User login
The Year Ahead
Rising pressure to contain healthcare costs, increasing demands for safety and quality improvement, more focus on institutional accountability: In 2010, healthcare experts expect several dominant themes to continue converging and moving hospitalists even more to the center of key policy debates.
Peter Pronovost, MD, PhD, medical director of the Center for Innovation in Quality Patient Care and director of the Quality and Safety Research Group at Johns Hopkins University in Baltimore, sees three big themes moving to the fore. One is a greater focus on outcome measurements and accountability for performance, and he expects both carrots and sticks to be wielded. “So, both payment reform and social humiliation, or making things public,” Dr. Pronovost says. “Two, I see a lot more focus on measures that are population-based rather than hospital-based, so looking more at episodes of care.” The shift will force hospitalists to expand their purview beyond the hospital and, he says, partner more with community physicians to develop and monitor performance in such areas as transitions of care and general benchmarks of care.
Dr. Pronovost also expects “significant pressure on both the provider organization and individual clinician being paid less for what they do.” Finding ways to minimize costs will be a priority as payors increase scrutiny on expenses like unnecessary hospital readmissions. But hospitalists, he says, are better positioned than many other physicians to play a key role in the drive toward efficiency while also improving healthcare quality and safety. “I think hospitalists’ roles are going to go up dramatically,” Dr. Pronovost adds, “and I hope the field responds by making sure they put out people who have the skills to lead.”
End-of-Life Issues
Nancy Berlinger, PhD, deputy director and research scholar at The Hastings Center in Garrison, N.Y., cites end-of-life care as another theme likely to gain traction in 2010. As project director of the center’s revised ethical guidelines for end-of-life care, Dr. Berlinger notes how often clinicians in her working group have invoked the hospitalist profession. It’s no accident. “Hospitalists are increasingly associated with the care of patients on Medicare,” she says, adding Medicare beneficiaries are far more likely to be nearing the end of life.
Demographics suggest that connection will continue to grow in 2010 and beyond. Dr. Berlinger points to a 2009 New England Journal of Medicine study showing that the odds of a hospitalized Medicare patient receiving care from a hospitalist increased at a brisk 29.2% annual clip from 1997 through 2006.1 And while the U.S. faces a shortage of geriatricians, HM is growing rapidly as a medical profession. “By default, whether or not hospitalists self-identify as caring for older Americans,” Dr. Berlinger says, “this is their area of practical specialization.”
With that specialization comes added responsibility to assist with advanced-care planning and helping patients to document their wishes. Similarly, she says, it means acknowledging that these patients are more likely to have comorbid conditions and identify with goals of care. “I don’t think there’s any way around this,” she says. “Medicare and hospitalists, whether by accident or design, are increasingly joined at the hip. That is something that hospitalists, as a profession, will always need to keep their eye on.”
A parallel trend is that other doctors increasingly view hospitalists as hospital specialists. “The hospitalist’s responsibilities are not just in terms of the patients they care for, but also in terms of the institution itself,” Dr. Berlinger says. Non-staff physicians, for example, expect hospitalists to know how a hospital’s in-patient care system works. Practically speaking, as electronic medical records (EMR) become more commonplace, hospitalists will be increasingly relied upon to understand a hospital’s information technology.
—Peter Pronovost, MD, PhD, medical director, Center for Innovation in Quality Patient Care, Johns Hopkins University, Baltimore
New Economy, New Hospital Landscape
Douglas Wood, MD, chair of the Division of Health Care Policy and Research at the Mayo Clinic in Rochester, Minn., points to language in the federal healthcare reform legislation as evidence that hospitals and hospitalists will need to be in sync in other ways to avoid future penalties. One provision, for example, would increase the penalties for hospital-acquired infections. Other language seeks to reduce unnecessary readmissions.
Likewise, Dr. Wood says, addressing geographical variations in healthcare payments driven largely by unnecessary overutilization—including excessive use of ICU care, in-patient care, imaging, and specialist services—might mean asking hospitalists to take on more aspects of patient care.
Meanwhile, increased interest in demonstration projects that might achieve savings (e.g., accountable care organizations and bundled payments) suggests that proactive hospitals should again look to hospitalists. The flurry of new proposals won’t fundamentally change hospitalists’ responsibilities to provide effective and efficient care, “but it will put more emphasis on what they’re doing,” Dr. Wood says, “to the degree that hospitalists could take a lead in demonstrating how you can provide better outcomes at a lower overall utilization of resources.”
Regardless of how slowly or quickly these initiatives proceed at the national level, he says, hospitalists should be mindful that several states are well ahead of the curve and are likely to be more aggressive in instituting policy changes.
The Bottom Line
If there’s a single, overriding theme for 2010, Bradley Flansbaum, DO, MPH, FACP, FHM, director of hospitalist services at Lenox Hill Hospital in New York City and a member of SHM’s Public Policy Committee, says it might be that of dealing with the unknown. Squeezing healthcare costs and more tightly regulating inflation will have a greater effect on a hospital’s bottom line and thus impact what’s required of hospitalists. Even so, the profession will have to wait and see whether and how various proposals are codified and implemented. “We don’t know exactly what things are going to look like,” he says.
Nor is there a good sense of how new standards for transparency, quality, and accountability might be measured. “While people want more measurement and they want more report-card-type information, the data that we can acquire right now and how we analyze that data are still fairly primitive,” Dr. Flansbaum says. Even current benchmarks are lacking in how to determine who’s doing a good job and who isn’t, he says.
One big question that must be answered, then: Are we even looking at the right measurements? “Or, do the right measurements exist, or do we have the databases, the registries, the sources, to make the decisions we need to make?” he says.
Any new proposals will require another round of such questions and filling-in of blanks to add workable details to vague and potentially confusing language.
“I think we know that change is afoot, and most smart hospitalists know that the system needs to run leaner,” Dr. Flansbaum says. “But how each one of us is going to function in our hospital, and the kinds of demands that will be placed on us, and what we’re going to need to do with the doctors in the community and the other nonphysician colleagues that we work with, is all really unknown.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Kuo YF, Sharma G, Freeman JL, Goodwin JS. Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11): 1102-1112.
Image Source: PAGADESIGN, OVERSNAP/ISTOCKPHOTO.COM
Rising pressure to contain healthcare costs, increasing demands for safety and quality improvement, more focus on institutional accountability: In 2010, healthcare experts expect several dominant themes to continue converging and moving hospitalists even more to the center of key policy debates.
Peter Pronovost, MD, PhD, medical director of the Center for Innovation in Quality Patient Care and director of the Quality and Safety Research Group at Johns Hopkins University in Baltimore, sees three big themes moving to the fore. One is a greater focus on outcome measurements and accountability for performance, and he expects both carrots and sticks to be wielded. “So, both payment reform and social humiliation, or making things public,” Dr. Pronovost says. “Two, I see a lot more focus on measures that are population-based rather than hospital-based, so looking more at episodes of care.” The shift will force hospitalists to expand their purview beyond the hospital and, he says, partner more with community physicians to develop and monitor performance in such areas as transitions of care and general benchmarks of care.
Dr. Pronovost also expects “significant pressure on both the provider organization and individual clinician being paid less for what they do.” Finding ways to minimize costs will be a priority as payors increase scrutiny on expenses like unnecessary hospital readmissions. But hospitalists, he says, are better positioned than many other physicians to play a key role in the drive toward efficiency while also improving healthcare quality and safety. “I think hospitalists’ roles are going to go up dramatically,” Dr. Pronovost adds, “and I hope the field responds by making sure they put out people who have the skills to lead.”
End-of-Life Issues
Nancy Berlinger, PhD, deputy director and research scholar at The Hastings Center in Garrison, N.Y., cites end-of-life care as another theme likely to gain traction in 2010. As project director of the center’s revised ethical guidelines for end-of-life care, Dr. Berlinger notes how often clinicians in her working group have invoked the hospitalist profession. It’s no accident. “Hospitalists are increasingly associated with the care of patients on Medicare,” she says, adding Medicare beneficiaries are far more likely to be nearing the end of life.
Demographics suggest that connection will continue to grow in 2010 and beyond. Dr. Berlinger points to a 2009 New England Journal of Medicine study showing that the odds of a hospitalized Medicare patient receiving care from a hospitalist increased at a brisk 29.2% annual clip from 1997 through 2006.1 And while the U.S. faces a shortage of geriatricians, HM is growing rapidly as a medical profession. “By default, whether or not hospitalists self-identify as caring for older Americans,” Dr. Berlinger says, “this is their area of practical specialization.”
With that specialization comes added responsibility to assist with advanced-care planning and helping patients to document their wishes. Similarly, she says, it means acknowledging that these patients are more likely to have comorbid conditions and identify with goals of care. “I don’t think there’s any way around this,” she says. “Medicare and hospitalists, whether by accident or design, are increasingly joined at the hip. That is something that hospitalists, as a profession, will always need to keep their eye on.”
A parallel trend is that other doctors increasingly view hospitalists as hospital specialists. “The hospitalist’s responsibilities are not just in terms of the patients they care for, but also in terms of the institution itself,” Dr. Berlinger says. Non-staff physicians, for example, expect hospitalists to know how a hospital’s in-patient care system works. Practically speaking, as electronic medical records (EMR) become more commonplace, hospitalists will be increasingly relied upon to understand a hospital’s information technology.
—Peter Pronovost, MD, PhD, medical director, Center for Innovation in Quality Patient Care, Johns Hopkins University, Baltimore
New Economy, New Hospital Landscape
Douglas Wood, MD, chair of the Division of Health Care Policy and Research at the Mayo Clinic in Rochester, Minn., points to language in the federal healthcare reform legislation as evidence that hospitals and hospitalists will need to be in sync in other ways to avoid future penalties. One provision, for example, would increase the penalties for hospital-acquired infections. Other language seeks to reduce unnecessary readmissions.
Likewise, Dr. Wood says, addressing geographical variations in healthcare payments driven largely by unnecessary overutilization—including excessive use of ICU care, in-patient care, imaging, and specialist services—might mean asking hospitalists to take on more aspects of patient care.
Meanwhile, increased interest in demonstration projects that might achieve savings (e.g., accountable care organizations and bundled payments) suggests that proactive hospitals should again look to hospitalists. The flurry of new proposals won’t fundamentally change hospitalists’ responsibilities to provide effective and efficient care, “but it will put more emphasis on what they’re doing,” Dr. Wood says, “to the degree that hospitalists could take a lead in demonstrating how you can provide better outcomes at a lower overall utilization of resources.”
Regardless of how slowly or quickly these initiatives proceed at the national level, he says, hospitalists should be mindful that several states are well ahead of the curve and are likely to be more aggressive in instituting policy changes.
The Bottom Line
If there’s a single, overriding theme for 2010, Bradley Flansbaum, DO, MPH, FACP, FHM, director of hospitalist services at Lenox Hill Hospital in New York City and a member of SHM’s Public Policy Committee, says it might be that of dealing with the unknown. Squeezing healthcare costs and more tightly regulating inflation will have a greater effect on a hospital’s bottom line and thus impact what’s required of hospitalists. Even so, the profession will have to wait and see whether and how various proposals are codified and implemented. “We don’t know exactly what things are going to look like,” he says.
Nor is there a good sense of how new standards for transparency, quality, and accountability might be measured. “While people want more measurement and they want more report-card-type information, the data that we can acquire right now and how we analyze that data are still fairly primitive,” Dr. Flansbaum says. Even current benchmarks are lacking in how to determine who’s doing a good job and who isn’t, he says.
One big question that must be answered, then: Are we even looking at the right measurements? “Or, do the right measurements exist, or do we have the databases, the registries, the sources, to make the decisions we need to make?” he says.
Any new proposals will require another round of such questions and filling-in of blanks to add workable details to vague and potentially confusing language.
“I think we know that change is afoot, and most smart hospitalists know that the system needs to run leaner,” Dr. Flansbaum says. “But how each one of us is going to function in our hospital, and the kinds of demands that will be placed on us, and what we’re going to need to do with the doctors in the community and the other nonphysician colleagues that we work with, is all really unknown.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Kuo YF, Sharma G, Freeman JL, Goodwin JS. Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11): 1102-1112.
Image Source: PAGADESIGN, OVERSNAP/ISTOCKPHOTO.COM
Rising pressure to contain healthcare costs, increasing demands for safety and quality improvement, more focus on institutional accountability: In 2010, healthcare experts expect several dominant themes to continue converging and moving hospitalists even more to the center of key policy debates.
Peter Pronovost, MD, PhD, medical director of the Center for Innovation in Quality Patient Care and director of the Quality and Safety Research Group at Johns Hopkins University in Baltimore, sees three big themes moving to the fore. One is a greater focus on outcome measurements and accountability for performance, and he expects both carrots and sticks to be wielded. “So, both payment reform and social humiliation, or making things public,” Dr. Pronovost says. “Two, I see a lot more focus on measures that are population-based rather than hospital-based, so looking more at episodes of care.” The shift will force hospitalists to expand their purview beyond the hospital and, he says, partner more with community physicians to develop and monitor performance in such areas as transitions of care and general benchmarks of care.
Dr. Pronovost also expects “significant pressure on both the provider organization and individual clinician being paid less for what they do.” Finding ways to minimize costs will be a priority as payors increase scrutiny on expenses like unnecessary hospital readmissions. But hospitalists, he says, are better positioned than many other physicians to play a key role in the drive toward efficiency while also improving healthcare quality and safety. “I think hospitalists’ roles are going to go up dramatically,” Dr. Pronovost adds, “and I hope the field responds by making sure they put out people who have the skills to lead.”
End-of-Life Issues
Nancy Berlinger, PhD, deputy director and research scholar at The Hastings Center in Garrison, N.Y., cites end-of-life care as another theme likely to gain traction in 2010. As project director of the center’s revised ethical guidelines for end-of-life care, Dr. Berlinger notes how often clinicians in her working group have invoked the hospitalist profession. It’s no accident. “Hospitalists are increasingly associated with the care of patients on Medicare,” she says, adding Medicare beneficiaries are far more likely to be nearing the end of life.
Demographics suggest that connection will continue to grow in 2010 and beyond. Dr. Berlinger points to a 2009 New England Journal of Medicine study showing that the odds of a hospitalized Medicare patient receiving care from a hospitalist increased at a brisk 29.2% annual clip from 1997 through 2006.1 And while the U.S. faces a shortage of geriatricians, HM is growing rapidly as a medical profession. “By default, whether or not hospitalists self-identify as caring for older Americans,” Dr. Berlinger says, “this is their area of practical specialization.”
With that specialization comes added responsibility to assist with advanced-care planning and helping patients to document their wishes. Similarly, she says, it means acknowledging that these patients are more likely to have comorbid conditions and identify with goals of care. “I don’t think there’s any way around this,” she says. “Medicare and hospitalists, whether by accident or design, are increasingly joined at the hip. That is something that hospitalists, as a profession, will always need to keep their eye on.”
A parallel trend is that other doctors increasingly view hospitalists as hospital specialists. “The hospitalist’s responsibilities are not just in terms of the patients they care for, but also in terms of the institution itself,” Dr. Berlinger says. Non-staff physicians, for example, expect hospitalists to know how a hospital’s in-patient care system works. Practically speaking, as electronic medical records (EMR) become more commonplace, hospitalists will be increasingly relied upon to understand a hospital’s information technology.
—Peter Pronovost, MD, PhD, medical director, Center for Innovation in Quality Patient Care, Johns Hopkins University, Baltimore
New Economy, New Hospital Landscape
Douglas Wood, MD, chair of the Division of Health Care Policy and Research at the Mayo Clinic in Rochester, Minn., points to language in the federal healthcare reform legislation as evidence that hospitals and hospitalists will need to be in sync in other ways to avoid future penalties. One provision, for example, would increase the penalties for hospital-acquired infections. Other language seeks to reduce unnecessary readmissions.
Likewise, Dr. Wood says, addressing geographical variations in healthcare payments driven largely by unnecessary overutilization—including excessive use of ICU care, in-patient care, imaging, and specialist services—might mean asking hospitalists to take on more aspects of patient care.
Meanwhile, increased interest in demonstration projects that might achieve savings (e.g., accountable care organizations and bundled payments) suggests that proactive hospitals should again look to hospitalists. The flurry of new proposals won’t fundamentally change hospitalists’ responsibilities to provide effective and efficient care, “but it will put more emphasis on what they’re doing,” Dr. Wood says, “to the degree that hospitalists could take a lead in demonstrating how you can provide better outcomes at a lower overall utilization of resources.”
Regardless of how slowly or quickly these initiatives proceed at the national level, he says, hospitalists should be mindful that several states are well ahead of the curve and are likely to be more aggressive in instituting policy changes.
The Bottom Line
If there’s a single, overriding theme for 2010, Bradley Flansbaum, DO, MPH, FACP, FHM, director of hospitalist services at Lenox Hill Hospital in New York City and a member of SHM’s Public Policy Committee, says it might be that of dealing with the unknown. Squeezing healthcare costs and more tightly regulating inflation will have a greater effect on a hospital’s bottom line and thus impact what’s required of hospitalists. Even so, the profession will have to wait and see whether and how various proposals are codified and implemented. “We don’t know exactly what things are going to look like,” he says.
Nor is there a good sense of how new standards for transparency, quality, and accountability might be measured. “While people want more measurement and they want more report-card-type information, the data that we can acquire right now and how we analyze that data are still fairly primitive,” Dr. Flansbaum says. Even current benchmarks are lacking in how to determine who’s doing a good job and who isn’t, he says.
One big question that must be answered, then: Are we even looking at the right measurements? “Or, do the right measurements exist, or do we have the databases, the registries, the sources, to make the decisions we need to make?” he says.
Any new proposals will require another round of such questions and filling-in of blanks to add workable details to vague and potentially confusing language.
“I think we know that change is afoot, and most smart hospitalists know that the system needs to run leaner,” Dr. Flansbaum says. “But how each one of us is going to function in our hospital, and the kinds of demands that will be placed on us, and what we’re going to need to do with the doctors in the community and the other nonphysician colleagues that we work with, is all really unknown.” TH
Bryn Nelson is a freelance medical writer based in Seattle.
Reference
- Kuo YF, Sharma G, Freeman JL, Goodwin JS. Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11): 1102-1112.
Image Source: PAGADESIGN, OVERSNAP/ISTOCKPHOTO.COM
A Pain in the Bone
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
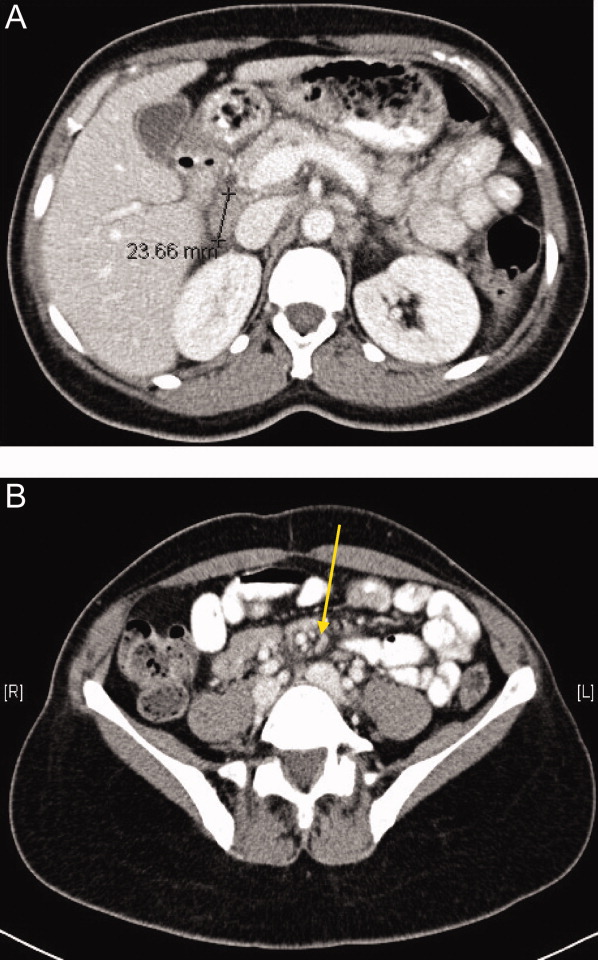
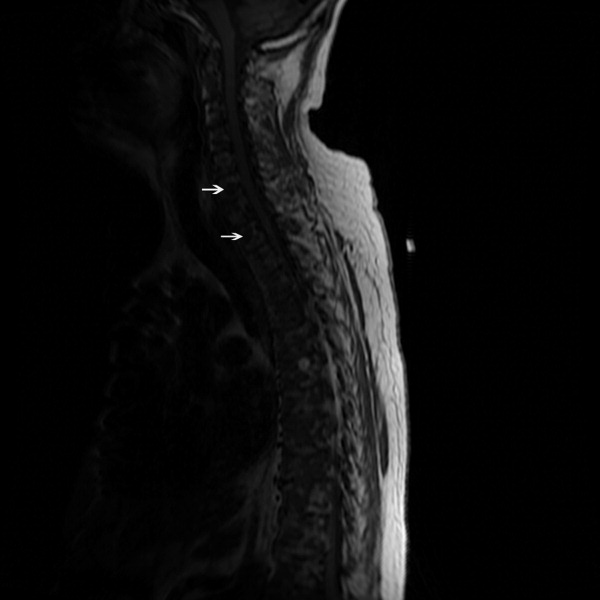
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.

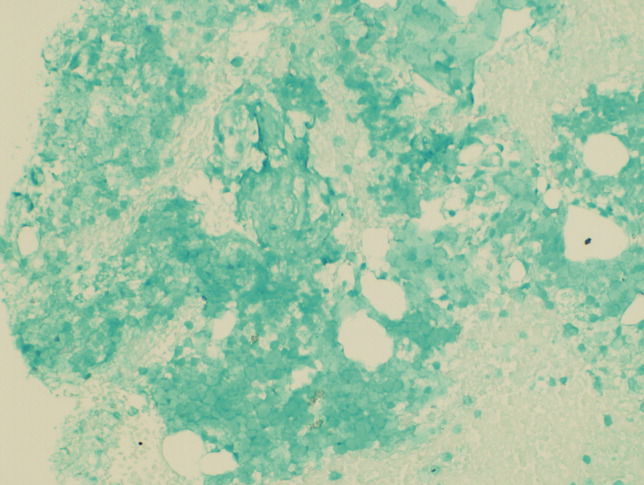
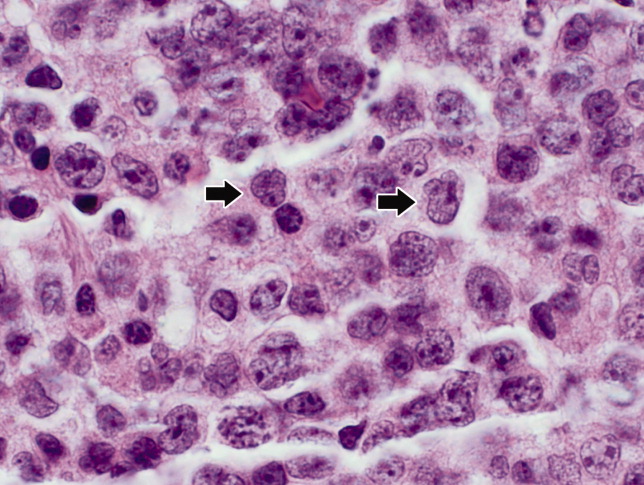
The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.
The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
A 71‐year‐old man presented to a hospital with a one week history of fatigue, polyuria, and polydipsia. He also reported pain in his back, hips, and ribs, in addition to frequent falls, intermittent confusion, constipation, and a weight loss of 10 pounds over the last 2 weeks. He denied cough, shortness of breath, chest pain, fever, night sweats, headache, and focal weakness.
Polyuria, which is often associated with polydipsia, can be arbitrarily defined as a urine output exceeding 3 L per day. After excluding osmotic diuresis due to uncontrolled diabetes mellitus, the 3 major causes of polyuria are primary polydipsia, central diabetes insipidus, and nephrogenic diabetes insipidus. Approximately 30% to 50% of cases of central diabetes insipidus are idiopathic; however, primary or secondary brain tumors or infiltrative diseases involving the hypothalamic‐pituitary region need to be considered in this 71‐year‐old man. The most common causes of nephrogenic diabetes insipidus in adults are chronic lithium ingestion, hypokalemia, and hypercalcemia. The patient describes symptoms that can result from severe hypercalcemia, including fatigue, confusion, constipation, polyuria, and polydipsia.
The patient's past medical history included long‐standing, insulin‐requiring type 2 diabetes with associated complications including coronary artery disease, transient ischemic attacks, proliferative retinopathy, peripheral diabetic neuropathy, and nephropathy. Seven years prior to presentation, he received a cadaveric renal transplant that was complicated by BK virus (polyomavirus) nephropathy and secondary hyperparathyroidism. Three years after his transplant surgery, he developed squamous cell carcinoma of the skin, which was treated with local surgical resection. Two years after that, he developed stage I laryngeal cancer of the glottis and received laser surgery, and since then he had been considered disease‐free. He also had a history of hypertension, hypercholesterolemia, osteoporosis, and depression. His medications included aspirin, amlodipine, metoprolol succinate, valsartan, furosemide, simvastatin, insulin, prednisone, sirolimus, and sulfamethoxazole/trimethoprim. He was a married psychiatrist. He denied tobacco use and reported occasional alcohol use.
The prolonged immunosuppressive therapy that is required following organ transplantation carries a markedly increased risk of the subsequent development of malignant tumors, including cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma. Primary brain lymphoma resulting in central diabetes insipidus would be unlikely in the absence of headache or focal weakness. An increased risk of lung cancer occurs in recipients of heart and lung transplants, and to a much lesser degree, recipients of kidney transplants. However, metastatic lung cancer is less likely in the absence of respiratory symptoms and smoking history (present in approximately 90% of all lung cancers). Nephrogenic diabetes insipidus, in its mild form, is relatively common in elderly patients with acute or chronic renal insufficiency because of a reduction in maximum urinary concentrating ability. On the other hand, this alone does not explain his remaining symptoms. The instinctive diagnosis in this case is tertiary hyperparathyroidism due to progression of untreated secondary hyperparathyroidism. This causes hypercalcemia, nephrogenic diabetes insipidus, and significant bone pain related to renal osteodystrophy.
On physical exam, the patient appeared chronically ill, but was in no acute distress. He weighed 197.6 pounds and his height was 70.5 inches. He was afebrile with a blood pressure of 146/82 mm Hg, a heart rate of 76 beats per minute, a respiratory rate of 12 breaths per minute, and an oxygen saturation of 97% while breathing room air. He had no generalized lymphadenopathy. Thyroid examination was unremarkable. Examination of the lungs, heart, abdomen, and lower extremities was normal. The rectal examination revealed no masses or prostate nodules; a test for fecal occult blood was negative. He had loss of sensation to light touch and vibration in the feet with absent Achilles deep tendon reflexes. He had a poorly healing surgical wound on his forehead at the site of his prior skin cancer, but no rash or other lesions. There was no joint swelling or erythema. There were tender points over the cervical, thoracic, and lumbar spine; on multiple ribs; and on the pelvic rims.
Perhaps of greatest importance is the lack of lymphadenopathy, organomegaly, or other findings suggestive of diffuse lymphoproliferative disease. His multifocal bone tenderness is concerning for renal osteodystrophy, multiple myeloma, or primary or metastatic bone disease. Cancers in men that metastasize to the bone usually originate from the prostate, lung, kidney, or thyroid gland. In any case, his physical examination did not reveal an enlarged, asymmetric, or nodular prostate or thyroid gland. I recommend a chest film to rule out primary lung malignancy and a basic laboratory evaluation to narrow down the differential diagnosis.
A complete blood count showed a normocytic anemia with a hemoglobin of 8.7 g/dL and a hematocrit of 25%. Other laboratory tests revealed the following values: sodium, 139 mmol/L; potassium, 4.1 mmol/L; blood urea nitrogen, 70 mg/dL; creatinine, 3.5 mg/dL (most recent value 2 months ago was 1.9 mg/dL); total calcium, 13.2 mg/dL (normal range, 8.5‐10.5 mg/dL); phosphate, 5.3 mg/dL; magnesium, 2.5 mg/dL; total bilirubin, 0.5 mg/dL; alkaline phosphatase, 130 U/L; aspartate aminotransferase, 28 U/L; alanine aminotransferase, 19 U/L; albumin, 3.5 g/dL; and lactate dehydrogenase (LDH), 1258 IU/L (normal range, 105‐333 IU/L). A chest radiograph was normal.
The most important laboratory findings are severe hypercalcemia, acute on chronic renal failure, and anemia. Hypercalcemia most commonly results from malignancy or hyperparathyroidism. Less frequently, hypercalcemia may result from sarcoidosis, vitamin D intoxication, or hyperthyroidism. The degree of hypercalcemia is useful diagnostically as hyperparathyroidism commonly results in mild hypercalcemia (serum calcium concentration often below 11 mg/dL). Values above 13 mg/dL are unusual in hyperparathyroidism and are most often due to malignancy. Malignancy is often evident clinically by the time it causes hypercalcemia, and patients with hypercalcemia of malignancy are more often symptomatic than those with hyperparathyroidism. Additionally, localized bone pain and weight loss do not result from hypercalcemia itself and their presence also raises concern for malignancy.
Nonmelanoma skin cancer is the most common cancer occurring after transplantation but does not cause hypercalcemia. Squamous cancers of the head and neck can rarely cause hypercalcemia due to secretion of parathyroid hormone‐related peptide; however, his early‐stage laryngeal cancer and the expected high likelihood of cure argue against this possibility. Osteolytic metastases account for approximately 20% of cases of hypercalcemia of malignancy (Table 1). Prostate cancer rarely results in hypercalcemia since bone metastases are predominantly osteoblastic, whereas metastatic non‐small‐cell lung cancer, thyroid cancer, and kidney cancer more commonly cause hypercalcemia due to osteolytic bone lesions. The total alkaline phosphatase has been traditionally used to assess the osteoblastic component of bone remodeling. Its normal level tends to predict a negative bone scan and supports the likelihood of lytic lesions. Posttransplantation lymphoproliferative disorders, which include a wide range of syndromes, can rarely result in hypercalcemia. I am also worried about the possibility of multiple myeloma as he has the classic triad of hypercalcemia, bone pain, and subacute kidney injury.
|
| Osteolytic metastases |
| Breast cancer |
| Multiple myeloma |
| Lymphoma |
| Leukemia |
| Humoral hypercalcemia (PTH‐related protein) |
| Squamous cell carcinomas |
| Renal carcinomas |
| Bladder carcinoma |
| Breast cancer |
| Ovarian carcinoma |
| Leukemia |
| Lymphoma |
| 1,25‐Dihydroxyvitamin D secretion |
| Lymphoma |
| Ovarian dysgerminomas |
| Ectopic PTH secretion (rare) |
| Ovarian carcinoma |
| Lung carcinomas |
| Neuroectodermal tumor |
| Thyroid papillary carcinoma |
| Rhabdomyosarcoma |
| Pancreatic cancer |
The first purpose of the laboratory evaluation is to differentiate parathyroid hormone (PTH)‐mediated hypercalcemia (primary and tertiary hyperparathyroidism) from non‐PTH‐mediated hypercalcemia (primarily malignancy, hyperthyroidism, vitamin D intoxication, and granulomatous disease). The production of vitamin D metabolites, PTH‐related protein, or hypercalcemia from osteolysis in these latter cases results in suppressed PTH levels.
In severe elevations of calcium, the initial goals of treatment are directed toward fluid resuscitation with normal saline and, unless contraindicated, the immediate institution of bisphosphonate therapy. A loop diuretic such as furosemide is often used, but a recent review concluded that there is little evidence to support its use in this setting.
The patient was admitted and treated with intravenous saline and furosemide. Additional laboratory evaluation revealed normal levels of prostate‐specific antigen and thyroid‐stimulating hormone. PTH was 44 pg/mL (the most recent value was 906 pg/mL eight years ago; normal range, 15‐65 pg/mL) and beta‐2 microglobulin (B2M) was 8 mg/L (normal range, 0.8‐2.2 mg/L).
The normal PTH level makes tertiary hyperparathyroidism unlikely and points toward non‐PTH‐related hypercalcemia. An elevated B2M level may occur in patients with chronic graft rejection, renal tubular dysfunction, dialysis‐related amyloidosis, multiple myeloma, or lymphoma. LDH is often elevated in patients with multiple myeloma and lymphoma, but this is not a specific finding. The next laboratory test would be measurement of PTH‐related protein and vitamin D metabolites, as these tests can differentiate between the causes of non‐PTH‐mediated hypercalcemia.
Serum concentrations of the vitamin D metabolites, 25‐hydroxyvitamin D (calcidiol) and 1,25‐dihydroxyvitamin D (calcitriol), were low‐normal. PTH‐related protein was not detected.
The marked elevation of serum LDH and B2M, the relatively suppressed PTH level, combined with undetectable PTH‐related protein suggest multiple myeloma or lymphoma as the likely cause of the patient's clinical presentation. The combination of hypercalcemia and multifocal bone pain makes multiple myeloma the leading diagnosis as hypercalcemia is uncommon in patients with lymphoma, especially at the time of initial clinical presentation.
I would proceed with serum and urine protein electrophoresis (SPEP and UPEP, respectively) and a skeletal survey. If these tests do not confirm the diagnosis of multiple myeloma, I would order a noncontrast computed tomography (CT) of the chest and abdomen and a magnetic resonance imaging (MRI) of the spine. In addition, I would like to monitor his response to the intravenous saline and furosemide.
Forty‐eight hours after presentation, repeat serum calcium and creatinine levels were 11.3 mg/dL and 2.9 mg/dL, respectively. He received salmon calcitonin 4 U/kg every 12 hours. Pamidronate was avoided because of his kidney disease. His confusion resolved. He received intravenous morphine intermittently to alleviate his bone pain.
The SPEP revealed a monoclonal immunoglobulin G (IgG) lambda (light chain) spike representing roughly 3% (200 mg/dL) of total protein. His serum Ig levels were normal. The UPEP was negative for monoclonal immunoglobulin and Bence‐Jones protein. The skeletal survey revealed marked osteopenia, and the bone scan was normal. An MRI of the spine showed multiple round lesions in the cervical, thoracic, and lumbar spine (Figure 1). A CT of the chest showed similar bone lesions in the ribs and pelvis. A CT of the abdomen and chest did not suggest any primary malignancy nor did it show thoracic or abdominal lymphadenopathy.
The lack of lymphadenopathy, splenomegaly, or a visceral mass by CT imaging and physical examination, along with the normal PSA level, exclude most common forms of non‐Hodgkin lymphoma and bone metastasis from solid tumors. In multiple myeloma, cytokines secreted by plasma cells suppress osteoblast activity; therefore, while discrete lytic bone lesions are apparent on skeletal survey, the bone scan is typically normal. The absence of lytic lesions, normal serum immunoglobulin levels, and unremarkable UPEP make multiple myeloma or light‐chain deposition disease a less likely diagnosis.
Typically, primary lymphoma of the bone produces increased uptake with bone scanning. However, because primary lymphoma of the bone is one of the least common primary skeletal malignancies and varies widely in appearance on imaging, confident diagnosis based on imaging alone usually is not possible.
Posttransplantation lymphoproliferative disorder (PTLD) refers to a syndrome that ranges from a self‐limited form of lymphoproliferation to an aggressive disseminated disease. Although the patient is at risk for PTLD, isolated bone involvement has only rarely been reported.
Primary lymphoma of the bone and PTLD are my leading diagnoses in this patient. At this point, I recommend a bone marrow biopsy and biopsy of an easily accessible representative bone lesion with special staining for Epstein‐Barr virus (EBV) (EBV‐encoded RNA [EBER] and latent membrane protein 1 [LMP1]). I expect this test to provide a definitive diagnosis. As 95% of PTLD cases are induced by infection with EBV, information regarding pretransplantation EBV status of the patient and the donor, current EBV status of the patient, and type and intensity of immunosuppression at the time of transplantation would be very helpful to determine their likelihood.
Seventy‐two hours after presentation, his serum calcium level normalized and most of his symptoms improved. Calcitonin was discontinued, and he was maintained on oral hydration. On hospital day number 5, he underwent CT‐guided bone biopsy of the L4 vertebral body, which showed large aggregates of atypical lymphoid cells (Figure 2). These cells were predominantly B‐cells interspersed with small reactive T‐cells. The cells did not express EBV LMP1 or EBER (Figure 3). On hospital day 7, he underwent a bone marrow biopsy, which revealed similar large atypical lymphoid cells that comprised the majority of marrow space (Figure 4). By immunohistochemistry, these cells brightly expressed the pan B cell marker, CD20, and coexpressed bcl‐2. EBER and LMP1 were also negative. A flow cytometry of the bone marrow demonstrated a lambda light chain restriction within the B lymphocytes.
The medical records indicated that the patient had positive pretransplantation EBV serologies. He received a regimen based on sirolimus, mycophenolate mofetil, and prednisone, and did not receive high doses of induction or maintenance immunosuppressive therapy.
The biopsy results establish a diagnosis of diffuse large B‐cell lymphoma of the bone. PTLD is unlikely given his positive pretransplantation EBV status, the late onset of his disease (6 years after transplantation), the isolated bone involvement, and the negative EBER and LMP1 tests.
The patient was discharged and was readmitted 1 week later for induction chemotherapy with etoposide, vincristine, doxorubicin, cyclophosphamide, and prednisone [EPOCH]Rituxan (rituximab). Over the next several months, he received 6 cycles of chemotherapy, his hypercalcemia resolved, and his back pain improved.
Commentary
Hypercalcemia is among the most common causes of nephrogenic diabetes insipidus in adults.1 A urinary concentrating defect usually becomes clinically apparent if the plasma calcium concentration is persistently above 11 mg/dL.1 This defect is generally reversible with correction of the hypercalcemia but may persist in patients in whom interstitial nephritis has induced permanent medullary damage. The mechanism by which the concentrating defect occurs is incompletely understood but may be related to impairments in sodium chloride reabsorption in the thick ascending limb and in the ability of antidiuretic hormone to increase water permeability in the collecting tubules.1
Although hypercalcemia in otherwise healthy outpatients is usually due to primary hyperparathyroidism, malignancy is more often responsible for hypercalcemia in hospitalized patients.2 While the signs and symptoms of hypercalcemia are similar regardless of the cause, several clinical features may help distinguish the etiology of hypercalcemia. For instance, the presence of tachycardia, warm skin, thinning of the hair, stare and lid lag, and widened pulse pressure points toward hypercalcemia related to hyperthyroidism. In addition, risk factors and comorbidities guide the diagnostic process. For example, low‐level hypercalcemia in an asymptomatic postmenopausal woman with a normal physical examination suggests primary hyperparathyroidism. In contrast, hypercalcemia in a transplant patient raises concern of malignancy including PTLDs.3, 4
PTLDs are uncommon causes of hypercalcemia but are among the most serious and potentially fatal complications of chronic immunosuppression in transplant recipients.5 They occur in 1.9% of patients after kidney transplantation. The lymphoproliferative disorders occurring after transplantation have different characteristics from those that occur in the general population. Non‐Hodgkin lymphoma accounts for 65% of lymphomas in the general population, compared to 93% in transplant recipients.5, 6 The pathogenesis of PTLD appears to be related to B cell proliferation induced by infection with EBV in the setting of chronic immunosuppression.6 Therefore, there is an increased frequency of PTLD among transplant recipients who are EBV seronegative at the time of operation. These patients, who have no preoperative immunity to EBV, usually acquire the infection from the donor. The level of immunosuppression (intensity and type) influences PTLD rates as well. The disease typically occurs within 12 months after transplantation and in two‐thirds of cases involves extranodal sites. Among these sites, the gastrointestinal tract is involved in about 26% of cases and central nervous system in about 27%. Isolated bone involvement is exceedingly rare.5, 6
Primary lymphoma of the bone is another rare cause of hypercalcemia and accounts for less than 5% of all primary bone tumors.7 The majority of cases are of the non‐Hodgkin's type, characterized as diffuse large B‐cell lymphomas, with peak occurrence in the sixth to seventh decades of life.8 The classic imaging findings of primary lymphoma of the bone are a solitary metadiaphyseal lesion with a layered periosteal reaction on plain radiographs, and corresponding surrounding soft‐tissue mass on MRI.9 Less commonly, primary lymphoma of the bone can be multifocal with diffuse osseous involvement and variable radiographic appearances, as in this case. Most series have reported that the long bones are affected most frequently (especially the femur), although a large series showed equal numbers of cases presenting in the long bones and the spine.712
In order to diagnose primary lymphoma of the bone, it is necessary to exclude nodal or disseminated disease by physical examination and imaging. As plain films are often normal, bone scan or MRI of clinically affected areas is necessary to establish disease extent.9 Distinguishing primary bone lymphomas (PLB) from other bone tumors is important because PLB has a better response to therapy and a better prognosis.10, 11
Randomized trials addressing treatment options for primary lymphoma of bone are not available. Historically, PLB was treated with radiotherapy alone with good local control. However, the rate of distant relapses was relatively high. Currently, chemotherapy with or without radiation therapy is preferred; 5‐year survival is approximately 70% after combined therapy.10, 11
In this case, symptomatic hypercalcemia, a history of transplantation, marked elevation of both LDH and B2M, and a normal PTH level all pointed toward the correct diagnosis of malignancy. Low or normal levels of vitamin D metabolites and PTH‐related protein occur in 20% of patients with hypercalcemia caused by malignancy.13, 14 Diffuse osteopenia on skeletal survey is a prominent feature of renal osteodystrophy or osteoporosis related to chronic corticosteroid use. However, in a patient with diffuse osteopenia and hypercalcemia, clinicians must consider multiple myeloma and other lymphoproliferative disorders; the absence of osteoblastic or osteolytic lesions and a normal alkaline phosphatase do not rule out these diagnoses. When the results of serum and urine protein electrophoresis exclude multiple myeloma, the next investigation should be a bone biopsy to exclude PLB, an uncommon cause of anemia, hypercalcemia, and osteopenic, painful bones.
Key Points for Hospitalists
-
Normal total alkaline phosphatase does not exclude primary or metastatic bone malignancy. While a normal level tends to predict a negative bone scan, further diagnostic tests are needed to exclude bone malignancy if high clinical suspicion exists.
-
The degree of hypercalcemia is useful diagnostically; values above 13 mg/dL are most often due to malignancy.
-
Hypercalcemia in transplant patients deserves special attention due to an increased risk of malignancy, including squamous cancers of the lips and skin, lymphoproliferative disorders, and bronchogenic carcinoma.
-
While rare, consider primary lymphoma of the bone in patients with hypercalcemia and bone pain, along with the more common diagnoses of multiple myeloma and metastatic bone disease.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.
- ,.Clinical Physiology of Acid‐Base and Electrolyte Disorders.5th ed.New York:McGraw‐Hill;2001:754–758.
- ,.Hypercalcemia: clinical manifestations, pathogenesis, diagnosis, and management. In: Favus MJ, ed.Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism.5th ed.Washington, DC:American Society for Bone and Mineral Research;2003:225–230.
- ,,, et al.Malignancy after renal transplantation: analysis of incidence and risk factors in 1700 patients followed during a 25‐year period.Transplant Proc.1997;29:831–833.
- ,.Malignancy‐associated hypercalcemia. In: DeGroot L, Jameson LJ, eds.Endocrinology.4th ed.Philadelphia, PA:Saunders;2001:1093–1100.
- ,.Diagnosis and management of posttransplant lymphoproliferative disorder in solid‐organ transplant recipients.Clin Infect Dis.2001;33(suppl 1):S38–S46.
- ,,, et al.Epstein‐Barr virus‐induced posttransplant lymphoproliferative disorders: ASTS/ASTP EBV‐PTLD Task Force and The Mayo Clinic Organized International Consensus Development Meeting.Transplantation.1999;68:1517–1525.
- ,,, et al.Primary bone lymphoma: a new and detailed characterization of 28 patients in a single‐institution study.Jpn J Clin Oncol.2007;37(3):216–223.
- ,,, et al.Diffuse large B‐cell lymphoma of bone. An analysis of differentiation‐associated antigens with clinical correlation.Am J Surg Pathol.2003;27:1269–1277.
- ,,,,,.Primary bone lymphoma: radiographic‐MR imaging correlation.Radiographics.2003;23:1371–1383.
- ,,, et al.Primary bone lymphoma in 24 patients treated between 1955 and 1999.Clin Orthop.2002;397:271–280.
- ,,, et al.A clinicopathological retrospective study of 131 patients with primary bone lymphoma: a population‐based study of successively treated cohorts from the British Columbia Cancer Agency.Ann Oncol.2007;18:129.
- ,,, et al.Malignant lymphoma of bone.Cancer.1986;58:2646–2655.
- .Hypercalcemia in malignant lymphoma and leukemia.Ann N Y Acad Sci.1974;230:240–246.
- .Incidence and prognostic significance of hypercalcemia in B‐cell non‐Hodgkin's lymphoma. [Letter]J Clin Pathol.2002;55:637–638.
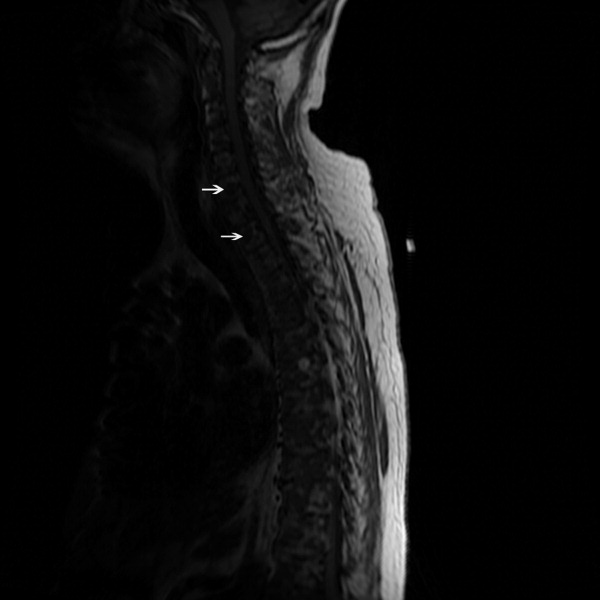
Aching for a Diagnosis
A 23‐year‐old Caucasian man presented to an outpatient clinic with a sore throat and associated subjective fevers. His evaluation included a negative rapid streptococcus test; nevertheless, he was empirically treated with amoxicillin. The following day, he experienced increasing sore throat and presented to the emergency department (ED). He was treated with prednisone and morphine sulfate and discharged home with azithromycin.
Initial considerations in a healthy young man who presents with fever and pharyngitis should focus on common infectious etiologies. Viral illnesses are the most frequent causes of sore throat and fever. These often manifest as mononucleosis‐like illnesses and include Epstein‐Barr virus (EBV) and cytomegalovirus (CMV). In this age group, it is also critical to consider sexually transmitted diseases (STDs) such as gonorrhea, human immunodeficiency virus (HIV), herpes simplex virus, and syphilis. Consideration of streptococcal pharyngitis is important. Since the rapid streptococcal antigen test is neither sensitive nor specific, confirmation of infection should be based on clinical findings and a culture of the pharynx for group A Streptococcus. Other common etiologies of fever and pharyngitis include acute or chronic sinusitis with postnasal drainage. Due to the progressive nature of the sore throat, there should be an evaluation for difficulty swallowing, problems phonating, or neck discomfort, any of which would be concerning for a retropharyngeal abscess. Additional history should be obtained with focus on sexual history, previous STDs, recent sick contacts, and other supporting signs and symptoms of viral illnesses.
Eight days after the initial onset of symptoms, the patient developed acute low back pain. The back pain was midline, severe, and constant around the lumbar spine. There was no saddle anesthesia, bowel or bladder dysfunction, or weakness or numbness in the extremities. He also noted swelling of the left fourth metacarpophalangeal joint and an erythematous rash on his right knee and anterior tibial region of the right leg. He continued to experience subjective fevers, sore throat, and swollen neck glands. Due to the severity and discomfort of symptoms, the patient returned to the ED.
With no history of trauma, the subsequent development of acute low back pain may be related to the patient's sore throat and fever. Monoarticular arthritis with contralateral skin lesions should raise suspicion for a systemic process, particularly infection or a rheumatologic syndrome. Infectious etiologies would include rheumatic fever, endocarditis with septic emboli, and osteomyelitis. Rheumatologic causes, such as ankylosing spondylitis and juvenile rheumatoid arthritis (RA), are also possibilities. The infectious evaluation should include an assessment of a history of intravenous drug use (IVDU) and underlying valvular disorders, which will increase the risk for endocarditis and therefore septic emboli. Acute HIV infection can be seen as early as 1 to 2 weeks postexposure and should be considered as well. Appropriate testing would include both conventional HIV antibody tests and HIV viral load assay. Lastly, in considering the patient's symptoms, obtaining his travel history to identify risk for Lyme disease would also be appropriate.
The patient did not report any further positive findings on review of systems. He did not have any significant past medical history and did not take any chronic medications. He had no sick contacts. He rarely drank alcohol and denied IVDU and sexual activity over the past year. He was previously involved in monogamous relationships with women. His last HIV test, 1 year prior, was negative. He did not have any history of STDs. He was a graduate student in computer science and lived in southern California. He had recently traveled to central California and France for 2 weeks, staying in larger cities. He had not been hiking during that time. His family history was significant for hypertension.
The travel history is provocative for 3 diseases of the reticuloendothelial system with possible systemic manifestations. First, toxoplasmosis, which is endemic in France where rare or raw beef and lamb are frequently consumed. It may present as a mononucleosis‐like illness and rarely as atypical pneumonia. Second, tuberculosis, which is also endemic in France, especially in major cities. Although most commonly a self‐limited respiratory disease, it may disseminate with systemic symptoms. Third, primary coccidioidomycosis, which is prevalent in the central valleys of California. The climate and wind patterns lead to aerosolization of the spores and make this a common respiratory pathogen.
The physical exam should include a detailed evaluation of the eyes for uveitis and iritis, seen in some rheumatologic disorders. A pharyngeal exam with assessment for exudate can support streptococcal pharyngitis or diphtheria. Evaluation for lymphadenopathy, while nonspecific, would be important for streptococcal pharyngitis, rheumatic fever, and juvenile RA. Further characterizing the rash is essential in distinguishing viral exanthems from the fleeting salmon‐colored maculopapular rash of juvenile RA. Assessment for peripheral stigmata of endocarditis should be done. A thorough joint exam should evaluate evidence of inflammatory or infectious joint disease.
On physical exam, he was a thin man who appeared anxious but in no acute distress. His temperature was 36.7C, blood pressure 111/68 mm Hg, heart rate 83 beats/minute, respiratory rate 16 breaths/minute, and oxygen saturation 99% on room air. Erythema was noted in the posterior oropharynx with no tonsillar exudate. There were several subcentimeter, nontender, and mobile lymph nodes in the anterior cervical chain bilaterally. The cardiovascular exam revealed normal sinus rhythm with a 2/6 systolic murmur at the apex, without radiation. His lungs were clear to auscultation. Skin exam revealed 2 blanching erythematous, indurated, and tender lesions on the right pretibial region, 2‐cm and 4‐cm in diameter. Two other similar, but smaller, lesions were noted on the left upper extremity and left ankle. His lumbar spine was slightly tender to touch. A complete joint exam was normal, including the left fourth metacarpophalangeal joint. Neurological exam, including bilateral strength, sensation, reflexes, and gait, was unremarkable.
Younger patients are subject to social‐acceptance bias and can deny sexual activity on initial inquiry. An objective evaluation for STDs with serologic workup should still be pursued. The cervical lymphadenopathy and tonsillar erythema continue to suggest a viral illness. While the systolic murmur may be physiologic, subjective fevers, disseminated cutaneous lesions, and arthritis warrant evaluation for bacterial endocarditis with blood cultures and an echocardiogram.
On exam, there is no evidence of true joint involvement and this decreases the likelihood of rheumatologic conditions, such as ankylosing spondylitis and juvenile RA. However, the skin lesions are suspicious for erythema nodosum (EN), which should prompt a biopsy and an evaluation for infectious etiologies. Serologies should include evaluation of Chlamydia, Mycoplasma, Coccidioides, and Histoplasma. I would also examine the feet carefully for potential transcutaneous inoculation by microorganisms that can produce a rash similar to EN. For instance, penetrating skin trauma can lead to pseudomonal infection. Brucella (from ingesting unpasteurized milk or milk products), Bartonella (from the scratches of feline animals), and Francisella tularensis (from rabbit exposure) can also produce skin lesions that mimic EN. These are best distinguished through a detailed history, concomitant serologic workup, and biopsy. Other noninfectious etiologies of EN can include inflammatory bowel disease, Behcet's, and sarcoidosis; however, the patient does not currently report any symptoms supporting these diagnoses. In addition to the above evaluation, complete blood count with differential, liver function tests, creatinine, and urinalysis should be obtained.
The patient's white blood cell (WBC) count was 12,100/L with 73% neutrophils, 14% lymphocytes, and 12% monocytes. Hemoglobin was 11.8 g/dL and platelet count 292,000/L. Chemistry panel and liver function tests were unremarkable. Erythrocyte sedimentation rate (ESR) was 71 mm/hour (range, 010). Urinalysis was negative for protein and red blood cells. Chest x‐ray did not illustrate any abnormalities. Computed tomography (CT) of the lumbar spine revealed a small posterior disc bulge at L4‐5 and L5‐S1.
The moderate leukocytosis with neutrophilic predominance and monocytosis raises concern for a systemic inflammatory process; the elevated ESR further supports this. Monocytosis can be seen in a number of infectious, autoimmune, and malignant conditions. Tuberculosis, brucellosis, bacterial endocarditis, syphilis, infectious mononucleosis, and viral illnesses are among the infections typically characterized by monocytosis. Autoimmune illnesses, such as systemic lupus erythematosus and RA can also have similar presentations. The patient does not have any features of an underlying malignancy, such as weight loss or night sweats; however, if the autoimmune and infectious evaluations are negative, Hodgkin's disease and certain leukemias should be considered. There is no evidence of osteomyelitis on the spine CT, which decreases the possibility of (but does not exclude) infectious or rheumatologic conditions of the spine. I would suggest a comprehensive laboratory evaluation for the discussed infectious and rheumatologic disorders.
The patient's back pain was controlled with antiinflammatory medications overnight. Due to the patient's stable condition and lack of a diagnosis, empiric antibiotics were not initiated. An extensive workup was sent, including antistreptolysin O, polymerase chain reaction for Chlamydia, Neisseria gonorrhoeae, EBV, and parvovirus B19 DNA, serologies for Coccidioides immunoglobulin G (IgG) and IgM, urinary antigen for Histoplasma, HIV enzyme‐linked immunosorbent assay (ELISA) and Western blot, serum angiotensin‐converting enzyme level, C‐reactive protein, rheumatoid factor, antinuclear antibody, and antidouble‐stranded DNA antibodies.
Without a clear diagnosis, I would recommend against treatment with empiric antibiotics. At this point, I agree with waiting for the results of the pending workup.
On hospital day 1, the patient developed severe acute left ankle pain. On examination, the joint was exquisitely tender with decreased range of motion. Arthrocentesis was promptly performed. The synovial fluid WBC count was 1370/L with a differential of 82% neutrophils and 18% monocytes. No crystals were identified and the bacterial Gram stain was negative. He was treated with antiinflammatory medications. Bacterial blood cultures, obtained from the day of admission, were negative.
The arthrocentesis reveals a polymorphonuclear‐predominant fluid; however, the WBC count in the fluid is only mildly elevated. While the elevated monocyte count could again be consistent with viral arthropathies or juvenile RA, there is currently no systemic evidence of either illness. It is important to await the results of the final cultures, but the low WBC count and negative Gram stain decrease the probability of a septic joint. Empiric antibiotics to cover Gram‐positive organisms and gonococci would not be unreasonable, pending joint fluid culture results. The monocytosis could also be consistent with a fungal arthritis.
On hospital day 2, the results of the rheumatologic and infectious evaluation were negative with the exception of C‐reactive protein, which was 11.8 mg/dL (normal, <0.8), antinuclear antibody titer of 1:160 (normal, <1:40), Coccidioides IgM enzyme immunoassay (EIA) 0.710 (negative, <0.150), and Coccidioides tube‐precipitin (TP) immunodiffusion (ID) antibody‐positive. Coccidioides IgG EIA was negative.
The serologic tests are consistent with primary coccidioidomycosis. This is often a challenging diagnosis due to the nonspecific signs and symptoms, such as cough, fever, myalgias, and fatigue. Since screening EIAs are sensitive but not specific, concern for coccidioidomycosis or abnormal EIA results should prompt confirmatory testing with complement fixation titers (CF) and TP ID. Treatment with fluconazole should be initiated. Since the patient does not have central nervous system (CNS) symptoms, I would not recommend lumbar puncture at this point. However, a bone scan should be done for assessment of the back pain.
The patient was diagnosed with primary coccidioidomycosis infection with immune‐complexmediated arthritis and EN. A bone scan was negative. The patient was treated with fluconazole and discharged with 3 months of therapy. At follow‐up clinic visits after completion of therapy, his symptoms had resolved and his titers had normalized.
Discussion
The diagnosis of coccidioidomycosis is often challenging due to its protean manifestations. Four clinical syndromes are commonly seen: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease involving the skin, lymph nodes, bones, joints, and meninges. The most common clinical manifestation, acute pneumonia, may be indistinguishable from other causes of community‐acquired pneumonia (CAP). In a study of CAP in Arizona, 29% of cases were positive for coccidioidal infection through serologic evaluation.1 Features suggestive of coccidioidal infection include fatigue, severe headache, and pleuritic chest pain. Adenopathy in the hilar or paratracheal regions can be seen in 25% of infections.2 Chronic progressive pneumonia refers to infections in which symptoms, including cough, hemoptysis, and weight loss, persist for longer than 3 months. Pulmonary nodules and cavities are residual manifestations of primary pulmonary infection and occur in 2% to 8% of cases. Extrapulmonary disease develops in less than 5% of immunocompetent patients with primary pulmonary infection, with higher prevalence in patients of African American and Filipino decent. Immunocompromised patients are at increased risk for extrapulmonary infection. The most serious site of extrapulmonary disease is the meninges. Coccidioidal meningitis carries nearly 100% mortality rate if left untreated. The presentation is variable with up to 75% of cases reporting headache. While coccidioidal pneumonia also frequently presents with headache, symptoms including altered mental status, focal neurological deficits, and persistent or progressive headache are more suggestive of meningeal disease.3
Patients with any presentation of coccidioidomycosis can display immune‐mediated manifestations such as EN, arthralgias (desert rheumatism), and in some cases mild conjunctivitis.4 It is hypothesized that these findings occur due to a hypersensitivity reaction to coccidioidomycosis.4 EN is an inflammatory process of the subcutaneous fat, which presents as tender and erythematous nodules typically on the lower extremities. EN is not a disease entity or site of metastatic infection, but a response to underlying illness. Its recognition should trigger a search for the primary etiology, as guided by the patient's history and clinical presentation. The differential diagnosis for EN is broad and includes rheumatologic, infectious, medication‐related, inflammatory, and idiopathic processes (Table 1). Coccidioidomycosis should be strongly considered based on geographical location, with the vast majority of cases seen in southern California, Arizona, Nevada, New Mexico, and Texas. While the pathophysiology of EN has not been completely elucidated, the lesions may reflect a vigorous immune response conferring a protective advantage. Interestingly, a study of pregnant women with coccidioidomycosis revealed a decreased incidence of disseminated disease in patients with EN.5, 6
| Rheumatologic/autoimmune |
| Systemic lupus erythematosus |
| Wegener's granulomatosis |
| Sarcoidosis |
| Infectious |
| Streptococcus pyogenes causing pharyngitis (most common) |
| Borrelia burgdorferi |
| Mycoplasma pneumoniae |
| Bartonella henselae |
| Shigella |
| Campylobacter jejuni |
| Salmonella |
| Yersinia enterocolitica |
| Chlamydia |
| Brucella |
| Escherichia coli |
| Treponema pallidum |
| Mycobacterium leprae |
| Neisseria gonorrhoeae |
| Mycobacterium tuberculosis |
| Human immunodeficiency virus |
| Epstein‐Barr virus |
| Cytomegalovirus |
| Influenza |
| Varicella Zoster virus |
| Coccidioides immitis |
| Histoplasma capsulatum |
| Blastomyces dermatitidis |
| Dermatophytic fungal infections (rare) |
| Gastrointestinal |
| Ulcerative colitis |
| Crohn's disease |
| Celiac disease |
| Behcet's disease |
| Medications |
| Oral contraceptives |
| Proton pump inhibitors |
| Sulfonamides |
| Leukotriene modifiers (montelukast) |
| Hepatitis B vaccine |
| Isoretinoin |
| Miscellaneous |
| Hodgkins lymphoma |
| Sweet's syndrome |
Coccidioidomycosis is also associated with immune‐mediated arthralgias and arthritis. These manifestations occur in up to one‐third of patients with concomitant EN. Arthritis may be monoarticular or polyarticular, often affecting large joints such as the knees or ankles. It is important to note that septic arthritis can also occur and should be differentiated from rheumatism by joint aspiration.
The diagnosis of coccidioidomycosis can be made by serologic testing, direct isolation of the organism on culture, or visualization on tissue biopsy. Of these methods, serologic testing is most commonly utilized. The 2007 Infectious Disease Society of America (IDSA) and American Thoracic Society guidelines recommend diagnostic testing in hospitalized patients with CAP who reside in or have recently traveled (within 2 weeks) to endemic areas.7 There are multiple approaches to serologic diagnosis based on identification of IgM or IgG antibodies to various coccidioidal antigens. During the early phase of infection, TP ID and EIA can be utilized to detect IgM antibodies. While EIA testing has 92% sensitivity, it has high rates of false‐positive results, and therefore confirmatory testing with ID is recommended. ID has variable sensitivity, but 90% of patients will test positive by 3 weeks of infection.8 During the later phase of the infection, IgG antibodies are detected either quantitatively by CF or qualitatively by ID and EIA. CF can provide information on the severity of illness and prognosis based on titer levels, as well as serving as a marker for response to treatment.2 Positive titers greater than 1:32 suggest disseminated disease. In addition, CF titer in the cerebrospinal fluid is the test of choice in diagnosis of coccidioidal meningitis. An evaluation for disseminated disease should be initiated if the patient has any risk factors or clinically concerning symptoms for bone or CNS involvement. This evaluation includes a bone scan and lumbar puncture. All patients should be assessed for immunocompromised status.
The management of coccidioidomycosis is based on the extent of infection, the severity of illness, and the immune status of the patient. In 95% of cases of uncomplicated pulmonary disease in an immunocompetent host, the symptoms will resolve without treatment with antifungal agents.9 The decision to treat uncomplicated pulmonary disease is based on severity of illness. While there is no consensus recommendation, commonly used indicators for treatment include persistent fever, age >55 years, symptoms greater than 2 months, hilar adenopathy, diffuse pulmonary infiltrates, weight loss, and inability to work.9 In patients with chronic progressive pneumonia or extrapulmonary involvement, treatment with antifungal medications should be initiated. While fluconazole remains the preferred treatment in coccidioidal pneumonia and meningitis, amphotericin B preparations should be considered for diffuse coccidioidal pneumonia and disseminated disease, including refractory meningitis.9 The use of newer azoles, particularly posaconazole, has been studied in a limited number of patients with refractory coccidioidomycosis with improvement in symptoms.10 Frequent follow‐up visits are recommended to detect progression of disease or to document resolution, with improving symptoms and decreasing titers. Duration of therapy in uncomplicated cases should be at least 3 months. Treatment of extrapulmonary disease can span years, and in the case of meningitis lifetime treatment is recommended given the high rate of relapse.
While the patient and the clinicians were aching for a diagnosis after the initial negative evaluation, recognition of the immunologic manifestations of coccidioidomycosis was essential in this case. Coccidioidomycosis should be considered in patients presenting with EN, regardless of presence of concurrent pulmonary symptoms; particularly in patients living in or with recent travel to endemic areas. Furthermore, the severity of symptoms can guide the decision and duration of treatment.
Teaching Points
-
Coccidioidomycosis has 4 main clinical presentations: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease.
-
Independent of pulmonary symptoms, coccidioidomycosis can present with immune‐mediated manifestations, such as EN and arthritis.
-
The diagnosis of coccidioidomycosis often relies on serologic testing for early and late infection.
-
Treatment of coccidioidomycosis is based on risk factors and severity of symptoms. High‐risk and symptomatic patients can be treated with fluconazole or amphotericin B.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,,, et al.Coccidioidomycosis as a common cause of community‐acquired pneumonia.Emerg Infect Dis.2006;12:958–962.
- ,.Coccidioidomycosis.Mayo Clin Proc.2008;83:343–349.
- ,.Coccidioidal meningitis.Clin Infect Dis.2006;42:103–107.
- ,.Coccidioidomycosis: host response and vaccine development.Clin Microbiol Rev.2004;17:804–839.
- ,,.Erythema nodosum in pregnant patients with coccidioidomycosis.Clin Infect Dis.1998;27:1201–1203.
- .Protective effects of erythema nodosum in coccidioidomycosis.Lancet.1999;353:168.
- ,,, et al.Infectious Disease Society of America/American Thoracic Society consensus guidelines on management of community acquired pneumonia in adults.Clin Infect Dis.2007;44:S27–S72
- .Laboratory aspects in the diagnosis of coccidioidomycosis.Ann N Y Acad Sci.2007;1111:301–314.
- ,,, et al.Coccidioidomycosis.Clin Infect Dis.2005;41:1217–1223.
- ,,,,.Refractory coccidioidomycosis treated with posaconazole.Clin Infect Dis.2005;40:1770–1776.
A 23‐year‐old Caucasian man presented to an outpatient clinic with a sore throat and associated subjective fevers. His evaluation included a negative rapid streptococcus test; nevertheless, he was empirically treated with amoxicillin. The following day, he experienced increasing sore throat and presented to the emergency department (ED). He was treated with prednisone and morphine sulfate and discharged home with azithromycin.
Initial considerations in a healthy young man who presents with fever and pharyngitis should focus on common infectious etiologies. Viral illnesses are the most frequent causes of sore throat and fever. These often manifest as mononucleosis‐like illnesses and include Epstein‐Barr virus (EBV) and cytomegalovirus (CMV). In this age group, it is also critical to consider sexually transmitted diseases (STDs) such as gonorrhea, human immunodeficiency virus (HIV), herpes simplex virus, and syphilis. Consideration of streptococcal pharyngitis is important. Since the rapid streptococcal antigen test is neither sensitive nor specific, confirmation of infection should be based on clinical findings and a culture of the pharynx for group A Streptococcus. Other common etiologies of fever and pharyngitis include acute or chronic sinusitis with postnasal drainage. Due to the progressive nature of the sore throat, there should be an evaluation for difficulty swallowing, problems phonating, or neck discomfort, any of which would be concerning for a retropharyngeal abscess. Additional history should be obtained with focus on sexual history, previous STDs, recent sick contacts, and other supporting signs and symptoms of viral illnesses.
Eight days after the initial onset of symptoms, the patient developed acute low back pain. The back pain was midline, severe, and constant around the lumbar spine. There was no saddle anesthesia, bowel or bladder dysfunction, or weakness or numbness in the extremities. He also noted swelling of the left fourth metacarpophalangeal joint and an erythematous rash on his right knee and anterior tibial region of the right leg. He continued to experience subjective fevers, sore throat, and swollen neck glands. Due to the severity and discomfort of symptoms, the patient returned to the ED.
With no history of trauma, the subsequent development of acute low back pain may be related to the patient's sore throat and fever. Monoarticular arthritis with contralateral skin lesions should raise suspicion for a systemic process, particularly infection or a rheumatologic syndrome. Infectious etiologies would include rheumatic fever, endocarditis with septic emboli, and osteomyelitis. Rheumatologic causes, such as ankylosing spondylitis and juvenile rheumatoid arthritis (RA), are also possibilities. The infectious evaluation should include an assessment of a history of intravenous drug use (IVDU) and underlying valvular disorders, which will increase the risk for endocarditis and therefore septic emboli. Acute HIV infection can be seen as early as 1 to 2 weeks postexposure and should be considered as well. Appropriate testing would include both conventional HIV antibody tests and HIV viral load assay. Lastly, in considering the patient's symptoms, obtaining his travel history to identify risk for Lyme disease would also be appropriate.
The patient did not report any further positive findings on review of systems. He did not have any significant past medical history and did not take any chronic medications. He had no sick contacts. He rarely drank alcohol and denied IVDU and sexual activity over the past year. He was previously involved in monogamous relationships with women. His last HIV test, 1 year prior, was negative. He did not have any history of STDs. He was a graduate student in computer science and lived in southern California. He had recently traveled to central California and France for 2 weeks, staying in larger cities. He had not been hiking during that time. His family history was significant for hypertension.
The travel history is provocative for 3 diseases of the reticuloendothelial system with possible systemic manifestations. First, toxoplasmosis, which is endemic in France where rare or raw beef and lamb are frequently consumed. It may present as a mononucleosis‐like illness and rarely as atypical pneumonia. Second, tuberculosis, which is also endemic in France, especially in major cities. Although most commonly a self‐limited respiratory disease, it may disseminate with systemic symptoms. Third, primary coccidioidomycosis, which is prevalent in the central valleys of California. The climate and wind patterns lead to aerosolization of the spores and make this a common respiratory pathogen.
The physical exam should include a detailed evaluation of the eyes for uveitis and iritis, seen in some rheumatologic disorders. A pharyngeal exam with assessment for exudate can support streptococcal pharyngitis or diphtheria. Evaluation for lymphadenopathy, while nonspecific, would be important for streptococcal pharyngitis, rheumatic fever, and juvenile RA. Further characterizing the rash is essential in distinguishing viral exanthems from the fleeting salmon‐colored maculopapular rash of juvenile RA. Assessment for peripheral stigmata of endocarditis should be done. A thorough joint exam should evaluate evidence of inflammatory or infectious joint disease.
On physical exam, he was a thin man who appeared anxious but in no acute distress. His temperature was 36.7C, blood pressure 111/68 mm Hg, heart rate 83 beats/minute, respiratory rate 16 breaths/minute, and oxygen saturation 99% on room air. Erythema was noted in the posterior oropharynx with no tonsillar exudate. There were several subcentimeter, nontender, and mobile lymph nodes in the anterior cervical chain bilaterally. The cardiovascular exam revealed normal sinus rhythm with a 2/6 systolic murmur at the apex, without radiation. His lungs were clear to auscultation. Skin exam revealed 2 blanching erythematous, indurated, and tender lesions on the right pretibial region, 2‐cm and 4‐cm in diameter. Two other similar, but smaller, lesions were noted on the left upper extremity and left ankle. His lumbar spine was slightly tender to touch. A complete joint exam was normal, including the left fourth metacarpophalangeal joint. Neurological exam, including bilateral strength, sensation, reflexes, and gait, was unremarkable.
Younger patients are subject to social‐acceptance bias and can deny sexual activity on initial inquiry. An objective evaluation for STDs with serologic workup should still be pursued. The cervical lymphadenopathy and tonsillar erythema continue to suggest a viral illness. While the systolic murmur may be physiologic, subjective fevers, disseminated cutaneous lesions, and arthritis warrant evaluation for bacterial endocarditis with blood cultures and an echocardiogram.
On exam, there is no evidence of true joint involvement and this decreases the likelihood of rheumatologic conditions, such as ankylosing spondylitis and juvenile RA. However, the skin lesions are suspicious for erythema nodosum (EN), which should prompt a biopsy and an evaluation for infectious etiologies. Serologies should include evaluation of Chlamydia, Mycoplasma, Coccidioides, and Histoplasma. I would also examine the feet carefully for potential transcutaneous inoculation by microorganisms that can produce a rash similar to EN. For instance, penetrating skin trauma can lead to pseudomonal infection. Brucella (from ingesting unpasteurized milk or milk products), Bartonella (from the scratches of feline animals), and Francisella tularensis (from rabbit exposure) can also produce skin lesions that mimic EN. These are best distinguished through a detailed history, concomitant serologic workup, and biopsy. Other noninfectious etiologies of EN can include inflammatory bowel disease, Behcet's, and sarcoidosis; however, the patient does not currently report any symptoms supporting these diagnoses. In addition to the above evaluation, complete blood count with differential, liver function tests, creatinine, and urinalysis should be obtained.
The patient's white blood cell (WBC) count was 12,100/L with 73% neutrophils, 14% lymphocytes, and 12% monocytes. Hemoglobin was 11.8 g/dL and platelet count 292,000/L. Chemistry panel and liver function tests were unremarkable. Erythrocyte sedimentation rate (ESR) was 71 mm/hour (range, 010). Urinalysis was negative for protein and red blood cells. Chest x‐ray did not illustrate any abnormalities. Computed tomography (CT) of the lumbar spine revealed a small posterior disc bulge at L4‐5 and L5‐S1.
The moderate leukocytosis with neutrophilic predominance and monocytosis raises concern for a systemic inflammatory process; the elevated ESR further supports this. Monocytosis can be seen in a number of infectious, autoimmune, and malignant conditions. Tuberculosis, brucellosis, bacterial endocarditis, syphilis, infectious mononucleosis, and viral illnesses are among the infections typically characterized by monocytosis. Autoimmune illnesses, such as systemic lupus erythematosus and RA can also have similar presentations. The patient does not have any features of an underlying malignancy, such as weight loss or night sweats; however, if the autoimmune and infectious evaluations are negative, Hodgkin's disease and certain leukemias should be considered. There is no evidence of osteomyelitis on the spine CT, which decreases the possibility of (but does not exclude) infectious or rheumatologic conditions of the spine. I would suggest a comprehensive laboratory evaluation for the discussed infectious and rheumatologic disorders.
The patient's back pain was controlled with antiinflammatory medications overnight. Due to the patient's stable condition and lack of a diagnosis, empiric antibiotics were not initiated. An extensive workup was sent, including antistreptolysin O, polymerase chain reaction for Chlamydia, Neisseria gonorrhoeae, EBV, and parvovirus B19 DNA, serologies for Coccidioides immunoglobulin G (IgG) and IgM, urinary antigen for Histoplasma, HIV enzyme‐linked immunosorbent assay (ELISA) and Western blot, serum angiotensin‐converting enzyme level, C‐reactive protein, rheumatoid factor, antinuclear antibody, and antidouble‐stranded DNA antibodies.
Without a clear diagnosis, I would recommend against treatment with empiric antibiotics. At this point, I agree with waiting for the results of the pending workup.
On hospital day 1, the patient developed severe acute left ankle pain. On examination, the joint was exquisitely tender with decreased range of motion. Arthrocentesis was promptly performed. The synovial fluid WBC count was 1370/L with a differential of 82% neutrophils and 18% monocytes. No crystals were identified and the bacterial Gram stain was negative. He was treated with antiinflammatory medications. Bacterial blood cultures, obtained from the day of admission, were negative.
The arthrocentesis reveals a polymorphonuclear‐predominant fluid; however, the WBC count in the fluid is only mildly elevated. While the elevated monocyte count could again be consistent with viral arthropathies or juvenile RA, there is currently no systemic evidence of either illness. It is important to await the results of the final cultures, but the low WBC count and negative Gram stain decrease the probability of a septic joint. Empiric antibiotics to cover Gram‐positive organisms and gonococci would not be unreasonable, pending joint fluid culture results. The monocytosis could also be consistent with a fungal arthritis.
On hospital day 2, the results of the rheumatologic and infectious evaluation were negative with the exception of C‐reactive protein, which was 11.8 mg/dL (normal, <0.8), antinuclear antibody titer of 1:160 (normal, <1:40), Coccidioides IgM enzyme immunoassay (EIA) 0.710 (negative, <0.150), and Coccidioides tube‐precipitin (TP) immunodiffusion (ID) antibody‐positive. Coccidioides IgG EIA was negative.
The serologic tests are consistent with primary coccidioidomycosis. This is often a challenging diagnosis due to the nonspecific signs and symptoms, such as cough, fever, myalgias, and fatigue. Since screening EIAs are sensitive but not specific, concern for coccidioidomycosis or abnormal EIA results should prompt confirmatory testing with complement fixation titers (CF) and TP ID. Treatment with fluconazole should be initiated. Since the patient does not have central nervous system (CNS) symptoms, I would not recommend lumbar puncture at this point. However, a bone scan should be done for assessment of the back pain.
The patient was diagnosed with primary coccidioidomycosis infection with immune‐complexmediated arthritis and EN. A bone scan was negative. The patient was treated with fluconazole and discharged with 3 months of therapy. At follow‐up clinic visits after completion of therapy, his symptoms had resolved and his titers had normalized.
Discussion
The diagnosis of coccidioidomycosis is often challenging due to its protean manifestations. Four clinical syndromes are commonly seen: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease involving the skin, lymph nodes, bones, joints, and meninges. The most common clinical manifestation, acute pneumonia, may be indistinguishable from other causes of community‐acquired pneumonia (CAP). In a study of CAP in Arizona, 29% of cases were positive for coccidioidal infection through serologic evaluation.1 Features suggestive of coccidioidal infection include fatigue, severe headache, and pleuritic chest pain. Adenopathy in the hilar or paratracheal regions can be seen in 25% of infections.2 Chronic progressive pneumonia refers to infections in which symptoms, including cough, hemoptysis, and weight loss, persist for longer than 3 months. Pulmonary nodules and cavities are residual manifestations of primary pulmonary infection and occur in 2% to 8% of cases. Extrapulmonary disease develops in less than 5% of immunocompetent patients with primary pulmonary infection, with higher prevalence in patients of African American and Filipino decent. Immunocompromised patients are at increased risk for extrapulmonary infection. The most serious site of extrapulmonary disease is the meninges. Coccidioidal meningitis carries nearly 100% mortality rate if left untreated. The presentation is variable with up to 75% of cases reporting headache. While coccidioidal pneumonia also frequently presents with headache, symptoms including altered mental status, focal neurological deficits, and persistent or progressive headache are more suggestive of meningeal disease.3
Patients with any presentation of coccidioidomycosis can display immune‐mediated manifestations such as EN, arthralgias (desert rheumatism), and in some cases mild conjunctivitis.4 It is hypothesized that these findings occur due to a hypersensitivity reaction to coccidioidomycosis.4 EN is an inflammatory process of the subcutaneous fat, which presents as tender and erythematous nodules typically on the lower extremities. EN is not a disease entity or site of metastatic infection, but a response to underlying illness. Its recognition should trigger a search for the primary etiology, as guided by the patient's history and clinical presentation. The differential diagnosis for EN is broad and includes rheumatologic, infectious, medication‐related, inflammatory, and idiopathic processes (Table 1). Coccidioidomycosis should be strongly considered based on geographical location, with the vast majority of cases seen in southern California, Arizona, Nevada, New Mexico, and Texas. While the pathophysiology of EN has not been completely elucidated, the lesions may reflect a vigorous immune response conferring a protective advantage. Interestingly, a study of pregnant women with coccidioidomycosis revealed a decreased incidence of disseminated disease in patients with EN.5, 6
| Rheumatologic/autoimmune |
| Systemic lupus erythematosus |
| Wegener's granulomatosis |
| Sarcoidosis |
| Infectious |
| Streptococcus pyogenes causing pharyngitis (most common) |
| Borrelia burgdorferi |
| Mycoplasma pneumoniae |
| Bartonella henselae |
| Shigella |
| Campylobacter jejuni |
| Salmonella |
| Yersinia enterocolitica |
| Chlamydia |
| Brucella |
| Escherichia coli |
| Treponema pallidum |
| Mycobacterium leprae |
| Neisseria gonorrhoeae |
| Mycobacterium tuberculosis |
| Human immunodeficiency virus |
| Epstein‐Barr virus |
| Cytomegalovirus |
| Influenza |
| Varicella Zoster virus |
| Coccidioides immitis |
| Histoplasma capsulatum |
| Blastomyces dermatitidis |
| Dermatophytic fungal infections (rare) |
| Gastrointestinal |
| Ulcerative colitis |
| Crohn's disease |
| Celiac disease |
| Behcet's disease |
| Medications |
| Oral contraceptives |
| Proton pump inhibitors |
| Sulfonamides |
| Leukotriene modifiers (montelukast) |
| Hepatitis B vaccine |
| Isoretinoin |
| Miscellaneous |
| Hodgkins lymphoma |
| Sweet's syndrome |
Coccidioidomycosis is also associated with immune‐mediated arthralgias and arthritis. These manifestations occur in up to one‐third of patients with concomitant EN. Arthritis may be monoarticular or polyarticular, often affecting large joints such as the knees or ankles. It is important to note that septic arthritis can also occur and should be differentiated from rheumatism by joint aspiration.
The diagnosis of coccidioidomycosis can be made by serologic testing, direct isolation of the organism on culture, or visualization on tissue biopsy. Of these methods, serologic testing is most commonly utilized. The 2007 Infectious Disease Society of America (IDSA) and American Thoracic Society guidelines recommend diagnostic testing in hospitalized patients with CAP who reside in or have recently traveled (within 2 weeks) to endemic areas.7 There are multiple approaches to serologic diagnosis based on identification of IgM or IgG antibodies to various coccidioidal antigens. During the early phase of infection, TP ID and EIA can be utilized to detect IgM antibodies. While EIA testing has 92% sensitivity, it has high rates of false‐positive results, and therefore confirmatory testing with ID is recommended. ID has variable sensitivity, but 90% of patients will test positive by 3 weeks of infection.8 During the later phase of the infection, IgG antibodies are detected either quantitatively by CF or qualitatively by ID and EIA. CF can provide information on the severity of illness and prognosis based on titer levels, as well as serving as a marker for response to treatment.2 Positive titers greater than 1:32 suggest disseminated disease. In addition, CF titer in the cerebrospinal fluid is the test of choice in diagnosis of coccidioidal meningitis. An evaluation for disseminated disease should be initiated if the patient has any risk factors or clinically concerning symptoms for bone or CNS involvement. This evaluation includes a bone scan and lumbar puncture. All patients should be assessed for immunocompromised status.
The management of coccidioidomycosis is based on the extent of infection, the severity of illness, and the immune status of the patient. In 95% of cases of uncomplicated pulmonary disease in an immunocompetent host, the symptoms will resolve without treatment with antifungal agents.9 The decision to treat uncomplicated pulmonary disease is based on severity of illness. While there is no consensus recommendation, commonly used indicators for treatment include persistent fever, age >55 years, symptoms greater than 2 months, hilar adenopathy, diffuse pulmonary infiltrates, weight loss, and inability to work.9 In patients with chronic progressive pneumonia or extrapulmonary involvement, treatment with antifungal medications should be initiated. While fluconazole remains the preferred treatment in coccidioidal pneumonia and meningitis, amphotericin B preparations should be considered for diffuse coccidioidal pneumonia and disseminated disease, including refractory meningitis.9 The use of newer azoles, particularly posaconazole, has been studied in a limited number of patients with refractory coccidioidomycosis with improvement in symptoms.10 Frequent follow‐up visits are recommended to detect progression of disease or to document resolution, with improving symptoms and decreasing titers. Duration of therapy in uncomplicated cases should be at least 3 months. Treatment of extrapulmonary disease can span years, and in the case of meningitis lifetime treatment is recommended given the high rate of relapse.
While the patient and the clinicians were aching for a diagnosis after the initial negative evaluation, recognition of the immunologic manifestations of coccidioidomycosis was essential in this case. Coccidioidomycosis should be considered in patients presenting with EN, regardless of presence of concurrent pulmonary symptoms; particularly in patients living in or with recent travel to endemic areas. Furthermore, the severity of symptoms can guide the decision and duration of treatment.
Teaching Points
-
Coccidioidomycosis has 4 main clinical presentations: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease.
-
Independent of pulmonary symptoms, coccidioidomycosis can present with immune‐mediated manifestations, such as EN and arthritis.
-
The diagnosis of coccidioidomycosis often relies on serologic testing for early and late infection.
-
Treatment of coccidioidomycosis is based on risk factors and severity of symptoms. High‐risk and symptomatic patients can be treated with fluconazole or amphotericin B.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
A 23‐year‐old Caucasian man presented to an outpatient clinic with a sore throat and associated subjective fevers. His evaluation included a negative rapid streptococcus test; nevertheless, he was empirically treated with amoxicillin. The following day, he experienced increasing sore throat and presented to the emergency department (ED). He was treated with prednisone and morphine sulfate and discharged home with azithromycin.
Initial considerations in a healthy young man who presents with fever and pharyngitis should focus on common infectious etiologies. Viral illnesses are the most frequent causes of sore throat and fever. These often manifest as mononucleosis‐like illnesses and include Epstein‐Barr virus (EBV) and cytomegalovirus (CMV). In this age group, it is also critical to consider sexually transmitted diseases (STDs) such as gonorrhea, human immunodeficiency virus (HIV), herpes simplex virus, and syphilis. Consideration of streptococcal pharyngitis is important. Since the rapid streptococcal antigen test is neither sensitive nor specific, confirmation of infection should be based on clinical findings and a culture of the pharynx for group A Streptococcus. Other common etiologies of fever and pharyngitis include acute or chronic sinusitis with postnasal drainage. Due to the progressive nature of the sore throat, there should be an evaluation for difficulty swallowing, problems phonating, or neck discomfort, any of which would be concerning for a retropharyngeal abscess. Additional history should be obtained with focus on sexual history, previous STDs, recent sick contacts, and other supporting signs and symptoms of viral illnesses.
Eight days after the initial onset of symptoms, the patient developed acute low back pain. The back pain was midline, severe, and constant around the lumbar spine. There was no saddle anesthesia, bowel or bladder dysfunction, or weakness or numbness in the extremities. He also noted swelling of the left fourth metacarpophalangeal joint and an erythematous rash on his right knee and anterior tibial region of the right leg. He continued to experience subjective fevers, sore throat, and swollen neck glands. Due to the severity and discomfort of symptoms, the patient returned to the ED.
With no history of trauma, the subsequent development of acute low back pain may be related to the patient's sore throat and fever. Monoarticular arthritis with contralateral skin lesions should raise suspicion for a systemic process, particularly infection or a rheumatologic syndrome. Infectious etiologies would include rheumatic fever, endocarditis with septic emboli, and osteomyelitis. Rheumatologic causes, such as ankylosing spondylitis and juvenile rheumatoid arthritis (RA), are also possibilities. The infectious evaluation should include an assessment of a history of intravenous drug use (IVDU) and underlying valvular disorders, which will increase the risk for endocarditis and therefore septic emboli. Acute HIV infection can be seen as early as 1 to 2 weeks postexposure and should be considered as well. Appropriate testing would include both conventional HIV antibody tests and HIV viral load assay. Lastly, in considering the patient's symptoms, obtaining his travel history to identify risk for Lyme disease would also be appropriate.
The patient did not report any further positive findings on review of systems. He did not have any significant past medical history and did not take any chronic medications. He had no sick contacts. He rarely drank alcohol and denied IVDU and sexual activity over the past year. He was previously involved in monogamous relationships with women. His last HIV test, 1 year prior, was negative. He did not have any history of STDs. He was a graduate student in computer science and lived in southern California. He had recently traveled to central California and France for 2 weeks, staying in larger cities. He had not been hiking during that time. His family history was significant for hypertension.
The travel history is provocative for 3 diseases of the reticuloendothelial system with possible systemic manifestations. First, toxoplasmosis, which is endemic in France where rare or raw beef and lamb are frequently consumed. It may present as a mononucleosis‐like illness and rarely as atypical pneumonia. Second, tuberculosis, which is also endemic in France, especially in major cities. Although most commonly a self‐limited respiratory disease, it may disseminate with systemic symptoms. Third, primary coccidioidomycosis, which is prevalent in the central valleys of California. The climate and wind patterns lead to aerosolization of the spores and make this a common respiratory pathogen.
The physical exam should include a detailed evaluation of the eyes for uveitis and iritis, seen in some rheumatologic disorders. A pharyngeal exam with assessment for exudate can support streptococcal pharyngitis or diphtheria. Evaluation for lymphadenopathy, while nonspecific, would be important for streptococcal pharyngitis, rheumatic fever, and juvenile RA. Further characterizing the rash is essential in distinguishing viral exanthems from the fleeting salmon‐colored maculopapular rash of juvenile RA. Assessment for peripheral stigmata of endocarditis should be done. A thorough joint exam should evaluate evidence of inflammatory or infectious joint disease.
On physical exam, he was a thin man who appeared anxious but in no acute distress. His temperature was 36.7C, blood pressure 111/68 mm Hg, heart rate 83 beats/minute, respiratory rate 16 breaths/minute, and oxygen saturation 99% on room air. Erythema was noted in the posterior oropharynx with no tonsillar exudate. There were several subcentimeter, nontender, and mobile lymph nodes in the anterior cervical chain bilaterally. The cardiovascular exam revealed normal sinus rhythm with a 2/6 systolic murmur at the apex, without radiation. His lungs were clear to auscultation. Skin exam revealed 2 blanching erythematous, indurated, and tender lesions on the right pretibial region, 2‐cm and 4‐cm in diameter. Two other similar, but smaller, lesions were noted on the left upper extremity and left ankle. His lumbar spine was slightly tender to touch. A complete joint exam was normal, including the left fourth metacarpophalangeal joint. Neurological exam, including bilateral strength, sensation, reflexes, and gait, was unremarkable.
Younger patients are subject to social‐acceptance bias and can deny sexual activity on initial inquiry. An objective evaluation for STDs with serologic workup should still be pursued. The cervical lymphadenopathy and tonsillar erythema continue to suggest a viral illness. While the systolic murmur may be physiologic, subjective fevers, disseminated cutaneous lesions, and arthritis warrant evaluation for bacterial endocarditis with blood cultures and an echocardiogram.
On exam, there is no evidence of true joint involvement and this decreases the likelihood of rheumatologic conditions, such as ankylosing spondylitis and juvenile RA. However, the skin lesions are suspicious for erythema nodosum (EN), which should prompt a biopsy and an evaluation for infectious etiologies. Serologies should include evaluation of Chlamydia, Mycoplasma, Coccidioides, and Histoplasma. I would also examine the feet carefully for potential transcutaneous inoculation by microorganisms that can produce a rash similar to EN. For instance, penetrating skin trauma can lead to pseudomonal infection. Brucella (from ingesting unpasteurized milk or milk products), Bartonella (from the scratches of feline animals), and Francisella tularensis (from rabbit exposure) can also produce skin lesions that mimic EN. These are best distinguished through a detailed history, concomitant serologic workup, and biopsy. Other noninfectious etiologies of EN can include inflammatory bowel disease, Behcet's, and sarcoidosis; however, the patient does not currently report any symptoms supporting these diagnoses. In addition to the above evaluation, complete blood count with differential, liver function tests, creatinine, and urinalysis should be obtained.
The patient's white blood cell (WBC) count was 12,100/L with 73% neutrophils, 14% lymphocytes, and 12% monocytes. Hemoglobin was 11.8 g/dL and platelet count 292,000/L. Chemistry panel and liver function tests were unremarkable. Erythrocyte sedimentation rate (ESR) was 71 mm/hour (range, 010). Urinalysis was negative for protein and red blood cells. Chest x‐ray did not illustrate any abnormalities. Computed tomography (CT) of the lumbar spine revealed a small posterior disc bulge at L4‐5 and L5‐S1.
The moderate leukocytosis with neutrophilic predominance and monocytosis raises concern for a systemic inflammatory process; the elevated ESR further supports this. Monocytosis can be seen in a number of infectious, autoimmune, and malignant conditions. Tuberculosis, brucellosis, bacterial endocarditis, syphilis, infectious mononucleosis, and viral illnesses are among the infections typically characterized by monocytosis. Autoimmune illnesses, such as systemic lupus erythematosus and RA can also have similar presentations. The patient does not have any features of an underlying malignancy, such as weight loss or night sweats; however, if the autoimmune and infectious evaluations are negative, Hodgkin's disease and certain leukemias should be considered. There is no evidence of osteomyelitis on the spine CT, which decreases the possibility of (but does not exclude) infectious or rheumatologic conditions of the spine. I would suggest a comprehensive laboratory evaluation for the discussed infectious and rheumatologic disorders.
The patient's back pain was controlled with antiinflammatory medications overnight. Due to the patient's stable condition and lack of a diagnosis, empiric antibiotics were not initiated. An extensive workup was sent, including antistreptolysin O, polymerase chain reaction for Chlamydia, Neisseria gonorrhoeae, EBV, and parvovirus B19 DNA, serologies for Coccidioides immunoglobulin G (IgG) and IgM, urinary antigen for Histoplasma, HIV enzyme‐linked immunosorbent assay (ELISA) and Western blot, serum angiotensin‐converting enzyme level, C‐reactive protein, rheumatoid factor, antinuclear antibody, and antidouble‐stranded DNA antibodies.
Without a clear diagnosis, I would recommend against treatment with empiric antibiotics. At this point, I agree with waiting for the results of the pending workup.
On hospital day 1, the patient developed severe acute left ankle pain. On examination, the joint was exquisitely tender with decreased range of motion. Arthrocentesis was promptly performed. The synovial fluid WBC count was 1370/L with a differential of 82% neutrophils and 18% monocytes. No crystals were identified and the bacterial Gram stain was negative. He was treated with antiinflammatory medications. Bacterial blood cultures, obtained from the day of admission, were negative.
The arthrocentesis reveals a polymorphonuclear‐predominant fluid; however, the WBC count in the fluid is only mildly elevated. While the elevated monocyte count could again be consistent with viral arthropathies or juvenile RA, there is currently no systemic evidence of either illness. It is important to await the results of the final cultures, but the low WBC count and negative Gram stain decrease the probability of a septic joint. Empiric antibiotics to cover Gram‐positive organisms and gonococci would not be unreasonable, pending joint fluid culture results. The monocytosis could also be consistent with a fungal arthritis.
On hospital day 2, the results of the rheumatologic and infectious evaluation were negative with the exception of C‐reactive protein, which was 11.8 mg/dL (normal, <0.8), antinuclear antibody titer of 1:160 (normal, <1:40), Coccidioides IgM enzyme immunoassay (EIA) 0.710 (negative, <0.150), and Coccidioides tube‐precipitin (TP) immunodiffusion (ID) antibody‐positive. Coccidioides IgG EIA was negative.
The serologic tests are consistent with primary coccidioidomycosis. This is often a challenging diagnosis due to the nonspecific signs and symptoms, such as cough, fever, myalgias, and fatigue. Since screening EIAs are sensitive but not specific, concern for coccidioidomycosis or abnormal EIA results should prompt confirmatory testing with complement fixation titers (CF) and TP ID. Treatment with fluconazole should be initiated. Since the patient does not have central nervous system (CNS) symptoms, I would not recommend lumbar puncture at this point. However, a bone scan should be done for assessment of the back pain.
The patient was diagnosed with primary coccidioidomycosis infection with immune‐complexmediated arthritis and EN. A bone scan was negative. The patient was treated with fluconazole and discharged with 3 months of therapy. At follow‐up clinic visits after completion of therapy, his symptoms had resolved and his titers had normalized.
Discussion
The diagnosis of coccidioidomycosis is often challenging due to its protean manifestations. Four clinical syndromes are commonly seen: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease involving the skin, lymph nodes, bones, joints, and meninges. The most common clinical manifestation, acute pneumonia, may be indistinguishable from other causes of community‐acquired pneumonia (CAP). In a study of CAP in Arizona, 29% of cases were positive for coccidioidal infection through serologic evaluation.1 Features suggestive of coccidioidal infection include fatigue, severe headache, and pleuritic chest pain. Adenopathy in the hilar or paratracheal regions can be seen in 25% of infections.2 Chronic progressive pneumonia refers to infections in which symptoms, including cough, hemoptysis, and weight loss, persist for longer than 3 months. Pulmonary nodules and cavities are residual manifestations of primary pulmonary infection and occur in 2% to 8% of cases. Extrapulmonary disease develops in less than 5% of immunocompetent patients with primary pulmonary infection, with higher prevalence in patients of African American and Filipino decent. Immunocompromised patients are at increased risk for extrapulmonary infection. The most serious site of extrapulmonary disease is the meninges. Coccidioidal meningitis carries nearly 100% mortality rate if left untreated. The presentation is variable with up to 75% of cases reporting headache. While coccidioidal pneumonia also frequently presents with headache, symptoms including altered mental status, focal neurological deficits, and persistent or progressive headache are more suggestive of meningeal disease.3
Patients with any presentation of coccidioidomycosis can display immune‐mediated manifestations such as EN, arthralgias (desert rheumatism), and in some cases mild conjunctivitis.4 It is hypothesized that these findings occur due to a hypersensitivity reaction to coccidioidomycosis.4 EN is an inflammatory process of the subcutaneous fat, which presents as tender and erythematous nodules typically on the lower extremities. EN is not a disease entity or site of metastatic infection, but a response to underlying illness. Its recognition should trigger a search for the primary etiology, as guided by the patient's history and clinical presentation. The differential diagnosis for EN is broad and includes rheumatologic, infectious, medication‐related, inflammatory, and idiopathic processes (Table 1). Coccidioidomycosis should be strongly considered based on geographical location, with the vast majority of cases seen in southern California, Arizona, Nevada, New Mexico, and Texas. While the pathophysiology of EN has not been completely elucidated, the lesions may reflect a vigorous immune response conferring a protective advantage. Interestingly, a study of pregnant women with coccidioidomycosis revealed a decreased incidence of disseminated disease in patients with EN.5, 6
| Rheumatologic/autoimmune |
| Systemic lupus erythematosus |
| Wegener's granulomatosis |
| Sarcoidosis |
| Infectious |
| Streptococcus pyogenes causing pharyngitis (most common) |
| Borrelia burgdorferi |
| Mycoplasma pneumoniae |
| Bartonella henselae |
| Shigella |
| Campylobacter jejuni |
| Salmonella |
| Yersinia enterocolitica |
| Chlamydia |
| Brucella |
| Escherichia coli |
| Treponema pallidum |
| Mycobacterium leprae |
| Neisseria gonorrhoeae |
| Mycobacterium tuberculosis |
| Human immunodeficiency virus |
| Epstein‐Barr virus |
| Cytomegalovirus |
| Influenza |
| Varicella Zoster virus |
| Coccidioides immitis |
| Histoplasma capsulatum |
| Blastomyces dermatitidis |
| Dermatophytic fungal infections (rare) |
| Gastrointestinal |
| Ulcerative colitis |
| Crohn's disease |
| Celiac disease |
| Behcet's disease |
| Medications |
| Oral contraceptives |
| Proton pump inhibitors |
| Sulfonamides |
| Leukotriene modifiers (montelukast) |
| Hepatitis B vaccine |
| Isoretinoin |
| Miscellaneous |
| Hodgkins lymphoma |
| Sweet's syndrome |
Coccidioidomycosis is also associated with immune‐mediated arthralgias and arthritis. These manifestations occur in up to one‐third of patients with concomitant EN. Arthritis may be monoarticular or polyarticular, often affecting large joints such as the knees or ankles. It is important to note that septic arthritis can also occur and should be differentiated from rheumatism by joint aspiration.
The diagnosis of coccidioidomycosis can be made by serologic testing, direct isolation of the organism on culture, or visualization on tissue biopsy. Of these methods, serologic testing is most commonly utilized. The 2007 Infectious Disease Society of America (IDSA) and American Thoracic Society guidelines recommend diagnostic testing in hospitalized patients with CAP who reside in or have recently traveled (within 2 weeks) to endemic areas.7 There are multiple approaches to serologic diagnosis based on identification of IgM or IgG antibodies to various coccidioidal antigens. During the early phase of infection, TP ID and EIA can be utilized to detect IgM antibodies. While EIA testing has 92% sensitivity, it has high rates of false‐positive results, and therefore confirmatory testing with ID is recommended. ID has variable sensitivity, but 90% of patients will test positive by 3 weeks of infection.8 During the later phase of the infection, IgG antibodies are detected either quantitatively by CF or qualitatively by ID and EIA. CF can provide information on the severity of illness and prognosis based on titer levels, as well as serving as a marker for response to treatment.2 Positive titers greater than 1:32 suggest disseminated disease. In addition, CF titer in the cerebrospinal fluid is the test of choice in diagnosis of coccidioidal meningitis. An evaluation for disseminated disease should be initiated if the patient has any risk factors or clinically concerning symptoms for bone or CNS involvement. This evaluation includes a bone scan and lumbar puncture. All patients should be assessed for immunocompromised status.
The management of coccidioidomycosis is based on the extent of infection, the severity of illness, and the immune status of the patient. In 95% of cases of uncomplicated pulmonary disease in an immunocompetent host, the symptoms will resolve without treatment with antifungal agents.9 The decision to treat uncomplicated pulmonary disease is based on severity of illness. While there is no consensus recommendation, commonly used indicators for treatment include persistent fever, age >55 years, symptoms greater than 2 months, hilar adenopathy, diffuse pulmonary infiltrates, weight loss, and inability to work.9 In patients with chronic progressive pneumonia or extrapulmonary involvement, treatment with antifungal medications should be initiated. While fluconazole remains the preferred treatment in coccidioidal pneumonia and meningitis, amphotericin B preparations should be considered for diffuse coccidioidal pneumonia and disseminated disease, including refractory meningitis.9 The use of newer azoles, particularly posaconazole, has been studied in a limited number of patients with refractory coccidioidomycosis with improvement in symptoms.10 Frequent follow‐up visits are recommended to detect progression of disease or to document resolution, with improving symptoms and decreasing titers. Duration of therapy in uncomplicated cases should be at least 3 months. Treatment of extrapulmonary disease can span years, and in the case of meningitis lifetime treatment is recommended given the high rate of relapse.
While the patient and the clinicians were aching for a diagnosis after the initial negative evaluation, recognition of the immunologic manifestations of coccidioidomycosis was essential in this case. Coccidioidomycosis should be considered in patients presenting with EN, regardless of presence of concurrent pulmonary symptoms; particularly in patients living in or with recent travel to endemic areas. Furthermore, the severity of symptoms can guide the decision and duration of treatment.
Teaching Points
-
Coccidioidomycosis has 4 main clinical presentations: (1) acute pneumonia, (2) chronic progressive pneumonia, (3) pulmonary cavities and nodules, and (4) extrapulmonary disease.
-
Independent of pulmonary symptoms, coccidioidomycosis can present with immune‐mediated manifestations, such as EN and arthritis.
-
The diagnosis of coccidioidomycosis often relies on serologic testing for early and late infection.
-
Treatment of coccidioidomycosis is based on risk factors and severity of symptoms. High‐risk and symptomatic patients can be treated with fluconazole or amphotericin B.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring the patient and the discussant.
- ,,, et al.Coccidioidomycosis as a common cause of community‐acquired pneumonia.Emerg Infect Dis.2006;12:958–962.
- ,.Coccidioidomycosis.Mayo Clin Proc.2008;83:343–349.
- ,.Coccidioidal meningitis.Clin Infect Dis.2006;42:103–107.
- ,.Coccidioidomycosis: host response and vaccine development.Clin Microbiol Rev.2004;17:804–839.
- ,,.Erythema nodosum in pregnant patients with coccidioidomycosis.Clin Infect Dis.1998;27:1201–1203.
- .Protective effects of erythema nodosum in coccidioidomycosis.Lancet.1999;353:168.
- ,,, et al.Infectious Disease Society of America/American Thoracic Society consensus guidelines on management of community acquired pneumonia in adults.Clin Infect Dis.2007;44:S27–S72
- .Laboratory aspects in the diagnosis of coccidioidomycosis.Ann N Y Acad Sci.2007;1111:301–314.
- ,,, et al.Coccidioidomycosis.Clin Infect Dis.2005;41:1217–1223.
- ,,,,.Refractory coccidioidomycosis treated with posaconazole.Clin Infect Dis.2005;40:1770–1776.
- ,,, et al.Coccidioidomycosis as a common cause of community‐acquired pneumonia.Emerg Infect Dis.2006;12:958–962.
- ,.Coccidioidomycosis.Mayo Clin Proc.2008;83:343–349.
- ,.Coccidioidal meningitis.Clin Infect Dis.2006;42:103–107.
- ,.Coccidioidomycosis: host response and vaccine development.Clin Microbiol Rev.2004;17:804–839.
- ,,.Erythema nodosum in pregnant patients with coccidioidomycosis.Clin Infect Dis.1998;27:1201–1203.
- .Protective effects of erythema nodosum in coccidioidomycosis.Lancet.1999;353:168.
- ,,, et al.Infectious Disease Society of America/American Thoracic Society consensus guidelines on management of community acquired pneumonia in adults.Clin Infect Dis.2007;44:S27–S72
- .Laboratory aspects in the diagnosis of coccidioidomycosis.Ann N Y Acad Sci.2007;1111:301–314.
- ,,, et al.Coccidioidomycosis.Clin Infect Dis.2005;41:1217–1223.
- ,,,,.Refractory coccidioidomycosis treated with posaconazole.Clin Infect Dis.2005;40:1770–1776.
Flushing Out the Diagnosis
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.

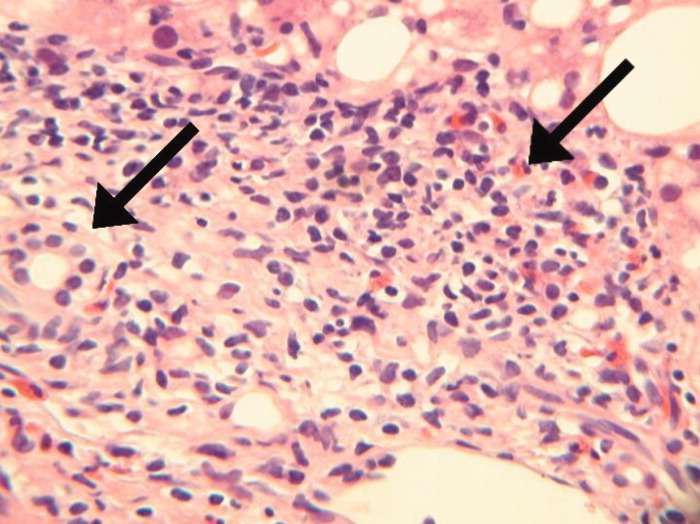
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 42‐year‐old woman with a history of mild asthma presented to the emergency department (ED) following 1 week of headache. She had been in her usual state of good health until 1 week prior to her presentation, when she noticed intermittent frontal headaches without neck stiffness or other neurologic symptoms. She then developed diffuse myalgias, fatigue, subjective fevers, and rigors for the 24 hours prior to presentation. On the morning of presentation, chest tightness, palpitations, and shortness of breath occurred. She used her albuterol metered‐dose inhaler without relief and went to the hospital.
Many of these features can be explained by a viral syndrome exacerbating underlying asthma or by a psychiatric condition such as anxiety or depression, but they may also be a harbinger of a systemic process, including infection, malignancy, or autoimmunity. Because the onset of headache is temporally distant from the other symptoms, I am more inclined to believe that it represents a primary intracranial process than I would if it were coincident with the onset of the other acute symptoms. If the fevers and rigors are verified, infection would be the initial concern. Failure to respond to her inhalers may either signify a severe asthma exacerbation or a nonbronchospastic cause of dyspnea.
She reported mild nausea, but denied photophobia, vomiting, abdominal pain, diarrhea, melena, or hematochezia. She did not have recent ill contacts, animal bites, or travel. Her medical history included asthma, diverticulitis, chronic right ankle pain, and obesity. She reported an allergic rash to amoxicillin. Her medications were sulindac and fluticasone/salmeterol, and albuterol metered‐dose inhalers. She worked as a preschool teacher and was married with 2 children. She denied any tobacco use and seldom drank alcoholic beverages. On exam, temperature was 36.7C, pulse was 107 beats per minute, blood pressure was 129/91 mm Hg, respiratory rate was 19 breaths per minute, and oxygen saturation was 98% while breathing ambient air. Her face and anterior neck were flushed and diaphoretic, and her sclerae were icteric. There was no nuchal rigidity. Her cardiac rhythm was regular without murmurs, lungs were clear to auscultation, and the abdomen was mildly tender to palpation in the epigastrium and right upper quadrant. The white blood cell (WBC) count was 9200/L, with 84% neutrophils, 3% lymphocytes, 6% monocytes, and 7% eosinophils. The hemoglobin was 14.8 g/dL and the platelet count was 166,000/L. Total serum bilirubin was 4.6 mg/dL, aspartate aminotransferase (AST) was 459 U/L (normal range, 8‐31), alanine aminotransferase (ALT) was 667 U/L (normal range, 7‐31), and alkaline phosphatase was 146 U/L (normal range, 39‐117). Serum electrolytes, creatinine, lactate, lipase, thyrotropin, coagulation studies, and cardiac enzymes were all normal. Urinalysis showed trace leukocyte esterase and bilirubin, as well as 3 WBCs and 2 red cells per high‐power field. Chest radiography and an electrocardiogram demonstrated no abnormalities.
The major findingwhich is critical to focusing problem‐solving in the face of a broad range of symptomsis her hepatitis. The common etiologies for hepatitis of this degree include viruses (hepatitis A and cytomegalovirus [CMV] should be considered given her work in preschool), toxins, autoimmunity, and vascular events. Liver disease in association with flushing raises the possibility of carcinoid syndrome with liver metastases. The lack of wheezing makes the bronchospasm of asthma or carcinoid less suitable explanations for her shortness of breath. Her eosinophilia is mild but probably is not accounted for alone by well‐controlled asthma in a person with no history of atopic disease. I would also ask her about any alternative and over‐the‐counter remedies. The paucity of lymphocytes raises the possibility of human immunodeficiency virus (HIV), Hodgkin's disease, or systemic lupus erythematosus. Although she does not have a documented fever or leukocytosis, she reported fevers and chills and is diaphoretic and tachycardic, so exclusion of biliary obstruction and cholangitis is the highest priority.
An abdominal ultrasound demonstrated hepatomegaly with moderate fatty infiltration and a normal gallbladder without pericholecystic fluid. The intrahepatic and extrahepatic biliary ducts were normal and the hepatic and portal veins were patent. Computed tomography of the abdomen showed slight thickening of the sigmoid colon wall. Ciprofloxacin and metronidazole were administered for possible diverticulitis. Over the first 48 hours of hospitalization her symptoms improved markedly. Her flushing resolved and she had no recorded fevers in the hospital. Serologies were negative for hepatitis A immunoglobulin M (IgM), hepatitis B surface antibody, hepatitis B surface antigen, and hepatitis C antibody. A monospot test was negative and the erythrocyte sedimentation rate was 11 mm/hour. Blood and urine cultures were negative. On the second hospital day the absolute eosinophil count rose to 855/L (15% of 5700 WBCs). On the fourth hospital day, the absolute eosinophil count was 1092/L, the total bilirubin was 1.9 mg/dL, and the AST and ALT were 174 U/L and 476 U/L, respectively. Antibiotics were stopped and she was discharged home.
Her prompt improvement suggests either a self‐limited condition or a response to the antibiotics. The rapid but incomplete resolution of her hepatitis is in keeping with a withdrawal of a toxin, relief of biliary obstruction, or a transient vascular event, and is less consistent with a viral hepatitis or an infiltrative process. With normal biliary system imaging, sterile blood cultures, and the absence of fever or leukocytosis, cholangitis is unlikely. Likewise, there is no suggestion of a vascular event, either obstructive or hemodynamic, that is impairing the liver.
A common cause of eosinophilia in hospitalized patients is medications, so it would be useful to monitor that count after the new antibiotics. At this point, I also wonder if the eosinophils are a feature of the underlying illness, as they were present to a modest degree on admission before any new medications were administered. The overlap of eosinophilia and hepatitis brings to mind a medication reaction (eg, to sulindac) or a hepatobiliary parasite, such as ascaris or clonorchis, for which she lacks a known exposure. Many patients experience flushing in the setting of fever or stress, but sustained flushing may suggest a systemic illness characterized by the release of vasoactive mediators such as carcinoid syndrome or mastocytosis. The latter might be considered more strongly if the eosinophilia is deemed to be primary (rather than reactive) after a thorough evaluation.
After 2 days at home, the patient had recurrence of subjective fevers, with chest, back, and abdominal pain, fatigue, loose stools, and rigors. She returned to the ED, where she was noted to have facial erythema and injected sclerae, but the remainder of her physical exam was normal. The total serum bilirubin was 1.1 mg/dL, AST was 156 U/L, ALT was 214 U/L, and alkaline phosphatase was 240 U/L. Serum lipase was normal. WBC count was 14,000/L, with 94% neutrophils, 3% lymphocytes, 2% monocytes, and 1% eosinophils. She was again treated empirically with ciprofloxacin and metronidazole. Endoscopic ultrasound was normal, with no evidence of gallbladder sludge or microlithiasis. Stool cultures, assay for Clostridium difficile, and examination for ova and parasites were negative. The 24‐hour urine demonstrated no elevation in 5‐hydroxyindoleacetic acid. An adrenocorticotropic hormone (ACTH) stimulation test was normal. HIV antibody was negative. Her symptoms improved within 2 days. The eosinophil count rose and peaked at 1541/L by the third hospital day, while the transaminase elevations resolved. Antibiotics were discontinued. A liver biopsy showed mixed macrovesicular and microvesicular fatty metamorphosis and steatohepatitis with eosinophils (Figures 1 and 2). She was discharged home on the sixth hospital day.
Her illness can now be characterized as relapsing inflammation, which given the frequency (over days) suggests either an indolent infectious focus that periodically causes systemic inflammation or reexposure to a toxic substance. The 2 most notable laboratory abnormalities, the hepatitis and the eosinophilia, persist but have differing trajectories. While the liver function tests have progressively normalized despite clinical relapses, the eosinophils have had a more fluctuating course characterized by increases during the hospitalization and higher levels during the second hospitalization. The absence of an infection, recurrent systemic inflammation, and eosinophilic hepatitis suggest a hypersensitivity reaction to a medication or other substance. She is most likely being reexposed at home, where her symptoms occur, and not in the hospital, where her symptoms resolve. Sulindac is a leading candidate, because nonsteroidal antiinflammatory drugs (NSAIDs) cause a number of hypersensitivity reactions and are frequently stopped when sick patients enter the hospital.
Seven days after discharge she developed acute onset of subjective fever, nausea, diffuse myalgias, and flushing, identical to the 2 prior episodes, and she again returned to the ED. Her temperature was 39.1C, heart rate was 120 beats per minute, blood pressure was 87/50 mm Hg, respiratory rate was 18 breaths per minute, and the oxygen saturation was 96% while breathing room air. She had diffuse flushing from her neck over her torso and was diaphoretic with injected sclera and conjunctiva. The WBC count was 11,400/L with 97% neutrophils and 3% lymphocytes. Total bilirubin was 0.8 mg/dL, AST was 134 U/L, ALT was 140 U/L, and alkaline phosphatase was 144 U/L. She was readmitted to the hospital. Following admission, she had no fevers, the flushing resolved, and AST and ALT levels decreased. The only treatment the patient received in the ED and during her hospital stay was acetaminophen as needed for pain or fever. The eosinophil count peaked at 1404/L by hospital day 4. Blood and urine cultures were negative. IgM antibodies to Epstein‐Barr virus were not detected, CMV DNA was not detected, and a rapid plasma reagin (RPR) test was nonreactive. Ferritin, ceruloplasmin, alpha‐1‐antitrypsin, and tryptase levels were normal. Antimitochondrial, antismooth muscle, antineutrophil cytoplasmic, and antinuclear antibodies were negative. There was no monoclonal band on serum protein electrophoresis. A blood smear for Borrelia detected no spirochetes.
A complete picture of the uncommon but classic flushing disorders, namely carcinoid, mastocytosis, and pheochromocytoma, has not emerged. The constellation of inflammation, mucosal and hepatic involvement, and eosinophilia are most consistent with a drug hypersensitivity reaction. Additionally, the recurrent inflammation is becoming more severe, as manifest by the fever and hemodynamic derangements, which suggests an increasing sensitization to the offending agent. I would review every drug she has received in the hospital, but given the recurrences after discharge her home medications are the most likely explanation. Of these, sulindac is the most likely culprit.
On further questioning, it was learned that the patient began taking sulindac 200 mg twice daily to treat her chronic ankle pain 6 weeks before the first admission. The medication had been stopped on each admission. She was instructed to discontinue sulindac. She has had no recurrences of symptoms and her hepatitis and eosinophilia have resolved.
DISCUSSION
This patient presented with recurrent skin findings, eosinophilia, hepatitis, and constitutional symptoms caused by hypersensitivity to sulindac. This drug‐induced hypersensitivity syndrome was originally described with anticonvulsant drugs (carbamazepine, phenytoin, and phenobarbitone) and named anticonvulsant hypersensitivity syndrome,1, 2 but has been observed with many other medications, including allopurinol, dapsone, minocycline, and nevirapine. The term drug rash with eosinophilia and systemic symptoms (DRESS) syndrome has been recently adopted to convey the cardinal features that characterize this disorder.3
DRESS syndrome is defined by rash, fever, and internal organ involvement.4 Also included in the diagnostic criteria are hematologic abnormalities (eosinophilia 1.5 109/mm3 or the presence of atypical lymphocytes) and lymphadenopathy.3 The multiorgan involvement distinguishes DRESS from other cutaneous drug eruptions. In a review of the French Pharmacovigilance Database for all cases of DRESS over a 15‐year period, 73% to 100% of patients were reported to have dermatologic abnormalities, most frequently a maculopapular rash or erythroderma. Less common skin findings include vesicles, bullae, pustules, erythroderma, and purpuric lesions. Liver abnormalities were observed in more than 60% of patients and were the most frequent systemic finding.5 Eosinophilia was the most common hematologic abnormality, present in more than 50% of cases. As this case underscores, DRESS syndrome typically begins 3 to 8 weeks after initiation of the drug because it is a delayed type IV hypersensitivity reaction.6 Fever can occur within hours on rechallenge because of the presence of memory T cells.
The main treatment for DRESS syndrome is withdrawal of the offending drug. Systemic corticosteroids have been recommended in cases with life‐threatening pulmonary or cardiac involvement, but have not been shown to be helpful in reversing renal or hepatic disease. Mortality, usually from end‐organ damage, occurs in about 10% of cases. The most common drugs are phenobarbital, carbamazepine, and phenytoin, with incidences of 1 in 5000 to 1 in 10,000. NSAIDs and antibiotics also have been implicated frequently. Human herpesvirus type 6 coinfection and genetically inherited slow acetylation have been associated with DRESS, although causal links have yet to be established.7, 8
The initial challenge in caring for a patient with multiple symptoms, exam findings, and test abnormalities is the coherent framing of the key clinical features that require explanation. This process, called problem representation, allows clinicians to search among a bounded list of possible diagnoses (or solutions) rather than invoking a differential diagnosis for every single abnormality. In searching for the proper diagnosis, this patient's clinical course required frequent reframing as more data became available.
Initially, the problem was framed as a 42‐year‐old woman with hepatitis. As the flushing and eosinophilia, which initially appeared to be transient and possibly nonspecific, became more prominent, the problem representation was revised to a 42‐year‐old woman with hepatitis, eosinophilia, and flushing. Since this triad did not immediately invoke a single diagnosis for the treating clinicians or the discussant, the differential diagnosis of hepatitis and eosinophilia and the differential diagnosis of flushing were considered in parallel.
Hepatitis and eosinophilia can occur coincidentally in the setting of parasitic infections, particularly helminths (ascaris, strongyloidiasis, and toxocaris) and liver flukes (opisthorchis and clonorchis), which invade the hepatobiliary system and induce a reactive eosinophilia. Some neoplasms, such as lymphomas and leukemias, and myeloproliferative disorders, including hypereosinophilic syndrome and mastocytosis, may have neoplastic cellular invasion of the liver and induce eosinophilia. Systemic drug hypersensitivity reactions are typically characterized by eosinophilia, transaminase elevations,9 and hepatitis on histology.10 As with this patient, liver biopsy in drug‐induced hepatitis shows a mixed microvesicular and macrovesicular steatosis, often with eosinophils.10
The flushing prompted a thorough but negative workup for the classic flushing disorders. The discussant's attempts to unify the flushing with the other clinical features illustrate how framing can affect our reasoning. Hepatitis, eosinophilia, and flushing defied an obvious single explanation and led the discussant down parallel diagnostic reasoning pathways. Although flushing is 1 of the well‐described dermatologic manifestations in drug‐hypersensitivity reactions, framing the central features as hepatitis, eosinophilia, and rash would have more readily suggested a drug reaction as a unifying diagnosis.11
The tempo and periodicity of this patient's illness provided the final formulation of a 42‐year‐old woman with hepatitis, eosinophilia, and flushing (or rash) that occurs every few days at home and resolves in the hospital. This formulation and the increasingly severe presentation, suggesting sensitization, were highly suggestive of an exogenous cause of her illness.
This case highlights how easily medication side effects can be overlooked during an extensive evaluation and how vigilant medication reconciliation coupled with an increased understanding of the spectrum of drug reactions can lead to early detection and prevention of potentially serious effects. In the case of DRESS, recognizing an association between a rash and organ involvement is central to making the diagnosis. Eosinophilia that accompanies a rash can further aid in narrowing the differential diagnosis.
The case also serves as a reminder of how framing with the slightest imprecision (eg, flushing instead of rash) can derail or delay the diagnostic process, yet is indispensable in tackling a complicated case. Finally, a time‐honored lesson in diagnosis is highlighted yet again: the diagnosis can usually be flushed out from the history.1214
Key Teaching Points
-
The constellation of skin findings, eosinophilia, organ involvement (particularly hepatitis), and constitutional symptoms should prompt consideration of DRESS syndrome and a hunt for a culprit drug.
-
Symptoms that resolve during hospitalization and repeatedly recur after discharge should prompt consideration of an exposure unique to the home, which may be environmental or pharmacologic.
-
Problem representation is critical in solving a complicated case, but adopting an inaccurate frame (representation) can derail or delay the diagnostic process.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
- ,.Anticonvulsant hypersensitivity syndrome. In vitro assessment of risk.J Clin Invest.1988;82:1826–1832.
- ,,.Anticonvulsant hypersensitivity syndrome: incidence, prevention, and management.Drug Saf.1999;21:489–501.
- ,,,.Drug rash with eosinophilia and systemic symptoms vs toxic epidermal necrolysis: the dilemma of classification.Clin Dermatol.2005;23:311–314.
- ,.Management of drug rash with eosinophilia and systemic symptoms (DRESS syndrome): an update.Dermatology.2003;206:353–356.
- ,,,,,Blayac J‐P; Network of the French Pharmacovigilance Centers. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist?Br J Dermatol.2006;155:422–428.
- ,.The immunological and clinical spectrum of delayed drug‐induced exanthems.Curr Opin Allergy Clin Immunol.2004;4:411–419.
- ,,.Association between anticonvulsant hypersensitivity syndrome and human herpesvirus 6 reactivation and hypogammaglobulinemia.Arch Dermatol.2004;140:183–188.
- ,.Treatment of severe drug reactions: Stevens‐Johnson Syndrome, toxic epidermal necrolysis and hypersensitivity syndrome.Dermatol Online J.2002;8:5.
- ,.Drug reactions. In: Bolognia JL, Jorizzo JL, Rapini RP, eds.Dermatology.2nd ed.Spain:Mosby Elsevier;2008:310–311.
- Pathology Outlines. Liver and intrahepatic bile ducts—non tumor. Hepatitis (non‐infectious). Drug/toxin‐induced hepatitis. Available at: http://www.pathologyoutlines.com/liver.html#drugtoxin. Accessed August2009.
- ,,.The flushing patient: differential diagnosis, workup and treatment.J Am Acad Dermatol.2006;55(2):193–208.
- ,,,,.Relative contributions of history‐taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients.Br Med J.1975;2(5969):486–489.
- ,,,,.Contributions to the history, physical examination, and laboratory investigation in making medical diagnosis.West J Med.1992;156(2):163–165.
- ,.A study on relative contributions of the history, physical examination and investigations in making medical diagnosis.J Assoc Physicians India.2000;48(8):771–775.
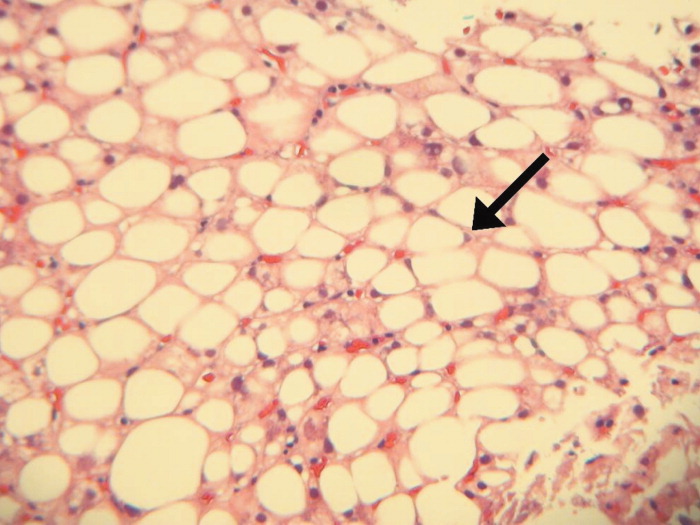
The Third Time's the Charm
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
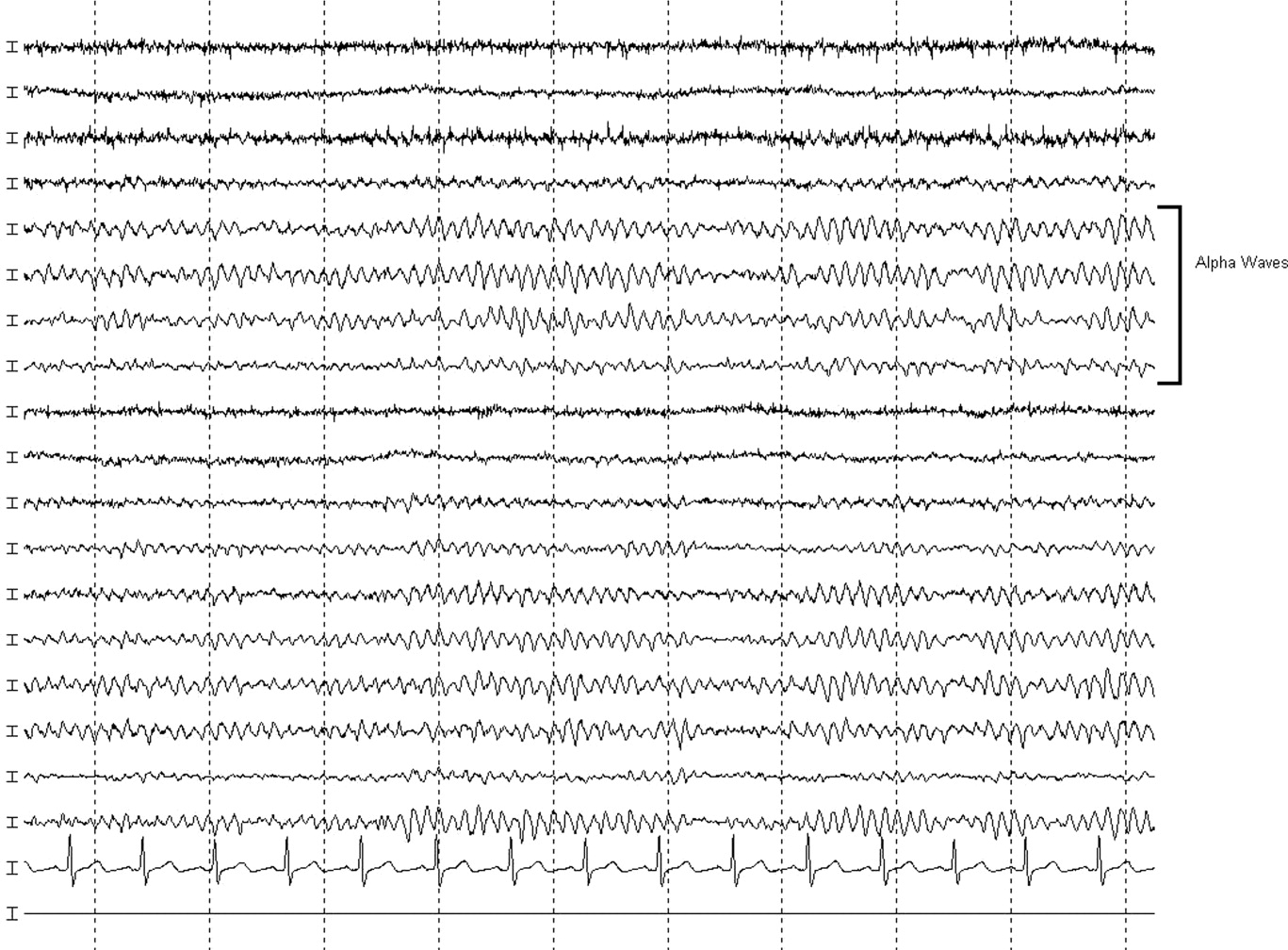
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, <5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, <10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
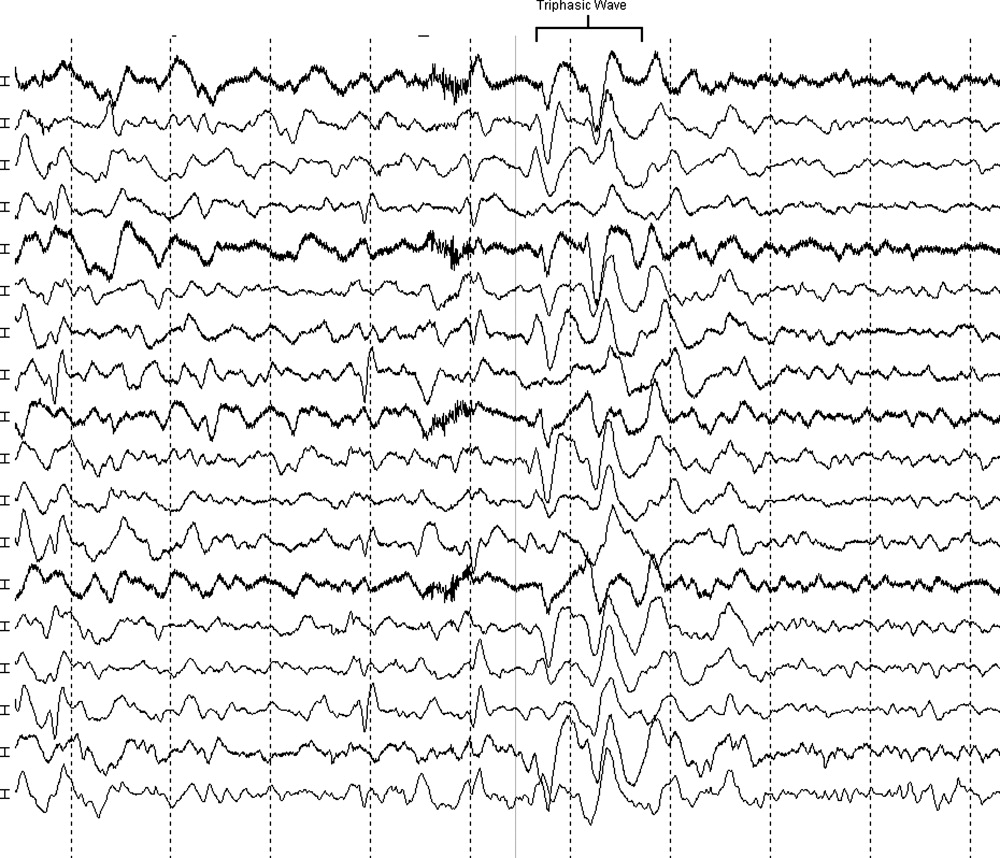
Advanced Measures in Palliative Care
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Resuscitation status and patient wishes in terms of advanced cardiopulmonary support must be addressed during inpatient hospital admissions. However, the lack of clarity of the patients' wishes and the variability in physicians' comfort addressing these issues often leads to ambiguity in an emergency setting. This may result in inappropriately aggressive management, and conversely, it may also lead to withholding potentially lifesaving therapy due to Do Not Resuscitate (DNR) designation. We report a case of hemodynamic instability due to acute supraventricular tachycardia (SVT) in a patient with a DNR designation. He was successfully treated according to the advanced cardiac life support (ACLS) protocol for SVT. We also discuss some of the ethical challenges of providing potential life‐sustaining interventions in palliative medicine, as well as the dilemma of whether or not to provide such interventions to patients who have DNR status.
Case Presentation
A 45‐year‐old man with advanced tonsillar cancer was admitted to an inpatient palliative care unit for evaluation and treatment of anorexia, progressive pain, and asthenia. He had undergone tumor debulking and neck dissection followed by adjuvant chemotherapy and external beam radiation therapy. Despite maximal therapy, the patient developed locally recurrent disease (leading to more surgery) and later, progressive metastatic disease (treated with palliative radiation therapy). With ongoing weight loss and failure to thrive, a percutaneous gastrostomy tube was placed for nutritional support. Still, the patient suffered from significant stomatitis, esophagitis, and diarrhea consistent with radiation‐induced injury, and had several admissions for dehydration and pain control.
During this and prior admissions, the patient clearly articulated his preference for DNR status. The patient was clinically declining, but was still functional, with and estimated survival of weeks to a few months. As with previous admissions, he was given intravenous fluids and parenteral opioids, and his electrolytes and vital signs normalized to his baseline. On the day of anticipated discharge, the patient was at his hemodynamic baseline (pulse of 100 beats per minute, blood pressure of 98/60 mmHg). Upon returning to bed after a shower, the patient developed acute dyspnea, weakness, and diaphoresis. Heart rate was 170 beats per minute and blood pressure was 70/50 mmHg. Intravenous normal saline boluses were given while electrocardiogram (EKG) was obtained. EKG revealed SVT with changes suggestive of demand myocardial ischemia. Carotid massage and Valsalva maneuvers were unsuccessful in converting the rhythm to sinus.
At that point, consideration was given to his DNR designation. The treating physician and patient briefly discussed the alternatives of no treatment of his arrhythmia, or alternatively, more aggressive treatment options on the Palliative Care Unit, including intravenous (IV) adenosine and direct current cardioversion. He did not have a detailed advanced directive discussing similar scenarios; he had only completed a commonly‐used, state‐issued Durable DNR form. All decided the SVT was potentially reversible and appeared to be causing many of the patient's acute symptoms; hence, aggressive treatment of the arrhythmia was in his best interest.
Despite absence of telemetry monitoring, consideration was given to IV diltiazem or metoprolol, either of which could precipitate worsening hypotension. However, the goals were to restore his previous rhythm, to relieve symptoms with a minimum of side effects and unintended effects, and to avoid intensive care unit (ICU) transfer. Intravenous adenosine and esmolol were also considered, given their shorter half‐life, potentially lower side effect profile, and ability to produce relief of the patient's distress without further complication. The pros and cons of the situation were discussed with the patient. While he desperately wanted to feel better, he wished to stay with his family where he was. He consented to a trial of adenosine, and agreed to remain on the Palliative Care Unit. The therapeutic plan was a trial of IV adenosine, and then metoprolol if necessary. He was assured that if this was unsuccessful, we would do all we could to keep him comfortable without ICU transfer. While the patient was monitored with a portable 12‐lead EKG machine, the Palliative Medicine fellow administered adenosine 6 mg IV. Predictably, the patient noted flushing, a sense of impending doom, and a short pause of asystole. This was followed by electrocardiographic conversion to sinus tachycardia at a rate of 100 beats per minute and hemodynamic and symptomatic improvement. The patient noted that his dyspnea and generalized sense of not feeling well resolved, and he was monitored for about 30 minutes without return of the SVT. The remainder of his hospitalization was uneventful, and he was discharged to home hospice the following day. He survived for another 3 weeks without return of symptoms of arrhythmia.
Discussion
Patient preferences in terms of advanced cardiopulmonary support must be addressed during hospital admission. This is in accord with recommendations from the Patient Self‐Determination Act of 1990, as well as the Joint Commission on Accreditation of Healthcare Organizations.1 Nevertheless, the number of U.S. adults with completed advance directives to guide care providers and families with preferences if personally unable to articulate them is estimated at 5% to 25%.2 Clearly‐documented wishes are particularly important in patients with advanced cancer; however, early studies show that this happens as little as 27% of the time3 in seriously ill cancer patients. In fact, oncology physicians report direct discussions about death with only 37% of their dying patients4 and cancer patients are found to have discussions at far lower rates than patients with amyotrophic lateral sclerosis despite worse survival.5
Cardiopulmonary resuscitation (CPR) and the advanced cardiac life support (ACLS) algorithms were established to treat life‐threatening arrhythmias (namely ventricular tachycardia/fibrillation) in otherwise healthy patients who experienced witnessed intraoperative arrest. Original reports of closed chest compressions were in the intraoperative or perioperative setting.6 However, benefits of rapid initiation of CPR in witnessed out‐of‐hospital cardiac arrest were later noted as providing the only reasonable hope for reduced mortality and improved neurologic outcomes.7, 8
While CPR has shown this marginal but significant difference in outcomes of witnessed out‐of‐hospital cardiac arrest, patient with advanced life‐limiting or life threatening illness tend to have even worse outcomes even if cardiac arrest is witnessed. Survival of all cardiac arrest patients to discharge has been estimated at 3% to 14% if cardiac arrest occurs outside of the hospital and 10% to 20% for witnessed, in‐hospital cardiac arrest.912 However, a recent meta‐analysis of resuscitation for cancer patients estimates overall survival to discharge at 6.2%, and less when factoring in metastatic disease (5.6%), or ICU care at time of arrest (2.2%).13
Multiple reasons have been cited regarding why patients choose to forego resuscitation or proceed with full resuscitation status despite advanced life‐threatening illness. Factors associated with refusal of CPR include being older, female, living in a nursing home and having a worsening functional status, depression, and/or an expected poor outcome.14, 15 One can speculate that fear of no longer being cared for or being abandoned may be inferred or directly stated, and this may or may not be related to socioeconomic factors, stressors outside of the medical system, or underlying depressive symptomatology, especially hopelessness. Alternatively, 1 study revealed that an unclear expectation of outcome and prognosis after cardiopulmonary arrest led some to proceed with full resuscitative measures.15
Reports differ regarding the advanced care trajectory based on patient wishes. One study of 872 critically ill cancer patients found no significant difference in application of life‐sustaining therapies regardless of presence of an advance directive.3 The SUPPORT study mentioned above was specifically designed to understand preferences for CPR.14 While SUPPORT found that foregoing CPR may be associated with a small reduction in intensity of care, there was no difference in overall hospital survival.14 Last, although advance directives are static in terms of patient's stated wishes, a patient with decision‐making capacity is able to request a shift in goals of care at any time. However, a case‐based survey of 241 responding physicians concluded that a DNR order may indeed be associated with less aggressive and/or life‐prolonging interventions, CPR notwithstanding.16 This concept of treating those with DNR status less aggressively is often born out in terms of popular perception.17 A recent study has demonstrated that patients who discuss these issues with physicians and elect a DNR status not only have fewer aggressive interventions, but also report a higher quality of life.4
A particular nidus for this confusion may be how one interprets the DNR directive. Although DNR is specifically associated with 3 basic tenets (no endotracheal intubation, no chest compressions, and no defibrillation in the setting of cardiopulmonary arrest), this designation does not substitute for intact patient decision‐making capacity in considering other supportive measures. Intermediate steps such as limited aggressive therapy orders have been suggested to provide time‐limited and goal‐limited advanced care.9 While this offers a broader array of scenarios to be considered prior to and during clinical encounters, this may also muddy the picture with impractical options and further lack of clarity in already complex situations. The Physician Orders for Life‐Sustaining Treatment (POLST) movement has taken roots in several states, targeting seriously ill patients such as the frail and elderly. The POLST provides more explicit information regarding limited advanced measures such as nutrition or antibiotics, and may be particularly useful as a prehospital decision aid.18 While the POLST, just as the traditional advance directive, may provide clinical guidance outside of situations described explicitly therein,, it may not provide further information about goals of care, (ie, Is there a situation when 1 of these measures may be acceptable?). To reiterate what was stated about traditional directives, the POLST also applies only in situations where a patient is lacking decision‐making capacity at the time of an acute event.
The designation of DNR may indeed allow for introduction of advanced care measures that may be in accord with the patient's overall wishes and clinical prognosis. Several interventions may be appropriate on a time‐limited basis. In addition to administration of adenosine or antiarrhythmics, as in the case of our patient, the use of broad‐spectrum antimicrobial therapy, vasoactive medications, and consideration for intensive monitoring may all be appropriate on a time‐limited basis. Nevertheless, without a clear understanding of the goals of limited aggressive therapy, some would argue there is always a slippery slope in terms of technology and the implementation of advanced care measures. Hence, expectations regarding perceived outcomes, goals to be achieved by the therapy, and reasonable time lines may further clarify the patient's wishes.
In this patient scenario, the administration of adenosine is generally safe, but may lead to prolonged asystole, atrial fibrillation, and ventricular tachyarrhythmias.1921 This may lead one to consider further downstream ACLS interventions, including defibrillation or atropine. From an ethical standpoint, it is valuable to consider what would have been the next step beyond this step, in terms of advanced care measures. In the case of our patient, these measures were considered, and all accepted the goals of our intervention and its limitations. While virtually all treatments provided by physicians may predispose patients to iatrogenesis, the risks and benefits of interventions are particularly important considerations in the seriously ill patient with limited life expectancy.
Iatrogenic adverse events can be serious and fatal, and occur in 4% to 9% of hospitalized patients.2224 There has been much debate about what to do for iatrogenic adverse events, particularly when patients have clearly articulated advanced directives and DNR requests. While some argue there is a higher moral duty to reverse complications resulting from physician error or treatment‐induced complication, others would feel that the fiduciary obligation is to the patient's request.25, 26 Again, in the setting of our clinical scenario, having clear, up‐front expectations about goals of care and limitation inherent were articulated as much as able.
With increasing complexity of inpatient care and team‐based models of care becoming the norm, discerning patient's wishes continuously throughout a hospital course is critical. While this responsibility previously would have fallen to the 1 coordinating clinician (ie, the primary care physician, or the patient's subspecialist), it is increasingly becoming the responsibility of all members of the team. While provider's level of prior education, exposure, and comfort may vary, several resources have attempted to address these concerns and attempted to lay a framework for overcoming barriers to these discussion and tips on empathetic and effective communication.17, 2729
Skills notwithstanding, hospitalists particularly face a challenge in communicating these tenuous issues with patients. While there is intrinsic value in having an standardized approach to these situations, hospitalists are often thrown into these difficult situations in a fragmented, nonlongitudinal fashion, further heightening the clinical and ethical tension.28, 30 However, hospitalists are also is an area where they can truly make an impact in these patients' lives at a critical juncture. Evidence suggests that regardless of the provider who broaches the subject, patients have a desire to talk about these issues.4, 14 Hospitalists may be in an advantageous position compared to their primary care or subspecialist colleagues, in that they can offer a fresh perspective and the ability to have a dialog with the patient about these issues.
Implications
While patients are entitled to die free from the intrusion of chest compression and endotracheal tubes, they are also entitled to have symptoms aggressively managed. Advanced care measures may be appropriate for symptom palliation in complex clinical situations. A careful understanding of the patient's wishes and goals of care, after thoughtful exploration, may include therapies that in isolation, appear to be extraordinary or excessive. SVT is often quickly and successfully treated at the bedside. Despite a firm DNR status, treatment with IV adenosine allowed our patient time to return home with his family.
Acknowledgements
Special thanks to Dr. Paul S. Mueller for his thoughtful review and commentary regarding this manuscript.
- 2006 Comprehensive Accreditation Manual for Hospitals: The Official Handbook (CAMH).Oak Brook Terrace, IL:Joint Commission Resources;2006.
- ,,.Advance directives and do‐not‐resuscitate orders on general medical wards versus the intensive care unit.Mil Med.204;169:433–436.
- ,,.Advance directives in critically ill cancer patients.Crit Care Nurs Clin North Am.2000;12:373–383.
- ,,, et al.Associations between end‐of‐life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment.JAMA.2008;300:1665–1673.
- ,,, et al.Decision‐making in patients with advanced cancer compared with amyotrophic lateral sclerosis.J Med Ethics.2008;34:664–668.
- ,,.Closed‐chest cardiac massage.JAMA.1960;173:1064–1067.
- ,,, et al.Modifiable factors associated with improved cardiac arrest survival in a multicenter basic life support/defibrillation system: OPALS Study Phase I results. Ontario prehospital advanced life support.Ann Emerg Med.1999;33:44–50.
- ,,, et al.Effect of bystander initiated cardiopulmonary resuscitation on ventricular fibrillation and survival after witnessed cardiac arrest outside hospital.Br Heart J.1994;72:408–412.
- ,,.CPR for patients labeled DNR: the role of the limited aggressive therapy order.Ann Intern Med.2003;138:65–68.
- ,,, et al.A comparison of repeated high doses and repeated standard doses of epinephrine for cardiac arrest outside the hospital. European Epinephrine Study Group.N Engl J Med.1998;339:1595–1601.
- ,,,.Does age affect outcomes of out‐of‐hospital cardiopulmonary resuscitation?JAMA.1990;264:2109–2110.
- ,,, et al.A comparison of standard cardiopulmonary resuscitation and active compression‐decompression resuscitation for out‐of‐hospital cardiac arrest. French Active Compression‐Decompression Cardiopulmonary Resuscitation Study Group.N Engl J Med.1999;341:569–575.
- ,,,,,.Survival in cancer patients undergoing in‐hospital cardiopulmonary resuscitation: a meta‐analysis.Resuscitation.2006;71:152–160.
- ,,, et al.Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. SUPPORT Investigators. Study to understand prognoses and preferences for outcomes and risks of treatments.Am J Med.1996:128–137.
- ,,,,,.Preferences for life‐sustaining treatments in advance care planning and surrogate decision making.J Palliat Med.2000;3(1):37–48.
- ,.The effect of do‐not‐resuscitate orders on physician‐making.J Am Geriatr Soc.2002;50:2057–2061.
- ,,, et al.A staff dialogue on do not resuscitate orders: psychosocial issues faced by patients, their families, and caregivers.Oncologist.1999;4:256–262.
- ,,,.Hope for the future: achieving the original intent of advance directives.Hastings Cent Rep.2005;12S:S26–S30.
- ,.Adenosine‐induced non‐sustained polymorphic ventricular tachycardia.Eur Heart J.1994;15:281–282.
- ,,,.Adenosine induced ventricular arrhythmias in the emergency room.Pacing Clin Electrophysiol.2001;24:450–455.
- ,.Torsades de pointes after intravenous adenosine in the presence of prolonged QT syndrome.Am Heart J.1992;123:794–796.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients—results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- .The hazards of hospitalization.Ann Intern Med.1964;60:100–110.
- ,,,.Iatrogenic illness on a general medical service at a university hospital.N Engl J Med.1981;304:638–642.
- ,.Overriding a patient's refusal of treatment after an iatrogenic complication.N Engl J Med.1997;336:1908–1910.
- ,,.Would physicians override a do‐not‐resuscitate order when a cardiac arrest is iatrogenic?J Gen Intern Med.1999;14:35–38.
- ,,.Discussing resuscitation preferences with patients: challenges and rewards.J Hosp Med.2006;1:231–240.
- .Decision making at a time of crisis near the end of life.JAMA.2004;292:1738–1743.
- ,,,.Advance care planning as a process: structuring the discussions in practice.J Am Geriatr Soc.1995;43:440–446.
- .Addressing end‐of‐life issues.JAMA.2005;293:162.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Resuscitation status and patient wishes in terms of advanced cardiopulmonary support must be addressed during inpatient hospital admissions. However, the lack of clarity of the patients' wishes and the variability in physicians' comfort addressing these issues often leads to ambiguity in an emergency setting. This may result in inappropriately aggressive management, and conversely, it may also lead to withholding potentially lifesaving therapy due to Do Not Resuscitate (DNR) designation. We report a case of hemodynamic instability due to acute supraventricular tachycardia (SVT) in a patient with a DNR designation. He was successfully treated according to the advanced cardiac life support (ACLS) protocol for SVT. We also discuss some of the ethical challenges of providing potential life‐sustaining interventions in palliative medicine, as well as the dilemma of whether or not to provide such interventions to patients who have DNR status.
Case Presentation
A 45‐year‐old man with advanced tonsillar cancer was admitted to an inpatient palliative care unit for evaluation and treatment of anorexia, progressive pain, and asthenia. He had undergone tumor debulking and neck dissection followed by adjuvant chemotherapy and external beam radiation therapy. Despite maximal therapy, the patient developed locally recurrent disease (leading to more surgery) and later, progressive metastatic disease (treated with palliative radiation therapy). With ongoing weight loss and failure to thrive, a percutaneous gastrostomy tube was placed for nutritional support. Still, the patient suffered from significant stomatitis, esophagitis, and diarrhea consistent with radiation‐induced injury, and had several admissions for dehydration and pain control.
During this and prior admissions, the patient clearly articulated his preference for DNR status. The patient was clinically declining, but was still functional, with and estimated survival of weeks to a few months. As with previous admissions, he was given intravenous fluids and parenteral opioids, and his electrolytes and vital signs normalized to his baseline. On the day of anticipated discharge, the patient was at his hemodynamic baseline (pulse of 100 beats per minute, blood pressure of 98/60 mmHg). Upon returning to bed after a shower, the patient developed acute dyspnea, weakness, and diaphoresis. Heart rate was 170 beats per minute and blood pressure was 70/50 mmHg. Intravenous normal saline boluses were given while electrocardiogram (EKG) was obtained. EKG revealed SVT with changes suggestive of demand myocardial ischemia. Carotid massage and Valsalva maneuvers were unsuccessful in converting the rhythm to sinus.
At that point, consideration was given to his DNR designation. The treating physician and patient briefly discussed the alternatives of no treatment of his arrhythmia, or alternatively, more aggressive treatment options on the Palliative Care Unit, including intravenous (IV) adenosine and direct current cardioversion. He did not have a detailed advanced directive discussing similar scenarios; he had only completed a commonly‐used, state‐issued Durable DNR form. All decided the SVT was potentially reversible and appeared to be causing many of the patient's acute symptoms; hence, aggressive treatment of the arrhythmia was in his best interest.
Despite absence of telemetry monitoring, consideration was given to IV diltiazem or metoprolol, either of which could precipitate worsening hypotension. However, the goals were to restore his previous rhythm, to relieve symptoms with a minimum of side effects and unintended effects, and to avoid intensive care unit (ICU) transfer. Intravenous adenosine and esmolol were also considered, given their shorter half‐life, potentially lower side effect profile, and ability to produce relief of the patient's distress without further complication. The pros and cons of the situation were discussed with the patient. While he desperately wanted to feel better, he wished to stay with his family where he was. He consented to a trial of adenosine, and agreed to remain on the Palliative Care Unit. The therapeutic plan was a trial of IV adenosine, and then metoprolol if necessary. He was assured that if this was unsuccessful, we would do all we could to keep him comfortable without ICU transfer. While the patient was monitored with a portable 12‐lead EKG machine, the Palliative Medicine fellow administered adenosine 6 mg IV. Predictably, the patient noted flushing, a sense of impending doom, and a short pause of asystole. This was followed by electrocardiographic conversion to sinus tachycardia at a rate of 100 beats per minute and hemodynamic and symptomatic improvement. The patient noted that his dyspnea and generalized sense of not feeling well resolved, and he was monitored for about 30 minutes without return of the SVT. The remainder of his hospitalization was uneventful, and he was discharged to home hospice the following day. He survived for another 3 weeks without return of symptoms of arrhythmia.
Discussion
Patient preferences in terms of advanced cardiopulmonary support must be addressed during hospital admission. This is in accord with recommendations from the Patient Self‐Determination Act of 1990, as well as the Joint Commission on Accreditation of Healthcare Organizations.1 Nevertheless, the number of U.S. adults with completed advance directives to guide care providers and families with preferences if personally unable to articulate them is estimated at 5% to 25%.2 Clearly‐documented wishes are particularly important in patients with advanced cancer; however, early studies show that this happens as little as 27% of the time3 in seriously ill cancer patients. In fact, oncology physicians report direct discussions about death with only 37% of their dying patients4 and cancer patients are found to have discussions at far lower rates than patients with amyotrophic lateral sclerosis despite worse survival.5
Cardiopulmonary resuscitation (CPR) and the advanced cardiac life support (ACLS) algorithms were established to treat life‐threatening arrhythmias (namely ventricular tachycardia/fibrillation) in otherwise healthy patients who experienced witnessed intraoperative arrest. Original reports of closed chest compressions were in the intraoperative or perioperative setting.6 However, benefits of rapid initiation of CPR in witnessed out‐of‐hospital cardiac arrest were later noted as providing the only reasonable hope for reduced mortality and improved neurologic outcomes.7, 8
While CPR has shown this marginal but significant difference in outcomes of witnessed out‐of‐hospital cardiac arrest, patient with advanced life‐limiting or life threatening illness tend to have even worse outcomes even if cardiac arrest is witnessed. Survival of all cardiac arrest patients to discharge has been estimated at 3% to 14% if cardiac arrest occurs outside of the hospital and 10% to 20% for witnessed, in‐hospital cardiac arrest.912 However, a recent meta‐analysis of resuscitation for cancer patients estimates overall survival to discharge at 6.2%, and less when factoring in metastatic disease (5.6%), or ICU care at time of arrest (2.2%).13
Multiple reasons have been cited regarding why patients choose to forego resuscitation or proceed with full resuscitation status despite advanced life‐threatening illness. Factors associated with refusal of CPR include being older, female, living in a nursing home and having a worsening functional status, depression, and/or an expected poor outcome.14, 15 One can speculate that fear of no longer being cared for or being abandoned may be inferred or directly stated, and this may or may not be related to socioeconomic factors, stressors outside of the medical system, or underlying depressive symptomatology, especially hopelessness. Alternatively, 1 study revealed that an unclear expectation of outcome and prognosis after cardiopulmonary arrest led some to proceed with full resuscitative measures.15
Reports differ regarding the advanced care trajectory based on patient wishes. One study of 872 critically ill cancer patients found no significant difference in application of life‐sustaining therapies regardless of presence of an advance directive.3 The SUPPORT study mentioned above was specifically designed to understand preferences for CPR.14 While SUPPORT found that foregoing CPR may be associated with a small reduction in intensity of care, there was no difference in overall hospital survival.14 Last, although advance directives are static in terms of patient's stated wishes, a patient with decision‐making capacity is able to request a shift in goals of care at any time. However, a case‐based survey of 241 responding physicians concluded that a DNR order may indeed be associated with less aggressive and/or life‐prolonging interventions, CPR notwithstanding.16 This concept of treating those with DNR status less aggressively is often born out in terms of popular perception.17 A recent study has demonstrated that patients who discuss these issues with physicians and elect a DNR status not only have fewer aggressive interventions, but also report a higher quality of life.4
A particular nidus for this confusion may be how one interprets the DNR directive. Although DNR is specifically associated with 3 basic tenets (no endotracheal intubation, no chest compressions, and no defibrillation in the setting of cardiopulmonary arrest), this designation does not substitute for intact patient decision‐making capacity in considering other supportive measures. Intermediate steps such as limited aggressive therapy orders have been suggested to provide time‐limited and goal‐limited advanced care.9 While this offers a broader array of scenarios to be considered prior to and during clinical encounters, this may also muddy the picture with impractical options and further lack of clarity in already complex situations. The Physician Orders for Life‐Sustaining Treatment (POLST) movement has taken roots in several states, targeting seriously ill patients such as the frail and elderly. The POLST provides more explicit information regarding limited advanced measures such as nutrition or antibiotics, and may be particularly useful as a prehospital decision aid.18 While the POLST, just as the traditional advance directive, may provide clinical guidance outside of situations described explicitly therein,, it may not provide further information about goals of care, (ie, Is there a situation when 1 of these measures may be acceptable?). To reiterate what was stated about traditional directives, the POLST also applies only in situations where a patient is lacking decision‐making capacity at the time of an acute event.
The designation of DNR may indeed allow for introduction of advanced care measures that may be in accord with the patient's overall wishes and clinical prognosis. Several interventions may be appropriate on a time‐limited basis. In addition to administration of adenosine or antiarrhythmics, as in the case of our patient, the use of broad‐spectrum antimicrobial therapy, vasoactive medications, and consideration for intensive monitoring may all be appropriate on a time‐limited basis. Nevertheless, without a clear understanding of the goals of limited aggressive therapy, some would argue there is always a slippery slope in terms of technology and the implementation of advanced care measures. Hence, expectations regarding perceived outcomes, goals to be achieved by the therapy, and reasonable time lines may further clarify the patient's wishes.
In this patient scenario, the administration of adenosine is generally safe, but may lead to prolonged asystole, atrial fibrillation, and ventricular tachyarrhythmias.1921 This may lead one to consider further downstream ACLS interventions, including defibrillation or atropine. From an ethical standpoint, it is valuable to consider what would have been the next step beyond this step, in terms of advanced care measures. In the case of our patient, these measures were considered, and all accepted the goals of our intervention and its limitations. While virtually all treatments provided by physicians may predispose patients to iatrogenesis, the risks and benefits of interventions are particularly important considerations in the seriously ill patient with limited life expectancy.
Iatrogenic adverse events can be serious and fatal, and occur in 4% to 9% of hospitalized patients.2224 There has been much debate about what to do for iatrogenic adverse events, particularly when patients have clearly articulated advanced directives and DNR requests. While some argue there is a higher moral duty to reverse complications resulting from physician error or treatment‐induced complication, others would feel that the fiduciary obligation is to the patient's request.25, 26 Again, in the setting of our clinical scenario, having clear, up‐front expectations about goals of care and limitation inherent were articulated as much as able.
With increasing complexity of inpatient care and team‐based models of care becoming the norm, discerning patient's wishes continuously throughout a hospital course is critical. While this responsibility previously would have fallen to the 1 coordinating clinician (ie, the primary care physician, or the patient's subspecialist), it is increasingly becoming the responsibility of all members of the team. While provider's level of prior education, exposure, and comfort may vary, several resources have attempted to address these concerns and attempted to lay a framework for overcoming barriers to these discussion and tips on empathetic and effective communication.17, 2729
Skills notwithstanding, hospitalists particularly face a challenge in communicating these tenuous issues with patients. While there is intrinsic value in having an standardized approach to these situations, hospitalists are often thrown into these difficult situations in a fragmented, nonlongitudinal fashion, further heightening the clinical and ethical tension.28, 30 However, hospitalists are also is an area where they can truly make an impact in these patients' lives at a critical juncture. Evidence suggests that regardless of the provider who broaches the subject, patients have a desire to talk about these issues.4, 14 Hospitalists may be in an advantageous position compared to their primary care or subspecialist colleagues, in that they can offer a fresh perspective and the ability to have a dialog with the patient about these issues.
Implications
While patients are entitled to die free from the intrusion of chest compression and endotracheal tubes, they are also entitled to have symptoms aggressively managed. Advanced care measures may be appropriate for symptom palliation in complex clinical situations. A careful understanding of the patient's wishes and goals of care, after thoughtful exploration, may include therapies that in isolation, appear to be extraordinary or excessive. SVT is often quickly and successfully treated at the bedside. Despite a firm DNR status, treatment with IV adenosine allowed our patient time to return home with his family.
Acknowledgements
Special thanks to Dr. Paul S. Mueller for his thoughtful review and commentary regarding this manuscript.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
Resuscitation status and patient wishes in terms of advanced cardiopulmonary support must be addressed during inpatient hospital admissions. However, the lack of clarity of the patients' wishes and the variability in physicians' comfort addressing these issues often leads to ambiguity in an emergency setting. This may result in inappropriately aggressive management, and conversely, it may also lead to withholding potentially lifesaving therapy due to Do Not Resuscitate (DNR) designation. We report a case of hemodynamic instability due to acute supraventricular tachycardia (SVT) in a patient with a DNR designation. He was successfully treated according to the advanced cardiac life support (ACLS) protocol for SVT. We also discuss some of the ethical challenges of providing potential life‐sustaining interventions in palliative medicine, as well as the dilemma of whether or not to provide such interventions to patients who have DNR status.
Case Presentation
A 45‐year‐old man with advanced tonsillar cancer was admitted to an inpatient palliative care unit for evaluation and treatment of anorexia, progressive pain, and asthenia. He had undergone tumor debulking and neck dissection followed by adjuvant chemotherapy and external beam radiation therapy. Despite maximal therapy, the patient developed locally recurrent disease (leading to more surgery) and later, progressive metastatic disease (treated with palliative radiation therapy). With ongoing weight loss and failure to thrive, a percutaneous gastrostomy tube was placed for nutritional support. Still, the patient suffered from significant stomatitis, esophagitis, and diarrhea consistent with radiation‐induced injury, and had several admissions for dehydration and pain control.
During this and prior admissions, the patient clearly articulated his preference for DNR status. The patient was clinically declining, but was still functional, with and estimated survival of weeks to a few months. As with previous admissions, he was given intravenous fluids and parenteral opioids, and his electrolytes and vital signs normalized to his baseline. On the day of anticipated discharge, the patient was at his hemodynamic baseline (pulse of 100 beats per minute, blood pressure of 98/60 mmHg). Upon returning to bed after a shower, the patient developed acute dyspnea, weakness, and diaphoresis. Heart rate was 170 beats per minute and blood pressure was 70/50 mmHg. Intravenous normal saline boluses were given while electrocardiogram (EKG) was obtained. EKG revealed SVT with changes suggestive of demand myocardial ischemia. Carotid massage and Valsalva maneuvers were unsuccessful in converting the rhythm to sinus.
At that point, consideration was given to his DNR designation. The treating physician and patient briefly discussed the alternatives of no treatment of his arrhythmia, or alternatively, more aggressive treatment options on the Palliative Care Unit, including intravenous (IV) adenosine and direct current cardioversion. He did not have a detailed advanced directive discussing similar scenarios; he had only completed a commonly‐used, state‐issued Durable DNR form. All decided the SVT was potentially reversible and appeared to be causing many of the patient's acute symptoms; hence, aggressive treatment of the arrhythmia was in his best interest.
Despite absence of telemetry monitoring, consideration was given to IV diltiazem or metoprolol, either of which could precipitate worsening hypotension. However, the goals were to restore his previous rhythm, to relieve symptoms with a minimum of side effects and unintended effects, and to avoid intensive care unit (ICU) transfer. Intravenous adenosine and esmolol were also considered, given their shorter half‐life, potentially lower side effect profile, and ability to produce relief of the patient's distress without further complication. The pros and cons of the situation were discussed with the patient. While he desperately wanted to feel better, he wished to stay with his family where he was. He consented to a trial of adenosine, and agreed to remain on the Palliative Care Unit. The therapeutic plan was a trial of IV adenosine, and then metoprolol if necessary. He was assured that if this was unsuccessful, we would do all we could to keep him comfortable without ICU transfer. While the patient was monitored with a portable 12‐lead EKG machine, the Palliative Medicine fellow administered adenosine 6 mg IV. Predictably, the patient noted flushing, a sense of impending doom, and a short pause of asystole. This was followed by electrocardiographic conversion to sinus tachycardia at a rate of 100 beats per minute and hemodynamic and symptomatic improvement. The patient noted that his dyspnea and generalized sense of not feeling well resolved, and he was monitored for about 30 minutes without return of the SVT. The remainder of his hospitalization was uneventful, and he was discharged to home hospice the following day. He survived for another 3 weeks without return of symptoms of arrhythmia.
Discussion
Patient preferences in terms of advanced cardiopulmonary support must be addressed during hospital admission. This is in accord with recommendations from the Patient Self‐Determination Act of 1990, as well as the Joint Commission on Accreditation of Healthcare Organizations.1 Nevertheless, the number of U.S. adults with completed advance directives to guide care providers and families with preferences if personally unable to articulate them is estimated at 5% to 25%.2 Clearly‐documented wishes are particularly important in patients with advanced cancer; however, early studies show that this happens as little as 27% of the time3 in seriously ill cancer patients. In fact, oncology physicians report direct discussions about death with only 37% of their dying patients4 and cancer patients are found to have discussions at far lower rates than patients with amyotrophic lateral sclerosis despite worse survival.5
Cardiopulmonary resuscitation (CPR) and the advanced cardiac life support (ACLS) algorithms were established to treat life‐threatening arrhythmias (namely ventricular tachycardia/fibrillation) in otherwise healthy patients who experienced witnessed intraoperative arrest. Original reports of closed chest compressions were in the intraoperative or perioperative setting.6 However, benefits of rapid initiation of CPR in witnessed out‐of‐hospital cardiac arrest were later noted as providing the only reasonable hope for reduced mortality and improved neurologic outcomes.7, 8
While CPR has shown this marginal but significant difference in outcomes of witnessed out‐of‐hospital cardiac arrest, patient with advanced life‐limiting or life threatening illness tend to have even worse outcomes even if cardiac arrest is witnessed. Survival of all cardiac arrest patients to discharge has been estimated at 3% to 14% if cardiac arrest occurs outside of the hospital and 10% to 20% for witnessed, in‐hospital cardiac arrest.912 However, a recent meta‐analysis of resuscitation for cancer patients estimates overall survival to discharge at 6.2%, and less when factoring in metastatic disease (5.6%), or ICU care at time of arrest (2.2%).13
Multiple reasons have been cited regarding why patients choose to forego resuscitation or proceed with full resuscitation status despite advanced life‐threatening illness. Factors associated with refusal of CPR include being older, female, living in a nursing home and having a worsening functional status, depression, and/or an expected poor outcome.14, 15 One can speculate that fear of no longer being cared for or being abandoned may be inferred or directly stated, and this may or may not be related to socioeconomic factors, stressors outside of the medical system, or underlying depressive symptomatology, especially hopelessness. Alternatively, 1 study revealed that an unclear expectation of outcome and prognosis after cardiopulmonary arrest led some to proceed with full resuscitative measures.15
Reports differ regarding the advanced care trajectory based on patient wishes. One study of 872 critically ill cancer patients found no significant difference in application of life‐sustaining therapies regardless of presence of an advance directive.3 The SUPPORT study mentioned above was specifically designed to understand preferences for CPR.14 While SUPPORT found that foregoing CPR may be associated with a small reduction in intensity of care, there was no difference in overall hospital survival.14 Last, although advance directives are static in terms of patient's stated wishes, a patient with decision‐making capacity is able to request a shift in goals of care at any time. However, a case‐based survey of 241 responding physicians concluded that a DNR order may indeed be associated with less aggressive and/or life‐prolonging interventions, CPR notwithstanding.16 This concept of treating those with DNR status less aggressively is often born out in terms of popular perception.17 A recent study has demonstrated that patients who discuss these issues with physicians and elect a DNR status not only have fewer aggressive interventions, but also report a higher quality of life.4
A particular nidus for this confusion may be how one interprets the DNR directive. Although DNR is specifically associated with 3 basic tenets (no endotracheal intubation, no chest compressions, and no defibrillation in the setting of cardiopulmonary arrest), this designation does not substitute for intact patient decision‐making capacity in considering other supportive measures. Intermediate steps such as limited aggressive therapy orders have been suggested to provide time‐limited and goal‐limited advanced care.9 While this offers a broader array of scenarios to be considered prior to and during clinical encounters, this may also muddy the picture with impractical options and further lack of clarity in already complex situations. The Physician Orders for Life‐Sustaining Treatment (POLST) movement has taken roots in several states, targeting seriously ill patients such as the frail and elderly. The POLST provides more explicit information regarding limited advanced measures such as nutrition or antibiotics, and may be particularly useful as a prehospital decision aid.18 While the POLST, just as the traditional advance directive, may provide clinical guidance outside of situations described explicitly therein,, it may not provide further information about goals of care, (ie, Is there a situation when 1 of these measures may be acceptable?). To reiterate what was stated about traditional directives, the POLST also applies only in situations where a patient is lacking decision‐making capacity at the time of an acute event.
The designation of DNR may indeed allow for introduction of advanced care measures that may be in accord with the patient's overall wishes and clinical prognosis. Several interventions may be appropriate on a time‐limited basis. In addition to administration of adenosine or antiarrhythmics, as in the case of our patient, the use of broad‐spectrum antimicrobial therapy, vasoactive medications, and consideration for intensive monitoring may all be appropriate on a time‐limited basis. Nevertheless, without a clear understanding of the goals of limited aggressive therapy, some would argue there is always a slippery slope in terms of technology and the implementation of advanced care measures. Hence, expectations regarding perceived outcomes, goals to be achieved by the therapy, and reasonable time lines may further clarify the patient's wishes.
In this patient scenario, the administration of adenosine is generally safe, but may lead to prolonged asystole, atrial fibrillation, and ventricular tachyarrhythmias.1921 This may lead one to consider further downstream ACLS interventions, including defibrillation or atropine. From an ethical standpoint, it is valuable to consider what would have been the next step beyond this step, in terms of advanced care measures. In the case of our patient, these measures were considered, and all accepted the goals of our intervention and its limitations. While virtually all treatments provided by physicians may predispose patients to iatrogenesis, the risks and benefits of interventions are particularly important considerations in the seriously ill patient with limited life expectancy.
Iatrogenic adverse events can be serious and fatal, and occur in 4% to 9% of hospitalized patients.2224 There has been much debate about what to do for iatrogenic adverse events, particularly when patients have clearly articulated advanced directives and DNR requests. While some argue there is a higher moral duty to reverse complications resulting from physician error or treatment‐induced complication, others would feel that the fiduciary obligation is to the patient's request.25, 26 Again, in the setting of our clinical scenario, having clear, up‐front expectations about goals of care and limitation inherent were articulated as much as able.
With increasing complexity of inpatient care and team‐based models of care becoming the norm, discerning patient's wishes continuously throughout a hospital course is critical. While this responsibility previously would have fallen to the 1 coordinating clinician (ie, the primary care physician, or the patient's subspecialist), it is increasingly becoming the responsibility of all members of the team. While provider's level of prior education, exposure, and comfort may vary, several resources have attempted to address these concerns and attempted to lay a framework for overcoming barriers to these discussion and tips on empathetic and effective communication.17, 2729
Skills notwithstanding, hospitalists particularly face a challenge in communicating these tenuous issues with patients. While there is intrinsic value in having an standardized approach to these situations, hospitalists are often thrown into these difficult situations in a fragmented, nonlongitudinal fashion, further heightening the clinical and ethical tension.28, 30 However, hospitalists are also is an area where they can truly make an impact in these patients' lives at a critical juncture. Evidence suggests that regardless of the provider who broaches the subject, patients have a desire to talk about these issues.4, 14 Hospitalists may be in an advantageous position compared to their primary care or subspecialist colleagues, in that they can offer a fresh perspective and the ability to have a dialog with the patient about these issues.
Implications
While patients are entitled to die free from the intrusion of chest compression and endotracheal tubes, they are also entitled to have symptoms aggressively managed. Advanced care measures may be appropriate for symptom palliation in complex clinical situations. A careful understanding of the patient's wishes and goals of care, after thoughtful exploration, may include therapies that in isolation, appear to be extraordinary or excessive. SVT is often quickly and successfully treated at the bedside. Despite a firm DNR status, treatment with IV adenosine allowed our patient time to return home with his family.
Acknowledgements
Special thanks to Dr. Paul S. Mueller for his thoughtful review and commentary regarding this manuscript.
- 2006 Comprehensive Accreditation Manual for Hospitals: The Official Handbook (CAMH).Oak Brook Terrace, IL:Joint Commission Resources;2006.
- ,,.Advance directives and do‐not‐resuscitate orders on general medical wards versus the intensive care unit.Mil Med.204;169:433–436.
- ,,.Advance directives in critically ill cancer patients.Crit Care Nurs Clin North Am.2000;12:373–383.
- ,,, et al.Associations between end‐of‐life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment.JAMA.2008;300:1665–1673.
- ,,, et al.Decision‐making in patients with advanced cancer compared with amyotrophic lateral sclerosis.J Med Ethics.2008;34:664–668.
- ,,.Closed‐chest cardiac massage.JAMA.1960;173:1064–1067.
- ,,, et al.Modifiable factors associated with improved cardiac arrest survival in a multicenter basic life support/defibrillation system: OPALS Study Phase I results. Ontario prehospital advanced life support.Ann Emerg Med.1999;33:44–50.
- ,,, et al.Effect of bystander initiated cardiopulmonary resuscitation on ventricular fibrillation and survival after witnessed cardiac arrest outside hospital.Br Heart J.1994;72:408–412.
- ,,.CPR for patients labeled DNR: the role of the limited aggressive therapy order.Ann Intern Med.2003;138:65–68.
- ,,, et al.A comparison of repeated high doses and repeated standard doses of epinephrine for cardiac arrest outside the hospital. European Epinephrine Study Group.N Engl J Med.1998;339:1595–1601.
- ,,,.Does age affect outcomes of out‐of‐hospital cardiopulmonary resuscitation?JAMA.1990;264:2109–2110.
- ,,, et al.A comparison of standard cardiopulmonary resuscitation and active compression‐decompression resuscitation for out‐of‐hospital cardiac arrest. French Active Compression‐Decompression Cardiopulmonary Resuscitation Study Group.N Engl J Med.1999;341:569–575.
- ,,,,,.Survival in cancer patients undergoing in‐hospital cardiopulmonary resuscitation: a meta‐analysis.Resuscitation.2006;71:152–160.
- ,,, et al.Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. SUPPORT Investigators. Study to understand prognoses and preferences for outcomes and risks of treatments.Am J Med.1996:128–137.
- ,,,,,.Preferences for life‐sustaining treatments in advance care planning and surrogate decision making.J Palliat Med.2000;3(1):37–48.
- ,.The effect of do‐not‐resuscitate orders on physician‐making.J Am Geriatr Soc.2002;50:2057–2061.
- ,,, et al.A staff dialogue on do not resuscitate orders: psychosocial issues faced by patients, their families, and caregivers.Oncologist.1999;4:256–262.
- ,,,.Hope for the future: achieving the original intent of advance directives.Hastings Cent Rep.2005;12S:S26–S30.
- ,.Adenosine‐induced non‐sustained polymorphic ventricular tachycardia.Eur Heart J.1994;15:281–282.
- ,,,.Adenosine induced ventricular arrhythmias in the emergency room.Pacing Clin Electrophysiol.2001;24:450–455.
- ,.Torsades de pointes after intravenous adenosine in the presence of prolonged QT syndrome.Am Heart J.1992;123:794–796.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients—results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- .The hazards of hospitalization.Ann Intern Med.1964;60:100–110.
- ,,,.Iatrogenic illness on a general medical service at a university hospital.N Engl J Med.1981;304:638–642.
- ,.Overriding a patient's refusal of treatment after an iatrogenic complication.N Engl J Med.1997;336:1908–1910.
- ,,.Would physicians override a do‐not‐resuscitate order when a cardiac arrest is iatrogenic?J Gen Intern Med.1999;14:35–38.
- ,,.Discussing resuscitation preferences with patients: challenges and rewards.J Hosp Med.2006;1:231–240.
- .Decision making at a time of crisis near the end of life.JAMA.2004;292:1738–1743.
- ,,,.Advance care planning as a process: structuring the discussions in practice.J Am Geriatr Soc.1995;43:440–446.
- .Addressing end‐of‐life issues.JAMA.2005;293:162.
- 2006 Comprehensive Accreditation Manual for Hospitals: The Official Handbook (CAMH).Oak Brook Terrace, IL:Joint Commission Resources;2006.
- ,,.Advance directives and do‐not‐resuscitate orders on general medical wards versus the intensive care unit.Mil Med.204;169:433–436.
- ,,.Advance directives in critically ill cancer patients.Crit Care Nurs Clin North Am.2000;12:373–383.
- ,,, et al.Associations between end‐of‐life discussions, patient mental health, medical care near death, and caregiver bereavement adjustment.JAMA.2008;300:1665–1673.
- ,,, et al.Decision‐making in patients with advanced cancer compared with amyotrophic lateral sclerosis.J Med Ethics.2008;34:664–668.
- ,,.Closed‐chest cardiac massage.JAMA.1960;173:1064–1067.
- ,,, et al.Modifiable factors associated with improved cardiac arrest survival in a multicenter basic life support/defibrillation system: OPALS Study Phase I results. Ontario prehospital advanced life support.Ann Emerg Med.1999;33:44–50.
- ,,, et al.Effect of bystander initiated cardiopulmonary resuscitation on ventricular fibrillation and survival after witnessed cardiac arrest outside hospital.Br Heart J.1994;72:408–412.
- ,,.CPR for patients labeled DNR: the role of the limited aggressive therapy order.Ann Intern Med.2003;138:65–68.
- ,,, et al.A comparison of repeated high doses and repeated standard doses of epinephrine for cardiac arrest outside the hospital. European Epinephrine Study Group.N Engl J Med.1998;339:1595–1601.
- ,,,.Does age affect outcomes of out‐of‐hospital cardiopulmonary resuscitation?JAMA.1990;264:2109–2110.
- ,,, et al.A comparison of standard cardiopulmonary resuscitation and active compression‐decompression resuscitation for out‐of‐hospital cardiac arrest. French Active Compression‐Decompression Cardiopulmonary Resuscitation Study Group.N Engl J Med.1999;341:569–575.
- ,,,,,.Survival in cancer patients undergoing in‐hospital cardiopulmonary resuscitation: a meta‐analysis.Resuscitation.2006;71:152–160.
- ,,, et al.Choices of seriously ill patients about cardiopulmonary resuscitation: correlates and outcomes. SUPPORT Investigators. Study to understand prognoses and preferences for outcomes and risks of treatments.Am J Med.1996:128–137.
- ,,,,,.Preferences for life‐sustaining treatments in advance care planning and surrogate decision making.J Palliat Med.2000;3(1):37–48.
- ,.The effect of do‐not‐resuscitate orders on physician‐making.J Am Geriatr Soc.2002;50:2057–2061.
- ,,, et al.A staff dialogue on do not resuscitate orders: psychosocial issues faced by patients, their families, and caregivers.Oncologist.1999;4:256–262.
- ,,,.Hope for the future: achieving the original intent of advance directives.Hastings Cent Rep.2005;12S:S26–S30.
- ,.Adenosine‐induced non‐sustained polymorphic ventricular tachycardia.Eur Heart J.1994;15:281–282.
- ,,,.Adenosine induced ventricular arrhythmias in the emergency room.Pacing Clin Electrophysiol.2001;24:450–455.
- ,.Torsades de pointes after intravenous adenosine in the presence of prolonged QT syndrome.Am Heart J.1992;123:794–796.
- ,,, et al.Incidence of adverse events and negligence in hospitalized patients—results of the Harvard Medical Practice Study I.N Engl J Med.1991;324:370–376.
- .The hazards of hospitalization.Ann Intern Med.1964;60:100–110.
- ,,,.Iatrogenic illness on a general medical service at a university hospital.N Engl J Med.1981;304:638–642.
- ,.Overriding a patient's refusal of treatment after an iatrogenic complication.N Engl J Med.1997;336:1908–1910.
- ,,.Would physicians override a do‐not‐resuscitate order when a cardiac arrest is iatrogenic?J Gen Intern Med.1999;14:35–38.
- ,,.Discussing resuscitation preferences with patients: challenges and rewards.J Hosp Med.2006;1:231–240.
- .Decision making at a time of crisis near the end of life.JAMA.2004;292:1738–1743.
- ,,,.Advance care planning as a process: structuring the discussions in practice.J Am Geriatr Soc.1995;43:440–446.
- .Addressing end‐of‐life issues.JAMA.2005;293:162.
The Devil is in the Details
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 47‐year‐old male presented to a community hospital with 5 weeks of daily fevers, accompanied by headache, myalgias, and malaise. He reported that his symptoms began abruptly 2 days after a weekend of camping in Connecticut.
This patient describes the onset of undifferentiated fever 2 days after a weekend of camping. Few infectious diseases have such short incubation periods, and either the accuracy of the history or the relationship of the camping trip to the present illness is thus questionable. However, more information about the onset and nature of the illness, and details about food, animal, water, mud, cave, wood chopping, and other environmental exposures during his trip is required. The exact dates of the camping trip may be helpful, as there is clear seasonality to vector‐borne diseases such as Lyme disease, babesiosis, ehrlichiosis, and rickettsial infections. Conditions unrelated to his camping trip, such as malignancies, rheumatologic conditions, and other infectious causes of prolonged fever, such as tuberculosis, endocarditis, or osteomyelitis, are more likely, given the duration of fever.
The fevers were accompanied by chills, without rigors, and subjectively worsened over the first 2 days. At that point, the patient began taking his temperature, and noted fevers of 38.5C to 40C occurring once or twice daily, generally in the afternoon or evening. The patient did not recall tick bites but did not carefully examine himself for ticks; he reported numerous mosquito bites during the trip. The patient camped in a tent and grilled meats and other food he had brought in a cooler. No family members or other travelers became ill. He denied spelunking, but had collected wood for camp fires, and acknowledged swimming in a freshwater pond during his trip, which occurred in August.
West Nile fever, St. Louis encephalitis, and eastern equine encephalitis are transmitted by mosquitoes in New England, but are unlikely causes of prolonged fever. Water exposure suggests the possibility of leptospirosis, and wood exposure suggests blastomycosis, but this usually presents with a pulmonary syndrome. Food‐borne illness seems unlikely. While no aspect of the history has pinpointed a specific diagnosis, exploring the progression of symptoms may offer a clue, and if he has undergone any previous evaluation, the results may significantly alter the differential diagnosis. For example, arthritis may develop weeks after fever in adult‐onset Still's disease, negative blood cultures would lower the probability of endocarditis, and common sites of pyrogenic malignancies (eg, liver, kidneys, and especially lymph nodes) may already have been imaged.
During the first 3 weeks of illness, the patient experienced daily fever and a gradual, 10‐pound weight loss. Over the next 10 days, he sought medical attention at 3 emergency departments. At one, a head computed tomography (CT) showed possible sinusitis, and he was prescribed a 7‐day course of clarithromycin, which he took without any improvement. At 2 others, he was told that his laboratory studies, and a CT of the abdomen, were normal, and that he had a viral syndrome. Several days later, and 5 weeks after the onset of symptoms, the development of dull right upper‐quadrant pain and mild nausea without vomiting prompted the current presentation to the community hospital. He reported several years of loose stools, but denied rash, arthritis, diarrhea, neck stiffness, cough, or other complaints.
A detailed past medical, social, and family history is required, with particular attention to ethnicity; immunocompromising conditions such as splenectomy or corticosteroid use; undiagnosed febrile diseases; severe, unusual, or recurrent infections; medication use; diet; sexual history; pet exposures; and any personal or family history of cancer. The development of right upper‐quadrant pain mandates attention to risk factors for viral hepatitis, known biliary pathology, or travel that might predispose the patient to pyogenic or amoebic liver abscess, and hematochezia, which could suggest a malignancy metastatic to the liver. Additionally, chronic diarrhea with new right upper‐quadrant pain may represent inflammatory bowel disease complicated by primary sclerosing cholangitis (PSC).
The patient was a Caucasian male of Mediterranean ancestry with thalassemia minor. He had undergone dilation of a benign esophageal stricture, but no surgical procedures, and he had never experienced unexplained fever or unusual infections. Medication exposure was limited to occasional use of acetaminophen for fever, and he had no known allergies. His diet was unremarkable and included no well water or unpasteurized dairy products. He denied risk factors for tuberculosis. He drank 2 to 10 beers a day, 5 times a week, had last smoked 10 years previously, and had never used illicit drugs. He denied any high‐risk sexual contacts and was monogamous with his wife, with whom he had 2 children. The family owned no pets and no relatives had suffered from malignant, rheumatologic, or febrile illness, with the exception of hand, foot, and mouth infection in an infant son, 1 year previously. The patient had never traveled outside of New England.
The history has uncovered several clues, but their relevance is doubtful. His ethnicity suggests possible familial Mediterranean fever, but recurrent abdominal pain and polyserositis, rather than a single prolonged episode, would be expected with this disease. A transfusion history should be obtained to explore the possibility of viral hepatitis. While iron overload can predispose patients to various infections including liver abscess, thalassemia minor should not require transfusion. Esophageal stricture could conceivably be due to histoplasmosis (complicated by mediastinal fibrosis) or tuberculosis, but is probably unrelated to his present illness. His excessive alcohol intake increases his risk for esophageal cancer and liver disease, but it is unlikely that metastatic disease to the liver would present with fever without preceding dysphagia, or that alcoholic hepatitis could have escaped detection after evaluations by several physicians.
We need to learn the details of the patient's physical examination. Given the development of right upper‐quadrant pain, I would particularly like to know if he had hepatosplenomegaly and if a Murphy's sign was present.
His temperature ranged from 36.9C to 39.8C, his pulse was 76 beats per minute with minimal elevations during fever spikes, and his respirations were 18 per minute. His blood pressure was 105/70 mm Hg. He was a well‐developed, overweight male with scleral icterus. He had good dentition and an oropharynx free of lesions. Cardiac examination demonstrated a regular rhythm with a normal S1 and S2, without murmurs or peripheral stigmata of infectious endocarditis. A smooth, minimally tender liver edge was palpable 2 cm below the costal margin; the spleen was nonpalpable. Murphy's sign was absent. There was no lymphadenopathy or rash. He had multiple, shallow, uninfected lacerations of both hands in various stages of healing. The remainder of his examination was normal.
The patient has obvious liver involvement. The pulse‐temperature dissociation suggests a variety of infections, including salmonellosis, psittacosis, typhoid fever, leptospirosis, tularemia, brucellosis, legionellosis, and mycoplasma pneumoniae infection. The patient should be asked how and when he injured his hands, as fresh water exposure can transmit leptospirosis across broken skin. However, while severe leptospirosis can cause fever and jaundice, the long duration of illness is not typical. The cryptogenic form of tularemiawhich can manifest as a typhoidal illnessshould be considered, given that tularemia is present in the area the patient visited; he should be asked about exposure to rabbits.
At this point, I would like to see a standard biochemical profile, a liver panel, a complete blood count and differential, urinalysis, chest X‐ray, and an electrocardiogram. I would examine thick and thin Wright‐Giemsa‐stained smears for evidence of babesiosis. Blood cultures should be held for at least 2 weeks to recover fastidious organisms like Francisella tularensis and Brucella sp. Bone marrow cultures should be obtained; they are more sensitive for mycobacteria and Brucella, and may also yield fungal pathogens. Serologies for a variety of infectious diseases, such as leptospirosis, typhoid fever, and tularemia, will be required if other diagnostic tests are unrevealing.
His white cell count was 8,100/L, with a normal differential, and his hemoglobin was 10 g/dL (normal range, 1417), with a mean corpuscular hemoglobin of 63 m3 (normal range, 8298). The platelet count was 303,000/L. Serum electrolytes were normal. His aspartate aminotransferase was 58 U/L and his alanine aminotransferase was 60 U/L (normal range for both, 1045). Bilirubin was 2.6 mg/dL (normal, <1.2); direct bilirubin was 0.9 mg/dL. Alkaline phosphatase was 150 U/L. Lactate dehydrogenase was 342 U/L (normal range, 2251). A lipase was 62 U/L. International normalized ratio (INR) was 1.4 with an activated partial thromboplastin time (aPTT) of 52 seconds (normal range, 2533). Erythrocyte sedimentation rate (ESR) was 50 mm/hour (normal range, 015). Iron studies showed a suppressed iron and iron‐binding capacity and elevated haptoglobin and ferritin (1878 ng/L; normal range, 22322). Several blood cultures obtained at admission showed no growth after 48 hours of incubation.
The anemia, low mean cell volume (MCV), and elevated ferritin and ESR are consistent with anemia of chronic disease, superimposed upon thalassemia minor. Transaminase elevations occur in a plethora of infectious processes. The elevated INR and aPTT are concerning, and may indicate a septic or malignant process with disseminated intravascular coagulation (DIC). While there is no mention of clinical DIC, it would be appropriate to obtain D‐dimers, fibrin degradation products, and a fibrinogen level. The platelet count is normal, which is reassuring.
Before initiating any empiric antimicrobials, I would obtain an abdominal ultrasound, and possibly an abdominal CT. Hepatitis (especially B and C), cytomegalovirus, and Epstein‐Barr virus serologies should be obtained. A variety of conditions including leptospirosis, tularemia, and babesiosis are possible; specific laboratory testing is required to guide therapy.
Ultrasound showed a thickened gallbladder; the liver was slightly enlarged with normal echotexture. Magnetic resonance cholangiopancreatography (MRCP) showed diffuse sequential beading and scarring of his extrahepatic biliary ducts.
There is no evidence of biliary stones, intrahepatic tumor, or abscess to explain the fever and hepatitis, although it would be helpful to know what other abdominal structures were imaged. The MRCP finding increases my suspicion of PSC, possibly complicated by infection, although the biliary abnormalities may be incidental, and an unrelated process may be responsible for the clinical presentation.
His physicians considered the possibilities of PSC and cholangiopathy due to as‐yet undiagnosed acquired immunodeficiency syndrome. Ampicillin‐sulbactam, ceftazidime, and gentamicin were administered for possible bacterial cholangitis, and endoscopic retrograde cholangiopancreatography was performed. This procedure showed only slight narrowing of his common bile duct, which was felt to be a normal variant. He felt no better after several days of antibiotic therapy, and was transferred to a tertiary care center for further evaluation. Repeat physical exam and laboratory studies were essentially unchanged. The patient explained that his hand lacerations were sustained during his work as a butcher who worked with lamb, beef, rabbit, and poultry. He rarely wore protective gloves because they induced contact dermatitis.
Tularemia becomes more likely given his history of rabbit butchering. Salmonellosis and leptospirosis also remain possible. Typhoid fever and brucellosis are unlikely unless the patient worked with imported exotic animals. At this point, given the systemic illness, empiric antibacterial therapy is reasonable. Of the chosen antimicrobials, only the gentamicin would reliably treat tularemia. I would stop ampicillin‐sulbactam and ceftazidime and replace gentamicin with ciprofloxacin, an effective and better‐tolerated agent for tularemia. Cultures of blood and bone marrow aspirate should be obtained. Stool should be cultured for Salmonella. Tularemia, leptospirosis, and typhoid serologies should be sent to a reference laboratory. At this point in the patient's illness, high‐titered antibodies should be present. However, it would be ideal to compare titers with those from previous serum sample, if possible.
The patient's antimicrobials were narrowed to doxycycline alone, for suspected zoonotic infection, but his fevers were unchanged after 1 week of treatment. Hepatitis serologies, human immunodeficiency virus (HIV) antibody, and smears for ehrlichiosis and babesiosis were negative. He had a positive immunoglobulin (Ig)G and a negative IgM for Epstein‐Barr virus and cytomegalovirus. Tularemia, ehrlichiosis, leptospirosis, brucellosis, and Query fever (Q fever) serologies were ordered. The elevated aPTT did not correct when his serum was mixed with normal serum. Thrombin was normal; factor VIII, von Willebrand (VW) factor, and VW cofactor were mildly elevated. Lupus anticoagulant was detected. A hepatologist declined to obtain a liver biopsy, citing the elevated aPTT and pending serologies. Given his clinical stability, the patient was discharged on doxycycline to await further results.
My highest suspicion is for tularemia, and I would switch antibiotic treatment to ciprofloxacin, awaiting serological results. Some in vitro studies have suggested that F. tularensis may often be resistant to doxycycline, and recent clinical experience has shown fluoroquinolones are superior to doxycycline in the treatment of tularemia.
His serologic results were as follows: tularemia, 1:32 (positive, 1:128); ehrlichia, 1:128 (granulocytic) and <1:64 (monocytic; normal for both, <1:64); leptospira, agglutinated nonspecifically; Brucella IgG and IgM 1 (negative, <9), Q fever (coxiella) IgG 1 + 2, IgM 1 + 2, all positive at 1:256 (<1:16). A transesophageal echocardiogram showed no evidence of endocarditis. The patient was treated with 10 weeks of doxycycline for Q fever hepatitis. His fever, headache, and laboratory abnormalities resolved, and he remained well after the completion of therapy.
The serologies suggest the patient had Coxiella burnetii hepatitis, and illustrate the value of a precise exposure history. Most butchers work only with muscle tissue and have a negligible risk of Q fever. In retrospect, it became clear that he worked part‐time in a slaughterhouse, where highly infectious reproductive tract fluids can dry and aerosolize.
Commentary
Q fever was proposed as the name for a febrile illness affecting Australian slaughterhouse workers in 1937.1 The etiologic agent, C. burnetii, is a small, gram‐negative, obligate intracellular proteobacterium that exists in 2 distinct phases, specializing either in entering or persisting in macrophage lysosomes.2 Additionally, spores are formed and can persist in soil.
Q fever is an uncommonly recognized disease, in part because most infected persons have no symptoms or mild symptoms.3 In the United States, the estimated annual incidence has been 0.28 per million (about 50 cases per year) since 1999, when Q fever became a reportable disease due to bioterrorism concerns. In France, more frequent farming of goats and sheep may be responsible for the much higher annual incidence of 500 per million.4 Spread is usually occupational, via aerosol contact with the dried reproductive tract secretions of animals (mainly cattle, sheep, and goats), in a slaughterhouse or farm setting. However, wind‐borne dust can carry spores long distances, and spread can occur from household pets, unpasteurized dairy products, laboratory work, and possibly ticks.3 More than 30 cases have been reported in military personnel deployed to Iraq and Afghanistan, several without obvious exposures.5 One review noted a single reported case of intradermal inoculation,3 making this patient's lacerations a possible site of infection, but he was also at risk for inhalational exposurewhen he was later asked about the details of his work, he acknowledged working at a slaughterhouse as well as a supermarket.
Symptomatic patients are male in 77% of cases, can usually identify an occupational exposure, and have a mean age of 50 years.4 Fever, which lasts 5 to 57 days, as well as fatigue and headaches, begin after a 1‐week to 3‐week incubation period. Atypical pneumonia or rash may occur; meningoencephalitis and myocarditis portend a worse prognosis. As with this patient, 45% to 85% of patients suffer from hepatitis, although few have an abnormal bilirubin.3 Liver biopsy usually reveals granulomas, which may have a classic doughnut hole appearance,2, 3 although this patient ultimately received a diagnosis without the procedure. Acute Q fever rarely (5%) requires hospitalization, and fatalities are extremely rare.3
Chronic infection (ie, lasting >6 months) most often occurs as endocarditis, although chronic hepatitis, osteomyelitis, and infections of other sites occur. Interestingly, this patient's lupus anticoagulant may have been related to his underlying illness, as autoantibodies frequently occur in Q fever, especially in patients with hepatitis, many of whom develop smooth muscle antibodies, a positive Coombs test, antiprothrombinase, or other autoantibodies,3 and there is a high incidence of antiphospholipid antibodies, particularly anticardiolipin and lupus anticoagulant antibodies.6
Because C. burnetii is an obligate intracellular pathogen, culture requires either tissue or live animal inoculation, and the diagnosis is usually made serologically. Paired sera demonstrating seroconversion or a 4‐fold increase in titers are most conclusive, but a single sample may be used. Anti‐phase II antibodies are detectable in 90% of patients within 3 weeks of infection3 and peak at 2 months5; this patient's phase II sera (IgG > 1:200, IgM > 1:50) are said to be 100% predictive for acute Q fever.3 High‐titer anti‐phase I antibodies, in contrast, indicate chronic infection, and a titer 1:800 is one of the modified Duke criteria for endocarditis.5
Acute Q fever is generally treated with doxycycline for 14 days, although prolonged therapy may be advisable to prevent endocarditis if preexisting valvular lesions are present.2, 5 Fluoroquinolones are another option and may be especially useful for meningoencephalitis.5 Because acute Q fever is generally self‐limited, demonstrating a clear benefit to antibiotic therapy is difficult. The available evidence, which was largely obtained from Q fever pneumonia patients, suggests that tetracycline therapy shortens fever duration.3 Patients with Q fever hepatitis may have a protracted course. On the basis of anecdotal reports, some experts add prednisone (tapered from 40 mg daily over a week) for patients with Q fever hepatitis who fail to respond to doxycycline promptly.3 While this patient's fever was unchanged after a week of therapy, he was well into his treatment course when his diagnosis was ultimately confirmed. His physicians felt that prednisone would be of uncertain benefit and opted not to administer it.
Treatment of Q fever endocarditis is often delayed by the combination of negative blood cultures and a low (12%) rate of vegetation formation, increasing the risk of morbidity and mortality.3 Tetracycline monotherapy is associated with a greater than 50% risk of death,5 and even 4 years of treatment may fail to sterilize valve tissue.3 However, if hydroxychloroquine is given with doxycycline for at least 18 months to alkalize lysosomes and improve bacterial killing, the mortality rate can be lowered to about 5%.3, 5 Patients should be warned of the risk of photosensitivity, and monitored for retinal toxicity2 and serologic evidence of relapse.5
Before serologic results confirmed the diagnosis of Q fever, both the patient's clinicians and the discussant had to craft an antibiotic regimen for a suspected zoonosis. The patient received doxycycline, a good choice for leptospirosis,7 brucellosis,8 tularemia,9 and Q fever,3 all possible after livestock exposure, as well as ehrlichiosis.10 The discussant, who suspected tularemia, worried about the possibility of doxycycline resistance and selected ciprofloxacin instead. While fluoroquinolones are probably superior to doxycycline for mild to moderate tularemia,11, 12 aminoglycosides would be preferred for severe disease,9 and ciprofloxacin experience in leptospirosis7 and ehrlichiosis10 is limited. Neither selection would be optimal for brucellosis, for which either doxycycline or ciprofloxacin should be combined with another agent such as rifampin.8 The most reasonable empiric regimen is debatable, but in the absence of pathognomonic findings of tularemia, his treating physicians favored the broader activity of doxycycline.
Ultimately, the choice of antibiotics in this case hinged on the details of the patient's occupational exposures. His first 2 courses of antibiotics were based not on his exposure history, but on radiographic findings that were later proven spurious. The regimens selected by the discussant and by physicians at the referral hospital both targeted pathogens suggested by the patient's occupational history instead, but both were missing parts of the puzzle as well. The discussant thought the patient performed commercial butcher‐shop work, which is only rarely13 mentioned in the context of Q fever transmission. Several of the admitting physicians at the referral hospital were unaware of the importance of the butcher/slaughterhouse‐worker distinction. Physicians need a detailed understanding of both the exposure history and the biology of possible pathogens to craft an optimal differential diagnosis and empiric antibiotic regimen.
On the other hand, in most patients with fever of unknown origin (FUO; ie, >3 weeks with temperature >38.3 on multiple occasions, without a diagnosis after a weeklong evaluation),14 empiric antibiotic therapy is rarely a wise intervention. Clinicians should avoid blind administration of antibiotics as a diagnostic tool, given the inability to distinguish clinical responses from spontaneous resolution, or pinpoint a specific cause and thus a precise treatment plan and duration. However, empiric tetracyclines have been employed when intracellular pathogens were a suspected cause of FUO, as in one series of French patients in which Q fever was common.15 In this patient's case, no specific finding pointed to Q fever before the serologies became available, but the rare infections considered in this case can be considered doxycycline‐deficient states, meaning that empiric tetracycline therapy often leads to improvement. Recognizing doxycycline deficiency can guide therapy while definitive results are pending, and empiric doxycycline is particularly important if potentially aggressive zoonoses, such as Rocky Mountain spotted fever, are suspected.
Teaching Points:
-
A detailed and precise exposure history is crucial for the diagnosis of Q fever and other zoonoses and for the individualized evaluation of FUO in general.
-
Q fever is a rare disease that most commonly causes undifferentiated fever, pneumonia, hepatitis, and when chronic, often reflects endovascular infection, which is frequently difficult to eradicate.
-
Doxycycline is effective for many, but not all zoonoses (babesia is a notable exception). Empiric therapy is reasonable if suspicion is high.
- .“Q” fever, new fever entity: clinical features, diagnosis and laboratory investigation.Med J Aust.1937;2:281–299.
- ,,.Q fever.Lancet.2006;367(9511):679–688.
- ,.Q fever.Clin Microbiol Rev.1999;12:518–553.
- ,,, et al.National surveillance and the epidemiology of human Q fever in the United States, 1978–2004.Am J Trop Med Hyg.2006;75:36–40.
- ,,,.Q fever: epidemiology, diagnosis, and treatment.Mayo Clin Proc.2008;83(5):574–579.
- ,,, et al.Prevalence, significance, and specificity of antibodies to phospholipids in Q fever.Clin Infect Dis.1994;18:213–218.
- ,,.Antimicrobial therapy of leptospirosis.Curr Opin Infect Dis.2006;19:533–537.
- ,,, et al.Treatment of human brucellosis with doxycycline plus rifampin or doxycycline plus streptomycin. A randomized, double‐blind study.Ann Intern Med.1992;117:25–30.
- ,,,.Tularemia: current epidemiology and disease management.Infect Dis Clin North Am.2006;20:289–311, ix.
- ,,,.Ehrlichioses in humans: epidemiology, clinical presentation, diagnosis, and treatment.Clin Infect Dis.2007;45(Suppl 1):S45–S51.
- ,.New approaches to diagnosis and therapy of tularemia.Ann NY Acad Sci.2007;1105:378–404.
- ,,, et al.Evaluation of clinical, laboratory, and therapeutic features of 145 tularemia cases: the role of quinolones in oropharyngeal tularemia.APMIS.2008;116:66–73.
- ,.A survey of Q fever antibodies in a high risk population in Panamá.Am J Trop Med Hyg.1980;29(5):1007–1011.
- ,.Fever of unknown origin.Lancet.1997;350:575–580.
- .Fever of unknown origin in adults: evaluation of 144 cases in a non‐university hospital.Scand J Infect Dis.2006;38:632–638.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 47‐year‐old male presented to a community hospital with 5 weeks of daily fevers, accompanied by headache, myalgias, and malaise. He reported that his symptoms began abruptly 2 days after a weekend of camping in Connecticut.
This patient describes the onset of undifferentiated fever 2 days after a weekend of camping. Few infectious diseases have such short incubation periods, and either the accuracy of the history or the relationship of the camping trip to the present illness is thus questionable. However, more information about the onset and nature of the illness, and details about food, animal, water, mud, cave, wood chopping, and other environmental exposures during his trip is required. The exact dates of the camping trip may be helpful, as there is clear seasonality to vector‐borne diseases such as Lyme disease, babesiosis, ehrlichiosis, and rickettsial infections. Conditions unrelated to his camping trip, such as malignancies, rheumatologic conditions, and other infectious causes of prolonged fever, such as tuberculosis, endocarditis, or osteomyelitis, are more likely, given the duration of fever.
The fevers were accompanied by chills, without rigors, and subjectively worsened over the first 2 days. At that point, the patient began taking his temperature, and noted fevers of 38.5C to 40C occurring once or twice daily, generally in the afternoon or evening. The patient did not recall tick bites but did not carefully examine himself for ticks; he reported numerous mosquito bites during the trip. The patient camped in a tent and grilled meats and other food he had brought in a cooler. No family members or other travelers became ill. He denied spelunking, but had collected wood for camp fires, and acknowledged swimming in a freshwater pond during his trip, which occurred in August.
West Nile fever, St. Louis encephalitis, and eastern equine encephalitis are transmitted by mosquitoes in New England, but are unlikely causes of prolonged fever. Water exposure suggests the possibility of leptospirosis, and wood exposure suggests blastomycosis, but this usually presents with a pulmonary syndrome. Food‐borne illness seems unlikely. While no aspect of the history has pinpointed a specific diagnosis, exploring the progression of symptoms may offer a clue, and if he has undergone any previous evaluation, the results may significantly alter the differential diagnosis. For example, arthritis may develop weeks after fever in adult‐onset Still's disease, negative blood cultures would lower the probability of endocarditis, and common sites of pyrogenic malignancies (eg, liver, kidneys, and especially lymph nodes) may already have been imaged.
During the first 3 weeks of illness, the patient experienced daily fever and a gradual, 10‐pound weight loss. Over the next 10 days, he sought medical attention at 3 emergency departments. At one, a head computed tomography (CT) showed possible sinusitis, and he was prescribed a 7‐day course of clarithromycin, which he took without any improvement. At 2 others, he was told that his laboratory studies, and a CT of the abdomen, were normal, and that he had a viral syndrome. Several days later, and 5 weeks after the onset of symptoms, the development of dull right upper‐quadrant pain and mild nausea without vomiting prompted the current presentation to the community hospital. He reported several years of loose stools, but denied rash, arthritis, diarrhea, neck stiffness, cough, or other complaints.
A detailed past medical, social, and family history is required, with particular attention to ethnicity; immunocompromising conditions such as splenectomy or corticosteroid use; undiagnosed febrile diseases; severe, unusual, or recurrent infections; medication use; diet; sexual history; pet exposures; and any personal or family history of cancer. The development of right upper‐quadrant pain mandates attention to risk factors for viral hepatitis, known biliary pathology, or travel that might predispose the patient to pyogenic or amoebic liver abscess, and hematochezia, which could suggest a malignancy metastatic to the liver. Additionally, chronic diarrhea with new right upper‐quadrant pain may represent inflammatory bowel disease complicated by primary sclerosing cholangitis (PSC).
The patient was a Caucasian male of Mediterranean ancestry with thalassemia minor. He had undergone dilation of a benign esophageal stricture, but no surgical procedures, and he had never experienced unexplained fever or unusual infections. Medication exposure was limited to occasional use of acetaminophen for fever, and he had no known allergies. His diet was unremarkable and included no well water or unpasteurized dairy products. He denied risk factors for tuberculosis. He drank 2 to 10 beers a day, 5 times a week, had last smoked 10 years previously, and had never used illicit drugs. He denied any high‐risk sexual contacts and was monogamous with his wife, with whom he had 2 children. The family owned no pets and no relatives had suffered from malignant, rheumatologic, or febrile illness, with the exception of hand, foot, and mouth infection in an infant son, 1 year previously. The patient had never traveled outside of New England.
The history has uncovered several clues, but their relevance is doubtful. His ethnicity suggests possible familial Mediterranean fever, but recurrent abdominal pain and polyserositis, rather than a single prolonged episode, would be expected with this disease. A transfusion history should be obtained to explore the possibility of viral hepatitis. While iron overload can predispose patients to various infections including liver abscess, thalassemia minor should not require transfusion. Esophageal stricture could conceivably be due to histoplasmosis (complicated by mediastinal fibrosis) or tuberculosis, but is probably unrelated to his present illness. His excessive alcohol intake increases his risk for esophageal cancer and liver disease, but it is unlikely that metastatic disease to the liver would present with fever without preceding dysphagia, or that alcoholic hepatitis could have escaped detection after evaluations by several physicians.
We need to learn the details of the patient's physical examination. Given the development of right upper‐quadrant pain, I would particularly like to know if he had hepatosplenomegaly and if a Murphy's sign was present.
His temperature ranged from 36.9C to 39.8C, his pulse was 76 beats per minute with minimal elevations during fever spikes, and his respirations were 18 per minute. His blood pressure was 105/70 mm Hg. He was a well‐developed, overweight male with scleral icterus. He had good dentition and an oropharynx free of lesions. Cardiac examination demonstrated a regular rhythm with a normal S1 and S2, without murmurs or peripheral stigmata of infectious endocarditis. A smooth, minimally tender liver edge was palpable 2 cm below the costal margin; the spleen was nonpalpable. Murphy's sign was absent. There was no lymphadenopathy or rash. He had multiple, shallow, uninfected lacerations of both hands in various stages of healing. The remainder of his examination was normal.
The patient has obvious liver involvement. The pulse‐temperature dissociation suggests a variety of infections, including salmonellosis, psittacosis, typhoid fever, leptospirosis, tularemia, brucellosis, legionellosis, and mycoplasma pneumoniae infection. The patient should be asked how and when he injured his hands, as fresh water exposure can transmit leptospirosis across broken skin. However, while severe leptospirosis can cause fever and jaundice, the long duration of illness is not typical. The cryptogenic form of tularemiawhich can manifest as a typhoidal illnessshould be considered, given that tularemia is present in the area the patient visited; he should be asked about exposure to rabbits.
At this point, I would like to see a standard biochemical profile, a liver panel, a complete blood count and differential, urinalysis, chest X‐ray, and an electrocardiogram. I would examine thick and thin Wright‐Giemsa‐stained smears for evidence of babesiosis. Blood cultures should be held for at least 2 weeks to recover fastidious organisms like Francisella tularensis and Brucella sp. Bone marrow cultures should be obtained; they are more sensitive for mycobacteria and Brucella, and may also yield fungal pathogens. Serologies for a variety of infectious diseases, such as leptospirosis, typhoid fever, and tularemia, will be required if other diagnostic tests are unrevealing.
His white cell count was 8,100/L, with a normal differential, and his hemoglobin was 10 g/dL (normal range, 1417), with a mean corpuscular hemoglobin of 63 m3 (normal range, 8298). The platelet count was 303,000/L. Serum electrolytes were normal. His aspartate aminotransferase was 58 U/L and his alanine aminotransferase was 60 U/L (normal range for both, 1045). Bilirubin was 2.6 mg/dL (normal, <1.2); direct bilirubin was 0.9 mg/dL. Alkaline phosphatase was 150 U/L. Lactate dehydrogenase was 342 U/L (normal range, 2251). A lipase was 62 U/L. International normalized ratio (INR) was 1.4 with an activated partial thromboplastin time (aPTT) of 52 seconds (normal range, 2533). Erythrocyte sedimentation rate (ESR) was 50 mm/hour (normal range, 015). Iron studies showed a suppressed iron and iron‐binding capacity and elevated haptoglobin and ferritin (1878 ng/L; normal range, 22322). Several blood cultures obtained at admission showed no growth after 48 hours of incubation.
The anemia, low mean cell volume (MCV), and elevated ferritin and ESR are consistent with anemia of chronic disease, superimposed upon thalassemia minor. Transaminase elevations occur in a plethora of infectious processes. The elevated INR and aPTT are concerning, and may indicate a septic or malignant process with disseminated intravascular coagulation (DIC). While there is no mention of clinical DIC, it would be appropriate to obtain D‐dimers, fibrin degradation products, and a fibrinogen level. The platelet count is normal, which is reassuring.
Before initiating any empiric antimicrobials, I would obtain an abdominal ultrasound, and possibly an abdominal CT. Hepatitis (especially B and C), cytomegalovirus, and Epstein‐Barr virus serologies should be obtained. A variety of conditions including leptospirosis, tularemia, and babesiosis are possible; specific laboratory testing is required to guide therapy.
Ultrasound showed a thickened gallbladder; the liver was slightly enlarged with normal echotexture. Magnetic resonance cholangiopancreatography (MRCP) showed diffuse sequential beading and scarring of his extrahepatic biliary ducts.
There is no evidence of biliary stones, intrahepatic tumor, or abscess to explain the fever and hepatitis, although it would be helpful to know what other abdominal structures were imaged. The MRCP finding increases my suspicion of PSC, possibly complicated by infection, although the biliary abnormalities may be incidental, and an unrelated process may be responsible for the clinical presentation.
His physicians considered the possibilities of PSC and cholangiopathy due to as‐yet undiagnosed acquired immunodeficiency syndrome. Ampicillin‐sulbactam, ceftazidime, and gentamicin were administered for possible bacterial cholangitis, and endoscopic retrograde cholangiopancreatography was performed. This procedure showed only slight narrowing of his common bile duct, which was felt to be a normal variant. He felt no better after several days of antibiotic therapy, and was transferred to a tertiary care center for further evaluation. Repeat physical exam and laboratory studies were essentially unchanged. The patient explained that his hand lacerations were sustained during his work as a butcher who worked with lamb, beef, rabbit, and poultry. He rarely wore protective gloves because they induced contact dermatitis.
Tularemia becomes more likely given his history of rabbit butchering. Salmonellosis and leptospirosis also remain possible. Typhoid fever and brucellosis are unlikely unless the patient worked with imported exotic animals. At this point, given the systemic illness, empiric antibacterial therapy is reasonable. Of the chosen antimicrobials, only the gentamicin would reliably treat tularemia. I would stop ampicillin‐sulbactam and ceftazidime and replace gentamicin with ciprofloxacin, an effective and better‐tolerated agent for tularemia. Cultures of blood and bone marrow aspirate should be obtained. Stool should be cultured for Salmonella. Tularemia, leptospirosis, and typhoid serologies should be sent to a reference laboratory. At this point in the patient's illness, high‐titered antibodies should be present. However, it would be ideal to compare titers with those from previous serum sample, if possible.
The patient's antimicrobials were narrowed to doxycycline alone, for suspected zoonotic infection, but his fevers were unchanged after 1 week of treatment. Hepatitis serologies, human immunodeficiency virus (HIV) antibody, and smears for ehrlichiosis and babesiosis were negative. He had a positive immunoglobulin (Ig)G and a negative IgM for Epstein‐Barr virus and cytomegalovirus. Tularemia, ehrlichiosis, leptospirosis, brucellosis, and Query fever (Q fever) serologies were ordered. The elevated aPTT did not correct when his serum was mixed with normal serum. Thrombin was normal; factor VIII, von Willebrand (VW) factor, and VW cofactor were mildly elevated. Lupus anticoagulant was detected. A hepatologist declined to obtain a liver biopsy, citing the elevated aPTT and pending serologies. Given his clinical stability, the patient was discharged on doxycycline to await further results.
My highest suspicion is for tularemia, and I would switch antibiotic treatment to ciprofloxacin, awaiting serological results. Some in vitro studies have suggested that F. tularensis may often be resistant to doxycycline, and recent clinical experience has shown fluoroquinolones are superior to doxycycline in the treatment of tularemia.
His serologic results were as follows: tularemia, 1:32 (positive, 1:128); ehrlichia, 1:128 (granulocytic) and <1:64 (monocytic; normal for both, <1:64); leptospira, agglutinated nonspecifically; Brucella IgG and IgM 1 (negative, <9), Q fever (coxiella) IgG 1 + 2, IgM 1 + 2, all positive at 1:256 (<1:16). A transesophageal echocardiogram showed no evidence of endocarditis. The patient was treated with 10 weeks of doxycycline for Q fever hepatitis. His fever, headache, and laboratory abnormalities resolved, and he remained well after the completion of therapy.
The serologies suggest the patient had Coxiella burnetii hepatitis, and illustrate the value of a precise exposure history. Most butchers work only with muscle tissue and have a negligible risk of Q fever. In retrospect, it became clear that he worked part‐time in a slaughterhouse, where highly infectious reproductive tract fluids can dry and aerosolize.
Commentary
Q fever was proposed as the name for a febrile illness affecting Australian slaughterhouse workers in 1937.1 The etiologic agent, C. burnetii, is a small, gram‐negative, obligate intracellular proteobacterium that exists in 2 distinct phases, specializing either in entering or persisting in macrophage lysosomes.2 Additionally, spores are formed and can persist in soil.
Q fever is an uncommonly recognized disease, in part because most infected persons have no symptoms or mild symptoms.3 In the United States, the estimated annual incidence has been 0.28 per million (about 50 cases per year) since 1999, when Q fever became a reportable disease due to bioterrorism concerns. In France, more frequent farming of goats and sheep may be responsible for the much higher annual incidence of 500 per million.4 Spread is usually occupational, via aerosol contact with the dried reproductive tract secretions of animals (mainly cattle, sheep, and goats), in a slaughterhouse or farm setting. However, wind‐borne dust can carry spores long distances, and spread can occur from household pets, unpasteurized dairy products, laboratory work, and possibly ticks.3 More than 30 cases have been reported in military personnel deployed to Iraq and Afghanistan, several without obvious exposures.5 One review noted a single reported case of intradermal inoculation,3 making this patient's lacerations a possible site of infection, but he was also at risk for inhalational exposurewhen he was later asked about the details of his work, he acknowledged working at a slaughterhouse as well as a supermarket.
Symptomatic patients are male in 77% of cases, can usually identify an occupational exposure, and have a mean age of 50 years.4 Fever, which lasts 5 to 57 days, as well as fatigue and headaches, begin after a 1‐week to 3‐week incubation period. Atypical pneumonia or rash may occur; meningoencephalitis and myocarditis portend a worse prognosis. As with this patient, 45% to 85% of patients suffer from hepatitis, although few have an abnormal bilirubin.3 Liver biopsy usually reveals granulomas, which may have a classic doughnut hole appearance,2, 3 although this patient ultimately received a diagnosis without the procedure. Acute Q fever rarely (5%) requires hospitalization, and fatalities are extremely rare.3
Chronic infection (ie, lasting >6 months) most often occurs as endocarditis, although chronic hepatitis, osteomyelitis, and infections of other sites occur. Interestingly, this patient's lupus anticoagulant may have been related to his underlying illness, as autoantibodies frequently occur in Q fever, especially in patients with hepatitis, many of whom develop smooth muscle antibodies, a positive Coombs test, antiprothrombinase, or other autoantibodies,3 and there is a high incidence of antiphospholipid antibodies, particularly anticardiolipin and lupus anticoagulant antibodies.6
Because C. burnetii is an obligate intracellular pathogen, culture requires either tissue or live animal inoculation, and the diagnosis is usually made serologically. Paired sera demonstrating seroconversion or a 4‐fold increase in titers are most conclusive, but a single sample may be used. Anti‐phase II antibodies are detectable in 90% of patients within 3 weeks of infection3 and peak at 2 months5; this patient's phase II sera (IgG > 1:200, IgM > 1:50) are said to be 100% predictive for acute Q fever.3 High‐titer anti‐phase I antibodies, in contrast, indicate chronic infection, and a titer 1:800 is one of the modified Duke criteria for endocarditis.5
Acute Q fever is generally treated with doxycycline for 14 days, although prolonged therapy may be advisable to prevent endocarditis if preexisting valvular lesions are present.2, 5 Fluoroquinolones are another option and may be especially useful for meningoencephalitis.5 Because acute Q fever is generally self‐limited, demonstrating a clear benefit to antibiotic therapy is difficult. The available evidence, which was largely obtained from Q fever pneumonia patients, suggests that tetracycline therapy shortens fever duration.3 Patients with Q fever hepatitis may have a protracted course. On the basis of anecdotal reports, some experts add prednisone (tapered from 40 mg daily over a week) for patients with Q fever hepatitis who fail to respond to doxycycline promptly.3 While this patient's fever was unchanged after a week of therapy, he was well into his treatment course when his diagnosis was ultimately confirmed. His physicians felt that prednisone would be of uncertain benefit and opted not to administer it.
Treatment of Q fever endocarditis is often delayed by the combination of negative blood cultures and a low (12%) rate of vegetation formation, increasing the risk of morbidity and mortality.3 Tetracycline monotherapy is associated with a greater than 50% risk of death,5 and even 4 years of treatment may fail to sterilize valve tissue.3 However, if hydroxychloroquine is given with doxycycline for at least 18 months to alkalize lysosomes and improve bacterial killing, the mortality rate can be lowered to about 5%.3, 5 Patients should be warned of the risk of photosensitivity, and monitored for retinal toxicity2 and serologic evidence of relapse.5
Before serologic results confirmed the diagnosis of Q fever, both the patient's clinicians and the discussant had to craft an antibiotic regimen for a suspected zoonosis. The patient received doxycycline, a good choice for leptospirosis,7 brucellosis,8 tularemia,9 and Q fever,3 all possible after livestock exposure, as well as ehrlichiosis.10 The discussant, who suspected tularemia, worried about the possibility of doxycycline resistance and selected ciprofloxacin instead. While fluoroquinolones are probably superior to doxycycline for mild to moderate tularemia,11, 12 aminoglycosides would be preferred for severe disease,9 and ciprofloxacin experience in leptospirosis7 and ehrlichiosis10 is limited. Neither selection would be optimal for brucellosis, for which either doxycycline or ciprofloxacin should be combined with another agent such as rifampin.8 The most reasonable empiric regimen is debatable, but in the absence of pathognomonic findings of tularemia, his treating physicians favored the broader activity of doxycycline.
Ultimately, the choice of antibiotics in this case hinged on the details of the patient's occupational exposures. His first 2 courses of antibiotics were based not on his exposure history, but on radiographic findings that were later proven spurious. The regimens selected by the discussant and by physicians at the referral hospital both targeted pathogens suggested by the patient's occupational history instead, but both were missing parts of the puzzle as well. The discussant thought the patient performed commercial butcher‐shop work, which is only rarely13 mentioned in the context of Q fever transmission. Several of the admitting physicians at the referral hospital were unaware of the importance of the butcher/slaughterhouse‐worker distinction. Physicians need a detailed understanding of both the exposure history and the biology of possible pathogens to craft an optimal differential diagnosis and empiric antibiotic regimen.
On the other hand, in most patients with fever of unknown origin (FUO; ie, >3 weeks with temperature >38.3 on multiple occasions, without a diagnosis after a weeklong evaluation),14 empiric antibiotic therapy is rarely a wise intervention. Clinicians should avoid blind administration of antibiotics as a diagnostic tool, given the inability to distinguish clinical responses from spontaneous resolution, or pinpoint a specific cause and thus a precise treatment plan and duration. However, empiric tetracyclines have been employed when intracellular pathogens were a suspected cause of FUO, as in one series of French patients in which Q fever was common.15 In this patient's case, no specific finding pointed to Q fever before the serologies became available, but the rare infections considered in this case can be considered doxycycline‐deficient states, meaning that empiric tetracycline therapy often leads to improvement. Recognizing doxycycline deficiency can guide therapy while definitive results are pending, and empiric doxycycline is particularly important if potentially aggressive zoonoses, such as Rocky Mountain spotted fever, are suspected.
Teaching Points:
-
A detailed and precise exposure history is crucial for the diagnosis of Q fever and other zoonoses and for the individualized evaluation of FUO in general.
-
Q fever is a rare disease that most commonly causes undifferentiated fever, pneumonia, hepatitis, and when chronic, often reflects endovascular infection, which is frequently difficult to eradicate.
-
Doxycycline is effective for many, but not all zoonoses (babesia is a notable exception). Empiric therapy is reasonable if suspicion is high.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 47‐year‐old male presented to a community hospital with 5 weeks of daily fevers, accompanied by headache, myalgias, and malaise. He reported that his symptoms began abruptly 2 days after a weekend of camping in Connecticut.
This patient describes the onset of undifferentiated fever 2 days after a weekend of camping. Few infectious diseases have such short incubation periods, and either the accuracy of the history or the relationship of the camping trip to the present illness is thus questionable. However, more information about the onset and nature of the illness, and details about food, animal, water, mud, cave, wood chopping, and other environmental exposures during his trip is required. The exact dates of the camping trip may be helpful, as there is clear seasonality to vector‐borne diseases such as Lyme disease, babesiosis, ehrlichiosis, and rickettsial infections. Conditions unrelated to his camping trip, such as malignancies, rheumatologic conditions, and other infectious causes of prolonged fever, such as tuberculosis, endocarditis, or osteomyelitis, are more likely, given the duration of fever.
The fevers were accompanied by chills, without rigors, and subjectively worsened over the first 2 days. At that point, the patient began taking his temperature, and noted fevers of 38.5C to 40C occurring once or twice daily, generally in the afternoon or evening. The patient did not recall tick bites but did not carefully examine himself for ticks; he reported numerous mosquito bites during the trip. The patient camped in a tent and grilled meats and other food he had brought in a cooler. No family members or other travelers became ill. He denied spelunking, but had collected wood for camp fires, and acknowledged swimming in a freshwater pond during his trip, which occurred in August.
West Nile fever, St. Louis encephalitis, and eastern equine encephalitis are transmitted by mosquitoes in New England, but are unlikely causes of prolonged fever. Water exposure suggests the possibility of leptospirosis, and wood exposure suggests blastomycosis, but this usually presents with a pulmonary syndrome. Food‐borne illness seems unlikely. While no aspect of the history has pinpointed a specific diagnosis, exploring the progression of symptoms may offer a clue, and if he has undergone any previous evaluation, the results may significantly alter the differential diagnosis. For example, arthritis may develop weeks after fever in adult‐onset Still's disease, negative blood cultures would lower the probability of endocarditis, and common sites of pyrogenic malignancies (eg, liver, kidneys, and especially lymph nodes) may already have been imaged.
During the first 3 weeks of illness, the patient experienced daily fever and a gradual, 10‐pound weight loss. Over the next 10 days, he sought medical attention at 3 emergency departments. At one, a head computed tomography (CT) showed possible sinusitis, and he was prescribed a 7‐day course of clarithromycin, which he took without any improvement. At 2 others, he was told that his laboratory studies, and a CT of the abdomen, were normal, and that he had a viral syndrome. Several days later, and 5 weeks after the onset of symptoms, the development of dull right upper‐quadrant pain and mild nausea without vomiting prompted the current presentation to the community hospital. He reported several years of loose stools, but denied rash, arthritis, diarrhea, neck stiffness, cough, or other complaints.
A detailed past medical, social, and family history is required, with particular attention to ethnicity; immunocompromising conditions such as splenectomy or corticosteroid use; undiagnosed febrile diseases; severe, unusual, or recurrent infections; medication use; diet; sexual history; pet exposures; and any personal or family history of cancer. The development of right upper‐quadrant pain mandates attention to risk factors for viral hepatitis, known biliary pathology, or travel that might predispose the patient to pyogenic or amoebic liver abscess, and hematochezia, which could suggest a malignancy metastatic to the liver. Additionally, chronic diarrhea with new right upper‐quadrant pain may represent inflammatory bowel disease complicated by primary sclerosing cholangitis (PSC).
The patient was a Caucasian male of Mediterranean ancestry with thalassemia minor. He had undergone dilation of a benign esophageal stricture, but no surgical procedures, and he had never experienced unexplained fever or unusual infections. Medication exposure was limited to occasional use of acetaminophen for fever, and he had no known allergies. His diet was unremarkable and included no well water or unpasteurized dairy products. He denied risk factors for tuberculosis. He drank 2 to 10 beers a day, 5 times a week, had last smoked 10 years previously, and had never used illicit drugs. He denied any high‐risk sexual contacts and was monogamous with his wife, with whom he had 2 children. The family owned no pets and no relatives had suffered from malignant, rheumatologic, or febrile illness, with the exception of hand, foot, and mouth infection in an infant son, 1 year previously. The patient had never traveled outside of New England.
The history has uncovered several clues, but their relevance is doubtful. His ethnicity suggests possible familial Mediterranean fever, but recurrent abdominal pain and polyserositis, rather than a single prolonged episode, would be expected with this disease. A transfusion history should be obtained to explore the possibility of viral hepatitis. While iron overload can predispose patients to various infections including liver abscess, thalassemia minor should not require transfusion. Esophageal stricture could conceivably be due to histoplasmosis (complicated by mediastinal fibrosis) or tuberculosis, but is probably unrelated to his present illness. His excessive alcohol intake increases his risk for esophageal cancer and liver disease, but it is unlikely that metastatic disease to the liver would present with fever without preceding dysphagia, or that alcoholic hepatitis could have escaped detection after evaluations by several physicians.
We need to learn the details of the patient's physical examination. Given the development of right upper‐quadrant pain, I would particularly like to know if he had hepatosplenomegaly and if a Murphy's sign was present.
His temperature ranged from 36.9C to 39.8C, his pulse was 76 beats per minute with minimal elevations during fever spikes, and his respirations were 18 per minute. His blood pressure was 105/70 mm Hg. He was a well‐developed, overweight male with scleral icterus. He had good dentition and an oropharynx free of lesions. Cardiac examination demonstrated a regular rhythm with a normal S1 and S2, without murmurs or peripheral stigmata of infectious endocarditis. A smooth, minimally tender liver edge was palpable 2 cm below the costal margin; the spleen was nonpalpable. Murphy's sign was absent. There was no lymphadenopathy or rash. He had multiple, shallow, uninfected lacerations of both hands in various stages of healing. The remainder of his examination was normal.
The patient has obvious liver involvement. The pulse‐temperature dissociation suggests a variety of infections, including salmonellosis, psittacosis, typhoid fever, leptospirosis, tularemia, brucellosis, legionellosis, and mycoplasma pneumoniae infection. The patient should be asked how and when he injured his hands, as fresh water exposure can transmit leptospirosis across broken skin. However, while severe leptospirosis can cause fever and jaundice, the long duration of illness is not typical. The cryptogenic form of tularemiawhich can manifest as a typhoidal illnessshould be considered, given that tularemia is present in the area the patient visited; he should be asked about exposure to rabbits.
At this point, I would like to see a standard biochemical profile, a liver panel, a complete blood count and differential, urinalysis, chest X‐ray, and an electrocardiogram. I would examine thick and thin Wright‐Giemsa‐stained smears for evidence of babesiosis. Blood cultures should be held for at least 2 weeks to recover fastidious organisms like Francisella tularensis and Brucella sp. Bone marrow cultures should be obtained; they are more sensitive for mycobacteria and Brucella, and may also yield fungal pathogens. Serologies for a variety of infectious diseases, such as leptospirosis, typhoid fever, and tularemia, will be required if other diagnostic tests are unrevealing.
His white cell count was 8,100/L, with a normal differential, and his hemoglobin was 10 g/dL (normal range, 1417), with a mean corpuscular hemoglobin of 63 m3 (normal range, 8298). The platelet count was 303,000/L. Serum electrolytes were normal. His aspartate aminotransferase was 58 U/L and his alanine aminotransferase was 60 U/L (normal range for both, 1045). Bilirubin was 2.6 mg/dL (normal, <1.2); direct bilirubin was 0.9 mg/dL. Alkaline phosphatase was 150 U/L. Lactate dehydrogenase was 342 U/L (normal range, 2251). A lipase was 62 U/L. International normalized ratio (INR) was 1.4 with an activated partial thromboplastin time (aPTT) of 52 seconds (normal range, 2533). Erythrocyte sedimentation rate (ESR) was 50 mm/hour (normal range, 015). Iron studies showed a suppressed iron and iron‐binding capacity and elevated haptoglobin and ferritin (1878 ng/L; normal range, 22322). Several blood cultures obtained at admission showed no growth after 48 hours of incubation.
The anemia, low mean cell volume (MCV), and elevated ferritin and ESR are consistent with anemia of chronic disease, superimposed upon thalassemia minor. Transaminase elevations occur in a plethora of infectious processes. The elevated INR and aPTT are concerning, and may indicate a septic or malignant process with disseminated intravascular coagulation (DIC). While there is no mention of clinical DIC, it would be appropriate to obtain D‐dimers, fibrin degradation products, and a fibrinogen level. The platelet count is normal, which is reassuring.
Before initiating any empiric antimicrobials, I would obtain an abdominal ultrasound, and possibly an abdominal CT. Hepatitis (especially B and C), cytomegalovirus, and Epstein‐Barr virus serologies should be obtained. A variety of conditions including leptospirosis, tularemia, and babesiosis are possible; specific laboratory testing is required to guide therapy.
Ultrasound showed a thickened gallbladder; the liver was slightly enlarged with normal echotexture. Magnetic resonance cholangiopancreatography (MRCP) showed diffuse sequential beading and scarring of his extrahepatic biliary ducts.
There is no evidence of biliary stones, intrahepatic tumor, or abscess to explain the fever and hepatitis, although it would be helpful to know what other abdominal structures were imaged. The MRCP finding increases my suspicion of PSC, possibly complicated by infection, although the biliary abnormalities may be incidental, and an unrelated process may be responsible for the clinical presentation.
His physicians considered the possibilities of PSC and cholangiopathy due to as‐yet undiagnosed acquired immunodeficiency syndrome. Ampicillin‐sulbactam, ceftazidime, and gentamicin were administered for possible bacterial cholangitis, and endoscopic retrograde cholangiopancreatography was performed. This procedure showed only slight narrowing of his common bile duct, which was felt to be a normal variant. He felt no better after several days of antibiotic therapy, and was transferred to a tertiary care center for further evaluation. Repeat physical exam and laboratory studies were essentially unchanged. The patient explained that his hand lacerations were sustained during his work as a butcher who worked with lamb, beef, rabbit, and poultry. He rarely wore protective gloves because they induced contact dermatitis.
Tularemia becomes more likely given his history of rabbit butchering. Salmonellosis and leptospirosis also remain possible. Typhoid fever and brucellosis are unlikely unless the patient worked with imported exotic animals. At this point, given the systemic illness, empiric antibacterial therapy is reasonable. Of the chosen antimicrobials, only the gentamicin would reliably treat tularemia. I would stop ampicillin‐sulbactam and ceftazidime and replace gentamicin with ciprofloxacin, an effective and better‐tolerated agent for tularemia. Cultures of blood and bone marrow aspirate should be obtained. Stool should be cultured for Salmonella. Tularemia, leptospirosis, and typhoid serologies should be sent to a reference laboratory. At this point in the patient's illness, high‐titered antibodies should be present. However, it would be ideal to compare titers with those from previous serum sample, if possible.
The patient's antimicrobials were narrowed to doxycycline alone, for suspected zoonotic infection, but his fevers were unchanged after 1 week of treatment. Hepatitis serologies, human immunodeficiency virus (HIV) antibody, and smears for ehrlichiosis and babesiosis were negative. He had a positive immunoglobulin (Ig)G and a negative IgM for Epstein‐Barr virus and cytomegalovirus. Tularemia, ehrlichiosis, leptospirosis, brucellosis, and Query fever (Q fever) serologies were ordered. The elevated aPTT did not correct when his serum was mixed with normal serum. Thrombin was normal; factor VIII, von Willebrand (VW) factor, and VW cofactor were mildly elevated. Lupus anticoagulant was detected. A hepatologist declined to obtain a liver biopsy, citing the elevated aPTT and pending serologies. Given his clinical stability, the patient was discharged on doxycycline to await further results.
My highest suspicion is for tularemia, and I would switch antibiotic treatment to ciprofloxacin, awaiting serological results. Some in vitro studies have suggested that F. tularensis may often be resistant to doxycycline, and recent clinical experience has shown fluoroquinolones are superior to doxycycline in the treatment of tularemia.
His serologic results were as follows: tularemia, 1:32 (positive, 1:128); ehrlichia, 1:128 (granulocytic) and <1:64 (monocytic; normal for both, <1:64); leptospira, agglutinated nonspecifically; Brucella IgG and IgM 1 (negative, <9), Q fever (coxiella) IgG 1 + 2, IgM 1 + 2, all positive at 1:256 (<1:16). A transesophageal echocardiogram showed no evidence of endocarditis. The patient was treated with 10 weeks of doxycycline for Q fever hepatitis. His fever, headache, and laboratory abnormalities resolved, and he remained well after the completion of therapy.
The serologies suggest the patient had Coxiella burnetii hepatitis, and illustrate the value of a precise exposure history. Most butchers work only with muscle tissue and have a negligible risk of Q fever. In retrospect, it became clear that he worked part‐time in a slaughterhouse, where highly infectious reproductive tract fluids can dry and aerosolize.
Commentary
Q fever was proposed as the name for a febrile illness affecting Australian slaughterhouse workers in 1937.1 The etiologic agent, C. burnetii, is a small, gram‐negative, obligate intracellular proteobacterium that exists in 2 distinct phases, specializing either in entering or persisting in macrophage lysosomes.2 Additionally, spores are formed and can persist in soil.
Q fever is an uncommonly recognized disease, in part because most infected persons have no symptoms or mild symptoms.3 In the United States, the estimated annual incidence has been 0.28 per million (about 50 cases per year) since 1999, when Q fever became a reportable disease due to bioterrorism concerns. In France, more frequent farming of goats and sheep may be responsible for the much higher annual incidence of 500 per million.4 Spread is usually occupational, via aerosol contact with the dried reproductive tract secretions of animals (mainly cattle, sheep, and goats), in a slaughterhouse or farm setting. However, wind‐borne dust can carry spores long distances, and spread can occur from household pets, unpasteurized dairy products, laboratory work, and possibly ticks.3 More than 30 cases have been reported in military personnel deployed to Iraq and Afghanistan, several without obvious exposures.5 One review noted a single reported case of intradermal inoculation,3 making this patient's lacerations a possible site of infection, but he was also at risk for inhalational exposurewhen he was later asked about the details of his work, he acknowledged working at a slaughterhouse as well as a supermarket.
Symptomatic patients are male in 77% of cases, can usually identify an occupational exposure, and have a mean age of 50 years.4 Fever, which lasts 5 to 57 days, as well as fatigue and headaches, begin after a 1‐week to 3‐week incubation period. Atypical pneumonia or rash may occur; meningoencephalitis and myocarditis portend a worse prognosis. As with this patient, 45% to 85% of patients suffer from hepatitis, although few have an abnormal bilirubin.3 Liver biopsy usually reveals granulomas, which may have a classic doughnut hole appearance,2, 3 although this patient ultimately received a diagnosis without the procedure. Acute Q fever rarely (5%) requires hospitalization, and fatalities are extremely rare.3
Chronic infection (ie, lasting >6 months) most often occurs as endocarditis, although chronic hepatitis, osteomyelitis, and infections of other sites occur. Interestingly, this patient's lupus anticoagulant may have been related to his underlying illness, as autoantibodies frequently occur in Q fever, especially in patients with hepatitis, many of whom develop smooth muscle antibodies, a positive Coombs test, antiprothrombinase, or other autoantibodies,3 and there is a high incidence of antiphospholipid antibodies, particularly anticardiolipin and lupus anticoagulant antibodies.6
Because C. burnetii is an obligate intracellular pathogen, culture requires either tissue or live animal inoculation, and the diagnosis is usually made serologically. Paired sera demonstrating seroconversion or a 4‐fold increase in titers are most conclusive, but a single sample may be used. Anti‐phase II antibodies are detectable in 90% of patients within 3 weeks of infection3 and peak at 2 months5; this patient's phase II sera (IgG > 1:200, IgM > 1:50) are said to be 100% predictive for acute Q fever.3 High‐titer anti‐phase I antibodies, in contrast, indicate chronic infection, and a titer 1:800 is one of the modified Duke criteria for endocarditis.5
Acute Q fever is generally treated with doxycycline for 14 days, although prolonged therapy may be advisable to prevent endocarditis if preexisting valvular lesions are present.2, 5 Fluoroquinolones are another option and may be especially useful for meningoencephalitis.5 Because acute Q fever is generally self‐limited, demonstrating a clear benefit to antibiotic therapy is difficult. The available evidence, which was largely obtained from Q fever pneumonia patients, suggests that tetracycline therapy shortens fever duration.3 Patients with Q fever hepatitis may have a protracted course. On the basis of anecdotal reports, some experts add prednisone (tapered from 40 mg daily over a week) for patients with Q fever hepatitis who fail to respond to doxycycline promptly.3 While this patient's fever was unchanged after a week of therapy, he was well into his treatment course when his diagnosis was ultimately confirmed. His physicians felt that prednisone would be of uncertain benefit and opted not to administer it.
Treatment of Q fever endocarditis is often delayed by the combination of negative blood cultures and a low (12%) rate of vegetation formation, increasing the risk of morbidity and mortality.3 Tetracycline monotherapy is associated with a greater than 50% risk of death,5 and even 4 years of treatment may fail to sterilize valve tissue.3 However, if hydroxychloroquine is given with doxycycline for at least 18 months to alkalize lysosomes and improve bacterial killing, the mortality rate can be lowered to about 5%.3, 5 Patients should be warned of the risk of photosensitivity, and monitored for retinal toxicity2 and serologic evidence of relapse.5
Before serologic results confirmed the diagnosis of Q fever, both the patient's clinicians and the discussant had to craft an antibiotic regimen for a suspected zoonosis. The patient received doxycycline, a good choice for leptospirosis,7 brucellosis,8 tularemia,9 and Q fever,3 all possible after livestock exposure, as well as ehrlichiosis.10 The discussant, who suspected tularemia, worried about the possibility of doxycycline resistance and selected ciprofloxacin instead. While fluoroquinolones are probably superior to doxycycline for mild to moderate tularemia,11, 12 aminoglycosides would be preferred for severe disease,9 and ciprofloxacin experience in leptospirosis7 and ehrlichiosis10 is limited. Neither selection would be optimal for brucellosis, for which either doxycycline or ciprofloxacin should be combined with another agent such as rifampin.8 The most reasonable empiric regimen is debatable, but in the absence of pathognomonic findings of tularemia, his treating physicians favored the broader activity of doxycycline.
Ultimately, the choice of antibiotics in this case hinged on the details of the patient's occupational exposures. His first 2 courses of antibiotics were based not on his exposure history, but on radiographic findings that were later proven spurious. The regimens selected by the discussant and by physicians at the referral hospital both targeted pathogens suggested by the patient's occupational history instead, but both were missing parts of the puzzle as well. The discussant thought the patient performed commercial butcher‐shop work, which is only rarely13 mentioned in the context of Q fever transmission. Several of the admitting physicians at the referral hospital were unaware of the importance of the butcher/slaughterhouse‐worker distinction. Physicians need a detailed understanding of both the exposure history and the biology of possible pathogens to craft an optimal differential diagnosis and empiric antibiotic regimen.
On the other hand, in most patients with fever of unknown origin (FUO; ie, >3 weeks with temperature >38.3 on multiple occasions, without a diagnosis after a weeklong evaluation),14 empiric antibiotic therapy is rarely a wise intervention. Clinicians should avoid blind administration of antibiotics as a diagnostic tool, given the inability to distinguish clinical responses from spontaneous resolution, or pinpoint a specific cause and thus a precise treatment plan and duration. However, empiric tetracyclines have been employed when intracellular pathogens were a suspected cause of FUO, as in one series of French patients in which Q fever was common.15 In this patient's case, no specific finding pointed to Q fever before the serologies became available, but the rare infections considered in this case can be considered doxycycline‐deficient states, meaning that empiric tetracycline therapy often leads to improvement. Recognizing doxycycline deficiency can guide therapy while definitive results are pending, and empiric doxycycline is particularly important if potentially aggressive zoonoses, such as Rocky Mountain spotted fever, are suspected.
Teaching Points:
-
A detailed and precise exposure history is crucial for the diagnosis of Q fever and other zoonoses and for the individualized evaluation of FUO in general.
-
Q fever is a rare disease that most commonly causes undifferentiated fever, pneumonia, hepatitis, and when chronic, often reflects endovascular infection, which is frequently difficult to eradicate.
-
Doxycycline is effective for many, but not all zoonoses (babesia is a notable exception). Empiric therapy is reasonable if suspicion is high.
- .“Q” fever, new fever entity: clinical features, diagnosis and laboratory investigation.Med J Aust.1937;2:281–299.
- ,,.Q fever.Lancet.2006;367(9511):679–688.
- ,.Q fever.Clin Microbiol Rev.1999;12:518–553.
- ,,, et al.National surveillance and the epidemiology of human Q fever in the United States, 1978–2004.Am J Trop Med Hyg.2006;75:36–40.
- ,,,.Q fever: epidemiology, diagnosis, and treatment.Mayo Clin Proc.2008;83(5):574–579.
- ,,, et al.Prevalence, significance, and specificity of antibodies to phospholipids in Q fever.Clin Infect Dis.1994;18:213–218.
- ,,.Antimicrobial therapy of leptospirosis.Curr Opin Infect Dis.2006;19:533–537.
- ,,, et al.Treatment of human brucellosis with doxycycline plus rifampin or doxycycline plus streptomycin. A randomized, double‐blind study.Ann Intern Med.1992;117:25–30.
- ,,,.Tularemia: current epidemiology and disease management.Infect Dis Clin North Am.2006;20:289–311, ix.
- ,,,.Ehrlichioses in humans: epidemiology, clinical presentation, diagnosis, and treatment.Clin Infect Dis.2007;45(Suppl 1):S45–S51.
- ,.New approaches to diagnosis and therapy of tularemia.Ann NY Acad Sci.2007;1105:378–404.
- ,,, et al.Evaluation of clinical, laboratory, and therapeutic features of 145 tularemia cases: the role of quinolones in oropharyngeal tularemia.APMIS.2008;116:66–73.
- ,.A survey of Q fever antibodies in a high risk population in Panamá.Am J Trop Med Hyg.1980;29(5):1007–1011.
- ,.Fever of unknown origin.Lancet.1997;350:575–580.
- .Fever of unknown origin in adults: evaluation of 144 cases in a non‐university hospital.Scand J Infect Dis.2006;38:632–638.
- .“Q” fever, new fever entity: clinical features, diagnosis and laboratory investigation.Med J Aust.1937;2:281–299.
- ,,.Q fever.Lancet.2006;367(9511):679–688.
- ,.Q fever.Clin Microbiol Rev.1999;12:518–553.
- ,,, et al.National surveillance and the epidemiology of human Q fever in the United States, 1978–2004.Am J Trop Med Hyg.2006;75:36–40.
- ,,,.Q fever: epidemiology, diagnosis, and treatment.Mayo Clin Proc.2008;83(5):574–579.
- ,,, et al.Prevalence, significance, and specificity of antibodies to phospholipids in Q fever.Clin Infect Dis.1994;18:213–218.
- ,,.Antimicrobial therapy of leptospirosis.Curr Opin Infect Dis.2006;19:533–537.
- ,,, et al.Treatment of human brucellosis with doxycycline plus rifampin or doxycycline plus streptomycin. A randomized, double‐blind study.Ann Intern Med.1992;117:25–30.
- ,,,.Tularemia: current epidemiology and disease management.Infect Dis Clin North Am.2006;20:289–311, ix.
- ,,,.Ehrlichioses in humans: epidemiology, clinical presentation, diagnosis, and treatment.Clin Infect Dis.2007;45(Suppl 1):S45–S51.
- ,.New approaches to diagnosis and therapy of tularemia.Ann NY Acad Sci.2007;1105:378–404.
- ,,, et al.Evaluation of clinical, laboratory, and therapeutic features of 145 tularemia cases: the role of quinolones in oropharyngeal tularemia.APMIS.2008;116:66–73.
- ,.A survey of Q fever antibodies in a high risk population in Panamá.Am J Trop Med Hyg.1980;29(5):1007–1011.
- ,.Fever of unknown origin.Lancet.1997;350:575–580.
- .Fever of unknown origin in adults: evaluation of 144 cases in a non‐university hospital.Scand J Infect Dis.2006;38:632–638.
The Tip of the Iceberg
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
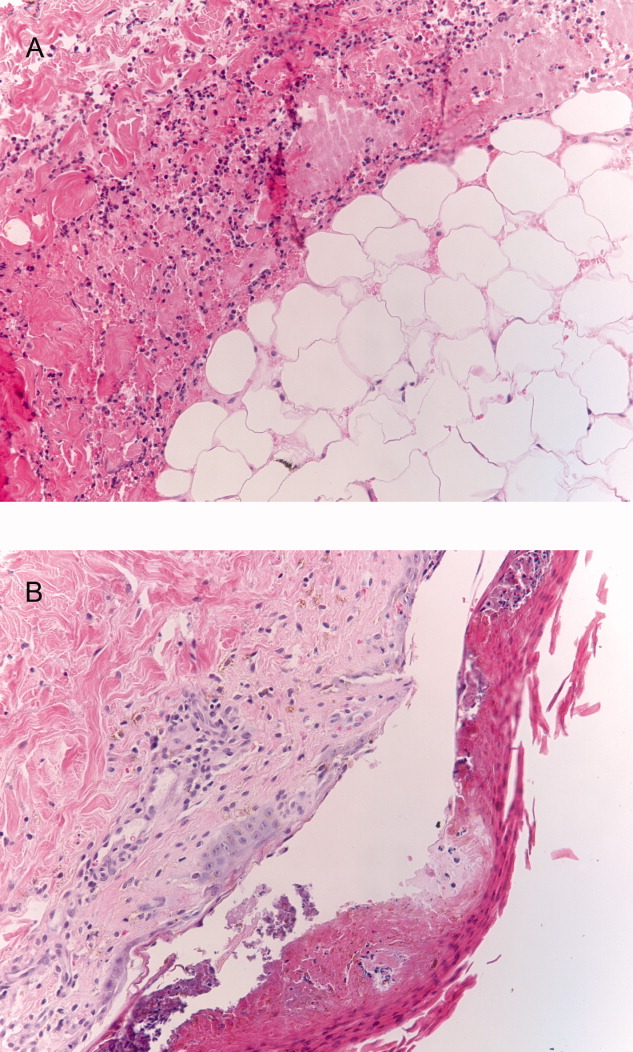
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 33‐year‐old African American man was seen in the emergency department for bilateral wrist pain and forearm swelling. He was a professional mover and had transported a piano 3 days before. Several hours after the move, he noticed wrist pain and a few small red, slightly pruritic bumps on the palmar aspect of both wrists. The following day, he developed nausea, vomiting, and watery diarrhea. His wrists and forearms became more swollen and painful.
The most distinctive aspect of the patient's symptom complex is forearm swelling. I am not certain if the primary pathology lies in the wrist or in the forearm. Examination should focus on the presence or absence of arthritis and whether the forearm swelling is simply adjacent to the wrist and rash or extends the entire length from the wrist to the elbow.
Strenuous lifting can lead to rhabdomyolysis and forearm compartment syndrome. However, transporting a piano is not an unusual task for a professional mover. More common causes of arm swelling such as fracture, cellulitis, deep vein thrombosis, and lymphatic obstruction are possible but are unexpected in this patient because of the bilateral findings. An inflammatory myopathy would not present in the distal extremities, but an infectious myositis, such as trichinosis (which can have early gastrointestinal symptoms), could.
Given the patient's age and the temporal correlation, I am inclined to pursue a unifying diagnosis between his gastrointestinal and upper extremity symptoms. The gastrointestinal symptoms could reflect vasculitis (eg, polyarteritis nodosa) or a nonspecific manifestation of systemic illness (eg, sepsis), whereas the rash could be due to infection with petechiae (eg, meningococcemia, gonococcemia, or endocarditis) or an infection with a predilection for peripheral skin lesions that progress centripetally as the illness progresses, such as Rocky Mountain spotted fever.
Although the patient had not seen spiders, he was concerned that his skin lesions might have resulted from spider bites. He had a history of atopic dermatitis but took no medications, did not smoke, and drank alcohol rarely. He lived in Denver, CO, had not traveled outside of the region, and had not visited rural areas. He was monogamous with a female partner and had no known exposures to human immunodeficiency virus. He had no family history of rheumatological disorders.
Patients and physicians frequently attribute papules, ulcers, or necrotic skin lesions to spider bites far out of proportion to their true prevalence. Bites by more innocuous arthropods such as ticks, fleas, bedbugs, or mites are much more common. The symmetry of the lesions and extensive swelling, however, make such bites unlikely.
Although the patient hails from the southwestern United States, there is no compelling evidence for endemic illness. Hantavirus infection causes a viral prodrome, but a severe pulmonary syndrome is its primary manifestation. Plague presents in bubonic, pulmonary, or septicemic forms, but other than the gastrointestinal symptoms, there is nothing to suggest such a systemic illness. The initial pulmonary infection of coccidioidomycosis is often unnoticed, and patients may present with extrapulmonary manifestations, including skin lesions and skeletal disease.
More common ailments remain on the differential. Disseminated gonococcemia must be considered in a sexually active adult with skin lesions and what may be tenosynovitis or arthritis of the distal extremities. The history of atopic dermatitis supports a diagnosis of allergic or contact dermatitis on the wrists (perhaps inoculated during the move), explaining the rash and adjacent swelling (but not the gastrointestinal symptoms).
The patient's temperature was 36.5C with a pulse of 103 beats per minute, a blood pressure of 103/67 mm Hg, and a respiratory rate of 24 breaths per minute. He appeared uncomfortable and was in moderate distress. His sclerae were injected, and his mucous membranes were dry. He had diffusely swollen fingers and firm nonpitting edema in both hands and forearms. His wrists and hands were tender and warm but had no appreciable redness. Two 1‐mm ulcerated lesions were present on his right wrist, and one 2‐mm ulcerated lesion was present on his left wrist. No lymphadenopathy was present.
The patient meets the criteria for systemic inflammatory response syndrome (SIRS), and considering his ill appearance, I am concerned that he may have an infectious process that has evolved into sepsis. The absence of cutaneous erythema rules out cellulitis, and although skin findings in necrotizing fasciitis can be modest in comparison with the underlying infection, an examination with nothing more than punctate ulcerations would be atypical. Plague and tularemia cause ulcerative skin lesions and systemic illness but usually have prominent lymphadenopathy. Cutaneous anthrax can cause intense local edema but is usually accompanied by some degree of necrosis.
I am struck by the degree of local edema. It would be a remarkable coincidence for spider bites to occur on both wrists; however, given the size of the lesions, this remains a consideration. Spider bites can cause severe local swelling and occasionally even SIRS.
The white blood cellcount was 6000/mm3 with a normal differential. The hemoglobin level was 16.6 g/dL, and the platelet count was 227,000/mm3. The serum sodium level was measured to be 135 mmol/L, the potassium level was 3.6 mmol/L, the chloride level was 94 mmol/L, and the bicarbonate level was 9 mmol/L. The urea nitrogen level was measured to be 48 mg/dL (normal, 622), and the serum creatinine level was 5.6 mg/dL (normal, 0.41.2). An arterial blood gas test on room air revealed a pH of 7.22, a partial pressure of carbon dioxide of 24 mm Hg, and a partial pressure of oxygen of 128 mm Hg. Liver enzymes were normal. The serum creatine kinase level was measured to be 194 U/L (normal, 0250). A chest radiograph revealed clear lung fields.
The anion gap is elevated and could be explained in part by renal failure, but it is quite pronounced, and I suspect that there is lactic acidosis either from SIRS and systemic hypoperfusion or from local underperfusion of the distal upper extremities (eg, compartment syndrome). Rhabdomyolysis can cause an anion gap acidosis and acute renal failure but is ruled out by a normal creatine kinase level.
Rheumatological diseases (such as polyarteritis nodosa and systemic scleroderma) can cause renal failure, gastrointestinal symptoms, and cutaneous lesions with skin ulceration but are often accompanied by hypertension. The combination of SIRS and volume depletion from nausea, vomiting, and diarrhea seems more likely, although an etiology for SIRS remains elusive. I suspect infectionmost likely of the upper extremitiesis the underlying cause in this patient despite his normal body temperature and white cell count. I would therefore start with plain films of the arms and hands. If these are unrevealing, I would proceed with an ultrasound of the arms to evaluate the soft tissues and particularly the vessels. Although imaging studies are unlikely to provide a diagnosis, they will provide guidance in choosing the next step, such as biopsy or culture.
The patient wasvolume‐resuscitated with saline and treated with clindamycin, pipercillin‐tazobactam, and vancomycin. Radiographs of the wrists and hands demonstrated edema but no subcutaneous gas and no abnormalities of the bones or joints. An ultrasound of the upper extremities was negative for superficial or deep venous thrombosis. Renal ultrasonography revealed normal‐sized kidneys and no hydronephrosis.
Short of superior vena cava thrombosis, which the ultrasound could not visualize, deep venous thrombosis can be ruled out with confidence. Superior vena cava syndrome could account for the bilateral upper extremity symptoms, but complete sparing of the face would be unusual, and the chest radiograph was normal.
Despite the bilateral nature, I doubt that this patient has an arthritis of the wrists and hands (eg, rheumatoid or psoriatic arthritis). The plain films did not show evidence of joint destruction, although that would not be expected in the first few days of most noninfectious arthropathies. Many rheumatological diseases and vasculitides have renal manifestations, but I do not find convincing evidence of such diseases yet.
Despite intravenous fluidsover the next 4 hours, the patient remained anuric. His upper extremity edema worsened, and he became increasingly tachycardic and hypotensive. Vasopressor support was required. Repeat laboratory studies demonstrated a serum creatinine level of 7.0 mg/dL, a bicarbonate level of 6 mmol/L, and a creatine kinase level of 1183 U/L. The serum lactate level was measured to be 6.5. Blood cultures obtained on admission were negative. An echocardiogram demonstrated marked impairment of left ventricular systolic dysfunction with an estimated ejection fraction of 35%. Repeat chest radiography revealed only mild pulmonary vascular congestion.
The patient has become progressively ill despite initial resuscitation. Any infection (cellulitis, fasciitis, or myositis) may progress to such a point (although to do so without fever or leukocytosis with this degree of illness is unusual) and would prompt me to ask a surgeon to explore the upper extremities for diagnostic and possibly therapeutic purposes. Among the spectrum of possible soft tissue infections, this clinical presentation is most consistent with pyomyositis (caused by Staphylococcus aureus) because of the relatively modest creatine kinase elevations that accompany it and the overall absence of cutaneous findings (save the punctuate lesions), which I would expect to be present with cellulitis or necrotizing fasciitis. A deep forearm infection and perhaps compartment syndrome leading to sepsis could explain lactic acidosis, decreased cardiac function, hypotension, and acute renal failure.
Because of the unusual characteristics noted so farparticularly the bilateral diseaseI continue to also consider systemic diseases that can cause skin lesions, cardiomyopathy, renal disease, ocular involvement (eg, keratoconjunctivitis or uveitis), and myositis. Sarcoidosis is possible, although in most of the aforementioned organs, histological disease is far more common than clinical disease, and sarcoidosis typically does not cause this degree of illness. Furthermore, over 90% of patients with sarcoidosis have pulmonary involvement. Polyarteritis nodosa could explain the multiorgan involvement and the brisk pace. If no infection is present on exploration, I would ask the surgeon to biopsy the muscle, particularly looking for granulomas or vasculitis. A progressive soft tissue infection leading to sepsis remains my leading consideration at this point.
Surgical consultation was obtained. A muscle biopsy of the left biceps and left forearm revealed group A streptococcus within the muscle and evidence of necrosis (Figure 1). Debridement of both arms and wrists was performed. The patient subsequently developed erythema of the palms and soles followed by diffuse sloughing of the skin. Streptococcal toxic shock syndrome with necrotizing myositis was diagnosed, and the antimicrobial regimen was changed to intravenous penicillin, clindamycin, and intravenous immunoglobulin G. Repeat debridement of the arms was required.
Even when a soft tissue infection is suspected, it can be challenging to preoperatively localize or characterize with precision. In retrospect, the overall severity of his illness and his previously good health status perhaps favor necrotizing myositis (and fasciitis) over pyomyositis.
I may have put undue emphasis on the absence of skin findings. Although the symmetric nature of the disease is unusual, the small skin lesions may have been portals of entry, and in retrospect, they represent the tip of the iceberg. The absence of fever and leukocytosisor hypothermia and leukopeniain a young, previously healthy patient along with the bilateral and symmetric findings made me hesitant to definitely label his illness as a deep soft tissue infection early on, but the gravity of the illness, the lack of a plausible alternative explanation, and his precipitous decline all made surgical exploration imperative.
The patient's skin sloughing progressed, and he was transferred to a regional burn unit. Three additional operations were required for debridement of both upper extremities. Despite apparent control of the initial infection, the patient continued to require significant hemodynamic and ventilator support. He subsequently developed neutropenia, thrombocytopenia, and Escherichia coli urosepsis. Necrosis of the lower extremities developed, and additional surgical debridement was recommended. After extensive discussion regarding prognosis, the family decided against further surgery and withdrew life support. The patient died shortly after extubation. The family refused an autopsy.
Commentary
Expert clinicians employ a variety of approaches to solve complex clinical problems. One of the most effective strategies is pattern recognition, in which the clinician divides the case into recognizable portions and compares these to previous cases that he or she has encountered.1, 2 If patterns from the new case appear similar or identical to those of previous cases, a diagnosis can be made quickly and without the need for unnecessary testing. However, when features of the case are unusual or atypical, pattern recognition may be disrupted. For example, although the discussant suspected a soft tissue infection, a number of features (including a normal white blood cell count, minimal skin findings, and a bilateral and symmetric distribution of swelling) did not match his illness script (a mental representation of a disease) of necrotizing fasciitis.
When pattern recognition fails, other strategies are available.3 Hypothetico‐deductive reasoning is a data‐to‐diagnosis method whereby the clinician uses the presenting information to construct a list of diagnostic possibilities.4 Additional testing and gathering of information are then used to continuously revise the diagnostic possibilities until confirmatory information is obtained and the diagnosis is established. After a pattern failed to materialize, the discussant employed this analytical strategy by noting the unusual characteristics of the case and incorporating laboratory and physiological data to revise his differential diagnosis. As a result, he requested the appropriate diagnostic test: surgical exploration of the forearms.
Necrotizing soft tissue infections are characterized by fulminant tissue destruction, rapid spread along tissue planes, and local vascular thrombosis. Mixed aerobic and anaerobic infections typically occur after penetrating skin injury or following surgery in patients with diabetes mellitus or vascular disease. In contrast, monomicrobial infections with S. aureus or group A streptococcus generally occur in healthy individuals. The prevalence of necrotizing group A streptococcal infections has increased dramatically in the last 15 to 20 years.5 Over one‐third of these cases are complicated by toxic shock syndrome.57 Mortality rates for necrotizing fasciitis with toxic shock exceed 30%, and early surgical consultation is directly associated with a reduction in morbidity and mortality.5, 8
Toxic shock is an inflammatory response syndrome caused by release of exotoxins from group A streptococcus and S. aureus.9, 10 In streptococcal toxic shock, Streptococcus pyogenes exotoxin A and Streptococcus pyogenes exotoxin B are the major toxins produced.10 These toxins activate the systemic production of inflammatory cytokines such as interleukin‐1, gamma‐interferon, and tumor necrosis factor, resulting in capillary leak, systemic hypotension, tissue hypoperfusion, and organ failure. The most common initial symptom is diffuse or localized pain that is severe and abrupt in onset and often precedes or is out of proportion to other physical findings of soft tissue infection.8 Up to 20% of individuals may also develop a viral‐like syndrome with myalgias, fever, nausea, vomiting, and diarrhea.11 Erythroderma of the skin and mucous membranes can be another early finding. The rash is diffuse, erythematous, and macular, resembling a sunburn. It involves the palms and soles but can be subtle and fleeting. Erythroderma may be particularly difficult to detect in dark‐skinned individuals.12 It is also important to consider that the absence of fever, erythroderma, or leukocytosis does not necessarily rule out the possibility of serious infection in necrotizing fasciitis patients, as the development of these signs and symptoms may occur later in the disease process.
The most common portals of entry for group A streptococcus are the skin, vagina, and pharynx. Predisposing factors include varicella infection, penetrating injuries, minor cuts, burns, splinters, and surgery. Interestingly, a portal of entry cannot be identified in 45% of cases.8 These patients in particular are at risk for developing severe necrotizing myositis or fasciitis at the site of a minor injury such as a strained muscle. Hematogenous translocation from the pharynx to the site of injury is the probable mechanism13 and would provide one scenario by which our piano mover developed bilateral and symmetric disease. An alternative explanation would be direct extension of bacteria from small breaks in the skin to adjacent areas of muscle strain. Regardless of the portal of entry, as the small skin lesions demonstrate in this patient, the smallest physical finding can represent the tip of the iceberg.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.
- ,,,,,.Fast and frugal models of clinical judgment in novice and expert physicians.Med Decis Making.2003;23(4):293–300.
- .What is it to be an expert? In: Chi M, Farr MJ, Glaser R, eds.The Nature of Expertise.Hillsdale, NJ:Lawrence Erlbaum;1988.
- .Clinical decision‐making: understanding how clinicians make a diagnosis. In: Saint S, Drazen JM, Solomon CG, eds.Clinical Problem‐Solving.New York, NY:McGraw‐Hill;2006.
- ,,.Medical Problem Solving: An Analysis of Clinical Reasoning.Cambridge, MA:Harvard University Press;1978.
- ,,,,.Population‐based surveillance for group A streptococcal necrotizing fasciitis: clinical features, prognostic indicators, and microbiologic analysis of seventy‐seven cases. Ontario Group A Streptococcal Study.Am J Med.1997;103(1):18–24.
- ,,, et al.Invasive group A streptococcal infections in Sweden in 1994 and 1995: epidemiology and clinical spectrum.Scand J Infect Dis.2000;32(6):609–614.
- ,,,.Reemergence of emm1 and a changed superantigen profile for group A streptococci causing invasive infections: results from a nationwide study.J Clin Microbiol.2005;43(4):1789–1796.
- ,,, et al.Severe group A streptococcal infections associated with a toxic shock‐like syndrome and scarlet fever toxin A.N Engl J Med.1989;321(1):1–7.
- ,,,.Rapidly progressive necrotizing fasciitis caused by Staphylococcus aureus.J Microbiol Immunol Infect.2005;38(5):361–364.
- ,.Streptococcal infections of skin and soft tissues.N Engl J Med.1996;334(4):240–245.
- ,.Streptococcal toxic shock syndrome after breast reconstruction.Ann Plast Surg.2005;54(5):553–556.
- ,.The influence of pigmentation and illumination on the perception of erythema.Photodermatol Photoimmunol Photomed.1992;9(2):45–47.
- .Streptococcal toxic‐shock syndrome: spectrum of disease, pathogenesis, and new concepts in treatment.Emerg Infect Dis.1995;1(3):69–78.
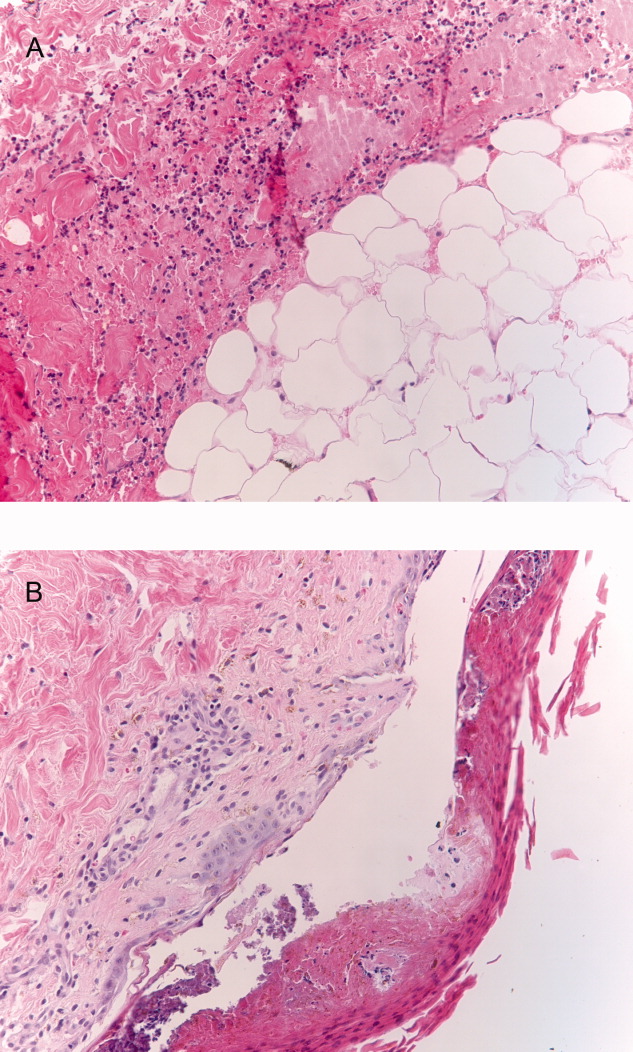
Caught in the Web: e‐Diagnosis
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 52‐year‐old woman presented with a 3‐month history of progressive bilateral leg edema and dyspnea while climbing a flight of stairs or while walking up a steep slope. She also complained of a tingling sensation in both hands and fingers, which started about 2 months prior to the onset of edema. She did not describe sensory problems in the lower extremities and did not have any other neurological complaints. She denied fever, cough, chest pain, palpitations, orthopnea, paroxysmal nocturnal dyspnea, and dark stools. She had no history of hypertension, diabetes, dyslipidemia, or asthma and had never been hospitalized. She did not smoke or consume alcohol and used no medications, including over‐the‐counter drugs or dietary supplements. The patient was born in Japan and had not traveled outside the country since her birth. She was a homemaker and had worked occasionally as a manual laborer in sugar cane agriculture. A review of systems revealed no history of polydipsia, polyuria, or cold or heat intolerance but did identify new hair growth, especially on the extremities.
This middle‐aged woman shows progressive changes in her general health status that are characterized by edema and dyspnea on effort. The differential diagnosis of edema includes a broad spectrum of illnesses, such as cardiac, lung, renal, endocrine, and hepatic diseases. Because of the life‐threatening potential, my first concern is cardiac disease, although the patient is not experiencing typical symptoms of ischemic heart disease or congestive failure. Bilateral and distal distribution of neuropathic symptoms is likely due to diseases of peripheral nerves rather than those of the central nervous system. Her complaint of a bilateral tingling sensation in the hands may suggest carpal tunnel syndrome as a result of her long‐term agricultural work. Other possible causes include radiculopathy of the cervical spine or polyneuropathy. Clues in the physical examination may help narrow the differential diagnosis to a cardiac, hepatic, or endocrine disorder.
The patient appeared ill. Her weight had increased from 48 to 61 kg since she was last weighed 6 months previously. Her blood pressure was 140/78 mm Hg, her heart rate was 72 beats/minute with a regular rhythm, her respiratory rate was 18/minute, and her temperature was 37.5C. The jugular venous pressure was elevated at 10 cm above the sternal angle. A grade III/VI systolic ejection murmur was evident at the second interspace along the left sternal border. The second heart sound was fixed and split. There were decreased breath sounds and complete dullness to percussion over both lower lung fields. Shifting dullness was noted on abdominal examination. There was pitting edema from the feet to the thighs, with slow pit‐recovery time in both legs, and she exhibited generalized hirsutism on the face, body, and extremities. There was no lymphadenopathy. On neurological examination, her mental status was normal. The cranial nerves were normal, as was coordination. There was mild generalized distal‐dominant motor weakness with generalized hyporeflexia. Sensory testing demonstrated glove‐and‐stocking type loss of sensation to pinpricks as well as dysesthesia in all extremities. Phalen and Tinel tests were negative.
The elevated venous pressure and pitting edema with slow recovery suggest high venous pressure edema rather than hypoproteinemic edema. Complete bilateral dullness of the chest and shifting dullness of the abdomen indicate the presence of bilateral pleural effusion and ascites. Edema from high venous pressure is usually caused by right, left, or biventricular cardiac failure. A fixed splitting of the second heart sound suggests an atrial septal defect, which is a rare cause of progressive right heart failure in adults. I recommend checking the patient's thyroid function to investigate the possibility of hypothyroidism, which is a common illness among middle‐aged women and could contribute to her edema as well as hirsutism. The neurological findings suggest a generalized polyneuropathy. The unusual combination of high venous pressure edema and polyneuropathy may indicate a rare multisystem disorder such as amyloidosis. Alternatively, the patient might have developed multiple diseases during the same time period. For instance, diabetic polyneuropathy is the most common cause of polyneuropathy among the middle‐aged. Finally, the differential diagnosis of hirsutism includes ovarian, adrenal, or pituitary sources of hyperandrogenism in addition to hypothyroidism. I would first evaluate for diabetes, thyroid disease, and cardiac disease and would like to see the results of laboratory tests for thyrotropin and plasma glucose as well as chest radiography and electrocardiography.
The white‐cell count was 5400/mm3 with a normal differential. Hemoglobin was 10.7 g/dL with normal red‐cell indices, and the platelet count was 276,000/mm3. The erythrocyte sedimentation rate was 29 mm/hour. Other laboratory tests revealed the following values: total protein, 6.2 g/dL; albumin, 3.3 g/dL; blood urea nitrogen, 12 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase, 6 U/L; alanine aminotransferase, 2 U/L; lactate dehydrogenase, 96 U/L; alkaline phosphatase, 115 U/L; creatine phosphokinase, 60 U/L; total bilirubin, 0.9 mg/dL; glucose, 96 mg/dL; hemoglobin A1c, 4.6%; total cholesterol, 111 mg/dL; and thyrotropin, 6.32 mIU/mL (normal range, 0.50‐5.00 mIU/mL). Serum free thyroxine, triiodothyronine, and urine testosterone were normal. Serum dehydroepiandrosterone sulfate was mildly elevated for her age (864 ng/mL: normal range, 180‐750 ng/mL). Serological studies for human immunodeficiency virus, human T‐lymphotrophic virus type 1, and syphilis were negative. Urinalysis was weakly positive for protein but negative for casts and occult blood. The stool was negative for occult blood.
A chest radiograph showed bilateral pleural effusions. Computed tomography demonstrated bilateral pleural effusions, ascites, mild hepatomegaly, and small, multiple, mediastinal lymph nodes. Her electrocardiogram was normal. A transesophageal echocardiogram with agitated saline contrast demonstrated normal ventricular systolic and diastolic function and no atrial septal defect. The inferior vena cava did not collapse with inspiration, and there was no evidence of infiltrative cardiomyopathy.
These laboratory results rule out diabetes as the cause of the polyneuropathy. The subclinical hypothyroidism would not explain profound edema and hirsutism. A serum albumin level of 3.3 g/dL confirms high venous pressure edema rather than hypoproteinemic edema. Normochromic, normocytic anemia and a mildly elevated sedimentation rate point to a chronic illness or inflammatory state. The mediastinal lymphadenopathy may reflect congestion as a result of the high venous pressure or reflect a systemic disease involving lymph nodes. Normal ventricular function with high venous pressure is suggestive of heart failure from diastolic dysfunction, although the patient does not have risk factors for diastolic dysfunction, such as hypertension, and has no other echocardiographic features of diastolic impairment. The combination of hyperandrogenism and neuropathy points to a systemic process, such as a paraneoplastic syndrome. I would next investigate the source of the excess androgens.
Because serum dehydroepiandrosterone sulfate was mildly elevated, I‐131 aldosterol scintigraphy was performed, and it was negative. Electromyography showed a pattern of generalized sensorimotor polyneuropathy.
At this point, it appears that cardiac, endocrine, hepatic, and renal diseases have been largely ruled out as a cause of her symptoms. Reframing and unifying the important clinical problems for this patient may be useful in resolving this diagnostic puzzle. They include (1) systemic high venous pressure edema; (2) generalized sensorimotor polyneuropathy; (3) hirsutism; (4) normocytic, normochromic anemia; (5) an elevated erythrocyte sedimentation rate; (6) mediastinal lymphadenopathy; and (7) subclinical hypothyroidism. At this point, I cannot unify these pieces of information into a single diagnosis. I would search the medical literature, focusing on these terms.
A general internist consultant performed MEDLINE and Google Scholar searches using the key words edema, polyneuropathy, and hirsutism. This search suggested the diagnosis of Crow‐Fukase syndrome, also known as POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) syndrome. Subsequent evaluations were performed. First, serum protein electrophoresis revealed the presence of monoclonal proteins, although hypergammaglobulinemia was not present. Second, a bone marrow examination demonstrated increased abnormal plasma cell proliferation (7%), although a radiographic skeletal survey found no lesions suggestive of plasmacytoma. Third, cerebrospinal fluid analysis showed normal cell counts but increased protein concentration (202 mg/dL). Fourth, a blood sample referred to an outside laboratory demonstrated elevated levels of vascular endothelial growth factor (3902 pg/mL: normal range, 150‐500 pg/mL). On the basis of these findings, the diagnosis of POEMS syndrome was made. After oral prednisolone (40 mg/day) was initiated, the systemic edema improved gradually, and she did well during the 2‐year follow‐up period.
Commentary
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder first described by Crow in 1956.1, 2 It is characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes, as indicated by the acronym. The diagnosis of POEMS syndrome is difficult as this syndrome is rare and requires high clinical suspicion. According to a nationwide cross‐sectional survey in Japan, the prevalence of POEMS syndrome is very low (about 3 patients per 1,000,000 persons),3 and its prevalence in Western countries is considered even lower than that in Japan. The average age at onset is around 45 to 50 years old, and men are twice as likely to have this syndrome as women.46 Table 1 shows the diagnostic criteria of POEMS syndrome, based on research by Dispenzieri and others at the Mayo Clinic, and Table 2 presents the relative frequency of these clinical features.6, 7 The initial symptomatology generally includes polyneuropathy, skin changes, and generalized edema, which are nonspecific symptoms, as are other well‐recognized associated conditions such as clubbing, weight loss, thrombocytosis, polycythemia, and hyperhidrosis. Thus, it is important to consider this syndrome when one is facing an undiagnosed illness involving multiple organ systems and to distinguish it from other conditions such as multiple myeloma, amyloidosis, and monoclonal gammopathy of undetermined significance. Vascular endothelial growth factor is thought to be involved in the edema of POEMS syndrome, as massive release from aggregated platelets increases vascular permeability and venous pressure.710
| |
| Major criteria | Polyneuropathy |
| Monoclonal plasma cell‐proliferative disorder | |
| Minor criteria | Sclerotic bone lesions |
| Castleman disease | |
| Organomegaly (splenomegaly, hepatomegaly, or lymphadenopathy) | |
| Edema (peripheral edema, pleural effusion, or ascites) | |
| Endocrinopathy (adrenal, thyroid, pituitary, gonadal, parathyroid, or pancreatic) | |
| Skin changes (hyperpigmentation, hirsutism, plethora, hemangiomata, and white nails) | |
| Papilledema | |
| Characteristic | % |
|---|---|
| |
| Peripheral neuropathy | 100 |
| Monoclonal plasma cell dyscrasia | 100 |
| Sclerotic bone lesions | 97 |
| Endocrinopathy | 71 |
| Skin changes | 68 |
| Organomegaly | 46 |
| Extravascular volume overload | 39 |
| Papilledema | 29 |
| Castleman disease | 11 |
Data regarding treatment and survival are largely observational. Overall mean survival from diagnosis in the 2003 Dispenzieri cohort was 13.7 years, with death often due to infection or cardiorespiratory failure.6 When a solitary plasmacytoma or osteosclerotic myeloma is present, radiation to the lesion can often lead to clinical remission. Other treatment options include alkylating agents and/or high‐dose chemotherapy with peripheral stem‐cell transplantation, corticosteroids, and supportive care.7
Clinicians frequently use the internet to aid in the clinical decision process. In a survey of the Royal New Zealand College of General Practitioners,11 half reported that they used the Internet to search for clinical information. Two well‐known resources are MEDLINE, which contains over 11 million references dating back to the 1960s, and internet search engines such as Google (and a more recent product, Google Scholar, which attempts to sort search results by including factors such as the author, the publication in which the article appears, and how often the article has been cited).
MEDLINE searches a well‐defined set of journals and uses the Medical Subject Headings (MeSH) vocabulary, which consists of sets of descriptive terms organized in a hierarchical structure to allow searching with various levels of specificity. For instance, entering the term heart attack will map to the MeSH term myocardial infarction and will also include more specific terms such as myocardial stunning and cardiogenic shock.
Google, in comparison, explores resources beyond journals without any clear boundary to its scope, and its advanced search functions can be occasionally unreliable. For instance, search results are occasionally marred by outdated citation information and may include materials that are not truly scholarly. However, search engines can search through the actual text of manuscripts and access the gray literature, which includes open‐source material that is usually original but not widely distributed or often easily available, such as technical reports and dissertations. A direct study comparing the results of searches in PubMed (one of the MEDLINE search engines) and Google Scholar is difficult, but the critical characteristics of each can be compared and contrasted (Table 3).
| Google Scholar | PubMed |
|---|---|
| 1. Database selection is clumped under subject areas, and it cannot be searched with unique identifiers: Con | 1. It allows one to choose a database at the outset and can search with a unique identifier (PubMed identifier): Pro |
| 2. Results cannot be filtered (ie, it does not allow multiple article selection): Con | 2. The single citation matcher allows retrieval of articles with pieces of information: Pro |
| 3. A search for related articles or similar pages is not available: Con | 3. It allows article selection by checkbox to reduce the number of articles relevant to the search query and to append the filter to search box: Pro |
| 4. It allows one to search by without words to exclude unwanted and confusing retrieved data: Pro | 4. It provides unique identifier (PubMed identifier) for each retrieved article for easy communicability: Pro |
| 5. It allows one to search a single journal/publication of interest: Pro | 5. Search are limited to journals only; it does not include the grey area of literature: Con |
| 6. Initial search results are those articles that are most cited by journals that themselves are the most cited: Pro | 6. It lists search results in chronological order and not by relevance: Con |
Internet searches may also suggest diagnoses from a compilation of clinical features, such as in this case. To be successful, such a search must complement the cognitive process; a search engine cannot completely replace clinical judgment. Clinicians must be able to identify salient clinical features and generate high‐yield search terms and then exercise skill in sifting through the citations to arrive at the appropriate diagnosis. A recent study found that Google searches revealed the correct diagnosis in 58% of the case records of the New England Journal of Medicine,12 although each search query resulted in many results, which then had to be manually reviewed for appropriateness within the case's context.
Like a traditional diagnostic test, a search can be described by sensitivity, specificity, and the number of articles needed to read.13 For example, in a study comparing the performance of search strategies to identify clinical practice guidelines in Google Scholar and SUMSearch (another freely accessible search engine), using the term guideline yielded the highest sensitivity, and using the term practice guideline generated the highest specificity and the lowest number of articles needed to read (Table 4).14
| Search Strategy | Sensitivity (%) | Specificity (%) | NNR |
|---|---|---|---|
| |||
| SUMSearch | |||
| Guideline* | 81.51 (74.5388.49) | 74.29 (72.6475.94) | 8.18 (6.9010.05) |
| Recommendation* | 60.50 (51.7269.28) | 76.28 (74.6777.89) | 9.93 (8.1412.72) |
| Practice guideline* | 40.34 (31.5249.16) | 89.45 (88.2990.61) | 6.96 (5.529.43) |
| Google Scholar | |||
| Guideline/s | 31.93 (23.5640.30) | 78.05 (76.5079.60) | 16.67 (12.7624.04) |
| Recommendation/s | 8.40 (3.4213.38) | 92.11 (91.0993.13) | 22.42 (13.9756.82) |
| Practice guideline/s | 11.76 (5.9817.54) | 95.72 (94.9696.48) | 9.29 (6.2118.38) |
Although there are several other popular hosts of web‐based search engines, a more robust decision‐support program may help physicians more efficiently consider relevant diagnoses. One program, named Isabel, has been developed through the indexing of a database of more than 11,000 diseases according to word patterns in journal articles associated with each disease, and it is updated as new and relevant articles emerge. One recent study demonstrated that the correct diagnosis was made in 48 of 50 cases (96%) with specific, key findings as search terms but in only 37 of the same 50 cases (74%) if the entire case history was simply pasted in, again emphasizing the importance of specific search terms.15
POEMS syndrome is a rare entity occasionally seen in middle‐aged individuals and marked by a multitude of nonspecific findings, particularly polyneuropathy and plasma cell dyscrasia. In this case, the diagnostic test was an internet search based on the most prominent clinical symptoms. Such a strategy can provide a powerful addition to traditional literature and MEDLINE resources. However, the efficiency of this process is heavily dependent on the quality of the search strategy and, therefore, the cognitive faculties of the treating physician to avoid the predictable shortcoming of low specificity. Garbage in, garbage out still applies whether the computer in question is the human mind or the desktop PC.
Teaching Points
-
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
-
Internet‐based searches, including Google and MEDLINE, are being used more frequently because they are widely available, quick, and freely accessed.
-
Internet searches appear most useful as adjuncts to PubMed and clinical reasoning in identifying case reports when a well‐constructed collection of symptoms and signs is used for searches.
- .Peripheral neuritis in myelomatosis.Br Med J.1956;2(4996):802–804.
- ,,,,,.Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature.Medicine (Baltimore).1980;59(4):311–322.
- .Nationwide Epidemiologic Survey of Crow‐Fukase Syndrome in 2004.Tokyo, Japan:Japanese Ministry of Health and Welfare Government Report, 2004.
- ,,, et al.The Crow‐Fukase syndrome: a study of 102 cases in Japan.Neurology.1984;34(6):712–720.
- ,,.POEMS syndrome: a study of 25 cases and a review of the literature. French Study Group on POEMS Syndrome.Am J Med.1994;97(6):543–553.
- ,,, et al.POEMS syndrome: definitions and long‐term outcome.Blood.2003;101(7):2496–2506.
- .POEMS syndrome.Hematology.2005;1(1):360–367.
- ,,,,.Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome.Lancet.1996;347(9002):702.
- ,.Assessment of hypoproteinaemic oedema: a simple physical sign.Br Med J.1978;1(6117):890–891.
- ,,, et al.Ratio of serum vascular endothelial growth factor to platelet count correlates with disease activity in a patient with POEMS syndrome.Eur J Intern Med.2002;13(1):70–74.
- .In search of evidence: family practitioners' use of the Internet for clinical information.J Med Libr Assoc.2002;90(4):370–379.
- ,.Googling for a diagnosis—use of Google as a diagnostic aid: internet based study.BMJ.2006;333(7579):1143–5114.
- ,,.The number needed to read—a new measure of journal value.Health Info Libr J.2005;22(2):81–82.
- ,,,.Developing search strategies for clinical practice guidelines in SUMSearch and Google Scholar and assessing their retrieval performance.BMC Med Res Methodol.2007;7:28.
- ,.Performance of a web‐based clinical diagnosis support system for internists.J Gen Intern Med.2008;23(suppl 1):37–40.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 52‐year‐old woman presented with a 3‐month history of progressive bilateral leg edema and dyspnea while climbing a flight of stairs or while walking up a steep slope. She also complained of a tingling sensation in both hands and fingers, which started about 2 months prior to the onset of edema. She did not describe sensory problems in the lower extremities and did not have any other neurological complaints. She denied fever, cough, chest pain, palpitations, orthopnea, paroxysmal nocturnal dyspnea, and dark stools. She had no history of hypertension, diabetes, dyslipidemia, or asthma and had never been hospitalized. She did not smoke or consume alcohol and used no medications, including over‐the‐counter drugs or dietary supplements. The patient was born in Japan and had not traveled outside the country since her birth. She was a homemaker and had worked occasionally as a manual laborer in sugar cane agriculture. A review of systems revealed no history of polydipsia, polyuria, or cold or heat intolerance but did identify new hair growth, especially on the extremities.
This middle‐aged woman shows progressive changes in her general health status that are characterized by edema and dyspnea on effort. The differential diagnosis of edema includes a broad spectrum of illnesses, such as cardiac, lung, renal, endocrine, and hepatic diseases. Because of the life‐threatening potential, my first concern is cardiac disease, although the patient is not experiencing typical symptoms of ischemic heart disease or congestive failure. Bilateral and distal distribution of neuropathic symptoms is likely due to diseases of peripheral nerves rather than those of the central nervous system. Her complaint of a bilateral tingling sensation in the hands may suggest carpal tunnel syndrome as a result of her long‐term agricultural work. Other possible causes include radiculopathy of the cervical spine or polyneuropathy. Clues in the physical examination may help narrow the differential diagnosis to a cardiac, hepatic, or endocrine disorder.
The patient appeared ill. Her weight had increased from 48 to 61 kg since she was last weighed 6 months previously. Her blood pressure was 140/78 mm Hg, her heart rate was 72 beats/minute with a regular rhythm, her respiratory rate was 18/minute, and her temperature was 37.5C. The jugular venous pressure was elevated at 10 cm above the sternal angle. A grade III/VI systolic ejection murmur was evident at the second interspace along the left sternal border. The second heart sound was fixed and split. There were decreased breath sounds and complete dullness to percussion over both lower lung fields. Shifting dullness was noted on abdominal examination. There was pitting edema from the feet to the thighs, with slow pit‐recovery time in both legs, and she exhibited generalized hirsutism on the face, body, and extremities. There was no lymphadenopathy. On neurological examination, her mental status was normal. The cranial nerves were normal, as was coordination. There was mild generalized distal‐dominant motor weakness with generalized hyporeflexia. Sensory testing demonstrated glove‐and‐stocking type loss of sensation to pinpricks as well as dysesthesia in all extremities. Phalen and Tinel tests were negative.
The elevated venous pressure and pitting edema with slow recovery suggest high venous pressure edema rather than hypoproteinemic edema. Complete bilateral dullness of the chest and shifting dullness of the abdomen indicate the presence of bilateral pleural effusion and ascites. Edema from high venous pressure is usually caused by right, left, or biventricular cardiac failure. A fixed splitting of the second heart sound suggests an atrial septal defect, which is a rare cause of progressive right heart failure in adults. I recommend checking the patient's thyroid function to investigate the possibility of hypothyroidism, which is a common illness among middle‐aged women and could contribute to her edema as well as hirsutism. The neurological findings suggest a generalized polyneuropathy. The unusual combination of high venous pressure edema and polyneuropathy may indicate a rare multisystem disorder such as amyloidosis. Alternatively, the patient might have developed multiple diseases during the same time period. For instance, diabetic polyneuropathy is the most common cause of polyneuropathy among the middle‐aged. Finally, the differential diagnosis of hirsutism includes ovarian, adrenal, or pituitary sources of hyperandrogenism in addition to hypothyroidism. I would first evaluate for diabetes, thyroid disease, and cardiac disease and would like to see the results of laboratory tests for thyrotropin and plasma glucose as well as chest radiography and electrocardiography.
The white‐cell count was 5400/mm3 with a normal differential. Hemoglobin was 10.7 g/dL with normal red‐cell indices, and the platelet count was 276,000/mm3. The erythrocyte sedimentation rate was 29 mm/hour. Other laboratory tests revealed the following values: total protein, 6.2 g/dL; albumin, 3.3 g/dL; blood urea nitrogen, 12 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase, 6 U/L; alanine aminotransferase, 2 U/L; lactate dehydrogenase, 96 U/L; alkaline phosphatase, 115 U/L; creatine phosphokinase, 60 U/L; total bilirubin, 0.9 mg/dL; glucose, 96 mg/dL; hemoglobin A1c, 4.6%; total cholesterol, 111 mg/dL; and thyrotropin, 6.32 mIU/mL (normal range, 0.50‐5.00 mIU/mL). Serum free thyroxine, triiodothyronine, and urine testosterone were normal. Serum dehydroepiandrosterone sulfate was mildly elevated for her age (864 ng/mL: normal range, 180‐750 ng/mL). Serological studies for human immunodeficiency virus, human T‐lymphotrophic virus type 1, and syphilis were negative. Urinalysis was weakly positive for protein but negative for casts and occult blood. The stool was negative for occult blood.
A chest radiograph showed bilateral pleural effusions. Computed tomography demonstrated bilateral pleural effusions, ascites, mild hepatomegaly, and small, multiple, mediastinal lymph nodes. Her electrocardiogram was normal. A transesophageal echocardiogram with agitated saline contrast demonstrated normal ventricular systolic and diastolic function and no atrial septal defect. The inferior vena cava did not collapse with inspiration, and there was no evidence of infiltrative cardiomyopathy.
These laboratory results rule out diabetes as the cause of the polyneuropathy. The subclinical hypothyroidism would not explain profound edema and hirsutism. A serum albumin level of 3.3 g/dL confirms high venous pressure edema rather than hypoproteinemic edema. Normochromic, normocytic anemia and a mildly elevated sedimentation rate point to a chronic illness or inflammatory state. The mediastinal lymphadenopathy may reflect congestion as a result of the high venous pressure or reflect a systemic disease involving lymph nodes. Normal ventricular function with high venous pressure is suggestive of heart failure from diastolic dysfunction, although the patient does not have risk factors for diastolic dysfunction, such as hypertension, and has no other echocardiographic features of diastolic impairment. The combination of hyperandrogenism and neuropathy points to a systemic process, such as a paraneoplastic syndrome. I would next investigate the source of the excess androgens.
Because serum dehydroepiandrosterone sulfate was mildly elevated, I‐131 aldosterol scintigraphy was performed, and it was negative. Electromyography showed a pattern of generalized sensorimotor polyneuropathy.
At this point, it appears that cardiac, endocrine, hepatic, and renal diseases have been largely ruled out as a cause of her symptoms. Reframing and unifying the important clinical problems for this patient may be useful in resolving this diagnostic puzzle. They include (1) systemic high venous pressure edema; (2) generalized sensorimotor polyneuropathy; (3) hirsutism; (4) normocytic, normochromic anemia; (5) an elevated erythrocyte sedimentation rate; (6) mediastinal lymphadenopathy; and (7) subclinical hypothyroidism. At this point, I cannot unify these pieces of information into a single diagnosis. I would search the medical literature, focusing on these terms.
A general internist consultant performed MEDLINE and Google Scholar searches using the key words edema, polyneuropathy, and hirsutism. This search suggested the diagnosis of Crow‐Fukase syndrome, also known as POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) syndrome. Subsequent evaluations were performed. First, serum protein electrophoresis revealed the presence of monoclonal proteins, although hypergammaglobulinemia was not present. Second, a bone marrow examination demonstrated increased abnormal plasma cell proliferation (7%), although a radiographic skeletal survey found no lesions suggestive of plasmacytoma. Third, cerebrospinal fluid analysis showed normal cell counts but increased protein concentration (202 mg/dL). Fourth, a blood sample referred to an outside laboratory demonstrated elevated levels of vascular endothelial growth factor (3902 pg/mL: normal range, 150‐500 pg/mL). On the basis of these findings, the diagnosis of POEMS syndrome was made. After oral prednisolone (40 mg/day) was initiated, the systemic edema improved gradually, and she did well during the 2‐year follow‐up period.
Commentary
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder first described by Crow in 1956.1, 2 It is characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes, as indicated by the acronym. The diagnosis of POEMS syndrome is difficult as this syndrome is rare and requires high clinical suspicion. According to a nationwide cross‐sectional survey in Japan, the prevalence of POEMS syndrome is very low (about 3 patients per 1,000,000 persons),3 and its prevalence in Western countries is considered even lower than that in Japan. The average age at onset is around 45 to 50 years old, and men are twice as likely to have this syndrome as women.46 Table 1 shows the diagnostic criteria of POEMS syndrome, based on research by Dispenzieri and others at the Mayo Clinic, and Table 2 presents the relative frequency of these clinical features.6, 7 The initial symptomatology generally includes polyneuropathy, skin changes, and generalized edema, which are nonspecific symptoms, as are other well‐recognized associated conditions such as clubbing, weight loss, thrombocytosis, polycythemia, and hyperhidrosis. Thus, it is important to consider this syndrome when one is facing an undiagnosed illness involving multiple organ systems and to distinguish it from other conditions such as multiple myeloma, amyloidosis, and monoclonal gammopathy of undetermined significance. Vascular endothelial growth factor is thought to be involved in the edema of POEMS syndrome, as massive release from aggregated platelets increases vascular permeability and venous pressure.710
| |
| Major criteria | Polyneuropathy |
| Monoclonal plasma cell‐proliferative disorder | |
| Minor criteria | Sclerotic bone lesions |
| Castleman disease | |
| Organomegaly (splenomegaly, hepatomegaly, or lymphadenopathy) | |
| Edema (peripheral edema, pleural effusion, or ascites) | |
| Endocrinopathy (adrenal, thyroid, pituitary, gonadal, parathyroid, or pancreatic) | |
| Skin changes (hyperpigmentation, hirsutism, plethora, hemangiomata, and white nails) | |
| Papilledema | |
| Characteristic | % |
|---|---|
| |
| Peripheral neuropathy | 100 |
| Monoclonal plasma cell dyscrasia | 100 |
| Sclerotic bone lesions | 97 |
| Endocrinopathy | 71 |
| Skin changes | 68 |
| Organomegaly | 46 |
| Extravascular volume overload | 39 |
| Papilledema | 29 |
| Castleman disease | 11 |
Data regarding treatment and survival are largely observational. Overall mean survival from diagnosis in the 2003 Dispenzieri cohort was 13.7 years, with death often due to infection or cardiorespiratory failure.6 When a solitary plasmacytoma or osteosclerotic myeloma is present, radiation to the lesion can often lead to clinical remission. Other treatment options include alkylating agents and/or high‐dose chemotherapy with peripheral stem‐cell transplantation, corticosteroids, and supportive care.7
Clinicians frequently use the internet to aid in the clinical decision process. In a survey of the Royal New Zealand College of General Practitioners,11 half reported that they used the Internet to search for clinical information. Two well‐known resources are MEDLINE, which contains over 11 million references dating back to the 1960s, and internet search engines such as Google (and a more recent product, Google Scholar, which attempts to sort search results by including factors such as the author, the publication in which the article appears, and how often the article has been cited).
MEDLINE searches a well‐defined set of journals and uses the Medical Subject Headings (MeSH) vocabulary, which consists of sets of descriptive terms organized in a hierarchical structure to allow searching with various levels of specificity. For instance, entering the term heart attack will map to the MeSH term myocardial infarction and will also include more specific terms such as myocardial stunning and cardiogenic shock.
Google, in comparison, explores resources beyond journals without any clear boundary to its scope, and its advanced search functions can be occasionally unreliable. For instance, search results are occasionally marred by outdated citation information and may include materials that are not truly scholarly. However, search engines can search through the actual text of manuscripts and access the gray literature, which includes open‐source material that is usually original but not widely distributed or often easily available, such as technical reports and dissertations. A direct study comparing the results of searches in PubMed (one of the MEDLINE search engines) and Google Scholar is difficult, but the critical characteristics of each can be compared and contrasted (Table 3).
| Google Scholar | PubMed |
|---|---|
| 1. Database selection is clumped under subject areas, and it cannot be searched with unique identifiers: Con | 1. It allows one to choose a database at the outset and can search with a unique identifier (PubMed identifier): Pro |
| 2. Results cannot be filtered (ie, it does not allow multiple article selection): Con | 2. The single citation matcher allows retrieval of articles with pieces of information: Pro |
| 3. A search for related articles or similar pages is not available: Con | 3. It allows article selection by checkbox to reduce the number of articles relevant to the search query and to append the filter to search box: Pro |
| 4. It allows one to search by without words to exclude unwanted and confusing retrieved data: Pro | 4. It provides unique identifier (PubMed identifier) for each retrieved article for easy communicability: Pro |
| 5. It allows one to search a single journal/publication of interest: Pro | 5. Search are limited to journals only; it does not include the grey area of literature: Con |
| 6. Initial search results are those articles that are most cited by journals that themselves are the most cited: Pro | 6. It lists search results in chronological order and not by relevance: Con |
Internet searches may also suggest diagnoses from a compilation of clinical features, such as in this case. To be successful, such a search must complement the cognitive process; a search engine cannot completely replace clinical judgment. Clinicians must be able to identify salient clinical features and generate high‐yield search terms and then exercise skill in sifting through the citations to arrive at the appropriate diagnosis. A recent study found that Google searches revealed the correct diagnosis in 58% of the case records of the New England Journal of Medicine,12 although each search query resulted in many results, which then had to be manually reviewed for appropriateness within the case's context.
Like a traditional diagnostic test, a search can be described by sensitivity, specificity, and the number of articles needed to read.13 For example, in a study comparing the performance of search strategies to identify clinical practice guidelines in Google Scholar and SUMSearch (another freely accessible search engine), using the term guideline yielded the highest sensitivity, and using the term practice guideline generated the highest specificity and the lowest number of articles needed to read (Table 4).14
| Search Strategy | Sensitivity (%) | Specificity (%) | NNR |
|---|---|---|---|
| |||
| SUMSearch | |||
| Guideline* | 81.51 (74.5388.49) | 74.29 (72.6475.94) | 8.18 (6.9010.05) |
| Recommendation* | 60.50 (51.7269.28) | 76.28 (74.6777.89) | 9.93 (8.1412.72) |
| Practice guideline* | 40.34 (31.5249.16) | 89.45 (88.2990.61) | 6.96 (5.529.43) |
| Google Scholar | |||
| Guideline/s | 31.93 (23.5640.30) | 78.05 (76.5079.60) | 16.67 (12.7624.04) |
| Recommendation/s | 8.40 (3.4213.38) | 92.11 (91.0993.13) | 22.42 (13.9756.82) |
| Practice guideline/s | 11.76 (5.9817.54) | 95.72 (94.9696.48) | 9.29 (6.2118.38) |
Although there are several other popular hosts of web‐based search engines, a more robust decision‐support program may help physicians more efficiently consider relevant diagnoses. One program, named Isabel, has been developed through the indexing of a database of more than 11,000 diseases according to word patterns in journal articles associated with each disease, and it is updated as new and relevant articles emerge. One recent study demonstrated that the correct diagnosis was made in 48 of 50 cases (96%) with specific, key findings as search terms but in only 37 of the same 50 cases (74%) if the entire case history was simply pasted in, again emphasizing the importance of specific search terms.15
POEMS syndrome is a rare entity occasionally seen in middle‐aged individuals and marked by a multitude of nonspecific findings, particularly polyneuropathy and plasma cell dyscrasia. In this case, the diagnostic test was an internet search based on the most prominent clinical symptoms. Such a strategy can provide a powerful addition to traditional literature and MEDLINE resources. However, the efficiency of this process is heavily dependent on the quality of the search strategy and, therefore, the cognitive faculties of the treating physician to avoid the predictable shortcoming of low specificity. Garbage in, garbage out still applies whether the computer in question is the human mind or the desktop PC.
Teaching Points
-
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
-
Internet‐based searches, including Google and MEDLINE, are being used more frequently because they are widely available, quick, and freely accessed.
-
Internet searches appear most useful as adjuncts to PubMed and clinical reasoning in identifying case reports when a well‐constructed collection of symptoms and signs is used for searches.
The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient's case in an approach typical of a morning report. Similarly to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A 52‐year‐old woman presented with a 3‐month history of progressive bilateral leg edema and dyspnea while climbing a flight of stairs or while walking up a steep slope. She also complained of a tingling sensation in both hands and fingers, which started about 2 months prior to the onset of edema. She did not describe sensory problems in the lower extremities and did not have any other neurological complaints. She denied fever, cough, chest pain, palpitations, orthopnea, paroxysmal nocturnal dyspnea, and dark stools. She had no history of hypertension, diabetes, dyslipidemia, or asthma and had never been hospitalized. She did not smoke or consume alcohol and used no medications, including over‐the‐counter drugs or dietary supplements. The patient was born in Japan and had not traveled outside the country since her birth. She was a homemaker and had worked occasionally as a manual laborer in sugar cane agriculture. A review of systems revealed no history of polydipsia, polyuria, or cold or heat intolerance but did identify new hair growth, especially on the extremities.
This middle‐aged woman shows progressive changes in her general health status that are characterized by edema and dyspnea on effort. The differential diagnosis of edema includes a broad spectrum of illnesses, such as cardiac, lung, renal, endocrine, and hepatic diseases. Because of the life‐threatening potential, my first concern is cardiac disease, although the patient is not experiencing typical symptoms of ischemic heart disease or congestive failure. Bilateral and distal distribution of neuropathic symptoms is likely due to diseases of peripheral nerves rather than those of the central nervous system. Her complaint of a bilateral tingling sensation in the hands may suggest carpal tunnel syndrome as a result of her long‐term agricultural work. Other possible causes include radiculopathy of the cervical spine or polyneuropathy. Clues in the physical examination may help narrow the differential diagnosis to a cardiac, hepatic, or endocrine disorder.
The patient appeared ill. Her weight had increased from 48 to 61 kg since she was last weighed 6 months previously. Her blood pressure was 140/78 mm Hg, her heart rate was 72 beats/minute with a regular rhythm, her respiratory rate was 18/minute, and her temperature was 37.5C. The jugular venous pressure was elevated at 10 cm above the sternal angle. A grade III/VI systolic ejection murmur was evident at the second interspace along the left sternal border. The second heart sound was fixed and split. There were decreased breath sounds and complete dullness to percussion over both lower lung fields. Shifting dullness was noted on abdominal examination. There was pitting edema from the feet to the thighs, with slow pit‐recovery time in both legs, and she exhibited generalized hirsutism on the face, body, and extremities. There was no lymphadenopathy. On neurological examination, her mental status was normal. The cranial nerves were normal, as was coordination. There was mild generalized distal‐dominant motor weakness with generalized hyporeflexia. Sensory testing demonstrated glove‐and‐stocking type loss of sensation to pinpricks as well as dysesthesia in all extremities. Phalen and Tinel tests were negative.
The elevated venous pressure and pitting edema with slow recovery suggest high venous pressure edema rather than hypoproteinemic edema. Complete bilateral dullness of the chest and shifting dullness of the abdomen indicate the presence of bilateral pleural effusion and ascites. Edema from high venous pressure is usually caused by right, left, or biventricular cardiac failure. A fixed splitting of the second heart sound suggests an atrial septal defect, which is a rare cause of progressive right heart failure in adults. I recommend checking the patient's thyroid function to investigate the possibility of hypothyroidism, which is a common illness among middle‐aged women and could contribute to her edema as well as hirsutism. The neurological findings suggest a generalized polyneuropathy. The unusual combination of high venous pressure edema and polyneuropathy may indicate a rare multisystem disorder such as amyloidosis. Alternatively, the patient might have developed multiple diseases during the same time period. For instance, diabetic polyneuropathy is the most common cause of polyneuropathy among the middle‐aged. Finally, the differential diagnosis of hirsutism includes ovarian, adrenal, or pituitary sources of hyperandrogenism in addition to hypothyroidism. I would first evaluate for diabetes, thyroid disease, and cardiac disease and would like to see the results of laboratory tests for thyrotropin and plasma glucose as well as chest radiography and electrocardiography.
The white‐cell count was 5400/mm3 with a normal differential. Hemoglobin was 10.7 g/dL with normal red‐cell indices, and the platelet count was 276,000/mm3. The erythrocyte sedimentation rate was 29 mm/hour. Other laboratory tests revealed the following values: total protein, 6.2 g/dL; albumin, 3.3 g/dL; blood urea nitrogen, 12 mg/dL; creatinine, 0.7 mg/dL; aspartate aminotransferase, 6 U/L; alanine aminotransferase, 2 U/L; lactate dehydrogenase, 96 U/L; alkaline phosphatase, 115 U/L; creatine phosphokinase, 60 U/L; total bilirubin, 0.9 mg/dL; glucose, 96 mg/dL; hemoglobin A1c, 4.6%; total cholesterol, 111 mg/dL; and thyrotropin, 6.32 mIU/mL (normal range, 0.50‐5.00 mIU/mL). Serum free thyroxine, triiodothyronine, and urine testosterone were normal. Serum dehydroepiandrosterone sulfate was mildly elevated for her age (864 ng/mL: normal range, 180‐750 ng/mL). Serological studies for human immunodeficiency virus, human T‐lymphotrophic virus type 1, and syphilis were negative. Urinalysis was weakly positive for protein but negative for casts and occult blood. The stool was negative for occult blood.
A chest radiograph showed bilateral pleural effusions. Computed tomography demonstrated bilateral pleural effusions, ascites, mild hepatomegaly, and small, multiple, mediastinal lymph nodes. Her electrocardiogram was normal. A transesophageal echocardiogram with agitated saline contrast demonstrated normal ventricular systolic and diastolic function and no atrial septal defect. The inferior vena cava did not collapse with inspiration, and there was no evidence of infiltrative cardiomyopathy.
These laboratory results rule out diabetes as the cause of the polyneuropathy. The subclinical hypothyroidism would not explain profound edema and hirsutism. A serum albumin level of 3.3 g/dL confirms high venous pressure edema rather than hypoproteinemic edema. Normochromic, normocytic anemia and a mildly elevated sedimentation rate point to a chronic illness or inflammatory state. The mediastinal lymphadenopathy may reflect congestion as a result of the high venous pressure or reflect a systemic disease involving lymph nodes. Normal ventricular function with high venous pressure is suggestive of heart failure from diastolic dysfunction, although the patient does not have risk factors for diastolic dysfunction, such as hypertension, and has no other echocardiographic features of diastolic impairment. The combination of hyperandrogenism and neuropathy points to a systemic process, such as a paraneoplastic syndrome. I would next investigate the source of the excess androgens.
Because serum dehydroepiandrosterone sulfate was mildly elevated, I‐131 aldosterol scintigraphy was performed, and it was negative. Electromyography showed a pattern of generalized sensorimotor polyneuropathy.
At this point, it appears that cardiac, endocrine, hepatic, and renal diseases have been largely ruled out as a cause of her symptoms. Reframing and unifying the important clinical problems for this patient may be useful in resolving this diagnostic puzzle. They include (1) systemic high venous pressure edema; (2) generalized sensorimotor polyneuropathy; (3) hirsutism; (4) normocytic, normochromic anemia; (5) an elevated erythrocyte sedimentation rate; (6) mediastinal lymphadenopathy; and (7) subclinical hypothyroidism. At this point, I cannot unify these pieces of information into a single diagnosis. I would search the medical literature, focusing on these terms.
A general internist consultant performed MEDLINE and Google Scholar searches using the key words edema, polyneuropathy, and hirsutism. This search suggested the diagnosis of Crow‐Fukase syndrome, also known as POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes) syndrome. Subsequent evaluations were performed. First, serum protein electrophoresis revealed the presence of monoclonal proteins, although hypergammaglobulinemia was not present. Second, a bone marrow examination demonstrated increased abnormal plasma cell proliferation (7%), although a radiographic skeletal survey found no lesions suggestive of plasmacytoma. Third, cerebrospinal fluid analysis showed normal cell counts but increased protein concentration (202 mg/dL). Fourth, a blood sample referred to an outside laboratory demonstrated elevated levels of vascular endothelial growth factor (3902 pg/mL: normal range, 150‐500 pg/mL). On the basis of these findings, the diagnosis of POEMS syndrome was made. After oral prednisolone (40 mg/day) was initiated, the systemic edema improved gradually, and she did well during the 2‐year follow‐up period.
Commentary
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder first described by Crow in 1956.1, 2 It is characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes, as indicated by the acronym. The diagnosis of POEMS syndrome is difficult as this syndrome is rare and requires high clinical suspicion. According to a nationwide cross‐sectional survey in Japan, the prevalence of POEMS syndrome is very low (about 3 patients per 1,000,000 persons),3 and its prevalence in Western countries is considered even lower than that in Japan. The average age at onset is around 45 to 50 years old, and men are twice as likely to have this syndrome as women.46 Table 1 shows the diagnostic criteria of POEMS syndrome, based on research by Dispenzieri and others at the Mayo Clinic, and Table 2 presents the relative frequency of these clinical features.6, 7 The initial symptomatology generally includes polyneuropathy, skin changes, and generalized edema, which are nonspecific symptoms, as are other well‐recognized associated conditions such as clubbing, weight loss, thrombocytosis, polycythemia, and hyperhidrosis. Thus, it is important to consider this syndrome when one is facing an undiagnosed illness involving multiple organ systems and to distinguish it from other conditions such as multiple myeloma, amyloidosis, and monoclonal gammopathy of undetermined significance. Vascular endothelial growth factor is thought to be involved in the edema of POEMS syndrome, as massive release from aggregated platelets increases vascular permeability and venous pressure.710
| |
| Major criteria | Polyneuropathy |
| Monoclonal plasma cell‐proliferative disorder | |
| Minor criteria | Sclerotic bone lesions |
| Castleman disease | |
| Organomegaly (splenomegaly, hepatomegaly, or lymphadenopathy) | |
| Edema (peripheral edema, pleural effusion, or ascites) | |
| Endocrinopathy (adrenal, thyroid, pituitary, gonadal, parathyroid, or pancreatic) | |
| Skin changes (hyperpigmentation, hirsutism, plethora, hemangiomata, and white nails) | |
| Papilledema | |
| Characteristic | % |
|---|---|
| |
| Peripheral neuropathy | 100 |
| Monoclonal plasma cell dyscrasia | 100 |
| Sclerotic bone lesions | 97 |
| Endocrinopathy | 71 |
| Skin changes | 68 |
| Organomegaly | 46 |
| Extravascular volume overload | 39 |
| Papilledema | 29 |
| Castleman disease | 11 |
Data regarding treatment and survival are largely observational. Overall mean survival from diagnosis in the 2003 Dispenzieri cohort was 13.7 years, with death often due to infection or cardiorespiratory failure.6 When a solitary plasmacytoma or osteosclerotic myeloma is present, radiation to the lesion can often lead to clinical remission. Other treatment options include alkylating agents and/or high‐dose chemotherapy with peripheral stem‐cell transplantation, corticosteroids, and supportive care.7
Clinicians frequently use the internet to aid in the clinical decision process. In a survey of the Royal New Zealand College of General Practitioners,11 half reported that they used the Internet to search for clinical information. Two well‐known resources are MEDLINE, which contains over 11 million references dating back to the 1960s, and internet search engines such as Google (and a more recent product, Google Scholar, which attempts to sort search results by including factors such as the author, the publication in which the article appears, and how often the article has been cited).
MEDLINE searches a well‐defined set of journals and uses the Medical Subject Headings (MeSH) vocabulary, which consists of sets of descriptive terms organized in a hierarchical structure to allow searching with various levels of specificity. For instance, entering the term heart attack will map to the MeSH term myocardial infarction and will also include more specific terms such as myocardial stunning and cardiogenic shock.
Google, in comparison, explores resources beyond journals without any clear boundary to its scope, and its advanced search functions can be occasionally unreliable. For instance, search results are occasionally marred by outdated citation information and may include materials that are not truly scholarly. However, search engines can search through the actual text of manuscripts and access the gray literature, which includes open‐source material that is usually original but not widely distributed or often easily available, such as technical reports and dissertations. A direct study comparing the results of searches in PubMed (one of the MEDLINE search engines) and Google Scholar is difficult, but the critical characteristics of each can be compared and contrasted (Table 3).
| Google Scholar | PubMed |
|---|---|
| 1. Database selection is clumped under subject areas, and it cannot be searched with unique identifiers: Con | 1. It allows one to choose a database at the outset and can search with a unique identifier (PubMed identifier): Pro |
| 2. Results cannot be filtered (ie, it does not allow multiple article selection): Con | 2. The single citation matcher allows retrieval of articles with pieces of information: Pro |
| 3. A search for related articles or similar pages is not available: Con | 3. It allows article selection by checkbox to reduce the number of articles relevant to the search query and to append the filter to search box: Pro |
| 4. It allows one to search by without words to exclude unwanted and confusing retrieved data: Pro | 4. It provides unique identifier (PubMed identifier) for each retrieved article for easy communicability: Pro |
| 5. It allows one to search a single journal/publication of interest: Pro | 5. Search are limited to journals only; it does not include the grey area of literature: Con |
| 6. Initial search results are those articles that are most cited by journals that themselves are the most cited: Pro | 6. It lists search results in chronological order and not by relevance: Con |
Internet searches may also suggest diagnoses from a compilation of clinical features, such as in this case. To be successful, such a search must complement the cognitive process; a search engine cannot completely replace clinical judgment. Clinicians must be able to identify salient clinical features and generate high‐yield search terms and then exercise skill in sifting through the citations to arrive at the appropriate diagnosis. A recent study found that Google searches revealed the correct diagnosis in 58% of the case records of the New England Journal of Medicine,12 although each search query resulted in many results, which then had to be manually reviewed for appropriateness within the case's context.
Like a traditional diagnostic test, a search can be described by sensitivity, specificity, and the number of articles needed to read.13 For example, in a study comparing the performance of search strategies to identify clinical practice guidelines in Google Scholar and SUMSearch (another freely accessible search engine), using the term guideline yielded the highest sensitivity, and using the term practice guideline generated the highest specificity and the lowest number of articles needed to read (Table 4).14
| Search Strategy | Sensitivity (%) | Specificity (%) | NNR |
|---|---|---|---|
| |||
| SUMSearch | |||
| Guideline* | 81.51 (74.5388.49) | 74.29 (72.6475.94) | 8.18 (6.9010.05) |
| Recommendation* | 60.50 (51.7269.28) | 76.28 (74.6777.89) | 9.93 (8.1412.72) |
| Practice guideline* | 40.34 (31.5249.16) | 89.45 (88.2990.61) | 6.96 (5.529.43) |
| Google Scholar | |||
| Guideline/s | 31.93 (23.5640.30) | 78.05 (76.5079.60) | 16.67 (12.7624.04) |
| Recommendation/s | 8.40 (3.4213.38) | 92.11 (91.0993.13) | 22.42 (13.9756.82) |
| Practice guideline/s | 11.76 (5.9817.54) | 95.72 (94.9696.48) | 9.29 (6.2118.38) |
Although there are several other popular hosts of web‐based search engines, a more robust decision‐support program may help physicians more efficiently consider relevant diagnoses. One program, named Isabel, has been developed through the indexing of a database of more than 11,000 diseases according to word patterns in journal articles associated with each disease, and it is updated as new and relevant articles emerge. One recent study demonstrated that the correct diagnosis was made in 48 of 50 cases (96%) with specific, key findings as search terms but in only 37 of the same 50 cases (74%) if the entire case history was simply pasted in, again emphasizing the importance of specific search terms.15
POEMS syndrome is a rare entity occasionally seen in middle‐aged individuals and marked by a multitude of nonspecific findings, particularly polyneuropathy and plasma cell dyscrasia. In this case, the diagnostic test was an internet search based on the most prominent clinical symptoms. Such a strategy can provide a powerful addition to traditional literature and MEDLINE resources. However, the efficiency of this process is heavily dependent on the quality of the search strategy and, therefore, the cognitive faculties of the treating physician to avoid the predictable shortcoming of low specificity. Garbage in, garbage out still applies whether the computer in question is the human mind or the desktop PC.
Teaching Points
-
POEMS syndrome, also known as Crow‐Fukase syndrome, is a rare multisystem disorder characterized by polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
-
Internet‐based searches, including Google and MEDLINE, are being used more frequently because they are widely available, quick, and freely accessed.
-
Internet searches appear most useful as adjuncts to PubMed and clinical reasoning in identifying case reports when a well‐constructed collection of symptoms and signs is used for searches.
- .Peripheral neuritis in myelomatosis.Br Med J.1956;2(4996):802–804.
- ,,,,,.Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature.Medicine (Baltimore).1980;59(4):311–322.
- .Nationwide Epidemiologic Survey of Crow‐Fukase Syndrome in 2004.Tokyo, Japan:Japanese Ministry of Health and Welfare Government Report, 2004.
- ,,, et al.The Crow‐Fukase syndrome: a study of 102 cases in Japan.Neurology.1984;34(6):712–720.
- ,,.POEMS syndrome: a study of 25 cases and a review of the literature. French Study Group on POEMS Syndrome.Am J Med.1994;97(6):543–553.
- ,,, et al.POEMS syndrome: definitions and long‐term outcome.Blood.2003;101(7):2496–2506.
- .POEMS syndrome.Hematology.2005;1(1):360–367.
- ,,,,.Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome.Lancet.1996;347(9002):702.
- ,.Assessment of hypoproteinaemic oedema: a simple physical sign.Br Med J.1978;1(6117):890–891.
- ,,, et al.Ratio of serum vascular endothelial growth factor to platelet count correlates with disease activity in a patient with POEMS syndrome.Eur J Intern Med.2002;13(1):70–74.
- .In search of evidence: family practitioners' use of the Internet for clinical information.J Med Libr Assoc.2002;90(4):370–379.
- ,.Googling for a diagnosis—use of Google as a diagnostic aid: internet based study.BMJ.2006;333(7579):1143–5114.
- ,,.The number needed to read—a new measure of journal value.Health Info Libr J.2005;22(2):81–82.
- ,,,.Developing search strategies for clinical practice guidelines in SUMSearch and Google Scholar and assessing their retrieval performance.BMC Med Res Methodol.2007;7:28.
- ,.Performance of a web‐based clinical diagnosis support system for internists.J Gen Intern Med.2008;23(suppl 1):37–40.
- .Peripheral neuritis in myelomatosis.Br Med J.1956;2(4996):802–804.
- ,,,,,.Plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes: the POEMS syndrome. Report on two cases and a review of the literature.Medicine (Baltimore).1980;59(4):311–322.
- .Nationwide Epidemiologic Survey of Crow‐Fukase Syndrome in 2004.Tokyo, Japan:Japanese Ministry of Health and Welfare Government Report, 2004.
- ,,, et al.The Crow‐Fukase syndrome: a study of 102 cases in Japan.Neurology.1984;34(6):712–720.
- ,,.POEMS syndrome: a study of 25 cases and a review of the literature. French Study Group on POEMS Syndrome.Am J Med.1994;97(6):543–553.
- ,,, et al.POEMS syndrome: definitions and long‐term outcome.Blood.2003;101(7):2496–2506.
- .POEMS syndrome.Hematology.2005;1(1):360–367.
- ,,,,.Greatly raised vascular endothelial growth factor (VEGF) in POEMS syndrome.Lancet.1996;347(9002):702.
- ,.Assessment of hypoproteinaemic oedema: a simple physical sign.Br Med J.1978;1(6117):890–891.
- ,,, et al.Ratio of serum vascular endothelial growth factor to platelet count correlates with disease activity in a patient with POEMS syndrome.Eur J Intern Med.2002;13(1):70–74.
- .In search of evidence: family practitioners' use of the Internet for clinical information.J Med Libr Assoc.2002;90(4):370–379.
- ,.Googling for a diagnosis—use of Google as a diagnostic aid: internet based study.BMJ.2006;333(7579):1143–5114.
- ,,.The number needed to read—a new measure of journal value.Health Info Libr J.2005;22(2):81–82.
- ,,,.Developing search strategies for clinical practice guidelines in SUMSearch and Google Scholar and assessing their retrieval performance.BMC Med Res Methodol.2007;7:28.
- ,.Performance of a web‐based clinical diagnosis support system for internists.J Gen Intern Med.2008;23(suppl 1):37–40.
Positively False
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
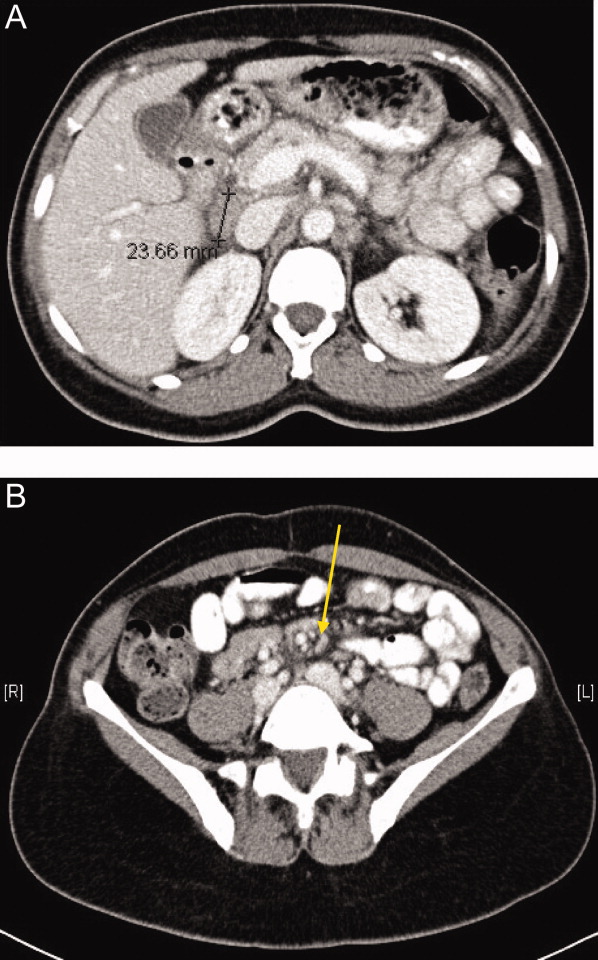
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
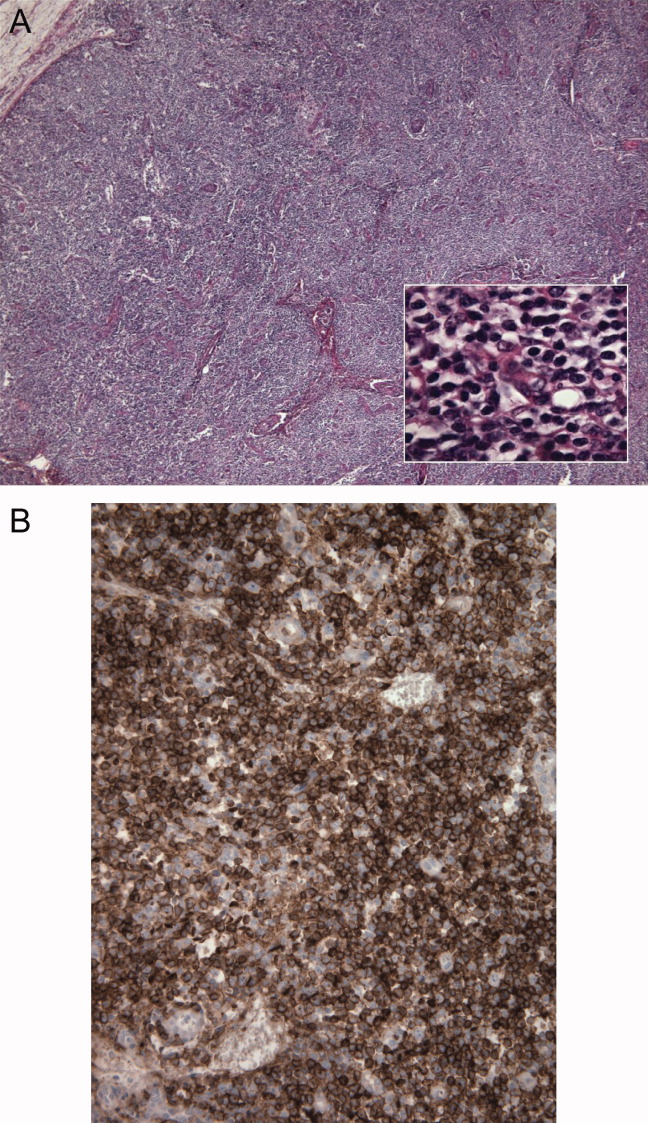
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
The approach to clinical conundrums by an expert clinician is revealed through presentation of an actual patient's case in an approach typical of morning report. Similar to patient care, sequential pieces of information are provided to the clinician who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
A51‐year‐old woman presented after 5 days of fever, rigors, anorexia, right‐sided abdominal pain, nausea, and dizziness. She had 2 loose stools the day before admission, without blood or mucus, but otherwise had recently been constipated. She denied cough, shortness of breath, chest pain, headache, sore throat, rash, arthritis, or dysuria.
In a 51‐year‐old woman with right‐sided abdominal pain and systemic symptoms, major concerns include biliary disease, liver abscess, or appendicitis. Right‐sided diverticulitis would be more unusual. Pyelonephritis infrequently presents with epigastric and lower quadrant pain, instead of flank pain. Basilar pneumonia may present with abdominal pain, but this is less likely in the absence of respiratory symptoms.
The patient used an albuterol inhaler for mild asthma and had experienced an episode of herpes zoster 7 years prior, but was otherwise well. Her surgical history was notable for a remote appendectomy. She was a native of the Dominican Republic who had lived in the United States for the past 20 years. She visited the Dominican Republic for 3 weeks every year, with her last visit occurring about 10 months before. She was a cleaning and maintenance worker. She had 2 adult children in good health, was divorced from her husband, and had not been sexually active for the past 8 years. The patient had no pets or other animal exposures. She did not smoke, drink alcohol, or use intravenous drugs.
The remote episode of shingles makes me a bit worried about chronic human immunodeficiency virus (HIV) infection. As a native of and traveler to the Dominican Republic, she is at risk for a variety of tropical pathogens. Hyperinfection syndrome from strongyloides can cause fever and bacteremia, but this is almost always associated with significant immunosuppression. Dengue fever has become very common in the Caribbean, but should occur within 2 weeks of travel. Her work in cleaning and maintenance might bring her into contact with rats and mice, putting her at risk for leptospirosis. This can present as a fairly nonspecific febrile syndrome, but this is unlikely without a major complaint of headache.
The patient appeared fatigued. Her temperature was 39.7C, her heart rate was 110 beats per minute, and her blood pressure 80/62 mm Hg. The oropharynx was normal. Mild cervical lymphadenopathy was present (less than 1 cm in diameter). The chest was clear and the cardiac examination unremarkable. Bowel sounds were present. Moderate right‐sided abdominal tenderness was noted, somewhat more marked in the right lower quadrant, without guarding or rebound. There was no hepatosplenomegaly. There was no rash. A bedside right upper quadrant ultrasound was negative for gallstones.
Her low blood pressure is concerning for bacterial sepsis. The negative right upper quadrant ultrasound makes cholecystitis or cholangitis less likely, but does not exclude diverticulitis or pelvic inflammatory disease. She lacks peritoneal signs, but they may be absent in these conditions. Another worrisome finding on her physical examination is cervical lymphadenopathy. In an older patient, this raises the specter of malignancy. In a younger patient, it could suggest a mononucleosis syndrome from Epstein‐Barr virus (EBV) or cytomegalovirus (CMV). In addition, HIV must be considered in any patient with unexplained lymphadenopathy.
She received intravenous levofloxacin and 1 L of intravenous normal saline, with a rise in her blood pressure to 100/59 mm Hg. Her white blood cell count was 5.0, with 71% polys and 9% bands. The hematocrit was 32%, with a normal mean corpuscular volume. The erythrocyte sedimentation rate (ESR) was 109 mm/hour. The platelet count, serum electrolytes, creatinine, aminotransferases, alkaline phosphatase, bilirubin, amylase, and lipase were normal. The serum level of lactate dehydrogenase (LDH) was 498 unit/L (normal range, 107231 units/L). Computed tomography (CT) scan of the abdomen showed multiple enlarged lymph nodes up to 1.2 cm in size along the gastrohepatic ligament and the mesentery, with mild associated fat stranding, consistent with mesenteric lymphadenitis.
Her blood pressure has responded to fluids; perhaps she was just volume‐depleted from not eating for several days. She has bandemia, which is consistent with acute bacterial infection, but might also signify a stress response. The low hematocrit and high ESR raise the possibility of anemia of chronic disease; perhaps her illness is more longstanding than her presentation suggests. Mesenteric lymphadenitis may be related to EBV and HIV, but in a patient originally from the Caribbean, it raises the possibility of gastrointestinal tuberculosis or histoplasmosis. Human T‐cell lymphotropic virus type 1 (HTLV‐1) is also endemic in the Caribbean, and may cause adult T‐cell leukemia/lymphoma (ATLL), which could explain her lymphadenopathy and elevated LDH. Mesenteric lymphadenitis is also characteristic of several bacterial infections, especially Yersinia, Salmonella, and Bartonella. The lack of diarrhea makes yersiniosis doubtful, and the absence of cat exposure makes bartonellosis unlikely. Salmonella infection is also associated with diarrhea, except for typhoid fever, in which patients have diarrhea, constipation, or normal stools.
A urine culture and 3 sets of blood cultures obtained prior to the initiation of antibiotics were negative. A blood smear showed no malaria or babesia parasites. The patient's fever continued for the first 2 days of her hospitalization, but subsequently abated. The patient had no loose stools during her hospitalization. Serologies for EBV, hepatitis A, CMV, and toxoplasmosis were indicative of remote infection. Serum rapid plasma reagin (RPR), Bartonella antibodies, antinuclear antibodies, hepatitis B surface antigen, and hepatitis C antibody were negative. Tuberculin skin testing and urinary histoplasma antigen were negative. By the fifth hospital day, she had been afebrile for over 48 hours, and her abdominal pain had improved, though it had not completely resolved. She was discharged to complete a 10‐day course of levofloxacin.
It is not clear to me whether she has just experienced a spontaneous remission in her illness, or whether her disease course has truly been modified with antibiotics. Could she have typhoid fever or another salmonellosis? The patient has not traveled abroad in several months. If she had typhoid fever, the source of infection would have to be imported food, or exposure to a family member who was a carrier. The negative tuberculin skin test makes tuberculosis less likely, but does not exclude it entirely. Similarly, while a negative histoplasma urinary antigen essentially rules out acute disseminated histoplasmosis, as would be seen in acquired immune deficiency syndrome (AIDS), it is less sensitive for more chronic forms of disseminated histoplasmosis, including gastrointestinal involvement.
One week later, the patient returned to the emergency department with recurrent abdominal pain, anorexia, fever, night sweats, and increased swelling of the lymph nodes in her neck. She had lost a total of 4.4 kg (10 lb) since the onset of her illness. Physical examination revealed moderate (up to 2 cm), slightly tender cervical and inguinal lymphadenopathy, and continued moderate right‐sided abdominal tenderness. Mesenteric, retroperitoneal, and inguinal lymphadenopathy was more prominent than on the prior CT scan (Figure 1).She was told by the emergency room physicians that the HIV test obtained during the prior hospitalization was positive. Further questioning elicited that her estranged husband had been promiscuous prior to their separation. She was transferred to a second hospital for further care. Blood cultures were sent for fungi and acid‐fast bacilli.
Everyone with fever of unknown origin (FUO) deserves an HIV test. Is this just lymphadenopathy from HIV, or is she suffering from an opportunistic infection? Mycobacterium avium infection is an attractive explanation for her fever, abdominal pain, and lymphadenopathy, but I would hold off on empiric treatment until the results of a CD4+ cell count were available. Could she have a secondary, HIV‐related lymphoma?
Full review of records from the outside hospital showed that the patient had a positive HIV enzyme immunoassay, with an indeterminate HIV Western blot. The patient's CD4 cell count was 313/cm3, with a CD4/CD8 ratio within the normal range. Her HIV enzyme immunoassay was repeated and found to be negative, and the HIV viral load was undetectable.
The HIV enzyme immunoassay is only a screening test, and must be confirmed with a positive HIV Western blot. (In most clinical laboratories, this is done automatically before the test is reported.) Indeterminate HIV Western blots are common in acute HIV infection, but outside of this setting, most patients with indeterminate HIV Western blots turn out not to have HIV infection. I am still very concerned about lymphoma, and would pursue a lymph node biopsy. Disseminated tuberculosis is still possible.
Biopsy of a right inguinal lymph node was performed on the third day of the second hospitalization. The patient was persistently febrile, developed swelling of the knees, ankles, and interphalangeal joints of the second and third digits of the left hand, and complained of pruritus. Anti‐double‐stranded DNA antibody, antineutrophilic cytoplasmic antibody, and Brucella antibody were negative. Antibody against cyclic citrullinated peptide (CCP) was weakly positive.
Now she has a more florid syndrome, with arthritic symptoms. Sarcoidosis is an attractive explanation for her fever, lymphadenopathy, and arthritis. Lupus could explain some features of her presentation, but the negative serology makes it unlikely. Antibody to cyclic citrullinated peptide is a newer diagnostic test for rheumatoid arthritis. Although anti‐CCP is more specific than rheumatoid factor for the diagnosis of rheumatoid arthritis, this result could still be a false positive, particularly given the low titer. As well, the patient has more impressive lymphadenopathy than is usual for rheumatoid arthritis. Reactive arthritis can follow enteric infection with Campylobacter, Salmonella, Yersinia, and Shigella, but lymphadenopathy is not a feature. Arthritis may be prominent in parvovirus B19, rubella, disseminated gonococcal infection, and Lyme disease, but mesenteric lymphadenitis and a prolonged, waxing and waning course would not be expected with any of these. I worry that the arthritis and pruritus are paraneoplastic manifestations of lymphoma.
The inguinal lymph node biopsy showed markedly distorted nodal architecture with an atypical proliferation of small‐sized to large‐sized lymphoid cells, with predominantly round nuclei, vesicular chromatin, small nucleoli, and variable amounts of clear to eosinophilic cytoplasm (Figure 2). Residual follicles and occasional apoptotic bodies and mitoses were seen. Large B‐cells were frequently present, which stained positive for EBV‐associated mRNA by in situ hybridization studies. Immunoperoxidase staining revealed that the atypical cell population was largely composed of CD4+ T‐cells. T‐cell receptor gene rearrangement studies demonstrated that the T‐cell population was monoclonal. These results were consistent with angioimmunoblastic T‐cell lymphoma (AITL). Positron emission tomography (PET) scanning was performed, showing diffuse fluorodeoxyglucose (FDG)‐avid lymphadenopathy involving cervical, axillary, mediastinal, retroperitoneal, mesenteric, and inguinal lymph nodes, up to 2 cm in diameter. The patient completed 6 cycles of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), as well as experimental treatment with denileukin diftitox. Her fever and arthritis subsided quickly, and she was clinically well and free of disease by CT scans and PET scans 1 year after diagnosis.
COMMENTARY
The differential diagnosis of FUO is one of the largest in medicine, encompassing a dizzying range of infectious, inflammatory, and neoplastic conditions. This has made the development of standardized diagnostic algorithms difficult.1 The workup of patients with FUO begins with a detailed history and physical examination, followed by a core set of microbiology cultures, imaging studies, and blood tests on all patients. Further testing is individualized, based on key clinical findings, also known as pivot points.2 Unfortunately, many of the diseases presenting as FUO have overlapping symptoms and signs, somewhat limiting the utility of pivot points. In a prospective study, 81% of these potentially diagnostic clues were misleading, although 19% of them contributed to the final diagnosis.3 Key clinical findings in patients with FUO may trigger a barrage of diagnostic tests, often leading to a large number of false‐positive results.4 Clinicians who investigate patients with FUO must remember that many clues are diagnostic dead ends, and should be wary of drawing positively‐false conclusions.
Fever pattern is usually not helpful in the diagnosis of FUO, with occasional exceptions, such as the tertian and quartan fevers in some forms of malaria. A minority of patients with Hodgkin's disease have Pel‐Ebstein fevers, in which 1 to 2 weeks of fever alternate with an afebrile period of similar or longer duration. More often, fever in lymphoma waxes and wanes unpredictably,5 a circumstance that may lead to the mistaken impression of response to antibiotics, as in this case.
Tissue biopsies are helpful in FUO when suggestive findings are present on physical examination or imaging studies. In patients with lymphadenopathy and FUO, lymph node biopsy is a high‐yield procedure, contributing to the final diagnosis 46% of the time. This is exceeded only by biopsies of skin lesions, which have a 63% diagnostic yield in FUO.3 In older patients with FUO, temporal artery biopsies have a significant diagnostic yield, in the range of 16% to 17%. Liver biopsies have a similar yield (14‐17%), but a higher risk of complications. Bone marrow biopsies have a low yield in most FUO patients.1
AITL makes up 1% of all non‐Hodgkin's lymphomas. AITL was once known as angioimmunoblastic lymphadenopathy, and was thought to be either a disorder of immune regulation or a premalignant lymphoid disease. However, molecular diagnostic techniques have established that monoclonal T‐cell populations and cytogenetic abnormalities are usually present at the time of diagnosis.6, 7 The prognosis in AITL is unfavorable. Disease is usually widespread at the time of diagnosis. Most patients achieve complete remission with anthracycline‐based chemotherapy, such as CHOP, but the duration of remission is often brief, and median survival after diagnosis is only 3 years.6 Novel treatments under investigation include denileukin diftitox,8 a fusion protein of interleukin‐2 (IL‐2) conjugated to diphtheria toxin, which leads to apoptosis of cells expressing the IL‐2 receptor; rituximab, which targets the reactive population of B‐cells in AITL, rather than the malignant clone of T‐cells; and antiangiogenic therapy, such as thalidomide.9
The diagnosis of AITL is usually elusive, and the average patient has symptoms for 4 months prior to diagnosis.6 This patient's clinical presentation, while certainly not specific, was typical of AITL. AITL usually presents as fever of unknown origin with generalized, nonbulky lymphadenopathy. Fever is present in 57% of patients with AITL, and up to 2% of FUO is caused by AITL.6, 10 Other features of this patient's illness, such as night sweats, weight loss, pruritus, and arthritis, are common in AITL.
T‐cell depletion and immune dysregulation are frequent in AITL, explaining why AITL shares a number of clinical features with HIV disease. These include a high incidence of drug rashes, immune thrombocytopenic purpura, polyclonal hypergammaglobulinemia, and autoantibodies.6, 7 As with HIV patients, death in AITL is often due to opportunistic infections or diffuse large B‐cell lymphomas.7 Over 95% of patients with AITL display a proliferation of EBV‐infected B cells, presumably from immune dysregulation, and the B‐cell lymphomas in these patients are usually EBV‐positive.11
Because AITL is a proinflammatory state, false‐positive antibody tests are fairly common. The occasional occurrence of positive HIV enzyme immunoassays, with indeterminate Western blot results, may be a particular source of diagnostic confusion.12, 13 The significance of indeterminate Western blots is often erroneously communicated to patients, as happened here. Indeterminate HIV Western blots may be seen in HIV seroconversion, HIV‐2 infection, or advanced HIV disease with loss of core antibody. They may also result from laboratory error, multiparity, syphilis, malaria, or cross‐reacting antibodies in autoimmune diseases. Indeterminate HIV Western blots in low‐risk patients usually do not represent true HIV infection.14
KEY POINTS FOR HOSPITALISTS
-
FUO is one of the most challenging diagnoses faced by hospitalists. Biopsies of new skin lesions and enlarged lymph nodes are particularly high‐yield diagnostic procedures in FUO. Fever pattern is generally not helpful in the diagnosis of FUO, with few exceptions, such as the tertian fevers of malaria.
-
Misleading and false‐positive tests results often occur in the course of FUO evaluation, due to the sheer number of tests ordered, and the higher likelihood of false‐positive serologic tests in the setting of inflammatory states.
-
AITL may be an elusive diagnosis in FUO with diverse clinical features, including weight loss, night sweats, rashes, arthritis, autoantibodies, immune dysregulation, and opportunistic infections. The prognosis traditionally has been guarded, but may be more hopeful in an era of emerging molecular therapies.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.
- ,,.A comprehensive evidence‐based approach to fever of unknown origin.Arch Intern Med.2003;163:545–551.
- ,.The art of diagnosis: solving the clinicopathological exercise.N Engl J Med.1982;306:1263–1268.
- ,,, et al.A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol.Medicine (Baltimore).2007;86:26–38.
- ,,.Fever of unknown origin (FUO). II. Diagnostic procedures in a prospective multicenter study of 167 patients. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:401–414.
- ,.Neoplastic diseases. In:Murray HW, ed.FUO: Fever of Undetermined Origin.New York, NY:Futura Publishing;1983:39–48.
- ,,, et al.Angioimmunoblastic T‐cell lymphoma: clinical and laboratory features at diagnosis in 77 patients.Medicine (Baltimore).2007;86:282–292.
- ,,.Angioimmunoblastic T‐cell lymphoma.Br J Haematol.2003;121:681–691.
- ,,, et al.Phase II trial of denileukin diftitox for relapsed/refractory T‐cell non‐Hodgkin lymphoma.Br J Haematol.2007;136:439–447.
- ,.Angioimmunoblastic T‐cell lymphoma: still a dismal prognosis with current treatment approaches.Leuk Lymphoma.2007;48:645–646.
- ,,.Fever of unknown origin (FUO). I. A prospective multicenter study of 167 patients with FUO, using fixed epidemiologic entry criteria. The Netherlands FUO Study Group.Medicine (Baltimore).1997;76:392–400.
- ,,, et al.Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression.Am J Surg Pathol.2007;31:1077–1088.
- ,,,.Angioimmunoblastic lymphadenopathy, immunoblastic lymphoma, and false‐positive seroconversion for human immunodeficiency virus.Ann Intern Med.1987;107:114.
- ,.Angioimmunoblastic T‐cell lymphoma associated with an antibody to human immunodeficiency virus protein.Int J Hematol.2003;78:160–162.
- ,,.Communicating indeterminate HIV Western blot test results to clients: an observational study of three community testing sites.AIDS Patient Care STDS.2006;20:620–627.