteristics including cigarette smoking, sex, age, and other factors.
Medical prevention of progression and rupture. Studies have assessed whether modification of risk factors can delay progression of growth of AAAs. In a small aneurysm trial in the United Kingdom, self-reported smoking status was associated with an incrementally increased growth rate of 0.4 mm per year.5 Each year of smoking increases the relative risk of AAA by 4%, and continued smoking leads to more rapid AAA expansion.6 There is no clear relationship between cholesterol levels and AAA expansion rate. Observational studies suggest that aneurysm expansion decreases with statin use, but there is not sufficient evidence to recommend statin therapy for AAA alone.6
Many patients with AAA, however, are candidates for statins because of concomitant coronary artery or peripheral vascular disease. Small, randomized controlled trials have shown that macrolides and tetracycline antibiotics might inhibit AAA growth, but prescribing them for this purpose is not currently the standard of care.7 Elevated mean blood pressure has been associated with rupture, but there is not good evidence showing delay of progression with treatment of hypertension.6 Early observational studies suggested that beta-blocker use would decrease AAA progression, but further evidence has not supported their benefit in slowing progression of size.8 Likewise, use of angiotensin-converting enzyme inhibitors has also shown no growth inhibition.7 An ongoing Cochrane review is evaluating evidence for these medical treatments of AAA.9
Surgical prevention of rupture. There are two surgical methods of AAA repair: open repair and endovascular aneurysm repair (EVAR). Both involve use of a prosthetic graft to prevent the aneurysm from enlarging. The EVAR procedure typically involves entry at the femoral artery, with use of catheters and guide wires to advance a graft to the desired location and anchor it in place. Because this utilizes an endovascular approach, regional rather than general anesthesia can be used.
Multiple investigators have evaluated for differences in outcomes between the two methods of surgical AAA repair. Studies have shown increased 30-day postoperative mortality with open repair, as well as significantly higher rates of postoperative cardiac, pulmonary, and renal complications. One randomized controlled study found 30-day operative mortality of 1.8% in the EVAR group and 4.3% in the open repair group.10 However, after a median six-year follow-up of patients after EVAR or open repair, there is no difference in total mortality or aneurysm-related mortality.10 Compared with open repair, the need for long-term surveillance and re-intervention post-EVAR is higher, with endoleak and graft migration the most common complications. This accounts for the loss of early survival advantage in post-EVAR patients. By two years post-operation, complication after repair with either technique is not statistically different. De Bruin et al found that six years after randomization for repair type, cumulative survival rates were 69.9% for patients after open repair and 68.9% with EVAR.11
Studies also have focused on subgroups of patients with a higher operative risk and shorter life expectancy, such as the elderly.12 A pooled analysis of 13,419 patients aged ≥80 years from six observational studies showed 8.6% immediate mortality after open repair and 2.3% after EVAR (risk difference 6.2%, 95% CI 5.4-7.0%).13 Pooled analysis of three longer-term studies showed similar overall survival at three years after EVAR and open repair.13 When EVAR is not available, open repair has acceptable short- and long-term survival in patients aged ≥80 years with an AAA at high risk of rupture.14
Screening. A Cochrane review evaluated the effect of ultrasound screening of asymptomatic AAA on mortality. In 127,891 men and 9,342 women aged 65 to 79, researchers found a significant decrease in mortality in men aged 65 to 79 who were screened (odds ratio 0.6, 95% CI 0.47-0.78) but no benefit to screening of women.15 The current U.S. Preventive Services Task Force (USPSTF) guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 who have any history of tobacco use. For men in this age group who have never smoked, the balance between benefits and harms of screening is too close for the USPSTF to make recommendations. Because of the lower prevalence in women, the USPSTF recommends against screening women for AAA.18
Timing of repair. Early repair of small AAAs (4 cm to 5.5 cm) has no long-term survival benefit compared to ultrasound surveillance without repair.16,17 Therefore, AAAs <5.5 cm should be followed with regular ultrasound surveillance every six months, with referral to surgery if the diameter reaches 5.5 cm, or grows >1 cm a year. The size at which surgery should be performed might be lower in women, given that their risk of rupture is higher than men. A thoughtful discussion of individual risks should take place in every case, but in many patients, even the elderly, repair of a large asymptomatic AAA is indicated. 5
Back to the Case
Our patient should have repeat imaging of his AAA in six months and regular surveillance afterward to monitor for growth every six months. When the AAA is >5.5 cm or if it grows >1 cm a year, he should be evaluated for EVAR or open repair.
Bottom Line
The current USPSTF guidelines recommend one-time ultrasound-guided (USG) screening for AAA in men aged 65 to 75 with a history of smoking. If an AAA >3 cm is found, the patient should undergo regular USG screening every six months. The AAA should be repaired if >5.5 cm or symptomatic, via either an endoscopic or open approach.
Dr. Best is a hospitalist at University of Washington Medicine at Harborview and associate program director of the internal-medicine residency program at the University of Washington, Seattle. Dr. Carpenter is a fellow in the division of geriatrics at the University of California at San Franscisco.
References
- Nordon IM, Hinchliffe RJ, Loftus IM, Thompson MM. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat Rev Cardiol. 2011;8, 92-102.
- Ernst CB. Abdominal aortic aneurysm. N Engl J Med. 1993;328(16):1167-1172.
- Lederle FA, Johnson GR, Wilson SE, et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA. 2002;287:2968-2972
- Brown PM, Zelt DT, Sobolev B. The risk of rupture in untreated aneurysms: the impact of size, gender, and expansion rate. J Vasc Surg. 2003;37:280-284.
- Powell JT, Sweeting MJ, Brown LC, Gotensparre SM, Fowkes FG, Thompson SG. Systematic review and meta-analysis of growth rates of small abdominal aortic aneurysms. British Journal of Surgery. 2011;98:609-618.
- Brown LC, Powell JT. Risk factors for aneurysm rupture in patients kept under ultrasound surveillance. UK Small Aneurysm Trial Participants. Ann Surg. 1999;230(3):289-297.
- Baxter BT, Terrin MC, Dalman RL. Medical management of small abdominal aortic aneurysms. Circulation. 2008;117:1883-1889.
- The Propranolol Aneurysm Trial Investigators. Propranolol for small abdominal aortic aneurysms: results of a randomized trial. J Vasc Surg. 2002:35:72-79.
- Ruhani G, Robertson L, Clarke M. Medical treatment for small abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012. Sep 12;9:CD009536. doi: 10.1002/14651858.
- The United Kingdom EVAR Trial Investigators. Endovascular versus open repair of abdominal aortic aneurysm. N Engl J Med. 2010; 362:1863-1871.
- De Bruin JL, Baas AF, Buth J, et al. Long-term outcome of open or endovascular repair of abdominal aortic aneurysm. N Engl J Med. 2010;362:1881-1889.
- Jackson RS, Chang DC, Freischlag JA. Comparison of long-term survival after open vs. endovascular repair of intact abdominal aortic aneurysm among Medicare beneficiaries. JAMA. 2012;307(15):1621-1628.
- Biancari F, Catania A, D’Andrea V. Elective endovascular vs. open repair of abdominal aortic aneurysm in patients aged 80 years and older: systematic review and meta-analysis. Eur J Vasc Endovasc Surg. 2011;42:571-576.
- Schermerhorn ML, O’Malley AJ, Jhaveri A, Cotterill P, Pomposelli F, Landon BE. Endovascular vs. open repair of abdominal aortic aneurysms in the Medicare population. N Engl J Med. 2008;358:464-474.
- Cosford PA, Leng GC, Tomas J. Screening for abdominal aortic aneurysm. Cochrane Database for Systematic Reviews 2007, Issue 2. Art. No.: CD002945. DOI: 10.1002/14651858.CD002945.pub2.
- Filardo G, Powell JT, Martinez MA, Ballard DJ. Surgery for small asymptomatic abdominal aortic aneurysms. Cochrane Database for Systematic Reviews 2012, Issue 3. Art. No.:CD001835. DOI: 10.1002/14651858.CD001835.pub3.
- The UK Small Aneurysm Trial Participants. Final 12-year follow-up of surgery versus surveillance in the UK small aneurysm trial. Brit J Surg. 2007;94:702-708.
- U.S. Preventive Services Task Force. Screening for Abdominal Aortic Aneurysm: Recommendation Statement. AHRQ Publication No. 05-0569-A, February 2005.
How is Acute Pericarditis Diagnosed and Treated?
Case
A 32-year-old female with no significant past medical history is evaluated for sharp, left-sided chest pain for five days. Her pain is intermittent, worse with deep inspiration and in the supine position. She denies any shortness of breath. Her temperature is 100.8ºF, but otherwise her vital signs are normal. The physical exam and chest radiograph are unremarkable, but an electrocardiogram shows diffuse ST-segment elevations. The initial troponin is mildly elevated at 0.35 ng/ml.
Could this patient have acute pericarditis? If so, how should she be managed?
Background
Pericarditis is the most common pericardial disease encountered by hospitalists. As many as 5% of chest pain cases unattributable to myocardial infarction (MI) are diagnosed with pericarditis.1 In immunocompetent individuals, as many as 90% of acute pericarditis cases are viral or idiopathic in etiology.1,2 Human immunodeficiency virus (HIV) and tuberculosis are common culprits in developing countries and immunocompromised hosts.3 Other specific etiologies of acute pericarditis include autoimmune diseases, neoplasms, chest irradiation, trauma, and metabolic disturbances (e.g. uremia). An etiologic classification of acute pericarditis is shown in Table 2 (p. 16).
Pericarditis primarily is a clinical diagnosis. Most patients present with chest pain.4 A pericardial friction rub may or may not be heard (sensitivity 16% to 85%), but when present is nearly 100% specific for pericarditis.2,5 Diffuse ST-segment elevation on electrocardiogram (EKG) is present in 60% to 90% of cases, but it can be difficult to differentiate from ST-segment elevations in acute MI.4,6
Uncomplicated acute pericarditis often is treated successfully as an outpatient.4 However, patients with high-risk features (see Table 1, right) should be hospitalized for identification and treatment of specific underlying etiology and for monitoring of complications, such as tamponade.7
Our patient has features consistent with pericarditis. In the following sections, we will review the diagnosis and treatment of acute pericarditis.

Review of the Data
How is acute pericarditis diagnosed?
Acute pericarditis is a clinical diagnosis supported by EKG and echocardiogram. At least two of the following four criteria must be present for the diagnosis: pleuritic chest pain, pericardial rub, diffuse ST-segment elevation on EKG, and pericardial effusion.8
History. Patients may report fever (46% in one small study of 69 patients) or a recent history of respiratory or gastrointestinal infection (40%).5 Most patients will report pleuritic chest pain. Typically, the pain is improved when sitting up and leaning forward, and gets worse when lying supine.4 Pain might radiate to the trapezius muscle ridge due to the common phrenic nerve innervation of pericardium and trapezius.9 However, pain might be minimal or absent in patients with uremic, neoplastic, tuberculous, or post-irradiation pericarditis.
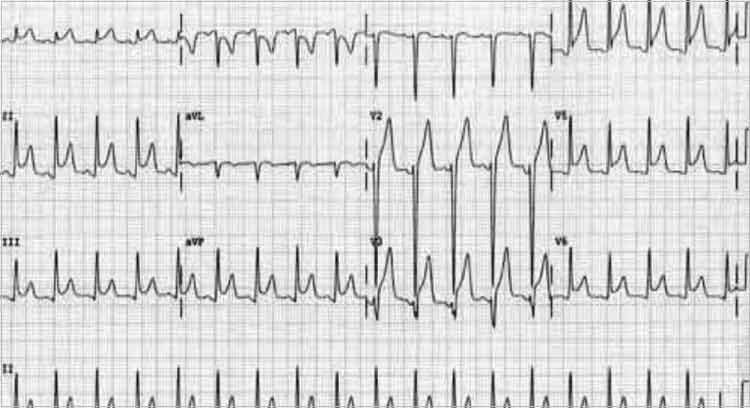
Physical exam. A pericardial friction rub is nearly 100% specific for a pericarditis diagnosis, but sensitivity can vary (16% to 85%) depending on the frequency of auscultation and underlying etiology.2,5 It is thought to be caused by friction between the parietal and visceral layers of inflamed pericardium. A pericardial rub classically is described as a superficial, high-pitched, scratchy, or squeaking sound best heard with the diaphragm of the stethoscope at the lower left sternal border with the patient leaning forward.
Laboratory data. A complete blood count, metabolic panel, and cardiac enzymes should be checked in all patients with suspected acute pericarditis. Troponin values are elevated in up to one-third of patients, indicating cardiac muscle injury or myopericarditis, but have not been shown to adversely impact hospital length of stay, readmission, or complication rates.5,10 Markers of inflammation (e.g. erythrocyte sedimentation rate or C-reactive protein) are frequently elevated but do not point to a specific underlying etiology. Routine viral cultures and antibody titers are not useful.11
Most cases of pericarditis are presumed idiopathic (viral); however, finding a specific etiology should be considered in patients who do not respond after one week of therapy. Anti-nuclear antibody, complement levels, and rheumatoid factor can serve as screening tests for autoimmune disease. Purified protein derivative or quantiferon testing and HIV testing might be indicated in patients with appropriate risk factors. In cases of suspected tuberculous or neoplastic pericarditis, pericardial fluid analysis and biopsy could be warranted.
Electrocardiography. The EKG is the most useful test in diagnosing acute pericarditis. EKG changes in acute pericarditis can progress over four stages:
- Stage 1: diffuse ST elevations with or without PR depressions, initially;
- Stage 2: normalization of ST and PR segments, typically after several days;
- Stage 3: diffuse T-wave inversions; and
- Stage 4: normalization of T-waves, typically after weeks or months.
While all four stages are unlikely to be present in a given case, 80% of patients with pericarditis will demonstrate diffuse ST-segment elevations and PR-segment depression (see Figure 2, above).12
Table 3 lists EKG features helpful in differentiating acute pericarditis from acute myocardial infarction.
Chest radiography. Because a pericardial effusion often accompanies pericarditis, a chest radiograph (CXR) should be performed in all suspected cases. The CXR might show enlargement of the cardiac silhouette if more than 250 ml of pericardial fluid is present.3 A CXR also is helpful to diagnose concomitant pulmonary infection, pleural effusion, or mediastinal mass—all findings that could point to an underlying specific etiology of pericarditis and/or pericardial effusion.
Echocardiography. An echocardiogram should be performed in all patients with suspected pericarditis to detect effusion, associated myocardial, or paracardial disease.13 The echocardiogram frequently is normal but could show an effusion in 60%, and tamponade (see Figure 1, p. 15) in 5%, of cases.4
Computed tomography (CT) and cardiac magnetic resonance imaging (CMR).CT or CMR are the imaging modalities of choice when an echocardiogram is inconclusive or in cases of pericarditis complicated by a hemorrhagic or localized effusion, pericardial thickening, or pericardial mass.14 They also help in precise imaging of neighboring structures, such as lungs or mediastinum.
Pericardial fluid analysis and pericardial biopsy. In cases of refractory pericarditis with effusion, pericardial fluid analysis might provide clues to the underlying etiology. Routine chemistry, cell count, gram and acid fast staining, culture, and cytology should be sent. In addition, acid-fast bacillus staining and culture, adenosine deaminase, and interferon-gamma testing should be ordered when tuberculous pericarditis is suspected. A pericardial biopsy may show granulomas or neoplastic cells. Overall, pericardial fluid analysis and biopsy reveal a diagnosis in roughly 20% of cases.11

How is acute pericarditis treated?
Most cases of uncomplicated acute pericarditis are viral and respond well to NSAID plus colchicine therapy.2,4 Failure to respond to NSAIDs plus colchicine—evidenced by persistent fever, pericardial chest pain, new pericardial effusion, or worsening of general illness—within a week of treatment should prompt a search for an underlying systemic illness. If found, treatment should be aimed at the causative illness.
Bacterial pericarditis usually requires surgical drainage in addition to treatment with appropriate antibiotics.11 Tuberculous pericarditis is treated with multidrug therapy; when underlying HIV is present, patients should receive highly active anti-retroviral therapy as well. Steroids and immunosuppressants should be considered in addition to NSAIDs and colchicine in autoimmune pericarditis.10 Neoplastic pericarditis may resolve with chemotherapy but it has a high recurrence rate.13 Uremic pericarditis requires intensified dialysis.
Treatment options for uncomplicated idiopathic or viral pericarditis include:
NSAIDs. It is important to adequately dose NSAIDs when treating acute pericarditis. Initial treatment options include ibuprofen (1,600 to 3,200 mg daily), indomethacin (75 to 150 mg daily) or aspirin (2 to 4 gm daily) for one week.11,15 Aspirin is preferred in patients with ischemic heart disease. For patients with symptoms that persist longer than a week, NSAIDS may be continued, but investigation for an underlying etiology is indicated. Concomitant proton-pump-inhibitor therapy should be considered in patients at high risk for peptic ulcer disease to minimize gastric side effects.
Colchicine. Colchicine has a favorable risk-benefit profile as an adjunct treatment for acute and recurrent pericarditis. Patients experience better symptom relief when treated with both colchicine and an NSAID, compared with NSAIDs alone (88% versus 63%). Recurrence rates are lower with combined therapy (11% versus 32%).16 Colchicine treatment (0.6 mg twice daily after a loading dose of up to 2 mg) is recommended for several months to greater than one year.13,16,17
Glucocorticoids. Routine glucocorticoid use should be avoided in the treatment of acute pericarditis, as it has been associated with an increased risk for recurrence (OR 4.3).16,18 Glucocorticoid use should be considered in cases of pericarditis refractory to NSAIDs and colchicine, cases in which NSAIDs and or colchicine are contraindicated, and in autoimmune or connective-tissue-disease-related pericarditis. Prednisone should be dosed up to 1 mg/kg/day for at least one month, depending on symptom resolution, then tapered after either NSAIDs or colchicine have been started.13 Smaller prednisone doses of up to 0.5 mg/kg/day could be as effective, with the added benefit of reduced side effects and recurrences.19
Invasive treatment. Pericardiocentesis and/or pericardiectomy should be considered when pericarditis is complicated by a large effusion or tamponade, constrictive physiology, or recurrent effusion.11 Pericardiocentesis is the least invasive option and helps provide immediate relief in cases of tamponade or large symptomatic effusions. It is the preferred modality for obtaining pericardial fluid for diagnostic analysis. However, effusions can recur and in those cases pericardial window is preferred, as it provides continued outflow of pericardial fluid. Pericardiectomy is recommended in cases of symptomatic constrictive pericarditis unresponsive to medical therapy.15
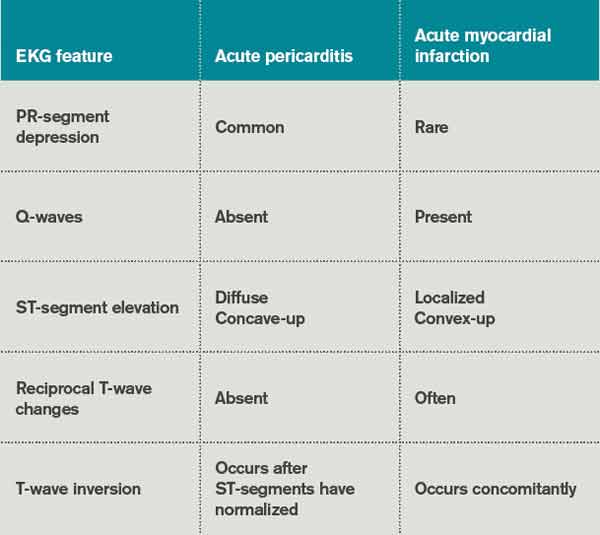
Back to the Case
The patient’s presentation—prodrome followed by fever and pleuritic chest pain—is characteristic of acute idiopathic pericarditis. No pericardial rub was heard, but EKG findings were typical. Troponin I elevation suggested underlying myopericarditis. An echocardiogram was unremarkable. Given the likely viral or idiopathic etiology, no further diagnostic tests were ordered to explore the possibility of an underlying systemic illness.
The patient was started on ibuprofen 600 mg every eight hours. She had significant relief of her symptoms within two days. A routine fever workup was negative. She was discharged the following day.
The patient was readmitted three months later with recurrent pleuritic chest pain, which did not improve with resumption of NSAID therapy. Initial troponin I was 0.22 ng/ml, electrocardiogram was unchanged, and an echocardiogram showed small effusion. She was started on ibuprofen 800 mg every eight hours, as well as colchicine 0.6 mg twice daily. Her symptoms resolved the next day and she was discharged with prescriptions for ibuprofen and colchicine. She was instructed to follow up with a primary-care doctor in one week.
At the clinic visit, ibuprofen was tapered but colchicine was continued for another six months. She remained asymptomatic at her six-month clinic follow-up.
Bottom Line
Acute pericarditis is a clinical diagnosis supported by EKG findings. Most cases are idiopathic or viral, and can be treated successfully with NSAIDs and colchicine. For cases that do not respond to initial therapy, or cases that present with high-risk features, a specific etiology should be sought.
Dr. Southern is chief of the division of hospital medicine at Montefiore Medical Center in Bronx, N.Y. Dr. Galhorta is an instructor and Drs. Martin, Korcak, and Stehlihova are assistant professors in the department of medicine at Albert Einstein.
References
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med. 2004;351:2195-2202.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol. 1995;75:378-382.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet. 2004;363:717-727.
- Imazio M, Demichelis B, Parrini I, et al. Day-hospital treatment of acute pericarditis: a management program for outpatient therapy. J Am Coll Cardiol. 2004;43:1042-1046.
- Bonnefoy E, Godon P, Kirkorian G, et al. Serum cardiac troponin I and ST-segment elevation in patients with acute pericarditis. Eur Heart J. 2000;21:832-836.
- Salisbury AC, Olalla-Gomez C, Rihal CS, et al. Frequency and predictors of urgent coronary angiography in patients with acute pericarditis. Mayo Clin Proc. 2009;84(1):11-15.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation. 2007;115:2739-2744.
- Imazio M, Spodick DH, Brucato A, et al. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract. 2010;64(10):1384-1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA. 2003;289:1150-1153.
- Imazio M, Demichelis B, Cecchi E. Cardiac troponin I in acute pericarditis. J Am Coll Cardiol. 2003;42(12):2144-2148.
- Sagristà Sauleda J, Permanyer Miralda G, Soler Soler J. Diagnosis and management of pericardial syndromes. Rev Esp Cardiol. 2005;58(7):830-841.
- Bruce MA, Spodick DH. Atypical electrocardiogram in acute pericarditis: characteristics and prevalence. J Electrocardiol. 1980;13:61-66.
- Maisch B, Seferovic PM, Ristic AD, et al. Guidelines on the diagnosis and management of pericardial diseases executive summary; the task force on the diagnosis and management of pericardial diseases of the European Society of Cardiology. Eur Heart J. 2004; 25(7):587-610.
- Verhaert D, Gabriel RS, Johnston D, et al. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging. 2010;3:333-343.
- Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121:916-928.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: results of the colchicine for acute pericarditis (COPE) trial. Circulation. 2005;112(13):2012-2016.
- Adler Y, Finkelstein Y, Guindo J, et al. Colchicine treatment for recurrent pericarditis: a decade of experience. Circulation. 1998;97:2183-185.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine as first-choice therapy for recurrent pericarditis: results of the colchicine for recurrent pericarditis (CORE) trial. Arch Intern Med. 2005;165:1987-1991.
- Imazio M, Brucato A, Cumetti D, et al. Corticosteroids for recurrent pericarditis: high versus low doses: a nonrandomized observation. Circulation. 2008;118:667-771.
Case
A 32-year-old female with no significant past medical history is evaluated for sharp, left-sided chest pain for five days. Her pain is intermittent, worse with deep inspiration and in the supine position. She denies any shortness of breath. Her temperature is 100.8ºF, but otherwise her vital signs are normal. The physical exam and chest radiograph are unremarkable, but an electrocardiogram shows diffuse ST-segment elevations. The initial troponin is mildly elevated at 0.35 ng/ml.
Could this patient have acute pericarditis? If so, how should she be managed?
Background
Pericarditis is the most common pericardial disease encountered by hospitalists. As many as 5% of chest pain cases unattributable to myocardial infarction (MI) are diagnosed with pericarditis.1 In immunocompetent individuals, as many as 90% of acute pericarditis cases are viral or idiopathic in etiology.1,2 Human immunodeficiency virus (HIV) and tuberculosis are common culprits in developing countries and immunocompromised hosts.3 Other specific etiologies of acute pericarditis include autoimmune diseases, neoplasms, chest irradiation, trauma, and metabolic disturbances (e.g. uremia). An etiologic classification of acute pericarditis is shown in Table 2 (p. 16).
Pericarditis primarily is a clinical diagnosis. Most patients present with chest pain.4 A pericardial friction rub may or may not be heard (sensitivity 16% to 85%), but when present is nearly 100% specific for pericarditis.2,5 Diffuse ST-segment elevation on electrocardiogram (EKG) is present in 60% to 90% of cases, but it can be difficult to differentiate from ST-segment elevations in acute MI.4,6
Uncomplicated acute pericarditis often is treated successfully as an outpatient.4 However, patients with high-risk features (see Table 1, right) should be hospitalized for identification and treatment of specific underlying etiology and for monitoring of complications, such as tamponade.7
Our patient has features consistent with pericarditis. In the following sections, we will review the diagnosis and treatment of acute pericarditis.
Review of the Data
How is acute pericarditis diagnosed?
Acute pericarditis is a clinical diagnosis supported by EKG and echocardiogram. At least two of the following four criteria must be present for the diagnosis: pleuritic chest pain, pericardial rub, diffuse ST-segment elevation on EKG, and pericardial effusion.8
History. Patients may report fever (46% in one small study of 69 patients) or a recent history of respiratory or gastrointestinal infection (40%).5 Most patients will report pleuritic chest pain. Typically, the pain is improved when sitting up and leaning forward, and gets worse when lying supine.4 Pain might radiate to the trapezius muscle ridge due to the common phrenic nerve innervation of pericardium and trapezius.9 However, pain might be minimal or absent in patients with uremic, neoplastic, tuberculous, or post-irradiation pericarditis.
Physical exam. A pericardial friction rub is nearly 100% specific for a pericarditis diagnosis, but sensitivity can vary (16% to 85%) depending on the frequency of auscultation and underlying etiology.2,5 It is thought to be caused by friction between the parietal and visceral layers of inflamed pericardium. A pericardial rub classically is described as a superficial, high-pitched, scratchy, or squeaking sound best heard with the diaphragm of the stethoscope at the lower left sternal border with the patient leaning forward.
Laboratory data. A complete blood count, metabolic panel, and cardiac enzymes should be checked in all patients with suspected acute pericarditis. Troponin values are elevated in up to one-third of patients, indicating cardiac muscle injury or myopericarditis, but have not been shown to adversely impact hospital length of stay, readmission, or complication rates.5,10 Markers of inflammation (e.g. erythrocyte sedimentation rate or C-reactive protein) are frequently elevated but do not point to a specific underlying etiology. Routine viral cultures and antibody titers are not useful.11
Most cases of pericarditis are presumed idiopathic (viral); however, finding a specific etiology should be considered in patients who do not respond after one week of therapy. Anti-nuclear antibody, complement levels, and rheumatoid factor can serve as screening tests for autoimmune disease. Purified protein derivative or quantiferon testing and HIV testing might be indicated in patients with appropriate risk factors. In cases of suspected tuberculous or neoplastic pericarditis, pericardial fluid analysis and biopsy could be warranted.
Electrocardiography. The EKG is the most useful test in diagnosing acute pericarditis. EKG changes in acute pericarditis can progress over four stages:
- Stage 1: diffuse ST elevations with or without PR depressions, initially;
- Stage 2: normalization of ST and PR segments, typically after several days;
- Stage 3: diffuse T-wave inversions; and
- Stage 4: normalization of T-waves, typically after weeks or months.
While all four stages are unlikely to be present in a given case, 80% of patients with pericarditis will demonstrate diffuse ST-segment elevations and PR-segment depression (see Figure 2, above).12
Table 3 lists EKG features helpful in differentiating acute pericarditis from acute myocardial infarction.
Chest radiography. Because a pericardial effusion often accompanies pericarditis, a chest radiograph (CXR) should be performed in all suspected cases. The CXR might show enlargement of the cardiac silhouette if more than 250 ml of pericardial fluid is present.3 A CXR also is helpful to diagnose concomitant pulmonary infection, pleural effusion, or mediastinal mass—all findings that could point to an underlying specific etiology of pericarditis and/or pericardial effusion.
Echocardiography. An echocardiogram should be performed in all patients with suspected pericarditis to detect effusion, associated myocardial, or paracardial disease.13 The echocardiogram frequently is normal but could show an effusion in 60%, and tamponade (see Figure 1, p. 15) in 5%, of cases.4
Computed tomography (CT) and cardiac magnetic resonance imaging (CMR).CT or CMR are the imaging modalities of choice when an echocardiogram is inconclusive or in cases of pericarditis complicated by a hemorrhagic or localized effusion, pericardial thickening, or pericardial mass.14 They also help in precise imaging of neighboring structures, such as lungs or mediastinum.
Pericardial fluid analysis and pericardial biopsy. In cases of refractory pericarditis with effusion, pericardial fluid analysis might provide clues to the underlying etiology. Routine chemistry, cell count, gram and acid fast staining, culture, and cytology should be sent. In addition, acid-fast bacillus staining and culture, adenosine deaminase, and interferon-gamma testing should be ordered when tuberculous pericarditis is suspected. A pericardial biopsy may show granulomas or neoplastic cells. Overall, pericardial fluid analysis and biopsy reveal a diagnosis in roughly 20% of cases.11
How is acute pericarditis treated?
Most cases of uncomplicated acute pericarditis are viral and respond well to NSAID plus colchicine therapy.2,4 Failure to respond to NSAIDs plus colchicine—evidenced by persistent fever, pericardial chest pain, new pericardial effusion, or worsening of general illness—within a week of treatment should prompt a search for an underlying systemic illness. If found, treatment should be aimed at the causative illness.
Bacterial pericarditis usually requires surgical drainage in addition to treatment with appropriate antibiotics.11 Tuberculous pericarditis is treated with multidrug therapy; when underlying HIV is present, patients should receive highly active anti-retroviral therapy as well. Steroids and immunosuppressants should be considered in addition to NSAIDs and colchicine in autoimmune pericarditis.10 Neoplastic pericarditis may resolve with chemotherapy but it has a high recurrence rate.13 Uremic pericarditis requires intensified dialysis.
Treatment options for uncomplicated idiopathic or viral pericarditis include:
NSAIDs. It is important to adequately dose NSAIDs when treating acute pericarditis. Initial treatment options include ibuprofen (1,600 to 3,200 mg daily), indomethacin (75 to 150 mg daily) or aspirin (2 to 4 gm daily) for one week.11,15 Aspirin is preferred in patients with ischemic heart disease. For patients with symptoms that persist longer than a week, NSAIDS may be continued, but investigation for an underlying etiology is indicated. Concomitant proton-pump-inhibitor therapy should be considered in patients at high risk for peptic ulcer disease to minimize gastric side effects.
Colchicine. Colchicine has a favorable risk-benefit profile as an adjunct treatment for acute and recurrent pericarditis. Patients experience better symptom relief when treated with both colchicine and an NSAID, compared with NSAIDs alone (88% versus 63%). Recurrence rates are lower with combined therapy (11% versus 32%).16 Colchicine treatment (0.6 mg twice daily after a loading dose of up to 2 mg) is recommended for several months to greater than one year.13,16,17
Glucocorticoids. Routine glucocorticoid use should be avoided in the treatment of acute pericarditis, as it has been associated with an increased risk for recurrence (OR 4.3).16,18 Glucocorticoid use should be considered in cases of pericarditis refractory to NSAIDs and colchicine, cases in which NSAIDs and or colchicine are contraindicated, and in autoimmune or connective-tissue-disease-related pericarditis. Prednisone should be dosed up to 1 mg/kg/day for at least one month, depending on symptom resolution, then tapered after either NSAIDs or colchicine have been started.13 Smaller prednisone doses of up to 0.5 mg/kg/day could be as effective, with the added benefit of reduced side effects and recurrences.19
Invasive treatment. Pericardiocentesis and/or pericardiectomy should be considered when pericarditis is complicated by a large effusion or tamponade, constrictive physiology, or recurrent effusion.11 Pericardiocentesis is the least invasive option and helps provide immediate relief in cases of tamponade or large symptomatic effusions. It is the preferred modality for obtaining pericardial fluid for diagnostic analysis. However, effusions can recur and in those cases pericardial window is preferred, as it provides continued outflow of pericardial fluid. Pericardiectomy is recommended in cases of symptomatic constrictive pericarditis unresponsive to medical therapy.15
Back to the Case
The patient’s presentation—prodrome followed by fever and pleuritic chest pain—is characteristic of acute idiopathic pericarditis. No pericardial rub was heard, but EKG findings were typical. Troponin I elevation suggested underlying myopericarditis. An echocardiogram was unremarkable. Given the likely viral or idiopathic etiology, no further diagnostic tests were ordered to explore the possibility of an underlying systemic illness.
The patient was started on ibuprofen 600 mg every eight hours. She had significant relief of her symptoms within two days. A routine fever workup was negative. She was discharged the following day.
The patient was readmitted three months later with recurrent pleuritic chest pain, which did not improve with resumption of NSAID therapy. Initial troponin I was 0.22 ng/ml, electrocardiogram was unchanged, and an echocardiogram showed small effusion. She was started on ibuprofen 800 mg every eight hours, as well as colchicine 0.6 mg twice daily. Her symptoms resolved the next day and she was discharged with prescriptions for ibuprofen and colchicine. She was instructed to follow up with a primary-care doctor in one week.
At the clinic visit, ibuprofen was tapered but colchicine was continued for another six months. She remained asymptomatic at her six-month clinic follow-up.
Bottom Line
Acute pericarditis is a clinical diagnosis supported by EKG findings. Most cases are idiopathic or viral, and can be treated successfully with NSAIDs and colchicine. For cases that do not respond to initial therapy, or cases that present with high-risk features, a specific etiology should be sought.
Dr. Southern is chief of the division of hospital medicine at Montefiore Medical Center in Bronx, N.Y. Dr. Galhorta is an instructor and Drs. Martin, Korcak, and Stehlihova are assistant professors in the department of medicine at Albert Einstein.
References
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med. 2004;351:2195-2202.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol. 1995;75:378-382.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet. 2004;363:717-727.
- Imazio M, Demichelis B, Parrini I, et al. Day-hospital treatment of acute pericarditis: a management program for outpatient therapy. J Am Coll Cardiol. 2004;43:1042-1046.
- Bonnefoy E, Godon P, Kirkorian G, et al. Serum cardiac troponin I and ST-segment elevation in patients with acute pericarditis. Eur Heart J. 2000;21:832-836.
- Salisbury AC, Olalla-Gomez C, Rihal CS, et al. Frequency and predictors of urgent coronary angiography in patients with acute pericarditis. Mayo Clin Proc. 2009;84(1):11-15.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation. 2007;115:2739-2744.
- Imazio M, Spodick DH, Brucato A, et al. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract. 2010;64(10):1384-1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA. 2003;289:1150-1153.
- Imazio M, Demichelis B, Cecchi E. Cardiac troponin I in acute pericarditis. J Am Coll Cardiol. 2003;42(12):2144-2148.
- Sagristà Sauleda J, Permanyer Miralda G, Soler Soler J. Diagnosis and management of pericardial syndromes. Rev Esp Cardiol. 2005;58(7):830-841.
- Bruce MA, Spodick DH. Atypical electrocardiogram in acute pericarditis: characteristics and prevalence. J Electrocardiol. 1980;13:61-66.
- Maisch B, Seferovic PM, Ristic AD, et al. Guidelines on the diagnosis and management of pericardial diseases executive summary; the task force on the diagnosis and management of pericardial diseases of the European Society of Cardiology. Eur Heart J. 2004; 25(7):587-610.
- Verhaert D, Gabriel RS, Johnston D, et al. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging. 2010;3:333-343.
- Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121:916-928.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: results of the colchicine for acute pericarditis (COPE) trial. Circulation. 2005;112(13):2012-2016.
- Adler Y, Finkelstein Y, Guindo J, et al. Colchicine treatment for recurrent pericarditis: a decade of experience. Circulation. 1998;97:2183-185.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine as first-choice therapy for recurrent pericarditis: results of the colchicine for recurrent pericarditis (CORE) trial. Arch Intern Med. 2005;165:1987-1991.
- Imazio M, Brucato A, Cumetti D, et al. Corticosteroids for recurrent pericarditis: high versus low doses: a nonrandomized observation. Circulation. 2008;118:667-771.
Case
A 32-year-old female with no significant past medical history is evaluated for sharp, left-sided chest pain for five days. Her pain is intermittent, worse with deep inspiration and in the supine position. She denies any shortness of breath. Her temperature is 100.8ºF, but otherwise her vital signs are normal. The physical exam and chest radiograph are unremarkable, but an electrocardiogram shows diffuse ST-segment elevations. The initial troponin is mildly elevated at 0.35 ng/ml.
Could this patient have acute pericarditis? If so, how should she be managed?
Background
Pericarditis is the most common pericardial disease encountered by hospitalists. As many as 5% of chest pain cases unattributable to myocardial infarction (MI) are diagnosed with pericarditis.1 In immunocompetent individuals, as many as 90% of acute pericarditis cases are viral or idiopathic in etiology.1,2 Human immunodeficiency virus (HIV) and tuberculosis are common culprits in developing countries and immunocompromised hosts.3 Other specific etiologies of acute pericarditis include autoimmune diseases, neoplasms, chest irradiation, trauma, and metabolic disturbances (e.g. uremia). An etiologic classification of acute pericarditis is shown in Table 2 (p. 16).
Pericarditis primarily is a clinical diagnosis. Most patients present with chest pain.4 A pericardial friction rub may or may not be heard (sensitivity 16% to 85%), but when present is nearly 100% specific for pericarditis.2,5 Diffuse ST-segment elevation on electrocardiogram (EKG) is present in 60% to 90% of cases, but it can be difficult to differentiate from ST-segment elevations in acute MI.4,6
Uncomplicated acute pericarditis often is treated successfully as an outpatient.4 However, patients with high-risk features (see Table 1, right) should be hospitalized for identification and treatment of specific underlying etiology and for monitoring of complications, such as tamponade.7
Our patient has features consistent with pericarditis. In the following sections, we will review the diagnosis and treatment of acute pericarditis.
Review of the Data
How is acute pericarditis diagnosed?
Acute pericarditis is a clinical diagnosis supported by EKG and echocardiogram. At least two of the following four criteria must be present for the diagnosis: pleuritic chest pain, pericardial rub, diffuse ST-segment elevation on EKG, and pericardial effusion.8
History. Patients may report fever (46% in one small study of 69 patients) or a recent history of respiratory or gastrointestinal infection (40%).5 Most patients will report pleuritic chest pain. Typically, the pain is improved when sitting up and leaning forward, and gets worse when lying supine.4 Pain might radiate to the trapezius muscle ridge due to the common phrenic nerve innervation of pericardium and trapezius.9 However, pain might be minimal or absent in patients with uremic, neoplastic, tuberculous, or post-irradiation pericarditis.
Physical exam. A pericardial friction rub is nearly 100% specific for a pericarditis diagnosis, but sensitivity can vary (16% to 85%) depending on the frequency of auscultation and underlying etiology.2,5 It is thought to be caused by friction between the parietal and visceral layers of inflamed pericardium. A pericardial rub classically is described as a superficial, high-pitched, scratchy, or squeaking sound best heard with the diaphragm of the stethoscope at the lower left sternal border with the patient leaning forward.
Laboratory data. A complete blood count, metabolic panel, and cardiac enzymes should be checked in all patients with suspected acute pericarditis. Troponin values are elevated in up to one-third of patients, indicating cardiac muscle injury or myopericarditis, but have not been shown to adversely impact hospital length of stay, readmission, or complication rates.5,10 Markers of inflammation (e.g. erythrocyte sedimentation rate or C-reactive protein) are frequently elevated but do not point to a specific underlying etiology. Routine viral cultures and antibody titers are not useful.11
Most cases of pericarditis are presumed idiopathic (viral); however, finding a specific etiology should be considered in patients who do not respond after one week of therapy. Anti-nuclear antibody, complement levels, and rheumatoid factor can serve as screening tests for autoimmune disease. Purified protein derivative or quantiferon testing and HIV testing might be indicated in patients with appropriate risk factors. In cases of suspected tuberculous or neoplastic pericarditis, pericardial fluid analysis and biopsy could be warranted.
Electrocardiography. The EKG is the most useful test in diagnosing acute pericarditis. EKG changes in acute pericarditis can progress over four stages:
- Stage 1: diffuse ST elevations with or without PR depressions, initially;
- Stage 2: normalization of ST and PR segments, typically after several days;
- Stage 3: diffuse T-wave inversions; and
- Stage 4: normalization of T-waves, typically after weeks or months.
While all four stages are unlikely to be present in a given case, 80% of patients with pericarditis will demonstrate diffuse ST-segment elevations and PR-segment depression (see Figure 2, above).12
Table 3 lists EKG features helpful in differentiating acute pericarditis from acute myocardial infarction.
Chest radiography. Because a pericardial effusion often accompanies pericarditis, a chest radiograph (CXR) should be performed in all suspected cases. The CXR might show enlargement of the cardiac silhouette if more than 250 ml of pericardial fluid is present.3 A CXR also is helpful to diagnose concomitant pulmonary infection, pleural effusion, or mediastinal mass—all findings that could point to an underlying specific etiology of pericarditis and/or pericardial effusion.
Echocardiography. An echocardiogram should be performed in all patients with suspected pericarditis to detect effusion, associated myocardial, or paracardial disease.13 The echocardiogram frequently is normal but could show an effusion in 60%, and tamponade (see Figure 1, p. 15) in 5%, of cases.4
Computed tomography (CT) and cardiac magnetic resonance imaging (CMR).CT or CMR are the imaging modalities of choice when an echocardiogram is inconclusive or in cases of pericarditis complicated by a hemorrhagic or localized effusion, pericardial thickening, or pericardial mass.14 They also help in precise imaging of neighboring structures, such as lungs or mediastinum.
Pericardial fluid analysis and pericardial biopsy. In cases of refractory pericarditis with effusion, pericardial fluid analysis might provide clues to the underlying etiology. Routine chemistry, cell count, gram and acid fast staining, culture, and cytology should be sent. In addition, acid-fast bacillus staining and culture, adenosine deaminase, and interferon-gamma testing should be ordered when tuberculous pericarditis is suspected. A pericardial biopsy may show granulomas or neoplastic cells. Overall, pericardial fluid analysis and biopsy reveal a diagnosis in roughly 20% of cases.11
How is acute pericarditis treated?
Most cases of uncomplicated acute pericarditis are viral and respond well to NSAID plus colchicine therapy.2,4 Failure to respond to NSAIDs plus colchicine—evidenced by persistent fever, pericardial chest pain, new pericardial effusion, or worsening of general illness—within a week of treatment should prompt a search for an underlying systemic illness. If found, treatment should be aimed at the causative illness.
Bacterial pericarditis usually requires surgical drainage in addition to treatment with appropriate antibiotics.11 Tuberculous pericarditis is treated with multidrug therapy; when underlying HIV is present, patients should receive highly active anti-retroviral therapy as well. Steroids and immunosuppressants should be considered in addition to NSAIDs and colchicine in autoimmune pericarditis.10 Neoplastic pericarditis may resolve with chemotherapy but it has a high recurrence rate.13 Uremic pericarditis requires intensified dialysis.
Treatment options for uncomplicated idiopathic or viral pericarditis include:
NSAIDs. It is important to adequately dose NSAIDs when treating acute pericarditis. Initial treatment options include ibuprofen (1,600 to 3,200 mg daily), indomethacin (75 to 150 mg daily) or aspirin (2 to 4 gm daily) for one week.11,15 Aspirin is preferred in patients with ischemic heart disease. For patients with symptoms that persist longer than a week, NSAIDS may be continued, but investigation for an underlying etiology is indicated. Concomitant proton-pump-inhibitor therapy should be considered in patients at high risk for peptic ulcer disease to minimize gastric side effects.
Colchicine. Colchicine has a favorable risk-benefit profile as an adjunct treatment for acute and recurrent pericarditis. Patients experience better symptom relief when treated with both colchicine and an NSAID, compared with NSAIDs alone (88% versus 63%). Recurrence rates are lower with combined therapy (11% versus 32%).16 Colchicine treatment (0.6 mg twice daily after a loading dose of up to 2 mg) is recommended for several months to greater than one year.13,16,17
Glucocorticoids. Routine glucocorticoid use should be avoided in the treatment of acute pericarditis, as it has been associated with an increased risk for recurrence (OR 4.3).16,18 Glucocorticoid use should be considered in cases of pericarditis refractory to NSAIDs and colchicine, cases in which NSAIDs and or colchicine are contraindicated, and in autoimmune or connective-tissue-disease-related pericarditis. Prednisone should be dosed up to 1 mg/kg/day for at least one month, depending on symptom resolution, then tapered after either NSAIDs or colchicine have been started.13 Smaller prednisone doses of up to 0.5 mg/kg/day could be as effective, with the added benefit of reduced side effects and recurrences.19
Invasive treatment. Pericardiocentesis and/or pericardiectomy should be considered when pericarditis is complicated by a large effusion or tamponade, constrictive physiology, or recurrent effusion.11 Pericardiocentesis is the least invasive option and helps provide immediate relief in cases of tamponade or large symptomatic effusions. It is the preferred modality for obtaining pericardial fluid for diagnostic analysis. However, effusions can recur and in those cases pericardial window is preferred, as it provides continued outflow of pericardial fluid. Pericardiectomy is recommended in cases of symptomatic constrictive pericarditis unresponsive to medical therapy.15
Back to the Case
The patient’s presentation—prodrome followed by fever and pleuritic chest pain—is characteristic of acute idiopathic pericarditis. No pericardial rub was heard, but EKG findings were typical. Troponin I elevation suggested underlying myopericarditis. An echocardiogram was unremarkable. Given the likely viral or idiopathic etiology, no further diagnostic tests were ordered to explore the possibility of an underlying systemic illness.
The patient was started on ibuprofen 600 mg every eight hours. She had significant relief of her symptoms within two days. A routine fever workup was negative. She was discharged the following day.
The patient was readmitted three months later with recurrent pleuritic chest pain, which did not improve with resumption of NSAID therapy. Initial troponin I was 0.22 ng/ml, electrocardiogram was unchanged, and an echocardiogram showed small effusion. She was started on ibuprofen 800 mg every eight hours, as well as colchicine 0.6 mg twice daily. Her symptoms resolved the next day and she was discharged with prescriptions for ibuprofen and colchicine. She was instructed to follow up with a primary-care doctor in one week.
At the clinic visit, ibuprofen was tapered but colchicine was continued for another six months. She remained asymptomatic at her six-month clinic follow-up.
Bottom Line
Acute pericarditis is a clinical diagnosis supported by EKG findings. Most cases are idiopathic or viral, and can be treated successfully with NSAIDs and colchicine. For cases that do not respond to initial therapy, or cases that present with high-risk features, a specific etiology should be sought.
Dr. Southern is chief of the division of hospital medicine at Montefiore Medical Center in Bronx, N.Y. Dr. Galhorta is an instructor and Drs. Martin, Korcak, and Stehlihova are assistant professors in the department of medicine at Albert Einstein.
References
- Lange RA, Hillis LD. Clinical practice. Acute pericarditis. N Engl J Med. 2004;351:2195-2202.
- Zayas R, Anguita M, Torres F, et al. Incidence of specific etiology and role of methods for specific etiologic diagnosis of primary acute pericarditis. Am J Cardiol. 1995;75:378-382.
- Troughton RW, Asher CR, Klein AL. Pericarditis. Lancet. 2004;363:717-727.
- Imazio M, Demichelis B, Parrini I, et al. Day-hospital treatment of acute pericarditis: a management program for outpatient therapy. J Am Coll Cardiol. 2004;43:1042-1046.
- Bonnefoy E, Godon P, Kirkorian G, et al. Serum cardiac troponin I and ST-segment elevation in patients with acute pericarditis. Eur Heart J. 2000;21:832-836.
- Salisbury AC, Olalla-Gomez C, Rihal CS, et al. Frequency and predictors of urgent coronary angiography in patients with acute pericarditis. Mayo Clin Proc. 2009;84(1):11-15.
- Imazio M, Cecchi E, Demichelis B, et al. Indicators of poor prognosis of acute pericarditis. Circulation. 2007;115:2739-2744.
- Imazio M, Spodick DH, Brucato A, et al. Diagnostic issues in the clinical management of pericarditis. Int J Clin Pract. 2010;64(10):1384-1392.
- Spodick DH. Acute pericarditis: current concepts and practice. JAMA. 2003;289:1150-1153.
- Imazio M, Demichelis B, Cecchi E. Cardiac troponin I in acute pericarditis. J Am Coll Cardiol. 2003;42(12):2144-2148.
- Sagristà Sauleda J, Permanyer Miralda G, Soler Soler J. Diagnosis and management of pericardial syndromes. Rev Esp Cardiol. 2005;58(7):830-841.
- Bruce MA, Spodick DH. Atypical electrocardiogram in acute pericarditis: characteristics and prevalence. J Electrocardiol. 1980;13:61-66.
- Maisch B, Seferovic PM, Ristic AD, et al. Guidelines on the diagnosis and management of pericardial diseases executive summary; the task force on the diagnosis and management of pericardial diseases of the European Society of Cardiology. Eur Heart J. 2004; 25(7):587-610.
- Verhaert D, Gabriel RS, Johnston D, et al. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovasc Imaging. 2010;3:333-343.
- Imazio M, Spodick DH, Brucato A, et al. Controversial issues in the management of pericardial diseases. Circulation. 2010;121:916-928.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine in addition to conventional therapy for acute pericarditis: results of the colchicine for acute pericarditis (COPE) trial. Circulation. 2005;112(13):2012-2016.
- Adler Y, Finkelstein Y, Guindo J, et al. Colchicine treatment for recurrent pericarditis: a decade of experience. Circulation. 1998;97:2183-185.
- Imazio M, Bobbio M, Cecchi E, et al. Colchicine as first-choice therapy for recurrent pericarditis: results of the colchicine for recurrent pericarditis (CORE) trial. Arch Intern Med. 2005;165:1987-1991.
- Imazio M, Brucato A, Cumetti D, et al. Corticosteroids for recurrent pericarditis: high versus low doses: a nonrandomized observation. Circulation. 2008;118:667-771.
When Is Testing for Thrombophilia Indicated?
The Case
A healthy 42-year-old woman presents to the hospital with acute-onset pleuritic chest pain and shortness of breath. She has not had any recent surgeries, takes no medications, and is very active. A lung ventilation-perfusion scan reveals a high probability of pulmonary embolism (PE). The patient’s history is notable for two second-trimester pregnancy losses. The patient is started on low-molecular heparin and warfarin (LMHW).
Should this patient be tested for thrombophilia?
Background
Thrombophilia can now be identified in more than half of all patients presenting with VTE, and testing for underlying causes of thrombophilia has become widespread.1 Physicians believe that thrombophilia testing frequently changes management of patients with VTE.2
Thrombophilias can be classified into three major categories: deficiency of natural inhibitors of coagulation, abnormal function or elevated level of coagulation factors, and acquired thrombophilias (see Table 1).
The prevalence of specific thrombophilias varies widely. For example, the prevalence of activated protein C resistance (the factor V Leiden mutation) is 3% to 7%. In comparison, the prevalence of antithrombin deficiency is estimated at 0.02%. Each thrombophilia is associated with an increased VTE risk, but the level of risk associated with a given thrombophilia varies greatly.1
Before testing for thrombophilia in acute VTE, assess the risk of recurrent VTE by determining if the thrombosis was provoked or unprovoked. A VTE event is considered provoked if it occurs in the setting of pregnancy within the previous three months; estrogen therapy; immobility from acute illness for more than one week; travel lasting for more than six hours; leg trauma, fracture, or surgery within the previous three months; or active malignancy (see Table 2,).3 Unprovoked VTE has a recurrence rate of 7.4% per patient year, compared with 3.3% per patient year for a provoked VTE; the risk is even lower (0.7% per patient year) if the risk factor for the provoked VTE was surgical.4
Testing for thrombophilia is indicated if the results would add significant prognostic information beyond the clinical history, or if it would change patient management—in particular, the intensity or the duration of anticoagulation.

Review of the Data
Does presence of thrombophilia alter the intensity of anticoagulation for VTE?
If thrombophilia increases the risk of VTE recurrence while on anticoagulation, then a more intense level of anticoagulation might prevent future VTE. There are no studies investigating higher intensity of anticoagulation, but if standard anticoagulation were insufficient for patients with identifiable thrombophilia, one might expect to observe increased recurrence rates among patients with thrombophilia treated with standard warfarin therapy.
In a substudy of the Extended Low-Intensity Anticoagulation for Unprovoked Venous ThromboEmbolism (ELATE) trial, the risk of recurrence of VTE among treated subjects was very low overall, and the presence of thrombophilic abnormalities was not associated with significantly higher risk.5 Observational studies have found VTE recurrence rates are low in patients treated with warfarin, with or without thrombophilia.6-8
The impact of the initial level of anticoagulation on recurrence after the completion of the treatment period has been evaluated. Although one study suggested that patients with substandard levels of anticoagulation were at an increased risk of subsequent VTE, this was not confirmed in the Leiden Thrombophilia Study (LETS). 9,10
In sum, the majority of data do not suggest a significantly increased risk of recurrent VTE in patients with thrombophilia treated with standard anticoagulation. Therefore, treatment with warfarin to a goal INR of 2 to 3 is sufficient.
Does presence of thrombophilia alter duration of VTE treatment?
A major decision clinicians face when caring for VTE patients is the duration of anticoagulation treatment. The current ACCP recommendation for treatment of a provoked VTE is three months, with treatment for an unprovoked VTE three months or lifelong.11 If the presence of thrombophilia increases the risk of recurrence after cessation of anticoagulation treatment, longer duration of treatment might be indicated. One of the goals of thrombophilia testing should be to identify those patients.
Overall, the recurrence rate after first VTE is high, with a cumulative incidence of 25% at five years, 30% at eight years, and 56% at 20 years.12,13
Deficiency of natural inhibitors of coagulation.
Deficiency of a natural inhibitor of coagulation has been associated with a risk of recurrence of VTE of as much as 10% per year, according to some studies.6,14 However, the estimates are based on studies that include individuals from thrombosis-prone families, and selection bias might have contributed to the high recurrence rates.1 In the unselected population represented in the LETS study, only a modest elevation was seen in the estimated risk of recurrence for patients with inhibitor deficiencies.15
Testing for deficiency of inhibitors offers little prognostic information beyond that obtained when determining whether a VTE event is provoked or unprovoked. In studies that have separately examined subjects with provoked vs. unprovoked VTE, deficiency of an inhibitor is not associated with increased risk of recurrence.15,16
Abnormal function or level of anticoagulation factors.
Factor V Leiden (FVL) is the most common cause of inherited thrombophilia and is associated with as much as a sixfold increase in VTE risk, while the prothrombin gene mutation is associated with a twofold increase.17,18
In contrast, the evidence associating these mutations with recurrent VTE risk is not as consistent. Although a study conducted at a referral center in Italy found an increased risk of recurrence with either Factor V Leiden or prothrombin gene mutation, a large meta-analysis of 23 studies found increased risk only with Factor V Leiden.19,20 Another meta-analysis demonstrated only a modest increased risk of recurrence in subjects with Factor V Leiden or prothrombin gene mutation, and a prospective study from Austria found no increased risk of recurrence with Factor V Leiden two years after discontinuation of anticoagulation.18,21 Additionally, when using patients with unprovoked VTE as reference, there was no increased risk of recurrence among patients homozygous for Factor V Leiden or the prothrombin gene mutation.22
In summary, although Factor V Leiden and prothrombin gene defects are associated with increased risk of recurrent VTE, the magnitude of the risk increase is modest and, therefore, should not alter duration of therapy.
Acquired thrombophilia.
It appears that the only thrombophilic state that might have a significant impact on the risk of recurrence is the antiphospholipid syndrome. The cessation of warfarin therapy in patients with thrombosis associated with antiphospholipid antibodies carries a 69% risk of recurrent thrombosis within a year.23 Some studies have suggested that the presence of specific antibodies (i.e. anticardiolipin antibodies) is associated with increased risk in patients with antiphospholipid syndrome.24
However, at present, all patients with VTE and antiphospholipid syndrome should be candidates for lifelong anticoagulation. Antiphospholipid antibody testing should be performed in patients with a suggestive history, including those with recurrent fetal loss or a single fetal loss after 10 weeks, or known collagen vascular disease.25

The role of provoked vs. unprovoked VTE.
Identifying whether a VTE is provoked or unprovoked has been shown to be an important predictor of recurrence. For example, one prospective, cohort study found two-year recurrence rates of zero in patients with a surgery or pregnancy-related VTE, 9% with other provoked VTE, and 19% with unprovoked VTE.26 In the same study, thrombophilia testing failed to reliably predict recurrence risk. Patients with unprovoked VTE who were tested and found to not have a defect were at equally high risk of recurrent VTE as those found to have a thrombophilia.27
The most significant predictor for VTE recurrence is whether the first event was provoked, and thrombophilia testing offers little additional prognostic information.28
VTE as a multifactorial disorder.
It is becoming increasingly clear that VTE is multifactorial disorder, caused by the interactions of genotypic, phenotypic, and environmental factors. In the case of an unprovoked VTE, the patient already carries a significantly elevated risk for recurrence, and further testing for known causes of thrombophilia appears to add very little additional information. The optimal duration of anticoagulation for unprovoked VTE is unclear, but current guidelines suggest at least three months—and clinicians should consider lifelong treatment.
In the vast majority of cases, testing for thrombophilia has no impact on the management of VTE and is not warranted. In patients with antiphospholipid-antibody syndrome, given the high risk of recurrence, long-term anticoagulation after a first VTE might be indicated. In select patients with a clinical picture suggestive of antiphospholipid-antibody syndrome, or a strong family history, testing should be considered.
Back to the Case
Our patient appears to have an unprovoked VTE. She should receive regular anticoagulation with warfarin, with a goal INR of 2 to 3, for at least three months. Lifelong anticoagulation therapy should be considered. Testing for heritable thrombophilia will not change the current management or treatment duration and, hence, is not indicated. However, the patient’s history is suggestive of antiphospholipid-antibody syndrome, so she should be tested. If the diagnosis of antiphospholipid syndrome is made, lifelong anticoagulation should be considered.
Bottom Line
Unprovoked VTE provides the strongest predictor for recurrence. Thrombophilia testing adds little in predicting recurrence and rarely is indicated.
Dr. Stehlikova is a clinical hospitalist in the division of hospital medicine, department of medicine, at Albert Einstein College of Medicine and Montefiore Medical Center in Bronx, N.Y. Dr. Martin is director of the Einstein Hospitalist Service. Dr. Janakiram is a fellow in the department of hematology at Einstein, and Dr. Korcak is an instructor at Einstein in the department of medicine and director of the Weiler Medical Service. Dr. Galhotra is associate director for inpatient quality in the department of medicine at Einstein; Dr. Averbukh is an academic hospitalist; and Dr. Southern is chief of the division of hospital medicine at Einstein.
References
- Middeldorp S, van Hylckama Vlieg A. Does thrombophilia testing help in the clinical management of patients? Br J Haematol. 2008;143:321-335.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5:1979-1981.
- Prandoni P, Noventa F, Ghirarduzzi A, et al. The risk of recurrent venous thromboembolism after discontinuing anticoagulation in patients with acute proximal deep vein thrombosis or pulmonary embolism. A prospective cohort study in 1,626 patients. Haematologica. 2007;92:199-205.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med. 2010;170:1710-1716.
- Kearon C, Julian JA, Kovacs MJ, et al. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood. 2008;112:4432-4436.
- Vossen CY, Walker ID, Svensson P, et al. Recurrence rate after a first venous thrombosis in patients with familial thrombophilia. Arterioscler Thromb Vasc Biol. 2005;25:1992-1997.
- Brown K, Luddington R, Williamson D, Baker P, Baglin T. Risk of venous thromboembolism associated with a G to A transition at position 20210 in the 3'-untranslated region of the prothrombin gene. Br J Haematol. 1997;98:907-909.
- Schulman S, Tengborn L. Treatment of venous thromboembolism in patients with congenital deficiency of antithrombin III. Thromb Haemost. 1992;68:634-636.
- Palareti G, Legnani C, Cosmi B, Guazzaloca G, Cini M, Mattarozzi S. Poor anticoagulation quality in the first 3 months after unprovoked venous thromboembolism is a risk factor for long-term recurrence. J Thromb Haemost. 2005;3:955-961.
- Gadisseur AP, Christiansen SC, van der Meer FJ, Rosendaal FR. The quality of oral anticoagulant therapy and recurrent venous thrombotic events in the Leiden Thrombophilia Study. J Thromb Haemost. 2007;5:931-936.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:454S-545S.
- Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1-7.
- Laczkovics C, Grafenhofer H, Kaider A, et al. Risk of recurrence after a first venous thromboembolic event in young women. Haematologica. 2007;92:1201-1207.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost. 2009;101:93-99.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293:2352-2361.
- De Stefano V, Simioni P, Rossi E, et al. The risk of recurrent venous thromboembolism in patients with inherited deficiency of natural anticoagulants antithrombin, protein C and protein S. Haematologica. 2006;91:695-698.
- Price DT, Ridker PM. Factor V Leiden mutation and the risks for thromboembolic disease: a clinical perspective. Ann Intern Med. 1997;127:895-903.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166:729-736.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood. 2000;96:3329-3333.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301:2472-2485.
- Eichinger S, Pabinger I, Stumpflen A, et al. The risk of recurrent venous thromboembolism in patients with and without factor V Leiden. Thromb Haemost. 1997;77:624-628.
- Lijfering WM, Middeldorp S, Veeger NJ, et al. Risk of recurrent venous thrombosis in homozygous carriers and double heterozygous carriers of factor V Leiden and prothrombin G20210A. Circulation. 2010;121(15):1706-1712.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med. 1998;104:332-338.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7:1737-1740.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet. 2003;362:523-526.
- Rosendaal FR. Once and only once. Circulation. 2010;121:1688-1690.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121:458-463.
The Case
A healthy 42-year-old woman presents to the hospital with acute-onset pleuritic chest pain and shortness of breath. She has not had any recent surgeries, takes no medications, and is very active. A lung ventilation-perfusion scan reveals a high probability of pulmonary embolism (PE). The patient’s history is notable for two second-trimester pregnancy losses. The patient is started on low-molecular heparin and warfarin (LMHW).
Should this patient be tested for thrombophilia?
Background
Thrombophilia can now be identified in more than half of all patients presenting with VTE, and testing for underlying causes of thrombophilia has become widespread.1 Physicians believe that thrombophilia testing frequently changes management of patients with VTE.2
Thrombophilias can be classified into three major categories: deficiency of natural inhibitors of coagulation, abnormal function or elevated level of coagulation factors, and acquired thrombophilias (see Table 1).
The prevalence of specific thrombophilias varies widely. For example, the prevalence of activated protein C resistance (the factor V Leiden mutation) is 3% to 7%. In comparison, the prevalence of antithrombin deficiency is estimated at 0.02%. Each thrombophilia is associated with an increased VTE risk, but the level of risk associated with a given thrombophilia varies greatly.1
Before testing for thrombophilia in acute VTE, assess the risk of recurrent VTE by determining if the thrombosis was provoked or unprovoked. A VTE event is considered provoked if it occurs in the setting of pregnancy within the previous three months; estrogen therapy; immobility from acute illness for more than one week; travel lasting for more than six hours; leg trauma, fracture, or surgery within the previous three months; or active malignancy (see Table 2,).3 Unprovoked VTE has a recurrence rate of 7.4% per patient year, compared with 3.3% per patient year for a provoked VTE; the risk is even lower (0.7% per patient year) if the risk factor for the provoked VTE was surgical.4
Testing for thrombophilia is indicated if the results would add significant prognostic information beyond the clinical history, or if it would change patient management—in particular, the intensity or the duration of anticoagulation.
Review of the Data
Does presence of thrombophilia alter the intensity of anticoagulation for VTE?
If thrombophilia increases the risk of VTE recurrence while on anticoagulation, then a more intense level of anticoagulation might prevent future VTE. There are no studies investigating higher intensity of anticoagulation, but if standard anticoagulation were insufficient for patients with identifiable thrombophilia, one might expect to observe increased recurrence rates among patients with thrombophilia treated with standard warfarin therapy.
In a substudy of the Extended Low-Intensity Anticoagulation for Unprovoked Venous ThromboEmbolism (ELATE) trial, the risk of recurrence of VTE among treated subjects was very low overall, and the presence of thrombophilic abnormalities was not associated with significantly higher risk.5 Observational studies have found VTE recurrence rates are low in patients treated with warfarin, with or without thrombophilia.6-8
The impact of the initial level of anticoagulation on recurrence after the completion of the treatment period has been evaluated. Although one study suggested that patients with substandard levels of anticoagulation were at an increased risk of subsequent VTE, this was not confirmed in the Leiden Thrombophilia Study (LETS). 9,10
In sum, the majority of data do not suggest a significantly increased risk of recurrent VTE in patients with thrombophilia treated with standard anticoagulation. Therefore, treatment with warfarin to a goal INR of 2 to 3 is sufficient.
Does presence of thrombophilia alter duration of VTE treatment?
A major decision clinicians face when caring for VTE patients is the duration of anticoagulation treatment. The current ACCP recommendation for treatment of a provoked VTE is three months, with treatment for an unprovoked VTE three months or lifelong.11 If the presence of thrombophilia increases the risk of recurrence after cessation of anticoagulation treatment, longer duration of treatment might be indicated. One of the goals of thrombophilia testing should be to identify those patients.
Overall, the recurrence rate after first VTE is high, with a cumulative incidence of 25% at five years, 30% at eight years, and 56% at 20 years.12,13
Deficiency of natural inhibitors of coagulation.
Deficiency of a natural inhibitor of coagulation has been associated with a risk of recurrence of VTE of as much as 10% per year, according to some studies.6,14 However, the estimates are based on studies that include individuals from thrombosis-prone families, and selection bias might have contributed to the high recurrence rates.1 In the unselected population represented in the LETS study, only a modest elevation was seen in the estimated risk of recurrence for patients with inhibitor deficiencies.15
Testing for deficiency of inhibitors offers little prognostic information beyond that obtained when determining whether a VTE event is provoked or unprovoked. In studies that have separately examined subjects with provoked vs. unprovoked VTE, deficiency of an inhibitor is not associated with increased risk of recurrence.15,16
Abnormal function or level of anticoagulation factors.
Factor V Leiden (FVL) is the most common cause of inherited thrombophilia and is associated with as much as a sixfold increase in VTE risk, while the prothrombin gene mutation is associated with a twofold increase.17,18
In contrast, the evidence associating these mutations with recurrent VTE risk is not as consistent. Although a study conducted at a referral center in Italy found an increased risk of recurrence with either Factor V Leiden or prothrombin gene mutation, a large meta-analysis of 23 studies found increased risk only with Factor V Leiden.19,20 Another meta-analysis demonstrated only a modest increased risk of recurrence in subjects with Factor V Leiden or prothrombin gene mutation, and a prospective study from Austria found no increased risk of recurrence with Factor V Leiden two years after discontinuation of anticoagulation.18,21 Additionally, when using patients with unprovoked VTE as reference, there was no increased risk of recurrence among patients homozygous for Factor V Leiden or the prothrombin gene mutation.22
In summary, although Factor V Leiden and prothrombin gene defects are associated with increased risk of recurrent VTE, the magnitude of the risk increase is modest and, therefore, should not alter duration of therapy.
Acquired thrombophilia.
It appears that the only thrombophilic state that might have a significant impact on the risk of recurrence is the antiphospholipid syndrome. The cessation of warfarin therapy in patients with thrombosis associated with antiphospholipid antibodies carries a 69% risk of recurrent thrombosis within a year.23 Some studies have suggested that the presence of specific antibodies (i.e. anticardiolipin antibodies) is associated with increased risk in patients with antiphospholipid syndrome.24
However, at present, all patients with VTE and antiphospholipid syndrome should be candidates for lifelong anticoagulation. Antiphospholipid antibody testing should be performed in patients with a suggestive history, including those with recurrent fetal loss or a single fetal loss after 10 weeks, or known collagen vascular disease.25
The role of provoked vs. unprovoked VTE.
Identifying whether a VTE is provoked or unprovoked has been shown to be an important predictor of recurrence. For example, one prospective, cohort study found two-year recurrence rates of zero in patients with a surgery or pregnancy-related VTE, 9% with other provoked VTE, and 19% with unprovoked VTE.26 In the same study, thrombophilia testing failed to reliably predict recurrence risk. Patients with unprovoked VTE who were tested and found to not have a defect were at equally high risk of recurrent VTE as those found to have a thrombophilia.27
The most significant predictor for VTE recurrence is whether the first event was provoked, and thrombophilia testing offers little additional prognostic information.28
VTE as a multifactorial disorder.
It is becoming increasingly clear that VTE is multifactorial disorder, caused by the interactions of genotypic, phenotypic, and environmental factors. In the case of an unprovoked VTE, the patient already carries a significantly elevated risk for recurrence, and further testing for known causes of thrombophilia appears to add very little additional information. The optimal duration of anticoagulation for unprovoked VTE is unclear, but current guidelines suggest at least three months—and clinicians should consider lifelong treatment.
In the vast majority of cases, testing for thrombophilia has no impact on the management of VTE and is not warranted. In patients with antiphospholipid-antibody syndrome, given the high risk of recurrence, long-term anticoagulation after a first VTE might be indicated. In select patients with a clinical picture suggestive of antiphospholipid-antibody syndrome, or a strong family history, testing should be considered.
Back to the Case
Our patient appears to have an unprovoked VTE. She should receive regular anticoagulation with warfarin, with a goal INR of 2 to 3, for at least three months. Lifelong anticoagulation therapy should be considered. Testing for heritable thrombophilia will not change the current management or treatment duration and, hence, is not indicated. However, the patient’s history is suggestive of antiphospholipid-antibody syndrome, so she should be tested. If the diagnosis of antiphospholipid syndrome is made, lifelong anticoagulation should be considered.
Bottom Line
Unprovoked VTE provides the strongest predictor for recurrence. Thrombophilia testing adds little in predicting recurrence and rarely is indicated.
Dr. Stehlikova is a clinical hospitalist in the division of hospital medicine, department of medicine, at Albert Einstein College of Medicine and Montefiore Medical Center in Bronx, N.Y. Dr. Martin is director of the Einstein Hospitalist Service. Dr. Janakiram is a fellow in the department of hematology at Einstein, and Dr. Korcak is an instructor at Einstein in the department of medicine and director of the Weiler Medical Service. Dr. Galhotra is associate director for inpatient quality in the department of medicine at Einstein; Dr. Averbukh is an academic hospitalist; and Dr. Southern is chief of the division of hospital medicine at Einstein.
References
- Middeldorp S, van Hylckama Vlieg A. Does thrombophilia testing help in the clinical management of patients? Br J Haematol. 2008;143:321-335.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5:1979-1981.
- Prandoni P, Noventa F, Ghirarduzzi A, et al. The risk of recurrent venous thromboembolism after discontinuing anticoagulation in patients with acute proximal deep vein thrombosis or pulmonary embolism. A prospective cohort study in 1,626 patients. Haematologica. 2007;92:199-205.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med. 2010;170:1710-1716.
- Kearon C, Julian JA, Kovacs MJ, et al. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood. 2008;112:4432-4436.
- Vossen CY, Walker ID, Svensson P, et al. Recurrence rate after a first venous thrombosis in patients with familial thrombophilia. Arterioscler Thromb Vasc Biol. 2005;25:1992-1997.
- Brown K, Luddington R, Williamson D, Baker P, Baglin T. Risk of venous thromboembolism associated with a G to A transition at position 20210 in the 3'-untranslated region of the prothrombin gene. Br J Haematol. 1997;98:907-909.
- Schulman S, Tengborn L. Treatment of venous thromboembolism in patients with congenital deficiency of antithrombin III. Thromb Haemost. 1992;68:634-636.
- Palareti G, Legnani C, Cosmi B, Guazzaloca G, Cini M, Mattarozzi S. Poor anticoagulation quality in the first 3 months after unprovoked venous thromboembolism is a risk factor for long-term recurrence. J Thromb Haemost. 2005;3:955-961.
- Gadisseur AP, Christiansen SC, van der Meer FJ, Rosendaal FR. The quality of oral anticoagulant therapy and recurrent venous thrombotic events in the Leiden Thrombophilia Study. J Thromb Haemost. 2007;5:931-936.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:454S-545S.
- Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1-7.
- Laczkovics C, Grafenhofer H, Kaider A, et al. Risk of recurrence after a first venous thromboembolic event in young women. Haematologica. 2007;92:1201-1207.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost. 2009;101:93-99.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293:2352-2361.
- De Stefano V, Simioni P, Rossi E, et al. The risk of recurrent venous thromboembolism in patients with inherited deficiency of natural anticoagulants antithrombin, protein C and protein S. Haematologica. 2006;91:695-698.
- Price DT, Ridker PM. Factor V Leiden mutation and the risks for thromboembolic disease: a clinical perspective. Ann Intern Med. 1997;127:895-903.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166:729-736.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood. 2000;96:3329-3333.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301:2472-2485.
- Eichinger S, Pabinger I, Stumpflen A, et al. The risk of recurrent venous thromboembolism in patients with and without factor V Leiden. Thromb Haemost. 1997;77:624-628.
- Lijfering WM, Middeldorp S, Veeger NJ, et al. Risk of recurrent venous thrombosis in homozygous carriers and double heterozygous carriers of factor V Leiden and prothrombin G20210A. Circulation. 2010;121(15):1706-1712.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med. 1998;104:332-338.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7:1737-1740.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet. 2003;362:523-526.
- Rosendaal FR. Once and only once. Circulation. 2010;121:1688-1690.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121:458-463.
The Case
A healthy 42-year-old woman presents to the hospital with acute-onset pleuritic chest pain and shortness of breath. She has not had any recent surgeries, takes no medications, and is very active. A lung ventilation-perfusion scan reveals a high probability of pulmonary embolism (PE). The patient’s history is notable for two second-trimester pregnancy losses. The patient is started on low-molecular heparin and warfarin (LMHW).
Should this patient be tested for thrombophilia?
Background
Thrombophilia can now be identified in more than half of all patients presenting with VTE, and testing for underlying causes of thrombophilia has become widespread.1 Physicians believe that thrombophilia testing frequently changes management of patients with VTE.2
Thrombophilias can be classified into three major categories: deficiency of natural inhibitors of coagulation, abnormal function or elevated level of coagulation factors, and acquired thrombophilias (see Table 1).
The prevalence of specific thrombophilias varies widely. For example, the prevalence of activated protein C resistance (the factor V Leiden mutation) is 3% to 7%. In comparison, the prevalence of antithrombin deficiency is estimated at 0.02%. Each thrombophilia is associated with an increased VTE risk, but the level of risk associated with a given thrombophilia varies greatly.1
Before testing for thrombophilia in acute VTE, assess the risk of recurrent VTE by determining if the thrombosis was provoked or unprovoked. A VTE event is considered provoked if it occurs in the setting of pregnancy within the previous three months; estrogen therapy; immobility from acute illness for more than one week; travel lasting for more than six hours; leg trauma, fracture, or surgery within the previous three months; or active malignancy (see Table 2,).3 Unprovoked VTE has a recurrence rate of 7.4% per patient year, compared with 3.3% per patient year for a provoked VTE; the risk is even lower (0.7% per patient year) if the risk factor for the provoked VTE was surgical.4
Testing for thrombophilia is indicated if the results would add significant prognostic information beyond the clinical history, or if it would change patient management—in particular, the intensity or the duration of anticoagulation.
Review of the Data
Does presence of thrombophilia alter the intensity of anticoagulation for VTE?
If thrombophilia increases the risk of VTE recurrence while on anticoagulation, then a more intense level of anticoagulation might prevent future VTE. There are no studies investigating higher intensity of anticoagulation, but if standard anticoagulation were insufficient for patients with identifiable thrombophilia, one might expect to observe increased recurrence rates among patients with thrombophilia treated with standard warfarin therapy.
In a substudy of the Extended Low-Intensity Anticoagulation for Unprovoked Venous ThromboEmbolism (ELATE) trial, the risk of recurrence of VTE among treated subjects was very low overall, and the presence of thrombophilic abnormalities was not associated with significantly higher risk.5 Observational studies have found VTE recurrence rates are low in patients treated with warfarin, with or without thrombophilia.6-8
The impact of the initial level of anticoagulation on recurrence after the completion of the treatment period has been evaluated. Although one study suggested that patients with substandard levels of anticoagulation were at an increased risk of subsequent VTE, this was not confirmed in the Leiden Thrombophilia Study (LETS). 9,10
In sum, the majority of data do not suggest a significantly increased risk of recurrent VTE in patients with thrombophilia treated with standard anticoagulation. Therefore, treatment with warfarin to a goal INR of 2 to 3 is sufficient.
Does presence of thrombophilia alter duration of VTE treatment?
A major decision clinicians face when caring for VTE patients is the duration of anticoagulation treatment. The current ACCP recommendation for treatment of a provoked VTE is three months, with treatment for an unprovoked VTE three months or lifelong.11 If the presence of thrombophilia increases the risk of recurrence after cessation of anticoagulation treatment, longer duration of treatment might be indicated. One of the goals of thrombophilia testing should be to identify those patients.
Overall, the recurrence rate after first VTE is high, with a cumulative incidence of 25% at five years, 30% at eight years, and 56% at 20 years.12,13
Deficiency of natural inhibitors of coagulation.
Deficiency of a natural inhibitor of coagulation has been associated with a risk of recurrence of VTE of as much as 10% per year, according to some studies.6,14 However, the estimates are based on studies that include individuals from thrombosis-prone families, and selection bias might have contributed to the high recurrence rates.1 In the unselected population represented in the LETS study, only a modest elevation was seen in the estimated risk of recurrence for patients with inhibitor deficiencies.15
Testing for deficiency of inhibitors offers little prognostic information beyond that obtained when determining whether a VTE event is provoked or unprovoked. In studies that have separately examined subjects with provoked vs. unprovoked VTE, deficiency of an inhibitor is not associated with increased risk of recurrence.15,16
Abnormal function or level of anticoagulation factors.
Factor V Leiden (FVL) is the most common cause of inherited thrombophilia and is associated with as much as a sixfold increase in VTE risk, while the prothrombin gene mutation is associated with a twofold increase.17,18
In contrast, the evidence associating these mutations with recurrent VTE risk is not as consistent. Although a study conducted at a referral center in Italy found an increased risk of recurrence with either Factor V Leiden or prothrombin gene mutation, a large meta-analysis of 23 studies found increased risk only with Factor V Leiden.19,20 Another meta-analysis demonstrated only a modest increased risk of recurrence in subjects with Factor V Leiden or prothrombin gene mutation, and a prospective study from Austria found no increased risk of recurrence with Factor V Leiden two years after discontinuation of anticoagulation.18,21 Additionally, when using patients with unprovoked VTE as reference, there was no increased risk of recurrence among patients homozygous for Factor V Leiden or the prothrombin gene mutation.22
In summary, although Factor V Leiden and prothrombin gene defects are associated with increased risk of recurrent VTE, the magnitude of the risk increase is modest and, therefore, should not alter duration of therapy.
Acquired thrombophilia.
It appears that the only thrombophilic state that might have a significant impact on the risk of recurrence is the antiphospholipid syndrome. The cessation of warfarin therapy in patients with thrombosis associated with antiphospholipid antibodies carries a 69% risk of recurrent thrombosis within a year.23 Some studies have suggested that the presence of specific antibodies (i.e. anticardiolipin antibodies) is associated with increased risk in patients with antiphospholipid syndrome.24
However, at present, all patients with VTE and antiphospholipid syndrome should be candidates for lifelong anticoagulation. Antiphospholipid antibody testing should be performed in patients with a suggestive history, including those with recurrent fetal loss or a single fetal loss after 10 weeks, or known collagen vascular disease.25
The role of provoked vs. unprovoked VTE.
Identifying whether a VTE is provoked or unprovoked has been shown to be an important predictor of recurrence. For example, one prospective, cohort study found two-year recurrence rates of zero in patients with a surgery or pregnancy-related VTE, 9% with other provoked VTE, and 19% with unprovoked VTE.26 In the same study, thrombophilia testing failed to reliably predict recurrence risk. Patients with unprovoked VTE who were tested and found to not have a defect were at equally high risk of recurrent VTE as those found to have a thrombophilia.27
The most significant predictor for VTE recurrence is whether the first event was provoked, and thrombophilia testing offers little additional prognostic information.28
VTE as a multifactorial disorder.
It is becoming increasingly clear that VTE is multifactorial disorder, caused by the interactions of genotypic, phenotypic, and environmental factors. In the case of an unprovoked VTE, the patient already carries a significantly elevated risk for recurrence, and further testing for known causes of thrombophilia appears to add very little additional information. The optimal duration of anticoagulation for unprovoked VTE is unclear, but current guidelines suggest at least three months—and clinicians should consider lifelong treatment.
In the vast majority of cases, testing for thrombophilia has no impact on the management of VTE and is not warranted. In patients with antiphospholipid-antibody syndrome, given the high risk of recurrence, long-term anticoagulation after a first VTE might be indicated. In select patients with a clinical picture suggestive of antiphospholipid-antibody syndrome, or a strong family history, testing should be considered.
Back to the Case
Our patient appears to have an unprovoked VTE. She should receive regular anticoagulation with warfarin, with a goal INR of 2 to 3, for at least three months. Lifelong anticoagulation therapy should be considered. Testing for heritable thrombophilia will not change the current management or treatment duration and, hence, is not indicated. However, the patient’s history is suggestive of antiphospholipid-antibody syndrome, so she should be tested. If the diagnosis of antiphospholipid syndrome is made, lifelong anticoagulation should be considered.
Bottom Line
Unprovoked VTE provides the strongest predictor for recurrence. Thrombophilia testing adds little in predicting recurrence and rarely is indicated.
Dr. Stehlikova is a clinical hospitalist in the division of hospital medicine, department of medicine, at Albert Einstein College of Medicine and Montefiore Medical Center in Bronx, N.Y. Dr. Martin is director of the Einstein Hospitalist Service. Dr. Janakiram is a fellow in the department of hematology at Einstein, and Dr. Korcak is an instructor at Einstein in the department of medicine and director of the Weiler Medical Service. Dr. Galhotra is associate director for inpatient quality in the department of medicine at Einstein; Dr. Averbukh is an academic hospitalist; and Dr. Southern is chief of the division of hospital medicine at Einstein.
References
- Middeldorp S, van Hylckama Vlieg A. Does thrombophilia testing help in the clinical management of patients? Br J Haematol. 2008;143:321-335.
- Coppens M, van Mourik JA, Eckmann CM, Büller HR, Middeldorp S. Current practise of testing for inherited thrombophilia. J Thromb Haemost. 2007;5:1979-1981.
- Prandoni P, Noventa F, Ghirarduzzi A, et al. The risk of recurrent venous thromboembolism after discontinuing anticoagulation in patients with acute proximal deep vein thrombosis or pulmonary embolism. A prospective cohort study in 1,626 patients. Haematologica. 2007;92:199-205.
- Iorio A, Kearon C, Filippucci E, et al. Risk of recurrence after a first episode of symptomatic venous thromboembolism provoked by a transient risk factor: a systematic review. Arch Intern Med. 2010;170:1710-1716.
- Kearon C, Julian JA, Kovacs MJ, et al. Influence of thrombophilia on risk of recurrent venous thromboembolism while on warfarin: results from a randomized trial. Blood. 2008;112:4432-4436.
- Vossen CY, Walker ID, Svensson P, et al. Recurrence rate after a first venous thrombosis in patients with familial thrombophilia. Arterioscler Thromb Vasc Biol. 2005;25:1992-1997.
- Brown K, Luddington R, Williamson D, Baker P, Baglin T. Risk of venous thromboembolism associated with a G to A transition at position 20210 in the 3'-untranslated region of the prothrombin gene. Br J Haematol. 1997;98:907-909.
- Schulman S, Tengborn L. Treatment of venous thromboembolism in patients with congenital deficiency of antithrombin III. Thromb Haemost. 1992;68:634-636.
- Palareti G, Legnani C, Cosmi B, Guazzaloca G, Cini M, Mattarozzi S. Poor anticoagulation quality in the first 3 months after unprovoked venous thromboembolism is a risk factor for long-term recurrence. J Thromb Haemost. 2005;3:955-961.
- Gadisseur AP, Christiansen SC, van der Meer FJ, Rosendaal FR. The quality of oral anticoagulant therapy and recurrent venous thrombotic events in the Leiden Thrombophilia Study. J Thromb Haemost. 2007;5:931-936.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Antithrombotic therapy for venous thromboembolic disease: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:454S-545S.
- Prandoni P, Lensing AW, Cogo A, et al. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996;125:1-7.
- Laczkovics C, Grafenhofer H, Kaider A, et al. Risk of recurrence after a first venous thromboembolic event in young women. Haematologica. 2007;92:1201-1207.
- Brouwer JL, Lijfering WM, Ten Kate MK, Kluin-Nelemans HC, Veeger NJ, van der Meer J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb Haemost. 2009;101:93-99.
- Christiansen SC, Cannegieter SC, Koster T, Vandenbroucke JP, Rosendaal FR. Thrombophilia, clinical factors, and recurrent venous thrombotic events. JAMA. 2005;293:2352-2361.
- De Stefano V, Simioni P, Rossi E, et al. The risk of recurrent venous thromboembolism in patients with inherited deficiency of natural anticoagulants antithrombin, protein C and protein S. Haematologica. 2006;91:695-698.
- Price DT, Ridker PM. Factor V Leiden mutation and the risks for thromboembolic disease: a clinical perspective. Ann Intern Med. 1997;127:895-903.
- Ho WK, Hankey GJ, Quinlan DJ, Eikelboom JW. Risk of recurrent venous thromboembolism in patients with common thrombophilia: a systematic review. Arch Intern Med. 2006;166:729-736.
- Simioni P, Prandoni P, Lensing AW, et al. Risk for subsequent venous thromboembolic complications in carriers of the prothrombin or the factor V gene mutation with a first episode of deep-vein thrombosis. Blood. 2000;96:3329-3333.
- Segal JB, Brotman DJ, Necochea AJ, et al. Predictive value of factor V Leiden and prothrombin G20210A in adults with venous thromboembolism and in family members of those with a mutation: a systematic review. JAMA. 2009;301:2472-2485.
- Eichinger S, Pabinger I, Stumpflen A, et al. The risk of recurrent venous thromboembolism in patients with and without factor V Leiden. Thromb Haemost. 1997;77:624-628.
- Lijfering WM, Middeldorp S, Veeger NJ, et al. Risk of recurrent venous thrombosis in homozygous carriers and double heterozygous carriers of factor V Leiden and prothrombin G20210A. Circulation. 2010;121(15):1706-1712.
- Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR. The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995;332:993-997.
- Schulman S, Svenungsson E, Granqvist S. Anticardiolipin antibodies predict early recurrence of thromboembolism and death among patients with venous thromboembolism following anticoagulant therapy. Duration of Anticoagulation Study Group. Am J Med. 1998;104:332-338.
- Pengo V, Tripodi A, Reber G, et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7:1737-1740.
- Baglin T, Luddington R, Brown K, Baglin C. Incidence of recurrent venous thromboembolism in relation to clinical and thrombophilic risk factors: prospective cohort study. Lancet. 2003;362:523-526.
- Rosendaal FR. Once and only once. Circulation. 2010;121:1688-1690.
- Dalen JE. Should patients with venous thromboembolism be screened for thrombophilia? Am J Med. 2008;121:458-463.
Chance Favors the Prepared Mind
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A previously healthy 18‐year‐old woman living in the Pacific Northwest was brought in by her parents to a local hospital with a 4‐day history of acting crazy. Two weeks prior to presentation, she complained of a new‐onset severe headache, diaphoresis, and chills. Four days prior to presentation, she became progressively more impulsive, which ultimately included jumping out of a moving vehicle and running away from home. She experienced unexplained emotional outbursts and was unable to identify familiar relatives or common objects. Additionally, she began having hyperventilation spells and auditory hallucinations.
In an adolescent presenting with erratic behavior, one should consider the possibility of substance abuse or a psychiatric disease such as bipolar disorder with manic features, psychotic manifestations of severe depression, or early schizophrenia. However, it is important to first rule out non‐psychiatric disease, with a diagnostic approach dependent on her human immunodeficiency virus (HIV) status. The presence of headache, diaphoresis, and chills raises concern for an infectious or noninfectious inflammatory central nervous system process. In addition to the effects of illicit drugs such as cocaine or methamphetamine, this presentation may be consistent with a medication‐ or herbal‐induced anticholinergic syndrome, which may present with confusion, ataxia, coma, and cardiopulmonary failure. Since this case originates in the Northwest, one should be aware of the regional outbreak of Cryptococcus gattii in immunocompetent hosts, and that local hallucinogenic plants, such as jimson weed or mushrooms (Amanita muscaria) can cause anticholinergic syndromes. At this point, the differential diagnosis is broad, and evaluation should focus on potentially reversible life‐threatening conditions; in particular, herpes encephalitis. In addition to a detailed history, examination, and routine laboratory studies including HIV serology, I would obtain a drug screen, and order a computed tomography (CT) scan of the brain before performing a lumbar puncture. I would also order a magnetic resonance imaging (MRI) study to evaluate for meningeal or cerebral enhancement suggestive of encephalitis.
The patient had no past medical, psychiatric, or surgical history and took no medications. She lived with her parents who thought she neither used illicit drugs or alcohol, nor was sexually active. She had recently graduated high school and was planning to attend college. Her family history was notable for a mother with bipolar and seizure disorders, and 2 healthy younger siblings. Her family had a healthy cat and dog, and reported a large number of bats living nearby. She had never traveled outside the western United States. The patient presented in late spring, but there was no obvious history of mosquito bites. Her last menstrual period was 4 months prior to presentation. Full review of systems was otherwise negative.
The family history of mood disorder supports continued consideration of bipolar disorder with psychotic manifestations. However, infectious or inflammatory processes remain highest on the differential at this point. The duration of symptoms makes common bacterial meningitis etiologies (Streptococcus, Neisseria, Haemophilus, Listeria) less likely, but would be consistent with herpes simplex encephalitis or lupus cerebritis. Additional infectious considerations would include other viral (eg, varicella zoster virus, Epstein‐Barr virus, enteroviruses, and the arthropod‐borne encephalitides) or unusual bacterial encephalitic syndromes. Although the health status of pets is rarely helpful, dogs can carry ticks that harbor Borrelia burgdorferi (the agent of Lyme disease), which may present with central nervous system (CNS) manifestations. Other conditions associated with pets (such as leptospirosis or cat scratch disease) seem unlikely. The exposure to bats raises the possibility of rabies infection. If she is HIV‐positive, one would need to consider the possibility of opportunistic infections such as cytomegalovirus (CMV), Cryptococcus, cerebral toxoplasmosis, and progressive multifocal leukoencephalopathy (PML) caused by JC virus reaction. Finally, regardless of history, given the patient's amenorrhea, we must perform a pregnancy test.
The patient's temperature was 97.3F, heart rate 129 beats per minute, respiratory rate 19 breaths per minute, and her blood pressure 144/97 mmHg. She was an obese, well‐developed young woman, who was drowsy but arousable, with marked speech latency. Her cranium and oropharynx were normal, and her neck was supple. Aside from tachycardia, her cardiopulmonary, musculoskeletal, and skin exams were normal. Her abdomen was obese and soft, without masses or organomegaly. A pelvic examination was not performed. On neurologic exam, her strength was symmetrically diminished throughout (3+/5). Otherwise, she was oriented to person and general location, but not to day of week, month, or year. Her cranial nerves, sensation, deep tendon reflexes, and muscle tone were normal. A cerebellar examination, plantar response, and gait test were not performed. A brain MRI revealed only a small subarachnoid cyst and possible subtle enhancement of temporal lobes. Initial laboratory studies demonstrated: white blood cell count 14,000/mm3 (72% neutrophils, 17% lymphocytes, 9% monocytes, 2% eosinophils); hemoglobin 14.0 g/dL (mean corpuscular volume 87.4 fL); platelet count 417,000/mm3. Serum electrolytes, liver function tests, coagulation studies, thyroid stimulating hormone, serum ammonia, and urinalysis were normal. Her serum pregnancy test and urine toxicology screen were negative. A room air arterial blood gas revealed a pH of 7.49, PaCO2 32 mmHg, PaO2 89 mmHg; and a bicarbonate 24 mmol/L. Cerebrospinal fluid demonstrated: red cell count of 2/mm3; white cell count 17/mm3 (88% lymphocytes, 3% neutrophils, 9% monocytes); protein 19 mg/dL (normal 1555 mg/dL); and glucose of 79 mg/dL (normal 4080 mg/dL). Gram stain, fungal and bacterial cultures, and HIV serology were negative, and herpes simplex virus was not detected via polymerase chain reaction (PCR).
The tachycardia, respiratory alkalosis, and leukocytosis continue to suggest an infection or inflammatory state. Her neurological deterioration without focal findings, cerebrospinal fluid (CSF) lymphocytic pleocytosis with normal glucose and protein, and temporal lobe enhancement on MRI strongly suggest a meningoencephalitis. This would be an unusual presentation for most bacterial pathogens, but Mycobacterium, Rickettsia, Listeria, Mycoplasma, and Bartonella may rarely mimic encephalitis. Autoimmune encephalitis secondary to lupus, vasculitis, or other autoimmune disorder remains possible, but at this point an infectious encephalitis, particularly herpes encephalitis, is my highest concern. West Nile virus must be considered, but usually produces a severe illness only in immunocompromised or elderly patients. Additionally, despite the rarity of rabies, the patient's exposure to bats and the rapid clinical deterioration, suggest this possibility. In addition to routine bacterial and viral analyses (eg, enteroviral panel), samples should be sent for rabies PCR and antibody testing, West Nile virus, Lyme disease, syphilis, and mycobacterial and fungal pathogens, such as the aforementioned Cryptococcus gattii. Finally, given her presenting syndrome and MRI, immediate treatment with acyclovir and antibiotics is indicated.
The patient was treated for presumed meningoencephalitis with acyclovir and ceftriaxone, but over the following several days became unresponsive to all stimuli and developed repetitive thrusting movements of her mouth, tongue, and jaw. On hospital day 10, with concern for seizures, pentobarbital coma was induced, and the patient was intubated and transferred to our facility. On arrival, her physical examination was essentially unchanged aside from being in a medical coma. Hematology, chemistries, and thyroid‐stimulating hormone (TSH) were again unremarkable with the exception of an elevated creatine kinase (414 U/L) and a new anemia (hemoglobin 8.9 g/dL; mean corpuscular volume 87.6 fL) without evidence of iron deficiency or hemolysis. Blood and urine cultures were negative. Repeat cerebrospinal fluid analysis was essentially unchanged, revealing a red cell count of 1/mm3; white cell count 20/mm3 (86% lymphocytes, 2% neutrophils, 12% monocytes); protein 14 mg/dL; glucose 63 mg/dL, and negative Gram stain. Continuous electroencephalography revealed diffuse generalized slowing, but no seizure activity. An extensive evaluation for viral, bacterial, autoimmune, and paraneoplastic disorders was negative, including tests for anti‐acetylcholine (ACh) receptor binding antibody, anti‐striated muscle antibody, anti‐N‐type calcium channel antibody, anti‐P/Q‐type calcium channel antibodies, anto‐cancer associated retinopathy (CAR) antibody (also known as anti‐recoverin antibody), and anti‐collapsin respons mediator protein (CRMP‐5). Without confirmatory results and continued deterioration, she was empirically treated with methylprednisolone for presumed autoimmune encephalitis from hospital days 16 to 21. The patient remained unresponsive and ventilator‐dependent, despite removal of all sedation. She experienced intermittent fevers as high as 40.5C, remained tachycardic, hypertensive, and exhibited orofacial dyskinesias and jaw clenching, ultimately requiring botulinum toxin injections to prevent tongue biting. Given the lack of improvement despite attempted therapies, a working diagnosis of viral encephalitis with lasting neuropsychiatric sequelae was made. A tracheostomy and percutaneous gastrostomy tube were placed, and a long‐term ventilator care facility was identified.
I continue to wonder if this may be an autoimmune encephalitis, and am concerned about her unexplained fevers. Neuroleptic malignant syndrome secondary to misuse of her parents' medications should be considered in light of the elevated creatine kinase, although the severity and duration of the syndrome seem more profound than I would anticipate. Tetanus could present with jaw dystonia, but the rest of the case does not seem to fit. At this point, considering the patient's young age and poor prognosis without identified etiology, prior to discharge I would argue for a brain biopsy looking for evidence of rabies, or other infectious or autoimmune etiologies of the patient's progressive neurologic deterioration.
On hospital day 25, due to the persistent fevers with concern for occult abscess, an abdominopelvic CT was obtained, which identified a complex 11.8 cm 9.0 cm adnexal mass consistent with a teratoma (Figure 1).

Given the size of the mass, it is surprising that the patient did not report abdominal symptoms and that the physicians were unable to palpate it on examination. The differential diagnosis of a complex adnexal mass in an adolescent should include an ectopic pregnancy, ovarian cysts, tubo‐ovarian abscess, rarely an ovarian carcinoma or leiomyosarcoma, and a teratoma or dermoid tumor. While I mentioned the possibility of a malignancy at the outset, I did not further consider it. Common neoplasms encountered in adolescent patients include lymphoma and leukemia, germ cell tumors (including teratomas), central nervous system tumors and sarcomas, many of which have been reported to cause paraneoplastic disorders. At this point, I now think her presumed teratoma is associated with a paraneoplastic syndrome resulting in her presentation of limbic encephalitis.
A literature search was performed by the managing clinicians who rapidly identified the association between teratoma and limbic encephalitis. The patient was initially treated with intravenous immune globulin (IVIG), with transient improvement in her mental status. Serology returned positive for the anti‐N‐methyl‐D‐aspartate receptor antibody, confirming the diagnosis of anti‐N‐methyl‐D‐aspartate receptor encephalitis. On hospital day 36, her mass was resected (Figure 2). Pathology was consistent with a mature teratoma. Postoperatively, the patient improved daily, and was discharged on hospital day 43 with a near complete neurologic recovery. Four months following discharge, the patient had enrolled full time in college.
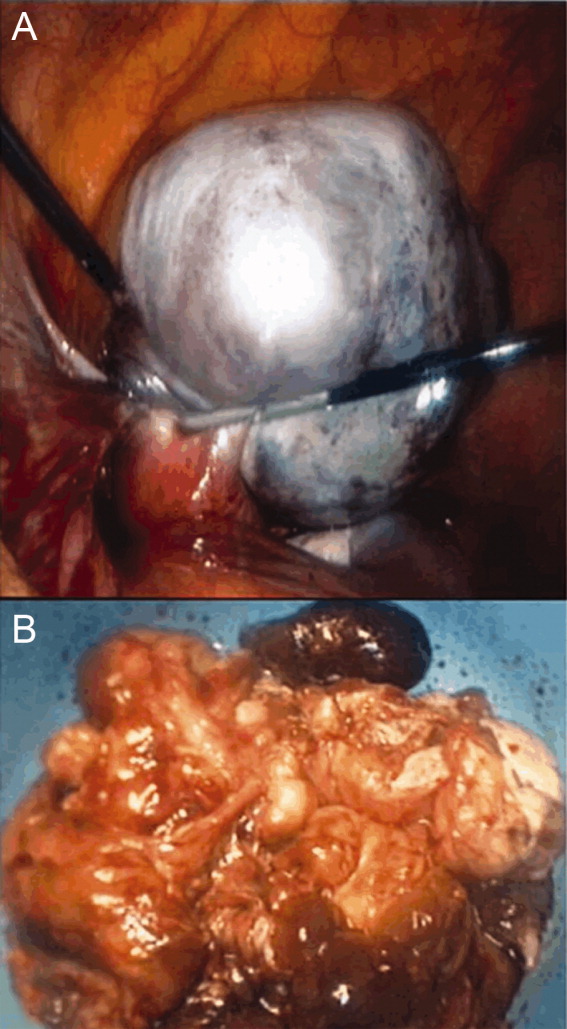
COMMENTARY
The N‐methyl‐D‐aspartate receptor (NMDAR) is an important regulator of synaptic transmission and memory within the CNS. Our patient's case illustrates the increasingly recognized syndrome of anti‐NMDAR encephalitis. NMDAR hypofunction is hypothesized to result in the cognitive and behavioral abnormalities of schizophrenia, and direct antagonism of the NMDAR by drugs such as phencyclidine (PCP) and ketamine results in symptoms such as psychosis, hallucinations, delusions, agitation, and dissociative amnesia.14 This constellation of symptoms is very similar to some of the initial neuropsychiatric symptoms observed in patients with anti‐NMDAR encephalitis.
Anti‐NMDAR encephalitis was first described in 2005 as a paraneoplastic limbic encephalitis associated with ovarian teratoma.5, 6 Characterized by the subacute onset (days to weeks) of short‐term memory loss, psychiatric symptoms, and sleep disturbances, limbic encephalitis is an inflammatory process caused by autoantibodies against intracellular or extracellar antigens in the limbic system and other brain structures. Limbic encephalitides associated with antibodies to intracellular antigens (such as Hu, Ma2, CV2/CRMP5, and Amphiphysin) are more often associated with malignancies, have worse outcomes (permanent neuropsychiatric sequelae and death), and are less responsive to immune therapy. Conversely, it appears that both the paraneoplastic and non‐paraneoplastic variants of limbic encephalitis associated with antibodies against cell membrane antigens (such as NMDAR and Voltage Gated Potassium Channels) respond more favorably to therapy.7
As with limbic encephalitis in general, anti‐NMDAR encephalitis can be non‐paraneoplastic as well as paraneoplastic in etiology. In a recently published series of 44 consecutive patients with anti‐NMDAR encephalitis, tumors were present in only 9 cases (8 teratomas).8 When associated with a teratoma, it has been postulated that anti‐NMDAR antibodies develop and cross the bloodbrain barrier to target central nervous system NMDA receptors. This process results in down‐regulation of the neuronal surface NMDAR which then causes the psychiatric and behavioral changes described.6 The mechanism by which these antibodies traverse the bloodbrain barrier is not completely understood, but likely requires some disruption of the barrier in order to trigger anti‐NMDAR encephalitis.8, 9 Non‐paraneoplastic cases evidently involve other unknown stimuli for NMDAR antibody synthesisone report has suggested that subunits of the NMDAR are expressed by normal ovarian tissue, something which may explain the female predilection even in the cohort unaffected by teratomas.10
Most patients with anti‐NMDAR encephalitis are female and young (median age 23 years), although men and children are also affected.8, 9, 11 While the exact incidence of anti‐NMDAR encephalitis is still unknown, the increasing number of case reports suggests that it may be more frequent than any other type of paraneoplastic encephalitis.12 The majority of patients with anti‐NMDAR encephalitis experience an antecedent infectious prodrome (eg, diarrheal illness or upper respiratory infection [URI]), followed 1020 days later by progressive neuropsychiatric and behavioral symptoms which include confusion, memory deficits, impaired responsiveness, seizures, central hypoventilation, and signs of autonomic instability (tachycardia, tachypnea, diaphoresis, cardiac dysrhythmia, blood pressure instability, and dysthermia). At this stage, patients may also manifest a unique constellation of choreoathetoid orofacial and limb movements such as lip licking, chewing, sustained jaw clenching, jaw opening dystonias, ocular deviation and disconjugation, grimacing, myoclonus, and bizarre arm movements. Due to cardiovascular complications and ventilator requirements, most patients require intensive care unit (ICU) level care. 8, 9, 11 As in our discussant's evaluation, other disorders to include in the differential diagnosis for this presentation includes paraneoplastic or autoimmune causes of limbic encephalitis, toxins, heavy metals, and viral causes of encephalitis; in particular, herpes simplex virus (HSV).7
The CNS imaging findings in this condition include brain MRI abnormalities in about 30%55% of patients, which can include increased signal on fluid‐attenuated inversion recovery (FLAIR) or T2 sequences of the cerebral cortex, overlying meninges, or basal ganglia. Abnormalities in the temporal lobes, corpus callosum, and brainstem have also been described. As in our patient, CSF lymphocytic pleocytosis has also been noted.6, 8, 9
Although many cases of limbic encephalitis portend a poor prognosis with permanent neuropsychiatric sequelae and death, anti‐NMDAR can be very responsive to treatment; particularly if diagnosed early. Successful treatment of anti‐NMDAR encephalitis involves immunotherapy and, preferably, early surgical resection of any tumor. 6, 8, 9 Non‐paraneoplastic cases appear to require more aggressive and prolonged immunotherapies to avoid relapse. In both groups, a trend towards improved outcome has been noted in patients treated early in disease course (40 days from symptom onset).8 There are no established guidelines for the treatment of anti‐NMDAR encephalitis, and no randomized controlled trials have evaluated anti‐NMDAR encephalitis treatment. Observational studies of immune‐modulating therapies have shown efficacy with high‐dose steroids and the addition of plasma exchange and/or intravenous immune globulin. Rituximab and cyclophosphamide can be considered if patients fail to improve on other immunotherapies.9 Data from case series seem to suggest a lower risk of relapse in patients treated with immunotherapy.13
Exploration of this patient's persistent high fevers ultimately led to the serendipitous diagnosis of the increasingly recognized syndrome of anti‐NMDAR encephalitis, although in retrospect nearly all of the features of her presentation fit well with this condition. Thus, it was only by a chance finding on her abdominal CT scan that this patient was ultimately diagnosed with a treatable, noninfectious encephalitis associated with an ovarian teratoma. This case reinforces the importance of thorough patient evaluations and being prepared to draw meaningful conclusions from unexpected findings. Given how close this patient was to being discharged to a long‐term care facility, we found this case a fascinating yet sobering reminder to guard against prematurely concluding a syndrome to be untreatable.
KEY TEACHING POINTS
-
Anti‐NMDAR encephalitis is an increasingly recognized cause of autoimmune limbic encephalitis, and thus should be considered in patients with new‐onset psychiatric symptoms accompanied by seizures, autonomic instability, hypoventilation, or dyskinesias.
-
A thorough history, examination, and evaluation of data is critical to make an early diagnosis of anti‐NMDAR encephalitis, because, unlike other forms of limbic encephalitis, this condition may be very responsive to early initiation of treatment.
- , . N‐methyl‐D‐aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49.
- , . Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308.
- , , . NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533.
- , , , et al. Ketamine‐induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118.
- , , , , , . Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58:594–604.
- , , , et al. Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
- , . Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13:261–271.
- , , , et al. N‐methyl‐D‐aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‐paraneoplastic disorder of both sexes. Brain. 2010;133:1655–1667.
- , . NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;11:298–304.
- , , , et al. Expression of various glutamate receptors including N‐methyl‐D‐aspartate receptor (NMDAR) in an ovarian teratoma removed from a young woman with anti‐NMDAR encephalitis. Intern Med. 2010;49:2167–2173.
- , , , et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1074–1075.
- , , , , . Clinical experience and laboratory investigations in patients with anti‐NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
- , , , et al. Analysis of relapses in anti‐NMDAR encephalitis. Neurology. 2011;77:996–999.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A previously healthy 18‐year‐old woman living in the Pacific Northwest was brought in by her parents to a local hospital with a 4‐day history of acting crazy. Two weeks prior to presentation, she complained of a new‐onset severe headache, diaphoresis, and chills. Four days prior to presentation, she became progressively more impulsive, which ultimately included jumping out of a moving vehicle and running away from home. She experienced unexplained emotional outbursts and was unable to identify familiar relatives or common objects. Additionally, she began having hyperventilation spells and auditory hallucinations.
In an adolescent presenting with erratic behavior, one should consider the possibility of substance abuse or a psychiatric disease such as bipolar disorder with manic features, psychotic manifestations of severe depression, or early schizophrenia. However, it is important to first rule out non‐psychiatric disease, with a diagnostic approach dependent on her human immunodeficiency virus (HIV) status. The presence of headache, diaphoresis, and chills raises concern for an infectious or noninfectious inflammatory central nervous system process. In addition to the effects of illicit drugs such as cocaine or methamphetamine, this presentation may be consistent with a medication‐ or herbal‐induced anticholinergic syndrome, which may present with confusion, ataxia, coma, and cardiopulmonary failure. Since this case originates in the Northwest, one should be aware of the regional outbreak of Cryptococcus gattii in immunocompetent hosts, and that local hallucinogenic plants, such as jimson weed or mushrooms (Amanita muscaria) can cause anticholinergic syndromes. At this point, the differential diagnosis is broad, and evaluation should focus on potentially reversible life‐threatening conditions; in particular, herpes encephalitis. In addition to a detailed history, examination, and routine laboratory studies including HIV serology, I would obtain a drug screen, and order a computed tomography (CT) scan of the brain before performing a lumbar puncture. I would also order a magnetic resonance imaging (MRI) study to evaluate for meningeal or cerebral enhancement suggestive of encephalitis.
The patient had no past medical, psychiatric, or surgical history and took no medications. She lived with her parents who thought she neither used illicit drugs or alcohol, nor was sexually active. She had recently graduated high school and was planning to attend college. Her family history was notable for a mother with bipolar and seizure disorders, and 2 healthy younger siblings. Her family had a healthy cat and dog, and reported a large number of bats living nearby. She had never traveled outside the western United States. The patient presented in late spring, but there was no obvious history of mosquito bites. Her last menstrual period was 4 months prior to presentation. Full review of systems was otherwise negative.
The family history of mood disorder supports continued consideration of bipolar disorder with psychotic manifestations. However, infectious or inflammatory processes remain highest on the differential at this point. The duration of symptoms makes common bacterial meningitis etiologies (Streptococcus, Neisseria, Haemophilus, Listeria) less likely, but would be consistent with herpes simplex encephalitis or lupus cerebritis. Additional infectious considerations would include other viral (eg, varicella zoster virus, Epstein‐Barr virus, enteroviruses, and the arthropod‐borne encephalitides) or unusual bacterial encephalitic syndromes. Although the health status of pets is rarely helpful, dogs can carry ticks that harbor Borrelia burgdorferi (the agent of Lyme disease), which may present with central nervous system (CNS) manifestations. Other conditions associated with pets (such as leptospirosis or cat scratch disease) seem unlikely. The exposure to bats raises the possibility of rabies infection. If she is HIV‐positive, one would need to consider the possibility of opportunistic infections such as cytomegalovirus (CMV), Cryptococcus, cerebral toxoplasmosis, and progressive multifocal leukoencephalopathy (PML) caused by JC virus reaction. Finally, regardless of history, given the patient's amenorrhea, we must perform a pregnancy test.
The patient's temperature was 97.3F, heart rate 129 beats per minute, respiratory rate 19 breaths per minute, and her blood pressure 144/97 mmHg. She was an obese, well‐developed young woman, who was drowsy but arousable, with marked speech latency. Her cranium and oropharynx were normal, and her neck was supple. Aside from tachycardia, her cardiopulmonary, musculoskeletal, and skin exams were normal. Her abdomen was obese and soft, without masses or organomegaly. A pelvic examination was not performed. On neurologic exam, her strength was symmetrically diminished throughout (3+/5). Otherwise, she was oriented to person and general location, but not to day of week, month, or year. Her cranial nerves, sensation, deep tendon reflexes, and muscle tone were normal. A cerebellar examination, plantar response, and gait test were not performed. A brain MRI revealed only a small subarachnoid cyst and possible subtle enhancement of temporal lobes. Initial laboratory studies demonstrated: white blood cell count 14,000/mm3 (72% neutrophils, 17% lymphocytes, 9% monocytes, 2% eosinophils); hemoglobin 14.0 g/dL (mean corpuscular volume 87.4 fL); platelet count 417,000/mm3. Serum electrolytes, liver function tests, coagulation studies, thyroid stimulating hormone, serum ammonia, and urinalysis were normal. Her serum pregnancy test and urine toxicology screen were negative. A room air arterial blood gas revealed a pH of 7.49, PaCO2 32 mmHg, PaO2 89 mmHg; and a bicarbonate 24 mmol/L. Cerebrospinal fluid demonstrated: red cell count of 2/mm3; white cell count 17/mm3 (88% lymphocytes, 3% neutrophils, 9% monocytes); protein 19 mg/dL (normal 1555 mg/dL); and glucose of 79 mg/dL (normal 4080 mg/dL). Gram stain, fungal and bacterial cultures, and HIV serology were negative, and herpes simplex virus was not detected via polymerase chain reaction (PCR).
The tachycardia, respiratory alkalosis, and leukocytosis continue to suggest an infection or inflammatory state. Her neurological deterioration without focal findings, cerebrospinal fluid (CSF) lymphocytic pleocytosis with normal glucose and protein, and temporal lobe enhancement on MRI strongly suggest a meningoencephalitis. This would be an unusual presentation for most bacterial pathogens, but Mycobacterium, Rickettsia, Listeria, Mycoplasma, and Bartonella may rarely mimic encephalitis. Autoimmune encephalitis secondary to lupus, vasculitis, or other autoimmune disorder remains possible, but at this point an infectious encephalitis, particularly herpes encephalitis, is my highest concern. West Nile virus must be considered, but usually produces a severe illness only in immunocompromised or elderly patients. Additionally, despite the rarity of rabies, the patient's exposure to bats and the rapid clinical deterioration, suggest this possibility. In addition to routine bacterial and viral analyses (eg, enteroviral panel), samples should be sent for rabies PCR and antibody testing, West Nile virus, Lyme disease, syphilis, and mycobacterial and fungal pathogens, such as the aforementioned Cryptococcus gattii. Finally, given her presenting syndrome and MRI, immediate treatment with acyclovir and antibiotics is indicated.
The patient was treated for presumed meningoencephalitis with acyclovir and ceftriaxone, but over the following several days became unresponsive to all stimuli and developed repetitive thrusting movements of her mouth, tongue, and jaw. On hospital day 10, with concern for seizures, pentobarbital coma was induced, and the patient was intubated and transferred to our facility. On arrival, her physical examination was essentially unchanged aside from being in a medical coma. Hematology, chemistries, and thyroid‐stimulating hormone (TSH) were again unremarkable with the exception of an elevated creatine kinase (414 U/L) and a new anemia (hemoglobin 8.9 g/dL; mean corpuscular volume 87.6 fL) without evidence of iron deficiency or hemolysis. Blood and urine cultures were negative. Repeat cerebrospinal fluid analysis was essentially unchanged, revealing a red cell count of 1/mm3; white cell count 20/mm3 (86% lymphocytes, 2% neutrophils, 12% monocytes); protein 14 mg/dL; glucose 63 mg/dL, and negative Gram stain. Continuous electroencephalography revealed diffuse generalized slowing, but no seizure activity. An extensive evaluation for viral, bacterial, autoimmune, and paraneoplastic disorders was negative, including tests for anti‐acetylcholine (ACh) receptor binding antibody, anti‐striated muscle antibody, anti‐N‐type calcium channel antibody, anti‐P/Q‐type calcium channel antibodies, anto‐cancer associated retinopathy (CAR) antibody (also known as anti‐recoverin antibody), and anti‐collapsin respons mediator protein (CRMP‐5). Without confirmatory results and continued deterioration, she was empirically treated with methylprednisolone for presumed autoimmune encephalitis from hospital days 16 to 21. The patient remained unresponsive and ventilator‐dependent, despite removal of all sedation. She experienced intermittent fevers as high as 40.5C, remained tachycardic, hypertensive, and exhibited orofacial dyskinesias and jaw clenching, ultimately requiring botulinum toxin injections to prevent tongue biting. Given the lack of improvement despite attempted therapies, a working diagnosis of viral encephalitis with lasting neuropsychiatric sequelae was made. A tracheostomy and percutaneous gastrostomy tube were placed, and a long‐term ventilator care facility was identified.
I continue to wonder if this may be an autoimmune encephalitis, and am concerned about her unexplained fevers. Neuroleptic malignant syndrome secondary to misuse of her parents' medications should be considered in light of the elevated creatine kinase, although the severity and duration of the syndrome seem more profound than I would anticipate. Tetanus could present with jaw dystonia, but the rest of the case does not seem to fit. At this point, considering the patient's young age and poor prognosis without identified etiology, prior to discharge I would argue for a brain biopsy looking for evidence of rabies, or other infectious or autoimmune etiologies of the patient's progressive neurologic deterioration.
On hospital day 25, due to the persistent fevers with concern for occult abscess, an abdominopelvic CT was obtained, which identified a complex 11.8 cm 9.0 cm adnexal mass consistent with a teratoma (Figure 1).
Given the size of the mass, it is surprising that the patient did not report abdominal symptoms and that the physicians were unable to palpate it on examination. The differential diagnosis of a complex adnexal mass in an adolescent should include an ectopic pregnancy, ovarian cysts, tubo‐ovarian abscess, rarely an ovarian carcinoma or leiomyosarcoma, and a teratoma or dermoid tumor. While I mentioned the possibility of a malignancy at the outset, I did not further consider it. Common neoplasms encountered in adolescent patients include lymphoma and leukemia, germ cell tumors (including teratomas), central nervous system tumors and sarcomas, many of which have been reported to cause paraneoplastic disorders. At this point, I now think her presumed teratoma is associated with a paraneoplastic syndrome resulting in her presentation of limbic encephalitis.
A literature search was performed by the managing clinicians who rapidly identified the association between teratoma and limbic encephalitis. The patient was initially treated with intravenous immune globulin (IVIG), with transient improvement in her mental status. Serology returned positive for the anti‐N‐methyl‐D‐aspartate receptor antibody, confirming the diagnosis of anti‐N‐methyl‐D‐aspartate receptor encephalitis. On hospital day 36, her mass was resected (Figure 2). Pathology was consistent with a mature teratoma. Postoperatively, the patient improved daily, and was discharged on hospital day 43 with a near complete neurologic recovery. Four months following discharge, the patient had enrolled full time in college.
COMMENTARY
The N‐methyl‐D‐aspartate receptor (NMDAR) is an important regulator of synaptic transmission and memory within the CNS. Our patient's case illustrates the increasingly recognized syndrome of anti‐NMDAR encephalitis. NMDAR hypofunction is hypothesized to result in the cognitive and behavioral abnormalities of schizophrenia, and direct antagonism of the NMDAR by drugs such as phencyclidine (PCP) and ketamine results in symptoms such as psychosis, hallucinations, delusions, agitation, and dissociative amnesia.14 This constellation of symptoms is very similar to some of the initial neuropsychiatric symptoms observed in patients with anti‐NMDAR encephalitis.
Anti‐NMDAR encephalitis was first described in 2005 as a paraneoplastic limbic encephalitis associated with ovarian teratoma.5, 6 Characterized by the subacute onset (days to weeks) of short‐term memory loss, psychiatric symptoms, and sleep disturbances, limbic encephalitis is an inflammatory process caused by autoantibodies against intracellular or extracellar antigens in the limbic system and other brain structures. Limbic encephalitides associated with antibodies to intracellular antigens (such as Hu, Ma2, CV2/CRMP5, and Amphiphysin) are more often associated with malignancies, have worse outcomes (permanent neuropsychiatric sequelae and death), and are less responsive to immune therapy. Conversely, it appears that both the paraneoplastic and non‐paraneoplastic variants of limbic encephalitis associated with antibodies against cell membrane antigens (such as NMDAR and Voltage Gated Potassium Channels) respond more favorably to therapy.7
As with limbic encephalitis in general, anti‐NMDAR encephalitis can be non‐paraneoplastic as well as paraneoplastic in etiology. In a recently published series of 44 consecutive patients with anti‐NMDAR encephalitis, tumors were present in only 9 cases (8 teratomas).8 When associated with a teratoma, it has been postulated that anti‐NMDAR antibodies develop and cross the bloodbrain barrier to target central nervous system NMDA receptors. This process results in down‐regulation of the neuronal surface NMDAR which then causes the psychiatric and behavioral changes described.6 The mechanism by which these antibodies traverse the bloodbrain barrier is not completely understood, but likely requires some disruption of the barrier in order to trigger anti‐NMDAR encephalitis.8, 9 Non‐paraneoplastic cases evidently involve other unknown stimuli for NMDAR antibody synthesisone report has suggested that subunits of the NMDAR are expressed by normal ovarian tissue, something which may explain the female predilection even in the cohort unaffected by teratomas.10
Most patients with anti‐NMDAR encephalitis are female and young (median age 23 years), although men and children are also affected.8, 9, 11 While the exact incidence of anti‐NMDAR encephalitis is still unknown, the increasing number of case reports suggests that it may be more frequent than any other type of paraneoplastic encephalitis.12 The majority of patients with anti‐NMDAR encephalitis experience an antecedent infectious prodrome (eg, diarrheal illness or upper respiratory infection [URI]), followed 1020 days later by progressive neuropsychiatric and behavioral symptoms which include confusion, memory deficits, impaired responsiveness, seizures, central hypoventilation, and signs of autonomic instability (tachycardia, tachypnea, diaphoresis, cardiac dysrhythmia, blood pressure instability, and dysthermia). At this stage, patients may also manifest a unique constellation of choreoathetoid orofacial and limb movements such as lip licking, chewing, sustained jaw clenching, jaw opening dystonias, ocular deviation and disconjugation, grimacing, myoclonus, and bizarre arm movements. Due to cardiovascular complications and ventilator requirements, most patients require intensive care unit (ICU) level care. 8, 9, 11 As in our discussant's evaluation, other disorders to include in the differential diagnosis for this presentation includes paraneoplastic or autoimmune causes of limbic encephalitis, toxins, heavy metals, and viral causes of encephalitis; in particular, herpes simplex virus (HSV).7
The CNS imaging findings in this condition include brain MRI abnormalities in about 30%55% of patients, which can include increased signal on fluid‐attenuated inversion recovery (FLAIR) or T2 sequences of the cerebral cortex, overlying meninges, or basal ganglia. Abnormalities in the temporal lobes, corpus callosum, and brainstem have also been described. As in our patient, CSF lymphocytic pleocytosis has also been noted.6, 8, 9
Although many cases of limbic encephalitis portend a poor prognosis with permanent neuropsychiatric sequelae and death, anti‐NMDAR can be very responsive to treatment; particularly if diagnosed early. Successful treatment of anti‐NMDAR encephalitis involves immunotherapy and, preferably, early surgical resection of any tumor. 6, 8, 9 Non‐paraneoplastic cases appear to require more aggressive and prolonged immunotherapies to avoid relapse. In both groups, a trend towards improved outcome has been noted in patients treated early in disease course (40 days from symptom onset).8 There are no established guidelines for the treatment of anti‐NMDAR encephalitis, and no randomized controlled trials have evaluated anti‐NMDAR encephalitis treatment. Observational studies of immune‐modulating therapies have shown efficacy with high‐dose steroids and the addition of plasma exchange and/or intravenous immune globulin. Rituximab and cyclophosphamide can be considered if patients fail to improve on other immunotherapies.9 Data from case series seem to suggest a lower risk of relapse in patients treated with immunotherapy.13
Exploration of this patient's persistent high fevers ultimately led to the serendipitous diagnosis of the increasingly recognized syndrome of anti‐NMDAR encephalitis, although in retrospect nearly all of the features of her presentation fit well with this condition. Thus, it was only by a chance finding on her abdominal CT scan that this patient was ultimately diagnosed with a treatable, noninfectious encephalitis associated with an ovarian teratoma. This case reinforces the importance of thorough patient evaluations and being prepared to draw meaningful conclusions from unexpected findings. Given how close this patient was to being discharged to a long‐term care facility, we found this case a fascinating yet sobering reminder to guard against prematurely concluding a syndrome to be untreatable.
KEY TEACHING POINTS
-
Anti‐NMDAR encephalitis is an increasingly recognized cause of autoimmune limbic encephalitis, and thus should be considered in patients with new‐onset psychiatric symptoms accompanied by seizures, autonomic instability, hypoventilation, or dyskinesias.
-
A thorough history, examination, and evaluation of data is critical to make an early diagnosis of anti‐NMDAR encephalitis, because, unlike other forms of limbic encephalitis, this condition may be very responsive to early initiation of treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A previously healthy 18‐year‐old woman living in the Pacific Northwest was brought in by her parents to a local hospital with a 4‐day history of acting crazy. Two weeks prior to presentation, she complained of a new‐onset severe headache, diaphoresis, and chills. Four days prior to presentation, she became progressively more impulsive, which ultimately included jumping out of a moving vehicle and running away from home. She experienced unexplained emotional outbursts and was unable to identify familiar relatives or common objects. Additionally, she began having hyperventilation spells and auditory hallucinations.
In an adolescent presenting with erratic behavior, one should consider the possibility of substance abuse or a psychiatric disease such as bipolar disorder with manic features, psychotic manifestations of severe depression, or early schizophrenia. However, it is important to first rule out non‐psychiatric disease, with a diagnostic approach dependent on her human immunodeficiency virus (HIV) status. The presence of headache, diaphoresis, and chills raises concern for an infectious or noninfectious inflammatory central nervous system process. In addition to the effects of illicit drugs such as cocaine or methamphetamine, this presentation may be consistent with a medication‐ or herbal‐induced anticholinergic syndrome, which may present with confusion, ataxia, coma, and cardiopulmonary failure. Since this case originates in the Northwest, one should be aware of the regional outbreak of Cryptococcus gattii in immunocompetent hosts, and that local hallucinogenic plants, such as jimson weed or mushrooms (Amanita muscaria) can cause anticholinergic syndromes. At this point, the differential diagnosis is broad, and evaluation should focus on potentially reversible life‐threatening conditions; in particular, herpes encephalitis. In addition to a detailed history, examination, and routine laboratory studies including HIV serology, I would obtain a drug screen, and order a computed tomography (CT) scan of the brain before performing a lumbar puncture. I would also order a magnetic resonance imaging (MRI) study to evaluate for meningeal or cerebral enhancement suggestive of encephalitis.
The patient had no past medical, psychiatric, or surgical history and took no medications. She lived with her parents who thought she neither used illicit drugs or alcohol, nor was sexually active. She had recently graduated high school and was planning to attend college. Her family history was notable for a mother with bipolar and seizure disorders, and 2 healthy younger siblings. Her family had a healthy cat and dog, and reported a large number of bats living nearby. She had never traveled outside the western United States. The patient presented in late spring, but there was no obvious history of mosquito bites. Her last menstrual period was 4 months prior to presentation. Full review of systems was otherwise negative.
The family history of mood disorder supports continued consideration of bipolar disorder with psychotic manifestations. However, infectious or inflammatory processes remain highest on the differential at this point. The duration of symptoms makes common bacterial meningitis etiologies (Streptococcus, Neisseria, Haemophilus, Listeria) less likely, but would be consistent with herpes simplex encephalitis or lupus cerebritis. Additional infectious considerations would include other viral (eg, varicella zoster virus, Epstein‐Barr virus, enteroviruses, and the arthropod‐borne encephalitides) or unusual bacterial encephalitic syndromes. Although the health status of pets is rarely helpful, dogs can carry ticks that harbor Borrelia burgdorferi (the agent of Lyme disease), which may present with central nervous system (CNS) manifestations. Other conditions associated with pets (such as leptospirosis or cat scratch disease) seem unlikely. The exposure to bats raises the possibility of rabies infection. If she is HIV‐positive, one would need to consider the possibility of opportunistic infections such as cytomegalovirus (CMV), Cryptococcus, cerebral toxoplasmosis, and progressive multifocal leukoencephalopathy (PML) caused by JC virus reaction. Finally, regardless of history, given the patient's amenorrhea, we must perform a pregnancy test.
The patient's temperature was 97.3F, heart rate 129 beats per minute, respiratory rate 19 breaths per minute, and her blood pressure 144/97 mmHg. She was an obese, well‐developed young woman, who was drowsy but arousable, with marked speech latency. Her cranium and oropharynx were normal, and her neck was supple. Aside from tachycardia, her cardiopulmonary, musculoskeletal, and skin exams were normal. Her abdomen was obese and soft, without masses or organomegaly. A pelvic examination was not performed. On neurologic exam, her strength was symmetrically diminished throughout (3+/5). Otherwise, she was oriented to person and general location, but not to day of week, month, or year. Her cranial nerves, sensation, deep tendon reflexes, and muscle tone were normal. A cerebellar examination, plantar response, and gait test were not performed. A brain MRI revealed only a small subarachnoid cyst and possible subtle enhancement of temporal lobes. Initial laboratory studies demonstrated: white blood cell count 14,000/mm3 (72% neutrophils, 17% lymphocytes, 9% monocytes, 2% eosinophils); hemoglobin 14.0 g/dL (mean corpuscular volume 87.4 fL); platelet count 417,000/mm3. Serum electrolytes, liver function tests, coagulation studies, thyroid stimulating hormone, serum ammonia, and urinalysis were normal. Her serum pregnancy test and urine toxicology screen were negative. A room air arterial blood gas revealed a pH of 7.49, PaCO2 32 mmHg, PaO2 89 mmHg; and a bicarbonate 24 mmol/L. Cerebrospinal fluid demonstrated: red cell count of 2/mm3; white cell count 17/mm3 (88% lymphocytes, 3% neutrophils, 9% monocytes); protein 19 mg/dL (normal 1555 mg/dL); and glucose of 79 mg/dL (normal 4080 mg/dL). Gram stain, fungal and bacterial cultures, and HIV serology were negative, and herpes simplex virus was not detected via polymerase chain reaction (PCR).
The tachycardia, respiratory alkalosis, and leukocytosis continue to suggest an infection or inflammatory state. Her neurological deterioration without focal findings, cerebrospinal fluid (CSF) lymphocytic pleocytosis with normal glucose and protein, and temporal lobe enhancement on MRI strongly suggest a meningoencephalitis. This would be an unusual presentation for most bacterial pathogens, but Mycobacterium, Rickettsia, Listeria, Mycoplasma, and Bartonella may rarely mimic encephalitis. Autoimmune encephalitis secondary to lupus, vasculitis, or other autoimmune disorder remains possible, but at this point an infectious encephalitis, particularly herpes encephalitis, is my highest concern. West Nile virus must be considered, but usually produces a severe illness only in immunocompromised or elderly patients. Additionally, despite the rarity of rabies, the patient's exposure to bats and the rapid clinical deterioration, suggest this possibility. In addition to routine bacterial and viral analyses (eg, enteroviral panel), samples should be sent for rabies PCR and antibody testing, West Nile virus, Lyme disease, syphilis, and mycobacterial and fungal pathogens, such as the aforementioned Cryptococcus gattii. Finally, given her presenting syndrome and MRI, immediate treatment with acyclovir and antibiotics is indicated.
The patient was treated for presumed meningoencephalitis with acyclovir and ceftriaxone, but over the following several days became unresponsive to all stimuli and developed repetitive thrusting movements of her mouth, tongue, and jaw. On hospital day 10, with concern for seizures, pentobarbital coma was induced, and the patient was intubated and transferred to our facility. On arrival, her physical examination was essentially unchanged aside from being in a medical coma. Hematology, chemistries, and thyroid‐stimulating hormone (TSH) were again unremarkable with the exception of an elevated creatine kinase (414 U/L) and a new anemia (hemoglobin 8.9 g/dL; mean corpuscular volume 87.6 fL) without evidence of iron deficiency or hemolysis. Blood and urine cultures were negative. Repeat cerebrospinal fluid analysis was essentially unchanged, revealing a red cell count of 1/mm3; white cell count 20/mm3 (86% lymphocytes, 2% neutrophils, 12% monocytes); protein 14 mg/dL; glucose 63 mg/dL, and negative Gram stain. Continuous electroencephalography revealed diffuse generalized slowing, but no seizure activity. An extensive evaluation for viral, bacterial, autoimmune, and paraneoplastic disorders was negative, including tests for anti‐acetylcholine (ACh) receptor binding antibody, anti‐striated muscle antibody, anti‐N‐type calcium channel antibody, anti‐P/Q‐type calcium channel antibodies, anto‐cancer associated retinopathy (CAR) antibody (also known as anti‐recoverin antibody), and anti‐collapsin respons mediator protein (CRMP‐5). Without confirmatory results and continued deterioration, she was empirically treated with methylprednisolone for presumed autoimmune encephalitis from hospital days 16 to 21. The patient remained unresponsive and ventilator‐dependent, despite removal of all sedation. She experienced intermittent fevers as high as 40.5C, remained tachycardic, hypertensive, and exhibited orofacial dyskinesias and jaw clenching, ultimately requiring botulinum toxin injections to prevent tongue biting. Given the lack of improvement despite attempted therapies, a working diagnosis of viral encephalitis with lasting neuropsychiatric sequelae was made. A tracheostomy and percutaneous gastrostomy tube were placed, and a long‐term ventilator care facility was identified.
I continue to wonder if this may be an autoimmune encephalitis, and am concerned about her unexplained fevers. Neuroleptic malignant syndrome secondary to misuse of her parents' medications should be considered in light of the elevated creatine kinase, although the severity and duration of the syndrome seem more profound than I would anticipate. Tetanus could present with jaw dystonia, but the rest of the case does not seem to fit. At this point, considering the patient's young age and poor prognosis without identified etiology, prior to discharge I would argue for a brain biopsy looking for evidence of rabies, or other infectious or autoimmune etiologies of the patient's progressive neurologic deterioration.
On hospital day 25, due to the persistent fevers with concern for occult abscess, an abdominopelvic CT was obtained, which identified a complex 11.8 cm 9.0 cm adnexal mass consistent with a teratoma (Figure 1).
Given the size of the mass, it is surprising that the patient did not report abdominal symptoms and that the physicians were unable to palpate it on examination. The differential diagnosis of a complex adnexal mass in an adolescent should include an ectopic pregnancy, ovarian cysts, tubo‐ovarian abscess, rarely an ovarian carcinoma or leiomyosarcoma, and a teratoma or dermoid tumor. While I mentioned the possibility of a malignancy at the outset, I did not further consider it. Common neoplasms encountered in adolescent patients include lymphoma and leukemia, germ cell tumors (including teratomas), central nervous system tumors and sarcomas, many of which have been reported to cause paraneoplastic disorders. At this point, I now think her presumed teratoma is associated with a paraneoplastic syndrome resulting in her presentation of limbic encephalitis.
A literature search was performed by the managing clinicians who rapidly identified the association between teratoma and limbic encephalitis. The patient was initially treated with intravenous immune globulin (IVIG), with transient improvement in her mental status. Serology returned positive for the anti‐N‐methyl‐D‐aspartate receptor antibody, confirming the diagnosis of anti‐N‐methyl‐D‐aspartate receptor encephalitis. On hospital day 36, her mass was resected (Figure 2). Pathology was consistent with a mature teratoma. Postoperatively, the patient improved daily, and was discharged on hospital day 43 with a near complete neurologic recovery. Four months following discharge, the patient had enrolled full time in college.
COMMENTARY
The N‐methyl‐D‐aspartate receptor (NMDAR) is an important regulator of synaptic transmission and memory within the CNS. Our patient's case illustrates the increasingly recognized syndrome of anti‐NMDAR encephalitis. NMDAR hypofunction is hypothesized to result in the cognitive and behavioral abnormalities of schizophrenia, and direct antagonism of the NMDAR by drugs such as phencyclidine (PCP) and ketamine results in symptoms such as psychosis, hallucinations, delusions, agitation, and dissociative amnesia.14 This constellation of symptoms is very similar to some of the initial neuropsychiatric symptoms observed in patients with anti‐NMDAR encephalitis.
Anti‐NMDAR encephalitis was first described in 2005 as a paraneoplastic limbic encephalitis associated with ovarian teratoma.5, 6 Characterized by the subacute onset (days to weeks) of short‐term memory loss, psychiatric symptoms, and sleep disturbances, limbic encephalitis is an inflammatory process caused by autoantibodies against intracellular or extracellar antigens in the limbic system and other brain structures. Limbic encephalitides associated with antibodies to intracellular antigens (such as Hu, Ma2, CV2/CRMP5, and Amphiphysin) are more often associated with malignancies, have worse outcomes (permanent neuropsychiatric sequelae and death), and are less responsive to immune therapy. Conversely, it appears that both the paraneoplastic and non‐paraneoplastic variants of limbic encephalitis associated with antibodies against cell membrane antigens (such as NMDAR and Voltage Gated Potassium Channels) respond more favorably to therapy.7
As with limbic encephalitis in general, anti‐NMDAR encephalitis can be non‐paraneoplastic as well as paraneoplastic in etiology. In a recently published series of 44 consecutive patients with anti‐NMDAR encephalitis, tumors were present in only 9 cases (8 teratomas).8 When associated with a teratoma, it has been postulated that anti‐NMDAR antibodies develop and cross the bloodbrain barrier to target central nervous system NMDA receptors. This process results in down‐regulation of the neuronal surface NMDAR which then causes the psychiatric and behavioral changes described.6 The mechanism by which these antibodies traverse the bloodbrain barrier is not completely understood, but likely requires some disruption of the barrier in order to trigger anti‐NMDAR encephalitis.8, 9 Non‐paraneoplastic cases evidently involve other unknown stimuli for NMDAR antibody synthesisone report has suggested that subunits of the NMDAR are expressed by normal ovarian tissue, something which may explain the female predilection even in the cohort unaffected by teratomas.10
Most patients with anti‐NMDAR encephalitis are female and young (median age 23 years), although men and children are also affected.8, 9, 11 While the exact incidence of anti‐NMDAR encephalitis is still unknown, the increasing number of case reports suggests that it may be more frequent than any other type of paraneoplastic encephalitis.12 The majority of patients with anti‐NMDAR encephalitis experience an antecedent infectious prodrome (eg, diarrheal illness or upper respiratory infection [URI]), followed 1020 days later by progressive neuropsychiatric and behavioral symptoms which include confusion, memory deficits, impaired responsiveness, seizures, central hypoventilation, and signs of autonomic instability (tachycardia, tachypnea, diaphoresis, cardiac dysrhythmia, blood pressure instability, and dysthermia). At this stage, patients may also manifest a unique constellation of choreoathetoid orofacial and limb movements such as lip licking, chewing, sustained jaw clenching, jaw opening dystonias, ocular deviation and disconjugation, grimacing, myoclonus, and bizarre arm movements. Due to cardiovascular complications and ventilator requirements, most patients require intensive care unit (ICU) level care. 8, 9, 11 As in our discussant's evaluation, other disorders to include in the differential diagnosis for this presentation includes paraneoplastic or autoimmune causes of limbic encephalitis, toxins, heavy metals, and viral causes of encephalitis; in particular, herpes simplex virus (HSV).7
The CNS imaging findings in this condition include brain MRI abnormalities in about 30%55% of patients, which can include increased signal on fluid‐attenuated inversion recovery (FLAIR) or T2 sequences of the cerebral cortex, overlying meninges, or basal ganglia. Abnormalities in the temporal lobes, corpus callosum, and brainstem have also been described. As in our patient, CSF lymphocytic pleocytosis has also been noted.6, 8, 9
Although many cases of limbic encephalitis portend a poor prognosis with permanent neuropsychiatric sequelae and death, anti‐NMDAR can be very responsive to treatment; particularly if diagnosed early. Successful treatment of anti‐NMDAR encephalitis involves immunotherapy and, preferably, early surgical resection of any tumor. 6, 8, 9 Non‐paraneoplastic cases appear to require more aggressive and prolonged immunotherapies to avoid relapse. In both groups, a trend towards improved outcome has been noted in patients treated early in disease course (40 days from symptom onset).8 There are no established guidelines for the treatment of anti‐NMDAR encephalitis, and no randomized controlled trials have evaluated anti‐NMDAR encephalitis treatment. Observational studies of immune‐modulating therapies have shown efficacy with high‐dose steroids and the addition of plasma exchange and/or intravenous immune globulin. Rituximab and cyclophosphamide can be considered if patients fail to improve on other immunotherapies.9 Data from case series seem to suggest a lower risk of relapse in patients treated with immunotherapy.13
Exploration of this patient's persistent high fevers ultimately led to the serendipitous diagnosis of the increasingly recognized syndrome of anti‐NMDAR encephalitis, although in retrospect nearly all of the features of her presentation fit well with this condition. Thus, it was only by a chance finding on her abdominal CT scan that this patient was ultimately diagnosed with a treatable, noninfectious encephalitis associated with an ovarian teratoma. This case reinforces the importance of thorough patient evaluations and being prepared to draw meaningful conclusions from unexpected findings. Given how close this patient was to being discharged to a long‐term care facility, we found this case a fascinating yet sobering reminder to guard against prematurely concluding a syndrome to be untreatable.
KEY TEACHING POINTS
-
Anti‐NMDAR encephalitis is an increasingly recognized cause of autoimmune limbic encephalitis, and thus should be considered in patients with new‐onset psychiatric symptoms accompanied by seizures, autonomic instability, hypoventilation, or dyskinesias.
-
A thorough history, examination, and evaluation of data is critical to make an early diagnosis of anti‐NMDAR encephalitis, because, unlike other forms of limbic encephalitis, this condition may be very responsive to early initiation of treatment.
- , . N‐methyl‐D‐aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49.
- , . Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308.
- , , . NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533.
- , , , et al. Ketamine‐induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118.
- , , , , , . Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58:594–604.
- , , , et al. Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
- , . Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13:261–271.
- , , , et al. N‐methyl‐D‐aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‐paraneoplastic disorder of both sexes. Brain. 2010;133:1655–1667.
- , . NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;11:298–304.
- , , , et al. Expression of various glutamate receptors including N‐methyl‐D‐aspartate receptor (NMDAR) in an ovarian teratoma removed from a young woman with anti‐NMDAR encephalitis. Intern Med. 2010;49:2167–2173.
- , , , et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1074–1075.
- , , , , . Clinical experience and laboratory investigations in patients with anti‐NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
- , , , et al. Analysis of relapses in anti‐NMDAR encephalitis. Neurology. 2011;77:996–999.
- , . N‐methyl‐D‐aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49.
- , . Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308.
- , , . NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533.
- , , , et al. Ketamine‐induced NMDA receptor hypofunction as a model of memory impairment and psychosis. Neuropsychopharmacology. 1999;20:106–118.
- , , , , , . Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58:594–604.
- , , , et al. Paraneoplastic anti‐N‐methyl‐D‐aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36.
- , . Limbic encephalitis and variants: classification, diagnosis and treatment. Neurologist. 2007;13:261–271.
- , , , et al. N‐methyl‐D‐aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‐paraneoplastic disorder of both sexes. Brain. 2010;133:1655–1667.
- , . NMDA receptor antibody encephalitis. Curr Neurol Neurosci Rep. 2011;11:298–304.
- , , , et al. Expression of various glutamate receptors including N‐methyl‐D‐aspartate receptor (NMDAR) in an ovarian teratoma removed from a young woman with anti‐NMDAR encephalitis. Intern Med. 2010;49:2167–2173.
- , , , et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1074–1075.
- , , , , . Clinical experience and laboratory investigations in patients with anti‐NMDAR encephalitis. Lancet Neurol. 2011;10:63–74.
- , , , et al. Analysis of relapses in anti‐NMDAR encephalitis. Neurology. 2011;77:996–999.
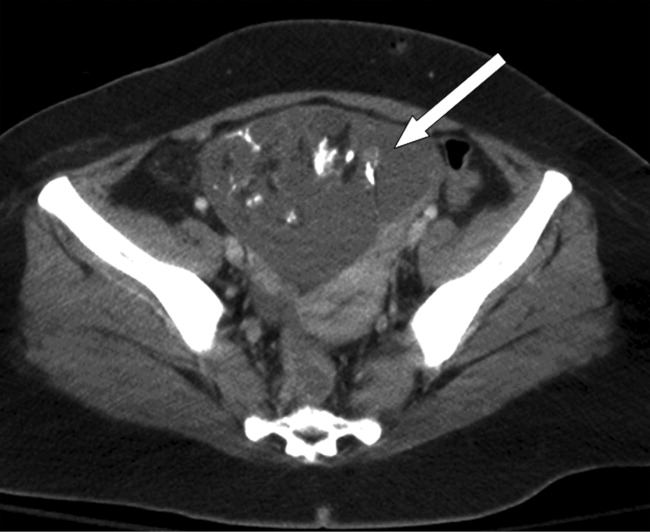
What Is the Best E&M of Fat Embolism Syndrome?
The Case
A 24-year-old white man with no past medical history is admitted after sustaining bilateral, closed femur fractures in a motor vehicle accident. Within hours of the trauma, he is taken to the operating room for open reduction and internal fixation. Of note, preoperatively, his hematocrit is 40%. After surgery, he is easily extubated and transferred to an unmonitored bed for further care. Approximately 30 hours after admission, he develops tachypnea with a respiratory rate of 35 breaths per minute and hypoxia with an oxygen saturation of 86% on room air. He is tachycardic (120 beats per minute) and febrile to 39.0oC. His blood pressure remains stable. He is somnolent, and when awake, he is confused. Notably, his hematocrit is now 22%. An electrocardiogram shows sinus tachycardia, an initial chest X-ray is normal, and a high-resolution CT scan is negative for a pulmonary embolism (PE).
Is this clinical picture consistent with fat embolism syndrome and, if so, how should he be managed?
Overview
“Fat embolism” refers to the presence of fat globules that obstruct the lung parenchyma and peripheral circulation. Fat embolism syndrome, on the other hand, is a more serious manifestation involving multiple organ systems. Specifically, it is a clinical diagnosis presenting with the classic triad of hypoxemia, neurologic abnormalities, and a petechial rash.
Fat embolism syndrome is usually associated with multiple traumas, including long-bone injuries and pelvic fractures. It is more frequently associated with closed fractures than open fractures, possibly due to the higher pressures associated with closed fractures. This syndrome has been less commonly associated with a variety of nontraumatic conditions (Table 1).

With an increased incidence of long-bone fractures in the younger demographic, fat embolism syndrome is most common in the second or third decade of life. While fat embolism occurs in up to 90% of patients with traumatic skeletal injuries, fat embolism syndrome occurs in 0.5% to 10% of patients following trauma, with a higher incidence in multiple fractures (5% to 10%) than in single long-bone fractures (0.5% to 2%).1-3
With the increasing role of hospitalists in assisting in the management of orthopedic patients, their knowledge of fat embolism syndrome is important so that it can be included in the differential diagnosis of acute respiratory failure in these orthopedic patients.
Review of the Data
Pathogenesis. Clinical manifestations of fat embolism syndrome have been acknowledged for more than 100 years. Since its first description in the 1860s, there has been speculation about the etiology of this condition. In the 1920s, two theories were proposed to explain the origin of the fat droplets: the mechanical and biochemical theories.2,4
Mechanical theory suggests that trauma to long bones disturbs fat cells within the bone marrow or adipose tissue, causing fat globules to mobilize.2,3 There is a rise in marrow pressure above venous pressure, which allows fat particles to enter the circulation through damaged venules surrounding the fracture site. Once lodged in the pulmonary microvasculature, embolized fat causes local ischemia and inflammation. Fat globules may pass into the arterial circulation either by paradoxical embolism through a patent foramen ovale, or by microemboli that pass through the lungs into the arterial circulation. This explains embolization to other organs, including the brain, retina, and skin.
Alternatively, biochemical theory hypothesizes that fat embolism syndrome is contingent on the production of toxic intermediaries from the breakdown of embolized fat.2,3 This theory suggests that the release of catecholamines after severe trauma can liberate free fatty acids from fat stores, or that acute-phase reactants at the trauma site affect fat solubility, causing agglutination and embolization. This theory helps to explain nontraumatic fat embolism syndrome, as well as the delay in development of the clinical syndrome after acute injury.
Clinical presentation. Most patients have a latent period after trauma of 12 to 72 hours before symptoms of fat embolism syndrome become apparent; however, clinical manifestations might occur immediately or up to one to two weeks following injury.2,4 As previously mentioned, the classic triad of symptoms includes respiratory compromise, neurological impairment, and a petechial rash.
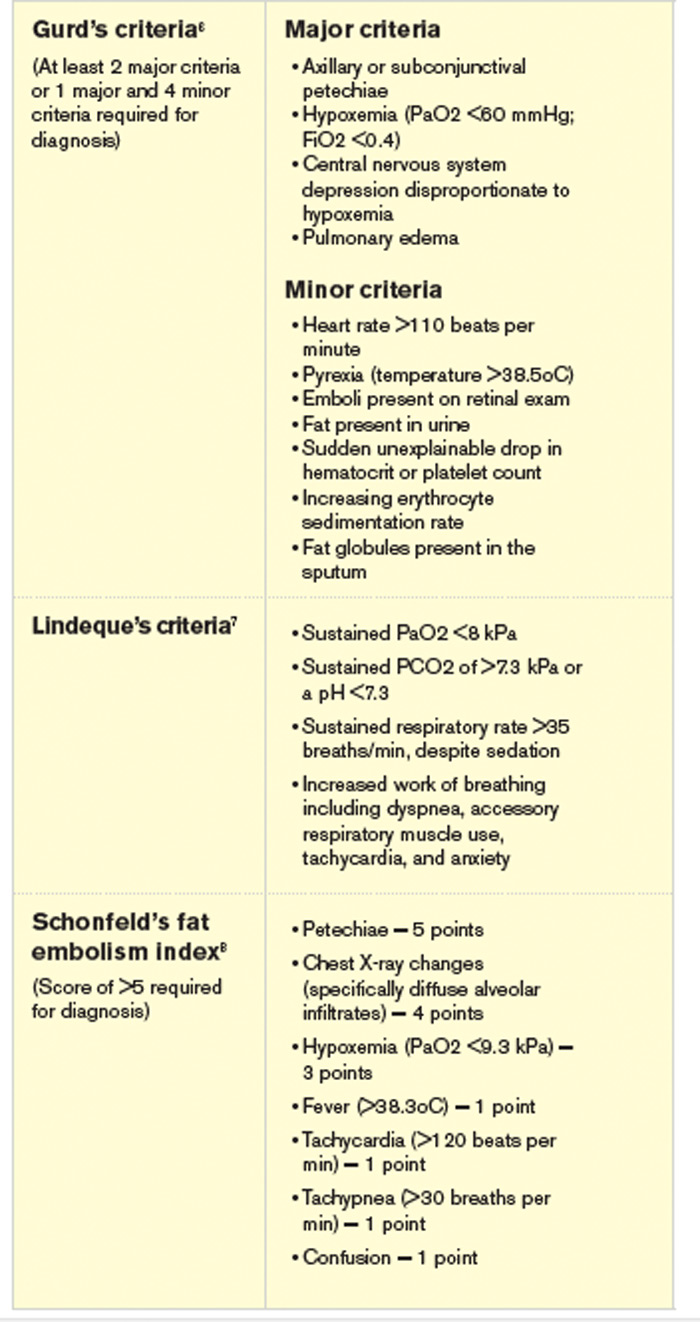
The most common and usually earliest manifestation is acute hypoxia, which must be distinguished from other treatable causes of hypoxia, including pneumothorax, hemothorax, PE, and pneumonia. Pulmonary changes might progress to respiratory failure similar to acute respiratory distress syndrome. Neurological manifestations are primarily nonspecific and include headache, irritability, delirium, seizures, and coma. Focal neurological deficits are rare but have been described.5 Almost all neurological symptoms are fully reversible. The petechial rash is distinctive and occurs on the chest, axilla, and subconjunctiva. Although the rash occurs in only 20% to 50% of patients and resolves fairly quickly, in the appropriate clinical setting, this rash is considered pathognomonic.1,2,4
A variety of other nonspecific signs and symptoms might also occur: pyrexia, tachycardia, fat in the urine or sputum, retinal changes, renal insufficiency, myocardial dysfunction, and an otherwise unexplained drop in hematocrit or platelet count.
Diagnosis. Fat embolism syndrome is a clinical diagnosis and a diagnosis of exclusion. There are no specific confirmatory tests. An arterial blood gas will usually reveal a PaO2 of <60 mmHg.3 Laboratory evaluation might also show fat globules in the urine or sputum on Sudan or Oil Red O staining, but these findings are nonspecific.3,4 Bronchoscopy with bronchial alveolar lavage (BAL) might similarly detect fat droplets in alveolar macrophages in the BAL fluid; however, the sensitivity and specificity for diagnosis of fat embolism syndrome are unknown.4 None of these tests can be used solely for the diagnosis of fat embolism syndrome.
Thrombocytopenia and anemia out of proportion to the expected drop from surgery are not uncommon in addition to other nonspecific laboratory findings, including hypocalcemia, elevated serum lipase level, and elevated erythrocyte sedimentation rate.4 Several radiological findings have been observed on lung and brain imaging, though the findings are nonspecific and none are diagnostic. A chest X-ray might be normal, but abnormalities are seen in 30% to 50% of cases.2 Typically, when abnormal, the chest X-ray shows diffuse interstitial and alveolar densities, as well as patchy perihilar and basilar infiltrates resembling pulmonary edema. These X-ray findings might not be seen for up to 12 to 24 hours following the onset of clinical symptoms.
The most commonly used diagnostic criteria for the diagnosis of fat embolism syndrome are published by Gurd et al.6 At least two major criteria or one major criterion and four minor criteria are required for the diagnosis of fat embolism syndrome. The major criteria are based on the three classic signs and symptoms of fat embolism syndrome; the minor criteria include the finding of fat globules in the urine and sputum as well as some of the previously mentioned nonspecific clinical signs and laboratory tests.
Other criteria for diagnosis have been suggested, including those published by Lindeque et al, which focuses primarily on the respiratory characteristics, and a more recent set of semiquantitative diagnostic criteria called the fat embolism index, published by Schonfeld et al.7,8 Schonfeld’s scoring index accounts for the major signs and symptoms of fat embolism syndrome and weighs them according to relative specificity. A score of 5 or more is required for diagnosis of fat embolism syndrome. Table 2 compares the three sets of criteria used for diagnosis of fat embolism syndrome.
Treatment. The treatment of fat embolism syndrome is supportive. Most often, this requires supplemental oxygen for hypoxia and, possibly, fluid resuscitation in the case of hypovolemia. Occasionally, though, these relatively minor supportive therapies need to be escalated to bipap or even full ventilatory support and vasopressors in the more severe cases.
Based on the premise that steroids will attenuate the inflammatory reaction to free fatty acids within the lung, steroids have been tried in the treatment of fat embolism syndrome. However, there are no studies that clearly show benefit with their use.
Prevention. Most of the methods of prevention involve surgical intervention rather than medical therapy. Because microscopic fat emboli are showered during manipulation of long-bone fragments, early immobilization of fractures is recommended, and operative correction rather than conservative management is the preferred method.2,3 One report estimates a 70% reduction in pulmonary complications from this intervention alone.9
Further, two surgical techniques are debated as possible means of preventing fat embolism syndrome. The first is “venting,” in which a hole is made distal to the site of intramedullary nail placement. This reduces intramedullary pressure elevation and, therefore, extravasation of fat into the circulation.10 The second technique is the use of a reamer, irrigator, aspirator (RIA) device. A reamer is a tool used to create an accurate-sized hole for an intramedullary nail. Reaming before intramedullary nail placement can release fat deposits into the circulation. The RIA device irrigates and aspirates resident fat deposits as it reams the canal, releasing fewer deposits into the circulation.11 At this time, these two techniques are considered but not used routinely by surgeons.
Corticosteroids remain a debated method of prevention of fat embolism syndrome. A number of smaller studies suggest steroid therapy might reduce the incidence of fat embolism syndrome and hypoxia; a 2009 meta-analysis pooling nearly 400 patients from these smaller studies found such results.12 Unfortunately, the included studies were noted to be of poor quality, and no change in mortality was found. These results, combined with the possibility of poor wound healing or infection as a complication of steroid use, keep steroids from being used routinely to prevent fat embolism syndrome.
Clinical course. The severity of fat embolism syndrome ranges from mild transient hypoxia with confusion to progressively worsening symptoms leading to acute respiratory distress syndrome and coma. Bulger et al found a 7% mortality rate in this population.1 Less commonly, patients have a fulminant presentation with symptom onset less than 12 hours after injury. With this presentation, patients have a higher rate of mortality—as high as 15%.13
Back to the Case
This young man with bilateral long-bone fractures was at high risk of developing fat embolism syndrome. As is recommended, he was quickly taken to the operating room for fracture stabilization with open reduction and internal fixation. In addition, a RIA device was used to decrease intramedullary pressure. Nonetheless, within the first two days of injury, he developed hypoxia and confusion. These clinical changes were associated with an unexpected drop in hematocrit.
Chest X-ray and high-resolution computed tomography did not reveal a cause of his hypoxia. Similarly, laboratory evaluation for a reversible cause of encephalopathy was negative. A Sudan stain of his urine revealed free fat globules. Though he did not develop axillary petechiae, this clinical picture is consistent with fat embolism syndrome based on Gurd’s criteria. He was supported with oxygen therapy, and he stabilized without further complications.
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132:435-439.
- Levy D. The fat embolism syndrome. Clin Orthop. 1990;261:281-286.
- Akhtar S. Fat embolism. Anes Clin. 2009;27:533-550.
- Gupta A, Reilly C. Fat embolism. Anaesth Crit Care Pain. 2007;7:148-151.
- Thomas JE, Ayyar DR. Systemic fat embolism. Arch Neurol. 1972;26:517-523.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56B:408-416.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br. 1987;69:128-131.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med. 1983;99:438-443.
- Robinson CM. Current concepts of respiratory insufficiency syndromes after fracture. J Bone Joint Surg Br. 2001;83:781-791.
- Kim YH, Oh SW, Kim JS. Prevalence of fat embolism following bilateral simultaneous and unilateral total hip arthroplasty performed with or without cement: a prospective, randomized clinical study. J Bone Joint Surg Am. 2002;84A:1372-1379.
- Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41 Suppl 2:S90-S93.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009;52:386-393.
- Bracco D, Favre JB, Joris F, Ravussin A. Fatal fat embolism syndrome: a case report. J Neurosurg Anesthesiol. 2000;12:221-224.
The Case
A 24-year-old white man with no past medical history is admitted after sustaining bilateral, closed femur fractures in a motor vehicle accident. Within hours of the trauma, he is taken to the operating room for open reduction and internal fixation. Of note, preoperatively, his hematocrit is 40%. After surgery, he is easily extubated and transferred to an unmonitored bed for further care. Approximately 30 hours after admission, he develops tachypnea with a respiratory rate of 35 breaths per minute and hypoxia with an oxygen saturation of 86% on room air. He is tachycardic (120 beats per minute) and febrile to 39.0oC. His blood pressure remains stable. He is somnolent, and when awake, he is confused. Notably, his hematocrit is now 22%. An electrocardiogram shows sinus tachycardia, an initial chest X-ray is normal, and a high-resolution CT scan is negative for a pulmonary embolism (PE).
Is this clinical picture consistent with fat embolism syndrome and, if so, how should he be managed?
Overview
“Fat embolism” refers to the presence of fat globules that obstruct the lung parenchyma and peripheral circulation. Fat embolism syndrome, on the other hand, is a more serious manifestation involving multiple organ systems. Specifically, it is a clinical diagnosis presenting with the classic triad of hypoxemia, neurologic abnormalities, and a petechial rash.
Fat embolism syndrome is usually associated with multiple traumas, including long-bone injuries and pelvic fractures. It is more frequently associated with closed fractures than open fractures, possibly due to the higher pressures associated with closed fractures. This syndrome has been less commonly associated with a variety of nontraumatic conditions (Table 1).
With an increased incidence of long-bone fractures in the younger demographic, fat embolism syndrome is most common in the second or third decade of life. While fat embolism occurs in up to 90% of patients with traumatic skeletal injuries, fat embolism syndrome occurs in 0.5% to 10% of patients following trauma, with a higher incidence in multiple fractures (5% to 10%) than in single long-bone fractures (0.5% to 2%).1-3
With the increasing role of hospitalists in assisting in the management of orthopedic patients, their knowledge of fat embolism syndrome is important so that it can be included in the differential diagnosis of acute respiratory failure in these orthopedic patients.
Review of the Data
Pathogenesis. Clinical manifestations of fat embolism syndrome have been acknowledged for more than 100 years. Since its first description in the 1860s, there has been speculation about the etiology of this condition. In the 1920s, two theories were proposed to explain the origin of the fat droplets: the mechanical and biochemical theories.2,4
Mechanical theory suggests that trauma to long bones disturbs fat cells within the bone marrow or adipose tissue, causing fat globules to mobilize.2,3 There is a rise in marrow pressure above venous pressure, which allows fat particles to enter the circulation through damaged venules surrounding the fracture site. Once lodged in the pulmonary microvasculature, embolized fat causes local ischemia and inflammation. Fat globules may pass into the arterial circulation either by paradoxical embolism through a patent foramen ovale, or by microemboli that pass through the lungs into the arterial circulation. This explains embolization to other organs, including the brain, retina, and skin.
Alternatively, biochemical theory hypothesizes that fat embolism syndrome is contingent on the production of toxic intermediaries from the breakdown of embolized fat.2,3 This theory suggests that the release of catecholamines after severe trauma can liberate free fatty acids from fat stores, or that acute-phase reactants at the trauma site affect fat solubility, causing agglutination and embolization. This theory helps to explain nontraumatic fat embolism syndrome, as well as the delay in development of the clinical syndrome after acute injury.
Clinical presentation. Most patients have a latent period after trauma of 12 to 72 hours before symptoms of fat embolism syndrome become apparent; however, clinical manifestations might occur immediately or up to one to two weeks following injury.2,4 As previously mentioned, the classic triad of symptoms includes respiratory compromise, neurological impairment, and a petechial rash.
The most common and usually earliest manifestation is acute hypoxia, which must be distinguished from other treatable causes of hypoxia, including pneumothorax, hemothorax, PE, and pneumonia. Pulmonary changes might progress to respiratory failure similar to acute respiratory distress syndrome. Neurological manifestations are primarily nonspecific and include headache, irritability, delirium, seizures, and coma. Focal neurological deficits are rare but have been described.5 Almost all neurological symptoms are fully reversible. The petechial rash is distinctive and occurs on the chest, axilla, and subconjunctiva. Although the rash occurs in only 20% to 50% of patients and resolves fairly quickly, in the appropriate clinical setting, this rash is considered pathognomonic.1,2,4
A variety of other nonspecific signs and symptoms might also occur: pyrexia, tachycardia, fat in the urine or sputum, retinal changes, renal insufficiency, myocardial dysfunction, and an otherwise unexplained drop in hematocrit or platelet count.
Diagnosis. Fat embolism syndrome is a clinical diagnosis and a diagnosis of exclusion. There are no specific confirmatory tests. An arterial blood gas will usually reveal a PaO2 of <60 mmHg.3 Laboratory evaluation might also show fat globules in the urine or sputum on Sudan or Oil Red O staining, but these findings are nonspecific.3,4 Bronchoscopy with bronchial alveolar lavage (BAL) might similarly detect fat droplets in alveolar macrophages in the BAL fluid; however, the sensitivity and specificity for diagnosis of fat embolism syndrome are unknown.4 None of these tests can be used solely for the diagnosis of fat embolism syndrome.
Thrombocytopenia and anemia out of proportion to the expected drop from surgery are not uncommon in addition to other nonspecific laboratory findings, including hypocalcemia, elevated serum lipase level, and elevated erythrocyte sedimentation rate.4 Several radiological findings have been observed on lung and brain imaging, though the findings are nonspecific and none are diagnostic. A chest X-ray might be normal, but abnormalities are seen in 30% to 50% of cases.2 Typically, when abnormal, the chest X-ray shows diffuse interstitial and alveolar densities, as well as patchy perihilar and basilar infiltrates resembling pulmonary edema. These X-ray findings might not be seen for up to 12 to 24 hours following the onset of clinical symptoms.
The most commonly used diagnostic criteria for the diagnosis of fat embolism syndrome are published by Gurd et al.6 At least two major criteria or one major criterion and four minor criteria are required for the diagnosis of fat embolism syndrome. The major criteria are based on the three classic signs and symptoms of fat embolism syndrome; the minor criteria include the finding of fat globules in the urine and sputum as well as some of the previously mentioned nonspecific clinical signs and laboratory tests.
Other criteria for diagnosis have been suggested, including those published by Lindeque et al, which focuses primarily on the respiratory characteristics, and a more recent set of semiquantitative diagnostic criteria called the fat embolism index, published by Schonfeld et al.7,8 Schonfeld’s scoring index accounts for the major signs and symptoms of fat embolism syndrome and weighs them according to relative specificity. A score of 5 or more is required for diagnosis of fat embolism syndrome. Table 2 compares the three sets of criteria used for diagnosis of fat embolism syndrome.
Treatment. The treatment of fat embolism syndrome is supportive. Most often, this requires supplemental oxygen for hypoxia and, possibly, fluid resuscitation in the case of hypovolemia. Occasionally, though, these relatively minor supportive therapies need to be escalated to bipap or even full ventilatory support and vasopressors in the more severe cases.
Based on the premise that steroids will attenuate the inflammatory reaction to free fatty acids within the lung, steroids have been tried in the treatment of fat embolism syndrome. However, there are no studies that clearly show benefit with their use.
Prevention. Most of the methods of prevention involve surgical intervention rather than medical therapy. Because microscopic fat emboli are showered during manipulation of long-bone fragments, early immobilization of fractures is recommended, and operative correction rather than conservative management is the preferred method.2,3 One report estimates a 70% reduction in pulmonary complications from this intervention alone.9
Further, two surgical techniques are debated as possible means of preventing fat embolism syndrome. The first is “venting,” in which a hole is made distal to the site of intramedullary nail placement. This reduces intramedullary pressure elevation and, therefore, extravasation of fat into the circulation.10 The second technique is the use of a reamer, irrigator, aspirator (RIA) device. A reamer is a tool used to create an accurate-sized hole for an intramedullary nail. Reaming before intramedullary nail placement can release fat deposits into the circulation. The RIA device irrigates and aspirates resident fat deposits as it reams the canal, releasing fewer deposits into the circulation.11 At this time, these two techniques are considered but not used routinely by surgeons.
Corticosteroids remain a debated method of prevention of fat embolism syndrome. A number of smaller studies suggest steroid therapy might reduce the incidence of fat embolism syndrome and hypoxia; a 2009 meta-analysis pooling nearly 400 patients from these smaller studies found such results.12 Unfortunately, the included studies were noted to be of poor quality, and no change in mortality was found. These results, combined with the possibility of poor wound healing or infection as a complication of steroid use, keep steroids from being used routinely to prevent fat embolism syndrome.
Clinical course. The severity of fat embolism syndrome ranges from mild transient hypoxia with confusion to progressively worsening symptoms leading to acute respiratory distress syndrome and coma. Bulger et al found a 7% mortality rate in this population.1 Less commonly, patients have a fulminant presentation with symptom onset less than 12 hours after injury. With this presentation, patients have a higher rate of mortality—as high as 15%.13
Back to the Case
This young man with bilateral long-bone fractures was at high risk of developing fat embolism syndrome. As is recommended, he was quickly taken to the operating room for fracture stabilization with open reduction and internal fixation. In addition, a RIA device was used to decrease intramedullary pressure. Nonetheless, within the first two days of injury, he developed hypoxia and confusion. These clinical changes were associated with an unexpected drop in hematocrit.
Chest X-ray and high-resolution computed tomography did not reveal a cause of his hypoxia. Similarly, laboratory evaluation for a reversible cause of encephalopathy was negative. A Sudan stain of his urine revealed free fat globules. Though he did not develop axillary petechiae, this clinical picture is consistent with fat embolism syndrome based on Gurd’s criteria. He was supported with oxygen therapy, and he stabilized without further complications.
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132:435-439.
- Levy D. The fat embolism syndrome. Clin Orthop. 1990;261:281-286.
- Akhtar S. Fat embolism. Anes Clin. 2009;27:533-550.
- Gupta A, Reilly C. Fat embolism. Anaesth Crit Care Pain. 2007;7:148-151.
- Thomas JE, Ayyar DR. Systemic fat embolism. Arch Neurol. 1972;26:517-523.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56B:408-416.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br. 1987;69:128-131.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med. 1983;99:438-443.
- Robinson CM. Current concepts of respiratory insufficiency syndromes after fracture. J Bone Joint Surg Br. 2001;83:781-791.
- Kim YH, Oh SW, Kim JS. Prevalence of fat embolism following bilateral simultaneous and unilateral total hip arthroplasty performed with or without cement: a prospective, randomized clinical study. J Bone Joint Surg Am. 2002;84A:1372-1379.
- Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41 Suppl 2:S90-S93.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009;52:386-393.
- Bracco D, Favre JB, Joris F, Ravussin A. Fatal fat embolism syndrome: a case report. J Neurosurg Anesthesiol. 2000;12:221-224.
The Case
A 24-year-old white man with no past medical history is admitted after sustaining bilateral, closed femur fractures in a motor vehicle accident. Within hours of the trauma, he is taken to the operating room for open reduction and internal fixation. Of note, preoperatively, his hematocrit is 40%. After surgery, he is easily extubated and transferred to an unmonitored bed for further care. Approximately 30 hours after admission, he develops tachypnea with a respiratory rate of 35 breaths per minute and hypoxia with an oxygen saturation of 86% on room air. He is tachycardic (120 beats per minute) and febrile to 39.0oC. His blood pressure remains stable. He is somnolent, and when awake, he is confused. Notably, his hematocrit is now 22%. An electrocardiogram shows sinus tachycardia, an initial chest X-ray is normal, and a high-resolution CT scan is negative for a pulmonary embolism (PE).
Is this clinical picture consistent with fat embolism syndrome and, if so, how should he be managed?
Overview
“Fat embolism” refers to the presence of fat globules that obstruct the lung parenchyma and peripheral circulation. Fat embolism syndrome, on the other hand, is a more serious manifestation involving multiple organ systems. Specifically, it is a clinical diagnosis presenting with the classic triad of hypoxemia, neurologic abnormalities, and a petechial rash.
Fat embolism syndrome is usually associated with multiple traumas, including long-bone injuries and pelvic fractures. It is more frequently associated with closed fractures than open fractures, possibly due to the higher pressures associated with closed fractures. This syndrome has been less commonly associated with a variety of nontraumatic conditions (Table 1).
With an increased incidence of long-bone fractures in the younger demographic, fat embolism syndrome is most common in the second or third decade of life. While fat embolism occurs in up to 90% of patients with traumatic skeletal injuries, fat embolism syndrome occurs in 0.5% to 10% of patients following trauma, with a higher incidence in multiple fractures (5% to 10%) than in single long-bone fractures (0.5% to 2%).1-3
With the increasing role of hospitalists in assisting in the management of orthopedic patients, their knowledge of fat embolism syndrome is important so that it can be included in the differential diagnosis of acute respiratory failure in these orthopedic patients.
Review of the Data
Pathogenesis. Clinical manifestations of fat embolism syndrome have been acknowledged for more than 100 years. Since its first description in the 1860s, there has been speculation about the etiology of this condition. In the 1920s, two theories were proposed to explain the origin of the fat droplets: the mechanical and biochemical theories.2,4
Mechanical theory suggests that trauma to long bones disturbs fat cells within the bone marrow or adipose tissue, causing fat globules to mobilize.2,3 There is a rise in marrow pressure above venous pressure, which allows fat particles to enter the circulation through damaged venules surrounding the fracture site. Once lodged in the pulmonary microvasculature, embolized fat causes local ischemia and inflammation. Fat globules may pass into the arterial circulation either by paradoxical embolism through a patent foramen ovale, or by microemboli that pass through the lungs into the arterial circulation. This explains embolization to other organs, including the brain, retina, and skin.
Alternatively, biochemical theory hypothesizes that fat embolism syndrome is contingent on the production of toxic intermediaries from the breakdown of embolized fat.2,3 This theory suggests that the release of catecholamines after severe trauma can liberate free fatty acids from fat stores, or that acute-phase reactants at the trauma site affect fat solubility, causing agglutination and embolization. This theory helps to explain nontraumatic fat embolism syndrome, as well as the delay in development of the clinical syndrome after acute injury.
Clinical presentation. Most patients have a latent period after trauma of 12 to 72 hours before symptoms of fat embolism syndrome become apparent; however, clinical manifestations might occur immediately or up to one to two weeks following injury.2,4 As previously mentioned, the classic triad of symptoms includes respiratory compromise, neurological impairment, and a petechial rash.
The most common and usually earliest manifestation is acute hypoxia, which must be distinguished from other treatable causes of hypoxia, including pneumothorax, hemothorax, PE, and pneumonia. Pulmonary changes might progress to respiratory failure similar to acute respiratory distress syndrome. Neurological manifestations are primarily nonspecific and include headache, irritability, delirium, seizures, and coma. Focal neurological deficits are rare but have been described.5 Almost all neurological symptoms are fully reversible. The petechial rash is distinctive and occurs on the chest, axilla, and subconjunctiva. Although the rash occurs in only 20% to 50% of patients and resolves fairly quickly, in the appropriate clinical setting, this rash is considered pathognomonic.1,2,4
A variety of other nonspecific signs and symptoms might also occur: pyrexia, tachycardia, fat in the urine or sputum, retinal changes, renal insufficiency, myocardial dysfunction, and an otherwise unexplained drop in hematocrit or platelet count.
Diagnosis. Fat embolism syndrome is a clinical diagnosis and a diagnosis of exclusion. There are no specific confirmatory tests. An arterial blood gas will usually reveal a PaO2 of <60 mmHg.3 Laboratory evaluation might also show fat globules in the urine or sputum on Sudan or Oil Red O staining, but these findings are nonspecific.3,4 Bronchoscopy with bronchial alveolar lavage (BAL) might similarly detect fat droplets in alveolar macrophages in the BAL fluid; however, the sensitivity and specificity for diagnosis of fat embolism syndrome are unknown.4 None of these tests can be used solely for the diagnosis of fat embolism syndrome.
Thrombocytopenia and anemia out of proportion to the expected drop from surgery are not uncommon in addition to other nonspecific laboratory findings, including hypocalcemia, elevated serum lipase level, and elevated erythrocyte sedimentation rate.4 Several radiological findings have been observed on lung and brain imaging, though the findings are nonspecific and none are diagnostic. A chest X-ray might be normal, but abnormalities are seen in 30% to 50% of cases.2 Typically, when abnormal, the chest X-ray shows diffuse interstitial and alveolar densities, as well as patchy perihilar and basilar infiltrates resembling pulmonary edema. These X-ray findings might not be seen for up to 12 to 24 hours following the onset of clinical symptoms.
The most commonly used diagnostic criteria for the diagnosis of fat embolism syndrome are published by Gurd et al.6 At least two major criteria or one major criterion and four minor criteria are required for the diagnosis of fat embolism syndrome. The major criteria are based on the three classic signs and symptoms of fat embolism syndrome; the minor criteria include the finding of fat globules in the urine and sputum as well as some of the previously mentioned nonspecific clinical signs and laboratory tests.
Other criteria for diagnosis have been suggested, including those published by Lindeque et al, which focuses primarily on the respiratory characteristics, and a more recent set of semiquantitative diagnostic criteria called the fat embolism index, published by Schonfeld et al.7,8 Schonfeld’s scoring index accounts for the major signs and symptoms of fat embolism syndrome and weighs them according to relative specificity. A score of 5 or more is required for diagnosis of fat embolism syndrome. Table 2 compares the three sets of criteria used for diagnosis of fat embolism syndrome.
Treatment. The treatment of fat embolism syndrome is supportive. Most often, this requires supplemental oxygen for hypoxia and, possibly, fluid resuscitation in the case of hypovolemia. Occasionally, though, these relatively minor supportive therapies need to be escalated to bipap or even full ventilatory support and vasopressors in the more severe cases.
Based on the premise that steroids will attenuate the inflammatory reaction to free fatty acids within the lung, steroids have been tried in the treatment of fat embolism syndrome. However, there are no studies that clearly show benefit with their use.
Prevention. Most of the methods of prevention involve surgical intervention rather than medical therapy. Because microscopic fat emboli are showered during manipulation of long-bone fragments, early immobilization of fractures is recommended, and operative correction rather than conservative management is the preferred method.2,3 One report estimates a 70% reduction in pulmonary complications from this intervention alone.9
Further, two surgical techniques are debated as possible means of preventing fat embolism syndrome. The first is “venting,” in which a hole is made distal to the site of intramedullary nail placement. This reduces intramedullary pressure elevation and, therefore, extravasation of fat into the circulation.10 The second technique is the use of a reamer, irrigator, aspirator (RIA) device. A reamer is a tool used to create an accurate-sized hole for an intramedullary nail. Reaming before intramedullary nail placement can release fat deposits into the circulation. The RIA device irrigates and aspirates resident fat deposits as it reams the canal, releasing fewer deposits into the circulation.11 At this time, these two techniques are considered but not used routinely by surgeons.
Corticosteroids remain a debated method of prevention of fat embolism syndrome. A number of smaller studies suggest steroid therapy might reduce the incidence of fat embolism syndrome and hypoxia; a 2009 meta-analysis pooling nearly 400 patients from these smaller studies found such results.12 Unfortunately, the included studies were noted to be of poor quality, and no change in mortality was found. These results, combined with the possibility of poor wound healing or infection as a complication of steroid use, keep steroids from being used routinely to prevent fat embolism syndrome.
Clinical course. The severity of fat embolism syndrome ranges from mild transient hypoxia with confusion to progressively worsening symptoms leading to acute respiratory distress syndrome and coma. Bulger et al found a 7% mortality rate in this population.1 Less commonly, patients have a fulminant presentation with symptom onset less than 12 hours after injury. With this presentation, patients have a higher rate of mortality—as high as 15%.13
Back to the Case
This young man with bilateral long-bone fractures was at high risk of developing fat embolism syndrome. As is recommended, he was quickly taken to the operating room for fracture stabilization with open reduction and internal fixation. In addition, a RIA device was used to decrease intramedullary pressure. Nonetheless, within the first two days of injury, he developed hypoxia and confusion. These clinical changes were associated with an unexpected drop in hematocrit.
Chest X-ray and high-resolution computed tomography did not reveal a cause of his hypoxia. Similarly, laboratory evaluation for a reversible cause of encephalopathy was negative. A Sudan stain of his urine revealed free fat globules. Though he did not develop axillary petechiae, this clinical picture is consistent with fat embolism syndrome based on Gurd’s criteria. He was supported with oxygen therapy, and he stabilized without further complications.
Drs. Smith and Rice are members of the Section of Hospital Medicine at Vanderbilt University in Nashville, Tenn.
References
- Bulger EM, Smith DG, Maier RV, Jurkovich GJ. Fat embolism syndrome. A 10-year review. Arch Surg. 1997;132:435-439.
- Levy D. The fat embolism syndrome. Clin Orthop. 1990;261:281-286.
- Akhtar S. Fat embolism. Anes Clin. 2009;27:533-550.
- Gupta A, Reilly C. Fat embolism. Anaesth Crit Care Pain. 2007;7:148-151.
- Thomas JE, Ayyar DR. Systemic fat embolism. Arch Neurol. 1972;26:517-523.
- Gurd AR, Wilson RI. The fat embolism syndrome. J Bone Joint Surg Br. 1974;56B:408-416.
- Lindeque BG, Schoeman HS, Dommisse GF, Boeyens MC, Vlok AL. Fat embolism and the fat embolism syndrome. A double-blind therapeutic study. J Bone Joint Surg Br. 1987;69:128-131.
- Schonfeld SA, Ploysongsang Y, DiLisio R, et al. Fat embolism prophylaxis with corticosteroids. A prospective study in high-risk patients. Ann Intern Med. 1983;99:438-443.
- Robinson CM. Current concepts of respiratory insufficiency syndromes after fracture. J Bone Joint Surg Br. 2001;83:781-791.
- Kim YH, Oh SW, Kim JS. Prevalence of fat embolism following bilateral simultaneous and unilateral total hip arthroplasty performed with or without cement: a prospective, randomized clinical study. J Bone Joint Surg Am. 2002;84A:1372-1379.
- Volgas DA, Burch T, Stannard JP, Ellis T, Bilotta J, Alonso JE. Fat embolus in femur fractures: a comparison of two reaming systems. Injury. 2010;41 Suppl 2:S90-S93.
- Bederman SS, Bhandari M, McKee MD, Schemitsch EH. Do corticosteroids reduce the risk of fat embolism syndrome in patients with long-bone fractures? A meta-analysis. Can J Surg. 2009;52:386-393.
- Bracco D, Favre JB, Joris F, Ravussin A. Fatal fat embolism syndrome: a case report. J Neurosurg Anesthesiol. 2000;12:221-224.
What Is the Optimal Therapy for Acute DVT?
The Case
A 55-year-old female undergoes cholecystectomy. On post-operative Day 2, she develops right-lower-extremity swelling and pain; venous ultrasound detects a proximal deep venous thrombosis (DVT). The patient denies smoking or use of hormonal medications. She has no history of venous thromboembolism (VTE), although her brother had a DVT at age 60. The hospitalist team is consulted for management of acute DVT.

Overview
VTE, including lower- and upper-extremity DVT and pulmonary embolism (PE), is one of the most common and preventable hospital diseases. DVT with PE is associated with a 10% mortality rate, and DVT with post-thrombotic syndrome can be associated with significant morbidity, including pain, edema, skin/pigment change, venous dilation, and ulcer development.1,2 Recognition of clinical symptoms and risk factors for DVT (see Table 1) in conjunction with validated clinical scoring predictors (such as the Wells Prediction Rule) and a high-sensitivity D-dimer assay can help diagnose the condition and determine the need for ultrasound.3-7
Pharmacologic Treatment
Anticoagulation should be initiated in all patients with VTE, regardless of patient symptoms. Anticoagulant options include:
- Intravenous (IV) or subcutaneous (SC) unfractionated heparin (UFH);
- SC low-molecular-weight heparins (LMWH), such as enoxaparin and dalteparin; and
- Fondaparinux (as effective as LMWH for acute treatment of VTE).8
These agents can be used while transitioning to oral vitamin K antagonists (VKA), such as warfarin.3
The 2012 American College of Chest Physicians (ACCP) guidelines on antithrombotic therapy for VTE recommend initial therapy with LMWH or fondaparinux (rather than IV or SC UFH). The guidelines suggest that LMWH once-daily dosing is favored over twice-daily dosing, based mainly on patient convenience, although this is a weak recommendation (2C) based on the overall quality of the data. The recommendation applies only if the daily dosing of the LMWH, including tinzaparin, dalteparin, and nadroparin, is equivalent to the twice-daily dosing (i.e., dalteparin may be dosed at 100 units/kg BID vs. 200 units/kg daily). Of importance, enoxaparin has not been studied at a once-daily dose (2 mg/kg), which is equivalent to the twice-daily dosing regimen (1 mg/kg twice daily). Additionally, one study suggests that once-daily dosing of enoxaparin 1.5mg/kg might be inferior to 1 mg/kg twice-daily dosing; therefore, caution must be exercised in applying this recommendation to the LMWH enoxaparin at this time.3,27,28 (updated Aug. 28, 2012)
Warfarin should be started simultaneously at a usual daily dose of 5 mg for the first two days, with subsequent doses adjusted to achieve a goal international normalized ratio (INR) of 2.0 to 3.0. Parenteral agents should be given for a minimum of five days and until the INR has been >2.0 for at least 24 hours.3
The new factor-Xa inhibitor rivaroxaban and the direct thrombin inhibitor dabigatran are promising oral alternatives to warfarin.9-11 However, neither drug is currently FDA-approved for the treatment of VTE, nor are they recommended by current guidelines (given limited data for DVT treatment and concerns of bleeding risk).3,12,13 See Table 2 (above) for comparisons of common anticoagulants.3,14-17
Duration of anticoagulation. Anticoagulant treatment of acute DVT should continue for at least three months, as shorter durations are associated with higher recurrence rates. Longer treatment may be indicated depending on the patient’s risk of recurrence.3
The ACCP guidelines estimate risk of recurrence using primary, secondary, and additional factors (see Table 3, p. 19) and recommend the following durations:
- First episode provoked: three months (proximal or distal, provoked by surgery or a nonsurgical transient risk factor);
- First episode unprovoked distal: three months (see “Considerations for isolated distal DVT,” below);
- First episode unprovoked proximal: Indefinite if low to moderate bleeding risk, three months if high bleeding risk;
- Recurrent unprovoked: Indefinite if low to moderate bleeding risk, three months if high bleeding risk; and
- With active cancer: Indefinite with LMWH due to higher risk of recurrence.3,18
These treatment duration guidelines might need to be individualized based on other factors including patient preference, ability to obtain accurate INR monitoring (for those on warfarin), treatment cost, and comorbidities.3
Considerations for isolated distal DVT. Patients with an initial episode of distal DVT, without significant symptoms or risk factors for extension (e.g. positive D-dimer, extensive clot near proximal veins, absence of a reversible provoking factor, active cancer, inpatient status, or previous VTE) might not need anticoagulation.
The DVT can be followed with serial ultrasounds for the first two weeks; anticoagulation is recommended only if the thrombus extends during that time period. The development of significant symptoms or risk factors of extension might indicate the need for anticoagulation.3
Considerations for upper-extremity DVT (UEDVT). Anticoagulation for an UEDVT is generally consistent with the above guidelines for lower-extremity DVT, with a few caveats. If an UEDVT is associated with a central venous catheter (CVC), the CVC should be removed if possible; there are no recommendations to determine whether CVC removal should be preceded by a period of anticoagulation.
A catheter-associated UEDVT requires a minimum of three months of anticoagulation; if the CVC remains in place beyond three months, anticoagulation should be continued until the catheter is removed. Unprovoked UEDVT has a lower risk of recurrence than lower-extremity DVT and three months of anticoagulation, rather than indefinite therapy, is recommended.3
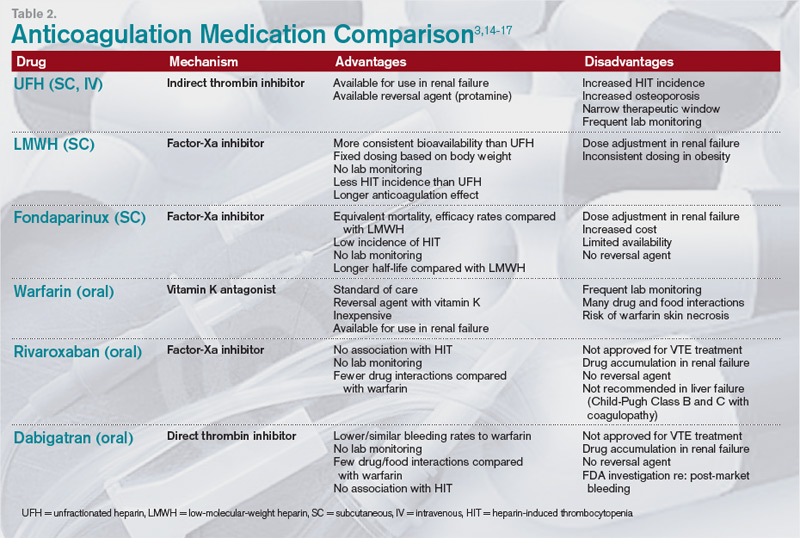
Mechanical Treatment
Non-pharmacologic therapies, such as knee-high graduated compression stockings with pressure of 30 mmHg to 40 mmHg at the ankle, can help reduce the morbidity of post-thrombotic syndrome (PTS) when combined with anticoagulation. Symptomatic patients who use compression stockings as soon as feasible and for a minimum of two years can reduce their incidence of PTS by 50%.3,19,20
Thigh-length stockings are not more effective than knee-high, and while multilayer compression bandages might relieve symptoms during the first-week post-DVT, they do not reduce the one-year incidence of PTS.21,22 Early mobilization is not associated with an increased risk of PE, extension of DVT, or death; patients should ambulate as soon as physically able.23,24
Pharmacomechanical Thrombolysis
For acute DVT, ACCP guidelines recommend anticoagulation alone over pharmacomechanical thrombolysis (either systemic or catheter-directed thrombolysis and mechanical thrombus fragmentation). The rare patient with impending venous gangrene despite anticoagulation is the only clinical scenario in which thrombolysis is clearly indicated. Patients who undergo pharmacomechanical thrombolysis still need a standard course of anticoagulation.3
Role for Inferior Vena Cava Filters
The optimal role of inferior vena cava (IVC) filters remains uncertain. Only one randomized trial found that IVC filters, in conjunction with systemic anticoagulation versus systemic anticoagulation alone, were associated with short-term reductions in the incidence of PE but long-term increases in recurrent DVT, with no differences in mortality or major bleeding. However, no trials have compared anticoagulation plus IVC filter placement with IVC filter placement alone.25,26
ACCP guidelines recommend IVC filter placement only in patients with acute, proximal DVT of the lower extremity, and a contraindication to anticoagulant therapy. If the contraindication resolves, a conventional course of anticoagulation can commence. Combining an IVC filter with an anticoagulant is not recommended. The risks and benefits of retrievable filters require further investigation.3
Back to the Case
Our patient has a provoked DVT secondary to a reversible risk factor (surgery) without additional clinical risk factors. Her family history of DVT is not significant (her brother was >age 50 when it occurred). This patient should be treated with LMWH or fondaparinux with initiation of warfarin with goal INR of 2.0 to 3.0 for at least three months. She does not need an IVC filter, and she should use compression stockings to reduce the risk of PTS.
Bottom Line
In hospitalized patients, treatment of DVT should include immediate anticoagulation with LMWH, fondaparinux, or IV heparin (in patients with renal failure) with transition to warfarin and a goal INR of 2.0 to 3.0. New oral anticoagulants could prove beneficial in acute treatment of DVT but require further testing. Duration of treatment is patient-specific, but most should be anticoagulated for at least three months; some warrant indefinite therapy based on risk factors.
Dr. Sebasky is an assistant professor and Dr. DeKorte is assistant professor of medicine in the division of hospital medicine at the University of California at San Diego.
References
- Agency for Healthcare Research and Quality. Talking Points to Attract Administration Support for Venous Thromboembolism Prevention Programs. U.S. Department of Health & Human Services website. Available at: http://www.ahrq.gov/qual/vtguide/vtguideapa.htm. Accessed Feb. 4, 2012.
- Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6:1105-1112.
- Kearon C, Akl E, Comerota AJ, et al. Antithrombotic Therapy for VTE Disease. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Hirsh J, Hull RD, Raskob GE. Clinical features and diagnosis of venous thrombosis. J Am Coll Cardiol. 1986;8(6 Suppl B):114B-127B.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Int Med. 2007;146:454-458.
- Tapson VF, Carroll BA, Davidson BL, et al. The diagnostic approach to acute venous thromboembolism. Clinical practice guideline. American Thoracic Society. Am J Respir Crit Care Med. 1999;160:1043-1066.
- Wells PS, Owen C, Doucette S, Fergusson D, Tran H. Does this patient have deep vein thrombosis? JAMA. 2006;295:199-207.
- Büller HR, Davidson BL, Decousus H, et al. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: a randomized trial. Ann Intern Med. 2004;140:867-873.
- EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499-25
- Garcia, D, Libby E, Crowther M. The new oral anticoagulants. Blood. 2010;115:15-20.
- Douketis JD. Pharmacologic properties of the new oral anticoagulants: a clinician-oriented review with a focus on perioperative management. Curr Pharm Des. 2010;16:3436-3441.
- U.S. Food and Drug Administration. Pradaxa (dabigatran etexilate mesylate): Drug Safety Communication—Safety Review of Post-Market Reports of Serious Bleeding Events. U.S. Food and Drug Administration website. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/
SafetyAlertsforHumanMedicalProducts/ucm282820.htm. Accessed March 12, 2012.
- Levi M, Erenberg E, Kamphuisen PW. Bleeding risk and reversal strategies for old and new anticoagulants and antiplatelet agents. J Thromb Haemost. 2011;9:1705.
- Erkens PM, Prins MH. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev. 2010;8(9);CD001100.Vardi M, Zittan E, Bitterman H. Subcutaneous unfractionated heparin for the initial treatment of venous thromboembolism. Cochrane Database Syst Rev. 2009;(4):CD006771.
- Hirsh J, Levine MN. Low molecular weight heparin. Blood. 1992;79:1-17.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361;2342-2352.
- Bauer KA. Long-term management of venous thromboembolism. JAMA. 2011;305:1336-1345.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the postthrombotic syndrome: a randomized, controlled trial. Ann Intern Med. 2004;141:249-256.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet. 1997;349:759-762.
- Prandoni P, Noventa F, Quintavalla R, et al. Thigh-length versus below-knee compression elastic stockings for prevention of the post-thrombotic syndrome in patients with proximal-venous thrombosis: a randomized trial. Blood. 2012;119:1561-1565.
- Roumen-Klappe EM, den Heijer M, van Rossum J, et al. Multilayer compression bandaging in the acute phase of deep-vein thrombosis has no effect on the development of the post-thrombotic syndrome. J Thromb Thrombolysis. 2009;27:400-405.
- Aissaoui N, Martins E, Mouly S, Weber S, Meune C. A meta-analysis of bed rest versus early ambulation in the management of pulmonary embolism, deep venous thrombosis, or both. Int J Cardiol. 2009;137:37-41.
- Anderson CM, Overend TJ, Godwin J, Sealy C, Sunderji A. Ambulation after deep vein thrombosis: a systematic review. Physiother Can. 2009;61:133-140.
- Hann CL, Streiff MB. The role of vena caval filters in the management of venous thromboembolism. Blood Rev. 2005;19:179-202.
- Decousus H, Leizorovicz A, Page Y, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338:409-415.
The Case
A 55-year-old female undergoes cholecystectomy. On post-operative Day 2, she develops right-lower-extremity swelling and pain; venous ultrasound detects a proximal deep venous thrombosis (DVT). The patient denies smoking or use of hormonal medications. She has no history of venous thromboembolism (VTE), although her brother had a DVT at age 60. The hospitalist team is consulted for management of acute DVT.
Overview
VTE, including lower- and upper-extremity DVT and pulmonary embolism (PE), is one of the most common and preventable hospital diseases. DVT with PE is associated with a 10% mortality rate, and DVT with post-thrombotic syndrome can be associated with significant morbidity, including pain, edema, skin/pigment change, venous dilation, and ulcer development.1,2 Recognition of clinical symptoms and risk factors for DVT (see Table 1) in conjunction with validated clinical scoring predictors (such as the Wells Prediction Rule) and a high-sensitivity D-dimer assay can help diagnose the condition and determine the need for ultrasound.3-7
Pharmacologic Treatment
Anticoagulation should be initiated in all patients with VTE, regardless of patient symptoms. Anticoagulant options include:
- Intravenous (IV) or subcutaneous (SC) unfractionated heparin (UFH);
- SC low-molecular-weight heparins (LMWH), such as enoxaparin and dalteparin; and
- Fondaparinux (as effective as LMWH for acute treatment of VTE).8
These agents can be used while transitioning to oral vitamin K antagonists (VKA), such as warfarin.3
The 2012 American College of Chest Physicians (ACCP) guidelines on antithrombotic therapy for VTE recommend initial therapy with LMWH or fondaparinux (rather than IV or SC UFH). The guidelines suggest that LMWH once-daily dosing is favored over twice-daily dosing, based mainly on patient convenience, although this is a weak recommendation (2C) based on the overall quality of the data. The recommendation applies only if the daily dosing of the LMWH, including tinzaparin, dalteparin, and nadroparin, is equivalent to the twice-daily dosing (i.e., dalteparin may be dosed at 100 units/kg BID vs. 200 units/kg daily). Of importance, enoxaparin has not been studied at a once-daily dose (2 mg/kg), which is equivalent to the twice-daily dosing regimen (1 mg/kg twice daily). Additionally, one study suggests that once-daily dosing of enoxaparin 1.5mg/kg might be inferior to 1 mg/kg twice-daily dosing; therefore, caution must be exercised in applying this recommendation to the LMWH enoxaparin at this time.3,27,28 (updated Aug. 28, 2012)
Warfarin should be started simultaneously at a usual daily dose of 5 mg for the first two days, with subsequent doses adjusted to achieve a goal international normalized ratio (INR) of 2.0 to 3.0. Parenteral agents should be given for a minimum of five days and until the INR has been >2.0 for at least 24 hours.3
The new factor-Xa inhibitor rivaroxaban and the direct thrombin inhibitor dabigatran are promising oral alternatives to warfarin.9-11 However, neither drug is currently FDA-approved for the treatment of VTE, nor are they recommended by current guidelines (given limited data for DVT treatment and concerns of bleeding risk).3,12,13 See Table 2 (above) for comparisons of common anticoagulants.3,14-17
Duration of anticoagulation. Anticoagulant treatment of acute DVT should continue for at least three months, as shorter durations are associated with higher recurrence rates. Longer treatment may be indicated depending on the patient’s risk of recurrence.3
The ACCP guidelines estimate risk of recurrence using primary, secondary, and additional factors (see Table 3, p. 19) and recommend the following durations:
- First episode provoked: three months (proximal or distal, provoked by surgery or a nonsurgical transient risk factor);
- First episode unprovoked distal: three months (see “Considerations for isolated distal DVT,” below);
- First episode unprovoked proximal: Indefinite if low to moderate bleeding risk, three months if high bleeding risk;
- Recurrent unprovoked: Indefinite if low to moderate bleeding risk, three months if high bleeding risk; and
- With active cancer: Indefinite with LMWH due to higher risk of recurrence.3,18
These treatment duration guidelines might need to be individualized based on other factors including patient preference, ability to obtain accurate INR monitoring (for those on warfarin), treatment cost, and comorbidities.3
Considerations for isolated distal DVT. Patients with an initial episode of distal DVT, without significant symptoms or risk factors for extension (e.g. positive D-dimer, extensive clot near proximal veins, absence of a reversible provoking factor, active cancer, inpatient status, or previous VTE) might not need anticoagulation.
The DVT can be followed with serial ultrasounds for the first two weeks; anticoagulation is recommended only if the thrombus extends during that time period. The development of significant symptoms or risk factors of extension might indicate the need for anticoagulation.3
Considerations for upper-extremity DVT (UEDVT). Anticoagulation for an UEDVT is generally consistent with the above guidelines for lower-extremity DVT, with a few caveats. If an UEDVT is associated with a central venous catheter (CVC), the CVC should be removed if possible; there are no recommendations to determine whether CVC removal should be preceded by a period of anticoagulation.
A catheter-associated UEDVT requires a minimum of three months of anticoagulation; if the CVC remains in place beyond three months, anticoagulation should be continued until the catheter is removed. Unprovoked UEDVT has a lower risk of recurrence than lower-extremity DVT and three months of anticoagulation, rather than indefinite therapy, is recommended.3
Mechanical Treatment
Non-pharmacologic therapies, such as knee-high graduated compression stockings with pressure of 30 mmHg to 40 mmHg at the ankle, can help reduce the morbidity of post-thrombotic syndrome (PTS) when combined with anticoagulation. Symptomatic patients who use compression stockings as soon as feasible and for a minimum of two years can reduce their incidence of PTS by 50%.3,19,20
Thigh-length stockings are not more effective than knee-high, and while multilayer compression bandages might relieve symptoms during the first-week post-DVT, they do not reduce the one-year incidence of PTS.21,22 Early mobilization is not associated with an increased risk of PE, extension of DVT, or death; patients should ambulate as soon as physically able.23,24
Pharmacomechanical Thrombolysis
For acute DVT, ACCP guidelines recommend anticoagulation alone over pharmacomechanical thrombolysis (either systemic or catheter-directed thrombolysis and mechanical thrombus fragmentation). The rare patient with impending venous gangrene despite anticoagulation is the only clinical scenario in which thrombolysis is clearly indicated. Patients who undergo pharmacomechanical thrombolysis still need a standard course of anticoagulation.3
Role for Inferior Vena Cava Filters
The optimal role of inferior vena cava (IVC) filters remains uncertain. Only one randomized trial found that IVC filters, in conjunction with systemic anticoagulation versus systemic anticoagulation alone, were associated with short-term reductions in the incidence of PE but long-term increases in recurrent DVT, with no differences in mortality or major bleeding. However, no trials have compared anticoagulation plus IVC filter placement with IVC filter placement alone.25,26
ACCP guidelines recommend IVC filter placement only in patients with acute, proximal DVT of the lower extremity, and a contraindication to anticoagulant therapy. If the contraindication resolves, a conventional course of anticoagulation can commence. Combining an IVC filter with an anticoagulant is not recommended. The risks and benefits of retrievable filters require further investigation.3
Back to the Case
Our patient has a provoked DVT secondary to a reversible risk factor (surgery) without additional clinical risk factors. Her family history of DVT is not significant (her brother was >age 50 when it occurred). This patient should be treated with LMWH or fondaparinux with initiation of warfarin with goal INR of 2.0 to 3.0 for at least three months. She does not need an IVC filter, and she should use compression stockings to reduce the risk of PTS.
Bottom Line
In hospitalized patients, treatment of DVT should include immediate anticoagulation with LMWH, fondaparinux, or IV heparin (in patients with renal failure) with transition to warfarin and a goal INR of 2.0 to 3.0. New oral anticoagulants could prove beneficial in acute treatment of DVT but require further testing. Duration of treatment is patient-specific, but most should be anticoagulated for at least three months; some warrant indefinite therapy based on risk factors.
Dr. Sebasky is an assistant professor and Dr. DeKorte is assistant professor of medicine in the division of hospital medicine at the University of California at San Diego.
References
- Agency for Healthcare Research and Quality. Talking Points to Attract Administration Support for Venous Thromboembolism Prevention Programs. U.S. Department of Health & Human Services website. Available at: http://www.ahrq.gov/qual/vtguide/vtguideapa.htm. Accessed Feb. 4, 2012.
- Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6:1105-1112.
- Kearon C, Akl E, Comerota AJ, et al. Antithrombotic Therapy for VTE Disease. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Hirsh J, Hull RD, Raskob GE. Clinical features and diagnosis of venous thrombosis. J Am Coll Cardiol. 1986;8(6 Suppl B):114B-127B.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Int Med. 2007;146:454-458.
- Tapson VF, Carroll BA, Davidson BL, et al. The diagnostic approach to acute venous thromboembolism. Clinical practice guideline. American Thoracic Society. Am J Respir Crit Care Med. 1999;160:1043-1066.
- Wells PS, Owen C, Doucette S, Fergusson D, Tran H. Does this patient have deep vein thrombosis? JAMA. 2006;295:199-207.
- Büller HR, Davidson BL, Decousus H, et al. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: a randomized trial. Ann Intern Med. 2004;140:867-873.
- EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499-25
- Garcia, D, Libby E, Crowther M. The new oral anticoagulants. Blood. 2010;115:15-20.
- Douketis JD. Pharmacologic properties of the new oral anticoagulants: a clinician-oriented review with a focus on perioperative management. Curr Pharm Des. 2010;16:3436-3441.
- U.S. Food and Drug Administration. Pradaxa (dabigatran etexilate mesylate): Drug Safety Communication—Safety Review of Post-Market Reports of Serious Bleeding Events. U.S. Food and Drug Administration website. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/
SafetyAlertsforHumanMedicalProducts/ucm282820.htm. Accessed March 12, 2012.
- Levi M, Erenberg E, Kamphuisen PW. Bleeding risk and reversal strategies for old and new anticoagulants and antiplatelet agents. J Thromb Haemost. 2011;9:1705.
- Erkens PM, Prins MH. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev. 2010;8(9);CD001100.Vardi M, Zittan E, Bitterman H. Subcutaneous unfractionated heparin for the initial treatment of venous thromboembolism. Cochrane Database Syst Rev. 2009;(4):CD006771.
- Hirsh J, Levine MN. Low molecular weight heparin. Blood. 1992;79:1-17.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361;2342-2352.
- Bauer KA. Long-term management of venous thromboembolism. JAMA. 2011;305:1336-1345.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the postthrombotic syndrome: a randomized, controlled trial. Ann Intern Med. 2004;141:249-256.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet. 1997;349:759-762.
- Prandoni P, Noventa F, Quintavalla R, et al. Thigh-length versus below-knee compression elastic stockings for prevention of the post-thrombotic syndrome in patients with proximal-venous thrombosis: a randomized trial. Blood. 2012;119:1561-1565.
- Roumen-Klappe EM, den Heijer M, van Rossum J, et al. Multilayer compression bandaging in the acute phase of deep-vein thrombosis has no effect on the development of the post-thrombotic syndrome. J Thromb Thrombolysis. 2009;27:400-405.
- Aissaoui N, Martins E, Mouly S, Weber S, Meune C. A meta-analysis of bed rest versus early ambulation in the management of pulmonary embolism, deep venous thrombosis, or both. Int J Cardiol. 2009;137:37-41.
- Anderson CM, Overend TJ, Godwin J, Sealy C, Sunderji A. Ambulation after deep vein thrombosis: a systematic review. Physiother Can. 2009;61:133-140.
- Hann CL, Streiff MB. The role of vena caval filters in the management of venous thromboembolism. Blood Rev. 2005;19:179-202.
- Decousus H, Leizorovicz A, Page Y, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338:409-415.
The Case
A 55-year-old female undergoes cholecystectomy. On post-operative Day 2, she develops right-lower-extremity swelling and pain; venous ultrasound detects a proximal deep venous thrombosis (DVT). The patient denies smoking or use of hormonal medications. She has no history of venous thromboembolism (VTE), although her brother had a DVT at age 60. The hospitalist team is consulted for management of acute DVT.
Overview
VTE, including lower- and upper-extremity DVT and pulmonary embolism (PE), is one of the most common and preventable hospital diseases. DVT with PE is associated with a 10% mortality rate, and DVT with post-thrombotic syndrome can be associated with significant morbidity, including pain, edema, skin/pigment change, venous dilation, and ulcer development.1,2 Recognition of clinical symptoms and risk factors for DVT (see Table 1) in conjunction with validated clinical scoring predictors (such as the Wells Prediction Rule) and a high-sensitivity D-dimer assay can help diagnose the condition and determine the need for ultrasound.3-7
Pharmacologic Treatment
Anticoagulation should be initiated in all patients with VTE, regardless of patient symptoms. Anticoagulant options include:
- Intravenous (IV) or subcutaneous (SC) unfractionated heparin (UFH);
- SC low-molecular-weight heparins (LMWH), such as enoxaparin and dalteparin; and
- Fondaparinux (as effective as LMWH for acute treatment of VTE).8
These agents can be used while transitioning to oral vitamin K antagonists (VKA), such as warfarin.3
The 2012 American College of Chest Physicians (ACCP) guidelines on antithrombotic therapy for VTE recommend initial therapy with LMWH or fondaparinux (rather than IV or SC UFH). The guidelines suggest that LMWH once-daily dosing is favored over twice-daily dosing, based mainly on patient convenience, although this is a weak recommendation (2C) based on the overall quality of the data. The recommendation applies only if the daily dosing of the LMWH, including tinzaparin, dalteparin, and nadroparin, is equivalent to the twice-daily dosing (i.e., dalteparin may be dosed at 100 units/kg BID vs. 200 units/kg daily). Of importance, enoxaparin has not been studied at a once-daily dose (2 mg/kg), which is equivalent to the twice-daily dosing regimen (1 mg/kg twice daily). Additionally, one study suggests that once-daily dosing of enoxaparin 1.5mg/kg might be inferior to 1 mg/kg twice-daily dosing; therefore, caution must be exercised in applying this recommendation to the LMWH enoxaparin at this time.3,27,28 (updated Aug. 28, 2012)
Warfarin should be started simultaneously at a usual daily dose of 5 mg for the first two days, with subsequent doses adjusted to achieve a goal international normalized ratio (INR) of 2.0 to 3.0. Parenteral agents should be given for a minimum of five days and until the INR has been >2.0 for at least 24 hours.3
The new factor-Xa inhibitor rivaroxaban and the direct thrombin inhibitor dabigatran are promising oral alternatives to warfarin.9-11 However, neither drug is currently FDA-approved for the treatment of VTE, nor are they recommended by current guidelines (given limited data for DVT treatment and concerns of bleeding risk).3,12,13 See Table 2 (above) for comparisons of common anticoagulants.3,14-17
Duration of anticoagulation. Anticoagulant treatment of acute DVT should continue for at least three months, as shorter durations are associated with higher recurrence rates. Longer treatment may be indicated depending on the patient’s risk of recurrence.3
The ACCP guidelines estimate risk of recurrence using primary, secondary, and additional factors (see Table 3, p. 19) and recommend the following durations:
- First episode provoked: three months (proximal or distal, provoked by surgery or a nonsurgical transient risk factor);
- First episode unprovoked distal: three months (see “Considerations for isolated distal DVT,” below);
- First episode unprovoked proximal: Indefinite if low to moderate bleeding risk, three months if high bleeding risk;
- Recurrent unprovoked: Indefinite if low to moderate bleeding risk, three months if high bleeding risk; and
- With active cancer: Indefinite with LMWH due to higher risk of recurrence.3,18
These treatment duration guidelines might need to be individualized based on other factors including patient preference, ability to obtain accurate INR monitoring (for those on warfarin), treatment cost, and comorbidities.3
Considerations for isolated distal DVT. Patients with an initial episode of distal DVT, without significant symptoms or risk factors for extension (e.g. positive D-dimer, extensive clot near proximal veins, absence of a reversible provoking factor, active cancer, inpatient status, or previous VTE) might not need anticoagulation.
The DVT can be followed with serial ultrasounds for the first two weeks; anticoagulation is recommended only if the thrombus extends during that time period. The development of significant symptoms or risk factors of extension might indicate the need for anticoagulation.3
Considerations for upper-extremity DVT (UEDVT). Anticoagulation for an UEDVT is generally consistent with the above guidelines for lower-extremity DVT, with a few caveats. If an UEDVT is associated with a central venous catheter (CVC), the CVC should be removed if possible; there are no recommendations to determine whether CVC removal should be preceded by a period of anticoagulation.
A catheter-associated UEDVT requires a minimum of three months of anticoagulation; if the CVC remains in place beyond three months, anticoagulation should be continued until the catheter is removed. Unprovoked UEDVT has a lower risk of recurrence than lower-extremity DVT and three months of anticoagulation, rather than indefinite therapy, is recommended.3
Mechanical Treatment
Non-pharmacologic therapies, such as knee-high graduated compression stockings with pressure of 30 mmHg to 40 mmHg at the ankle, can help reduce the morbidity of post-thrombotic syndrome (PTS) when combined with anticoagulation. Symptomatic patients who use compression stockings as soon as feasible and for a minimum of two years can reduce their incidence of PTS by 50%.3,19,20
Thigh-length stockings are not more effective than knee-high, and while multilayer compression bandages might relieve symptoms during the first-week post-DVT, they do not reduce the one-year incidence of PTS.21,22 Early mobilization is not associated with an increased risk of PE, extension of DVT, or death; patients should ambulate as soon as physically able.23,24
Pharmacomechanical Thrombolysis
For acute DVT, ACCP guidelines recommend anticoagulation alone over pharmacomechanical thrombolysis (either systemic or catheter-directed thrombolysis and mechanical thrombus fragmentation). The rare patient with impending venous gangrene despite anticoagulation is the only clinical scenario in which thrombolysis is clearly indicated. Patients who undergo pharmacomechanical thrombolysis still need a standard course of anticoagulation.3
Role for Inferior Vena Cava Filters
The optimal role of inferior vena cava (IVC) filters remains uncertain. Only one randomized trial found that IVC filters, in conjunction with systemic anticoagulation versus systemic anticoagulation alone, were associated with short-term reductions in the incidence of PE but long-term increases in recurrent DVT, with no differences in mortality or major bleeding. However, no trials have compared anticoagulation plus IVC filter placement with IVC filter placement alone.25,26
ACCP guidelines recommend IVC filter placement only in patients with acute, proximal DVT of the lower extremity, and a contraindication to anticoagulant therapy. If the contraindication resolves, a conventional course of anticoagulation can commence. Combining an IVC filter with an anticoagulant is not recommended. The risks and benefits of retrievable filters require further investigation.3
Back to the Case
Our patient has a provoked DVT secondary to a reversible risk factor (surgery) without additional clinical risk factors. Her family history of DVT is not significant (her brother was >age 50 when it occurred). This patient should be treated with LMWH or fondaparinux with initiation of warfarin with goal INR of 2.0 to 3.0 for at least three months. She does not need an IVC filter, and she should use compression stockings to reduce the risk of PTS.
Bottom Line
In hospitalized patients, treatment of DVT should include immediate anticoagulation with LMWH, fondaparinux, or IV heparin (in patients with renal failure) with transition to warfarin and a goal INR of 2.0 to 3.0. New oral anticoagulants could prove beneficial in acute treatment of DVT but require further testing. Duration of treatment is patient-specific, but most should be anticoagulated for at least three months; some warrant indefinite therapy based on risk factors.
Dr. Sebasky is an assistant professor and Dr. DeKorte is assistant professor of medicine in the division of hospital medicine at the University of California at San Diego.
References
- Agency for Healthcare Research and Quality. Talking Points to Attract Administration Support for Venous Thromboembolism Prevention Programs. U.S. Department of Health & Human Services website. Available at: http://www.ahrq.gov/qual/vtguide/vtguideapa.htm. Accessed Feb. 4, 2012.
- Kahn SR, Shbaklo H, Lamping DL, et al. Determinants of health-related quality of life during the 2 years following deep vein thrombosis. J Thromb Haemost. 2008;6:1105-1112.
- Kearon C, Akl E, Comerota AJ, et al. Antithrombotic Therapy for VTE Disease. Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e419S-e494S.
- Hirsh J, Hull RD, Raskob GE. Clinical features and diagnosis of venous thrombosis. J Am Coll Cardiol. 1986;8(6 Suppl B):114B-127B.
- Qaseem A, Snow V, Barry P, et al. Current diagnosis of venous thromboembolism in primary care: a clinical practice guideline from the American Academy of Family Physicians and the American College of Physicians. Ann Int Med. 2007;146:454-458.
- Tapson VF, Carroll BA, Davidson BL, et al. The diagnostic approach to acute venous thromboembolism. Clinical practice guideline. American Thoracic Society. Am J Respir Crit Care Med. 1999;160:1043-1066.
- Wells PS, Owen C, Doucette S, Fergusson D, Tran H. Does this patient have deep vein thrombosis? JAMA. 2006;295:199-207.
- Büller HR, Davidson BL, Decousus H, et al. Fondaparinux or enoxaparin for the initial treatment of symptomatic deep venous thrombosis: a randomized trial. Ann Intern Med. 2004;140:867-873.
- EINSTEIN Investigators, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499-25
- Garcia, D, Libby E, Crowther M. The new oral anticoagulants. Blood. 2010;115:15-20.
- Douketis JD. Pharmacologic properties of the new oral anticoagulants: a clinician-oriented review with a focus on perioperative management. Curr Pharm Des. 2010;16:3436-3441.
- U.S. Food and Drug Administration. Pradaxa (dabigatran etexilate mesylate): Drug Safety Communication—Safety Review of Post-Market Reports of Serious Bleeding Events. U.S. Food and Drug Administration website. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/
SafetyAlertsforHumanMedicalProducts/ucm282820.htm. Accessed March 12, 2012.
- Levi M, Erenberg E, Kamphuisen PW. Bleeding risk and reversal strategies for old and new anticoagulants and antiplatelet agents. J Thromb Haemost. 2011;9:1705.
- Erkens PM, Prins MH. Fixed dose subcutaneous low molecular weight heparins versus adjusted dose unfractionated heparin for venous thromboembolism. Cochrane Database Syst Rev. 2010;8(9);CD001100.Vardi M, Zittan E, Bitterman H. Subcutaneous unfractionated heparin for the initial treatment of venous thromboembolism. Cochrane Database Syst Rev. 2009;(4):CD006771.
- Hirsh J, Levine MN. Low molecular weight heparin. Blood. 1992;79:1-17.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361;2342-2352.
- Bauer KA. Long-term management of venous thromboembolism. JAMA. 2011;305:1336-1345.
- Prandoni P, Lensing AW, Prins MH, et al. Below-knee elastic compression stockings to prevent the postthrombotic syndrome: a randomized, controlled trial. Ann Intern Med. 2004;141:249-256.
- Brandjes DP, Büller HR, Heijboer H, et al. Randomised trial of effect of compression stockings in patients with symptomatic proximal-vein thrombosis. Lancet. 1997;349:759-762.
- Prandoni P, Noventa F, Quintavalla R, et al. Thigh-length versus below-knee compression elastic stockings for prevention of the post-thrombotic syndrome in patients with proximal-venous thrombosis: a randomized trial. Blood. 2012;119:1561-1565.
- Roumen-Klappe EM, den Heijer M, van Rossum J, et al. Multilayer compression bandaging in the acute phase of deep-vein thrombosis has no effect on the development of the post-thrombotic syndrome. J Thromb Thrombolysis. 2009;27:400-405.
- Aissaoui N, Martins E, Mouly S, Weber S, Meune C. A meta-analysis of bed rest versus early ambulation in the management of pulmonary embolism, deep venous thrombosis, or both. Int J Cardiol. 2009;137:37-41.
- Anderson CM, Overend TJ, Godwin J, Sealy C, Sunderji A. Ambulation after deep vein thrombosis: a systematic review. Physiother Can. 2009;61:133-140.
- Hann CL, Streiff MB. The role of vena caval filters in the management of venous thromboembolism. Blood Rev. 2005;19:179-202.
- Decousus H, Leizorovicz A, Page Y, et al. A clinical trial of vena caval filters in the prevention of pulmonary embolism in patients with proximal deep-vein thrombosis. N Engl J Med. 1998;338:409-415.
Collaboration Prevents Identification Band Errors

Clinical question: Can a quality-improvement (QI) collaborative decrease patient identification (ID) band errors?
Background: ID band errors often result in medication errors and unsafe care. Consequently, correct patient identification, through the use of at least two identifiers, has been an ongoing Joint Commission National Patient Safety Goal. Although individual sites have demonstrated improvement in accuracy of patient identification, there have not been reports of dissemination of successful practices.
Study design: Collaborative quality-improvement initiative.
Setting: Six hospitals.
Synopsis: ID band audits in 11,377 patients were performed in the learning collaborative’s six participating hospitals.
The audits were organized primarily around monthly conference calls. The hospital settings were diverse: community hospitals, hospitals within an academic medical center, and freestanding children’s hospitals. The aim of the collaborative was to reduce ID band errors by 50% within a one-year time frame across the collective sites.
Key interventions included transparent data collection and reporting; engagement of staff, families and leadership; voluntary event reporting; and auditing of failures. The mean combined ID band failure rate decreased to 4% from 22% within 13 months, representing a 77% relative reduction (P<0.001).
QI collaboratives are not designed to specifically result in generalizable knowledge, yet they might produce widespread improvement, as this effort demonstrates. The careful documentation of iterative factors implemented across sites in this initiative provides a blueprint for hospitals looking to replicate this success. Additionally, the interventions represent feasible and logical concepts within the basic constructs of improvement science methodology.
Bottom line: A QI collaborative might result in rapid and significant reductions in ID band errors.
Citation: Phillips SC, Saysana M, Worley S, Hain PD. Reduction in pediatric identification band errors: a quality collaborative. Pediatrics. 2012;129(6):e1587-e1593.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Can a quality-improvement (QI) collaborative decrease patient identification (ID) band errors?
Background: ID band errors often result in medication errors and unsafe care. Consequently, correct patient identification, through the use of at least two identifiers, has been an ongoing Joint Commission National Patient Safety Goal. Although individual sites have demonstrated improvement in accuracy of patient identification, there have not been reports of dissemination of successful practices.
Study design: Collaborative quality-improvement initiative.
Setting: Six hospitals.
Synopsis: ID band audits in 11,377 patients were performed in the learning collaborative’s six participating hospitals.
The audits were organized primarily around monthly conference calls. The hospital settings were diverse: community hospitals, hospitals within an academic medical center, and freestanding children’s hospitals. The aim of the collaborative was to reduce ID band errors by 50% within a one-year time frame across the collective sites.
Key interventions included transparent data collection and reporting; engagement of staff, families and leadership; voluntary event reporting; and auditing of failures. The mean combined ID band failure rate decreased to 4% from 22% within 13 months, representing a 77% relative reduction (P<0.001).
QI collaboratives are not designed to specifically result in generalizable knowledge, yet they might produce widespread improvement, as this effort demonstrates. The careful documentation of iterative factors implemented across sites in this initiative provides a blueprint for hospitals looking to replicate this success. Additionally, the interventions represent feasible and logical concepts within the basic constructs of improvement science methodology.
Bottom line: A QI collaborative might result in rapid and significant reductions in ID band errors.
Citation: Phillips SC, Saysana M, Worley S, Hain PD. Reduction in pediatric identification band errors: a quality collaborative. Pediatrics. 2012;129(6):e1587-e1593.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Can a quality-improvement (QI) collaborative decrease patient identification (ID) band errors?
Background: ID band errors often result in medication errors and unsafe care. Consequently, correct patient identification, through the use of at least two identifiers, has been an ongoing Joint Commission National Patient Safety Goal. Although individual sites have demonstrated improvement in accuracy of patient identification, there have not been reports of dissemination of successful practices.
Study design: Collaborative quality-improvement initiative.
Setting: Six hospitals.
Synopsis: ID band audits in 11,377 patients were performed in the learning collaborative’s six participating hospitals.
The audits were organized primarily around monthly conference calls. The hospital settings were diverse: community hospitals, hospitals within an academic medical center, and freestanding children’s hospitals. The aim of the collaborative was to reduce ID band errors by 50% within a one-year time frame across the collective sites.
Key interventions included transparent data collection and reporting; engagement of staff, families and leadership; voluntary event reporting; and auditing of failures. The mean combined ID band failure rate decreased to 4% from 22% within 13 months, representing a 77% relative reduction (P<0.001).
QI collaboratives are not designed to specifically result in generalizable knowledge, yet they might produce widespread improvement, as this effort demonstrates. The careful documentation of iterative factors implemented across sites in this initiative provides a blueprint for hospitals looking to replicate this success. Additionally, the interventions represent feasible and logical concepts within the basic constructs of improvement science methodology.
Bottom line: A QI collaborative might result in rapid and significant reductions in ID band errors.
Citation: Phillips SC, Saysana M, Worley S, Hain PD. Reduction in pediatric identification band errors: a quality collaborative. Pediatrics. 2012;129(6):e1587-e1593.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Is Hospitalist Proficiency in Bedside Procedures in Decline?
It’s 3:30 p.m. You’ve seen your old patients, holdovers, and an admission, but you haven’t finished your notes yet. Lunch was an afterthought between emails about schedule changes for the upcoming year. Two pages ring happily from your belt, the first from you-know-who in the ED, and the next from a nurse: “THORA SUPPLIES AT BEDSIDE SINCE THIS AM—WHEN WILL THIS HAPPEN?” The phone number on the wall for the on-call radiologist beckons...
An all-too-familiar situation for hospitalists across the country, this awkward moment raises a series of difficult questions:
Should I set aside time from my day to perform a procedure that could be time-consuming?
- Do I feel confident I can perform this procedure safely?
- Am I really the best physician to provide this service?
- As hospitalists are tasked with an ever-increasing array of responsibilities, answering the call of duty for bedside procedures is becoming more difficult for some.
A Core Competency
“The Core Competencies in Hospital Medicine,” authored by a group of HM thought leaders, was published as a supplement to the January/February 2006 issue of the Journal of Hospital Medicine. The core competencies include such bedside procedures as arthrocentesis, paracentesis, thoracentesis, lumbar puncture, and vascular (arterial and central venous) access (see “Core Competencies in Hospital Medicine: Procedures,” below). Although the authors stressed that the core competencies are to be viewed as a resource rather than as a set of requirements, the inclusion of bedside procedures emphasized the importance of procedural skills for future hospitalists.
“[Hospitalists] are in a perfect spot to continue to perform procedures in a structured manner,” says Joshua Lenchus, DO, RPh, FACP, FHM, associate director of the University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety. “As agents of quality and safety, hospitalists should continue to perform this clinically necessary service.”
Jeffrey Barsuk, MD, FHM, associate professor of medicine at Northwestern Feinberg School of Medicine in Chicago and an academic hospitalist at Northwestern Memorial Hospital (NMH), not only agrees that bedside procedures should be a core competency, but he also says hospitalists are the most appropriate providers of these services.
“I think this is part of hospital medicine. We’re in the hospital, [and] that’s what we do,” Dr. Barsuk says. Other providers, such as interventional radiologists, “really don’t understand why I’m doing [a procedure]. They understand it’s safe to do it, but they might not understand all the indications for it, and they certainly don’t understand the interpretation of the tests they’re sending.”
Despite the goals set forth by the core competencies and authorities in procedural safety, the reality of who actually performs bedside procedures is somewhat murky and varies greatly by institution. Many point to HM program setting (urban vs. rural) or structure (academic vs. community) to explain variance, but often it is other factors that determine whether hospitalists are actually preforming bedside procedures regularly.

Where Does HM Perform Procedures?
Community hospitalists, with strong support from interventional radiologists and subspecialists, often find it more efficient—even necessary, considering their patient volumes—to leave procedures to others. Community hospitalists with ICU admitting privileges, intensivists, and other HM subgroups say that being able to perform procedures should be a prerequisite for employment. Hospitalists in rural communities say they are doing procedures because they are “the only game in town.”
“Sometimes you are the only one available, and you are called upon to stretch your abilities,” says Beatrice Szantyr, MD, FAAP, a community hospitalist and pediatrician in Lincoln, Maine, who has practiced most of her career in rural settings.
Academic hospitalists in large, research-based HM programs can, paradoxically, find themselves performing fewer procedures as residents often take the lead on the majority of such cases. Conversely, academic hospitalists in large, nonteaching programs often find themselves called on to perform more bedside procedures.

No matter the setting, the simplicity of being the physician to recognize the need for a procedure, perform it, and interpret the results is undeniably efficient and “clean,” according to authorities on inpatient bedside procedures. Having to consult other physicians, optimize the patient’s lab values to their standards (a common issue with interventional radiologists), and adhere to their work schedules can often delay procedures unnecessarily.
—Joshua Lenchus, DO, RPh, FACP, FHM, associate director, University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety
“Hospitalists care for floor and ICU patients in many hospitals, and the inability to perform bedside procedures delays patient care,” says Dr. Nilam Soni, an academic hospitalist at the University of Chicago and a recognized expert on procedural safety.
Dr. Soni notes that when it comes to current techniques, many hospitalists suffer from a knowledge deficit. “The introduction of ultrasound for guidance of bedside procedures has been shown to improve the success and safety of certain procedures,” he says, “but the majority of practicing hospitalists did not learn how to use ultrasound for procedure guidance during residency.”
Heterogeneity of Training, Experience, and Skill
While all hospitalists draw upon different bases of training and experience, the heterogeneity of training, confidence, and inherent skill is greatest when it comes to bedside procedures. Mirroring the heterogeneity at the individual level, hospitalist programs vary greatly on the requirements placed on their staffs in regards to procedural skill and privileging.
Such research-driven programs as Brigham and Women’s Hospital (BWH) in Boston often find requiring maintenance of privileges in bedside procedures to be difficult, says Sally Wang, MD, FHM, director of procedural education at BWH. In fact, a new procedure service being created there will be staffed mainly with ED physicians. On the flipside, most community hospitalist programs leave the task of procedural “policing” to the hospital’s medical staff affairs office.
At the University of California at San Diego (UCSD) Medical Center, the HM group is instituting a division standard in which hospitalists maintain privileging and proficiency in a core group of bedside procedures. Other large hospitalist groups have created “proceduralist” subgroups that shoulder the burden of trainee education, as well as provide a resource for less skilled or less experienced inpatient providers.

“If you have a big group, you could have a dedicated procedure service and have a core group of hospitalists who are experts in procedure,” Dr. Barsuk says. “But it needs to be self-sustaining.” Once started, Dr. Barsuk says, proceduralist groups would continue to provide hospitals with ongoing return-on-investment (ROI) benefit.
Variability in procedure volume and payor mix, however, can make it hard for HM groups to demonstrate to hospital leadership a satisfactory ROI for a proceduralist program. Financial backing from grant support or a high-volume procedure—such as paracentesis in hospitals with large hepatology programs—can nurture starting proceduralist programs until all procedural revenues can justify the costs. Lower ROI can also be justified by showing improvement in quality indices—such as CLABSI rates—reduced time to procedures, and reduced costs compared to other subspecialists offering similar services.
“I’m of the firm belief that we can reduce costs by doing the procedures at the bedside rather than referring them to departments such as interventional radiology (IR),” Dr. Barsuk says. “What you would have to do is show the institution that it costs more money to have IR do [bedside procedures].”
National Response

—Jeffrey Barsuk, MD, MS, FHM, associate professor of medicine, Northwestern Feinberg School of Medicine, academic hospitalist, Northwestern Memorial Hospital, Chicago
Filling in the procedural training gaps found on the local level, such national organizations as SHM have stepped in to provide education and support for hospitalists yearning for training. Since its inception, an SHM annual meeting pre-course that focuses on hand-held ultrasound and invasive procedures has consistently been one of the first to sell out. Other national organizations, such as ACP and its annual meeting, have seen similar interest in their courses on ultrasound-guided procedures.
The popularity of this continuing education bears out a worrisome trend: Hospitalists feel they are losing their procedural skills. An online survey conducted by The Hospitalist in May 2011 found that a majority of respondents (62%) had experienced deterioration of their procedural skills in the past five years; only 25% said they experienced improvement over the same period.
Historically, general internists have claimed bedside procedures as their domain. As stated dispassionately in the 1978 book The House of God, “There is no body cavity that cannot be reached with a #14G needle and a good strong arm.”1 Yet much has changed since Samuel Shem’s apocryphal description of medical residency training.
Most notably, the Accreditation Council of Graduate Medical Education (ACGME) has not only progressively restricted inpatient hours and patient loads for residents, but also increased the requirements for outpatient training. Some feel the balance of inpatient and outpatient training has tipped too far toward the latter in medicine residency programs, especially in light of the growing popularity of the hospitalist career path amongst new residency program graduates. This stands in contrast to ED training programs, which have embraced focused procedures training more readily.
“Adult care appears to be diverging into two career tacks as a result of external forces, of which we have limited control over, “ says Michael Beck, MD, a pediatric and adult hospitalist at Milton S. Hershey Medical Center in Hershey, Pa. “With new career choices emerging for graduates, the same square-peg, round-hole residency training should not exist.”

Dr. Beck advocates continuing an ongoing trend of “track” creation in residency programs, which allow trainees to focus training on their planned career path. Hospitalist tracks already exist in many medicine programs, including those at Cleveland Clinic and Northwestern. But many other factors limit the opportunity for trainees to obtain experience with bedside procedures, including competition with nurse practitioners and physician assistants. Even the increasing availability of ancillary phlebotomy and IV-start teams can increase a resident’s anxiety about procedures.
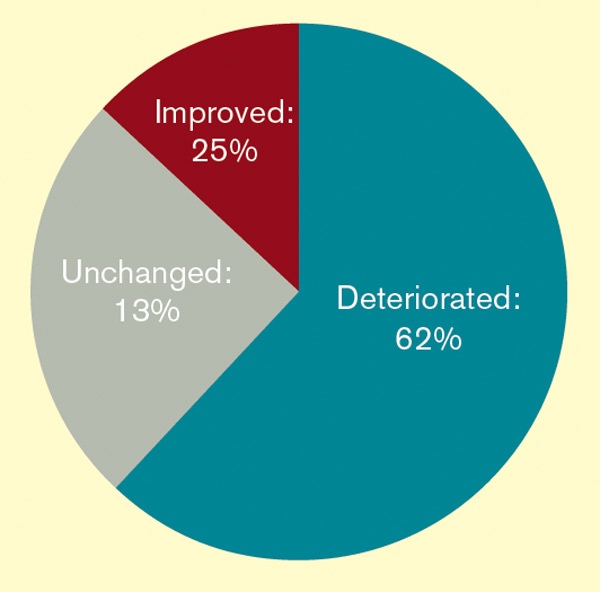

“By the time my residency was over [in 1993] and the work restrictions were beginning, hospital employees were doing all these tasks, making the residents less comfortable with hurting a patient when it was therapeutically necessary,” says Katharine Deiss, MD, assistant clinical professor of medicine at the University of Rochester Medical Center in New York. Interns who came from medical schools without extensive ancillary services in their teaching hospitals, she adds, were more comfortable with invasive procedures.

ACGME has sent a subtle message by decreasing emphasis on procedural skills by eradicating the requirement of showing manual proficiency in most bedside procedures as a requirement for certification. The omission has left individual residency programs and hospitalist groups to determine training and proficiency requirements for more invasive bedside procedures without a national standard.
In an editorial in the March 2007 issue of the Annals of Internal Medicine, F. Daniel Duffy, MD, and Eric Holmboe, MD, wrote that the American Board of Internal Medicine (ABIM) could only give a “qualified ‘yes’” to the question of whether residents should be trained in procedures they may not perform in practice. Although the authors asserted that the relaxed ABIM policy was “an important but small step toward revamping procedure skill training during residency,” others say it portrays an image of the ABIM de-emphasizing the importance of procedural training.
In addition, the recently established Focused Practice in Hospital Medicine (FPHM) pathway to ABIM Maintenance of Certification (MOC) has no requirement to show proficiency in bedside procedures.
“The absence of the procedural requirement in no way constitutes a statement that procedural skills are not important,” says Jeff Wiese, MD, FACP, SFHM, associate professor of medicine and residency program director at Tulane University Health Sciences Center in New Orleans, chair of the ABIM Hospital Medicine MOC Question Writing Committee, and former SHM president. “Rather, it is merely a practical issue with respect to making the MOC process applicable to all physicians engaged in hospital medicine (i.e. many hospitalists do not do procedures) while still making the MOC focused on the skill sets that are common for physicians doing hospital medicine.”
Once released into the world, even if trained well in residency, hospitalists can find it difficult to maintain their skills. In community and nonteaching settings, the pressure to admit and discharge in a timely manner can make procedures seem like the easiest corner to cut. Before long, it has been months since they have laid eyes on a needle of any sort. Many begin to develop performance anxiety.
In teaching hospitals, academic hospitalists often are called upon to participate in quality improvement (QI) and research efforts, which take time away from clinical rotations. Once there, it can be easy for a ward attending to rely upon a well-trained resident to supervise interns doing procedures. The lack of first-hand or even supervisory experience can lead to many academic hospitalists losing facility with procedures, with potentially disastrous results.
“In order to supervise a group of residents, the attending needs to be technically proficient and able to salvage a botched, or failed, procedure,” UM-JMH’s Dr. Lenchus says. “To this end, we strictly limit who can attend on the service.”
So what’s a residency or HM program director to do in the face of wavering support nationally, and sometimes locally, for maintaining procedural skills for hospitalists and trainees? Many hospitalists in teaching hospitals say it’s critical for clinicians to “get their own house in order,” to maintain procedural standards of proficiency with ongoing training, education, and verification.
“The profession now needs to redesign procedural training across the continuum of education and a lifetime of practice,” Drs. Duffy and Holmboe editorialized in the March 2007 Annals paper. “This approach would recognize the varied settings of internal-medicine practice and offer manual skills training to those whose practice settings require such skills.” Hospitalists can partner with medicine residency program leaders to provide procedural education and training to residents, either as a standalone elective or as a more general resource.
Hospitalists in such teaching hospitals as UCSD, Brigham and Women’s, UM-JMH, and Northwestern are leading efforts to provide procedural education to medical students, residents, and attendings. Training takes many forms, including formal procedural electives, required procedure rotations, or even brief one- or two-day courses in procedural skills at a simulation center.
Utilizing simulation training has been shown in many studies to be helpful in establishing procedural skills in learners of all training levels. Dr. Barsuk and his colleagues at Northwestern published studies in the Journal of Hospital Medicine in 2008 and 2009 showing that simulation training of residents was effective in improving skills in thoracentesis and central venous catheterization, respectively.3,4
In the community hospital setting, requirements for procedural skills can vary greatly based on the institution. For those community programs requiring procedural skills of their hospitalists, the clear definition of procedural training and requirements at the time of hiring is critical. Even after vetting a hospitalist’s procedural skills at hire, however, community programs should consider monitoring procedural skills and provide ongoing time and money for CME focused on procedural skills.
Currently, most hospitals depend on the honesty of individual physicians during the privileging process for bedside procedures. Even when the skills of physicians begin to wane, most are reluctant to voluntarily give up their procedure privileges.
“I think it would be pretty unusual for a hospitalist to relinquish their privileges,” Dr. Barsuk admits. But ideally, physicians who relinquish their privileges due to lack of experience could get retrained in simulation centers, then reproctored in order to regain their privileges. Northwestern established the Center for Simulation Technology and Immersive Learning as a resource for simulation training both locally and nationally.
Establishing an environment that supports hospitalists performing bedside procedures is critical. This includes the need to limit hospitalist workload to ensure adequate time to meet the procedural needs of patients. Providing easy access to the tools necessary to perform bedside procedures (e.g. portable ultrasound and pre-packaged procedure trays) helps avoid additional hurdles.
Academic hospitalist programs can serve as a regional resource by developing ongoing procedure mastery programs for hospitalists in their communities, as many smaller institutions do not have the resources to provide ongoing training in bedside procedures. This process can be tedious, but it should not be humiliating.
If the popularity of the SHM pre-course in bedside ultrasound and procedures is any indication, when given the opportunity to receive protected time for procedure training, most hospitalists will likely jump at the chance.
Dr. Chang is an associate clinical professor of medicine in the division of hospital medicine at Diego Medical Center. He is also a member of Team Hospitalist.
References
- Shem S. The House of God. New York: Dell Publishing; 1978.
- Duffy FD, Holmboe ES. What procedures should internists do? Ann Intern Med. 2007;146(5):392-393.
- Wayne DB, Barsuk JH, O’Leary KJ, Fudala MJ, McGaghie WC. Mastery learning of thoracentesis skills by internal medicine residents using simulation technology and deliberate practice. J Hosp Med. 2008;3(1):48-54.
- Barsuk JH, McGaghie WC, Cohen ER, Balachandran JS, Wayne DB. Use of simulation-based mastery learning to improve the quality of central venous catheter placement in a medical intensive care unit. J Hosp Med. 2009;4(7):397–403.
It’s 3:30 p.m. You’ve seen your old patients, holdovers, and an admission, but you haven’t finished your notes yet. Lunch was an afterthought between emails about schedule changes for the upcoming year. Two pages ring happily from your belt, the first from you-know-who in the ED, and the next from a nurse: “THORA SUPPLIES AT BEDSIDE SINCE THIS AM—WHEN WILL THIS HAPPEN?” The phone number on the wall for the on-call radiologist beckons...
An all-too-familiar situation for hospitalists across the country, this awkward moment raises a series of difficult questions:
Should I set aside time from my day to perform a procedure that could be time-consuming?
- Do I feel confident I can perform this procedure safely?
- Am I really the best physician to provide this service?
- As hospitalists are tasked with an ever-increasing array of responsibilities, answering the call of duty for bedside procedures is becoming more difficult for some.
A Core Competency
“The Core Competencies in Hospital Medicine,” authored by a group of HM thought leaders, was published as a supplement to the January/February 2006 issue of the Journal of Hospital Medicine. The core competencies include such bedside procedures as arthrocentesis, paracentesis, thoracentesis, lumbar puncture, and vascular (arterial and central venous) access (see “Core Competencies in Hospital Medicine: Procedures,” below). Although the authors stressed that the core competencies are to be viewed as a resource rather than as a set of requirements, the inclusion of bedside procedures emphasized the importance of procedural skills for future hospitalists.
“[Hospitalists] are in a perfect spot to continue to perform procedures in a structured manner,” says Joshua Lenchus, DO, RPh, FACP, FHM, associate director of the University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety. “As agents of quality and safety, hospitalists should continue to perform this clinically necessary service.”
Jeffrey Barsuk, MD, FHM, associate professor of medicine at Northwestern Feinberg School of Medicine in Chicago and an academic hospitalist at Northwestern Memorial Hospital (NMH), not only agrees that bedside procedures should be a core competency, but he also says hospitalists are the most appropriate providers of these services.
“I think this is part of hospital medicine. We’re in the hospital, [and] that’s what we do,” Dr. Barsuk says. Other providers, such as interventional radiologists, “really don’t understand why I’m doing [a procedure]. They understand it’s safe to do it, but they might not understand all the indications for it, and they certainly don’t understand the interpretation of the tests they’re sending.”
Despite the goals set forth by the core competencies and authorities in procedural safety, the reality of who actually performs bedside procedures is somewhat murky and varies greatly by institution. Many point to HM program setting (urban vs. rural) or structure (academic vs. community) to explain variance, but often it is other factors that determine whether hospitalists are actually preforming bedside procedures regularly.
Where Does HM Perform Procedures?
Community hospitalists, with strong support from interventional radiologists and subspecialists, often find it more efficient—even necessary, considering their patient volumes—to leave procedures to others. Community hospitalists with ICU admitting privileges, intensivists, and other HM subgroups say that being able to perform procedures should be a prerequisite for employment. Hospitalists in rural communities say they are doing procedures because they are “the only game in town.”
“Sometimes you are the only one available, and you are called upon to stretch your abilities,” says Beatrice Szantyr, MD, FAAP, a community hospitalist and pediatrician in Lincoln, Maine, who has practiced most of her career in rural settings.
Academic hospitalists in large, research-based HM programs can, paradoxically, find themselves performing fewer procedures as residents often take the lead on the majority of such cases. Conversely, academic hospitalists in large, nonteaching programs often find themselves called on to perform more bedside procedures.
No matter the setting, the simplicity of being the physician to recognize the need for a procedure, perform it, and interpret the results is undeniably efficient and “clean,” according to authorities on inpatient bedside procedures. Having to consult other physicians, optimize the patient’s lab values to their standards (a common issue with interventional radiologists), and adhere to their work schedules can often delay procedures unnecessarily.
—Joshua Lenchus, DO, RPh, FACP, FHM, associate director, University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety
“Hospitalists care for floor and ICU patients in many hospitals, and the inability to perform bedside procedures delays patient care,” says Dr. Nilam Soni, an academic hospitalist at the University of Chicago and a recognized expert on procedural safety.
Dr. Soni notes that when it comes to current techniques, many hospitalists suffer from a knowledge deficit. “The introduction of ultrasound for guidance of bedside procedures has been shown to improve the success and safety of certain procedures,” he says, “but the majority of practicing hospitalists did not learn how to use ultrasound for procedure guidance during residency.”
Heterogeneity of Training, Experience, and Skill
While all hospitalists draw upon different bases of training and experience, the heterogeneity of training, confidence, and inherent skill is greatest when it comes to bedside procedures. Mirroring the heterogeneity at the individual level, hospitalist programs vary greatly on the requirements placed on their staffs in regards to procedural skill and privileging.
Such research-driven programs as Brigham and Women’s Hospital (BWH) in Boston often find requiring maintenance of privileges in bedside procedures to be difficult, says Sally Wang, MD, FHM, director of procedural education at BWH. In fact, a new procedure service being created there will be staffed mainly with ED physicians. On the flipside, most community hospitalist programs leave the task of procedural “policing” to the hospital’s medical staff affairs office.
At the University of California at San Diego (UCSD) Medical Center, the HM group is instituting a division standard in which hospitalists maintain privileging and proficiency in a core group of bedside procedures. Other large hospitalist groups have created “proceduralist” subgroups that shoulder the burden of trainee education, as well as provide a resource for less skilled or less experienced inpatient providers.
“If you have a big group, you could have a dedicated procedure service and have a core group of hospitalists who are experts in procedure,” Dr. Barsuk says. “But it needs to be self-sustaining.” Once started, Dr. Barsuk says, proceduralist groups would continue to provide hospitals with ongoing return-on-investment (ROI) benefit.
Variability in procedure volume and payor mix, however, can make it hard for HM groups to demonstrate to hospital leadership a satisfactory ROI for a proceduralist program. Financial backing from grant support or a high-volume procedure—such as paracentesis in hospitals with large hepatology programs—can nurture starting proceduralist programs until all procedural revenues can justify the costs. Lower ROI can also be justified by showing improvement in quality indices—such as CLABSI rates—reduced time to procedures, and reduced costs compared to other subspecialists offering similar services.
“I’m of the firm belief that we can reduce costs by doing the procedures at the bedside rather than referring them to departments such as interventional radiology (IR),” Dr. Barsuk says. “What you would have to do is show the institution that it costs more money to have IR do [bedside procedures].”
National Response
—Jeffrey Barsuk, MD, MS, FHM, associate professor of medicine, Northwestern Feinberg School of Medicine, academic hospitalist, Northwestern Memorial Hospital, Chicago
Filling in the procedural training gaps found on the local level, such national organizations as SHM have stepped in to provide education and support for hospitalists yearning for training. Since its inception, an SHM annual meeting pre-course that focuses on hand-held ultrasound and invasive procedures has consistently been one of the first to sell out. Other national organizations, such as ACP and its annual meeting, have seen similar interest in their courses on ultrasound-guided procedures.
The popularity of this continuing education bears out a worrisome trend: Hospitalists feel they are losing their procedural skills. An online survey conducted by The Hospitalist in May 2011 found that a majority of respondents (62%) had experienced deterioration of their procedural skills in the past five years; only 25% said they experienced improvement over the same period.
Historically, general internists have claimed bedside procedures as their domain. As stated dispassionately in the 1978 book The House of God, “There is no body cavity that cannot be reached with a #14G needle and a good strong arm.”1 Yet much has changed since Samuel Shem’s apocryphal description of medical residency training.
Most notably, the Accreditation Council of Graduate Medical Education (ACGME) has not only progressively restricted inpatient hours and patient loads for residents, but also increased the requirements for outpatient training. Some feel the balance of inpatient and outpatient training has tipped too far toward the latter in medicine residency programs, especially in light of the growing popularity of the hospitalist career path amongst new residency program graduates. This stands in contrast to ED training programs, which have embraced focused procedures training more readily.
“Adult care appears to be diverging into two career tacks as a result of external forces, of which we have limited control over, “ says Michael Beck, MD, a pediatric and adult hospitalist at Milton S. Hershey Medical Center in Hershey, Pa. “With new career choices emerging for graduates, the same square-peg, round-hole residency training should not exist.”
Dr. Beck advocates continuing an ongoing trend of “track” creation in residency programs, which allow trainees to focus training on their planned career path. Hospitalist tracks already exist in many medicine programs, including those at Cleveland Clinic and Northwestern. But many other factors limit the opportunity for trainees to obtain experience with bedside procedures, including competition with nurse practitioners and physician assistants. Even the increasing availability of ancillary phlebotomy and IV-start teams can increase a resident’s anxiety about procedures.
“By the time my residency was over [in 1993] and the work restrictions were beginning, hospital employees were doing all these tasks, making the residents less comfortable with hurting a patient when it was therapeutically necessary,” says Katharine Deiss, MD, assistant clinical professor of medicine at the University of Rochester Medical Center in New York. Interns who came from medical schools without extensive ancillary services in their teaching hospitals, she adds, were more comfortable with invasive procedures.
ACGME has sent a subtle message by decreasing emphasis on procedural skills by eradicating the requirement of showing manual proficiency in most bedside procedures as a requirement for certification. The omission has left individual residency programs and hospitalist groups to determine training and proficiency requirements for more invasive bedside procedures without a national standard.
In an editorial in the March 2007 issue of the Annals of Internal Medicine, F. Daniel Duffy, MD, and Eric Holmboe, MD, wrote that the American Board of Internal Medicine (ABIM) could only give a “qualified ‘yes’” to the question of whether residents should be trained in procedures they may not perform in practice. Although the authors asserted that the relaxed ABIM policy was “an important but small step toward revamping procedure skill training during residency,” others say it portrays an image of the ABIM de-emphasizing the importance of procedural training.
In addition, the recently established Focused Practice in Hospital Medicine (FPHM) pathway to ABIM Maintenance of Certification (MOC) has no requirement to show proficiency in bedside procedures.
“The absence of the procedural requirement in no way constitutes a statement that procedural skills are not important,” says Jeff Wiese, MD, FACP, SFHM, associate professor of medicine and residency program director at Tulane University Health Sciences Center in New Orleans, chair of the ABIM Hospital Medicine MOC Question Writing Committee, and former SHM president. “Rather, it is merely a practical issue with respect to making the MOC process applicable to all physicians engaged in hospital medicine (i.e. many hospitalists do not do procedures) while still making the MOC focused on the skill sets that are common for physicians doing hospital medicine.”
Once released into the world, even if trained well in residency, hospitalists can find it difficult to maintain their skills. In community and nonteaching settings, the pressure to admit and discharge in a timely manner can make procedures seem like the easiest corner to cut. Before long, it has been months since they have laid eyes on a needle of any sort. Many begin to develop performance anxiety.
In teaching hospitals, academic hospitalists often are called upon to participate in quality improvement (QI) and research efforts, which take time away from clinical rotations. Once there, it can be easy for a ward attending to rely upon a well-trained resident to supervise interns doing procedures. The lack of first-hand or even supervisory experience can lead to many academic hospitalists losing facility with procedures, with potentially disastrous results.
“In order to supervise a group of residents, the attending needs to be technically proficient and able to salvage a botched, or failed, procedure,” UM-JMH’s Dr. Lenchus says. “To this end, we strictly limit who can attend on the service.”
So what’s a residency or HM program director to do in the face of wavering support nationally, and sometimes locally, for maintaining procedural skills for hospitalists and trainees? Many hospitalists in teaching hospitals say it’s critical for clinicians to “get their own house in order,” to maintain procedural standards of proficiency with ongoing training, education, and verification.
“The profession now needs to redesign procedural training across the continuum of education and a lifetime of practice,” Drs. Duffy and Holmboe editorialized in the March 2007 Annals paper. “This approach would recognize the varied settings of internal-medicine practice and offer manual skills training to those whose practice settings require such skills.” Hospitalists can partner with medicine residency program leaders to provide procedural education and training to residents, either as a standalone elective or as a more general resource.
Hospitalists in such teaching hospitals as UCSD, Brigham and Women’s, UM-JMH, and Northwestern are leading efforts to provide procedural education to medical students, residents, and attendings. Training takes many forms, including formal procedural electives, required procedure rotations, or even brief one- or two-day courses in procedural skills at a simulation center.
Utilizing simulation training has been shown in many studies to be helpful in establishing procedural skills in learners of all training levels. Dr. Barsuk and his colleagues at Northwestern published studies in the Journal of Hospital Medicine in 2008 and 2009 showing that simulation training of residents was effective in improving skills in thoracentesis and central venous catheterization, respectively.3,4
In the community hospital setting, requirements for procedural skills can vary greatly based on the institution. For those community programs requiring procedural skills of their hospitalists, the clear definition of procedural training and requirements at the time of hiring is critical. Even after vetting a hospitalist’s procedural skills at hire, however, community programs should consider monitoring procedural skills and provide ongoing time and money for CME focused on procedural skills.
Currently, most hospitals depend on the honesty of individual physicians during the privileging process for bedside procedures. Even when the skills of physicians begin to wane, most are reluctant to voluntarily give up their procedure privileges.
“I think it would be pretty unusual for a hospitalist to relinquish their privileges,” Dr. Barsuk admits. But ideally, physicians who relinquish their privileges due to lack of experience could get retrained in simulation centers, then reproctored in order to regain their privileges. Northwestern established the Center for Simulation Technology and Immersive Learning as a resource for simulation training both locally and nationally.
Establishing an environment that supports hospitalists performing bedside procedures is critical. This includes the need to limit hospitalist workload to ensure adequate time to meet the procedural needs of patients. Providing easy access to the tools necessary to perform bedside procedures (e.g. portable ultrasound and pre-packaged procedure trays) helps avoid additional hurdles.
Academic hospitalist programs can serve as a regional resource by developing ongoing procedure mastery programs for hospitalists in their communities, as many smaller institutions do not have the resources to provide ongoing training in bedside procedures. This process can be tedious, but it should not be humiliating.
If the popularity of the SHM pre-course in bedside ultrasound and procedures is any indication, when given the opportunity to receive protected time for procedure training, most hospitalists will likely jump at the chance.
Dr. Chang is an associate clinical professor of medicine in the division of hospital medicine at Diego Medical Center. He is also a member of Team Hospitalist.
References
- Shem S. The House of God. New York: Dell Publishing; 1978.
- Duffy FD, Holmboe ES. What procedures should internists do? Ann Intern Med. 2007;146(5):392-393.
- Wayne DB, Barsuk JH, O’Leary KJ, Fudala MJ, McGaghie WC. Mastery learning of thoracentesis skills by internal medicine residents using simulation technology and deliberate practice. J Hosp Med. 2008;3(1):48-54.
- Barsuk JH, McGaghie WC, Cohen ER, Balachandran JS, Wayne DB. Use of simulation-based mastery learning to improve the quality of central venous catheter placement in a medical intensive care unit. J Hosp Med. 2009;4(7):397–403.
It’s 3:30 p.m. You’ve seen your old patients, holdovers, and an admission, but you haven’t finished your notes yet. Lunch was an afterthought between emails about schedule changes for the upcoming year. Two pages ring happily from your belt, the first from you-know-who in the ED, and the next from a nurse: “THORA SUPPLIES AT BEDSIDE SINCE THIS AM—WHEN WILL THIS HAPPEN?” The phone number on the wall for the on-call radiologist beckons...
An all-too-familiar situation for hospitalists across the country, this awkward moment raises a series of difficult questions:
Should I set aside time from my day to perform a procedure that could be time-consuming?
- Do I feel confident I can perform this procedure safely?
- Am I really the best physician to provide this service?
- As hospitalists are tasked with an ever-increasing array of responsibilities, answering the call of duty for bedside procedures is becoming more difficult for some.
A Core Competency
“The Core Competencies in Hospital Medicine,” authored by a group of HM thought leaders, was published as a supplement to the January/February 2006 issue of the Journal of Hospital Medicine. The core competencies include such bedside procedures as arthrocentesis, paracentesis, thoracentesis, lumbar puncture, and vascular (arterial and central venous) access (see “Core Competencies in Hospital Medicine: Procedures,” below). Although the authors stressed that the core competencies are to be viewed as a resource rather than as a set of requirements, the inclusion of bedside procedures emphasized the importance of procedural skills for future hospitalists.
“[Hospitalists] are in a perfect spot to continue to perform procedures in a structured manner,” says Joshua Lenchus, DO, RPh, FACP, FHM, associate director of the University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety. “As agents of quality and safety, hospitalists should continue to perform this clinically necessary service.”
Jeffrey Barsuk, MD, FHM, associate professor of medicine at Northwestern Feinberg School of Medicine in Chicago and an academic hospitalist at Northwestern Memorial Hospital (NMH), not only agrees that bedside procedures should be a core competency, but he also says hospitalists are the most appropriate providers of these services.
“I think this is part of hospital medicine. We’re in the hospital, [and] that’s what we do,” Dr. Barsuk says. Other providers, such as interventional radiologists, “really don’t understand why I’m doing [a procedure]. They understand it’s safe to do it, but they might not understand all the indications for it, and they certainly don’t understand the interpretation of the tests they’re sending.”
Despite the goals set forth by the core competencies and authorities in procedural safety, the reality of who actually performs bedside procedures is somewhat murky and varies greatly by institution. Many point to HM program setting (urban vs. rural) or structure (academic vs. community) to explain variance, but often it is other factors that determine whether hospitalists are actually preforming bedside procedures regularly.
Where Does HM Perform Procedures?
Community hospitalists, with strong support from interventional radiologists and subspecialists, often find it more efficient—even necessary, considering their patient volumes—to leave procedures to others. Community hospitalists with ICU admitting privileges, intensivists, and other HM subgroups say that being able to perform procedures should be a prerequisite for employment. Hospitalists in rural communities say they are doing procedures because they are “the only game in town.”
“Sometimes you are the only one available, and you are called upon to stretch your abilities,” says Beatrice Szantyr, MD, FAAP, a community hospitalist and pediatrician in Lincoln, Maine, who has practiced most of her career in rural settings.
Academic hospitalists in large, research-based HM programs can, paradoxically, find themselves performing fewer procedures as residents often take the lead on the majority of such cases. Conversely, academic hospitalists in large, nonteaching programs often find themselves called on to perform more bedside procedures.
No matter the setting, the simplicity of being the physician to recognize the need for a procedure, perform it, and interpret the results is undeniably efficient and “clean,” according to authorities on inpatient bedside procedures. Having to consult other physicians, optimize the patient’s lab values to their standards (a common issue with interventional radiologists), and adhere to their work schedules can often delay procedures unnecessarily.
—Joshua Lenchus, DO, RPh, FACP, FHM, associate director, University of Miami-Jackson Memorial Hospital (UM-JMH) Center for Patient Safety
“Hospitalists care for floor and ICU patients in many hospitals, and the inability to perform bedside procedures delays patient care,” says Dr. Nilam Soni, an academic hospitalist at the University of Chicago and a recognized expert on procedural safety.
Dr. Soni notes that when it comes to current techniques, many hospitalists suffer from a knowledge deficit. “The introduction of ultrasound for guidance of bedside procedures has been shown to improve the success and safety of certain procedures,” he says, “but the majority of practicing hospitalists did not learn how to use ultrasound for procedure guidance during residency.”
Heterogeneity of Training, Experience, and Skill
While all hospitalists draw upon different bases of training and experience, the heterogeneity of training, confidence, and inherent skill is greatest when it comes to bedside procedures. Mirroring the heterogeneity at the individual level, hospitalist programs vary greatly on the requirements placed on their staffs in regards to procedural skill and privileging.
Such research-driven programs as Brigham and Women’s Hospital (BWH) in Boston often find requiring maintenance of privileges in bedside procedures to be difficult, says Sally Wang, MD, FHM, director of procedural education at BWH. In fact, a new procedure service being created there will be staffed mainly with ED physicians. On the flipside, most community hospitalist programs leave the task of procedural “policing” to the hospital’s medical staff affairs office.
At the University of California at San Diego (UCSD) Medical Center, the HM group is instituting a division standard in which hospitalists maintain privileging and proficiency in a core group of bedside procedures. Other large hospitalist groups have created “proceduralist” subgroups that shoulder the burden of trainee education, as well as provide a resource for less skilled or less experienced inpatient providers.
“If you have a big group, you could have a dedicated procedure service and have a core group of hospitalists who are experts in procedure,” Dr. Barsuk says. “But it needs to be self-sustaining.” Once started, Dr. Barsuk says, proceduralist groups would continue to provide hospitals with ongoing return-on-investment (ROI) benefit.
Variability in procedure volume and payor mix, however, can make it hard for HM groups to demonstrate to hospital leadership a satisfactory ROI for a proceduralist program. Financial backing from grant support or a high-volume procedure—such as paracentesis in hospitals with large hepatology programs—can nurture starting proceduralist programs until all procedural revenues can justify the costs. Lower ROI can also be justified by showing improvement in quality indices—such as CLABSI rates—reduced time to procedures, and reduced costs compared to other subspecialists offering similar services.
“I’m of the firm belief that we can reduce costs by doing the procedures at the bedside rather than referring them to departments such as interventional radiology (IR),” Dr. Barsuk says. “What you would have to do is show the institution that it costs more money to have IR do [bedside procedures].”
National Response
—Jeffrey Barsuk, MD, MS, FHM, associate professor of medicine, Northwestern Feinberg School of Medicine, academic hospitalist, Northwestern Memorial Hospital, Chicago
Filling in the procedural training gaps found on the local level, such national organizations as SHM have stepped in to provide education and support for hospitalists yearning for training. Since its inception, an SHM annual meeting pre-course that focuses on hand-held ultrasound and invasive procedures has consistently been one of the first to sell out. Other national organizations, such as ACP and its annual meeting, have seen similar interest in their courses on ultrasound-guided procedures.
The popularity of this continuing education bears out a worrisome trend: Hospitalists feel they are losing their procedural skills. An online survey conducted by The Hospitalist in May 2011 found that a majority of respondents (62%) had experienced deterioration of their procedural skills in the past five years; only 25% said they experienced improvement over the same period.
Historically, general internists have claimed bedside procedures as their domain. As stated dispassionately in the 1978 book The House of God, “There is no body cavity that cannot be reached with a #14G needle and a good strong arm.”1 Yet much has changed since Samuel Shem’s apocryphal description of medical residency training.
Most notably, the Accreditation Council of Graduate Medical Education (ACGME) has not only progressively restricted inpatient hours and patient loads for residents, but also increased the requirements for outpatient training. Some feel the balance of inpatient and outpatient training has tipped too far toward the latter in medicine residency programs, especially in light of the growing popularity of the hospitalist career path amongst new residency program graduates. This stands in contrast to ED training programs, which have embraced focused procedures training more readily.
“Adult care appears to be diverging into two career tacks as a result of external forces, of which we have limited control over, “ says Michael Beck, MD, a pediatric and adult hospitalist at Milton S. Hershey Medical Center in Hershey, Pa. “With new career choices emerging for graduates, the same square-peg, round-hole residency training should not exist.”
Dr. Beck advocates continuing an ongoing trend of “track” creation in residency programs, which allow trainees to focus training on their planned career path. Hospitalist tracks already exist in many medicine programs, including those at Cleveland Clinic and Northwestern. But many other factors limit the opportunity for trainees to obtain experience with bedside procedures, including competition with nurse practitioners and physician assistants. Even the increasing availability of ancillary phlebotomy and IV-start teams can increase a resident’s anxiety about procedures.
“By the time my residency was over [in 1993] and the work restrictions were beginning, hospital employees were doing all these tasks, making the residents less comfortable with hurting a patient when it was therapeutically necessary,” says Katharine Deiss, MD, assistant clinical professor of medicine at the University of Rochester Medical Center in New York. Interns who came from medical schools without extensive ancillary services in their teaching hospitals, she adds, were more comfortable with invasive procedures.
ACGME has sent a subtle message by decreasing emphasis on procedural skills by eradicating the requirement of showing manual proficiency in most bedside procedures as a requirement for certification. The omission has left individual residency programs and hospitalist groups to determine training and proficiency requirements for more invasive bedside procedures without a national standard.
In an editorial in the March 2007 issue of the Annals of Internal Medicine, F. Daniel Duffy, MD, and Eric Holmboe, MD, wrote that the American Board of Internal Medicine (ABIM) could only give a “qualified ‘yes’” to the question of whether residents should be trained in procedures they may not perform in practice. Although the authors asserted that the relaxed ABIM policy was “an important but small step toward revamping procedure skill training during residency,” others say it portrays an image of the ABIM de-emphasizing the importance of procedural training.
In addition, the recently established Focused Practice in Hospital Medicine (FPHM) pathway to ABIM Maintenance of Certification (MOC) has no requirement to show proficiency in bedside procedures.
“The absence of the procedural requirement in no way constitutes a statement that procedural skills are not important,” says Jeff Wiese, MD, FACP, SFHM, associate professor of medicine and residency program director at Tulane University Health Sciences Center in New Orleans, chair of the ABIM Hospital Medicine MOC Question Writing Committee, and former SHM president. “Rather, it is merely a practical issue with respect to making the MOC process applicable to all physicians engaged in hospital medicine (i.e. many hospitalists do not do procedures) while still making the MOC focused on the skill sets that are common for physicians doing hospital medicine.”
Once released into the world, even if trained well in residency, hospitalists can find it difficult to maintain their skills. In community and nonteaching settings, the pressure to admit and discharge in a timely manner can make procedures seem like the easiest corner to cut. Before long, it has been months since they have laid eyes on a needle of any sort. Many begin to develop performance anxiety.
In teaching hospitals, academic hospitalists often are called upon to participate in quality improvement (QI) and research efforts, which take time away from clinical rotations. Once there, it can be easy for a ward attending to rely upon a well-trained resident to supervise interns doing procedures. The lack of first-hand or even supervisory experience can lead to many academic hospitalists losing facility with procedures, with potentially disastrous results.
“In order to supervise a group of residents, the attending needs to be technically proficient and able to salvage a botched, or failed, procedure,” UM-JMH’s Dr. Lenchus says. “To this end, we strictly limit who can attend on the service.”
So what’s a residency or HM program director to do in the face of wavering support nationally, and sometimes locally, for maintaining procedural skills for hospitalists and trainees? Many hospitalists in teaching hospitals say it’s critical for clinicians to “get their own house in order,” to maintain procedural standards of proficiency with ongoing training, education, and verification.
“The profession now needs to redesign procedural training across the continuum of education and a lifetime of practice,” Drs. Duffy and Holmboe editorialized in the March 2007 Annals paper. “This approach would recognize the varied settings of internal-medicine practice and offer manual skills training to those whose practice settings require such skills.” Hospitalists can partner with medicine residency program leaders to provide procedural education and training to residents, either as a standalone elective or as a more general resource.
Hospitalists in such teaching hospitals as UCSD, Brigham and Women’s, UM-JMH, and Northwestern are leading efforts to provide procedural education to medical students, residents, and attendings. Training takes many forms, including formal procedural electives, required procedure rotations, or even brief one- or two-day courses in procedural skills at a simulation center.
Utilizing simulation training has been shown in many studies to be helpful in establishing procedural skills in learners of all training levels. Dr. Barsuk and his colleagues at Northwestern published studies in the Journal of Hospital Medicine in 2008 and 2009 showing that simulation training of residents was effective in improving skills in thoracentesis and central venous catheterization, respectively.3,4
In the community hospital setting, requirements for procedural skills can vary greatly based on the institution. For those community programs requiring procedural skills of their hospitalists, the clear definition of procedural training and requirements at the time of hiring is critical. Even after vetting a hospitalist’s procedural skills at hire, however, community programs should consider monitoring procedural skills and provide ongoing time and money for CME focused on procedural skills.
Currently, most hospitals depend on the honesty of individual physicians during the privileging process for bedside procedures. Even when the skills of physicians begin to wane, most are reluctant to voluntarily give up their procedure privileges.
“I think it would be pretty unusual for a hospitalist to relinquish their privileges,” Dr. Barsuk admits. But ideally, physicians who relinquish their privileges due to lack of experience could get retrained in simulation centers, then reproctored in order to regain their privileges. Northwestern established the Center for Simulation Technology and Immersive Learning as a resource for simulation training both locally and nationally.
Establishing an environment that supports hospitalists performing bedside procedures is critical. This includes the need to limit hospitalist workload to ensure adequate time to meet the procedural needs of patients. Providing easy access to the tools necessary to perform bedside procedures (e.g. portable ultrasound and pre-packaged procedure trays) helps avoid additional hurdles.
Academic hospitalist programs can serve as a regional resource by developing ongoing procedure mastery programs for hospitalists in their communities, as many smaller institutions do not have the resources to provide ongoing training in bedside procedures. This process can be tedious, but it should not be humiliating.
If the popularity of the SHM pre-course in bedside ultrasound and procedures is any indication, when given the opportunity to receive protected time for procedure training, most hospitalists will likely jump at the chance.
Dr. Chang is an associate clinical professor of medicine in the division of hospital medicine at Diego Medical Center. He is also a member of Team Hospitalist.
References
- Shem S. The House of God. New York: Dell Publishing; 1978.
- Duffy FD, Holmboe ES. What procedures should internists do? Ann Intern Med. 2007;146(5):392-393.
- Wayne DB, Barsuk JH, O’Leary KJ, Fudala MJ, McGaghie WC. Mastery learning of thoracentesis skills by internal medicine residents using simulation technology and deliberate practice. J Hosp Med. 2008;3(1):48-54.
- Barsuk JH, McGaghie WC, Cohen ER, Balachandran JS, Wayne DB. Use of simulation-based mastery learning to improve the quality of central venous catheter placement in a medical intensive care unit. J Hosp Med. 2009;4(7):397–403.