User login
Prophylaxis and Treatment of Venous Thromboembolism in Cancer Patients
The Case
A 62-year-old woman with a past medical history significant for metastatic adenocarcinoma of the lung presents to the ED with complaints of fever and shortness of breath. She has recently completed her first cycle of carboplatin, pemetrexed, and bevacizumab. Upon admission, she is found to have an absolute neutrophil count of 800 and a platelet count of 48,000. She is admitted for neutropenic fever and placed on IV antimicrobials. Sequential compression devices are initiated for DVT prophylaxis.
Key Clinical Questions
What risk do cancer patients have for VTE?
Patients with cancer have a risk of clinically significant VTE that is four to seven times that of patients without malignancy.1 This is due to a number of reasons:
- Tumor cells produce procoagulant activity inducing thrombin formation;2
- The cancer itself can compress or invade deep veins; and3
- Some cancer therapies such L-asparaginase and thalidomide/lenalidomide, plus high-dose steroids, or anti-estrogen medications such as tamoxifen can also increase patients’ risk of VTE.3,4,5
What inpatients with cancer need VTE prophylaxis?
Much like other hospitalized medical patients, patients with cancer who have reduced mobility and are not on therapeutic anticoagulation should receive pharmacologic prophylaxis unless there is a contraindication.3,6,7,8 Cancer patients with acute medical illnesses should also likely receive prophylaxis if there are no contraindications, because the vast majority of these have factors increasing their VTE risk, including infection, kidney disease, or pulmonary disease.3,6,7,8 Patients undergoing major cancer surgery should also receive pharmacologic prophylaxis prior to surgery and for at least seven to 10 days post-operatively.3,6,7,8
For ambulatory cancer patients who are admitted for short courses of chemotherapy or for minor procedures, however, there is not enough evidence to recommend routine VTE prophylaxis.6,7 An exception to this is patients with multiple myeloma receiving thalidomide-based or lenalidomide-based chemotherapy, who should receive pharmacologic prophylaxis.6,7
What are the options available for VTE in hospitalized cancer patients?
The guidelines for VTE prophylaxis in hospitalized cancer patients recommend either unfractionated heparin (UFH) or low molecular weight heparin (LMWH) for prophylaxis when no contraindications exist.5 The only two LMWH that have been FDA approved for prophylaxis are enoxaparin and dalteparin. When deciding between UFH and LMWH, no evidence shows that one is better than the other in preventing VTE in hospitalized cancer patients.9 There is evidence that the use of LMWH results in a lower incidence of major hemorrhage when compared to UFH.10
What are the contraindications to pharmacologic VTE prophylaxis in cancer patients?
Contraindications for pharmacologic VTE prophylaxis in cancer patients include active major bleeding, thrombocytopenia (platelet count <50,000/µL), severe coagulopathy, inherited bleeding disorder, and at the time of surgery or invasive procedures (including lumbar puncture and epidural or spinal anesthesia).3,6,7 Those with contraindications to pharmacologic VTE prophylaxis should have mechanical prophylaxis instead.
What is the recommended treatment of VTE in cancer patients?
After the diagnosis of pulmonary embolism (PE) or DVT is found, LMWH is the preferred initial anticoagulant instead of UFH unless the patient has severe renal impairment (CrCl of less than 30 ml/min).6,7,8 LMWH is also preferred over warfarin for long-term anticoagulation during the initial six months of therapy.6,7,8 Following the initial six months, continued anticoagulation with either LMWH or warfarin could be considered in patients with active cancer, metastatic disease, or ongoing chemotherapy.6,7,8
When should IVC filters be considered in treating VTE in cancer patients?
IVC filter insertion should be reserved for those patients found to have a DVT or PE who have a contraindication to pharmacologic anticoagulation.3,6 It can be considered in patients who have recurrent VTE despite the appropriate use of optimally dosed LMWH therapy.6,8
What about the new oral anticoagulants?
At this point, because the majority of the major trials looking at the new oral anticoagulants (dabigatran, rivaroxaban, and apixaban) excluded cancer patients or included them only in small numbers, there is not enough evidence to support their use in cancer patients diagnosed with VTE.6,7,8
Back to the Case
On hospital day three, the patient is clinically improved. She is afebrile, her neutropenia has resolved, and her platelet count is up to 80,000. Her only complaint is pain and swelling of her left leg. A lower extremity Doppler is performed. She is found to have an acute left femoral DVT. The patient is then started on enoxaparin 1 mg/kg every 12 hours. Her left leg swelling and pain begin to improve, and she is discharged on enoxaparin and follows up with her oncologist in the next week. TH
Drs. Bell and O’Rourke are assistant professor of medicine in the division of hospital medicine at the University of California San Diego.
References
1. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712-1723.
2. Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715-722.
3. Streiff MB, Bockenstedt PL, Cataland SR, et al. Venous thromboembolic disease. J Natl Compr Canc Netw. 2013;11(11):1402-1429.
4. Payne JH, Vora AJ. Thrombosis and acute lymphoblastic leukaemia. Br J Haematol. 2007;138(4):430-445.
5. Amir E, Seruga B, Niraula S, Carlsson L, Ocaña A. Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst. 2011;103(17):1299-1309.
6. Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013;31(17):2189-2204.
7. Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: american society of clinical oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654-656.
8. Farge D, Debourdeau P, Beckers M, et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J Thromb Haemost. 2013;11(1):56-70.
9. Khorana AA. The NCCN clinical practice guidelines on venous thromboembolic disease: strategies for improving VTE prophylaxis in hospitalized cancer patients. Oncologist. 2007;12(11):1361-1370.
10. Mismetti P, Laporte-Simitisidis S, Tardy B, et al. Prevention of venous thromboembolism in internal medicine with unfractionated or low-molecular-weight heparins: a meta-analysis of randomized clinical trials. Thromb Haemost. 2000;83(1):14-19.
The Case
A 62-year-old woman with a past medical history significant for metastatic adenocarcinoma of the lung presents to the ED with complaints of fever and shortness of breath. She has recently completed her first cycle of carboplatin, pemetrexed, and bevacizumab. Upon admission, she is found to have an absolute neutrophil count of 800 and a platelet count of 48,000. She is admitted for neutropenic fever and placed on IV antimicrobials. Sequential compression devices are initiated for DVT prophylaxis.
Key Clinical Questions
What risk do cancer patients have for VTE?
Patients with cancer have a risk of clinically significant VTE that is four to seven times that of patients without malignancy.1 This is due to a number of reasons:
- Tumor cells produce procoagulant activity inducing thrombin formation;2
- The cancer itself can compress or invade deep veins; and3
- Some cancer therapies such L-asparaginase and thalidomide/lenalidomide, plus high-dose steroids, or anti-estrogen medications such as tamoxifen can also increase patients’ risk of VTE.3,4,5
What inpatients with cancer need VTE prophylaxis?
Much like other hospitalized medical patients, patients with cancer who have reduced mobility and are not on therapeutic anticoagulation should receive pharmacologic prophylaxis unless there is a contraindication.3,6,7,8 Cancer patients with acute medical illnesses should also likely receive prophylaxis if there are no contraindications, because the vast majority of these have factors increasing their VTE risk, including infection, kidney disease, or pulmonary disease.3,6,7,8 Patients undergoing major cancer surgery should also receive pharmacologic prophylaxis prior to surgery and for at least seven to 10 days post-operatively.3,6,7,8
For ambulatory cancer patients who are admitted for short courses of chemotherapy or for minor procedures, however, there is not enough evidence to recommend routine VTE prophylaxis.6,7 An exception to this is patients with multiple myeloma receiving thalidomide-based or lenalidomide-based chemotherapy, who should receive pharmacologic prophylaxis.6,7
What are the options available for VTE in hospitalized cancer patients?
The guidelines for VTE prophylaxis in hospitalized cancer patients recommend either unfractionated heparin (UFH) or low molecular weight heparin (LMWH) for prophylaxis when no contraindications exist.5 The only two LMWH that have been FDA approved for prophylaxis are enoxaparin and dalteparin. When deciding between UFH and LMWH, no evidence shows that one is better than the other in preventing VTE in hospitalized cancer patients.9 There is evidence that the use of LMWH results in a lower incidence of major hemorrhage when compared to UFH.10
What are the contraindications to pharmacologic VTE prophylaxis in cancer patients?
Contraindications for pharmacologic VTE prophylaxis in cancer patients include active major bleeding, thrombocytopenia (platelet count <50,000/µL), severe coagulopathy, inherited bleeding disorder, and at the time of surgery or invasive procedures (including lumbar puncture and epidural or spinal anesthesia).3,6,7 Those with contraindications to pharmacologic VTE prophylaxis should have mechanical prophylaxis instead.
What is the recommended treatment of VTE in cancer patients?
After the diagnosis of pulmonary embolism (PE) or DVT is found, LMWH is the preferred initial anticoagulant instead of UFH unless the patient has severe renal impairment (CrCl of less than 30 ml/min).6,7,8 LMWH is also preferred over warfarin for long-term anticoagulation during the initial six months of therapy.6,7,8 Following the initial six months, continued anticoagulation with either LMWH or warfarin could be considered in patients with active cancer, metastatic disease, or ongoing chemotherapy.6,7,8
When should IVC filters be considered in treating VTE in cancer patients?
IVC filter insertion should be reserved for those patients found to have a DVT or PE who have a contraindication to pharmacologic anticoagulation.3,6 It can be considered in patients who have recurrent VTE despite the appropriate use of optimally dosed LMWH therapy.6,8
What about the new oral anticoagulants?
At this point, because the majority of the major trials looking at the new oral anticoagulants (dabigatran, rivaroxaban, and apixaban) excluded cancer patients or included them only in small numbers, there is not enough evidence to support their use in cancer patients diagnosed with VTE.6,7,8
Back to the Case
On hospital day three, the patient is clinically improved. She is afebrile, her neutropenia has resolved, and her platelet count is up to 80,000. Her only complaint is pain and swelling of her left leg. A lower extremity Doppler is performed. She is found to have an acute left femoral DVT. The patient is then started on enoxaparin 1 mg/kg every 12 hours. Her left leg swelling and pain begin to improve, and she is discharged on enoxaparin and follows up with her oncologist in the next week. TH
Drs. Bell and O’Rourke are assistant professor of medicine in the division of hospital medicine at the University of California San Diego.
References
1. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712-1723.
2. Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715-722.
3. Streiff MB, Bockenstedt PL, Cataland SR, et al. Venous thromboembolic disease. J Natl Compr Canc Netw. 2013;11(11):1402-1429.
4. Payne JH, Vora AJ. Thrombosis and acute lymphoblastic leukaemia. Br J Haematol. 2007;138(4):430-445.
5. Amir E, Seruga B, Niraula S, Carlsson L, Ocaña A. Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst. 2011;103(17):1299-1309.
6. Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013;31(17):2189-2204.
7. Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: american society of clinical oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654-656.
8. Farge D, Debourdeau P, Beckers M, et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J Thromb Haemost. 2013;11(1):56-70.
9. Khorana AA. The NCCN clinical practice guidelines on venous thromboembolic disease: strategies for improving VTE prophylaxis in hospitalized cancer patients. Oncologist. 2007;12(11):1361-1370.
10. Mismetti P, Laporte-Simitisidis S, Tardy B, et al. Prevention of venous thromboembolism in internal medicine with unfractionated or low-molecular-weight heparins: a meta-analysis of randomized clinical trials. Thromb Haemost. 2000;83(1):14-19.
The Case
A 62-year-old woman with a past medical history significant for metastatic adenocarcinoma of the lung presents to the ED with complaints of fever and shortness of breath. She has recently completed her first cycle of carboplatin, pemetrexed, and bevacizumab. Upon admission, she is found to have an absolute neutrophil count of 800 and a platelet count of 48,000. She is admitted for neutropenic fever and placed on IV antimicrobials. Sequential compression devices are initiated for DVT prophylaxis.
Key Clinical Questions
What risk do cancer patients have for VTE?
Patients with cancer have a risk of clinically significant VTE that is four to seven times that of patients without malignancy.1 This is due to a number of reasons:
- Tumor cells produce procoagulant activity inducing thrombin formation;2
- The cancer itself can compress or invade deep veins; and3
- Some cancer therapies such L-asparaginase and thalidomide/lenalidomide, plus high-dose steroids, or anti-estrogen medications such as tamoxifen can also increase patients’ risk of VTE.3,4,5
What inpatients with cancer need VTE prophylaxis?
Much like other hospitalized medical patients, patients with cancer who have reduced mobility and are not on therapeutic anticoagulation should receive pharmacologic prophylaxis unless there is a contraindication.3,6,7,8 Cancer patients with acute medical illnesses should also likely receive prophylaxis if there are no contraindications, because the vast majority of these have factors increasing their VTE risk, including infection, kidney disease, or pulmonary disease.3,6,7,8 Patients undergoing major cancer surgery should also receive pharmacologic prophylaxis prior to surgery and for at least seven to 10 days post-operatively.3,6,7,8
For ambulatory cancer patients who are admitted for short courses of chemotherapy or for minor procedures, however, there is not enough evidence to recommend routine VTE prophylaxis.6,7 An exception to this is patients with multiple myeloma receiving thalidomide-based or lenalidomide-based chemotherapy, who should receive pharmacologic prophylaxis.6,7
What are the options available for VTE in hospitalized cancer patients?
The guidelines for VTE prophylaxis in hospitalized cancer patients recommend either unfractionated heparin (UFH) or low molecular weight heparin (LMWH) for prophylaxis when no contraindications exist.5 The only two LMWH that have been FDA approved for prophylaxis are enoxaparin and dalteparin. When deciding between UFH and LMWH, no evidence shows that one is better than the other in preventing VTE in hospitalized cancer patients.9 There is evidence that the use of LMWH results in a lower incidence of major hemorrhage when compared to UFH.10
What are the contraindications to pharmacologic VTE prophylaxis in cancer patients?
Contraindications for pharmacologic VTE prophylaxis in cancer patients include active major bleeding, thrombocytopenia (platelet count <50,000/µL), severe coagulopathy, inherited bleeding disorder, and at the time of surgery or invasive procedures (including lumbar puncture and epidural or spinal anesthesia).3,6,7 Those with contraindications to pharmacologic VTE prophylaxis should have mechanical prophylaxis instead.
What is the recommended treatment of VTE in cancer patients?
After the diagnosis of pulmonary embolism (PE) or DVT is found, LMWH is the preferred initial anticoagulant instead of UFH unless the patient has severe renal impairment (CrCl of less than 30 ml/min).6,7,8 LMWH is also preferred over warfarin for long-term anticoagulation during the initial six months of therapy.6,7,8 Following the initial six months, continued anticoagulation with either LMWH or warfarin could be considered in patients with active cancer, metastatic disease, or ongoing chemotherapy.6,7,8
When should IVC filters be considered in treating VTE in cancer patients?
IVC filter insertion should be reserved for those patients found to have a DVT or PE who have a contraindication to pharmacologic anticoagulation.3,6 It can be considered in patients who have recurrent VTE despite the appropriate use of optimally dosed LMWH therapy.6,8
What about the new oral anticoagulants?
At this point, because the majority of the major trials looking at the new oral anticoagulants (dabigatran, rivaroxaban, and apixaban) excluded cancer patients or included them only in small numbers, there is not enough evidence to support their use in cancer patients diagnosed with VTE.6,7,8
Back to the Case
On hospital day three, the patient is clinically improved. She is afebrile, her neutropenia has resolved, and her platelet count is up to 80,000. Her only complaint is pain and swelling of her left leg. A lower extremity Doppler is performed. She is found to have an acute left femoral DVT. The patient is then started on enoxaparin 1 mg/kg every 12 hours. Her left leg swelling and pain begin to improve, and she is discharged on enoxaparin and follows up with her oncologist in the next week. TH
Drs. Bell and O’Rourke are assistant professor of medicine in the division of hospital medicine at the University of California San Diego.
References
1. Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712-1723.
2. Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715-722.
3. Streiff MB, Bockenstedt PL, Cataland SR, et al. Venous thromboembolic disease. J Natl Compr Canc Netw. 2013;11(11):1402-1429.
4. Payne JH, Vora AJ. Thrombosis and acute lymphoblastic leukaemia. Br J Haematol. 2007;138(4):430-445.
5. Amir E, Seruga B, Niraula S, Carlsson L, Ocaña A. Toxicity of adjuvant endocrine therapy in postmenopausal breast cancer patients: a systematic review and meta-analysis. J Natl Cancer Inst. 2011;103(17):1299-1309.
6. Lyman GH, Khorana AA, Kuderer NM, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2013;31(17):2189-2204.
7. Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: american society of clinical oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654-656.
8. Farge D, Debourdeau P, Beckers M, et al. International clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. J Thromb Haemost. 2013;11(1):56-70.
9. Khorana AA. The NCCN clinical practice guidelines on venous thromboembolic disease: strategies for improving VTE prophylaxis in hospitalized cancer patients. Oncologist. 2007;12(11):1361-1370.
10. Mismetti P, Laporte-Simitisidis S, Tardy B, et al. Prevention of venous thromboembolism in internal medicine with unfractionated or low-molecular-weight heparins: a meta-analysis of randomized clinical trials. Thromb Haemost. 2000;83(1):14-19.
What Should Hospitalists Know about Surgical Tubes and Drains?
Case
A 45-year-old woman was admitted with choledocholithiasis. Two days prior, following endoscopic retrograde cholangiopancreatography (ERCP), she had gone to the OR for cholecystectomy. The procedure was completed laparoscopically, though the surgeon reported a difficult dissection. The surgeon left a Blake drain in the gallbladder fossa, which initially contained punch-colored fluid. Today, there is bilious fluid in the drain.
Overview
Surgical drains are used to monitor for postoperative leaks or abscesses, to collect normal physiologic fluid, or to minimize dead space. A hospitalist caring for surgical patients may be the first provider to note when something changes in the color or volume of surgical drains. Table 1 lists various types of drains with their indications for use.
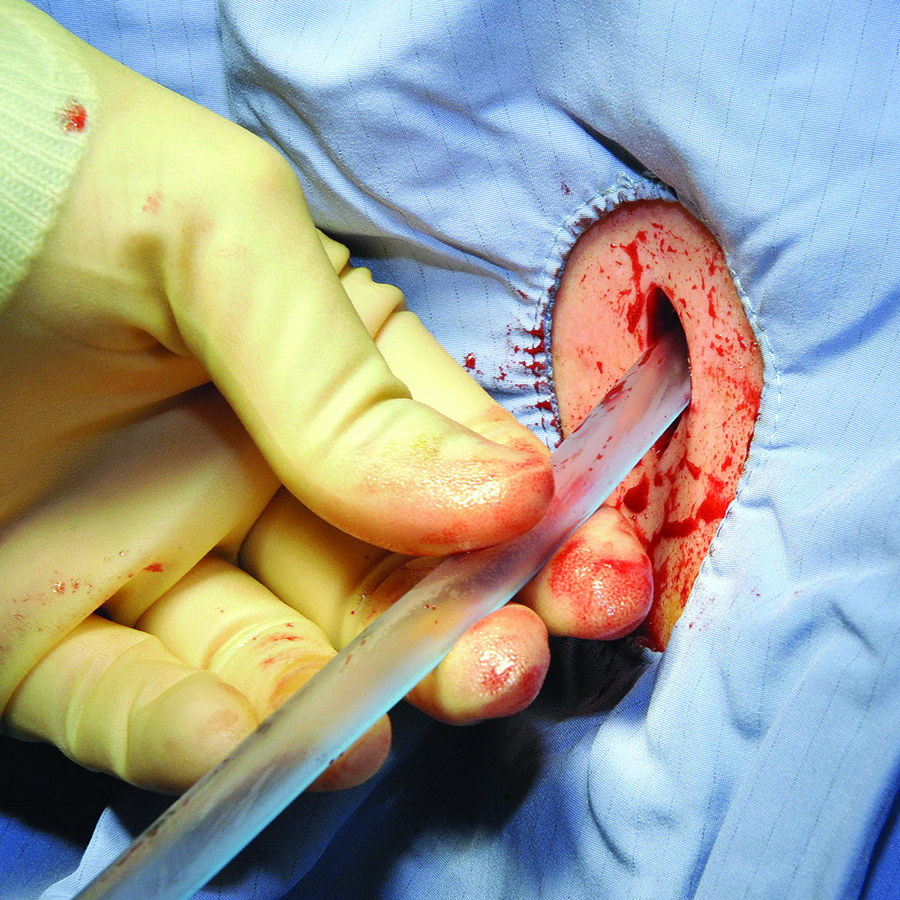
Surgical Tubes and Drains
Chest tubes. Chest tubes are placed in the pleural space to evacuate air or fluid. They can be as thin as 20 French or as thick as 40 French (for adults). Chest tubes are typically placed between the fourth and fifth intercostal spaces in the anterior axillary or mid-axillary line; however, the location may vary according to the indication for placement. The tubes can be straight or angled.
The tubes are connected to a collecting system with a three-way chamber. The water chamber holds a column of water, which prevents air from being sucked into the pleural space with inhalation. The suction chamber can be attached to continuous wall suction to remove air or fluid, or it can be placed on “water seal” with no active suction mechanism. The third chamber is the collection chamber for fluid drainage.
Indications for a chest tube include pneumothorax, hemothorax, or a persistent or large pleural effusion. Pneumothorax and hemothorax usually require immediate chest tube placement. Chest tubes are also commonly placed at the end of thoracic surgeries to allow for appropriate re-expansion of the lung tissue.
A chest X-ray should be obtained after any chest tube insertion to ensure appropriate placement. Chest tubes are equipped with a radiopaque line along the longitudinal axis, which should be visible on X-ray. Respiratory variation in the fluid in the collecting tube, called “tidling,” should also be seen in a correctly placed chest tube, and should be monitored at the bedside to reassure continued appropriate location. The interventional radiologist or surgeon who placed the tube should determine the subsequent frequency of serial chest X-rays required to monitor the location of the chest tube.
If the patient has a pneumothorax, air bubbles will be visible in the water chamber; called an air leak, these are often more apparent when the patient coughs. The chest tube should initially be set to continuous suction at -20 mmHg to evacuate the air. Once the air leak has stopped, the chest tube should be placed on water seal to confirm resolution of the pneumothorax (water seal mimics normal physiology). If, after the transition from suction to water seal, resumption of the air leak is noted, it may indicate recurrence of the patient’s pneumothorax. A stat chest X-ray should be obtained, and the chest tube should be placed back on continuous suction. In general, a chest X-ray should be obtained any time the chest tube is changed from suction to water seal or vice versa.
If the patient experiences ongoing or worsening pain, fever, or inadequate drainage, a chest computed tomographic (CT) scan may be warranted to identify inappropriate positioning or other complications, such as occlusion or effusion of the tube. Blood or other debris might clog chest tubes; the surgical team may be able to evacuate the tube with suction tubing at the bedside. If unsuccessful, the tube may need to be removed and reinserted.
The team that placed the tube should help the hospitalist determine the timing of the chest tube removal. If the patient has a pleural effusion, the chest tube can usually be removed when the output is less than 100-200 mL per day and the lung is expanded. The tube should usually be taken off suction and placed on water seal to rule out pneumothorax prior to tube removal.
Penrose drains. Penrose drains are often used to drain fluid or to keep a space open for drainage. Surgeons may use sutures to anchor Penrose drains to skin. Common indications include:
- Ventral hernia repair;
- Debridement of infected pancreatitis; and
- Drainage of superficial abscess cavities.
Penrose drains are simple, flexible tubes that are open at both ends; in contrast to closed drains, they permit ingress as well as egress, facilitating colonization.
Closed suction drains. Closed suction drains with a plastic bulb attachment (i.e., Jackson-Pratt, Blake, Hemovac) are used to collect fluid from a postoperative cavity. Common indications include:
- Post-mastectomy to drain subcutaneous fluid;
- Abdominal surgery;
- Plastic surgery to prevent seroma formation and promote tissue apposition;
- Cholecystectomy if there is concern for damage to ducts of Luschka or other source of bile leak;
- Inadvertent postoperative leakage following a difficult rectal anastomosis; and
- Post-pancreatic surgery.
The quality and quantity of fluid drained should always be carefully noted and recorded. Changes in the fluid can imply development of bleed, leak, or other complications. The surgical team should be contacted immediately if changes are noted.
Typically, closed suction drains will be left in place until the drainage is less than 20 mL per day. These drains can be left in for weeks if necessary and will often be removed during the patient’s scheduled surgical follow-up. Rare complications include erosion into surrounding tissues and inadvertent suturing of the drain in place, such that reexploration is required to remove it. If a closed suction drain becomes occluded, contact the team that placed the drain for further recommendations on adjustment, replacement, or removal.
Nasogastric and duodenal tubes. Nasogastric tubes (NGTs) are often used in the nonoperative management of small bowel obstruction or ileus. They should be placed in the most dependent portion of the gastric lumen and confirmed by chest or abdominal X-ray. NGTs are sump pumps and have a double lumen, which includes an air port to assure flow. The air port should be patent for optimal functioning. The tube may be connected to continuous wall suction or intermittent suction, set to low (less than 60 mmHg) to avoid mucosal avulsion.
NGT output should decrease during the resolution of obstruction or ileus, and symptoms of nausea, vomiting, and abdominal distention should concomitantly improve. Persistently high output in a patient with other indicators of bowel function (flatus, for example) may suggest postpyloric placement (and placement should be checked by X-ray). The timing of NGT removal depends on resumption of bowel function.
Gastrostomy and jejunostomy tubes. Gastrostomy tubes are most commonly used for feeding but may also be used for decompression of functional or anatomic gastric outlet obstruction. They are indicated when patients need prolonged enteral access, such as those with prolonged mechanical ventilation or head and neck pathology that prohibits oral feeding. They are also rarely used for gastropexy to tack an atonic or patulous stomach to the abdominal wall or to prevent recurrence of paraesophageal hernias. These tubes can be placed percutaneously by interventional radiologists, endoscopically by surgeons and gastroenterologists, or laparoscopically or laparotomally by surgeons. This last option is often reserved for patients with difficult anatomy or those who are having laparotomy for another reason.
Because of the stomach’s generous lumen, gastrostomy tubes rarely clog. In the event that they do get clogged, carbonated liquids, meat tenderizer, or enzymes can help dissolve the obstruction. If a gastrostomy tube is left to drain, the patient may experience significant fluid and electrolyte losses, so these need to be carefully monitored.
Jejunostomy tubes are used exclusively for feeding and are usually placed 10-20 cm distal to the ligament of Treitz. These tubes are indicated in patients who require distal feedings due to gastric dysfunction or in those who have undergone a surgery in which a proximal anastomosis requires time to heal. These tubes are more apt to clog and can be more difficult to manage because the lumen of the small bowel is smaller than the stomach. Some prefer not to put pills down the tube to mitigate this risk. Routine flushes with water or saline (30 mL every four to six hours) are also helpful in mitigating the risk of clogging. In the event that they do get clogged, they may be treated like gastrostomy tubes, using carbonated liquids, meat tenderizer, or enzymes to help dissolve the obstruction.
Percutaneous tube sites should be examined frequently for signs of infection. Though gastrostomy and jejunostomy tubes are typically well secured intraabdominally, they can become dislodged. If a gastrostomy or jejunostomy tube has been in place for more than two weeks, it can easily be replaced at the bedside with a tube of comparable caliber by a member of the surgical team or by an experienced hospitalist. If the tube has been in place less than two weeks, it requires replacement with radiographic guidance, as the risk of creating a false lumen is high. Over time, tubes can become loose and fall out. If they need replacement, the preceding guidelines apply.
Back to the Case
A potential major complication of cholecystectomy is severance of the common bile duct, which necessitates significant further surgery. Less severe complications include injuries to the cholecystohepatic ducts (otherwise known as the ducts of Luschka), which can result in leakage of bile into the peritoneal cavity. A bile leak can lead to abscess and systemic infection if left undrained.
Surgeons who are concerned for such a complication intraoperatively may opt to leave a closed suction drain in the gallbladder fossa, such as a Blake drain, for monitoring and subsequent drainage. The drain will remain in place at least until the patient’s diet has been advanced fully, because digestion promotes the secretion of bile and may elucidate a leak. Bilious fluid in the Blake drain is suspicious for a leak.
The surgeon should be notified, and imaging should be obtained to find the nature of the injury to the biliary tree (CT scan with IV contrast, hepatobiliary iminodiacetic acid scan, or endoscopic retrograde cholangiopancreatography). If injury to major biliary structures (the cystic duct stump, the hepatic ducts, or the common bile duct) is diagnosed, a stent may be placed in order to restore ductile continuity.
Minor leaks, with damage to the cystic duct stump, hepatic ducts, and common bile duct ruled out, more often resolve on their own over time, and thus the patient’s closed suction drain will be left in place until biliary drainage ceases, without further initial intervention.
Bottom Line
Surgical tubes and drains have several placement indications. Alterations in quality and quantity of output can indicate changes in clinical status, and hospitalists should be able to handle initial troubleshooting. TH
Dr. Columbus is a general surgery resident at Brigham and Women’s Hospital in Boston. Dr. Havens is an instructor for the department of surgery at Brigham and Women’s Hospital. Dr. Peetz is an instructor for the department of surgery at University Hospital Case Medical Center in Cleveland.
Case
A 45-year-old woman was admitted with choledocholithiasis. Two days prior, following endoscopic retrograde cholangiopancreatography (ERCP), she had gone to the OR for cholecystectomy. The procedure was completed laparoscopically, though the surgeon reported a difficult dissection. The surgeon left a Blake drain in the gallbladder fossa, which initially contained punch-colored fluid. Today, there is bilious fluid in the drain.
Overview
Surgical drains are used to monitor for postoperative leaks or abscesses, to collect normal physiologic fluid, or to minimize dead space. A hospitalist caring for surgical patients may be the first provider to note when something changes in the color or volume of surgical drains. Table 1 lists various types of drains with their indications for use.
Surgical Tubes and Drains
Chest tubes. Chest tubes are placed in the pleural space to evacuate air or fluid. They can be as thin as 20 French or as thick as 40 French (for adults). Chest tubes are typically placed between the fourth and fifth intercostal spaces in the anterior axillary or mid-axillary line; however, the location may vary according to the indication for placement. The tubes can be straight or angled.
The tubes are connected to a collecting system with a three-way chamber. The water chamber holds a column of water, which prevents air from being sucked into the pleural space with inhalation. The suction chamber can be attached to continuous wall suction to remove air or fluid, or it can be placed on “water seal” with no active suction mechanism. The third chamber is the collection chamber for fluid drainage.
Indications for a chest tube include pneumothorax, hemothorax, or a persistent or large pleural effusion. Pneumothorax and hemothorax usually require immediate chest tube placement. Chest tubes are also commonly placed at the end of thoracic surgeries to allow for appropriate re-expansion of the lung tissue.
A chest X-ray should be obtained after any chest tube insertion to ensure appropriate placement. Chest tubes are equipped with a radiopaque line along the longitudinal axis, which should be visible on X-ray. Respiratory variation in the fluid in the collecting tube, called “tidling,” should also be seen in a correctly placed chest tube, and should be monitored at the bedside to reassure continued appropriate location. The interventional radiologist or surgeon who placed the tube should determine the subsequent frequency of serial chest X-rays required to monitor the location of the chest tube.
If the patient has a pneumothorax, air bubbles will be visible in the water chamber; called an air leak, these are often more apparent when the patient coughs. The chest tube should initially be set to continuous suction at -20 mmHg to evacuate the air. Once the air leak has stopped, the chest tube should be placed on water seal to confirm resolution of the pneumothorax (water seal mimics normal physiology). If, after the transition from suction to water seal, resumption of the air leak is noted, it may indicate recurrence of the patient’s pneumothorax. A stat chest X-ray should be obtained, and the chest tube should be placed back on continuous suction. In general, a chest X-ray should be obtained any time the chest tube is changed from suction to water seal or vice versa.
If the patient experiences ongoing or worsening pain, fever, or inadequate drainage, a chest computed tomographic (CT) scan may be warranted to identify inappropriate positioning or other complications, such as occlusion or effusion of the tube. Blood or other debris might clog chest tubes; the surgical team may be able to evacuate the tube with suction tubing at the bedside. If unsuccessful, the tube may need to be removed and reinserted.
The team that placed the tube should help the hospitalist determine the timing of the chest tube removal. If the patient has a pleural effusion, the chest tube can usually be removed when the output is less than 100-200 mL per day and the lung is expanded. The tube should usually be taken off suction and placed on water seal to rule out pneumothorax prior to tube removal.
Penrose drains. Penrose drains are often used to drain fluid or to keep a space open for drainage. Surgeons may use sutures to anchor Penrose drains to skin. Common indications include:
- Ventral hernia repair;
- Debridement of infected pancreatitis; and
- Drainage of superficial abscess cavities.
Penrose drains are simple, flexible tubes that are open at both ends; in contrast to closed drains, they permit ingress as well as egress, facilitating colonization.
Closed suction drains. Closed suction drains with a plastic bulb attachment (i.e., Jackson-Pratt, Blake, Hemovac) are used to collect fluid from a postoperative cavity. Common indications include:
- Post-mastectomy to drain subcutaneous fluid;
- Abdominal surgery;
- Plastic surgery to prevent seroma formation and promote tissue apposition;
- Cholecystectomy if there is concern for damage to ducts of Luschka or other source of bile leak;
- Inadvertent postoperative leakage following a difficult rectal anastomosis; and
- Post-pancreatic surgery.
The quality and quantity of fluid drained should always be carefully noted and recorded. Changes in the fluid can imply development of bleed, leak, or other complications. The surgical team should be contacted immediately if changes are noted.
Typically, closed suction drains will be left in place until the drainage is less than 20 mL per day. These drains can be left in for weeks if necessary and will often be removed during the patient’s scheduled surgical follow-up. Rare complications include erosion into surrounding tissues and inadvertent suturing of the drain in place, such that reexploration is required to remove it. If a closed suction drain becomes occluded, contact the team that placed the drain for further recommendations on adjustment, replacement, or removal.
Nasogastric and duodenal tubes. Nasogastric tubes (NGTs) are often used in the nonoperative management of small bowel obstruction or ileus. They should be placed in the most dependent portion of the gastric lumen and confirmed by chest or abdominal X-ray. NGTs are sump pumps and have a double lumen, which includes an air port to assure flow. The air port should be patent for optimal functioning. The tube may be connected to continuous wall suction or intermittent suction, set to low (less than 60 mmHg) to avoid mucosal avulsion.
NGT output should decrease during the resolution of obstruction or ileus, and symptoms of nausea, vomiting, and abdominal distention should concomitantly improve. Persistently high output in a patient with other indicators of bowel function (flatus, for example) may suggest postpyloric placement (and placement should be checked by X-ray). The timing of NGT removal depends on resumption of bowel function.
Gastrostomy and jejunostomy tubes. Gastrostomy tubes are most commonly used for feeding but may also be used for decompression of functional or anatomic gastric outlet obstruction. They are indicated when patients need prolonged enteral access, such as those with prolonged mechanical ventilation or head and neck pathology that prohibits oral feeding. They are also rarely used for gastropexy to tack an atonic or patulous stomach to the abdominal wall or to prevent recurrence of paraesophageal hernias. These tubes can be placed percutaneously by interventional radiologists, endoscopically by surgeons and gastroenterologists, or laparoscopically or laparotomally by surgeons. This last option is often reserved for patients with difficult anatomy or those who are having laparotomy for another reason.
Because of the stomach’s generous lumen, gastrostomy tubes rarely clog. In the event that they do get clogged, carbonated liquids, meat tenderizer, or enzymes can help dissolve the obstruction. If a gastrostomy tube is left to drain, the patient may experience significant fluid and electrolyte losses, so these need to be carefully monitored.
Jejunostomy tubes are used exclusively for feeding and are usually placed 10-20 cm distal to the ligament of Treitz. These tubes are indicated in patients who require distal feedings due to gastric dysfunction or in those who have undergone a surgery in which a proximal anastomosis requires time to heal. These tubes are more apt to clog and can be more difficult to manage because the lumen of the small bowel is smaller than the stomach. Some prefer not to put pills down the tube to mitigate this risk. Routine flushes with water or saline (30 mL every four to six hours) are also helpful in mitigating the risk of clogging. In the event that they do get clogged, they may be treated like gastrostomy tubes, using carbonated liquids, meat tenderizer, or enzymes to help dissolve the obstruction.
Percutaneous tube sites should be examined frequently for signs of infection. Though gastrostomy and jejunostomy tubes are typically well secured intraabdominally, they can become dislodged. If a gastrostomy or jejunostomy tube has been in place for more than two weeks, it can easily be replaced at the bedside with a tube of comparable caliber by a member of the surgical team or by an experienced hospitalist. If the tube has been in place less than two weeks, it requires replacement with radiographic guidance, as the risk of creating a false lumen is high. Over time, tubes can become loose and fall out. If they need replacement, the preceding guidelines apply.
Back to the Case
A potential major complication of cholecystectomy is severance of the common bile duct, which necessitates significant further surgery. Less severe complications include injuries to the cholecystohepatic ducts (otherwise known as the ducts of Luschka), which can result in leakage of bile into the peritoneal cavity. A bile leak can lead to abscess and systemic infection if left undrained.
Surgeons who are concerned for such a complication intraoperatively may opt to leave a closed suction drain in the gallbladder fossa, such as a Blake drain, for monitoring and subsequent drainage. The drain will remain in place at least until the patient’s diet has been advanced fully, because digestion promotes the secretion of bile and may elucidate a leak. Bilious fluid in the Blake drain is suspicious for a leak.
The surgeon should be notified, and imaging should be obtained to find the nature of the injury to the biliary tree (CT scan with IV contrast, hepatobiliary iminodiacetic acid scan, or endoscopic retrograde cholangiopancreatography). If injury to major biliary structures (the cystic duct stump, the hepatic ducts, or the common bile duct) is diagnosed, a stent may be placed in order to restore ductile continuity.
Minor leaks, with damage to the cystic duct stump, hepatic ducts, and common bile duct ruled out, more often resolve on their own over time, and thus the patient’s closed suction drain will be left in place until biliary drainage ceases, without further initial intervention.
Bottom Line
Surgical tubes and drains have several placement indications. Alterations in quality and quantity of output can indicate changes in clinical status, and hospitalists should be able to handle initial troubleshooting. TH
Dr. Columbus is a general surgery resident at Brigham and Women’s Hospital in Boston. Dr. Havens is an instructor for the department of surgery at Brigham and Women’s Hospital. Dr. Peetz is an instructor for the department of surgery at University Hospital Case Medical Center in Cleveland.
Case
A 45-year-old woman was admitted with choledocholithiasis. Two days prior, following endoscopic retrograde cholangiopancreatography (ERCP), she had gone to the OR for cholecystectomy. The procedure was completed laparoscopically, though the surgeon reported a difficult dissection. The surgeon left a Blake drain in the gallbladder fossa, which initially contained punch-colored fluid. Today, there is bilious fluid in the drain.
Overview
Surgical drains are used to monitor for postoperative leaks or abscesses, to collect normal physiologic fluid, or to minimize dead space. A hospitalist caring for surgical patients may be the first provider to note when something changes in the color or volume of surgical drains. Table 1 lists various types of drains with their indications for use.
Surgical Tubes and Drains
Chest tubes. Chest tubes are placed in the pleural space to evacuate air or fluid. They can be as thin as 20 French or as thick as 40 French (for adults). Chest tubes are typically placed between the fourth and fifth intercostal spaces in the anterior axillary or mid-axillary line; however, the location may vary according to the indication for placement. The tubes can be straight or angled.
The tubes are connected to a collecting system with a three-way chamber. The water chamber holds a column of water, which prevents air from being sucked into the pleural space with inhalation. The suction chamber can be attached to continuous wall suction to remove air or fluid, or it can be placed on “water seal” with no active suction mechanism. The third chamber is the collection chamber for fluid drainage.
Indications for a chest tube include pneumothorax, hemothorax, or a persistent or large pleural effusion. Pneumothorax and hemothorax usually require immediate chest tube placement. Chest tubes are also commonly placed at the end of thoracic surgeries to allow for appropriate re-expansion of the lung tissue.
A chest X-ray should be obtained after any chest tube insertion to ensure appropriate placement. Chest tubes are equipped with a radiopaque line along the longitudinal axis, which should be visible on X-ray. Respiratory variation in the fluid in the collecting tube, called “tidling,” should also be seen in a correctly placed chest tube, and should be monitored at the bedside to reassure continued appropriate location. The interventional radiologist or surgeon who placed the tube should determine the subsequent frequency of serial chest X-rays required to monitor the location of the chest tube.
If the patient has a pneumothorax, air bubbles will be visible in the water chamber; called an air leak, these are often more apparent when the patient coughs. The chest tube should initially be set to continuous suction at -20 mmHg to evacuate the air. Once the air leak has stopped, the chest tube should be placed on water seal to confirm resolution of the pneumothorax (water seal mimics normal physiology). If, after the transition from suction to water seal, resumption of the air leak is noted, it may indicate recurrence of the patient’s pneumothorax. A stat chest X-ray should be obtained, and the chest tube should be placed back on continuous suction. In general, a chest X-ray should be obtained any time the chest tube is changed from suction to water seal or vice versa.
If the patient experiences ongoing or worsening pain, fever, or inadequate drainage, a chest computed tomographic (CT) scan may be warranted to identify inappropriate positioning or other complications, such as occlusion or effusion of the tube. Blood or other debris might clog chest tubes; the surgical team may be able to evacuate the tube with suction tubing at the bedside. If unsuccessful, the tube may need to be removed and reinserted.
The team that placed the tube should help the hospitalist determine the timing of the chest tube removal. If the patient has a pleural effusion, the chest tube can usually be removed when the output is less than 100-200 mL per day and the lung is expanded. The tube should usually be taken off suction and placed on water seal to rule out pneumothorax prior to tube removal.
Penrose drains. Penrose drains are often used to drain fluid or to keep a space open for drainage. Surgeons may use sutures to anchor Penrose drains to skin. Common indications include:
- Ventral hernia repair;
- Debridement of infected pancreatitis; and
- Drainage of superficial abscess cavities.
Penrose drains are simple, flexible tubes that are open at both ends; in contrast to closed drains, they permit ingress as well as egress, facilitating colonization.
Closed suction drains. Closed suction drains with a plastic bulb attachment (i.e., Jackson-Pratt, Blake, Hemovac) are used to collect fluid from a postoperative cavity. Common indications include:
- Post-mastectomy to drain subcutaneous fluid;
- Abdominal surgery;
- Plastic surgery to prevent seroma formation and promote tissue apposition;
- Cholecystectomy if there is concern for damage to ducts of Luschka or other source of bile leak;
- Inadvertent postoperative leakage following a difficult rectal anastomosis; and
- Post-pancreatic surgery.
The quality and quantity of fluid drained should always be carefully noted and recorded. Changes in the fluid can imply development of bleed, leak, or other complications. The surgical team should be contacted immediately if changes are noted.
Typically, closed suction drains will be left in place until the drainage is less than 20 mL per day. These drains can be left in for weeks if necessary and will often be removed during the patient’s scheduled surgical follow-up. Rare complications include erosion into surrounding tissues and inadvertent suturing of the drain in place, such that reexploration is required to remove it. If a closed suction drain becomes occluded, contact the team that placed the drain for further recommendations on adjustment, replacement, or removal.
Nasogastric and duodenal tubes. Nasogastric tubes (NGTs) are often used in the nonoperative management of small bowel obstruction or ileus. They should be placed in the most dependent portion of the gastric lumen and confirmed by chest or abdominal X-ray. NGTs are sump pumps and have a double lumen, which includes an air port to assure flow. The air port should be patent for optimal functioning. The tube may be connected to continuous wall suction or intermittent suction, set to low (less than 60 mmHg) to avoid mucosal avulsion.
NGT output should decrease during the resolution of obstruction or ileus, and symptoms of nausea, vomiting, and abdominal distention should concomitantly improve. Persistently high output in a patient with other indicators of bowel function (flatus, for example) may suggest postpyloric placement (and placement should be checked by X-ray). The timing of NGT removal depends on resumption of bowel function.
Gastrostomy and jejunostomy tubes. Gastrostomy tubes are most commonly used for feeding but may also be used for decompression of functional or anatomic gastric outlet obstruction. They are indicated when patients need prolonged enteral access, such as those with prolonged mechanical ventilation or head and neck pathology that prohibits oral feeding. They are also rarely used for gastropexy to tack an atonic or patulous stomach to the abdominal wall or to prevent recurrence of paraesophageal hernias. These tubes can be placed percutaneously by interventional radiologists, endoscopically by surgeons and gastroenterologists, or laparoscopically or laparotomally by surgeons. This last option is often reserved for patients with difficult anatomy or those who are having laparotomy for another reason.
Because of the stomach’s generous lumen, gastrostomy tubes rarely clog. In the event that they do get clogged, carbonated liquids, meat tenderizer, or enzymes can help dissolve the obstruction. If a gastrostomy tube is left to drain, the patient may experience significant fluid and electrolyte losses, so these need to be carefully monitored.
Jejunostomy tubes are used exclusively for feeding and are usually placed 10-20 cm distal to the ligament of Treitz. These tubes are indicated in patients who require distal feedings due to gastric dysfunction or in those who have undergone a surgery in which a proximal anastomosis requires time to heal. These tubes are more apt to clog and can be more difficult to manage because the lumen of the small bowel is smaller than the stomach. Some prefer not to put pills down the tube to mitigate this risk. Routine flushes with water or saline (30 mL every four to six hours) are also helpful in mitigating the risk of clogging. In the event that they do get clogged, they may be treated like gastrostomy tubes, using carbonated liquids, meat tenderizer, or enzymes to help dissolve the obstruction.
Percutaneous tube sites should be examined frequently for signs of infection. Though gastrostomy and jejunostomy tubes are typically well secured intraabdominally, they can become dislodged. If a gastrostomy or jejunostomy tube has been in place for more than two weeks, it can easily be replaced at the bedside with a tube of comparable caliber by a member of the surgical team or by an experienced hospitalist. If the tube has been in place less than two weeks, it requires replacement with radiographic guidance, as the risk of creating a false lumen is high. Over time, tubes can become loose and fall out. If they need replacement, the preceding guidelines apply.
Back to the Case
A potential major complication of cholecystectomy is severance of the common bile duct, which necessitates significant further surgery. Less severe complications include injuries to the cholecystohepatic ducts (otherwise known as the ducts of Luschka), which can result in leakage of bile into the peritoneal cavity. A bile leak can lead to abscess and systemic infection if left undrained.
Surgeons who are concerned for such a complication intraoperatively may opt to leave a closed suction drain in the gallbladder fossa, such as a Blake drain, for monitoring and subsequent drainage. The drain will remain in place at least until the patient’s diet has been advanced fully, because digestion promotes the secretion of bile and may elucidate a leak. Bilious fluid in the Blake drain is suspicious for a leak.
The surgeon should be notified, and imaging should be obtained to find the nature of the injury to the biliary tree (CT scan with IV contrast, hepatobiliary iminodiacetic acid scan, or endoscopic retrograde cholangiopancreatography). If injury to major biliary structures (the cystic duct stump, the hepatic ducts, or the common bile duct) is diagnosed, a stent may be placed in order to restore ductile continuity.
Minor leaks, with damage to the cystic duct stump, hepatic ducts, and common bile duct ruled out, more often resolve on their own over time, and thus the patient’s closed suction drain will be left in place until biliary drainage ceases, without further initial intervention.
Bottom Line
Surgical tubes and drains have several placement indications. Alterations in quality and quantity of output can indicate changes in clinical status, and hospitalists should be able to handle initial troubleshooting. TH
Dr. Columbus is a general surgery resident at Brigham and Women’s Hospital in Boston. Dr. Havens is an instructor for the department of surgery at Brigham and Women’s Hospital. Dr. Peetz is an instructor for the department of surgery at University Hospital Case Medical Center in Cleveland.
What Are the Strategies for Secondary Stroke Prevention after Transient Ischemic Attack?
Case
Mr. G is an 80-year-old man with a pacemaker, peripheral artery disease, atrial fibrillation (AF) on warfarin, and tachy-brady syndrome. He presented after experiencing episodes in which he was unable to speak and had weakness on his right side. He had a normal neurological exam upon arrival to the ED, and his blood pressure was 160/80 mm Hg.
Overview
Transient ischemic attacks (TIAs) are brief interruptions in brain perfusion that do not result in permanent neurologic damage. Up to half a million TIAs occur each year in the U.S., and they account for one third of acute cerebrovascular disease.1 While the term suggests that TIAs are benign, they are in fact an important warning sign of impending stroke and are essentially analogous to unstable angina. Some 10% of TIAs convert to full strokes within 90 days, but growing evidence suggests appropriate interventions can decrease this risk to 3%.2
Unfortunately, the symptoms of TIA have usually resolved by the time patients arrive at the hospital, which makes them challenging to diagnose. This article provides a summary of how to diagnose TIA accurately, using a focused history informed by cerebrovascular localization; how to triage, evaluate, and risk stratify patients; and how to implement preventative strategies.
Review of the Data
Classically, TIAs are defined as lasting less than 24 hours; however, 24 hours is an arbitrary number, and most TIAs last less than one hour.1 Furthermore, this definition has evolved with advances in neuroimaging that reveal that up to 50% of classically defined TIAs have evidence of infarct on MRI.1 There is no absolute temporal cut-off after which infarct is always seen on MRI, but longer duration of symptoms correlates with a higher likelihood of infarct. To reconcile these observations, a recently proposed definition stipulates that a true TIA lasts no more than one hour and does not show evidence of infarct on MRI.3
The causes of TIA are identical to those for ischemic stroke. Cerebral ischemia can result from an embolus, arterial thrombosis, or hypoperfusion due to arterial stenosis. Emboli can be cardiac, most commonly due to AF, or non-cardiac, stemming from a ruptured atherosclerotic plaque in the aortic arch, the carotid or vertebral artery, or an intracranial vessel. Atherosclerotic disease in the carotid arteries or intracranial vessels can also lead to thrombosis and occlusion or flow-related TIAs as a result of severe stenosis.
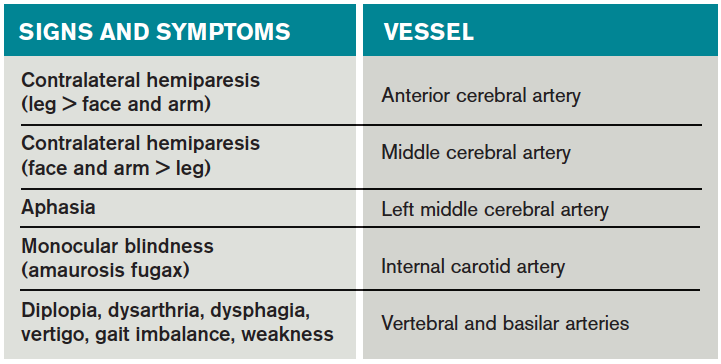
Risk factors for TIA mirror those for heart disease. Non-modifiable risk factors include older age, black race, male sex, and family history of stroke. Modifiable factors include hypertension, hyperlipidemia, tobacco smoking, diabetes, and AF.4
Most of the time, patients’ symptoms will have resolved by the time they are evaluated by a physician. Therefore, the diagnosis of TIA relies almost exclusively on the patient history. Eliciting a good history helps physicians determine whether the episode of transient neurologic dysfunction was caused by cerebral ischemia, as opposed to another mechanism, such as migraine or seizure. This calls for a basic understanding of cerebrovascular anatomy (see Table 1).
Types of Ischemia
Anterior cerebral artery ischemia causes contralateral leg weakness because it supplies the medial frontal and parietal lobes, where the legs in the sensorimotor homunculus are represented. Middle cerebral artery (MCA) ischemia causes contralateral face and arm weakness out of proportion to leg weakness. Ischemia in Broca’s area of the brain, which is supplied by the left MCA, may also cause expressive aphasia. Transient monocular blindness is a TIA of the retina due to atheroemboli originating from the internal carotid artery. Vertebrobasilar TIA is less common than anterior circulation TIA and manifests with brainstem symptoms that include diplopia, dysarthria, dysphagia, vertigo, gait imbalance, and weakness. In general, language and motor symptoms are more specific for cerebral ischemia and therefore more worrisome for TIA than sensory symptoms.5
Once a clinical diagnosis of TIA is made, an ABCD2 score (age, blood pressure, clinical features, duration of TIA, presence of diabetes) can be used to predict the short-term risk of subsequent stroke (see Table 2).6,7 A general rule of thumb is to admit patients who present within 72 hours of the event and have an ABCD2 score of three or higher for observation, work-up, and initiation of secondary prevention.1
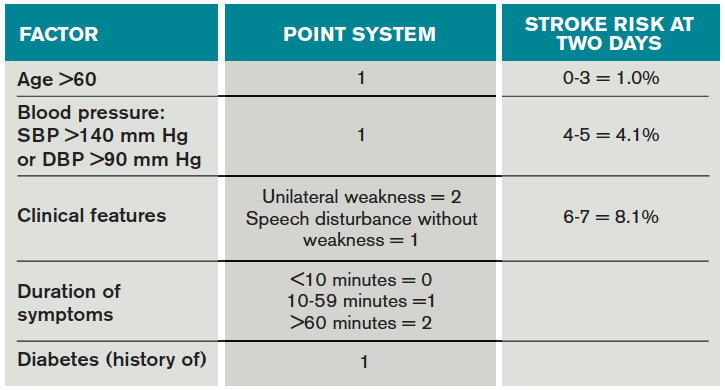
Although only a small percentage of patients with TIA will have a stroke during the period of observation in the hospital, this approach may be cost effective based on the assumption that hospitalized patients are more likely to receive intravenous tissue plasminogen activator.8 The decision should also be guided by clinical judgment. It is reasonable to admit a patient whose diagnostic workup cannot be rapidly completed.1
The workup for TIA includes routine labs, EKG with cardiac monitoring, and brain imaging. Labs are useful to evaluate for other mimics of TIA such as hyponatremia and glucose abnormalities. In addition, risk factors such as hyperlipidemia and diabetes should be evaluated with fasting lipid panel and blood glucose. The purpose of EKG and telemetry is to identify MI and capture paroxysmal AF. The goal of imaging is to ascertain the presence of vascular disease and to exclude a non-ischemic etiology. While less likely to cause transient neurologic symptoms, a hemorrhagic event must be ruled out, as it would trigger a different management pathway.
Imaging for TIA
There are two primary modes of brain imaging: computed tomography (CT) and MRI. Most patients who are suspected to have had a TIA undergo CT scan, and an infarct is seen about 20% of the time.1 The presence of an infarct usually correlates with the duration of symptoms and has prognostic value. In one study, a new infarct was associated with four times higher risk of stroke in the subsequent 90 days.9 Diffusion-weighted imaging, an MR-based technique, is the preferred modality when it is available because of its higher sensitivity and specificity for identifying acute lesions.1 In an international and multicenter study, incorporating imaging data increased the discriminatory power of stroke prediction.10
Extracranial imaging is mandatory to rule out carotid stenosis as a potential etiology of TIA. The least invasive modality is ultrasound, which can detect carotid stenosis with a sensitivity and specificity approaching 80%.1 While both the intra- and extracranial vasculature can be concurrently assessed using MR- or CT-angiography (CTA), this is not usually necessary in the acute setting, because only detecting carotid stenosis will result in a management change.1
Carotid endarterectomy is standard for symptomatic patients with greater than 70% stenosis and is a consideration for symptomatic patients with greater than 50% stenosis if it is the most probable explanation for the ischemic event.11 Despite a comprehensive workup, about 50% of TIA cases remain cryptogenic.12 In some of these patients, AF can be detected using extended ambulatory cardiac monitoring.12
The goal of admitting high-risk patients is to expedite workup and initiate therapy. Two studies have shown that immediate initiation of preventative treatment significantly reduces the risk of stroke by as much as 80%.13,14 Unless there is a specific indication for anticoagulation, all TIA patients should be started on an antiplatelet agent such as aspirin or clopidogrel. A large randomized trial conducted in China and published in 2013 demonstrated that dual antiplatelet therapy with aspirin and clopidogrel for 21 days, followed by clopidogrel monotherapy, reduced the risk of stroke compared to aspirin monotherapy. An international multicenter trial designed to test the efficacy of short-term dual antiplatelet therapy is ongoing, and if the benefit of this approach is confirmed, this will likely become the standard of care. Evidence-based indications for anticoagulation after TIA are restricted to AF and mural thrombus in the setting of recent MI. Patients with implanted mechanical devices, including left ventricular assist devices and metal heart valves, should also receive anticoagulation.15
Risk factors should also be targeted in every case. Hypertension should be treated with a goal of lower than 140/90 mm Hg (or 130/80 mm Hg in diabetics and those with renal disease). Studies have shown that patients who are discharged with a blood pressure lower than 140/90 mm Hg are more likely to maintain this blood pressure at one-year follow-up.16 The choice of medication is less well studied, but drugs that act on the renin-angiotensin-aldosterone system and thiazides are generally preferred.15 Treatment with a statin is recommended after cerebrovascular ischemic events, with a goal LDL under 100. This reduces risk of secondary stroke by about 20%.17
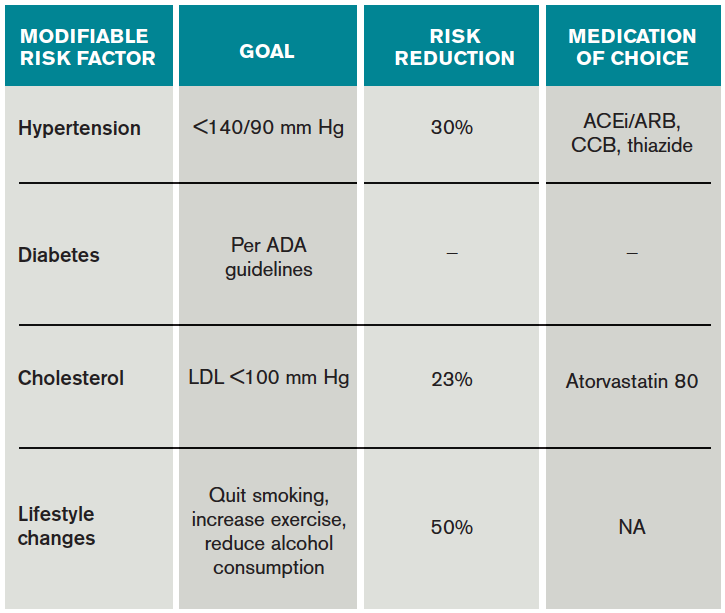
At discharge, it is also important to counsel patients on their role in preventing strokes. As with many diseases, making lifestyle changes is key to stroke prevention. Encourage smoking cessation and an increase in physical activity, and discourage heavy alcohol use. The association between smoking and the risk for first stroke is well established. Moderate to high-intensity exercise can reduce secondary stroke risk by as much as 50%18 (see Table 3). While light alcohol consumption can be protective against strokes, heavy use is strongly discouraged. Emerging data suggest obstructive sleep apnea (OSA) may be another modifiable risk factor for stroke and TIA, so screening for potential OSA and referral may be needed.15
Back to the Case
When Mr. G arrived at the ED, his symptoms had resolved. Based on the history of expressive aphasia and right-sided weakness, he most likely had a TIA in the left MCA territory. Hemorrhage was ruled out with a non-contrast head CT. His pacemaker precluded obtaining an MRI. CTA revealed diffuse atherosclerotic disease without evidence of carotid stenosis. His ABCD2 score was six given his age, blood pressure, weakness, and symptom duration, and he was admitted for an expedited workup. His sodium and glucose were within normal limits. His hemoglobin A1c was 6.5%, his LDL was 120, and his international normalized ratio (INR) was therapeutic at 2.1. His TIA may have been due to AF, despite a therapeutic INR, because warfarin does not fully eliminate the stroke risk. It might also have been caused by intracranial atherosclerosis.
Two days later, the patient was discharged on atorvastatin at 80 mg, and his lisinopril was increased for blood pressure control. For his age group, A1c of 6.5% was acceptable, and he was not initiated on glycemic control.
Bottom Line
TIAs are diagnosed based on patient history. Urgent initiation of secondary prevention is important to reduce the short-term risk of stroke and should be implemented by the time of discharge from the hospital.
Dr. Zeng is a hospitalist in the department of internal medicine at Vanderbilt University Medical Center in Nashville, and Dr. Douglas is associate professor in the department of neurology at the University of California at San Francisco.
References
- Easton JD, Saver JL, Albers GW, et al. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009;40(6):2276-2293.
- Sundararajan V, Thrift AG, Phan TG, Choi PM, Clissold B, Srikanth VK. Trends over time in the risk of stroke after an incident transient ischemic attack. Stroke. 2014;45(11):3214-3218.
- Albers GW, Caplan LR, Easton JD, et al. Transient ischemic attack–proposal for a new definition. N Engl J Med. 2002;347(21):1713-1716.
- Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin. 2008;26(4):871-895, vii.
- Johnston SC, Sidney S, Bernstein AL, Gress DR. A comparison of risk factors for recurrent TIA and stroke in patients diagnosed with TIA. Neurology. 2003;60(2):280-285.
- Tsivgoulis G, Stamboulis E, Sharma VK, et al. Multicenter external validation of the ABCD2 score in triaging TIA patients. Neurology. 2010;74(17):1351-1357.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet. 2007;369(9558):283-292.
- Nguyen-Huynh MN, Johnston SC. Is hospitalization after TIA cost-effective on the basis of treatment with tPA? Neurology. 2005;65(11):1799-1801.
- Douglas VC, Johnston CM, Elkins J, Sidney S, Gress DR, Johnston SC. Head computed tomography findings predict short-term stroke risk after transient ischemic attack. Stroke. 2003;34(12):2894-2898.
- Giles MF, Albers GW, Amarenco P, et al. Addition of brain infarction to the ABCD2 Score (ABCD2I): a collaborative analysis of unpublished data on 4574 patients. Stroke. 2010;41(9):1907-1913.
- Lanzino G, Rabinstein AA, Brown RD Jr. Treatment of carotid artery stenosis: medical therapy, surgery, or stenting? Mayo Clin Proc. 2009;84(4):362-387; quiz 367-368.
- Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370(26):2467-2477.
- Lavallée PC, Meseguer E, Abboud H, et al. A transient ischaemic attack clinic with round-the-clock access (SOS-TIA): feasibility and effects. Lancet Neurol. 2007;6(11):953-960.
- Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370(9596):1432-1442.
- Kernan WN, Ovbiagele B, Black HR, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160-2236.
- Roumie CL, Zillich AJ, Bravata DM, et al. Hypertension treatment intensification among stroke survivors with uncontrolled blood pressure. Stroke. 2015;46(2):465-470.
- Amarenco P, Bogousslavsky J, Callahan A, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355(6):549-559.
- Lennon O, Galvin R, Smith K, Doody C, Blake C. Lifestyle interventions for secondary disease prevention in stroke and transient ischaemic attack: a systematic review. Eur J Prev Cardiol. 2014;21(8):1026-1039.
Case
Mr. G is an 80-year-old man with a pacemaker, peripheral artery disease, atrial fibrillation (AF) on warfarin, and tachy-brady syndrome. He presented after experiencing episodes in which he was unable to speak and had weakness on his right side. He had a normal neurological exam upon arrival to the ED, and his blood pressure was 160/80 mm Hg.
Overview
Transient ischemic attacks (TIAs) are brief interruptions in brain perfusion that do not result in permanent neurologic damage. Up to half a million TIAs occur each year in the U.S., and they account for one third of acute cerebrovascular disease.1 While the term suggests that TIAs are benign, they are in fact an important warning sign of impending stroke and are essentially analogous to unstable angina. Some 10% of TIAs convert to full strokes within 90 days, but growing evidence suggests appropriate interventions can decrease this risk to 3%.2
Unfortunately, the symptoms of TIA have usually resolved by the time patients arrive at the hospital, which makes them challenging to diagnose. This article provides a summary of how to diagnose TIA accurately, using a focused history informed by cerebrovascular localization; how to triage, evaluate, and risk stratify patients; and how to implement preventative strategies.
Review of the Data
Classically, TIAs are defined as lasting less than 24 hours; however, 24 hours is an arbitrary number, and most TIAs last less than one hour.1 Furthermore, this definition has evolved with advances in neuroimaging that reveal that up to 50% of classically defined TIAs have evidence of infarct on MRI.1 There is no absolute temporal cut-off after which infarct is always seen on MRI, but longer duration of symptoms correlates with a higher likelihood of infarct. To reconcile these observations, a recently proposed definition stipulates that a true TIA lasts no more than one hour and does not show evidence of infarct on MRI.3
The causes of TIA are identical to those for ischemic stroke. Cerebral ischemia can result from an embolus, arterial thrombosis, or hypoperfusion due to arterial stenosis. Emboli can be cardiac, most commonly due to AF, or non-cardiac, stemming from a ruptured atherosclerotic plaque in the aortic arch, the carotid or vertebral artery, or an intracranial vessel. Atherosclerotic disease in the carotid arteries or intracranial vessels can also lead to thrombosis and occlusion or flow-related TIAs as a result of severe stenosis.
Risk factors for TIA mirror those for heart disease. Non-modifiable risk factors include older age, black race, male sex, and family history of stroke. Modifiable factors include hypertension, hyperlipidemia, tobacco smoking, diabetes, and AF.4
Most of the time, patients’ symptoms will have resolved by the time they are evaluated by a physician. Therefore, the diagnosis of TIA relies almost exclusively on the patient history. Eliciting a good history helps physicians determine whether the episode of transient neurologic dysfunction was caused by cerebral ischemia, as opposed to another mechanism, such as migraine or seizure. This calls for a basic understanding of cerebrovascular anatomy (see Table 1).
Types of Ischemia
Anterior cerebral artery ischemia causes contralateral leg weakness because it supplies the medial frontal and parietal lobes, where the legs in the sensorimotor homunculus are represented. Middle cerebral artery (MCA) ischemia causes contralateral face and arm weakness out of proportion to leg weakness. Ischemia in Broca’s area of the brain, which is supplied by the left MCA, may also cause expressive aphasia. Transient monocular blindness is a TIA of the retina due to atheroemboli originating from the internal carotid artery. Vertebrobasilar TIA is less common than anterior circulation TIA and manifests with brainstem symptoms that include diplopia, dysarthria, dysphagia, vertigo, gait imbalance, and weakness. In general, language and motor symptoms are more specific for cerebral ischemia and therefore more worrisome for TIA than sensory symptoms.5
Once a clinical diagnosis of TIA is made, an ABCD2 score (age, blood pressure, clinical features, duration of TIA, presence of diabetes) can be used to predict the short-term risk of subsequent stroke (see Table 2).6,7 A general rule of thumb is to admit patients who present within 72 hours of the event and have an ABCD2 score of three or higher for observation, work-up, and initiation of secondary prevention.1
Although only a small percentage of patients with TIA will have a stroke during the period of observation in the hospital, this approach may be cost effective based on the assumption that hospitalized patients are more likely to receive intravenous tissue plasminogen activator.8 The decision should also be guided by clinical judgment. It is reasonable to admit a patient whose diagnostic workup cannot be rapidly completed.1
The workup for TIA includes routine labs, EKG with cardiac monitoring, and brain imaging. Labs are useful to evaluate for other mimics of TIA such as hyponatremia and glucose abnormalities. In addition, risk factors such as hyperlipidemia and diabetes should be evaluated with fasting lipid panel and blood glucose. The purpose of EKG and telemetry is to identify MI and capture paroxysmal AF. The goal of imaging is to ascertain the presence of vascular disease and to exclude a non-ischemic etiology. While less likely to cause transient neurologic symptoms, a hemorrhagic event must be ruled out, as it would trigger a different management pathway.
Imaging for TIA
There are two primary modes of brain imaging: computed tomography (CT) and MRI. Most patients who are suspected to have had a TIA undergo CT scan, and an infarct is seen about 20% of the time.1 The presence of an infarct usually correlates with the duration of symptoms and has prognostic value. In one study, a new infarct was associated with four times higher risk of stroke in the subsequent 90 days.9 Diffusion-weighted imaging, an MR-based technique, is the preferred modality when it is available because of its higher sensitivity and specificity for identifying acute lesions.1 In an international and multicenter study, incorporating imaging data increased the discriminatory power of stroke prediction.10
Extracranial imaging is mandatory to rule out carotid stenosis as a potential etiology of TIA. The least invasive modality is ultrasound, which can detect carotid stenosis with a sensitivity and specificity approaching 80%.1 While both the intra- and extracranial vasculature can be concurrently assessed using MR- or CT-angiography (CTA), this is not usually necessary in the acute setting, because only detecting carotid stenosis will result in a management change.1
Carotid endarterectomy is standard for symptomatic patients with greater than 70% stenosis and is a consideration for symptomatic patients with greater than 50% stenosis if it is the most probable explanation for the ischemic event.11 Despite a comprehensive workup, about 50% of TIA cases remain cryptogenic.12 In some of these patients, AF can be detected using extended ambulatory cardiac monitoring.12
The goal of admitting high-risk patients is to expedite workup and initiate therapy. Two studies have shown that immediate initiation of preventative treatment significantly reduces the risk of stroke by as much as 80%.13,14 Unless there is a specific indication for anticoagulation, all TIA patients should be started on an antiplatelet agent such as aspirin or clopidogrel. A large randomized trial conducted in China and published in 2013 demonstrated that dual antiplatelet therapy with aspirin and clopidogrel for 21 days, followed by clopidogrel monotherapy, reduced the risk of stroke compared to aspirin monotherapy. An international multicenter trial designed to test the efficacy of short-term dual antiplatelet therapy is ongoing, and if the benefit of this approach is confirmed, this will likely become the standard of care. Evidence-based indications for anticoagulation after TIA are restricted to AF and mural thrombus in the setting of recent MI. Patients with implanted mechanical devices, including left ventricular assist devices and metal heart valves, should also receive anticoagulation.15
Risk factors should also be targeted in every case. Hypertension should be treated with a goal of lower than 140/90 mm Hg (or 130/80 mm Hg in diabetics and those with renal disease). Studies have shown that patients who are discharged with a blood pressure lower than 140/90 mm Hg are more likely to maintain this blood pressure at one-year follow-up.16 The choice of medication is less well studied, but drugs that act on the renin-angiotensin-aldosterone system and thiazides are generally preferred.15 Treatment with a statin is recommended after cerebrovascular ischemic events, with a goal LDL under 100. This reduces risk of secondary stroke by about 20%.17
At discharge, it is also important to counsel patients on their role in preventing strokes. As with many diseases, making lifestyle changes is key to stroke prevention. Encourage smoking cessation and an increase in physical activity, and discourage heavy alcohol use. The association between smoking and the risk for first stroke is well established. Moderate to high-intensity exercise can reduce secondary stroke risk by as much as 50%18 (see Table 3). While light alcohol consumption can be protective against strokes, heavy use is strongly discouraged. Emerging data suggest obstructive sleep apnea (OSA) may be another modifiable risk factor for stroke and TIA, so screening for potential OSA and referral may be needed.15
Back to the Case
When Mr. G arrived at the ED, his symptoms had resolved. Based on the history of expressive aphasia and right-sided weakness, he most likely had a TIA in the left MCA territory. Hemorrhage was ruled out with a non-contrast head CT. His pacemaker precluded obtaining an MRI. CTA revealed diffuse atherosclerotic disease without evidence of carotid stenosis. His ABCD2 score was six given his age, blood pressure, weakness, and symptom duration, and he was admitted for an expedited workup. His sodium and glucose were within normal limits. His hemoglobin A1c was 6.5%, his LDL was 120, and his international normalized ratio (INR) was therapeutic at 2.1. His TIA may have been due to AF, despite a therapeutic INR, because warfarin does not fully eliminate the stroke risk. It might also have been caused by intracranial atherosclerosis.
Two days later, the patient was discharged on atorvastatin at 80 mg, and his lisinopril was increased for blood pressure control. For his age group, A1c of 6.5% was acceptable, and he was not initiated on glycemic control.
Bottom Line
TIAs are diagnosed based on patient history. Urgent initiation of secondary prevention is important to reduce the short-term risk of stroke and should be implemented by the time of discharge from the hospital.
Dr. Zeng is a hospitalist in the department of internal medicine at Vanderbilt University Medical Center in Nashville, and Dr. Douglas is associate professor in the department of neurology at the University of California at San Francisco.
References
- Easton JD, Saver JL, Albers GW, et al. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009;40(6):2276-2293.
- Sundararajan V, Thrift AG, Phan TG, Choi PM, Clissold B, Srikanth VK. Trends over time in the risk of stroke after an incident transient ischemic attack. Stroke. 2014;45(11):3214-3218.
- Albers GW, Caplan LR, Easton JD, et al. Transient ischemic attack–proposal for a new definition. N Engl J Med. 2002;347(21):1713-1716.
- Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin. 2008;26(4):871-895, vii.
- Johnston SC, Sidney S, Bernstein AL, Gress DR. A comparison of risk factors for recurrent TIA and stroke in patients diagnosed with TIA. Neurology. 2003;60(2):280-285.
- Tsivgoulis G, Stamboulis E, Sharma VK, et al. Multicenter external validation of the ABCD2 score in triaging TIA patients. Neurology. 2010;74(17):1351-1357.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet. 2007;369(9558):283-292.
- Nguyen-Huynh MN, Johnston SC. Is hospitalization after TIA cost-effective on the basis of treatment with tPA? Neurology. 2005;65(11):1799-1801.
- Douglas VC, Johnston CM, Elkins J, Sidney S, Gress DR, Johnston SC. Head computed tomography findings predict short-term stroke risk after transient ischemic attack. Stroke. 2003;34(12):2894-2898.
- Giles MF, Albers GW, Amarenco P, et al. Addition of brain infarction to the ABCD2 Score (ABCD2I): a collaborative analysis of unpublished data on 4574 patients. Stroke. 2010;41(9):1907-1913.
- Lanzino G, Rabinstein AA, Brown RD Jr. Treatment of carotid artery stenosis: medical therapy, surgery, or stenting? Mayo Clin Proc. 2009;84(4):362-387; quiz 367-368.
- Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370(26):2467-2477.
- Lavallée PC, Meseguer E, Abboud H, et al. A transient ischaemic attack clinic with round-the-clock access (SOS-TIA): feasibility and effects. Lancet Neurol. 2007;6(11):953-960.
- Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370(9596):1432-1442.
- Kernan WN, Ovbiagele B, Black HR, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160-2236.
- Roumie CL, Zillich AJ, Bravata DM, et al. Hypertension treatment intensification among stroke survivors with uncontrolled blood pressure. Stroke. 2015;46(2):465-470.
- Amarenco P, Bogousslavsky J, Callahan A, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355(6):549-559.
- Lennon O, Galvin R, Smith K, Doody C, Blake C. Lifestyle interventions for secondary disease prevention in stroke and transient ischaemic attack: a systematic review. Eur J Prev Cardiol. 2014;21(8):1026-1039.
Case
Mr. G is an 80-year-old man with a pacemaker, peripheral artery disease, atrial fibrillation (AF) on warfarin, and tachy-brady syndrome. He presented after experiencing episodes in which he was unable to speak and had weakness on his right side. He had a normal neurological exam upon arrival to the ED, and his blood pressure was 160/80 mm Hg.
Overview
Transient ischemic attacks (TIAs) are brief interruptions in brain perfusion that do not result in permanent neurologic damage. Up to half a million TIAs occur each year in the U.S., and they account for one third of acute cerebrovascular disease.1 While the term suggests that TIAs are benign, they are in fact an important warning sign of impending stroke and are essentially analogous to unstable angina. Some 10% of TIAs convert to full strokes within 90 days, but growing evidence suggests appropriate interventions can decrease this risk to 3%.2
Unfortunately, the symptoms of TIA have usually resolved by the time patients arrive at the hospital, which makes them challenging to diagnose. This article provides a summary of how to diagnose TIA accurately, using a focused history informed by cerebrovascular localization; how to triage, evaluate, and risk stratify patients; and how to implement preventative strategies.
Review of the Data
Classically, TIAs are defined as lasting less than 24 hours; however, 24 hours is an arbitrary number, and most TIAs last less than one hour.1 Furthermore, this definition has evolved with advances in neuroimaging that reveal that up to 50% of classically defined TIAs have evidence of infarct on MRI.1 There is no absolute temporal cut-off after which infarct is always seen on MRI, but longer duration of symptoms correlates with a higher likelihood of infarct. To reconcile these observations, a recently proposed definition stipulates that a true TIA lasts no more than one hour and does not show evidence of infarct on MRI.3
The causes of TIA are identical to those for ischemic stroke. Cerebral ischemia can result from an embolus, arterial thrombosis, or hypoperfusion due to arterial stenosis. Emboli can be cardiac, most commonly due to AF, or non-cardiac, stemming from a ruptured atherosclerotic plaque in the aortic arch, the carotid or vertebral artery, or an intracranial vessel. Atherosclerotic disease in the carotid arteries or intracranial vessels can also lead to thrombosis and occlusion or flow-related TIAs as a result of severe stenosis.
Risk factors for TIA mirror those for heart disease. Non-modifiable risk factors include older age, black race, male sex, and family history of stroke. Modifiable factors include hypertension, hyperlipidemia, tobacco smoking, diabetes, and AF.4
Most of the time, patients’ symptoms will have resolved by the time they are evaluated by a physician. Therefore, the diagnosis of TIA relies almost exclusively on the patient history. Eliciting a good history helps physicians determine whether the episode of transient neurologic dysfunction was caused by cerebral ischemia, as opposed to another mechanism, such as migraine or seizure. This calls for a basic understanding of cerebrovascular anatomy (see Table 1).
Types of Ischemia
Anterior cerebral artery ischemia causes contralateral leg weakness because it supplies the medial frontal and parietal lobes, where the legs in the sensorimotor homunculus are represented. Middle cerebral artery (MCA) ischemia causes contralateral face and arm weakness out of proportion to leg weakness. Ischemia in Broca’s area of the brain, which is supplied by the left MCA, may also cause expressive aphasia. Transient monocular blindness is a TIA of the retina due to atheroemboli originating from the internal carotid artery. Vertebrobasilar TIA is less common than anterior circulation TIA and manifests with brainstem symptoms that include diplopia, dysarthria, dysphagia, vertigo, gait imbalance, and weakness. In general, language and motor symptoms are more specific for cerebral ischemia and therefore more worrisome for TIA than sensory symptoms.5
Once a clinical diagnosis of TIA is made, an ABCD2 score (age, blood pressure, clinical features, duration of TIA, presence of diabetes) can be used to predict the short-term risk of subsequent stroke (see Table 2).6,7 A general rule of thumb is to admit patients who present within 72 hours of the event and have an ABCD2 score of three or higher for observation, work-up, and initiation of secondary prevention.1
Although only a small percentage of patients with TIA will have a stroke during the period of observation in the hospital, this approach may be cost effective based on the assumption that hospitalized patients are more likely to receive intravenous tissue plasminogen activator.8 The decision should also be guided by clinical judgment. It is reasonable to admit a patient whose diagnostic workup cannot be rapidly completed.1
The workup for TIA includes routine labs, EKG with cardiac monitoring, and brain imaging. Labs are useful to evaluate for other mimics of TIA such as hyponatremia and glucose abnormalities. In addition, risk factors such as hyperlipidemia and diabetes should be evaluated with fasting lipid panel and blood glucose. The purpose of EKG and telemetry is to identify MI and capture paroxysmal AF. The goal of imaging is to ascertain the presence of vascular disease and to exclude a non-ischemic etiology. While less likely to cause transient neurologic symptoms, a hemorrhagic event must be ruled out, as it would trigger a different management pathway.
Imaging for TIA
There are two primary modes of brain imaging: computed tomography (CT) and MRI. Most patients who are suspected to have had a TIA undergo CT scan, and an infarct is seen about 20% of the time.1 The presence of an infarct usually correlates with the duration of symptoms and has prognostic value. In one study, a new infarct was associated with four times higher risk of stroke in the subsequent 90 days.9 Diffusion-weighted imaging, an MR-based technique, is the preferred modality when it is available because of its higher sensitivity and specificity for identifying acute lesions.1 In an international and multicenter study, incorporating imaging data increased the discriminatory power of stroke prediction.10
Extracranial imaging is mandatory to rule out carotid stenosis as a potential etiology of TIA. The least invasive modality is ultrasound, which can detect carotid stenosis with a sensitivity and specificity approaching 80%.1 While both the intra- and extracranial vasculature can be concurrently assessed using MR- or CT-angiography (CTA), this is not usually necessary in the acute setting, because only detecting carotid stenosis will result in a management change.1
Carotid endarterectomy is standard for symptomatic patients with greater than 70% stenosis and is a consideration for symptomatic patients with greater than 50% stenosis if it is the most probable explanation for the ischemic event.11 Despite a comprehensive workup, about 50% of TIA cases remain cryptogenic.12 In some of these patients, AF can be detected using extended ambulatory cardiac monitoring.12
The goal of admitting high-risk patients is to expedite workup and initiate therapy. Two studies have shown that immediate initiation of preventative treatment significantly reduces the risk of stroke by as much as 80%.13,14 Unless there is a specific indication for anticoagulation, all TIA patients should be started on an antiplatelet agent such as aspirin or clopidogrel. A large randomized trial conducted in China and published in 2013 demonstrated that dual antiplatelet therapy with aspirin and clopidogrel for 21 days, followed by clopidogrel monotherapy, reduced the risk of stroke compared to aspirin monotherapy. An international multicenter trial designed to test the efficacy of short-term dual antiplatelet therapy is ongoing, and if the benefit of this approach is confirmed, this will likely become the standard of care. Evidence-based indications for anticoagulation after TIA are restricted to AF and mural thrombus in the setting of recent MI. Patients with implanted mechanical devices, including left ventricular assist devices and metal heart valves, should also receive anticoagulation.15
Risk factors should also be targeted in every case. Hypertension should be treated with a goal of lower than 140/90 mm Hg (or 130/80 mm Hg in diabetics and those with renal disease). Studies have shown that patients who are discharged with a blood pressure lower than 140/90 mm Hg are more likely to maintain this blood pressure at one-year follow-up.16 The choice of medication is less well studied, but drugs that act on the renin-angiotensin-aldosterone system and thiazides are generally preferred.15 Treatment with a statin is recommended after cerebrovascular ischemic events, with a goal LDL under 100. This reduces risk of secondary stroke by about 20%.17
At discharge, it is also important to counsel patients on their role in preventing strokes. As with many diseases, making lifestyle changes is key to stroke prevention. Encourage smoking cessation and an increase in physical activity, and discourage heavy alcohol use. The association between smoking and the risk for first stroke is well established. Moderate to high-intensity exercise can reduce secondary stroke risk by as much as 50%18 (see Table 3). While light alcohol consumption can be protective against strokes, heavy use is strongly discouraged. Emerging data suggest obstructive sleep apnea (OSA) may be another modifiable risk factor for stroke and TIA, so screening for potential OSA and referral may be needed.15
Back to the Case
When Mr. G arrived at the ED, his symptoms had resolved. Based on the history of expressive aphasia and right-sided weakness, he most likely had a TIA in the left MCA territory. Hemorrhage was ruled out with a non-contrast head CT. His pacemaker precluded obtaining an MRI. CTA revealed diffuse atherosclerotic disease without evidence of carotid stenosis. His ABCD2 score was six given his age, blood pressure, weakness, and symptom duration, and he was admitted for an expedited workup. His sodium and glucose were within normal limits. His hemoglobin A1c was 6.5%, his LDL was 120, and his international normalized ratio (INR) was therapeutic at 2.1. His TIA may have been due to AF, despite a therapeutic INR, because warfarin does not fully eliminate the stroke risk. It might also have been caused by intracranial atherosclerosis.
Two days later, the patient was discharged on atorvastatin at 80 mg, and his lisinopril was increased for blood pressure control. For his age group, A1c of 6.5% was acceptable, and he was not initiated on glycemic control.
Bottom Line
TIAs are diagnosed based on patient history. Urgent initiation of secondary prevention is important to reduce the short-term risk of stroke and should be implemented by the time of discharge from the hospital.
Dr. Zeng is a hospitalist in the department of internal medicine at Vanderbilt University Medical Center in Nashville, and Dr. Douglas is associate professor in the department of neurology at the University of California at San Francisco.
References
- Easton JD, Saver JL, Albers GW, et al. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009;40(6):2276-2293.
- Sundararajan V, Thrift AG, Phan TG, Choi PM, Clissold B, Srikanth VK. Trends over time in the risk of stroke after an incident transient ischemic attack. Stroke. 2014;45(11):3214-3218.
- Albers GW, Caplan LR, Easton JD, et al. Transient ischemic attack–proposal for a new definition. N Engl J Med. 2002;347(21):1713-1716.
- Grysiewicz RA, Thomas K, Pandey DK. Epidemiology of ischemic and hemorrhagic stroke: incidence, prevalence, mortality, and risk factors. Neurol Clin. 2008;26(4):871-895, vii.
- Johnston SC, Sidney S, Bernstein AL, Gress DR. A comparison of risk factors for recurrent TIA and stroke in patients diagnosed with TIA. Neurology. 2003;60(2):280-285.
- Tsivgoulis G, Stamboulis E, Sharma VK, et al. Multicenter external validation of the ABCD2 score in triaging TIA patients. Neurology. 2010;74(17):1351-1357.
- Johnston SC, Rothwell PM, Nguyen-Huynh MN, et al. Validation and refinement of scores to predict very early stroke risk after transient ischaemic attack. Lancet. 2007;369(9558):283-292.
- Nguyen-Huynh MN, Johnston SC. Is hospitalization after TIA cost-effective on the basis of treatment with tPA? Neurology. 2005;65(11):1799-1801.
- Douglas VC, Johnston CM, Elkins J, Sidney S, Gress DR, Johnston SC. Head computed tomography findings predict short-term stroke risk after transient ischemic attack. Stroke. 2003;34(12):2894-2898.
- Giles MF, Albers GW, Amarenco P, et al. Addition of brain infarction to the ABCD2 Score (ABCD2I): a collaborative analysis of unpublished data on 4574 patients. Stroke. 2010;41(9):1907-1913.
- Lanzino G, Rabinstein AA, Brown RD Jr. Treatment of carotid artery stenosis: medical therapy, surgery, or stenting? Mayo Clin Proc. 2009;84(4):362-387; quiz 367-368.
- Gladstone DJ, Spring M, Dorian P, et al. Atrial fibrillation in patients with cryptogenic stroke. N Engl J Med. 2014;370(26):2467-2477.
- Lavallée PC, Meseguer E, Abboud H, et al. A transient ischaemic attack clinic with round-the-clock access (SOS-TIA): feasibility and effects. Lancet Neurol. 2007;6(11):953-960.
- Rothwell PM, Giles MF, Chandratheva A, et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): a prospective population-based sequential comparison. Lancet. 2007;370(9596):1432-1442.
- Kernan WN, Ovbiagele B, Black HR, et al. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45(7):2160-2236.
- Roumie CL, Zillich AJ, Bravata DM, et al. Hypertension treatment intensification among stroke survivors with uncontrolled blood pressure. Stroke. 2015;46(2):465-470.
- Amarenco P, Bogousslavsky J, Callahan A, et al. High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med. 2006;355(6):549-559.
- Lennon O, Galvin R, Smith K, Doody C, Blake C. Lifestyle interventions for secondary disease prevention in stroke and transient ischaemic attack: a systematic review. Eur J Prev Cardiol. 2014;21(8):1026-1039.
When Should Hospitalists Order Continuous Cardiac Monitoring?
Case
Two patients on continuous cardiac monitoring (CCM) are admitted to the hospital. One is a 56-year-old man with hemodynamically stable sepsis secondary to pneumonia. There is no sign of arrhythmia on initial evaluation. The second patient is a 67-year-old man with a history of coronary artery disease (CAD) admitted with chest pain. Should these patients be admitted with CCM?
Overview
CCM was first introduced in hospitals in the early 1960s for heart rate and rhythm monitoring in coronary ICUs. Since that time, CCM has been widely used in the hospital setting among critically and noncritically ill patients. Some hospitals have a limited capacity for monitoring, which is dictated by bed or technology availability. Other hospitals have the ability to monitor any patient.
Guidelines from the American College of Cardiology (ACC) in 1991 and the American Heart Association (AHA) in 2004 guide inpatient use of CCM. These guidelines make recommendations based on the likelihood of patient benefit—will likely benefit, may benefit, unlikely to benefit—and are primarily based on expert opinion; rigorous clinical trial data is not available.1,2 Based on these guidelines, patients with primary cardiac diagnoses, including acute coronary syndrome (ACS), post-cardiac surgery, and arrhythmia, are the most likely to benefit from monitoring.2,3
In practical use, many hospitalists use CCM to detect signs of hemodynamic instability.3 Currently there is no data to support the idea that CCM is a safe or equivalent method of detecting hemodynamic instability compared to close clinical evaluation and frequent vital sign measurement. In fact, physicians overestimate the utility of CCM in guiding management decisions, and witnessed clinical deterioration is a more frequent factor in the decision to escalate the level of care of a patient.3,4
Guideline Recommendations

CCM is intended to identify life-threatening arrhythmias, ischemia, and QT prolongation (see Figure 1). The AHA guidelines address which patients will benefit from CCM; the main indications include an acute cardiac diagnosis or critical illness.1
In addition, the AHA guidelines provide recommendations for the duration of monitoring. These recommendations vary from time-limited monitoring (e.g. unexplained syncope) to a therapeutic-based recommendation (e.g. high-grade atrioventricular block requiring pacemaker placement).
The guidelines also identify a subset of patients who are unlikely to benefit from monitoring (Class III), including low-risk post-operative patients, patients with rate-controlled atrial fibrillation, and patients undergoing hemodialysis without other indications for monitoring.
Several studies have examined the frequency of CCM use. In one study of 236 admissions to a community hospital general ward population, approximately 50% of the 745 monitoring days were not indicated by ACC/AHA guidelines.5 In this study, only 5% of telemetry events occurred in patients without indications, and none of these events required any specific therapy.5 Thus, improved adherence to the ACC/AHA guidelines can decrease CCM use in patients who are unlikely to benefit.
Life-threatening arrhythmia detection. Cleverley and colleagues reported that patients who suffered a cardiac arrest on noncritical care units had a higher survival to hospital discharge if they were on CCM during the event.6 However, a similar study recently showed no benefit to cardiac monitoring for in-hospital arrest if patients were monitored remotely.7 Patients who experience a cardiac arrest in a noncritical care area may benefit from direct cardiac monitoring, though larger studies are needed to assess all potential confounding effects, including nurse-to-patient ratios, location of monitoring (remote or unit-based), advanced cardiac life support response times, and whether the event was witnessed.

Bottom line: AHA guidelines recommend use of CCM in patients with a higher likelihood of developing a life-threatening arrhythmia, including those with an ACS, those experiencing post-cardiac arrest, or those who are critically ill. Medical ward patients who should be monitored include those with acute or subacute congestive heart failure, syncope of unknown etiology, and uncontrolled atrial fibrillation.1
Ischemia surveillance. Computerized ST-segment monitoring has been available for high-risk post-operative patients and those with acute cardiac events since the mid-1980s. When properly used, it offers the ability to detect “silent” ischemia, which is associated with increased in-hospital complications and worse patient outcomes.
Computerized ST-segment monitoring is often associated with a high rate of false positive alarms, however, and has not been universally adopted. Recommendations for its use are based on expert opinion, because no randomized trial has shown that increasing the sensitivity of ischemia detection improves patient outcomes.
Bottom line: AHA guidelines recommend ST-segment monitoring in patients with early ACS and post-acute MI as well as in patients at high risk for silent ischemia, including high-risk post-operative patients.1
QT-interval monitoring. A corrected QT-interval (QTc) greater than 0.50 milliseconds correlates with a higher risk for torsades de pointes and is associated with higher mortality. In critically ill patients in a large academic medical center, guideline-based QT-interval monitoring showed poor specificity for predicting the development of QTc prolongation; however, the risk of QTc prolongation increased with the presence of multiple risk factors.8
Bottom line: AHA guidelines recommend QT-interval monitoring in patients with risk factors for QTc-prolongation, including those starting QTc-prolonging drugs, those with overdose of pro-arrhythmic drugs, those with new-onset bradyarrhythmias, those with severe hypokalemia or hypomagnesemia, and those who have experienced acute neurologic events.1
Recommendations Outside of Guidelines
Patients admitted to medical services for noncardiac diagnoses have a high rate of telemetry use and a perceived benefit associated with cardiac monitoring.3 Although guidelines for noncardiac patients to direct hospitalists on when to use this technology are lacking, there may be some utility in monitoring certain subsets of inpatients.
Sepsis. Patients with hemodynamically stable sepsis develop atrial fibrillation at a higher rate than patients without sepsis and have higher in-hospital mortality. Patients at highest risk are those who are elderly or have severe sepsis.7 CCM can identify atrial fibrillation in real time, which may allow for earlier intervention; however, it is important to consider that other modalities, such as patient symptoms, physical exam, and standard EKG, are potentially as effective at detecting atrial fibrillation as CCM.
Bottom line: Our recommendation is to use CCM in patients who are at higher risk, including elderly patients and those with severe sepsis, until sepsis has resolved and/or the patient is hemodynamically stable for 24 hours.
Alcohol withdrawal. Patients with severe alcohol withdrawal have an increased incidence of arrhythmia and ischemia during the detoxification process. Specifically, patients with delirium tremens and seizures are at higher risk for significant QTc prolongation and tachyarrhythmias.9
Bottom line: Our recommendation is to use CCM in patients with severe alcohol withdrawal and to discontinue monitoring once withdrawal has resolved.
COPD. Patients with COPD exacerbations have a high risk of in-hospital and long-term mortality. The highest risk for mortality appears to be in patients presenting with atrial or ventricular arrhythmias and those over 65 years old.10 There is no clear evidence that beta-agonist use in COPD exacerbations increases arrhythmias other than sinus tachycardia or is associated with worse outcomes.11
Bottom line: Our recommendation is to use CCM only in patients with COPD exacerbation who have other indications as described in the AHA guidelines.
CCM Disadvantages
Alarm fatigue. Alarm fatigue is defined as the desensitization of a clinician to an alarm stimulus, resulting from sensory overload and causing the response of an alarm to be delayed or dismissed.12 In 2014, the Emergency Care Research Institute named alarm hazards as the number one health technology hazard, noting that numerous alarms on a daily basis can lead to desensitization and “alarm fatigue.”
CCM, and the overuse of CCM in particular, contribute to alarm fatigue, which can lead to patient safety issues, including delays in treatment, medication errors, and potentially death.
Increased cost. Because telemetry requires specialized equipment and trained monitoring staff, cost can be significant. In addition to equipment, cost includes time spent by providers, nurses, and technicians interpreting the images and discussing findings with consultants, as well as the additional studies obtained as a result of identified arrhythmias.
Studies on CCM cost vary widely, with conservative estimates of approximately $53 to as much as $1,400 per patient per day in some hospitals.13
Lack of specificity. Because of the high sensitivity and low specificity of CCM, use of CCM in low-risk patients without indications increases the risk of misinterpreting false-positive findings as clinically significant. This can lead to errors in management, including overtesting, unnecessary consultation with subspecialists, and the potential for inappropriate invasive procedures.1
High-Value CCM Use
Because of the low value associated with cardiac monitoring in many patients and the high sensitivity of the guidelines to capture patients at high risk for cardiac events, many hospitals have sought to limit the overuse of this technology. The most successful interventions have targeted the electronic ordering system by requiring an indication and hardwiring an order duration based on guideline recommendations. In a recent study, this intervention led to a 70% decrease in usage and reported $4.8 million cost savings without increasing the rate of in-hospital rapid response or cardiac arrest.14
Systems-level interventions to decrease inappropriate initiation and facilitate discontinuation of cardiac monitoring are a proven way to increase compliance with guidelines and decrease the overuse of CCM.
Back to the Case
According to AHA guidelines, the only patient who has an indication for CCM is the 67-year-old man with known CAD and chest pain, and, accordingly, the patient was placed on CCM. The patient underwent evaluation for ACS, and monitoring was discontinued after 24 hours when ACS was ruled out. The 56-year-old man with sepsis responded to treatment of pneumonia and was not placed on CCM.
In general, patients admitted with acute cardiac-related diseases should be placed on CCM. Guidelines are lacking with respect to many noncardiac diseases, and we recommend a time-limited duration (typically 24 hours) if CCM is ordered for a patient with a special circumstance outside of guidelines (see Figure 3).
Key Takeaway
Hospitalists should use continuous cardiac monitoring for specific indications and not routinely for all patients.
Drs. Lacy and Rendon are hospitalists in the department of internal medicine at the University of New Mexico School of Medicine in Albuquerque. Dr. Davis is a resident in internal medicine at UNM, and Dr. Tolstrup is a cardiologist at UNM.
References
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation. 2004;110(17):2721-2746. doi:10.1161/01.CIR.0000145144.56673.59.
- Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. Emergency Cardiac Care Committee members. J Am Coll Cardiol. 1991;18(6):1431-1433.
- Najafi N, Auerbach A. Use and outcomes of telemetry monitoring on a medicine service. Arch Intern Med. 2012;172(17):1349-1350. doi:10.1001/archinternmed.2012.3163.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol. 1995;76(12):960-965.
- Curry JP, Hanson CW III, Russell MW, Hanna C, Devine G, Ochroch EA. The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97(5):1483-1487.
- Cleverley K, Mousavi N, Stronger L, et al. The impact of telemetry on survival of in-hospital cardiac arrests in non-critical care patients. Resuscitation. 2013;84(7):878-882. doi:10.1016/j.resuscitation.2013.01.038.
- Walkey AJ, Greiner MA, Heckbert SR, et al. Atrial fibrillation among Medicare beneficiaries hospitalized with sepsis: incidence and risk factors. Am Heart J. 2013;165(6):949-955.e3. doi:10.1016/j.ahj.2013.03.020.
- Pickham D, Helfenbein E, Shinn JA, Chan G, Funk M, Drew BJ. How many patients need QT interval monitoring in critical care units? Preliminary report of the QT in Practice study. J Electrocardiol. 2010;43(6):572-576. doi:10.1016/j.jelectrocard.2010.05.016.
- Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227. doi:2006/13/smw-11319.
- Fuso L, Incalzi RA, Pistelli R, et al. Predicting mortality of patients hospitalized for acutely exacerbated chronic obstructive pulmonary disease. Am J Med. 1995;98(3):272-277.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardiovascular effects of beta-agonists in patients with asthma and COPD: a meta-analysis. Chest. 2004;125(6):2309-2321.
- McCartney PR. Clinical alarm management. MCN Am J Matern Child Nurs. 2012;37(3):202. doi:10.1097/NMC.0b013e31824c5b4a.
- Benjamin EM, Klugman RA, Luckmann R, Fairchild DG, Abookire SA. Impact of cardiac telemetry on patient safety and cost. Am J Manag Care. 2013;19(6):e225-e232.
- Dressler R, Dryer MM, Coletti C, Mahoney D, Doorey AJ. Altering overuse of cardiac telemetry in non-intensive care unit settings by hardwiring the use of American Heart Association guidelines. JAMA Intern Med. 2014;174(11):1852-1854. doi:10.1001/jamainternmed.2014.4491.
Case
Two patients on continuous cardiac monitoring (CCM) are admitted to the hospital. One is a 56-year-old man with hemodynamically stable sepsis secondary to pneumonia. There is no sign of arrhythmia on initial evaluation. The second patient is a 67-year-old man with a history of coronary artery disease (CAD) admitted with chest pain. Should these patients be admitted with CCM?
Overview
CCM was first introduced in hospitals in the early 1960s for heart rate and rhythm monitoring in coronary ICUs. Since that time, CCM has been widely used in the hospital setting among critically and noncritically ill patients. Some hospitals have a limited capacity for monitoring, which is dictated by bed or technology availability. Other hospitals have the ability to monitor any patient.
Guidelines from the American College of Cardiology (ACC) in 1991 and the American Heart Association (AHA) in 2004 guide inpatient use of CCM. These guidelines make recommendations based on the likelihood of patient benefit—will likely benefit, may benefit, unlikely to benefit—and are primarily based on expert opinion; rigorous clinical trial data is not available.1,2 Based on these guidelines, patients with primary cardiac diagnoses, including acute coronary syndrome (ACS), post-cardiac surgery, and arrhythmia, are the most likely to benefit from monitoring.2,3
In practical use, many hospitalists use CCM to detect signs of hemodynamic instability.3 Currently there is no data to support the idea that CCM is a safe or equivalent method of detecting hemodynamic instability compared to close clinical evaluation and frequent vital sign measurement. In fact, physicians overestimate the utility of CCM in guiding management decisions, and witnessed clinical deterioration is a more frequent factor in the decision to escalate the level of care of a patient.3,4
Guideline Recommendations
CCM is intended to identify life-threatening arrhythmias, ischemia, and QT prolongation (see Figure 1). The AHA guidelines address which patients will benefit from CCM; the main indications include an acute cardiac diagnosis or critical illness.1
In addition, the AHA guidelines provide recommendations for the duration of monitoring. These recommendations vary from time-limited monitoring (e.g. unexplained syncope) to a therapeutic-based recommendation (e.g. high-grade atrioventricular block requiring pacemaker placement).
The guidelines also identify a subset of patients who are unlikely to benefit from monitoring (Class III), including low-risk post-operative patients, patients with rate-controlled atrial fibrillation, and patients undergoing hemodialysis without other indications for monitoring.
Several studies have examined the frequency of CCM use. In one study of 236 admissions to a community hospital general ward population, approximately 50% of the 745 monitoring days were not indicated by ACC/AHA guidelines.5 In this study, only 5% of telemetry events occurred in patients without indications, and none of these events required any specific therapy.5 Thus, improved adherence to the ACC/AHA guidelines can decrease CCM use in patients who are unlikely to benefit.
Life-threatening arrhythmia detection. Cleverley and colleagues reported that patients who suffered a cardiac arrest on noncritical care units had a higher survival to hospital discharge if they were on CCM during the event.6 However, a similar study recently showed no benefit to cardiac monitoring for in-hospital arrest if patients were monitored remotely.7 Patients who experience a cardiac arrest in a noncritical care area may benefit from direct cardiac monitoring, though larger studies are needed to assess all potential confounding effects, including nurse-to-patient ratios, location of monitoring (remote or unit-based), advanced cardiac life support response times, and whether the event was witnessed.
Bottom line: AHA guidelines recommend use of CCM in patients with a higher likelihood of developing a life-threatening arrhythmia, including those with an ACS, those experiencing post-cardiac arrest, or those who are critically ill. Medical ward patients who should be monitored include those with acute or subacute congestive heart failure, syncope of unknown etiology, and uncontrolled atrial fibrillation.1
Ischemia surveillance. Computerized ST-segment monitoring has been available for high-risk post-operative patients and those with acute cardiac events since the mid-1980s. When properly used, it offers the ability to detect “silent” ischemia, which is associated with increased in-hospital complications and worse patient outcomes.
Computerized ST-segment monitoring is often associated with a high rate of false positive alarms, however, and has not been universally adopted. Recommendations for its use are based on expert opinion, because no randomized trial has shown that increasing the sensitivity of ischemia detection improves patient outcomes.
Bottom line: AHA guidelines recommend ST-segment monitoring in patients with early ACS and post-acute MI as well as in patients at high risk for silent ischemia, including high-risk post-operative patients.1
QT-interval monitoring. A corrected QT-interval (QTc) greater than 0.50 milliseconds correlates with a higher risk for torsades de pointes and is associated with higher mortality. In critically ill patients in a large academic medical center, guideline-based QT-interval monitoring showed poor specificity for predicting the development of QTc prolongation; however, the risk of QTc prolongation increased with the presence of multiple risk factors.8
Bottom line: AHA guidelines recommend QT-interval monitoring in patients with risk factors for QTc-prolongation, including those starting QTc-prolonging drugs, those with overdose of pro-arrhythmic drugs, those with new-onset bradyarrhythmias, those with severe hypokalemia or hypomagnesemia, and those who have experienced acute neurologic events.1
Recommendations Outside of Guidelines
Patients admitted to medical services for noncardiac diagnoses have a high rate of telemetry use and a perceived benefit associated with cardiac monitoring.3 Although guidelines for noncardiac patients to direct hospitalists on when to use this technology are lacking, there may be some utility in monitoring certain subsets of inpatients.
Sepsis. Patients with hemodynamically stable sepsis develop atrial fibrillation at a higher rate than patients without sepsis and have higher in-hospital mortality. Patients at highest risk are those who are elderly or have severe sepsis.7 CCM can identify atrial fibrillation in real time, which may allow for earlier intervention; however, it is important to consider that other modalities, such as patient symptoms, physical exam, and standard EKG, are potentially as effective at detecting atrial fibrillation as CCM.
Bottom line: Our recommendation is to use CCM in patients who are at higher risk, including elderly patients and those with severe sepsis, until sepsis has resolved and/or the patient is hemodynamically stable for 24 hours.
Alcohol withdrawal. Patients with severe alcohol withdrawal have an increased incidence of arrhythmia and ischemia during the detoxification process. Specifically, patients with delirium tremens and seizures are at higher risk for significant QTc prolongation and tachyarrhythmias.9
Bottom line: Our recommendation is to use CCM in patients with severe alcohol withdrawal and to discontinue monitoring once withdrawal has resolved.
COPD. Patients with COPD exacerbations have a high risk of in-hospital and long-term mortality. The highest risk for mortality appears to be in patients presenting with atrial or ventricular arrhythmias and those over 65 years old.10 There is no clear evidence that beta-agonist use in COPD exacerbations increases arrhythmias other than sinus tachycardia or is associated with worse outcomes.11
Bottom line: Our recommendation is to use CCM only in patients with COPD exacerbation who have other indications as described in the AHA guidelines.
CCM Disadvantages
Alarm fatigue. Alarm fatigue is defined as the desensitization of a clinician to an alarm stimulus, resulting from sensory overload and causing the response of an alarm to be delayed or dismissed.12 In 2014, the Emergency Care Research Institute named alarm hazards as the number one health technology hazard, noting that numerous alarms on a daily basis can lead to desensitization and “alarm fatigue.”
CCM, and the overuse of CCM in particular, contribute to alarm fatigue, which can lead to patient safety issues, including delays in treatment, medication errors, and potentially death.
Increased cost. Because telemetry requires specialized equipment and trained monitoring staff, cost can be significant. In addition to equipment, cost includes time spent by providers, nurses, and technicians interpreting the images and discussing findings with consultants, as well as the additional studies obtained as a result of identified arrhythmias.
Studies on CCM cost vary widely, with conservative estimates of approximately $53 to as much as $1,400 per patient per day in some hospitals.13
Lack of specificity. Because of the high sensitivity and low specificity of CCM, use of CCM in low-risk patients without indications increases the risk of misinterpreting false-positive findings as clinically significant. This can lead to errors in management, including overtesting, unnecessary consultation with subspecialists, and the potential for inappropriate invasive procedures.1
High-Value CCM Use
Because of the low value associated with cardiac monitoring in many patients and the high sensitivity of the guidelines to capture patients at high risk for cardiac events, many hospitals have sought to limit the overuse of this technology. The most successful interventions have targeted the electronic ordering system by requiring an indication and hardwiring an order duration based on guideline recommendations. In a recent study, this intervention led to a 70% decrease in usage and reported $4.8 million cost savings without increasing the rate of in-hospital rapid response or cardiac arrest.14
Systems-level interventions to decrease inappropriate initiation and facilitate discontinuation of cardiac monitoring are a proven way to increase compliance with guidelines and decrease the overuse of CCM.
Back to the Case
According to AHA guidelines, the only patient who has an indication for CCM is the 67-year-old man with known CAD and chest pain, and, accordingly, the patient was placed on CCM. The patient underwent evaluation for ACS, and monitoring was discontinued after 24 hours when ACS was ruled out. The 56-year-old man with sepsis responded to treatment of pneumonia and was not placed on CCM.
In general, patients admitted with acute cardiac-related diseases should be placed on CCM. Guidelines are lacking with respect to many noncardiac diseases, and we recommend a time-limited duration (typically 24 hours) if CCM is ordered for a patient with a special circumstance outside of guidelines (see Figure 3).
Key Takeaway
Hospitalists should use continuous cardiac monitoring for specific indications and not routinely for all patients.
Drs. Lacy and Rendon are hospitalists in the department of internal medicine at the University of New Mexico School of Medicine in Albuquerque. Dr. Davis is a resident in internal medicine at UNM, and Dr. Tolstrup is a cardiologist at UNM.
References
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation. 2004;110(17):2721-2746. doi:10.1161/01.CIR.0000145144.56673.59.
- Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. Emergency Cardiac Care Committee members. J Am Coll Cardiol. 1991;18(6):1431-1433.
- Najafi N, Auerbach A. Use and outcomes of telemetry monitoring on a medicine service. Arch Intern Med. 2012;172(17):1349-1350. doi:10.1001/archinternmed.2012.3163.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol. 1995;76(12):960-965.
- Curry JP, Hanson CW III, Russell MW, Hanna C, Devine G, Ochroch EA. The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97(5):1483-1487.
- Cleverley K, Mousavi N, Stronger L, et al. The impact of telemetry on survival of in-hospital cardiac arrests in non-critical care patients. Resuscitation. 2013;84(7):878-882. doi:10.1016/j.resuscitation.2013.01.038.
- Walkey AJ, Greiner MA, Heckbert SR, et al. Atrial fibrillation among Medicare beneficiaries hospitalized with sepsis: incidence and risk factors. Am Heart J. 2013;165(6):949-955.e3. doi:10.1016/j.ahj.2013.03.020.
- Pickham D, Helfenbein E, Shinn JA, Chan G, Funk M, Drew BJ. How many patients need QT interval monitoring in critical care units? Preliminary report of the QT in Practice study. J Electrocardiol. 2010;43(6):572-576. doi:10.1016/j.jelectrocard.2010.05.016.
- Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227. doi:2006/13/smw-11319.
- Fuso L, Incalzi RA, Pistelli R, et al. Predicting mortality of patients hospitalized for acutely exacerbated chronic obstructive pulmonary disease. Am J Med. 1995;98(3):272-277.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardiovascular effects of beta-agonists in patients with asthma and COPD: a meta-analysis. Chest. 2004;125(6):2309-2321.
- McCartney PR. Clinical alarm management. MCN Am J Matern Child Nurs. 2012;37(3):202. doi:10.1097/NMC.0b013e31824c5b4a.
- Benjamin EM, Klugman RA, Luckmann R, Fairchild DG, Abookire SA. Impact of cardiac telemetry on patient safety and cost. Am J Manag Care. 2013;19(6):e225-e232.
- Dressler R, Dryer MM, Coletti C, Mahoney D, Doorey AJ. Altering overuse of cardiac telemetry in non-intensive care unit settings by hardwiring the use of American Heart Association guidelines. JAMA Intern Med. 2014;174(11):1852-1854. doi:10.1001/jamainternmed.2014.4491.
Case
Two patients on continuous cardiac monitoring (CCM) are admitted to the hospital. One is a 56-year-old man with hemodynamically stable sepsis secondary to pneumonia. There is no sign of arrhythmia on initial evaluation. The second patient is a 67-year-old man with a history of coronary artery disease (CAD) admitted with chest pain. Should these patients be admitted with CCM?
Overview
CCM was first introduced in hospitals in the early 1960s for heart rate and rhythm monitoring in coronary ICUs. Since that time, CCM has been widely used in the hospital setting among critically and noncritically ill patients. Some hospitals have a limited capacity for monitoring, which is dictated by bed or technology availability. Other hospitals have the ability to monitor any patient.
Guidelines from the American College of Cardiology (ACC) in 1991 and the American Heart Association (AHA) in 2004 guide inpatient use of CCM. These guidelines make recommendations based on the likelihood of patient benefit—will likely benefit, may benefit, unlikely to benefit—and are primarily based on expert opinion; rigorous clinical trial data is not available.1,2 Based on these guidelines, patients with primary cardiac diagnoses, including acute coronary syndrome (ACS), post-cardiac surgery, and arrhythmia, are the most likely to benefit from monitoring.2,3
In practical use, many hospitalists use CCM to detect signs of hemodynamic instability.3 Currently there is no data to support the idea that CCM is a safe or equivalent method of detecting hemodynamic instability compared to close clinical evaluation and frequent vital sign measurement. In fact, physicians overestimate the utility of CCM in guiding management decisions, and witnessed clinical deterioration is a more frequent factor in the decision to escalate the level of care of a patient.3,4
Guideline Recommendations
CCM is intended to identify life-threatening arrhythmias, ischemia, and QT prolongation (see Figure 1). The AHA guidelines address which patients will benefit from CCM; the main indications include an acute cardiac diagnosis or critical illness.1
In addition, the AHA guidelines provide recommendations for the duration of monitoring. These recommendations vary from time-limited monitoring (e.g. unexplained syncope) to a therapeutic-based recommendation (e.g. high-grade atrioventricular block requiring pacemaker placement).
The guidelines also identify a subset of patients who are unlikely to benefit from monitoring (Class III), including low-risk post-operative patients, patients with rate-controlled atrial fibrillation, and patients undergoing hemodialysis without other indications for monitoring.
Several studies have examined the frequency of CCM use. In one study of 236 admissions to a community hospital general ward population, approximately 50% of the 745 monitoring days were not indicated by ACC/AHA guidelines.5 In this study, only 5% of telemetry events occurred in patients without indications, and none of these events required any specific therapy.5 Thus, improved adherence to the ACC/AHA guidelines can decrease CCM use in patients who are unlikely to benefit.
Life-threatening arrhythmia detection. Cleverley and colleagues reported that patients who suffered a cardiac arrest on noncritical care units had a higher survival to hospital discharge if they were on CCM during the event.6 However, a similar study recently showed no benefit to cardiac monitoring for in-hospital arrest if patients were monitored remotely.7 Patients who experience a cardiac arrest in a noncritical care area may benefit from direct cardiac monitoring, though larger studies are needed to assess all potential confounding effects, including nurse-to-patient ratios, location of monitoring (remote or unit-based), advanced cardiac life support response times, and whether the event was witnessed.
Bottom line: AHA guidelines recommend use of CCM in patients with a higher likelihood of developing a life-threatening arrhythmia, including those with an ACS, those experiencing post-cardiac arrest, or those who are critically ill. Medical ward patients who should be monitored include those with acute or subacute congestive heart failure, syncope of unknown etiology, and uncontrolled atrial fibrillation.1
Ischemia surveillance. Computerized ST-segment monitoring has been available for high-risk post-operative patients and those with acute cardiac events since the mid-1980s. When properly used, it offers the ability to detect “silent” ischemia, which is associated with increased in-hospital complications and worse patient outcomes.
Computerized ST-segment monitoring is often associated with a high rate of false positive alarms, however, and has not been universally adopted. Recommendations for its use are based on expert opinion, because no randomized trial has shown that increasing the sensitivity of ischemia detection improves patient outcomes.
Bottom line: AHA guidelines recommend ST-segment monitoring in patients with early ACS and post-acute MI as well as in patients at high risk for silent ischemia, including high-risk post-operative patients.1
QT-interval monitoring. A corrected QT-interval (QTc) greater than 0.50 milliseconds correlates with a higher risk for torsades de pointes and is associated with higher mortality. In critically ill patients in a large academic medical center, guideline-based QT-interval monitoring showed poor specificity for predicting the development of QTc prolongation; however, the risk of QTc prolongation increased with the presence of multiple risk factors.8
Bottom line: AHA guidelines recommend QT-interval monitoring in patients with risk factors for QTc-prolongation, including those starting QTc-prolonging drugs, those with overdose of pro-arrhythmic drugs, those with new-onset bradyarrhythmias, those with severe hypokalemia or hypomagnesemia, and those who have experienced acute neurologic events.1
Recommendations Outside of Guidelines
Patients admitted to medical services for noncardiac diagnoses have a high rate of telemetry use and a perceived benefit associated with cardiac monitoring.3 Although guidelines for noncardiac patients to direct hospitalists on when to use this technology are lacking, there may be some utility in monitoring certain subsets of inpatients.
Sepsis. Patients with hemodynamically stable sepsis develop atrial fibrillation at a higher rate than patients without sepsis and have higher in-hospital mortality. Patients at highest risk are those who are elderly or have severe sepsis.7 CCM can identify atrial fibrillation in real time, which may allow for earlier intervention; however, it is important to consider that other modalities, such as patient symptoms, physical exam, and standard EKG, are potentially as effective at detecting atrial fibrillation as CCM.
Bottom line: Our recommendation is to use CCM in patients who are at higher risk, including elderly patients and those with severe sepsis, until sepsis has resolved and/or the patient is hemodynamically stable for 24 hours.
Alcohol withdrawal. Patients with severe alcohol withdrawal have an increased incidence of arrhythmia and ischemia during the detoxification process. Specifically, patients with delirium tremens and seizures are at higher risk for significant QTc prolongation and tachyarrhythmias.9
Bottom line: Our recommendation is to use CCM in patients with severe alcohol withdrawal and to discontinue monitoring once withdrawal has resolved.
COPD. Patients with COPD exacerbations have a high risk of in-hospital and long-term mortality. The highest risk for mortality appears to be in patients presenting with atrial or ventricular arrhythmias and those over 65 years old.10 There is no clear evidence that beta-agonist use in COPD exacerbations increases arrhythmias other than sinus tachycardia or is associated with worse outcomes.11
Bottom line: Our recommendation is to use CCM only in patients with COPD exacerbation who have other indications as described in the AHA guidelines.
CCM Disadvantages
Alarm fatigue. Alarm fatigue is defined as the desensitization of a clinician to an alarm stimulus, resulting from sensory overload and causing the response of an alarm to be delayed or dismissed.12 In 2014, the Emergency Care Research Institute named alarm hazards as the number one health technology hazard, noting that numerous alarms on a daily basis can lead to desensitization and “alarm fatigue.”
CCM, and the overuse of CCM in particular, contribute to alarm fatigue, which can lead to patient safety issues, including delays in treatment, medication errors, and potentially death.
Increased cost. Because telemetry requires specialized equipment and trained monitoring staff, cost can be significant. In addition to equipment, cost includes time spent by providers, nurses, and technicians interpreting the images and discussing findings with consultants, as well as the additional studies obtained as a result of identified arrhythmias.
Studies on CCM cost vary widely, with conservative estimates of approximately $53 to as much as $1,400 per patient per day in some hospitals.13
Lack of specificity. Because of the high sensitivity and low specificity of CCM, use of CCM in low-risk patients without indications increases the risk of misinterpreting false-positive findings as clinically significant. This can lead to errors in management, including overtesting, unnecessary consultation with subspecialists, and the potential for inappropriate invasive procedures.1
High-Value CCM Use
Because of the low value associated with cardiac monitoring in many patients and the high sensitivity of the guidelines to capture patients at high risk for cardiac events, many hospitals have sought to limit the overuse of this technology. The most successful interventions have targeted the electronic ordering system by requiring an indication and hardwiring an order duration based on guideline recommendations. In a recent study, this intervention led to a 70% decrease in usage and reported $4.8 million cost savings without increasing the rate of in-hospital rapid response or cardiac arrest.14
Systems-level interventions to decrease inappropriate initiation and facilitate discontinuation of cardiac monitoring are a proven way to increase compliance with guidelines and decrease the overuse of CCM.
Back to the Case
According to AHA guidelines, the only patient who has an indication for CCM is the 67-year-old man with known CAD and chest pain, and, accordingly, the patient was placed on CCM. The patient underwent evaluation for ACS, and monitoring was discontinued after 24 hours when ACS was ruled out. The 56-year-old man with sepsis responded to treatment of pneumonia and was not placed on CCM.
In general, patients admitted with acute cardiac-related diseases should be placed on CCM. Guidelines are lacking with respect to many noncardiac diseases, and we recommend a time-limited duration (typically 24 hours) if CCM is ordered for a patient with a special circumstance outside of guidelines (see Figure 3).
Key Takeaway
Hospitalists should use continuous cardiac monitoring for specific indications and not routinely for all patients.
Drs. Lacy and Rendon are hospitalists in the department of internal medicine at the University of New Mexico School of Medicine in Albuquerque. Dr. Davis is a resident in internal medicine at UNM, and Dr. Tolstrup is a cardiologist at UNM.
References
- Drew BJ, Califf RM, Funk M, et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical-Care Nurses. Circulation. 2004;110(17):2721-2746. doi:10.1161/01.CIR.0000145144.56673.59.
- Recommended guidelines for in-hospital cardiac monitoring of adults for detection of arrhythmia. Emergency Cardiac Care Committee members. J Am Coll Cardiol. 1991;18(6):1431-1433.
- Najafi N, Auerbach A. Use and outcomes of telemetry monitoring on a medicine service. Arch Intern Med. 2012;172(17):1349-1350. doi:10.1001/archinternmed.2012.3163.
- Estrada CA, Rosman HS, Prasad NK, et al. Role of telemetry monitoring in the non-intensive care unit. Am J Cardiol. 1995;76(12):960-965.
- Curry JP, Hanson CW III, Russell MW, Hanna C, Devine G, Ochroch EA. The use and effectiveness of electrocardiographic telemetry monitoring in a community hospital general care setting. Anesth Analg. 2003;97(5):1483-1487.
- Cleverley K, Mousavi N, Stronger L, et al. The impact of telemetry on survival of in-hospital cardiac arrests in non-critical care patients. Resuscitation. 2013;84(7):878-882. doi:10.1016/j.resuscitation.2013.01.038.
- Walkey AJ, Greiner MA, Heckbert SR, et al. Atrial fibrillation among Medicare beneficiaries hospitalized with sepsis: incidence and risk factors. Am Heart J. 2013;165(6):949-955.e3. doi:10.1016/j.ahj.2013.03.020.
- Pickham D, Helfenbein E, Shinn JA, Chan G, Funk M, Drew BJ. How many patients need QT interval monitoring in critical care units? Preliminary report of the QT in Practice study. J Electrocardiol. 2010;43(6):572-576. doi:10.1016/j.jelectrocard.2010.05.016.
- Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227. doi:2006/13/smw-11319.
- Fuso L, Incalzi RA, Pistelli R, et al. Predicting mortality of patients hospitalized for acutely exacerbated chronic obstructive pulmonary disease. Am J Med. 1995;98(3):272-277.
- Salpeter SR, Ormiston TM, Salpeter EE. Cardiovascular effects of beta-agonists in patients with asthma and COPD: a meta-analysis. Chest. 2004;125(6):2309-2321.
- McCartney PR. Clinical alarm management. MCN Am J Matern Child Nurs. 2012;37(3):202. doi:10.1097/NMC.0b013e31824c5b4a.
- Benjamin EM, Klugman RA, Luckmann R, Fairchild DG, Abookire SA. Impact of cardiac telemetry on patient safety and cost. Am J Manag Care. 2013;19(6):e225-e232.
- Dressler R, Dryer MM, Coletti C, Mahoney D, Doorey AJ. Altering overuse of cardiac telemetry in non-intensive care unit settings by hardwiring the use of American Heart Association guidelines. JAMA Intern Med. 2014;174(11):1852-1854. doi:10.1001/jamainternmed.2014.4491.
Of Mice and Men
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
Fever in an elderly man is a nonspecific finding, occurring most commonly with infections but also with certain malignancies, rheumatologic disorders, and drug exposures. The complaint of rigors with diaphoresis makes an infection most likely. The acuity of his illness makes infections with more chronic presentations such as tuberculosis or actinomycosis less likely. The presence of frontal headache might suggest a sinus or brain source, but headache also occurs in generalized infections such as pneumonia, bacteremia from any cause, malaria, rickettsial infections, viral illnesses, and others. Additional history should include detailed inquiry into travel, vocational, and avocational exposures.
The patient's difficulty standing implies the development of lower extremity weakness and infections associated with neurological syndromes. His leg weakness may be related to early Guillain‐Barre syndrome, which is associated most commonly with Campylobacter jejuni, but also other bacteria and viruses such as Haemophilus influenza, Mycoplasma pneumonia, Influenza virus, Cytomegalovirus and hepatitis E. Other viral infections associated with pure motor deficits include echovirus, coxsackie virus, enterovirus, and West Nile virus (WNV). The paralytic syndrome associated with enteroviruses is more common in children, whereas the neuroinvasive variant of WNV more often affects the elderly and can be associated with encephalitis as well as a flaccid paralysis. Although acute paralytic shellfish poisoning could account for both his weakness and his acute gastrointestinal syndrome, this diagnosis is unlikely because the symptoms often have a prominent sensory component, and there is usually the history of recent ingestion of the suspect bivalves. Like all adults presenting for medical care, he should be screened for human immunodeficiency virus (HIV) infection; if testing is positive, the differential diagnosis for his current illness broadens significantly. Finally, he may have a spinal cord disorder or infection such as an epidural abscess, or transverse myelitis, which would present with lower extremity weakness and fever. It would be helpful to know the time of year of his illness, exposure to mosquito bites, his neurological exam findings, and results of blood and stool cultures. If the patient had signs of meningitis or encephalitis, cerebral spinal fluid analysis would be helpful. If his neurological exam was suggestive of cord involvement, it would be helpful to know the results of magnetic resonance imaging of the spinal cord.
The patient's past medical history includes relatively common problems for a 73‐year‐old man and does not substantially influence the differential diagnosis of his current illness. His travel history to Uganda a year previously may be relevant, because malaria (Plasmodium vivax) could present with fever and weakness. Less commonly, African trypanosomiasis (Trypanosoma brucei gambiense) can, in the late phase, present with fever and malaise, but also typically includes symptoms of encephalitis, including depressed mental status, confusion, ataxia, and possibly personality changes. His travel to Zurich should not impose any particular infection risk, unless he was hiking in the mountains around Zurich, where he could have contracted tick‐borne encephalitis; however, his travel more than 6 months prior to presentation makes this unlikely. Lyme disease due to Borrelia burgdorferi is also a potential exposure in the Swiss mountains, and can present with fever in the acute phase, as well as arthritis with chronic disease, but should not cause fever, rigor, diaphoresis, and headache many months later. Summering in Cape Cod puts him at risk for babesiosis, but an incubation period of 5 months is too long. Keeping chickens places him at risk for Salmonella exposure and typhoid fever. Ingesting raw oysters carries a risk for shellfish poisoning and Vibrio infections, but the incubation period (1 month) again seems too long to cause his current symptoms.
Notable physical findings are an ill‐appearing man with injected sclera and a high fever but normal blood pressure and heart rate. He also demonstrates proximal lower extremity weakness manifested by difficulty rising from a chair and a slow gait with short strides and deliberate (possibly on‐block) turning. His neurological exam is most consistent with Parkinsonian symptoms that have been described in patients with severe influenza A, which would explain all of his other symptoms as well. Pulse‐temperature dissociation is classically described with typhoid fever but usually occurs later in the disease course, and could be masked by the patient's metoprolol. Typhoid fever can also be associated with neurological symptoms including meningitis and movement disorders.
The patient has a remarkable bandemia, suggesting a bacterial infection, as well as a slight reduction in hematocrit and platelet count. Additionally, his labs revealed a mild transaminitis, but with significantly elevated alkaline phosphatase and GGT, and microscopic hematuria. His ferritin is significantly elevated, which may simply represent an acute phase reactant. Infections associated with hepatitis, cytopenias, and hematuria include sepsis with disseminated intravascular coagulation, previously mentioned malaria, leptospirosis, dengue, ehrlichiosis, and rickettsial diseases, but he has no special risks for these infections, and other aspects of his illness (Parkinsonian features, bandemia) do not fit. His lung findings with hematuria might suggest a pulmonary/renal syndrome, but, once again, other features of his illness are not typical of these syndromes. Salmonella (typhoid fever) or influenza, now complicated by an early bacterial pneumonia, are viable possibilities.
The patient's ongoing clinical course is notable for a nontoxic (non‐SIRS) appearance but continued high‐grade fever with blood and urine cultures that are sterile. This argues against a common bacteremia with sepsis, and for either relapsing malaria (P vivax), influenza with a Parkinsonian‐like illness, typhoid fever, leptospirosis, dengue, or a rickettsial infection. Mycoplasma pneumonia is also possible given the atypical chest x‐ray appearance, slightly low hematocrit with elevated bilirubin, and neurological symptoms that may represent ataxia.
The subsequent negative laboratory tests listed are helpful in likely excluding many of the diagnoses suggested such as malaria, Babesia, common bacteremias, viral hepatitis, HIV, and WNV. Furthermore, the new history of mouse exposure brings to the forefront rodent‐associated infections, specifically exposure to mouse urine, a vehicle for leptospirosis. The patient's hepatitis, anemia, thrombocytopenia, scleral injection, along with the rest of his symptoms in the context of exposure to mouse urine makes leptospirosis the likely diagnosis. A negative Leptospira antibody early in his illness does not rule out the disease, and a convalescent titer should be obtained to confirm the diagnosis.
COMMENTARY
This case describes an elderly man who presented with a fever of unknown origin (FUO), and was eventually diagnosed with leptospirosis. FUO presents slightly differently in elderly patients, as elderly patients are less likely to mount a high fever, and when they do, the etiology is more likely to indicate a serious bacterial or viral infection. Additionally, an etiology for FUO in the elderly is found in over 70% of presenting cases, compared to 51% in patients under the age of 65 years.[1] A detailed, comprehensive social, travel, and exposure history and physical examination remains the cornerstone of elucidating the diagnosis for FUO. The exposure to mouse urine in this case was an unusual and a helpful piece of the history to further focus the differential diagnosis.
Leptospirosis is an emerging bacterial zoonosis, and causes both endemic and epidemic severe multisystem disease. The Leptospira spirochete is maintained in nature through a chronic renal infection in mammalian reservoir hosts, such as mice,[2, 4] and is transmitted through direct or aerosolized contact with infected urine or tissue. After a mean incubation period of 10 days, a variety of clinical manifestations may be seen. In this case, the patient's clinical presentation revealed many classic symptoms of leptospirosis, including fevers, rigors, headache, lower extremity myalgias, nausea, vomiting, and diarrhea; however, these symptoms are nonspecific. The presence of a conjunctival suffusion in leptospirosis infection had a specificity of 98% in a high‐incidence cohort of febrile patients in Sri Lanka,[3] and was an important diagnostic clue in this case. Leptospirosis is a self‐limited illness in most patients, with an initial septicemic, febrile phase followed by an immune phase. A more severe presentation may be seen in the immune phase of the illness, which includes renal and hepatic dysfunction (known as Weil's disease), as well as cardiac, pulmonary, and central nervous system abnormalities. With a 14% case fatality rate, the risk of death has been shown to be higher in patients over 40 years old, with altered mental status and multiorgan failure.[4]
The early diagnosis of leptospirosis relies heavily on physical exam findings and epidemiologic history. In this case, the patient's laboratory abnormalities, including immature granulocytes, thrombocytopenia, hyponatremia, hypokalemia, mild hepatitis, and pyuria with granular casts are all reported with leptospirosis infection2; however, independently, these laboratory findings are nonspecific. Patients may not have a detectable antibody levels in the acute phase of the disease. In this case, given the strong clinical suspicion based on the findings of conjunctival suffusion and exposure to mouse urine history, the lack of a more plausible alternate diagnosis, and known delay in antibody positivity, the patient was treated empirically with doxycycline for presumed leptospirosis.[5] Forthcoming novel diagnostic strategies such as next‐generation DNA sequencing techniques may provide real‐time diagnosis of this zoonotic infection, thus decreasing the window period between empirical antimicrobial coverage and diagnostic confirmation.[6]
Leptospirosis is prevalent in tropical climates and has been associated with impoverished communities.[7] Urban slums, with poor sanitation and high rodent density, are an ideal environment for leptospirosis. The reported risk of infection in a Brazilian slum was as high as 3% per year.[8] Additionally, rodent sightings, as well as the presence of chickens, were risk factors for leptospirosis transmission in urban slums.[9] Correspondingly in this case, we hypothesize that the patient's interest in urban farming, specifically the chickens he kept, likely attracted the mice infected with leptospirosis. Urban chicken farming is becoming increasingly popular in the United States,[10] and may be a developing risk factor for human leptospirosis infection. Leptospirosis is one of many emerging zoonoses, such as avian influenza, tick‐borne illness, and ebola, resulting from changing human ecology. Thus, when considering infectious etiologies, clinicians should ask patients about vocational and avocational exposures, including new trends such as urban farming, which may expose them to previously underappreciated illnesses.
TEACHING POINTS
- Elderly patients with a FUO are more likely to be diagnosed with an underlying serious bacterial or viral infection when compared to a younger cohort of FUO patients.
- The diagnosis of leptospirosis may initially be based on clinical suspicion in patients with classic features and exposures, noting the high specificity of conjunctival suffusion, and initial titers may be nondiagnostic; therefore, empiric treatment should be considered when clinical suspicion is high.
- Increased interest in urban chicken farming in the United States, with associated higher rodent density, may represent a newly recognized risk factor for human leptospirosis infection.
Disclosures
The authors report no conflicts of interest.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
- , , . Fever of unknown origin in older persons. Infect Dis Clin North Am. 2007;21(4):937–945.
- . Leptospirosis. Clin Microbiol Rev. 2011;14(2):296–326.
- , , , et al. Leptospirosis as frequent cause of acute febrile illness in southern Sri Lanka. Emerg Infect Dis. 2011;17(9):1678–1684.
- , , , et al. Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases. Philadelphia, PA: Elsevier; 2010.
- , . Antibiotics for leptospirosis. The Cochrane Database Syst Rev. 2012;2:CD008264.
- , , , et al. Actionable diagnosis of neuroleptospirosis by next‐generation sequencing. N Engl J Med. 2014;370(25):2408–2417.
- , , , . Cases distribution of leptospirosis in City of Manaus, State of Amazonas, Brazil, 2000–2010. Rev Soc Bras Med Trop. 2012;45(6):713–716.
- , , , et al. Prospective study of leptospirosis transmission in an urban slum community: role of poor environment in repeated exposures to the leptospira agent. PLoS Negl Trop Dis. 2014;8(5):e2927.
- , , , et al. Impact of environment and social gradient on leptospira infection in urban slums. PLoS Negl Trop Dis. 2008;2(4):e228.
- Urban chicken ownership in four U.S. cities. United States Department of Agriculture website. Available at: http://www.aphis.usda.gov/animal_health/nahms/poultry/downloads/poultry10/Poultry10_dr_Urban_Chicken_Four.pdf. Published April 2013. Accessed June 9, 2015.
Taking the Detour
A 60‐year‐old woman presented to a community hospital's emergency department with 4 days of right‐sided abdominal pain and multiple episodes of black stools. She reported nausea without vomiting. She denied light‐headedness, chest pain, or shortness of breath. She also denied difficulty in swallowing, weight loss, jaundice, or other bleeding.
The first priority when assessing a patient with gastrointestinal (GI) bleeding is to ensure hemodynamic stability. Next, it is important to carefully characterize the stools to help narrow the differential diagnosis. As blood is a cathartic, frequent, loose, and black stools suggest vigorous bleeding. It is essential to establish that the stools are actually black, as some patients will mistake dark brown stools for melena. Using a visual aid like a black pen or shoes as a point of reference can help the patient differentiate between dark stool and melena. It is also important to obtain a thorough medication history because iron supplements or bismuth‐containing remedies can turn stool black. The use of any antiplatelet agents or anticoagulants should also be noted. The right‐sided abdominal pain should be characterized by establishing the frequency, severity, and association with eating, movement, and position. For this patient's presentation, increased pain with eating would rapidly heighten concern for mesenteric ischemia.
The patient reported having 1 to 2 semiformed, tarry, black bowel movements per day. The night prior to admission she had passed some bright red blood along with the melena. The abdominal pain had increased gradually over 4 days, was dull, constant, did not radiate, and there were no evident aggravating or relieving factors. She rated the pain as 4 out of 10 in intensity, worst in her right upper quadrant.
Her past medical history was notable for recurrent deep venous thromboses and pulmonary emboli that had occurred even while on oral anticoagulation. Inferior vena cava (IVC) filters had twice been placed many years prior; anticoagulation had been subsequently discontinued. Additionally, she was known to have chronic superior vena cava (SVC) occlusion, presumably related to hypercoagulability. Previous evaluation had identified only hyperhomocysteinemia as a risk factor for recurrent thromboses. Other medical problems included hemorrhoids, gastroesophageal reflux disease, and asthma. Her only surgical history was an abdominal hysterectomy and bilateral oophorectomy many years ago for nonmalignant disease. Home medications were omeprazole, ranitidine, albuterol, and fluticasone‐salmeterol. She denied using nonsteroidal anti‐inflammatory drugs, aspirin, or any dietary supplements. She denied smoking, alcohol, or recreational drug use.
Because melena is confirmed, an upper GI tract bleeding source is most likely. The more recent appearance of bright red blood is concerning for acceleration of bleeding, or may point to a distal small bowel or right colonic source. Given the history of thromboembolic disease and likely underlying hypercoagulability, vascular occlusion is a leading possibility. Thus, mesenteric arterial insufficiency or mesenteric venous thrombosis should be considered, even though the patient does not report the characteristic postprandial exacerbation of pain. Ischemic colitis due to arterial insufficiency typically presents with severe, acute pain, with or without hematochezia. This syndrome is typically manifested in vascular watershed areas such as the splenic flexure, but can also affect the right colon. Mesenteric venous thrombosis is a rare condition that most often occurs in patients with hypercoagulability. Patients present with variable degrees of abdominal pain and often with GI bleeding. Finally, portal venous thrombosis may be seen alongside thromboses of other mesenteric veins or may occur independently. Portal hypertension due to portal vein thrombosis can result in esophageal and/or gastric varices. Although variceal bleeding classically presents with dramatic hematemesis, the absence of hematemesis does not rule out a variceal bleed in this patient.
On physical examination, the patient had a temperature of 37.1C with a pulse of 90 beats per minute and blood pressure of 161/97 mm Hg. Orthostatics were not performed. No blood was seen on nasal and oropharyngeal exam. Respiratory and cardiovascular exams were normal. On abdominal exam, there was tenderness to palpation of the right upper quadrant without rebound or guarding. The spleen and the liver were not palpable. There was a lower midline incisional scar. Rectal exam revealed nonbleeding hemorrhoids and heme‐positive stool without gross blood. Bilateral lower extremities had trace pitting edema, hyperpigmentation, and superficial venous varicosities. On skin exam, there were distended subcutaneous veins radiating outward from around the umbilicus as well as prominent subcutaneous venous collaterals over the chest and lateral abdomen.
The collateral veins over the chest and lateral abdomen are consistent with central venous obstruction from the patient's known SVC thrombus. However, the presence of paraumbilical venous collaterals (caput medusa) is highly suggestive of portal hypertension. This evidence, in addition to the known central venous occlusion and history of thromboembolic disease, raises the suspicion for mesenteric thrombosis as a cause of her bleeding and pain. The first diagnostic procedure should be an esophagogastroduodenoscopy (EGD) to identify and potentially treat the source of bleeding, whether it is portal hypertension related (portal gastropathy, variceal bleed) or from a more common cause (peptic ulcer disease, stress gastritis). If the EGD is not diagnostic, the next step should be to obtain computed tomography (CT) of the abdomen and pelvis with intravenous (IV) and oral contrast. In many patients with GI bleed, a colonoscopy would typically be performed as the next diagnostic study after EGD. However, in this patient, a CT scan is likely to be of higher yield because it could help assess the mesenteric and portal vessels for patency and characterize the appearance of the small intestine and colon. Depending on the findings of the CT, additional dedicated vascular diagnostics might be needed.
Hemoglobin was 8.5 g/dL (12.4 g/dL 6 weeks prior) with a normal mean corpuscular volume and red cell distribution. The white cell count was normal, and the platelet count was 142,000/mm3. The blood urea nitrogen was 27 mg/dL, with a creatinine of 1.1 mg/dL. Routine chemistries, liver enzymes, bilirubin, and coagulation parameters were normal. Ferritin was 15 ng/mL (normal: 15200 ng/mL).
The patient was admitted to the intensive care unit. An EGD revealed a hiatal hernia and grade II nonbleeding esophageal varices with normal=appearing stomach and duodenum. The varices did not have stigmata of a recent bleed and were not ligated. The patient continued to bleed and received 2 U of packed red blood cells (RBCs), as her hemoglobin had decreased to 7.3 g/dL. On hospital day 3, a colonoscopy was done that showed blood clots in the ascending colon but was otherwise normal. The patient had ongoing abdominal pain, melena, and hematochezia, and continued to require blood transfusions every other day.
Esophageal varices were confirmed on EGD. However, no high‐risk stigmata were seen. Findings that suggest either recent bleeding or are risk factors for subsequent bleeding include large size of the varices, nipple sign referring to a protruding vessel from an underlying varix, or red wale sign, referring to a longitudinal red streak on a varix. The lack of evidence for an esophageal, gastric, or duodenal bleeding source correlates with lack of clinical signs of upper GI tract hemorrhage such as hematemesis or coffee ground emesis. Because the colonoscopy also did not identify a bleeding source, the bleeding remains unexplained. The absence of significant abnormalities in liver function or liver inflammation labs suggests that the patient does not have advanced cirrhosis and supports the suspicion of a vascular cause of the portal hypertension. At this point, it would be most useful to obtain a CT scan of the abdomen and pelvis.
The patient continued to bleed, requiring a total of 7 U of packed RBCs over 7 days. On hospital day 4, a repeat EGD showed nonbleeding varices with a red wale sign that were banded. Despite this, the hemoglobin continued to drop. A technetium‐tagged RBC study showed a small area of subumbilical activity, which appeared to indicate transverse colonic or small bowel bleeding (Figure 1). A subsequent mesenteric angiogram failed to show active bleeding.

A red wale sign confers a higher risk of bleeding from esophageal varices. However, this finding can be subjective, and the endoscopist must individualize the decision for banding based on the size and appearance of the varices. It was reasonable to proceed with banding this time because the varices were large, had a red wale sign, and there was otherwise unexplained ongoing bleeding. Because her hemoglobin continued to drop after the banding and a tagged RBC study best localized the bleeding to the small intestine or transverse colon, it is unlikely that the varices are the primary source of bleeding. It is not surprising that the mesenteric angiogram did not show a source of bleeding, because this study requires active bleeding at a sufficient rate to radiographically identify the source.
The leading diagnosis remains an as yet uncharacterized small bowel bleeding source related to mesenteric thrombotic disease. Cross‐sectional imaging with IV contrast to identify significant vascular occlusion should be the next diagnostic step. Capsule endoscopy would be a more expensive and time‐consuming option, and although this could reveal the source of bleeding, it might not characterize the underlying vascular nature of the problem.
Due to persistent abdominal pain, a CT without intravenous contrast was done on hospital day 10. This showed extensive collateral vessels along the chest and abdominal wall with a distended azygos vein. The study was otherwise unrevealing. Her bloody stools cleared, so she was discharged with a plan for capsule endoscopy and outpatient follow‐up with her gastroenterologist. On the day of discharge (hospital day 11), hemoglobin was 7.5 g/dL and she received an eighth unit of packed RBCs. Overt bleeding was absent.
As an outpatient, intermittent hematochezia and melena recurred. The capsule endoscopy showed active bleeding approximately 45 minutes after the capsule exited the stomach. The lesion was not precisely located or characterized, but was believed to be in the distal small bowel.
The capsule finding supports the growing body of evidence implicating a small bowel source of bleeding. Furthermore, the ongoing but slow rate of blood loss makes a venous bleed more likely than an arterial bleed. A CT scan was performed prior to capsule study, but this was done without intravenous contrast. The brief description of the CT findings emphasizes the subcutaneous venous changes; a contraindication to IV contrast is not mentioned. Certainly IV contrast would have been very helpful to characterize the mesenteric arterial and venous vasculature. If there is no contraindication, a repeat CT scan with IV contrast should be performed. If there is a contraindication to IV contrast, it would be beneficial to revisit the noncontrast study with the specific purpose of searching for clues suggesting mesenteric or portal thrombosis. If the source still remains unclear, the next steps should be to perform push enteroscopy to assess the small intestine from the luminal side and magnetic resonance angiogram with venous phase imaging (or CT venogram if there is no contraindication to contrast) to evaluate the venous circulation.
The patient was readmitted 9 days after discharge with persistent melena and hematochezia. Her hemoglobin was 7.2 g/dL. Given the lack of a diagnosis, the patient was transferred to a tertiary care hospital, where a second colonoscopy and mesenteric angiogram were negative for bleeding. Small bowel enteroscopy showed no source of bleeding up to 60 cm past the pylorus. A third colonoscopy was performed due to recurrent bleeding; this showed a large amount of dark blood and clots throughout the entire colon including the cecum (Figure 2). After copious irrigation, the underlying mucosa was seen to be normal. At this point, a CT angiogram with both venous and arterial phases was done due to the high suspicion for a distal jejunal bleeding source. The CT angiogram showed numerous venous collaterals encasing a loop of midsmall bowel demonstrating progressive submucosal venous enhancement. In addition, a venous collateral ran down the right side of the sternum to the infraumbilical area and drained through the encasing collaterals into the portal venous system (Figure 3). The CT scan also revealed IVC obstruction below the distal IVC filter and an enlarged portal vein measuring 18 mm (normal <12 mm).
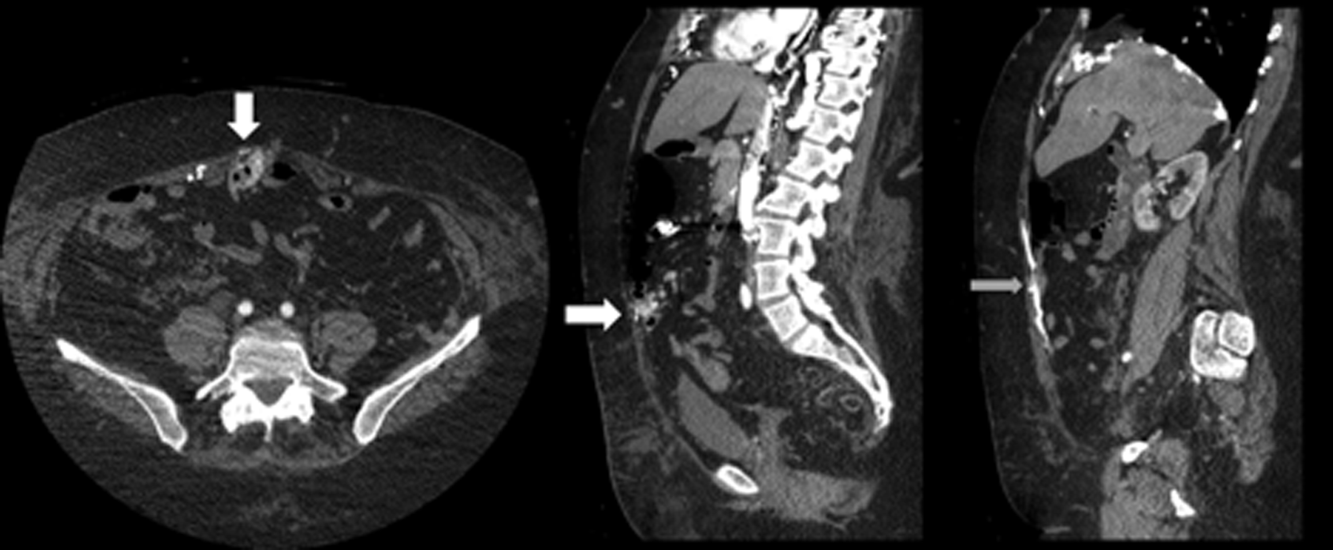
The CT angiogram provides much‐needed clarity. The continued bleeding is likely due to ectopic varices in the small bowel. The venous phase of the CT angiogram shows thrombosis of key venous structures and evidence of a dilated portal vein (indicating portal hypertension) leading to ectopic varices in the abdominal wall and jejunum. Given the prior studies that suggest a small bowel source of bleeding, jejunal varices are the most likely cause of recurrent GI bleeding in this patient.
The patient underwent exploratory laparotomy. Loops of small bowel were found to be adherent to the hysterectomy scar. There were many venous collaterals from the abdominal wall to these loops of bowel, dilating the veins both in intestinal walls and those in the adjacent mesentery. After clamping these veins, the small bowel was detached from the abdominal wall. On unclamping, the collaterals bled with a high venous pressure. Because these systemic‐portal shunts were responsible for the bleeding, the collaterals were sutured, stopping the bleeding. Thus, partial small bowel resection was not necessary. Postoperatively, her bleeding resolved completely and she maintained normal hemoglobin at 1‐year follow‐up.
COMMENTARY
The axiom common ailments are encountered most frequently underpins the classical stepwise approach to GI bleeding. First, a focused history helps localize the source of bleeding to the upper or lower GI tract. Next, endoscopy is performed to identify and treat the cause of bleeding. Finally, advanced tests such as angiography and capsule endoscopy are performed if needed. For this patient, following the usual algorithm failed to make the diagnosis or stop the bleeding. Despite historical and examination features suggesting that her case fell outside of the common patterns of GI bleeding, this patient underwent 3 upper endoscopies, 3 colonoscopies, a capsule endoscopy, a technetium‐tagged RBC study, 2 mesenteric angiograms, and a noncontrast CT scan before the study that was ultimately diagnostic was performed. The clinicians caring for this patient struggled to incorporate the atypical features of her history and presentation and failed to take an earlier detour from the usual algorithm. Instead, the same studies that had not previously led to the diagnosis were repeated multiple times.
Ectopic varices are enlarged portosystemic venous collaterals located anywhere outside the gastroesophageal region.[1] They occur in the setting of portal hypertension, surgical procedures involving abdominal viscera and vasculature, and venous occlusion. Ectopic varices account for 4% to 5% of all variceal bleeding episodes.[1] The most common sites include the anorectal junction (44%), duodenum (17%33%), jejunum/emleum (5%17%), colon (3.5%14%), and sites of previous abdominal surgery.[2, 3] Ectopic varices can cause either luminal or extraluminal (i.e., peritoneal) bleeding.[3] Luminal bleeding, seen in this case, is caused by venous protrusion into the submucosa. Ectopic varices present as a slow venous ooze, which explains this patient's ongoing requirement for recurrent blood transfusions.[4]
In this patient, submucosal ectopic varices developed as a result of a combination of known risk factors: portal hypertension in the setting of chronic venous occlusion from her hypercoagulability and a history of abdominal surgery (hysterectomy). [5] The apposition of her abdominal wall structures (drained by the systemic veins) to the bowel (drained by the portal veins) resulted in adhesion formation, detour of venous flow, collateralization, and submucosal varix formation.[1, 2, 6]
The key diagnostic study for this patient was a CT angiogram, with both arterial and venous phases. The prior 2 mesenteric angiograms had been limited to the arterial phase, which had missed identifying the venous abnormalities altogether. This highlights an important lesson from this case: contrast‐enhanced CT may have a higher yield in diagnosing ectopic varices compared to repeated endoscopiesespecially when captured in the late venous phaseand should strongly be considered for unexplained bleeding in patients with stigmata of liver disease or portal hypertension.[7, 8] Another clue for ectopic varices in a bleeding patient are nonbleeding esophageal or gastric varices, as was the case in this patient.[9]
The initial management of ectopic varices is similar to bleeding secondary to esophageal varices.[1] Definitive treatment includes endoscopic embolization or ligation, interventional radiological procedures such as portosystemic shunting or percutaneous embolization, and exploratory laparotomy to either resect the segment of bowel that is the source of bleeding or to decompress the collaterals surgically.[9] Although endoscopic ligation has been shown to have a lower rebleeding rate and mortality compared to endoscopic injection sclerotherapy in patients with esophageal varices, the data are too sparse in jejunal varices to recommend 1 treatment over another. Both have been used successfully either alone or in combination with each other, and can be useful alternatives for patients who are unable to undergo laparotomy.[9]
Diagnostic errors due to cognitive biases can be avoided by following diagnostic algorithms. However, over‐reliance on algorithms can result in vertical line failure, a form of cognitive bias in which the clinician subconsciously adheres to an inflexible diagnostic approach.[10] To overcome this bias, clinicians need to think laterally and consider alternative diagnoses when algorithms do not lead to expected outcomes. This case highlights the challenges of knowing when to break free of conventional approaches and the rewards of taking a well‐chosen detour that leads to the diagnosis.
KEY POINTS
- Recurrent, occult gastrointestinal bleeding should raise concern for a small bowel source, and clinicians may need to take a detour away from the usual workup to arrive at a diagnosis.
- CT angiography of the abdomen and pelvis may miss venous sources of bleeding, unless a venous phase is specifically requested.
- Ectopic varices can occur in patients with portal hypertension who have had a history of abdominal surgery; these patients can develop venous collaterals for decompression into the systemic circulation through the abdominal wall.
Disclosure
Nothing to report.
- , , . Updates in the pathogenesis, diagnosis and management of ectopic varices. Hepatol Int. 2008;2:322–334.
- , , . Management of ectopic varices. Hepatology. 1998;28:1154–1158.
- , , , et al. Current status of ectopic varices in Japan: results of a survey by the Japan Society for Portal Hypertension. Hepatol Res. 2010;40:763–766.
- , , . Stomal Varices: Management with decompression TIPS and transvenous obliteration or sclerosis. Tech Vasc Interv Radiol. 2013;16:126–134.
- , , , et al. Jejunal varices as a cause of massive gastrointestinal bleeding. Am J Gastroenterol. 1992;87:514–517.
- , . Ectopic varices in portal hypertension. Clin Gastroenterol. 1985;14:105–121.
- , , , et al. Ectopic varices in portal hypertension: computed tomographic angiography instead of repeated endoscopies for diagnosis. Eur J Gastroenterol Hepatol. 2011;23:620–622.
- , , , et al. ACR appropriateness criteria. Radiologic management of lower gastrointestinal tract bleeding. Reston, VA: American College of Radiology; 2011. Available at: http://www.acr.org/Quality‐Safety/Appropriateness‐Criteria/∼/media/5F9CB95C164E4DA19DCBCFBBA790BB3C.pdf. Accessed January 28, 2015.
- , . Diagnosis and management of ectopic varices. Gastrointest Interv. 2012;1:3–10.
- . Achieving quality in clinical decision making: cognitive strategies and detection of bias. Acad Emerg Med. 2002;9:1184–1204.
A 60‐year‐old woman presented to a community hospital's emergency department with 4 days of right‐sided abdominal pain and multiple episodes of black stools. She reported nausea without vomiting. She denied light‐headedness, chest pain, or shortness of breath. She also denied difficulty in swallowing, weight loss, jaundice, or other bleeding.
The first priority when assessing a patient with gastrointestinal (GI) bleeding is to ensure hemodynamic stability. Next, it is important to carefully characterize the stools to help narrow the differential diagnosis. As blood is a cathartic, frequent, loose, and black stools suggest vigorous bleeding. It is essential to establish that the stools are actually black, as some patients will mistake dark brown stools for melena. Using a visual aid like a black pen or shoes as a point of reference can help the patient differentiate between dark stool and melena. It is also important to obtain a thorough medication history because iron supplements or bismuth‐containing remedies can turn stool black. The use of any antiplatelet agents or anticoagulants should also be noted. The right‐sided abdominal pain should be characterized by establishing the frequency, severity, and association with eating, movement, and position. For this patient's presentation, increased pain with eating would rapidly heighten concern for mesenteric ischemia.
The patient reported having 1 to 2 semiformed, tarry, black bowel movements per day. The night prior to admission she had passed some bright red blood along with the melena. The abdominal pain had increased gradually over 4 days, was dull, constant, did not radiate, and there were no evident aggravating or relieving factors. She rated the pain as 4 out of 10 in intensity, worst in her right upper quadrant.
Her past medical history was notable for recurrent deep venous thromboses and pulmonary emboli that had occurred even while on oral anticoagulation. Inferior vena cava (IVC) filters had twice been placed many years prior; anticoagulation had been subsequently discontinued. Additionally, she was known to have chronic superior vena cava (SVC) occlusion, presumably related to hypercoagulability. Previous evaluation had identified only hyperhomocysteinemia as a risk factor for recurrent thromboses. Other medical problems included hemorrhoids, gastroesophageal reflux disease, and asthma. Her only surgical history was an abdominal hysterectomy and bilateral oophorectomy many years ago for nonmalignant disease. Home medications were omeprazole, ranitidine, albuterol, and fluticasone‐salmeterol. She denied using nonsteroidal anti‐inflammatory drugs, aspirin, or any dietary supplements. She denied smoking, alcohol, or recreational drug use.
Because melena is confirmed, an upper GI tract bleeding source is most likely. The more recent appearance of bright red blood is concerning for acceleration of bleeding, or may point to a distal small bowel or right colonic source. Given the history of thromboembolic disease and likely underlying hypercoagulability, vascular occlusion is a leading possibility. Thus, mesenteric arterial insufficiency or mesenteric venous thrombosis should be considered, even though the patient does not report the characteristic postprandial exacerbation of pain. Ischemic colitis due to arterial insufficiency typically presents with severe, acute pain, with or without hematochezia. This syndrome is typically manifested in vascular watershed areas such as the splenic flexure, but can also affect the right colon. Mesenteric venous thrombosis is a rare condition that most often occurs in patients with hypercoagulability. Patients present with variable degrees of abdominal pain and often with GI bleeding. Finally, portal venous thrombosis may be seen alongside thromboses of other mesenteric veins or may occur independently. Portal hypertension due to portal vein thrombosis can result in esophageal and/or gastric varices. Although variceal bleeding classically presents with dramatic hematemesis, the absence of hematemesis does not rule out a variceal bleed in this patient.
On physical examination, the patient had a temperature of 37.1C with a pulse of 90 beats per minute and blood pressure of 161/97 mm Hg. Orthostatics were not performed. No blood was seen on nasal and oropharyngeal exam. Respiratory and cardiovascular exams were normal. On abdominal exam, there was tenderness to palpation of the right upper quadrant without rebound or guarding. The spleen and the liver were not palpable. There was a lower midline incisional scar. Rectal exam revealed nonbleeding hemorrhoids and heme‐positive stool without gross blood. Bilateral lower extremities had trace pitting edema, hyperpigmentation, and superficial venous varicosities. On skin exam, there were distended subcutaneous veins radiating outward from around the umbilicus as well as prominent subcutaneous venous collaterals over the chest and lateral abdomen.
The collateral veins over the chest and lateral abdomen are consistent with central venous obstruction from the patient's known SVC thrombus. However, the presence of paraumbilical venous collaterals (caput medusa) is highly suggestive of portal hypertension. This evidence, in addition to the known central venous occlusion and history of thromboembolic disease, raises the suspicion for mesenteric thrombosis as a cause of her bleeding and pain. The first diagnostic procedure should be an esophagogastroduodenoscopy (EGD) to identify and potentially treat the source of bleeding, whether it is portal hypertension related (portal gastropathy, variceal bleed) or from a more common cause (peptic ulcer disease, stress gastritis). If the EGD is not diagnostic, the next step should be to obtain computed tomography (CT) of the abdomen and pelvis with intravenous (IV) and oral contrast. In many patients with GI bleed, a colonoscopy would typically be performed as the next diagnostic study after EGD. However, in this patient, a CT scan is likely to be of higher yield because it could help assess the mesenteric and portal vessels for patency and characterize the appearance of the small intestine and colon. Depending on the findings of the CT, additional dedicated vascular diagnostics might be needed.
Hemoglobin was 8.5 g/dL (12.4 g/dL 6 weeks prior) with a normal mean corpuscular volume and red cell distribution. The white cell count was normal, and the platelet count was 142,000/mm3. The blood urea nitrogen was 27 mg/dL, with a creatinine of 1.1 mg/dL. Routine chemistries, liver enzymes, bilirubin, and coagulation parameters were normal. Ferritin was 15 ng/mL (normal: 15200 ng/mL).
The patient was admitted to the intensive care unit. An EGD revealed a hiatal hernia and grade II nonbleeding esophageal varices with normal=appearing stomach and duodenum. The varices did not have stigmata of a recent bleed and were not ligated. The patient continued to bleed and received 2 U of packed red blood cells (RBCs), as her hemoglobin had decreased to 7.3 g/dL. On hospital day 3, a colonoscopy was done that showed blood clots in the ascending colon but was otherwise normal. The patient had ongoing abdominal pain, melena, and hematochezia, and continued to require blood transfusions every other day.
Esophageal varices were confirmed on EGD. However, no high‐risk stigmata were seen. Findings that suggest either recent bleeding or are risk factors for subsequent bleeding include large size of the varices, nipple sign referring to a protruding vessel from an underlying varix, or red wale sign, referring to a longitudinal red streak on a varix. The lack of evidence for an esophageal, gastric, or duodenal bleeding source correlates with lack of clinical signs of upper GI tract hemorrhage such as hematemesis or coffee ground emesis. Because the colonoscopy also did not identify a bleeding source, the bleeding remains unexplained. The absence of significant abnormalities in liver function or liver inflammation labs suggests that the patient does not have advanced cirrhosis and supports the suspicion of a vascular cause of the portal hypertension. At this point, it would be most useful to obtain a CT scan of the abdomen and pelvis.
The patient continued to bleed, requiring a total of 7 U of packed RBCs over 7 days. On hospital day 4, a repeat EGD showed nonbleeding varices with a red wale sign that were banded. Despite this, the hemoglobin continued to drop. A technetium‐tagged RBC study showed a small area of subumbilical activity, which appeared to indicate transverse colonic or small bowel bleeding (Figure 1). A subsequent mesenteric angiogram failed to show active bleeding.
A red wale sign confers a higher risk of bleeding from esophageal varices. However, this finding can be subjective, and the endoscopist must individualize the decision for banding based on the size and appearance of the varices. It was reasonable to proceed with banding this time because the varices were large, had a red wale sign, and there was otherwise unexplained ongoing bleeding. Because her hemoglobin continued to drop after the banding and a tagged RBC study best localized the bleeding to the small intestine or transverse colon, it is unlikely that the varices are the primary source of bleeding. It is not surprising that the mesenteric angiogram did not show a source of bleeding, because this study requires active bleeding at a sufficient rate to radiographically identify the source.
The leading diagnosis remains an as yet uncharacterized small bowel bleeding source related to mesenteric thrombotic disease. Cross‐sectional imaging with IV contrast to identify significant vascular occlusion should be the next diagnostic step. Capsule endoscopy would be a more expensive and time‐consuming option, and although this could reveal the source of bleeding, it might not characterize the underlying vascular nature of the problem.
Due to persistent abdominal pain, a CT without intravenous contrast was done on hospital day 10. This showed extensive collateral vessels along the chest and abdominal wall with a distended azygos vein. The study was otherwise unrevealing. Her bloody stools cleared, so she was discharged with a plan for capsule endoscopy and outpatient follow‐up with her gastroenterologist. On the day of discharge (hospital day 11), hemoglobin was 7.5 g/dL and she received an eighth unit of packed RBCs. Overt bleeding was absent.
As an outpatient, intermittent hematochezia and melena recurred. The capsule endoscopy showed active bleeding approximately 45 minutes after the capsule exited the stomach. The lesion was not precisely located or characterized, but was believed to be in the distal small bowel.
The capsule finding supports the growing body of evidence implicating a small bowel source of bleeding. Furthermore, the ongoing but slow rate of blood loss makes a venous bleed more likely than an arterial bleed. A CT scan was performed prior to capsule study, but this was done without intravenous contrast. The brief description of the CT findings emphasizes the subcutaneous venous changes; a contraindication to IV contrast is not mentioned. Certainly IV contrast would have been very helpful to characterize the mesenteric arterial and venous vasculature. If there is no contraindication, a repeat CT scan with IV contrast should be performed. If there is a contraindication to IV contrast, it would be beneficial to revisit the noncontrast study with the specific purpose of searching for clues suggesting mesenteric or portal thrombosis. If the source still remains unclear, the next steps should be to perform push enteroscopy to assess the small intestine from the luminal side and magnetic resonance angiogram with venous phase imaging (or CT venogram if there is no contraindication to contrast) to evaluate the venous circulation.
The patient was readmitted 9 days after discharge with persistent melena and hematochezia. Her hemoglobin was 7.2 g/dL. Given the lack of a diagnosis, the patient was transferred to a tertiary care hospital, where a second colonoscopy and mesenteric angiogram were negative for bleeding. Small bowel enteroscopy showed no source of bleeding up to 60 cm past the pylorus. A third colonoscopy was performed due to recurrent bleeding; this showed a large amount of dark blood and clots throughout the entire colon including the cecum (Figure 2). After copious irrigation, the underlying mucosa was seen to be normal. At this point, a CT angiogram with both venous and arterial phases was done due to the high suspicion for a distal jejunal bleeding source. The CT angiogram showed numerous venous collaterals encasing a loop of midsmall bowel demonstrating progressive submucosal venous enhancement. In addition, a venous collateral ran down the right side of the sternum to the infraumbilical area and drained through the encasing collaterals into the portal venous system (Figure 3). The CT scan also revealed IVC obstruction below the distal IVC filter and an enlarged portal vein measuring 18 mm (normal <12 mm).
The CT angiogram provides much‐needed clarity. The continued bleeding is likely due to ectopic varices in the small bowel. The venous phase of the CT angiogram shows thrombosis of key venous structures and evidence of a dilated portal vein (indicating portal hypertension) leading to ectopic varices in the abdominal wall and jejunum. Given the prior studies that suggest a small bowel source of bleeding, jejunal varices are the most likely cause of recurrent GI bleeding in this patient.
The patient underwent exploratory laparotomy. Loops of small bowel were found to be adherent to the hysterectomy scar. There were many venous collaterals from the abdominal wall to these loops of bowel, dilating the veins both in intestinal walls and those in the adjacent mesentery. After clamping these veins, the small bowel was detached from the abdominal wall. On unclamping, the collaterals bled with a high venous pressure. Because these systemic‐portal shunts were responsible for the bleeding, the collaterals were sutured, stopping the bleeding. Thus, partial small bowel resection was not necessary. Postoperatively, her bleeding resolved completely and she maintained normal hemoglobin at 1‐year follow‐up.
COMMENTARY
The axiom common ailments are encountered most frequently underpins the classical stepwise approach to GI bleeding. First, a focused history helps localize the source of bleeding to the upper or lower GI tract. Next, endoscopy is performed to identify and treat the cause of bleeding. Finally, advanced tests such as angiography and capsule endoscopy are performed if needed. For this patient, following the usual algorithm failed to make the diagnosis or stop the bleeding. Despite historical and examination features suggesting that her case fell outside of the common patterns of GI bleeding, this patient underwent 3 upper endoscopies, 3 colonoscopies, a capsule endoscopy, a technetium‐tagged RBC study, 2 mesenteric angiograms, and a noncontrast CT scan before the study that was ultimately diagnostic was performed. The clinicians caring for this patient struggled to incorporate the atypical features of her history and presentation and failed to take an earlier detour from the usual algorithm. Instead, the same studies that had not previously led to the diagnosis were repeated multiple times.
Ectopic varices are enlarged portosystemic venous collaterals located anywhere outside the gastroesophageal region.[1] They occur in the setting of portal hypertension, surgical procedures involving abdominal viscera and vasculature, and venous occlusion. Ectopic varices account for 4% to 5% of all variceal bleeding episodes.[1] The most common sites include the anorectal junction (44%), duodenum (17%33%), jejunum/emleum (5%17%), colon (3.5%14%), and sites of previous abdominal surgery.[2, 3] Ectopic varices can cause either luminal or extraluminal (i.e., peritoneal) bleeding.[3] Luminal bleeding, seen in this case, is caused by venous protrusion into the submucosa. Ectopic varices present as a slow venous ooze, which explains this patient's ongoing requirement for recurrent blood transfusions.[4]
In this patient, submucosal ectopic varices developed as a result of a combination of known risk factors: portal hypertension in the setting of chronic venous occlusion from her hypercoagulability and a history of abdominal surgery (hysterectomy). [5] The apposition of her abdominal wall structures (drained by the systemic veins) to the bowel (drained by the portal veins) resulted in adhesion formation, detour of venous flow, collateralization, and submucosal varix formation.[1, 2, 6]
The key diagnostic study for this patient was a CT angiogram, with both arterial and venous phases. The prior 2 mesenteric angiograms had been limited to the arterial phase, which had missed identifying the venous abnormalities altogether. This highlights an important lesson from this case: contrast‐enhanced CT may have a higher yield in diagnosing ectopic varices compared to repeated endoscopiesespecially when captured in the late venous phaseand should strongly be considered for unexplained bleeding in patients with stigmata of liver disease or portal hypertension.[7, 8] Another clue for ectopic varices in a bleeding patient are nonbleeding esophageal or gastric varices, as was the case in this patient.[9]
The initial management of ectopic varices is similar to bleeding secondary to esophageal varices.[1] Definitive treatment includes endoscopic embolization or ligation, interventional radiological procedures such as portosystemic shunting or percutaneous embolization, and exploratory laparotomy to either resect the segment of bowel that is the source of bleeding or to decompress the collaterals surgically.[9] Although endoscopic ligation has been shown to have a lower rebleeding rate and mortality compared to endoscopic injection sclerotherapy in patients with esophageal varices, the data are too sparse in jejunal varices to recommend 1 treatment over another. Both have been used successfully either alone or in combination with each other, and can be useful alternatives for patients who are unable to undergo laparotomy.[9]
Diagnostic errors due to cognitive biases can be avoided by following diagnostic algorithms. However, over‐reliance on algorithms can result in vertical line failure, a form of cognitive bias in which the clinician subconsciously adheres to an inflexible diagnostic approach.[10] To overcome this bias, clinicians need to think laterally and consider alternative diagnoses when algorithms do not lead to expected outcomes. This case highlights the challenges of knowing when to break free of conventional approaches and the rewards of taking a well‐chosen detour that leads to the diagnosis.
KEY POINTS
- Recurrent, occult gastrointestinal bleeding should raise concern for a small bowel source, and clinicians may need to take a detour away from the usual workup to arrive at a diagnosis.
- CT angiography of the abdomen and pelvis may miss venous sources of bleeding, unless a venous phase is specifically requested.
- Ectopic varices can occur in patients with portal hypertension who have had a history of abdominal surgery; these patients can develop venous collaterals for decompression into the systemic circulation through the abdominal wall.
Disclosure
Nothing to report.
A 60‐year‐old woman presented to a community hospital's emergency department with 4 days of right‐sided abdominal pain and multiple episodes of black stools. She reported nausea without vomiting. She denied light‐headedness, chest pain, or shortness of breath. She also denied difficulty in swallowing, weight loss, jaundice, or other bleeding.
The first priority when assessing a patient with gastrointestinal (GI) bleeding is to ensure hemodynamic stability. Next, it is important to carefully characterize the stools to help narrow the differential diagnosis. As blood is a cathartic, frequent, loose, and black stools suggest vigorous bleeding. It is essential to establish that the stools are actually black, as some patients will mistake dark brown stools for melena. Using a visual aid like a black pen or shoes as a point of reference can help the patient differentiate between dark stool and melena. It is also important to obtain a thorough medication history because iron supplements or bismuth‐containing remedies can turn stool black. The use of any antiplatelet agents or anticoagulants should also be noted. The right‐sided abdominal pain should be characterized by establishing the frequency, severity, and association with eating, movement, and position. For this patient's presentation, increased pain with eating would rapidly heighten concern for mesenteric ischemia.
The patient reported having 1 to 2 semiformed, tarry, black bowel movements per day. The night prior to admission she had passed some bright red blood along with the melena. The abdominal pain had increased gradually over 4 days, was dull, constant, did not radiate, and there were no evident aggravating or relieving factors. She rated the pain as 4 out of 10 in intensity, worst in her right upper quadrant.
Her past medical history was notable for recurrent deep venous thromboses and pulmonary emboli that had occurred even while on oral anticoagulation. Inferior vena cava (IVC) filters had twice been placed many years prior; anticoagulation had been subsequently discontinued. Additionally, she was known to have chronic superior vena cava (SVC) occlusion, presumably related to hypercoagulability. Previous evaluation had identified only hyperhomocysteinemia as a risk factor for recurrent thromboses. Other medical problems included hemorrhoids, gastroesophageal reflux disease, and asthma. Her only surgical history was an abdominal hysterectomy and bilateral oophorectomy many years ago for nonmalignant disease. Home medications were omeprazole, ranitidine, albuterol, and fluticasone‐salmeterol. She denied using nonsteroidal anti‐inflammatory drugs, aspirin, or any dietary supplements. She denied smoking, alcohol, or recreational drug use.
Because melena is confirmed, an upper GI tract bleeding source is most likely. The more recent appearance of bright red blood is concerning for acceleration of bleeding, or may point to a distal small bowel or right colonic source. Given the history of thromboembolic disease and likely underlying hypercoagulability, vascular occlusion is a leading possibility. Thus, mesenteric arterial insufficiency or mesenteric venous thrombosis should be considered, even though the patient does not report the characteristic postprandial exacerbation of pain. Ischemic colitis due to arterial insufficiency typically presents with severe, acute pain, with or without hematochezia. This syndrome is typically manifested in vascular watershed areas such as the splenic flexure, but can also affect the right colon. Mesenteric venous thrombosis is a rare condition that most often occurs in patients with hypercoagulability. Patients present with variable degrees of abdominal pain and often with GI bleeding. Finally, portal venous thrombosis may be seen alongside thromboses of other mesenteric veins or may occur independently. Portal hypertension due to portal vein thrombosis can result in esophageal and/or gastric varices. Although variceal bleeding classically presents with dramatic hematemesis, the absence of hematemesis does not rule out a variceal bleed in this patient.
On physical examination, the patient had a temperature of 37.1C with a pulse of 90 beats per minute and blood pressure of 161/97 mm Hg. Orthostatics were not performed. No blood was seen on nasal and oropharyngeal exam. Respiratory and cardiovascular exams were normal. On abdominal exam, there was tenderness to palpation of the right upper quadrant without rebound or guarding. The spleen and the liver were not palpable. There was a lower midline incisional scar. Rectal exam revealed nonbleeding hemorrhoids and heme‐positive stool without gross blood. Bilateral lower extremities had trace pitting edema, hyperpigmentation, and superficial venous varicosities. On skin exam, there were distended subcutaneous veins radiating outward from around the umbilicus as well as prominent subcutaneous venous collaterals over the chest and lateral abdomen.
The collateral veins over the chest and lateral abdomen are consistent with central venous obstruction from the patient's known SVC thrombus. However, the presence of paraumbilical venous collaterals (caput medusa) is highly suggestive of portal hypertension. This evidence, in addition to the known central venous occlusion and history of thromboembolic disease, raises the suspicion for mesenteric thrombosis as a cause of her bleeding and pain. The first diagnostic procedure should be an esophagogastroduodenoscopy (EGD) to identify and potentially treat the source of bleeding, whether it is portal hypertension related (portal gastropathy, variceal bleed) or from a more common cause (peptic ulcer disease, stress gastritis). If the EGD is not diagnostic, the next step should be to obtain computed tomography (CT) of the abdomen and pelvis with intravenous (IV) and oral contrast. In many patients with GI bleed, a colonoscopy would typically be performed as the next diagnostic study after EGD. However, in this patient, a CT scan is likely to be of higher yield because it could help assess the mesenteric and portal vessels for patency and characterize the appearance of the small intestine and colon. Depending on the findings of the CT, additional dedicated vascular diagnostics might be needed.
Hemoglobin was 8.5 g/dL (12.4 g/dL 6 weeks prior) with a normal mean corpuscular volume and red cell distribution. The white cell count was normal, and the platelet count was 142,000/mm3. The blood urea nitrogen was 27 mg/dL, with a creatinine of 1.1 mg/dL. Routine chemistries, liver enzymes, bilirubin, and coagulation parameters were normal. Ferritin was 15 ng/mL (normal: 15200 ng/mL).
The patient was admitted to the intensive care unit. An EGD revealed a hiatal hernia and grade II nonbleeding esophageal varices with normal=appearing stomach and duodenum. The varices did not have stigmata of a recent bleed and were not ligated. The patient continued to bleed and received 2 U of packed red blood cells (RBCs), as her hemoglobin had decreased to 7.3 g/dL. On hospital day 3, a colonoscopy was done that showed blood clots in the ascending colon but was otherwise normal. The patient had ongoing abdominal pain, melena, and hematochezia, and continued to require blood transfusions every other day.
Esophageal varices were confirmed on EGD. However, no high‐risk stigmata were seen. Findings that suggest either recent bleeding or are risk factors for subsequent bleeding include large size of the varices, nipple sign referring to a protruding vessel from an underlying varix, or red wale sign, referring to a longitudinal red streak on a varix. The lack of evidence for an esophageal, gastric, or duodenal bleeding source correlates with lack of clinical signs of upper GI tract hemorrhage such as hematemesis or coffee ground emesis. Because the colonoscopy also did not identify a bleeding source, the bleeding remains unexplained. The absence of significant abnormalities in liver function or liver inflammation labs suggests that the patient does not have advanced cirrhosis and supports the suspicion of a vascular cause of the portal hypertension. At this point, it would be most useful to obtain a CT scan of the abdomen and pelvis.
The patient continued to bleed, requiring a total of 7 U of packed RBCs over 7 days. On hospital day 4, a repeat EGD showed nonbleeding varices with a red wale sign that were banded. Despite this, the hemoglobin continued to drop. A technetium‐tagged RBC study showed a small area of subumbilical activity, which appeared to indicate transverse colonic or small bowel bleeding (Figure 1). A subsequent mesenteric angiogram failed to show active bleeding.
A red wale sign confers a higher risk of bleeding from esophageal varices. However, this finding can be subjective, and the endoscopist must individualize the decision for banding based on the size and appearance of the varices. It was reasonable to proceed with banding this time because the varices were large, had a red wale sign, and there was otherwise unexplained ongoing bleeding. Because her hemoglobin continued to drop after the banding and a tagged RBC study best localized the bleeding to the small intestine or transverse colon, it is unlikely that the varices are the primary source of bleeding. It is not surprising that the mesenteric angiogram did not show a source of bleeding, because this study requires active bleeding at a sufficient rate to radiographically identify the source.
The leading diagnosis remains an as yet uncharacterized small bowel bleeding source related to mesenteric thrombotic disease. Cross‐sectional imaging with IV contrast to identify significant vascular occlusion should be the next diagnostic step. Capsule endoscopy would be a more expensive and time‐consuming option, and although this could reveal the source of bleeding, it might not characterize the underlying vascular nature of the problem.
Due to persistent abdominal pain, a CT without intravenous contrast was done on hospital day 10. This showed extensive collateral vessels along the chest and abdominal wall with a distended azygos vein. The study was otherwise unrevealing. Her bloody stools cleared, so she was discharged with a plan for capsule endoscopy and outpatient follow‐up with her gastroenterologist. On the day of discharge (hospital day 11), hemoglobin was 7.5 g/dL and she received an eighth unit of packed RBCs. Overt bleeding was absent.
As an outpatient, intermittent hematochezia and melena recurred. The capsule endoscopy showed active bleeding approximately 45 minutes after the capsule exited the stomach. The lesion was not precisely located or characterized, but was believed to be in the distal small bowel.
The capsule finding supports the growing body of evidence implicating a small bowel source of bleeding. Furthermore, the ongoing but slow rate of blood loss makes a venous bleed more likely than an arterial bleed. A CT scan was performed prior to capsule study, but this was done without intravenous contrast. The brief description of the CT findings emphasizes the subcutaneous venous changes; a contraindication to IV contrast is not mentioned. Certainly IV contrast would have been very helpful to characterize the mesenteric arterial and venous vasculature. If there is no contraindication, a repeat CT scan with IV contrast should be performed. If there is a contraindication to IV contrast, it would be beneficial to revisit the noncontrast study with the specific purpose of searching for clues suggesting mesenteric or portal thrombosis. If the source still remains unclear, the next steps should be to perform push enteroscopy to assess the small intestine from the luminal side and magnetic resonance angiogram with venous phase imaging (or CT venogram if there is no contraindication to contrast) to evaluate the venous circulation.
The patient was readmitted 9 days after discharge with persistent melena and hematochezia. Her hemoglobin was 7.2 g/dL. Given the lack of a diagnosis, the patient was transferred to a tertiary care hospital, where a second colonoscopy and mesenteric angiogram were negative for bleeding. Small bowel enteroscopy showed no source of bleeding up to 60 cm past the pylorus. A third colonoscopy was performed due to recurrent bleeding; this showed a large amount of dark blood and clots throughout the entire colon including the cecum (Figure 2). After copious irrigation, the underlying mucosa was seen to be normal. At this point, a CT angiogram with both venous and arterial phases was done due to the high suspicion for a distal jejunal bleeding source. The CT angiogram showed numerous venous collaterals encasing a loop of midsmall bowel demonstrating progressive submucosal venous enhancement. In addition, a venous collateral ran down the right side of the sternum to the infraumbilical area and drained through the encasing collaterals into the portal venous system (Figure 3). The CT scan also revealed IVC obstruction below the distal IVC filter and an enlarged portal vein measuring 18 mm (normal <12 mm).
The CT angiogram provides much‐needed clarity. The continued bleeding is likely due to ectopic varices in the small bowel. The venous phase of the CT angiogram shows thrombosis of key venous structures and evidence of a dilated portal vein (indicating portal hypertension) leading to ectopic varices in the abdominal wall and jejunum. Given the prior studies that suggest a small bowel source of bleeding, jejunal varices are the most likely cause of recurrent GI bleeding in this patient.
The patient underwent exploratory laparotomy. Loops of small bowel were found to be adherent to the hysterectomy scar. There were many venous collaterals from the abdominal wall to these loops of bowel, dilating the veins both in intestinal walls and those in the adjacent mesentery. After clamping these veins, the small bowel was detached from the abdominal wall. On unclamping, the collaterals bled with a high venous pressure. Because these systemic‐portal shunts were responsible for the bleeding, the collaterals were sutured, stopping the bleeding. Thus, partial small bowel resection was not necessary. Postoperatively, her bleeding resolved completely and she maintained normal hemoglobin at 1‐year follow‐up.
COMMENTARY
The axiom common ailments are encountered most frequently underpins the classical stepwise approach to GI bleeding. First, a focused history helps localize the source of bleeding to the upper or lower GI tract. Next, endoscopy is performed to identify and treat the cause of bleeding. Finally, advanced tests such as angiography and capsule endoscopy are performed if needed. For this patient, following the usual algorithm failed to make the diagnosis or stop the bleeding. Despite historical and examination features suggesting that her case fell outside of the common patterns of GI bleeding, this patient underwent 3 upper endoscopies, 3 colonoscopies, a capsule endoscopy, a technetium‐tagged RBC study, 2 mesenteric angiograms, and a noncontrast CT scan before the study that was ultimately diagnostic was performed. The clinicians caring for this patient struggled to incorporate the atypical features of her history and presentation and failed to take an earlier detour from the usual algorithm. Instead, the same studies that had not previously led to the diagnosis were repeated multiple times.
Ectopic varices are enlarged portosystemic venous collaterals located anywhere outside the gastroesophageal region.[1] They occur in the setting of portal hypertension, surgical procedures involving abdominal viscera and vasculature, and venous occlusion. Ectopic varices account for 4% to 5% of all variceal bleeding episodes.[1] The most common sites include the anorectal junction (44%), duodenum (17%33%), jejunum/emleum (5%17%), colon (3.5%14%), and sites of previous abdominal surgery.[2, 3] Ectopic varices can cause either luminal or extraluminal (i.e., peritoneal) bleeding.[3] Luminal bleeding, seen in this case, is caused by venous protrusion into the submucosa. Ectopic varices present as a slow venous ooze, which explains this patient's ongoing requirement for recurrent blood transfusions.[4]
In this patient, submucosal ectopic varices developed as a result of a combination of known risk factors: portal hypertension in the setting of chronic venous occlusion from her hypercoagulability and a history of abdominal surgery (hysterectomy). [5] The apposition of her abdominal wall structures (drained by the systemic veins) to the bowel (drained by the portal veins) resulted in adhesion formation, detour of venous flow, collateralization, and submucosal varix formation.[1, 2, 6]
The key diagnostic study for this patient was a CT angiogram, with both arterial and venous phases. The prior 2 mesenteric angiograms had been limited to the arterial phase, which had missed identifying the venous abnormalities altogether. This highlights an important lesson from this case: contrast‐enhanced CT may have a higher yield in diagnosing ectopic varices compared to repeated endoscopiesespecially when captured in the late venous phaseand should strongly be considered for unexplained bleeding in patients with stigmata of liver disease or portal hypertension.[7, 8] Another clue for ectopic varices in a bleeding patient are nonbleeding esophageal or gastric varices, as was the case in this patient.[9]
The initial management of ectopic varices is similar to bleeding secondary to esophageal varices.[1] Definitive treatment includes endoscopic embolization or ligation, interventional radiological procedures such as portosystemic shunting or percutaneous embolization, and exploratory laparotomy to either resect the segment of bowel that is the source of bleeding or to decompress the collaterals surgically.[9] Although endoscopic ligation has been shown to have a lower rebleeding rate and mortality compared to endoscopic injection sclerotherapy in patients with esophageal varices, the data are too sparse in jejunal varices to recommend 1 treatment over another. Both have been used successfully either alone or in combination with each other, and can be useful alternatives for patients who are unable to undergo laparotomy.[9]
Diagnostic errors due to cognitive biases can be avoided by following diagnostic algorithms. However, over‐reliance on algorithms can result in vertical line failure, a form of cognitive bias in which the clinician subconsciously adheres to an inflexible diagnostic approach.[10] To overcome this bias, clinicians need to think laterally and consider alternative diagnoses when algorithms do not lead to expected outcomes. This case highlights the challenges of knowing when to break free of conventional approaches and the rewards of taking a well‐chosen detour that leads to the diagnosis.
KEY POINTS
- Recurrent, occult gastrointestinal bleeding should raise concern for a small bowel source, and clinicians may need to take a detour away from the usual workup to arrive at a diagnosis.
- CT angiography of the abdomen and pelvis may miss venous sources of bleeding, unless a venous phase is specifically requested.
- Ectopic varices can occur in patients with portal hypertension who have had a history of abdominal surgery; these patients can develop venous collaterals for decompression into the systemic circulation through the abdominal wall.
Disclosure
Nothing to report.
- , , . Updates in the pathogenesis, diagnosis and management of ectopic varices. Hepatol Int. 2008;2:322–334.
- , , . Management of ectopic varices. Hepatology. 1998;28:1154–1158.
- , , , et al. Current status of ectopic varices in Japan: results of a survey by the Japan Society for Portal Hypertension. Hepatol Res. 2010;40:763–766.
- , , . Stomal Varices: Management with decompression TIPS and transvenous obliteration or sclerosis. Tech Vasc Interv Radiol. 2013;16:126–134.
- , , , et al. Jejunal varices as a cause of massive gastrointestinal bleeding. Am J Gastroenterol. 1992;87:514–517.
- , . Ectopic varices in portal hypertension. Clin Gastroenterol. 1985;14:105–121.
- , , , et al. Ectopic varices in portal hypertension: computed tomographic angiography instead of repeated endoscopies for diagnosis. Eur J Gastroenterol Hepatol. 2011;23:620–622.
- , , , et al. ACR appropriateness criteria. Radiologic management of lower gastrointestinal tract bleeding. Reston, VA: American College of Radiology; 2011. Available at: http://www.acr.org/Quality‐Safety/Appropriateness‐Criteria/∼/media/5F9CB95C164E4DA19DCBCFBBA790BB3C.pdf. Accessed January 28, 2015.
- , . Diagnosis and management of ectopic varices. Gastrointest Interv. 2012;1:3–10.
- . Achieving quality in clinical decision making: cognitive strategies and detection of bias. Acad Emerg Med. 2002;9:1184–1204.
- , , . Updates in the pathogenesis, diagnosis and management of ectopic varices. Hepatol Int. 2008;2:322–334.
- , , . Management of ectopic varices. Hepatology. 1998;28:1154–1158.
- , , , et al. Current status of ectopic varices in Japan: results of a survey by the Japan Society for Portal Hypertension. Hepatol Res. 2010;40:763–766.
- , , . Stomal Varices: Management with decompression TIPS and transvenous obliteration or sclerosis. Tech Vasc Interv Radiol. 2013;16:126–134.
- , , , et al. Jejunal varices as a cause of massive gastrointestinal bleeding. Am J Gastroenterol. 1992;87:514–517.
- , . Ectopic varices in portal hypertension. Clin Gastroenterol. 1985;14:105–121.
- , , , et al. Ectopic varices in portal hypertension: computed tomographic angiography instead of repeated endoscopies for diagnosis. Eur J Gastroenterol Hepatol. 2011;23:620–622.
- , , , et al. ACR appropriateness criteria. Radiologic management of lower gastrointestinal tract bleeding. Reston, VA: American College of Radiology; 2011. Available at: http://www.acr.org/Quality‐Safety/Appropriateness‐Criteria/∼/media/5F9CB95C164E4DA19DCBCFBBA790BB3C.pdf. Accessed January 28, 2015.
- , . Diagnosis and management of ectopic varices. Gastrointest Interv. 2012;1:3–10.
- . Achieving quality in clinical decision making: cognitive strategies and detection of bias. Acad Emerg Med. 2002;9:1184–1204.
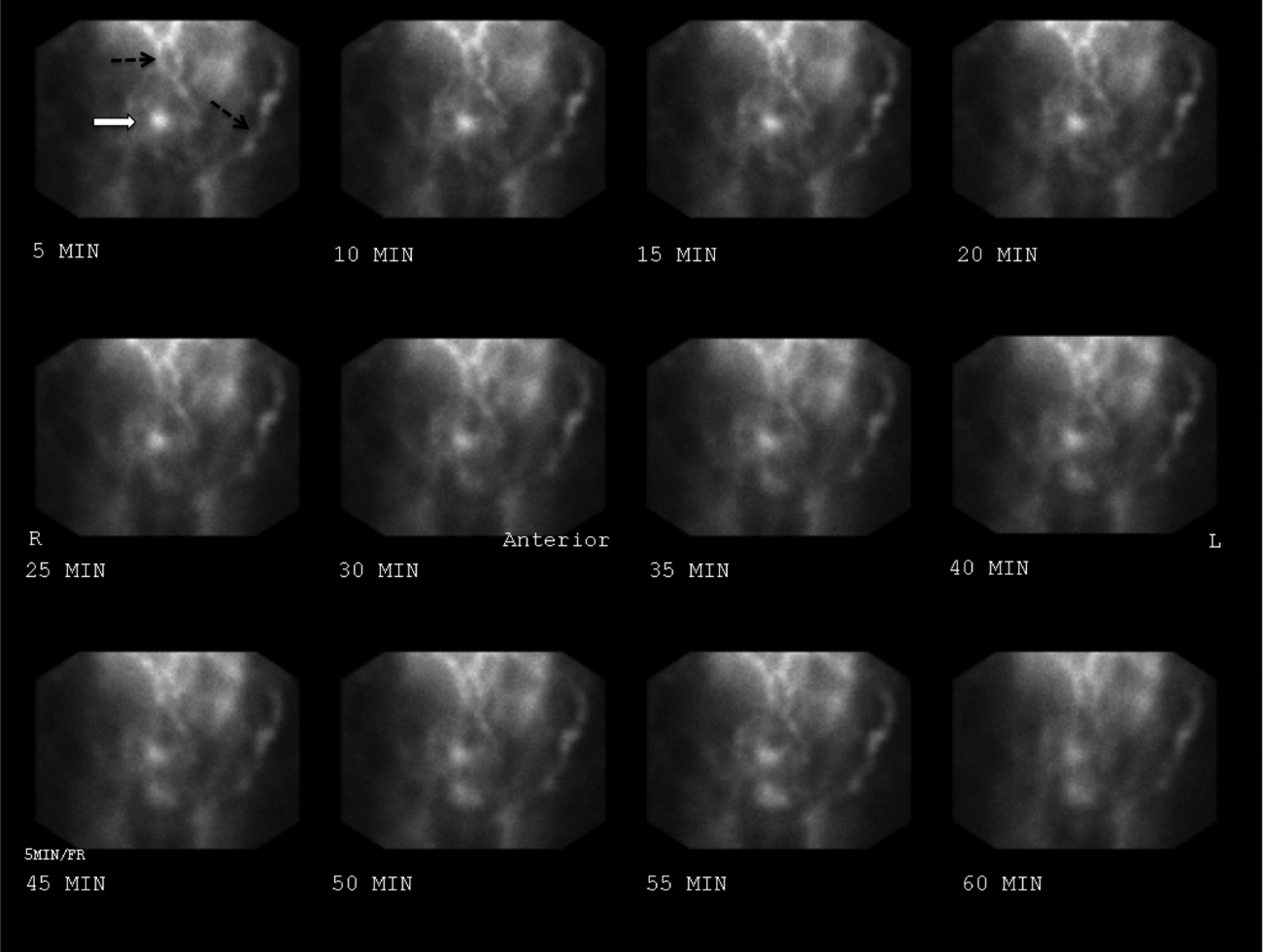
What Should You Do If You Get a Needlestick?
EDITOR’S NOTE: This month’s KCQ first appeared in October 2010 and since that time has been one of our website’s most-read articles, generating nearly 35,000-plus page views. Enjoy it again this month!
Case
While placing a central line, you sustain a needlestick. You’ve washed the area thoroughly with soap and water, but you are concerned about contracting a bloodborne pathogen. What is the risk of contracting such a pathogen, and what can be done to reduce this risk?
Overview
Needlestick injuries are a common occupational hazard in the hospital setting. According to the International Health Care Worker Safety Center, approximately 295,000 hospital-based healthcare workers experience occupational percutaneous injuries annually. In 1991, Mangione and colleagues surveyed internal medicine house staff and found an annual incidence of 674 needlestick injuries per 1,000 participants.1 Other retrospective data estimate this risk to be as high as 839 per 1,000 healthcare workers annually.2 Evidence from the CDC in 2004 suggests that because these numbers represent only self-reported injuries, the annual incidence of such injuries is much higher than the current estimates suggest.2,3,4
More than 20 bloodborne pathogens (see Table 1) might be transmitted from contaminated needles or sharps, including human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV). A quick and appropriate response to a needlestick injury can greatly decrease the risk of disease transmission following an occupational exposure to potentially infectious materials.

Review of the Data
After any needlestick injury, an affected healthcare worker should wash the area with soap and water immediately. There is no contraindication to using antiseptic solutions, but there is also no evidence to suggest that this reduces the rates of disease transmission.
Because decisions for post-exposure prophylaxis often need to be made within hours, a healthcare worker should seek care in the facility areas responsible for managing occupational exposures. Healthcare providers should be encouraged and supported in reporting all sharps-related injuries to such departments.
The source patient should be identified and evaluated for potentially transmissible diseases, including HIV, HBV, and HCV. If indicated, the source patient should then undergo appropriate serological testing, and any indicated antiviral prophylaxis should be initiated (see Table 2).
Risk of Seroconversion
For all bloodborne pathogens, a needlestick injury carries a greater risk for transmission than other occupational exposures (e.g. mucous membrane exposure). If a needlestick injury occurs in the setting of an infected patient source, the risk of disease transmission varies for HIV, HBV, and HCV (see Table 3). In general, risk for seroconversion is increased with a deep injury, an injury with a device visibly contaminated with the source patient’s blood, or an injury involving a needle placed in the source patient’s artery or vein.3,5,6

Human immunodeficiency virus. Contracting HIV after needlestick injury is rare. From 1981 to 2006, the CDC documented only 57 cases of HIV/AIDS in healthcare workers following occupational exposure and identified an additional “possible” 140 cases post-exposure.5,6 Of the 57 documented cases, 48 sustained a percutaneous injury.
Following needlestick injury involving a known HIV-positive source, the one-year risk of seroconversion has been estimated to be 0.3%.5,6 In 1997, Cardo and colleagues identified four factors associated with increased risk for seroconversion after a needlestick/sharps injury from a known positive-HIV source:
- Deep injury;
- Injury with a device visibly contaminated with the source patient’s blood;
- A procedure involving a needle placed in the source patient’s artery or vein; and
- Exposure to a source patient who died of AIDS in the two months following the occupational exposure.5
Hepatitis B virus. Widespread immunization of healthcare workers has led to a dramatic decline in occupationally acquired HBV. The CDC estimated that in 1985, approximately 12,500 new HBV infections occurred in healthcare workers.3 This estimate plummeted to approximately 500 new occupationally acquired HBV infections in 1997.3
Despite this improvement, hospital-based healthcare personnel remain at risk for HBV transmission after a needlestick injury from a known positive patient source. Few studies have evaluated the occupational risk of HBV transmission after a needlestick injury. Buergler and colleagues reported that, following a needlestick injury involving a known HBV-positive source, the one-year risk of seroconversion was 0.76% to 7.35% for nonimmunized surgeons and 0.23% to 2.28% for nonimmunized anesthesiologists.7
In the absence of post-exposure prophylaxis (PEP), an exposed healthcare worker has a 6% to 30% risk of becoming infected with HBV.3,8 The risk is greatest if the patient source is known to be hepatitis B e antigen-positive, a marker for greater disease infectivity. When given within one week of injury, PEP with multiple doses of hepatitis B immune globulin (HBIG) provides an estimated 75% protection from transmission.

Healthcare workers who have received the hepatitis B vaccine and developed immunity have virtually no risk for infection.6,7
Hepatitis C virus. Prospective evaluation has demonstrated that the average risk of HCV transmission after percutaneous exposure to a known HCV-positive source ranges from 0% to 7%.3 The Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections evaluated HCV seroconversion within six months of a reported exposure with enzyme immunoassay and immunoblot assay. In this study, the authors found a seroconversion rate of 1.2%.9
Further, they suggested that HCV seroconversion only occurred from hollow-bore needles, because no seroconversions were noted in healthcare workers who sustained injuries with solid sharp objects.
Post-Exposure Management
The CDC does not recommend prophylaxis when source fluids make contact with intact skin; however, if a percutaneous occupational exposure has occurred, PEPs exist for HIV and HBV but not for HCV.3,6 If a source patient’s HIV, HBV, and HCV statuses are unknown, occupational health personnel can interview the patient to evaluate his or her risks and initiate testing. Specific information about the time and nature of exposure should be documented.

When testing is indicated, it should be done following institutional and state-specific exposure control policies and informed consent guidelines. In all situations, the decision to begin antiviral PEP should be carefully considered, weighing the benefits of PEP versus the risks and toxicity of treatment.
Human immunodeficiency virus. If a source patient is known to be HIV-positive, has a positive rapid HIV test, or if HIV status cannot be quickly determined, PEP is indicated and should be started as quickly as possible.3,8,10
The 2013 U.S. Public Health Service recommendations for PEP call for initiating three (or more) antiretroviral drugs for all occupational exposures. Current recommendations indicate that PEP should be continued for four weeks, with concurrent clinical and laboratory evaluation for drug toxicity.10
Although the combination of HBIG and the hepatitis vaccine B series has not been evaluated as PEP in the occupational setting, evidence in the perinatal setting suggests this regimen is more effective than HBIG alone.3,6,8
Hepatitis C virus. No PEP exists for HCV, and current recommendations for post-exposure management focus on early identification and treatment of chronic disease. There are insufficient data for a treatment recommendation for patients with acute HCV infection with no evidence of disease; the appropriate dosing of such a regimen is unknown. Further, evidence suggests that treatment started early in the course of chronic infection could be just as effective and might eliminate the need to treat persons whose infection will spontaneously resolve.7
Back to the Case
Your needlestick occurred while using a hollow-bore needle to cannulate a source patient’s vein, placing you at higher risk for seroconversion. You immediately reported the exposure to the department of occupational health at your hospital. The source patient’s HIV, HBV, and HCV serological statuses were tested, and the patient was found to be HBV-positive. After appropriate counseling, you decide to receive HBIG prophylaxis to reduce your chances of becoming infected with HBV infection.
Bottom Line
Healthcare workers who suffer occupational needlestick injuries require immediate identification and attention to avoid transmission of such infectious diseases as HIV, HBV, and HCV. Source patients should undergo rapid serological testing to determine appropriate PEP.
Dr. Zehnder is a hospitalist and assistant professor of medicine at the University of Colorado Denver in Aurora.
References
- Mangione CM, Gerberding JL, Cummings SR. Occupational exposure to HIV: Frequency and rates of underreporting of percutaneous and mucocutaneous exposures by medical housestaff. Am J Med. 1991;90(1):85-90.
- Lee JM, Botteman MF, Nicklasson L, Cobden D, Pashos CL. Needlestick injury in acute care nurses caring for patients with diabetes mellitus: a retrospective study. Curr Med Res Opin. 2005;21(5):741-747.
- Centers for Disease Control and Prevention. Workbook for designing, implementing, and evaluating a sharps injury prevention program. CDC website. Accessed May 31, 2015.
- Lee JM, Botteman MF, Xanthakos N, Nicklasson L. Needlestick injuries in the United States. Epidemiologic, economic, and quality of life issues. AAOHN J. 2005;53(3):117-133.
- Cardo DM, Culver DH, Ciesielski CA, et al. A case-control study of HIV seroconversion in health care workers after percutaneous exposure. Centers for Disease Control and Prevention Needlestick Surveillance Group. N Engl J Med. 1997;337(21):1485-1490.
- Centers for Disease Control and Prevention. Exposure to blood: What healthcare personnel need to know. CDC website. Accessed May 31, 2015.
- Buergler JM, Kim R, Thisted RA, Cohn SJ, Lichtor JL, Roizen MF. Risk of human immunodeficiency virus in surgeons, anesthesiologists, and medical students. Anesth Analg. 1992;75(1):118-124.
- Centers for Disease Control and Prevention. Updated U.S. Public Health Service guidelines for the management of occupational exposures to HBV, HCV, and HIV and recommendations for postexposure prophylaxis. CDC website. Accessed May 31, 2015.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposure in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control. 1995;23(5):273-277.
- Updated US Public Health Service Guidelines for the management of occupational exposures to Human Immunodeficiency Virus and Recommendations for Postexposure Prophylaxis. Accessed May 31, 2015.
EDITOR’S NOTE: This month’s KCQ first appeared in October 2010 and since that time has been one of our website’s most-read articles, generating nearly 35,000-plus page views. Enjoy it again this month!
Case
While placing a central line, you sustain a needlestick. You’ve washed the area thoroughly with soap and water, but you are concerned about contracting a bloodborne pathogen. What is the risk of contracting such a pathogen, and what can be done to reduce this risk?
Overview
Needlestick injuries are a common occupational hazard in the hospital setting. According to the International Health Care Worker Safety Center, approximately 295,000 hospital-based healthcare workers experience occupational percutaneous injuries annually. In 1991, Mangione and colleagues surveyed internal medicine house staff and found an annual incidence of 674 needlestick injuries per 1,000 participants.1 Other retrospective data estimate this risk to be as high as 839 per 1,000 healthcare workers annually.2 Evidence from the CDC in 2004 suggests that because these numbers represent only self-reported injuries, the annual incidence of such injuries is much higher than the current estimates suggest.2,3,4
More than 20 bloodborne pathogens (see Table 1) might be transmitted from contaminated needles or sharps, including human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV). A quick and appropriate response to a needlestick injury can greatly decrease the risk of disease transmission following an occupational exposure to potentially infectious materials.
Review of the Data
After any needlestick injury, an affected healthcare worker should wash the area with soap and water immediately. There is no contraindication to using antiseptic solutions, but there is also no evidence to suggest that this reduces the rates of disease transmission.
Because decisions for post-exposure prophylaxis often need to be made within hours, a healthcare worker should seek care in the facility areas responsible for managing occupational exposures. Healthcare providers should be encouraged and supported in reporting all sharps-related injuries to such departments.
The source patient should be identified and evaluated for potentially transmissible diseases, including HIV, HBV, and HCV. If indicated, the source patient should then undergo appropriate serological testing, and any indicated antiviral prophylaxis should be initiated (see Table 2).
Risk of Seroconversion
For all bloodborne pathogens, a needlestick injury carries a greater risk for transmission than other occupational exposures (e.g. mucous membrane exposure). If a needlestick injury occurs in the setting of an infected patient source, the risk of disease transmission varies for HIV, HBV, and HCV (see Table 3). In general, risk for seroconversion is increased with a deep injury, an injury with a device visibly contaminated with the source patient’s blood, or an injury involving a needle placed in the source patient’s artery or vein.3,5,6
Human immunodeficiency virus. Contracting HIV after needlestick injury is rare. From 1981 to 2006, the CDC documented only 57 cases of HIV/AIDS in healthcare workers following occupational exposure and identified an additional “possible” 140 cases post-exposure.5,6 Of the 57 documented cases, 48 sustained a percutaneous injury.
Following needlestick injury involving a known HIV-positive source, the one-year risk of seroconversion has been estimated to be 0.3%.5,6 In 1997, Cardo and colleagues identified four factors associated with increased risk for seroconversion after a needlestick/sharps injury from a known positive-HIV source:
- Deep injury;
- Injury with a device visibly contaminated with the source patient’s blood;
- A procedure involving a needle placed in the source patient’s artery or vein; and
- Exposure to a source patient who died of AIDS in the two months following the occupational exposure.5
Hepatitis B virus. Widespread immunization of healthcare workers has led to a dramatic decline in occupationally acquired HBV. The CDC estimated that in 1985, approximately 12,500 new HBV infections occurred in healthcare workers.3 This estimate plummeted to approximately 500 new occupationally acquired HBV infections in 1997.3
Despite this improvement, hospital-based healthcare personnel remain at risk for HBV transmission after a needlestick injury from a known positive patient source. Few studies have evaluated the occupational risk of HBV transmission after a needlestick injury. Buergler and colleagues reported that, following a needlestick injury involving a known HBV-positive source, the one-year risk of seroconversion was 0.76% to 7.35% for nonimmunized surgeons and 0.23% to 2.28% for nonimmunized anesthesiologists.7
In the absence of post-exposure prophylaxis (PEP), an exposed healthcare worker has a 6% to 30% risk of becoming infected with HBV.3,8 The risk is greatest if the patient source is known to be hepatitis B e antigen-positive, a marker for greater disease infectivity. When given within one week of injury, PEP with multiple doses of hepatitis B immune globulin (HBIG) provides an estimated 75% protection from transmission.
Healthcare workers who have received the hepatitis B vaccine and developed immunity have virtually no risk for infection.6,7
Hepatitis C virus. Prospective evaluation has demonstrated that the average risk of HCV transmission after percutaneous exposure to a known HCV-positive source ranges from 0% to 7%.3 The Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections evaluated HCV seroconversion within six months of a reported exposure with enzyme immunoassay and immunoblot assay. In this study, the authors found a seroconversion rate of 1.2%.9
Further, they suggested that HCV seroconversion only occurred from hollow-bore needles, because no seroconversions were noted in healthcare workers who sustained injuries with solid sharp objects.
Post-Exposure Management
The CDC does not recommend prophylaxis when source fluids make contact with intact skin; however, if a percutaneous occupational exposure has occurred, PEPs exist for HIV and HBV but not for HCV.3,6 If a source patient’s HIV, HBV, and HCV statuses are unknown, occupational health personnel can interview the patient to evaluate his or her risks and initiate testing. Specific information about the time and nature of exposure should be documented.
When testing is indicated, it should be done following institutional and state-specific exposure control policies and informed consent guidelines. In all situations, the decision to begin antiviral PEP should be carefully considered, weighing the benefits of PEP versus the risks and toxicity of treatment.
Human immunodeficiency virus. If a source patient is known to be HIV-positive, has a positive rapid HIV test, or if HIV status cannot be quickly determined, PEP is indicated and should be started as quickly as possible.3,8,10
The 2013 U.S. Public Health Service recommendations for PEP call for initiating three (or more) antiretroviral drugs for all occupational exposures. Current recommendations indicate that PEP should be continued for four weeks, with concurrent clinical and laboratory evaluation for drug toxicity.10
Although the combination of HBIG and the hepatitis vaccine B series has not been evaluated as PEP in the occupational setting, evidence in the perinatal setting suggests this regimen is more effective than HBIG alone.3,6,8
Hepatitis C virus. No PEP exists for HCV, and current recommendations for post-exposure management focus on early identification and treatment of chronic disease. There are insufficient data for a treatment recommendation for patients with acute HCV infection with no evidence of disease; the appropriate dosing of such a regimen is unknown. Further, evidence suggests that treatment started early in the course of chronic infection could be just as effective and might eliminate the need to treat persons whose infection will spontaneously resolve.7
Back to the Case
Your needlestick occurred while using a hollow-bore needle to cannulate a source patient’s vein, placing you at higher risk for seroconversion. You immediately reported the exposure to the department of occupational health at your hospital. The source patient’s HIV, HBV, and HCV serological statuses were tested, and the patient was found to be HBV-positive. After appropriate counseling, you decide to receive HBIG prophylaxis to reduce your chances of becoming infected with HBV infection.
Bottom Line
Healthcare workers who suffer occupational needlestick injuries require immediate identification and attention to avoid transmission of such infectious diseases as HIV, HBV, and HCV. Source patients should undergo rapid serological testing to determine appropriate PEP.
Dr. Zehnder is a hospitalist and assistant professor of medicine at the University of Colorado Denver in Aurora.
References
- Mangione CM, Gerberding JL, Cummings SR. Occupational exposure to HIV: Frequency and rates of underreporting of percutaneous and mucocutaneous exposures by medical housestaff. Am J Med. 1991;90(1):85-90.
- Lee JM, Botteman MF, Nicklasson L, Cobden D, Pashos CL. Needlestick injury in acute care nurses caring for patients with diabetes mellitus: a retrospective study. Curr Med Res Opin. 2005;21(5):741-747.
- Centers for Disease Control and Prevention. Workbook for designing, implementing, and evaluating a sharps injury prevention program. CDC website. Accessed May 31, 2015.
- Lee JM, Botteman MF, Xanthakos N, Nicklasson L. Needlestick injuries in the United States. Epidemiologic, economic, and quality of life issues. AAOHN J. 2005;53(3):117-133.
- Cardo DM, Culver DH, Ciesielski CA, et al. A case-control study of HIV seroconversion in health care workers after percutaneous exposure. Centers for Disease Control and Prevention Needlestick Surveillance Group. N Engl J Med. 1997;337(21):1485-1490.
- Centers for Disease Control and Prevention. Exposure to blood: What healthcare personnel need to know. CDC website. Accessed May 31, 2015.
- Buergler JM, Kim R, Thisted RA, Cohn SJ, Lichtor JL, Roizen MF. Risk of human immunodeficiency virus in surgeons, anesthesiologists, and medical students. Anesth Analg. 1992;75(1):118-124.
- Centers for Disease Control and Prevention. Updated U.S. Public Health Service guidelines for the management of occupational exposures to HBV, HCV, and HIV and recommendations for postexposure prophylaxis. CDC website. Accessed May 31, 2015.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposure in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control. 1995;23(5):273-277.
- Updated US Public Health Service Guidelines for the management of occupational exposures to Human Immunodeficiency Virus and Recommendations for Postexposure Prophylaxis. Accessed May 31, 2015.
EDITOR’S NOTE: This month’s KCQ first appeared in October 2010 and since that time has been one of our website’s most-read articles, generating nearly 35,000-plus page views. Enjoy it again this month!
Case
While placing a central line, you sustain a needlestick. You’ve washed the area thoroughly with soap and water, but you are concerned about contracting a bloodborne pathogen. What is the risk of contracting such a pathogen, and what can be done to reduce this risk?
Overview
Needlestick injuries are a common occupational hazard in the hospital setting. According to the International Health Care Worker Safety Center, approximately 295,000 hospital-based healthcare workers experience occupational percutaneous injuries annually. In 1991, Mangione and colleagues surveyed internal medicine house staff and found an annual incidence of 674 needlestick injuries per 1,000 participants.1 Other retrospective data estimate this risk to be as high as 839 per 1,000 healthcare workers annually.2 Evidence from the CDC in 2004 suggests that because these numbers represent only self-reported injuries, the annual incidence of such injuries is much higher than the current estimates suggest.2,3,4
More than 20 bloodborne pathogens (see Table 1) might be transmitted from contaminated needles or sharps, including human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV). A quick and appropriate response to a needlestick injury can greatly decrease the risk of disease transmission following an occupational exposure to potentially infectious materials.
Review of the Data
After any needlestick injury, an affected healthcare worker should wash the area with soap and water immediately. There is no contraindication to using antiseptic solutions, but there is also no evidence to suggest that this reduces the rates of disease transmission.
Because decisions for post-exposure prophylaxis often need to be made within hours, a healthcare worker should seek care in the facility areas responsible for managing occupational exposures. Healthcare providers should be encouraged and supported in reporting all sharps-related injuries to such departments.
The source patient should be identified and evaluated for potentially transmissible diseases, including HIV, HBV, and HCV. If indicated, the source patient should then undergo appropriate serological testing, and any indicated antiviral prophylaxis should be initiated (see Table 2).
Risk of Seroconversion
For all bloodborne pathogens, a needlestick injury carries a greater risk for transmission than other occupational exposures (e.g. mucous membrane exposure). If a needlestick injury occurs in the setting of an infected patient source, the risk of disease transmission varies for HIV, HBV, and HCV (see Table 3). In general, risk for seroconversion is increased with a deep injury, an injury with a device visibly contaminated with the source patient’s blood, or an injury involving a needle placed in the source patient’s artery or vein.3,5,6
Human immunodeficiency virus. Contracting HIV after needlestick injury is rare. From 1981 to 2006, the CDC documented only 57 cases of HIV/AIDS in healthcare workers following occupational exposure and identified an additional “possible” 140 cases post-exposure.5,6 Of the 57 documented cases, 48 sustained a percutaneous injury.
Following needlestick injury involving a known HIV-positive source, the one-year risk of seroconversion has been estimated to be 0.3%.5,6 In 1997, Cardo and colleagues identified four factors associated with increased risk for seroconversion after a needlestick/sharps injury from a known positive-HIV source:
- Deep injury;
- Injury with a device visibly contaminated with the source patient’s blood;
- A procedure involving a needle placed in the source patient’s artery or vein; and
- Exposure to a source patient who died of AIDS in the two months following the occupational exposure.5
Hepatitis B virus. Widespread immunization of healthcare workers has led to a dramatic decline in occupationally acquired HBV. The CDC estimated that in 1985, approximately 12,500 new HBV infections occurred in healthcare workers.3 This estimate plummeted to approximately 500 new occupationally acquired HBV infections in 1997.3
Despite this improvement, hospital-based healthcare personnel remain at risk for HBV transmission after a needlestick injury from a known positive patient source. Few studies have evaluated the occupational risk of HBV transmission after a needlestick injury. Buergler and colleagues reported that, following a needlestick injury involving a known HBV-positive source, the one-year risk of seroconversion was 0.76% to 7.35% for nonimmunized surgeons and 0.23% to 2.28% for nonimmunized anesthesiologists.7
In the absence of post-exposure prophylaxis (PEP), an exposed healthcare worker has a 6% to 30% risk of becoming infected with HBV.3,8 The risk is greatest if the patient source is known to be hepatitis B e antigen-positive, a marker for greater disease infectivity. When given within one week of injury, PEP with multiple doses of hepatitis B immune globulin (HBIG) provides an estimated 75% protection from transmission.
Healthcare workers who have received the hepatitis B vaccine and developed immunity have virtually no risk for infection.6,7
Hepatitis C virus. Prospective evaluation has demonstrated that the average risk of HCV transmission after percutaneous exposure to a known HCV-positive source ranges from 0% to 7%.3 The Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections evaluated HCV seroconversion within six months of a reported exposure with enzyme immunoassay and immunoblot assay. In this study, the authors found a seroconversion rate of 1.2%.9
Further, they suggested that HCV seroconversion only occurred from hollow-bore needles, because no seroconversions were noted in healthcare workers who sustained injuries with solid sharp objects.
Post-Exposure Management
The CDC does not recommend prophylaxis when source fluids make contact with intact skin; however, if a percutaneous occupational exposure has occurred, PEPs exist for HIV and HBV but not for HCV.3,6 If a source patient’s HIV, HBV, and HCV statuses are unknown, occupational health personnel can interview the patient to evaluate his or her risks and initiate testing. Specific information about the time and nature of exposure should be documented.
When testing is indicated, it should be done following institutional and state-specific exposure control policies and informed consent guidelines. In all situations, the decision to begin antiviral PEP should be carefully considered, weighing the benefits of PEP versus the risks and toxicity of treatment.
Human immunodeficiency virus. If a source patient is known to be HIV-positive, has a positive rapid HIV test, or if HIV status cannot be quickly determined, PEP is indicated and should be started as quickly as possible.3,8,10
The 2013 U.S. Public Health Service recommendations for PEP call for initiating three (or more) antiretroviral drugs for all occupational exposures. Current recommendations indicate that PEP should be continued for four weeks, with concurrent clinical and laboratory evaluation for drug toxicity.10
Although the combination of HBIG and the hepatitis vaccine B series has not been evaluated as PEP in the occupational setting, evidence in the perinatal setting suggests this regimen is more effective than HBIG alone.3,6,8
Hepatitis C virus. No PEP exists for HCV, and current recommendations for post-exposure management focus on early identification and treatment of chronic disease. There are insufficient data for a treatment recommendation for patients with acute HCV infection with no evidence of disease; the appropriate dosing of such a regimen is unknown. Further, evidence suggests that treatment started early in the course of chronic infection could be just as effective and might eliminate the need to treat persons whose infection will spontaneously resolve.7
Back to the Case
Your needlestick occurred while using a hollow-bore needle to cannulate a source patient’s vein, placing you at higher risk for seroconversion. You immediately reported the exposure to the department of occupational health at your hospital. The source patient’s HIV, HBV, and HCV serological statuses were tested, and the patient was found to be HBV-positive. After appropriate counseling, you decide to receive HBIG prophylaxis to reduce your chances of becoming infected with HBV infection.
Bottom Line
Healthcare workers who suffer occupational needlestick injuries require immediate identification and attention to avoid transmission of such infectious diseases as HIV, HBV, and HCV. Source patients should undergo rapid serological testing to determine appropriate PEP.
Dr. Zehnder is a hospitalist and assistant professor of medicine at the University of Colorado Denver in Aurora.
References
- Mangione CM, Gerberding JL, Cummings SR. Occupational exposure to HIV: Frequency and rates of underreporting of percutaneous and mucocutaneous exposures by medical housestaff. Am J Med. 1991;90(1):85-90.
- Lee JM, Botteman MF, Nicklasson L, Cobden D, Pashos CL. Needlestick injury in acute care nurses caring for patients with diabetes mellitus: a retrospective study. Curr Med Res Opin. 2005;21(5):741-747.
- Centers for Disease Control and Prevention. Workbook for designing, implementing, and evaluating a sharps injury prevention program. CDC website. Accessed May 31, 2015.
- Lee JM, Botteman MF, Xanthakos N, Nicklasson L. Needlestick injuries in the United States. Epidemiologic, economic, and quality of life issues. AAOHN J. 2005;53(3):117-133.
- Cardo DM, Culver DH, Ciesielski CA, et al. A case-control study of HIV seroconversion in health care workers after percutaneous exposure. Centers for Disease Control and Prevention Needlestick Surveillance Group. N Engl J Med. 1997;337(21):1485-1490.
- Centers for Disease Control and Prevention. Exposure to blood: What healthcare personnel need to know. CDC website. Accessed May 31, 2015.
- Buergler JM, Kim R, Thisted RA, Cohn SJ, Lichtor JL, Roizen MF. Risk of human immunodeficiency virus in surgeons, anesthesiologists, and medical students. Anesth Analg. 1992;75(1):118-124.
- Centers for Disease Control and Prevention. Updated U.S. Public Health Service guidelines for the management of occupational exposures to HBV, HCV, and HIV and recommendations for postexposure prophylaxis. CDC website. Accessed May 31, 2015.
- Puro V, Petrosillo N, Ippolito G. Risk of hepatitis C seroconversion after occupational exposure in health care workers. Italian Study Group on Occupational Risk of HIV and Other Bloodborne Infections. Am J Infect Control. 1995;23(5):273-277.
- Updated US Public Health Service Guidelines for the management of occupational exposures to Human Immunodeficiency Virus and Recommendations for Postexposure Prophylaxis. Accessed May 31, 2015.
How Should a Patient with Pulmonary Hypertension Be Evaluated, Managed?
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
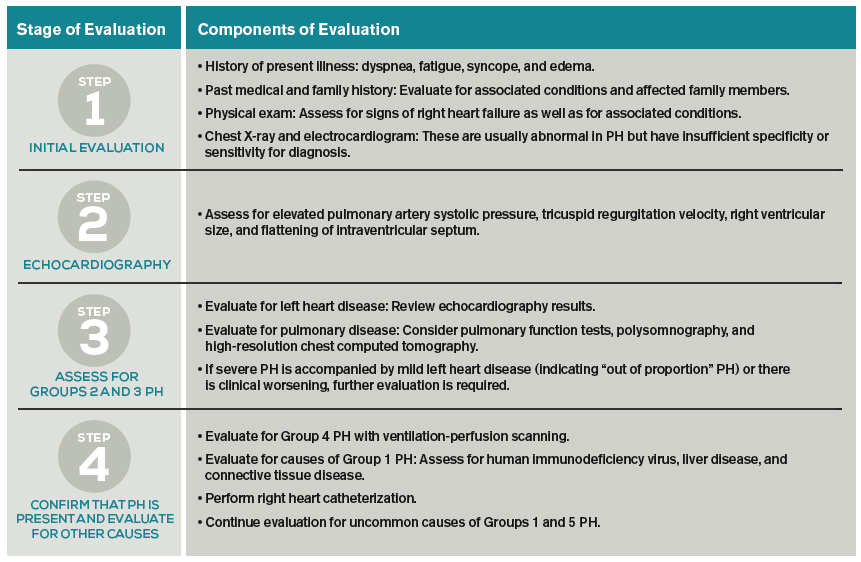
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.

All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.
All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.
Case
A 62-year-old female with no significant past medical history presents with three weeks of progressive dyspnea on exertion and bilateral lower extremity edema. Family members report that the patient often snores and “gasps for air” during sleep. B-type natriuretic peptide is elevated at 2,261 pg/ml. Due to concern for congestive heart failure, transthoracic echocardiography (TTE) is performed and shows normal left ventricular systolic function, mild left ventricular diastolic dysfunction, severely elevated right ventricular systolic pressure of 74 mm Hg, and right ventricular dilatation and hypokinesis.
How should this patient with newfound pulmonary hypertension (PH) be evaluated and managed?
Background
PH is a progressive disease that presents with nonspecific signs and symptoms and can be fatal if untreated. Ernst von Romberg first identified the disease in 1891, and efforts have been made through the last century to understand its etiology and mechanisms.1
PH is defined as an elevated mean pulmonary arterial pressure (mPAP) of ≥25 mmHg at rest; a mPAP of ≤20 mmHg is considered normal, and a mPAP of 21-24 mmHg is borderline.2 This elevation of the mPAP can be due to a primary elevation of pressures in the pulmonary arterial system alone (pulmonary arterial hypertension) or secondary to elevation in pressures in the pulmonary venous and pulmonary capillary systems (pulmonary venous hypertension).
PH classification has endured many modifications through the years with better understanding of its pathophysiology. Currently, the World Health Organization (WHO) classification system includes five groups based on etiology (see Table 1):3,4
- Group 1: Pulmonary arterial hypertension (PAH);
- Group 2: PH due to left heart disease;
- Group 3: PH due to chronic lung disease and hypoxemia;
- Group 4: Chronic thromboembolic PH (CTEPH); and
- Group 5: PH due to unclear multifactorial mechanisms.
The pathophysiology differs among the groups, and much of what is known has come from studies performed in patients with idiopathic PAH. It is a proliferative vasculopathy characterized by vasoconstriction, cell proliferation, fibrosis, and thrombosis. Both genetic predisposition and modifiers that include drugs and toxins, human immunodeficiency virus (HIV), congenital heart disease with left-to-right shunting, and potassium channel dysfunction play a role in the pathogenesis.3,5,6 Although many processes underlying the pathophysiology of PH groups 2, 3, 4, and 5 are not fully understood, vascular remodeling and increased vascular resistance are common to all of them.
PH affects both genders and all age groups and races. Due to its broad classification and multiple etiologies, it is difficult to assess PH prevalence in the general population. There are wide ranges among different populations, with PH prevalence in sickle cell disease ranging from 20% to 40%, in systemic sclerosis from 10% to 15%, and in portal hypertension from 2% to 16%.7,8,9 PH in COPD is usually mild to moderate, with preserved cardiac output, although a minority of patients develop severe PH.10-12 PH is present in approximately 20% of patients with moderate to severe sleep apnea.13 The prevalence of PH in left heart disease is unknown due to variability in populations assessed and methods used in various studies; estimates have ranged from 25-100%.14
Evaluation
Initial evaluation: A thorough history and physical examination can help determine PH etiology, identify associated conditions, and determine the severity of disease. Dyspnea on exertion is the most common presenting complaint; weakness, fatigue, and angina may be present.15 Lower extremity edema and ascites are indicative of more advanced disease.
A patient’s symptoms may suggest the presence of undiagnosed conditions that are associated with PH, and past medical history should evaluate for previous diagnoses of these conditions (see Table 1).
Family history may reveal relatives with PH, given the genetic predisposition to development of Group 1 PH. Physical exam findings include a prominent pulmonic valve closure during the second heart sound, a palpable left parasternal heave, and a tricuspid regurgitation murmur.
Electrocardiogram (ECG) and chest X-ray (CXR) are not sufficiently sensitive or specific to diagnose PH but may provide initial supporting evidence that prompts further testing. Signs of right ventricular hypertrophy and right atrial enlargement may be present on ECG. The CXR may show pruning (prominent hilar vasculature with reduced vasculature peripherally) and right ventricular hypertrophy, as evidenced by shrinking of the retrosternal window on lateral CXR. An unremarkable ECG or normal CXR does not rule out PH.
Echocardiography: TTE allows estimation of pulmonary artery systolic pressure (PASP) via measurement of tricuspid regurgitation jet velocity and estimation of right atrial pressure. Although results of TTE do correlate with measurements from right heart catheterization (RHC), underestimation and overestimation commonly occur. PASP thresholds for diagnosing or ruling out PH cannot thus be defined easily. An elevated PASP less than 36 mmHg, tricuspid regurgitation velocity <2.8 m/s, and no additional echocardiographic variables suggestive of PH may indicate that PH is unlikely, based on arbitrary criteria from one clinical practice guideline.16
The guideline suggested that tricuspid regurgitation velocity >3.4 m/s or estimated PASP >50 mmHg indicated that PH was likely. Other echocardiographic variables that may suggest the presence of PH include right ventricular enlargement or intraventricular septal flattening. Finally, TTE should also be used to assess for possible causes of PH, such as left heart disease or cardiac shunts.
Further evaluation: Following identification of PH via TTE, further testing can confirm the diagnosis, determine the etiology of the PH, and allow appropriate treatment (see Table 2). Much of this evaluation may occur after hospital discharge and, in cases of unexplained PH, referral to a pulmonologist for further evaluation and management is appropriate. Depending on patient stability, test availability, and patient ability to follow up, some testing may be reasonable during the inpatient stay.
Patients should undergo a stepwise series of testing that initially may be guided by clinical suspicion for underlying conditions.15-19 Polysomnography can identify sleep-disordered breathing, and pulmonary function tests and high-resolution chest CT can assess for chronic pulmonary diseases. Patients with groups 2 and 3 PH, whose PH can be explained by left heart disease or lung disease, do not necessarily require RHC or extensive evaluation for other etiologies of PH.2,17 These patients may be monitored while their underlying conditions are managed.
Patients with worsening clinical course or PH that is “out of proportion” to their lung disease or heart disease, however, do require further evaluation, including RHC. “Out of proportion” has not been consistently defined but generally refers to severe PH observed in patients with mild left heart or lung disease.18 More precise terminology and criteria to define patients with out of proportion PH have been proposed.14
Ventilation-perfusion scanning is required in all cases of PH of unknown etiology to evaluate for CTEPH (Group 4 PH). CT angiography, while appropriate to use in testing for acute pulmonary embolism, is not sufficiently sensitive to evaluate for CTEPH. Tests for liver function, HIV, and connective tissue disease may identify conditions associated with Group 1 PH. Ultimately, RHC is required to confirm the diagnosis of PH, given the shortcomings of TTE. A vasodilator study during RHC allows identification of candidates for advanced therapies, such as patients with Group 1 PH.
Management
The prognosis and treatment of PH varies by WHO Group. The hospitalist will often undertake initial management of symptomatic patients (see Table 3). Intravenous loop diuretics will successfully treat peripheral edema and hepatic congestion in all PH patients.20 Due to the possibility of decreased cardiac output or worsened hypotension in some PH groups, patients should be monitored closely during initial diuresis.
All patients with PH should be assessed for hypoxia during rest, ambulation, and sleep during their hospitalization. Supplemental oxygen therapy should be initiated in all patients with evidence of persistent hypoxia (arterial oxygen blood pressure <60 mmHg).20 Vaccination against pneumococcus and influenza should also be performed during the initial hospitalization. Pregnant patients diagnosed with PH require urgent maternal-fetal medicine consultation.
Further management should be guided by the underlying etiology of the PH:17,18
- Group 1 PH. These patients should be evaluated by a pulmonology consultant, if one is available, as they require intense outpatient follow-up with a pulmonologist. Specialized treatment regimens include calcium channel blockers, phosphodiesterase inhibitors, prostanoids, endothelin receptor antagonists, or newly approved guanylate cyclase stimulants. In previously diagnosed patients, these medications should be continued during a patient’s admission unless the medication is clearly causing the patient harm (such as worsening hypotension) or preventing improvement. Many of these patients are placed on chronic anticoagulation with warfarin, with a goal international normalized ratio (INR) of 1.5 to 2.5.
- Group 2 PH. Patients with left heart or valvular dysfunction and PH have a worse prognosis than similar patients without PH. Management of these patients should focus on treating the underlying etiology. Use of prostanoids may be harmful in this patient population.18
- Group 3 PH. Patients whose PH is fully explained by pulmonary disease should be started on continuous oxygen therapy to treat persistent hypoxemia, and their underlying disorder should be treated, with pulmonologist consultation and referral if necessary.
- Group 4 PH. Patients with newly diagnosed CTEPH should be initiated on warfarin with a goal INR of 2.0 to 3.0. They should undergo evaluation by a pulmonologist for thromboendarterectomy and possibly advanced medical therapies.
- Group 5 PH. Patients with sarcoidosis as the cause of their PH may benefit from prostanoid or endothelin receptor antagonist therapy and should undergo evaluation by a pulmonologist.21,22
Patients with sickle cell anemia, metabolic disorders, and other causes should undergo further subspecialist evaluation prior to initiating therapy to treat their PH.
Back to the Case
The patient underwent diuresis with intravenous furosemide over several days, with gradual improvement in her lower extremity edema and dyspnea. She was placed on oxygen therapy for persistent hypoxemia. As her highly elevated pulmonary artery pressure appeared to be “out of proportion” to her mild left ventricular diastolic dysfunction, further evaluation was pursued. Ventilation-perfusion scanning was performed and showed no mismatch of perfusion and ventilation, effectively ruling out CTEPH. Liver function, HIV, and connective tissue disease testing yielded unremarkable results.
The patient was euvolemic after one week of diuresis and was discharged home with plans for PH specialist follow-up, polysomnography to evaluate for sleep-disordered breathing, and likely RHC. The etiology of her PH was not clear at discharge.
Bottom Line
Evaluation of PH is a step-wise process that starts with history and physical exam and may require extensive evaluation, including right heart catheterization to confirm the diagnosis and define the etiology. A primary goal of evaluation is to define the appropriate therapy for a given patient, which may include advanced therapies in some cases.
Dr. Griffith is a quality improvement fellow and instructor of medicine in the Hospital Medicine Division at the University of Colorado Denver. Drs. McFarland and Smolkin are hospitalists and instructors of medicine at the University of Colorado Denver.
References
- von Romberg E. Über sklerose der lungenarterie. Dtsch Arch Klin Med. 1891;48:197-206.
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D42-D50.
- Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34-D41.
- Rich S, Rubin L, Abenhail L, et al. Executive summary from the World Symposium on primary pulmonary hypertension. Evian, France: The World Health Organization; 1998.
- Newman JH, Wheeler L, Lane KB, et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. New Engl J Med. 2001;345(5):319-24.'
- Petitpretz P, Brenot F, Azarian R, et al. Pulmonary hypertension in patients with human immunodeficiency virus infection. Comparison with primary pulmonary hypertension. Circulation. 1994;89(6):2722-2727.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. New Engl J Med. 2008;359(21):2254-2265.
- Wigley FM, Lima JA, Mayes M, McLain D, Chapin JL, Ward-Able C. The prevalence of undiagnosed pulmonary arterial hypertension in subjects with connective tissue disease at the secondary health care level of community-based rheumatologists (the UNCOVER study). Arthritis Rheum. 2005;52(7):2125-2132.
- Ramsay MA, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transpl Surg. 1997;3(5):494-500.
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219-24.
- Chaouat A, Bugnet AS, Kadaoui N, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189-94.
- Thabut G, Dauriat G, Stern JB, et al. Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest. 2005;127(5):1531-1536.
- Yamakawa H, Shiomi T, Sasanabe R, et al. Pulmonary hypertension in patients with severe obstructive sleep apnea. Psychiatry Clin Neurosci. 2002;56(3):311-312.
- Vachiery JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100-D108.
- McGoon M, Gutterman D, Steen V, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(1 Suppl):14S-34S.
- Grünig E, Barner A, Bell M, et al. Non-invasive diagnosis of pulmonary hypertension: ESC/ERS Guidelines with Updated Commentary of the Cologne Consensus Conference 2011. Int J Cardiol. 2011;154 Suppl 1:S3-12.
- Galiè N, Hoeper MM, Humbert M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J. 2009;30(20):2493-2537.
- McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250-2294.
- Brown K, Gutierrez AJ, Mohammed TL, et al. ACR Appropriateness Criteria(R) pulmonary hypertension. J Thorac Imaging. 2013;28(4):W57-60.
- Galiè N, Corris PA, Frost A, et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D60-72.
- Fisher KA, Serlin DM, Wilson KC, Walter RE, Berman JS, Farber HW. Sarcoidosis-associated pulmonary hypertension: outcome with long-term epoprostenol treatment. Chest. 2006;130(5):1481-1488.
- Steiner MK, Preston IR, Klinger JR, et al. Conversion to bosentan from prostacyclin infusion therapy in pulmonary arterial hypertension: a pilot study. Chest. 2006;130(5):1471-1480.
More Than a Mnemonic
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
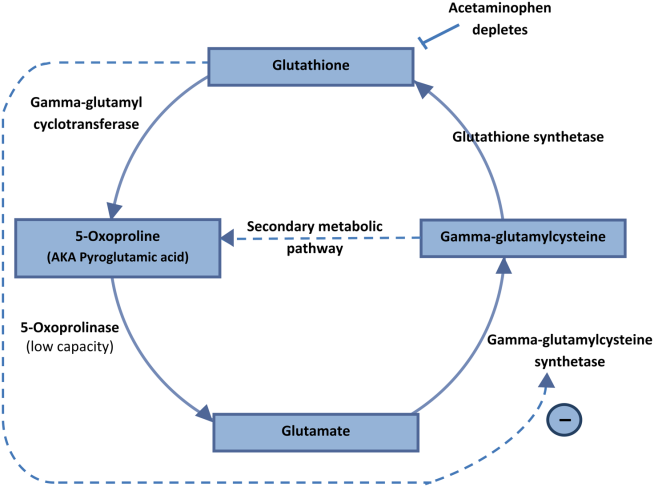
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
A 51‐year‐old man presented to the emergency department after 1 day of progressive dyspnea and increasing confusion.
Acute dyspnea most commonly stems from a cardiac or pulmonary disorder such as heart failure, acute coronary syndrome, pneumonia, pulmonary embolism, or exacerbations of asthma or chronic obstructive pulmonary disease. Less frequent cardiopulmonary considerations include pericardial or pleural effusion, pneumothorax, aspiration, and upper airway obstruction. Dyspnea might also be the initial manifestation of profound anemia or metabolic acidosis.
The presence of confusion suggests either a severe presentation of any of the aforementioned possibilities (with confusion resulting from hypoxia, hypercapnia, or hypotension); a multiorgan illness such as sepsis, malignancy, thromboembolic disease, vasculitis, thyroid dysfunction, or toxic ingestion; or a metabolic derangement related to the underlying cause of dyspnea (for example, hypercalcemia or hyponatremia associated with lung cancer).
Twelve hours prior to presentation, he started to have visual hallucinations. He denied fever, chills, cough, chest discomfort, palpitations, weight gain, headache, neck pain, or weakness.
Visual hallucinations could result from a toxic‐metabolic encephalopathy, such as drug overdose or withdrawal, liver or kidney failure, or hypoxia. A structural brain abnormality may also manifest with visual hallucination. Acute onset at age 51 and the absence of auditory hallucinations argue against a neurodegenerative illness and a primary psychiatric disturbance, respectively.
Episodic hallucinations would support the possibility of seizures, monocular hallucinations would point to a retinal or ocular problem, and a description of yellow‐green hue would suggest a side effect of digoxin.
His past medical history was remarkable for diet‐controlled type 2 diabetes mellitus, hypertension, hyperlipidemia, and chronic low back pain. His medications included metoprolol tartrate 25 mg twice daily, omeprazole 40 mg daily, baclofen 15 mg twice daily, oxycodone 30 mg 3 times daily, and hydrocodone 10 mg/acetaminophen 325 mg, 2 tablets 3 times daily as needed for back pain. He was a smoker with a 30 pack‐year history. He had a history of alcohol and cocaine use, but denied any recent substance use. He had no known history of obstructive pulmonary disease.
The patient takes 3 medications well known to cause confusion and hallucinations (oxycodone, hydrocodone, and baclofen), especially when they accumulate due to excessive ingestion or impaired clearance. Although these medications may suppress ventilatory drive, dyspnea would not be a common presenting complaint. He has risk factors for ischemic heart disease and cardiomyopathy, and his smoking history raises the possibility of malignancy.
On exam, the patient's temperature was 94.4C, heart rate 128 beats per minute, respiration rate 28 breaths per minute, blood pressure 155/63 mm Hg, and oxygen saturation 100% while breathing ambient air. The patient was cachectic and appeared in moderate respiratory distress. His pupils were equal and reactive to light, and extraocular movements were intact. He did not have scleral icterus, or cervical or clavicular lymphadenopathy. His oropharynx was negative for erythema, edema, or exudate. His cardiovascular exam revealed a regular tachycardia without rubs or diastolic gallops. There was a 2/6 systolic murmur heard best at left sternal border, without radiation. He did not have jugular venous distention. His pulmonary exam was notable for tachypnea but with normal vesicular breath sounds throughout. He did not have stridor, wheezing, rhonchi, or rales. His abdomen had normal bowel tones and was soft without tenderness, distention, or organomegaly. His extremities were warm, revealed normal pulses, and no edema was present. His joints were cool to palpation, without effusion. On neurologic exam, he was oriented to person and place and able to answer yes/no questions, but unable to provide detailed history. His speech was fluent. His motor exam was without focal deficits. His skin was without any notable lesions.
The constellation of findings does not point to a specific toxidrome. The finding of warm extremities in a hypothermic patient suggests heat loss due to inappropriate peripheral vasodilation. In the absence of vasodilators or features of aortic insufficiency, sepsis becomes a leading consideration. Infection could result in hypothermia and altered sensorium, and accompanying lactic acidosis could trigger tachypnea.
Shortly after admission, he became more somnolent and developed progressive respiratory distress, requiring intubation. Arterial blood gas revealed a pH of 6.93, PaCO2<20 mm Hg, PaO2 127 mm Hg, and HCO3<5 mEq/L. Other laboratory results included a lactate of 4.1 mmol/L, blood urea nitrogen 49 mg/dL, creatinine 2.3 mg/dL (0.8 at 1 month prior), sodium level of 143 mmol/L, chloride of 106 mmol/L, and bicarbonate level of <5 mg/dL. His aspartate aminotransferase was 34 IU/L, alanine transaminase was 28 IU/L, total bilirubin was 0.6 mg/dL, International Normalized Ratio was 1.3. A complete blood count revealed a white blood cell count of 23,000/L, hemoglobin of 10.6 g/dL, and platelet count of 454,000/L. A urinalysis was unremarkable. Cultures of blood, urine, and sputum were collected. Head computed tomography was negative.
This patient has a combined anion gap and nongap metabolic acidosis, as well as respiratory alkalosis. Although his acute kidney failure could produce these 2 types of metabolic acidosis, the modest elevation of the serum creatinine is not commensurate with such profound acidosis. Similarly, sepsis without hypotension or more striking elevation in lactate levels would not account for the entirety of the acidosis. Severe diabetic ketoacidosis can result in profound metabolic acidosis, and marked hyperglycemia or hyperosmolarity could result in somnolence; however, his diabetes has been controlled without medication and there is no obvious precipitant for an episode of ketoacidosis.
Remaining causes of anion gap acidosis include ingestion of methanol, ethylene glycol, ethanol, or salicylates. A careful history of ingestions and medications from witnesses including any prehospital personnel might suggest a source of intoxication. Absent this information, the hypothermia favors an ingestion of an alcohol over salicylates, and the lack of urine crystals and the presence of prominent visual hallucinations would point more toward methanol poisoning than ethylene glycol. A serum osmolarity measurement would allow determination of the osmolar gap, which would be elevated in the setting of methanol or ethylene glycol poisoning. If he were this ill from ethanol, I would have expected to see evidence of hepatotoxicity.
I would administer sodium bicarbonate to reverse the acidosis and to promote renal clearance of salicylates, methanol, ethylene glycol, and their metabolites. Orogastric decontamination with activated charcoal should be considered. If the osmolar gap is elevated, I would also administer intravenous fomepizole to attempt to reverse methanol or ethylene glycol poisoning. I would not delay treatment while waiting for these serum levels to return.
Initial serologic toxicology performed in the emergency department revealed negative ethanol, salicylates, and ketones. His osmolar gap was 13 mOsm/kg. His acetaminophen level was 69 g/mL (normal <120 g/mL). A creatinine phosphokinase was 84 IU/L and myoglobin was 93 ng/mL. His subsequent serum toxicology screen was negative for methanol, ethylene glycol, isopropranol, and hippuric acid. Urine toxicology was positive for opiates, but negative for amphetamine, benzodiazepine, cannabinoid, and cocaine.
Serum and urine ketone assays typically involve the nitroprusside reaction and detect acetoacetate, but not ‐hydroxybutyrate, and can lead to negative test results early in diabetic or alcoholic ketoacidosis. However, the normal ethanol level argues against alcoholic ketoacidosis. Rare causes of elevated anion gap acidosis include toluene toxicity, acetaminophen poisoning, and ingestion of other alcohols. Toluene is metabolized to hippuric acid, and acetaminophen toxicity and associated glutathione depletion can lead to 5‐oxoproline accumulation, producing an anion gap. Patients who abuse alcohol are at risk for acetaminophen toxicity even at doses considered normal. However, this degree of encephalopathy would be unusual for acetaminophen toxicity unless liver failure had developed or unless there was another ingestion that might alter sensorium. Furthermore, the elevated osmolar gap is not a feature of acetaminophen poisoning. I would monitor liver enzyme tests and consider a serum ammonia level, but would not attribute the entire picture to acetaminophen.
The combination of elevated anion gap with an elevated osmolar gap narrows the diagnostic possibilities. Ingestion of several alcohols (ethanol, methanol, ethylene glycol, diethylene glycol) or toluene could produce these abnormalities. Of note, the osmolar gap is typically most markedly elevated early in methanol and ethylene glycol ingestions, and then as the parent compound is metabolized, the osmolar gap closes and the accumulation of metabolites produces the anion gap. Hallucinations are more common with methanol and toluene, and renal failure is more typical of ethylene glycol or toluene. The lack of oxalate crystalluria does not exclude ethylene glycol poisoning. Unfortunately, urine testing for oxalate crystals or fluorescein examination are neither sensitive nor specific enough to diagnosis ethylene glycol toxicity reliably. In most hospitals, assays used for serum testing for alcohols are insensitive, and require confirmation with gas chromatography performed at a specialty lab.
Additional history might reveal the likely culprit or culprits. Inhalant abuse including huffing would point to toluene or organic acid exposure. Solvent ingestion (eg, antifreeze, brake fluid) would suggest methanol or ethylene glycol. Absent this history, I remain suspicious for poisoning with methanol or ethylene glycol and would consider empiric treatment after urgent consultation with a medical toxicologist. A careful ophthalmologic exam might demonstrate characteristic features of methanol poisoning. Serum samples should be sent to a regional lab for analysis for alcohols and organic acids.
He was admitted to the intensive care unit, and empiric antibiotics started. He was empirically started on N‐acetylcysteine and sodium bicarbonate drips. However, his acidemia persisted and he required hemodialysis, which was initiated 12 hours after initial presentation. His acidemia and mental status quickly improved after hemodialysis. He was extubated on hospital day 2 and no longer required hemodialysis.
The differential diagnosis at this point consists of 3 main possibilities: ingestion of methanol, ethylene glycol, or inhalant abuse such as from toluene. The normal hippuric acid level points away from toluene, whereas serum levels can be misleading in the alcohol poisonings. Other discriminating features to consider include exposure history and unique clinical aspects. In this patient, an exposure history is lacking, but 4 clinical features stand out: visual hallucinations, acute kidney injury, mild lactic acidosis, and rapid improvement with hemodialysis. Both ethylene glycol and methanol toxicity may produce a mild lactic acidosis by increasing hepatic metabolism of pyruvate to lactate, and both are rapidly cleared by dialysis. Although it is tempting to place methanol at the top of the list of possibilities due to the report of visual hallucinations, the subjective visual complaints without objective exam corollaries (loss of visual acuity, abnormal pupillary reflexes, or optic disc hyperemia) are nonspecific and might be provoked by alcohol or an inhalant. Furthermore, the acute renal failure is much more typical of ethylene glycol, and thus I would consider ethylene glycol as being the more likely of the ingestions. Coingestion of multiple alcohols is a possibility, but it would be statistically less likely. Confirmation of ethylene glycol poisoning would consist of further insight into his exposures and measurement of levels using gas chromatography.
A urine sample from his emergency department presentation was sent to an outside lab for organic acid levels. Based on high clinical suspicion for 5‐oxoprolinemia (pyroglutamic acidemia) the patient was counseled to avoid any acetaminophen. His primary care provider was informed of this and acetaminophen was added as an adverse drug reaction. The patient left against medical advice soon after extubation. Following discharge, his 5‐oxoproline (pyroglutamic acid) level returned markedly elevated at greater than 10,000 mmol/mol creatinine (200 times the upper limit of normal).
Elevations in 5‐oxoproline levels in this patient most likely stem from glutathione depletion related to chronic acetaminophen use. Alcohol use and malnutrition may have heightened this patient's susceptibility. Despite the common occurrence of acetaminophen use in alcohol abusers or the malnourished, the rarity of severe 5‐oxoproline toxicity suggests unknown factors may be present in predisposed individuals, or under‐recognition. Although acetaminophen‐induced hepatotoxicity may occur along with 5‐oxoprolinemia, this does not always occur.
Several features led me away from this syndrome. First, its rarity lowered my pretest probability. Second, the lack of exposure history and details about the serum assays, specifically whether the measurements were confirmed by gas chromatography, reduced my confidence in eliminating more common ingestions. Third, several aspects proved to be less useful discriminating features: the mild elevation in osmolar gap, renal failure, and hallucinations, which in retrospect proved to be nonspecific.
The patient admitted that he had a longstanding use of acetaminophen in addition to using his girlfriend's acetaminophen‐hydrocodone. He had significant weight loss of over 50 pounds over the previous year, which he attributed to poor appetite. On further chart review, he had been admitted 3 times with a similar clinical presentation and recovered quickly with intensive and supportive care, with no etiology found at those times. He had 2 subsequent hospital admissions for altered mental status and respiratory failure, and his final hospitalization resulted in cardiac arrest and death.
DISCUSSION
5‐Oxoprolinemia is a rare, but potentially lethal cause of severe anion gap metabolic acidosis.[1, 2] The mechanism is thought to be impairment of glutathione metabolism, in the context of other predisposing factors. This can be a congenital error of metabolism, or can be acquired and exacerbated by acetaminophen use. Ingestion of acetaminophen leads to glutathione depletion, which in turn may precipitate accumulation of pyroglutamic acid and subsequent anion gap metabolic acidosis (Figure 1). Additional risk factors that may predispose patients to this condition include malnutrition, renal insufficiency, concurrent infection, and female gender.[1, 2, 3]
The diagnosis of 5‐oxoprolinemia is made via urine or serum organic acid analysis, testing routinely performed in pediatric populations when screening for congenital metabolic disorders. The pathophysiology suggests that obtaining a urine sample early in presentation, when acidosis is greatest, would lead to the highest 5‐oxoproline levels and best chance for diagnosis. Case patients have had normal levels prior to and in convalescent phases after the acute episode.[4] Given the long turnaround time for lab testing, presumptive diagnosis and treatment may be necessary.
Treatment of 5‐oxoprolinemia is primarily supportive, aimed at the metabolic acidosis. Fluid resuscitation and bicarbonate therapy are reasonable temporizing measures. Hemodialysis can clear 5‐oxoproline and may be indicated in severe acidosis.[5] Furthermore, the proposed pathophysiology suggests that administration of N‐acetylcysteine (NAC) may help to address the underlying process, but there are no trials to support a specific dosing regimen. However, given the fulminant presentation and common competing concern for acetaminophen toxicity, it is reasonable to initiate NAC aimed at treatment for possible acetaminophen overdose. Prevention of recurrence includes avoidance of acetaminophen, and counseling the patient to avoid acetaminophen in prescription combination medications and over‐the‐counter preparations.
Recent regulatory changes regarding acetaminophen/opioid combinations may reduce the incidence of 5‐oxoprolinemia. The US Food and Drug Administration has taken action to reduce adverse effects from acetaminophen exposure by limiting the amount of acetaminophen in opioid combination pills from 500 mg to a maximum of 325 mg per pill. This is aimed at preventing hepatotoxicity from ingestion of higher‐than‐recommended doses. However, clinicians should remember that 5‐oxoprolinemia can result from ingestion of acetaminophen at therapeutic levels.
Given its rare incidence, low clinical suspicion, and transient nature of confirmatory testing, it is likely this remains an underdiagnosed syndrome. In the case discussed, subsequent chart review demonstrated 5 previous admissions in multiple hospitals for severe transient anion gap acidosis. The likelihood that 5‐oxoprolinemia was missed in each of these cases supports a lack of awareness of this syndrome. In this patient, the discussant appropriately identified the possibility of 5‐oxoproline toxicity, but felt ethylene glycol ingestion was more likely. As this case underscores, a cornerstone in the management of suspected ingestions is empiric treatment for the most likely etiologies. Here, treatment for acetaminophen overdose and for methanol or ethylene glycol were warranted, and fortunately also addressed the rarer possibility of 5‐oxoproline toxicity.
The mnemonic MUDPILES is commonly used to identify possible causes of life‐threatening anion gap metabolic acidosis, as such heuristics have benefits in rapidly generating a differential diagnosis to guide initial evaluation. Given the fact that the traditional letter P (paraldehyde) in MUDPILES is no longer clinically utilized, some authors have suggested replacing this with pyroglutamic acid (a synonym of 5‐oxoproline). Such a change may help providers who have ruled out other causes of a high anion gap metabolic acidosis, facilitating diagnosis of this life‐threatening syndrome. In any case, clinicians must be mindful that simple memory aids may mislead clinicians, and a complete differential diagnosis may require more than a mnemonic.
TEACHING POINTS
- Acetaminophen use, even at therapeutic levels, can lead to 5‐oxoprolinemia, a potentially lethal anion gap metabolic acidosis.
- 5‐oxoprolinemia is likely related to glutathione depletion, worsened by acetaminophen, malnutrition, renal insufficiency, female gender, and infection. This implies theoretical benefit from administration of NAC for glutathione repletion.
- Mnemonics can be useful, but have limitations by way of oversimplification. This case suggests that changing the letter P in MUDPILES from paraldehyde to pyroglutamic acid could reduce underdiagnosis.
Disclosure: Nothing to report.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
- , , , , . 5‐oxoprolinemia causing elevated anion gap metabolic acidosis in the setting of acetaminophen use. J Emerg Med. 2012;43(1):54–57.
- , , , . What is the clinical significance of 5‐oxoproline (pyroglutamic acid) in high anion gap metabolic acidosis following paracetamol (acetaminophen) exposure? Clin Toxicol (Phila). 2013;51(9):817–827.
- , , , , . Increased anion gap metabolic acidosis as a result of 5‐oxoproline (pyroglutamic acid): a role for acetaminophen. Clin J Am Soc Nephrol. 2006;1(3):441–447.
- , , , et al. Recurrent high anion gap metabolic acidosis secondary to 5‐oxoproline (pyroglutamic acid). Am J Kidney Dis. 2005;46(1):e4–e10.
- , , . Profound metabolic acidosis from pyroglutamic acidemia: an underappreciated cause of high anion gap metabolic acidosis. CJEM. 2010;12(5):449–452.
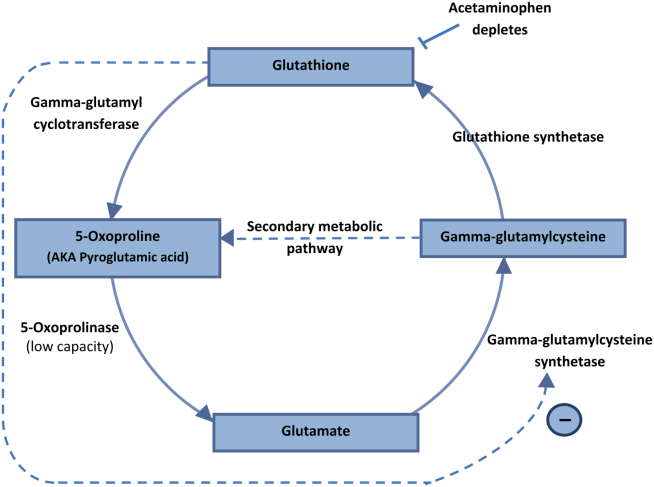
A coat with a clue
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
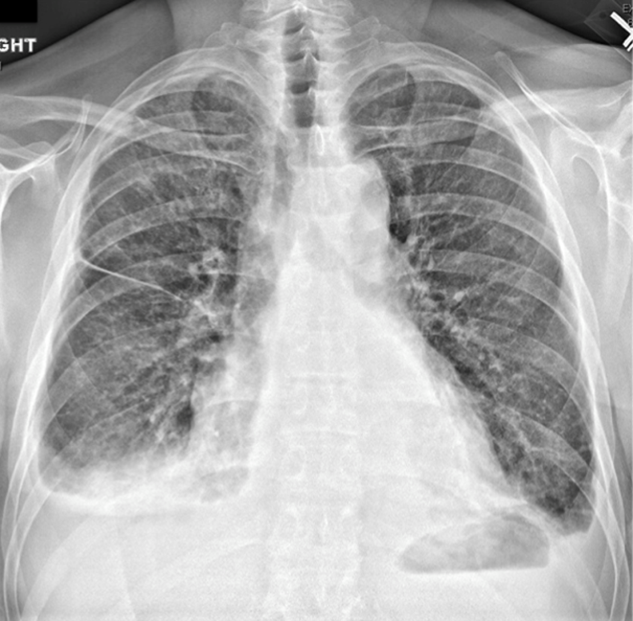
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
A 59‐year‐old man with a history of hypertension was admitted with 6 months of shortness of breath, night sweats, and debilitating fatigue. His symptoms were initially mild and would persist for weeks at a time, after which he would feel better for several days. Over the 2 weeks prior to admission his symptoms had progressed, and he had become dyspneic with minimal exertion.
Progressive dyspnea has a broad differential that includes diseases of the heart (eg, congestive heart failure, aortic stenosis, constrictive pericarditis), lung (eg, chronic obstructive pulmonary disease, interstitial lung disease, pulmonary hypertension, pleural effusion), and blood (eg, anemia).
Night sweats suggest an inflammatory condition, but do not help prioritize infection, malignancy, or autoimmunity. Any of those conditions can be relapsing and remitting, at least in their early phases, but the return to normalcy raises the possibility of hypersensitivity pneumonitis from a periodic exposure.
The 6‐month duration makes typical bacterial and viral infections less likely and suggests indolent infections such as mycobacteria, fungi, or human immunodeficiency virus. Lymphoma or chronic leukemia could cause dyspnea through pleural or pulmonary involvement or from anemia. Autoimmune conditions such as systemic lupus erythematosus or adult Still's disease could also present with this course.
On admission, he described progressive orthopnea, lower extremity edema, and a 15‐lb weight gain. He denied chest pain or palpitations. His symptoms did not correlate with environmental or occupational exposures. He had been diagnosed with essential hypertension a few years earlier but was not taking any medications. He worked as an editor for a newspaper and had traveled throughout California. He never used tobacco and drank alcohol in moderation. He previously smoked marijuana. His father died of Alzheimer's disease, and his mother and 2 siblings were healthy.
Orthopnea, lower extremity edema, and weight gain suggest volume overload, which can result from heart failure, cirrhosis, renal failure, or nephrotic syndrome. The untreated hypertension is a principal risk factor for heart failure. Subacute bacterial endocarditis is an important consideration in a patient with suspected heart failure and night sweats. Travel through the central valley of California may have exposed him to coccidiodomycosis, which can cause chronic pulmonary and extrapulmonary infection.
Physical examination revealed a chronically ill‐appearing man in mild respiratory distress. His temperature was 37.2C, heart rate was 83 bpm, and blood pressure was 168/81 mm Hg. His oxygen saturation was 97% with a respiratory rate of 17 while breathing ambient air. Bilateral chemosis was present. He had crackles at the lung bases. There was a 2/6 systolic murmur loudest at the left lower sternal border with apical radiation. His jugular venous pressure was 2 cm above the sternal angle at 45. He had mild pitting edema of both lower extremities. His abdomen was soft and nondistended. He demonstrated full range of motion of all extremities and had no rashes. He was alert and oriented to person, place, and time. There were no cranial nerve deficits. His strength, sensation and coordination were intact, and he had a normal gait.
Chemosis (conjunctival edema) usually represents conjunctival irritation from an allergic, infectious, or toxic process. It can also be seen in cases of increased ophthalmic venous pressure such as hyperthyroid ophthalmopathy, superior vena cava syndrome, or carotid‐cavernous sinus fistula. The crackles, weight gain, borderline jugular venous distention, and edema suggest some systemic volume overload, but not enough to produce chemosis.
The location and timing of the murmur suggests regurgitation through the mitral or tricuspid valve, a ventricular septal defect, or hypertrophic cardiomyopathy. Tricuspid regurgitation may indicate pulmonary hypertension with right ventricular failure. Despite the absence of fever, subacute bacterial endocarditis remains a concern.
Laboratory evaluation revealed a white blood cell count of 9600/L, hemoglobin of 8.7 g/dL, and platelet count of 522,000/L. Mean corpuscular volume was 88 fL. Serum chemistries were normal; serum creatinine was 1.2 mg/dL. Serum albumin was 2.6 g/dL. A urinalysis was normal. An electrocardiogram demonstrated normal sinus rhythm and left ventricular hypertrophy (LVH). A chest x‐ray revealed interstitial edema and small bilateral pleural effusions. A transthoracic echocardiogram demonstrated normal left ventricular systolic function, an ejection fraction of 65%, mild LVH, and mild diastolic dysfunction. Mild mitral regurgitation, a mildly dilated left atrium, and a minimal pericardial effusion were also noted. A renal ultrasound revealed an atrophic left kidney without arterial flow. He was treated with diuretics for presumed heart failure related to diastolic dysfunction. His dyspnea partially improved, and he was discharged.
Heart failure with preserved ejection fraction may be contributing to his dyspnea but is unlikely to be entirely explanatory given the laboratory abnormalities. The absence of valvular vegetations on transthoracic echocardiogram lowers the probability of bacterial endocarditis. The interstitial pulmonary markings may represent pulmonary edema but alternatively could reflect interstitial lung disease, lymphangitic spread of cancer, infection (eg, Pneumocystis jiroveci), or diffuse alveolar hemorrhage.
Anemia may also be contributing to his dyspnea. There is no evidence of bleeding on history, examination, or imaging. Hemolysis is unlikely given the absence of jaundice, splenomegaly, or a known predisposing condition. The normocytic anemia may also arise from chronic inflammation. Severe anemia can cause high output heart failure, but usually the hemoglobin level is much lower and the echocardiogram would have suggestive findings. Thrombocytosis suggests inflammation, a primary myeloproliferative disorder, or severe iron deficiency (not suspected here). His hypoalbuminemia is further evidence of chronic inflammation especially in the absence of nephropathy, hepatopathy, or a protein‐losing enteropathy.
An atrophic kidney may be congenital or result from long‐standing unilateral renal ischemia, infection, or obstruction. Diminished arterial flow in a middle‐aged man with hypertension may simply reflect atherosclerotic renal artery stenosis, but mass effect within the left renal artery from thrombus, infection, or cancer cannot be ruled out.
Four weeks later he was readmitted for progressive dyspnea and persistent night sweats. He was afebrile, fatigued, and in marked respiratory distress. The remainder of his physical examination was unchanged. Laboratory evaluation revealed a white blood cell count of 20,000/L with neutrophilic predominance, hemoglobin of 11 g/dL, and platelet count of 614,000/L. Creatinine was 1.4 mg/dL. Erythrocyte sedimentation rate (ESR) was greater than 100 mm/h, and C‐reactive protein (CRP) was 44 mg/L. Blood cultures were negative. Chest x‐ray (Figure 1) revealed persistent interstitial edema and increased bilateral pleural effusions.
Although clinical and radiologic features continue to suggest heart failure, the marked respiratory distress and persistent chest x‐ray abnormalities imply that a superimposed process is affecting the lungs. The night sweats, neutrophilia, and elevated ESR and CRP strongly suggest an inflammatory state from infection, malignancy, or autoimmunity.
A computed tomography (CT) scan of the lungs would help assess for interstitial lung disease, lymphangitic carcinomatosis and septic emboli. Blood cultures should be repeated to definitively exclude subacute endocarditis. A peripheral blood smear is needed to evaluate for hematologic malignancy. Finally, human immunodeficiency virus antibody testing is indicated.
CT of the abdomen and pelvis demonstrated left renal artery stenosis, an atrophic left kidney, right kidney edema with mild perinephric stranding, and mild‐to‐moderate right hydroureter without an obstructing mass or calculus. There was mild splenomegaly and mesenteric lymphadenopathy up to 3 cm in diameter. The distal thoracic and suprarenal abdominal aorta had crescentic high‐density wall thickening. There were small sclerotic densities of the proximal femora, pelvic girdle, and thoracolumbar spine (Figure 2). Contrast chest CT demonstrated severe wall thickening of his entire thoracic aorta. There was also cardiomegaly, mild interlobular septal thickening, small bilateral pleural effusions, a 3.2‐cm right upper lobe paratracheal lymph node, and nodular pleural thickening (Figure 3).
Diffuse aortopathy is caused by inflammatory, infectious, or infiltrative processes. Large vessel vasculitides such as Behet's disease, giant cell arteritis, and Takayasu's arteritis are unlikely, as the patient lacks the associated clinical findings or epidemiology. Imaging does not reveal preexisting aortic pathology, such as an aneurysm or atherosclerotic plaque, which could predispose him to bacterial endovascular infection.
Urinary system dilation without an obvious obstruction could be explained by retroperitoneal fibrosis. Generalized lymphadenopathy, (suspected) retroperitoneal fibrosis, sclerotic bone lesions, and cardiopulmonary disease collectively suggest a widespread infiltrative process. Lymphoma may lead to lymphadenopathy and bone lesions but would not explain the aortopathy. He lacks risk factors for infections like tuberculosis or tertiary syphilis, a well‐known cause of aortopathy in the past.
Widespread multisystem involvement invites consideration of nonmalignant, noninfectious infiltrative disorders such as immunoglobulin G4‐related disease (IgG4‐RD), histiocytoses such as Erdheim‐Chester disease (ECD), systemic mastocytosis (SM), and sarcoidosis. ECD is a disorder of non‐Langerhans histiocytes that infiltrate the aorta, bones, retroperitoneum, lungs, myocardium, and periorbital structures. Perinephric stranding is sometimes seen in this condition. The lymphoplasmacytes in IgG4‐RD and noncaseating granulomas of sarcoidosis infiltrate many of the same organs. Common sites infiltrated by mast cells in SM include the bone and lymph nodes. Among these diseases, ECD and IgG4‐RD more commonly manifest with aortic and retroperitoneal infiltration and thus are prioritized on this differential diagnosis.
A positron emission tomography (PET) scan revealed abnormal fluordeoxyglucose uptake involving the thoracic aorta, right apical pleural surface, perinephric soft tissue, and various marrow spaces. Core needle biopsy of a sclerotic lesion in the right ischium demonstrated focal marrow replacement by a fibrohistiocytic process. No malignant cells or pathogenic organisms were identified. Biopsy of the right kidney revealed chronic interstitial nephritis with features of megalocytic interstitial nephritis (histiocytic inflammation) and arteriolar nephrosclerosis. A transbronchial biopsy demonstrated alveolar tissue with focal intra‐alveolar hemorrhage and organization, but no malignancy, atypia, or pathogenic organisms.
The biopsy results do not support infection, lymphoma, or carcinoma. The absence of noncaseating granulomas and mastocytes on multiple biopsies essentially rules out sarcoidosis and SM, respectively. None of the characteristic pathologic features of IgG4‐RDlymphoplasmacytic infiltrate, obliterative phlebitis, and fibrosiswere observed. The pulmonary pathology points to injury, but not the underlying cause. The bone and kidney tissue samples reveal histiocytic infiltration.
The abnormalities of the aorta, bone, lung, kidney, and retroperitoneum can be explained by the diffuse histiocytic involvement seen in ECD. The perinephric stranding detected on CT and perinephric inflammation on the PET scan may reflect the hairy kidney of ECD, which is a result of histiocytic infiltration. It is possible that the chemosis relates to exophthalmos from histiocytic orbital infiltrates. Sensitivity for detecting orbital pathology on a PET scan is limited because of the high signal from the adjacent brain.
ECD should be distinguished from Langerhans cell histiocytosis (LCH) by immunohistologic staining and microscopic characteristics of the histiocytes. LCH usually does not involve the aorta, and it more commonly involves the skin.
Serum IgG4 was within normal limits, and immunohistochemical staining of pathology specimens for IgG4 was negative. The BRAF V600E mutation, which is present in the majority of patients with ECD, was detected in a subsequent right perinephric biopsy specimen. The patient was diagnosed with ECD.
Prednisone and pegylated interferon‐ led to a rapid improvement in his symptoms. As the prednisone was tapered, he developed bilateral periorbital swelling. Magnetic resonance imaging (MRI) revealed well‐circumscribed, intraorbital soft tissue masses with partial encasement of his optic nerves and superior ophthalmic veins, as well as infiltration of his transverse sinuses, consistent with intracranial manifestations of ECD. There was no evidence of pituitary, hypothalamic, or other brain parenchymal infiltration. His dyspnea, night sweats, and hypertension improved; however, 3 months into therapy he developed an extensive rash. Interferon was discontinued. Vemurafinib, a serine kinase inhibitor that targets the BRAF mutation, was prescribed with subsequent resolution of the rash.
COMMENTARY
This patient suffered from a chronic, progressive, inflammatory illness. Although the disease initially appeared to be confined to the heart and lungs, laboratory testing signaled a more systemic condition, and subsequent imaging demonstrated involvement of a disparate group of organs. Subacute disease processes with elevated markers of inflammation and diffuse organ involvement often fall into 1 of 3 categories: infectious, autoimmune, or neoplastic. The histiocytoses inhabit a fourth and easily overlooked category that can be described as infiltrative. Infiltrative diseases are a heterogeneous group of conditions that cause illness when cells or substances not normally found in tissues lead to organ dysfunction.
Although traditional teaching has focused on sarcoidosis, amyloidosis, and hemochromatosis as the primary representatives of this category, the medical literature describes a number of other infiltrative disease processes. IgG4‐RD is a fibroinflammatory disorder characterized by space‐occupying lesions, a lymphoplasmacytic infiltrate of IgG4‐positive plasma cells, and storiform (matted and irregularly whorled microscopic pattern) fibrosis.[1] IgG4‐RD, like sarcoidosis, blurs the categorical line between infiltrative and autoimmune diseases. Other infiltrative cellular disorders, such as histiocytosis and mastocytosis, exist on a spectrum between monoclonal proliferation and neoplastic invasion.
The histiocytoses represent a diverse group of disorders with an evolving nomenclature, characterized by localized or diffuse infiltration of macrophages, monocytes, and dendritic cells (Table 1). ECD is a rare, non‐Langerhans histiocytosis characterized by excessive recruitment and activation of histiocytes through kinase signaling pathways.[2, 3] Immunohistochemical staining for CD68, CD163, and Factor XIIIa, with lack of staining for CD1a, S100, and CD207, supports the diagnosis.[3] Mutations in the BRAF V600E gene (a protein kinase involved in cell proliferation) represent the most likely etiology of this overactivation. An estimated 38% to 100% of patients with ECD harbor this mutation, with detection rates influenced by the sensitivity of testing techniques.[3] The serine kinase inhibitor vemurafinib targets this mutation, and early experience with this agent in ECD demonstrates encouraging results.[4]
| Dendritic cell disorders |
| Langerhans cell histiocytosis |
| Secondary dendritic cell processes |
| Juvenile xanthogranuloma and related disorders (including Erdheim‐Chester disease) |
| Solitary histiocytomas with a dendritic phenotype |
| Macrophage‐related disorders |
| Primary hemophagocytic lymphohistiocytosis (familial and sporadic) |
| Secondary hemophagocytic syndromes |
| Sinus histiocytosis with massive lymphadenopathy (Rosai‐Dorfman disease) |
| Solitary histiocytoma with a macrophage phenotype |
| Malignant histiocytic disorders |
| Monocyte‐related leukemias |
| Extramedullary monocytic tumor or sarcoma |
| Dendritic cell‐related histiocytic sarcoma |
| Macrophage‐related histiocytic sarcoma |
ECD presents heterogeneously, occurring most commonly between the ages of 40 and 70 years. Nonspecific symptoms include weakness, fatigue, fever, chills, weight loss, and night sweats. Typical sites of involvement include the bone, central nervous system, cardiovascular system, lungs, and retroperitoneum. Bone involvement is nearly universal, and bone pain is the most common presenting symptom. Symmetric diaphyseal and metaphyseal osteosclerotic lesions may be seen on x‐rays, bone scan, PET, CT, and MRI.[3] Approximately 50% of patients have extraskeletal involvement at diagnosis.[5] Neurologic manifestations may result from invasion of histiocytes into the facial bones, orbits, meninges, and intracranial vessels, as eventually developed in this patient. Diabetes insipidus is the most common neurologic manifestation of ECD, followed by exophthalmos, cerebellar ataxia, panhypopituitarism, and papilledema.[6, 7] Approximately 75% of patients eventually suffer from cardiovascular disease, including hypertension, congestive heart failure, acute myocardial infarction, valvular dysfunction, pericardial infiltration, and cardiac tamponade.[8] Vascular involvement includes perivascular infiltration and periaortic fibrosis, resulting in the coated aorta seen in 20% of patients with ECD.[3] Pulmonary manifestations of ECD include interstitial, pleural, and consolidative lung disease. A review of high‐resolution chest CTs of patients with ECD demonstrated that greater than half had evidence of parenchymal lung disease, with interlobular septal thickening being the most common finding.[9] Infiltration and fibrosis of retroperitoneal structures is common. Infiltration of perinephric fat creates irregular renal borders, appearing radiographically as hairy kidneys.
Arriving at the diagnosis in this case proved to be challenging because the early presentation was consistent with congestive heart failure. As the patient's conditioned deteriorated, imaging suggested multisystem involvement. It was the extensive aortopathy in particularnot the less specific bone, kidney, lymph node, or pulmonary findingsthat allowed the clinicians to hone the extensive differential diagnosis. The coated aorta is a finding that has been strongly associated with ECD; few other conditions coat the aorta in a similar fashion.[10] In most mysteries, the perpetrator's coat conceals his identity; however, in this story the coat gave it away.
KEY LEARNING POINTS
- Subacute, inflammatory, multiorgan disease is usually explained by 3 categoriesinfection, autoimmunity, and neoplasiabut a fourth category, infiltrative disorders, sometimes warrants consideration.
- ECD presents heterogeneously, ranging from localized disease to widespread organ infiltration. The classic presentation includes bone pain, diabetes insipidus, and exophthalmos.
- Characteristic radiological findings that suggest ECD include long bone osteosclerosis, a coated aorta from periaortic infiltration, and hairy kidneys from perinephric infiltration.
Disclosure
Nothing to report
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.
- , , IgG4‐related disease. N Engl J Med. 2012;366(6):539–551.
- , , , Diagnosing Erdheim‐Chester disease. Ann Rheum Dis. 2013;72(7):e19.
- , , , et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood. 2014;124(4):483–492.
- , , , et al. The efficacy of vemurafenib in Erdheim‐Chester Disease and Langerhans Cell Histiocytosis: preliminary results from VE‐Basket Study. Blood. 2014;124(21):635.
- , , , et al. Erdheim‐Chester disease. Clinical and radiologic characteristics of 59 cases. Medicine (Baltimore). 1996;75(3):157–169.
- , , , et al. Neurological manifestations and neuroradiological presentation of Erdheim‐Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253(10):1267–1277.
- , , , et al. Cerebral, facial, and orbital involvement in Erdheim‐Chester disease: CT and MR imaging findings. Radiology. 2010;255(2):586–594.
- , , , et al. Images in cardiovascular medicine. Cardiac involvement in Erdheim‐Chester disease: magnetic resonance and computed tomographic scan imaging in a monocentric series of 37 patients. Circulation. 2009;119(25):e597–e598.
- , , , et al. Pulmonary involvement in Erdheim‐Chester disease: a single‐center study of thirty‐four patients and a review of the literature. Arthritis Rheum. 2010;62(11):3504–3512.
- , , , et al. “Coated aorta”: a new sign of Erdheim‐Chester disease. J Rheumatol. 2000;27(6):1550–1553.