User login
What Is the Role of Steroids in Septic Shock?
The Case
An 81-year-old woman with diabetes mellitus presents with a three-day history of fever, chills, left-side flank pain, and dysuria. Her blood pressure upon presentation is 75/45 mm/Hg, her heart rate is 120 beats per minute, and she has a temperature of 103.1°`F and a respiratory rate of 22 breaths/minute. On physical examination, she is an ill-appearing elderly woman, with dry oral mucosa and left costo-vertebral angle tenderness. Lab work shows leukocytosis of 18,000 mg/dL with 88% polymorphonuclear leukocyte (PMN), the urine analysis is consistent with a urinary tract infection, and a chemistry panel reveals elevated BUN and creatinine levels of 52 mg/dL and 2.4 mg/dL, respectively. In the emergency department, she is given a bolus of 2 liters normal saline, but her blood pressure remains 78/49 mm/Hg. She is then started on broad-spectrum antibiotics and a norepinephrine drip, and is admitted to the ICU.
What role would steroids add to her management?
Background
Sepsis is the clinical syndrome defined by the presence of systemic inflammatory response syndrome (SIRS) in the setting of an infection. SIRS is defined by the presence of at least two of the following: fever or hypothermia; leukocytosis, leukopenia, or bandemia; heart rate >90 bpm; or tachypnea or hypocapnia.
When acute organ dysfunction, such as acute renal failure, altered mental status, or acute lung injury (hypoxemia), is present, sepsis is classified as severe.
Septic shock is a state of sepsis associated with acute circulatory collapse characterized by persistent arterial hypotension (defined as a systolic blood pressure <90 mmHg, a mean arterial pressure <60 mmHg, or a reduction in systolic blood pressure of >40 mmHg from baseline) despite fluid resuscitation attempts.1
The incidence and mortality due to sepsis and septic shock is directly related to the age of the patient, many of whom require ICU hospitalization.2 Clinically, this portends a great challenge, as the incidence of sepsis is likely to increase as the U.S. population ages.
Initial management of a patient with sepsis/septic shock is goal-directed therapy, which consists of early administration of broad-spectrum antibiotics, crystalloid or colloid fluid resuscitation, and use of vasopressor support to improve hemodynamics and maintain a mean arterial
pressure ≥65 mmHg. Patients with acute lung injury may also require prompt ventilator support.
The role of steroids in sepsis is controversial.
Review of Steroids
Steroids have long been known for their anti-inflammatory properties. From the 1950s to the 1980s, high-dose steroids (methylprednisolone 30mg/kg and dexamethasone 3 mg/kg to 6 mg/kg in divided doses) were used in the management of sepsis. This was based on a study by Schumer that showed steroids reduced mortality to 10% from 38%.3
Later, Sprung and colleagues demonstrated reversal of shock and improved short-term survival with high-dose steroids in patients with sepsis, but subsequent prospective randomized trials did not support this beneficial effect of high-dose steroids.4-6 In fact, two meta-analyses in 1995 concluded that high-dose steroids are ineffective and potentially harmful, and associated with higher mortality, secondary infections, and renal and hepatic dysfunction.7,8 Thereafter, the use of high-dose steroids fell into disfavor.
In the early 2000s, there was an emergence in the use of low-dose steroids in patients with sepsis. This was based on various trials showing the benefit of the use of low-dose steroids in the reversal of septic shock without significant side effects, discussed further below.
Pathophysiology
Steroids improve hemodynamic parameters. In an animal model, Hinshaw and colleagues induced septic shock in adrenalectomized dogs by infusing lethal doses of E. coli. The untreated dogs died within hours, whereas the dogs treated with antibiotics and steroids had a complete recovery from shock, and survived more than 100 hours.9
During sepsis, endotoxins induce nitric oxide synthase, which produces relaxation of vascular smooth muscle tone, with resultant hypotension and reduced contractility response to norepinephrine.10 Corticosteroids prevent induction of nitric oxide synthase and enhance the vaso-active response to catecholamines through the glucocorticoid receptors. In vascular endothelial cells, glucocorticoids also inhibit serum phospholipase A2, reducing the production of vasodilators, such as prostacyclin and prostaglandin E1.11,12
Steroids reduce inflammation. Sepsis is driven by a systemic inflammatory response, in which components of the outer-cell membrane of both gram-positive and gram-negative bacteria and endotoxins induce the production of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-alpha) and interleukin-1 (IL-1).13 These cytokines have a direct toxic effect on various tissues. In addition, inflammatory cytokines suppress adrenal response to adrenocorticotropic hormone (ACTH), which results in decreased endogenous cortisol production, and compete with glucocorticoids for their receptors, inducing resistance to the action of steroids at the tissue level.14
In healthy volunteers challenged with bacterial endotoxins, low-dose steroids (~10 mg of prednisolone) suppress the release of proinflammatory cytokines and prevent the activation of endothelial cells and neutrophils.15 Steroids also inhibit the release of toxic enzymes, such as lysozymes and superoxides from neutrophils.16,17
Data Supporting Steroid Use in Septic Shock
Mortality data. Two major studies evaluated the effect of low-dose steroids in patients with septic shock. Annane and colleagues conducted a placebo-controlled, blinded trial and divided the study population into “responders” and “nonresponders” based on their response to ACTH stimulation test. Within the “nonresponder” group, steroids reduced the risk of mortality by 16% (63% mortality in the placebo group and 53% mortality in the corticosteroid group, P=0.02).16 Steroids also significantly reduced ICU mortality (70% versus 58%), hospital mortality (72% versus 61%) and one-year mortality (77% versus 68%) compared with placebo. No statistically significant difference in mortality between steroids and placebo was seen in the “responder” group.16
The CORTICUS trial, a multicenter, randomized, double-blind, placebo-controlled trial, showed no significant difference in 28-day mortality between those treated with corticosteroids (39.2%) and those receiving a placebo (36%, P=0.069).17 There was also no significant difference in either hospital or ICU mortality in this study.
A recent meta-analysis demonstrated no significant effect of corticosteroid treatment on 28-day mortality, ICU mortality, or hospital mortality in septic shock. However, subanalysis of trials using a prolonged course (>5 days) of low-dose steroids (300 mg of hydrocortisone or equivalent) showed a significant reduction in 28-day all-cause mortality (P=0.02) and hospital mortality (P =0.05), and a decrease in ICU length of stay.18
Reversal of shock. Various studies have shown a decrease in the time necessary to reverse septic shock with the use of low-dose steroids. Annane and colleagues showed the median time to vasopressor therapy withdrawal was seven days in the group treated with steroids versus nine days in the placebo group (P=0.01).16 The CORTICUS study demonstrated significantly shorter times to reversal of shock in the group treated with hydrocortisone compared with the placebo group—3.3 days versus 5.8 days (P<0.001).17 In a smaller study, 68% of hydrocortisone-treated patients achieved seven-day shock reversal compared with 21% in the placebo group, a difference of 47% in the rate of reversal of shock.18
Guidelines for the Use of Steroids in Septic Shock
In which sepsis patients should I use steroids? The large clinical trials that found a benefit to low-dose steroids included patients with a systolic blood pressure <90 mm/Hg for more than one hour, despite aggressive fluid and vasopressor therapy. Based on these and other smaller trials, the Surviving Sepsis Campaign recommends the addition of IV steroids to those patients with septic shock who don’t respond to adequate fluid and vasopressor resuscitation.19
Should I obtain ACTH stimulation test in these patients? Although the Annane study showed that “nonresponders”—those who did not achieve ≥9 mcg/dL increase in cortisol level after 30 to 60 minutes of ACTH administration—were more likely to benefit from steroids, the overall trial population appeared to benefit regardless of the ACTH response.16
Furthermore, the CORTICUS study showed no difference between the corticotropin responder and nonresponder group. Also, most cortisol immunoassays measure total cortisol (free and protein-bound), whereas the free cortisol level is the more clinically relevant measurement. Hence, current guidelines from the American College of Critical Care Medicine and Surviving Sepsis Campaign do not recommend performing an ACTH stimulation test prior to administration of steroids.20
What type of steroids should I use, and at what dose? Current guidelines recommend IV hydrocortisone in a dose of 200 mg/day to 300 mg/day given as 50 mg every six hours or 100 mg every eight hours for at least seven days before tapering. Alternatively, IV hydrocortisone can be given as a bolus of 100 mg followed by a continuous infusion at 10 mg/hr (240 mg/day). Hydrocortisone at this dose has intrinsic mineralocorticoid activity, obviating the need for adding fludrocortisone. Fludrocortisone may otherwise be added as 50 mcg daily, if using a corticosteroid without significant mineralocorticoid activity. Patients with septic shock should not be treated with dexamethasone, which causes immediate and prolonged suppression of the hypothalamic-pituitary-adrenal axis.21
Do I need to taper off the steroids? It is recommended to wean the steroids after seven or more days of use, when vasopressors are no longer required. Keh and colleagues noted a 30% recurrence of shock in patients when the steroids were not tapered.22 There was also evidence of immunologic rebound after abrupt cessation of steroids, with an increase in inflammatory markers.23 The taper should decrease the dose every two or three days in small steps.
What potential side effects should I be concerned about? Overall, the use of higher dose corticosteroids is associated with significant potential side effects, including a worsening of the underlying infection, new infection, hyperglycemia, hypernatremia, and gastrointestinal bleeding. In a meta-analysis of nine clinical trials with high-dose corticosteroids (a starting dose of ~30mg/kg/day of methylprednisolone), Cronin and colleagues found a trend toward increased mortality due to secondary infections (relative risk 1.70; 95% confidence interval, 0.70 to 4.12).24 A recent meta-analysis of 15 trials found low-dose corticosteroids reduced ICU mortality and increased the proportion of shock reversal by Day 7 and by Day 28 without increasing the rate of gastroduodenal bleeding, super-infection, or hyperglycemia.25
Back to the Case
Our patient was admitted to the medical ICU. After obtaining urine and blood cultures, she was started on IV levofloxacin. She remained hypotensive despite IV fluids and IV norepinephrine. She was started on IV hydrocortisone 50 mg every six hours. Over the next 48 hours, her hemodynamic parameters improved. Urine and blood cultures came back positive for E. coli. Her BUN and creatinine decreased to 24 mg/dL and 1.4 mg/dL, respectively.
Later, she was weaned off norepinephrine and transferred out of the ICU. On hospital Day 7, a slow taper of her hydrocortisone initiated, and antibiotics were switched to oral levofloxacin. She was later discharged home in stable condition.
Bottom Line
In patients with septic shock that is unresponsive to IV fluid resuscitation and vasopressors, the addition of low-dose corticosteroids is relatively safe and can improve rate of reversal of shock, reduce time to reversal of shock, decrease ICU length of stay, and potentially lower mortality.
Drs. Gandhi and Asudani are health science assistant professors of medicine in the Division of Hospital Medicine at the University of California at San Diego.
References
- Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530-538.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303-1310.
- Schumer W. Steroids in the treatment of clinical septic shock. Ann Surg. 1976;184:333-341.
- Sprung CL, Caralis PV, Marcial EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311:1137-1143.
- The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317:659-665.
- Bone RC, Fisher CJ Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653-658.
- Lefering R, Neugebauer EA. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294-1303.
- Cronin L, Cook DJ, Cartlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;24:1430-1439.
- Hinshaw LB, Beller BK, Chang AC, et al. Corticosteroid/antibiotic treatment of adrenalectomized dogs challenged with lethal E. coli. Circ Shock. 1985;16:265-277.
- Rees DD, Cellek S, Palmer RM, Moncada S. Dexamethasone prevents the induction of NO synthase and the associated effects on vascular tone, an insight into endotoxin shock. BioChem BioPhy Res Comm. 1990;173:541-547.
- Axelrod L. Inhibition of prostacyclin production mediates permissive effect of glucocorticoids on vascular tone. Lancet. 1983;1:904-906.
- Annane D, Bellissant E, Sebille V, et al. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenaline reserve. Br J Clin Pharmacol. 1991;46:589-597.
- DeKruif MD, Lemaire LL, Giebelen IA, et al. Prednisolone dose dependently influences inflammation and coagulation during human endotoxemia. J Immunol. 2007;178:1845-1851.
- Goldstein IM, Roos D, Weissman, G et al. Influence of corticosteroids on human polymorphonuclear leukocyte function in vitro. Inflammation. 1976;1:305-316.
- Briegel J, Kellerman W, Forst H, et al. Low-dose hydrocortisone infusion attenuates the SIRS. The Phospolipase A2 Study Group. Clin Invest. 1994;72:782-787.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisones on mortality in patients with septic shock. JAMA. 2002;288:862-871.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy in patients with septic shock. N Engl J Med. 2008;358:111-124.
- Annane D, Bellissant E, Bollaert P. Corticosteroids in the treatment of severe sepsis and septic shock in adults. A systematic review. JAMA. 2009;301:2362-2375.
- Dellinger DP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17-60.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937-1949.
- Reincke M, Allolio B, Würth G, et al. The hypothalamic-pituitary-adrenal axis in critical illness: Response to dexamethasone and corticotropin-releasing hormone. J Clin Endocrinol Metab. 1993;77:151-156
- Keh D, Weber-Carstens S, Ahlers O. Adjunctive therapies in severe sepsis and septic shock: current place of steroids. Curr Infect Dis Rep. 2008;10:354-361.
- Keh D, Boehnke T, Weber-Cartens S, et al. Immunologic and hemodynamic effects of “low dose” hydrocortisone in septic shock: a double blind study, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med. 2003;167:512-520.
- Cronin L, Cook DJ, Carlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23;1430-1439.
- Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids for treating severe sepsis and septic shock. Cochrane Database Syst Rev. 2004;1:CD002243.
The Case
An 81-year-old woman with diabetes mellitus presents with a three-day history of fever, chills, left-side flank pain, and dysuria. Her blood pressure upon presentation is 75/45 mm/Hg, her heart rate is 120 beats per minute, and she has a temperature of 103.1°`F and a respiratory rate of 22 breaths/minute. On physical examination, she is an ill-appearing elderly woman, with dry oral mucosa and left costo-vertebral angle tenderness. Lab work shows leukocytosis of 18,000 mg/dL with 88% polymorphonuclear leukocyte (PMN), the urine analysis is consistent with a urinary tract infection, and a chemistry panel reveals elevated BUN and creatinine levels of 52 mg/dL and 2.4 mg/dL, respectively. In the emergency department, she is given a bolus of 2 liters normal saline, but her blood pressure remains 78/49 mm/Hg. She is then started on broad-spectrum antibiotics and a norepinephrine drip, and is admitted to the ICU.
What role would steroids add to her management?
Background
Sepsis is the clinical syndrome defined by the presence of systemic inflammatory response syndrome (SIRS) in the setting of an infection. SIRS is defined by the presence of at least two of the following: fever or hypothermia; leukocytosis, leukopenia, or bandemia; heart rate >90 bpm; or tachypnea or hypocapnia.
When acute organ dysfunction, such as acute renal failure, altered mental status, or acute lung injury (hypoxemia), is present, sepsis is classified as severe.
Septic shock is a state of sepsis associated with acute circulatory collapse characterized by persistent arterial hypotension (defined as a systolic blood pressure <90 mmHg, a mean arterial pressure <60 mmHg, or a reduction in systolic blood pressure of >40 mmHg from baseline) despite fluid resuscitation attempts.1
The incidence and mortality due to sepsis and septic shock is directly related to the age of the patient, many of whom require ICU hospitalization.2 Clinically, this portends a great challenge, as the incidence of sepsis is likely to increase as the U.S. population ages.
Initial management of a patient with sepsis/septic shock is goal-directed therapy, which consists of early administration of broad-spectrum antibiotics, crystalloid or colloid fluid resuscitation, and use of vasopressor support to improve hemodynamics and maintain a mean arterial
pressure ≥65 mmHg. Patients with acute lung injury may also require prompt ventilator support.
The role of steroids in sepsis is controversial.
Review of Steroids
Steroids have long been known for their anti-inflammatory properties. From the 1950s to the 1980s, high-dose steroids (methylprednisolone 30mg/kg and dexamethasone 3 mg/kg to 6 mg/kg in divided doses) were used in the management of sepsis. This was based on a study by Schumer that showed steroids reduced mortality to 10% from 38%.3
Later, Sprung and colleagues demonstrated reversal of shock and improved short-term survival with high-dose steroids in patients with sepsis, but subsequent prospective randomized trials did not support this beneficial effect of high-dose steroids.4-6 In fact, two meta-analyses in 1995 concluded that high-dose steroids are ineffective and potentially harmful, and associated with higher mortality, secondary infections, and renal and hepatic dysfunction.7,8 Thereafter, the use of high-dose steroids fell into disfavor.
In the early 2000s, there was an emergence in the use of low-dose steroids in patients with sepsis. This was based on various trials showing the benefit of the use of low-dose steroids in the reversal of septic shock without significant side effects, discussed further below.
Pathophysiology
Steroids improve hemodynamic parameters. In an animal model, Hinshaw and colleagues induced septic shock in adrenalectomized dogs by infusing lethal doses of E. coli. The untreated dogs died within hours, whereas the dogs treated with antibiotics and steroids had a complete recovery from shock, and survived more than 100 hours.9
During sepsis, endotoxins induce nitric oxide synthase, which produces relaxation of vascular smooth muscle tone, with resultant hypotension and reduced contractility response to norepinephrine.10 Corticosteroids prevent induction of nitric oxide synthase and enhance the vaso-active response to catecholamines through the glucocorticoid receptors. In vascular endothelial cells, glucocorticoids also inhibit serum phospholipase A2, reducing the production of vasodilators, such as prostacyclin and prostaglandin E1.11,12
Steroids reduce inflammation. Sepsis is driven by a systemic inflammatory response, in which components of the outer-cell membrane of both gram-positive and gram-negative bacteria and endotoxins induce the production of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-alpha) and interleukin-1 (IL-1).13 These cytokines have a direct toxic effect on various tissues. In addition, inflammatory cytokines suppress adrenal response to adrenocorticotropic hormone (ACTH), which results in decreased endogenous cortisol production, and compete with glucocorticoids for their receptors, inducing resistance to the action of steroids at the tissue level.14
In healthy volunteers challenged with bacterial endotoxins, low-dose steroids (~10 mg of prednisolone) suppress the release of proinflammatory cytokines and prevent the activation of endothelial cells and neutrophils.15 Steroids also inhibit the release of toxic enzymes, such as lysozymes and superoxides from neutrophils.16,17
Data Supporting Steroid Use in Septic Shock
Mortality data. Two major studies evaluated the effect of low-dose steroids in patients with septic shock. Annane and colleagues conducted a placebo-controlled, blinded trial and divided the study population into “responders” and “nonresponders” based on their response to ACTH stimulation test. Within the “nonresponder” group, steroids reduced the risk of mortality by 16% (63% mortality in the placebo group and 53% mortality in the corticosteroid group, P=0.02).16 Steroids also significantly reduced ICU mortality (70% versus 58%), hospital mortality (72% versus 61%) and one-year mortality (77% versus 68%) compared with placebo. No statistically significant difference in mortality between steroids and placebo was seen in the “responder” group.16
The CORTICUS trial, a multicenter, randomized, double-blind, placebo-controlled trial, showed no significant difference in 28-day mortality between those treated with corticosteroids (39.2%) and those receiving a placebo (36%, P=0.069).17 There was also no significant difference in either hospital or ICU mortality in this study.
A recent meta-analysis demonstrated no significant effect of corticosteroid treatment on 28-day mortality, ICU mortality, or hospital mortality in septic shock. However, subanalysis of trials using a prolonged course (>5 days) of low-dose steroids (300 mg of hydrocortisone or equivalent) showed a significant reduction in 28-day all-cause mortality (P=0.02) and hospital mortality (P =0.05), and a decrease in ICU length of stay.18
Reversal of shock. Various studies have shown a decrease in the time necessary to reverse septic shock with the use of low-dose steroids. Annane and colleagues showed the median time to vasopressor therapy withdrawal was seven days in the group treated with steroids versus nine days in the placebo group (P=0.01).16 The CORTICUS study demonstrated significantly shorter times to reversal of shock in the group treated with hydrocortisone compared with the placebo group—3.3 days versus 5.8 days (P<0.001).17 In a smaller study, 68% of hydrocortisone-treated patients achieved seven-day shock reversal compared with 21% in the placebo group, a difference of 47% in the rate of reversal of shock.18
Guidelines for the Use of Steroids in Septic Shock
In which sepsis patients should I use steroids? The large clinical trials that found a benefit to low-dose steroids included patients with a systolic blood pressure <90 mm/Hg for more than one hour, despite aggressive fluid and vasopressor therapy. Based on these and other smaller trials, the Surviving Sepsis Campaign recommends the addition of IV steroids to those patients with septic shock who don’t respond to adequate fluid and vasopressor resuscitation.19
Should I obtain ACTH stimulation test in these patients? Although the Annane study showed that “nonresponders”—those who did not achieve ≥9 mcg/dL increase in cortisol level after 30 to 60 minutes of ACTH administration—were more likely to benefit from steroids, the overall trial population appeared to benefit regardless of the ACTH response.16
Furthermore, the CORTICUS study showed no difference between the corticotropin responder and nonresponder group. Also, most cortisol immunoassays measure total cortisol (free and protein-bound), whereas the free cortisol level is the more clinically relevant measurement. Hence, current guidelines from the American College of Critical Care Medicine and Surviving Sepsis Campaign do not recommend performing an ACTH stimulation test prior to administration of steroids.20
What type of steroids should I use, and at what dose? Current guidelines recommend IV hydrocortisone in a dose of 200 mg/day to 300 mg/day given as 50 mg every six hours or 100 mg every eight hours for at least seven days before tapering. Alternatively, IV hydrocortisone can be given as a bolus of 100 mg followed by a continuous infusion at 10 mg/hr (240 mg/day). Hydrocortisone at this dose has intrinsic mineralocorticoid activity, obviating the need for adding fludrocortisone. Fludrocortisone may otherwise be added as 50 mcg daily, if using a corticosteroid without significant mineralocorticoid activity. Patients with septic shock should not be treated with dexamethasone, which causes immediate and prolonged suppression of the hypothalamic-pituitary-adrenal axis.21
Do I need to taper off the steroids? It is recommended to wean the steroids after seven or more days of use, when vasopressors are no longer required. Keh and colleagues noted a 30% recurrence of shock in patients when the steroids were not tapered.22 There was also evidence of immunologic rebound after abrupt cessation of steroids, with an increase in inflammatory markers.23 The taper should decrease the dose every two or three days in small steps.
What potential side effects should I be concerned about? Overall, the use of higher dose corticosteroids is associated with significant potential side effects, including a worsening of the underlying infection, new infection, hyperglycemia, hypernatremia, and gastrointestinal bleeding. In a meta-analysis of nine clinical trials with high-dose corticosteroids (a starting dose of ~30mg/kg/day of methylprednisolone), Cronin and colleagues found a trend toward increased mortality due to secondary infections (relative risk 1.70; 95% confidence interval, 0.70 to 4.12).24 A recent meta-analysis of 15 trials found low-dose corticosteroids reduced ICU mortality and increased the proportion of shock reversal by Day 7 and by Day 28 without increasing the rate of gastroduodenal bleeding, super-infection, or hyperglycemia.25
Back to the Case
Our patient was admitted to the medical ICU. After obtaining urine and blood cultures, she was started on IV levofloxacin. She remained hypotensive despite IV fluids and IV norepinephrine. She was started on IV hydrocortisone 50 mg every six hours. Over the next 48 hours, her hemodynamic parameters improved. Urine and blood cultures came back positive for E. coli. Her BUN and creatinine decreased to 24 mg/dL and 1.4 mg/dL, respectively.
Later, she was weaned off norepinephrine and transferred out of the ICU. On hospital Day 7, a slow taper of her hydrocortisone initiated, and antibiotics were switched to oral levofloxacin. She was later discharged home in stable condition.
Bottom Line
In patients with septic shock that is unresponsive to IV fluid resuscitation and vasopressors, the addition of low-dose corticosteroids is relatively safe and can improve rate of reversal of shock, reduce time to reversal of shock, decrease ICU length of stay, and potentially lower mortality.
Drs. Gandhi and Asudani are health science assistant professors of medicine in the Division of Hospital Medicine at the University of California at San Diego.
References
- Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530-538.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303-1310.
- Schumer W. Steroids in the treatment of clinical septic shock. Ann Surg. 1976;184:333-341.
- Sprung CL, Caralis PV, Marcial EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311:1137-1143.
- The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317:659-665.
- Bone RC, Fisher CJ Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653-658.
- Lefering R, Neugebauer EA. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294-1303.
- Cronin L, Cook DJ, Cartlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;24:1430-1439.
- Hinshaw LB, Beller BK, Chang AC, et al. Corticosteroid/antibiotic treatment of adrenalectomized dogs challenged with lethal E. coli. Circ Shock. 1985;16:265-277.
- Rees DD, Cellek S, Palmer RM, Moncada S. Dexamethasone prevents the induction of NO synthase and the associated effects on vascular tone, an insight into endotoxin shock. BioChem BioPhy Res Comm. 1990;173:541-547.
- Axelrod L. Inhibition of prostacyclin production mediates permissive effect of glucocorticoids on vascular tone. Lancet. 1983;1:904-906.
- Annane D, Bellissant E, Sebille V, et al. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenaline reserve. Br J Clin Pharmacol. 1991;46:589-597.
- DeKruif MD, Lemaire LL, Giebelen IA, et al. Prednisolone dose dependently influences inflammation and coagulation during human endotoxemia. J Immunol. 2007;178:1845-1851.
- Goldstein IM, Roos D, Weissman, G et al. Influence of corticosteroids on human polymorphonuclear leukocyte function in vitro. Inflammation. 1976;1:305-316.
- Briegel J, Kellerman W, Forst H, et al. Low-dose hydrocortisone infusion attenuates the SIRS. The Phospolipase A2 Study Group. Clin Invest. 1994;72:782-787.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisones on mortality in patients with septic shock. JAMA. 2002;288:862-871.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy in patients with septic shock. N Engl J Med. 2008;358:111-124.
- Annane D, Bellissant E, Bollaert P. Corticosteroids in the treatment of severe sepsis and septic shock in adults. A systematic review. JAMA. 2009;301:2362-2375.
- Dellinger DP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17-60.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937-1949.
- Reincke M, Allolio B, Würth G, et al. The hypothalamic-pituitary-adrenal axis in critical illness: Response to dexamethasone and corticotropin-releasing hormone. J Clin Endocrinol Metab. 1993;77:151-156
- Keh D, Weber-Carstens S, Ahlers O. Adjunctive therapies in severe sepsis and septic shock: current place of steroids. Curr Infect Dis Rep. 2008;10:354-361.
- Keh D, Boehnke T, Weber-Cartens S, et al. Immunologic and hemodynamic effects of “low dose” hydrocortisone in septic shock: a double blind study, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med. 2003;167:512-520.
- Cronin L, Cook DJ, Carlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23;1430-1439.
- Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids for treating severe sepsis and septic shock. Cochrane Database Syst Rev. 2004;1:CD002243.
The Case
An 81-year-old woman with diabetes mellitus presents with a three-day history of fever, chills, left-side flank pain, and dysuria. Her blood pressure upon presentation is 75/45 mm/Hg, her heart rate is 120 beats per minute, and she has a temperature of 103.1°`F and a respiratory rate of 22 breaths/minute. On physical examination, she is an ill-appearing elderly woman, with dry oral mucosa and left costo-vertebral angle tenderness. Lab work shows leukocytosis of 18,000 mg/dL with 88% polymorphonuclear leukocyte (PMN), the urine analysis is consistent with a urinary tract infection, and a chemistry panel reveals elevated BUN and creatinine levels of 52 mg/dL and 2.4 mg/dL, respectively. In the emergency department, she is given a bolus of 2 liters normal saline, but her blood pressure remains 78/49 mm/Hg. She is then started on broad-spectrum antibiotics and a norepinephrine drip, and is admitted to the ICU.
What role would steroids add to her management?
Background
Sepsis is the clinical syndrome defined by the presence of systemic inflammatory response syndrome (SIRS) in the setting of an infection. SIRS is defined by the presence of at least two of the following: fever or hypothermia; leukocytosis, leukopenia, or bandemia; heart rate >90 bpm; or tachypnea or hypocapnia.
When acute organ dysfunction, such as acute renal failure, altered mental status, or acute lung injury (hypoxemia), is present, sepsis is classified as severe.
Septic shock is a state of sepsis associated with acute circulatory collapse characterized by persistent arterial hypotension (defined as a systolic blood pressure <90 mmHg, a mean arterial pressure <60 mmHg, or a reduction in systolic blood pressure of >40 mmHg from baseline) despite fluid resuscitation attempts.1
The incidence and mortality due to sepsis and septic shock is directly related to the age of the patient, many of whom require ICU hospitalization.2 Clinically, this portends a great challenge, as the incidence of sepsis is likely to increase as the U.S. population ages.
Initial management of a patient with sepsis/septic shock is goal-directed therapy, which consists of early administration of broad-spectrum antibiotics, crystalloid or colloid fluid resuscitation, and use of vasopressor support to improve hemodynamics and maintain a mean arterial
pressure ≥65 mmHg. Patients with acute lung injury may also require prompt ventilator support.
The role of steroids in sepsis is controversial.
Review of Steroids
Steroids have long been known for their anti-inflammatory properties. From the 1950s to the 1980s, high-dose steroids (methylprednisolone 30mg/kg and dexamethasone 3 mg/kg to 6 mg/kg in divided doses) were used in the management of sepsis. This was based on a study by Schumer that showed steroids reduced mortality to 10% from 38%.3
Later, Sprung and colleagues demonstrated reversal of shock and improved short-term survival with high-dose steroids in patients with sepsis, but subsequent prospective randomized trials did not support this beneficial effect of high-dose steroids.4-6 In fact, two meta-analyses in 1995 concluded that high-dose steroids are ineffective and potentially harmful, and associated with higher mortality, secondary infections, and renal and hepatic dysfunction.7,8 Thereafter, the use of high-dose steroids fell into disfavor.
In the early 2000s, there was an emergence in the use of low-dose steroids in patients with sepsis. This was based on various trials showing the benefit of the use of low-dose steroids in the reversal of septic shock without significant side effects, discussed further below.
Pathophysiology
Steroids improve hemodynamic parameters. In an animal model, Hinshaw and colleagues induced septic shock in adrenalectomized dogs by infusing lethal doses of E. coli. The untreated dogs died within hours, whereas the dogs treated with antibiotics and steroids had a complete recovery from shock, and survived more than 100 hours.9
During sepsis, endotoxins induce nitric oxide synthase, which produces relaxation of vascular smooth muscle tone, with resultant hypotension and reduced contractility response to norepinephrine.10 Corticosteroids prevent induction of nitric oxide synthase and enhance the vaso-active response to catecholamines through the glucocorticoid receptors. In vascular endothelial cells, glucocorticoids also inhibit serum phospholipase A2, reducing the production of vasodilators, such as prostacyclin and prostaglandin E1.11,12
Steroids reduce inflammation. Sepsis is driven by a systemic inflammatory response, in which components of the outer-cell membrane of both gram-positive and gram-negative bacteria and endotoxins induce the production of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-alpha) and interleukin-1 (IL-1).13 These cytokines have a direct toxic effect on various tissues. In addition, inflammatory cytokines suppress adrenal response to adrenocorticotropic hormone (ACTH), which results in decreased endogenous cortisol production, and compete with glucocorticoids for their receptors, inducing resistance to the action of steroids at the tissue level.14
In healthy volunteers challenged with bacterial endotoxins, low-dose steroids (~10 mg of prednisolone) suppress the release of proinflammatory cytokines and prevent the activation of endothelial cells and neutrophils.15 Steroids also inhibit the release of toxic enzymes, such as lysozymes and superoxides from neutrophils.16,17
Data Supporting Steroid Use in Septic Shock
Mortality data. Two major studies evaluated the effect of low-dose steroids in patients with septic shock. Annane and colleagues conducted a placebo-controlled, blinded trial and divided the study population into “responders” and “nonresponders” based on their response to ACTH stimulation test. Within the “nonresponder” group, steroids reduced the risk of mortality by 16% (63% mortality in the placebo group and 53% mortality in the corticosteroid group, P=0.02).16 Steroids also significantly reduced ICU mortality (70% versus 58%), hospital mortality (72% versus 61%) and one-year mortality (77% versus 68%) compared with placebo. No statistically significant difference in mortality between steroids and placebo was seen in the “responder” group.16
The CORTICUS trial, a multicenter, randomized, double-blind, placebo-controlled trial, showed no significant difference in 28-day mortality between those treated with corticosteroids (39.2%) and those receiving a placebo (36%, P=0.069).17 There was also no significant difference in either hospital or ICU mortality in this study.
A recent meta-analysis demonstrated no significant effect of corticosteroid treatment on 28-day mortality, ICU mortality, or hospital mortality in septic shock. However, subanalysis of trials using a prolonged course (>5 days) of low-dose steroids (300 mg of hydrocortisone or equivalent) showed a significant reduction in 28-day all-cause mortality (P=0.02) and hospital mortality (P =0.05), and a decrease in ICU length of stay.18
Reversal of shock. Various studies have shown a decrease in the time necessary to reverse septic shock with the use of low-dose steroids. Annane and colleagues showed the median time to vasopressor therapy withdrawal was seven days in the group treated with steroids versus nine days in the placebo group (P=0.01).16 The CORTICUS study demonstrated significantly shorter times to reversal of shock in the group treated with hydrocortisone compared with the placebo group—3.3 days versus 5.8 days (P<0.001).17 In a smaller study, 68% of hydrocortisone-treated patients achieved seven-day shock reversal compared with 21% in the placebo group, a difference of 47% in the rate of reversal of shock.18
Guidelines for the Use of Steroids in Septic Shock
In which sepsis patients should I use steroids? The large clinical trials that found a benefit to low-dose steroids included patients with a systolic blood pressure <90 mm/Hg for more than one hour, despite aggressive fluid and vasopressor therapy. Based on these and other smaller trials, the Surviving Sepsis Campaign recommends the addition of IV steroids to those patients with septic shock who don’t respond to adequate fluid and vasopressor resuscitation.19
Should I obtain ACTH stimulation test in these patients? Although the Annane study showed that “nonresponders”—those who did not achieve ≥9 mcg/dL increase in cortisol level after 30 to 60 minutes of ACTH administration—were more likely to benefit from steroids, the overall trial population appeared to benefit regardless of the ACTH response.16
Furthermore, the CORTICUS study showed no difference between the corticotropin responder and nonresponder group. Also, most cortisol immunoassays measure total cortisol (free and protein-bound), whereas the free cortisol level is the more clinically relevant measurement. Hence, current guidelines from the American College of Critical Care Medicine and Surviving Sepsis Campaign do not recommend performing an ACTH stimulation test prior to administration of steroids.20
What type of steroids should I use, and at what dose? Current guidelines recommend IV hydrocortisone in a dose of 200 mg/day to 300 mg/day given as 50 mg every six hours or 100 mg every eight hours for at least seven days before tapering. Alternatively, IV hydrocortisone can be given as a bolus of 100 mg followed by a continuous infusion at 10 mg/hr (240 mg/day). Hydrocortisone at this dose has intrinsic mineralocorticoid activity, obviating the need for adding fludrocortisone. Fludrocortisone may otherwise be added as 50 mcg daily, if using a corticosteroid without significant mineralocorticoid activity. Patients with septic shock should not be treated with dexamethasone, which causes immediate and prolonged suppression of the hypothalamic-pituitary-adrenal axis.21
Do I need to taper off the steroids? It is recommended to wean the steroids after seven or more days of use, when vasopressors are no longer required. Keh and colleagues noted a 30% recurrence of shock in patients when the steroids were not tapered.22 There was also evidence of immunologic rebound after abrupt cessation of steroids, with an increase in inflammatory markers.23 The taper should decrease the dose every two or three days in small steps.
What potential side effects should I be concerned about? Overall, the use of higher dose corticosteroids is associated with significant potential side effects, including a worsening of the underlying infection, new infection, hyperglycemia, hypernatremia, and gastrointestinal bleeding. In a meta-analysis of nine clinical trials with high-dose corticosteroids (a starting dose of ~30mg/kg/day of methylprednisolone), Cronin and colleagues found a trend toward increased mortality due to secondary infections (relative risk 1.70; 95% confidence interval, 0.70 to 4.12).24 A recent meta-analysis of 15 trials found low-dose corticosteroids reduced ICU mortality and increased the proportion of shock reversal by Day 7 and by Day 28 without increasing the rate of gastroduodenal bleeding, super-infection, or hyperglycemia.25
Back to the Case
Our patient was admitted to the medical ICU. After obtaining urine and blood cultures, she was started on IV levofloxacin. She remained hypotensive despite IV fluids and IV norepinephrine. She was started on IV hydrocortisone 50 mg every six hours. Over the next 48 hours, her hemodynamic parameters improved. Urine and blood cultures came back positive for E. coli. Her BUN and creatinine decreased to 24 mg/dL and 1.4 mg/dL, respectively.
Later, she was weaned off norepinephrine and transferred out of the ICU. On hospital Day 7, a slow taper of her hydrocortisone initiated, and antibiotics were switched to oral levofloxacin. She was later discharged home in stable condition.
Bottom Line
In patients with septic shock that is unresponsive to IV fluid resuscitation and vasopressors, the addition of low-dose corticosteroids is relatively safe and can improve rate of reversal of shock, reduce time to reversal of shock, decrease ICU length of stay, and potentially lower mortality.
Drs. Gandhi and Asudani are health science assistant professors of medicine in the Division of Hospital Medicine at the University of California at San Diego.
References
- Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Intensive Care Med. 2003;29:530-538.
- Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303-1310.
- Schumer W. Steroids in the treatment of clinical septic shock. Ann Surg. 1976;184:333-341.
- Sprung CL, Caralis PV, Marcial EH, et al. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med. 1984;311:1137-1143.
- The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317:659-665.
- Bone RC, Fisher CJ Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653-658.
- Lefering R, Neugebauer EA. Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med. 1995;23:1294-1303.
- Cronin L, Cook DJ, Cartlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;24:1430-1439.
- Hinshaw LB, Beller BK, Chang AC, et al. Corticosteroid/antibiotic treatment of adrenalectomized dogs challenged with lethal E. coli. Circ Shock. 1985;16:265-277.
- Rees DD, Cellek S, Palmer RM, Moncada S. Dexamethasone prevents the induction of NO synthase and the associated effects on vascular tone, an insight into endotoxin shock. BioChem BioPhy Res Comm. 1990;173:541-547.
- Axelrod L. Inhibition of prostacyclin production mediates permissive effect of glucocorticoids on vascular tone. Lancet. 1983;1:904-906.
- Annane D, Bellissant E, Sebille V, et al. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenaline reserve. Br J Clin Pharmacol. 1991;46:589-597.
- DeKruif MD, Lemaire LL, Giebelen IA, et al. Prednisolone dose dependently influences inflammation and coagulation during human endotoxemia. J Immunol. 2007;178:1845-1851.
- Goldstein IM, Roos D, Weissman, G et al. Influence of corticosteroids on human polymorphonuclear leukocyte function in vitro. Inflammation. 1976;1:305-316.
- Briegel J, Kellerman W, Forst H, et al. Low-dose hydrocortisone infusion attenuates the SIRS. The Phospolipase A2 Study Group. Clin Invest. 1994;72:782-787.
- Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisones on mortality in patients with septic shock. JAMA. 2002;288:862-871.
- Sprung CL, Annane D, Keh D, et al. Hydrocortisone therapy in patients with septic shock. N Engl J Med. 2008;358:111-124.
- Annane D, Bellissant E, Bollaert P. Corticosteroids in the treatment of severe sepsis and septic shock in adults. A systematic review. JAMA. 2009;301:2362-2375.
- Dellinger DP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008;34:17-60.
- Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937-1949.
- Reincke M, Allolio B, Würth G, et al. The hypothalamic-pituitary-adrenal axis in critical illness: Response to dexamethasone and corticotropin-releasing hormone. J Clin Endocrinol Metab. 1993;77:151-156
- Keh D, Weber-Carstens S, Ahlers O. Adjunctive therapies in severe sepsis and septic shock: current place of steroids. Curr Infect Dis Rep. 2008;10:354-361.
- Keh D, Boehnke T, Weber-Cartens S, et al. Immunologic and hemodynamic effects of “low dose” hydrocortisone in septic shock: a double blind study, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med. 2003;167:512-520.
- Cronin L, Cook DJ, Carlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23;1430-1439.
- Annane D, Bellissant E, Bollaert PE, et al. Corticosteroids for treating severe sepsis and septic shock. Cochrane Database Syst Rev. 2004;1:CD002243.
The Hand That Feeds You
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
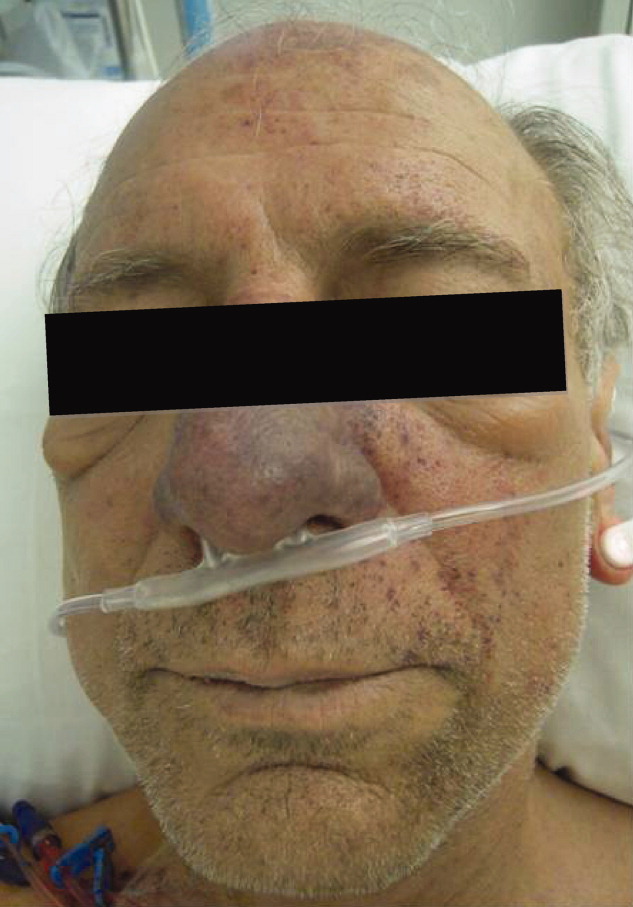
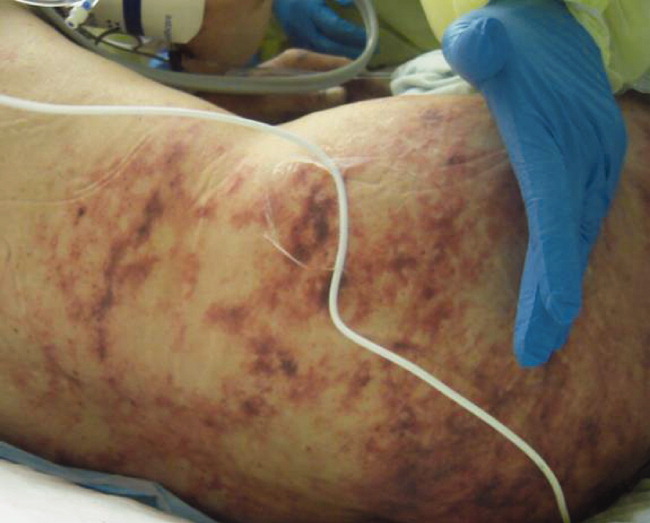
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
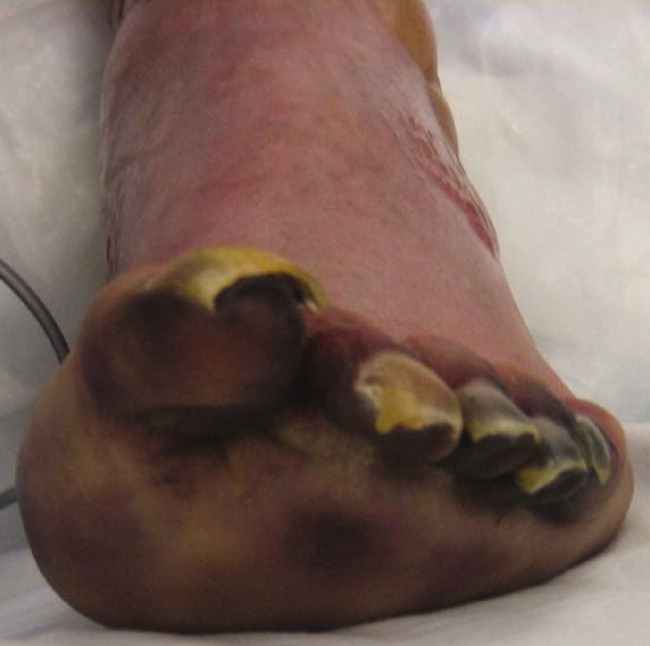
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
A 66‐year‐old man presented to the Emergency Department (ED) with rash and malaise in early April. He was in his usual state of good health until the morning of presentation, when he awoke feeling lethargic. Over the course of the day, his hands and feet grew cold and numb, his nose became dark red, and he developed a diffuse, net‐like red rash over his legs, hands, buttocks, and trunk. He had multiple maroon bowel movements. His wife noted that he became incoherent and brought him to the ED.
This apparently previously healthy man presented with an acute episode of fatigue and altered mental status accompanied by a prominent cutaneous eruption. The differential diagnosis will ultimately be guided by the morphology of the rash. At this stage, infectious diseases, drug or toxin exposure, and allergic processes including anaphylaxis must all be considered in this patient with rash and acute illness. The maroon bowel movements likely represent a gastrointestinal bleed that may be part of a unifying diagnosisa hematologic disorder, a vasculitis, or liver disease.
In the ED, the patient was reportedly febrile (exact temperature not recorded) with a blood pressure of 96/54 mmHg. He had pulse oximetry of 88% on room air and a diffuse purpuric rash. The patient was noted to have a leukocytosis, thrombocytopenia, coagulopathy, and an elevation of his creatinine and cardiac enzymes. He was given fluids, fresh frozen plasma, and broad‐spectrum antibiotics, and transferred directly to the intensive care unit of a tertiary medical center for further management.
Upon arrival to the intensive care unit, he complained of fatigue, progression of his nonpruritic, nonpainful rash, and worsening numbness and tingling of his extremities. He denied headache, nuchal rigidity, photophobia, vision or hearing changes, chest pain, cough, abdominal pain, myalgias, or arthralgias. While being interviewed, he had dark brown emesis and a bloody bowel movement.
The patient's past medical history included bacterial pericarditis as a teenager and remote hepatitis of unclear etiology. He rarely saw a physician, took no medications, and had no known medication allergies.
The patient worked as president of a software company and lived with his wife. He had smoked 1 to 2 packs of cigarettes a day for the past 30 years. He endorsed 2 of 4 CAGE criteria (need to Cut down, Annoyed when asked about alcohol, feel Guilty about drinking, need for an Eye opener), and his wife and had never been tested for human immunodeficiency virus (HIV). Family history was unremarkable.
The patient's presentation is concerning for a life‐threatening disease process with a rapid course. In the setting of the laboratory abnormalities demonstrating multi‐organ dysfunction, aggressive volume resuscitation and prompt initiation of broad‐spectrum antibiotics are indicated. The history does not reveal an obvious source of infection or exposure to a new drug, toxin, or allergen. His apparent gastrointestinal bleed could be explained by complications of liver disease from chronic alcohol use. For example, he could have variceal bleeding or gastropathy from portal hypertension. Alternatively, he may have bleeding secondary to a coagulopathy from decreased synthetic function of clotting factors. Other possibilities include a perforated viscus (eg, peptic ulcer) leading to bleeding and peritonitis or mesenteric ischemia, though the absence of abdominal pain makes these unlikely.
At this point, the overall presentation is most concerning for infection, especially given his chronic alcohol use and the vague history of hepatitis. The acute onset and severity of the illness are consistent with an aggressive, suppurative bacterial infection. The most likely causative organisms include gram‐negative bacteria, especially Neisseria meningitidis (with or without meningitis), as well as Staphylococcus aureus, Streptococcus pyogenes, and Rickettsia rickettsii (Rocky Mountain spotted fever).
Several months prior to presentation, he had traveled to Mexico. Two months prior to presentation, he made a trip to North Carolina and Ohio to visit his brother, who subsequently died of pneumonia. One month prior to presentation, he had traveled to urban China for work.
Because the presentation is so acute and the patient's travel took place over 1 month ago, this is unlikely to be a travel‐associated illness. Furthermore, the course is too acute to be consistent with endemic diseases of Central America and the midwestern United States, such as tuberculosis, brucellosis, and histoplasmosis.
He had a temperature of 38.7C. His heart rate was 110 beats per minute. His blood pressure was 115/78 mmHg, respiratory rate was 24 breaths per minute, and oxygen saturation was 99% on 6 liters via nasal cannula. The patient was a well‐nourished, middle‐aged man who appeared uncomfortable. He was in mild respiratory distress, though able to speak in full sentences. He was alert, coherent, and oriented to self, place, date, and time.
Skin examination revealed nonblanching purpuric papules coalescing into stellate plaques on his scalp, forehead, nose, cheeks, bilateral ears, hands, and feet (Figure 1). Acral surfaces, including hands and feet, were cyanotic without evidence of gangrene. He had nonblanching retiform purpuric plaques on his right flank, lower abdomen, low back, buttock, penis, scrotum, thighs, and legs (Figure 2). His right dorsal hand had 3 healing erosions of 3 to 10 mm in size without associated edema, erythema, or drainage.
Mucous membranes were dry without lesions. Cardiac examination demonstrated tachycardia without appreciable murmur. He was mildly tachypneic and his lungs were clear to auscultation without adventitious breath sounds. His abdominal examination was unremarkable. His hands and feet were cool with decreased sensation to touch. He had full range of motion and intact muscle strength, but mild bilateral dysmetria with finger‐nose‐finger testing. His radial and dorsalis pedis pulses were symmetric and brisk. Rectal exam revealed guaiac‐positive stool.
The patient's vital signs are compatible with the systemic inflammatory response syndrome. The presence of retiform purpura raises concerns for a systemic vasculitis with destruction of the vessel wall, or intravascular occlusion with thrombosis or emboli. Absence of murmur does not rule out endocarditis but makes it less likely. He has no risk factors for vasculitis, so the purpura, in conjunction with both bleeding and thrombosis, is much more suggestive of disseminated intravascular coagulation (DIC). This clotting disorder can result from a noninfectious trigger, such as acute pancreatitis or malignancy, but his presentation is more worrisome for a severe infection leading to DIC and complicated by purpura fulminans. He does not show signs of hepatic encephalopathy or cirrhosis, making decompensated liver disease a less likely inciting factor of his presentation.
Further exposure history was obtained: The patient often spent time outdoors near his rural home and used a weed‐whacker in his yard the day before admission. He owned 3 horses which he fed and often rode. He had 3 healthy dogs and had been bitten in attempts to break up fights among them, most recently 3 days prior to admission. He lived in mountain lion territory but had no direct exposure to lions. He had no known insect bites. He regularly drank well water, and consumed medium‐rare hamburgers 4 days prior to admission. One week prior to admission, a child with possible streptococcal pharyngitis visited his home.
With this history, the patient was treated with aggressive intravenous fluids and meningeal doses of ceftriaxone, vancomycin, and metronidazole.
In the summer, outdoor exposure to brush confers a risk of tick‐borne infections, including rickettsial diseases, ehrlichiosis, and spirochetal relapsing fever. However, this patient presented in the spring, and apart from rickettsial spotted fever, these illnesses tend to be indolent. It is conceivable, though unlikely, that the weed‐cutting device may have aerosolized fulminant zoonotic pathogens such as Francisella tularensis or plague that can be found in mountain lion territory.
Well water exposure suggests leptospirosis, which can present in a fulminant fashion with multi‐organ dysfunction, but is more often a subacute illness (developing over many days to a week or two). His ingestion of potentially undercooked meat raises the possibility of enterohemorrhagic infection complicated by the hemolytic uremic syndrome (HUS). However, while the purpuric rash and renal failure are compatible with HUS, the pace of illness and accompanying hypotension once again favor alternative infectious diagnoses.
The incubation period and presentation is concerning overwhelming bacterial infection related to the dog bite. Microbiological considerations include streptococcal species, Staphylococcus aureus, and gram‐negative organisms including Pasteurella species and Capnocytophaga canimorsus. The latter 2 organisms are of particular interest since they tend to cause severe sepsis in patients with alcoholism.
The antibiotic selection in this case is not straightforward. In general, empiric therapy for infections related to dog bites should include treatment for beta‐lactamaseproducing bacteria and anaerobes (eg, piperacillin/tazobactam). Yet, given the clinical presentation, severity of illness, and possible DIC, it is appropriate to be concerned about meningococcemia. Unfortunately, the tazobactam in piperacillin/tazobactam has poor central nervous system penetration so would be suboptimal treatment for meningitis. At this point, ceftriaxone, vancomycin, and metronidazole is a reasonable regimen.
Laboratory results were notable for blood urea nitrogen 50 mg/dL, creatinine 3.47 mg/dL, white cell count 21,800/L, with an absolute neutrophil count of 20,690/L, hematocrit 35.9%, platelet count 34,000/L, International Normalized Ratio 1.5, and partial thromboplastin time 44.0 seconds. His alanine aminotransferase was 356 U/L (1641 U/L), aspartate aminotransferase 959 U/L (1259 U/L), alkaline phosphatase 50 U/L (29111 U/L), and total bilirubin 1.7 mg/dL (0.31.3 mg/dL). Fibrinogen was 283 g/L (202430 g/L), lactate dehydrogenase was 1883 U/L (91185 IU/L), and uric acid was 10.5 mg/dL (3.77.7 mg/dL). His troponin I was 1.18 ng/mL (<0.05 ng/ml), and his electrocardiogram showed sinus tachycardia but no evidence of myocardial ischemia. Chest x‐ray showed no infiltrate or evidence of volume overload. Lumbar puncture was deferred out of concern for ongoing disseminated intravascular coagulation.
Transthoracic echocardiogram revealed global hypokinesis and reduced left ventricular systolic function with ejection fraction of 35%. There was no evidence of vegetations or thrombus.
The patient's thrombocytopenia and prolonged coagulation parameters further support the presence of DIC. A peripheral blood smear should be examined. If microangiopathic changes are found, other diagnoses such as thrombotic thrombocytopenic purpura might be considered, although the rapid pace of illness and presence of hypotension still make sepsis with DIC more likely.
While septic shock often causes multi‐organ system failure secondary to hypoperfusion, the presumed rapid onset of hepatic and renal abnormalities suggests that microvascular thrombosis is playing a larger role in his organ system dysfunction. Microvascular thrombosis could also contribute to his myocardial injury, though globally depressed ejection fraction and elevated troponin might also be explained by infectious myocarditis. A third possibility is that his severe sepsis caused his myocardial dysfunction. Regardless of its etiology, the patient has no clinical evidence of congestive heart failure, so no specific therapy is required at this time. However, his cardiopulmonary exam should be monitored closely, and if he survives, he should have repeat echocardiography to monitor for resolution of the global hypokinesis.
Further evaluation revealed creatine kinase of 45,000 ng/ml (55380 ng/ml) and repeat troponin of >22 ng/ml. Protein C level was low at 30%. Testing for HIV was negative. Blood smear from time of transfer had few schistocytes. Urinalysis showed muddy brown casts but no dysmorphic red blood cells or red cell casts. The patient was placed on continuous veno‐venous hemofiltration (CVVH) for worsening renal failure and oliguria from presumed acute tubular necrosis in the setting of rhabdomyolysis and sepsis.
The patient has severe rhabdomyolysis that cannot fully be explained by his initial hypoperfusion and is more likely related to the overwhelming infection and microthrombosis. Rhabdomyolysis probably contributed to his acute tubular necrosis and renal failure.
Dermatology consultation identified the rash as likely purpura fulminans. They recommended a skin biopsy to rule out vasculitis. Three skin biopsies revealed micro‐vascular thrombosis; direct immunofluorescence test was negative for vasculitis; his skin tissue culture was negative for bacterial, mycobacterial, and fungal organisms.
Input from the dermatology service was key in identifying the rash. Purpura fulminans has a limited differential that includes severe infection from gram‐negative organisms and protein C and S deficiency. Since the biopsy results made vasculitis unlikely, the team was able to focus greater attention on potential pathogens such as Pasteurella species and C. canimorsus.
The biopsy also confirms the clinical suspicion that microvascular thrombosis is causing the patient's acute kidney injury, rhabdomyolysis, and myocardial ischemia. The presence of microvascular thrombosis prompts consideration of antithrombotic therapy such as heparin, but benefits of this therapy must be weighed against contraindications including bleeding and thrombocytopenia.
Ultimately out of concerns for recurrent gastrointestinal bleeding, the primary team decided not to treat with heparin or other antithrombotic therapy.
After several days of supportive care with antibiotics and renal replacement therapy, the patient showed gradual improvement of his retiform purpura, sensory neuropathy, laboratory data, and other markers of end‐organ dysfunction. Purpura of his fingertips, feet, and toes progressed to dry gangrene (Figure 3), which was monitored for potential need for amputation. He remained dependent on intermittent hemodialysis.
His initial antibiotic regimen was narrowed to ceftriaxone monotherapy. Five days after initial presentation, blood cultures drawn from the outside emergency department grew a gram‐negative rod in the anaerobic broth. Ten days later, this gram‐negative rod was identified as Capnocytophaga canimorsus. He was ultimately discharged to a skilled nursing facility.
Generally growth of an organism in broth only suggests either a very low inoculum or that the isolate is a contaminant. In this case, it was because the causative organism, C. canimorsus, is an obligate anaerobe and quite fastidious, so unlikely to grow easily. The identification of C. canimorsus from the initial blood culture is not surprising in this patient who presented with severe sepsis, DIC, and purpura fulminans after a recent dog bite. While the patient's chronic alcohol use may explain his fulminant infection from an atypical organism, one should always consider occult underlying malignancy as a predisposing factor, particularly in patients of this age group.
With the appropriate course of antibiotics, C. canimorsus infection should be completely cured. However, recovery of kidney and cardiac function could take weeks to months, and his dry gangrene may or may not resolve.
COMMENTARY
Capnocytophaga canimorsis sepsis is a rare and potentially deadly complication of dog bites that can present with rash, cellulitis, purpura fulminans, arthritis, meningitis, and endocarditis. The discussant considered a broad differential for the presentation of fever, rash, and acute illness. While the travel history was intriguing, the severity and pace of illness allowed him to focus attention on more recent infectious exposures. The ultimate key to the diagnosis was the patient's history of dog bite, an important but underrecognized source of serious infection in the United States.
According to the Centers for Disease Control and Prevention, there are approximately 4 million dog bites in the country each year. Of these, 300,000 bite victims seek care in the emergency department, resulting in 13,000 hospitalizations and 20 deaths annually.1 Infected dog bite wounds often grow polymicrobial flora. Pasteurella species are the most frequently found organisms in both dog and cat bite wounds. However, other aerobes such as streptococci, staphylococci, Moraxella, and Neisseria, as well as anaerobes including Fusobacterium and Bacteroides species, are also common.2
C. canimorsis is a facultative, fastidious gram‐negative bacillus found in the mouth flora of not only dogs but also cats and humans. It is often mistaken for other gram‐negative rod species.3 As with the patient described in this report, systemic infection from C. canimorsis can follow even superficial or well‐healed bite wounds.
Since this bacterium was first described in the literature 30 years ago, more than 100 cases of C. canimorsus infection have been described, with a mortality rate of nearly 30%.4 C. canimorsus occurs more frequently in males and in patients 50 to 70 years of age. Traditional risk factors include alcohol abuse, asplenia, immunosuppression, and corticosteroid treatment. However, in a case series of 56 isolates in California, only 10% of patients with Capnocytophaga sepsis were asplenic and none had alcohol abuse reported in their medical charts. In this series, median time from dog bite to the onset of symptoms was 3 days. Eighty‐five percent of patients presented with fever, while 32% had sepsis and 13% had DIC or septic shock.3
While C. canimorsus was once susceptible to a range of antibiotics, several reports from Canada and Europe document rising rates of beta‐lactamaseproducing strains that have caused clinically significant disease.5, 6 Individual susceptibility data take days to obtain, so it is important to start with empiric therapy. In general, empiric therapy for all serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes, for example, with amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam. If the patient is allergic to penicillin, clindamycin plus a fluoroquinolone can be used instead.
There are previous reports of purpura fulminans and symmetric peripheral gangrene following Capnocytophaga infection from dog bites.7, 8 Purpura fulminans is defined as rapidly progressive skin necrosis due to dermal vascular thrombosis, often in the setting of DIC. Early involvement occurs at acral sites, such as the nose, ears, fingers, and toes. Purpuric lesions often progress to skin necrosis or dry gangrene within 24 to 48 hours. In a review of 12 patients with purpura fulminans, only 9 survived. Eight of the 9 survivors required amputation of at least 1 limb, and 4 of them required 4‐limb amputation.7
In this patient who presented with fever and rash, the discussant recognized early on an underlying infectious etiology. Although the patient's exposure history led the discussant to consider a host of possibilities, the recognition of purpura fulminans allowed him to narrow his differential. Ultimately, the dog's bite clinched the diagnosis.
KEY TEACHING POINTS
-
Sepsis caused by C. canimorsus is often characterized by rash, cellulitis, arthritis, meningitis, and endocarditis. In some instances, infection can progress to purpura fulminans.
-
In cases where fastidious organisms are suspected as an infectious source, microbiology labs should be notified of suspected organisms so they can extend incubation periods or use special media to maximize culture yield and the likelihood of accurate identification.
-
Empiric therapy for serious dog bites should cover beta‐lactamaseproducing bacteria and anaerobes. Consider using amoxicillin/clavulanate, ampicillin/sulbactam, or piperacillin/tazobactam.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors thank Snigdha Vallabhaneni, MD, from the UCSF Division of Infectious Diseases, for her contributions to the discussion on C. canimorsus. They also thank Kanade Shinkai, MD, PhD, from the UCSF Department of Dermatology, and Heather Nye, MD, PhD, from the UCSF Division of Hospital Medicine, for their review of the manuscript.
Disclosure: Nothing to report.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
- ,,.Incidence of dog bite injuries treated in emergency departments.JAMA.1998;279:51–53.
- ,,,,.Bacteriologic analysis of infected dog and cat bites. Emergency Medicine Animal Bite Infection Study Group.N Engl J Med.1999;340:85–92.
- ,,,.Diagnosing Capnocytophaga canimorsus infections.Emerg Infect Dis.2006;12:340–342.
- ,,.Capnocytophaga canimorsus infections in human: review of the literature and cases report.Eur J Epidemiol.1996;12:521–533.
- ,,,,.Antimicrobial susceptibilities and beta‐lactamase characterization of Capnocytophaga species.Antimicrob Agents Chemother.1992;36:2197–2200.
- ,,,,,.Bacteremia due to Capnocytophaga species in patients with neutropenia: high frequency of beta‐lactamase‐producing strains.Clin Infect Dis.1999;28:1172–1174.
- ,,.Presentation and outcome of purpura fulminans associated with peripheral gangrene in 12 patients at Mayo Clinic.J Am Acad Dermatol.2007;57:944–956.
- ,,,.Capnocytophaga canimorsus sepsis with purpura fulminans and symmetrical gangrene following a dog bite in a shelter employee.Am J Med Sci.2004:327:369–372.
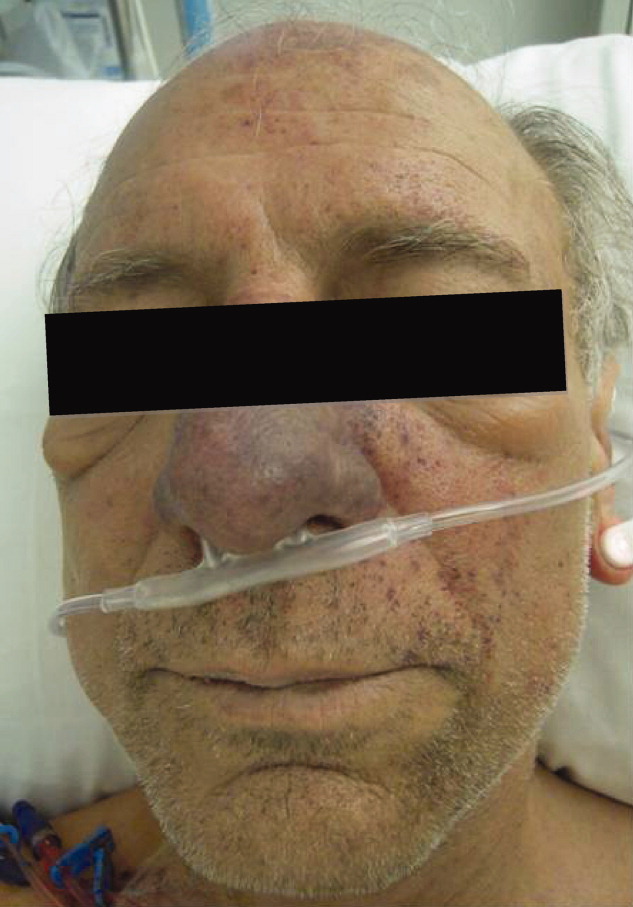
Overcome by Weakness
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.

Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
An 89‐year‐old man presented to the emergency department with progressive fatigue, confusion, and generalized weakness over 2 months, worsening in the prior few days.
Four categories of disease account for most cases of confusion in the elderly: metabolic derangements; infection (both within and outside of the central nervous system); structural brain disorder (eg, bleed or tumor); and toxins (generally medications). It will be important early on to determine if weakness refers to true loss of motor function, reflecting a neuromuscular lesion.
At baseline, the patient had normal cognition, ambulated without assistance, and was independent in activities of daily living. Over the preceding 2 months, general functional decline, unsteady gait, balance problems, and word‐finding difficulty developed. He also needed a front‐wheel walker to avoid falling. One month prior to presentation, the patient's children noticed he was markedly fatigued and was requiring a nightly sedative‐hypnotic in order to fall asleep.
He denied any recent travel, sick contacts, or recent illness. He denied vertigo, dizziness, or syncope. He reported occasional urinary incontinence which he attributed to being too weak to get to the bathroom promptly.
This rapid progression over 2 months is not consistent with the time course of the more common neurodegenerative causes of dementia, such as Alzheimer's or Parkinson's disease. In Parkinson's, cognitive impairment is a late feature, occurring years after gait and motor disturbances develop. Normal pressure hydrocephalus, which causes the classic triad of incontinence, ataxia, and confusion, would also be unlikely to develop so abruptly. Although we do not think of vascular (multi‐infarct) dementia as having such a short time course, on occasion a seemingly rapid presentation is the postscript to a more insidious progression that has been underway for years. A subdural hematoma, which may have occurred with any of his falls, must also be considered, as should neoplastic and paraneoplastic processes.
His past medical history included paroxysmal atrial fibrillation, diabetes mellitus, hypertension, hyperlipidemia, coronary artery disease complicated by prior myocardial infarction for which he underwent coronary artery bypass grafting 7 years prior, mild aortic sclerosis and insufficiency, mild mitral regurgitation, anemia, recurrent low‐grade bladder cancer treated with serial local resections over the last 8 years, low‐grade prostate cancer which had not required treatment, hypothyroidism, chronic kidney disease, and lumbar spinal stenosis.
His atrial fibrillation and valvular disease put him at risk for thrombotic and infective embolic phenomena causing multiple cerebral infarcts. He has all the requisite underlying conditions for vascular dementia. Untreated hypothyroidism could explain his decline and sedation. Prostate and bladder cancers would be unusual causes of subacute central nervous system (CNS) disease. Finally, his chronic kidney disease may have progressed to uremia.
One year prior to admission, the patient developed bilateral shoulder pain, right‐sided headache with loss of vision in his right eye, fevers, and an elevated erythrocyte sedimentation rate (ESR). Although temporal artery biopsy specimens did not reveal arterial inflammation, he was started on high‐dose prednisone for polymyalgia rheumatica and giant cell arteritis (GCA); he experienced improvement in his ESR and in all symptoms, with the exception of permanent right eye blindness. Maintenance prednisone was continued for disease suppression.
Even without confirmatory biopsy results, the clinical case for GCA was compelling and the rationale for starting steroids strong; his sustained response over 1 year further supports the diagnosis. GCA is almost always confined to extracranial vessels, and altered sensorium would be an unusual manifestation. His extended treatment with prednisone expands the list of CNS and systemic infections, particularly opportunistic ones, for which he is now at risk.
Outpatient medications were prednisone at doses fluctuating between 10 and 20 mg daily, furosemide 20 mg daily, amiodarone 200 mg daily, levothyroxine 50 mcg daily, alendronate 70 mg weekly, eszopiclone 1 mg nightly, losartan 50 mg daily, and warfarin. The patient was an accomplished professor and had published a book 1 year prior to admission. He quit smoking over 30 years ago, and he occasionally drank wine. He denied any drug use.
Three months prior to the current presentation, the patient was hospitalized for right upper‐lobe pneumonia for which he received a course of doxycycline, and his symptoms improved. Follow‐up chest x‐ray, 4 weeks later (2 months prior to admission), showed only slight improvement of the right upper‐lobe opacity.
Leading possibilities for the persistent lung opacity are cancer and untreated infection. After 3 decades of being tobacco‐free, his smoking‐related risk of cancer is low, but remains above baseline population risk. There are at least 4 ways untreated lung cancer may render patients confused: direct metastases to the brain, carcinomatous or lymphomatous meningitis, paraneoplastic phenomenon (eg, limbic encephalitis), and metabolic derangements (eg, syndrome of inappropriate antidiuretic hormone secretion, hypercalcemia).
The upper‐lobe infiltrate that failed to improve with doxycycline could also reflect an aspiration pneumonia that evolved into an abscess, or an infection with mycobacteria or endemic fungi.
In the emergency department, the patient's temperature was 38.5C, blood pressure 139/56 mmHg, heart rate 92 beats per minute, respiratory rate 18 breaths per minute, and oxygen saturation while breathing ambient room air was 98%.
He was alert and well‐appearing. Jugular venous pressure was normal. The thyroid was normal. He had rhonchi in his right anterior upper chest and right lower lung base. Cardiac exam demonstrated a regular rhythm, with a 3/6 systolic murmur at the second right intercostal space that radiated to the carotids, and a 2/6 nonradiating holosystolic murmur at the apex. Abdomen was soft with no organomegaly or masses. There was no lymphadenopathy, and his extremities showed no clubbing or edema. There were multiple contusions in various stages of healing on his legs.
He was confused, had word‐finding difficulty, and frequently would lose his train of thought, stopping in mid‐sentence. He had no dysarthria. Cranial nerves were normal, except for reduced visual acuity and diminished pupillary response to light in his right pupil, which had been previously documented. Finger‐to‐nose testing was slow bilaterally, but was more sluggish on the right. Rapid alternating hand movements were intact. He was unable to perform heel‐to‐shin testing. Sensation was intact. Plantar reflexes were flexor bilaterally. Strength in his limbs was preserved both distally and proximally, and deep tendon reflexes were normal. However, he was unable to sit up or stand on his own due to weakness.
The fever on prednisone is a red flag for infection. The infection may be the primary diagnosis (eg, meningoencephalitis) or may reflect an additional superimposed insult (eg, urinary tract infection) on the underlying encephalopathy. Two murmurs in a febrile patient with the multifocal CNS findings suggest endocarditis. The abnormalities on chest examination could indicate a lung infection complicated by hematogenous spread to the brain, such as a lung abscess (secondary to the aspiration event), tuberculosis (TB), or endemic fungal infection.
Serum chemistries were normal, and the serum creatinine was 1.1 mg/dL. White blood cell count was 20,100 per mm3 with 90% neutrophils, 9% lymphocytes, and 1% monocytes. Hemoglobin was 13.7 g/dL, platelet count was 464,000 per mm3. Thyroid stimulating hormone (TSH) was 6.0 IU/mL (normal, <5.5). International normalized ratio (INR) was 2.2. Urinalysis was normal. Transaminases, bilirubin, and alkaline phosphatase were normal. Lactate was 1.9 mmol/L.
Electrocardiogram (EKG) was unchanged from his baseline. ESR was >120 mm/hr (the maximum reportable value); his ESR measurements had been gradually rising during the previous 4 months. Chest x‐ray demonstrated a right upper‐lobe opacity, slightly more pronounced in comparison with chest x‐ray 2 months earlier.
His fever, leukocytosis, elevated ESR, and thrombocytosis all reflect severe inflammation. While infection and then malignancy remain the primary considerations, a third category of inflammatory diseaseautoimmunitywarrants mention. For instance, Wegener's granulomatosis can cause pulmonary and CNS disease in the elderly.
Intravenous ceftriaxone and oral doxycycline were administered. Chest computed tomography (CT) (Figure 1) demonstrated dense right upper‐lobe mass‐like consolidation with associated adenopathy and pleural effusion; in addition, several nodules were present in the left and right lower lobes, the largest of which was 10 mm. CT of the chest 10 months prior to current admission had been normal. CT of the brain, performed without contrast, demonstrated multiple areas of abnormal vasogenic edema with suggestion of underlying masses.
The imaging provides evidence of a combined pulmonaryCNS syndrome. It is far more common for disease to originate in the lungs (a common portal of entry and environmental exposure) and spread to the brain than vice versa. The list of diseases and pathogens that affect the lungs and spread to the brain includes: primary lung cancer, lymphoma, bacteria, mycobacteria, fungi, molds (eg, Aspergillus), Wegener's granulomatosis, and lymphomatoid granulomatosis. Bacterial lung abscess, such as that caused by Streptococcus milleri group, may spread to the brain. Nocardia, a ubiquitous soil organism, infects immunocompromised patients and causes a similar pattern. Actinomycosis is an atypical infection that may mimic cancer, particularly in the lungs; while head and neck disease is characteristic, CNS involvement is less so. Overall, the imaging does not specifically pinpoint 1 entity, but infection remains heavily favored over malignancy, with autoimmunity a distant third.
Respiratory cultures showed normal respiratory flora. Blood cultures grew no organisms. Two samples of induced sputum were negative for acid‐fast bacilli (AFB) on smear examination. Forty‐eight hours after a purified protein derivative (PPD) skin test was placed, there was 0 mm of induration. Magnetic resonance imaging (MRI) of the brain (Figure 2) demonstrated 8 ring‐enhancing supratentorial lesions at the graywhite junction.
Negative blood cultures substantially lower the probability of bacterial endocarditis; there are no epidemiologic risk factors for the rare causes of culture‐negative endocarditis (eg, farm exposure, homelessness). Two negative smears for AFB with dense pulmonary or cavitary disease signify a low probability of tuberculosis.
In the setting of depressed cell‐mediated immunity (eg, human immunodeficiency virus [HIV] infection or chronic prednisone use), multiple ring‐enhancing CNS lesions are a classic appearance of toxoplasmosis, but they also are typical of bacterial brain abscesses and Nocardia. Brain metastases are usually solid, but as central necrosis develops, peripheral enhancement may appear. The diffuse distribution and the localization at the graywhite junction further support a hematogenously disseminated process, but do not differentiate infection from metastases.
Transthoracic echocardiogram demonstrated normal left ventricular ejection fraction, clinically insignificant aortic sclerosis and mitral regurgitation, and no evidence of vegetations. Results of a CT‐guided fine‐needle aspiration of the lung were nondiagnostic, showing necropurulent material and benign lung parenchyma with fibrosis. A core biopsy of the lung showed alveolar tissue with patchy mild deposition of fibrinous material and rare scattered acute and chronic inflammatory cells without granulomas. Pleural fluid cytology showed reactive mesothelial cells with mixed inflammatory cells. There were no fungal elements or malignant cells.
The failure to detect malignancy after 2 biopsies and 1 thoracentesis lowers the suspicion of cancer, and thereby bolsters the probability of atypical infections which may elude diagnosis on routine cultures and biopsy. A detailed history, with attention to geographic exposures, is warranted to see which endemic mycosis would put him most at risk. Based on his California residency, disseminated coccidiomycosis or the ubiquitous Cryptococcus are conceivable. Nocardia remains a strong consideration because of his chronic immunosuppression and the lung‐CNS pattern.
Fungal stains and cultures from the biopsies and pleural fluid were negative. Serum antibodies to coccidiomycosis and serum cryptococcal antigen tests were negative. On the eighth hospital day, the microbiology lab reported a few acid‐fast bacilli from a third induced sputum sample. RNA amplification testing for Mycobacterium tuberculosis was negative.
Due to his continued decline, the patient met with the palliative care team and expressed his desire to go home with hospice. While arrangements were being made, he died later that day in the hospital.
There is reasonable evidence that tuberculosis is not the culprit pathogen here: negative PPD, 2 negative sputa in the setting of a massive necrotic lesion, and a negative RNA amplification test. Nontuberculous mycobacteria such as Mycobacterium avium complex (MAC) and M. kansasii may cause disease similar to TB, but they are usually not this difficult to identify. Nocardia is classically a weakly acid‐fast positive bacteria and fits this patient's clinical picture best.
Four colonies of Nocardia (not further speciated) were identified postmortem from the patient's sputum.
DISCUSSION
Nocardia species are ubiquitous soil‐dwelling, Gram‐positive, branching rods which are weakly positive with acid‐fast staining.1 Almost all Nocardia infections occur in patients with immune systems compromised by chronic disease (HIV, malignancy, alcoholism, chronic lung or kidney disease) or by medications. Corticosteroid treatment is the most frequent risk factor. In cases of nocardiosis in patients taking steroids, the median daily prednisone dose was 25 mg (range, 1080 mg) for a median duration of 3 months.2, 3
Nocardia should be considered in any patient with unexplained pulmonary, CNS, or cutaneous disease and appropriate risk factors. Pulmonary disease is most common, seen in approximately two‐thirds of patients, and is typically bilateral. Chest radiographic findings include infiltrates (59%), nodules (35%), effusions, and cavities.2 Up to half of all cases of pulmonary nocardiosis are associated with hematogenous dissemination, most commonly to the CNS, where manifestations include incidentally discovered asymptomatic lesions, headache, confusion, and focal neurologic deficits; meningitis is rare.1 CNS involvement and severe predisposing illness are adverse prognostic markers.
Diagnosis of nocardiosis is typically delayed by 6 weeks to 1 year.4, 5 This has been attributed to its rarity, its nonspecific and indolent presentation, its slow growth, and the difficulty isolating Nocardia from clinical specimens. Although Nocardia may disseminate widely to almost any site, isolation of Nocardia from blood cultures is rare. Clinicians must rely on sputum or tissue samples to demonstrate the characteristic Gram‐positive rods which stain weakly on acid‐fast preparations. Polymerase chain reaction (PCR)‐based tests improve the yield but are not routinely available.
The standard antibiotic for the treatment of Nocardia infections is trimethoprim‐sulfamethoxazole (TMP‐SMX) which has excellent CNS penetration. In patients with pulmonary disease or CNS dissemination, a second parenteral antimicrobial (usually amikacin or imipenem) is typically added to TMP‐SMX, and treatment is extended to 12 months or longer.6, 7 Prophylaxis with TMP‐SMX, which is usually prescribed to prevent Pneumocystis jirovecii in susceptible hosts, also reduces the incidence of Nocardia.2, 3, 6 Nocardia's restricted susceptibility pattern presents a challenge for hospitalists, as TMP‐SMX and aminoglycosides are rarely administered empirically for cases of suspected pneumonia or atypical pulmonary infections (other than P. jirovecii).
When confronted with the pattern of simultaneous pulmonary and CNS lesions, hospitalists must consider infections (lung abscess, mycobacteria, fungi, Nocardia), malignancies, and autoimmune conditions (sarcoidosis, Wegener's granulomatosis). This patient's weakness was a direct result of his weakened immune system, which allowed this weakly acid‐fast organism to flourish. Only by recognizing the possibility of nocardiosis (eg, a patient receiving steroids who develops pulmonary and CNS lesions) is there hope for early diagnosis and treatment.
TEACHING POINTS
-
Suspect disseminated nocardiosis in immunocompromised patients with unexplained pulmonary disease and CNS disease characterized by multiple ring‐enhancing abscesses.
-
Corticosteroid treatment is the most common risk factor for Nocardia infections. Patients taking prednisone at doses in excess of 10 mg daily for greater than 3 months should receive P. jirovecii prophylaxis with TMP‐SMX, which also reduces the incidence of Nocardia.
-
Prolonged courses of TMP‐SMX combined with at least 1 other agent for at least 612 months are typically required to treat disseminated Nocardia.
Acknowledgements
Disclosure: Dr Thomas E. Baudendistel is a former Deputy Editor at the Journal of Hospital Medicine and received a stipend for this work.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
- ,.Nocardia species: host‐parasite relationships.Clin Microbiol Rev.1994;7:213–264.
- ,,,,,.Nocardiosis at the turn of the century.Medicine (Baltimore).2009;88:250–261.
- ,.A case series and focused review of nocardiosis: clinical and microbiologic aspects.Medicine (Baltimore).2004;83:300–313.
- ,,, et al.Pulmonary nocardiosis: risk factors and outcomes.Respirology.2007;12:394–400.
- ,.Infection with Nocardia species in Queensland. A review of 102 clinical isolates.Med J Aust.1992;156:692–697.
- .Nocardia in solid organ transplant recipients.Am J Transplant.2009;9:S70–S77.
- .Nocardiosis.Clin Infect Dis.1996;22:891–905.
Rounding up the usual suspects
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
A 76‐year‐old white male presented to his primary care physician with a 40‐pound weight loss and gradual decline in function over the prior 6 months. In addition, over the previous 2 months, he had begun to suffer a constant, non‐bloody, and non‐productive cough accompanied by night sweats. Associated complaints included a decline in physical activity, increased sleep needs, decreased appetite, irritability, and generalized body aches.
The patient, an elderly man, presents with a subacute, progressive systemic illness, which appears to have a pulmonary component. Broad disease categories meriting consideration include infections such as tuberculosis, endemic fungi, and infectious endocarditis; malignancies including bronchogenic carcinoma, as well as a variety of other neoplasms; and rheumatologic conditions including temporal arteritis/polymyalgia rheumatica and Wegener's granulomatosis. His complaints of anhedonia, somnolence, and irritability, while decidedly nonspecific, raise the possibility of central nervous system involvement.
His past medical history was notable for coronary artery disease, moderate aortic stenosis, hypertension, hyperlipidemia, and chronic sinusitis. Two years ago, he had unexplained kidney failure. Anti‐neutrophilic cytoplasmic antibodies (ANCA) were present, and indirect immunoflorescence revealed a peri‐nuclear (P‐ANCA) pattern on kidney biopsy. The patient had been empirically placed on azathioprine for presumed focal segmental glomerulosclerosis (FSGS), and his renal function remained stable at an estimated glomerular filtrate rate ranging from 15 to 30 mL/min/1.73 m2. His other medications included nifedipine, metoprolol, aspirin, isosorbide mononitrate, atorvastatin, calcitriol, and docusate. His family and social histories were unremarkable, including no history of tobacco. He had no pets and denied illicit drug use. He admitted to spending a considerable amount of time gardening, including working in his yard in bare feet.
The associations of focal segmental glomerulosclerosis, if indeed this diagnosis is correct, include lupus, vasculitis, and human immunodeficiency virus (HIV) infection. The nephrotic syndrome is a frequent manifestation of this entity, although, based on limited information, this patient does not appear to be clinically nephrotic. If possible, the biopsy pathology should be reviewed by a pathologist with interest in the kidney. The report of a positive P‐ANCA may not be particularly helpful here, given the frequency of false‐positive results, and in any event, P‐ANCAs have been associated with a host of conditions other than vasculitis.
The patient's gardening exposure, in bare feet no less, is intriguing. This potentially places him at risk for fungal infections including blastomycosis, histoplasmosis, cryptococcosis, and sporotrichosis. Gardening without shoes is a somewhat different enterprise in northeast Ohio than, say, Mississippi, and it will be helpful to know where this took place. Exposure in Appalachia or the South should prompt consideration of disseminated strongyloidiasis, given his azathioprine use.
Vital signs were as follows: blood pressure 151/76 mmHg, pulse 67 beats per minute, respiratory rate 20 breaths per minute, temperature 35.6C, and oxygen saturation 98% on room air. On examination, he appeared very thin but not in distress. Examination of the skin did not reveal rashes or lesions, and there was no lymphadenopathy. His thyroid was symmetric and normal in size. Lungs were clear to auscultation, and cardiac exam revealed a regular rate with a previously documented III/VI holosystolic murmur over the aortic auscultatory area. Abdominal exam revealed no organomegaly or tenderness. Joints were noted to be non‐inflamed, and extremities non‐edematous. Radial, brachial, popliteal, and dorsalis pedis pulses were normal bilaterally. A neurological exam revealed no focal deficits.
The physical examination does not help to substantively narrow or redirect the differential diagnosis. Although he appears to be tachypneic, this may simply reflect charting artifact. At this point, I would like to proceed with a number of basic diagnostic studies. In addition to complete blood count with differential, chemistries, and liver function panel, I would also obtain a thyroid stimulating hormone (TSH) assay, urinalysis, blood cultures, erythrocyte sedimentation rate/C‐reactive protein, a HIV enzyme‐linked immunosorbent assay (ELISA), chest radiograph, and a repeat ANCA panel. A purified protein derivative (PPD) skin test should be placed.
Blood chemistries were as follows: glucose 88 mg/dL, blood urea nitrogen (BUN) 48 mg/dL, creatinine 2.71 mg/dL, sodium 139 mmol/L, potassium 5.5 mmol/L, chloride 103 mmol/L, CO2 28 mmol/L, and anion gap 8 mmol/L. TSH, urinalysis, and PPD tests were unremarkable. His white blood cell count (WBC) was 33.62 K/L with 94% eosinophils and an absolute eosinophil count of 31.6 K/L. His platelet count was 189 K/L, hemoglobin 12.1 g/dL, and hematocrit 36.9%. A chest x‐ray revealed reticular opacities in the mid‐to‐lower lungs, and subsequent computed tomography (CT) scan of the chest demonstrated multiple bilateral indeterminate nodules and right axillary adenopathy.
The patient's strikingly elevated absolute eosinophil count is a very important clue that helps to significantly focus the diagnostic possibilities. In general, an eosinophilia this pronounced signifies one of several possibilities, including primary hypereosinophilic syndrome, ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, eosinophilic leukemia, and perhaps chronic eosinophilic pneumonia. In addition, Wegener's granulomatosis still merits consideration, although an eosinophil count this high would certainly be unusual.
Of the above possibilities, ChurgStrauss seems less likely given his apparent absence of a history of asthma. Parasitic infections, particularly ascariasis but also strongyloidiasis, hookworm, and even visceral larva migrans are possible, although we have not been told whether geographical exposure exists to support the first 3 of these. Hypereosinophilic syndrome remains a strong consideration, although the patient does not yet clearly meet criteria for this diagnosis.
At this juncture, I would send stool and sputum for ova and parasite exam, and order Strongyloides serology, have the peripheral smear reviewed by a pathologist, await the repeat ANCA studies, and consider obtaining hematology consultation.
Tests for anti‐Smith, anti‐ribonuclear (RNP), anti‐SSA, anti‐SSB, anti‐centromere, anti‐Scl 70, and anti‐Jo antibodies were negative. Repeat ANCA testing was positive with P‐ANCA pattern on indirect immunofluorescence. His erythrocyte sedimentation rate and C‐reactive Protein (CRP) were mildly elevated at 29 mm/hr and 1.1 mg/dL, respectively. An immunodeficiency panel work‐up consisting of CD3, CD4, CD8, CD19, T‐cell, B‐cell, and natural killer (NK) cell differential counts demonstrated CD8 T‐cell depletion. Blood cultures demonstrated no growth at 72 hours. No definite M protein was identified on serum and urine protein electrophoresis. Strongyloides IgG was negative. HIV ELISA was negative. A serologic fungal battery to measure antibodies against Aspergillus, Blastomyces, Histoplasma, and Coccidiodes was negative. A microscopic examination of stool and sputum for ova and parasites was also negative. A peripheral blood smear showed anisocytosis and confirmed the elevated eosinophil count.
The preceding wealth of information helps to further refine the picture. The positive P‐ANCA by ELISA as well as immunofluorescence suggests this is a real phenomenon, and makes ChurgStrauss syndrome more likely, despite the absence of preceding or concurrent asthma. I am not aware of an association between P‐ANCA and hypereosinophilic syndrome, nor of a similar link to either chronic eosinophilic pneumonia or hematological malignancies. Although I would like to see 2 additional stool studies for ova and parasites performed by an experienced laboratory technician before discarding the diagnosis of parasitic infection entirely, I am increasingly suspicious that this patient has a prednisone‐deficient state, most likely ChurgStrauss syndrome. I am uncertain of the relationship between his more recent symptoms and his pre‐existing kidney disease, but proceeding to lung biopsy appears to be appropriate.
Bronchoscopic examination with accompanying bronchoalveolar lavage (BAL) and transbronchial biopsy were performed. The BAL showed many Aspergillus fumigatus as well as hemosiderin‐laden macrophages, and the biopsy demonstrated an eosinophilic infiltrate throughout the interstitia, alveolar spaces, and bronchiolar walls. However, the airways did not show features of asthma, capillaritis, vasculitis, or granulomas. A bone marrow biopsy showed no evidence of clonal hematologic disease.
The Aspergillus recovered from BAL, although unexpected, probably does not adequately explain the picture. I am not convinced that the patient has invasive aspergillosis, and although components of the case are consistent with allergic bronchopulmonary aspergillosis, the absence of an asthma history and the extreme degree of peripheral eosinophilia seem to speak against this diagnosis. The biopsy does not corroborate a vasculitic process, but the yield of transbronchial biopsy is relatively low in this setting, and the pulmonary vasculitides remain in play unless a more substantial biopsy specimen is obtained. It is worth noting that high‐dose corticosteroids are a risk factor for the conversion of Aspergillus colonization to invasive aspergillosis, and treatment with voriconazole would certainly be appropriate if prednisone was to be initiated.
I believe ChurgStrauss syndrome, hypereosinophilic syndrome, and chronic eosinophilic pneumonia remain the leading diagnostic possibilities, with the P‐ANCA likely serving as a red herring if the diagnosis turns out to be one of the latter entities. An open lung biopsy would be an appropriate next step, after first obtaining those additional ova and parasite exams for completeness.
An infectious diseases specialist recommended that the patient be discharged on voriconazole 300 mg PO bid for Aspergillus colonization with an underlying lung disease and likely allergic bronchopulmonary aspergillosis or invasive aspergillosis. Steroid therapy was contemplated but not initiated.
Three weeks later, the patient re‐presented with worsening of fatigue and cognitive deterioration marked by episodes of confusion and word‐finding difficulties. His WBC had increased to 45.67 K/L (94% eosinophils). He had now lost a total of 70 pounds, and an increase in generalized weakness was apparent. His blood pressure on presentation was 120/63 mmHg, pulse rate 75 beats per minute, respiratory rate 18 breaths per minute, temperature 35.8C, and oxygen saturation 97% on room air. He appeared cachectic, but not in overt distress. His skin, head, neck, chest, cardiac, abdominal, peripheral vascular, and neurological exam demonstrated no change from the last admission. A follow‐up chest x‐ray showed mild pulmonary edema and new poorly defined pulmonary nodules in the right upper lobe. A repeat CT scan of the thorax demonstrated interval progression of ground‐glass attenuation nodules, which were now more solid‐appearing and increased in number, and present in all lobes of the lung. A CT of the brain did not reveal acute processes such as intracranial hemorrhage, infarction, or mass lesions. Lumbar puncture was performed, with a normal opening pressure. Analysis of the clear and colorless cerebrospinal fluid (CSF) showed 1 red blood cell count (RBC)/L, 2 WBC/L with 92% lymphocytes, glucose 68 mg/dL, and protein 39 mg/dL. CSF fungal cultures, routine cultures, venereal disease reaction level (VDRL), and cryptococcal antigen were negative. CSF cytology did not demonstrate malignant cells. Multiple ova and parasite exams obtained from the previous admission were confirmed to be negative.
The patient's continued deterioration points to either ChurgStrauss syndrome or hypereosinophilic syndrome, I believe. His renal function and P‐ANCA (if related) support the former possibility, while the development of what now appear to be clear encephalopathic symptoms are more in favor of the latter. I would initiate steroid therapy while proceeding to an open lung biopsy in an effort to secure a definitive diagnosis, again under the cover of voriconazole, and would ask for hematology input if this had not already been obtained.
A video‐assisted right thoracoscopy with wedge resection of 2 visible nodules in the right lower lobe was performed. The biopsy conclusively diagnosed a peripheral T‐cell lymphoma. The patient's condition deteriorated, and ultimately he and his family chose a palliative approach.
COMMENTARY
Eosinophils are cells of myeloid lineage that contain cationic‐rich protein granules that mediate allergic response, reaction to parasitic infections, tissue inflammation, and immune modulation.1, 2 Eosinophilia (absolute eosinophil count 600 cells/L) suggests the possibility of a wide array of disorders. The degree of eosinophilia can be categorized as mild (6001500 cells/L), moderate (15005000 cells/L), or severe (>5000 cells/L).3 It may signify a reactive phenomenon (secondary) or, less commonly, either an underlying hematological neoplasm (primary) or an idiopathic process.2 Clinicians faced with an unexplained eosinophilia should seek the most frequent causes first.
Initial investigation should include a careful travel history; consideration of both prescription and over‐the‐counter medications, especially non‐steroidal anti‐inflammatory drugs (NSAIDs), with withdrawal of non‐essential agents; serology for Strongyloides stercoralis antibodies (and possibly other helminths, depending on potential exposure) should be assessed; and stool examinations for ova and parasites should be obtained. The possibility of a wide variety of other potential causes of eosinophilia (Table 1) should be entertained,413 and a careful search for end‐organ damage related to eosinophilic infiltration should be performed if eosinophilia is moderate or severe.1
| Differential Diagnoses | Comments |
|---|---|
| Asthma and common allergic diseases (atopic dermatitis, allergic rhinitis) | Levels >1500 cell/l are uncommon |
| Paraneoplastic eosinophilia | Associated with adenocarcinomas, Hodgkin disease, T‐cell lymphomas, and systemic mastocytosis |
| Drugs and drug‐associated eosinophilic syndromes | Commonly associated with antibiotics (especially B‐lactams) and anti‐epileptic drugs |
| Immunodeficiency disorders | Hyper‐IgE syndrome and Omenn syndrome are rare causes of eosinophilia |
| Adrenal insufficiency | Important consideration in the critical care setting because endogenous glucocorticoids are involved in the stimulation of eosinophil apoptosis |
| Organ‐specific eosinophilic disorders | Examples: acute and chronic eosinophilic pneumonia, gastrointestinal eosinophilic disorders (esophagitis, colitis) |
| Primary eosinophilia: clonal or idiopathic | Clonal eosinophilia has histologic, cytogenetic, or molecular evidence of an underlying myeloid malignancy |
| Helminthic infections | An active tissue migration phase may manifest with hypereosinophilia |
| Hypereosinophilic syndrome | Classic criteria: hypereosinophilia for at least 6 mo, exclusion of both secondary and clonal eosinophilia, and evidence of organ involvement |
| ChurgStrauss syndrome | Hypereosinophilia with asthma, systemic vasculitis, migratory pulmonary infiltrates, sinusitis, and extravascular eosinophils |
| Allergic bronchopulmonary aspergillosis (ABPA) | Major criteria: history of asthma, central bronchiectasis, immediate skin reactivity to Aspergillus, elevated total serum IgE (>1000 ng/mL), elevated IgE or IgG to Aspergillus |
Hypereosinophilia is defined as an eosinophil level greater than 1500 cells/L. These levels may be associated with end‐organ damage regardless of the underlying etiology, although the degree of eosinophilia frequently does not correlate closely with eosinophilic tissue infiltration. As a result, relatively modest degrees of peripheral eosinophilia may be seen in association with end‐organ damage, while severe eosinophilia may be tolerated well for prolonged periods in other cases.1 The most serious complications of hypereosinophilia are myocardial damage with ultimate development of cardiac fibrosis and refractory heart failure; pulmonary involvement with hypoxia; and involvement of both the central and peripheral nervous systems including stroke, encephalopathy, and mononeuritis multiplex. A number of studies should be considered to help evaluate for the possibility of end‐organ damage as well as to assess for the presence of primary and idiopathic causes of hypereosinophilia. These include peripheral blood smear looking particularly for dysplastic eosinophils or blasts, serum tryptase, serum vitamin B12, serum IgE, cardiac troponin levels, anti‐neutrophil cytoplasmic antibody, electrocardiography, echocardiography, pulmonary function tests, and thoracoabdominal CT scanning. Endoscopic studies with esophageal, duodenal, and colonic biopsy should be performed if eosinophilic gastroenteritis is suspected.1, 7, 10
While more modest degrees of eosinophilia are associated with a plethora of conditions, severe eosinophilia, especially that approaching the levels displayed by this patient, suggests a much more circumscribed differential diagnosis. This should prompt consideration of ChurgStrauss syndrome, parasitic infection with an active tissue migration phase, and hypereosinophilic syndrome (HES).4 HES has classically been characterized by hypereosinophilia for at least 6 months, exclusion of both secondary and clonal eosinophilia, and evidence of end‐organ involvement. More recently, however, a revised definition consisting of marked eosinophilia with reasonable exclusion of other causes has gained favor.1, 7, 10, 1416 While perhaps as many as 75% of cases of HES continue to be considered idiopathic at present, 2 subtypes have now been recognized, with important prognostic and therapeutic implications. Myeloproliferative HES has a strong male predominance, is frequently associated with elevated serum tryptase and B12 levels, often manifests with hepatosplenomegaly, and displays a characteristic gene mutation, FIP1L1/PDGFRA. Lymphocytic HES is typified by polyclonal eosinophilic expansion in response to elevated IL‐5 levels, is associated with less cardiac involvement and a somewhat more favorable prognosis in the absence of therapy, and has been associated with transformation into T‐cell lymphoma.1, 1417 We suspect, though we are unable to prove, that our patient was finally diagnosed at the end of a journey that began as lymphocytic HES and ultimately progressed to T‐cell lymphoma. T‐cell lymphoma has rarely been associated with profound eosinophilia. This appears to reflect disordered production of IL‐5, as was true of this patient, and many of these cases may represent transformed lymphocytic HES.14
Specific therapy exists for the myeloproliferative subtype of HES, consisting of the tyrosine kinase inhibitor imatinib, with excellent response in typical cases. Initial treatment of most other extreme eosinophilic syndromes not caused by parasitic infection, including lymphocytic and idiopathic HES as well as ChurgStrauss syndrome, consists of high‐dose corticosteroids, with a variety of other agents used as second‐line and steroid‐sparing treatments. The urgency of therapy is dictated by the presence and severity of end‐organ damage, and in some instances corticosteroids may need to be given before the diagnosis is fully secure. When S. stercoralis infection has not been ruled out, concurrent therapy with ivermectin should be given to prevent triggering Strongyloides hyperinfection. Hematology input is critical when HES is under serious consideration, with bone marrow examination, cytogenetic studies, T‐cell phenotyping and T‐cell receptor rearrangement studies essential in helping to establish the correct diagnosis.10, 17
The differential diagnosis of peripheral eosinophilia is broad and requires a thorough, stepwise approach. Although profound eosinophilia is usually caused by a limited number of diseases, this patient reminds us that Captain Renault's advice in the film Casablanca to round up the usual suspects does not always suffice, as the diagnosis of T‐cell lymphoma was not considered by either the clinicians or the discussant until lung biopsy results became available. Most patients with hypereosinophilia not caused by parasitic infection will ultimately require an invasive procedure to establish a diagnosis, which is essential before embarking on an often‐toxic course of therapy, as well as for providing an accurate prognosis.
TEACHING POINTS
-
The most common causes of eosinophilia include helminthic infections (the leading cause worldwide), asthma, allergic conditions (the leading cause in the United States), malignancies, and drugs.
-
Hypereosinophilia may lead to end‐organ damage. The most important etiologies include ChurgStrauss Syndrome, HES, or a helminthic infection in the larval migration phase.
-
The mainstay of therapy for most cases of HES is corticosteroids. The goal of therapy is to prevent, or ameliorate, end‐organ damage.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.
- ,.Practical approach to the patient with hypereosinophilia.J Allergy Clin Immunol.2010;126(1):39–44.
- ,,.Eosinophilia: secondary, clonal and idiopathic.Br J Haematol.2006;133(5):468–492.
- .Blood eosinophilia: a new paradigm in disease classification, diagnosis, and treatment.Mayo Clin Proc.2005;80(1):75–83.
- ,,,.Clinical manifestations and treatment of Churg‐Strauss syndrome.Rheum Dis Clin North Am.2010;36(3):527–543.
- ,,,.Relative eosinophilia and functional adrenal insufficiency in critically ill patients.Lancet.1999;353(9165):1675–1676.
- ,,,.Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes.J Immunol.1996;156(10):4422–4428.
- ,.Eosinophilic disorders.J Allergy Clin Immunol.2007;119(6):1291–1300; quiz 1301–1302.
- ,.Pulmonary eosinophilia.Clin Rev Allergy Immunol.2008;34(3):367–371.
- .Eosinophilic diseases of the gastrointestinal tract.Scand J Gastroenterol.2010;45(9):1013–1021.
- ,,.Hypereosinophilic syndrome and clonal eosinophilia: point‐of‐care diagnostic algorithm and treatment update.Mayo Clin Proc.2010;85(2):158–164.
- ,,,,,.Eosinophilia as a predictor of food allergy in atopic dermatitis.Allergy Asthma Proc.2010;31(2):e18–e24.
- ,,, et al.The American College of Rheumatology 1990 criteria for the classification of Churg‐Strauss syndrome (allergic granulomatosis and angiitis).Arthritis Rheum.1990;33(8):1094–1100.
- .Allergic bronchopulmonary aspergillosis. In: Adkinson NF, Yunginger JW, Busse WW, et al, eds. Middleton's Allergy Principles 2003:1353–1371.
- ,,, et al.TARC and IL‐5 expression correlates with tissue eosinophilia in peripheral T‐cell lymphomas.Leuk Res.2008;32(9):1431–1438.
- ,,.Hypereosinophilic syndrome and proliferative diseases.Acta Dermatovenerol Croat.2009;17(4):323–330.
- ,.The hypereosinophilic syndromes: current concepts and treatments.Br J Haematol.2009;145(3):271–285.
- ,,.Lymphocytic variant hypereosinophilic syndromes.Immunol Allergy Clin North Am.2007;27(3):389–413.
Different Strokes for Different Folks
A 35‐year‐old woman presented to her primary care physician complaining of left post‐auricular pain, swelling, and redness. She described the pain as 8 out of 10, constant, sharp, and nonradiating. She denied fever or chills. A presumptive diagnosis of cellulitis led to a prescription for oral trimethoprim‐sulfamethoxazole. Left facial swelling worsened despite 4 days of antibiotics, so she came to the emergency department.
Noninfectious causes of this woman's symptoms include trauma, or an inflammatory condition such as polychondritis. Key infectious considerations are mastoiditis or a mastoid abscess. Herpes zoster with involvement of the pinna and auditory canal may also present with pain and redness. In the absence of findings suggestive of an infection arising from the auditory canal, cellulitis is a reasonable consideration. With the growing incidence of community‐acquired methicillin‐resistant Staphylococcus aureus infections, an agent effective against this pathogen such as trimethoprim‐sulfamethoxazole may be used, usually in combination with an antibiotic that provides more reliable coverage for group A streptococcus.
Her past medical history included poorly controlled type II diabetes mellitus and asthma. She reported no previous surgical history. Her current medications were insulin, albuterol inhaler, and trimethoprim‐sulfamethoxazole, although she had a history of noncompliance with her insulin. She was married with 1 child and was unemployed. She smoked 1 pack of cigarettes daily, drank up to 6 beers daily, and denied use of illicit drugs.
Her history of diabetes increases her risk of malignant otitis externa. Both diabetes and excess alcohol consumption are risk factors for herpes zoster. Smoking has been shown to increase the risk of otitis media and carriage by S. pneumoniae, a common pathogen in ear infections.
She was ill‐appearing and in moderate respiratory distress. Her temperature was 39C, blood pressure 149/93 mmHg, pulse 95 beats per minute, respiratory rate of 26 times per minute, with an oxygen saturation of 96% while breathing ambient air. She had swelling of the left side of the face extending to the left forehead and lateral neck. Examination of the external ear and auditory canal were unremarkable. The swelling had no associated erythema, tenderness, or lymphadenopathy. She had no oropharyngeal or nasal ulcers present. Her pupils were equal, round, and reactive to light and accommodation with normal sclera. Her trachea was midline; thyroid exam was normal. The heart sounds included normal S1 and S2 without murmurs, rubs, or gallops. Her lung exam was remarkable for inspiratory stridor. The abdominal examination revealed no distention, tenderness, organomegaly, or masses. Cranial nerve testing revealed a left‐sided central seventh nerve palsy along with decreased visual acuity of the left eye. Strength, sensation, and deep tendon reflexes were normal.
While there are many causes of facial nerve palsy, distinguishing between a peripheral palsy (which causes paralysis of the entire ipsilateral side of the face) and a central palsy (which spares the musculature of the forehead) is important. The most common type of peripheral facial nerve palsy is Bell's palsy. Infections such as meningitis or tumors of the central nervous system can cause central facial nerve or other cranial nerve palsy. Important infections to consider in this case would be viral such as herpes zoster or simplex, or atypical bacteria such as Mycoplasma and Rickettsia, which may explain the neurologic but not all of the other clinical findings in this case. It is also critical to determine whether she has an isolated seventh cranial nerve palsy or if other cranial nerves are involved such as may occur with basilar meningitis, which has a myriad of infectious and noninfectious causes. The decreased visual acuity may be a result of corneal dryness and abrasions from inability to close the eye but may also represent optic nerve problems, so detailed ophthalmologic exam is essential. Her ill appearance coupled with facial and neck swelling leads me to at least consider Lemierre's syndrome with central nervous system involvement. Finally, facial swelling and the inspiratory stridor may represent angioedema, although one‐sided involvement of the face would be unusual.
The results of initial laboratory testing were as follows: sodium, 138 mmol/L; potassium, 3.4 mmol/L; chloride, 109 mmol/L; bicarbonate, 14 mmol/L; blood urea nitrogen level, 19 mg/dL; creatinine, 1.1 mg/dL; white cell count, 23,510/mm3; differential, 90% neutrophils, 1% bands, 7% lymphocytes, 2% monocytes; hemoglobin level, 12.5 g/dL; platelet count, 566,000/mm3; hemoglobin A1c, 11%; albumin, 1.6 g/dL; total protein, 6.2 g/dL; total bilirubin, 0.8 mg/dL; alkaline phosphatase, 103 U/L; alanine aminotransferase level, 14 U/L; international normalized ratio of 1.2; partial thromboplastin time, 29 seconds (normal value, 2434 seconds); erythrocyte sedimentation rate, 121 mm/hr; creatine kinase, 561 U/L (normal value 25190). Arterial blood gas measurements with the patient breathing 50% oxygen revealed a pH of 7.34, a partial pressure of carbon dioxide of 28 mmHg, and a partial pressure of oxygen of 228 mmHg.
I am concerned that this patient has sepsis, likely due to an infectious trigger. With her clinical presentation localized to the head and neck, her history of diabetes, and the accelerated sedimentation rate, malignant otitis externa would explain many of her findings. Empiric anti‐infective therapy directed toward Pseudomonas aeruginosa should be initiated, and imaging of the head and ear should be undertaken.
The patient required intubation due to increased respiratory distress and stridor. Her physicians used intravenous vancomycin, clindamycin, and piperacillin/tazobactam to treat presumed cellulitis. Her abnormal neurologic exam led to magnetic resonance (MR) imaging and MR angiography of her neck and brain, which showed evidence of multiple regions of ischemia in the left occipital and inferior parietal distributions, as well as bilateral cerebellar distributions and enhancement of the parotid gland and mastoid air cells (Figure 1). A cerebral angiogram revealed irregularity and caliber reduction in multiple cervical and intracranial arteries, associated with intraluminal thrombi within the left intracranial vertebral artery, consistent with either vasculitis or infectious angioinvasion (Figure 2).

The angioinvasive nature of the findings on imaging leads me to suspect fungal infection. The patient's history of diabetes mellitus and acidosis are risk factors for mucormycosis. Aspergillus and Fusarium may also be angioinvasive but would be much more likely in neutropenic or severely immunocompromised patients. S. aureus may cause septic emboli mimicking angioinvasion but should be readily detected in conventional blood cultures. At this point, I would empirically begin amphotericin B; tissue, however, is needed for definitive diagnosis and a surgical consult should be requested.
After reviewing her imaging studies, an investigation for vasculitis and hypercoagulable states including antinuclear antibody, anti‐deoxyribonucleic acid, anti‐Smith antibody, anti‐SSA antibody level, anti‐SSB level, antineutrophil cytoplasmic antibody, activated protein C resistance level, factor VIII level, human immunodeficiency virus antibody, homocysteine level, cardiolipin antibody testing, lupus anticoagulant, prothrombin 20210 mutation, and protein C level was done, and all tests were normal. Protein S level was slightly low at 64% (normal value 65%140%). Given the enlarged parotid gland and the enhancement of the left parotid bed on magnetic resonance imaging, she underwent a parotid biopsy that revealed sialadenitis.
Systemic vasculitides can result in tissue damage, mediated by the release of endogenous cellular contents from dying cells, known as damage‐associated molecular patterns, sufficient to cause systemic inflammatory response syndrome (SIRS). This patient presented with acute symptoms but has negative laboratory studies for autoantibodies. The parotid biopsy also did not reveal evidence of vasculitis. All these findings make the diagnosis of vasculitis much less likely.
She remained in the medical intensive care unit on mechanical ventilation, with minimal symptomatic improvement. On hospital day 10, the patient developed necrosis of the left external ear. A punch biopsy of the necrotic area of her left pinna was performed; the pathology report read: Sections of punch biopsy of skin show an unremarkable epidermis. There is dermal necrosis involving the stroma and adnexal structures. Intravascular thrombi within the deep dermis are seen. Within superficial dermis there are broad, elongated, nonseptated hyaline structures reminiscent of Mucor. Special stains (periodic acid‐Schiff stain and Grocott Gomori methenamine silver stain [GMS]) performed with appropriately reactive controls fail to highlight these structures (Figure 3). The infectious disease team reviewed the pathology slides with the pathologist. As there was inconclusive evidence for zygomycosis, ie, only a few hyaline structures which failed to stain with GMS stain, the consultants recommended no change in the patient's management.
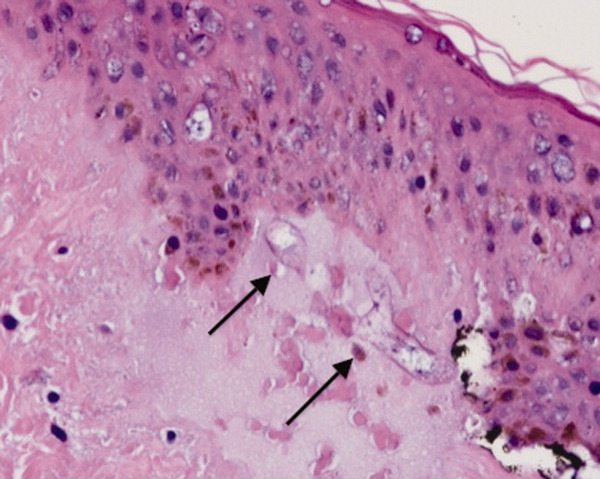
The gross and microscopic evidence of necrosis and areas of intravascular thrombi are nonspecific but compatible with a fungal infection in a patient with risk factors for zygomycosis. The GMS stain is a very sensitive stain for fungal structures, so a negative stain in this case is surprising, but additional testing such as immunohistochemistry should be pursued to confirm or refute this diagnosis. While Rhizopus species can be contaminants, the laboratory finding of these organisms in specimens from patients with risk factors for zygomycosis should not be ignored.
On hospital day 12, the patient was noted to have increased facial swelling. A computed tomographic (CT) angiogram of the neck revealed necrosis of the anterior and posterior paraspinal muscles from the skull base to C34, marked swelling of the left parotid gland, and left inferior parieto‐occipital enhancing lesion. An incisional parotid biopsy was performed. Special stains were positive for broad‐based fungal hyphae consistent with mucormycosis (Figure 4).
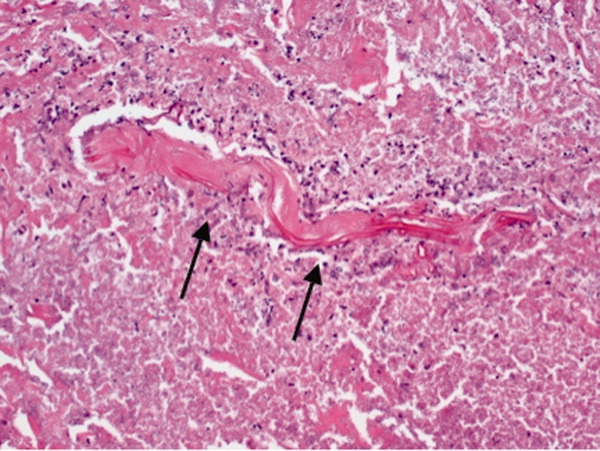
Given these findings, the patient should be started on amphotericin B immediately. Medical therapy alone generally does not suffice, and aggressive surgical debridement combined with intravenous antifungal therapy results in better outcomes. The longer the duration of symptoms and the greater the progression of disease, the less favorable the prognosis.
The patient was started on amphotericin B lipid complex and micafungin. However, after 16 days of therapy, repeat imaging of the neck showed worsening necrosis of the neck muscles. At this time, she underwent extensive debridement of face and neck, and posaconazole was added. After prolonged hospitalization, she was discharged to a rehabilitation facility on posaconazole. She resided in a nursing facility for 6 months. One year after her hospitalization, she is living at home and is able to ambulate independently, but requires feeding through a percutaneous endoscopic gastrostomy (PEG) tube because she remains dysphagic.
COMMENTARY
Infections caused by the ubiquitous fungi of the class Zygomycetes typically take 1 of 5 forms: rhinocerebral, pulmonary, gastrointestinal, disseminated, and cutaneous. The presentation varies widely, ranging from plaques, skin swelling, pustules, cellulitis, blisters, nodules, ulcerations, and ecthyma gangrenosum‐like lesions to deeper infections such as necrotizing fasciitis, osteomyelitis, and disseminated infection.1 Infections typically occur in immunocompromised hosts, including transplant recipients and patients with hematologic malignancy, but also occur in patients with diabetes mellitus, intravenous drug users, and patients on deferoxamine therapy.2 Deferoxamine and other iron‐binding therapy is thought to predispose to zygomycetes infections because of improved iron uptake of the fungal species and, thus, stimulation of growth.3 Pulmonary and rhinocerebral infections are the most common clinically encountered forms, and 44% of cutaneous infections are complicated by deep extension or dissemination.4
The articles cited above describe the more typical presentations of this rare disease. However, this patient had an unusual presentation, as parotid involvement due to zygomycosis has only been described once previously.5 Her inflammatory vasculitis and ensuing strokes from involvement of the carotid artery are recognized complications of zygomycosis, and in 1 case series of 41 patients with rhinocerebral mucormycosis, carotid involvement was seen in 31% of patients.6 After the punch biopsy of the patient's pinna showing nonseptated hyphae reminiscent of Mucor, why did her physicians delay administering amphotericin?
There are 2 likely possibilities: anchoring bias or error in medical decision‐making due to inaccurate probability estimates. Anchoring bias describes a heuristic where the initial diagnosis or gestalt biases the physician's process for assigning a final diagnosis.7, 8 This bias creates cognitive errors by limiting creativity in diagnosis. In this case, the infectious disease team carefully weighed the information obtained from the first biopsy. Given their low pretest estimate of this virtually unreported presentation of a rare disease, they decided to evaluate further without beginning antifungal therapy. Of note, there were few hyaline structures, and those structures lacked uptake of GMS. Since they considered the diagnosis yet rejected the diagnosis due to insufficient evidence, it is unlikely that anchoring bias played a role.
Was there an error in medical decision‐making? The physicians in this case faced a very common medical dilemma: whether or not to start a toxic medication empirically or wait for diagnostic confirmation prior to treatment.9 To solve this dilemma, one can apply decision analysis. Moskowitz et al described 5 phases of medical decision analysis by which a probabilistic right answer to clinical scenarios can be deduced mathematically.10 To solve this problem, probabilities must be assigned to the risk of giving a drug to a patient without the disease versus the risk of not giving a drug to a patient with the disease. For example, amphotericin deoxycholate causes acute renal failure in 30% to 47% of patients. Newer formulations of amphotericin, such as liposomal amphotericin and lipid complex, result in lower rates of nephrotoxicity (27% vs 47%). The risk of not giving amphotericin to a patient with zygomycosis is death. Even in patients treated with amphotericin, the mortality rate has been shown to be 66%, and up to 100% in those with strokes related to zygomycosis.2, 6, 11 Simply looking at these probabilities, decision analysis would favor empiric treatment.
The physicians caring for this patient did not have the luxury of retrospective speculation. After looking at all of the data, the equivocal skin biopsy and rare clinical presentation, the question to ask would change: What is the risk of giving amphotericin empirically to someone who, based on available information, has a very low probability of having zygomycosis? When phrased in this manner, there is a 47% chance of nephrotoxicity with amphotericin versus the very small probability that you have diagnosed a case of zygomycosis that has only been described once in the literature. Mathematically andmore importantlyclinically, this question becomes more difficult to answer. However, no value can be placed on the possibility of death in suspected zygomycosis, and the risk of short‐term amphotericin use is much less than that of a course of treatment. As such, empiric therapy should always be given.
Physicians are not mathematicians, and dynamic clinical scenarios are not so easily made into static math problems. Disease presentations evolve over time towards a diagnosable clinical pattern, as was the case with this patient. Two days after the aforementioned biopsy, she worsened and in less time than it would have taken to isolate zygomycosis from the first biopsy, a second biopsy revealed the typical nonseptated hyphae demarcated with the GMS stain. Even appropriate diagnostic testing, thoughtful interpretation, and avoidance of certain cognitive errors can result in incorrect diagnoses and delayed treatment. It is monitoring the progression of disease and collecting additional data that allows physicians to mold a diagnosis and create a treatment plan.
The primary treatment of zygomycosis should include amphotericin. However, there are limited data to support combination therapy with an echinocandin in severe cases, as in this patient.12 Posaconazole is not recommended for monotherapy as an initial therapy, but there is data for its use as salvage therapy in zygomycosis.13 This case highlights the difficulties that physicians face in the diagnosis and treatment of rare diseases. Cerebral infarction in a hematologic malignancy, uncontrolled diabetes, or iron chelation therapy could be the initial presentation of rhinocerebral zygomycosis. There truly are different strokes for different folks. Recognizing this and similar presentations may lead to a more rapid diagnosis and treatment of zygomycosis.
TEACHING POINTS
-
Zygomycosis has a wide range of clinical presentations ranging from skin lesions to deep tissue infections. As it is an angioinvasive organism, it can also present as cerebral infarcts and brain abscesses.
-
Zygomycosis infections should be suspected in patients with uncontrolled diabetes, hematologic or oncologic malignancies, and patients on iron chelation therapy with a potentially compatible clinical picture.
-
If zygomycosis infection is suspected, rapid histologic diagnosis should be attempted. However, as histologic diagnosis can take time, empiric therapy with amphotericin should always be administered.
-
Amphotericin remains the primary medical therapy for this disease; however, there is limited emerging evidence to suggest that echinocandins can be used in combination with amphotericin for improved treatment of severe rhinocerebral zygomyocosis. Posaconazole has a role as salvage therapy in zygomycosis, but should not be used as the sole primary treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors are indebted to Dr Glenn Roberson at the Department of Radiology, University of Alabama at Birmingham, for providing the radiographic images; to Dr Aleodor Andea at the Department of Pathology, University of Alabama at Birmingham, for providing the pathology images; and to Dr. Crysten Brinkley at the Department of Neurology at the University of Alabama at Birmingham for her assistance with this case presentation.
Disclosure: Nothing to report.
- ,,,.Mucormycosis: emerging prominence of cutaneous infections.Clin Infect Dis.1994;19:67–76.
- ,,,.Zygomycosis in the 1990s in a tertiary‐care cancer center.Clin Infect Dis.2000;30:851–856.
- ,,, et al.Mucormycosis during deferoxamine therapy is a siderophore‐mediated infection. In vitro and in vivo animal studies.J Clin Invest.1993;91:1979–1986.
- ,,, et al.Epidemiology and outcome of zygomycosis: a review of 929 reported cases.Clin Infect Dis.2005;41:634–653.
- ,,,,,.Cutaneous mucormycosis of the head and neck with parotid gland involvement: first report of a case.Ear Nose Throat J.2004;83:282–286.
- ,,,,,.A successful combined endovascular and surgical treatment of a cranial base mucormycosis with an associated internal carotid artery pseudoaneurysm.Neurosurgery.2009;65:733–740.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,,,.Clinical problem‐solving. Anchors away.N Engl J Med.2007;356:504–509.
- ,,,.Clinical problem‐solving. Empirically incorrect.N Engl J Med.2006;354:509–514.
- ,,.Dealing with uncertainty, risks, and tradeoffs in clinical decisions. A cognitive science approach.Ann Intern Med.1988;108:435–449.
- ,,.Fatal strokes in patients with rhino‐orbito‐cerebral mucormycosis and associated vasculopathy.Scand J Infect Dis.2004;36:643–648.
- ,,, et al.Combination polyene‐caspofungin treatment of rhino‐orbital‐cerebral mucormycosis.Clin Infect Dis.2008;47:364–371.
- ,,,,.Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases.Clin Infect Dis.2006;42:e61–e65.
A 35‐year‐old woman presented to her primary care physician complaining of left post‐auricular pain, swelling, and redness. She described the pain as 8 out of 10, constant, sharp, and nonradiating. She denied fever or chills. A presumptive diagnosis of cellulitis led to a prescription for oral trimethoprim‐sulfamethoxazole. Left facial swelling worsened despite 4 days of antibiotics, so she came to the emergency department.
Noninfectious causes of this woman's symptoms include trauma, or an inflammatory condition such as polychondritis. Key infectious considerations are mastoiditis or a mastoid abscess. Herpes zoster with involvement of the pinna and auditory canal may also present with pain and redness. In the absence of findings suggestive of an infection arising from the auditory canal, cellulitis is a reasonable consideration. With the growing incidence of community‐acquired methicillin‐resistant Staphylococcus aureus infections, an agent effective against this pathogen such as trimethoprim‐sulfamethoxazole may be used, usually in combination with an antibiotic that provides more reliable coverage for group A streptococcus.
Her past medical history included poorly controlled type II diabetes mellitus and asthma. She reported no previous surgical history. Her current medications were insulin, albuterol inhaler, and trimethoprim‐sulfamethoxazole, although she had a history of noncompliance with her insulin. She was married with 1 child and was unemployed. She smoked 1 pack of cigarettes daily, drank up to 6 beers daily, and denied use of illicit drugs.
Her history of diabetes increases her risk of malignant otitis externa. Both diabetes and excess alcohol consumption are risk factors for herpes zoster. Smoking has been shown to increase the risk of otitis media and carriage by S. pneumoniae, a common pathogen in ear infections.
She was ill‐appearing and in moderate respiratory distress. Her temperature was 39C, blood pressure 149/93 mmHg, pulse 95 beats per minute, respiratory rate of 26 times per minute, with an oxygen saturation of 96% while breathing ambient air. She had swelling of the left side of the face extending to the left forehead and lateral neck. Examination of the external ear and auditory canal were unremarkable. The swelling had no associated erythema, tenderness, or lymphadenopathy. She had no oropharyngeal or nasal ulcers present. Her pupils were equal, round, and reactive to light and accommodation with normal sclera. Her trachea was midline; thyroid exam was normal. The heart sounds included normal S1 and S2 without murmurs, rubs, or gallops. Her lung exam was remarkable for inspiratory stridor. The abdominal examination revealed no distention, tenderness, organomegaly, or masses. Cranial nerve testing revealed a left‐sided central seventh nerve palsy along with decreased visual acuity of the left eye. Strength, sensation, and deep tendon reflexes were normal.
While there are many causes of facial nerve palsy, distinguishing between a peripheral palsy (which causes paralysis of the entire ipsilateral side of the face) and a central palsy (which spares the musculature of the forehead) is important. The most common type of peripheral facial nerve palsy is Bell's palsy. Infections such as meningitis or tumors of the central nervous system can cause central facial nerve or other cranial nerve palsy. Important infections to consider in this case would be viral such as herpes zoster or simplex, or atypical bacteria such as Mycoplasma and Rickettsia, which may explain the neurologic but not all of the other clinical findings in this case. It is also critical to determine whether she has an isolated seventh cranial nerve palsy or if other cranial nerves are involved such as may occur with basilar meningitis, which has a myriad of infectious and noninfectious causes. The decreased visual acuity may be a result of corneal dryness and abrasions from inability to close the eye but may also represent optic nerve problems, so detailed ophthalmologic exam is essential. Her ill appearance coupled with facial and neck swelling leads me to at least consider Lemierre's syndrome with central nervous system involvement. Finally, facial swelling and the inspiratory stridor may represent angioedema, although one‐sided involvement of the face would be unusual.
The results of initial laboratory testing were as follows: sodium, 138 mmol/L; potassium, 3.4 mmol/L; chloride, 109 mmol/L; bicarbonate, 14 mmol/L; blood urea nitrogen level, 19 mg/dL; creatinine, 1.1 mg/dL; white cell count, 23,510/mm3; differential, 90% neutrophils, 1% bands, 7% lymphocytes, 2% monocytes; hemoglobin level, 12.5 g/dL; platelet count, 566,000/mm3; hemoglobin A1c, 11%; albumin, 1.6 g/dL; total protein, 6.2 g/dL; total bilirubin, 0.8 mg/dL; alkaline phosphatase, 103 U/L; alanine aminotransferase level, 14 U/L; international normalized ratio of 1.2; partial thromboplastin time, 29 seconds (normal value, 2434 seconds); erythrocyte sedimentation rate, 121 mm/hr; creatine kinase, 561 U/L (normal value 25190). Arterial blood gas measurements with the patient breathing 50% oxygen revealed a pH of 7.34, a partial pressure of carbon dioxide of 28 mmHg, and a partial pressure of oxygen of 228 mmHg.
I am concerned that this patient has sepsis, likely due to an infectious trigger. With her clinical presentation localized to the head and neck, her history of diabetes, and the accelerated sedimentation rate, malignant otitis externa would explain many of her findings. Empiric anti‐infective therapy directed toward Pseudomonas aeruginosa should be initiated, and imaging of the head and ear should be undertaken.
The patient required intubation due to increased respiratory distress and stridor. Her physicians used intravenous vancomycin, clindamycin, and piperacillin/tazobactam to treat presumed cellulitis. Her abnormal neurologic exam led to magnetic resonance (MR) imaging and MR angiography of her neck and brain, which showed evidence of multiple regions of ischemia in the left occipital and inferior parietal distributions, as well as bilateral cerebellar distributions and enhancement of the parotid gland and mastoid air cells (Figure 1). A cerebral angiogram revealed irregularity and caliber reduction in multiple cervical and intracranial arteries, associated with intraluminal thrombi within the left intracranial vertebral artery, consistent with either vasculitis or infectious angioinvasion (Figure 2).
The angioinvasive nature of the findings on imaging leads me to suspect fungal infection. The patient's history of diabetes mellitus and acidosis are risk factors for mucormycosis. Aspergillus and Fusarium may also be angioinvasive but would be much more likely in neutropenic or severely immunocompromised patients. S. aureus may cause septic emboli mimicking angioinvasion but should be readily detected in conventional blood cultures. At this point, I would empirically begin amphotericin B; tissue, however, is needed for definitive diagnosis and a surgical consult should be requested.
After reviewing her imaging studies, an investigation for vasculitis and hypercoagulable states including antinuclear antibody, anti‐deoxyribonucleic acid, anti‐Smith antibody, anti‐SSA antibody level, anti‐SSB level, antineutrophil cytoplasmic antibody, activated protein C resistance level, factor VIII level, human immunodeficiency virus antibody, homocysteine level, cardiolipin antibody testing, lupus anticoagulant, prothrombin 20210 mutation, and protein C level was done, and all tests were normal. Protein S level was slightly low at 64% (normal value 65%140%). Given the enlarged parotid gland and the enhancement of the left parotid bed on magnetic resonance imaging, she underwent a parotid biopsy that revealed sialadenitis.
Systemic vasculitides can result in tissue damage, mediated by the release of endogenous cellular contents from dying cells, known as damage‐associated molecular patterns, sufficient to cause systemic inflammatory response syndrome (SIRS). This patient presented with acute symptoms but has negative laboratory studies for autoantibodies. The parotid biopsy also did not reveal evidence of vasculitis. All these findings make the diagnosis of vasculitis much less likely.
She remained in the medical intensive care unit on mechanical ventilation, with minimal symptomatic improvement. On hospital day 10, the patient developed necrosis of the left external ear. A punch biopsy of the necrotic area of her left pinna was performed; the pathology report read: Sections of punch biopsy of skin show an unremarkable epidermis. There is dermal necrosis involving the stroma and adnexal structures. Intravascular thrombi within the deep dermis are seen. Within superficial dermis there are broad, elongated, nonseptated hyaline structures reminiscent of Mucor. Special stains (periodic acid‐Schiff stain and Grocott Gomori methenamine silver stain [GMS]) performed with appropriately reactive controls fail to highlight these structures (Figure 3). The infectious disease team reviewed the pathology slides with the pathologist. As there was inconclusive evidence for zygomycosis, ie, only a few hyaline structures which failed to stain with GMS stain, the consultants recommended no change in the patient's management.
The gross and microscopic evidence of necrosis and areas of intravascular thrombi are nonspecific but compatible with a fungal infection in a patient with risk factors for zygomycosis. The GMS stain is a very sensitive stain for fungal structures, so a negative stain in this case is surprising, but additional testing such as immunohistochemistry should be pursued to confirm or refute this diagnosis. While Rhizopus species can be contaminants, the laboratory finding of these organisms in specimens from patients with risk factors for zygomycosis should not be ignored.
On hospital day 12, the patient was noted to have increased facial swelling. A computed tomographic (CT) angiogram of the neck revealed necrosis of the anterior and posterior paraspinal muscles from the skull base to C34, marked swelling of the left parotid gland, and left inferior parieto‐occipital enhancing lesion. An incisional parotid biopsy was performed. Special stains were positive for broad‐based fungal hyphae consistent with mucormycosis (Figure 4).
Given these findings, the patient should be started on amphotericin B immediately. Medical therapy alone generally does not suffice, and aggressive surgical debridement combined with intravenous antifungal therapy results in better outcomes. The longer the duration of symptoms and the greater the progression of disease, the less favorable the prognosis.
The patient was started on amphotericin B lipid complex and micafungin. However, after 16 days of therapy, repeat imaging of the neck showed worsening necrosis of the neck muscles. At this time, she underwent extensive debridement of face and neck, and posaconazole was added. After prolonged hospitalization, she was discharged to a rehabilitation facility on posaconazole. She resided in a nursing facility for 6 months. One year after her hospitalization, she is living at home and is able to ambulate independently, but requires feeding through a percutaneous endoscopic gastrostomy (PEG) tube because she remains dysphagic.
COMMENTARY
Infections caused by the ubiquitous fungi of the class Zygomycetes typically take 1 of 5 forms: rhinocerebral, pulmonary, gastrointestinal, disseminated, and cutaneous. The presentation varies widely, ranging from plaques, skin swelling, pustules, cellulitis, blisters, nodules, ulcerations, and ecthyma gangrenosum‐like lesions to deeper infections such as necrotizing fasciitis, osteomyelitis, and disseminated infection.1 Infections typically occur in immunocompromised hosts, including transplant recipients and patients with hematologic malignancy, but also occur in patients with diabetes mellitus, intravenous drug users, and patients on deferoxamine therapy.2 Deferoxamine and other iron‐binding therapy is thought to predispose to zygomycetes infections because of improved iron uptake of the fungal species and, thus, stimulation of growth.3 Pulmonary and rhinocerebral infections are the most common clinically encountered forms, and 44% of cutaneous infections are complicated by deep extension or dissemination.4
The articles cited above describe the more typical presentations of this rare disease. However, this patient had an unusual presentation, as parotid involvement due to zygomycosis has only been described once previously.5 Her inflammatory vasculitis and ensuing strokes from involvement of the carotid artery are recognized complications of zygomycosis, and in 1 case series of 41 patients with rhinocerebral mucormycosis, carotid involvement was seen in 31% of patients.6 After the punch biopsy of the patient's pinna showing nonseptated hyphae reminiscent of Mucor, why did her physicians delay administering amphotericin?
There are 2 likely possibilities: anchoring bias or error in medical decision‐making due to inaccurate probability estimates. Anchoring bias describes a heuristic where the initial diagnosis or gestalt biases the physician's process for assigning a final diagnosis.7, 8 This bias creates cognitive errors by limiting creativity in diagnosis. In this case, the infectious disease team carefully weighed the information obtained from the first biopsy. Given their low pretest estimate of this virtually unreported presentation of a rare disease, they decided to evaluate further without beginning antifungal therapy. Of note, there were few hyaline structures, and those structures lacked uptake of GMS. Since they considered the diagnosis yet rejected the diagnosis due to insufficient evidence, it is unlikely that anchoring bias played a role.
Was there an error in medical decision‐making? The physicians in this case faced a very common medical dilemma: whether or not to start a toxic medication empirically or wait for diagnostic confirmation prior to treatment.9 To solve this dilemma, one can apply decision analysis. Moskowitz et al described 5 phases of medical decision analysis by which a probabilistic right answer to clinical scenarios can be deduced mathematically.10 To solve this problem, probabilities must be assigned to the risk of giving a drug to a patient without the disease versus the risk of not giving a drug to a patient with the disease. For example, amphotericin deoxycholate causes acute renal failure in 30% to 47% of patients. Newer formulations of amphotericin, such as liposomal amphotericin and lipid complex, result in lower rates of nephrotoxicity (27% vs 47%). The risk of not giving amphotericin to a patient with zygomycosis is death. Even in patients treated with amphotericin, the mortality rate has been shown to be 66%, and up to 100% in those with strokes related to zygomycosis.2, 6, 11 Simply looking at these probabilities, decision analysis would favor empiric treatment.
The physicians caring for this patient did not have the luxury of retrospective speculation. After looking at all of the data, the equivocal skin biopsy and rare clinical presentation, the question to ask would change: What is the risk of giving amphotericin empirically to someone who, based on available information, has a very low probability of having zygomycosis? When phrased in this manner, there is a 47% chance of nephrotoxicity with amphotericin versus the very small probability that you have diagnosed a case of zygomycosis that has only been described once in the literature. Mathematically andmore importantlyclinically, this question becomes more difficult to answer. However, no value can be placed on the possibility of death in suspected zygomycosis, and the risk of short‐term amphotericin use is much less than that of a course of treatment. As such, empiric therapy should always be given.
Physicians are not mathematicians, and dynamic clinical scenarios are not so easily made into static math problems. Disease presentations evolve over time towards a diagnosable clinical pattern, as was the case with this patient. Two days after the aforementioned biopsy, she worsened and in less time than it would have taken to isolate zygomycosis from the first biopsy, a second biopsy revealed the typical nonseptated hyphae demarcated with the GMS stain. Even appropriate diagnostic testing, thoughtful interpretation, and avoidance of certain cognitive errors can result in incorrect diagnoses and delayed treatment. It is monitoring the progression of disease and collecting additional data that allows physicians to mold a diagnosis and create a treatment plan.
The primary treatment of zygomycosis should include amphotericin. However, there are limited data to support combination therapy with an echinocandin in severe cases, as in this patient.12 Posaconazole is not recommended for monotherapy as an initial therapy, but there is data for its use as salvage therapy in zygomycosis.13 This case highlights the difficulties that physicians face in the diagnosis and treatment of rare diseases. Cerebral infarction in a hematologic malignancy, uncontrolled diabetes, or iron chelation therapy could be the initial presentation of rhinocerebral zygomycosis. There truly are different strokes for different folks. Recognizing this and similar presentations may lead to a more rapid diagnosis and treatment of zygomycosis.
TEACHING POINTS
-
Zygomycosis has a wide range of clinical presentations ranging from skin lesions to deep tissue infections. As it is an angioinvasive organism, it can also present as cerebral infarcts and brain abscesses.
-
Zygomycosis infections should be suspected in patients with uncontrolled diabetes, hematologic or oncologic malignancies, and patients on iron chelation therapy with a potentially compatible clinical picture.
-
If zygomycosis infection is suspected, rapid histologic diagnosis should be attempted. However, as histologic diagnosis can take time, empiric therapy with amphotericin should always be administered.
-
Amphotericin remains the primary medical therapy for this disease; however, there is limited emerging evidence to suggest that echinocandins can be used in combination with amphotericin for improved treatment of severe rhinocerebral zygomyocosis. Posaconazole has a role as salvage therapy in zygomycosis, but should not be used as the sole primary treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors are indebted to Dr Glenn Roberson at the Department of Radiology, University of Alabama at Birmingham, for providing the radiographic images; to Dr Aleodor Andea at the Department of Pathology, University of Alabama at Birmingham, for providing the pathology images; and to Dr. Crysten Brinkley at the Department of Neurology at the University of Alabama at Birmingham for her assistance with this case presentation.
Disclosure: Nothing to report.
A 35‐year‐old woman presented to her primary care physician complaining of left post‐auricular pain, swelling, and redness. She described the pain as 8 out of 10, constant, sharp, and nonradiating. She denied fever or chills. A presumptive diagnosis of cellulitis led to a prescription for oral trimethoprim‐sulfamethoxazole. Left facial swelling worsened despite 4 days of antibiotics, so she came to the emergency department.
Noninfectious causes of this woman's symptoms include trauma, or an inflammatory condition such as polychondritis. Key infectious considerations are mastoiditis or a mastoid abscess. Herpes zoster with involvement of the pinna and auditory canal may also present with pain and redness. In the absence of findings suggestive of an infection arising from the auditory canal, cellulitis is a reasonable consideration. With the growing incidence of community‐acquired methicillin‐resistant Staphylococcus aureus infections, an agent effective against this pathogen such as trimethoprim‐sulfamethoxazole may be used, usually in combination with an antibiotic that provides more reliable coverage for group A streptococcus.
Her past medical history included poorly controlled type II diabetes mellitus and asthma. She reported no previous surgical history. Her current medications were insulin, albuterol inhaler, and trimethoprim‐sulfamethoxazole, although she had a history of noncompliance with her insulin. She was married with 1 child and was unemployed. She smoked 1 pack of cigarettes daily, drank up to 6 beers daily, and denied use of illicit drugs.
Her history of diabetes increases her risk of malignant otitis externa. Both diabetes and excess alcohol consumption are risk factors for herpes zoster. Smoking has been shown to increase the risk of otitis media and carriage by S. pneumoniae, a common pathogen in ear infections.
She was ill‐appearing and in moderate respiratory distress. Her temperature was 39C, blood pressure 149/93 mmHg, pulse 95 beats per minute, respiratory rate of 26 times per minute, with an oxygen saturation of 96% while breathing ambient air. She had swelling of the left side of the face extending to the left forehead and lateral neck. Examination of the external ear and auditory canal were unremarkable. The swelling had no associated erythema, tenderness, or lymphadenopathy. She had no oropharyngeal or nasal ulcers present. Her pupils were equal, round, and reactive to light and accommodation with normal sclera. Her trachea was midline; thyroid exam was normal. The heart sounds included normal S1 and S2 without murmurs, rubs, or gallops. Her lung exam was remarkable for inspiratory stridor. The abdominal examination revealed no distention, tenderness, organomegaly, or masses. Cranial nerve testing revealed a left‐sided central seventh nerve palsy along with decreased visual acuity of the left eye. Strength, sensation, and deep tendon reflexes were normal.
While there are many causes of facial nerve palsy, distinguishing between a peripheral palsy (which causes paralysis of the entire ipsilateral side of the face) and a central palsy (which spares the musculature of the forehead) is important. The most common type of peripheral facial nerve palsy is Bell's palsy. Infections such as meningitis or tumors of the central nervous system can cause central facial nerve or other cranial nerve palsy. Important infections to consider in this case would be viral such as herpes zoster or simplex, or atypical bacteria such as Mycoplasma and Rickettsia, which may explain the neurologic but not all of the other clinical findings in this case. It is also critical to determine whether she has an isolated seventh cranial nerve palsy or if other cranial nerves are involved such as may occur with basilar meningitis, which has a myriad of infectious and noninfectious causes. The decreased visual acuity may be a result of corneal dryness and abrasions from inability to close the eye but may also represent optic nerve problems, so detailed ophthalmologic exam is essential. Her ill appearance coupled with facial and neck swelling leads me to at least consider Lemierre's syndrome with central nervous system involvement. Finally, facial swelling and the inspiratory stridor may represent angioedema, although one‐sided involvement of the face would be unusual.
The results of initial laboratory testing were as follows: sodium, 138 mmol/L; potassium, 3.4 mmol/L; chloride, 109 mmol/L; bicarbonate, 14 mmol/L; blood urea nitrogen level, 19 mg/dL; creatinine, 1.1 mg/dL; white cell count, 23,510/mm3; differential, 90% neutrophils, 1% bands, 7% lymphocytes, 2% monocytes; hemoglobin level, 12.5 g/dL; platelet count, 566,000/mm3; hemoglobin A1c, 11%; albumin, 1.6 g/dL; total protein, 6.2 g/dL; total bilirubin, 0.8 mg/dL; alkaline phosphatase, 103 U/L; alanine aminotransferase level, 14 U/L; international normalized ratio of 1.2; partial thromboplastin time, 29 seconds (normal value, 2434 seconds); erythrocyte sedimentation rate, 121 mm/hr; creatine kinase, 561 U/L (normal value 25190). Arterial blood gas measurements with the patient breathing 50% oxygen revealed a pH of 7.34, a partial pressure of carbon dioxide of 28 mmHg, and a partial pressure of oxygen of 228 mmHg.
I am concerned that this patient has sepsis, likely due to an infectious trigger. With her clinical presentation localized to the head and neck, her history of diabetes, and the accelerated sedimentation rate, malignant otitis externa would explain many of her findings. Empiric anti‐infective therapy directed toward Pseudomonas aeruginosa should be initiated, and imaging of the head and ear should be undertaken.
The patient required intubation due to increased respiratory distress and stridor. Her physicians used intravenous vancomycin, clindamycin, and piperacillin/tazobactam to treat presumed cellulitis. Her abnormal neurologic exam led to magnetic resonance (MR) imaging and MR angiography of her neck and brain, which showed evidence of multiple regions of ischemia in the left occipital and inferior parietal distributions, as well as bilateral cerebellar distributions and enhancement of the parotid gland and mastoid air cells (Figure 1). A cerebral angiogram revealed irregularity and caliber reduction in multiple cervical and intracranial arteries, associated with intraluminal thrombi within the left intracranial vertebral artery, consistent with either vasculitis or infectious angioinvasion (Figure 2).
The angioinvasive nature of the findings on imaging leads me to suspect fungal infection. The patient's history of diabetes mellitus and acidosis are risk factors for mucormycosis. Aspergillus and Fusarium may also be angioinvasive but would be much more likely in neutropenic or severely immunocompromised patients. S. aureus may cause septic emboli mimicking angioinvasion but should be readily detected in conventional blood cultures. At this point, I would empirically begin amphotericin B; tissue, however, is needed for definitive diagnosis and a surgical consult should be requested.
After reviewing her imaging studies, an investigation for vasculitis and hypercoagulable states including antinuclear antibody, anti‐deoxyribonucleic acid, anti‐Smith antibody, anti‐SSA antibody level, anti‐SSB level, antineutrophil cytoplasmic antibody, activated protein C resistance level, factor VIII level, human immunodeficiency virus antibody, homocysteine level, cardiolipin antibody testing, lupus anticoagulant, prothrombin 20210 mutation, and protein C level was done, and all tests were normal. Protein S level was slightly low at 64% (normal value 65%140%). Given the enlarged parotid gland and the enhancement of the left parotid bed on magnetic resonance imaging, she underwent a parotid biopsy that revealed sialadenitis.
Systemic vasculitides can result in tissue damage, mediated by the release of endogenous cellular contents from dying cells, known as damage‐associated molecular patterns, sufficient to cause systemic inflammatory response syndrome (SIRS). This patient presented with acute symptoms but has negative laboratory studies for autoantibodies. The parotid biopsy also did not reveal evidence of vasculitis. All these findings make the diagnosis of vasculitis much less likely.
She remained in the medical intensive care unit on mechanical ventilation, with minimal symptomatic improvement. On hospital day 10, the patient developed necrosis of the left external ear. A punch biopsy of the necrotic area of her left pinna was performed; the pathology report read: Sections of punch biopsy of skin show an unremarkable epidermis. There is dermal necrosis involving the stroma and adnexal structures. Intravascular thrombi within the deep dermis are seen. Within superficial dermis there are broad, elongated, nonseptated hyaline structures reminiscent of Mucor. Special stains (periodic acid‐Schiff stain and Grocott Gomori methenamine silver stain [GMS]) performed with appropriately reactive controls fail to highlight these structures (Figure 3). The infectious disease team reviewed the pathology slides with the pathologist. As there was inconclusive evidence for zygomycosis, ie, only a few hyaline structures which failed to stain with GMS stain, the consultants recommended no change in the patient's management.
The gross and microscopic evidence of necrosis and areas of intravascular thrombi are nonspecific but compatible with a fungal infection in a patient with risk factors for zygomycosis. The GMS stain is a very sensitive stain for fungal structures, so a negative stain in this case is surprising, but additional testing such as immunohistochemistry should be pursued to confirm or refute this diagnosis. While Rhizopus species can be contaminants, the laboratory finding of these organisms in specimens from patients with risk factors for zygomycosis should not be ignored.
On hospital day 12, the patient was noted to have increased facial swelling. A computed tomographic (CT) angiogram of the neck revealed necrosis of the anterior and posterior paraspinal muscles from the skull base to C34, marked swelling of the left parotid gland, and left inferior parieto‐occipital enhancing lesion. An incisional parotid biopsy was performed. Special stains were positive for broad‐based fungal hyphae consistent with mucormycosis (Figure 4).
Given these findings, the patient should be started on amphotericin B immediately. Medical therapy alone generally does not suffice, and aggressive surgical debridement combined with intravenous antifungal therapy results in better outcomes. The longer the duration of symptoms and the greater the progression of disease, the less favorable the prognosis.
The patient was started on amphotericin B lipid complex and micafungin. However, after 16 days of therapy, repeat imaging of the neck showed worsening necrosis of the neck muscles. At this time, she underwent extensive debridement of face and neck, and posaconazole was added. After prolonged hospitalization, she was discharged to a rehabilitation facility on posaconazole. She resided in a nursing facility for 6 months. One year after her hospitalization, she is living at home and is able to ambulate independently, but requires feeding through a percutaneous endoscopic gastrostomy (PEG) tube because she remains dysphagic.
COMMENTARY
Infections caused by the ubiquitous fungi of the class Zygomycetes typically take 1 of 5 forms: rhinocerebral, pulmonary, gastrointestinal, disseminated, and cutaneous. The presentation varies widely, ranging from plaques, skin swelling, pustules, cellulitis, blisters, nodules, ulcerations, and ecthyma gangrenosum‐like lesions to deeper infections such as necrotizing fasciitis, osteomyelitis, and disseminated infection.1 Infections typically occur in immunocompromised hosts, including transplant recipients and patients with hematologic malignancy, but also occur in patients with diabetes mellitus, intravenous drug users, and patients on deferoxamine therapy.2 Deferoxamine and other iron‐binding therapy is thought to predispose to zygomycetes infections because of improved iron uptake of the fungal species and, thus, stimulation of growth.3 Pulmonary and rhinocerebral infections are the most common clinically encountered forms, and 44% of cutaneous infections are complicated by deep extension or dissemination.4
The articles cited above describe the more typical presentations of this rare disease. However, this patient had an unusual presentation, as parotid involvement due to zygomycosis has only been described once previously.5 Her inflammatory vasculitis and ensuing strokes from involvement of the carotid artery are recognized complications of zygomycosis, and in 1 case series of 41 patients with rhinocerebral mucormycosis, carotid involvement was seen in 31% of patients.6 After the punch biopsy of the patient's pinna showing nonseptated hyphae reminiscent of Mucor, why did her physicians delay administering amphotericin?
There are 2 likely possibilities: anchoring bias or error in medical decision‐making due to inaccurate probability estimates. Anchoring bias describes a heuristic where the initial diagnosis or gestalt biases the physician's process for assigning a final diagnosis.7, 8 This bias creates cognitive errors by limiting creativity in diagnosis. In this case, the infectious disease team carefully weighed the information obtained from the first biopsy. Given their low pretest estimate of this virtually unreported presentation of a rare disease, they decided to evaluate further without beginning antifungal therapy. Of note, there were few hyaline structures, and those structures lacked uptake of GMS. Since they considered the diagnosis yet rejected the diagnosis due to insufficient evidence, it is unlikely that anchoring bias played a role.
Was there an error in medical decision‐making? The physicians in this case faced a very common medical dilemma: whether or not to start a toxic medication empirically or wait for diagnostic confirmation prior to treatment.9 To solve this dilemma, one can apply decision analysis. Moskowitz et al described 5 phases of medical decision analysis by which a probabilistic right answer to clinical scenarios can be deduced mathematically.10 To solve this problem, probabilities must be assigned to the risk of giving a drug to a patient without the disease versus the risk of not giving a drug to a patient with the disease. For example, amphotericin deoxycholate causes acute renal failure in 30% to 47% of patients. Newer formulations of amphotericin, such as liposomal amphotericin and lipid complex, result in lower rates of nephrotoxicity (27% vs 47%). The risk of not giving amphotericin to a patient with zygomycosis is death. Even in patients treated with amphotericin, the mortality rate has been shown to be 66%, and up to 100% in those with strokes related to zygomycosis.2, 6, 11 Simply looking at these probabilities, decision analysis would favor empiric treatment.
The physicians caring for this patient did not have the luxury of retrospective speculation. After looking at all of the data, the equivocal skin biopsy and rare clinical presentation, the question to ask would change: What is the risk of giving amphotericin empirically to someone who, based on available information, has a very low probability of having zygomycosis? When phrased in this manner, there is a 47% chance of nephrotoxicity with amphotericin versus the very small probability that you have diagnosed a case of zygomycosis that has only been described once in the literature. Mathematically andmore importantlyclinically, this question becomes more difficult to answer. However, no value can be placed on the possibility of death in suspected zygomycosis, and the risk of short‐term amphotericin use is much less than that of a course of treatment. As such, empiric therapy should always be given.
Physicians are not mathematicians, and dynamic clinical scenarios are not so easily made into static math problems. Disease presentations evolve over time towards a diagnosable clinical pattern, as was the case with this patient. Two days after the aforementioned biopsy, she worsened and in less time than it would have taken to isolate zygomycosis from the first biopsy, a second biopsy revealed the typical nonseptated hyphae demarcated with the GMS stain. Even appropriate diagnostic testing, thoughtful interpretation, and avoidance of certain cognitive errors can result in incorrect diagnoses and delayed treatment. It is monitoring the progression of disease and collecting additional data that allows physicians to mold a diagnosis and create a treatment plan.
The primary treatment of zygomycosis should include amphotericin. However, there are limited data to support combination therapy with an echinocandin in severe cases, as in this patient.12 Posaconazole is not recommended for monotherapy as an initial therapy, but there is data for its use as salvage therapy in zygomycosis.13 This case highlights the difficulties that physicians face in the diagnosis and treatment of rare diseases. Cerebral infarction in a hematologic malignancy, uncontrolled diabetes, or iron chelation therapy could be the initial presentation of rhinocerebral zygomycosis. There truly are different strokes for different folks. Recognizing this and similar presentations may lead to a more rapid diagnosis and treatment of zygomycosis.
TEACHING POINTS
-
Zygomycosis has a wide range of clinical presentations ranging from skin lesions to deep tissue infections. As it is an angioinvasive organism, it can also present as cerebral infarcts and brain abscesses.
-
Zygomycosis infections should be suspected in patients with uncontrolled diabetes, hematologic or oncologic malignancies, and patients on iron chelation therapy with a potentially compatible clinical picture.
-
If zygomycosis infection is suspected, rapid histologic diagnosis should be attempted. However, as histologic diagnosis can take time, empiric therapy with amphotericin should always be administered.
-
Amphotericin remains the primary medical therapy for this disease; however, there is limited emerging evidence to suggest that echinocandins can be used in combination with amphotericin for improved treatment of severe rhinocerebral zygomyocosis. Posaconazole has a role as salvage therapy in zygomycosis, but should not be used as the sole primary treatment.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
This icon represents the patient's case. Each paragraph that follows represents the discussant's thoughts.
Acknowledgements
The authors are indebted to Dr Glenn Roberson at the Department of Radiology, University of Alabama at Birmingham, for providing the radiographic images; to Dr Aleodor Andea at the Department of Pathology, University of Alabama at Birmingham, for providing the pathology images; and to Dr. Crysten Brinkley at the Department of Neurology at the University of Alabama at Birmingham for her assistance with this case presentation.
Disclosure: Nothing to report.
- ,,,.Mucormycosis: emerging prominence of cutaneous infections.Clin Infect Dis.1994;19:67–76.
- ,,,.Zygomycosis in the 1990s in a tertiary‐care cancer center.Clin Infect Dis.2000;30:851–856.
- ,,, et al.Mucormycosis during deferoxamine therapy is a siderophore‐mediated infection. In vitro and in vivo animal studies.J Clin Invest.1993;91:1979–1986.
- ,,, et al.Epidemiology and outcome of zygomycosis: a review of 929 reported cases.Clin Infect Dis.2005;41:634–653.
- ,,,,,.Cutaneous mucormycosis of the head and neck with parotid gland involvement: first report of a case.Ear Nose Throat J.2004;83:282–286.
- ,,,,,.A successful combined endovascular and surgical treatment of a cranial base mucormycosis with an associated internal carotid artery pseudoaneurysm.Neurosurgery.2009;65:733–740.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,,,.Clinical problem‐solving. Anchors away.N Engl J Med.2007;356:504–509.
- ,,,.Clinical problem‐solving. Empirically incorrect.N Engl J Med.2006;354:509–514.
- ,,.Dealing with uncertainty, risks, and tradeoffs in clinical decisions. A cognitive science approach.Ann Intern Med.1988;108:435–449.
- ,,.Fatal strokes in patients with rhino‐orbito‐cerebral mucormycosis and associated vasculopathy.Scand J Infect Dis.2004;36:643–648.
- ,,, et al.Combination polyene‐caspofungin treatment of rhino‐orbital‐cerebral mucormycosis.Clin Infect Dis.2008;47:364–371.
- ,,,,.Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases.Clin Infect Dis.2006;42:e61–e65.
- ,,,.Mucormycosis: emerging prominence of cutaneous infections.Clin Infect Dis.1994;19:67–76.
- ,,,.Zygomycosis in the 1990s in a tertiary‐care cancer center.Clin Infect Dis.2000;30:851–856.
- ,,, et al.Mucormycosis during deferoxamine therapy is a siderophore‐mediated infection. In vitro and in vivo animal studies.J Clin Invest.1993;91:1979–1986.
- ,,, et al.Epidemiology and outcome of zygomycosis: a review of 929 reported cases.Clin Infect Dis.2005;41:634–653.
- ,,,,,.Cutaneous mucormycosis of the head and neck with parotid gland involvement: first report of a case.Ear Nose Throat J.2004;83:282–286.
- ,,,,,.A successful combined endovascular and surgical treatment of a cranial base mucormycosis with an associated internal carotid artery pseudoaneurysm.Neurosurgery.2009;65:733–740.
- ,.Judgment under uncertainty: heuristics and biases.Science.1974;185:1124–1131.
- ,,,,.Clinical problem‐solving. Anchors away.N Engl J Med.2007;356:504–509.
- ,,,.Clinical problem‐solving. Empirically incorrect.N Engl J Med.2006;354:509–514.
- ,,.Dealing with uncertainty, risks, and tradeoffs in clinical decisions. A cognitive science approach.Ann Intern Med.1988;108:435–449.
- ,,.Fatal strokes in patients with rhino‐orbito‐cerebral mucormycosis and associated vasculopathy.Scand J Infect Dis.2004;36:643–648.
- ,,, et al.Combination polyene‐caspofungin treatment of rhino‐orbital‐cerebral mucormycosis.Clin Infect Dis.2008;47:364–371.
- ,,,,.Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases.Clin Infect Dis.2006;42:e61–e65.
What Are the Clinical Indications for Noninvasive Positive Pressure Ventilation?
Case
A 63-year-old man with severe chronic obstructive pulmonary disease (COPD) presents with one week of increasing sputum, cough, and dyspnea. His respiratory rate is 26/minute and oxygen saturation is 86% on room air (RA). He is lethargic and appears mildly uncomfortable, but he responds appropriately to questions in three- to four-word sentences. He is tachypneic with accessory muscle use and has diffuse wheezes throughout his bilateral lung fields. His initial room air arterial blood gas (ABG) is 7.32/68/86/32. Chest radiograph is notable for flattened hemidiaphragms without focal opacity. The patient is placed on oxygen and receives prednisone with nebulized albuterol and ipratropium, but his dyspnea and tachypnea persist. Due to his respiratory distress, bilevel positive airway pressure (BiPAP) is considered.
What are the clinical indications for noninvasive positive pressure ventilation (NPPV)?
Overview
NPPV assists ventilation by delivering positive expiratory and/or inspiratory pressure without the use of an endotracheal tube. Theoretically, NPPV is a preferred method of ventilation as it may eliminate the need for endotracheal intubation and its associated morbidity and mortality, including airway trauma, loss of airway defense mechanisms (ventilator-associated pneumonia), mechanical ventilation (barotrauma), and disruption of speech and swallowing.1
NPPV is generally delivered via full-face mask or nasal mask. Nasal mask is often preferred for patient comfort, though air leaks occur with mouth breathing. There is no difference between nasal and full-face masks in outcomes including intubation rates and mortality.2,3,4 NPPV can be delivered via a portable or standard ventilator using the same modes available for endotracheal intubation, though pressure-cycled ventilators utilizing continuous positive airway pressure (CPAP) and BiPAP are most common. CPAP delivers air at a continuous fixed pressure throughout the respiratory cycle. BiPAP delivers positive pressure at alternating levels—higher for inspiration and lower for expiration. Guidelines suggest choosing a mode based on the etiology and pathophysiology of the respiratory failure and leveraging local comfort and expertise.2,3
In general, good candidates for NPPV display signs of tachypnea and dyspnea due to hypoxic or hypercapnic respiratory failure but are hemodynamically stable, without excessive secretions, and can protect their airway and achieve a proper seal with the mask.3 Difficulty may arise due to patient intolerance, claustrophobia, gastric distention, and poor fit that leads to air leak or skin erosion. With initiation of NPPV, patients should be followed in a care setting with the capacity for frequent monitoring and, if needed, quick access to invasive airway management. Monitoring should include patient comfort and ability to tolerate the device, vital signs, breathing pattern, oxygen saturation, ABG, and mental status. This initial evaluation may help predict the success of NPPV (see Table 2). Appropriately chosen candidates who do well with NPPV often demonstrate respiratory turnaround in a relatively brief interval.2,3
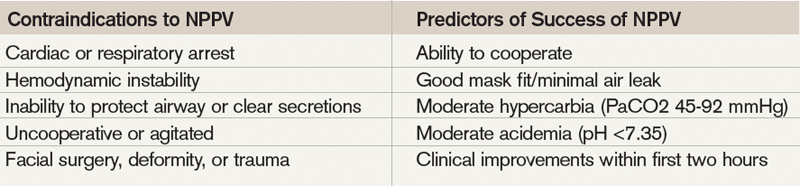
Review of the Data
NPPV is increasingly utilized in a variety of clinical situations. In 2000, the American Thoracic Society published consensus guidelines on the use of NPPV in acute respiratory failure.2 More recently, the Canadian Medical Association developed clinical guidelines for the use of NPPV in the acute-care setting.4 Clinical scenarios in which there is evidence for the efficacy of NPPV include severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia; it can also be used as a bridge to extubation in COPD patients.1-4
Acute exacerbation of COPD. Several randomized controlled trials (RCT) and meta-analyses have assessed the potential benefits of NPPV in patients with acute exacerbations of COPD. In COPD, NPPV improves gas exchange and facilitates respiratory muscle rest to decrease the work of breathing, which allows for respiratory recovery and time to effectiveness of standard therapies.5 Multiple trials have demonstrated that the addition of NPPV to usual care decreases intubation and mortality rates, as well as hospital lengths of stay (LOS).5-8
A Cochrane review of eight RCTs comparing NPPV with usual care noted a greater than 50% reduction in risk of intubation, and a number needed to treat (NNT) of eight patients to prevent one death.5 Quon and colleagues also compared NPPV to usual care in a meta-analysis of 14 trials.6 Eleven of these trials evaluated hospital mortality, which was decreased by 55% in patients receiving NPPV. Twelve trials assessed need for intubation, which decreased by 65%. In these trials, BiPAP was the most commonly used modality (see Table 3, for a comparison of NPPV modalities). Study patients had an average pH of 7.31 with an average PaCO2 of 68 mmHg. It was noted that the beneficial effects of NPPV increased as pH decreased. An earlier meta-analysis from Keenan and colleagues supported this notion, noting that the subgroup of patients with pH <7.3 benefited most in terms of decreased rates of intubation, hospital LOS, and hospital mortality.7 In this 2003 study, patients with relatively mild exacerbations of COPD did not benefit from the addition of NPPV to usual care. Based on the amount of positive evidence, NPPV is recommended in patients experiencing severe exacerbations of COPD as evidenced by a pH <7.35 and relative hypercarbia.1,2,4,7
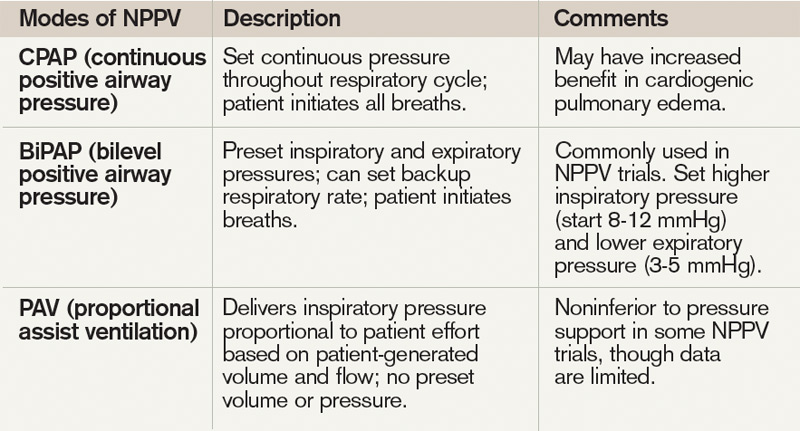
Cardiogenic pulmonary edema. In patients with acute cardiogenic pulmonary edema, NPPV has been found to be beneficial, decreasing mortality, rates of intubation, and hospital LOS. Physiologically, NPPV augments cardiac output, improves respiratory mechanics, and decreases afterload.10 Cardiogenic edema is variably defined and has a number of causes elucidated in an analysis of 11 RCTs conducted by Masip and colleagues. These causes included acute coronary syndrome (31%), hypertension (27%), congestive heart failure (14%), and a combination of respiratory infection, arrhythmia, volume overload, and treatment noncompliance (28%).9 In this analysis, CPAP and BiPAP demonstrated a combined 43% reduction in mortality and a 57% reduction in intubation. More recently, Peter and colleagues described a statistically significant reduction in hospital mortality and the need for intubation with CPAP, while BiPAP only demonstrated a statistically significant decrease in need for intubation.10 Thus, there appears to be some evidence that CPAP is the preferred NPPV mode in patients with acute cardiogenic pulmonary edema. Despite inclusion of a recent, large RCT showing no benefit of NPPV versus usual care in cardiogenic pulmonary edema, the overall positive effect of NPPV persisted, particularly when the cause of pulmonary edema was acute coronary syndrome.11
Weaning after intubation. NPPV has been evaluated as a method to facilitate early extubation, as a measure to prevent extubation failure, and as a treatment modality for respiratory failure following extubation, with mixed results.12,13 In 1998, a small trial compared the use of NPPV in COPD patients to facilitate early extubation with a standard weaning protocol. In this population, early NPPV resulted in better weaning rates with shorter times of mechanical ventilation (10 vs. 16 days), fewer days in the ICU, and improved 60-day survival rates (92% vs. 72%).3,14 In another RCT not limited to COPD patients, Grault and colleagues found that NPPV reduced the duration of intubation (4.5 vs. 7.6 days) but was not associated with benefits in ICU length of stay or survival previously described.3,15 Thus, though NPPV may be beneficial in facilitating early extubation in COPD patients, it is not recommended in other patient populations.4
NPPV has also been evaluated as a measure to prevent respiratory failure in patients at high risk for extubation failure. When applied immediately after extubation in patients with COPD and obesity, NPPV reduced reintubation rates and ICU mortality.3,4 In 2004, Esteban and colleagues examined NPPV in patients who had respiratory failure following extubation. In this setting, NPPV was ineffective at preventing reintubation and had no survival benefit.
In summary, NPPV may facilitate early extubation and prevent extubation failure in appropriate patients, such as those with COPD, but is unlikely to be beneficial and is not recommended in patients with existing respiratory failure after extubation.4,15
Immunosuppressed patients. A 2001 single-center, randomized-controlled trial by Holbert and colleagues demonstrated decreased intubation rates and mortality with the application of NPPV in immunosuppressed patients with hypoxemic respiratory failure, fever, and pulmonary infiltrates.16
In this study, immunosuppression occurred most commonly as a result of malignancy. In the group receiving NPPV alternating with oxygen (at least 45 minutes of NPPV alternated every three hours with periods of spontaneous breathing), the rate of subsequent intubation decreased to 46%, compared with 77% in those receiving oxygen alone. The mortality rate was 38% in the NPPV group, as compared with 69% in the standard treatment group.
Though the outcomes in immunocompromised patients with hypoxemia, fever, and pulmonary infiltrates were very poor (38% mortality even with NPPV), this small study and recent guidelines suggest a trial of NPPV in this population.4,16
Other indications. NPPV has been applied in multiple other clinical scenarios, including exacerbation of asthma, community-acquired pneumonia, acute lung injury, and bronchoscopy in hypoxemic patients. It has also been evaluated in the postsurgical period and in chest trauma. There are mixed and less robust data in these various applications, and larger controlled trials are lacking.
In asthma exacerbation, NPPV may improve dyspnea, but data regarding outcomes (intubation, mortality) are lacking. A 2005 Cochrane review concluded that data remain controversial due to insufficient evidence, and guidelines make no recommendations concerning NPPV in asthma exacerbation.4,17 Similarly, in community-acquired pneumonia without prior history of COPD, there is no major role for NPPV.1,3,4 Limited data suggest that NPPV lacks efficacy in preventing post-surgical respiratory failure, though it may be useful in treating existing respiratory failure or preventing intubation in patients following lung resection or abdominal surgery.1,4 In hypoxemic patients undergoing bronchoscopy, NPPV may improve oxygenation (lower respiratory rates and improved PaO2 to FiO2 ratios, compared with oxygen alone) as well as hemodynamics (minimizing the drop in mean arterial pressure). However, outcome data are lacking and the data set is small.4,18 In acute lung injury/acute respiratory distress syndrome, data are also limited, but NPPV appears to have a high failure rate and confers little benefit.1,4
Back to the Case
The patient was admitted to the hospital and placed on BiPAP for approximately 1.5 hours. The patient’s respiratory rate improved to 20/minute and he appeared increasingly comfortable and alert. A repeat ABG revealed improved hypercarbia and acidosis. He was continued on steroids and antibiotics and eventually was weaned from BiPAP and discharged home.
Bottom Line
NPPV is an effective method to decrease mortality, intubation rates, and duration of ICU stay in severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia, and as a bridge to extubation in COPD patients.
Dr. Kraynek is an internal medicine resident in the Department of Medicine at the University of Washington School of Medicine in Seattle. Dr. Best is assistant professor of medicine in the Division of General Internal Medicine at the University of Washington School of Medicine.
References
- Ambrosino N, Vagheggini G. Noninvasive positive pressure ventilation in the acute care setting: where are we? Euro Resp J. 2008;31:874-856.
- American Thoracic Society. International Consensus Conferences in Intensive Care Medicine: Noninvasive Positive Pressure Ventilation in Acute Respiratory Failure. Am J Respir Crit Care Med. 2001;163:283-291.
- Liesching T, Kwok H, Hill N. Acute applications of noninvasive positive pressure ventilation. Chest. 2003;124:699-713.
- Keenan S, Sinuff T, Burns K, et al. Clinical practice guidelines for the use of noninvasive positive pressure ventilation and noninvasive continuous positive airway pressure in the acute care setting. CMAJ. 2001;183:E195-E214.
- Lightowler J, Wedzicha J, Elliot M, Ram F. Noninvasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ. 2003;326:185.
- Quon B, Gan W, Sin D. Contemporary management of acute exacerbations of COPD: a systematic review of the metaanalysis. Chest. 2008;133:756-766.
- Keenan S, Sinuff T, Cook D, Hill, N. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive pressure ventilation? Ann Intern Med. 2003;138:861-870.
- Scala R, Naldi M, Archinucci I, Conigilo G, Nava S. Noninvasive positive pressure ventilation in patients with acute exacerbations of COPD and varying levels of consciousness. Chest. 2005;128:1657-1666.
- Masip J, Roque M, Sanchez B, Fernandez R, Subirana M, Exposito J. Noninvasive ventilation in acute cardiogenic pulmonary edema. JAMA. 2005;294:3124-3130.
- Peter J, Moran J, Phillips-Hughes J, Graham P, Bersten A. Effect of non-invasive positive pressure ventilation (NIPPV) on mortality in patients with acute cardiogenic pulmonary oedema: a meta-anaylsis. Lancet. 2006;367:1155-1163.
- Weng C, Zhao Y, Liu Q, et al. Meta-analysis: noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med. 2010;152:560-600.
- Keenan S, Sinuff T, Cook D, Hill N. Does noninvasive positive pressure ventilation improve outcome in acute hypoxemic respiratory failure? A systematic review. Crit Care Med. 2004;32:2516-2523.
- Esteban A, Frutos-Vivar F, Fergusun N, et al. Noninvasive positive pressure ventilation for respiratory failure after extubation. N Engl J Med. 2004;350:2452-2460.
- Nava S, Ambrosino N, Clinie E, et al. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease: a randomized, controlled trial. Ann Intern Med. 1998;128:721-728.
- Grault C, Daudenthun I, Chevron V, et al. Noninvasive ventilation as a systematic extubation and weaning technique in acute on chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med. 1999;160:86-92.
- Hilbert G, Gruson D, Vargas F, et al. Noninvasive ventilation in immunosuppressed patients with pulmonary infiltrates, fever, and acute respiratory failure. N Engl J Med. 2001;344:481-487.
- Ram FSF, Wellington SR, Rowe BH, Wedzicha JA. Non-invasive positive pressure ventilation for treatment of respiratory failure due to severe acute exacerbations of asthma. Cochrane Database of Systematic Reviews. 2005, Issue 3.
- Antonelli M, Conti G, Rocco M, et al. Noninvasive positive pressure ventilation vs. conventional oxygen supplementation in hypoxemic patients undergoing diagnostic bronchoscopy. Chest. 2002;121:1149-1154.
Case
A 63-year-old man with severe chronic obstructive pulmonary disease (COPD) presents with one week of increasing sputum, cough, and dyspnea. His respiratory rate is 26/minute and oxygen saturation is 86% on room air (RA). He is lethargic and appears mildly uncomfortable, but he responds appropriately to questions in three- to four-word sentences. He is tachypneic with accessory muscle use and has diffuse wheezes throughout his bilateral lung fields. His initial room air arterial blood gas (ABG) is 7.32/68/86/32. Chest radiograph is notable for flattened hemidiaphragms without focal opacity. The patient is placed on oxygen and receives prednisone with nebulized albuterol and ipratropium, but his dyspnea and tachypnea persist. Due to his respiratory distress, bilevel positive airway pressure (BiPAP) is considered.
What are the clinical indications for noninvasive positive pressure ventilation (NPPV)?
Overview
NPPV assists ventilation by delivering positive expiratory and/or inspiratory pressure without the use of an endotracheal tube. Theoretically, NPPV is a preferred method of ventilation as it may eliminate the need for endotracheal intubation and its associated morbidity and mortality, including airway trauma, loss of airway defense mechanisms (ventilator-associated pneumonia), mechanical ventilation (barotrauma), and disruption of speech and swallowing.1
NPPV is generally delivered via full-face mask or nasal mask. Nasal mask is often preferred for patient comfort, though air leaks occur with mouth breathing. There is no difference between nasal and full-face masks in outcomes including intubation rates and mortality.2,3,4 NPPV can be delivered via a portable or standard ventilator using the same modes available for endotracheal intubation, though pressure-cycled ventilators utilizing continuous positive airway pressure (CPAP) and BiPAP are most common. CPAP delivers air at a continuous fixed pressure throughout the respiratory cycle. BiPAP delivers positive pressure at alternating levels—higher for inspiration and lower for expiration. Guidelines suggest choosing a mode based on the etiology and pathophysiology of the respiratory failure and leveraging local comfort and expertise.2,3
In general, good candidates for NPPV display signs of tachypnea and dyspnea due to hypoxic or hypercapnic respiratory failure but are hemodynamically stable, without excessive secretions, and can protect their airway and achieve a proper seal with the mask.3 Difficulty may arise due to patient intolerance, claustrophobia, gastric distention, and poor fit that leads to air leak or skin erosion. With initiation of NPPV, patients should be followed in a care setting with the capacity for frequent monitoring and, if needed, quick access to invasive airway management. Monitoring should include patient comfort and ability to tolerate the device, vital signs, breathing pattern, oxygen saturation, ABG, and mental status. This initial evaluation may help predict the success of NPPV (see Table 2). Appropriately chosen candidates who do well with NPPV often demonstrate respiratory turnaround in a relatively brief interval.2,3
Review of the Data
NPPV is increasingly utilized in a variety of clinical situations. In 2000, the American Thoracic Society published consensus guidelines on the use of NPPV in acute respiratory failure.2 More recently, the Canadian Medical Association developed clinical guidelines for the use of NPPV in the acute-care setting.4 Clinical scenarios in which there is evidence for the efficacy of NPPV include severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia; it can also be used as a bridge to extubation in COPD patients.1-4
Acute exacerbation of COPD. Several randomized controlled trials (RCT) and meta-analyses have assessed the potential benefits of NPPV in patients with acute exacerbations of COPD. In COPD, NPPV improves gas exchange and facilitates respiratory muscle rest to decrease the work of breathing, which allows for respiratory recovery and time to effectiveness of standard therapies.5 Multiple trials have demonstrated that the addition of NPPV to usual care decreases intubation and mortality rates, as well as hospital lengths of stay (LOS).5-8
A Cochrane review of eight RCTs comparing NPPV with usual care noted a greater than 50% reduction in risk of intubation, and a number needed to treat (NNT) of eight patients to prevent one death.5 Quon and colleagues also compared NPPV to usual care in a meta-analysis of 14 trials.6 Eleven of these trials evaluated hospital mortality, which was decreased by 55% in patients receiving NPPV. Twelve trials assessed need for intubation, which decreased by 65%. In these trials, BiPAP was the most commonly used modality (see Table 3, for a comparison of NPPV modalities). Study patients had an average pH of 7.31 with an average PaCO2 of 68 mmHg. It was noted that the beneficial effects of NPPV increased as pH decreased. An earlier meta-analysis from Keenan and colleagues supported this notion, noting that the subgroup of patients with pH <7.3 benefited most in terms of decreased rates of intubation, hospital LOS, and hospital mortality.7 In this 2003 study, patients with relatively mild exacerbations of COPD did not benefit from the addition of NPPV to usual care. Based on the amount of positive evidence, NPPV is recommended in patients experiencing severe exacerbations of COPD as evidenced by a pH <7.35 and relative hypercarbia.1,2,4,7
Cardiogenic pulmonary edema. In patients with acute cardiogenic pulmonary edema, NPPV has been found to be beneficial, decreasing mortality, rates of intubation, and hospital LOS. Physiologically, NPPV augments cardiac output, improves respiratory mechanics, and decreases afterload.10 Cardiogenic edema is variably defined and has a number of causes elucidated in an analysis of 11 RCTs conducted by Masip and colleagues. These causes included acute coronary syndrome (31%), hypertension (27%), congestive heart failure (14%), and a combination of respiratory infection, arrhythmia, volume overload, and treatment noncompliance (28%).9 In this analysis, CPAP and BiPAP demonstrated a combined 43% reduction in mortality and a 57% reduction in intubation. More recently, Peter and colleagues described a statistically significant reduction in hospital mortality and the need for intubation with CPAP, while BiPAP only demonstrated a statistically significant decrease in need for intubation.10 Thus, there appears to be some evidence that CPAP is the preferred NPPV mode in patients with acute cardiogenic pulmonary edema. Despite inclusion of a recent, large RCT showing no benefit of NPPV versus usual care in cardiogenic pulmonary edema, the overall positive effect of NPPV persisted, particularly when the cause of pulmonary edema was acute coronary syndrome.11
Weaning after intubation. NPPV has been evaluated as a method to facilitate early extubation, as a measure to prevent extubation failure, and as a treatment modality for respiratory failure following extubation, with mixed results.12,13 In 1998, a small trial compared the use of NPPV in COPD patients to facilitate early extubation with a standard weaning protocol. In this population, early NPPV resulted in better weaning rates with shorter times of mechanical ventilation (10 vs. 16 days), fewer days in the ICU, and improved 60-day survival rates (92% vs. 72%).3,14 In another RCT not limited to COPD patients, Grault and colleagues found that NPPV reduced the duration of intubation (4.5 vs. 7.6 days) but was not associated with benefits in ICU length of stay or survival previously described.3,15 Thus, though NPPV may be beneficial in facilitating early extubation in COPD patients, it is not recommended in other patient populations.4
NPPV has also been evaluated as a measure to prevent respiratory failure in patients at high risk for extubation failure. When applied immediately after extubation in patients with COPD and obesity, NPPV reduced reintubation rates and ICU mortality.3,4 In 2004, Esteban and colleagues examined NPPV in patients who had respiratory failure following extubation. In this setting, NPPV was ineffective at preventing reintubation and had no survival benefit.
In summary, NPPV may facilitate early extubation and prevent extubation failure in appropriate patients, such as those with COPD, but is unlikely to be beneficial and is not recommended in patients with existing respiratory failure after extubation.4,15
Immunosuppressed patients. A 2001 single-center, randomized-controlled trial by Holbert and colleagues demonstrated decreased intubation rates and mortality with the application of NPPV in immunosuppressed patients with hypoxemic respiratory failure, fever, and pulmonary infiltrates.16
In this study, immunosuppression occurred most commonly as a result of malignancy. In the group receiving NPPV alternating with oxygen (at least 45 minutes of NPPV alternated every three hours with periods of spontaneous breathing), the rate of subsequent intubation decreased to 46%, compared with 77% in those receiving oxygen alone. The mortality rate was 38% in the NPPV group, as compared with 69% in the standard treatment group.
Though the outcomes in immunocompromised patients with hypoxemia, fever, and pulmonary infiltrates were very poor (38% mortality even with NPPV), this small study and recent guidelines suggest a trial of NPPV in this population.4,16
Other indications. NPPV has been applied in multiple other clinical scenarios, including exacerbation of asthma, community-acquired pneumonia, acute lung injury, and bronchoscopy in hypoxemic patients. It has also been evaluated in the postsurgical period and in chest trauma. There are mixed and less robust data in these various applications, and larger controlled trials are lacking.
In asthma exacerbation, NPPV may improve dyspnea, but data regarding outcomes (intubation, mortality) are lacking. A 2005 Cochrane review concluded that data remain controversial due to insufficient evidence, and guidelines make no recommendations concerning NPPV in asthma exacerbation.4,17 Similarly, in community-acquired pneumonia without prior history of COPD, there is no major role for NPPV.1,3,4 Limited data suggest that NPPV lacks efficacy in preventing post-surgical respiratory failure, though it may be useful in treating existing respiratory failure or preventing intubation in patients following lung resection or abdominal surgery.1,4 In hypoxemic patients undergoing bronchoscopy, NPPV may improve oxygenation (lower respiratory rates and improved PaO2 to FiO2 ratios, compared with oxygen alone) as well as hemodynamics (minimizing the drop in mean arterial pressure). However, outcome data are lacking and the data set is small.4,18 In acute lung injury/acute respiratory distress syndrome, data are also limited, but NPPV appears to have a high failure rate and confers little benefit.1,4
Back to the Case
The patient was admitted to the hospital and placed on BiPAP for approximately 1.5 hours. The patient’s respiratory rate improved to 20/minute and he appeared increasingly comfortable and alert. A repeat ABG revealed improved hypercarbia and acidosis. He was continued on steroids and antibiotics and eventually was weaned from BiPAP and discharged home.
Bottom Line
NPPV is an effective method to decrease mortality, intubation rates, and duration of ICU stay in severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia, and as a bridge to extubation in COPD patients.
Dr. Kraynek is an internal medicine resident in the Department of Medicine at the University of Washington School of Medicine in Seattle. Dr. Best is assistant professor of medicine in the Division of General Internal Medicine at the University of Washington School of Medicine.
References
- Ambrosino N, Vagheggini G. Noninvasive positive pressure ventilation in the acute care setting: where are we? Euro Resp J. 2008;31:874-856.
- American Thoracic Society. International Consensus Conferences in Intensive Care Medicine: Noninvasive Positive Pressure Ventilation in Acute Respiratory Failure. Am J Respir Crit Care Med. 2001;163:283-291.
- Liesching T, Kwok H, Hill N. Acute applications of noninvasive positive pressure ventilation. Chest. 2003;124:699-713.
- Keenan S, Sinuff T, Burns K, et al. Clinical practice guidelines for the use of noninvasive positive pressure ventilation and noninvasive continuous positive airway pressure in the acute care setting. CMAJ. 2001;183:E195-E214.
- Lightowler J, Wedzicha J, Elliot M, Ram F. Noninvasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ. 2003;326:185.
- Quon B, Gan W, Sin D. Contemporary management of acute exacerbations of COPD: a systematic review of the metaanalysis. Chest. 2008;133:756-766.
- Keenan S, Sinuff T, Cook D, Hill, N. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive pressure ventilation? Ann Intern Med. 2003;138:861-870.
- Scala R, Naldi M, Archinucci I, Conigilo G, Nava S. Noninvasive positive pressure ventilation in patients with acute exacerbations of COPD and varying levels of consciousness. Chest. 2005;128:1657-1666.
- Masip J, Roque M, Sanchez B, Fernandez R, Subirana M, Exposito J. Noninvasive ventilation in acute cardiogenic pulmonary edema. JAMA. 2005;294:3124-3130.
- Peter J, Moran J, Phillips-Hughes J, Graham P, Bersten A. Effect of non-invasive positive pressure ventilation (NIPPV) on mortality in patients with acute cardiogenic pulmonary oedema: a meta-anaylsis. Lancet. 2006;367:1155-1163.
- Weng C, Zhao Y, Liu Q, et al. Meta-analysis: noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med. 2010;152:560-600.
- Keenan S, Sinuff T, Cook D, Hill N. Does noninvasive positive pressure ventilation improve outcome in acute hypoxemic respiratory failure? A systematic review. Crit Care Med. 2004;32:2516-2523.
- Esteban A, Frutos-Vivar F, Fergusun N, et al. Noninvasive positive pressure ventilation for respiratory failure after extubation. N Engl J Med. 2004;350:2452-2460.
- Nava S, Ambrosino N, Clinie E, et al. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease: a randomized, controlled trial. Ann Intern Med. 1998;128:721-728.
- Grault C, Daudenthun I, Chevron V, et al. Noninvasive ventilation as a systematic extubation and weaning technique in acute on chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med. 1999;160:86-92.
- Hilbert G, Gruson D, Vargas F, et al. Noninvasive ventilation in immunosuppressed patients with pulmonary infiltrates, fever, and acute respiratory failure. N Engl J Med. 2001;344:481-487.
- Ram FSF, Wellington SR, Rowe BH, Wedzicha JA. Non-invasive positive pressure ventilation for treatment of respiratory failure due to severe acute exacerbations of asthma. Cochrane Database of Systematic Reviews. 2005, Issue 3.
- Antonelli M, Conti G, Rocco M, et al. Noninvasive positive pressure ventilation vs. conventional oxygen supplementation in hypoxemic patients undergoing diagnostic bronchoscopy. Chest. 2002;121:1149-1154.
Case
A 63-year-old man with severe chronic obstructive pulmonary disease (COPD) presents with one week of increasing sputum, cough, and dyspnea. His respiratory rate is 26/minute and oxygen saturation is 86% on room air (RA). He is lethargic and appears mildly uncomfortable, but he responds appropriately to questions in three- to four-word sentences. He is tachypneic with accessory muscle use and has diffuse wheezes throughout his bilateral lung fields. His initial room air arterial blood gas (ABG) is 7.32/68/86/32. Chest radiograph is notable for flattened hemidiaphragms without focal opacity. The patient is placed on oxygen and receives prednisone with nebulized albuterol and ipratropium, but his dyspnea and tachypnea persist. Due to his respiratory distress, bilevel positive airway pressure (BiPAP) is considered.
What are the clinical indications for noninvasive positive pressure ventilation (NPPV)?
Overview
NPPV assists ventilation by delivering positive expiratory and/or inspiratory pressure without the use of an endotracheal tube. Theoretically, NPPV is a preferred method of ventilation as it may eliminate the need for endotracheal intubation and its associated morbidity and mortality, including airway trauma, loss of airway defense mechanisms (ventilator-associated pneumonia), mechanical ventilation (barotrauma), and disruption of speech and swallowing.1
NPPV is generally delivered via full-face mask or nasal mask. Nasal mask is often preferred for patient comfort, though air leaks occur with mouth breathing. There is no difference between nasal and full-face masks in outcomes including intubation rates and mortality.2,3,4 NPPV can be delivered via a portable or standard ventilator using the same modes available for endotracheal intubation, though pressure-cycled ventilators utilizing continuous positive airway pressure (CPAP) and BiPAP are most common. CPAP delivers air at a continuous fixed pressure throughout the respiratory cycle. BiPAP delivers positive pressure at alternating levels—higher for inspiration and lower for expiration. Guidelines suggest choosing a mode based on the etiology and pathophysiology of the respiratory failure and leveraging local comfort and expertise.2,3
In general, good candidates for NPPV display signs of tachypnea and dyspnea due to hypoxic or hypercapnic respiratory failure but are hemodynamically stable, without excessive secretions, and can protect their airway and achieve a proper seal with the mask.3 Difficulty may arise due to patient intolerance, claustrophobia, gastric distention, and poor fit that leads to air leak or skin erosion. With initiation of NPPV, patients should be followed in a care setting with the capacity for frequent monitoring and, if needed, quick access to invasive airway management. Monitoring should include patient comfort and ability to tolerate the device, vital signs, breathing pattern, oxygen saturation, ABG, and mental status. This initial evaluation may help predict the success of NPPV (see Table 2). Appropriately chosen candidates who do well with NPPV often demonstrate respiratory turnaround in a relatively brief interval.2,3
Review of the Data
NPPV is increasingly utilized in a variety of clinical situations. In 2000, the American Thoracic Society published consensus guidelines on the use of NPPV in acute respiratory failure.2 More recently, the Canadian Medical Association developed clinical guidelines for the use of NPPV in the acute-care setting.4 Clinical scenarios in which there is evidence for the efficacy of NPPV include severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia; it can also be used as a bridge to extubation in COPD patients.1-4
Acute exacerbation of COPD. Several randomized controlled trials (RCT) and meta-analyses have assessed the potential benefits of NPPV in patients with acute exacerbations of COPD. In COPD, NPPV improves gas exchange and facilitates respiratory muscle rest to decrease the work of breathing, which allows for respiratory recovery and time to effectiveness of standard therapies.5 Multiple trials have demonstrated that the addition of NPPV to usual care decreases intubation and mortality rates, as well as hospital lengths of stay (LOS).5-8
A Cochrane review of eight RCTs comparing NPPV with usual care noted a greater than 50% reduction in risk of intubation, and a number needed to treat (NNT) of eight patients to prevent one death.5 Quon and colleagues also compared NPPV to usual care in a meta-analysis of 14 trials.6 Eleven of these trials evaluated hospital mortality, which was decreased by 55% in patients receiving NPPV. Twelve trials assessed need for intubation, which decreased by 65%. In these trials, BiPAP was the most commonly used modality (see Table 3, for a comparison of NPPV modalities). Study patients had an average pH of 7.31 with an average PaCO2 of 68 mmHg. It was noted that the beneficial effects of NPPV increased as pH decreased. An earlier meta-analysis from Keenan and colleagues supported this notion, noting that the subgroup of patients with pH <7.3 benefited most in terms of decreased rates of intubation, hospital LOS, and hospital mortality.7 In this 2003 study, patients with relatively mild exacerbations of COPD did not benefit from the addition of NPPV to usual care. Based on the amount of positive evidence, NPPV is recommended in patients experiencing severe exacerbations of COPD as evidenced by a pH <7.35 and relative hypercarbia.1,2,4,7
Cardiogenic pulmonary edema. In patients with acute cardiogenic pulmonary edema, NPPV has been found to be beneficial, decreasing mortality, rates of intubation, and hospital LOS. Physiologically, NPPV augments cardiac output, improves respiratory mechanics, and decreases afterload.10 Cardiogenic edema is variably defined and has a number of causes elucidated in an analysis of 11 RCTs conducted by Masip and colleagues. These causes included acute coronary syndrome (31%), hypertension (27%), congestive heart failure (14%), and a combination of respiratory infection, arrhythmia, volume overload, and treatment noncompliance (28%).9 In this analysis, CPAP and BiPAP demonstrated a combined 43% reduction in mortality and a 57% reduction in intubation. More recently, Peter and colleagues described a statistically significant reduction in hospital mortality and the need for intubation with CPAP, while BiPAP only demonstrated a statistically significant decrease in need for intubation.10 Thus, there appears to be some evidence that CPAP is the preferred NPPV mode in patients with acute cardiogenic pulmonary edema. Despite inclusion of a recent, large RCT showing no benefit of NPPV versus usual care in cardiogenic pulmonary edema, the overall positive effect of NPPV persisted, particularly when the cause of pulmonary edema was acute coronary syndrome.11
Weaning after intubation. NPPV has been evaluated as a method to facilitate early extubation, as a measure to prevent extubation failure, and as a treatment modality for respiratory failure following extubation, with mixed results.12,13 In 1998, a small trial compared the use of NPPV in COPD patients to facilitate early extubation with a standard weaning protocol. In this population, early NPPV resulted in better weaning rates with shorter times of mechanical ventilation (10 vs. 16 days), fewer days in the ICU, and improved 60-day survival rates (92% vs. 72%).3,14 In another RCT not limited to COPD patients, Grault and colleagues found that NPPV reduced the duration of intubation (4.5 vs. 7.6 days) but was not associated with benefits in ICU length of stay or survival previously described.3,15 Thus, though NPPV may be beneficial in facilitating early extubation in COPD patients, it is not recommended in other patient populations.4
NPPV has also been evaluated as a measure to prevent respiratory failure in patients at high risk for extubation failure. When applied immediately after extubation in patients with COPD and obesity, NPPV reduced reintubation rates and ICU mortality.3,4 In 2004, Esteban and colleagues examined NPPV in patients who had respiratory failure following extubation. In this setting, NPPV was ineffective at preventing reintubation and had no survival benefit.
In summary, NPPV may facilitate early extubation and prevent extubation failure in appropriate patients, such as those with COPD, but is unlikely to be beneficial and is not recommended in patients with existing respiratory failure after extubation.4,15
Immunosuppressed patients. A 2001 single-center, randomized-controlled trial by Holbert and colleagues demonstrated decreased intubation rates and mortality with the application of NPPV in immunosuppressed patients with hypoxemic respiratory failure, fever, and pulmonary infiltrates.16
In this study, immunosuppression occurred most commonly as a result of malignancy. In the group receiving NPPV alternating with oxygen (at least 45 minutes of NPPV alternated every three hours with periods of spontaneous breathing), the rate of subsequent intubation decreased to 46%, compared with 77% in those receiving oxygen alone. The mortality rate was 38% in the NPPV group, as compared with 69% in the standard treatment group.
Though the outcomes in immunocompromised patients with hypoxemia, fever, and pulmonary infiltrates were very poor (38% mortality even with NPPV), this small study and recent guidelines suggest a trial of NPPV in this population.4,16
Other indications. NPPV has been applied in multiple other clinical scenarios, including exacerbation of asthma, community-acquired pneumonia, acute lung injury, and bronchoscopy in hypoxemic patients. It has also been evaluated in the postsurgical period and in chest trauma. There are mixed and less robust data in these various applications, and larger controlled trials are lacking.
In asthma exacerbation, NPPV may improve dyspnea, but data regarding outcomes (intubation, mortality) are lacking. A 2005 Cochrane review concluded that data remain controversial due to insufficient evidence, and guidelines make no recommendations concerning NPPV in asthma exacerbation.4,17 Similarly, in community-acquired pneumonia without prior history of COPD, there is no major role for NPPV.1,3,4 Limited data suggest that NPPV lacks efficacy in preventing post-surgical respiratory failure, though it may be useful in treating existing respiratory failure or preventing intubation in patients following lung resection or abdominal surgery.1,4 In hypoxemic patients undergoing bronchoscopy, NPPV may improve oxygenation (lower respiratory rates and improved PaO2 to FiO2 ratios, compared with oxygen alone) as well as hemodynamics (minimizing the drop in mean arterial pressure). However, outcome data are lacking and the data set is small.4,18 In acute lung injury/acute respiratory distress syndrome, data are also limited, but NPPV appears to have a high failure rate and confers little benefit.1,4
Back to the Case
The patient was admitted to the hospital and placed on BiPAP for approximately 1.5 hours. The patient’s respiratory rate improved to 20/minute and he appeared increasingly comfortable and alert. A repeat ABG revealed improved hypercarbia and acidosis. He was continued on steroids and antibiotics and eventually was weaned from BiPAP and discharged home.
Bottom Line
NPPV is an effective method to decrease mortality, intubation rates, and duration of ICU stay in severe exacerbations of COPD, cardiogenic pulmonary edema, immunosuppressed patients with pulmonary infiltrates, and hypoxia, and as a bridge to extubation in COPD patients.
Dr. Kraynek is an internal medicine resident in the Department of Medicine at the University of Washington School of Medicine in Seattle. Dr. Best is assistant professor of medicine in the Division of General Internal Medicine at the University of Washington School of Medicine.
References
- Ambrosino N, Vagheggini G. Noninvasive positive pressure ventilation in the acute care setting: where are we? Euro Resp J. 2008;31:874-856.
- American Thoracic Society. International Consensus Conferences in Intensive Care Medicine: Noninvasive Positive Pressure Ventilation in Acute Respiratory Failure. Am J Respir Crit Care Med. 2001;163:283-291.
- Liesching T, Kwok H, Hill N. Acute applications of noninvasive positive pressure ventilation. Chest. 2003;124:699-713.
- Keenan S, Sinuff T, Burns K, et al. Clinical practice guidelines for the use of noninvasive positive pressure ventilation and noninvasive continuous positive airway pressure in the acute care setting. CMAJ. 2001;183:E195-E214.
- Lightowler J, Wedzicha J, Elliot M, Ram F. Noninvasive positive pressure ventilation to treat respiratory failure resulting from exacerbations of chronic obstructive pulmonary disease: Cochrane systematic review and meta-analysis. BMJ. 2003;326:185.
- Quon B, Gan W, Sin D. Contemporary management of acute exacerbations of COPD: a systematic review of the metaanalysis. Chest. 2008;133:756-766.
- Keenan S, Sinuff T, Cook D, Hill, N. Which patients with acute exacerbation of chronic obstructive pulmonary disease benefit from noninvasive positive pressure ventilation? Ann Intern Med. 2003;138:861-870.
- Scala R, Naldi M, Archinucci I, Conigilo G, Nava S. Noninvasive positive pressure ventilation in patients with acute exacerbations of COPD and varying levels of consciousness. Chest. 2005;128:1657-1666.
- Masip J, Roque M, Sanchez B, Fernandez R, Subirana M, Exposito J. Noninvasive ventilation in acute cardiogenic pulmonary edema. JAMA. 2005;294:3124-3130.
- Peter J, Moran J, Phillips-Hughes J, Graham P, Bersten A. Effect of non-invasive positive pressure ventilation (NIPPV) on mortality in patients with acute cardiogenic pulmonary oedema: a meta-anaylsis. Lancet. 2006;367:1155-1163.
- Weng C, Zhao Y, Liu Q, et al. Meta-analysis: noninvasive ventilation in acute cardiogenic pulmonary edema. Ann Intern Med. 2010;152:560-600.
- Keenan S, Sinuff T, Cook D, Hill N. Does noninvasive positive pressure ventilation improve outcome in acute hypoxemic respiratory failure? A systematic review. Crit Care Med. 2004;32:2516-2523.
- Esteban A, Frutos-Vivar F, Fergusun N, et al. Noninvasive positive pressure ventilation for respiratory failure after extubation. N Engl J Med. 2004;350:2452-2460.
- Nava S, Ambrosino N, Clinie E, et al. Noninvasive mechanical ventilation in the weaning of patients with respiratory failure due to chronic obstructive pulmonary disease: a randomized, controlled trial. Ann Intern Med. 1998;128:721-728.
- Grault C, Daudenthun I, Chevron V, et al. Noninvasive ventilation as a systematic extubation and weaning technique in acute on chronic respiratory failure: a prospective, randomized controlled study. Am J Respir Crit Care Med. 1999;160:86-92.
- Hilbert G, Gruson D, Vargas F, et al. Noninvasive ventilation in immunosuppressed patients with pulmonary infiltrates, fever, and acute respiratory failure. N Engl J Med. 2001;344:481-487.
- Ram FSF, Wellington SR, Rowe BH, Wedzicha JA. Non-invasive positive pressure ventilation for treatment of respiratory failure due to severe acute exacerbations of asthma. Cochrane Database of Systematic Reviews. 2005, Issue 3.
- Antonelli M, Conti G, Rocco M, et al. Noninvasive positive pressure ventilation vs. conventional oxygen supplementation in hypoxemic patients undergoing diagnostic bronchoscopy. Chest. 2002;121:1149-1154.
Diagnosis by Treatment
A 46‐year‐old Mexican woman with acquired immune deficiency syndrome (AIDS), admitted for 6 months of diarrhea and failure to thrive, developed acute shortness of breath following colonoscopy. She reported dyspnea in the recumbent position, associated with a nonproductive cough, which improved with elevation of the head of the bed. She denied chest pain, palpitations, lightheadedness, hemoptysis, abdominal pain, nausea, and fever.
The approach to acute shortness of breath in hospitalized patients should include evaluation for life‐threatening cardiopulmonary processes. The patient should be assessed for cardiopulmonary process, including myocardial infarction, pulmonary embolism, aortic dissection, congestive heart failure, unstable arrhythmias, cardiac tamponade, and pneumothorax. The presence of orthopnea does suggest pulmonary congestion and a cardiac process. Given the timing of her symptoms, there is also concern for complications related to the colonoscopy, including aspiration pneumonitis, bronchospasm due to ethylene glycol, and methemoglobinemia from benzocaine used during the procedure.
The patient had been admitted the prior day for 6 months of diarrhea, weight loss, and failure to thrive. On admission, she was afebrile with a blood pressure of 110/50 mmHg and a pulse of 110 beats per minute; electrocardiogram (EKG) at the time revealed normal sinus rhythm. Her oxygen saturation was 100% on ambient air, and she had no complaints of cough, fevers, or dyspnea.
On admission, a peripherally inserted central catheter (PICC) was placed and total parenteral nutrition (TPN) was initiated. A gastroenterology consult was obtained, and colonoscopy was recommended to evaluate the cause of her chronic diarrhea. Overnight, the patient was started on polyethylene glycol electrolyte solution, with nothing else by mouth, and initiation of maintenance intravenous normal saline at 50 ml/hr in addition to her TPN. The patient expressed difficulty completing the colonoscopy preparation, but her preparation was acceptable to proceed with the procedure. She denied fever, chills, abdominal pain, and respiratory symptoms. She was taken down to endoscopy where she underwent conscious sedation, followed by an uneventful colonoscopy with mucosal biopsies. She subsequently was transported back to her hospital room in the supine position and almost immediately began to complain of mild shortness of breath. No aspiration event was witnessed following her procedure and transport.
Considering the patient's chronic diarrhea, there may be a unifying cause of both the gastrointestinal (GI) and pulmonary symptoms. Possibilities include infectious causes (Toxoplasma gondii and Trypanosoma cruzi), infiltrative diseases (amyloidosis), and metabolic processes (hyperthyroidism). More specifically, T. cruzi can cause dilated cardiomyopathy, with subsequent congestive heart failure and associated pulmonary symptoms; furthermore, it can lead to a dilated colon with abnormal bowel movements. Opportunistic infections, including Microsporidia, Cryptosporidium, Mycobacterium avium complex (MAC), and cytomegalovirus (CMV) should be considered. MAC and CMV can present with non‐bloody diarrhea and evolve into respiratory illnesses. Lastly, human immunodeficiency virus (HIV) is known to involve multiple organ systems, including the heart and gastrointestinal tract. History of prior cardiac or pulmonary disease, CD4 count and viral load, use of antiretroviral and prophylactic medications, and recent travel should be obtained.
Thirteen years previously, the patient was diagnosed with HIV, and subsequently developed AIDS with thrush and uncomplicated CMV viremia. At that time, highly active antiretroviral therapy (HAART)was initiated, but she was intolerant of her medications and received therapy intermittently. Her past medical history included multiple fractures secondary to osteoporosis. She denied any history of respiratory or cardiac symptoms. The patient was born in rural Mexico and immigrated to the United States 20 years prior. Her last visit to Mexico occurred 2 months prior to admission, and 4 months following the development of chronic diarrhea. Previously, she worked as a housekeeper and was not aware of any toxic exposures during cleaning. She denied a history of alcohol or recreational drug use. Despite her generalized weakness, her baseline functional status included performing all activities of daily living without symptoms.
Over the prior 6 months, the patient had developed diffuse watery diarrhea, associated with a 20‐pound weight loss. Stool evaluation, 1 week prior, was negative for Clostridium difficile, Microsporidia, Isospora, Cryptosporidium, Escherichia coli, Campylobacter, and ova and parasites. Her CD4 count was 8 cells per cubic millimeter.
The low CD4 count predisposes the patient to all opportunistic infections. Considering the history of CMV viremia, there is likelihood of reactivation with viremia and colitis, leading to chronic diarrhea and pneumonia. Disseminated MAC infection is also a consideration and would account for wasting, diarrhea, and dyspnea. However, it is important to note that the acute onset of dyspnea is atypical for CMV and MAC infections.
On physical exam, she was a thin woman with temporal wasting, in mild respiratory distress. Her temperature was 37.3C, blood pressure 133/55 mmHg, heart rate 140 beats/min, respiratory rate 22 breaths/min, and oxygen saturation 89% on room air. Her oropharynx was clear and without acral cyanosis. Use of accessory muscles for breathing was noted. The trachea was midline and no lymphadenopathy or thyromegaly were present. Her jugular venous pulse was normal. Cardiac exam revealed tachycardia with a new S4 gallop. A prominent apical impulse was noted. No murmurs or rubs were appreciated. There was no pulsus paradoxus. Her radial, femoral, and dorsalis pedis pulses were 2+ without delay. Her lung exam revealed inspiratory crackles involving the lower one‐third of both lungs. The lower extremities revealed 2+ pitting edema to the knees. The rest of her exam, including her neurologic evaluation, was unremarkable.
These clinical findings are consistent with left‐sided heart failure, concerning for ischemic injury or structural disorders of the heart. It is possible that the patient has had progressive heart failure, which is now unmasked by the volume received with TPN and endoscopy. If the heart failure has been longstanding, one has to consider potential non‐ischemic causes of cardiomyopathy, including infectious etiologies such as HIV, Epstein‐Barr virus (EBV), coxsackie virus, CMV, Toxoplasma gondii, and Trypanosoma cruzi; alcohol‐associated; and pericardial disease with Mycobacterium tuberculosis (MTB). Toxoplasma should be evaluated if the patient has exposure to cats. Considering her country of origin and travel history, risk factors for trypanosomiasis and MTB should be assessed.
At this point, the patient's respiratory failure should be aggressively addressed. Supplemental oxygen should be administered. She should be evaluated for acute coronary syndrome with an EKG and serial cardiac enzymes. A chest x‐ray should be obtained to grossly evaluate for pulmonary, pericardial, and aortic illnesses. Brain natriuretic peptide (BNP) levels should also be sent. Considering the evidence of volume overload and her HIV status, liver function tests, serum electrolytes, and urinalysis should be sent to exclude liver and renal involvement.
The patient was placed on 2 liters of oxygen by nasal cannula with resolution of her symptoms and improvement in her oxygen saturation to 95%. An EKG demonstrated sinus tachycardia, without evidence of ischemia. Metabolic panel revealed sodium 134 mmol/L; potassium 4.3 mmol/L; chloride 105 mmol/L; bicarbonate 16 mmol/L; creatinine 0.6 mg/dL, and liver function tests were within normal limits. Her troponin level was within the normal range for a negative value, and BNP was 823 pg/ml (normal <100). The complete blood count demonstrated leukopenia and anemia (hemoglobin 9.8 g/dL), which were unchanged from admission. Urinalysis was negative. A portable chest x‐ray demonstrated vascular congestion and mild pulmonary edema, without evidence of pneumothorax or pleural effusion.
The significantly elevated BNP and pulmonary vascular congestion seen on chest x‐ray confirm the clinical diagnosis of heart failure. However, the negative troponin and unremarkable EKG suggest a non‐ischemic cause for her symptoms. An echocardiogram should be obtained with specific emphasis on the presence of valve regurgitation, pericardial effusion, and ventricular/atrial thickening consistent with infiltrative disorders. Thyroid stimulating hormone (TSH) and serologies for infectious agents, including, T. cruzi, HIV, CMV, and Toxoplasmosis gondii, should also be sent. The patient should receive intravenous loop diuretics to improve her cardiac dynamics and pulmonary edema.
Intravenous furosemide was administered. Her symptoms improved and oxygen saturation on room air was 92%. An echocardiogram revealed global hypokinesis with left ventricular ejection fraction (LVEF) of 35% to 40%. There was no evidence of an underlying valvular or infiltrative process. TSH was normal. T. cruzi antibodies were sent.
The echocardiogram did not reveal an underlying structural heart abnormality. Infiltrative cardiomyopathies do not typically demonstrate global hypokinesis on echocardiogram, particularly without evidence of ventricular wall thickening or increased echogenicity, that can be seen in amyloid and sarcoid cardiomyopathies. Therefore, infiltrative cardiomyopathy is unlikely to be a cause of this patient's heart failure. The rapid improvement of her symptoms with furosemide decreases the likelihood of infectious causes for her acute decompensation. In reviewing the patient's history, she had developed severe chronic diarrhea associated with poor oral intake and a 20‐pound weight loss prior to hospitalization. These symptoms, along with a history of osteoporosis at an early age without traditional risk factors, indicate a state of severe malnutrition, placing her at risk for thiamine deficiency. Checking the thiamine level would be appropriate.
Considering the patient's long history of malnutrition and negative infectious and ischemic evaluation, she was empirically treated for wet beriberi with thiamine supplementation through her TPN. A serum thiamine B1 was obtained prior to supplementation. A vitamin D 25OH level was also sent, which was 15 ng/mL (normal >30 ng/mL), further suggesting malnutrition.
The patient continued to improve and furosemide was discontinued. Her initial serum thiamine level was 49 nmol/L (reference range: 70‐180 nmol/L). A repeat echocardiogram 5 days later revealed resolution of her systolic dysfunction and regional wall motion abnormality. The LVEF improved to 60%. Her colonoscopy biopsies revealed evidence of HIV enteropathy and CMV inclusion bodies. Her CMV viral load was 1223 genomes/mL. The T. cruzi antibodies were negative. She was restarted on HAART and ganciclovir. She continued to have diarrhea and was discharged home with TPN. Her serum thiamine level at discharge was 123 nmol/L.
Heart failure due to thiamine deficiency, or wet beriberi, was diagnosed considering the rapid clinical improvement in cardiac function after initiating thiamine therapy. While HIV cardiomyopathy could have contributed to heart failure in this patient, it is unlikely to improve so significantly over such a brief period of time.
DISCUSSION
Beriberi is a disease caused by severe thiamine deficiency. In fact, thiamine, also known as vitamin B1, was first named the anti‐beriberi factor in 1926. However, the earliest descriptions of beriberi can be found in Chinese medical texts dating back to 2697 BC.1 Beriberi is most commonly seen in Asia, where the diet is high in polished rice and the thiamine‐containing rice germs and husks have been removed. In the United States, thiamine‐enriched bread has virtually abolished the disease, except in severely malnourished populations such as alcoholics, those on fad diets, and patients with chronic diarrhea. Beriberi may also occur in patients with altered intestinal absorption such as post‐bariatric surgery patients.2 In 1985, the first case of beriberi as a complication of TPN without vitamin supplementation was reported.3 Subsequent cases of Wernicke's encephalopathy and beriberi have been noted in patients with gastrointestinal diseases and malabsorption on chronic TPN. More recently, thiamine deficiency has also been recognized in patients on long‐term diuretic therapy, as diuretics increase urinary excretion of this water‐soluble vitamin.4, 5 Since there is limited tissue storage of thiamine and its biologic half‐life is 10 to 20 days, high‐risk patients can develop thiamine deficiency within 4 weeks of initiation of diuretic therapy.6
Beriberi is classically divided into 2 types: wet, characterized by congestive heart failure, and dry, manifested as a symmetric peripheral neuropathy with both sensory and motor impairments.7 These 2 types of beriberi can coexist in the same patient; however, it is unclear why both types occur in some patients and not in others. Wet beriberi, also known as beriberi cardiomyopathy, typically presents as high‐output heart failure secondary to vasodilation, with a compensatory increase in blood volume and tachycardia.8 This state eventually leads to myocardial injury with systolic dysfunction and development of a low‐output state.8 Patients experience hypotension, lactic acidosis, and eventually fulminant vascular collapse. Although minor EKG changes such as sinus tachycardia, low‐voltage ventricular complexes, QT prolongation, and biphasic or inverted T waves are not uncommon in beriberi cardiomyopathy, major EKG changes, such as ST segment elevations and tall or deeply inverted T waves, are rare. Similarly, troponin elevation in beriberi cardiomyopathy is uncommon, but has been described.6
The pathogenesis of heart failure in beriberi is multifactorial. Thiamine is required for glucose to enter the Krebs cycle for aerobic metabolism, serving as a catalyst in the conversion of pyruvate to acetyl‐CoA. Without thiamine, anaerobic metabolism occurs, leading to the development of lactic acidosis and cellular malfunction. In fact, severe metabolic acidosis with serum pH values as low as 6.70 have been reported in cases of fulminant beriberi (although it is unclear if the lactic acidosis is mostly from anaerobic metabolism or from the low‐output state ultimately caused by thiamine deficiency).3
Laboratory diagnosis of thiamine deficiency, based on measurements of thiamine stores and metabolites, is often fraught with error and therefore unreliable. Serum pyruvate and lactate levels are commonly measured, and while elevated levels may be sensitive for thiamine deficiency, they are nonspecific. Measurement of whole blood thiamine is easy and the test is widely available; however, a low blood thiamine concentration is not always a sensitive indicator of deficiency since less than 1% of total body thiamine is found in whole blood.9 Additionally, this value may also be artificially elevated by thiamine intake immediately preceding the measurement. Urinary thiamine excretion has been proposed as a more accurate measurement, but this laboratory test is also problematic since urinary thiamine excretion reflects dietary intake more than total body stores.9 Erythrocyte transketolase activity (ETKA) is a functional enzyme test in which transketolase uses thiamine pyrophosphate as a catalyzer. This may be a more reliable measurement since red blood cells are among the first cells to be affected by thiamine depletion.9 Although a low ETKA level often indicates thiamine deficiency, this test is influenced by the hemoglobin concentration, and it is not widely available. Thus, the diagnosis of wet beriberi is usually made on the basis of rapid response to thiamine replacement.
Similar to the patient discussed, the clinical improvement in wet beriberi occurs within hours of treatment. There is an initial elevation in blood pressure and resolution of acidosis, followed by decrease in heart rate and normalization of cardiac output. Overall cardiac function improves within 24 to 48 hours after treatment, and return to a normal hemodynamic condition often occurs within 2 weeks of the start of treatment.10
There are no well‐established guidelines for the treatment of patients with beriberi, but general recommendations are an initial loading dose of intravenous thiamine 100 to 500 mg followed by 25 to 100 mg orally for 7 to 14 days.1 Thereafter, the daily thiamine requirement can be calculated based upon total caloric intake. The current recommendations in the United States are 0.5 mg of thiamine per 1000 kcal.1 However, one must consider whether a patient has impaired intestinal absorption or increased urinary losses when determining an appropriate maintenance dose.
Chronic malnutrition can lead to significant morbidity and mortality. Prior to admission, this patient already exhibited signs of severe malnutrition with a history of multiple pathologic fractures and diagnosis of osteoporosis. Considering her age and lack of risk factors for bone disease, osteoporosis suggests vitamin D deficiency. In this patient with chronic diarrhea caused by CMV, it is unlikely that a selective absorptive deficiency would occur. When the common causes of acute heart failure following volume challenge were excluded, the diagnosis of thiamine deficiency became more likely. Fortunately, an empiric trial of intravenous thiamine resulted in diagnosis by treatment and improvement of her cardiac function.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
KEY TEACHING POINTS
-
Hospitalists should consider vitamin B1 deficiency in patients with chronic illness and malnutrition.
-
Diagnosis of wet beriberi based on laboratory values can be challenging and, therefore, high clinical suspicion should prompt immediate treatment with thiamine.
-
Congestive heart failure due to thiamine deficiency can be reversed with thiamine replacement.
- ,,, et al.Thiamin. Modern Nutrition in Health and Disease.9th ed.1999:381–389.
- ,,, et al.One‐year outcomes of Roux‐en‐Y gastric bypass for morbidly obese adolescents: a multicenter study from the Pediatric Bariatric Study Group.J Pediatr Surg.2006;41:137–143.
- ,,.Severe acute metabolic acidosis (acute beriberi): an avoidable complication of total parenteral nutrition.J Parenter Enteral Nutr.1985;9(2):216–219.
- ,,, et al.Urinary thiamine excretion in the rat: effects of furosemide, other diuretics, and volume load.J Lab Clin Med.1999;134:232–237.
- ,,, et al.Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers.J Lab ClinMed.1999;134:238–243.
- .Increased troponin I in “wet” beriberi.J Clin Pathol.2006;59(5):555.
- ,,, et al.Shoshin syndrome: two case reports representing opposite ends of the same disease spectrum.Acta Cardiol.1998;53:195.
- ,.The challenge of cardiomyopathy.J Am Coll Cardiol.1989;13:1219–1239.
- .Loop diuretic therapy, thiamine balance, and heart failure.Congest Heart Fail.2007;13(4):244–247.
- ,,.Cardiovascular beriberi.Am J Cardiol.1972;30:418–422.
A 46‐year‐old Mexican woman with acquired immune deficiency syndrome (AIDS), admitted for 6 months of diarrhea and failure to thrive, developed acute shortness of breath following colonoscopy. She reported dyspnea in the recumbent position, associated with a nonproductive cough, which improved with elevation of the head of the bed. She denied chest pain, palpitations, lightheadedness, hemoptysis, abdominal pain, nausea, and fever.
The approach to acute shortness of breath in hospitalized patients should include evaluation for life‐threatening cardiopulmonary processes. The patient should be assessed for cardiopulmonary process, including myocardial infarction, pulmonary embolism, aortic dissection, congestive heart failure, unstable arrhythmias, cardiac tamponade, and pneumothorax. The presence of orthopnea does suggest pulmonary congestion and a cardiac process. Given the timing of her symptoms, there is also concern for complications related to the colonoscopy, including aspiration pneumonitis, bronchospasm due to ethylene glycol, and methemoglobinemia from benzocaine used during the procedure.
The patient had been admitted the prior day for 6 months of diarrhea, weight loss, and failure to thrive. On admission, she was afebrile with a blood pressure of 110/50 mmHg and a pulse of 110 beats per minute; electrocardiogram (EKG) at the time revealed normal sinus rhythm. Her oxygen saturation was 100% on ambient air, and she had no complaints of cough, fevers, or dyspnea.
On admission, a peripherally inserted central catheter (PICC) was placed and total parenteral nutrition (TPN) was initiated. A gastroenterology consult was obtained, and colonoscopy was recommended to evaluate the cause of her chronic diarrhea. Overnight, the patient was started on polyethylene glycol electrolyte solution, with nothing else by mouth, and initiation of maintenance intravenous normal saline at 50 ml/hr in addition to her TPN. The patient expressed difficulty completing the colonoscopy preparation, but her preparation was acceptable to proceed with the procedure. She denied fever, chills, abdominal pain, and respiratory symptoms. She was taken down to endoscopy where she underwent conscious sedation, followed by an uneventful colonoscopy with mucosal biopsies. She subsequently was transported back to her hospital room in the supine position and almost immediately began to complain of mild shortness of breath. No aspiration event was witnessed following her procedure and transport.
Considering the patient's chronic diarrhea, there may be a unifying cause of both the gastrointestinal (GI) and pulmonary symptoms. Possibilities include infectious causes (Toxoplasma gondii and Trypanosoma cruzi), infiltrative diseases (amyloidosis), and metabolic processes (hyperthyroidism). More specifically, T. cruzi can cause dilated cardiomyopathy, with subsequent congestive heart failure and associated pulmonary symptoms; furthermore, it can lead to a dilated colon with abnormal bowel movements. Opportunistic infections, including Microsporidia, Cryptosporidium, Mycobacterium avium complex (MAC), and cytomegalovirus (CMV) should be considered. MAC and CMV can present with non‐bloody diarrhea and evolve into respiratory illnesses. Lastly, human immunodeficiency virus (HIV) is known to involve multiple organ systems, including the heart and gastrointestinal tract. History of prior cardiac or pulmonary disease, CD4 count and viral load, use of antiretroviral and prophylactic medications, and recent travel should be obtained.
Thirteen years previously, the patient was diagnosed with HIV, and subsequently developed AIDS with thrush and uncomplicated CMV viremia. At that time, highly active antiretroviral therapy (HAART)was initiated, but she was intolerant of her medications and received therapy intermittently. Her past medical history included multiple fractures secondary to osteoporosis. She denied any history of respiratory or cardiac symptoms. The patient was born in rural Mexico and immigrated to the United States 20 years prior. Her last visit to Mexico occurred 2 months prior to admission, and 4 months following the development of chronic diarrhea. Previously, she worked as a housekeeper and was not aware of any toxic exposures during cleaning. She denied a history of alcohol or recreational drug use. Despite her generalized weakness, her baseline functional status included performing all activities of daily living without symptoms.
Over the prior 6 months, the patient had developed diffuse watery diarrhea, associated with a 20‐pound weight loss. Stool evaluation, 1 week prior, was negative for Clostridium difficile, Microsporidia, Isospora, Cryptosporidium, Escherichia coli, Campylobacter, and ova and parasites. Her CD4 count was 8 cells per cubic millimeter.
The low CD4 count predisposes the patient to all opportunistic infections. Considering the history of CMV viremia, there is likelihood of reactivation with viremia and colitis, leading to chronic diarrhea and pneumonia. Disseminated MAC infection is also a consideration and would account for wasting, diarrhea, and dyspnea. However, it is important to note that the acute onset of dyspnea is atypical for CMV and MAC infections.
On physical exam, she was a thin woman with temporal wasting, in mild respiratory distress. Her temperature was 37.3C, blood pressure 133/55 mmHg, heart rate 140 beats/min, respiratory rate 22 breaths/min, and oxygen saturation 89% on room air. Her oropharynx was clear and without acral cyanosis. Use of accessory muscles for breathing was noted. The trachea was midline and no lymphadenopathy or thyromegaly were present. Her jugular venous pulse was normal. Cardiac exam revealed tachycardia with a new S4 gallop. A prominent apical impulse was noted. No murmurs or rubs were appreciated. There was no pulsus paradoxus. Her radial, femoral, and dorsalis pedis pulses were 2+ without delay. Her lung exam revealed inspiratory crackles involving the lower one‐third of both lungs. The lower extremities revealed 2+ pitting edema to the knees. The rest of her exam, including her neurologic evaluation, was unremarkable.
These clinical findings are consistent with left‐sided heart failure, concerning for ischemic injury or structural disorders of the heart. It is possible that the patient has had progressive heart failure, which is now unmasked by the volume received with TPN and endoscopy. If the heart failure has been longstanding, one has to consider potential non‐ischemic causes of cardiomyopathy, including infectious etiologies such as HIV, Epstein‐Barr virus (EBV), coxsackie virus, CMV, Toxoplasma gondii, and Trypanosoma cruzi; alcohol‐associated; and pericardial disease with Mycobacterium tuberculosis (MTB). Toxoplasma should be evaluated if the patient has exposure to cats. Considering her country of origin and travel history, risk factors for trypanosomiasis and MTB should be assessed.
At this point, the patient's respiratory failure should be aggressively addressed. Supplemental oxygen should be administered. She should be evaluated for acute coronary syndrome with an EKG and serial cardiac enzymes. A chest x‐ray should be obtained to grossly evaluate for pulmonary, pericardial, and aortic illnesses. Brain natriuretic peptide (BNP) levels should also be sent. Considering the evidence of volume overload and her HIV status, liver function tests, serum electrolytes, and urinalysis should be sent to exclude liver and renal involvement.
The patient was placed on 2 liters of oxygen by nasal cannula with resolution of her symptoms and improvement in her oxygen saturation to 95%. An EKG demonstrated sinus tachycardia, without evidence of ischemia. Metabolic panel revealed sodium 134 mmol/L; potassium 4.3 mmol/L; chloride 105 mmol/L; bicarbonate 16 mmol/L; creatinine 0.6 mg/dL, and liver function tests were within normal limits. Her troponin level was within the normal range for a negative value, and BNP was 823 pg/ml (normal <100). The complete blood count demonstrated leukopenia and anemia (hemoglobin 9.8 g/dL), which were unchanged from admission. Urinalysis was negative. A portable chest x‐ray demonstrated vascular congestion and mild pulmonary edema, without evidence of pneumothorax or pleural effusion.
The significantly elevated BNP and pulmonary vascular congestion seen on chest x‐ray confirm the clinical diagnosis of heart failure. However, the negative troponin and unremarkable EKG suggest a non‐ischemic cause for her symptoms. An echocardiogram should be obtained with specific emphasis on the presence of valve regurgitation, pericardial effusion, and ventricular/atrial thickening consistent with infiltrative disorders. Thyroid stimulating hormone (TSH) and serologies for infectious agents, including, T. cruzi, HIV, CMV, and Toxoplasmosis gondii, should also be sent. The patient should receive intravenous loop diuretics to improve her cardiac dynamics and pulmonary edema.
Intravenous furosemide was administered. Her symptoms improved and oxygen saturation on room air was 92%. An echocardiogram revealed global hypokinesis with left ventricular ejection fraction (LVEF) of 35% to 40%. There was no evidence of an underlying valvular or infiltrative process. TSH was normal. T. cruzi antibodies were sent.
The echocardiogram did not reveal an underlying structural heart abnormality. Infiltrative cardiomyopathies do not typically demonstrate global hypokinesis on echocardiogram, particularly without evidence of ventricular wall thickening or increased echogenicity, that can be seen in amyloid and sarcoid cardiomyopathies. Therefore, infiltrative cardiomyopathy is unlikely to be a cause of this patient's heart failure. The rapid improvement of her symptoms with furosemide decreases the likelihood of infectious causes for her acute decompensation. In reviewing the patient's history, she had developed severe chronic diarrhea associated with poor oral intake and a 20‐pound weight loss prior to hospitalization. These symptoms, along with a history of osteoporosis at an early age without traditional risk factors, indicate a state of severe malnutrition, placing her at risk for thiamine deficiency. Checking the thiamine level would be appropriate.
Considering the patient's long history of malnutrition and negative infectious and ischemic evaluation, she was empirically treated for wet beriberi with thiamine supplementation through her TPN. A serum thiamine B1 was obtained prior to supplementation. A vitamin D 25OH level was also sent, which was 15 ng/mL (normal >30 ng/mL), further suggesting malnutrition.
The patient continued to improve and furosemide was discontinued. Her initial serum thiamine level was 49 nmol/L (reference range: 70‐180 nmol/L). A repeat echocardiogram 5 days later revealed resolution of her systolic dysfunction and regional wall motion abnormality. The LVEF improved to 60%. Her colonoscopy biopsies revealed evidence of HIV enteropathy and CMV inclusion bodies. Her CMV viral load was 1223 genomes/mL. The T. cruzi antibodies were negative. She was restarted on HAART and ganciclovir. She continued to have diarrhea and was discharged home with TPN. Her serum thiamine level at discharge was 123 nmol/L.
Heart failure due to thiamine deficiency, or wet beriberi, was diagnosed considering the rapid clinical improvement in cardiac function after initiating thiamine therapy. While HIV cardiomyopathy could have contributed to heart failure in this patient, it is unlikely to improve so significantly over such a brief period of time.
DISCUSSION
Beriberi is a disease caused by severe thiamine deficiency. In fact, thiamine, also known as vitamin B1, was first named the anti‐beriberi factor in 1926. However, the earliest descriptions of beriberi can be found in Chinese medical texts dating back to 2697 BC.1 Beriberi is most commonly seen in Asia, where the diet is high in polished rice and the thiamine‐containing rice germs and husks have been removed. In the United States, thiamine‐enriched bread has virtually abolished the disease, except in severely malnourished populations such as alcoholics, those on fad diets, and patients with chronic diarrhea. Beriberi may also occur in patients with altered intestinal absorption such as post‐bariatric surgery patients.2 In 1985, the first case of beriberi as a complication of TPN without vitamin supplementation was reported.3 Subsequent cases of Wernicke's encephalopathy and beriberi have been noted in patients with gastrointestinal diseases and malabsorption on chronic TPN. More recently, thiamine deficiency has also been recognized in patients on long‐term diuretic therapy, as diuretics increase urinary excretion of this water‐soluble vitamin.4, 5 Since there is limited tissue storage of thiamine and its biologic half‐life is 10 to 20 days, high‐risk patients can develop thiamine deficiency within 4 weeks of initiation of diuretic therapy.6
Beriberi is classically divided into 2 types: wet, characterized by congestive heart failure, and dry, manifested as a symmetric peripheral neuropathy with both sensory and motor impairments.7 These 2 types of beriberi can coexist in the same patient; however, it is unclear why both types occur in some patients and not in others. Wet beriberi, also known as beriberi cardiomyopathy, typically presents as high‐output heart failure secondary to vasodilation, with a compensatory increase in blood volume and tachycardia.8 This state eventually leads to myocardial injury with systolic dysfunction and development of a low‐output state.8 Patients experience hypotension, lactic acidosis, and eventually fulminant vascular collapse. Although minor EKG changes such as sinus tachycardia, low‐voltage ventricular complexes, QT prolongation, and biphasic or inverted T waves are not uncommon in beriberi cardiomyopathy, major EKG changes, such as ST segment elevations and tall or deeply inverted T waves, are rare. Similarly, troponin elevation in beriberi cardiomyopathy is uncommon, but has been described.6
The pathogenesis of heart failure in beriberi is multifactorial. Thiamine is required for glucose to enter the Krebs cycle for aerobic metabolism, serving as a catalyst in the conversion of pyruvate to acetyl‐CoA. Without thiamine, anaerobic metabolism occurs, leading to the development of lactic acidosis and cellular malfunction. In fact, severe metabolic acidosis with serum pH values as low as 6.70 have been reported in cases of fulminant beriberi (although it is unclear if the lactic acidosis is mostly from anaerobic metabolism or from the low‐output state ultimately caused by thiamine deficiency).3
Laboratory diagnosis of thiamine deficiency, based on measurements of thiamine stores and metabolites, is often fraught with error and therefore unreliable. Serum pyruvate and lactate levels are commonly measured, and while elevated levels may be sensitive for thiamine deficiency, they are nonspecific. Measurement of whole blood thiamine is easy and the test is widely available; however, a low blood thiamine concentration is not always a sensitive indicator of deficiency since less than 1% of total body thiamine is found in whole blood.9 Additionally, this value may also be artificially elevated by thiamine intake immediately preceding the measurement. Urinary thiamine excretion has been proposed as a more accurate measurement, but this laboratory test is also problematic since urinary thiamine excretion reflects dietary intake more than total body stores.9 Erythrocyte transketolase activity (ETKA) is a functional enzyme test in which transketolase uses thiamine pyrophosphate as a catalyzer. This may be a more reliable measurement since red blood cells are among the first cells to be affected by thiamine depletion.9 Although a low ETKA level often indicates thiamine deficiency, this test is influenced by the hemoglobin concentration, and it is not widely available. Thus, the diagnosis of wet beriberi is usually made on the basis of rapid response to thiamine replacement.
Similar to the patient discussed, the clinical improvement in wet beriberi occurs within hours of treatment. There is an initial elevation in blood pressure and resolution of acidosis, followed by decrease in heart rate and normalization of cardiac output. Overall cardiac function improves within 24 to 48 hours after treatment, and return to a normal hemodynamic condition often occurs within 2 weeks of the start of treatment.10
There are no well‐established guidelines for the treatment of patients with beriberi, but general recommendations are an initial loading dose of intravenous thiamine 100 to 500 mg followed by 25 to 100 mg orally for 7 to 14 days.1 Thereafter, the daily thiamine requirement can be calculated based upon total caloric intake. The current recommendations in the United States are 0.5 mg of thiamine per 1000 kcal.1 However, one must consider whether a patient has impaired intestinal absorption or increased urinary losses when determining an appropriate maintenance dose.
Chronic malnutrition can lead to significant morbidity and mortality. Prior to admission, this patient already exhibited signs of severe malnutrition with a history of multiple pathologic fractures and diagnosis of osteoporosis. Considering her age and lack of risk factors for bone disease, osteoporosis suggests vitamin D deficiency. In this patient with chronic diarrhea caused by CMV, it is unlikely that a selective absorptive deficiency would occur. When the common causes of acute heart failure following volume challenge were excluded, the diagnosis of thiamine deficiency became more likely. Fortunately, an empiric trial of intravenous thiamine resulted in diagnosis by treatment and improvement of her cardiac function.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
KEY TEACHING POINTS
-
Hospitalists should consider vitamin B1 deficiency in patients with chronic illness and malnutrition.
-
Diagnosis of wet beriberi based on laboratory values can be challenging and, therefore, high clinical suspicion should prompt immediate treatment with thiamine.
-
Congestive heart failure due to thiamine deficiency can be reversed with thiamine replacement.
A 46‐year‐old Mexican woman with acquired immune deficiency syndrome (AIDS), admitted for 6 months of diarrhea and failure to thrive, developed acute shortness of breath following colonoscopy. She reported dyspnea in the recumbent position, associated with a nonproductive cough, which improved with elevation of the head of the bed. She denied chest pain, palpitations, lightheadedness, hemoptysis, abdominal pain, nausea, and fever.
The approach to acute shortness of breath in hospitalized patients should include evaluation for life‐threatening cardiopulmonary processes. The patient should be assessed for cardiopulmonary process, including myocardial infarction, pulmonary embolism, aortic dissection, congestive heart failure, unstable arrhythmias, cardiac tamponade, and pneumothorax. The presence of orthopnea does suggest pulmonary congestion and a cardiac process. Given the timing of her symptoms, there is also concern for complications related to the colonoscopy, including aspiration pneumonitis, bronchospasm due to ethylene glycol, and methemoglobinemia from benzocaine used during the procedure.
The patient had been admitted the prior day for 6 months of diarrhea, weight loss, and failure to thrive. On admission, she was afebrile with a blood pressure of 110/50 mmHg and a pulse of 110 beats per minute; electrocardiogram (EKG) at the time revealed normal sinus rhythm. Her oxygen saturation was 100% on ambient air, and she had no complaints of cough, fevers, or dyspnea.
On admission, a peripherally inserted central catheter (PICC) was placed and total parenteral nutrition (TPN) was initiated. A gastroenterology consult was obtained, and colonoscopy was recommended to evaluate the cause of her chronic diarrhea. Overnight, the patient was started on polyethylene glycol electrolyte solution, with nothing else by mouth, and initiation of maintenance intravenous normal saline at 50 ml/hr in addition to her TPN. The patient expressed difficulty completing the colonoscopy preparation, but her preparation was acceptable to proceed with the procedure. She denied fever, chills, abdominal pain, and respiratory symptoms. She was taken down to endoscopy where she underwent conscious sedation, followed by an uneventful colonoscopy with mucosal biopsies. She subsequently was transported back to her hospital room in the supine position and almost immediately began to complain of mild shortness of breath. No aspiration event was witnessed following her procedure and transport.
Considering the patient's chronic diarrhea, there may be a unifying cause of both the gastrointestinal (GI) and pulmonary symptoms. Possibilities include infectious causes (Toxoplasma gondii and Trypanosoma cruzi), infiltrative diseases (amyloidosis), and metabolic processes (hyperthyroidism). More specifically, T. cruzi can cause dilated cardiomyopathy, with subsequent congestive heart failure and associated pulmonary symptoms; furthermore, it can lead to a dilated colon with abnormal bowel movements. Opportunistic infections, including Microsporidia, Cryptosporidium, Mycobacterium avium complex (MAC), and cytomegalovirus (CMV) should be considered. MAC and CMV can present with non‐bloody diarrhea and evolve into respiratory illnesses. Lastly, human immunodeficiency virus (HIV) is known to involve multiple organ systems, including the heart and gastrointestinal tract. History of prior cardiac or pulmonary disease, CD4 count and viral load, use of antiretroviral and prophylactic medications, and recent travel should be obtained.
Thirteen years previously, the patient was diagnosed with HIV, and subsequently developed AIDS with thrush and uncomplicated CMV viremia. At that time, highly active antiretroviral therapy (HAART)was initiated, but she was intolerant of her medications and received therapy intermittently. Her past medical history included multiple fractures secondary to osteoporosis. She denied any history of respiratory or cardiac symptoms. The patient was born in rural Mexico and immigrated to the United States 20 years prior. Her last visit to Mexico occurred 2 months prior to admission, and 4 months following the development of chronic diarrhea. Previously, she worked as a housekeeper and was not aware of any toxic exposures during cleaning. She denied a history of alcohol or recreational drug use. Despite her generalized weakness, her baseline functional status included performing all activities of daily living without symptoms.
Over the prior 6 months, the patient had developed diffuse watery diarrhea, associated with a 20‐pound weight loss. Stool evaluation, 1 week prior, was negative for Clostridium difficile, Microsporidia, Isospora, Cryptosporidium, Escherichia coli, Campylobacter, and ova and parasites. Her CD4 count was 8 cells per cubic millimeter.
The low CD4 count predisposes the patient to all opportunistic infections. Considering the history of CMV viremia, there is likelihood of reactivation with viremia and colitis, leading to chronic diarrhea and pneumonia. Disseminated MAC infection is also a consideration and would account for wasting, diarrhea, and dyspnea. However, it is important to note that the acute onset of dyspnea is atypical for CMV and MAC infections.
On physical exam, she was a thin woman with temporal wasting, in mild respiratory distress. Her temperature was 37.3C, blood pressure 133/55 mmHg, heart rate 140 beats/min, respiratory rate 22 breaths/min, and oxygen saturation 89% on room air. Her oropharynx was clear and without acral cyanosis. Use of accessory muscles for breathing was noted. The trachea was midline and no lymphadenopathy or thyromegaly were present. Her jugular venous pulse was normal. Cardiac exam revealed tachycardia with a new S4 gallop. A prominent apical impulse was noted. No murmurs or rubs were appreciated. There was no pulsus paradoxus. Her radial, femoral, and dorsalis pedis pulses were 2+ without delay. Her lung exam revealed inspiratory crackles involving the lower one‐third of both lungs. The lower extremities revealed 2+ pitting edema to the knees. The rest of her exam, including her neurologic evaluation, was unremarkable.
These clinical findings are consistent with left‐sided heart failure, concerning for ischemic injury or structural disorders of the heart. It is possible that the patient has had progressive heart failure, which is now unmasked by the volume received with TPN and endoscopy. If the heart failure has been longstanding, one has to consider potential non‐ischemic causes of cardiomyopathy, including infectious etiologies such as HIV, Epstein‐Barr virus (EBV), coxsackie virus, CMV, Toxoplasma gondii, and Trypanosoma cruzi; alcohol‐associated; and pericardial disease with Mycobacterium tuberculosis (MTB). Toxoplasma should be evaluated if the patient has exposure to cats. Considering her country of origin and travel history, risk factors for trypanosomiasis and MTB should be assessed.
At this point, the patient's respiratory failure should be aggressively addressed. Supplemental oxygen should be administered. She should be evaluated for acute coronary syndrome with an EKG and serial cardiac enzymes. A chest x‐ray should be obtained to grossly evaluate for pulmonary, pericardial, and aortic illnesses. Brain natriuretic peptide (BNP) levels should also be sent. Considering the evidence of volume overload and her HIV status, liver function tests, serum electrolytes, and urinalysis should be sent to exclude liver and renal involvement.
The patient was placed on 2 liters of oxygen by nasal cannula with resolution of her symptoms and improvement in her oxygen saturation to 95%. An EKG demonstrated sinus tachycardia, without evidence of ischemia. Metabolic panel revealed sodium 134 mmol/L; potassium 4.3 mmol/L; chloride 105 mmol/L; bicarbonate 16 mmol/L; creatinine 0.6 mg/dL, and liver function tests were within normal limits. Her troponin level was within the normal range for a negative value, and BNP was 823 pg/ml (normal <100). The complete blood count demonstrated leukopenia and anemia (hemoglobin 9.8 g/dL), which were unchanged from admission. Urinalysis was negative. A portable chest x‐ray demonstrated vascular congestion and mild pulmonary edema, without evidence of pneumothorax or pleural effusion.
The significantly elevated BNP and pulmonary vascular congestion seen on chest x‐ray confirm the clinical diagnosis of heart failure. However, the negative troponin and unremarkable EKG suggest a non‐ischemic cause for her symptoms. An echocardiogram should be obtained with specific emphasis on the presence of valve regurgitation, pericardial effusion, and ventricular/atrial thickening consistent with infiltrative disorders. Thyroid stimulating hormone (TSH) and serologies for infectious agents, including, T. cruzi, HIV, CMV, and Toxoplasmosis gondii, should also be sent. The patient should receive intravenous loop diuretics to improve her cardiac dynamics and pulmonary edema.
Intravenous furosemide was administered. Her symptoms improved and oxygen saturation on room air was 92%. An echocardiogram revealed global hypokinesis with left ventricular ejection fraction (LVEF) of 35% to 40%. There was no evidence of an underlying valvular or infiltrative process. TSH was normal. T. cruzi antibodies were sent.
The echocardiogram did not reveal an underlying structural heart abnormality. Infiltrative cardiomyopathies do not typically demonstrate global hypokinesis on echocardiogram, particularly without evidence of ventricular wall thickening or increased echogenicity, that can be seen in amyloid and sarcoid cardiomyopathies. Therefore, infiltrative cardiomyopathy is unlikely to be a cause of this patient's heart failure. The rapid improvement of her symptoms with furosemide decreases the likelihood of infectious causes for her acute decompensation. In reviewing the patient's history, she had developed severe chronic diarrhea associated with poor oral intake and a 20‐pound weight loss prior to hospitalization. These symptoms, along with a history of osteoporosis at an early age without traditional risk factors, indicate a state of severe malnutrition, placing her at risk for thiamine deficiency. Checking the thiamine level would be appropriate.
Considering the patient's long history of malnutrition and negative infectious and ischemic evaluation, she was empirically treated for wet beriberi with thiamine supplementation through her TPN. A serum thiamine B1 was obtained prior to supplementation. A vitamin D 25OH level was also sent, which was 15 ng/mL (normal >30 ng/mL), further suggesting malnutrition.
The patient continued to improve and furosemide was discontinued. Her initial serum thiamine level was 49 nmol/L (reference range: 70‐180 nmol/L). A repeat echocardiogram 5 days later revealed resolution of her systolic dysfunction and regional wall motion abnormality. The LVEF improved to 60%. Her colonoscopy biopsies revealed evidence of HIV enteropathy and CMV inclusion bodies. Her CMV viral load was 1223 genomes/mL. The T. cruzi antibodies were negative. She was restarted on HAART and ganciclovir. She continued to have diarrhea and was discharged home with TPN. Her serum thiamine level at discharge was 123 nmol/L.
Heart failure due to thiamine deficiency, or wet beriberi, was diagnosed considering the rapid clinical improvement in cardiac function after initiating thiamine therapy. While HIV cardiomyopathy could have contributed to heart failure in this patient, it is unlikely to improve so significantly over such a brief period of time.
DISCUSSION
Beriberi is a disease caused by severe thiamine deficiency. In fact, thiamine, also known as vitamin B1, was first named the anti‐beriberi factor in 1926. However, the earliest descriptions of beriberi can be found in Chinese medical texts dating back to 2697 BC.1 Beriberi is most commonly seen in Asia, where the diet is high in polished rice and the thiamine‐containing rice germs and husks have been removed. In the United States, thiamine‐enriched bread has virtually abolished the disease, except in severely malnourished populations such as alcoholics, those on fad diets, and patients with chronic diarrhea. Beriberi may also occur in patients with altered intestinal absorption such as post‐bariatric surgery patients.2 In 1985, the first case of beriberi as a complication of TPN without vitamin supplementation was reported.3 Subsequent cases of Wernicke's encephalopathy and beriberi have been noted in patients with gastrointestinal diseases and malabsorption on chronic TPN. More recently, thiamine deficiency has also been recognized in patients on long‐term diuretic therapy, as diuretics increase urinary excretion of this water‐soluble vitamin.4, 5 Since there is limited tissue storage of thiamine and its biologic half‐life is 10 to 20 days, high‐risk patients can develop thiamine deficiency within 4 weeks of initiation of diuretic therapy.6
Beriberi is classically divided into 2 types: wet, characterized by congestive heart failure, and dry, manifested as a symmetric peripheral neuropathy with both sensory and motor impairments.7 These 2 types of beriberi can coexist in the same patient; however, it is unclear why both types occur in some patients and not in others. Wet beriberi, also known as beriberi cardiomyopathy, typically presents as high‐output heart failure secondary to vasodilation, with a compensatory increase in blood volume and tachycardia.8 This state eventually leads to myocardial injury with systolic dysfunction and development of a low‐output state.8 Patients experience hypotension, lactic acidosis, and eventually fulminant vascular collapse. Although minor EKG changes such as sinus tachycardia, low‐voltage ventricular complexes, QT prolongation, and biphasic or inverted T waves are not uncommon in beriberi cardiomyopathy, major EKG changes, such as ST segment elevations and tall or deeply inverted T waves, are rare. Similarly, troponin elevation in beriberi cardiomyopathy is uncommon, but has been described.6
The pathogenesis of heart failure in beriberi is multifactorial. Thiamine is required for glucose to enter the Krebs cycle for aerobic metabolism, serving as a catalyst in the conversion of pyruvate to acetyl‐CoA. Without thiamine, anaerobic metabolism occurs, leading to the development of lactic acidosis and cellular malfunction. In fact, severe metabolic acidosis with serum pH values as low as 6.70 have been reported in cases of fulminant beriberi (although it is unclear if the lactic acidosis is mostly from anaerobic metabolism or from the low‐output state ultimately caused by thiamine deficiency).3
Laboratory diagnosis of thiamine deficiency, based on measurements of thiamine stores and metabolites, is often fraught with error and therefore unreliable. Serum pyruvate and lactate levels are commonly measured, and while elevated levels may be sensitive for thiamine deficiency, they are nonspecific. Measurement of whole blood thiamine is easy and the test is widely available; however, a low blood thiamine concentration is not always a sensitive indicator of deficiency since less than 1% of total body thiamine is found in whole blood.9 Additionally, this value may also be artificially elevated by thiamine intake immediately preceding the measurement. Urinary thiamine excretion has been proposed as a more accurate measurement, but this laboratory test is also problematic since urinary thiamine excretion reflects dietary intake more than total body stores.9 Erythrocyte transketolase activity (ETKA) is a functional enzyme test in which transketolase uses thiamine pyrophosphate as a catalyzer. This may be a more reliable measurement since red blood cells are among the first cells to be affected by thiamine depletion.9 Although a low ETKA level often indicates thiamine deficiency, this test is influenced by the hemoglobin concentration, and it is not widely available. Thus, the diagnosis of wet beriberi is usually made on the basis of rapid response to thiamine replacement.
Similar to the patient discussed, the clinical improvement in wet beriberi occurs within hours of treatment. There is an initial elevation in blood pressure and resolution of acidosis, followed by decrease in heart rate and normalization of cardiac output. Overall cardiac function improves within 24 to 48 hours after treatment, and return to a normal hemodynamic condition often occurs within 2 weeks of the start of treatment.10
There are no well‐established guidelines for the treatment of patients with beriberi, but general recommendations are an initial loading dose of intravenous thiamine 100 to 500 mg followed by 25 to 100 mg orally for 7 to 14 days.1 Thereafter, the daily thiamine requirement can be calculated based upon total caloric intake. The current recommendations in the United States are 0.5 mg of thiamine per 1000 kcal.1 However, one must consider whether a patient has impaired intestinal absorption or increased urinary losses when determining an appropriate maintenance dose.
Chronic malnutrition can lead to significant morbidity and mortality. Prior to admission, this patient already exhibited signs of severe malnutrition with a history of multiple pathologic fractures and diagnosis of osteoporosis. Considering her age and lack of risk factors for bone disease, osteoporosis suggests vitamin D deficiency. In this patient with chronic diarrhea caused by CMV, it is unlikely that a selective absorptive deficiency would occur. When the common causes of acute heart failure following volume challenge were excluded, the diagnosis of thiamine deficiency became more likely. Fortunately, an empiric trial of intravenous thiamine resulted in diagnosis by treatment and improvement of her cardiac function.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
KEY TEACHING POINTS
-
Hospitalists should consider vitamin B1 deficiency in patients with chronic illness and malnutrition.
-
Diagnosis of wet beriberi based on laboratory values can be challenging and, therefore, high clinical suspicion should prompt immediate treatment with thiamine.
-
Congestive heart failure due to thiamine deficiency can be reversed with thiamine replacement.
- ,,, et al.Thiamin. Modern Nutrition in Health and Disease.9th ed.1999:381–389.
- ,,, et al.One‐year outcomes of Roux‐en‐Y gastric bypass for morbidly obese adolescents: a multicenter study from the Pediatric Bariatric Study Group.J Pediatr Surg.2006;41:137–143.
- ,,.Severe acute metabolic acidosis (acute beriberi): an avoidable complication of total parenteral nutrition.J Parenter Enteral Nutr.1985;9(2):216–219.
- ,,, et al.Urinary thiamine excretion in the rat: effects of furosemide, other diuretics, and volume load.J Lab Clin Med.1999;134:232–237.
- ,,, et al.Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers.J Lab ClinMed.1999;134:238–243.
- .Increased troponin I in “wet” beriberi.J Clin Pathol.2006;59(5):555.
- ,,, et al.Shoshin syndrome: two case reports representing opposite ends of the same disease spectrum.Acta Cardiol.1998;53:195.
- ,.The challenge of cardiomyopathy.J Am Coll Cardiol.1989;13:1219–1239.
- .Loop diuretic therapy, thiamine balance, and heart failure.Congest Heart Fail.2007;13(4):244–247.
- ,,.Cardiovascular beriberi.Am J Cardiol.1972;30:418–422.
- ,,, et al.Thiamin. Modern Nutrition in Health and Disease.9th ed.1999:381–389.
- ,,, et al.One‐year outcomes of Roux‐en‐Y gastric bypass for morbidly obese adolescents: a multicenter study from the Pediatric Bariatric Study Group.J Pediatr Surg.2006;41:137–143.
- ,,.Severe acute metabolic acidosis (acute beriberi): an avoidable complication of total parenteral nutrition.J Parenter Enteral Nutr.1985;9(2):216–219.
- ,,, et al.Urinary thiamine excretion in the rat: effects of furosemide, other diuretics, and volume load.J Lab Clin Med.1999;134:232–237.
- ,,, et al.Urinary loss of thiamine is increased by low doses of furosemide in healthy volunteers.J Lab ClinMed.1999;134:238–243.
- .Increased troponin I in “wet” beriberi.J Clin Pathol.2006;59(5):555.
- ,,, et al.Shoshin syndrome: two case reports representing opposite ends of the same disease spectrum.Acta Cardiol.1998;53:195.
- ,.The challenge of cardiomyopathy.J Am Coll Cardiol.1989;13:1219–1239.
- .Loop diuretic therapy, thiamine balance, and heart failure.Congest Heart Fail.2007;13(4):244–247.
- ,,.Cardiovascular beriberi.Am J Cardiol.1972;30:418–422.
What Is the Best Approach for the Evaluation and Management of Endocrine Incidentalomas?
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
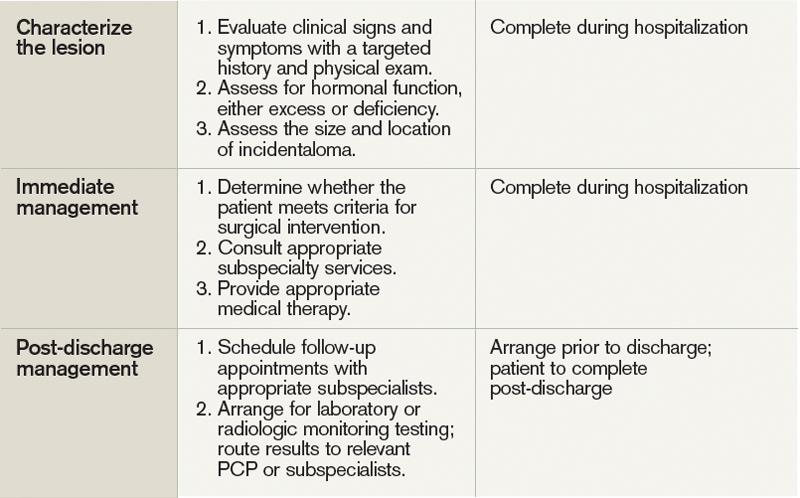
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
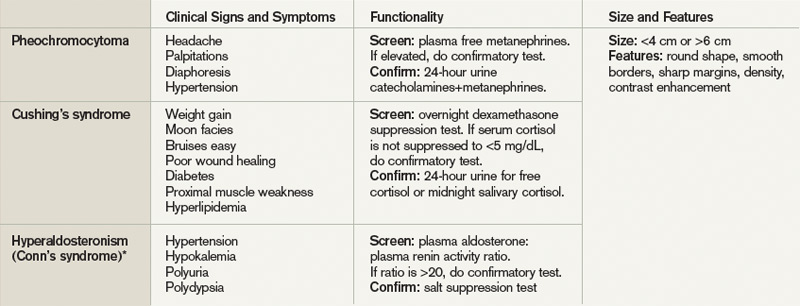
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Case
A 54-year-old man with a history of hypertension treated with hydrocholorothiazide and Type 2 diabetes mellitus is admitted with abdominal pain and found to have an incidental 2.1-cm left adrenal mass on CT scan of the abdomen. He denies symptoms of headache, palpitations, weight gain, or muscle weakness. His exam is significant for mildly elevated blood pressure. What is the best approach for evaluation and management of this incidental finding?
Overview
Incidentalomas are mass lesions that are inadvertently discovered during radiolographic diagnostic testing or treatment for other clinical conditions that are unrelated to the incidental mass. In recent decades, improvements in radiographic diagnostic techniques and sensitivity have led to increasing discovery of incidental lesions that are often in the absence of clinical signs or symptoms.1 Three commonly discovered lesions by hospitalists are pituitary, thyroid, and adrenal incidentalomas.2 The concerns associated with these findings relate to the potential for dysfunctional hormone secretion or malignancy.
Patients found with pituitary incidentalomas can be susceptible to several types of adverse outcomes: hormonal hypersecretion, hypopituitarism, neurologic morbidity due to tumor size, and malignancy in rare cases. Thyroid incidentalomas are impalpable nodules discovered in the setting of ultrasound or cross-sectional neck scans, such as positron emission tomography (PET) scans. Discovery of a thyroid incidentaloma raises concern for thyroid malignancy.3 The increased use of abdominal ultrasound, CT scans, and MRI has fueled the growing incidence of adrenal incidentalomas (AIs).
The discovery of an endocrine incidentaloma in the inpatient setting warrants a systematic approach that includes both diagnostic and potentially therapeutic management. A hospitalist should consider an approach that includes (see Table 1):
- Characterization of the incidentaloma, including clinical signs and symptoms, size, hormonal function, and malignant potential;
- Immediate management, including medical versus surgical treatment; and
- Post-discharge management, including monitoring.
Review of the Data
Pituitary incidentalomas. The prevalence of pituitary incidentalomas found by CT ranges from 3.7% to 20%, while the prevalence found by MRI approximates 10%. Autopsy studies have revealed a prevalence ranging from 1.5% to 26.7% for adenomas less than 10 mm, considered to be microadenomas. Broad categories of etiologies should be considered: pituitary adenoma, nonpituitary tumors, vascular lesions, infiltrative disorders, and others (see Table 2). The majority of pituitary adenomas secrete prolactin (30% to 40%) or are nonsecreting (30% to 40%). Adenomas secreting adrenocorticotropin hormone (ACTH, 2% to 10%), growth hormone (GH, 2% to 10%), thyroid-stimulating hormone (TSH, <1%), follicle-stimulating hormone (FSH), and luteinizing hormone (LH) are much less common.2 Significant morbidity and premature mortality are associated with hyperprolactinemia, acromegaly (growth hormone excess), Cushing’s syndrome, and hyperthyroidism. Additionally, up to 41% of patients with macroadenomas were found to have varying degrees of hypopituitarism due to compression of the hypothalamus, the hypothalamic-pituitary stalk, or the pituitary itself.4
Recently, the Endocrine Society released consensus recommendations to guide the evaluation and treatment of pituitary incidentalomas, which are included in the approach outlined below.5 A detailed history and physical examination should be obtained with specific inquiry as to signs and symptoms of hormonal excess and mass effect from the tumor. Examples of symptoms of hormone excess can include:
- Prolactin: menstrual irregularity, anovulation, infertility, decreased libido, impotence, osteoporosis;
- Growth hormone: high frequency of colonic polyps and colon cancer (chronic excess);
- TSH: thyrotoxicosis, atrial fibrillation; and
- ACTH: hypertension, osteoporosis, accelerated vascular disease.
Symptoms related to the mass effect of the tumor include visual field defects and hypopituitarism related to the deficient hormone, including:
- FSH/LH: oligomenorrhea, decreased libido, infertility;
- TSH: hypothyroidism (weight gain, constipation, cold intolerance);
- ACTH: adrenal insufficiency (hypotension, hypoglycemia, weight loss); and
- ADH: polyuria, polydypsia.
The size and location of the pituitary lesion must be assessed. Lesions greater than 10 mm are considered macroademonas, and their size will affect their management. If the lesion was initially identified by CT scan, an MRI is recommended to better evaluate it.5 If the MRI locates the incidentaloma abutting the optic nerve or chiasm, then the patient should undergo a formal visual field examination.
Indications for an inpatient surgical referral for treatment include: a lesion larger than 2 cm, evidence of mass effect such as visual field defects, neurologic compromise, opthalmoplegia, hypopituitarism, a tumor abutting the optic nerve or chiasm, pituitary apoplexy, and hypersecretion of hormones other than prolactin. Patients with prolactinomas warrant an inpatient endo-crinology consult and may need medical management with a dopamine agonist. Hormone replacement therapy can also be provided for patients with hypopituitarism.2,5
For patients who do not meet the criteria for inpatient surgical therapy, follow-up management must be arranged at the time of discharge. Clinical, laboratory assessment, and an MRI should be scheduled six months after the initial finding of the incidentaloma with the patient’s PCP or with an endocrinologist.5
Thyroid incidentalomas. The prevalence of thyroid nodules based on ultrasound studies ranges from 19% to 46%, with autopsy studies estimating an incidence of approximately 50%.2,6 Incidence of thyroid nodules also increases with age, as almost 60% of people over the age of 60 harbor a thyroid incidentaloma. The rate of malignancy in the general population has ranged between 8% and 24%; however, in the last decade, the rates have increased by 2.4 times as more sophisticated ultrasound techniques and liberal use of fine-needle aspiration (FNA) biopsies have detected subclinical disease.7,8
Etiologies for incidental thyroid nodules can be divided into benign and malignant causes. Benign etiologies include thyroid cyst (simple or complex), multinodular goiter, and Hashimoto’s thryoiditis, while malignant causes include papillary, medullary, follicular, Hurthle cell, and anaplastic carcinomas, thyroid lymphomas, and rare instances of metastatic cancers.2,3
Targeted history and physical examination helps to characterize the thyroid incidentaloma. Historical features, such as palpitations, weight loss, anxiety, new onset atrial fibrillation, or menstrual irregularities, coupled with tachycardia, tremors, proximal muscle weakness, and a palpable nodule aid in the diagnosis of hyperthyroidism. Findings such as a family history of thyroid cancer, symptoms of hoarseness or dysphagia, rapid growth of the nodule, environmental or history of head or neck irradiation along with physical findings of a hard, fixed nodule, or cervical lymphadenopathy increase the suspicion for malignancy.2,7
The functionality of the nodule can be assessed by checking TSH, free T3, and free T4 levels. Suppression of TSH (< 0.1 mU/L) with elevated levels of free T3 and T4 indicates nodule production of excess thyroid hormone and warrants thyroid scintography. Thyroid scintography will identify the nodule as “hot” (hyperfunctioning) or “cold” (nonfunctioning).2
Regardless of the radiographic modality that initially identified the thyroid incidentaloma, a dedicated thyroid high-resolution ultrasound should be ordered to assess the size, multiplicity (single or multinodular), location, and character (solid, cystic, or mixed).7
Recommendations for proceeding to FNA to evaluate for malignancy differ among subspecialty societies. Generally, nodules larger than 1 cm or nodules smaller than 1 cm with risk factors for malignancy should be referred for FNA.2,7
If diagnostic workup identifies a patient with hyperthyroidism due to an autonomously functional nodule or a nodule that may be at high risk for malignancy, it is appropriate to involve an endocrinologist and possibly a surgical subspecialist prior to discharge. Management of hyperthyroidism can include starting antithyroid agents (methimazole or propylthiouracil), radioactive iodine ablation, or referral for surgery.
Preparation for discharge of the patient whose incidentaloma is nonfunctional or does not appear to be malignant should include appointments to recheck thyroid hormone levels, including TSH as well as a thyroid ultrasound within one year of the initial discovery.
Adrenal incidentaloma. The prevalence of AIs found by CT of the abdomen ranges from 0.4% to 4%, while autopsy studies have found a prevalence of 1.4% to 9% with increasing prevalence with age.2,9,10 The majority of AIs are benign and nonfunctioning adenomas, in the absence of known malignancy. Other differential diagnoses include Cushing’s syndrome, pheochromocytoma, adrenocortical adenoma, aldosteronoma, and metastatic lesions.
Because functioning adrenal incidentalomas may be clinically silent, any patient found with an AI must undergo biochemical workup as part of their evaluation to assess for pheochromocytoma, Cushing’s syndrome, and if he or she has a history of hypertension or hyperaldosteronism (Conn’s syndrome). Table 3 outlines the approach for characterizing adrenal incidentalomas.2,11,12 An important point is that imaging studies are not useful in distinguishing a functioning versus nonfunctioning tumor but rather can help to discriminate malignant lesions.11
Inpatient surgical consult for resection is indicated if the patient is found to have pheochromocytoma, clinically apparent functioning adrenocortical adenoma, or a tumor size greater than 4 cm. Consultation with an endocrinologist is also recommended if biochemical tests are positive. If the diagnostic workup leads to suspicion for infection or metastatic disease, the patient should be referred for FNA.2,12
For patients whose lesions do not require surgical resection, repeat CT scan of the abdomen is recommended six months from the initial finding. Hospitalists should also arrange for the patient to repeat biochemical testing, including an overnight dexamethasone test.12,13
Back to the Case
The patient underwent biochemical testing and was found to have normal levels of plasma-free metanephrines, a plasma aldosterone, plasma renin activity ratio less than 20, and a serum cortisol level of 7 mg/dL after his overnight dexamethasone suppression test. The 24-hour urine collection for free cortisol revealed elevated levels of cortisol in the urine, and the ACTH level was low.
Endocrinology and endocrine surgery teams were consulted, and recommended surgical resection. After surgical resection of his tumor, the patient was started on glucocorticoid replacement and was discharged with a follow-up appointment with endocrinology.
Bottom Line
An inpatient approach to endocrine incidentalomas should include characterization of the clinical signs and symptoms, size, function, and malignant potential of the lesion. Based on this, inpatient surgical or medical management can be determined. Post-discharge management should include arrangements for surveillance testing and follow-up with appropriate subspecialists.
Dr. Tad-y is assistant professor of medicine and a hospitalist at the University of Colorado Denver.
References
- Aron DC, Howlett TA. Pituitary incidentalomas. Endocrinol Metab Clin North Am. 2000;29:205-221.
- Shirodkar M, Jabbour SA. Endocrine incidentalomas. Int J Clin Pract. 2008;62:1423-1431.
- Burguera B, Gharib H. Thyroid incidentalomas. Prevalence, diagnosis, significance, and management. Endocrinol Metab Clin North Am. 2000;29:187-203.
- Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinol Metab Clin North Am. 2008;37:151-171, xi.
- Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:894-904.
- Gough J, Scott-Coombes D, Fausto Palazzo F. Thyroid incidentaloma: an evidence-based assessment of management strategy. World J Surg. 2008;32:1264-1268.
- Iyer NG, Shaha AR, Silver CE, et al. Thyroid incidentalomas: to treat or not to treat. Eur Arch Otorhinolaryngol. 2010;267:1019-1026.
- Jin J, Wilhelm SM, McHenry CR. Incidental thyroid nodule: patterns of diagnosis and rate of malignancy. Am J Surg. 2009;197:320-324.
- Davenport C, Liew L, Doherty B, et al. The prevalence of adrenal incidentaloma in routine clinical practice. Endocrine. 2011;40:80-83.
- Zeiger MA, Siegelman SS, Hamrahian AH. Medical and surgical evaluation and treatment of adrenal incidentalomas. J Clin Endocrinol Metab. 2011;96: 2004-2015.
- Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450-453.
- NIH state-of-the-science statement on management of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19:1-25.
- Young WF. Clinical practice. The incidentally discovered adrenal mass. N Engl J Med. 2007;356:601-610.
- Chidiac RM, Aron DC. Incidentalomas. A disease of modern technology. Endocrinol Metab Clin North Am. 1997;26:233-253.
Cracking the Case
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
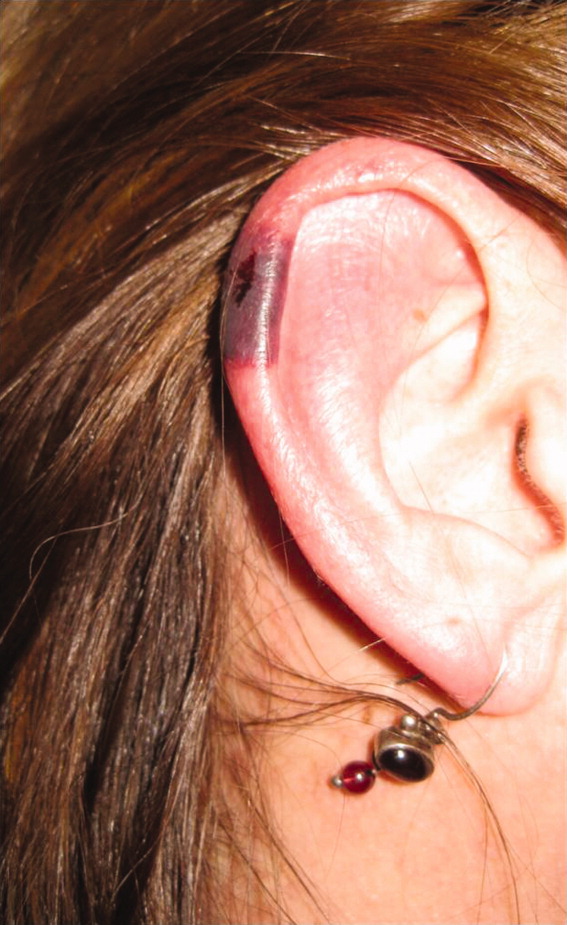
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
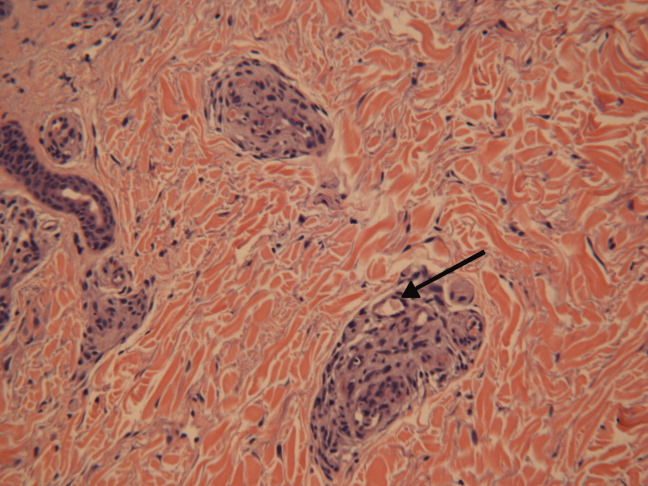
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 43‐year‐old woman presented to an outside hospital with painful plaques and patches on her bilateral lower extremities. Two weeks prior to presentation, she had noticed a single red lesion on her left ankle. Over the next two weeks, the lesion enlarged to involve the lower half of her posterior calf and subsequently turned purple and became exquisitely tender. Similar but smaller purple, tender lesions simultaneously appeared, first over her right shin and then on her bilateral thighs and hips. She also reported fatigue as well as diffuse joint pains in her hands and wrists bilaterally for the past month. She denied any swelling of these joints or functional impairment. She denied fevers, weight loss, headache, sinus symptoms, difficulty breathing, or abdominal pain.
Although we do not yet have a physical exam, the tempo, pattern of spread, and accompanying features allow some early hypotheses to be considered. Distal lower extremity lesions which darkened and spread could be erythema nodosum or erythema induratum. Malignancies rarely have such prominent skin manifestations, although leukemia cutis or an aggressive cutaneous T cell lymphoma might present with disseminated and darkened plaques, and Kaposi's sarcoma is characteristically purple and multifocal. Autoimmune disorders such as sarcoidosis, cutaneous lupus, and psoriasis may similarly present with widespread plaques. Most disseminated infections that start with patches evolve to pustules, ulcers, bullae, or other forms that reflect the invasive nature of the infection; syphilis warrants consideration for any widespread eruption of unknown etiology. Antecedent arthralgias with fatigue suggest an autoimmune condition, although infections such as hepatitis or parvovirus can do the same. Systemic lupus erythematosus (SLE) or rheumatoid arthritis (RA) would be favored initially on account of her demographics and the hand and wrist involvement, and each can be associated with vasculitis.
The significant pain as described is not compatible with most of the aforementioned diagnoses. Its presence, coupled with potential autoimmune symptoms, suggests a vasculitis such as polyarteritis nodosa (which can have prominent diffuse skin involvement), Henoch Schonlein purpura (with its predilection for the lower extremities, including extension to the hips and buttocks), cryoglobulinemia, or SLE‐ or RA‐associated vasculitis. Calciphylaxis is another ischemic vascular disorder that can cause diffuse dark painful lesions, but this only warrants consideration if advanced renal disease is present.
A skin biopsy ofher right hip was taken at the outside hospital. She was discharged on a two‐week course of prednisone for suspected vasculitis while biopsy results were pending. Over the next two weeks, none of the skin lesions improved, despite compliance with this treatment, and the skin over her left posterior calf and right shin lesions began to erode and bleed. In addition, small purple, tender lesions appeared over the pinnae of both ears. Three weeks after her initial evaluation, she presented to another emergency department for ulcerating skin lesions and worsening pain. At that point, the initial skin biopsy result was available and revealed vasculopathy of the small vessels with thrombi but no vasculitis.
The patient had no children,and denied a history of miscarriages. Her past medical history was unremarkable. She did not report any history of thrombotic events. She started a new job as a software engineer one month ago and was feeling stressed about her new responsibilities. She denied any high‐risk sexual behavior and any history of intravenous drug use. She had not traveled recently and did not own any pets. There was no family history of rheumatologic disorders, hypercoagulable states, or thrombotic events.
This picture of occluded but noninflamed vessels shifts the diagnosis away from vasculitis and focuses attention on hypercoagulable states with prominent dermal manifestations, including antiphospholipid antibody syndrome (APLS) and livedoid vasculopathy. In this young woman with arthralgias, consideration of SLE and APLS is warranted. Her recent increase in stress and widespread purpuric and ulcerative lesions could bring to mind a factitious disorder, but the histology results eliminate this possibility.
The patient's temperaturewas 36.5C, her blood pressure was 110/70 mmHg, respiratory rate was 16 breaths per minute, and her heart rate was 65 beats per minute. She was well‐appearing but in moderate pain. She did not have any oral lesions. Her cardiac, respiratory, and abdominal exams were normal. Skin exam revealed a 10‐cm by 4‐cm area of bloody granulation tissue draining serosanguinous fluid, surrounded by stellate palpable netlike purpura on her left posterior calf. There was a similar 4‐cm by 2‐cm ulcerated lesion on her right shin. Both lesions were exquisitely tender to palpation. On her bilateral thighs and hips, there were multiple stellate purpuric patches, all 4 cm in diameter or less, and only minimally tender to palpation. She also had 1‐cm purpuric bullae on the helices of both ears (Figure 1) which were slightly tender to palpation. Splinter hemorrhages were also noted on multiple nail beds bilaterally. Musculoskeletal exam did not reveal any synovitis.
The original purpura on her calf and ear demonstrate a clear demarcation corresponding to cutaneous vascular insufficiency. The development of bullae (ear) and ulceration (calf) are compatible with ischemia. Despite the presence of multiple splinter hemorrhages, the distribution of lesions is very unusual for an embolic phenomenon (eg, endocarditis, cholesterol emboli, or atrial myxoma). The multifocal nature of the skin lesions with progression to well‐demarcated cutaneous necrosis is reminiscent of calciphylaxis or warfarin‐induced skin necrosis, although she lacks the relevant risk factors. A toxin such as cocaine or methamphetamine mediating multifocal vasoconstriction or hypercoagulability should be excluded.
The bilateral ear involvement remains decidedly unusual and makes me wonder if there is something about the ear, such as the nature of its circulation or its potentially lower temperature (as an acral organ) that might render it particularly susceptible, for instance, to cryoglobulinemia or cryofibrinogenemia‐mediated ischemia.
Laboratory studiesdemonstrated: white blood cell count of 1500/mm3 (37.3% neutrophils, 5.1% lymphocytes, 6.7% monocytes, and 1.3% eosinophils); hemoglobin 9.3 g/dl (mean corpuscular volume 91 fL); platelet count 212/mm3; erythrocyte sedimentation rate 62 mm/hr; C‐reactive protein 14.6 mg/L. Serum electrolytes, liver tests, coagulation studies, and urinalysis were normal. Fecal occult blood test was negative.
Her neutropenia and anemia suggest decreased production in the marrow by infection, malignancy, or toxin, or increased destruction, perhaps from an autoimmune process. The associated infections are usually viral, such as human immunodeficiency virus (HIV) and Epstein‐Barr virus (EBV), although their linkage with her cutaneous disease is tenuous. It is possible that malignancy could be present in the marrow with resultant dermal hypercoagulability and ischemia, but this seems unlikely. We do not know about any toxins that she has been exposed to, but these hematologic findings would mandate directed inquiry along those lines. In this young woman with cutaneous ulcers secondary to thrombotic vasculopathy, bicytopenia, antecedent arthralgias without synovitis, and elevated inflammatory markers, I favor an autoimmune process such as SLE, which I would evaluate with an antinuclear antibody (ANA) and antiphospholipid antibody studies.
She was admittedto the hospital and received hydromorphone for pain control. Corticosteroids were not administered. Peripheral blood morphology was normal. Antibodies against HIV1 and 2 were negative, as were antibodies against cytomegalovirus, EBV, parvovirus B19, mycoplasma pneumoniae, and hepatitis C virus. Bilateral lower extremity ultrasound was negative for deep vein thrombosis. Transthoracic echocardiogram was normal. Repeat skin biopsy confirmed small vessel vasculopathy without vasculitis (Figure 2). The results of the following investigations were also negative: ANA, rheumatoid factor, double‐stranded DNA (dsDNA), cyclic citrullinated peptide, ribonucleoprotein (RNP), and anti‐Smith antibodies. C3 and C4 complement levels were normal.
Given how much the histology is driving the clinical reasoning and focusing the differential diagnosis in this case, I agree with the decision to repeat the biopsy. In complex or undiagnosed cases, repeat histology samples allow for confirmation of the original interpretation (often with the perspective of new clinicians and pathologists) and sometimes reveal pathognomonic or additional findings that only appear after the disease has evolved over time. HIV seronegativity helps constrain the differential diagnosis, and parvovirus is another excellent consideration for arthralgias and cytopenias (with the predilection to involve cells lines other than RBCs particularly seen in HIV), although ulcers are not seen with this condition. Herpes simplex virus (HSV) is another viral infection that can cause painful skin ulcerations and cytopenias, although the duration and distribution are highly atypical. The negative ANA and dsDNA and normal complement levels make SLE unlikely. The negative lower extremity ultrasound helps frame the thromboses as a local cutaneous process rather than a systemic hypercoagulable state. Although the peripheral blood smear is normal, a bone marrow biopsy will be necessary to exclude a marrow invasive process, such as leukemia or lymphoma. A bone marrow biopsy would also provide another opportunity to examine tissue for mycobacteria or fungi which can cause ulcerations and cytopenias, although there is little reason to suspect she is susceptible to those pathogens. As this clinical picture fails to fit clearly with an infectious, autoimmune, or neoplastic disorder, I would revisit the possibility of toxinsprescription, complementary, over‐the‐counter, or illegal (eg, cocaine) at this time.
In further discussionwith the patient, she reported using cocaine intranasally for the past three months. Her urine toxicology was positive for cocaine. She was found to have positive perinuclear antineutrophil cytoplasmic antibodies (p‐ANCA), antimyeloperoxidase (MPO) antibodies, anticardiolipin (ACL) antibodies, and lupus anticoagulant (LAC). By hospital day 3, her lesions had significantly improved without any intervention, and her absolute neutrophil count increased to 1080/mm3.
The presence of widespread cutaneous ischemia (with bland thrombosis) and detectable ACL and LAC antibodies is compatible with APLS; the APLS could be deemed primary, because there is no clear associated rheumatologic or other systemic disease. However, neutropenia is not a characteristic of APLS, which has thrombocytopenia as its more frequently associated hematologic abnormality. Livedoid vasculopathy, a related disorder, is also supported by the ACL and LAC results, but also does not feature neutropenia. While the presence of diffuse thrombosis could be attributed to a widespread secondary effect of cocaine vasoconstriction, the appearance of ANCA (which can be drug‐induced, eg, propylthiouracil [PTU]) and the slowly resolving neutropenia during hospitalization without specific treatment is very suggestive of a toxin. The demographic, diffuse skin ulcers, and hematologic and serologic profile is compatible with the recently described toxidrome related to levamisole adulteration of cocaine.
A send‐out studyof a urine sample returned positive for levamisole. Based on purpuric skin lesions with a predilection for the ears, agranulocytosis, and skin biopsy revealing thrombotic vasculopathy, she was diagnosed with levamisole‐adulterated cocaine exposure. One week after discharge, her lower extremity pain and ulcerations were significantly improved. Her absolute neutrophil count increased to 2820/mm.3 Her urine toxicology screen was negative for cocaine.
DISCUSSION
Levamisole was initially developed in 1964 as an antihelminthic agent. Its incidentally discovered immunomodulatory effects led to trials for the treatment of chronic infections, inflammatory bowel disease, rheumatic diseases,1 and nephrotic syndrome in children.2 By 1990, 3 major studies supported levamisole as an adjunctive therapy in melanoma3 and colon cancer.4
Although levamisole appeared to be nontoxic at single or low doses, long‐term use in clinical trials demonstrated that 2.5%‐13% of patients developed life‐threatening agranulocytosis, and up to 10% of those instances resulted in death.5 A distinctive cutaneous pseudovasculitis was noted in children on therapeutic levamisole. They presented with purpura that had a predilection for the ears, cheeks, and thighs,6 and positive serologic markers for ANCA and antiphospholipid antibodies. Skin biopsies of the purpuric lesions revealed leukocytoclastic vasculitis, thrombotic vasculitis, and/or vascular occlusions.
Levamisole was withdrawn from the market in 2000 in the United States due to its side effects,7 but quickly found its way onto the black market. It was first detected in cocaine in 2002, and the percentage of cocaine containing levamisole has steadily been increasing since then. In July 2009, over 70% of cocaine seized by the Drug Enforcement Administration was found to contain levamisole.8 It is unclear exactly why this drug is used as an adulterant in cocaine. Theories include potentiation of the euphoric effects of cocaine, serving as a bulking agent, or functioning as a chemical signature to track distribution.9
The resurgence of levamisole has brought a new face to a problem seen over a decade ago. Current reports of levamisole toxicity describe adults presenting with purpura preferentially involving the ears, neutropenia, positive ANCA, and positive antiphospholipid antibodies.1012 Since 2002, there have been at least 20 confirmed cases of agranulocytosis and two deaths associated with levamisole‐adulterated cocaine.8, 13, 14 In September 2009, the Department of Health and Human Services issued a public health alert warning of an impending increase in levamisole‐related illness.
Levamisole is not detected on routine toxicology screens, but can be tested for using gas chromatography and mass spectrometry. Most laboratories do not offer testing for levamisole and send‐out testing is required. Given its half‐life of 5.6 hours, levamisole can only be detected in the blood within 24 hours, and in the urine within 48‐72 hours of exposure.15, 16 Urine samples are preferred over blood samples, since blood levels decline more rapidly and have lower sensitivity. Cocaine can also be sent out to local or state forensics laboratories to be tested for levamisole. The only definitive treatment for levamisole‐induced cutaneous pseudovasculitis and neutropenia is cessation of toxin exposure.
Although the discussant had familiarity with this toxidrome from local and published cases, he was only able to settle on levamisole toxicity after a series of competing hypotheses were ruled out on the basis of irreconcilable features (vasculitis and histology results; APLS and neutropenia; SLE and negative ANA with no visceral involvement) and by using analogical reasoning (eg, to infer the presence of a toxin on the basis of neutropenia [as seen with chemotherapy and other drugs] and ANCA induction [as seen with PTU]). It was a laborious process of hypothesis testing, but one that ultimately allowed him to crack the case.
Key Teaching Points
-
In patients presenting with neutropenia and purpuric skin lesionsparticularly with a predilection for the earsconsider levamisole‐adulterated cocaine exposure.
-
Tests supporting this diagnosis include positive serologies for ANCA and antiphospholipid antibodies, and skin biopsies that show leukocytoclastic vasculitis, thrombotic vasculitis, or vascular occlusion. Urine studies for levamisole are definitive if sent within 48 to 72 hours of exposure.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
- ,.Levamisole, the story and the lessons.Int J Immunopharmocol.1992;14(3):481–486.
- British Association for Paediatric Nephrology.Levamisole for corticosteroid‐dependent nephrotic syndrome in childhood.Lancet.1991;337:1555–1557.
- ,,,,.Improved survival in patients with poor‐prognosis malignant melanoma treated with adjuvant levamisole: a phase III study by the National Cancer Institute of Canada Clinical Trials Group.J Clin Oncol.1991;9:729–735.
- ,,.Levamisole and fluorouracil for adjuvant therapy of resected colon carcinoma.N Engl J Med.1990;322:352–358.
- ,,.Studies on levamisole‐induced agranulocytosis.Blood.1980;56(3):388–396.
- ,,.Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long‐term treatment with levamisole in children.Br J Dermatol.1999;140:948–951.
- . Janssen Discontinues Ergamisol. Available at: http://findarticles.com/p/articles/mi_m3374/is_18_22/ai_68536218/. Accessed July 25,2010.
- SAMHSA. Nationwide Public Health Alert Issued Concerning Life‐Threatening Risk Posed by Cocaine Laced with Veterinary Anti‐Parasite Drug. Available at: http://www.samhsa.gov/newsroom/advisories/090921vet5101.aspx. Accessed July 20,2010.
- .Unusual adulterants in cocaine seized on Italian clandestine market.Forensic Sci Int.2007;172(2–3):e1.
- ,,.Levamisole‐induced occlusive necrotizing vasculitis of the ears after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Jun 12.
- ,,,.Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole.Ann Intern Med.2010;1;152(11):758–759.
- ,,,,.Cocaine‐associated retiform purpura and neutropenia: is levamisole the culprit?J Am Acad Dermatol.2010;Mar 19.
- ,,,.A confirmed case of agranulocytosis after use of cocaine contaminated with levamisole.J Med Toxicol.2010;Apr 1.
- Centers for Disease Control and Prevention.Agranulocytosis associated with cocaine use—four States, March 2008–November 2009.MMWR.2009;58(49):1381–1385.
- ,,.Levamisole as a contaminant of illicit cocaine.Journal of the Clandestine Laboratory Investigating Chemists Association.2006;16:6–11. Available at: http://www.tiaft2006.org/proceedings/pdf/PT‐p‐06.pdf. Accessed July 20, 2010.
- . Cocaine Cutting Agents—A Discussion. Laboratory Medicine and Pathology, University of Alberta. Available at: http://www.vandu.org/documents/Levamisole_Cocaine.pdf. Accessed July 20,2010.
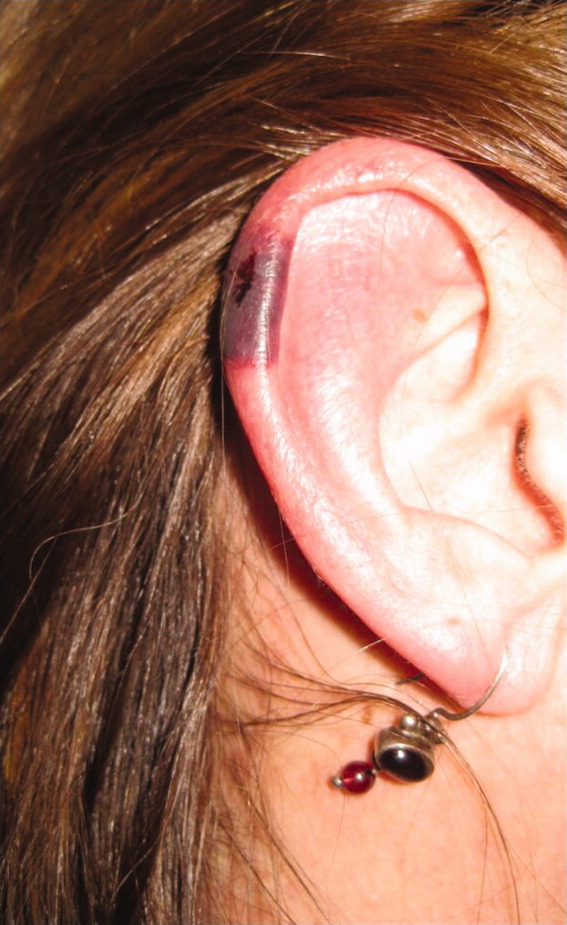
What is the best approach to treat an upper-extremity DVT?
Case
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
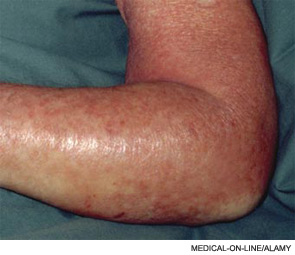
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.

Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.
Case
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.
Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.
Case
A 45-year-old female with a history of cellulitis requiring peripheral inserted central catheter (PICC) line placement for intravenous antibiotics presents two weeks after line removal with persistent, dull, aching pain in her right shoulder and difficulty removing the rings on her right hand. The pain worsens with exercise and is relieved with rest. The physical exam reveals nonpitting edema of her hand. The ultrasound shows subclavian vein thrombosis. What is the best approach to treating her upper extremity deep venous thrombosis (UEDVT)?
Background
DVT and pulmonary embolism (PE) have been subject to increased publicity recently, and both conditions are recognized as serious entities with life-threatening consequences. In fact, more people die annually from blood clots than breast cancer and AIDS combined.1,2 Still, the increased DVT and PE awareness is primarily focused on lower extremity DVT (LEDVT), while UEDVT is thought of as a more benign entity. However, current data suggest that UEDVT is associated with equally significant morbidity and mortality.
UEDVT prevalence has increased in step with the increased use of central venous catheters (CVCs) and pacemakers. Although most patients present with pain, swelling, parathesias, and prominent veins throughout the arm or shoulder, many patients will not display any local DVT symptoms. For example, Kabani et al recently presented data for 1,275 patients admitted to the surgical ICU over a 12-month period. They found the incidence of UEDVT was higher than that of LEDVT (17% vs. 11%; P=0.11). They also determined that scanning all four extremities diagnosed more DVT than two-extremity scans (33% vs. 7%; P<0.001).3
While current medical literature has pushed for increased UEDVT attention, there is no consensus on its treatment. Recent American College of Chest Physicians (ACCP) guidelines addressed UEDVT treatment specifically and recommended analogous treatment to LEDVT with heparin and warfarin.4 This follows prospective studies that have shown patients with UEDVT and LEDVT have similar three-month clinical outcomes. The ACCP guidelines do not specifically recommend different treatment courses based on whether the UEDVT is catheter-related or not. Furthermore, while one might assume that removal of an associated catheter might reduce the treatment duration, there is limited data to support shorter courses in this scenario.
Review of the Data
Incidence: UEDVT is becoming more common secondary to increased interventions in the upper extremity (CVC, pacemaker), and is more easily recognized due to improvement in noninvasive ultrasound technology. UEDVT accounts for up to 10% of all DVT, with an incidence of approximately three per 100,000 persons in the general population.5-8 Because UEDVT can also be asymptomatic, it is believed that the incidence likely is higher than previously reported, but prospective data are lacking.
Risk factors: UEDVT is further categorized as either primary or secondary, depending upon the cause. First described in the late 1800s, spontaneous primary thrombosis of the upper extremity, or Paget-Schroetter syndrome, accounts for approximately 20% of UEDVT.9 Primary UEDVT includes both idiopathic and “effort-related” thrombosis. Effort-related thrombosis usually develops among young people after strenuous or repetitive exercise, such as pitching a baseball. Some hypothesize that effort-related thrombosis is related to a hypercoaguable state or anatomic abnormalities, although a specific cause, such as thoracic outlet syndrome, is found in only 5% of these cases.10,11
Secondary UEDVT characterizes thrombosis in which an endogenous or exogenous risk factor is present. Endogenous risk factors include coagulation abnormalities, such as antithrombin, protein C and protein S deficiencies; factor V Leiden gene mutation; hyperhomocysteinemia; and antiphospholipid antibody syndrome. Exogenous risk factors include CVC pacemakers, intracardiac defibrillators, malignancy, previous or concurrent LEDVT, oral contraceptives, some artificial reproductive technologies (women can develop ovarian hyperstimulation syndrome, which is associated with increased hypercoaguability), trauma, and IV drug use (especially cocaine).5,12-14
Clinical presentation and diagnosis: Swelling (80% of patients) and pain (40% of patients) are the most common UEDVT symptoms at presentation.2 Other clinical features include new, prominent veins throughout the shoulder girdle, erythema, increased warmth, functional impairment, parathesias, and non-specific feelings of arm heaviness or discomfort. Symptoms typically worsen with arm use and improve with rest and elevation.15 Patients with UEDVT related to CVC are more likely to be asymptomatic and may present only with PE.16 The differential diagnosis includes superficial phlebitis, lymphatic edema, hematoma, contusions, venous compression, and muscle tears.17
Contrast venography is the gold standard for the UEDVT diagnosis. However, it is more expensive and invasive than ultrasound, and thus serial compression ultrasound is now the standard test in UEDVT evaluation. Then again, contrast venography remains the test of choice in patients with high pre-test probability and negative ultrasound results.18,19
Prevention: Nearly 70% of secondary UEDVT is associated with a CVC.5 Further, CVC use is the most powerful predictor of UEDVT (adjusted odds ratio (OR), 9.7; 95% CI, 7.8 to 12.2).2 Despite the association between CVCs and UEDVT, anticoagulant prophylaxis is not recommended. Studies evaluating the results of 1-mg warfarin conflict and include small populations. Warfarin’s potential interaction with antibiotics and dosing variance based on nutritional intake logically prompted studies on the potential benefit of low-weight molecular heparain (LWMH); however, these studies have failed to show benefit.20,21
Treatment: ACCP guidelines recommend treating UEDVT patients with unfractionated heparin (UFH) or LMWH and warfarin, with an INR goal of 2 to 3 for at least three months depending upon the overall clinical scenario. Two small studies evaluating catheter-related thrombosis (15 patients in each trial) reported no subsequent embolic phenomenon.22,23 Some authors interpreted this data to mean UEDVT was not as morbid as LEDVT and, subsequently, that catheter-related UEDVTs require only one month of therapy. Since the small studies were published, the increasing incidence and relevance of UEDVT have become more widely recognized, and most authors are recommending three months of treatment.
Still, it’s important to note that there aren’t any published data directly comparing the one-month and the three-month anticoagulation therapies. The RIETE registry, which is the largest ongoing published registry of patients with confirmed DVT or PE, reports similar three-month clinical outcomes between those with UEDVT and LEDVT.
Small, single-center trials have shown that such active interventions as thrombolysis, surgery, or multi-staged approaches are associated with increased vein patency and decreased rates of post-thrombotic syndrome.24,25 However, ACCP has withheld general recommendations for these interventions based on a lack of sufficient data to comment on their overall safety and efficacy, as well as comparable rates of post-thrombotic syndrome (15% to 50%) in studies that directly compared surgical and medical intervention. In fact, the ACCP recommends against interventional treatments unless the patient has failed anticoagulation therapy, has severe symptoms, and expertise is available.4
Superior vena cava filters are available at some centers for patients in whom anticoagulation is contraindicated, but efficacy data is limited. While the data for filter use in UEDVT is limited, its use should be considered in patients who have a contraindication to anticoagulation and remain high risk for UEDVT (e.g., prolonged central line placement).
Complications: Post-thrombotic syndrome (PTS) is the most significant local complication of UEDVT. PTS characteristics are edema, pain, venous ulcers, and skin pigmentation changes, and it is the result of chronic venous insufficiency due to the clot. A meta-analysis of clinical studies on UEDVT noted that PTS occurs in 7% to 46% (mean 15%) of patients.26 One hypothesis for the wide range in frequency is the lack of clear diagnostic criteria for PTS.27 No clear beneficial treatment or prevention for PTS exists, but many recommend graduated compression stockings for the arm.
Residual and recurrent thrombosis are associated with increased PTS risk, which emphasizes the need for further study of interventional treatment because preliminary work has shown increased rates of vein patency in comparison to anticoagulants alone. Recurrent venous thromboembolism (VTE), another local complication, appears to occur less often than it does in patients with LEDVTs, but reaches 8% after five years of followup.28
PE is less common on presentation among patients with UEDVT when compared to patients with LEDVT, but when PE occurs, the three-month outcome is similar.3 PE appears to be more frequent in patients who have a CVC, with an incidence as high as 36% of DVT patients.4,13,21,29
Increased mortality: The mortality among UEDVT patients has been described as 10% to 50% in the 12 months after diagnosis, which is much higher than the ratio in LEDVT patients.21,30 This in part is due to sicker cohorts getting UEDVT. For example, patients with distant metastasis are more likely to develop UEDVT than those with confined malignancy (adjusted OR 11.5; 95% CI, 1.6 to 80.2).31
Occult malignancy, most commonly lung cancer or lymphoma, has been found in as many as 24% of UEDVT patients.32 The high rate of mortality associated with UEDVT appears to be related more with the patient's overall poor clinical condition rather than directly related to complications from the DVT.
However, its presence should alert hospitalists to the patient's potentially poorer prognosis and prompt evaluation for occult malignancy if no risk factor is present.
Back to the Case
This patient should be started on either UFH or LMWH while simultaneously beginning warfarin. She should continue warfarin treatment for at least three months, with a goal INR of 2.0 to 3.0, similar to treatment for LEDVT. The ultimate treatment duration with warfarin follows the same guidelines as treatment with a LEDVT. Although prophylaxis is not routinely recommended, dosing 1 mg of warfarin beginning three days before subsequent CVC placement should be considered if this patient requires a future CVC.
Additionally, an evaluation for occult malignancy should be considered in this patient.
Bottom Line
Upper extremity DVT is not a benign condition, and is associated with a general increase in mortality. It should be treated similarly to LEDVT in order to decrease PTS, recurrent DVT, and pumonary embolism.
Dr. Hollberg is an assistant professor of medicine, Emory University School of Medicine, Atlanta, and medical director for information services, Emory Healthcare.
References
- Hirsh J, Hoak J. Management of deep vein thrombosis and pulmonary embolism. A statement for healthcare professionals. Council on Thrombosis, American Heart Association. Circulation. 1996;93(12):2212-2245.
- Gerotziafas GT, Samama MM. Prophylaxis of venous thromboembolism in medical patients. Curr Opin Pulm Med. 2004;10(5):356-365.
- Kabani L, et al. Upper extremity DVT as prevalent as lower extremity DVT in ICU patients. Society of Critical Care Medicine (SCCM) 38th annual Critical Care Congress: Abstract 305. Presented Feb. 2, 2009.
- Kearon C, Kahn SR, Agnelli G, Goldhaber S, Raskob GE, Comerota AJ. Therapy for venous thromboembolic disease: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest. 2008;133(6Suppl):454S-545S.
- Joffe HV, Kucher N, Tapson VF, Goldhaber SZ. Upper extremity deep vein thrombosis: a prospective registry of 592 patients. Circulation. 2004;110:1605.
- Munoz FJ, Mismetti P, Poggio R, et al. Clinical outcome of patients with an upper-extremity deep vein thrombosis: results from the RIETE registry. Chest. 2008,133:143-148.
- Coon WW, Willis PW. Thrombosis of axillary and subclavian veins. Arch Surg. 1967;94(5):657-663.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—a report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Piccioli A, Marchiori A, Girolami B, Prandoni P. Upper extremity deep vein thrombosis: risk factors, diagnosis, and management. Semin Vasc Med. 2001;1(1):105;110.
- Heron E, Lozinguez O, Alhenc-Gelas M, Emmerich J, Flessinger JN. Hypercoagulable states in primary upper-extremity deep vein thrombosis. Arch Intern Med. 2000;160:382-386.
- Ninet J, Demolombe-Rague S, Bureau Du Colombier P, Coppere B. Les thromboses veineuses profondes des members superieurs. Sang Thromb Vaisseaux. 1994;6:103-114.
- Painter TD, Kerpf M. Deep venous thrombosis of the upper extremity five years experience at a university hospital. Angiology. 1984;35(35):743-749.
- Chan WS, Ginsberg JS. A review of upper extremity deep vein thrombosis in pregnancy: unmasking the “ART” behind the clot. J Thromb Haemost. 2006; 4(8):1673-1677.
- Hughes MJ, D’Agostino JC. Upper extremity deep vein thrombosis: a case report and review of current diagnostic/therapeutic modalities. Am J Emerg Med. 1994;12(6):631-635.
- Prandoni P, Polistena P, Bernardi E, et al. Upper extremity deep vein thrombosis. Risk factors, diagnosis, and complications. Arch Intern Med. 1997;157:57-62.
- Van Rooden CJ, Tesslar ME, Osanto S, Rosendal FR, Huisman MV. Deep vein thrombosis associated with central venous catheters—a review. J Thromb Haemost. 2005;3:2049-2419.
- Horattas MC, Wright DJ, Fenton AH, et al. Changing concepts of deep venous thrombosis of the upper extremity—report of a series and review of the literature. Surgery. 1988;104(3):561-567.
- Bernardi E, Pesavento R, Prandoni P. Upper extremity deep venous thrombosis. Semin Thromb Hemost. 2006;32(7):729-736.
- Baxter GM, McKechnie S, Duffy P. Colour Doppler ultrasound in deep venous thrombosis: a comparison with venography. Clin Radiol. 1990;42(1):32-36.
- Bern MM, Lokich JJ, Wallach SR, et al. Very low doses of warfarin can prevent thrombosis in central venous catheters. A randomized prospective trial. Ann Intern Med. 1990;112(6):423-428.
- Couban S, Goodyear M, Burnell M, et al. Randomized placebo-controlled study of low-dose warfarin for the prevention of central venous catheter-associated thrombosis in patients with cancer. J Clin Oncol. 2005;23(18):4063-4069.
- Lokich JJ, Both A, Benotti P. Complications and management of implanted central venous catheters. J Clin Oncol. 1985;3:710-717.
- Moss JF, Wagman LD, Rijhmaki DU, Terz JJ. Central venous thrombosis related to the silastic Hickman-Broviac catheter in an oncologic population. J Parenter Enteral Nutr. 1989;13:397.
- Machleder HI. Evaluation of a new treatment strategy for Paget-Schroetter syndrome: spontaneous thrombosis of the axillary-subclavian vein. J Vasc Surg. 1993;17:305-315.
- Malcynski J, O’Donnell TF, Mackey WC. Long-term results of treatment for axillary subclavian vein thrombosis. Can J Surg. 1993;36:365-371.
- Elman EE, Kahn SR. The post-thrombotic syndrome after upper extremity deep vein thrombosis in adults: a systematic review. Thromb Res. 2006;117(6):609-614.
- Baarslag HJ, Koopman MM, Hutten BA, et al. Long-term follow up of patients with suspected deep vein thrombosis of the upper extremity: survival, risk factors and post-thrombotic syndrome. Eur J Intern Med. 2004;15:503-507.
- Prandoni P, Bernardi E, Marchiori A, et al. The long term clinical consequence of acute deep venous thrombosis of the arm: prospective cohort study. BMJ. 2004;329:484-485.
- Monreal M, Raventos A, Lerma R, et al. Pulmonary embolism in patients with upper extremity DVT associated to venous central lines—a prospective study. Thromb Haemost. 1994;72(4):548-550.
- Hingorani A, Ascher E, Lorenson E, et al. Upper extremity deep venous thrombosis and its impact on morbidity and mortality rates in a hospital-based population. J Vasc Surg. 1997;26:853-860.
- Blom JW, Doggen CM, Osanto S, Rosendaal FR. Old and new risk factors for upper extremity deep vein thrombosis. J Thromb Haemost. 2005;3:2471-2478.
- Girolami A, Prandoni P, Zanon E, Bagatella P, Girolami B. Venous thromboses of upper limbs are more frequently associated with occult cancer as compared with those of lower limbs. Blood Coagul Fibrinolysis. 1999;10(8):455-457.