User login
The Third Time's the Charm
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
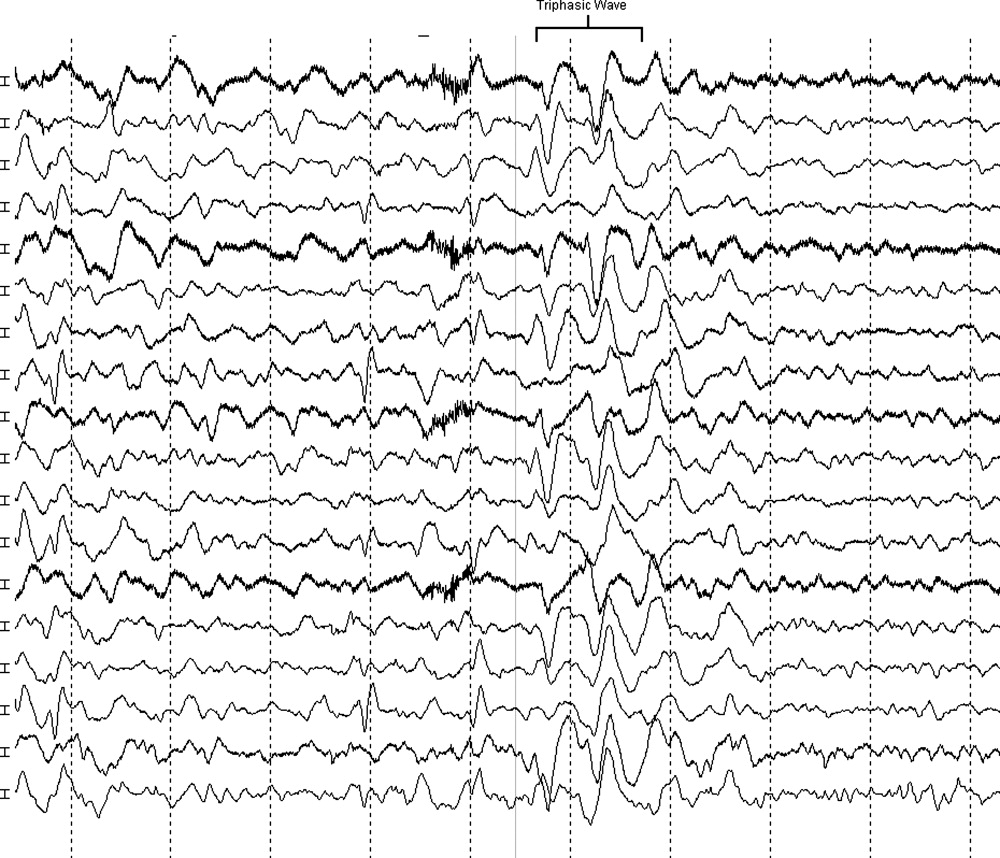
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
A 58‐year old woman was brought to the emergency department with confusion. Her husband stated that for several hours she had been drifting in and out at home, and that he had to shout to get her attention. He described no seizure activity, weakness, incontinence, or difficulty speaking, and had noted no complaints of headache, fevers, chest pain, shortness of breath, or gastrointestinal complaints.
Altered mental status in a middle‐aged woman can result from a diverse set of etiologies. A key distinction in the neurological examination will be to assure that the complaint of confusion is accurate as opposed to aphasia; the former is usually indicative of diffuse cerebral dysfunction while the latter suggests a focal lesion in the dominant hemisphere.
The acuity of the change in mental status is important, as are the fluctuations described by the husband. Unwitnessed or nonconvulsive seizure activity can present this way. Toxic/metabolic etiologies, infectious and inflammatory disorders of the central nervous system (CNS), and vascular diseases are also important considerations. Although stroke does not typically present with global encephalopathy, intermittent large vessel occlusion, especially in the posterior circulation, can disrupt cognition in this manner. Following a physical examination, initial workup should focus on toxic/metabolic etiologies, followed rapidly by head imaging if no cause is identified.
Her past medical history was notable for type 2 diabetes mellitus, coronary artery disease, hyperlipidemia, and an unspecified seizure disorder, which according to her husband was diagnosed during a recent hospitalization for a similar presentation. She also had a remote history of venous thromboembolism and antithrombin‐III deficiency. She was unemployed, lived with her husband, and spent most of her time at home. She never smoked, and rarely drank alcohol. Her family history was unobtainable, and her husband denied that she used any illicit drugs. Her medications included pioglitazone, aspirin, simvastatin, pregabalin, ferrous sulfate, levetiracetam, warfarin, and magnesium oxide, and she was allergic to sulfa.
While the differential diagnosis remains broad, 3 elements of the history are potentially relevant. The history of epilepsy based on a similar prior presentation increases the likelihood that the current spell is ictal in nature; examination of previous records would be important in order to document whether these spells have indeed been proven to be epileptic, as many conditions can mimic seizures. Given the history of venous thromboembolism and hypercoagulability, one must consider cerebral venous sinus thrombosis, which can present with global neurologic dysfunction and seizures. Prompt identification, usually via computed tomography (CT) or magnetic resonance angiography, is vital, because anticoagulation can mitigate this potentially life‐threatening illness. Finally, although many medications can cause encephalopathy in overdose, levetiracetam has well‐described cognitive side effects even at usual doses, including encephalopathy, irritability, and depression.
The records from that recent hospitalization remarked that she had presented confused and stuporous. Her potassium had been 2.7 mmol/L, international normalized ration (INR) 3.4, and hemoglobin 8 g/dL; other routine laboratory studies were normal. CT and magnetic resonance imaging (MRI) of the brain had been negative, and electroencephalogram (EEG) reportedly was performed but specific results were unknown. She was discharged alert and oriented 1 week prior to the current presentation on the above medications, including levetiracetam for this newly‐diagnosed seizure disorder.
Previous records confirm that the current presentation is that of a relapsing acute alteration in mental status. Regardless of the EEG findings or response to antiepileptic medications, a seizure disorder should remain a primary consideration, although relapsing inflammatory, toxic/metabolic conditions, and, rarely, vascular disorders can also present in this manner.
The neurologic manifestations of hypokalemia are usually peripheral in nature, including periodic paralysis; confusion accompanying hypokalemia is usually not a result of the low potassium itself but rather due to an underlying toxic or endocrinologic cause. Various causes of anemia can lead to mental status changes; the mean corpuscular volume (MCV) will be particularly helpful given known associations between megaloblastic anemia and confusional states.
On examination, she appeared to be in good health and in no distress. She was afebrile. Her blood pressure was 93/57, pulse 90 beats per minute, respiratory rate 16 per minute, and room air oxygen saturation 100%. She was oriented to her surroundings, but slow in her responses to questioning. There were no cranial nerve, motor, or sensory deficits, or abnormal reflexes or movements. Examination of the head, skin, chest, cardiovascular system, abdomen, and extremities was normal. Serum sodium was 136 mmol/L, creatinine 1.2 mg/dL, calcium 9.3 mg/dL, and glucose 81 mg/dL; other routine blood chemistries were normal. Her white blood cell (WBC) count was 7100/L, hemoglobin 9.2 g/dL with normal MCV, and platelet count 275,000/L. INR was 3.4, and liver function tests were normal. CT of the brain demonstrated no evidence of acute pathology.
Given that her laboratory results (aside from the hemoglobin) and CT were essentially normal, the most common etiology of a recurrent encephalopathy would be a toxic exposure including drugs, alcohol, and environmental toxins or poisons. A comprehensive serum drug screen, including heavy metals, could follow a basic urinary screen for drugs of abuse; specific etiologies may be suggested by patterns of injury seen on MRI such as those seen with carbon monoxide or methanol exposure. Other recurrent metabolic processes include the porphyrias and relapsing inflammatory disorders, which could be entertained if further diagnostics are unrevealing.
An EEG is warranted at this point and is a test that is underutilized in the workup of altered mental status. Patients who have a spell and do not quickly awaken should be considered to be in nonconvulsive status epilepticus until proven otherwise. This can be easily identified on the EEG and is an important entity to recognize quickly. Additional findings on EEG may suggest focal cerebral dysfunction (such as that following a seizure or acute unilateral injury), diffuse encephalopathy (eg, triphasic waves), or fairly specific diagnoses (eg, periodic lateralized epileptiform discharges from the temporal lobes in suspected herpes simplex meningoencephalitis). While the CT of the brain is a reasonable initial screen, MRI is more sensitive for structural disease and should be obtained if no etiology is rapidly identified.
Finally, acute infectious etiologies such as abscess, encephalitis, or meningoencephalitis need to be excluded via lumbar puncture. Spinal fluid examination also can be helpful in the consideration of inflammatory and autoimmune disorders.
Over the next several hours, while still in the emergency department, she became increasingly obtunded, to the point that she was unresponsive to all stimuli. No seizure activity was witnessed, her vital signs were unchanged, and no medications had been administered. She was urgently transferred to a tertiary care center, where, at the time of arrival, she was obtunded and nonverbal, and opened her eyes only to noxious stimuli. She would withdraw all 4 extremities in response to pain. Pupils were 2 mm and symmetrically reactive. Corneal reflexes were normal, and her gag reflex was diminished. Motor tone was decreased in all 4 extremities. No fasciculations were noted. Deep tendon reflexes were present but symmetrically diminished throughout, and Babinski testing demonstrated a withdrawal response bilaterally.
Coma is a state of profound unconsciousness where the patient is unarousable and unaware of her surroundings. Coma can result either from bihemispheric dysfunction or diffuse injury to the reticular activating system in the brainstem, and the physical examination should focus on distinguishing between these 2 sites. Because the nuclei of cranial nerves III through XII (excepting XI) reside in the brainstem, the coma examination emphasizes testing the cranial nerves; although all cranial nerves are not tested in this patient, the ones that are appear to be normal, making bihemispheric dysfunction most likely. Bihemispheric coma most commonly results from diffuse toxic or metabolic etiologies such as intoxication or hepatic encephalopathy, but it can also be caused by bilateral structural lesions (including the bilateral thalami) or ongoing seizure activity.
Although an EEG remains the key test in this patient given her past history and an MRI would prove extremely useful, her deterioration warrants a workup for CNS infection. Since the head CT was negative, it would be prudent to proceed with urgent lumbar puncture (although it should never be performed in a patient with significant coagulopathy due to risks of hemorrhage leading to spinal cord injury). She should be covered empirically with broad spectrum meningeal‐dose antibiotics, including acyclovir, until the results of the spinal fluid examination are known, given that bacterial meningitis and herpes meningoencephalitis carry a high morbidity and mortality if not treated promptly.
Routine blood tests were similar to her labs at the referring emergency room. Ammonia level was 10 mol/L. Urine toxicology screen was negative, and blood tests for ethanol, salicylates, lithium, and acetaminophen were negative. Chest X‐ray and urinalysis were normal, and electrocardiogram was notable only for a sinus tachycardia. Cultures of the blood were obtained and the patient was admitted to the intensive care unit.
Levetiracetam, vancomycin, piperacillin‐tazobactam, and acyclovir were initiated. A lumbar puncture was performed without reversing the anticoagulation, and the procedure was traumatic. The cerebrospinal fluid was bloody, with a clear supernatant. Cell count demonstrated a red blood cell (RBC) count of 1250/L and a WBC count of 9/L, with a WBC differential of 42% neutrophils, 48% lymphocytes, and 8% monocytes. The cerebrospinal fluid (CSF) glucose was 62 mg/dL (with a serum glucose of 74 mg/dL) and protein 41 mg/dL. The CSF Gram stain demonstrated no organisms, and fluid was sent for routine culture and polymerase chain reaction (PCR) to detect herpes simplex virus (HSV). A neurology consultation was urgently requested.
As mentioned, it would have been more appropriate to reverse the patient's anticoagulation prior to lumbar puncture. The absence of xanthochromia suggests that the RBCs seen in the sample were introduced at the time of the lumbar puncture, arguing against a hemorrhagic disorder of the CNS (occasionally seen with herpes simplex encephalitis) or spinal fluid (eg, subarachnoid hemorrhage).
A reasonable rule of thumb to correct for the number of RBCs in a traumatic lumbar puncture is to allow 1 WBC for every 700 RBCs/L. Given this conversion, there are still too many WBCs in this sample, indicating a mild pleocytosis that is approximately one‐half neutrophilic and one‐half lymphocytic. This profile is nonspecific and can occur with a variety of conditions including stroke, seizure, inflammatory disorders, and infections, including viruses such as West Nile virus.
While coverage with acyclovir and broad‐spectrum antibacterials is appropriate, it should be noted that piperacillin‐tazobactam has poor CSF penetration and therefore is not a good choice for empiric coverage of CNS infections.
The neurologist's examination additionally noted multifocal myoclonus with noxious stimuli, most prominent in the face and toes. An urgent EEG demonstrated continuous, slow, generalized triphasic wave activity (Figures 1 and 2); no epileptiform discharges were seen. The erythrocyte sedimentation rate (ESR) was 66 mm/hour (normal, 0‐30), and tests for antinuclear antibodies, serum levetiracetam level, and thyroid function studies were ordered.
Stimulus‐evoked multifocal myoclonus is a general marker of encephalopathy found in many conditions, including hepatic and renal failure, drug intoxication (eg, opiates), neurodegenerative disorders (eg, Creutzfeldt‐Jakob disease [CJD]), and postanoxic injury, the latter of which is termed the Lance‐Adams syndrome.
Triphasic waves on EEG, while commonly associated with hepatic encephalopathy, have a similarly broad differential diagnosis, although in a comatose patient, they must first and foremost be distinguished from the repetitive discharges characteristic of nonconvulsive status epilepticus. In addition to hepatic and renal failure, triphasic waves have also been described in medication toxicity (especially with anticonvulsants, lithium, and cephalosporins), CNS infections (including Lyme disease and West Nile virus), strokes involving the bilateral thalami (usually from deep venous thrombosis), inflammatory disorders (such as Hashimoto's encephalopathy [HE]), and neurodegenerative diseases. It is important to remember that a single EEG does not exclude the possibility of an episodic ictal disorder and longer‐term monitoring would be required to definitively exclude seizures.
At this point, although the myoclonus and triphasic waves most commonly would indicate a toxic/metabolic process, the elevated ESR and CSF pleocytosis argue for an inflammatory or infectious condition. An MRI remains the next most useful test to guide further workup because many such conditions have distinct signatures on MRI.
The following day, she was noted to have periods of alertnessopening her eyes and following some commandsbut at other times she was difficult to arouse or obtunded. Tremulous movements and sporadic myoclonic jerks continued but no focal neurologic signs were found. Although there was increased muscle tone throughout, she was intermittently seen moving her limbs spontaneously, but not to command. No new findings were appreciated on routine laboratory tests. Antinuclear antibody testing was negative. Serum levetiracetam level was 23.5 g/mL (reference range, 545). Serum thyroid‐stimulating hormone was less than 0.005 U/mL, but free T3 was 3.5 pg/mL (normal, 1.8‐4.6) and free T4 was 2.0 ng/dL (normal, 0.71.8). An MRI of the brain was compromised by motion artifact but no significant abnormalities were appreciated.
At this point, a family member in another state disclosed that the patient had also been hospitalized 2 months previously while visiting him. Her chief complaint had been shortness of breath. The records were obtained; a cardiac catheterization had revealed nonobstructive coronary disease, and medical management was recommended. The notes mentioned that during the hospitalization she developed altered mental status with disorientation and shaking. CT and MRI of the brain had been unremarkable. The confusion was not explained, but she was discharged in good condition, alert and fully‐oriented.
The additional history confirms a relapsing encephalopathy, now with at least 3 occurrences. The most common etiologies in the face of a normal MRI and basic labs would be recurrent intoxication or exposures, but the inflammatory CSF profile and elevated ESR are not consistent with this. A variety of inflammatory disorders can present with recurrent encephalopathy, including demyelinating diseases and neurosarcoidosis. Some systemic rheumatologic conditions, such as systemic lupus erythematosus, can present with relapsing neurologic symptoms due to seizures, vasculitis, or cerebritis. Vasculitis would fit this picture as well, except for the normal findings on 2 MRIs. In a patient with such dramatic symptoms of neurologic dysfunction, one would expect to see changes on the MRI of cerebral inflammation with probable ischemia.
Therefore, given the CSF, ESR, clinical course, and unrevealing MRI and EEG, the most likely group of disorders responsible would be the nonvasculitic autoimmune meningoencephalitides, which present with recurrent encephalopathy and feature spontaneous remissions and/or often‐dramatic responses to corticosteroids. Key disorders in this category include Sjogren's disease, lupus, and steroid responsive encephalopathy associated with autoimmune thyroiditis (Hashimoto's encephalopathy). The latter condition is the most common of the group and is suggested by the abnormal thyroid‐stimulating hormone testing, although it may occur in the setting of normal thyroid function. The diagnosis can be confirmed with thyroperoxidase and thyroglobulin antibody testing.
Three days into the hospitalization, her mental status had gradually improved such that she was more consistently awake and oriented to person and place, and she was transferred to a regular nursing unit. Final results from the CSF and blood cultures were negative, as was PCR for HSV. The antimicrobials were discontinued. Routine serum chemistries continued to be unremarkable. Additional studies recommended by the neurologist demonstrated an antithyroperoxidase antibody concentration of 587.1 IU/mL (normal, 5), and antithyroglobulin antibody level of 52.2 IU/mL (normal, 10).
These results confirm the diagnosis of HE which, in addition to its presentation as a recurrent illness, is an important treatable cause of dementia and should be considered in young patients, those with autoimmune and thyroid disorders, and those whose dementia is rapidly progressive. Most cases are thought to be steroid‐responsive, but some studies have defined the disorder based on this responsiveness, resulting in some nonresponders likely being overlooked.
A trial of corticosteroids should be considered if the patient does not quickly return to baseline given the potential morbidities associated with prolonged altered mental status to this degree. Whether initiation of chronic immunosuppression could prevent these attacks in the future is unclear from the literature but should be considered given the recurrent, dramatic presentation in this patient.
A diagnosis of HE was made, and she was prescribed corticosteroids. Twenty‐four hours later, she was alert and fully‐oriented. She was discharged to home on prednisone and seen in follow‐up in neurology clinic 1 month later. She had had no further episodes of confusion or stupor, but because of steroid‐induced hyperglycemia, her corticosteroids were decreased and mycophenolate mofetil added for chronic immunosuppression. Four months after discharge she was neurologically stable but continued to struggle with the adverse effects of chronic corticosteroid treatment.
COMMENTARY
HE is an uncommon condition that can present with a rapidly progressive decline and should be considered in patients who present with recurrent mental status change in the setting of normal imaging studies and routine laboratory results. The entity was initially described by Lord William Russell Brain in 1966, and in the most recent terminology is known as steroid‐responsive encephalopathy associated with autoimmune thyroiditis (SREAT).1 It is characterized by an acute or subacute encephalopathy associated with thyroid autoimmunity. Patients typically present with fluctuating symptoms, episodes of confusion, alterations of consciousness, and rapid cognitive decline.2 Common features include myoclonus, tremor, ataxia, speech disturbance, stroke‐like episodes, increased muscle tone, neuropsychiatric manifestations, and seizures, that in some cases may progress to status epilepticus.3, 4
Although serum antithyroglobulin and antithyroperoxidase antibodies are elevated in HE, their presence is thought to be an epiphenomenon of the condition rather than the direct cause. Supporting this are the facts that the incidence of encephalopathy is not increased in patients with established autoimmune thyroiditis, and the presence of antithyroid antibodies ranges from 5% to 20% in the general population.2, 5 There is also no evidence that thyroid antibodies directly react with brain tissue, and the levels of these antibodies do not correlate with either neurologic manifestations or clinical improvement.2, 4, 5 As HE has been reported in patients with euthyroidism, hypothyroidism, and hyperthyroidism (with hypothyroidismeither subclinical or activemost common), it is also unlikely that the level of thyroid hormones play a role in the etiology of this disease.2, 4, 6
The etiology and pathogenesis of HE are unclear, although an immune‐mediated process is generally implicated, either from an inflammatory vasculitis or as a form of acute disseminated encephalomyelitis.7‐9 Global hypoperfusion on single‐photon emission computed tomography (SPECT) studies has also been reported.10, 11 Patients with HE may have nonspecific evidence of inflammation, including an elevated ESR, CRP, and CSF protein.12 Other laboratory abnormalities may include a mild elevation of liver aminotransferase levels; renal impairment has also been reported in a few cases of HE in the form of glomerulonephritis, and may be related to deposition of immune complexes containing thyroglobulin antigen.6, 12‐14 MRI of the brain is normal or nonspecific in most cases, and the EEG most commonly shows diffuse slowing.
The differential for a rapidly progressive cognitive decline includes CJD, CNS vasculitis, paraneoplastic syndromes, and autoimmune and subacute infectious encephalopathies. In patients with CJD, T2‐weighted imaging may show hyperintense signals in the basal ganglia, while diffusion‐weighted sequences may reveal changes in the cortical ribbon and bilateral thalami.15 In CNS vasculitis, the imaging findings are variable and range from discrete areas of vascular infarcts to hemorrhagic lesions.16 In paraneoplastic and autoimmune encephalopathies (excluding HE), MRI often shows nonenhancing signal intensity changes in the mesial temporal lobes.12 This patient had repeatedly normal MRI studies of the brain, which in combination with the history of tremor, myoclonus, seizures, and interval return to baseline status, helped point to the diagnosis of HE.
Different approaches to treatment of HE have been recommended. As the acronym SREAT suggests, patients typically respond dramatically to high‐dose steroid therapy. Although a number of patients also improve spontaneously, up to 60% of patients experience a relapsing course and require chronic immunosuppressive agents for maintenance therapy, including long‐term steroids and azathioprine.2, 17 Treatment with plasma exchange and intravenous immune globulin have also been reported, but with mixed results.18, 19 Due to her history of multiple relapses, the patient was placed on mycophenolate mofetil for additional maintenance immunosuppression, as her corticosteroid dose was reduced due to adverse effects.
Acute mental status change is a potentially emergent situation that must be evaluated with careful history and studies to exclude life‐threatening metabolic, infectious, and vascular conditions. This patient presented similarly on 2 prior occasions, and each time her physician team evaluated what appeared to be a new onset of altered consciousness, reaching a plausible but ultimately incorrect diagnosis. The patient's third presentation was finally the charm, as her physicians learned of the repeated history of a confusional state, and in particular the return to baseline status, allowing them to create a differential that focused on etiologies of relapsing encephalopathy and make the correct diagnosis.
Key Points
-
Recurrent acute or subacute cognitive deterioration invokes a differential diagnosis of toxic/metabolic disorders and unusual inflammatory conditions.
-
The nonvasculitic autoimmune encephalopathies are a group of uncommon conditions characterized by nonspecific findings of inflammation and generally unremarkable CNS imaging studies.
-
HE, or SREAT, is the most common of these conditions, and is notable for mental status changes, various findings of increased muscular tone, thyroid autoimmunity, and generally a dramatic response to corticosteroids.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
- , , .Hashimoto's disease and encephalopathy.Lancet.1966;2:512–514.
- , , .Hashimoto encephalopathy: syndrome or myth?Arch Neurol.2003;60:164–171.
- , , .Pisani F. Recurrent status epilepticus as the main feature of Hashimoto's encephalopathy.Epilepsy Behav.2006;8:328–330.
- , , , et al.Steroid‐responsive encephalopathy associated with autoimmune thyroiditis.Arch Neurol.2006;63:197–202.
- , , , , .Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment.J Neurol.1996;243:585–593.
- , , , , .Hashimoto's encephalopathy: a steroid‐responsive disorder associated with high anti‐thyroid antibody titers‐report of 5 cases.Neurology.1991;41:228–233.
- , , , , .Hashimoto encephalopathy: a brainstem vasculitis?Neurology.2000;54:769–770.
- , , , , .Nonvasculitic autoimmune inflammatory meningoencephalitis (NAIM): A reversible form of encephalopathy.Neurology.1999;53:1579–1581.
- , , , .Hashimoto's encephalopathy: postmortem findings after fatal status epilepticus.Neurology.2003;61:1124–1126.
- , , .Autoimmune thyroiditis and a rapidly progressive dementia: global hypoperfusion on SPECT scanning suggests a possible mechanism.Neurology.1997;49:623–626.
- , , , , .Hashimoto's encephalopathy: clinical, SPECT and neurophysiologic data.QJM.2003;96:455–457.
- , , .Autoimmune Encephalopathies.The Neurologist.2007;13:140–147.
- , , , , , .Thyroid antigen‐antibody nephritis.Clin Immunol Immunopathol1976;6:341–346.
- , , .Immune complex glomerulonephritis mediated by thyroid antigens.Arch Pathol Lab Med1978;102:530–533.
- , , , et al.Diffusion‐weighted MR imaging of early‐stage Creutzfeldt‐Jakob disease: typical and atypical manifestations.Radiographics.2006;26:S191–S204.
- , , , , .CNS vasculitis in autoimmune disease: MR imaging findings and correlation with angiography.AJNR Am J Neuroradiol.1999;20:75–85.
- , .Long‐Term Treatment of Hashimoto's Encephalopathy.J Neuropsychiatry Clin Neurosci.2006;18:14–20.
- , .Hashimoto's encephalopathy: steroid resistance and response to intravenouc immunoglobulins.J Neurol Neurosurg Psychiatry.2005;76:455–456.
- , .Hashimoto's encephalopathy responding to plasmapheresis.J Neurol Neurosurg Psychiatry.2001;70:132.
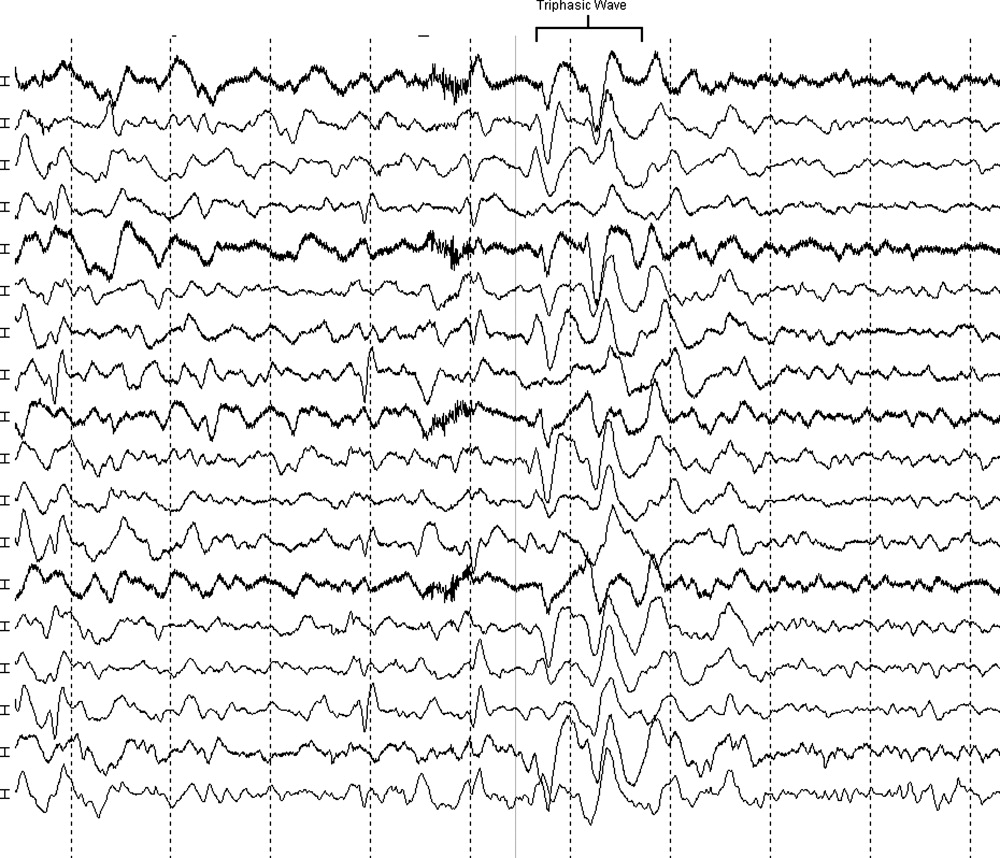
Small fiber neuropathy: A burning problem
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
In many of these patients, the findings on neurologic examination, nerve conduction studies, and electromyography are normal, although some may show signs of mild distal sensory loss on physical examination. The lack of objective findings on routine nerve conduction studies and electromyography may lead many physicians to attribute the symptoms to other disorders such as plantar fasciitis, vascular insufficiency, or degenerative lumbosacral spine disease.
The past 2 decades have seen the development of specialized tests that have greatly facilitated the diagnosis of small fiber neuropathy; these include skin biopsy to evaluate the density of nerve fibers in the epidermis and studies of autonomic nerve function. Common etiologies have been identified for small fiber neuropathy and can be specifically treated, which is critical for controlling progression of the disease. Pain management is becoming easier with more available options but is still quite challenging.
WHAT IS SMALL FIBER NEUROPATHY?
Peripheral nerve fibers can be classified according to size, which correlates with the degree of myelination.
- Large nerve fibers are heavily myelinated and include A-alpha fibers, which mediate motor strength, and A-beta fibers, which mediate vibratory and touch sensation.
- Medium-sized fibers, known as A-gamma fibers, are also myelinated and carry information to muscle spindles.
- Small fibers include myelinated A-delta fibers and unmyelinated C fibers, which innervate skin (somatic fibers) and involuntary muscles, including cardiac and smooth muscles (autonomic fibers). Together, they mediate pain, thermal sensation, and autonomic function.
Small fiber neuropathy results from selective impairment of small myelinated A-delta and unmyelinated C fibers.
Sensory symptoms: Pain, burning, tingling, numbness
Damage to or loss of small somatic nerve fibers results in pain, burning, tingling, or numbness that typically affects the limbs in a distal-to-proximal gradient. In rare cases, small fiber neuropathy follows a non-length-dependent distribution in which symptoms may be manifested predominantly in the arms, face, or trunk.
Symptoms may be mild initially, with some patients complaining of vague discomfort in one or both feet similar to the sensation of a sock gathering at the end of a shoe. Others report a wooden quality in their feet, numbness in their toes, or a feeling as if they are walking on pebbles, sand, or golf balls. The most bothersome and fairly typical symptom is burning pain in the feet that extends proximally in a stocking-glove distribution and is often accompanied by stabbing or aching pains, electric shock-like or pins-and-needles sensations, or cramping of the feet and calves.
Symptoms are usually worse at night and often affect sleep. Some patients say that their feet have become so exquisitely tender that they cannot bear having the bed sheets touch them, and so they sleep with their feet uncovered. A small number of patients do not have pain but report a feeling of tightness and swelling in their feet (even though the feet appear normal).
Examination often reveals allodynia (perception of nonpainful stimuli as being painful), hyperalgesia (perception of painful stimuli as being more painful than expected), or reduced pinprick and thermal sensation in the affected area. Vibratory sensation can be mildly reduced at the toes. Motor strength, tendon reflexes, and proprioception, however, are preserved because they are functions of large nerve fibers.
Autonomic symptoms
When autonomic fibers are affected, patients may experience dry eyes, dry mouth, orthostatic dizziness, constipation, bladder incontinence, sexual dysfunction, trouble sweating, or red or white skin discoloration.2 Examination may show orthostatic hypotension and skin changes. The skin over the affected area may appear atrophic, dry, shiny, discolored, or mildly edematous as the result of sudomotor and vasomotor abnormalities.
WHAT CAUSES SMALL FIBER NEUROPATHY?
Small fiber neuropathy has been associated with many medical conditions, including glucose dysmetabolism,3 connective tissue disease,4,5 dysthyroidism,6 vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus (HIV) infection,7 hepatitis C virus infection, celiac disease,8 restless legs syndrome,9 neurotoxic drug exposure, hereditary diseases, and paraneoplastic syndrome. While most of these conditions cause a length-dependent small fiber neuropathy, others (Sjögren disease, celiac disease, and paraneoplastic syndrome) can cause a form of small fiber neuropathy that is not length-dependent.4,8,10
Diabetes and prediabetes
Glucose dysmetabolism, including diabetes and prediabetes with impaired oral glucose tolerance (a glucose level 140–199 mg/dL 2 hours after a 75-g oral dextrose load), is the most common identifiable associated condition, present in about one-third of patients with painful sensory neuropathy11 and in nearly half of those with otherwise idiopathic small fiber neuropathy.12–14
Research findings strongly suggest that even prediabetes is a risk factor for small fiber neuropathy, and that so-called “impaired glucose tolerance neuropathy” may represent the earliest stage of diabetic neuropathy. Several recent studies have found a high prevalence of impaired glucose tolerance in patients with sensory peripheral neuropathy,12–14 with a rate of up to 42% in cases initially thought to be idiopathic14 compared with 14% in the general population.15 Also, a dose-response relationship between the severity of hyperglycemia and the degree of neuropathy was demonstrated in one study, in which patients with impaired glucose tolerance more often had small fiber neuropathy, whereas those with diabetes more often had polyneuropathy involving both small and large fibers.14 And studies in animals and cell cultures have shown that intermittent hyperglycemia, which can be seen in patients with impaired glucose tolerance, caused sensory neuron and nerve fiber damage and increased spontaneous C-fiber firing, resulting in neuropathic pain.8,16,17
Metabolic syndrome
Insulin resistance with prediabetes and diabetes is a part of the metabolic syndrome, which also consists of hypertension, hyperlipidemia, and obesity. The individual components of the metabolic syndrome have been implicated as risk factors not only for cardiovascular and cerebrovascular disease but also for small fiber neuropathy.
One study in 548 patients with type 2 diabetes showed that those with the metabolic syndrome were twice as likely to have neuropathy as those without.18 Another study showed that in 1,200 patients with type 1 diabetes without neuropathy at baseline, hypertension, hyperlipidemia, and increased body mass index were each independently associated with a higher risk of developing neuropathy.19
A recent study of 219 patients with idiopathic distal symmetrical peripheral neuropathy and 175 diabetic patients without neuropathy found a higher prevalence of metabolic syndrome in patients with neuropathy than in normal populations. The prevalence of dyslipidemia (high levels of total and low-density lipoprotein cholesterol and triglycerides and low levels of high-density lipoprotein cholesterol), but not hypertension or obesity, was higher in patients with neuropathy than in patients with diabetes but no neuropathy.20 The findings linked dyslipidemia to neuropathy and showed the need for further studies of the potential pathogenic role of dyslipidemia in neuropathy.
Hereditary causes
Hereditary causes of small fiber neuropathy are rare and include Fabry disease, Tangier disease, hereditary sensory autonomic neuropathy, and hereditary amyloidosis.
HOW DO YOU EVALUATE PATIENTS WITH SUSPECTED SMALL FIBER NEUROPATHY?
A thorough history should be taken to obtain details regarding onset and features of neuropathy symptoms, exacerbating factors, and progression. It is also important to ascertain whether the patient has any associated conditions as mentioned above, a family history of neuropathy, risk factors for HIV or hepatitis C virus infection, or a history of neurotoxic drug exposure.
Clinical suspicion of small fiber neuropathy should be high if a patient presents with predominant small fiber symptoms and signs with preserved large fiber functions.
Nerve conduction studies and electromyography
For diagnostic testing, routine nerve conduction studies and electromyography assess the function of large nerve fibers only and are thus normal in small fiber neuropathy. These tests should still be ordered to rule out subclinical involvement of large fibers, which may affect the diagnostic evaluation, prognosis, and treatment plan. However, if the results of these tests are normal, specialized studies are needed to evaluate small fibers.
Although several tests are available to evaluate somatic and autonomic small fibers, the two that have the highest diagnostic efficiency for small fiber neuropathy and that are used most often are skin biopsy, to evaluate intraepidermal nerve fiber density, and quantitative sudomotor axon reflex testing (QSART), to assess sudomotor autonomic function.21–23
Skin biopsy
Skin biopsy is a minimally invasive procedure in which 3-mm-diameter punch biopsy specimens are taken from the distal leg, distal thigh, and proximal thigh of one lower limb. The procedure takes only 10 to 15 minutes.
Biopsy specimens are immunostained using an antibody against protein gene product 9.5, which is a panaxonal marker. Small nerve fibers in the epidermis are counted under a microscope, and intraepithelial nerve fiber densities are calculated and compared with established normative values. The diagnosis of small fiber neuropathy can be established if the intraepidermal nerve fiber density is lower than normal (Figure 1). Nerve fiber density may be normal in the early stage of small fiber neuropathy, but in this setting skin biopsy often shows abnormal morphologic changes in the small fibers, especially large swellings,24 and repeat biopsy in 6 to 12 months may be considered.
The diagnostic efficiency of skin biopsy is about 88%.21,23 For diagnosing small fiber neuropathy, it is more sensitive than quantitative sensory testing21,25 and more sensitive and less invasive than sural nerve biopsy.26 Intraepidermal nerve fiber density also correlates well with a variety of measures of severity of HIV distal sensory neuropathy and thus may be used to measure the severity and treatment response of small fiber neuropathy.27
Quantitative sudomotor axon reflex testing
QSART is an autonomic study that measures sweat output in response to acetylcholine, which reflects the function of postganglionic sympathetic unmyelinated sudomotor nerve fibers. Electrodes are placed on the arms and legs to record the volume of sweat produced by acetylcholine iontophoresis, in which a mild electrical stimulation on the skin allows acetylcholine to stimulate the sweat glands. The output is compared with normative values.
One prospective study showed that 67 (72.8%) of 92 patients with painful feet had abnormal results on QSART, ie, low sweat output.28 A retrospective study found that 77 (62%) of 125 patients with clinical features of distal small fiber neuropathy had a length-dependent pattern of QSART abnormalities.22 QSART abnormalities were detected in some patients without autonomic symptoms.
If these tests are not available
Skin biopsy and QSART are objective, reproducible, sensitive, and complementary in diagnosing small fiber neuropathy. One or both can be ordered, depending on whether the patient has somatic symptoms, autonomic symptoms, or both. However, these two tests are not widely available. Only a few laboratories in the country can process skin biopsy specimens to evaluate intraepidermal nerve fiber density. Nevertheless, it is easy to learn the skin punch biopsy procedure, and primary care physicians and neurologists can perform it after appropriate training. (A concern is avoiding damage to the epidermis.) They can then send specimens to one of the cutaneous nerve laboratories (but not to a routine reference laboratory).
A special technique, including unique fixative and cryoprotectant, is used to fix and process the biopsy specimens, because routine techniques for processing dermatologic punch biopsy specimens often result in lower intraepidermal nerve fiber densities. Therefore, it is very important to contact the laboratory regarding fixative and processing before performing a biopsy.
QSART requires specialized equipment and must be performed on site. In addition, the test is very sensitive to drugs that can affect sweating, such as antihistamines and antidepressants, and such drugs must be discontinued 48 hours before the study.
Basic laboratory tests to find the cause
Once the diagnosis of small fiber neuropathy is established, the next important step is to order a battery of laboratory tests to search for an underlying cause. The tests should include the following:
- Complete blood cell count
- Comprehensive metabolic panel
- Lipid panel
- Erythrocyte sedimentation rate
- Thyroid-stimulating hormone level
- Free thyroxine (T4) level
- Antinuclear antibody
- Extractable nuclear antigens
- Angiotensin-converting enzyme (ACE) level
- Serum and urine immunofixation tests
- Vitamin B12 level
- 2-hour oral glucose tolerance test.
Oral glucose tolerance testing is much more sensitive than measuring the hemoglobin A1c and fasting glucose levels in detecting diabetes and prediabetes. These two conditions were detected by oral glucose tolerance testing in more than 50% of patients with otherwise idiopathic sensory-predominant peripheral neuropathy and normal hemoglobin A1c and fasting glucose levels.13,14 Therefore, every patient with small fiber neuropathy without a known history of diabetes or prediabetes should have an oral glucose tolerance test.
Special laboratory tests in special cases
- If there is a history of gastrointestinal symptoms or herpetiform-like rash, then testing for gliadin antibody and tissue transglutaminase antibodies as well as small-bowel biopsy may be pursued to evaluate for celiac sprue.
- Serologic tests for HIV or hepatitis C should be ordered if the patient has risk factors.
- If there is a significant family history, further genetic testing should be considered.
- Lip biopsy or bone marrow biopsy should be considered if clinical suspicion is high for Sjögren disease, seronegative sicca syndrome, or amyloidosis.
- The serum ACE level has a low sensitivity and specificity; therefore, if sarcoid is suspected clinically, additional confirmatory testing, such as computed tomography of the chest, should be ordered despite a normal ACE value.
HOW DO YOU TREAT SMALL FIBER NEUROPATHY?
Treatment of small fiber neuropathy should target the underlying cause and neuropathic pain. Cause-specific treatment is a key in preventing small fiber neuropathy or slowing its progression.
Glucose control, weight control, and regular exercise
As glucose dysmetabolism is the condition most often associated with small fiber neuropathy (and since individual components of the metabolic syndrome are potential risk factors for it), tight glycemic control and lifestyle modification with diet control, weight control, and regular exercise are of paramount importance in patients with these conditions.
The Diabetic Prevention Program,29 a study in 3,234 people with prediabetes, found that diet and exercise were more effective than metformin (Glucophage) in preventing full-blown diabetes. At an average of 2.8 years of follow-up, the incidence of diabetes was 11.0 cases per 100 patient-years in a group assigned to receive placebo, compared with 7.8 in those assigned to receive metformin (31% lower), and 4.8 (58% lower) in those who were assigned to undergo a lifestyle intervention that included at least 150 minutes of physical activity per week with a weight-loss goal of 7%. Put another way, to prevent one case of diabetes over 3 years, 6.9 patients would have to undergo the lifestyle intervention program, or 13.9 would have to receive metformin. Since impaired glucose tolerance neuropathy may represent the earliest stage of diabetic neuropathy, the neuropathy at this stage may be reversible with lifestyle intervention and improvement of impaired glucose tolerance.
This concept is supported by a 3-year study in 31 people, which showed that lifestyle intervention significantly improved impaired glucose tolerance, reduced the body mass index, and lowered total serum cholesterol levels.30 Changes in these metabolic variables were accompanied by significant improvement of neuropathy as evidenced by significantly increased intraepidermal nerve fiber density, increased foot sweat volume, and decreased neuropathic pain.30
Treatment of other diseases
It has also been reported that treatment of sarcoidosis, autoimmune diseases, and celiac disease improved the symptoms of small fiber neuropathy resulting from these conditions.8,31 Therefore, it is important to identify the cause and treat it to prevent and slow the progression of small fiber neuropathy, and doing so may improve the disease in some mild cases.
Pain management
First-line choices of pain medications are the anticonvulsants gabapentin (Neurontin) and pregabalin (Lyrica), the tricyclic antidepressants amitriptyline (Elavil) and nortriptyline (Aventyl), a 5% lidocaine patch (Lidoderm), and the semisynthetic opioid analgesic tramadol (Ultram). These can be used alone or in combination.
Gabapentin is relatively well tolerated, but drowsiness can occur, especially with high starting doses. We usually start with 300 mg daily and increase it by 300 mg every week up to 1,200 mg three times a day as tolerated. Most patients need 600 to 900 mg three times a day.
Pregabalin is a newer antiepileptic drug, similar to gabapentin but less sedating. It can be started at 75 mg twice a day and gradually increased to 300 mg twice a day as needed. Weight gain and, rarely, swelling of the lower extremities may limit the use of both of these drugs.
Tricyclic antidepressants, such as amitriptyline, nortriptyline, and desipramine (Norpramin), are proven effective in controlling neuropathic pain, although no response with amitriptyline was seen in patients with painful HIV distal sensory neuropathy.34
Lidocaine patch is preferred if the painful area is small. Patients should be instructed to use the patch to cover the painful area 12 hours on and 12 hours off. If it does not provide relief within 1 week, it should be discontinued.
Tramadol is also helpful in treating neuropathic pain. It can be started at 50 mg two to four times a day as needed.
Nonsteroidal anti-inflammatory drugs and selective serotonin reuptake inhibitors are typically less effective than the other drugs mentioned.
Opioids should be reserved for refractory cases, given the potential for addiction, but they are sometimes necessary in patients with disabling pain that does not respond to other drugs.
TENS may be of benefit. The patient controls a pocket-size device that sends electrical signals to leads placed on affected areas.
Alternative therapies for small fiber neuropathy, such as meditation, yoga, and acupuncture, have yet to be studied.
It is also important to explain to patients that the typical course of small fiber neuropathy is relatively benign, as many patients worry about developing weakness and eventually not being able to walk. These concerns and fears can aggravate pain and depression, which can make treatment difficult.
WHAT IS THE PROGNOSIS OF SMALL FIBER NEUROPATHY?
Most patients with small fiber neuropathy experience a slowly progressive course, with symptoms and signs spreading proximally over time.
In one study, only 13% of 124 patients with small fiber neuropathy showed evidence of large-fiber involvement over a 2-year period. 21 None went on to develop Charcot joints, foot ulcers, weakness, or sensory ataxia, as is often seen in patients with long-standing or severe large fiber neuropathy. Neuropathic pain worsened in 30% and resolved spontaneously in 11%.21
Most patients with small fiber neuropathy require chronic pain management. Again, treatment of the underlying cause is important and can improve the prognosis.
We believe that the overall progression of small fiber neuropathy is slow. A longitudinal study with a follow-up longer than 2 years would be useful to confirm this.
TAKE-HOME POINTS
As the population continues to age and as more patients develop diabetes and the metabolic syndrome, the prevalence of small fiber neuropathy will rise. Patients who present to their primary care physicians with painful, burning feet require a thorough diagnostic evaluation, which may include referral for specialized neurodiagnostic testing. Aggressive cause-specific treatment, lifestyle modification, and pain control are key elements of a team approach to managing small fiber neuropathy.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
- Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract 2007; 77:485–488.
- Lacomis D. Small fiber neuropathy. Muscle Nerve 2002; 26:173–188.
- Smith AG, Singleton JR. Impaired glucose tolerance and neuropathy. Neurologist 2008; 14:23–29.
- Chai J, Herrmann DN, Stanton M, Barbano RL, Logigian EL. Painful small-fiber neuropathy in Sjogren syndrome. Neurology 2005; 65:925–927.
- Goransson LG, Tjensvoll AB, Herigstad A, Mellgren SI, Omdal R. Small-diameter nerve fiber neuropathy in systemic lupus erythematosus. Arch Neurol 2006; 63:401–404.
- Orstavik K, Norheim I, Jorum E. Pain and small-fiber neuropathy in patients with hypothyroidism. Neurology 2006; 67:786–791.
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol 2005; 4:543–555.
- Brannagan TH, Hays AP, Chin SS, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol 2005; 62:1574–1578.
- Polydefkis M, Allen RP, Hauer P, Earley CJ, Griffin JW, McArthur JC. Subclinical sensory neuropathy in late-onset restless legs syndrome. Neurology 2000; 55:1115–1121.
- Gorson KC, Herrmann DN, Thiagarajan R, et al. Non-length dependent small fibre neuropathy/ganglionopathy. J Neurol Neurosurg Psychiatry 2008; 79:163–169.
- Singleton JR, Smith AG, Bromberg MB. Increased prevalence of impaired glucose tolerance in patients with painful sensory neuropathy. Diabetes Care 2001; 24:1448–1453.
- Novella SP, Inzucchi SE, Goldstein JM. The frequency of undiagnosed diabetes and impaired glucose tolerance in patients with idiopathic sensory neuropathy. Muscle Nerve 2001; 24:1229–1231.
- Smith AG, Singleton JR. The diagnostic yield of a standardized approach to idiopathic sensory-predominant neuropathy. Arch Intern Med 2004; 164:1021–1025.
- Sumner CJ, Sheth S, Griffin JW, Cornblath DR, Polydefkis M. The spectrum of neuropathy in diabetes and impaired glucose tolerance. Neurology 2003; 60:108–111.
- Gregg EW, Sorlie P, Paulose-Ram R, et al. Prevalence of lower-extremity disease in the US adult population >=40 years of age with and without diabetes: 1999–2000 National Health and Nutrition Examination Survey. Diabetes Care 2004; 27:1591–1597.
- Boulton A. What causes neuropathic pain? J Diabetes Complications 1992; 6:58–63.
- Russell JW, Sullivan KA, Windebank AJ, Herrmann DN, Feldman EL. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol Dis 1999; 6:347–363.
- Costa LA, Canani LH, Lisboa HR, Tres GS, Gross JL. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabet Med 2004; 21:252–255.
- Tesfaye S, Chaturvedi N, Eaton SE, et al. Vascular risk factors and diabetic neuropathy. N Engl J Med 2005; 352:341–350.
- Smith A, Rose K, Singleton J. Idiopathic neuropathy patients are at high risk for metabolic syndrome. J Neurol Sci 2008; 273:25–28.
- Devigili G, Tugnoli V, Penza P, et al. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain 2008; 131:1912– 1925.
- Low VA, Sandroni P, Fealey RD, Low PA. Detection of small-fiber neuropathy by sudomotor testing. Muscle Nerve 2006; 34:57–61.
- McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998; 55:1513–1520.
- Gibbons CH, Griffin JW, Polydefkis M, et al. The utility of skin biopsy for prediction of progression in suspected small fiber neuropathy. Neurology 2006; 66:256–258.
- Polydefkis M, Yiannoutsos CT, Cohen BA, et al. Reduced intraepidermal nerve fiber density in HIV-associated sensory neuropathy. Neurology 2002; 58:115–119.
- Herrmann DN, Griffin JW, Hauer P, Cornblath DR, McArthur JC. Epidermal nerve fiber density and sural nerve morphometry in peripheral neuropathies. Neurology 1999; 53:1634–1640.
- Zhou L, Kitch DW, Evans SR, et al. Correlates of epidermal nerve fiber densities in HIV-associated distal sensory polyneuropathy. Neurology 2007; 68:2113–2119.
- Novak V, Freimer ML, Kissel JT, et al. Autonomic impairment in painful neuropathy. Neurology 2001; 56:861–868.
- Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002; 346:393–403.
- Smith AG, Russell J, Feldman EL, et al. Lifestyle intervention for prediabetic neuropathy. Diabetes Care 2006; 29:1294–1299.
- Hoitsma E, Faber CG, van Santen-Hoeufft M, De Vries J, Reulen JP, Drent M. Improvement of small fiber neuropathy in a sarcoidosis patient after treatment with infliximab. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23:73–77.
- Chen H, Lamer TJ, Rho RH, et al. Contemporary management of neuropathic pain for the primary care physician. Mayo Clin Proc 2004; 79:1533–1545.
- Galluzzi KE. Managing neuropathic pain. J Am Osteopath Assoc 2007; 107( suppl 6):ES39–ES48.
- Kieburtz K, Simpson D, Yiannoutsos C, et al. A randomized trial of amitriptyline and mexiletine for painful neuropathy in HIV infection. AIDS Clinical Trial Group 242 Protocol Team. Neurology 1998; 51:1682–1688.
KEY POINTS
- Symptoms of small fiber neuropathy typically start with burning feet and numb toes.
- Causes and associated conditions can be found in over 50% of cases. These include glucose dysmetabolism, connective tissue diseases, sarcoidosis, dysthyroidism, vitamin B12 deficiency, paraproteinemia, human immunodeficiency virus infection, celiac disease, neurotoxic drug exposure, and paraneoplastic syndrome.
- Findings on routine nerve conduction studies and electromyography are typically normal in this disease.
- Management includes aggressively identifying and treating the underlying cause, advising lifestyle modifications, and alleviating pain.