User login
Prediction Model Identifies Potentially Avoidable 30-Day Readmissions
Clinical question: Can a prediction model based on administrative and clinical data identify potentially avoidable 30-day readmissions in medical patients prior to discharge?
Background: An estimated 18% of Medicare beneficiaries are readmitted to the hospital within 30 days of discharge, costing nearly $17 billion per year. Interventions to reduce readmission rates are costly and should be focused on high-risk patients. To date, using models to predict 30-day readmission has been problematic and unreliable.
Study design: Retrospective cohort.
Setting: Academic medical center in Boston.
Synopsis: Using consecutive discharges from all medical services of Brigham and Women’s Hospital occurring over one year, this study derived and internally validated a prediction model for potentially avoidable 30-day readmissions. Of 10,731 discharges, there were 2,399 (22%) 30-day readmissions, and 879 (8.5%) were deemed potentially avoidable. Seven independent predictors for readmission were identified and used to create a predictor score referred to as the HOSPITAL score. Predictors included hemoglobin and sodium levels at discharge, number of hospitalizations in the past year, and four features of the index hospitalization, including type, discharge from an oncology service, presence of procedures, and length of stay. The score was internally validated and found to predict potentially avoidable 30-day readmission in medical patients with fair discriminatory power and good calibration.
This study is unique in that none of the classic comorbidities (e.g. congestive heart failure) were associated with a higher risk of 30-day readmission. Previously unrecognized predictors, including hemoglobin, sodium, and number of procedures performed, were incorporated. This suggests that comorbidities are not as important as illness severity or clinical instability. Hospitalists should await studies that externally validate the HOSPITAL score before incorporating it into practice.
Bottom line: A unique and simple seven-item prediction model identifies potentially avoidable 30-day readmissions but needs to be externally validated before being widely utilized.
Citation: Donze J, Drahomir A, Williams D, Schnipper JL. Potentially avoidable 30-day hospital readmissions in medical patients. JAMA Intern Med. 2013;137(8):632-638.
Clinical question: Can a prediction model based on administrative and clinical data identify potentially avoidable 30-day readmissions in medical patients prior to discharge?
Background: An estimated 18% of Medicare beneficiaries are readmitted to the hospital within 30 days of discharge, costing nearly $17 billion per year. Interventions to reduce readmission rates are costly and should be focused on high-risk patients. To date, using models to predict 30-day readmission has been problematic and unreliable.
Study design: Retrospective cohort.
Setting: Academic medical center in Boston.
Synopsis: Using consecutive discharges from all medical services of Brigham and Women’s Hospital occurring over one year, this study derived and internally validated a prediction model for potentially avoidable 30-day readmissions. Of 10,731 discharges, there were 2,399 (22%) 30-day readmissions, and 879 (8.5%) were deemed potentially avoidable. Seven independent predictors for readmission were identified and used to create a predictor score referred to as the HOSPITAL score. Predictors included hemoglobin and sodium levels at discharge, number of hospitalizations in the past year, and four features of the index hospitalization, including type, discharge from an oncology service, presence of procedures, and length of stay. The score was internally validated and found to predict potentially avoidable 30-day readmission in medical patients with fair discriminatory power and good calibration.
This study is unique in that none of the classic comorbidities (e.g. congestive heart failure) were associated with a higher risk of 30-day readmission. Previously unrecognized predictors, including hemoglobin, sodium, and number of procedures performed, were incorporated. This suggests that comorbidities are not as important as illness severity or clinical instability. Hospitalists should await studies that externally validate the HOSPITAL score before incorporating it into practice.
Bottom line: A unique and simple seven-item prediction model identifies potentially avoidable 30-day readmissions but needs to be externally validated before being widely utilized.
Citation: Donze J, Drahomir A, Williams D, Schnipper JL. Potentially avoidable 30-day hospital readmissions in medical patients. JAMA Intern Med. 2013;137(8):632-638.
Clinical question: Can a prediction model based on administrative and clinical data identify potentially avoidable 30-day readmissions in medical patients prior to discharge?
Background: An estimated 18% of Medicare beneficiaries are readmitted to the hospital within 30 days of discharge, costing nearly $17 billion per year. Interventions to reduce readmission rates are costly and should be focused on high-risk patients. To date, using models to predict 30-day readmission has been problematic and unreliable.
Study design: Retrospective cohort.
Setting: Academic medical center in Boston.
Synopsis: Using consecutive discharges from all medical services of Brigham and Women’s Hospital occurring over one year, this study derived and internally validated a prediction model for potentially avoidable 30-day readmissions. Of 10,731 discharges, there were 2,399 (22%) 30-day readmissions, and 879 (8.5%) were deemed potentially avoidable. Seven independent predictors for readmission were identified and used to create a predictor score referred to as the HOSPITAL score. Predictors included hemoglobin and sodium levels at discharge, number of hospitalizations in the past year, and four features of the index hospitalization, including type, discharge from an oncology service, presence of procedures, and length of stay. The score was internally validated and found to predict potentially avoidable 30-day readmission in medical patients with fair discriminatory power and good calibration.
This study is unique in that none of the classic comorbidities (e.g. congestive heart failure) were associated with a higher risk of 30-day readmission. Previously unrecognized predictors, including hemoglobin, sodium, and number of procedures performed, were incorporated. This suggests that comorbidities are not as important as illness severity or clinical instability. Hospitalists should await studies that externally validate the HOSPITAL score before incorporating it into practice.
Bottom line: A unique and simple seven-item prediction model identifies potentially avoidable 30-day readmissions but needs to be externally validated before being widely utilized.
Citation: Donze J, Drahomir A, Williams D, Schnipper JL. Potentially avoidable 30-day hospital readmissions in medical patients. JAMA Intern Med. 2013;137(8):632-638.
Surgical-Site Infection Risk Not Associated with Prophylactic Antibiotic Timing
Clinical question: How does timing of surgical antibiotic prophylaxis affect risk of postoperative surgical-site infections (SSIs)?
Background: Antibiotic prophylaxis for major surgical procedures has been proven in clinical trials to reduce rates of SSI. The Centers for Medicare & Medicaid Services’ (CMS) Surgical Care Improvement Project (SCIP) has implemented quality metrics to ensure antibiotics are administered within 60 minutes of incision; however, studies have failed to show that a 60-minute pre-incision window is advantageous.
Study design: Retrospective cohort.
Setting: Veterans Affairs hospitals.
Synopsis: Using SCIP and VA Surgical Quality Improvement Program data from 112 VA hospitals, 32,459 cases of hip or knee arthroplasty, colorectal surgery, arterial vascular surgery, and hysterectomy from 2005-2009 were reviewed. A postoperative SSI occurred in 1,497 cases (4.6%). Using several statistical methods, the relationship between timing of prophylactic antibiotic administration and postoperative SSI within 30 days was evaluated. In unadjusted models, higher SSI rates were observed with antibiotic administration more than 60 minutes prior to incision (OR 1.34, 95% CI 1.08-1.66) but not after incision (OR 1.26, 95% CI 0.92-1.72), compared with procedures with antibiotics administered within 60 minutes pre-incision. However, after adjustment for patient, procedure, and antibiotic variables, no significant relationship between timing and SSI was observed (P=0.50 for all specialties).
The study sample was comprised primarily of older men and did not include patients who underwent cardiac procedures, limiting the generalizability of the findings. Nonetheless, the study is the largest of its kind and confirms previous studies that suggest there is no significant relationship between timing of antibiotics and SSI. Prophylactic antibiotics should still be used when indicated; however, using timing of prophylactic antibiotics as a quality measure is unlikely to improve outcomes.
Bottom line: Adherence to the empiric 60-minute window metric for timing of prophylactic antibiotics is not significantly associated with risk of SSI.
Citation: Hawn MT, Richman JS, Vick CC, et al. Timing of surgical antibiotic prophylaxis and the risk of surgical site infection. JAMA Surg. 2013 March 20:1-8. doi: 10.1001/jamasurg.2013.134 [Epub ahead of print].
Clinical question: How does timing of surgical antibiotic prophylaxis affect risk of postoperative surgical-site infections (SSIs)?
Background: Antibiotic prophylaxis for major surgical procedures has been proven in clinical trials to reduce rates of SSI. The Centers for Medicare & Medicaid Services’ (CMS) Surgical Care Improvement Project (SCIP) has implemented quality metrics to ensure antibiotics are administered within 60 minutes of incision; however, studies have failed to show that a 60-minute pre-incision window is advantageous.
Study design: Retrospective cohort.
Setting: Veterans Affairs hospitals.
Synopsis: Using SCIP and VA Surgical Quality Improvement Program data from 112 VA hospitals, 32,459 cases of hip or knee arthroplasty, colorectal surgery, arterial vascular surgery, and hysterectomy from 2005-2009 were reviewed. A postoperative SSI occurred in 1,497 cases (4.6%). Using several statistical methods, the relationship between timing of prophylactic antibiotic administration and postoperative SSI within 30 days was evaluated. In unadjusted models, higher SSI rates were observed with antibiotic administration more than 60 minutes prior to incision (OR 1.34, 95% CI 1.08-1.66) but not after incision (OR 1.26, 95% CI 0.92-1.72), compared with procedures with antibiotics administered within 60 minutes pre-incision. However, after adjustment for patient, procedure, and antibiotic variables, no significant relationship between timing and SSI was observed (P=0.50 for all specialties).
The study sample was comprised primarily of older men and did not include patients who underwent cardiac procedures, limiting the generalizability of the findings. Nonetheless, the study is the largest of its kind and confirms previous studies that suggest there is no significant relationship between timing of antibiotics and SSI. Prophylactic antibiotics should still be used when indicated; however, using timing of prophylactic antibiotics as a quality measure is unlikely to improve outcomes.
Bottom line: Adherence to the empiric 60-minute window metric for timing of prophylactic antibiotics is not significantly associated with risk of SSI.
Citation: Hawn MT, Richman JS, Vick CC, et al. Timing of surgical antibiotic prophylaxis and the risk of surgical site infection. JAMA Surg. 2013 March 20:1-8. doi: 10.1001/jamasurg.2013.134 [Epub ahead of print].
Clinical question: How does timing of surgical antibiotic prophylaxis affect risk of postoperative surgical-site infections (SSIs)?
Background: Antibiotic prophylaxis for major surgical procedures has been proven in clinical trials to reduce rates of SSI. The Centers for Medicare & Medicaid Services’ (CMS) Surgical Care Improvement Project (SCIP) has implemented quality metrics to ensure antibiotics are administered within 60 minutes of incision; however, studies have failed to show that a 60-minute pre-incision window is advantageous.
Study design: Retrospective cohort.
Setting: Veterans Affairs hospitals.
Synopsis: Using SCIP and VA Surgical Quality Improvement Program data from 112 VA hospitals, 32,459 cases of hip or knee arthroplasty, colorectal surgery, arterial vascular surgery, and hysterectomy from 2005-2009 were reviewed. A postoperative SSI occurred in 1,497 cases (4.6%). Using several statistical methods, the relationship between timing of prophylactic antibiotic administration and postoperative SSI within 30 days was evaluated. In unadjusted models, higher SSI rates were observed with antibiotic administration more than 60 minutes prior to incision (OR 1.34, 95% CI 1.08-1.66) but not after incision (OR 1.26, 95% CI 0.92-1.72), compared with procedures with antibiotics administered within 60 minutes pre-incision. However, after adjustment for patient, procedure, and antibiotic variables, no significant relationship between timing and SSI was observed (P=0.50 for all specialties).
The study sample was comprised primarily of older men and did not include patients who underwent cardiac procedures, limiting the generalizability of the findings. Nonetheless, the study is the largest of its kind and confirms previous studies that suggest there is no significant relationship between timing of antibiotics and SSI. Prophylactic antibiotics should still be used when indicated; however, using timing of prophylactic antibiotics as a quality measure is unlikely to improve outcomes.
Bottom line: Adherence to the empiric 60-minute window metric for timing of prophylactic antibiotics is not significantly associated with risk of SSI.
Citation: Hawn MT, Richman JS, Vick CC, et al. Timing of surgical antibiotic prophylaxis and the risk of surgical site infection. JAMA Surg. 2013 March 20:1-8. doi: 10.1001/jamasurg.2013.134 [Epub ahead of print].
One-Year Survival Nearly 60% for Elderly Survivors of In-Hospital Cardiac Arrest
Clinical question: What is the long-term outcome of elderly survivors of in-hospital cardiac arrest?
Background: Previous studies have examined in-hospital survival from in-hospital cardiac arrest but have not looked at long-term outcomes and readmission of in-hospital cardiac arrest survivors.
Study design: Retrospective cohort.
Setting: Acute-care hospitals that submitted data to the Get with the Guidelines—Resuscitation registry between 2000 and 2008.
Synopsis: Using the Get with the Guidelines—Resuscitation registry from 401 acute-care hospitals, data from 6,972 Medicare patients aged 65 years or older who had a pulseless in-hospital cardiac arrest and survived to discharge were analyzed. Survival rates were 82% at 30 days, 72% at three months, 58.5% at one year, and 49.6% at two years. Survival at three years was 43.5%, similar to patients discharged with heart failure.
One-year survival decreased with increasing age. Survival also decreased with black race (52.5% vs. 60.4% for white patients, P=0.001) and male sex (58.6% vs. 60.9% for women, P=0.03). Patients with mild or no neurologic disability at discharge had a higher survival rate at one year than patients with moderate, severe, or coma state. Readmission rates at one year after discharge were 65.6% and 76.2% at two years. Black patients, women, and patients with neurologic disability at discharge were more likely to be readmitted.
Because this is an observational study looking at a quality database of Medicare patients, it excludes patients at VA hospitals and non-Medicare facilities. This data excludes assessments of quality of life after discharge and health status among those with long-term survival, and does not include cause of death.
Bottom line: One-year survival following in-hospital cardiac arrest for patients over age 65 approaches 60% and decreases with increasing age, male sex, and black race.
Citation: Chan PS, Nallamothu BK, Krumholz HM, et al. Long-term outcomes in elderly survivors of in-hospital cardiac arrest. N Engl J Med. 2013;368:1019-1026.
Clinical question: What is the long-term outcome of elderly survivors of in-hospital cardiac arrest?
Background: Previous studies have examined in-hospital survival from in-hospital cardiac arrest but have not looked at long-term outcomes and readmission of in-hospital cardiac arrest survivors.
Study design: Retrospective cohort.
Setting: Acute-care hospitals that submitted data to the Get with the Guidelines—Resuscitation registry between 2000 and 2008.
Synopsis: Using the Get with the Guidelines—Resuscitation registry from 401 acute-care hospitals, data from 6,972 Medicare patients aged 65 years or older who had a pulseless in-hospital cardiac arrest and survived to discharge were analyzed. Survival rates were 82% at 30 days, 72% at three months, 58.5% at one year, and 49.6% at two years. Survival at three years was 43.5%, similar to patients discharged with heart failure.
One-year survival decreased with increasing age. Survival also decreased with black race (52.5% vs. 60.4% for white patients, P=0.001) and male sex (58.6% vs. 60.9% for women, P=0.03). Patients with mild or no neurologic disability at discharge had a higher survival rate at one year than patients with moderate, severe, or coma state. Readmission rates at one year after discharge were 65.6% and 76.2% at two years. Black patients, women, and patients with neurologic disability at discharge were more likely to be readmitted.
Because this is an observational study looking at a quality database of Medicare patients, it excludes patients at VA hospitals and non-Medicare facilities. This data excludes assessments of quality of life after discharge and health status among those with long-term survival, and does not include cause of death.
Bottom line: One-year survival following in-hospital cardiac arrest for patients over age 65 approaches 60% and decreases with increasing age, male sex, and black race.
Citation: Chan PS, Nallamothu BK, Krumholz HM, et al. Long-term outcomes in elderly survivors of in-hospital cardiac arrest. N Engl J Med. 2013;368:1019-1026.
Clinical question: What is the long-term outcome of elderly survivors of in-hospital cardiac arrest?
Background: Previous studies have examined in-hospital survival from in-hospital cardiac arrest but have not looked at long-term outcomes and readmission of in-hospital cardiac arrest survivors.
Study design: Retrospective cohort.
Setting: Acute-care hospitals that submitted data to the Get with the Guidelines—Resuscitation registry between 2000 and 2008.
Synopsis: Using the Get with the Guidelines—Resuscitation registry from 401 acute-care hospitals, data from 6,972 Medicare patients aged 65 years or older who had a pulseless in-hospital cardiac arrest and survived to discharge were analyzed. Survival rates were 82% at 30 days, 72% at three months, 58.5% at one year, and 49.6% at two years. Survival at three years was 43.5%, similar to patients discharged with heart failure.
One-year survival decreased with increasing age. Survival also decreased with black race (52.5% vs. 60.4% for white patients, P=0.001) and male sex (58.6% vs. 60.9% for women, P=0.03). Patients with mild or no neurologic disability at discharge had a higher survival rate at one year than patients with moderate, severe, or coma state. Readmission rates at one year after discharge were 65.6% and 76.2% at two years. Black patients, women, and patients with neurologic disability at discharge were more likely to be readmitted.
Because this is an observational study looking at a quality database of Medicare patients, it excludes patients at VA hospitals and non-Medicare facilities. This data excludes assessments of quality of life after discharge and health status among those with long-term survival, and does not include cause of death.
Bottom line: One-year survival following in-hospital cardiac arrest for patients over age 65 approaches 60% and decreases with increasing age, male sex, and black race.
Citation: Chan PS, Nallamothu BK, Krumholz HM, et al. Long-term outcomes in elderly survivors of in-hospital cardiac arrest. N Engl J Med. 2013;368:1019-1026.
Peer Benchmarking Network May Reduce Overutilization in Pediatric Bronchiolitis

Clinical question: What is the impact of a peer benchmarking network on resource utilization in acute bronchiolitis?
Background: Acute bronchiolitis is the most common illness requiring hospitalization in children. Despite the publication of national evidence-based guidelines, variation and overuse of common therapies remains. Despite one report of successful implementation of evidence-based guidelines in a collaborative of freestanding children’s hospitals, most children are hospitalized outside of such institutions, and large-scale, lower-resource efforts have not been described.
Study design: Voluntary, quality-improvement (QI), and benchmarking collaborative.
Setting: Seventeen hospitals, including both community and freestanding children’s facilities.
Synopsis: Over a four-year period, data on 11,568 bronchiolitis hospitalizations were collected. The collaborative facilitated sharing of resources (e.g. scoring tools, guidelines), celebrated high performers on an annual basis, and encouraged regular data collection, primarily via conference calls and email. Notably, a common bundle of interventions were not used; groups worked on local improvement cycles, with only a few groups forming a small subcollaborative utilizing a shared pathway. A significant decrease in bronchodilator utilization and chest physiotherapy was seen over the course of the collaborative, although no change in chest radiography, steroid utilization, and RSV testing was noted.
This voluntary and low-resource effort by similarly motivated peers across a variety of inpatient settings demonstrated improvement over time. It is particularly notable as inpatient collaboratives with face-to-face meeting requirements, and annual fees, become more commonplace.
Study limitations include the lack of a conceptual model for studying contextual factors that might have led to improvement in the varied settings and secular changes over this time period. Additionally, EDs were not included in this initiative, which likely accounted for the lack of improvement in chest radiography and RSV testing. Nonetheless, scalable innovations such as this will become increasingly important as hospitalists search for value in health care.
Bottom line: Creating a national community of practice may reduce overutilization in bronchiolitis.
Citation: Ralston S, Garber M, Narang S, et al. Decreasing unnecessary utilization in acute bronchiolitis care: results from the Value in Inpatient Pediatrics Network. J Hosp Med. 2013;8(1):25-30.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What is the impact of a peer benchmarking network on resource utilization in acute bronchiolitis?
Background: Acute bronchiolitis is the most common illness requiring hospitalization in children. Despite the publication of national evidence-based guidelines, variation and overuse of common therapies remains. Despite one report of successful implementation of evidence-based guidelines in a collaborative of freestanding children’s hospitals, most children are hospitalized outside of such institutions, and large-scale, lower-resource efforts have not been described.
Study design: Voluntary, quality-improvement (QI), and benchmarking collaborative.
Setting: Seventeen hospitals, including both community and freestanding children’s facilities.
Synopsis: Over a four-year period, data on 11,568 bronchiolitis hospitalizations were collected. The collaborative facilitated sharing of resources (e.g. scoring tools, guidelines), celebrated high performers on an annual basis, and encouraged regular data collection, primarily via conference calls and email. Notably, a common bundle of interventions were not used; groups worked on local improvement cycles, with only a few groups forming a small subcollaborative utilizing a shared pathway. A significant decrease in bronchodilator utilization and chest physiotherapy was seen over the course of the collaborative, although no change in chest radiography, steroid utilization, and RSV testing was noted.
This voluntary and low-resource effort by similarly motivated peers across a variety of inpatient settings demonstrated improvement over time. It is particularly notable as inpatient collaboratives with face-to-face meeting requirements, and annual fees, become more commonplace.
Study limitations include the lack of a conceptual model for studying contextual factors that might have led to improvement in the varied settings and secular changes over this time period. Additionally, EDs were not included in this initiative, which likely accounted for the lack of improvement in chest radiography and RSV testing. Nonetheless, scalable innovations such as this will become increasingly important as hospitalists search for value in health care.
Bottom line: Creating a national community of practice may reduce overutilization in bronchiolitis.
Citation: Ralston S, Garber M, Narang S, et al. Decreasing unnecessary utilization in acute bronchiolitis care: results from the Value in Inpatient Pediatrics Network. J Hosp Med. 2013;8(1):25-30.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: What is the impact of a peer benchmarking network on resource utilization in acute bronchiolitis?
Background: Acute bronchiolitis is the most common illness requiring hospitalization in children. Despite the publication of national evidence-based guidelines, variation and overuse of common therapies remains. Despite one report of successful implementation of evidence-based guidelines in a collaborative of freestanding children’s hospitals, most children are hospitalized outside of such institutions, and large-scale, lower-resource efforts have not been described.
Study design: Voluntary, quality-improvement (QI), and benchmarking collaborative.
Setting: Seventeen hospitals, including both community and freestanding children’s facilities.
Synopsis: Over a four-year period, data on 11,568 bronchiolitis hospitalizations were collected. The collaborative facilitated sharing of resources (e.g. scoring tools, guidelines), celebrated high performers on an annual basis, and encouraged regular data collection, primarily via conference calls and email. Notably, a common bundle of interventions were not used; groups worked on local improvement cycles, with only a few groups forming a small subcollaborative utilizing a shared pathway. A significant decrease in bronchodilator utilization and chest physiotherapy was seen over the course of the collaborative, although no change in chest radiography, steroid utilization, and RSV testing was noted.
This voluntary and low-resource effort by similarly motivated peers across a variety of inpatient settings demonstrated improvement over time. It is particularly notable as inpatient collaboratives with face-to-face meeting requirements, and annual fees, become more commonplace.
Study limitations include the lack of a conceptual model for studying contextual factors that might have led to improvement in the varied settings and secular changes over this time period. Additionally, EDs were not included in this initiative, which likely accounted for the lack of improvement in chest radiography and RSV testing. Nonetheless, scalable innovations such as this will become increasingly important as hospitalists search for value in health care.
Bottom line: Creating a national community of practice may reduce overutilization in bronchiolitis.
Citation: Ralston S, Garber M, Narang S, et al. Decreasing unnecessary utilization in acute bronchiolitis care: results from the Value in Inpatient Pediatrics Network. J Hosp Med. 2013;8(1):25-30.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
How Can Tumor Lysis Syndrome Be Prevented and Managed in Cancer Patients?
Case
A 25-year-old male with HIV/AIDS and a CD4 count of 65 cells/μL presents to the ED with intractable nausea and vomiting for one week. Laboratory evaluation revealed a white blood cell of 67,000 cells/mm3. An extended chemistry panel reveals creatinine 3.5 mg/dL, potassium 3.0 mmol/L, LDH 250 IU/L, and uric acid 5mg/dL. Calcium and phosphorus were both normal. The patient was admitted for further evaluation and management, and was later diagnosed with Burkitt’s lymphoma.
Overview
Tumor lysis syndrome (TLS) is an acute cell lysis of tumor cells with the release of cell content into circulation either spontaneously or in response to therapy, leading to hyperurecemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.1-3
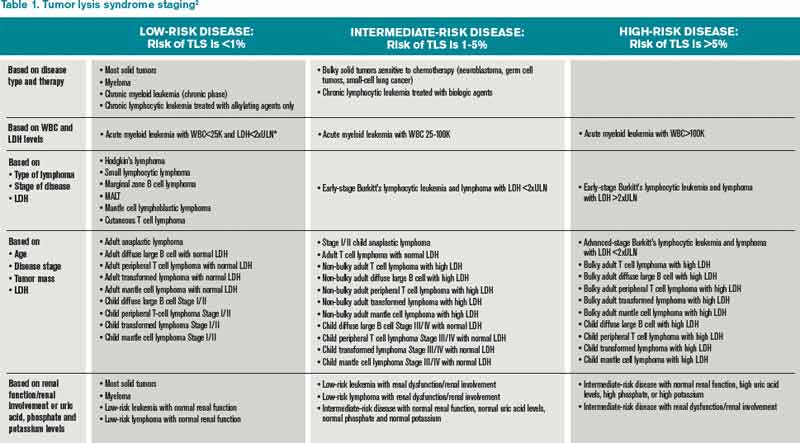
TLS is one of the most common oncology emergencies encountered by hospitalists caring for patients with hematologic malignancies. The incidence and severity of TLS depend on the cell burden, cell proliferation rate, potential for cell lysis or chemo sensitivity, baseline clinical characteristics, and preventive measures taken (see Table 1).2,4
TLS is classified as laboratory or clinical. Laboratory TLS is described as the presence of two or more of the following serum abnormalities at the same time, present within three days before or seven days after the start of therapy.5
- Uric acid >8 mg/dL (475.8 micromole/L) or 25% increase;
- Potassium >6 mEq/L (6 mmol/L) or 25% increase;
- Phosphorus >6.5 mg/dL (2.1 mmol/L) for children or >4.5 mg/dl (1.45 mmol/L) for adults or 25% increase; and
- Calcium >7 mg/dL (1.75 mmol/L) or 25% increase.
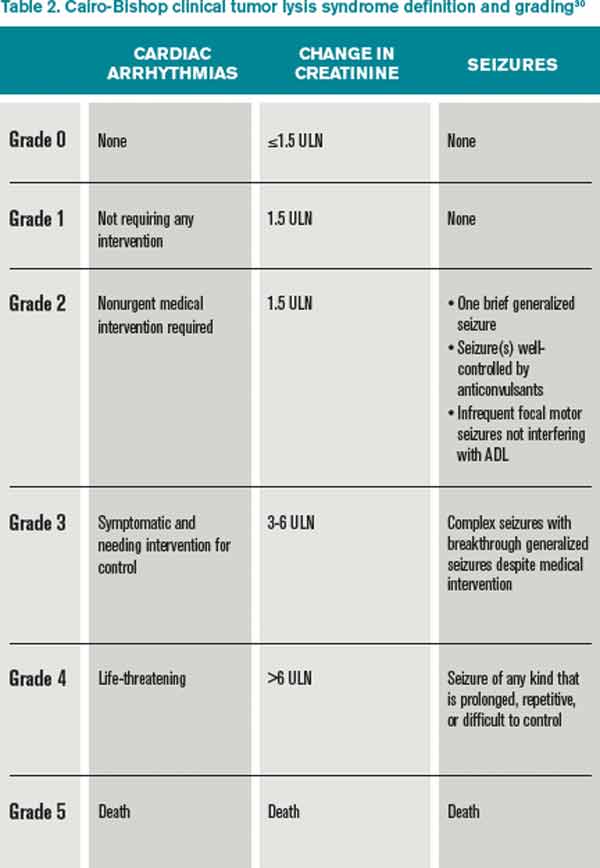
Clinical TLS is defined as laboratory TLS in association with increased creatinine levels, seizures, cardiac arrhythmias, or death (see Table 2).5
Pathogenesis
Tumor cell lysis releases DNA, cytokines, phosphate, and potassium. DNA is metabolized into adenosine and guanosine, which are then converted into xanthines. Xanthines are oxidized by xanthine oxidase into uric acid, which is then excreted through the kidneys.
TLS develops when the accumulation of xanthine, uric acid, potassium, and phosphorus exceeds the kidney’s capacity to excrete them. Cytokines cause hypotension, inflammation, and kidney injury, and worsen the kidney’s excretory capacity. Damage to the kidneys also occurs by renal precipitation of uric acid, xanthine, and calcium phosphate.4
Phosphorus concentrations in tumor cells are four times higher than in normal cells. When the calcium phosphorus product exceeds 60 mg2/dL2, there is an increased risk of calcium phosphate precipitation in the kidney tubules, which could lead to kidney failure. Accumulation of calcium phosphate product may also be cardiotoxic and can lead to cardiac arrhythmias. In addition, hyperphosphatemia can cause secondary hypocalcemia, which may lead to parasthesias, tetany, and cardiac arrhythmias.2,4
TLS is most common in tumors with high proliferative rates and high tumor burden, such as acute lymphoblastic leukemia and Burkitt’s lymphoma, but it can occur with other hematologic malignancies, such as T-cell precursor acute lymphocytic leukemia (ALL), B-cell precursor ALL, acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), anaplastic large cell lymphoma, and plasma cell disorders (e.g. multiple myeloma and plasmacytoma).6,7 TLS has also been reported with the treatment of solid organ nonhematologic tumors (see Table 3).
In hematologic tumors, TLS frequently is associated with cytotoxic chemotherapy, and less frequently with glucocorticoid treatment, monoclonal antibodies (eg, rituximab, bortezomab, imatinib), and radiation therapy.25-29
Patient factors, such as baseline kidney disease or lack of prophylactic/preventive measures for TLS, also increase the risk.4 TLS, however, can develop in patients classified as low risk (see Table 1.
TLS Prevention
Intravenous fluids. Every patient at intermediate or high risk of TLS should receive intravenous fluids (IVF) prior to cancer treatment; those at low risk may receive IVF based on the provider’s clinical judgment.30 The purpose of administering IVF is to generate high urine output to reduce the risk of precipitation of uric acid in the renal tubules.30 Both adults and children should receive approximately 2 to 3 L/m2 per day of IVF,30 and urine output should be maintained at 2 ml/kg/hr (or 4 to 6 ml/kg/hr for children <10kg).30 IVF should be cautiously administered in patients with renal insufficiency or heart failure, and diuretics may be used to maintain goal urine output. Recommended initial fluids are D51/4 normal saline, or normal saline for patients who are dehydrated or hyponatremic.30
Allopurinol. Allopurinol is usually also administered to patients at risk for developing TLS.30 Allopurinol inhibits the metabolism of hypoxanthine and xanthine to uric acid, which decreases the accumulation of uric acid in the renal tubules, thus preventing obstructive renal disease from precipitation of uric acid.4 The recommended dose of allopurinol is 100 mg/m2 every eight hours, and should not exceed 800 mg per day in adults. It should be started one to two days prior to induction chemotherapy and continued for three to seven days after the treatment and until uric acid levels and other electrolyte levels have returned to normal. The dose is adjusted to 50 mg/m2 every eight hours in patients with kidney failure.30
In some cases, allopurinol can lead to increased levels of xanthine crystals in the renal tubules, leading to acute kidney injury. Also, allopurinol does not have any effect on uric acid that has already been formed, so patients with elevated uric acid levels prior to the initiation of cancer therapy will not have any reduction in the levels of uric acid. Allopurinol reduces the degradation of other purines, so it can cause toxicity in patients on azathioprine and 6-mercaptopurine if the doses of these medications are not adjusted.
Rasburicase. Rasburicase is a recombinant urate oxidase, derived from aspergillus favus, which catalyzes the breakdown of uric acid to allantoin, which is a water-soluble product. Rasburicase is recommended as a first-line treatment for patients at high risk for clinical TLS.30 Rasburicase has an earlier onset than allopurinol and rapidly decreases serum levels of uric acid within four hours of administration.30,31 The recommended dose is 0.10 to 0.20 mg/kg once a day for five days in adults.30
A Phase III trial compared the efficiency and safety of rasburicase to rasburicase with allopurinol or allopurinol alone.32 A significantly higher normalization of uric acid was found in patients on rasburicase compared to allopurinol alone. The incidence of laboratory TLS was also significantly lower with rasburicase alone compared to allopurinol alone, and was even lower with allopurinol plus rasburicase. The incidence of acute kidney injury was the same with rasburicase alone or allopurinol alone but was higher with rasburicase plus allopurinol.
Serum uric acid, phosphorus, potassium, and calcium need to be monitored every four hours for 24 hours after the completion of chemotherapy in patients on rasburicase.4 The sample of blood drawn to check the uric acid levels has to be placed on ice and processed within four hours in order to avoid falsely lower levels of uric acid due to the conversion of uric acid to allantoin. Rasburicase is contraindicated in patients with G6PD deficiency and pregnant women, because one of the byproducts of uric acid breakdown is hydrogen peroxide, which can cause severe hemolysis and the formation of methemoglobin in these patients.30
Rasburicase has been approved for use in both children and adults, but there is more evidence for the use in children. Rasburicase has a black-box label for patients with anaphylaxis, methemoglobinemia, hemolysis, and hemoglobinuria, and there is a recommendation to check G6PD deficiency before use in high-risk patients.30
TLS Treatment
Alkalinization. Alkalinization of urine is controversial in the management of TLS. Urine alkalinization increases uric acid solubility but causes hyperphosphatemia and decreases calcium phosphate solubility, which can then deposit in the kidney once cancer treatment starts. Of note, hyperphosphatemia is much more difficult to correct than high levels of uric acid, and there are no clinical trials proving the superiority of urine alkalinization over normal saline.
Normalization of electrolytes. Electrolyte abnormalities should be corrected to avoid arrhythmias and seizures. Phosphorus levels >6.5 mg/dl (2.1 mmol/L) should be managed by restricting phosphorus intake, and by the use of phosphate binders (calcium acetate, calcium carbonate, sevelamer, lanthanum, or aluminum hydroxide). Aluminum hydroxide should be avoided in patients with renal insufficiency. In severe cases of hyperphosphatemia, dialysis should be considered.
Symptomatic hypocalcemia should be treated with calcium gluconate if changes are present on the electrocardiography (ECG). Hypocalcemia in the presence of hyperphosphatemia should be treated only in patients with tetany or cardiac arrhythmias; otherwise, hypocalcemia should not be treated until hyperphosphatemia has been corrected.
In cases of hyperkalemia, patients should be placed on a cardiac monitor and stabilized with calcium gluconate; kayexalate should be administered to reduce total body potassium. Other interventions, such as intravenous insulin given with dextrose, sodium bicarbonate, and albuterol, have a temporary effect on hyperkalemia and can be used as adjunct treatments in patients with severe hyperkalemia (>7). Hemodialysis should be strongly considered in severe cases of hyperkalemia, particularly in patients with persistently elevated potassium levels despite other treatments.
Back to the Case
Our patient was started on IVFs with close monitoring of his urine output. He was considered intermediate risk for developing TLS. Allopurinol, renally dosed, was administered for two days prior to initiating treatment with rituximab plus chemotherapy. His chemistry panel was monitored daily and he did not develop any form of TLS.
Bottom Line
TLS is a common oncology emergency in patients with hematologic malignancies. Preventative measures include starting IVF prior to cancer treatment, and administering allopurinol and/or rasburicase to patients at risk of developing TLS. Treatment should include normalizing electrolytes to avoid arrhythmias and seizures.
Dr. Akwe is assistant professor of medicine at the Emory University School of Medicine and a clinical instructor of medicine at the Morehouse School of Medicine, both in Atlanta. Dr. Smith is an assistant director for education in the division of hospital medicine at Emory. Both work as hospitalists at the Atlanta VA Medical Center.
References
- Abu-Alfa AK, Younes A. Tumor lysis syndrome and acute kidney injury: evaluation, prevention, and management. Am J Kidney Dis. 2010;55:Suppl 3:S1-S13.
- Cairo MS, Coiffier B, Reiter A, Younes A. Recommendations for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
- Gertz MA. Managing tumor lysis syndrome in 2010. Leuk Lymphoma. 2010;51:179-180.
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011;364:1844.
- Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3.
- Wössmann W, Schrappe M, Meyer U, et al. Incidence of tumor lysis syndrome in children with advanced stage Burkitt’s lymphoma/leukemia before and after introduction of prophylactic use of urate oxidase. Ann Hematol. 2003;82:160.
- Hussain K, Mazza JJ, Clouse LH. Tumor lysis syndrome (TLS) following fludarabine therapy Gemici C. Tumor lysis syndrome in solid tumors. J Clin Oncol. 2009;27:2738-2739
- Rostom AY, El-Hussainy G, Kandil A, Allam A. Tumor lysis syndrome following hemi-body irradiation for metastatic breast cancer. Ann Oncol. 2000;11:1349.
- Drakos P, Bar-Ziv J, Catane R. Tumor lysis syndrome in nonhematologic malignancies. Report of a case and review of the literature. Am J Clin Oncol. 1994;17:502.
- Baeksgaard L, Sørensen JB. Acute tumor lysis syndrome in solid tumors—a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187.
- Kalemkerian GP, Darwish B, Varterasian ML. Tumor lysis syndrome in small cell carcinoma and other solid tumors. Am J Med. 1997;103:363.
- Noh GY, Choe DH, Kim CH, Lee JC. Fatal tumor lysis syndrome during radiotherapy for non-small-cell lung cancer. J Clin Oncol. 2008;26:6005-6006.
- Pentheroudakis G, O’Neill VJ, Vasey P, Kaye SB. Spontaneous acute tumour lysis syndrome in patients with metastatic germ cell tumours. Report of two cases. Support Care Cancer. 2001;9:554.
- Joshita S, Yoshizawa K, Sano K, et al., A patient with advanced hepatocellular carcinoma treated with sorafenib tosylate showed massive tumor lysis with avoidance of tumor lysis syndrome. Intern Med. 2010;49:991-994.
- Huang WS, Yang CH. Sorafenib-induced tumor lysis syndrome in an advanced hepatocellular carcinoma patient. World J Gastroenterol. 2009;15:4464-4466.
- Bilgrami SF, Fallon BG. Tumor lysis syndrome after combination chemotherapy for ovarian cancer. Med Pediatr Oncol. 1993;21:521.
- Chan JK, Lin SS, McMeekin DS, Berman ML. Patients with malignancy requiring urgent therapy: CASE 3. Tumor lysis syndrome associated with chemotherapy in ovarian cancer. J Clin Oncol. 2005;23:6794.
- Godoy H, Kesterson JP, Lele S. Tumor lysis syndrome associated with carboplatin and paclitaxel in a woman with recurrent endometrial cancer. Int J Gynaecol Obstet. 2010;109:254.
- Shamseddine AI, Khalil AM, Wehbeh MH. Acute tumor lysis syndrome with squamous cell carcinoma of the vulva. Gynecol Oncol 1993;51:258
- Pinder EM, Atwal GS, Ayantunde AA, et al. Tumour lysis syndrome occurring in a patient with metastatic gastrointestinal stromal tumour treated with Glivec (imatinib mesylate, Gleevec, STI571). Sarcoma. 2007;2007:82012.
- Krishnan G, D’Silva K, Al-Janadi A. Cetuximab-related tumor lysis syndrome in metastatic colon carcinoma. J Clin Oncol. 2008;26:2406-2408.
- Oztop I, Demirkan B, Yaren A, et al. Rapid tumor lysis syndrome in a patient with metastatic colon cancer as a complication of treatment with 5-fluorouracil/leucoverin and irinotecan. Tumori. 2004;90:514.
- Lin CJ, Lim KH, Cheng YC, et al. Tumor lysis syndrome after treatment with gemcitabine for metastatic transitional cell carcinoma. Med Oncol. 2007;24:455.
- Malik IA, Abubakar S, Alam F, Khan A. Dexamethasone-induced tumor lysis syndrome in high-grade non-Hodgkin’s lymphoma. South Med J. 1994;87:409.
- Jabr FI. Acute tumor lysis syndrome induced by rituximab in diffuse large B-cell lymphoma. Int J Hematol. 2005;82:312.
- Sezer O, Vesole DH, Singhal S, et al. Bortezomib-induced tumor lysis syndrome in multiple myeloma. Clin Lymphoma Myeloma. 2006;7:233.
- Jensen M, Winkler U, Manzke O, et al. Rapid tumor lysis in a patient with B-cell chronic lymphocytic leukemia and lymphocytosis treated with an anti-CD20 monoclonal antibody (IDEC-C2B8, rituximab). Ann Hematol. 1998;77:89.
- Linck D, Basara N, Tran V, et al. Peracute onset of severe tumor lysis syndrome immediately after 4 Gy fractionated TBI as part of reduced intensity preparative regimen in a patient with T-ALL with high tumor burden. Bone Marrow Transplant. 2003;31:935.
- Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26(16):2767-2778. [Erratum, J Clin Oncol. 2010;28:708.]
- Cheuk DK, Chiang AK, Chan GC, Ha SY. Urate oxidase for the prevention and treatment of tumor lysis syndrome in children with cancer. Cochrane Database Syst Rev. 2010;(6):CD006945.
- Cortes J, Moore JO, Maziarz RT, et al. Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone—results of a multicenter phase III study. J Clin Oncol. 2010;28:4207.
Case
A 25-year-old male with HIV/AIDS and a CD4 count of 65 cells/μL presents to the ED with intractable nausea and vomiting for one week. Laboratory evaluation revealed a white blood cell of 67,000 cells/mm3. An extended chemistry panel reveals creatinine 3.5 mg/dL, potassium 3.0 mmol/L, LDH 250 IU/L, and uric acid 5mg/dL. Calcium and phosphorus were both normal. The patient was admitted for further evaluation and management, and was later diagnosed with Burkitt’s lymphoma.
Overview
Tumor lysis syndrome (TLS) is an acute cell lysis of tumor cells with the release of cell content into circulation either spontaneously or in response to therapy, leading to hyperurecemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.1-3
TLS is one of the most common oncology emergencies encountered by hospitalists caring for patients with hematologic malignancies. The incidence and severity of TLS depend on the cell burden, cell proliferation rate, potential for cell lysis or chemo sensitivity, baseline clinical characteristics, and preventive measures taken (see Table 1).2,4
TLS is classified as laboratory or clinical. Laboratory TLS is described as the presence of two or more of the following serum abnormalities at the same time, present within three days before or seven days after the start of therapy.5
- Uric acid >8 mg/dL (475.8 micromole/L) or 25% increase;
- Potassium >6 mEq/L (6 mmol/L) or 25% increase;
- Phosphorus >6.5 mg/dL (2.1 mmol/L) for children or >4.5 mg/dl (1.45 mmol/L) for adults or 25% increase; and
- Calcium >7 mg/dL (1.75 mmol/L) or 25% increase.
Clinical TLS is defined as laboratory TLS in association with increased creatinine levels, seizures, cardiac arrhythmias, or death (see Table 2).5
Pathogenesis
Tumor cell lysis releases DNA, cytokines, phosphate, and potassium. DNA is metabolized into adenosine and guanosine, which are then converted into xanthines. Xanthines are oxidized by xanthine oxidase into uric acid, which is then excreted through the kidneys.
TLS develops when the accumulation of xanthine, uric acid, potassium, and phosphorus exceeds the kidney’s capacity to excrete them. Cytokines cause hypotension, inflammation, and kidney injury, and worsen the kidney’s excretory capacity. Damage to the kidneys also occurs by renal precipitation of uric acid, xanthine, and calcium phosphate.4
Phosphorus concentrations in tumor cells are four times higher than in normal cells. When the calcium phosphorus product exceeds 60 mg2/dL2, there is an increased risk of calcium phosphate precipitation in the kidney tubules, which could lead to kidney failure. Accumulation of calcium phosphate product may also be cardiotoxic and can lead to cardiac arrhythmias. In addition, hyperphosphatemia can cause secondary hypocalcemia, which may lead to parasthesias, tetany, and cardiac arrhythmias.2,4
TLS is most common in tumors with high proliferative rates and high tumor burden, such as acute lymphoblastic leukemia and Burkitt’s lymphoma, but it can occur with other hematologic malignancies, such as T-cell precursor acute lymphocytic leukemia (ALL), B-cell precursor ALL, acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), anaplastic large cell lymphoma, and plasma cell disorders (e.g. multiple myeloma and plasmacytoma).6,7 TLS has also been reported with the treatment of solid organ nonhematologic tumors (see Table 3).
In hematologic tumors, TLS frequently is associated with cytotoxic chemotherapy, and less frequently with glucocorticoid treatment, monoclonal antibodies (eg, rituximab, bortezomab, imatinib), and radiation therapy.25-29
Patient factors, such as baseline kidney disease or lack of prophylactic/preventive measures for TLS, also increase the risk.4 TLS, however, can develop in patients classified as low risk (see Table 1.
TLS Prevention
Intravenous fluids. Every patient at intermediate or high risk of TLS should receive intravenous fluids (IVF) prior to cancer treatment; those at low risk may receive IVF based on the provider’s clinical judgment.30 The purpose of administering IVF is to generate high urine output to reduce the risk of precipitation of uric acid in the renal tubules.30 Both adults and children should receive approximately 2 to 3 L/m2 per day of IVF,30 and urine output should be maintained at 2 ml/kg/hr (or 4 to 6 ml/kg/hr for children <10kg).30 IVF should be cautiously administered in patients with renal insufficiency or heart failure, and diuretics may be used to maintain goal urine output. Recommended initial fluids are D51/4 normal saline, or normal saline for patients who are dehydrated or hyponatremic.30
Allopurinol. Allopurinol is usually also administered to patients at risk for developing TLS.30 Allopurinol inhibits the metabolism of hypoxanthine and xanthine to uric acid, which decreases the accumulation of uric acid in the renal tubules, thus preventing obstructive renal disease from precipitation of uric acid.4 The recommended dose of allopurinol is 100 mg/m2 every eight hours, and should not exceed 800 mg per day in adults. It should be started one to two days prior to induction chemotherapy and continued for three to seven days after the treatment and until uric acid levels and other electrolyte levels have returned to normal. The dose is adjusted to 50 mg/m2 every eight hours in patients with kidney failure.30
In some cases, allopurinol can lead to increased levels of xanthine crystals in the renal tubules, leading to acute kidney injury. Also, allopurinol does not have any effect on uric acid that has already been formed, so patients with elevated uric acid levels prior to the initiation of cancer therapy will not have any reduction in the levels of uric acid. Allopurinol reduces the degradation of other purines, so it can cause toxicity in patients on azathioprine and 6-mercaptopurine if the doses of these medications are not adjusted.
Rasburicase. Rasburicase is a recombinant urate oxidase, derived from aspergillus favus, which catalyzes the breakdown of uric acid to allantoin, which is a water-soluble product. Rasburicase is recommended as a first-line treatment for patients at high risk for clinical TLS.30 Rasburicase has an earlier onset than allopurinol and rapidly decreases serum levels of uric acid within four hours of administration.30,31 The recommended dose is 0.10 to 0.20 mg/kg once a day for five days in adults.30
A Phase III trial compared the efficiency and safety of rasburicase to rasburicase with allopurinol or allopurinol alone.32 A significantly higher normalization of uric acid was found in patients on rasburicase compared to allopurinol alone. The incidence of laboratory TLS was also significantly lower with rasburicase alone compared to allopurinol alone, and was even lower with allopurinol plus rasburicase. The incidence of acute kidney injury was the same with rasburicase alone or allopurinol alone but was higher with rasburicase plus allopurinol.
Serum uric acid, phosphorus, potassium, and calcium need to be monitored every four hours for 24 hours after the completion of chemotherapy in patients on rasburicase.4 The sample of blood drawn to check the uric acid levels has to be placed on ice and processed within four hours in order to avoid falsely lower levels of uric acid due to the conversion of uric acid to allantoin. Rasburicase is contraindicated in patients with G6PD deficiency and pregnant women, because one of the byproducts of uric acid breakdown is hydrogen peroxide, which can cause severe hemolysis and the formation of methemoglobin in these patients.30
Rasburicase has been approved for use in both children and adults, but there is more evidence for the use in children. Rasburicase has a black-box label for patients with anaphylaxis, methemoglobinemia, hemolysis, and hemoglobinuria, and there is a recommendation to check G6PD deficiency before use in high-risk patients.30
TLS Treatment
Alkalinization. Alkalinization of urine is controversial in the management of TLS. Urine alkalinization increases uric acid solubility but causes hyperphosphatemia and decreases calcium phosphate solubility, which can then deposit in the kidney once cancer treatment starts. Of note, hyperphosphatemia is much more difficult to correct than high levels of uric acid, and there are no clinical trials proving the superiority of urine alkalinization over normal saline.
Normalization of electrolytes. Electrolyte abnormalities should be corrected to avoid arrhythmias and seizures. Phosphorus levels >6.5 mg/dl (2.1 mmol/L) should be managed by restricting phosphorus intake, and by the use of phosphate binders (calcium acetate, calcium carbonate, sevelamer, lanthanum, or aluminum hydroxide). Aluminum hydroxide should be avoided in patients with renal insufficiency. In severe cases of hyperphosphatemia, dialysis should be considered.
Symptomatic hypocalcemia should be treated with calcium gluconate if changes are present on the electrocardiography (ECG). Hypocalcemia in the presence of hyperphosphatemia should be treated only in patients with tetany or cardiac arrhythmias; otherwise, hypocalcemia should not be treated until hyperphosphatemia has been corrected.
In cases of hyperkalemia, patients should be placed on a cardiac monitor and stabilized with calcium gluconate; kayexalate should be administered to reduce total body potassium. Other interventions, such as intravenous insulin given with dextrose, sodium bicarbonate, and albuterol, have a temporary effect on hyperkalemia and can be used as adjunct treatments in patients with severe hyperkalemia (>7). Hemodialysis should be strongly considered in severe cases of hyperkalemia, particularly in patients with persistently elevated potassium levels despite other treatments.
Back to the Case
Our patient was started on IVFs with close monitoring of his urine output. He was considered intermediate risk for developing TLS. Allopurinol, renally dosed, was administered for two days prior to initiating treatment with rituximab plus chemotherapy. His chemistry panel was monitored daily and he did not develop any form of TLS.
Bottom Line
TLS is a common oncology emergency in patients with hematologic malignancies. Preventative measures include starting IVF prior to cancer treatment, and administering allopurinol and/or rasburicase to patients at risk of developing TLS. Treatment should include normalizing electrolytes to avoid arrhythmias and seizures.
Dr. Akwe is assistant professor of medicine at the Emory University School of Medicine and a clinical instructor of medicine at the Morehouse School of Medicine, both in Atlanta. Dr. Smith is an assistant director for education in the division of hospital medicine at Emory. Both work as hospitalists at the Atlanta VA Medical Center.
References
- Abu-Alfa AK, Younes A. Tumor lysis syndrome and acute kidney injury: evaluation, prevention, and management. Am J Kidney Dis. 2010;55:Suppl 3:S1-S13.
- Cairo MS, Coiffier B, Reiter A, Younes A. Recommendations for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
- Gertz MA. Managing tumor lysis syndrome in 2010. Leuk Lymphoma. 2010;51:179-180.
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011;364:1844.
- Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3.
- Wössmann W, Schrappe M, Meyer U, et al. Incidence of tumor lysis syndrome in children with advanced stage Burkitt’s lymphoma/leukemia before and after introduction of prophylactic use of urate oxidase. Ann Hematol. 2003;82:160.
- Hussain K, Mazza JJ, Clouse LH. Tumor lysis syndrome (TLS) following fludarabine therapy Gemici C. Tumor lysis syndrome in solid tumors. J Clin Oncol. 2009;27:2738-2739
- Rostom AY, El-Hussainy G, Kandil A, Allam A. Tumor lysis syndrome following hemi-body irradiation for metastatic breast cancer. Ann Oncol. 2000;11:1349.
- Drakos P, Bar-Ziv J, Catane R. Tumor lysis syndrome in nonhematologic malignancies. Report of a case and review of the literature. Am J Clin Oncol. 1994;17:502.
- Baeksgaard L, Sørensen JB. Acute tumor lysis syndrome in solid tumors—a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187.
- Kalemkerian GP, Darwish B, Varterasian ML. Tumor lysis syndrome in small cell carcinoma and other solid tumors. Am J Med. 1997;103:363.
- Noh GY, Choe DH, Kim CH, Lee JC. Fatal tumor lysis syndrome during radiotherapy for non-small-cell lung cancer. J Clin Oncol. 2008;26:6005-6006.
- Pentheroudakis G, O’Neill VJ, Vasey P, Kaye SB. Spontaneous acute tumour lysis syndrome in patients with metastatic germ cell tumours. Report of two cases. Support Care Cancer. 2001;9:554.
- Joshita S, Yoshizawa K, Sano K, et al., A patient with advanced hepatocellular carcinoma treated with sorafenib tosylate showed massive tumor lysis with avoidance of tumor lysis syndrome. Intern Med. 2010;49:991-994.
- Huang WS, Yang CH. Sorafenib-induced tumor lysis syndrome in an advanced hepatocellular carcinoma patient. World J Gastroenterol. 2009;15:4464-4466.
- Bilgrami SF, Fallon BG. Tumor lysis syndrome after combination chemotherapy for ovarian cancer. Med Pediatr Oncol. 1993;21:521.
- Chan JK, Lin SS, McMeekin DS, Berman ML. Patients with malignancy requiring urgent therapy: CASE 3. Tumor lysis syndrome associated with chemotherapy in ovarian cancer. J Clin Oncol. 2005;23:6794.
- Godoy H, Kesterson JP, Lele S. Tumor lysis syndrome associated with carboplatin and paclitaxel in a woman with recurrent endometrial cancer. Int J Gynaecol Obstet. 2010;109:254.
- Shamseddine AI, Khalil AM, Wehbeh MH. Acute tumor lysis syndrome with squamous cell carcinoma of the vulva. Gynecol Oncol 1993;51:258
- Pinder EM, Atwal GS, Ayantunde AA, et al. Tumour lysis syndrome occurring in a patient with metastatic gastrointestinal stromal tumour treated with Glivec (imatinib mesylate, Gleevec, STI571). Sarcoma. 2007;2007:82012.
- Krishnan G, D’Silva K, Al-Janadi A. Cetuximab-related tumor lysis syndrome in metastatic colon carcinoma. J Clin Oncol. 2008;26:2406-2408.
- Oztop I, Demirkan B, Yaren A, et al. Rapid tumor lysis syndrome in a patient with metastatic colon cancer as a complication of treatment with 5-fluorouracil/leucoverin and irinotecan. Tumori. 2004;90:514.
- Lin CJ, Lim KH, Cheng YC, et al. Tumor lysis syndrome after treatment with gemcitabine for metastatic transitional cell carcinoma. Med Oncol. 2007;24:455.
- Malik IA, Abubakar S, Alam F, Khan A. Dexamethasone-induced tumor lysis syndrome in high-grade non-Hodgkin’s lymphoma. South Med J. 1994;87:409.
- Jabr FI. Acute tumor lysis syndrome induced by rituximab in diffuse large B-cell lymphoma. Int J Hematol. 2005;82:312.
- Sezer O, Vesole DH, Singhal S, et al. Bortezomib-induced tumor lysis syndrome in multiple myeloma. Clin Lymphoma Myeloma. 2006;7:233.
- Jensen M, Winkler U, Manzke O, et al. Rapid tumor lysis in a patient with B-cell chronic lymphocytic leukemia and lymphocytosis treated with an anti-CD20 monoclonal antibody (IDEC-C2B8, rituximab). Ann Hematol. 1998;77:89.
- Linck D, Basara N, Tran V, et al. Peracute onset of severe tumor lysis syndrome immediately after 4 Gy fractionated TBI as part of reduced intensity preparative regimen in a patient with T-ALL with high tumor burden. Bone Marrow Transplant. 2003;31:935.
- Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26(16):2767-2778. [Erratum, J Clin Oncol. 2010;28:708.]
- Cheuk DK, Chiang AK, Chan GC, Ha SY. Urate oxidase for the prevention and treatment of tumor lysis syndrome in children with cancer. Cochrane Database Syst Rev. 2010;(6):CD006945.
- Cortes J, Moore JO, Maziarz RT, et al. Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone—results of a multicenter phase III study. J Clin Oncol. 2010;28:4207.
Case
A 25-year-old male with HIV/AIDS and a CD4 count of 65 cells/μL presents to the ED with intractable nausea and vomiting for one week. Laboratory evaluation revealed a white blood cell of 67,000 cells/mm3. An extended chemistry panel reveals creatinine 3.5 mg/dL, potassium 3.0 mmol/L, LDH 250 IU/L, and uric acid 5mg/dL. Calcium and phosphorus were both normal. The patient was admitted for further evaluation and management, and was later diagnosed with Burkitt’s lymphoma.
Overview
Tumor lysis syndrome (TLS) is an acute cell lysis of tumor cells with the release of cell content into circulation either spontaneously or in response to therapy, leading to hyperurecemia, hyperkalemia, hyperphosphatemia, and hypocalcemia.1-3
TLS is one of the most common oncology emergencies encountered by hospitalists caring for patients with hematologic malignancies. The incidence and severity of TLS depend on the cell burden, cell proliferation rate, potential for cell lysis or chemo sensitivity, baseline clinical characteristics, and preventive measures taken (see Table 1).2,4
TLS is classified as laboratory or clinical. Laboratory TLS is described as the presence of two or more of the following serum abnormalities at the same time, present within three days before or seven days after the start of therapy.5
- Uric acid >8 mg/dL (475.8 micromole/L) or 25% increase;
- Potassium >6 mEq/L (6 mmol/L) or 25% increase;
- Phosphorus >6.5 mg/dL (2.1 mmol/L) for children or >4.5 mg/dl (1.45 mmol/L) for adults or 25% increase; and
- Calcium >7 mg/dL (1.75 mmol/L) or 25% increase.
Clinical TLS is defined as laboratory TLS in association with increased creatinine levels, seizures, cardiac arrhythmias, or death (see Table 2).5
Pathogenesis
Tumor cell lysis releases DNA, cytokines, phosphate, and potassium. DNA is metabolized into adenosine and guanosine, which are then converted into xanthines. Xanthines are oxidized by xanthine oxidase into uric acid, which is then excreted through the kidneys.
TLS develops when the accumulation of xanthine, uric acid, potassium, and phosphorus exceeds the kidney’s capacity to excrete them. Cytokines cause hypotension, inflammation, and kidney injury, and worsen the kidney’s excretory capacity. Damage to the kidneys also occurs by renal precipitation of uric acid, xanthine, and calcium phosphate.4
Phosphorus concentrations in tumor cells are four times higher than in normal cells. When the calcium phosphorus product exceeds 60 mg2/dL2, there is an increased risk of calcium phosphate precipitation in the kidney tubules, which could lead to kidney failure. Accumulation of calcium phosphate product may also be cardiotoxic and can lead to cardiac arrhythmias. In addition, hyperphosphatemia can cause secondary hypocalcemia, which may lead to parasthesias, tetany, and cardiac arrhythmias.2,4
TLS is most common in tumors with high proliferative rates and high tumor burden, such as acute lymphoblastic leukemia and Burkitt’s lymphoma, but it can occur with other hematologic malignancies, such as T-cell precursor acute lymphocytic leukemia (ALL), B-cell precursor ALL, acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), anaplastic large cell lymphoma, and plasma cell disorders (e.g. multiple myeloma and plasmacytoma).6,7 TLS has also been reported with the treatment of solid organ nonhematologic tumors (see Table 3).
In hematologic tumors, TLS frequently is associated with cytotoxic chemotherapy, and less frequently with glucocorticoid treatment, monoclonal antibodies (eg, rituximab, bortezomab, imatinib), and radiation therapy.25-29
Patient factors, such as baseline kidney disease or lack of prophylactic/preventive measures for TLS, also increase the risk.4 TLS, however, can develop in patients classified as low risk (see Table 1.
TLS Prevention
Intravenous fluids. Every patient at intermediate or high risk of TLS should receive intravenous fluids (IVF) prior to cancer treatment; those at low risk may receive IVF based on the provider’s clinical judgment.30 The purpose of administering IVF is to generate high urine output to reduce the risk of precipitation of uric acid in the renal tubules.30 Both adults and children should receive approximately 2 to 3 L/m2 per day of IVF,30 and urine output should be maintained at 2 ml/kg/hr (or 4 to 6 ml/kg/hr for children <10kg).30 IVF should be cautiously administered in patients with renal insufficiency or heart failure, and diuretics may be used to maintain goal urine output. Recommended initial fluids are D51/4 normal saline, or normal saline for patients who are dehydrated or hyponatremic.30
Allopurinol. Allopurinol is usually also administered to patients at risk for developing TLS.30 Allopurinol inhibits the metabolism of hypoxanthine and xanthine to uric acid, which decreases the accumulation of uric acid in the renal tubules, thus preventing obstructive renal disease from precipitation of uric acid.4 The recommended dose of allopurinol is 100 mg/m2 every eight hours, and should not exceed 800 mg per day in adults. It should be started one to two days prior to induction chemotherapy and continued for three to seven days after the treatment and until uric acid levels and other electrolyte levels have returned to normal. The dose is adjusted to 50 mg/m2 every eight hours in patients with kidney failure.30
In some cases, allopurinol can lead to increased levels of xanthine crystals in the renal tubules, leading to acute kidney injury. Also, allopurinol does not have any effect on uric acid that has already been formed, so patients with elevated uric acid levels prior to the initiation of cancer therapy will not have any reduction in the levels of uric acid. Allopurinol reduces the degradation of other purines, so it can cause toxicity in patients on azathioprine and 6-mercaptopurine if the doses of these medications are not adjusted.
Rasburicase. Rasburicase is a recombinant urate oxidase, derived from aspergillus favus, which catalyzes the breakdown of uric acid to allantoin, which is a water-soluble product. Rasburicase is recommended as a first-line treatment for patients at high risk for clinical TLS.30 Rasburicase has an earlier onset than allopurinol and rapidly decreases serum levels of uric acid within four hours of administration.30,31 The recommended dose is 0.10 to 0.20 mg/kg once a day for five days in adults.30
A Phase III trial compared the efficiency and safety of rasburicase to rasburicase with allopurinol or allopurinol alone.32 A significantly higher normalization of uric acid was found in patients on rasburicase compared to allopurinol alone. The incidence of laboratory TLS was also significantly lower with rasburicase alone compared to allopurinol alone, and was even lower with allopurinol plus rasburicase. The incidence of acute kidney injury was the same with rasburicase alone or allopurinol alone but was higher with rasburicase plus allopurinol.
Serum uric acid, phosphorus, potassium, and calcium need to be monitored every four hours for 24 hours after the completion of chemotherapy in patients on rasburicase.4 The sample of blood drawn to check the uric acid levels has to be placed on ice and processed within four hours in order to avoid falsely lower levels of uric acid due to the conversion of uric acid to allantoin. Rasburicase is contraindicated in patients with G6PD deficiency and pregnant women, because one of the byproducts of uric acid breakdown is hydrogen peroxide, which can cause severe hemolysis and the formation of methemoglobin in these patients.30
Rasburicase has been approved for use in both children and adults, but there is more evidence for the use in children. Rasburicase has a black-box label for patients with anaphylaxis, methemoglobinemia, hemolysis, and hemoglobinuria, and there is a recommendation to check G6PD deficiency before use in high-risk patients.30
TLS Treatment
Alkalinization. Alkalinization of urine is controversial in the management of TLS. Urine alkalinization increases uric acid solubility but causes hyperphosphatemia and decreases calcium phosphate solubility, which can then deposit in the kidney once cancer treatment starts. Of note, hyperphosphatemia is much more difficult to correct than high levels of uric acid, and there are no clinical trials proving the superiority of urine alkalinization over normal saline.
Normalization of electrolytes. Electrolyte abnormalities should be corrected to avoid arrhythmias and seizures. Phosphorus levels >6.5 mg/dl (2.1 mmol/L) should be managed by restricting phosphorus intake, and by the use of phosphate binders (calcium acetate, calcium carbonate, sevelamer, lanthanum, or aluminum hydroxide). Aluminum hydroxide should be avoided in patients with renal insufficiency. In severe cases of hyperphosphatemia, dialysis should be considered.
Symptomatic hypocalcemia should be treated with calcium gluconate if changes are present on the electrocardiography (ECG). Hypocalcemia in the presence of hyperphosphatemia should be treated only in patients with tetany or cardiac arrhythmias; otherwise, hypocalcemia should not be treated until hyperphosphatemia has been corrected.
In cases of hyperkalemia, patients should be placed on a cardiac monitor and stabilized with calcium gluconate; kayexalate should be administered to reduce total body potassium. Other interventions, such as intravenous insulin given with dextrose, sodium bicarbonate, and albuterol, have a temporary effect on hyperkalemia and can be used as adjunct treatments in patients with severe hyperkalemia (>7). Hemodialysis should be strongly considered in severe cases of hyperkalemia, particularly in patients with persistently elevated potassium levels despite other treatments.
Back to the Case
Our patient was started on IVFs with close monitoring of his urine output. He was considered intermediate risk for developing TLS. Allopurinol, renally dosed, was administered for two days prior to initiating treatment with rituximab plus chemotherapy. His chemistry panel was monitored daily and he did not develop any form of TLS.
Bottom Line
TLS is a common oncology emergency in patients with hematologic malignancies. Preventative measures include starting IVF prior to cancer treatment, and administering allopurinol and/or rasburicase to patients at risk of developing TLS. Treatment should include normalizing electrolytes to avoid arrhythmias and seizures.
Dr. Akwe is assistant professor of medicine at the Emory University School of Medicine and a clinical instructor of medicine at the Morehouse School of Medicine, both in Atlanta. Dr. Smith is an assistant director for education in the division of hospital medicine at Emory. Both work as hospitalists at the Atlanta VA Medical Center.
References
- Abu-Alfa AK, Younes A. Tumor lysis syndrome and acute kidney injury: evaluation, prevention, and management. Am J Kidney Dis. 2010;55:Suppl 3:S1-S13.
- Cairo MS, Coiffier B, Reiter A, Younes A. Recommendations for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
- Gertz MA. Managing tumor lysis syndrome in 2010. Leuk Lymphoma. 2010;51:179-180.
- Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011;364:1844.
- Cairo MS, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3.
- Wössmann W, Schrappe M, Meyer U, et al. Incidence of tumor lysis syndrome in children with advanced stage Burkitt’s lymphoma/leukemia before and after introduction of prophylactic use of urate oxidase. Ann Hematol. 2003;82:160.
- Hussain K, Mazza JJ, Clouse LH. Tumor lysis syndrome (TLS) following fludarabine therapy Gemici C. Tumor lysis syndrome in solid tumors. J Clin Oncol. 2009;27:2738-2739
- Rostom AY, El-Hussainy G, Kandil A, Allam A. Tumor lysis syndrome following hemi-body irradiation for metastatic breast cancer. Ann Oncol. 2000;11:1349.
- Drakos P, Bar-Ziv J, Catane R. Tumor lysis syndrome in nonhematologic malignancies. Report of a case and review of the literature. Am J Clin Oncol. 1994;17:502.
- Baeksgaard L, Sørensen JB. Acute tumor lysis syndrome in solid tumors—a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187.
- Kalemkerian GP, Darwish B, Varterasian ML. Tumor lysis syndrome in small cell carcinoma and other solid tumors. Am J Med. 1997;103:363.
- Noh GY, Choe DH, Kim CH, Lee JC. Fatal tumor lysis syndrome during radiotherapy for non-small-cell lung cancer. J Clin Oncol. 2008;26:6005-6006.
- Pentheroudakis G, O’Neill VJ, Vasey P, Kaye SB. Spontaneous acute tumour lysis syndrome in patients with metastatic germ cell tumours. Report of two cases. Support Care Cancer. 2001;9:554.
- Joshita S, Yoshizawa K, Sano K, et al., A patient with advanced hepatocellular carcinoma treated with sorafenib tosylate showed massive tumor lysis with avoidance of tumor lysis syndrome. Intern Med. 2010;49:991-994.
- Huang WS, Yang CH. Sorafenib-induced tumor lysis syndrome in an advanced hepatocellular carcinoma patient. World J Gastroenterol. 2009;15:4464-4466.
- Bilgrami SF, Fallon BG. Tumor lysis syndrome after combination chemotherapy for ovarian cancer. Med Pediatr Oncol. 1993;21:521.
- Chan JK, Lin SS, McMeekin DS, Berman ML. Patients with malignancy requiring urgent therapy: CASE 3. Tumor lysis syndrome associated with chemotherapy in ovarian cancer. J Clin Oncol. 2005;23:6794.
- Godoy H, Kesterson JP, Lele S. Tumor lysis syndrome associated with carboplatin and paclitaxel in a woman with recurrent endometrial cancer. Int J Gynaecol Obstet. 2010;109:254.
- Shamseddine AI, Khalil AM, Wehbeh MH. Acute tumor lysis syndrome with squamous cell carcinoma of the vulva. Gynecol Oncol 1993;51:258
- Pinder EM, Atwal GS, Ayantunde AA, et al. Tumour lysis syndrome occurring in a patient with metastatic gastrointestinal stromal tumour treated with Glivec (imatinib mesylate, Gleevec, STI571). Sarcoma. 2007;2007:82012.
- Krishnan G, D’Silva K, Al-Janadi A. Cetuximab-related tumor lysis syndrome in metastatic colon carcinoma. J Clin Oncol. 2008;26:2406-2408.
- Oztop I, Demirkan B, Yaren A, et al. Rapid tumor lysis syndrome in a patient with metastatic colon cancer as a complication of treatment with 5-fluorouracil/leucoverin and irinotecan. Tumori. 2004;90:514.
- Lin CJ, Lim KH, Cheng YC, et al. Tumor lysis syndrome after treatment with gemcitabine for metastatic transitional cell carcinoma. Med Oncol. 2007;24:455.
- Malik IA, Abubakar S, Alam F, Khan A. Dexamethasone-induced tumor lysis syndrome in high-grade non-Hodgkin’s lymphoma. South Med J. 1994;87:409.
- Jabr FI. Acute tumor lysis syndrome induced by rituximab in diffuse large B-cell lymphoma. Int J Hematol. 2005;82:312.
- Sezer O, Vesole DH, Singhal S, et al. Bortezomib-induced tumor lysis syndrome in multiple myeloma. Clin Lymphoma Myeloma. 2006;7:233.
- Jensen M, Winkler U, Manzke O, et al. Rapid tumor lysis in a patient with B-cell chronic lymphocytic leukemia and lymphocytosis treated with an anti-CD20 monoclonal antibody (IDEC-C2B8, rituximab). Ann Hematol. 1998;77:89.
- Linck D, Basara N, Tran V, et al. Peracute onset of severe tumor lysis syndrome immediately after 4 Gy fractionated TBI as part of reduced intensity preparative regimen in a patient with T-ALL with high tumor burden. Bone Marrow Transplant. 2003;31:935.
- Coiffier B, Altman A, Pui CH, Younes A, Cairo MS. Guidelines for the management of pediatric and adult tumor lysis syndrome: an evidence-based review. J Clin Oncol. 2008;26(16):2767-2778. [Erratum, J Clin Oncol. 2010;28:708.]
- Cheuk DK, Chiang AK, Chan GC, Ha SY. Urate oxidase for the prevention and treatment of tumor lysis syndrome in children with cancer. Cochrane Database Syst Rev. 2010;(6):CD006945.
- Cortes J, Moore JO, Maziarz RT, et al. Control of plasma uric acid in adults at risk for tumor Lysis syndrome: efficacy and safety of rasburicase alone and rasburicase followed by allopurinol compared with allopurinol alone—results of a multicenter phase III study. J Clin Oncol. 2010;28:4207.
What Is the Best Management of Hereditary Angioedema?
Case
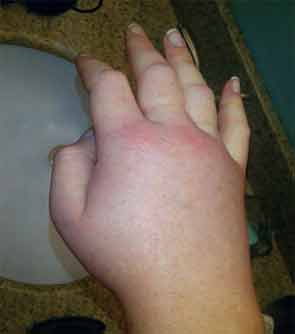
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
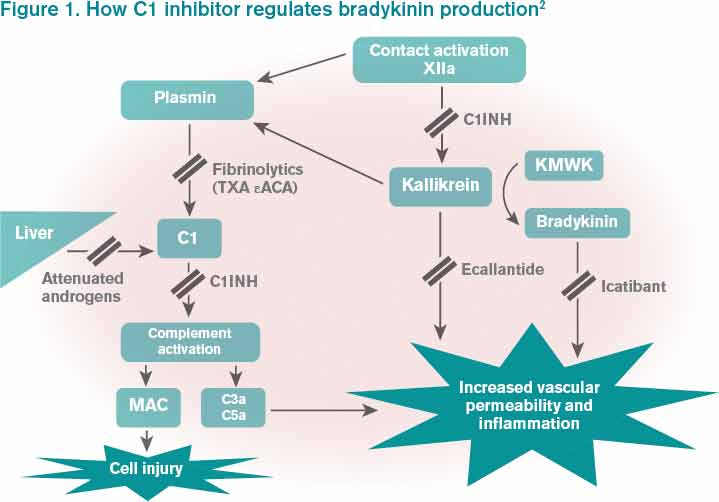
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
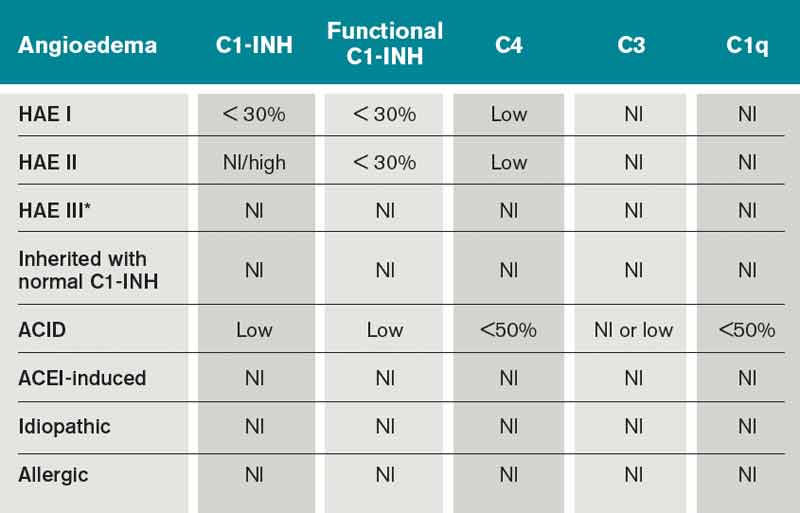
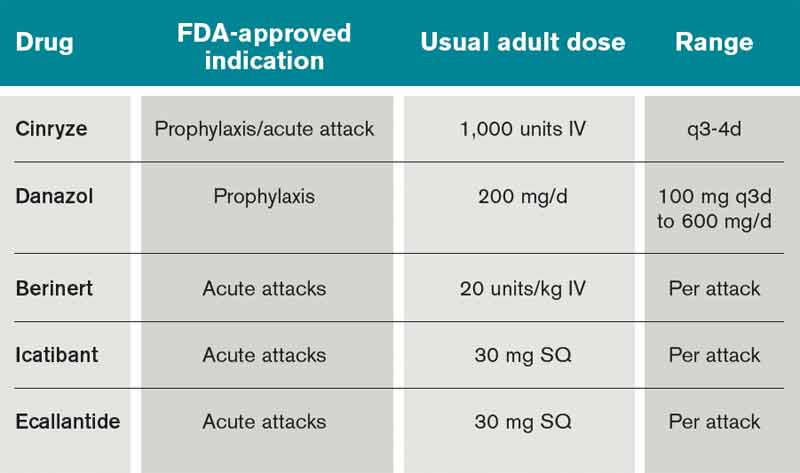
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.
Case
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.
Case
A 36-year-old man with a known history of hereditary angioedema (HAE) presents with severe orofacial swelling and laryngeal angioedema, requiring expectant management, including endotracheal intubation. His previous angioedema (AE) episodes involved his hands, feet, and genitalia; episodes generally occurred after physical trauma. Ten years prior to admission, he had an episode of secondary small bowel obstruction. The patient had been prescribed prophylactic danazol (Danacrine) 100 mg BID but he had gradually been reducing the dosage due to mood changes; at the time of presentation, he had already tapered to 100 mg danazol three times per week (Monday, Wednesday, and Friday).
Overview
HAE is an autosomal dominant condition characterized by localized, episodic swelling of the deeper dermal layers and/or mucosal tissue. Its acute presentation can vary in severity; presentations can be lethal.
HAE is generally unresponsive to conventional treatments used for other causes of AE (e.g. food or drug reactions) including glucocorticoids, antihistamines, and epinephrine. The pharmacologic treatment of acute attacks, as well as for short- and long-term prophylaxis of HAE, has evolved significantly in recent years and now includes several forms of C1 inhibitor (C1INH) protein replacement, as well as a bradykinin antagonist, and a kallikrein inhibitor.
Review of the Data
Epidemiology. HAE is an autosomal dominant disease with prevalence in the U.S. of 1 in 10,000 to 1 in 50,000 patients. All ethnic groups are equally affected, with no gender predilection. In most cases, a positive family history is present; however, in 25% of cases, spontaneous mutations occur such that an unremarkable family history does not rule out the diagnosis.1
Pathophysiology. In the past decade, there has been substantial advancement in our understanding of HAE pathophysiology. HAE occurs as a result of functional or quantitative C1 esterase inhibitor (C1INH) deficiency.
C1INH belongs to a group of proteins known as serpins (serine protease inhibitors). The C1INH gene is located on chromosome 11, and has several polymorphic sites, which predispose to spontaneous mutations.1
Bradykinin is the core bioactive mediator, which causes vasodilation, smooth muscle contraction, and subsequent edema.1 C1INH regulates bradykinin production by blocking kallikrein’s conversion of factor XII into XIIa, prekallikrein to kallikrein, and cleavage of high-molecular-weight kininogen by activated kallikrein to form bradykinin (see Figure 1).1,2
Clinical Manifestations
HAE is characterized by recurrent episodes of swelling, the frequency and severity of which are quite variable. Virtually all HAE patients have abdominal- and extremity-swelling episodes, and 50% will have episodes of laryngeal swelling; other involved areas might include the face, oropharynx, and genitalia.4 These episodes are usually unilateral; edema is nonpruritic, nonpitting, and often painless. Episodes involving the oropharynx, larynx, and abdomen can be associated with potentially serious morbidity and mortality.1, 3
HAE episodes usually commence during late childhood and early puberty (on average at age 11). Approximately half of HAE patients will have oropharyngeal involvement that might occur many years, even decades, after the initial onset of the disease. The annual rate of severe, life-threatening laryngeal edema was 0.9% in a recent retrospective study.4
Severity of the disease is variable. Attacks are episodic, and occur on average every 10 to 20 days in untreated patients. These attacks typically peak over 24 hours, then usually resolve after 48 to 72 hours. However, the complete resolution of signs and symptoms can last for up to one week after the attacks.5
There is no concomitant pruritus or urticaria that accompanies the AE. However, erythema marginatum, an evanescent nonpruritic rash with serpiginous borders involving the trunk and inner surface of extremities but sparing the face, might herald the onset of an episode. This rash usually has central pallor that blanches with pressure and worsens with heat.
HAE can be triggered by stressful events, including trauma, surgery, menstruation, and viral infections. However, in many instances, HAE attacks occur without an identifiable cause.5
Differential Diagnosis from Other Causes of Angioedema
Type I HAE is characterized by a quantitative C1INH deficiency (which is functionally abnormal as well), and occurs in 85% of patients. Type II HAE occurs in 15% of patients, and results from a functionally abnormal C1INH.
In patients with Type I and II HAE, as well as acquired C1 inhibitor deficiency (ACID), C4 levels are low during and between attacks. C2 levels are also low during acute attacks. In ACID, levels of C1q are also reduced; these patients require further workup to rule out an undiagnosed malignancy or an autoimmune process. In contrast, patients with ACE-induced, idiopathic, and allergic AE have normal complement profiles.3,6
Type III is a more recently described type of HAE that is rare, not well understood, and generally affects women.3,6 Clinically, it resembles Type I and Type II HAE but complement levels, including C1 inhibitor, are normal (see Table 1).
Treatment
HAE types I, II, III, and ACID are generally unresponsive to glucocorticoids, antihistamines, and epinephrine. These forms of AE may be exacerbated by exogenous estrogen.1,8 For this reason, HAE patients should avoid oral hormonal contraception and estrogen replacement therapy. In addition, ACE inhibitors should also be avoided based on their effect on bradykinin degradation.
Until the introduction of newer therapeutic choices, as noted in our case, the treatment of acute attacks of AE was essentially supportive. Patients with impending laryngeal obstruction were managed with intubation prior to progression of the AE to limit airway patency. Prior to the modern era, a substantial proportion of HAE patients died of asphyxiation.
Fresh frozen plasma (FFP) has been used to treat acute HAE attacks, but given its content of contact system proteins (in addition to C1INH), FFP might also pose a risk for worsening of HAE; for this reason, it must be given cautiously to patients who are symptomatic.9
In the past decade, there has been significant progress in the available treatments for HAE. Currently in the U.S., there are several agents recently approved by, or have pending approvals from, the FDA, including several forms of C1INH replacement, a bradykinin antagonist, and a kallikrein inhibitor.
The C1 esterase inhibitor (human) drugs are administered intravenously; both have been shown to be efficacious and safe. Nanofiltered C1 inhibitor provided relief in a median time of two hours when used acutely; when used as prophylaxis, it decreased the number of attacks in a three-month period by 50% (six vs. 12 with placebo, P<0.001).11
The other C1INH is rhucin, still not approved in U.S. This drug is characterized by a short half-life (approximately two to four hours) compared with the plasma-derived C1INH agents (24 to 48 hours). It is contraindicated in patients with rabbit hypersensitivity, as it is purified from rabbit breast milk.10
Ecallantide is a kallikrein inhibitor for acute therapy that is administered via three subcutaneous injections. This agent has been linked to allergic/anaphylactic reactions in a minority of patients (approximately 4%); therefore, it should be administered cautiously, by a health-care provider, and in a setting where anaphylaxis can be successfully managed.12 Icatibant is a bradykinin antagonist recently approved in the U.S. and administered SC via a single injection.10
In light of the development of these new agents, there is a need for updated guidelines for the long- and short-term prophylaxis and acute management of HAE. A recent guideline focused on the management of HAE in gynecologic and obstetric patients recommended the use of plasma-derived C1INH C1 esterase inhibitor (human) (Cinryze) for short- and long-term prophylaxis and acute treatment of HAE.13 The effect of pregnancy on HAE is variable: Some women worsen and other women have less swelling during their pregnancy. Swelling at the time of parturition is rare; however, the risk rises during the post-partum period.
Type III HAE. An additional form of HAE has been recognized with a pattern of AE episodes that mimics Type I or Type II HAE but with unremarkable laboratory studies of the complement cascade, including C1 inhibitor level and function. At this time, there is no laboratory test with which a diagnosis of Type III HAE can be confirmed. The diagnosis should be suspected in patients with a strong family history of AE reflecting autosomal dominant inheritance. In some, but not all, cases, the condition is manifest in association with high estrogen levels (e.g. pregnancy or administration of oral contraceptives). Type III HAE patients have a salutary response to the same agents that are efficacious for Type I and II HAE.
Acquired C1 inhibitor deficiency (ACID). ACID generally occurs in adults and is clinically indistinguishable from HAE. ACID is not associated with a remarkable family history of AE. In contrast to HAE, this is a consumptive deficiency of C1 inhibitor and results from enhanced catabolism that exceeds the capacity for regenerating C1 inhibitor protein. It is often associated with neoplastic (usually lymphoproliferative) or autoimmune disorders; treatment of the underlying condition frequently leads to improvement in ACID. Although its management is similar to HAE, it tends to be more responsive to anti-fibrinolytics. A salutary response to C1INH replacement therapy might not occur in patients with autoantibodies to C1 inhibitor, but efficacy of ecallantide and icatibant for the treatment of acquired AE has been reported.14, 15
ACEI angioedema. Treatment with angiotensin-converting enzyme inhibitors (ACE-I) has been associated with recurrent AE without urticaria in 0.1 to 0.7% of patients exposed to these drugs.16 Angioedema from ACE-I more frequently occurs within the first few months of therapy, but it might occur even after years of continuous therapy. ACEI-induced AE is secondary to impaired degradation of bradykinin. The main treatment is to discontinue the offending agent and avoid all other ACE-I, as this is a class-specific reaction.17
Angiotensin receptor blockers (ARBs) have been associated less commonly with AE. The mechanism for ARB-associated AE has not been elucidated. A meta-analysis showed that in 2% to 17% of patients who were switched to ARBs, recurrence of AE was observed.18 From the pooling of these data with two randomized controlled trials, it is estimated that approximately 10% or less of patients with ACEI-associated AE who switched to ARBs will develop AE.19 In the majority of cases, patients can be switched to ARBs with no recurrence of AE; however, the decision to prescribe an ARB to a patient who has had AE while receiving ACEI should be made carefully on an individualized risk/benefit basis.19
Preventive Treatment
The 17 α-alkylated androgens that can be used for treatment of HAE are danazol (Danacrine), stanozolol (Winstrol), oxandralone (Oxandrine) and methyltestosterone (Android). In patients with HAE, attenuated androgens can significantly reduce the frequency and severity of attacks; however, their use is limited by risk for untoward effects (virilization, abnormal liver function tests, change in libido, anxiety, etc.).21 There is also a risk for hepatotoxicity, including development of hepatic adenomas and hepatic carcinoma.
Antifibrinolytics also may have efficacy for HAE, but these agents have been associated with a variety of adverse effects, including nausea and diarrhea, postural hypotension, fatigue, enhanced thrombosis, retinal changes, and teratogenicity.8, 22, 23
In 2009, long-term prophylaxis with C1-INH concentrate was recommended for patients with HAE with frequent or disabling attacks, a history of laryngeal attacks, and poor quality of life. The 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of HAE recommended long-term prophylaxis in patients with more than one monthly severe HAE attack, more than five days of disability per month, or any history of airway compromise.24, 25
The decision to prescribe long-term prophylaxis, and the dose/frequency of medication required, should be individualized based on clinical parameters, such as frequency and severity of attacks, and not on C1 INH or C4 levels.
Perioperative Considerations
It is well established that any trauma, including dental procedures or surgery, can precipitate HAE attacks. For this reason, short-term prophylactic treatment in HAE patients undergoing procedures is recommended. Ideally, avoiding endotracheal intubation is the best approach; however, if intubation cannot be avoided, then adequate prophylaxis should be administered.2
Attenuated androgens can be given up to seven days before a procedure, or C1 INH can be administered 24 hours in advance. If C1 INH is unavailable, FFP can be given six to 12 hours in advance in patients who are not symptomatic; in case of endotracheal intubation, either FFP or C1 INH should be administered immediately before.2
Several case reports in multiple specialty surgical patients (abdominal surgery, cardiopulmonary bypass, orthopedic surgery, etc.) have confirmed the successful use of C1 INH in the prevention of acute attacks with favorable outcomes.2
There is no need to follow C1 INH levels, as it has no clinical relevance.
Back to the Case
The patient was admitted to the ICU and received a total of eight units of FFP. He was transferred to our institution and was able to be extubated three days after initial presentation. Laboratory studies revealed C4 10mg/dL and C1 esterase inhibitor 10mg/dL (both low).
Danazol was resumed. However, within several months after discharge, Cinryze became available in the U.S. market and was eventually prescribed. The patient has not had further significant attacks requiring inpatient management.
Dr. Auron is an assistant professor of medicine and pediatrics at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. Dr. Lang is co-director of the Asthma Center and director of the Allergy/Immunology Fellowship Training Program at the Cleveland Clinic.
References
- Bernstein, JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-412.
- Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110(5):1271-1280.
- Busse PJ. Angioedema: Differential diagnosis and treatment. Allergy Asthma Proc. 2011;32:Suppl 1:S3-S11.
- Khan DA. Hereditary angioedema: historical aspects, classification, pathophysiology, clinical presentation, and laboratory diagnosis. Allergy Asthma Proc. 2011;32(1):1-10.
- Bork K, Meng G, Staubach P, Hardt, J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-274.
- Zuraw BL, Christiansen SC. Pathogenesis and laboratory diagnosis of hereditary angioedema. Allergy Asthma Proc. 2009;30:487-492.
- Frazer-Abel A, Giclas PC. Update on laboratory tests for the diagnosis and differentiation of hereditary angioedema and acquired angioedema. Allergy Asthma Proc. 2011;32:Suppl 1:S17-S21.
- Banerjee A. Current treatment of hereditary angioedema: an update on clinical studies. Allergy Asthma Proc. 2010;31:398-406.
- Donaldson VH. Therapy of "the neurotic edema." N Engl J Med. 1972;286(15):835-836.
- Riedl MA. Update on the acute treatment of hereditary angioedema. Allergy Asthma Proc. 2011;32:11-16.
- Zuraw BL, Busse PJ, White M, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-522.
- Cicardi M, Levy RJ, McNeil DL. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-531.
- Caballero T, Farkas H, Bouillet L, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-320.
- Cicardi M, Zanichelli A. Acquired angioedema. J Allergy Clin Immunol. 2010;6(1):14.
- Zanichelli A, Badini M, Nataloni I, Montano N, Cicardi M. Treatment of acquired angioedema with icatibant: a case report. Intern Emerg Med. 2011;6(3):279-280.
- Byrd JB, Adam A, Brown NJ. Angiotensin-converting enzyme inhibitor-associated angioedema. Immunol Allergy Clin North Am. 2006;26(4):725-737.
- Haymore BR, Yoon J, Mikita CP, Klote MM, DeZee KJ. Risk of angioedema with angiotensin receptor blockers in patients with prior angioedema associated with angiotensin-converting enzyme inhibitors: a meta-analysis. Ann Allergy Asthma Immunol. 2008;101(5):495-499.
- Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45(4):520-524.
- Nzeako UC. Diagnosis and management of angioedema with abdominal involvement: a gastroenterology perspective. World J Gastroenterol. 2010; 16(39):4913-4921.
- Banerji A, Sloane DE, Sheffer AL. Hereditary angioedema: a current state-of-the-art review, V: attenuated androgens for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S19-22.
- Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008; 359(10):1027-1036.
- Zuraw BL. Hereditary angioedema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1) (Suppl 2):S13-S18.
- Bowen T, Cicardi M, Bork K, et al. Hereditary angioedema: a current state-of-the-art review, VII: Canadian Hungarian 2007 International Consensus Algorithm for the Diagnosis, Therapy, and Management of Hereditary Angioedema. Ann Allergy Asthma Immunol. 2008;100(1)(Suppl 2):S30-40.
- Craig T, Riedl M, Dykewicz M, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009.102(5):366-372.
- Frank MM. Update on preventive therapy (prophylaxis) of hereditary angioedema. Allergy Asthma Proc. 2011;32(1):17-21.
Nonurgent Pediatric Admissions on Weekends Bump Up Hospital Costs

Clinical question: Do weekend admissions for failure to thrive (FTT) result in higher costs and length of stay (LOS)?
Background: FTT accounts for up to 5% of all admissions for children younger than 2 years of age. The optimal approach to inpatient or outpatient care is not well defined. Hospitalizations sometimes are used to facilitate costly and intense workups for organic disease. Given the nonurgent nature of this condition and expected barriers to efficient workup on weekends, it is likely that weekend admissions for FTT might not add much value.
Study design: Retrospective cohort study.
Setting: Forty-two tertiary-care pediatric hospitals.
Synopsis: A total of 23,332 children younger than 2 were studied over an eight-year period. Saturday and Sunday admissions resulted in an average increase in LOS by 1.93 days and an increase in cost by $2,785 when compared with weekday admissions. Patients admitted on weekends were more likely to have imaging studies and lab tests performed, but were less likely to have a discharge diagnosis of FTT. The authors estimate that if one-half of the weekend admissions from 2010 with a consistent FTT diagnosis at admission and discharge were converted to a Monday admission, $534,145 in savings to the health-care system would result.
One notable limitation of the authors’ conclusions is that patients admitted on weekends appeared to have more organic diagnoses documented and might in fact have been more acutely ill, requiring more workup and intervention. Researchers were not able to further explore this using the administrative data. Nonetheless, a subset of weekend admissions with a consistent FTT diagnosis appeared to represent no value added to the system, and potentially could have resulted in a $3.5 million cost savings had they simply been admitted instead on a weekday.
Bottom line: Nonurgent weekend admissions for FTT are inefficient.
Citation: Thompson RT, Bennett WE, Finnell SME, Downs SM. Increased length of stay and costs associated with weekend admissions for failure to thrive. Pediatrics. 2012;131:e805-e810.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Do weekend admissions for failure to thrive (FTT) result in higher costs and length of stay (LOS)?
Background: FTT accounts for up to 5% of all admissions for children younger than 2 years of age. The optimal approach to inpatient or outpatient care is not well defined. Hospitalizations sometimes are used to facilitate costly and intense workups for organic disease. Given the nonurgent nature of this condition and expected barriers to efficient workup on weekends, it is likely that weekend admissions for FTT might not add much value.
Study design: Retrospective cohort study.
Setting: Forty-two tertiary-care pediatric hospitals.
Synopsis: A total of 23,332 children younger than 2 were studied over an eight-year period. Saturday and Sunday admissions resulted in an average increase in LOS by 1.93 days and an increase in cost by $2,785 when compared with weekday admissions. Patients admitted on weekends were more likely to have imaging studies and lab tests performed, but were less likely to have a discharge diagnosis of FTT. The authors estimate that if one-half of the weekend admissions from 2010 with a consistent FTT diagnosis at admission and discharge were converted to a Monday admission, $534,145 in savings to the health-care system would result.
One notable limitation of the authors’ conclusions is that patients admitted on weekends appeared to have more organic diagnoses documented and might in fact have been more acutely ill, requiring more workup and intervention. Researchers were not able to further explore this using the administrative data. Nonetheless, a subset of weekend admissions with a consistent FTT diagnosis appeared to represent no value added to the system, and potentially could have resulted in a $3.5 million cost savings had they simply been admitted instead on a weekday.
Bottom line: Nonurgent weekend admissions for FTT are inefficient.
Citation: Thompson RT, Bennett WE, Finnell SME, Downs SM. Increased length of stay and costs associated with weekend admissions for failure to thrive. Pediatrics. 2012;131:e805-e810.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Clinical question: Do weekend admissions for failure to thrive (FTT) result in higher costs and length of stay (LOS)?
Background: FTT accounts for up to 5% of all admissions for children younger than 2 years of age. The optimal approach to inpatient or outpatient care is not well defined. Hospitalizations sometimes are used to facilitate costly and intense workups for organic disease. Given the nonurgent nature of this condition and expected barriers to efficient workup on weekends, it is likely that weekend admissions for FTT might not add much value.
Study design: Retrospective cohort study.
Setting: Forty-two tertiary-care pediatric hospitals.
Synopsis: A total of 23,332 children younger than 2 were studied over an eight-year period. Saturday and Sunday admissions resulted in an average increase in LOS by 1.93 days and an increase in cost by $2,785 when compared with weekday admissions. Patients admitted on weekends were more likely to have imaging studies and lab tests performed, but were less likely to have a discharge diagnosis of FTT. The authors estimate that if one-half of the weekend admissions from 2010 with a consistent FTT diagnosis at admission and discharge were converted to a Monday admission, $534,145 in savings to the health-care system would result.
One notable limitation of the authors’ conclusions is that patients admitted on weekends appeared to have more organic diagnoses documented and might in fact have been more acutely ill, requiring more workup and intervention. Researchers were not able to further explore this using the administrative data. Nonetheless, a subset of weekend admissions with a consistent FTT diagnosis appeared to represent no value added to the system, and potentially could have resulted in a $3.5 million cost savings had they simply been admitted instead on a weekday.
Bottom line: Nonurgent weekend admissions for FTT are inefficient.
Citation: Thompson RT, Bennett WE, Finnell SME, Downs SM. Increased length of stay and costs associated with weekend admissions for failure to thrive. Pediatrics. 2012;131:e805-e810.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
The Heart of the Matter
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
A 52 year‐old male presented to the emergency department with a 2‐month history of a sensation of fluttering in his chest and rapid heartbeat. The symptoms occurred episodically 6 to 8 times per day and lasted 15 to 60 minutes without associated chest pain, lightheadedness, or syncope. Over the past 2 weeks, he also began to experience dyspnea with minimal exertion.
These symptoms strongly hint at a cardiac dysrhythmia. Premature atrial and ventricular beats are frequent causes of palpitations in outpatients; however, the associated dyspnea on exertion indicates a more serious etiology. The 2‐month duration and absence of more severe sequelae up until now are points against life‐threatening ventricular tachycardia. A supraventricular arrhythmia would be most likely, especially atrial fibrillation, atrial flutter, atrioventricular nodal re‐entrant (AVNRT) or atrioventricular re‐entrant tachycardia (AVRT).
Evaluation should proceed along 2 parallel paths: to diagnose the specific type of arrhythmia and to uncover predisposing conditions. Etiologies of supraventricular arrhythmias include hypertensive heart disease, other structural heart disease including cardiomyopathy, pulmonary disease (eg, chronic obstructive pulmonary disease, pulmonary hypertension, or pulmonary embolism), pericardial disease, hyperthyroidism, sympathomimetic drug use, and in the case of AVRT, an underlying accessory pathway.
The patient's past medical history included hyperlipidemia. Two years prior, his electrocardiogram (ECG) at the time of a health insurance screening had demonstrated sinus rhythm with Q waves in leads III and aVF, and T wave inversions in the inferolateral and anterior leads. An exercise treadmill thallium test at that time demonstrated an area of reversibility in the inferior wall of the left ventricle and a normal ejection fraction. Coronary angiography revealed focal inferior and apical hypokinesis, with frequent premature ventricular contractions (PVCs) and normal coronary arteries.
These prior studies reveal an underlying cardiomyopathy. An ischemic etiology is less likely in the face of normal‐appearing coronary arteries, and he lacks a history of hypertension. Hypertrophic and restrictive cardiomyopathies are possibilities, and tachycardia‐induced cardiomyopathy is an uncommon cause to consider. The pattern of wall‐motion abnormalities is not classic for the Takotsubo phenomenon of apical ballooning, which is typically transient, related to stress, and more common in women. Frequent PVCs are associated with an increased risk of sudden death. I would inquire about illicit drug use and family history of sudden death or cardiac disease.
The patient was a married Caucasian male who reported significant stress related to his career at a software company. He drank 4 glasses of red wine weekly and never smoked cigarettes. He last used cocaine 30 years previously and denied ever using intravenous drugs. Prior to this illness he exercised regularly and traveled frequently to Europe, China, and Japan. He had no family history of cardiac disease or sudden cardiac death. On review of systems, he endorsed a dry cough for 3 weeks without fever, chills, or sweats, and he denied rashes or joint pains. Medications included aspirin, metoprolol, ezetimibe/simvastatin, fish oil, vitamin E, saw palmetto, glucosamine, chondroitin, and a multivitamin.
His remote cocaine use may have predisposed him to cardiomyopathy and hints at ongoing unacknowledged use, but otherwise the additional history is not helpful.
On physical examination, the patient appeared ill. His heart rate was 86 beats per minute, blood pressure 114/67 mm Hg, temperature 36.4C, respiratory rate 18 breaths per minute, and oxygen saturation was 95% while breathing ambient air. There was no conjunctival erythema or pallor, and the oropharynx was moist. The jugular venous pressure (JVP) was not elevated. The heart rhythm was irregular, with a variable intensity of the first heart sound; there were no murmurs or gallops. The apical impulse was normal. The lungs were clear to auscultation. The abdomen was soft, nontender, and nondistended without hepatosplenomegaly. The extremities were without clubbing, cyanosis, or edema. There was no joint swelling. Neurological examination was normal.
In this ill‐appearing patient with 2 months of palpitations, dry cough, and dyspnea on exertion, 2 diagnostic possibilities leap to the front: primary cardiac disease or a primary pulmonary disorder producing a cardiac arrhythmia. The normal JVP, apical impulse, clear lungs, and absence of edema indicate he does not have decompensated heart failure. However, based on prior studies that demonstrated structural heart disease, a cardiac etiology remains more probable. An oxygen saturation of 95% is not normal in a 52‐year‐old nonsmoker and needs to be investigated.
The white blood cell count was 10,000/mm3 with a normal differential, hemoglobin was 15 g/dL, and platelets were 250,000/mm3. Chemistries including sodium, potassium, chloride, bicarbonate, blood urea nitrogen, creatinine, glucose, calcium, magnesium, total protein, albumin, liver enzymes, and troponin‐I were all normal.
ECG (Figure 11) demonstrated sinus rhythm with an incomplete right bundle branch block, right axis deviation, low voltage, premature atrial contractions, and frequent multiform PVCs with couplets and triplets. Chest radiographs (Figure 22) demonstrated bilateral pleural effusions and a borderline enlarged cardiac silhouette.
The review of systems, physical exam, and laboratory tests provided no evidence of widespread systemic disease, promoting the hypothesis that a primary cardiac or pulmonary disorder is responsible for this patient's illness. The markedly abnormal ECG with conduction disturbance and ventricular ectopy provide further evidence of cardiomyopathy. Cardiomyopathies can be categorized as restrictive, dilated, hypertrophic, arrhythmogenic right ventricular, and miscellaneous causes. Transthoracic echocardiogram is the next key diagnostic test.
The patient was admitted to the hospital. Over the first 24 hours, serial ECGs and telemetry demonstrated runs of ventricular tachycardia at a rate of 169 beats per minute, frequent multiform PVCs, bifascicular block, and runs of supraventricular tachycardia.
Transthoracic echocardiogram showed right and left atrial enlargement, 2+ mitral regurgitation, an estimated right ventricular peak pressure of 35 mm Hg, severe left ventricular global hypokinesis with ejection fraction of 20% to 25%, and moderate right ventricular global hypokinesis. Oral amiodarone was administered, and subsequently an internal cardiac defibrillator (ICD) was placed.
I suspect the pulmonary hypertension and mitral regurgitation are consequences of left ventricular impairment, and therefore are not useful diagnostic clues. By contrast, the presence of severe biventricular failure narrows the diagnostic possibilities considerably. I would attempt to obtain the prior coronary angiography films to confirm the presence of normal coronary arteries. In the absence of coronary artery disease, biventricular failure suggests an advanced infiltrative or dilated cardiomyopathy, because hypertrophic cardiomyopathies are less likely to impair the right ventricle this profoundly.
Causes of restrictive cardiomyopathy in adults include amyloidosis, hemochromatosis, sarcoidosis, and the hypereosinophilic syndrome. Dilated cardiomyopathy may arise from antecedent myocarditis from numerous viruses including parvovirus B19, human herpesvirus 6, coxsackievirus, influenza, human immunodeficiency virus (HIV), or from other infections such as Chagas and Lyme disease, toxins (including alcohol and cocaine), autoimmune disease, hypothyroidism, peripartum, genetic causes, nutritional deficiency, or may be idiopathic.
I would check for antibodies to HIV, serum thyrotropin, transferrin saturation, and ferritin, test for serum and urine light chains (looking for evidence of AL amyloid), and obtain a toxicology screen. I would also obtain a computed tomography (CT) scan of the chest to look for supportive evidence of sarcoidosis in this mildly hypoxic patient.
Prior coronary angiography films were unobtainable. Repeat cardiac catheterization demonstrated normal coronary arteries, mildly enlarged left ventricle with ejection fraction of 35%. The mean right atrial, right ventricular end‐diastolic, and left ventricular end‐diastolic pressures were equal at 11 mm Hg, pulmonary capillary wedge pressure was 8 mm Hg. Serologies for coxsackie B, HIV, syphilis, cytomegalovirus, Epstein‐Barr virus, and hepatitis B and C were negative. A purified protein derivative was placed and was nonreactive 48 hours later. Erythrocyte sedimentation rate, C‐reactive protein, antinuclear antibodies, rheumatoid factor, and antibodies to citrullinated peptide were negative. Serum angiotensin‐converting enzyme (ACE) level was normal, lysozyme was elevated at 27 g/mL (normal range, 917), and interleukin (IL)6 was elevated at 27 pg/mL (normal range, 05). Serum protein electrophoresis, serum thyrotropin, transferrin saturation, and ferritin were normal.
The finding of equalization of diastolic pressures at catheterization suggests either constrictive or restrictive physiology; pressure measurements alone cannot distinguish the 2. In the absence of an obvious etiology of constrictive pericarditis (eg, tuberculosis, prior radiation therapy, or cardiac surgery), I remain concerned about infiltrative diseases. Normal iron studies rule out hemochromatosis, and the absence of peripheral eosinophilia removes hypereosinophilic syndrome as a diagnostic consideration. Sarcoidosis can definitely manifest with conduction block as well as biventricular failure, as can amyloidosis. By the time cardiac involvement manifests in sarcoidosis, pulmonary disease is often present, although it may be subclinical. Chest radiography and serum ACE levels are neither sensitive nor specific for screening for pulmonary sarcoidosis. Lysozyme and IL‐6 levels may be elevated in sarcoid, but these too are not specific.
Cardiac involvement in amyloidosis is typically due to AL amyloid light chain deposition associated with a plasma cell dyscrasia. I would expect evidence of organ involvement elsewhere, such as the liver, intestinal tract, tongue, peripheral nerves, or kidneys, none of which are evident in this man. Furthermore, lung involvement in amyloidosis is much less common than in sarcoid. If chest CT fails to demonstrate evidence of sarcoidosis, assays for light chains in the serum and urine might be warranted, as serum protein electrophoresis may fail to detect the abnormal paraprotein.
Chest CT demonstrated bronchial thickening and peribronchovascular bundle ground‐glass opacification, predominantly in the apical lobes with diffuse nodules, and mediastinal lymphadenopathy.
Taken together with the rest of this patient's illness, the CT findings are highly suspicious for sarcoidosis. Biopsy confirmation is essential prior to initiating immunosuppressive therapy. Endomyocardial biopsy and transbronchial biopsy would both be reasonable options; I would discuss these possibilities with pulmonary and cardiology consultants.
An endomyocardial biopsy was performed. The results (Figure 33) revealed the presence of noncaseating granulomas. A diagnosis of cardiac and pulmonary sarcoidosis was made, and treatment with corticosteroids was initiated. At follow‐up 3 years later, he was stable with New York Heart Association class II symptoms and an ejection fraction of 40% to 45%.
DISCUSSION
In outpatient medical practice, up to 16% of individuals report palpitations.[1] In 1 study, primary cardiac disorders accounted for 43% of palpitations, and clinically significant arrhythmias were found in 19% of patients.[2] A history of cardiac disease substantially raises the probability of an arrhythmic etiology of palpitations; over 90% of cases of palpitations in patients with prior cardiac disease are due to arrhythmias.[3]
In patients with palpitations, the history and physical examination do not reliably differentiate patients with significant arrhythmias from those without arrhythmias or those with benign arrhythmias (PVCs and sinus tachycardia). In a recent systematic review, palpitations awakening patients from sleep or occurring while at work, or a known history of cardiac disease, modestly increase the probability of a cardiac arrhythmia, with positive likelihood ratios of 2.03 to 2.29. On the other hand, palpitations lasting 5 minutes and a known history of panic disorder make an arrhythmia much less likely. Interestingly, palpitations associated with a regular rapid‐pounding sensation in the neck (as opposed to neck fullness) substantially increase the probability of AVNRT with an impressive likelihood ratio of 177.[3]
Sarcoidosis is a rare cause of palpitations and arrhythmias. Most commonly seen in young and middle‐aged adults, sarcoidosis is a disorder of unknown cause characterized by the formation of granulomas in multiple organs. Cardiac involvement is detected in 20% to 30% of sarcoidosis patients at autopsy, but only 5% of patients have clinically significant cardiac involvement.[4] Cardiac involvement can be the presenting and lone feature of sarcoidosis or may occur later in a patient with multisystem disease.
Within the heart, sarcoid granulomas are most abundant in the myocardium of the left ventricular free wall followed by the interventricular septum, right ventricle, and atria. The diffuse cardiac involvement explains the protean clinical and electrocardiographic manifestations seen in cardiac sarcoid. Symptoms of cardiac disease include palpitations, syncope, sudden death, or heart failure. The most common ECG manifestations are heart blocks of all types, followed by ventricular arrhythmias and then supraventricular arrhythmias, the latter attributed to secondary atrial enlargement or direct atrial infiltration by granuloma.[5]
The diagnosis of sarcoidosis is challenging. Presenting clinical features, physical exam, routine laboratory tests, ECG, and echocardiography are neither sensitive nor specific. Among the noninvasive tests, serum ACE has been commonly used, but its low sensitivity ranging from 60% to 77%[6, 7, 8] and 50% specificity[8] limit its usefulness in the diagnosis of sarcoid. IL‐6 and lysozyme are other serum markers sometimes obtained in cases of suspected sarcoid, but they too lack adequate sensitivity and specificity to be useful diagnostic tools.[8, 9]
When available, cardiac magnetic resonance imaging (MRI) can enhance clinicians' ability to diagnose cardiac sarcoidosis. It demonstrates zones of thinning and segmental myocardial wall motion abnormalities with increased signal intensity, more pronounced on T2‐weighted images due to inflammation and granulomatous edema. One study reported 100% sensitivity and 78% specificity of MRI in diagnosing cardiac sarcoid.[10]
Because of the limitations of noninvasive tests, tissue biopsy is necessary to diagnose sarcoidosis. If an accessible extracardiac site, such as an enlarged lymph node or skin lesion, is unavailable, a more invasive biopsy is recommended. Transbronchial biopsy is an option if there is obvious thoracic disease. Another alternative is to obtain a 18‐fluorodeoxyglucose positron emission tomography (18FDG‐PET) scan to identify hypermetabolic granulomas, which can be targeted for biopsy. For cardiac sarcoidosis, endomyocardial biopsy is often performed. This procedure is generally quite safe, with severe complications such as right ventricular perforation occurring in fewer than 1% of procedures.[11] However, the patchy nature of heart involvement in sarcoidosis results in a sensitivity as low as 20%.[12] Despite its low yield, according to guidelines from the American College of Cardiology and the American Heart Association, patients with unexplained heart failure of 3 months' duration associated with heart block or ventricular arrhythmias have a class I indication for endomyocardial biopsy.[11]
The prognosis of sarcoidosis is generally favorable, with fewer than 5% of patients dying from the disease. Although the impact of cardiac involvement is poorly established, the available literature indicate a worse prognosis for patients with symptomatic heart disease due to sarcoidosis. In 1 series, over half of 19 patients with cardiac involvement were either dead or required an ICD or pacemaker within 2 years of detection, as opposed to none of 82 sarcoid patients without clinically apparent cardiac involvement.[13]
The mainstay of treatment of cardiac sarcoidosis is corticosteroids, which may halt disease progression and improve survival, but do not reduce the incidence of ventricular arrhythmias. Initially, 1 mg/kg doses of prednisone dose are administered daily. Patients should be reassessed for response to treatment, and repeat ejection fraction measurement by echocardiogram should be obtained if symptoms worsen. The use of serial serum ACE levels to monitor disease activity is controversial. For patients responding to prednisone, the dose can be tapered over a period of 6 months to a maintenance daily dose of 10 to 15 mg, with a goal of eventually stopping therapy if disease is quiescent.[14] For patients who do not respond to glucocorticoids or who experience intolerable side effects, other immunosuppressive agents have been tried with reported success based on limited data. Options include methotrexate, azathioprine, hydroxychloroquine, cyclophosphamide, and infliximab.[5] Treatment of asymptomatic or minimally symptomatic patients with corticosteroids remains controversial.[14]
Adjunctive treatments are often necessary in cardiac sarcoidosis. Permanent pacemaker implantation is indicated if there is complete atrioventricular block or other high‐grade conduction system disease. Survivors of sudden cardiac death, individuals with refractory ventricular arrhythmias, and those with severely impaired systolic function are candidates for ICDs.[15] Catheter radiofrequency ablation may be effective in patients with ventricular tachyarrhythmias.[16]
Cardiac sarcoidosis is important to suspect in a patient with unexplained cardiomyopathy associated with conduction blocks or tachyarrhythmias because it is potentially reversible. Diagnosis can be elusive, as noninvasive tests lack sufficient sensitivity and specificity to establish the presence or absence of the disorder. Biopsy of affected organs is essential to identify the noncaseating granulomas that characterize the disease. When no extracardiac target exists, clinicians may need an endomyocardial biopsy to get to the heart of the matter.
CLINICAL TEACHING POINTS
- A history of cardiac disease substantially raises the possibility of an arrhythmic etiology of palpitations.
- Cardiac involvement in sarcoidosis can be asymptomatic or include conduction blocks, supraventricular and ventricular tachyarrhythmias, or cardiomyopathy.
- Cardiac sarcoid can be an elusive diagnosis to establish, because both noninvasive tests and endomyocardial biopsy demonstrate low sensitivity.
- Cardiac sarcoidosis portends a worse prognosis than sarcoid in general, but is a potentially reversible condition that therefore warrants an aggressive approach to establishing a diagnosis.
Acknowledgments
The authors thank Ellen Killebrew, MD, for help with the formal interpretation of the admission ECG.
Disclosures
Dr. Baudendistel is a former Deputy Editor and CME Editor of the Journal of Hospital Medicine, a position he ended in 2011. He received a stipend of less than $2000 for this work in 2010 and 2011. The authors are not aware of any conflicts of interest related to this article. The initial oral part of this presentation was presented at the University of California Davis Grand Rounds on August 16, 2010.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
- , , , . Predictors of persistent palpitations and continued medical utilization. J Fam Pract. 1996;42:465–472.
- , . Evaluation and outcomes of patients with palpitations. Am J Med. 1996;100:138–148.
- , , , , . Does this patient with palpitations have a cardiac arrhythmia? JAMA. 2009;302:2135–2143.
- , . Myocardial sarcoidosis in forensic medicine. Am J Forensic Med Pathol. 1999;20:52–56.
- , , , et al. Cardiac sarcoidosis. Am Heart J. 2009;157:9–21.
- , , . Sarcoidosis. N Engl J Med. 2007;357:2153–2165.
- , . An angiotensin‐converting enzyme (ACE) inhibitor in human serum. Increased sensitivity of the serum ACE assay for detecting active sarcoidosis. Chest. 1986;90:869–875.
- , , , et al. Comparative evaluation of serum markers in pulmonary sarcoidosis. Chest. 2010;137:1391–1397.
- , , . Cardiac sarcoidosis: cytokine patterns in the course of the disease. Arch Pathol Lab Med. 2003;127:1207–1210.
- , , , et al. Evaluation of the accuracy of gadolinium‐enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. J Am Coll Cardiol. 2005;45:1683–1690.
- , , . Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86:1095–1102.
- , , , , , . Histologic diagnostic rate of cardiac sarcoidosis: evaluation of endomyocardial biopsies. Am Heart J. 1999;138:299–302.
- , , , et al. Cardiac involvement in patients with pulmonary sarcoidosis assessed at two university medical centers in the Netherlands. Chest. 2005;128(1):30–35.
- , , , et al. Prognostic determinants of long‐term survival in Japanese patients with cardiac sarcoidosis treated with prednisone. Am J Cardiol. 2001;88:1006–1010.
- , , , . The automated implantable cardiac defibrillator. Prophylaxis in cardiac sarcoidosis. Chest. 1994;106:1603–1607.
- , , , et al. Ventricular tachycardia in cardiac sarcoidosis controlled by radiofrequency catheter ablation. Intern Med. 2011;50:1201–1206.
How Should Common Symptoms at the End of Life be Managed?
Case
A 58-year-old male with colon cancer metastatic to the liver and lungs presents with vomiting, dyspnea, and abdominal pain. His disease has progressed through third-line chemotherapy and his care is now focused entirely on symptom management. He has not had a bowel movement in five days and he began vomiting two days ago.
Overview
The majority of patients in the United States die in acute-care hospitals. The Study to Understand Prognosis and Preferences for Outcomes and Risks of Treatments (SUPPORT), which evaluated the courses of close to 10,000 hospitalized patients with serious and life-limiting illnesses, illustrated that patients’ end-of-life (EOL) experiences often are characterized by poor symptom management and invasive care that is not congruent with the patients’ overall goals of care.1 Studies of factors identified as priorities in EOL care have consistently shown that excellent pain and symptom management are highly valued by patients and families. As the hospitalist movement continues to grow, hospitalists will play a large role in caring for patients at EOL and will need to know how to provide adequate pain and symptom management so that high-quality care can be achieved.
Pain: A Basic Tenet
A basic tenet of palliative medicine is to evaluate and treat all types of suffering.2 Physical pain at EOL is frequently accompanied by other types of pain, such as psychological, social, religious, or existential pain. However, this review will focus on the pharmacologic management of physical pain.
Pain management must begin with a thorough evaluation of the severity, location, and characteristics of the discomfort to assess which therapies are most likely to be beneficial (see Table 1).3 The consistent use of one scale of pain severity (such as 0-10, or mild/moderate/severe) assists in the choice of initial dose of pain medication, in determining the response to the medication, and in assessing the need for change in dose.4
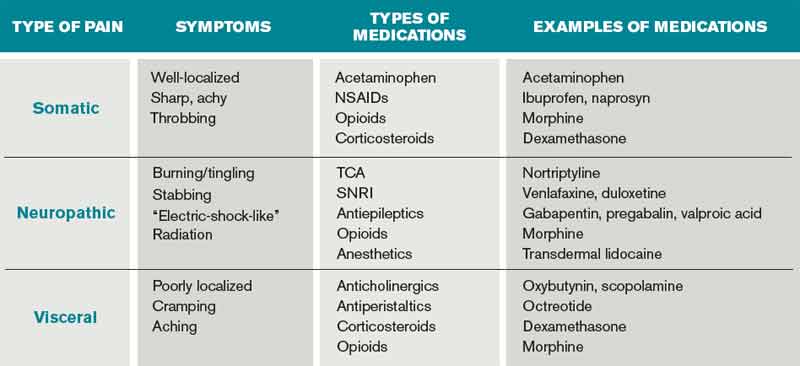
Opioids are the foundation of pain management in advanced diseases because they are available in a number of formulations and, when dosed appropriately, they are effective and safe. Starting doses and equianalgesic doses of common opioids are presented in Table 2. Guidelines recommend the use of short-acting opioids for dose titration to gain control of poorly controlled pain.3 If a patient is experiencing mild pain on a specific regimen, the medication dose can be increased up to 25%; by 25% to 50%, if pain is moderate; and 50% to 100%, if severe.5 When the pain is better-controlled, the total amount of pain medication used in 24 hours (24-hour dose) can be converted to a long-acting formulation for more consistent pain management. Because there is a constant component to most advanced pain syndromes, it is recommended that pain medication is given on a standing basis, with as-needed (prn) doses available for exacerbations of pain.3 Prn doses of short-acting medication (equivalent to approximately 10% of the 24-hour dose of medication) should be available at one- or two-hour intervals prn (longer if hepatic or renal impairment is present) for IV or PO medications, respectively.
Opioids often are categorized as low potency (i.e. codeine, hydrocodone) and high-potency (i.e. oxycodone, morphine, hydromorphone, fentanyl). When given in “equianalgesic doses,” the analgesic effect and common side effects (nausea/vomiting, constipation, sedation, confusion, pruritis) of different opioids can vary in different patients. Due to differences in levels of expressed subtypes of opioid receptors, a given patient might be more sensitive to the analgesic effect or side effects of a specific medication. Therefore, if dose escalation of one opioid is inadequate to control pain and further increases in dose are limited by intolerable side effects, rotation to another opioid is recommended.4 Tables documenting equianalgesic doses of different opioids are based on only moderate evidence from equivalency trials performed in healthy volunteers.6 Due to interpatient differences in responses, it is recommended that the equianalgesic dose of the new medication be decreased by 25% to 50% for initial dosing.5
Certain treatments are indicated for specific pain syndromes. Bony metastases respond to NSAIDs, bisphosphonates, and radiation therapy in addition to opioid medications. As focal back pain is the first symptom of spinal cord compression, clinicians should have a high index of suspicion for compression in any patient with malignancy and new back pain. Steroids and radiation therapy are considered emergent treatments for pain control and to prevent paralysis in this circumstance. Pain due to bowel obstruction is usually colicky in nature and responds well to octreotide as discussed in the section on nausea and vomiting. Steroids (such as dexamethasone 4 mg PO bid-tid) might be an effective adjuvant medication in bone pain, tumor pain, or inflammation.
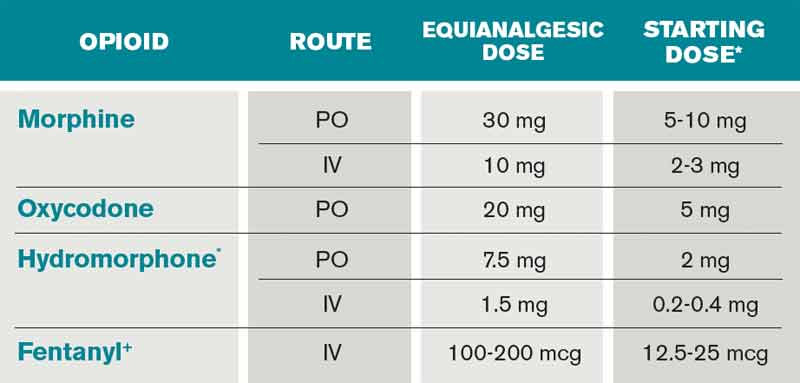
*Half this dose should be used in renal or liver dysfunction and in the elderly.
Preferred in renal dysfunction.
SOURCES: Adapted from Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008, and Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
Back to the Case
At home, the patient was taking 60 mg of extended-release morphine twice daily and six doses per day of 15-mg immediate-release morphine for breakthrough pain. This is the equivalent of 210 mg of oral morphine in 24 hours. His pain is severe on this regimen, but it is unclear how much of this medication he is absorbing due to his vomiting. Using the IV route of administration and a patient-controlled analgesia (PCA) system will enable rapid dose titration and pain control. The equivalent of the 24-hour dose of 210 mg oral morphine is 70 mg IV morphine, which is equivalent to a drip basal rate of approximately 3 mg IV morphine per hour. This basal rate with a bolus dose of 7 mg (10% of the 24-hour dose) IV morphine q1 hour prn is reasonable as a starting point.
Review of the Data: Nausea and Vomiting
Nausea and vomiting affect 40% to 70% of patients in a palliative setting.7 A thorough history and physical exam can enable one to determine the most likely causes, pathways, and receptors involved in the process of nausea and vomiting. It is important to review the timing, frequency, and triggers of vomiting. The oral, abdominal, neurologic, and rectal exams, in addition to a complete chemistry panel, offer helpful information. The most common etiologies and recommended medications are included in Table 3. It is worthwhile to note that serotonin-antagonists (i.e. ondansetron) are first-line therapies only for chemotherapy and radiation-therapy-induced emesis. If a 24-hour trial of one antiemetic therapy is ineffective, one should reassess the etiology and escalate the antiemetic dose, or add a second therapy with a different (pertinent) mechanism of action. Although most studies of antiemetic therapy are case series, there is good evidence for this mechanistic approach.8
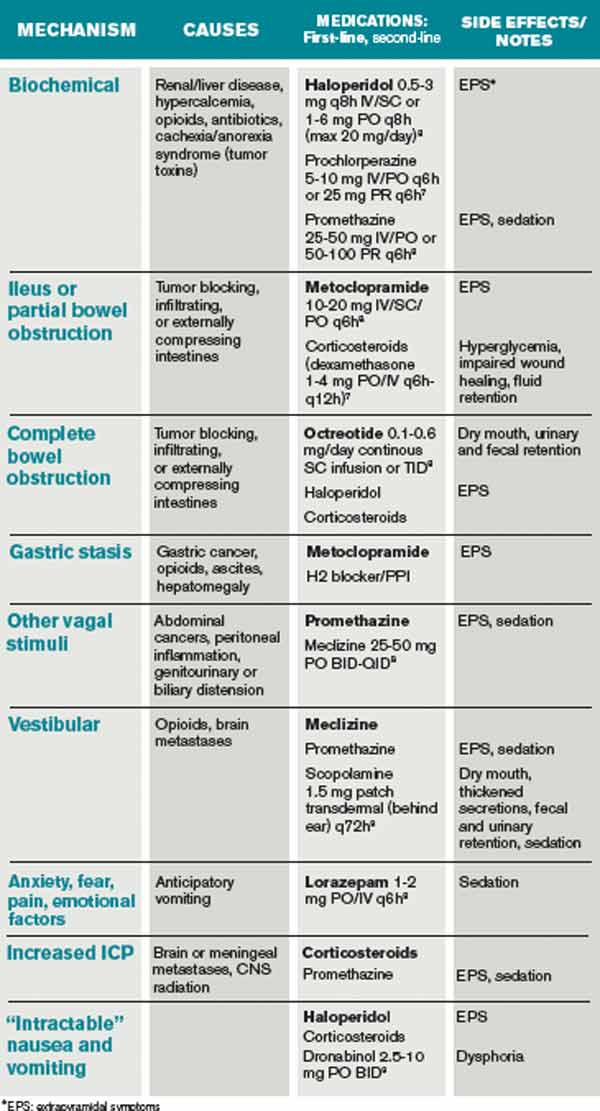
*EPS: extrapyramidal symptoms
The various insults and pathways that can cause vomiting are quite complex. The medullary vomiting center (VC) receives vestibular, peripheral (via splanchnic and vagal nerves), and higher cortical inputs and is the final common pathway in the vomiting reflex. The chemoreceptor trigger zone (CTZ) near the fourth ventricle receives input from the vagal and splanchnic nerves, and generates output to the VC.
General dietary recommendations are to avoid sweet, fatty, and highly salted or spiced foods. Small portions of bland foods without strong odors are best tolerated.7 Constipation commonly contributes to nausea and vomiting and should be managed with disimpaction, enemas, and laxatives as tolerated. Imaging may be required to make the important distinction between partial and complete bowel obstruction, as the treatments differ. Surgical procedures, such as colostomy or placement of a venting gastrostomy tube, can relieve pain and vomiting associated with complete bowel obstruction.
Back to the Case
The patient is found to have a fecal impaction on rectal exam, but vomiting persists after disimpaction and enema use. Imaging documents a complete bowel obstruction at the site of a palpable mass in the right upper quadrant and multiple large hepatic metastases. Octreotide is initiated to decrease intestinal secretions and peristalsis. Steroids are given to decrease tumor burden and associated inflammation in the intestine and liver, as well as to relieve distension of the hepatic capsule. Haloperidol is used in low doses to control episodes of nausea.
Review of the Data: Dyspnea
Dyspnea is a common symptom faced by patients at EOL. An estimated 50% of patients who are evaluated in acute-care hospitals seek treatment for the management of this often-crippling symptom.10 Unfortunately, as disease burden progresses, the incidence of dyspnea increases towards EOL, and the presence and severity of dyspnea is strongly correlated with mortality.
It is imperative for providers to appreciate that dyspnea is a subjective symptom, similar to pain. The presence and severity of dyspnea, therefore, depends on patient report. Given its subjective nature, the degree of dyspnea experienced by a patient might not correlate with objective laboratory findings or test results. In practice, the severity of dyspnea is commonly assessed with a numeric rating scale (0-10), verbal analogue scale, or with verbal descriptors (mild, moderate, severe). It is important to determine the underlying etiology of the dyspnea and, if possible, to target interventions to relieve the underlying cause. However, at the end of life, the burdens of invasive studies to determine the exact cause of dyspnea might outweigh the benefits, and invasive testing might not correlate with patients’ and families’ goals of care. In that instance, the goal of treatment should be aggressive symptom management and providers should use clinical judgment to tailor therapies based on the patient’s underlying illness, physical examination, and perhaps on noninvasive radiological or laboratory findings. Below are nonpharmacological and pharmacological interventions that can be employed to help alleviate dyspnea in the actively dying patient.
Nonpharmacological Management
A handheld fan aimed near the patient’s face has been shown to reduce the sensation of dyspnea.11 This relatively safe and inexpensive intervention has no major side effects and can provide improvement in this distressing symptom.
Often, the first line of therapy in the hospital setting for a patient reporting dyspnea is the administration of oxygen therapy. However, recent evidence does not show superiority of oxygen over air inhalation via nasal prongs for dyspnea in patients with advanced cancer or heart failure.12,13
Pharmacological Management
Opioids are first-line therapy for alleviating dyspnea in patients at EOL. The administration of opioids has been shown in systematic reviews to provide effective management of dyspnea.14,15 Practice guidelines by leading expert groups advocate for the use of opioids in the management of dyspnea for patients with advanced malignant and noncancer diseases.10,16 Fear of causing unintended respiratory sedation with opioids limits the prescription of opioids for dyspnea. However, studies have not found a change in mortality with the use of opioids appropriately titrated to control dyspnea.17
Studies examining the role of benzodiazepines in dyspnea management are conflicting. Anecdotal clinical evidence in actively dying patients supports treating dyspnea with benzodiazepines in conjunction with opioid therapy. Benzodiazepines are most beneficial when there is an anxiety-related component to the dyspnea.
Many patients with advanced disease and evidence of airflow obstruction will benefit from nebulized bronchodilator therapy for dyspnea. Patients with dyspnea from fluid overload (i.e. end-stage congestive heart failure or renal disease) might benefit from systemic diuretics. An increasing number of trials are under way to evaluate the efficacy of nebulized furosemide in the symptomatic management of dyspnea.
Back to the Case
The patient’s clinical course decompensates, and he begins to report worsening dyspnea in addition to his underlying pain. He becomes increasingly anxious about what this new symptom means. In addition to having a discussion about disease progression and prognosis, you increase his PCA basal dose to morphine 4 mg/hour to help him with this new symptom. You also add low-dose lorazepam 0.5 mg IV q8 hours as an adjunct agent for his dyspnea. The patient reports improvement of his symptom burden.
Review of the Data: Secretions
Physiological changes occur as a patient enters the active phase of dying. Two such changes are the loss of the ability to swallow and a reduced cough reflex. These changes culminate in an inability to clear secretions, which pool in the oropharynx and the airways. As the patient breathes, air moves over the pooled secretions and produces a gurgling sound that is referred to as the “death rattle.” The onset of this clinical marker has been shown to have significant prognostic significance for predicting imminent death within a period of hours to days. Proposed treatments for the symptom are listed below.
Nonpharmacological Management
Nonpharmacological options include repositioning the patient in a manner that facilitates postural draining.18 Careful and gentle oral suctioning might help reduce secretions if they are salivary in origin. This will not help to clear deeper bronchial secretions. Suctioning of deeper secretions often causes more burden than benefit, as this can cause repeated trauma and possible bleeding.
Family and caregivers at the bedside can find the “death rattle” quite disturbing and often fear that their loved one is “drowning.” Education and counseling that this is not the case, and that the development of secretions is a natural part of the dying process, can help alleviate this concern. Explaining that pharmacological agents can be titrated to decrease secretions is also reassuring to caregivers.
Pharmacological Management
Pharmacological options for secretion management include utilizing anticholinergic medications to prevent the formation of further secretions. These medications are standard of care for managing the death rattle and have been found to be most efficacious if started earlier in the actively dying phase.19,20 Anticholinergic medications include glycopyrrolate (0.2 mg IV q8 hours), atropine sulfate ophthalmological drops (1% solution, 1-2 drops SL q6 hours), hyoscyamine (0.125 mg one to four times a day), and scopolamine (1.5 mg patch q72 hours). These medications all have possible side effects typical of anticholinergic agents, including delirium, constipation, blurred vision, and urinary retention.
Back to the Case
The patient becomes increasingly lethargic. You meet with his family and explain that he is actively dying. His family reiterates that the goals of medical care should focus on maximizing symptom management. His family is concerned about the “gurgly” sound they hear and want to know if that means he is suffering. You educate the family about expected changes that occur with the dying process and inform them that glycopyrrolate 0.2 mg IV q8 hour will be started to minimize further secretions.
Bottom Line
Pain, nausea, dyspnea, and secretions are common end-of-life symptoms that hospitalists should be competent in treating.
Dr. Litrivis is an associate director and assistant professor at the Mount Sinai School of Medicine in New York, and Dr. Neale is an assistant professor at the University of New Mexico School of Medicine in Albuquerque.
References
- The SUPPORT Principal Investigators. A controlled trial to improve the care for seriously ill hospitalized patients. The study to understand prognoses and preferences for outcomes and risks of treatments (SUPPORT). JAMA. 1995;274(20):1591-1598.
- World Health Organization Definition of Palliative Care. World Health Organization website. Available at: http://www.who.int/cancer/palliative/definition/en/. Accessed April 12, 2012.
- NCCN Guidelines Version 2. 2011 Adult Cancer Pain. National Comprehensive Cancer Network website. Available at: http://www.nccn.org/professionals/physician_gls/pdf/pain.pdf. Accessed April 12, 2012.
- Whitecar PS, Jonas AP, Clasen ME. Managing pain in the dying patient. Am Fam Physician. 2000;61(3):755-764.
- Bial A, Levine S. Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Sydney M, et al. Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
- Mannix KA. Gastrointestinal symptoms. In: Doyle D, Hanks G, Cherny N, Calman K, eds. Oxford Textbook of Palliative Medicine. 3rd ed. New York, NY: Oxford University Press; 2005.
- Tyler LS. Nausea and vomiting in palliative care. In: Lipman AG, Jackson KC, Tyler LS, eds. Evidence-Based Symptom Control in Palliative Care. New York, NY: The Hawthorn Press; 2000.
- Policzer JS, Sobel J. Management of Selected Nonpain Symptoms of Life-Limiting Illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 4. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Parshall MB, Schwartzstein RM, Adams L, et al. An official American Thoracic Society statement: update on the mechanisms, assessment, and management of dyspnea. Am J Respir Crit Care Med. 2012;185(4): 435-452.
- Galbraith S, Fagan P, Perkins P, Lynch A, Booth S. Does the use of a handheld fan improve chronic dyspnea? A randomized controlled, crossover trial. J Pain Symptom Manage. 2010;39(5): 831-838.
- Philip J, Gold M, Milner A, Di Iulio J, Miller B, Spruyt O. A randomized, double-blind, crossover trial of the effect of oxygen on dyspnea in patients with advanced cancer. J Pain Symptom Manage. 2006;32(6):541-550.
- Cranston JM, Crockett A, Currow D. Oxygen therapy for dyspnea in adults. Cochrane Database Syst Rev. 2008;(3):CD004769.
- Jennings AL, Davies AN, Higgins JP, Broadley K. Opioids for the palliation of breathlessness in terminal illness. Cochrane Database Syst Rev. 2001;(4):CD002066.
- Ben-Aharon I, Gafter-Gvili A, Paul M, Leibovici, L, Stemmer, SM. Interventions for alleviating cancer-related dyspnea. A systematic review. J Clin Oncol. 2008;26(14): 2396-2404.
- Qaseem A, Snow V, Shekelle P, et al. Evidence-based interventions to improve the palliative care of pain, dyspnea, and depression at the end of life: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148(2):141-146
- Booth S, Moosavi SH, Higginson IJ. The etiology and management of intractable breathlessness in patients with advanced cancer: a systematic review of pharmacological therapy. Nat Clin Pract Oncol. 2008;5(2):90–100.
- Bickel K, Arnold R. EPERC Fast Facts Documents #109 Death Rattle and Oral Secretions, 2nd ed. Available at: http://www.eperc.mcw.edu/EPERC/FastFactsIndex/ff_109.htm. Accessed April 15, 2012.
- Wildiers H, Dhaenekint C, Demeulenaere P, et al. Atropine, hyoscine butylbromide, or scopalamine are equally effective for the treatment of death rattle in terminal care. J Pain Symptom Manage. 2009;38(1):124-133.
- Hugel H, Ellershaw J, Gambles M. Respiratory tract secretions in the dying patient: a comparison between glycopyrronium and hyoscine hydrobromide. J Palliat Med. 2006;9(2):279-285.
Case
A 58-year-old male with colon cancer metastatic to the liver and lungs presents with vomiting, dyspnea, and abdominal pain. His disease has progressed through third-line chemotherapy and his care is now focused entirely on symptom management. He has not had a bowel movement in five days and he began vomiting two days ago.
Overview
The majority of patients in the United States die in acute-care hospitals. The Study to Understand Prognosis and Preferences for Outcomes and Risks of Treatments (SUPPORT), which evaluated the courses of close to 10,000 hospitalized patients with serious and life-limiting illnesses, illustrated that patients’ end-of-life (EOL) experiences often are characterized by poor symptom management and invasive care that is not congruent with the patients’ overall goals of care.1 Studies of factors identified as priorities in EOL care have consistently shown that excellent pain and symptom management are highly valued by patients and families. As the hospitalist movement continues to grow, hospitalists will play a large role in caring for patients at EOL and will need to know how to provide adequate pain and symptom management so that high-quality care can be achieved.
Pain: A Basic Tenet
A basic tenet of palliative medicine is to evaluate and treat all types of suffering.2 Physical pain at EOL is frequently accompanied by other types of pain, such as psychological, social, religious, or existential pain. However, this review will focus on the pharmacologic management of physical pain.
Pain management must begin with a thorough evaluation of the severity, location, and characteristics of the discomfort to assess which therapies are most likely to be beneficial (see Table 1).3 The consistent use of one scale of pain severity (such as 0-10, or mild/moderate/severe) assists in the choice of initial dose of pain medication, in determining the response to the medication, and in assessing the need for change in dose.4
Opioids are the foundation of pain management in advanced diseases because they are available in a number of formulations and, when dosed appropriately, they are effective and safe. Starting doses and equianalgesic doses of common opioids are presented in Table 2. Guidelines recommend the use of short-acting opioids for dose titration to gain control of poorly controlled pain.3 If a patient is experiencing mild pain on a specific regimen, the medication dose can be increased up to 25%; by 25% to 50%, if pain is moderate; and 50% to 100%, if severe.5 When the pain is better-controlled, the total amount of pain medication used in 24 hours (24-hour dose) can be converted to a long-acting formulation for more consistent pain management. Because there is a constant component to most advanced pain syndromes, it is recommended that pain medication is given on a standing basis, with as-needed (prn) doses available for exacerbations of pain.3 Prn doses of short-acting medication (equivalent to approximately 10% of the 24-hour dose of medication) should be available at one- or two-hour intervals prn (longer if hepatic or renal impairment is present) for IV or PO medications, respectively.
Opioids often are categorized as low potency (i.e. codeine, hydrocodone) and high-potency (i.e. oxycodone, morphine, hydromorphone, fentanyl). When given in “equianalgesic doses,” the analgesic effect and common side effects (nausea/vomiting, constipation, sedation, confusion, pruritis) of different opioids can vary in different patients. Due to differences in levels of expressed subtypes of opioid receptors, a given patient might be more sensitive to the analgesic effect or side effects of a specific medication. Therefore, if dose escalation of one opioid is inadequate to control pain and further increases in dose are limited by intolerable side effects, rotation to another opioid is recommended.4 Tables documenting equianalgesic doses of different opioids are based on only moderate evidence from equivalency trials performed in healthy volunteers.6 Due to interpatient differences in responses, it is recommended that the equianalgesic dose of the new medication be decreased by 25% to 50% for initial dosing.5
Certain treatments are indicated for specific pain syndromes. Bony metastases respond to NSAIDs, bisphosphonates, and radiation therapy in addition to opioid medications. As focal back pain is the first symptom of spinal cord compression, clinicians should have a high index of suspicion for compression in any patient with malignancy and new back pain. Steroids and radiation therapy are considered emergent treatments for pain control and to prevent paralysis in this circumstance. Pain due to bowel obstruction is usually colicky in nature and responds well to octreotide as discussed in the section on nausea and vomiting. Steroids (such as dexamethasone 4 mg PO bid-tid) might be an effective adjuvant medication in bone pain, tumor pain, or inflammation.
*Half this dose should be used in renal or liver dysfunction and in the elderly.
Preferred in renal dysfunction.
SOURCES: Adapted from Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008, and Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
Back to the Case
At home, the patient was taking 60 mg of extended-release morphine twice daily and six doses per day of 15-mg immediate-release morphine for breakthrough pain. This is the equivalent of 210 mg of oral morphine in 24 hours. His pain is severe on this regimen, but it is unclear how much of this medication he is absorbing due to his vomiting. Using the IV route of administration and a patient-controlled analgesia (PCA) system will enable rapid dose titration and pain control. The equivalent of the 24-hour dose of 210 mg oral morphine is 70 mg IV morphine, which is equivalent to a drip basal rate of approximately 3 mg IV morphine per hour. This basal rate with a bolus dose of 7 mg (10% of the 24-hour dose) IV morphine q1 hour prn is reasonable as a starting point.
Review of the Data: Nausea and Vomiting
Nausea and vomiting affect 40% to 70% of patients in a palliative setting.7 A thorough history and physical exam can enable one to determine the most likely causes, pathways, and receptors involved in the process of nausea and vomiting. It is important to review the timing, frequency, and triggers of vomiting. The oral, abdominal, neurologic, and rectal exams, in addition to a complete chemistry panel, offer helpful information. The most common etiologies and recommended medications are included in Table 3. It is worthwhile to note that serotonin-antagonists (i.e. ondansetron) are first-line therapies only for chemotherapy and radiation-therapy-induced emesis. If a 24-hour trial of one antiemetic therapy is ineffective, one should reassess the etiology and escalate the antiemetic dose, or add a second therapy with a different (pertinent) mechanism of action. Although most studies of antiemetic therapy are case series, there is good evidence for this mechanistic approach.8
*EPS: extrapyramidal symptoms
The various insults and pathways that can cause vomiting are quite complex. The medullary vomiting center (VC) receives vestibular, peripheral (via splanchnic and vagal nerves), and higher cortical inputs and is the final common pathway in the vomiting reflex. The chemoreceptor trigger zone (CTZ) near the fourth ventricle receives input from the vagal and splanchnic nerves, and generates output to the VC.
General dietary recommendations are to avoid sweet, fatty, and highly salted or spiced foods. Small portions of bland foods without strong odors are best tolerated.7 Constipation commonly contributes to nausea and vomiting and should be managed with disimpaction, enemas, and laxatives as tolerated. Imaging may be required to make the important distinction between partial and complete bowel obstruction, as the treatments differ. Surgical procedures, such as colostomy or placement of a venting gastrostomy tube, can relieve pain and vomiting associated with complete bowel obstruction.
Back to the Case
The patient is found to have a fecal impaction on rectal exam, but vomiting persists after disimpaction and enema use. Imaging documents a complete bowel obstruction at the site of a palpable mass in the right upper quadrant and multiple large hepatic metastases. Octreotide is initiated to decrease intestinal secretions and peristalsis. Steroids are given to decrease tumor burden and associated inflammation in the intestine and liver, as well as to relieve distension of the hepatic capsule. Haloperidol is used in low doses to control episodes of nausea.
Review of the Data: Dyspnea
Dyspnea is a common symptom faced by patients at EOL. An estimated 50% of patients who are evaluated in acute-care hospitals seek treatment for the management of this often-crippling symptom.10 Unfortunately, as disease burden progresses, the incidence of dyspnea increases towards EOL, and the presence and severity of dyspnea is strongly correlated with mortality.
It is imperative for providers to appreciate that dyspnea is a subjective symptom, similar to pain. The presence and severity of dyspnea, therefore, depends on patient report. Given its subjective nature, the degree of dyspnea experienced by a patient might not correlate with objective laboratory findings or test results. In practice, the severity of dyspnea is commonly assessed with a numeric rating scale (0-10), verbal analogue scale, or with verbal descriptors (mild, moderate, severe). It is important to determine the underlying etiology of the dyspnea and, if possible, to target interventions to relieve the underlying cause. However, at the end of life, the burdens of invasive studies to determine the exact cause of dyspnea might outweigh the benefits, and invasive testing might not correlate with patients’ and families’ goals of care. In that instance, the goal of treatment should be aggressive symptom management and providers should use clinical judgment to tailor therapies based on the patient’s underlying illness, physical examination, and perhaps on noninvasive radiological or laboratory findings. Below are nonpharmacological and pharmacological interventions that can be employed to help alleviate dyspnea in the actively dying patient.
Nonpharmacological Management
A handheld fan aimed near the patient’s face has been shown to reduce the sensation of dyspnea.11 This relatively safe and inexpensive intervention has no major side effects and can provide improvement in this distressing symptom.
Often, the first line of therapy in the hospital setting for a patient reporting dyspnea is the administration of oxygen therapy. However, recent evidence does not show superiority of oxygen over air inhalation via nasal prongs for dyspnea in patients with advanced cancer or heart failure.12,13
Pharmacological Management
Opioids are first-line therapy for alleviating dyspnea in patients at EOL. The administration of opioids has been shown in systematic reviews to provide effective management of dyspnea.14,15 Practice guidelines by leading expert groups advocate for the use of opioids in the management of dyspnea for patients with advanced malignant and noncancer diseases.10,16 Fear of causing unintended respiratory sedation with opioids limits the prescription of opioids for dyspnea. However, studies have not found a change in mortality with the use of opioids appropriately titrated to control dyspnea.17
Studies examining the role of benzodiazepines in dyspnea management are conflicting. Anecdotal clinical evidence in actively dying patients supports treating dyspnea with benzodiazepines in conjunction with opioid therapy. Benzodiazepines are most beneficial when there is an anxiety-related component to the dyspnea.
Many patients with advanced disease and evidence of airflow obstruction will benefit from nebulized bronchodilator therapy for dyspnea. Patients with dyspnea from fluid overload (i.e. end-stage congestive heart failure or renal disease) might benefit from systemic diuretics. An increasing number of trials are under way to evaluate the efficacy of nebulized furosemide in the symptomatic management of dyspnea.
Back to the Case
The patient’s clinical course decompensates, and he begins to report worsening dyspnea in addition to his underlying pain. He becomes increasingly anxious about what this new symptom means. In addition to having a discussion about disease progression and prognosis, you increase his PCA basal dose to morphine 4 mg/hour to help him with this new symptom. You also add low-dose lorazepam 0.5 mg IV q8 hours as an adjunct agent for his dyspnea. The patient reports improvement of his symptom burden.
Review of the Data: Secretions
Physiological changes occur as a patient enters the active phase of dying. Two such changes are the loss of the ability to swallow and a reduced cough reflex. These changes culminate in an inability to clear secretions, which pool in the oropharynx and the airways. As the patient breathes, air moves over the pooled secretions and produces a gurgling sound that is referred to as the “death rattle.” The onset of this clinical marker has been shown to have significant prognostic significance for predicting imminent death within a period of hours to days. Proposed treatments for the symptom are listed below.
Nonpharmacological Management
Nonpharmacological options include repositioning the patient in a manner that facilitates postural draining.18 Careful and gentle oral suctioning might help reduce secretions if they are salivary in origin. This will not help to clear deeper bronchial secretions. Suctioning of deeper secretions often causes more burden than benefit, as this can cause repeated trauma and possible bleeding.
Family and caregivers at the bedside can find the “death rattle” quite disturbing and often fear that their loved one is “drowning.” Education and counseling that this is not the case, and that the development of secretions is a natural part of the dying process, can help alleviate this concern. Explaining that pharmacological agents can be titrated to decrease secretions is also reassuring to caregivers.
Pharmacological Management
Pharmacological options for secretion management include utilizing anticholinergic medications to prevent the formation of further secretions. These medications are standard of care for managing the death rattle and have been found to be most efficacious if started earlier in the actively dying phase.19,20 Anticholinergic medications include glycopyrrolate (0.2 mg IV q8 hours), atropine sulfate ophthalmological drops (1% solution, 1-2 drops SL q6 hours), hyoscyamine (0.125 mg one to four times a day), and scopolamine (1.5 mg patch q72 hours). These medications all have possible side effects typical of anticholinergic agents, including delirium, constipation, blurred vision, and urinary retention.
Back to the Case
The patient becomes increasingly lethargic. You meet with his family and explain that he is actively dying. His family reiterates that the goals of medical care should focus on maximizing symptom management. His family is concerned about the “gurgly” sound they hear and want to know if that means he is suffering. You educate the family about expected changes that occur with the dying process and inform them that glycopyrrolate 0.2 mg IV q8 hour will be started to minimize further secretions.
Bottom Line
Pain, nausea, dyspnea, and secretions are common end-of-life symptoms that hospitalists should be competent in treating.
Dr. Litrivis is an associate director and assistant professor at the Mount Sinai School of Medicine in New York, and Dr. Neale is an assistant professor at the University of New Mexico School of Medicine in Albuquerque.
References
- The SUPPORT Principal Investigators. A controlled trial to improve the care for seriously ill hospitalized patients. The study to understand prognoses and preferences for outcomes and risks of treatments (SUPPORT). JAMA. 1995;274(20):1591-1598.
- World Health Organization Definition of Palliative Care. World Health Organization website. Available at: http://www.who.int/cancer/palliative/definition/en/. Accessed April 12, 2012.
- NCCN Guidelines Version 2. 2011 Adult Cancer Pain. National Comprehensive Cancer Network website. Available at: http://www.nccn.org/professionals/physician_gls/pdf/pain.pdf. Accessed April 12, 2012.
- Whitecar PS, Jonas AP, Clasen ME. Managing pain in the dying patient. Am Fam Physician. 2000;61(3):755-764.
- Bial A, Levine S. Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Sydney M, et al. Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
- Mannix KA. Gastrointestinal symptoms. In: Doyle D, Hanks G, Cherny N, Calman K, eds. Oxford Textbook of Palliative Medicine. 3rd ed. New York, NY: Oxford University Press; 2005.
- Tyler LS. Nausea and vomiting in palliative care. In: Lipman AG, Jackson KC, Tyler LS, eds. Evidence-Based Symptom Control in Palliative Care. New York, NY: The Hawthorn Press; 2000.
- Policzer JS, Sobel J. Management of Selected Nonpain Symptoms of Life-Limiting Illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 4. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Parshall MB, Schwartzstein RM, Adams L, et al. An official American Thoracic Society statement: update on the mechanisms, assessment, and management of dyspnea. Am J Respir Crit Care Med. 2012;185(4): 435-452.
- Galbraith S, Fagan P, Perkins P, Lynch A, Booth S. Does the use of a handheld fan improve chronic dyspnea? A randomized controlled, crossover trial. J Pain Symptom Manage. 2010;39(5): 831-838.
- Philip J, Gold M, Milner A, Di Iulio J, Miller B, Spruyt O. A randomized, double-blind, crossover trial of the effect of oxygen on dyspnea in patients with advanced cancer. J Pain Symptom Manage. 2006;32(6):541-550.
- Cranston JM, Crockett A, Currow D. Oxygen therapy for dyspnea in adults. Cochrane Database Syst Rev. 2008;(3):CD004769.
- Jennings AL, Davies AN, Higgins JP, Broadley K. Opioids for the palliation of breathlessness in terminal illness. Cochrane Database Syst Rev. 2001;(4):CD002066.
- Ben-Aharon I, Gafter-Gvili A, Paul M, Leibovici, L, Stemmer, SM. Interventions for alleviating cancer-related dyspnea. A systematic review. J Clin Oncol. 2008;26(14): 2396-2404.
- Qaseem A, Snow V, Shekelle P, et al. Evidence-based interventions to improve the palliative care of pain, dyspnea, and depression at the end of life: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148(2):141-146
- Booth S, Moosavi SH, Higginson IJ. The etiology and management of intractable breathlessness in patients with advanced cancer: a systematic review of pharmacological therapy. Nat Clin Pract Oncol. 2008;5(2):90–100.
- Bickel K, Arnold R. EPERC Fast Facts Documents #109 Death Rattle and Oral Secretions, 2nd ed. Available at: http://www.eperc.mcw.edu/EPERC/FastFactsIndex/ff_109.htm. Accessed April 15, 2012.
- Wildiers H, Dhaenekint C, Demeulenaere P, et al. Atropine, hyoscine butylbromide, or scopalamine are equally effective for the treatment of death rattle in terminal care. J Pain Symptom Manage. 2009;38(1):124-133.
- Hugel H, Ellershaw J, Gambles M. Respiratory tract secretions in the dying patient: a comparison between glycopyrronium and hyoscine hydrobromide. J Palliat Med. 2006;9(2):279-285.
Case
A 58-year-old male with colon cancer metastatic to the liver and lungs presents with vomiting, dyspnea, and abdominal pain. His disease has progressed through third-line chemotherapy and his care is now focused entirely on symptom management. He has not had a bowel movement in five days and he began vomiting two days ago.
Overview
The majority of patients in the United States die in acute-care hospitals. The Study to Understand Prognosis and Preferences for Outcomes and Risks of Treatments (SUPPORT), which evaluated the courses of close to 10,000 hospitalized patients with serious and life-limiting illnesses, illustrated that patients’ end-of-life (EOL) experiences often are characterized by poor symptom management and invasive care that is not congruent with the patients’ overall goals of care.1 Studies of factors identified as priorities in EOL care have consistently shown that excellent pain and symptom management are highly valued by patients and families. As the hospitalist movement continues to grow, hospitalists will play a large role in caring for patients at EOL and will need to know how to provide adequate pain and symptom management so that high-quality care can be achieved.
Pain: A Basic Tenet
A basic tenet of palliative medicine is to evaluate and treat all types of suffering.2 Physical pain at EOL is frequently accompanied by other types of pain, such as psychological, social, religious, or existential pain. However, this review will focus on the pharmacologic management of physical pain.
Pain management must begin with a thorough evaluation of the severity, location, and characteristics of the discomfort to assess which therapies are most likely to be beneficial (see Table 1).3 The consistent use of one scale of pain severity (such as 0-10, or mild/moderate/severe) assists in the choice of initial dose of pain medication, in determining the response to the medication, and in assessing the need for change in dose.4
Opioids are the foundation of pain management in advanced diseases because they are available in a number of formulations and, when dosed appropriately, they are effective and safe. Starting doses and equianalgesic doses of common opioids are presented in Table 2. Guidelines recommend the use of short-acting opioids for dose titration to gain control of poorly controlled pain.3 If a patient is experiencing mild pain on a specific regimen, the medication dose can be increased up to 25%; by 25% to 50%, if pain is moderate; and 50% to 100%, if severe.5 When the pain is better-controlled, the total amount of pain medication used in 24 hours (24-hour dose) can be converted to a long-acting formulation for more consistent pain management. Because there is a constant component to most advanced pain syndromes, it is recommended that pain medication is given on a standing basis, with as-needed (prn) doses available for exacerbations of pain.3 Prn doses of short-acting medication (equivalent to approximately 10% of the 24-hour dose of medication) should be available at one- or two-hour intervals prn (longer if hepatic or renal impairment is present) for IV or PO medications, respectively.
Opioids often are categorized as low potency (i.e. codeine, hydrocodone) and high-potency (i.e. oxycodone, morphine, hydromorphone, fentanyl). When given in “equianalgesic doses,” the analgesic effect and common side effects (nausea/vomiting, constipation, sedation, confusion, pruritis) of different opioids can vary in different patients. Due to differences in levels of expressed subtypes of opioid receptors, a given patient might be more sensitive to the analgesic effect or side effects of a specific medication. Therefore, if dose escalation of one opioid is inadequate to control pain and further increases in dose are limited by intolerable side effects, rotation to another opioid is recommended.4 Tables documenting equianalgesic doses of different opioids are based on only moderate evidence from equivalency trials performed in healthy volunteers.6 Due to interpatient differences in responses, it is recommended that the equianalgesic dose of the new medication be decreased by 25% to 50% for initial dosing.5
Certain treatments are indicated for specific pain syndromes. Bony metastases respond to NSAIDs, bisphosphonates, and radiation therapy in addition to opioid medications. As focal back pain is the first symptom of spinal cord compression, clinicians should have a high index of suspicion for compression in any patient with malignancy and new back pain. Steroids and radiation therapy are considered emergent treatments for pain control and to prevent paralysis in this circumstance. Pain due to bowel obstruction is usually colicky in nature and responds well to octreotide as discussed in the section on nausea and vomiting. Steroids (such as dexamethasone 4 mg PO bid-tid) might be an effective adjuvant medication in bone pain, tumor pain, or inflammation.
*Half this dose should be used in renal or liver dysfunction and in the elderly.
Preferred in renal dysfunction.
SOURCES: Adapted from Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008, and Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
Back to the Case
At home, the patient was taking 60 mg of extended-release morphine twice daily and six doses per day of 15-mg immediate-release morphine for breakthrough pain. This is the equivalent of 210 mg of oral morphine in 24 hours. His pain is severe on this regimen, but it is unclear how much of this medication he is absorbing due to his vomiting. Using the IV route of administration and a patient-controlled analgesia (PCA) system will enable rapid dose titration and pain control. The equivalent of the 24-hour dose of 210 mg oral morphine is 70 mg IV morphine, which is equivalent to a drip basal rate of approximately 3 mg IV morphine per hour. This basal rate with a bolus dose of 7 mg (10% of the 24-hour dose) IV morphine q1 hour prn is reasonable as a starting point.
Review of the Data: Nausea and Vomiting
Nausea and vomiting affect 40% to 70% of patients in a palliative setting.7 A thorough history and physical exam can enable one to determine the most likely causes, pathways, and receptors involved in the process of nausea and vomiting. It is important to review the timing, frequency, and triggers of vomiting. The oral, abdominal, neurologic, and rectal exams, in addition to a complete chemistry panel, offer helpful information. The most common etiologies and recommended medications are included in Table 3. It is worthwhile to note that serotonin-antagonists (i.e. ondansetron) are first-line therapies only for chemotherapy and radiation-therapy-induced emesis. If a 24-hour trial of one antiemetic therapy is ineffective, one should reassess the etiology and escalate the antiemetic dose, or add a second therapy with a different (pertinent) mechanism of action. Although most studies of antiemetic therapy are case series, there is good evidence for this mechanistic approach.8
*EPS: extrapyramidal symptoms
The various insults and pathways that can cause vomiting are quite complex. The medullary vomiting center (VC) receives vestibular, peripheral (via splanchnic and vagal nerves), and higher cortical inputs and is the final common pathway in the vomiting reflex. The chemoreceptor trigger zone (CTZ) near the fourth ventricle receives input from the vagal and splanchnic nerves, and generates output to the VC.
General dietary recommendations are to avoid sweet, fatty, and highly salted or spiced foods. Small portions of bland foods without strong odors are best tolerated.7 Constipation commonly contributes to nausea and vomiting and should be managed with disimpaction, enemas, and laxatives as tolerated. Imaging may be required to make the important distinction between partial and complete bowel obstruction, as the treatments differ. Surgical procedures, such as colostomy or placement of a venting gastrostomy tube, can relieve pain and vomiting associated with complete bowel obstruction.
Back to the Case
The patient is found to have a fecal impaction on rectal exam, but vomiting persists after disimpaction and enema use. Imaging documents a complete bowel obstruction at the site of a palpable mass in the right upper quadrant and multiple large hepatic metastases. Octreotide is initiated to decrease intestinal secretions and peristalsis. Steroids are given to decrease tumor burden and associated inflammation in the intestine and liver, as well as to relieve distension of the hepatic capsule. Haloperidol is used in low doses to control episodes of nausea.
Review of the Data: Dyspnea
Dyspnea is a common symptom faced by patients at EOL. An estimated 50% of patients who are evaluated in acute-care hospitals seek treatment for the management of this often-crippling symptom.10 Unfortunately, as disease burden progresses, the incidence of dyspnea increases towards EOL, and the presence and severity of dyspnea is strongly correlated with mortality.
It is imperative for providers to appreciate that dyspnea is a subjective symptom, similar to pain. The presence and severity of dyspnea, therefore, depends on patient report. Given its subjective nature, the degree of dyspnea experienced by a patient might not correlate with objective laboratory findings or test results. In practice, the severity of dyspnea is commonly assessed with a numeric rating scale (0-10), verbal analogue scale, or with verbal descriptors (mild, moderate, severe). It is important to determine the underlying etiology of the dyspnea and, if possible, to target interventions to relieve the underlying cause. However, at the end of life, the burdens of invasive studies to determine the exact cause of dyspnea might outweigh the benefits, and invasive testing might not correlate with patients’ and families’ goals of care. In that instance, the goal of treatment should be aggressive symptom management and providers should use clinical judgment to tailor therapies based on the patient’s underlying illness, physical examination, and perhaps on noninvasive radiological or laboratory findings. Below are nonpharmacological and pharmacological interventions that can be employed to help alleviate dyspnea in the actively dying patient.
Nonpharmacological Management
A handheld fan aimed near the patient’s face has been shown to reduce the sensation of dyspnea.11 This relatively safe and inexpensive intervention has no major side effects and can provide improvement in this distressing symptom.
Often, the first line of therapy in the hospital setting for a patient reporting dyspnea is the administration of oxygen therapy. However, recent evidence does not show superiority of oxygen over air inhalation via nasal prongs for dyspnea in patients with advanced cancer or heart failure.12,13
Pharmacological Management
Opioids are first-line therapy for alleviating dyspnea in patients at EOL. The administration of opioids has been shown in systematic reviews to provide effective management of dyspnea.14,15 Practice guidelines by leading expert groups advocate for the use of opioids in the management of dyspnea for patients with advanced malignant and noncancer diseases.10,16 Fear of causing unintended respiratory sedation with opioids limits the prescription of opioids for dyspnea. However, studies have not found a change in mortality with the use of opioids appropriately titrated to control dyspnea.17
Studies examining the role of benzodiazepines in dyspnea management are conflicting. Anecdotal clinical evidence in actively dying patients supports treating dyspnea with benzodiazepines in conjunction with opioid therapy. Benzodiazepines are most beneficial when there is an anxiety-related component to the dyspnea.
Many patients with advanced disease and evidence of airflow obstruction will benefit from nebulized bronchodilator therapy for dyspnea. Patients with dyspnea from fluid overload (i.e. end-stage congestive heart failure or renal disease) might benefit from systemic diuretics. An increasing number of trials are under way to evaluate the efficacy of nebulized furosemide in the symptomatic management of dyspnea.
Back to the Case
The patient’s clinical course decompensates, and he begins to report worsening dyspnea in addition to his underlying pain. He becomes increasingly anxious about what this new symptom means. In addition to having a discussion about disease progression and prognosis, you increase his PCA basal dose to morphine 4 mg/hour to help him with this new symptom. You also add low-dose lorazepam 0.5 mg IV q8 hours as an adjunct agent for his dyspnea. The patient reports improvement of his symptom burden.
Review of the Data: Secretions
Physiological changes occur as a patient enters the active phase of dying. Two such changes are the loss of the ability to swallow and a reduced cough reflex. These changes culminate in an inability to clear secretions, which pool in the oropharynx and the airways. As the patient breathes, air moves over the pooled secretions and produces a gurgling sound that is referred to as the “death rattle.” The onset of this clinical marker has been shown to have significant prognostic significance for predicting imminent death within a period of hours to days. Proposed treatments for the symptom are listed below.
Nonpharmacological Management
Nonpharmacological options include repositioning the patient in a manner that facilitates postural draining.18 Careful and gentle oral suctioning might help reduce secretions if they are salivary in origin. This will not help to clear deeper bronchial secretions. Suctioning of deeper secretions often causes more burden than benefit, as this can cause repeated trauma and possible bleeding.
Family and caregivers at the bedside can find the “death rattle” quite disturbing and often fear that their loved one is “drowning.” Education and counseling that this is not the case, and that the development of secretions is a natural part of the dying process, can help alleviate this concern. Explaining that pharmacological agents can be titrated to decrease secretions is also reassuring to caregivers.
Pharmacological Management
Pharmacological options for secretion management include utilizing anticholinergic medications to prevent the formation of further secretions. These medications are standard of care for managing the death rattle and have been found to be most efficacious if started earlier in the actively dying phase.19,20 Anticholinergic medications include glycopyrrolate (0.2 mg IV q8 hours), atropine sulfate ophthalmological drops (1% solution, 1-2 drops SL q6 hours), hyoscyamine (0.125 mg one to four times a day), and scopolamine (1.5 mg patch q72 hours). These medications all have possible side effects typical of anticholinergic agents, including delirium, constipation, blurred vision, and urinary retention.
Back to the Case
The patient becomes increasingly lethargic. You meet with his family and explain that he is actively dying. His family reiterates that the goals of medical care should focus on maximizing symptom management. His family is concerned about the “gurgly” sound they hear and want to know if that means he is suffering. You educate the family about expected changes that occur with the dying process and inform them that glycopyrrolate 0.2 mg IV q8 hour will be started to minimize further secretions.
Bottom Line
Pain, nausea, dyspnea, and secretions are common end-of-life symptoms that hospitalists should be competent in treating.
Dr. Litrivis is an associate director and assistant professor at the Mount Sinai School of Medicine in New York, and Dr. Neale is an assistant professor at the University of New Mexico School of Medicine in Albuquerque.
References
- The SUPPORT Principal Investigators. A controlled trial to improve the care for seriously ill hospitalized patients. The study to understand prognoses and preferences for outcomes and risks of treatments (SUPPORT). JAMA. 1995;274(20):1591-1598.
- World Health Organization Definition of Palliative Care. World Health Organization website. Available at: http://www.who.int/cancer/palliative/definition/en/. Accessed April 12, 2012.
- NCCN Guidelines Version 2. 2011 Adult Cancer Pain. National Comprehensive Cancer Network website. Available at: http://www.nccn.org/professionals/physician_gls/pdf/pain.pdf. Accessed April 12, 2012.
- Whitecar PS, Jonas AP, Clasen ME. Managing pain in the dying patient. Am Fam Physician. 2000;61(3):755-764.
- Bial A, Levine S. Assessment and treatment of physical pain associated with life-limiting illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 3. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Sydney M, et al. Evidence-based standards for cancer pain management. J Clin Oncol. 2008;26(23):3879-3885.
- Mannix KA. Gastrointestinal symptoms. In: Doyle D, Hanks G, Cherny N, Calman K, eds. Oxford Textbook of Palliative Medicine. 3rd ed. New York, NY: Oxford University Press; 2005.
- Tyler LS. Nausea and vomiting in palliative care. In: Lipman AG, Jackson KC, Tyler LS, eds. Evidence-Based Symptom Control in Palliative Care. New York, NY: The Hawthorn Press; 2000.
- Policzer JS, Sobel J. Management of Selected Nonpain Symptoms of Life-Limiting Illness. Hospice and Palliative Care Training for Physicians: UNIPAC. Vol 4. 3rd ed. Glenview, IL: American Academy of Hospice and Palliative Medicine; 2008.
- Parshall MB, Schwartzstein RM, Adams L, et al. An official American Thoracic Society statement: update on the mechanisms, assessment, and management of dyspnea. Am J Respir Crit Care Med. 2012;185(4): 435-452.
- Galbraith S, Fagan P, Perkins P, Lynch A, Booth S. Does the use of a handheld fan improve chronic dyspnea? A randomized controlled, crossover trial. J Pain Symptom Manage. 2010;39(5): 831-838.
- Philip J, Gold M, Milner A, Di Iulio J, Miller B, Spruyt O. A randomized, double-blind, crossover trial of the effect of oxygen on dyspnea in patients with advanced cancer. J Pain Symptom Manage. 2006;32(6):541-550.
- Cranston JM, Crockett A, Currow D. Oxygen therapy for dyspnea in adults. Cochrane Database Syst Rev. 2008;(3):CD004769.
- Jennings AL, Davies AN, Higgins JP, Broadley K. Opioids for the palliation of breathlessness in terminal illness. Cochrane Database Syst Rev. 2001;(4):CD002066.
- Ben-Aharon I, Gafter-Gvili A, Paul M, Leibovici, L, Stemmer, SM. Interventions for alleviating cancer-related dyspnea. A systematic review. J Clin Oncol. 2008;26(14): 2396-2404.
- Qaseem A, Snow V, Shekelle P, et al. Evidence-based interventions to improve the palliative care of pain, dyspnea, and depression at the end of life: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;148(2):141-146
- Booth S, Moosavi SH, Higginson IJ. The etiology and management of intractable breathlessness in patients with advanced cancer: a systematic review of pharmacological therapy. Nat Clin Pract Oncol. 2008;5(2):90–100.
- Bickel K, Arnold R. EPERC Fast Facts Documents #109 Death Rattle and Oral Secretions, 2nd ed. Available at: http://www.eperc.mcw.edu/EPERC/FastFactsIndex/ff_109.htm. Accessed April 15, 2012.
- Wildiers H, Dhaenekint C, Demeulenaere P, et al. Atropine, hyoscine butylbromide, or scopalamine are equally effective for the treatment of death rattle in terminal care. J Pain Symptom Manage. 2009;38(1):124-133.
- Hugel H, Ellershaw J, Gambles M. Respiratory tract secretions in the dying patient: a comparison between glycopyrronium and hyoscine hydrobromide. J Palliat Med. 2006;9(2):279-285.
Pediatric Readmissions Vary Significantly Across Children’s Hospitals

Pediatric Readmissions Vary Significantly across Children’s Hospitals
Clinical question: What are the characteristics of readmissions to children’s hospitals?
Background: Thirty-day readmissions in adult Medicare beneficiaries are common and thought to represent potential for significant improvements in the quality of care. Penalties will be levied upon hospitals with excessively high readmission rates in adults. The stage is set for a translation of this practice to pediatric readmissions. However, the characteristics of readmissions to children’s hospitals are not well-defined.
Study design: Retrospective review.
Setting: National Association of Children’s Hospitals and Related Institutions (NACHRI) Case Mix data set.
Synopsis: Of 568,845 readmissions examined across 72 children’s hospitals, the 30-day readmission rate was 6.5%. Readmission rates varied by many factors: age, chronic conditions, insurance type, race/ethnicity, length of stay, number of annual hospital admissions, and hospital type. Rates varied significantly across hospitals, even after adjustment for age and chronic conditions. Anemia or neutropenia, ventricular shunt procedures, and sickle cell crisis had the highest unadjusted, 30-day, condition-specific readmission rates.
This study is notable for its large sample size but limited by the administrative data, which might, for example, underestimate readmissions that went to another hospital. Additionally, the majority of children in the U.S. are hospitalized outside of children’s hospitals, which are overrepresented in this study.
However, this study paints a clear picture of the differences between adult readmissions and pediatric readmissions—rates are lower than in elderly adults, and the top three conditions are distinctly different. Anemia or neutropenia likely are due to effects of chemotherapy; ventricular shunt readmissions often reflect surgery-related issues; and sickle cell disease is a lifelong, chronic condition. The significant variation between hospitals after case-mix adjustment offers an opportunity for further investigation and improvement.
Bottom line: Pediatric readmissions differ from adult readmissions and vary significantly across children’s hospitals.
Citation: Berry JG, Toomey SL, Zaslavsky AM, et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372-380.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Pediatric Readmissions Vary Significantly across Children’s Hospitals
Clinical question: What are the characteristics of readmissions to children’s hospitals?
Background: Thirty-day readmissions in adult Medicare beneficiaries are common and thought to represent potential for significant improvements in the quality of care. Penalties will be levied upon hospitals with excessively high readmission rates in adults. The stage is set for a translation of this practice to pediatric readmissions. However, the characteristics of readmissions to children’s hospitals are not well-defined.
Study design: Retrospective review.
Setting: National Association of Children’s Hospitals and Related Institutions (NACHRI) Case Mix data set.
Synopsis: Of 568,845 readmissions examined across 72 children’s hospitals, the 30-day readmission rate was 6.5%. Readmission rates varied by many factors: age, chronic conditions, insurance type, race/ethnicity, length of stay, number of annual hospital admissions, and hospital type. Rates varied significantly across hospitals, even after adjustment for age and chronic conditions. Anemia or neutropenia, ventricular shunt procedures, and sickle cell crisis had the highest unadjusted, 30-day, condition-specific readmission rates.
This study is notable for its large sample size but limited by the administrative data, which might, for example, underestimate readmissions that went to another hospital. Additionally, the majority of children in the U.S. are hospitalized outside of children’s hospitals, which are overrepresented in this study.
However, this study paints a clear picture of the differences between adult readmissions and pediatric readmissions—rates are lower than in elderly adults, and the top three conditions are distinctly different. Anemia or neutropenia likely are due to effects of chemotherapy; ventricular shunt readmissions often reflect surgery-related issues; and sickle cell disease is a lifelong, chronic condition. The significant variation between hospitals after case-mix adjustment offers an opportunity for further investigation and improvement.
Bottom line: Pediatric readmissions differ from adult readmissions and vary significantly across children’s hospitals.
Citation: Berry JG, Toomey SL, Zaslavsky AM, et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372-380.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.
Pediatric Readmissions Vary Significantly across Children’s Hospitals
Clinical question: What are the characteristics of readmissions to children’s hospitals?
Background: Thirty-day readmissions in adult Medicare beneficiaries are common and thought to represent potential for significant improvements in the quality of care. Penalties will be levied upon hospitals with excessively high readmission rates in adults. The stage is set for a translation of this practice to pediatric readmissions. However, the characteristics of readmissions to children’s hospitals are not well-defined.
Study design: Retrospective review.
Setting: National Association of Children’s Hospitals and Related Institutions (NACHRI) Case Mix data set.
Synopsis: Of 568,845 readmissions examined across 72 children’s hospitals, the 30-day readmission rate was 6.5%. Readmission rates varied by many factors: age, chronic conditions, insurance type, race/ethnicity, length of stay, number of annual hospital admissions, and hospital type. Rates varied significantly across hospitals, even after adjustment for age and chronic conditions. Anemia or neutropenia, ventricular shunt procedures, and sickle cell crisis had the highest unadjusted, 30-day, condition-specific readmission rates.
This study is notable for its large sample size but limited by the administrative data, which might, for example, underestimate readmissions that went to another hospital. Additionally, the majority of children in the U.S. are hospitalized outside of children’s hospitals, which are overrepresented in this study.
However, this study paints a clear picture of the differences between adult readmissions and pediatric readmissions—rates are lower than in elderly adults, and the top three conditions are distinctly different. Anemia or neutropenia likely are due to effects of chemotherapy; ventricular shunt readmissions often reflect surgery-related issues; and sickle cell disease is a lifelong, chronic condition. The significant variation between hospitals after case-mix adjustment offers an opportunity for further investigation and improvement.
Bottom line: Pediatric readmissions differ from adult readmissions and vary significantly across children’s hospitals.
Citation: Berry JG, Toomey SL, Zaslavsky AM, et al. Pediatric readmission prevalence and variability across hospitals. JAMA. 2013;309(4):372-380.
Reviewed by Pediatric Editor Mark Shen, MD, SFHM, medical director of hospital medicine at Dell Children's Medical Center, Austin, Texas.