User login
‘Supercharge’ antidepressants by adding thyroid hormones
Prescribing thyroid hormones with antidepressants—whether to augment the antidepressant effect or accelerate patient response—is a well-researched strategy for treatment-resistant major depressive disorder (MDD). Thyroid hormones are known to boost response to tricyclics, and preliminary evidence shows they may be useful adjuvants to selective serotonin reuptake inhibitors (SSRIs) as well.
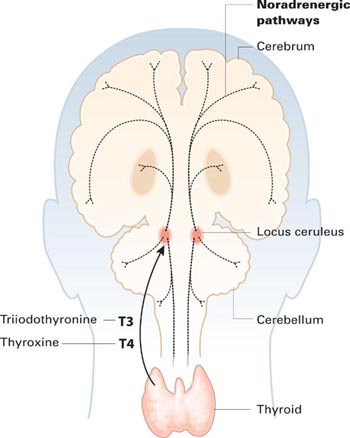
Thyroid hormones enter the brain slowly across the blood-brain barrier and choroid plexus. They accumulate in the locus ceruleus and other structures and are distributed widely along noradrenergic pathways.
Effective treatments are available for MDD, although 30% to 40% of patients do not respond to one or more antidepressant trials (Box).1-4 This article offers:
- new information about why triiodothyronine (T3) and thyroxine (T4) can “super-charge” antidepressant response
- tips on how to use thyroid hormones in patients with MDD, including effective dosages, patient monitoring, and treatment durations.
- 30% to 40% of patients with major depressive disorder (MDD) do not respond sufficiently to usual antidepressant treatment1
- Even under optimal treatment conditions, only one-third of patients achieve remission2
- Among patients who fail to respond to two pharmacologic interventions, remission rates with the next antidepressant are as low as 12%3
- A patient becomes less likely to respond clinically with each additional nonresponse to antidepressant treatment4
Why thyroid hormones?
Thyroid hormones enter the brain slowly across the blood-brain barrier and the choroid plexus—cerebrospinal fluid barriers. T4 is the main source of brain T3—after attack by 5’deiodinase—but circulating T3 also crosses the blood-brain barrier through active transport.
Thyroid hormones accumulate in the locus ceruleus and other central noradrenergic structures and are distributed widely in the brain along noradrenergic pathways.5-7 The mechanism of their therapeutic effect for MDD is not well understood, and various hypotheses have been proposed.
Subclinical hypothyroidism. Early studies such as by Howland8 of treatment-refractory MDD suggested that thyroid hormone augmentation might correct a hypothyroid state. However, blood thyroid hormone levels are not associated with resistance to antidepressant treatment, according to studies of MDD populations.9-10 Also, thyroid hormones’ therapeutic action in MDD appears unlikely to be related to treating subclinical hypothyroidism because patients’ euthyroid status was verified in all adjuvant studies since 1980.
Joffe et al11 proposed that MDD is characterized by a relative excess of T4 versus T3—probably related to a deficit in converting T4 to T3 in the periphery—and administering T3 would therefore correct this imbalance. They offered no strong evidence for an increased T4 level, however, and later studies failed to detect the postulated blood T3 abnormalities in MDD.9-10 Also—as suggested by studies with high-dose T4 augmentation12,13—the adjuvant antidepressant effect of thyroid hormones is not restricted to T3, although T3 may be more efficacious than T4.14
Neurotransmitter effects. Thyroid hormones’ role in increasing serotonin (5-HT) release could partially explain the benefit of adjuvant thyroid hormone therapy in MDD. Researchers found:
- T3 increased cortical 5-HT levels, probably by reducing the autoinhibitory effect of the presynaptic 5-HT1A receptor15
- adding T3 to clomipramine therapy increased 5-HT levels to a greater extent than T3 or clomipramine used alone16
- Low 5-HT activity, shown in hypothyroid patients, increased after T4 replacement.17
This 5-HT release theory cannot explain why thyroid hormones have a rapid clinical effect in MDD. Most studies report the hormones have clinical efficacy in MDD within 4 weeks.18 Similarly, selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels within hours of treatment onset, but the clinical effect occurs 4 to 6 weeks later.19 Therefore, increased 5-HT levels cannot fully explain thyroid hormones’ early effect.
Close interaction between thyroid hormones and the noradrenergic system also has been examined. Brain T3 is primarily localized in the central noradrenergic systems, with axonal anterograde transport of T3 from the locus ceruleus. T3 is processed and accumulated in the noradrenergic system, carried via axonal transport, then delivered from nerve cell bodies to its neuronal targets.5,7 T3 thus functions as a coneurotransmitter with norepinephrine.
Cellular energy metabolism. We recently reported that thyroid hormones’ antidepressant effect may be related to brain cellular energy metabolism. Thyroid hormones increase cellular levels of adenosine triphosphate (ATP) and phosphocreatine (PCr) in the hypothyroid brain.20 Brain imaging—phosphorus-31 nuclear magnetic resonance spectroscopy (31P-MRS)—of subjects with MDD shows decreased brain levels of ATP and increased PCr.21
Our group showed that the antidepressant effect of T3 augmentation of SSRIs is correlated with significant increases in ATP levels and decreases in PCr. This effect—which appears to represent re-normalization of brain bioenergetics in treatment responders—did not occur in nonresponders (Iosifescu et al, presented at APA, 2004).
The effect of thyroid hormones on bioenergetic metabolism is compatible with the hypothesized effects on noradrenergic and serotonergic systems.5,7 These mechanisms may represent different links in the same chain of events.
Antidepressant boosters
Thyroid hormones have been used extensively to treat MDD since the 1950s, when researchers reported that T3 monotherapy was efficacious for treating depression. These early studies had important methodologic limitations, including open designs and poorly defined diagnostic criteria and response.
For MDD, the most extensively researched uses of thyroid hormones are to augment therapy for antidepressant nonresponders and to accelerate partial response to antidepressants.
Tricyclics. Open studies primarily among outpatients in the 1970s and ‘80s suggested that thyroid hormones are a valid augmentation strategy for nonresponders to tricyclic antidepressants. Most—but not all—reported response rates >50% with T3 dosages of 20 to 50 mcg/d.22,23
Compared with outpatient studies, however, an open trial of T3 augmentation by Birkenhager et al24 found no evidence of efficacy in 14 severely depressed inpatients who had not responded to 6 weeks of tricyclics. These patients—mainly with melancholic and/or psychotic depression—showed greater response to a monoamine oxidase inhibitor (MAOI) or electroconvulsive therapy than to thyroid hormone.
Three of five double-blind controlled studies of thyroid hormone augmentation of tricyclics reported response rates of 50 to 65% (Table).14,18,25 Earlier controlled studies, including two negative studies26,27 had important methodologic limitations: all included few subjects (≤16), and two studies included both unipolar and bipolar patients. Joffe et al partially addressed these problems with two randomized, double-blind studies that were controlled with placebo or T4:
- In a 3-week trial, T3 was more effective than T4 when added to imipramine or desipramine.14
- In a 2-week trial, T3 was significantly more effective than placebo when added to imipramine or desipramine.25
In the latter study, T3 and lithium augmentation appeared equally effective. Conversely, T4 was more effective than lithium as the first augmentation strategy in a double-blind, crossover study by Spoov and Lahdelma.28
In conclusion, depressed patients given T3 with tricyclic antidepressants may be twice as likely as controls to respond to treatment, according to a meta-analysis of eight studies totaling 292 patients. This analysis by Aronson et al29 supports T3 augmentation while addressing the surveyed studies’ limitations.
Table
Treatment-resistant MDD: Controlled studies of thyroid hormone augmentation
| Study authors/design | Initial therapy | Augmentation | Results |
|---|---|---|---|
| Steiner et al, 1978 Randomized double-blind; 8 patients* | Several TCAs (6 weeks) | T3, 25 mcg/d, or placebo (35 days) | T3: 75% responders (3/4) Placebo: 75% (3/4) |
| Goodwin et al, 1982 Double-blind, mirror design; 12 patients* | Desipramine or imipramine (4 weeks) | T3, 25 to 50 mcg/d, or placebo (21 days) | T3: 33% responders (4/12) Placebo: 0% (0/6) |
| Gitlin et al, 1987 Double-blind with crossover; 16 patients | Imipramine (4 weeks) | T3, 25 mcg/d, or placebo (2 weeks); crossover (2 weeks) | No difference between T3 and placebo |
| Joffe and Singer, 1990 Randomized double-blind; 38 patients | Desipramine or imipramine (4 weeks) | T3, 37.5 mcg/d, or T4, 150 mcg/d (21 days) | T3: 53% responders (9/17) T4: 19% (4/21) |
| Joffe et al, 1993 Randomized double-blind; 33 patients | Desipramine or imipramine (5 weeks) | T3, 37.5 mcg/d, or placebo (14 days) | T3: 59% responders (10/17) Placebo: 19% (3/16) |
| Sopov and Lahdelma, 1998 Randomized double-blind with crossover; 22 patients* | TCA, MAOI antidepressants | T4, 200 mcg/d, or lithium, 500 mg/d, (4 weeks); then crossover (4 weeks) | T4: 64% responders (7/11) Lithium: 18% responders (2/11) |
| *Included patients with unipolar or bipolar depression | |||
| MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; T3: triiodothyronine; T4: thyroxine; TCA: tricyclic antidepressant. | |||
MAO inhibitors. One small report has addressed the efficacy of thyroid hormones as adjuvants to MAOIs.30 In two patients, adding T3 to phenelzine enhanced the antidepressant response.
High-dose T4. Two open studies of patients with treatment-resistant bipolar or unipolar depression12,13 have examined the efficacy of high-dose T4 augmentation. These patients were taking a variety of antidepressants, including TCAs and SSRIs.
- In the study by Baurer et al,12 clinical remission (Hamilton Depression Scale [HAM-D] score ≤10) occurred in 4 of 5 patients with severe treatment-resistant unipolar depression who received adjunctive T4, mean 482±72 mcg/d, with antidepressants.
- In the study by Rudas et al,13 clinical remission (HAM-D ≤9) occurred in 7 of 9 patients with treatment-resistant MDD after T4, 150 to 300 mcg/d, was added to their antidepressant therapy. Side effects may limit this high-dose strategy, however, because 2 of the patients dropped out with thyrotoxicosis symptoms.
SSRIs. Three open trials to date have investigated using thyroid hormones to augment SSRIs in treatment-resistant MDD. In a prospective study by Agid and Lerer,31 10 of 25 (40%) patients who did not respond to SSRI treatment did so after T3 was added. No men improved, however, which led the authors to suggest that men and women might respond differently to T3 augmentation of SSRIs.
In our study, 7 of 20 patients (35%) with MDD who did not respond to 8 weeks of SSRI therapy did so when we added T3, 50 mcg/d, for 4 weeks (Figure). Response rates were high (5/5, 100%) in patients with atypical features by DSMIV criteria and low (1/8, 12.5%) in those with melancholic features.32
Abraham et al33 added T3, 50 mcg/d, to the regimens of 12 patients with MDD who did not respond to SSRIs alone. One patient dropped out with side effects. After 4 weeks of T3 augmentation, 5 patients (42%) showed 50% or greater improvement in HAM-D scores from baseline.
Figure T3 augmentation of SSRIs in 20 patients with resistant major depressive disorder
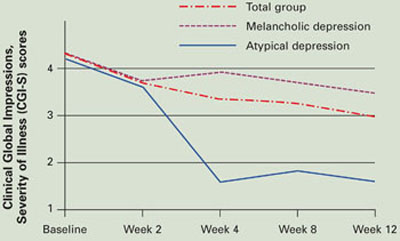
Open T3 augmentation, 50 mcg/d, given to 20 nonresponders to 8 weeks of selective serotonin reuptake inhibitors (SSRIs) improved baseline CGI-S scores significantly (P=0.006) at 4 weeks in those with atypical depression and modestly (P>0.05) in those with melancholic depression.
Source: Reference 32Antidepressant accelerators. Five of seven early double-blind, controlled studies indicated that adding small doses of thyroid hormones at the beginning of antidepressant treatment accelerated treatment response. All were limited by small sample sizes and other methodologic problems. A more-recent meta-analysis of six studies totalling 125 patients by Altshuler et al34 found:
- T3 was significantly more effective than placebo in accelerating clinical response to tricyclics
- the acceleration effect was more pronounced for women than for men.
Clinical recommendations
Thyroid hormones can be useful to augment and accelerate treatment of MDD. Evidence strongly supports their use with tricyclic antidepressants and suggests they also can be effective adjuvants for patients who do not respond to SSRIs.
Either T3 (up to 50 mcg/d) or T4 (up to 150 mcg/d) can be used as augmentation. T3’s antidepressant properties are considered more effective than those of T4, but the only head-to-head study supporting this conclusion was small (38 patients).14 Some T4 augmentation studies used very high dosages (300 to 600 mcg/d),12 which increase the risk of acute overdose.
Start T3 augmentation at 25 mcg/d and increase, if tolerated, to 50 mcg/d after 1 week. Measure baseline serum thyroid-stimulating hormone (TSH), and do not treat patients with TSH <0.5 mIU/L). Baseline TSH, T4, or T3 levels do not predict response to T3 augmentation in euthyroid MDD patients.32
Common side effects. Adjuvant T3, 25 to 50 mcg/d, was well-tolerated in our study of 20 patients also taking SSRIs:
- 2 (10%) experienced fatigue and diaphoresis
- 1 each (5%) had tremor, dry mouth, headaches, muscle aches, and vivid dreams.32
We saw no significant changes in blood pressure, but heart rates increased significantly in our 4-week study—from 76±12 bpm (range 60 to 96) to 82±9 bpm (range 66 to 96). Thus, T3 augmentation may not be indicated for patients with coronary artery disease or chronic heart failure. Patients’ weight decreased an average 2.5±6.6 lbs (range –20 to +7).
Thyroid hormones may cause hypoglycemia and change insulin requirements in patients with diabetes. High doses of T3 or T4 may be associated with hyperthyroidism, weight loss, nervousness, sweating, tachycardia, insomnia, heat intolerance, menstrual irregularities, palpitations, psychosis, or fever. Discontinue treatment if these symptoms develop.
Onset of antidepressant effect. Assess patient response in 4 to 6 weeks, whether using augmentation to address antidepressant nonresponse14,25 or to accelerate response.33 If you detect only partial improvement, studies support continuing treatment up to 8 weeks.
Treatment duration. No guidelines exist on how long to continue thyroid hormones after the initial antidepressant response. TSH levels become suppressed (TSH <0.1 mIU/L) after 4 weeks of T3, 50 mcg/d, in patients with normal baseline thyroid function.32 This suggests thyroid hormone’s booster effect is self limited, and augmentation may not need to continue after 2 to 3 months—even in responders.
T3 augmentation at 25 mcg/d can be discontinued immediately. For 50 to 75 mcg/d, taper across 1 to 2 weeks. The hypothalamic-pituitary-thyroid axis returns to normal function 6 to 8 weeks after T3 augmentation is stopped.
In this model, thyroid hormone augmentation can be used to boost antidepressant efficacy several months at a time. If effective, the same strategy could be tried again for subsequent MDD episodes.
Related resources
- DeBattista C. Augmentation and combination strategies for depression. J Psychopharmacol 2006;20(3):11-18.
- Massachusetts General Hospital Psychiatric Academy (including web-casts on treatment-resistant depression. www.mghcme.com
Drug brand names
- Desipramine • Norpramin
- Imipramine • Tofranil
- Levothyroxine (T4) • Levoxyl, Levothroid, Synthroid, others
- Liothyronine (synthetic T3) • Cytomel
Disclosures
The author receives research support from Aspect Medical Systems, Forest Laboratories, and Janssen Pharmaceutica; is a consultant to Pfizer, Inc., and Forest Laboratories; and a speaker for Eli Lilly and Co., Pfizer, Inc., and Cephalon.
1. Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996;19:179-200.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Nierenberg AA, McLean NE, Alpert JE, et al. Early nonresponse to fluoxetine as a predictor of poor 8-week outcome. Am J Psychiatry 1995;152:1500-3.
4. Nierenberg AA, Papakostas GI, Petersen T, et al. Nortriptyline for treatment-resistant depression. J Clin Psychiatry 2003;64(1):35-9.
5. Rozanov CB, Dratman MB. Immunohistochemical mapping of brain triiodothyronine reveals prominent localization in central noradrenergic systems. Neuroscience 1996;74:897-915.
6. Cheng LY, Outterbridge LV, Covatta ND, et al. Film autoradiography identifies unique features of [125I]3,3’5’-(reverse) triiodothyronine transport from blood to brain. J Neurophysiol 1994;72:380-91.
7. Gordon JT, Kaminski DM, Rozanov CB, Dratman MB. Evidence that 3,3’,5-triiodothyronine is concentrated in and delivered from the locus coeruleus to its noradrenergic targets via anterograde axonal transport. Neuroscience 1999;93:943-54.
8. Howland RH. Thyroid dysfunction in refractory depression: implications for pathophysiology and treatment. J Clin Psychiatry 1993;54:47-54.
9. Joffe RT. Peripheral thyroid hormone levels in treatment resistant depression. Biol Psychiatry 1999;45:1053-5.
10. Iosifescu DV, Howarth S, Alpert JE, et al. T3 blood levels and treatment outcome in depression. Int J Psychiatry Med 2001;31:367-73.
11. Joffe RT, Roy-Byrne PP, Udhe TW, Post RM. Thyroid function and affective illness: a reappraisal. Biol Psychiatry 1984;19:1685-91.
12. Bauer M, Hellweg R, Graf KJ, Baumgartner A. Treatment of refractory depression with high-dose thyroxine. Neuropsychopharmacology 1998;18:444-55.
13. Rudas S, Schmitz M, Pichler P, Baumgartner A. Treatment of refractory chronic depression and dysthymia with high-dose thyroxine. Biol Psychiatry 1999;45:229-33.
14. Joffe RT, Singer W. A comparison of triiodothyronine and thyroxine in the potentiation of tricyclic antidepressants. Psychiatry Res 1990;32:241-51.
15. Sandrini M, Vitale G, Vergoni AV, et al. Effect of acute and chronic treatment with triiodothyronine on serotonin levels and serotonergic receptor subtypes in the rat brain. Life Sci 1996;58:1551-9.
16. Gur E, Lerer B, Newman ME. Chronic clomipramine and triiodothyronine increase serotonin levels in rat frontal cortex in vivo: relationship to serotonin autoreceptor activity. J Pharmacol Exp Ther 1999;288:81-7.
17. Cleare AJ, McGregor A, Chambers SM, et al. Thyroxine replacement increases central 5-hydroxytryptamine activity and reduces depressive symptoms in hypothyroidism. Neuroendocrinology 1996;64:65-9.
18. Goodwin FK, Prange AJ, Post RM, et al. Potentiation of antidepressant effects by l-triiodothyronine in tricyclic nonresponders. Am J Psychiatry 1982;139:34-8.
19. Blier P. Pharmacology of rapid-onset antidepressant treatment strategies. J Clin Psychiatry 2001;62(suppl 15):12-7.
20. Smith CD, Ain KB. Brain metabolism in hypothyroidism studied with 31P magnetic-resonance spectroscopy. Lancet 1995;345:619-20.
21. Kato T, Murashita J, Shioiri T, et al. Effect of photic stimulation on energy metabolism in the human brain measured by 31P-MR spectroscopy. J Neuropsychiatry Clin Neurosci 1996;8:417-22.
22. Earle BV. Thyroid hormone and tricyclic antidepressants in resistant depressions. Am J Psychiatry 1970;126:1667-9.
23. Thase ME, Kupfer DJ, Jarrett DB. Treatment of imipramine-resistant recurrent depression: I. An open clinical trial of adjunctive l-triiodothyronine. J Clin Psychiatry 1989;50:385-8.
24. Birkenhager TK, Vegt M, Nolen WA. An open study of triiodothyronine augmentation of tricyclic antidepressants in inpatients with refractory depression. Pharmacopsychiatry 1997;30:23-6.
25. Joffe RT, Singer W, Levitt AJ, MacDonald C. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-93.
26. Steiner M, Radwan M, Elizur A, et al. Failure of l-triiodothyronine to potentiate tricyclic antidepressant response. Curr Ther Res 1978;23:655-9.
27. Gitlin MJ, Weiner H, Fairbanks L, et al. Failure of T3 to potentiate tricyclic antidepressant response. J Affective Disord 1987;13:267-72.
28. Spoov J, Lahdelma L. Should thyroid augmentation precede lithium augmentation—a pilot study. J Affect Disord 1998;49:235-9.
29. Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996;53:842-8.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry 1988;49:409-10.
31. Agid O, Lerer B. Algorithm-based treatment of major depression in an outpatient clinic: clinical correlates of response to a specific serotonin reuptake inhibitor and to triiodothyronine augmentation. Int J Neuropsychopharmacol 2003;6(1):41-9.
32. Iosifescu DV, Nierenberg AA, Mischoulon D, et al. An open study of triiodothyronine augmentation of selective serotonin reuptake inhibitors in treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66:1038-42.
33. Abraham G, Milev R, Stuart Lawson J. T3 augmentation of SSRI resistant depression. J Affect Disord 2006;91(2-3):211-15.
34. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry 2001;158:1617-22.
Prescribing thyroid hormones with antidepressants—whether to augment the antidepressant effect or accelerate patient response—is a well-researched strategy for treatment-resistant major depressive disorder (MDD). Thyroid hormones are known to boost response to tricyclics, and preliminary evidence shows they may be useful adjuvants to selective serotonin reuptake inhibitors (SSRIs) as well.
Thyroid hormones enter the brain slowly across the blood-brain barrier and choroid plexus. They accumulate in the locus ceruleus and other structures and are distributed widely along noradrenergic pathways.
Effective treatments are available for MDD, although 30% to 40% of patients do not respond to one or more antidepressant trials (Box).1-4 This article offers:
- new information about why triiodothyronine (T3) and thyroxine (T4) can “super-charge” antidepressant response
- tips on how to use thyroid hormones in patients with MDD, including effective dosages, patient monitoring, and treatment durations.
- 30% to 40% of patients with major depressive disorder (MDD) do not respond sufficiently to usual antidepressant treatment1
- Even under optimal treatment conditions, only one-third of patients achieve remission2
- Among patients who fail to respond to two pharmacologic interventions, remission rates with the next antidepressant are as low as 12%3
- A patient becomes less likely to respond clinically with each additional nonresponse to antidepressant treatment4
Why thyroid hormones?
Thyroid hormones enter the brain slowly across the blood-brain barrier and the choroid plexus—cerebrospinal fluid barriers. T4 is the main source of brain T3—after attack by 5’deiodinase—but circulating T3 also crosses the blood-brain barrier through active transport.
Thyroid hormones accumulate in the locus ceruleus and other central noradrenergic structures and are distributed widely in the brain along noradrenergic pathways.5-7 The mechanism of their therapeutic effect for MDD is not well understood, and various hypotheses have been proposed.
Subclinical hypothyroidism. Early studies such as by Howland8 of treatment-refractory MDD suggested that thyroid hormone augmentation might correct a hypothyroid state. However, blood thyroid hormone levels are not associated with resistance to antidepressant treatment, according to studies of MDD populations.9-10 Also, thyroid hormones’ therapeutic action in MDD appears unlikely to be related to treating subclinical hypothyroidism because patients’ euthyroid status was verified in all adjuvant studies since 1980.
Joffe et al11 proposed that MDD is characterized by a relative excess of T4 versus T3—probably related to a deficit in converting T4 to T3 in the periphery—and administering T3 would therefore correct this imbalance. They offered no strong evidence for an increased T4 level, however, and later studies failed to detect the postulated blood T3 abnormalities in MDD.9-10 Also—as suggested by studies with high-dose T4 augmentation12,13—the adjuvant antidepressant effect of thyroid hormones is not restricted to T3, although T3 may be more efficacious than T4.14
Neurotransmitter effects. Thyroid hormones’ role in increasing serotonin (5-HT) release could partially explain the benefit of adjuvant thyroid hormone therapy in MDD. Researchers found:
- T3 increased cortical 5-HT levels, probably by reducing the autoinhibitory effect of the presynaptic 5-HT1A receptor15
- adding T3 to clomipramine therapy increased 5-HT levels to a greater extent than T3 or clomipramine used alone16
- Low 5-HT activity, shown in hypothyroid patients, increased after T4 replacement.17
This 5-HT release theory cannot explain why thyroid hormones have a rapid clinical effect in MDD. Most studies report the hormones have clinical efficacy in MDD within 4 weeks.18 Similarly, selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels within hours of treatment onset, but the clinical effect occurs 4 to 6 weeks later.19 Therefore, increased 5-HT levels cannot fully explain thyroid hormones’ early effect.
Close interaction between thyroid hormones and the noradrenergic system also has been examined. Brain T3 is primarily localized in the central noradrenergic systems, with axonal anterograde transport of T3 from the locus ceruleus. T3 is processed and accumulated in the noradrenergic system, carried via axonal transport, then delivered from nerve cell bodies to its neuronal targets.5,7 T3 thus functions as a coneurotransmitter with norepinephrine.
Cellular energy metabolism. We recently reported that thyroid hormones’ antidepressant effect may be related to brain cellular energy metabolism. Thyroid hormones increase cellular levels of adenosine triphosphate (ATP) and phosphocreatine (PCr) in the hypothyroid brain.20 Brain imaging—phosphorus-31 nuclear magnetic resonance spectroscopy (31P-MRS)—of subjects with MDD shows decreased brain levels of ATP and increased PCr.21
Our group showed that the antidepressant effect of T3 augmentation of SSRIs is correlated with significant increases in ATP levels and decreases in PCr. This effect—which appears to represent re-normalization of brain bioenergetics in treatment responders—did not occur in nonresponders (Iosifescu et al, presented at APA, 2004).
The effect of thyroid hormones on bioenergetic metabolism is compatible with the hypothesized effects on noradrenergic and serotonergic systems.5,7 These mechanisms may represent different links in the same chain of events.
Antidepressant boosters
Thyroid hormones have been used extensively to treat MDD since the 1950s, when researchers reported that T3 monotherapy was efficacious for treating depression. These early studies had important methodologic limitations, including open designs and poorly defined diagnostic criteria and response.
For MDD, the most extensively researched uses of thyroid hormones are to augment therapy for antidepressant nonresponders and to accelerate partial response to antidepressants.
Tricyclics. Open studies primarily among outpatients in the 1970s and ‘80s suggested that thyroid hormones are a valid augmentation strategy for nonresponders to tricyclic antidepressants. Most—but not all—reported response rates >50% with T3 dosages of 20 to 50 mcg/d.22,23
Compared with outpatient studies, however, an open trial of T3 augmentation by Birkenhager et al24 found no evidence of efficacy in 14 severely depressed inpatients who had not responded to 6 weeks of tricyclics. These patients—mainly with melancholic and/or psychotic depression—showed greater response to a monoamine oxidase inhibitor (MAOI) or electroconvulsive therapy than to thyroid hormone.
Three of five double-blind controlled studies of thyroid hormone augmentation of tricyclics reported response rates of 50 to 65% (Table).14,18,25 Earlier controlled studies, including two negative studies26,27 had important methodologic limitations: all included few subjects (≤16), and two studies included both unipolar and bipolar patients. Joffe et al partially addressed these problems with two randomized, double-blind studies that were controlled with placebo or T4:
- In a 3-week trial, T3 was more effective than T4 when added to imipramine or desipramine.14
- In a 2-week trial, T3 was significantly more effective than placebo when added to imipramine or desipramine.25
In the latter study, T3 and lithium augmentation appeared equally effective. Conversely, T4 was more effective than lithium as the first augmentation strategy in a double-blind, crossover study by Spoov and Lahdelma.28
In conclusion, depressed patients given T3 with tricyclic antidepressants may be twice as likely as controls to respond to treatment, according to a meta-analysis of eight studies totaling 292 patients. This analysis by Aronson et al29 supports T3 augmentation while addressing the surveyed studies’ limitations.
Table
Treatment-resistant MDD: Controlled studies of thyroid hormone augmentation
| Study authors/design | Initial therapy | Augmentation | Results |
|---|---|---|---|
| Steiner et al, 1978 Randomized double-blind; 8 patients* | Several TCAs (6 weeks) | T3, 25 mcg/d, or placebo (35 days) | T3: 75% responders (3/4) Placebo: 75% (3/4) |
| Goodwin et al, 1982 Double-blind, mirror design; 12 patients* | Desipramine or imipramine (4 weeks) | T3, 25 to 50 mcg/d, or placebo (21 days) | T3: 33% responders (4/12) Placebo: 0% (0/6) |
| Gitlin et al, 1987 Double-blind with crossover; 16 patients | Imipramine (4 weeks) | T3, 25 mcg/d, or placebo (2 weeks); crossover (2 weeks) | No difference between T3 and placebo |
| Joffe and Singer, 1990 Randomized double-blind; 38 patients | Desipramine or imipramine (4 weeks) | T3, 37.5 mcg/d, or T4, 150 mcg/d (21 days) | T3: 53% responders (9/17) T4: 19% (4/21) |
| Joffe et al, 1993 Randomized double-blind; 33 patients | Desipramine or imipramine (5 weeks) | T3, 37.5 mcg/d, or placebo (14 days) | T3: 59% responders (10/17) Placebo: 19% (3/16) |
| Sopov and Lahdelma, 1998 Randomized double-blind with crossover; 22 patients* | TCA, MAOI antidepressants | T4, 200 mcg/d, or lithium, 500 mg/d, (4 weeks); then crossover (4 weeks) | T4: 64% responders (7/11) Lithium: 18% responders (2/11) |
| *Included patients with unipolar or bipolar depression | |||
| MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; T3: triiodothyronine; T4: thyroxine; TCA: tricyclic antidepressant. | |||
MAO inhibitors. One small report has addressed the efficacy of thyroid hormones as adjuvants to MAOIs.30 In two patients, adding T3 to phenelzine enhanced the antidepressant response.
High-dose T4. Two open studies of patients with treatment-resistant bipolar or unipolar depression12,13 have examined the efficacy of high-dose T4 augmentation. These patients were taking a variety of antidepressants, including TCAs and SSRIs.
- In the study by Baurer et al,12 clinical remission (Hamilton Depression Scale [HAM-D] score ≤10) occurred in 4 of 5 patients with severe treatment-resistant unipolar depression who received adjunctive T4, mean 482±72 mcg/d, with antidepressants.
- In the study by Rudas et al,13 clinical remission (HAM-D ≤9) occurred in 7 of 9 patients with treatment-resistant MDD after T4, 150 to 300 mcg/d, was added to their antidepressant therapy. Side effects may limit this high-dose strategy, however, because 2 of the patients dropped out with thyrotoxicosis symptoms.
SSRIs. Three open trials to date have investigated using thyroid hormones to augment SSRIs in treatment-resistant MDD. In a prospective study by Agid and Lerer,31 10 of 25 (40%) patients who did not respond to SSRI treatment did so after T3 was added. No men improved, however, which led the authors to suggest that men and women might respond differently to T3 augmentation of SSRIs.
In our study, 7 of 20 patients (35%) with MDD who did not respond to 8 weeks of SSRI therapy did so when we added T3, 50 mcg/d, for 4 weeks (Figure). Response rates were high (5/5, 100%) in patients with atypical features by DSMIV criteria and low (1/8, 12.5%) in those with melancholic features.32
Abraham et al33 added T3, 50 mcg/d, to the regimens of 12 patients with MDD who did not respond to SSRIs alone. One patient dropped out with side effects. After 4 weeks of T3 augmentation, 5 patients (42%) showed 50% or greater improvement in HAM-D scores from baseline.
Figure T3 augmentation of SSRIs in 20 patients with resistant major depressive disorder
Open T3 augmentation, 50 mcg/d, given to 20 nonresponders to 8 weeks of selective serotonin reuptake inhibitors (SSRIs) improved baseline CGI-S scores significantly (P=0.006) at 4 weeks in those with atypical depression and modestly (P>0.05) in those with melancholic depression.
Source: Reference 32Antidepressant accelerators. Five of seven early double-blind, controlled studies indicated that adding small doses of thyroid hormones at the beginning of antidepressant treatment accelerated treatment response. All were limited by small sample sizes and other methodologic problems. A more-recent meta-analysis of six studies totalling 125 patients by Altshuler et al34 found:
- T3 was significantly more effective than placebo in accelerating clinical response to tricyclics
- the acceleration effect was more pronounced for women than for men.
Clinical recommendations
Thyroid hormones can be useful to augment and accelerate treatment of MDD. Evidence strongly supports their use with tricyclic antidepressants and suggests they also can be effective adjuvants for patients who do not respond to SSRIs.
Either T3 (up to 50 mcg/d) or T4 (up to 150 mcg/d) can be used as augmentation. T3’s antidepressant properties are considered more effective than those of T4, but the only head-to-head study supporting this conclusion was small (38 patients).14 Some T4 augmentation studies used very high dosages (300 to 600 mcg/d),12 which increase the risk of acute overdose.
Start T3 augmentation at 25 mcg/d and increase, if tolerated, to 50 mcg/d after 1 week. Measure baseline serum thyroid-stimulating hormone (TSH), and do not treat patients with TSH <0.5 mIU/L). Baseline TSH, T4, or T3 levels do not predict response to T3 augmentation in euthyroid MDD patients.32
Common side effects. Adjuvant T3, 25 to 50 mcg/d, was well-tolerated in our study of 20 patients also taking SSRIs:
- 2 (10%) experienced fatigue and diaphoresis
- 1 each (5%) had tremor, dry mouth, headaches, muscle aches, and vivid dreams.32
We saw no significant changes in blood pressure, but heart rates increased significantly in our 4-week study—from 76±12 bpm (range 60 to 96) to 82±9 bpm (range 66 to 96). Thus, T3 augmentation may not be indicated for patients with coronary artery disease or chronic heart failure. Patients’ weight decreased an average 2.5±6.6 lbs (range –20 to +7).
Thyroid hormones may cause hypoglycemia and change insulin requirements in patients with diabetes. High doses of T3 or T4 may be associated with hyperthyroidism, weight loss, nervousness, sweating, tachycardia, insomnia, heat intolerance, menstrual irregularities, palpitations, psychosis, or fever. Discontinue treatment if these symptoms develop.
Onset of antidepressant effect. Assess patient response in 4 to 6 weeks, whether using augmentation to address antidepressant nonresponse14,25 or to accelerate response.33 If you detect only partial improvement, studies support continuing treatment up to 8 weeks.
Treatment duration. No guidelines exist on how long to continue thyroid hormones after the initial antidepressant response. TSH levels become suppressed (TSH <0.1 mIU/L) after 4 weeks of T3, 50 mcg/d, in patients with normal baseline thyroid function.32 This suggests thyroid hormone’s booster effect is self limited, and augmentation may not need to continue after 2 to 3 months—even in responders.
T3 augmentation at 25 mcg/d can be discontinued immediately. For 50 to 75 mcg/d, taper across 1 to 2 weeks. The hypothalamic-pituitary-thyroid axis returns to normal function 6 to 8 weeks after T3 augmentation is stopped.
In this model, thyroid hormone augmentation can be used to boost antidepressant efficacy several months at a time. If effective, the same strategy could be tried again for subsequent MDD episodes.
Related resources
- DeBattista C. Augmentation and combination strategies for depression. J Psychopharmacol 2006;20(3):11-18.
- Massachusetts General Hospital Psychiatric Academy (including web-casts on treatment-resistant depression. www.mghcme.com
Drug brand names
- Desipramine • Norpramin
- Imipramine • Tofranil
- Levothyroxine (T4) • Levoxyl, Levothroid, Synthroid, others
- Liothyronine (synthetic T3) • Cytomel
Disclosures
The author receives research support from Aspect Medical Systems, Forest Laboratories, and Janssen Pharmaceutica; is a consultant to Pfizer, Inc., and Forest Laboratories; and a speaker for Eli Lilly and Co., Pfizer, Inc., and Cephalon.
Prescribing thyroid hormones with antidepressants—whether to augment the antidepressant effect or accelerate patient response—is a well-researched strategy for treatment-resistant major depressive disorder (MDD). Thyroid hormones are known to boost response to tricyclics, and preliminary evidence shows they may be useful adjuvants to selective serotonin reuptake inhibitors (SSRIs) as well.
Thyroid hormones enter the brain slowly across the blood-brain barrier and choroid plexus. They accumulate in the locus ceruleus and other structures and are distributed widely along noradrenergic pathways.
Effective treatments are available for MDD, although 30% to 40% of patients do not respond to one or more antidepressant trials (Box).1-4 This article offers:
- new information about why triiodothyronine (T3) and thyroxine (T4) can “super-charge” antidepressant response
- tips on how to use thyroid hormones in patients with MDD, including effective dosages, patient monitoring, and treatment durations.
- 30% to 40% of patients with major depressive disorder (MDD) do not respond sufficiently to usual antidepressant treatment1
- Even under optimal treatment conditions, only one-third of patients achieve remission2
- Among patients who fail to respond to two pharmacologic interventions, remission rates with the next antidepressant are as low as 12%3
- A patient becomes less likely to respond clinically with each additional nonresponse to antidepressant treatment4
Why thyroid hormones?
Thyroid hormones enter the brain slowly across the blood-brain barrier and the choroid plexus—cerebrospinal fluid barriers. T4 is the main source of brain T3—after attack by 5’deiodinase—but circulating T3 also crosses the blood-brain barrier through active transport.
Thyroid hormones accumulate in the locus ceruleus and other central noradrenergic structures and are distributed widely in the brain along noradrenergic pathways.5-7 The mechanism of their therapeutic effect for MDD is not well understood, and various hypotheses have been proposed.
Subclinical hypothyroidism. Early studies such as by Howland8 of treatment-refractory MDD suggested that thyroid hormone augmentation might correct a hypothyroid state. However, blood thyroid hormone levels are not associated with resistance to antidepressant treatment, according to studies of MDD populations.9-10 Also, thyroid hormones’ therapeutic action in MDD appears unlikely to be related to treating subclinical hypothyroidism because patients’ euthyroid status was verified in all adjuvant studies since 1980.
Joffe et al11 proposed that MDD is characterized by a relative excess of T4 versus T3—probably related to a deficit in converting T4 to T3 in the periphery—and administering T3 would therefore correct this imbalance. They offered no strong evidence for an increased T4 level, however, and later studies failed to detect the postulated blood T3 abnormalities in MDD.9-10 Also—as suggested by studies with high-dose T4 augmentation12,13—the adjuvant antidepressant effect of thyroid hormones is not restricted to T3, although T3 may be more efficacious than T4.14
Neurotransmitter effects. Thyroid hormones’ role in increasing serotonin (5-HT) release could partially explain the benefit of adjuvant thyroid hormone therapy in MDD. Researchers found:
- T3 increased cortical 5-HT levels, probably by reducing the autoinhibitory effect of the presynaptic 5-HT1A receptor15
- adding T3 to clomipramine therapy increased 5-HT levels to a greater extent than T3 or clomipramine used alone16
- Low 5-HT activity, shown in hypothyroid patients, increased after T4 replacement.17
This 5-HT release theory cannot explain why thyroid hormones have a rapid clinical effect in MDD. Most studies report the hormones have clinical efficacy in MDD within 4 weeks.18 Similarly, selective serotonin reuptake inhibitors (SSRIs) increase 5-HT levels within hours of treatment onset, but the clinical effect occurs 4 to 6 weeks later.19 Therefore, increased 5-HT levels cannot fully explain thyroid hormones’ early effect.
Close interaction between thyroid hormones and the noradrenergic system also has been examined. Brain T3 is primarily localized in the central noradrenergic systems, with axonal anterograde transport of T3 from the locus ceruleus. T3 is processed and accumulated in the noradrenergic system, carried via axonal transport, then delivered from nerve cell bodies to its neuronal targets.5,7 T3 thus functions as a coneurotransmitter with norepinephrine.
Cellular energy metabolism. We recently reported that thyroid hormones’ antidepressant effect may be related to brain cellular energy metabolism. Thyroid hormones increase cellular levels of adenosine triphosphate (ATP) and phosphocreatine (PCr) in the hypothyroid brain.20 Brain imaging—phosphorus-31 nuclear magnetic resonance spectroscopy (31P-MRS)—of subjects with MDD shows decreased brain levels of ATP and increased PCr.21
Our group showed that the antidepressant effect of T3 augmentation of SSRIs is correlated with significant increases in ATP levels and decreases in PCr. This effect—which appears to represent re-normalization of brain bioenergetics in treatment responders—did not occur in nonresponders (Iosifescu et al, presented at APA, 2004).
The effect of thyroid hormones on bioenergetic metabolism is compatible with the hypothesized effects on noradrenergic and serotonergic systems.5,7 These mechanisms may represent different links in the same chain of events.
Antidepressant boosters
Thyroid hormones have been used extensively to treat MDD since the 1950s, when researchers reported that T3 monotherapy was efficacious for treating depression. These early studies had important methodologic limitations, including open designs and poorly defined diagnostic criteria and response.
For MDD, the most extensively researched uses of thyroid hormones are to augment therapy for antidepressant nonresponders and to accelerate partial response to antidepressants.
Tricyclics. Open studies primarily among outpatients in the 1970s and ‘80s suggested that thyroid hormones are a valid augmentation strategy for nonresponders to tricyclic antidepressants. Most—but not all—reported response rates >50% with T3 dosages of 20 to 50 mcg/d.22,23
Compared with outpatient studies, however, an open trial of T3 augmentation by Birkenhager et al24 found no evidence of efficacy in 14 severely depressed inpatients who had not responded to 6 weeks of tricyclics. These patients—mainly with melancholic and/or psychotic depression—showed greater response to a monoamine oxidase inhibitor (MAOI) or electroconvulsive therapy than to thyroid hormone.
Three of five double-blind controlled studies of thyroid hormone augmentation of tricyclics reported response rates of 50 to 65% (Table).14,18,25 Earlier controlled studies, including two negative studies26,27 had important methodologic limitations: all included few subjects (≤16), and two studies included both unipolar and bipolar patients. Joffe et al partially addressed these problems with two randomized, double-blind studies that were controlled with placebo or T4:
- In a 3-week trial, T3 was more effective than T4 when added to imipramine or desipramine.14
- In a 2-week trial, T3 was significantly more effective than placebo when added to imipramine or desipramine.25
In the latter study, T3 and lithium augmentation appeared equally effective. Conversely, T4 was more effective than lithium as the first augmentation strategy in a double-blind, crossover study by Spoov and Lahdelma.28
In conclusion, depressed patients given T3 with tricyclic antidepressants may be twice as likely as controls to respond to treatment, according to a meta-analysis of eight studies totaling 292 patients. This analysis by Aronson et al29 supports T3 augmentation while addressing the surveyed studies’ limitations.
Table
Treatment-resistant MDD: Controlled studies of thyroid hormone augmentation
| Study authors/design | Initial therapy | Augmentation | Results |
|---|---|---|---|
| Steiner et al, 1978 Randomized double-blind; 8 patients* | Several TCAs (6 weeks) | T3, 25 mcg/d, or placebo (35 days) | T3: 75% responders (3/4) Placebo: 75% (3/4) |
| Goodwin et al, 1982 Double-blind, mirror design; 12 patients* | Desipramine or imipramine (4 weeks) | T3, 25 to 50 mcg/d, or placebo (21 days) | T3: 33% responders (4/12) Placebo: 0% (0/6) |
| Gitlin et al, 1987 Double-blind with crossover; 16 patients | Imipramine (4 weeks) | T3, 25 mcg/d, or placebo (2 weeks); crossover (2 weeks) | No difference between T3 and placebo |
| Joffe and Singer, 1990 Randomized double-blind; 38 patients | Desipramine or imipramine (4 weeks) | T3, 37.5 mcg/d, or T4, 150 mcg/d (21 days) | T3: 53% responders (9/17) T4: 19% (4/21) |
| Joffe et al, 1993 Randomized double-blind; 33 patients | Desipramine or imipramine (5 weeks) | T3, 37.5 mcg/d, or placebo (14 days) | T3: 59% responders (10/17) Placebo: 19% (3/16) |
| Sopov and Lahdelma, 1998 Randomized double-blind with crossover; 22 patients* | TCA, MAOI antidepressants | T4, 200 mcg/d, or lithium, 500 mg/d, (4 weeks); then crossover (4 weeks) | T4: 64% responders (7/11) Lithium: 18% responders (2/11) |
| *Included patients with unipolar or bipolar depression | |||
| MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; T3: triiodothyronine; T4: thyroxine; TCA: tricyclic antidepressant. | |||
MAO inhibitors. One small report has addressed the efficacy of thyroid hormones as adjuvants to MAOIs.30 In two patients, adding T3 to phenelzine enhanced the antidepressant response.
High-dose T4. Two open studies of patients with treatment-resistant bipolar or unipolar depression12,13 have examined the efficacy of high-dose T4 augmentation. These patients were taking a variety of antidepressants, including TCAs and SSRIs.
- In the study by Baurer et al,12 clinical remission (Hamilton Depression Scale [HAM-D] score ≤10) occurred in 4 of 5 patients with severe treatment-resistant unipolar depression who received adjunctive T4, mean 482±72 mcg/d, with antidepressants.
- In the study by Rudas et al,13 clinical remission (HAM-D ≤9) occurred in 7 of 9 patients with treatment-resistant MDD after T4, 150 to 300 mcg/d, was added to their antidepressant therapy. Side effects may limit this high-dose strategy, however, because 2 of the patients dropped out with thyrotoxicosis symptoms.
SSRIs. Three open trials to date have investigated using thyroid hormones to augment SSRIs in treatment-resistant MDD. In a prospective study by Agid and Lerer,31 10 of 25 (40%) patients who did not respond to SSRI treatment did so after T3 was added. No men improved, however, which led the authors to suggest that men and women might respond differently to T3 augmentation of SSRIs.
In our study, 7 of 20 patients (35%) with MDD who did not respond to 8 weeks of SSRI therapy did so when we added T3, 50 mcg/d, for 4 weeks (Figure). Response rates were high (5/5, 100%) in patients with atypical features by DSMIV criteria and low (1/8, 12.5%) in those with melancholic features.32
Abraham et al33 added T3, 50 mcg/d, to the regimens of 12 patients with MDD who did not respond to SSRIs alone. One patient dropped out with side effects. After 4 weeks of T3 augmentation, 5 patients (42%) showed 50% or greater improvement in HAM-D scores from baseline.
Figure T3 augmentation of SSRIs in 20 patients with resistant major depressive disorder
Open T3 augmentation, 50 mcg/d, given to 20 nonresponders to 8 weeks of selective serotonin reuptake inhibitors (SSRIs) improved baseline CGI-S scores significantly (P=0.006) at 4 weeks in those with atypical depression and modestly (P>0.05) in those with melancholic depression.
Source: Reference 32Antidepressant accelerators. Five of seven early double-blind, controlled studies indicated that adding small doses of thyroid hormones at the beginning of antidepressant treatment accelerated treatment response. All were limited by small sample sizes and other methodologic problems. A more-recent meta-analysis of six studies totalling 125 patients by Altshuler et al34 found:
- T3 was significantly more effective than placebo in accelerating clinical response to tricyclics
- the acceleration effect was more pronounced for women than for men.
Clinical recommendations
Thyroid hormones can be useful to augment and accelerate treatment of MDD. Evidence strongly supports their use with tricyclic antidepressants and suggests they also can be effective adjuvants for patients who do not respond to SSRIs.
Either T3 (up to 50 mcg/d) or T4 (up to 150 mcg/d) can be used as augmentation. T3’s antidepressant properties are considered more effective than those of T4, but the only head-to-head study supporting this conclusion was small (38 patients).14 Some T4 augmentation studies used very high dosages (300 to 600 mcg/d),12 which increase the risk of acute overdose.
Start T3 augmentation at 25 mcg/d and increase, if tolerated, to 50 mcg/d after 1 week. Measure baseline serum thyroid-stimulating hormone (TSH), and do not treat patients with TSH <0.5 mIU/L). Baseline TSH, T4, or T3 levels do not predict response to T3 augmentation in euthyroid MDD patients.32
Common side effects. Adjuvant T3, 25 to 50 mcg/d, was well-tolerated in our study of 20 patients also taking SSRIs:
- 2 (10%) experienced fatigue and diaphoresis
- 1 each (5%) had tremor, dry mouth, headaches, muscle aches, and vivid dreams.32
We saw no significant changes in blood pressure, but heart rates increased significantly in our 4-week study—from 76±12 bpm (range 60 to 96) to 82±9 bpm (range 66 to 96). Thus, T3 augmentation may not be indicated for patients with coronary artery disease or chronic heart failure. Patients’ weight decreased an average 2.5±6.6 lbs (range –20 to +7).
Thyroid hormones may cause hypoglycemia and change insulin requirements in patients with diabetes. High doses of T3 or T4 may be associated with hyperthyroidism, weight loss, nervousness, sweating, tachycardia, insomnia, heat intolerance, menstrual irregularities, palpitations, psychosis, or fever. Discontinue treatment if these symptoms develop.
Onset of antidepressant effect. Assess patient response in 4 to 6 weeks, whether using augmentation to address antidepressant nonresponse14,25 or to accelerate response.33 If you detect only partial improvement, studies support continuing treatment up to 8 weeks.
Treatment duration. No guidelines exist on how long to continue thyroid hormones after the initial antidepressant response. TSH levels become suppressed (TSH <0.1 mIU/L) after 4 weeks of T3, 50 mcg/d, in patients with normal baseline thyroid function.32 This suggests thyroid hormone’s booster effect is self limited, and augmentation may not need to continue after 2 to 3 months—even in responders.
T3 augmentation at 25 mcg/d can be discontinued immediately. For 50 to 75 mcg/d, taper across 1 to 2 weeks. The hypothalamic-pituitary-thyroid axis returns to normal function 6 to 8 weeks after T3 augmentation is stopped.
In this model, thyroid hormone augmentation can be used to boost antidepressant efficacy several months at a time. If effective, the same strategy could be tried again for subsequent MDD episodes.
Related resources
- DeBattista C. Augmentation and combination strategies for depression. J Psychopharmacol 2006;20(3):11-18.
- Massachusetts General Hospital Psychiatric Academy (including web-casts on treatment-resistant depression. www.mghcme.com
Drug brand names
- Desipramine • Norpramin
- Imipramine • Tofranil
- Levothyroxine (T4) • Levoxyl, Levothroid, Synthroid, others
- Liothyronine (synthetic T3) • Cytomel
Disclosures
The author receives research support from Aspect Medical Systems, Forest Laboratories, and Janssen Pharmaceutica; is a consultant to Pfizer, Inc., and Forest Laboratories; and a speaker for Eli Lilly and Co., Pfizer, Inc., and Cephalon.
1. Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996;19:179-200.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Nierenberg AA, McLean NE, Alpert JE, et al. Early nonresponse to fluoxetine as a predictor of poor 8-week outcome. Am J Psychiatry 1995;152:1500-3.
4. Nierenberg AA, Papakostas GI, Petersen T, et al. Nortriptyline for treatment-resistant depression. J Clin Psychiatry 2003;64(1):35-9.
5. Rozanov CB, Dratman MB. Immunohistochemical mapping of brain triiodothyronine reveals prominent localization in central noradrenergic systems. Neuroscience 1996;74:897-915.
6. Cheng LY, Outterbridge LV, Covatta ND, et al. Film autoradiography identifies unique features of [125I]3,3’5’-(reverse) triiodothyronine transport from blood to brain. J Neurophysiol 1994;72:380-91.
7. Gordon JT, Kaminski DM, Rozanov CB, Dratman MB. Evidence that 3,3’,5-triiodothyronine is concentrated in and delivered from the locus coeruleus to its noradrenergic targets via anterograde axonal transport. Neuroscience 1999;93:943-54.
8. Howland RH. Thyroid dysfunction in refractory depression: implications for pathophysiology and treatment. J Clin Psychiatry 1993;54:47-54.
9. Joffe RT. Peripheral thyroid hormone levels in treatment resistant depression. Biol Psychiatry 1999;45:1053-5.
10. Iosifescu DV, Howarth S, Alpert JE, et al. T3 blood levels and treatment outcome in depression. Int J Psychiatry Med 2001;31:367-73.
11. Joffe RT, Roy-Byrne PP, Udhe TW, Post RM. Thyroid function and affective illness: a reappraisal. Biol Psychiatry 1984;19:1685-91.
12. Bauer M, Hellweg R, Graf KJ, Baumgartner A. Treatment of refractory depression with high-dose thyroxine. Neuropsychopharmacology 1998;18:444-55.
13. Rudas S, Schmitz M, Pichler P, Baumgartner A. Treatment of refractory chronic depression and dysthymia with high-dose thyroxine. Biol Psychiatry 1999;45:229-33.
14. Joffe RT, Singer W. A comparison of triiodothyronine and thyroxine in the potentiation of tricyclic antidepressants. Psychiatry Res 1990;32:241-51.
15. Sandrini M, Vitale G, Vergoni AV, et al. Effect of acute and chronic treatment with triiodothyronine on serotonin levels and serotonergic receptor subtypes in the rat brain. Life Sci 1996;58:1551-9.
16. Gur E, Lerer B, Newman ME. Chronic clomipramine and triiodothyronine increase serotonin levels in rat frontal cortex in vivo: relationship to serotonin autoreceptor activity. J Pharmacol Exp Ther 1999;288:81-7.
17. Cleare AJ, McGregor A, Chambers SM, et al. Thyroxine replacement increases central 5-hydroxytryptamine activity and reduces depressive symptoms in hypothyroidism. Neuroendocrinology 1996;64:65-9.
18. Goodwin FK, Prange AJ, Post RM, et al. Potentiation of antidepressant effects by l-triiodothyronine in tricyclic nonresponders. Am J Psychiatry 1982;139:34-8.
19. Blier P. Pharmacology of rapid-onset antidepressant treatment strategies. J Clin Psychiatry 2001;62(suppl 15):12-7.
20. Smith CD, Ain KB. Brain metabolism in hypothyroidism studied with 31P magnetic-resonance spectroscopy. Lancet 1995;345:619-20.
21. Kato T, Murashita J, Shioiri T, et al. Effect of photic stimulation on energy metabolism in the human brain measured by 31P-MR spectroscopy. J Neuropsychiatry Clin Neurosci 1996;8:417-22.
22. Earle BV. Thyroid hormone and tricyclic antidepressants in resistant depressions. Am J Psychiatry 1970;126:1667-9.
23. Thase ME, Kupfer DJ, Jarrett DB. Treatment of imipramine-resistant recurrent depression: I. An open clinical trial of adjunctive l-triiodothyronine. J Clin Psychiatry 1989;50:385-8.
24. Birkenhager TK, Vegt M, Nolen WA. An open study of triiodothyronine augmentation of tricyclic antidepressants in inpatients with refractory depression. Pharmacopsychiatry 1997;30:23-6.
25. Joffe RT, Singer W, Levitt AJ, MacDonald C. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-93.
26. Steiner M, Radwan M, Elizur A, et al. Failure of l-triiodothyronine to potentiate tricyclic antidepressant response. Curr Ther Res 1978;23:655-9.
27. Gitlin MJ, Weiner H, Fairbanks L, et al. Failure of T3 to potentiate tricyclic antidepressant response. J Affective Disord 1987;13:267-72.
28. Spoov J, Lahdelma L. Should thyroid augmentation precede lithium augmentation—a pilot study. J Affect Disord 1998;49:235-9.
29. Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996;53:842-8.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry 1988;49:409-10.
31. Agid O, Lerer B. Algorithm-based treatment of major depression in an outpatient clinic: clinical correlates of response to a specific serotonin reuptake inhibitor and to triiodothyronine augmentation. Int J Neuropsychopharmacol 2003;6(1):41-9.
32. Iosifescu DV, Nierenberg AA, Mischoulon D, et al. An open study of triiodothyronine augmentation of selective serotonin reuptake inhibitors in treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66:1038-42.
33. Abraham G, Milev R, Stuart Lawson J. T3 augmentation of SSRI resistant depression. J Affect Disord 2006;91(2-3):211-15.
34. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry 2001;158:1617-22.
1. Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am 1996;19:179-200.
2. Trivedi MH, Rush AJ, Wisniewski SR, et al. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006;163(1):28-40.
3. Nierenberg AA, McLean NE, Alpert JE, et al. Early nonresponse to fluoxetine as a predictor of poor 8-week outcome. Am J Psychiatry 1995;152:1500-3.
4. Nierenberg AA, Papakostas GI, Petersen T, et al. Nortriptyline for treatment-resistant depression. J Clin Psychiatry 2003;64(1):35-9.
5. Rozanov CB, Dratman MB. Immunohistochemical mapping of brain triiodothyronine reveals prominent localization in central noradrenergic systems. Neuroscience 1996;74:897-915.
6. Cheng LY, Outterbridge LV, Covatta ND, et al. Film autoradiography identifies unique features of [125I]3,3’5’-(reverse) triiodothyronine transport from blood to brain. J Neurophysiol 1994;72:380-91.
7. Gordon JT, Kaminski DM, Rozanov CB, Dratman MB. Evidence that 3,3’,5-triiodothyronine is concentrated in and delivered from the locus coeruleus to its noradrenergic targets via anterograde axonal transport. Neuroscience 1999;93:943-54.
8. Howland RH. Thyroid dysfunction in refractory depression: implications for pathophysiology and treatment. J Clin Psychiatry 1993;54:47-54.
9. Joffe RT. Peripheral thyroid hormone levels in treatment resistant depression. Biol Psychiatry 1999;45:1053-5.
10. Iosifescu DV, Howarth S, Alpert JE, et al. T3 blood levels and treatment outcome in depression. Int J Psychiatry Med 2001;31:367-73.
11. Joffe RT, Roy-Byrne PP, Udhe TW, Post RM. Thyroid function and affective illness: a reappraisal. Biol Psychiatry 1984;19:1685-91.
12. Bauer M, Hellweg R, Graf KJ, Baumgartner A. Treatment of refractory depression with high-dose thyroxine. Neuropsychopharmacology 1998;18:444-55.
13. Rudas S, Schmitz M, Pichler P, Baumgartner A. Treatment of refractory chronic depression and dysthymia with high-dose thyroxine. Biol Psychiatry 1999;45:229-33.
14. Joffe RT, Singer W. A comparison of triiodothyronine and thyroxine in the potentiation of tricyclic antidepressants. Psychiatry Res 1990;32:241-51.
15. Sandrini M, Vitale G, Vergoni AV, et al. Effect of acute and chronic treatment with triiodothyronine on serotonin levels and serotonergic receptor subtypes in the rat brain. Life Sci 1996;58:1551-9.
16. Gur E, Lerer B, Newman ME. Chronic clomipramine and triiodothyronine increase serotonin levels in rat frontal cortex in vivo: relationship to serotonin autoreceptor activity. J Pharmacol Exp Ther 1999;288:81-7.
17. Cleare AJ, McGregor A, Chambers SM, et al. Thyroxine replacement increases central 5-hydroxytryptamine activity and reduces depressive symptoms in hypothyroidism. Neuroendocrinology 1996;64:65-9.
18. Goodwin FK, Prange AJ, Post RM, et al. Potentiation of antidepressant effects by l-triiodothyronine in tricyclic nonresponders. Am J Psychiatry 1982;139:34-8.
19. Blier P. Pharmacology of rapid-onset antidepressant treatment strategies. J Clin Psychiatry 2001;62(suppl 15):12-7.
20. Smith CD, Ain KB. Brain metabolism in hypothyroidism studied with 31P magnetic-resonance spectroscopy. Lancet 1995;345:619-20.
21. Kato T, Murashita J, Shioiri T, et al. Effect of photic stimulation on energy metabolism in the human brain measured by 31P-MR spectroscopy. J Neuropsychiatry Clin Neurosci 1996;8:417-22.
22. Earle BV. Thyroid hormone and tricyclic antidepressants in resistant depressions. Am J Psychiatry 1970;126:1667-9.
23. Thase ME, Kupfer DJ, Jarrett DB. Treatment of imipramine-resistant recurrent depression: I. An open clinical trial of adjunctive l-triiodothyronine. J Clin Psychiatry 1989;50:385-8.
24. Birkenhager TK, Vegt M, Nolen WA. An open study of triiodothyronine augmentation of tricyclic antidepressants in inpatients with refractory depression. Pharmacopsychiatry 1997;30:23-6.
25. Joffe RT, Singer W, Levitt AJ, MacDonald C. A placebo-controlled comparison of lithium and triiodothyronine augmentation of tricyclic antidepressants in unipolar refractory depression. Arch Gen Psychiatry 1993;50:387-93.
26. Steiner M, Radwan M, Elizur A, et al. Failure of l-triiodothyronine to potentiate tricyclic antidepressant response. Curr Ther Res 1978;23:655-9.
27. Gitlin MJ, Weiner H, Fairbanks L, et al. Failure of T3 to potentiate tricyclic antidepressant response. J Affective Disord 1987;13:267-72.
28. Spoov J, Lahdelma L. Should thyroid augmentation precede lithium augmentation—a pilot study. J Affect Disord 1998;49:235-9.
29. Aronson R, Offman HJ, Joffe RT, Naylor CD. Triiodothyronine augmentation in the treatment of refractory depression. A meta-analysis. Arch Gen Psychiatry 1996;53:842-8.
30. Joffe RT. Triiodothyronine potentiation of the antidepressant effect of phenelzine. J Clin Psychiatry 1988;49:409-10.
31. Agid O, Lerer B. Algorithm-based treatment of major depression in an outpatient clinic: clinical correlates of response to a specific serotonin reuptake inhibitor and to triiodothyronine augmentation. Int J Neuropsychopharmacol 2003;6(1):41-9.
32. Iosifescu DV, Nierenberg AA, Mischoulon D, et al. An open study of triiodothyronine augmentation of selective serotonin reuptake inhibitors in treatment-resistant major depressive disorder. J Clin Psychiatry 2005;66:1038-42.
33. Abraham G, Milev R, Stuart Lawson J. T3 augmentation of SSRI resistant depression. J Affect Disord 2006;91(2-3):211-15.
34. Altshuler LL, Bauer M, Frye MA, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry 2001;158:1617-22.
Compulsive shopping: When spending begins to consume the consumer
Ms. A has been compulsively shopping and spending since age 19 when she first obtained credit cards. After years of intense urges to shop and remorse over the financial consequences, she seeks psychiatric help. Now age 37 and divorced, she has controlled her spending only for two 1- to 2-year periods that coincided with bankruptcy proceedings.
With easy access to credit, many persons such as Ms. A develop what is variously called compulsive buying, compulsive shopping, addictive shopping, or shopaholism. Although “medicalizing” excessive shopping may seem to obscure its broader cultural and social causes,1 increasing evidence points to a discrete shopping disorder.
Our group has contributed to compulsive buying research and continues to evaluate potential treatments. We offer evidence and practical advice to help you:
- identify compulsive shopping disorder using the patient’s history and three screening questions
- differentiate compulsive shopping from manic or hypomanic shopping sprees
- educate patients about four steps to control compulsive shopping.
Table 1
Compulsive shopping disorder’s clinical signs
| Onset in late adolescence to early adulthood |
| Female-to-male ratio may be 9:1 |
| Behaviors include shopping frequently, spending inappropriately, and fantasizing about future purchases |
| Psychiatric comorbidity—mood disorders, substance abuse, eating disorders—is common among patients and first-degree relatives |
| Chronic symptoms wax and wane, with widely varying severity |
| Irresistible urges prompt spending by some patients |
| Shopping is intensely exciting, with transitory feelings of happiness and power |
| Feelings of distress and guilt develop after shopping; patients often hide purchases |
| Patients may be in denial or feel embarrassed to disclose symptoms |
An Evolving Picture
Ms. A says shopping is her primary social activity and entertainment. Though she works full time, she shops three or more times a week, cruising expensive department stores and discount outlets on evenings and weekends. She buys clothing, shoes, makeup, jewelry, antiques, household electronics, and other items.
She says her shopping is spontaneous and impulsive. Shopping gives her an emotional “rush” that is frequently followed by periods of guilt, and she often returns or gives away purchased items. She is disappointed at her inability to control her shopping behavior and ashamed of the financial crises she has caused.
Compulsive buying is characterized by persistent or poorly controlled preoccupations, urges, or behaviors regarding shopping or spending, leading to adverse consequences.2 Onset in late adolescence to early adulthood is the usual pattern, and the disorder is thought to be chronic or recurrent. It is not listed in DSM-IV-TR but is considered an example of an impulse control disorder not otherwise specified. For this paper, we use the terms compulsive shopping and compulsive buying interchangeably.
The disorder’s tentative classification reflects debate about its conceptualization. Some clinicians and researchers consider compulsive buying an addiction similar to drug or alcohol misuse; others have linked it to depression or anxiety. Hollander3 and others have commented on its similarities with obsessive-compulsive disorder (OCD), and a recent study noted that compulsive buying is more common in patients with OCD than in matched controls.4 Still others—drawing on Kraepelin’s and Bleuler’s early work—consider compulsive buying an impulse control disorder, having features in common with pathological gambling and kleptomania.5
Prevalence. One survey estimates 2% to 8% of U.S. adults meet criteria for a compulsive shopping disorder, and community-based and clinical surveys suggest that 86% to 95% of them are women.5 The reported gender difference may be artifactual; women readily acknowledge that they enjoy shopping, whereas men are more likely to report that they “collect.”
Behavior patterns. No careful, longitudinal studies have examined compulsive buying disorder, but case reports suggest the condition is chronic, with a waxing and waning course and wide variance in symptom severity. In 20 consecutive patients with compulsive buying symptoms, one-half reported that irresistible urges prompted spending and three-quarters preferred to shop alone.6
Compulsive shoppers tend to shop frequently and spend inappropriately:
- at department and discount stores, specialty shops, and boutiques
- from mail order, television, and online merchants.
While shopping, compulsive shoppers may report feeling intensely excited, happy, and powerful. These emotions are frequently followed by distress or guilt. They may return purchases or hide them in closets or attics, never to be used.
Low-income persons who shop compulsively may do so at consignment shops or garage sales. In one of our studies, the most severe compulsive buyers had the lowest incomes,6 suggesting that:
- lack of money does not prevent compulsive shopping disorder from developing
- severe compulsive shoppers lack the ability to delay their shopping.
Psychiatric comorbidity
Compulsive buyers differ from matched controls when dimensional scales are used to measure psychopathology. One study found that compulsive buyers had elevated scores on the Beck Depression Inventory, the Spielberger Trait Anxiety Scale, and the Maudsley Obsessive Compulsive Inventory.2
Compulsive buyers and their first-degree relatives often have comorbid psychiatric disorders, particularly mood, anxiety, substance use, and eating disorders.5 Axis II disorders are also common; no particular type predominates, but the obsessive-compulsive, borderline, and avoidant personality types are seen most frequently.
McElroy et al7 defined compulsive buying disorder as:
- uncontrollable
- markedly distressing, time-consuming, and/or resulting in family, social, vocational, and/or financial difficulties
- not occurring only in the context of hypomanic or manic symptoms.
In a larger controlled study, our group8 compared 33 individuals who met the McElroy et al criteria for compulsive buying disorder and 22 control patients. The 137 first-degree relatives of the compulsive shoppers were significantly more likely than the controls’ relatives to have histories of depression, alcoholism, substance use, or multiple psychiatric diagnoses (as measured by the Family History Research Diagnostic Criteria).
Identifying a patient’s psychiatric comorbidities can help you develop:
- a biopsychosocial counseling plan—such as for a patient with borderline personality disorder who shops to relieve tension from relationship stress
- pharmacologic treatment strategies—such as prescribing a selective serotonin reuptake inhibitor (SSRI) for patients with comorbid major depression.
Manic versus compulsive behavior
Manic and hypomanic symptoms may be associated with impulsive and reckless spending. Thus, when evaluating excessive spending, always carefully evaluate patients for bipolar disorder.
Bipolar mania and excessive spending related to a compulsive buying disorder are relatively easy to differentiate:
- The manic patient’s unrestrained spending sprees correspond to manic episodes and are accompanied by euphoric mood, grandiosity, unrealistic plans, and often a giddy, overly bright affect.
- The compulsive shopper’s spending occurs year-round in a pattern suggesting ongoing preoccupation.
Not so for the manic, who may boast of his or her spending, display the evidence, and try to convince family and friends that the purchase is necessary or fits into some grandiose scheme. “Who doesn’t need two BMWs?” a manic patient said to one of the authors [DWB].
Screening and diagnosis
As with any psychiatric disorder, gathering an accurate history through a careful interview is important. This can be challenging with compulsive shopping disorder, however, because the patient may minimize symptoms out of embarrassment or denial. Your goal is to identify the shopping problem through nonjudgmental inquiries.
Diagnostic instruments. Researchers use assessment tools such as Faber and O’Guinn’s 7-item Compulsive Buying Scale9 to help diagnose this disorder. Our group developed a shopping version of the Yale-Brown Obsessive Compulsive Scale (YBOCS-SV) to help rate severity and change during clinical trials.10
Formal instruments may help in the clinical setting, but you can often elicit compulsive buying symptoms with a few screening questions (Table 2). If screening indicates a positive response, move to more detailed questions about:
- frequency of excessive shopping
- time spent shopping
- factors that trigger or worsen the shopping behavior
- amount of money spent.
Table 2
Is your patient a compulsive shopper? Ask these screening questions
| Do you feel preoccupied with shopping and spending? |
| Do you ever feel that your shopping behavior is excessive, inappropriate, or uncontrolled? |
Have your shopping desires, urges, fantasies, or behaviors ever:
|
| Source: Black DW. Assessment of compulsive buying. In: Benson AL, ed. I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216. |
Stopping uncontrolled shopping
Compulsive shopping has no standard treatment, but evidence shows benefit from some SSRIs and psychotherapies.
Fluvoxamine. An early case series suggested antidepressants could curb compulsive buying,5 but later research has yielded mixed results.
Ms. A entered an experimental drug trial. She was randomly assigned to receive fluvoxamine and—despite difficulties with oversedation—tolerated a sustained dosage of 100 mg/d. After the 9-week trial, Ms. A said she thought less frequently about shopping, felt less compulsion to shop, and was spending less money and time shopping.
This open-label trial we conducted indicated that fluvoxamine, up to 300 mg/d, could be an effective treatment for compulsive buying.11 Two subsequent randomized controlled trials, however, found fluvoxamine did no better than placebo when treating compulsive shoppers.12,13
Citalopram. In an open-label trial,14 23 women and 1 man who met diagnostic criteria for compulsive shopping disorder (YBOCS-SV scores ≥17) received citalopram for 7 weeks. Dosages started at 20 mg/d and were increased as tolerated to 60 mg/d. Fifteen patients (63%) met response criteria—“much improved” or “very much improved” as measured by the Clinical Global Impressions-Improvement scale and a ≥50% decrease in YBOCS-SV score. Three patients (13%) discontinued treatment because of adverse effects (headache, rash, insomnia).
The 15 responders were then enrolled in a 9-week double-blind, placebo-controlled trial. Compulsive shopping symptoms recurred in 5 of 8 patients (63%) assigned to placebo, compared with none of the 7 who continued taking citalopram.
By comparison, escitalopram, 10 to 20 mg/d, showed little effect for compulsive shopping symptoms in an identically designed discontinuation trial by the same investigators. During the 7-week, open-label trial, 19 of 26 patients met response criteria. In the 9-week double-blind, controlled phase, however, 63% of initial responders relapsed while taking escitalopram, compared with 67% of those randomized to placebo.15
A naturalistic follow-up study of 24 patients treated with citalopram, 20 to 60 mg/d, noted that patients who responded at 3 months were more likely to be symptom-free after 1 year than those who did not respond to acute treatment.16 Responders’ mean 2-week compulsive spending declined from $773 before treatment to $351 at 12 months, and their mean total debt declined from $17,833 to $16,752.
Because remission was not significantly associated with taking citalopram, however, the authors concluded that the mechanisms responsible for maintaining remission were unclear.
Psychotherapy. Cognitive-behavioral therapy (CBT) may help, but few therapists are familiar with this disorder. CBT challenges the patient’s cognitive distortions and faulty schemas about shopping, such as:
- “Having the latest fashions will make me more popular.”
- “Having 5 pair of new shoes will make me a happier and better person.”
Our recommendations. Medication—such as an antidepressant for major depression or a mood stabilizer for bipolar disorder—may improve compulsive shopping in patients with a comorbid psychiatric disorder. For other compulsive shoppers, however, medication trials provide little guidance for treatment.
We inform patients such as Ms. A that they cannot rely on medication to control their behavior. Instead, we recommend a four-step approach to break the compulsive shopping habit (Table 3).
Financial counseling, provided free of charge by many banks, benefits some patients. Self-help books describe strategies to overcome compulsive spending (Related resources). Debtors Anonymous, a 12-step program patterned after Alcoholics Anonymous, also can help by offering acceptance, belonging, forgiveness, and understanding.
In the most severe cases we recommend appointing a financial conservator to control the patient’s finances. We rarely advise this strategy but have encountered cases in which there seemed to be no other option. Having a conservator controls the patient’s spending but does not reverse the preoccupation with shopping.
Table 3
Patient education: 4 steps to control compulsive spending
|
For clinicians
- Black DW. Assessment of compulsive buying. In: Benson AL (ed). I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216.
- Arenson G. Born to spend: how to overcome compulsive spending. Blue Ridge Summit, PA: Tab Books, 1991.
- Benson AL. Stopping Overshopping. A site for shopaholics and the people who love them. www.stoppingovershopping.com.
- Mellan O. Money harmony: resolving money conflicts in your life and relationships. New York: Walker, 2005.
Drug brand names
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluvoxamine • Luvox
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives grant/research support or is a consultant or speaker for Forest Laboratories and Shire Pharmaceuticals
1. Lee S, Mysyk A. The medicalization of compulsive buying. Soc Sci Med 2004;58(9):1709-18.
2. Black DW. Compulsive buying disorder: definition, assessment, epidemiology and clinical management. CNS Drugs 2001;15(1):17-27.
3. Hollander E. Obsessive compulsive related disorders. Washington, DC: American Psychiatric Press; 1993.
4. Lejoyeux M, Bailly F, Moula H, Loi S, Ades J. Study of compulsive buying in patients presenting with obsessive-compulsive disorder. Compr Psychiatry 2005;46:105-10.
5. Black DW. Compulsive shopping. In: Hollander E, Stein D (eds). Clinical manual of impulse control disorders. Washington, DC: American Psychiatric Publishing; 2003;203–27.
6. Black DW, Monahan P, Schlosser S, Repertinger S. Compulsive buying severity: an analysis of Compulsive Buying Scale results in 44 subjects. J Nerv Ment Dis 2001;189:123-7.
7. McElroy SL, Keck PE, Jr, Pope HG, Jr, et al. Compulsive buying: a report of 20 cases. J Clin Psychiatry 1994;55:242-8.
8. Black DW, Repertinger S, Gaffney GR, Gabel J. Family history and psychiatric comorbidity in persons with compulsive buying: preliminary findings. Am J Psychiatry 1998;155:960-3.
9. Faber RJ, O’Guinn TC. A clinical screener for compulsive buying. Consum Res 1992;19:459-69.
10. Monahan P, Black DW, Gabel J. Reliability and validity of a scale to measure change in persons with compulsive buying. Psychiatr Res 1996;64:59-67.
11. Black DW, Monahan P, Gabel J. Fluvoxamine in the treatment of compulsive buying. J Clin Psychiatry 1997;58:159-63.
12. Ninan PT, McElroy SL, Kane CP, et al. Placebo-controlled study of fluvoxamine in the treatment of patients with compulsive buying. J Clin Psychopharmacol 2000;20:362-6.
13. Black DW, Gabel J, Hansen J, Schlosser S. A double-blind comparison of fluvoxamine versus placebo in the treatment of compulsive buying disorder. Ann Clin Psychiatry 2000;12:205-11.
14. Koran LM, Chuong HW, Bullock KD, Smith SC. Citalopram for compulsive shopping disorder: an open-label study followed by double-blind discontinuation. J Clin Psychiatry 2003;64:793-8.
15. Koran LM. Escitalopram treatment evaluated in patients with compulsive shopping disorder. Primary Psychiatry 2005;12(12):13.-
16. Aboujaoude E, Gamel N, Koran LM. A 1-year naturalistic follow-up of patients with compulsive shopping disorder. J Clin Psychiatry 2003;64:946-50.
Ms. A has been compulsively shopping and spending since age 19 when she first obtained credit cards. After years of intense urges to shop and remorse over the financial consequences, she seeks psychiatric help. Now age 37 and divorced, she has controlled her spending only for two 1- to 2-year periods that coincided with bankruptcy proceedings.
With easy access to credit, many persons such as Ms. A develop what is variously called compulsive buying, compulsive shopping, addictive shopping, or shopaholism. Although “medicalizing” excessive shopping may seem to obscure its broader cultural and social causes,1 increasing evidence points to a discrete shopping disorder.
Our group has contributed to compulsive buying research and continues to evaluate potential treatments. We offer evidence and practical advice to help you:
- identify compulsive shopping disorder using the patient’s history and three screening questions
- differentiate compulsive shopping from manic or hypomanic shopping sprees
- educate patients about four steps to control compulsive shopping.
Table 1
Compulsive shopping disorder’s clinical signs
| Onset in late adolescence to early adulthood |
| Female-to-male ratio may be 9:1 |
| Behaviors include shopping frequently, spending inappropriately, and fantasizing about future purchases |
| Psychiatric comorbidity—mood disorders, substance abuse, eating disorders—is common among patients and first-degree relatives |
| Chronic symptoms wax and wane, with widely varying severity |
| Irresistible urges prompt spending by some patients |
| Shopping is intensely exciting, with transitory feelings of happiness and power |
| Feelings of distress and guilt develop after shopping; patients often hide purchases |
| Patients may be in denial or feel embarrassed to disclose symptoms |
An Evolving Picture
Ms. A says shopping is her primary social activity and entertainment. Though she works full time, she shops three or more times a week, cruising expensive department stores and discount outlets on evenings and weekends. She buys clothing, shoes, makeup, jewelry, antiques, household electronics, and other items.
She says her shopping is spontaneous and impulsive. Shopping gives her an emotional “rush” that is frequently followed by periods of guilt, and she often returns or gives away purchased items. She is disappointed at her inability to control her shopping behavior and ashamed of the financial crises she has caused.
Compulsive buying is characterized by persistent or poorly controlled preoccupations, urges, or behaviors regarding shopping or spending, leading to adverse consequences.2 Onset in late adolescence to early adulthood is the usual pattern, and the disorder is thought to be chronic or recurrent. It is not listed in DSM-IV-TR but is considered an example of an impulse control disorder not otherwise specified. For this paper, we use the terms compulsive shopping and compulsive buying interchangeably.
The disorder’s tentative classification reflects debate about its conceptualization. Some clinicians and researchers consider compulsive buying an addiction similar to drug or alcohol misuse; others have linked it to depression or anxiety. Hollander3 and others have commented on its similarities with obsessive-compulsive disorder (OCD), and a recent study noted that compulsive buying is more common in patients with OCD than in matched controls.4 Still others—drawing on Kraepelin’s and Bleuler’s early work—consider compulsive buying an impulse control disorder, having features in common with pathological gambling and kleptomania.5
Prevalence. One survey estimates 2% to 8% of U.S. adults meet criteria for a compulsive shopping disorder, and community-based and clinical surveys suggest that 86% to 95% of them are women.5 The reported gender difference may be artifactual; women readily acknowledge that they enjoy shopping, whereas men are more likely to report that they “collect.”
Behavior patterns. No careful, longitudinal studies have examined compulsive buying disorder, but case reports suggest the condition is chronic, with a waxing and waning course and wide variance in symptom severity. In 20 consecutive patients with compulsive buying symptoms, one-half reported that irresistible urges prompted spending and three-quarters preferred to shop alone.6
Compulsive shoppers tend to shop frequently and spend inappropriately:
- at department and discount stores, specialty shops, and boutiques
- from mail order, television, and online merchants.
While shopping, compulsive shoppers may report feeling intensely excited, happy, and powerful. These emotions are frequently followed by distress or guilt. They may return purchases or hide them in closets or attics, never to be used.
Low-income persons who shop compulsively may do so at consignment shops or garage sales. In one of our studies, the most severe compulsive buyers had the lowest incomes,6 suggesting that:
- lack of money does not prevent compulsive shopping disorder from developing
- severe compulsive shoppers lack the ability to delay their shopping.
Psychiatric comorbidity
Compulsive buyers differ from matched controls when dimensional scales are used to measure psychopathology. One study found that compulsive buyers had elevated scores on the Beck Depression Inventory, the Spielberger Trait Anxiety Scale, and the Maudsley Obsessive Compulsive Inventory.2
Compulsive buyers and their first-degree relatives often have comorbid psychiatric disorders, particularly mood, anxiety, substance use, and eating disorders.5 Axis II disorders are also common; no particular type predominates, but the obsessive-compulsive, borderline, and avoidant personality types are seen most frequently.
McElroy et al7 defined compulsive buying disorder as:
- uncontrollable
- markedly distressing, time-consuming, and/or resulting in family, social, vocational, and/or financial difficulties
- not occurring only in the context of hypomanic or manic symptoms.
In a larger controlled study, our group8 compared 33 individuals who met the McElroy et al criteria for compulsive buying disorder and 22 control patients. The 137 first-degree relatives of the compulsive shoppers were significantly more likely than the controls’ relatives to have histories of depression, alcoholism, substance use, or multiple psychiatric diagnoses (as measured by the Family History Research Diagnostic Criteria).
Identifying a patient’s psychiatric comorbidities can help you develop:
- a biopsychosocial counseling plan—such as for a patient with borderline personality disorder who shops to relieve tension from relationship stress
- pharmacologic treatment strategies—such as prescribing a selective serotonin reuptake inhibitor (SSRI) for patients with comorbid major depression.
Manic versus compulsive behavior
Manic and hypomanic symptoms may be associated with impulsive and reckless spending. Thus, when evaluating excessive spending, always carefully evaluate patients for bipolar disorder.
Bipolar mania and excessive spending related to a compulsive buying disorder are relatively easy to differentiate:
- The manic patient’s unrestrained spending sprees correspond to manic episodes and are accompanied by euphoric mood, grandiosity, unrealistic plans, and often a giddy, overly bright affect.
- The compulsive shopper’s spending occurs year-round in a pattern suggesting ongoing preoccupation.
Not so for the manic, who may boast of his or her spending, display the evidence, and try to convince family and friends that the purchase is necessary or fits into some grandiose scheme. “Who doesn’t need two BMWs?” a manic patient said to one of the authors [DWB].
Screening and diagnosis
As with any psychiatric disorder, gathering an accurate history through a careful interview is important. This can be challenging with compulsive shopping disorder, however, because the patient may minimize symptoms out of embarrassment or denial. Your goal is to identify the shopping problem through nonjudgmental inquiries.
Diagnostic instruments. Researchers use assessment tools such as Faber and O’Guinn’s 7-item Compulsive Buying Scale9 to help diagnose this disorder. Our group developed a shopping version of the Yale-Brown Obsessive Compulsive Scale (YBOCS-SV) to help rate severity and change during clinical trials.10
Formal instruments may help in the clinical setting, but you can often elicit compulsive buying symptoms with a few screening questions (Table 2). If screening indicates a positive response, move to more detailed questions about:
- frequency of excessive shopping
- time spent shopping
- factors that trigger or worsen the shopping behavior
- amount of money spent.
Table 2
Is your patient a compulsive shopper? Ask these screening questions
| Do you feel preoccupied with shopping and spending? |
| Do you ever feel that your shopping behavior is excessive, inappropriate, or uncontrolled? |
Have your shopping desires, urges, fantasies, or behaviors ever:
|
| Source: Black DW. Assessment of compulsive buying. In: Benson AL, ed. I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216. |
Stopping uncontrolled shopping
Compulsive shopping has no standard treatment, but evidence shows benefit from some SSRIs and psychotherapies.
Fluvoxamine. An early case series suggested antidepressants could curb compulsive buying,5 but later research has yielded mixed results.
Ms. A entered an experimental drug trial. She was randomly assigned to receive fluvoxamine and—despite difficulties with oversedation—tolerated a sustained dosage of 100 mg/d. After the 9-week trial, Ms. A said she thought less frequently about shopping, felt less compulsion to shop, and was spending less money and time shopping.
This open-label trial we conducted indicated that fluvoxamine, up to 300 mg/d, could be an effective treatment for compulsive buying.11 Two subsequent randomized controlled trials, however, found fluvoxamine did no better than placebo when treating compulsive shoppers.12,13
Citalopram. In an open-label trial,14 23 women and 1 man who met diagnostic criteria for compulsive shopping disorder (YBOCS-SV scores ≥17) received citalopram for 7 weeks. Dosages started at 20 mg/d and were increased as tolerated to 60 mg/d. Fifteen patients (63%) met response criteria—“much improved” or “very much improved” as measured by the Clinical Global Impressions-Improvement scale and a ≥50% decrease in YBOCS-SV score. Three patients (13%) discontinued treatment because of adverse effects (headache, rash, insomnia).
The 15 responders were then enrolled in a 9-week double-blind, placebo-controlled trial. Compulsive shopping symptoms recurred in 5 of 8 patients (63%) assigned to placebo, compared with none of the 7 who continued taking citalopram.
By comparison, escitalopram, 10 to 20 mg/d, showed little effect for compulsive shopping symptoms in an identically designed discontinuation trial by the same investigators. During the 7-week, open-label trial, 19 of 26 patients met response criteria. In the 9-week double-blind, controlled phase, however, 63% of initial responders relapsed while taking escitalopram, compared with 67% of those randomized to placebo.15
A naturalistic follow-up study of 24 patients treated with citalopram, 20 to 60 mg/d, noted that patients who responded at 3 months were more likely to be symptom-free after 1 year than those who did not respond to acute treatment.16 Responders’ mean 2-week compulsive spending declined from $773 before treatment to $351 at 12 months, and their mean total debt declined from $17,833 to $16,752.
Because remission was not significantly associated with taking citalopram, however, the authors concluded that the mechanisms responsible for maintaining remission were unclear.
Psychotherapy. Cognitive-behavioral therapy (CBT) may help, but few therapists are familiar with this disorder. CBT challenges the patient’s cognitive distortions and faulty schemas about shopping, such as:
- “Having the latest fashions will make me more popular.”
- “Having 5 pair of new shoes will make me a happier and better person.”
Our recommendations. Medication—such as an antidepressant for major depression or a mood stabilizer for bipolar disorder—may improve compulsive shopping in patients with a comorbid psychiatric disorder. For other compulsive shoppers, however, medication trials provide little guidance for treatment.
We inform patients such as Ms. A that they cannot rely on medication to control their behavior. Instead, we recommend a four-step approach to break the compulsive shopping habit (Table 3).
Financial counseling, provided free of charge by many banks, benefits some patients. Self-help books describe strategies to overcome compulsive spending (Related resources). Debtors Anonymous, a 12-step program patterned after Alcoholics Anonymous, also can help by offering acceptance, belonging, forgiveness, and understanding.
In the most severe cases we recommend appointing a financial conservator to control the patient’s finances. We rarely advise this strategy but have encountered cases in which there seemed to be no other option. Having a conservator controls the patient’s spending but does not reverse the preoccupation with shopping.
Table 3
Patient education: 4 steps to control compulsive spending
|
For clinicians
- Black DW. Assessment of compulsive buying. In: Benson AL (ed). I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216.
- Arenson G. Born to spend: how to overcome compulsive spending. Blue Ridge Summit, PA: Tab Books, 1991.
- Benson AL. Stopping Overshopping. A site for shopaholics and the people who love them. www.stoppingovershopping.com.
- Mellan O. Money harmony: resolving money conflicts in your life and relationships. New York: Walker, 2005.
Drug brand names
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluvoxamine • Luvox
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives grant/research support or is a consultant or speaker for Forest Laboratories and Shire Pharmaceuticals
Ms. A has been compulsively shopping and spending since age 19 when she first obtained credit cards. After years of intense urges to shop and remorse over the financial consequences, she seeks psychiatric help. Now age 37 and divorced, she has controlled her spending only for two 1- to 2-year periods that coincided with bankruptcy proceedings.
With easy access to credit, many persons such as Ms. A develop what is variously called compulsive buying, compulsive shopping, addictive shopping, or shopaholism. Although “medicalizing” excessive shopping may seem to obscure its broader cultural and social causes,1 increasing evidence points to a discrete shopping disorder.
Our group has contributed to compulsive buying research and continues to evaluate potential treatments. We offer evidence and practical advice to help you:
- identify compulsive shopping disorder using the patient’s history and three screening questions
- differentiate compulsive shopping from manic or hypomanic shopping sprees
- educate patients about four steps to control compulsive shopping.
Table 1
Compulsive shopping disorder’s clinical signs
| Onset in late adolescence to early adulthood |
| Female-to-male ratio may be 9:1 |
| Behaviors include shopping frequently, spending inappropriately, and fantasizing about future purchases |
| Psychiatric comorbidity—mood disorders, substance abuse, eating disorders—is common among patients and first-degree relatives |
| Chronic symptoms wax and wane, with widely varying severity |
| Irresistible urges prompt spending by some patients |
| Shopping is intensely exciting, with transitory feelings of happiness and power |
| Feelings of distress and guilt develop after shopping; patients often hide purchases |
| Patients may be in denial or feel embarrassed to disclose symptoms |
An Evolving Picture
Ms. A says shopping is her primary social activity and entertainment. Though she works full time, she shops three or more times a week, cruising expensive department stores and discount outlets on evenings and weekends. She buys clothing, shoes, makeup, jewelry, antiques, household electronics, and other items.
She says her shopping is spontaneous and impulsive. Shopping gives her an emotional “rush” that is frequently followed by periods of guilt, and she often returns or gives away purchased items. She is disappointed at her inability to control her shopping behavior and ashamed of the financial crises she has caused.
Compulsive buying is characterized by persistent or poorly controlled preoccupations, urges, or behaviors regarding shopping or spending, leading to adverse consequences.2 Onset in late adolescence to early adulthood is the usual pattern, and the disorder is thought to be chronic or recurrent. It is not listed in DSM-IV-TR but is considered an example of an impulse control disorder not otherwise specified. For this paper, we use the terms compulsive shopping and compulsive buying interchangeably.
The disorder’s tentative classification reflects debate about its conceptualization. Some clinicians and researchers consider compulsive buying an addiction similar to drug or alcohol misuse; others have linked it to depression or anxiety. Hollander3 and others have commented on its similarities with obsessive-compulsive disorder (OCD), and a recent study noted that compulsive buying is more common in patients with OCD than in matched controls.4 Still others—drawing on Kraepelin’s and Bleuler’s early work—consider compulsive buying an impulse control disorder, having features in common with pathological gambling and kleptomania.5
Prevalence. One survey estimates 2% to 8% of U.S. adults meet criteria for a compulsive shopping disorder, and community-based and clinical surveys suggest that 86% to 95% of them are women.5 The reported gender difference may be artifactual; women readily acknowledge that they enjoy shopping, whereas men are more likely to report that they “collect.”
Behavior patterns. No careful, longitudinal studies have examined compulsive buying disorder, but case reports suggest the condition is chronic, with a waxing and waning course and wide variance in symptom severity. In 20 consecutive patients with compulsive buying symptoms, one-half reported that irresistible urges prompted spending and three-quarters preferred to shop alone.6
Compulsive shoppers tend to shop frequently and spend inappropriately:
- at department and discount stores, specialty shops, and boutiques
- from mail order, television, and online merchants.
While shopping, compulsive shoppers may report feeling intensely excited, happy, and powerful. These emotions are frequently followed by distress or guilt. They may return purchases or hide them in closets or attics, never to be used.
Low-income persons who shop compulsively may do so at consignment shops or garage sales. In one of our studies, the most severe compulsive buyers had the lowest incomes,6 suggesting that:
- lack of money does not prevent compulsive shopping disorder from developing
- severe compulsive shoppers lack the ability to delay their shopping.
Psychiatric comorbidity
Compulsive buyers differ from matched controls when dimensional scales are used to measure psychopathology. One study found that compulsive buyers had elevated scores on the Beck Depression Inventory, the Spielberger Trait Anxiety Scale, and the Maudsley Obsessive Compulsive Inventory.2
Compulsive buyers and their first-degree relatives often have comorbid psychiatric disorders, particularly mood, anxiety, substance use, and eating disorders.5 Axis II disorders are also common; no particular type predominates, but the obsessive-compulsive, borderline, and avoidant personality types are seen most frequently.
McElroy et al7 defined compulsive buying disorder as:
- uncontrollable
- markedly distressing, time-consuming, and/or resulting in family, social, vocational, and/or financial difficulties
- not occurring only in the context of hypomanic or manic symptoms.
In a larger controlled study, our group8 compared 33 individuals who met the McElroy et al criteria for compulsive buying disorder and 22 control patients. The 137 first-degree relatives of the compulsive shoppers were significantly more likely than the controls’ relatives to have histories of depression, alcoholism, substance use, or multiple psychiatric diagnoses (as measured by the Family History Research Diagnostic Criteria).
Identifying a patient’s psychiatric comorbidities can help you develop:
- a biopsychosocial counseling plan—such as for a patient with borderline personality disorder who shops to relieve tension from relationship stress
- pharmacologic treatment strategies—such as prescribing a selective serotonin reuptake inhibitor (SSRI) for patients with comorbid major depression.
Manic versus compulsive behavior
Manic and hypomanic symptoms may be associated with impulsive and reckless spending. Thus, when evaluating excessive spending, always carefully evaluate patients for bipolar disorder.
Bipolar mania and excessive spending related to a compulsive buying disorder are relatively easy to differentiate:
- The manic patient’s unrestrained spending sprees correspond to manic episodes and are accompanied by euphoric mood, grandiosity, unrealistic plans, and often a giddy, overly bright affect.
- The compulsive shopper’s spending occurs year-round in a pattern suggesting ongoing preoccupation.
Not so for the manic, who may boast of his or her spending, display the evidence, and try to convince family and friends that the purchase is necessary or fits into some grandiose scheme. “Who doesn’t need two BMWs?” a manic patient said to one of the authors [DWB].
Screening and diagnosis
As with any psychiatric disorder, gathering an accurate history through a careful interview is important. This can be challenging with compulsive shopping disorder, however, because the patient may minimize symptoms out of embarrassment or denial. Your goal is to identify the shopping problem through nonjudgmental inquiries.
Diagnostic instruments. Researchers use assessment tools such as Faber and O’Guinn’s 7-item Compulsive Buying Scale9 to help diagnose this disorder. Our group developed a shopping version of the Yale-Brown Obsessive Compulsive Scale (YBOCS-SV) to help rate severity and change during clinical trials.10
Formal instruments may help in the clinical setting, but you can often elicit compulsive buying symptoms with a few screening questions (Table 2). If screening indicates a positive response, move to more detailed questions about:
- frequency of excessive shopping
- time spent shopping
- factors that trigger or worsen the shopping behavior
- amount of money spent.
Table 2
Is your patient a compulsive shopper? Ask these screening questions
| Do you feel preoccupied with shopping and spending? |
| Do you ever feel that your shopping behavior is excessive, inappropriate, or uncontrolled? |
Have your shopping desires, urges, fantasies, or behaviors ever:
|
| Source: Black DW. Assessment of compulsive buying. In: Benson AL, ed. I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216. |
Stopping uncontrolled shopping
Compulsive shopping has no standard treatment, but evidence shows benefit from some SSRIs and psychotherapies.
Fluvoxamine. An early case series suggested antidepressants could curb compulsive buying,5 but later research has yielded mixed results.
Ms. A entered an experimental drug trial. She was randomly assigned to receive fluvoxamine and—despite difficulties with oversedation—tolerated a sustained dosage of 100 mg/d. After the 9-week trial, Ms. A said she thought less frequently about shopping, felt less compulsion to shop, and was spending less money and time shopping.
This open-label trial we conducted indicated that fluvoxamine, up to 300 mg/d, could be an effective treatment for compulsive buying.11 Two subsequent randomized controlled trials, however, found fluvoxamine did no better than placebo when treating compulsive shoppers.12,13
Citalopram. In an open-label trial,14 23 women and 1 man who met diagnostic criteria for compulsive shopping disorder (YBOCS-SV scores ≥17) received citalopram for 7 weeks. Dosages started at 20 mg/d and were increased as tolerated to 60 mg/d. Fifteen patients (63%) met response criteria—“much improved” or “very much improved” as measured by the Clinical Global Impressions-Improvement scale and a ≥50% decrease in YBOCS-SV score. Three patients (13%) discontinued treatment because of adverse effects (headache, rash, insomnia).
The 15 responders were then enrolled in a 9-week double-blind, placebo-controlled trial. Compulsive shopping symptoms recurred in 5 of 8 patients (63%) assigned to placebo, compared with none of the 7 who continued taking citalopram.
By comparison, escitalopram, 10 to 20 mg/d, showed little effect for compulsive shopping symptoms in an identically designed discontinuation trial by the same investigators. During the 7-week, open-label trial, 19 of 26 patients met response criteria. In the 9-week double-blind, controlled phase, however, 63% of initial responders relapsed while taking escitalopram, compared with 67% of those randomized to placebo.15
A naturalistic follow-up study of 24 patients treated with citalopram, 20 to 60 mg/d, noted that patients who responded at 3 months were more likely to be symptom-free after 1 year than those who did not respond to acute treatment.16 Responders’ mean 2-week compulsive spending declined from $773 before treatment to $351 at 12 months, and their mean total debt declined from $17,833 to $16,752.
Because remission was not significantly associated with taking citalopram, however, the authors concluded that the mechanisms responsible for maintaining remission were unclear.
Psychotherapy. Cognitive-behavioral therapy (CBT) may help, but few therapists are familiar with this disorder. CBT challenges the patient’s cognitive distortions and faulty schemas about shopping, such as:
- “Having the latest fashions will make me more popular.”
- “Having 5 pair of new shoes will make me a happier and better person.”
Our recommendations. Medication—such as an antidepressant for major depression or a mood stabilizer for bipolar disorder—may improve compulsive shopping in patients with a comorbid psychiatric disorder. For other compulsive shoppers, however, medication trials provide little guidance for treatment.
We inform patients such as Ms. A that they cannot rely on medication to control their behavior. Instead, we recommend a four-step approach to break the compulsive shopping habit (Table 3).
Financial counseling, provided free of charge by many banks, benefits some patients. Self-help books describe strategies to overcome compulsive spending (Related resources). Debtors Anonymous, a 12-step program patterned after Alcoholics Anonymous, also can help by offering acceptance, belonging, forgiveness, and understanding.
In the most severe cases we recommend appointing a financial conservator to control the patient’s finances. We rarely advise this strategy but have encountered cases in which there seemed to be no other option. Having a conservator controls the patient’s spending but does not reverse the preoccupation with shopping.
Table 3
Patient education: 4 steps to control compulsive spending
|
For clinicians
- Black DW. Assessment of compulsive buying. In: Benson AL (ed). I shop, therefore I am: Compulsive buying and the search for self. Northvale, NJ: Jason Aronson; 2000:191-216.
- Arenson G. Born to spend: how to overcome compulsive spending. Blue Ridge Summit, PA: Tab Books, 1991.
- Benson AL. Stopping Overshopping. A site for shopaholics and the people who love them. www.stoppingovershopping.com.
- Mellan O. Money harmony: resolving money conflicts in your life and relationships. New York: Walker, 2005.
Drug brand names
- Citalopram • Celexa
- Escitalopram • Lexapro
- Fluvoxamine • Luvox
Dr. Kuzma reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Black receives grant/research support or is a consultant or speaker for Forest Laboratories and Shire Pharmaceuticals
1. Lee S, Mysyk A. The medicalization of compulsive buying. Soc Sci Med 2004;58(9):1709-18.
2. Black DW. Compulsive buying disorder: definition, assessment, epidemiology and clinical management. CNS Drugs 2001;15(1):17-27.
3. Hollander E. Obsessive compulsive related disorders. Washington, DC: American Psychiatric Press; 1993.
4. Lejoyeux M, Bailly F, Moula H, Loi S, Ades J. Study of compulsive buying in patients presenting with obsessive-compulsive disorder. Compr Psychiatry 2005;46:105-10.
5. Black DW. Compulsive shopping. In: Hollander E, Stein D (eds). Clinical manual of impulse control disorders. Washington, DC: American Psychiatric Publishing; 2003;203–27.
6. Black DW, Monahan P, Schlosser S, Repertinger S. Compulsive buying severity: an analysis of Compulsive Buying Scale results in 44 subjects. J Nerv Ment Dis 2001;189:123-7.
7. McElroy SL, Keck PE, Jr, Pope HG, Jr, et al. Compulsive buying: a report of 20 cases. J Clin Psychiatry 1994;55:242-8.
8. Black DW, Repertinger S, Gaffney GR, Gabel J. Family history and psychiatric comorbidity in persons with compulsive buying: preliminary findings. Am J Psychiatry 1998;155:960-3.
9. Faber RJ, O’Guinn TC. A clinical screener for compulsive buying. Consum Res 1992;19:459-69.
10. Monahan P, Black DW, Gabel J. Reliability and validity of a scale to measure change in persons with compulsive buying. Psychiatr Res 1996;64:59-67.
11. Black DW, Monahan P, Gabel J. Fluvoxamine in the treatment of compulsive buying. J Clin Psychiatry 1997;58:159-63.
12. Ninan PT, McElroy SL, Kane CP, et al. Placebo-controlled study of fluvoxamine in the treatment of patients with compulsive buying. J Clin Psychopharmacol 2000;20:362-6.
13. Black DW, Gabel J, Hansen J, Schlosser S. A double-blind comparison of fluvoxamine versus placebo in the treatment of compulsive buying disorder. Ann Clin Psychiatry 2000;12:205-11.
14. Koran LM, Chuong HW, Bullock KD, Smith SC. Citalopram for compulsive shopping disorder: an open-label study followed by double-blind discontinuation. J Clin Psychiatry 2003;64:793-8.
15. Koran LM. Escitalopram treatment evaluated in patients with compulsive shopping disorder. Primary Psychiatry 2005;12(12):13.-
16. Aboujaoude E, Gamel N, Koran LM. A 1-year naturalistic follow-up of patients with compulsive shopping disorder. J Clin Psychiatry 2003;64:946-50.
1. Lee S, Mysyk A. The medicalization of compulsive buying. Soc Sci Med 2004;58(9):1709-18.
2. Black DW. Compulsive buying disorder: definition, assessment, epidemiology and clinical management. CNS Drugs 2001;15(1):17-27.
3. Hollander E. Obsessive compulsive related disorders. Washington, DC: American Psychiatric Press; 1993.
4. Lejoyeux M, Bailly F, Moula H, Loi S, Ades J. Study of compulsive buying in patients presenting with obsessive-compulsive disorder. Compr Psychiatry 2005;46:105-10.
5. Black DW. Compulsive shopping. In: Hollander E, Stein D (eds). Clinical manual of impulse control disorders. Washington, DC: American Psychiatric Publishing; 2003;203–27.
6. Black DW, Monahan P, Schlosser S, Repertinger S. Compulsive buying severity: an analysis of Compulsive Buying Scale results in 44 subjects. J Nerv Ment Dis 2001;189:123-7.
7. McElroy SL, Keck PE, Jr, Pope HG, Jr, et al. Compulsive buying: a report of 20 cases. J Clin Psychiatry 1994;55:242-8.
8. Black DW, Repertinger S, Gaffney GR, Gabel J. Family history and psychiatric comorbidity in persons with compulsive buying: preliminary findings. Am J Psychiatry 1998;155:960-3.
9. Faber RJ, O’Guinn TC. A clinical screener for compulsive buying. Consum Res 1992;19:459-69.
10. Monahan P, Black DW, Gabel J. Reliability and validity of a scale to measure change in persons with compulsive buying. Psychiatr Res 1996;64:59-67.
11. Black DW, Monahan P, Gabel J. Fluvoxamine in the treatment of compulsive buying. J Clin Psychiatry 1997;58:159-63.
12. Ninan PT, McElroy SL, Kane CP, et al. Placebo-controlled study of fluvoxamine in the treatment of patients with compulsive buying. J Clin Psychopharmacol 2000;20:362-6.
13. Black DW, Gabel J, Hansen J, Schlosser S. A double-blind comparison of fluvoxamine versus placebo in the treatment of compulsive buying disorder. Ann Clin Psychiatry 2000;12:205-11.
14. Koran LM, Chuong HW, Bullock KD, Smith SC. Citalopram for compulsive shopping disorder: an open-label study followed by double-blind discontinuation. J Clin Psychiatry 2003;64:793-8.
15. Koran LM. Escitalopram treatment evaluated in patients with compulsive shopping disorder. Primary Psychiatry 2005;12(12):13.-
16. Aboujaoude E, Gamel N, Koran LM. A 1-year naturalistic follow-up of patients with compulsive shopping disorder. J Clin Psychiatry 2003;64:946-50.
More adolescents are gambling—with addiction
Matt, age 17, grew up in a household of gamblers. He learned to play poker from his father at age 8 and bet on sports with his friends in the 5th grade. For 2 years he has been playing poker three to four nights per week and watching a lot of televised poker. His parents worry he might be “addicted” to gambling and bring him for evaluation.
Easy access to gambling through casinos, lotteries, and Internet games has affected many social groups, particularly adolescents.1 Teens such as Matt are more likely than adults to become pathological gamblers,2 and they often have psychiatric disorders and antisocial behavior patterns that interfere with normal development. This article:
- suggests screening tools to identify problem teen gambling
- discusses how to use psychotherapy, medications, and other options to help gambling-obsessed adolescents change their high-risk behavior.
Why adolescents gamble
Gambling activates the same neural reward pathways affected by cocaine and amphetamines.3 Even so, many adolescents view gambling as less harmful than drugs4 and consider it a rite of passage:
- 90% report having gambled for money
- 75% have gambled at home for money
- 85% of parents do not object to gambling behaviors, teens say.4,5
Adolescent gambling behavior. For approximately 85% of adolescents, gambling becomes no more than a social activity. For others, it can become problematic or even pathological (Table 1).5,7 Approximately 4 to 8% of adolescents meet criteria for pathological gambling, compared with 1% of adults.
Adolescents’ higher rate might reflect pathological gambling’s natural course—peaking during adolescence, then tapering during adulthood. Some adolescents may adapt their gambling behavior over time. Problem gambling tends to be more transient and episodic than pathological gambling, remitting when adolescents take on new responsibilities (such as with college, marriage, employment, or death of parents).8
Table 1
Adolescent gambling: From entertainment to mental illness
| Behavior | Prevalence in U.S. teens (%) | Definition |
|---|---|---|
| Social gambling | 80 to 85 | Gambling socially for a limited amount of time with predetermined, acceptable losses |
| Problem gambling | 10 to 14 | Gambling for recreation at the expense of other developmental activities; may interfere with time management, productivity, and relationships |
| Pathological gambling | 4 to 8 | Persistent and recurrent gambling that disrupts personal, family, or vocational pursuits |
| Source: References 3, 10. | ||
Case continued: ‘Up big-time’
Matt says he loves to gamble and believes he is more gifted at poker than his peers. He also recognizes his behavior could be addictive and admits lying to his parents about his gambling. He plays Internet poker 3 to 4 hours per day and claims he wins more often than he loses. “I’m up big time,” he boasts.
Matt denies going to casinos or using bookies. He says he is managing school and home responsibilities without difficulty. He denies mood or anxiety symptoms but admits using methylphenidate while gambling. He obtains the stimulant from friends at school and usually snorts it to get “a bigger rush.”
Matt is clearly at risk for problem gambling. He was exposed to gambling early, is becoming preoccupied with it, lies about how much he gambles, and combines gambling with substance abuse. progress more rapidly and can become problem gamblers within 12 to 14 months.2
Neurobiologic differences between adolescent and adult pathological gamblers are not well-defined. Adults show evidence of dysregulated dopamine, norepinephrine, and serotonin neuro-transmission.9 Neuroimaging in adult pathological gamblers shows perturbations in reward processing centers and frontal lobe structures that control inhibition. These factors have been examined little in adolescents.
Identifying problem gambling in teens
Diagnostic “red flags” for problem gambling in adolescents include declining school performance, sleep disturbance, generalized anxiety or irritability, or possibly lack of response to general psychiatric treatment. Three tools can be useful for screening adolescents:
The Lie-Bet Questionnaire10 is a 2-question screen for problem gambling:
- Have you ever lied to anyone important about how often you gamble?
- Have you ever had to increase your bet to get the same excitement from gambling?
The South-Oaks Gambling Screen11 is the standard pathological gambling screen for adults. Like the (SOGS-RA) is based and validated using DSM-III criteria (see Related resources). A score of 2 to 5 indicates at-risk gambling behaviors, and ≥6 indicate need for treatment.
The Gamblers Anonymous questionnaire12 comprises 20 questions that identify negative social, physical, and emotional consequences of gambling behaviors (Box). Seven or more positive responses indicate probable pathological gambling. This screen has shown reliability in adolescents.
Use one or more of these quick screens with every adolescent presenting for treatment—especially in substance abuse treatment settings. When results are positive, probe for gambling behaviors and consequences. Rely on DSM-IV-TR criteria and clinical presentation to differentiate social gambling from pathological gambling.
Most compulsive gamblers will answer “yes” to at least 7 of these questions:
- Did you ever lose time from work or school due to gambling?
- Has gambling ever made your home life unhappy?
- Did gambling affect your reputation?
- Have you ever felt remorse after gambling?
- Did you ever gamble to get money with which to pay debts or otherwise solve financial difficulties?
- Did gambling cause a decrease in your ambition or efficiency?
- After losing did you feel you must return as soon as possible and win back your losses?
- After a win did you have a strong urge to return and win more?
- Did you often gamble until your last dollar was gone?
- Did you ever borrow to finance your gambling?
- Have you ever sold anything to finance gambling?
- Were you reluctant to use “gambling money” for normal expenditures?
- Did gambling make you careless of the welfare of yourself or your family?
- Did you ever gamble longer than you had planned?
- Have you ever gambled to escape worry or trouble?
- Have you ever committed, or considered committing, an illegal act to finance
- Did gambling cause you to have difficulty in sleeping?
- Do arguments, disappointments, or frustrations create an urge to gamble?
- Did you ever have an urge to celebrate any good fortune by a few hours of gambling?
- Have you ever considered self-destruction or suicide as a result of your gambling?
Behaviors and comorbidities
Negative consequences. Pathological gambling often consumes 10 to 20 hours per week of the adolescent’s time,13 hurting school performance and delaying developmental milestones. Teen gamblers may abandon extracurricular school activities, and their few friends often gamble, too. They are at risk for delinquency, criminal activity, and antisocial behaviors (such as selling drugs, engaging in prostitution)14 unprotected sexual activity, drug use, reckless driving, and carrying weapons.15,16
Psychiatric comorbidity is the rule and often what brings adolescent gamblers to treatment. Substance abuse, major depression, attention-deficit/hyperactivity disorder, and personality disorders are most common (Table 2) Adolescent substance abuse at least triples the risk of pathological gambling.18
Adolescent pathological gamblers have increased rates of suicidal ideation and suicide attempts.17 They are at risk for other impulsive behaviors as well,19 although they are unlikely to volunteer this information. The Minnesota Impulsive Disorders Interview help identify comorbid pathological gambling, trichotillomania, kleptomania, pyromania, intermittent explosive disorder, compulsive buying, and compulsive sexual behaviors.19 Screen for these comorbidities during the first sessions or if a patient does not respond to treatment of gambling behavior.
Table 2
Risk factors for pathological adolescent gambling
| Component | Risk factors |
|---|---|
| Family history | Family history of gambling problems; possible genetic influences (neurotransmitter activity, risk-taking behaviors, risk perception, heightened physical response to rewards) |
| Psychiatric comorbidity | Substance abuse, mood/anxiety disorders, ADHD |
| Personality traits | Low self-esteem, competitiveness, sensitivity to stress or rejection, peer influences, immaturity, suicidal tendencies |
| Social factors | Early-age exposure to gambling, having peers who gamble regularly, increased access to gambling (such as via the Internet), chaotic home environment (divorce, neglect, abuse) |
| ADHD: attention-deficit/hyperactivity disorder | |
Treating adolescent gamblers
Matt’s answers to the gambling screening questionnaires (one “yes” to the Lie-Bet Questionnaire, a SOGS-RA score of 4, and a GA-20 Questions score of 5) indicate problem gambling but not pathological gambling. You recommend that he attend Gamblers Anonymous, but he refuses. He also rejects individual therapy or taking medications.
Matt acknowledges misusing methylphenidate, however, and agrees to consider an outpatient substance abuse program. He also agrees to six sessions with a gambling treatment specialist to learn about problem gambling’s signs and symptoms, how to cope with betting loses, and how to reduce his preoccupation with gambling.
No guidelines exist for treating adolescent pathological gamblers, and specialized teen treatment programs are rare. Most services are provided in mental health or substance abuse settings, using adult treatments modified for adolescents.
Cognitive behavioral therapy (CBT) can be successful for highly motivated gamblers, although adolescents might not want to change their pathological behaviors. In case reports, four adolescents achieved remission after 6 months of CBT.20,21 CBT appears to have long-term benefits for adults, but this has not been evaluated in teens. Even so, individual CBT may be ideal for adolescent gamblers because side effects are minimal.
Gamblers Anonymous (GA) in adults has shown a low retention rate and a 1-year abstinence rate of 22 Its effectiveness for adolescents lacks empiric support, but the 12-step program’s availability, structure, and fellowship may be useful.
Medications. Consider medication as first-line treatment for adolescents with psychiatric comorbidity. Try psychosocial treatment first for those without psychiatric comorbidity; consider medication as second-line therapy if response to psychosocial treatment is inadequate.
No medications are FDA-approved for pathological gambling, and no studies have examined medication use for adolescent gamblers. Controlled trials with adults suggest some medications may reduce urges and cravings or decrease gambling behaviors. These include:
- selective serotonin reuptake inhibitors
- opioid antagonists such as naltrexone or its analogue nalmefene
- or mood stabilizers such as lithium, divalproex sodium, or possibly topiramate.23
Working with families. Many parents are aware of the destructive potential of substance abuse but not of gambling. They may feel shame that they did not recognize their teen’s gambling problem or “control” their child’s behavior. The adolescent may feel remorse for having bet with the parents’ money.
Even when the family recognizes the teen’s problem, denial or enabling can perpetuate the behavior. For teens such as Matt who learned to gamble at a home, advise the parents to abstain from gambling as well. Consider screening the parents for pathological gambling, given its high rate of heritability.
Address guilt and shame by acknowledging that pathological gambling is a psychiatric disease caused by biological, psychological, and social factors—not dysfunctional family relations. Emphasize that treatment works. Because gambling is easily hidden, educate families about relapse signs, such as preoccupation with money, personality changes, or failing to fulfill family responsibilities.25
- Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-88.
- Gamblers Anonymous. www.gamblersanonymous.org.
- National Council on Problem Gambling. www.ncpgambling.org.
- International Centre for Youth Gambling Problems and High-Risk Behaviors. www.youthgambling.com.
- Nalmefene • Revex
- Naltrexone • ReVia
- Lithium • Lithobid, others
- Divalproex sodium • Depakote, others
- Topiramate • Topamax
Dr. Fong receives grant/research support from Ortho McNeil Pharmaceutical and Somaxon Pharmaceuticals and is a speaker for Forest Pharmaceuticals.
1. Gupta R, Derevensky JL. Adolescents with gambling problems: from research to treatment. J Gambl Stud 2000;16(2-3):315-42.
2. Evans RI. Some theoretical models and constructs generic to substance abuse prevention programs for adolescents: possible relevance and limitations for problem gambling. J Gambl Stud 2003;19(3):287-302.
3. Crockford DN, Goodyear B, Edwards J, et al. Cue-induced brain activity in pathological gamblers. Biol Psychiatry 2005;58(10):787-95.
4. Gupta R, Derevensky JL. Adolescent gambling behavior: A prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;14(4):319-45.
5. Derevensky JL, Gupta R, Winters K. Prevalence rates of youth gambling problems: are the current rates inflated? J Gambl Stud 2003;19(4):405-25.
6. Griffiths M. Adolescent gambling. London: Routledge; 1995.
7. Pietrzak RH, Ladd GT, Petry NM. Disordered gambling in adolescents: epidemiology, diagnosis, and treatment. Paediatr Drugs 2003;5(9):583-95.
8. Slutske WS, Jackson KM, Sher KJ. The natural history of problem gambling from age 18 to 29. J Abnorm Psychol 2003;112(2):263-74.
9. Goudriaan AE, Oosterlaan J, de Beurs E, Van den Brink W. Pathological gambling: a comprehensive review of biobehavioral findings. Neurosci Biobehav Rev 2004;28(2):123-41.
10. Johnson EE, Hamer RM, Nora RM. The Lie/Bet Questionnaire for screening pathological gamblers: a follow-up study. Psychol Rep 1998;83(3 Pt 2):1219-24.
11. Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-8.
12. Ursua MP, Uribelarrea LL. 20 questions of Gamblers Anonymous: a psychometric study with population of Spain. J Gambl Stud 1998;14:3-15.
13. Ladouceur R, Dube D. Gambling among primary school students in the Quebec Metropolitan area. J Gambl Stud 1994;10:363-70.
14. Shaffer HJ, Korn DA. Gambling and related mental disorders: a public health analysis. Annu Rev Public Health 2002;23:171-212.
15. Winters KC, Anderson N. Gambling involvement and drug use among adolescents. J Gambl Stud 2000;16(2-3):175-98.
16. Proimos J, DuRant RH, Pierce JD, Goodman E. Gambling and other risk behaviors among 8th- to 12th-grade students. Pediatrics 1998;102(2):e23.-
17. Gupta R, Derevensky JL. Adolescent gambling behavior: a prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;(4):319-45.
18. Kaminer Y, Burleson JA, Jadamec A. Gambling behavior in adolescent substance abuse. Subst Abus 2002;23(3):191-8.
19. Grant JE, Kim SW. Comorbidity of impulse control disorders in pathological gamblers. Acta Psychiatr Scand 2003;108(3):203-7.
20. Ladouceur R, Dube D, Bujold A. Prevalence of pathological gambling and related problems among college students in the Quebec metropolitan area. Can J Psychiatry 1994;39(5):289-93.
21. Ladouceur R, Boisvert JM, Dumont J. Cognitive-behavioral treatment for adolescent pathological gamblers. Behav Modif 1994;18(2):230-42.
22. Stewart RM, Brown RI. An outcome study of Gamblers Anonymous. Br J Psychiatry 1988;152:284-8.
23. Grant JE, Kim SW. Pharmacotherapy of pathological gambling. Psychiatr Ann 2002;32:186-91.
24. Carlton PL, Manowitz P, McBride H, et al. Attention deficit disorder and pathological gambling. J Clin Psychiatry 1987;48(12):487-8.
25. Langhinrichsen-Rohling J, Rohde P, Seeley JR, Rohling ML. Individual, family, and peer correlates of adolescent gambling. J Gambl Stud 2004;20(1):23-46.
Matt, age 17, grew up in a household of gamblers. He learned to play poker from his father at age 8 and bet on sports with his friends in the 5th grade. For 2 years he has been playing poker three to four nights per week and watching a lot of televised poker. His parents worry he might be “addicted” to gambling and bring him for evaluation.
Easy access to gambling through casinos, lotteries, and Internet games has affected many social groups, particularly adolescents.1 Teens such as Matt are more likely than adults to become pathological gamblers,2 and they often have psychiatric disorders and antisocial behavior patterns that interfere with normal development. This article:
- suggests screening tools to identify problem teen gambling
- discusses how to use psychotherapy, medications, and other options to help gambling-obsessed adolescents change their high-risk behavior.
Why adolescents gamble
Gambling activates the same neural reward pathways affected by cocaine and amphetamines.3 Even so, many adolescents view gambling as less harmful than drugs4 and consider it a rite of passage:
- 90% report having gambled for money
- 75% have gambled at home for money
- 85% of parents do not object to gambling behaviors, teens say.4,5
Adolescent gambling behavior. For approximately 85% of adolescents, gambling becomes no more than a social activity. For others, it can become problematic or even pathological (Table 1).5,7 Approximately 4 to 8% of adolescents meet criteria for pathological gambling, compared with 1% of adults.
Adolescents’ higher rate might reflect pathological gambling’s natural course—peaking during adolescence, then tapering during adulthood. Some adolescents may adapt their gambling behavior over time. Problem gambling tends to be more transient and episodic than pathological gambling, remitting when adolescents take on new responsibilities (such as with college, marriage, employment, or death of parents).8
Table 1
Adolescent gambling: From entertainment to mental illness
| Behavior | Prevalence in U.S. teens (%) | Definition |
|---|---|---|
| Social gambling | 80 to 85 | Gambling socially for a limited amount of time with predetermined, acceptable losses |
| Problem gambling | 10 to 14 | Gambling for recreation at the expense of other developmental activities; may interfere with time management, productivity, and relationships |
| Pathological gambling | 4 to 8 | Persistent and recurrent gambling that disrupts personal, family, or vocational pursuits |
| Source: References 3, 10. | ||
Case continued: ‘Up big-time’
Matt says he loves to gamble and believes he is more gifted at poker than his peers. He also recognizes his behavior could be addictive and admits lying to his parents about his gambling. He plays Internet poker 3 to 4 hours per day and claims he wins more often than he loses. “I’m up big time,” he boasts.
Matt denies going to casinos or using bookies. He says he is managing school and home responsibilities without difficulty. He denies mood or anxiety symptoms but admits using methylphenidate while gambling. He obtains the stimulant from friends at school and usually snorts it to get “a bigger rush.”
Matt is clearly at risk for problem gambling. He was exposed to gambling early, is becoming preoccupied with it, lies about how much he gambles, and combines gambling with substance abuse. progress more rapidly and can become problem gamblers within 12 to 14 months.2
Neurobiologic differences between adolescent and adult pathological gamblers are not well-defined. Adults show evidence of dysregulated dopamine, norepinephrine, and serotonin neuro-transmission.9 Neuroimaging in adult pathological gamblers shows perturbations in reward processing centers and frontal lobe structures that control inhibition. These factors have been examined little in adolescents.
Identifying problem gambling in teens
Diagnostic “red flags” for problem gambling in adolescents include declining school performance, sleep disturbance, generalized anxiety or irritability, or possibly lack of response to general psychiatric treatment. Three tools can be useful for screening adolescents:
The Lie-Bet Questionnaire10 is a 2-question screen for problem gambling:
- Have you ever lied to anyone important about how often you gamble?
- Have you ever had to increase your bet to get the same excitement from gambling?
The South-Oaks Gambling Screen11 is the standard pathological gambling screen for adults. Like the (SOGS-RA) is based and validated using DSM-III criteria (see Related resources). A score of 2 to 5 indicates at-risk gambling behaviors, and ≥6 indicate need for treatment.
The Gamblers Anonymous questionnaire12 comprises 20 questions that identify negative social, physical, and emotional consequences of gambling behaviors (Box). Seven or more positive responses indicate probable pathological gambling. This screen has shown reliability in adolescents.
Use one or more of these quick screens with every adolescent presenting for treatment—especially in substance abuse treatment settings. When results are positive, probe for gambling behaviors and consequences. Rely on DSM-IV-TR criteria and clinical presentation to differentiate social gambling from pathological gambling.
Most compulsive gamblers will answer “yes” to at least 7 of these questions:
- Did you ever lose time from work or school due to gambling?
- Has gambling ever made your home life unhappy?
- Did gambling affect your reputation?
- Have you ever felt remorse after gambling?
- Did you ever gamble to get money with which to pay debts or otherwise solve financial difficulties?
- Did gambling cause a decrease in your ambition or efficiency?
- After losing did you feel you must return as soon as possible and win back your losses?
- After a win did you have a strong urge to return and win more?
- Did you often gamble until your last dollar was gone?
- Did you ever borrow to finance your gambling?
- Have you ever sold anything to finance gambling?
- Were you reluctant to use “gambling money” for normal expenditures?
- Did gambling make you careless of the welfare of yourself or your family?
- Did you ever gamble longer than you had planned?
- Have you ever gambled to escape worry or trouble?
- Have you ever committed, or considered committing, an illegal act to finance
- Did gambling cause you to have difficulty in sleeping?
- Do arguments, disappointments, or frustrations create an urge to gamble?
- Did you ever have an urge to celebrate any good fortune by a few hours of gambling?
- Have you ever considered self-destruction or suicide as a result of your gambling?
Behaviors and comorbidities
Negative consequences. Pathological gambling often consumes 10 to 20 hours per week of the adolescent’s time,13 hurting school performance and delaying developmental milestones. Teen gamblers may abandon extracurricular school activities, and their few friends often gamble, too. They are at risk for delinquency, criminal activity, and antisocial behaviors (such as selling drugs, engaging in prostitution)14 unprotected sexual activity, drug use, reckless driving, and carrying weapons.15,16
Psychiatric comorbidity is the rule and often what brings adolescent gamblers to treatment. Substance abuse, major depression, attention-deficit/hyperactivity disorder, and personality disorders are most common (Table 2) Adolescent substance abuse at least triples the risk of pathological gambling.18
Adolescent pathological gamblers have increased rates of suicidal ideation and suicide attempts.17 They are at risk for other impulsive behaviors as well,19 although they are unlikely to volunteer this information. The Minnesota Impulsive Disorders Interview help identify comorbid pathological gambling, trichotillomania, kleptomania, pyromania, intermittent explosive disorder, compulsive buying, and compulsive sexual behaviors.19 Screen for these comorbidities during the first sessions or if a patient does not respond to treatment of gambling behavior.
Table 2
Risk factors for pathological adolescent gambling
| Component | Risk factors |
|---|---|
| Family history | Family history of gambling problems; possible genetic influences (neurotransmitter activity, risk-taking behaviors, risk perception, heightened physical response to rewards) |
| Psychiatric comorbidity | Substance abuse, mood/anxiety disorders, ADHD |
| Personality traits | Low self-esteem, competitiveness, sensitivity to stress or rejection, peer influences, immaturity, suicidal tendencies |
| Social factors | Early-age exposure to gambling, having peers who gamble regularly, increased access to gambling (such as via the Internet), chaotic home environment (divorce, neglect, abuse) |
| ADHD: attention-deficit/hyperactivity disorder | |
Treating adolescent gamblers
Matt’s answers to the gambling screening questionnaires (one “yes” to the Lie-Bet Questionnaire, a SOGS-RA score of 4, and a GA-20 Questions score of 5) indicate problem gambling but not pathological gambling. You recommend that he attend Gamblers Anonymous, but he refuses. He also rejects individual therapy or taking medications.
Matt acknowledges misusing methylphenidate, however, and agrees to consider an outpatient substance abuse program. He also agrees to six sessions with a gambling treatment specialist to learn about problem gambling’s signs and symptoms, how to cope with betting loses, and how to reduce his preoccupation with gambling.
No guidelines exist for treating adolescent pathological gamblers, and specialized teen treatment programs are rare. Most services are provided in mental health or substance abuse settings, using adult treatments modified for adolescents.
Cognitive behavioral therapy (CBT) can be successful for highly motivated gamblers, although adolescents might not want to change their pathological behaviors. In case reports, four adolescents achieved remission after 6 months of CBT.20,21 CBT appears to have long-term benefits for adults, but this has not been evaluated in teens. Even so, individual CBT may be ideal for adolescent gamblers because side effects are minimal.
Gamblers Anonymous (GA) in adults has shown a low retention rate and a 1-year abstinence rate of 22 Its effectiveness for adolescents lacks empiric support, but the 12-step program’s availability, structure, and fellowship may be useful.
Medications. Consider medication as first-line treatment for adolescents with psychiatric comorbidity. Try psychosocial treatment first for those without psychiatric comorbidity; consider medication as second-line therapy if response to psychosocial treatment is inadequate.
No medications are FDA-approved for pathological gambling, and no studies have examined medication use for adolescent gamblers. Controlled trials with adults suggest some medications may reduce urges and cravings or decrease gambling behaviors. These include:
- selective serotonin reuptake inhibitors
- opioid antagonists such as naltrexone or its analogue nalmefene
- or mood stabilizers such as lithium, divalproex sodium, or possibly topiramate.23
Working with families. Many parents are aware of the destructive potential of substance abuse but not of gambling. They may feel shame that they did not recognize their teen’s gambling problem or “control” their child’s behavior. The adolescent may feel remorse for having bet with the parents’ money.
Even when the family recognizes the teen’s problem, denial or enabling can perpetuate the behavior. For teens such as Matt who learned to gamble at a home, advise the parents to abstain from gambling as well. Consider screening the parents for pathological gambling, given its high rate of heritability.
Address guilt and shame by acknowledging that pathological gambling is a psychiatric disease caused by biological, psychological, and social factors—not dysfunctional family relations. Emphasize that treatment works. Because gambling is easily hidden, educate families about relapse signs, such as preoccupation with money, personality changes, or failing to fulfill family responsibilities.25
- Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-88.
- Gamblers Anonymous. www.gamblersanonymous.org.
- National Council on Problem Gambling. www.ncpgambling.org.
- International Centre for Youth Gambling Problems and High-Risk Behaviors. www.youthgambling.com.
- Nalmefene • Revex
- Naltrexone • ReVia
- Lithium • Lithobid, others
- Divalproex sodium • Depakote, others
- Topiramate • Topamax
Dr. Fong receives grant/research support from Ortho McNeil Pharmaceutical and Somaxon Pharmaceuticals and is a speaker for Forest Pharmaceuticals.
Matt, age 17, grew up in a household of gamblers. He learned to play poker from his father at age 8 and bet on sports with his friends in the 5th grade. For 2 years he has been playing poker three to four nights per week and watching a lot of televised poker. His parents worry he might be “addicted” to gambling and bring him for evaluation.
Easy access to gambling through casinos, lotteries, and Internet games has affected many social groups, particularly adolescents.1 Teens such as Matt are more likely than adults to become pathological gamblers,2 and they often have psychiatric disorders and antisocial behavior patterns that interfere with normal development. This article:
- suggests screening tools to identify problem teen gambling
- discusses how to use psychotherapy, medications, and other options to help gambling-obsessed adolescents change their high-risk behavior.
Why adolescents gamble
Gambling activates the same neural reward pathways affected by cocaine and amphetamines.3 Even so, many adolescents view gambling as less harmful than drugs4 and consider it a rite of passage:
- 90% report having gambled for money
- 75% have gambled at home for money
- 85% of parents do not object to gambling behaviors, teens say.4,5
Adolescent gambling behavior. For approximately 85% of adolescents, gambling becomes no more than a social activity. For others, it can become problematic or even pathological (Table 1).5,7 Approximately 4 to 8% of adolescents meet criteria for pathological gambling, compared with 1% of adults.
Adolescents’ higher rate might reflect pathological gambling’s natural course—peaking during adolescence, then tapering during adulthood. Some adolescents may adapt their gambling behavior over time. Problem gambling tends to be more transient and episodic than pathological gambling, remitting when adolescents take on new responsibilities (such as with college, marriage, employment, or death of parents).8
Table 1
Adolescent gambling: From entertainment to mental illness
| Behavior | Prevalence in U.S. teens (%) | Definition |
|---|---|---|
| Social gambling | 80 to 85 | Gambling socially for a limited amount of time with predetermined, acceptable losses |
| Problem gambling | 10 to 14 | Gambling for recreation at the expense of other developmental activities; may interfere with time management, productivity, and relationships |
| Pathological gambling | 4 to 8 | Persistent and recurrent gambling that disrupts personal, family, or vocational pursuits |
| Source: References 3, 10. | ||
Case continued: ‘Up big-time’
Matt says he loves to gamble and believes he is more gifted at poker than his peers. He also recognizes his behavior could be addictive and admits lying to his parents about his gambling. He plays Internet poker 3 to 4 hours per day and claims he wins more often than he loses. “I’m up big time,” he boasts.
Matt denies going to casinos or using bookies. He says he is managing school and home responsibilities without difficulty. He denies mood or anxiety symptoms but admits using methylphenidate while gambling. He obtains the stimulant from friends at school and usually snorts it to get “a bigger rush.”
Matt is clearly at risk for problem gambling. He was exposed to gambling early, is becoming preoccupied with it, lies about how much he gambles, and combines gambling with substance abuse. progress more rapidly and can become problem gamblers within 12 to 14 months.2
Neurobiologic differences between adolescent and adult pathological gamblers are not well-defined. Adults show evidence of dysregulated dopamine, norepinephrine, and serotonin neuro-transmission.9 Neuroimaging in adult pathological gamblers shows perturbations in reward processing centers and frontal lobe structures that control inhibition. These factors have been examined little in adolescents.
Identifying problem gambling in teens
Diagnostic “red flags” for problem gambling in adolescents include declining school performance, sleep disturbance, generalized anxiety or irritability, or possibly lack of response to general psychiatric treatment. Three tools can be useful for screening adolescents:
The Lie-Bet Questionnaire10 is a 2-question screen for problem gambling:
- Have you ever lied to anyone important about how often you gamble?
- Have you ever had to increase your bet to get the same excitement from gambling?
The South-Oaks Gambling Screen11 is the standard pathological gambling screen for adults. Like the (SOGS-RA) is based and validated using DSM-III criteria (see Related resources). A score of 2 to 5 indicates at-risk gambling behaviors, and ≥6 indicate need for treatment.
The Gamblers Anonymous questionnaire12 comprises 20 questions that identify negative social, physical, and emotional consequences of gambling behaviors (Box). Seven or more positive responses indicate probable pathological gambling. This screen has shown reliability in adolescents.
Use one or more of these quick screens with every adolescent presenting for treatment—especially in substance abuse treatment settings. When results are positive, probe for gambling behaviors and consequences. Rely on DSM-IV-TR criteria and clinical presentation to differentiate social gambling from pathological gambling.
Most compulsive gamblers will answer “yes” to at least 7 of these questions:
- Did you ever lose time from work or school due to gambling?
- Has gambling ever made your home life unhappy?
- Did gambling affect your reputation?
- Have you ever felt remorse after gambling?
- Did you ever gamble to get money with which to pay debts or otherwise solve financial difficulties?
- Did gambling cause a decrease in your ambition or efficiency?
- After losing did you feel you must return as soon as possible and win back your losses?
- After a win did you have a strong urge to return and win more?
- Did you often gamble until your last dollar was gone?
- Did you ever borrow to finance your gambling?
- Have you ever sold anything to finance gambling?
- Were you reluctant to use “gambling money” for normal expenditures?
- Did gambling make you careless of the welfare of yourself or your family?
- Did you ever gamble longer than you had planned?
- Have you ever gambled to escape worry or trouble?
- Have you ever committed, or considered committing, an illegal act to finance
- Did gambling cause you to have difficulty in sleeping?
- Do arguments, disappointments, or frustrations create an urge to gamble?
- Did you ever have an urge to celebrate any good fortune by a few hours of gambling?
- Have you ever considered self-destruction or suicide as a result of your gambling?
Behaviors and comorbidities
Negative consequences. Pathological gambling often consumes 10 to 20 hours per week of the adolescent’s time,13 hurting school performance and delaying developmental milestones. Teen gamblers may abandon extracurricular school activities, and their few friends often gamble, too. They are at risk for delinquency, criminal activity, and antisocial behaviors (such as selling drugs, engaging in prostitution)14 unprotected sexual activity, drug use, reckless driving, and carrying weapons.15,16
Psychiatric comorbidity is the rule and often what brings adolescent gamblers to treatment. Substance abuse, major depression, attention-deficit/hyperactivity disorder, and personality disorders are most common (Table 2) Adolescent substance abuse at least triples the risk of pathological gambling.18
Adolescent pathological gamblers have increased rates of suicidal ideation and suicide attempts.17 They are at risk for other impulsive behaviors as well,19 although they are unlikely to volunteer this information. The Minnesota Impulsive Disorders Interview help identify comorbid pathological gambling, trichotillomania, kleptomania, pyromania, intermittent explosive disorder, compulsive buying, and compulsive sexual behaviors.19 Screen for these comorbidities during the first sessions or if a patient does not respond to treatment of gambling behavior.
Table 2
Risk factors for pathological adolescent gambling
| Component | Risk factors |
|---|---|
| Family history | Family history of gambling problems; possible genetic influences (neurotransmitter activity, risk-taking behaviors, risk perception, heightened physical response to rewards) |
| Psychiatric comorbidity | Substance abuse, mood/anxiety disorders, ADHD |
| Personality traits | Low self-esteem, competitiveness, sensitivity to stress or rejection, peer influences, immaturity, suicidal tendencies |
| Social factors | Early-age exposure to gambling, having peers who gamble regularly, increased access to gambling (such as via the Internet), chaotic home environment (divorce, neglect, abuse) |
| ADHD: attention-deficit/hyperactivity disorder | |
Treating adolescent gamblers
Matt’s answers to the gambling screening questionnaires (one “yes” to the Lie-Bet Questionnaire, a SOGS-RA score of 4, and a GA-20 Questions score of 5) indicate problem gambling but not pathological gambling. You recommend that he attend Gamblers Anonymous, but he refuses. He also rejects individual therapy or taking medications.
Matt acknowledges misusing methylphenidate, however, and agrees to consider an outpatient substance abuse program. He also agrees to six sessions with a gambling treatment specialist to learn about problem gambling’s signs and symptoms, how to cope with betting loses, and how to reduce his preoccupation with gambling.
No guidelines exist for treating adolescent pathological gamblers, and specialized teen treatment programs are rare. Most services are provided in mental health or substance abuse settings, using adult treatments modified for adolescents.
Cognitive behavioral therapy (CBT) can be successful for highly motivated gamblers, although adolescents might not want to change their pathological behaviors. In case reports, four adolescents achieved remission after 6 months of CBT.20,21 CBT appears to have long-term benefits for adults, but this has not been evaluated in teens. Even so, individual CBT may be ideal for adolescent gamblers because side effects are minimal.
Gamblers Anonymous (GA) in adults has shown a low retention rate and a 1-year abstinence rate of 22 Its effectiveness for adolescents lacks empiric support, but the 12-step program’s availability, structure, and fellowship may be useful.
Medications. Consider medication as first-line treatment for adolescents with psychiatric comorbidity. Try psychosocial treatment first for those without psychiatric comorbidity; consider medication as second-line therapy if response to psychosocial treatment is inadequate.
No medications are FDA-approved for pathological gambling, and no studies have examined medication use for adolescent gamblers. Controlled trials with adults suggest some medications may reduce urges and cravings or decrease gambling behaviors. These include:
- selective serotonin reuptake inhibitors
- opioid antagonists such as naltrexone or its analogue nalmefene
- or mood stabilizers such as lithium, divalproex sodium, or possibly topiramate.23
Working with families. Many parents are aware of the destructive potential of substance abuse but not of gambling. They may feel shame that they did not recognize their teen’s gambling problem or “control” their child’s behavior. The adolescent may feel remorse for having bet with the parents’ money.
Even when the family recognizes the teen’s problem, denial or enabling can perpetuate the behavior. For teens such as Matt who learned to gamble at a home, advise the parents to abstain from gambling as well. Consider screening the parents for pathological gambling, given its high rate of heritability.
Address guilt and shame by acknowledging that pathological gambling is a psychiatric disease caused by biological, psychological, and social factors—not dysfunctional family relations. Emphasize that treatment works. Because gambling is easily hidden, educate families about relapse signs, such as preoccupation with money, personality changes, or failing to fulfill family responsibilities.25
- Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-88.
- Gamblers Anonymous. www.gamblersanonymous.org.
- National Council on Problem Gambling. www.ncpgambling.org.
- International Centre for Youth Gambling Problems and High-Risk Behaviors. www.youthgambling.com.
- Nalmefene • Revex
- Naltrexone • ReVia
- Lithium • Lithobid, others
- Divalproex sodium • Depakote, others
- Topiramate • Topamax
Dr. Fong receives grant/research support from Ortho McNeil Pharmaceutical and Somaxon Pharmaceuticals and is a speaker for Forest Pharmaceuticals.
1. Gupta R, Derevensky JL. Adolescents with gambling problems: from research to treatment. J Gambl Stud 2000;16(2-3):315-42.
2. Evans RI. Some theoretical models and constructs generic to substance abuse prevention programs for adolescents: possible relevance and limitations for problem gambling. J Gambl Stud 2003;19(3):287-302.
3. Crockford DN, Goodyear B, Edwards J, et al. Cue-induced brain activity in pathological gamblers. Biol Psychiatry 2005;58(10):787-95.
4. Gupta R, Derevensky JL. Adolescent gambling behavior: A prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;14(4):319-45.
5. Derevensky JL, Gupta R, Winters K. Prevalence rates of youth gambling problems: are the current rates inflated? J Gambl Stud 2003;19(4):405-25.
6. Griffiths M. Adolescent gambling. London: Routledge; 1995.
7. Pietrzak RH, Ladd GT, Petry NM. Disordered gambling in adolescents: epidemiology, diagnosis, and treatment. Paediatr Drugs 2003;5(9):583-95.
8. Slutske WS, Jackson KM, Sher KJ. The natural history of problem gambling from age 18 to 29. J Abnorm Psychol 2003;112(2):263-74.
9. Goudriaan AE, Oosterlaan J, de Beurs E, Van den Brink W. Pathological gambling: a comprehensive review of biobehavioral findings. Neurosci Biobehav Rev 2004;28(2):123-41.
10. Johnson EE, Hamer RM, Nora RM. The Lie/Bet Questionnaire for screening pathological gamblers: a follow-up study. Psychol Rep 1998;83(3 Pt 2):1219-24.
11. Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-8.
12. Ursua MP, Uribelarrea LL. 20 questions of Gamblers Anonymous: a psychometric study with population of Spain. J Gambl Stud 1998;14:3-15.
13. Ladouceur R, Dube D. Gambling among primary school students in the Quebec Metropolitan area. J Gambl Stud 1994;10:363-70.
14. Shaffer HJ, Korn DA. Gambling and related mental disorders: a public health analysis. Annu Rev Public Health 2002;23:171-212.
15. Winters KC, Anderson N. Gambling involvement and drug use among adolescents. J Gambl Stud 2000;16(2-3):175-98.
16. Proimos J, DuRant RH, Pierce JD, Goodman E. Gambling and other risk behaviors among 8th- to 12th-grade students. Pediatrics 1998;102(2):e23.-
17. Gupta R, Derevensky JL. Adolescent gambling behavior: a prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;(4):319-45.
18. Kaminer Y, Burleson JA, Jadamec A. Gambling behavior in adolescent substance abuse. Subst Abus 2002;23(3):191-8.
19. Grant JE, Kim SW. Comorbidity of impulse control disorders in pathological gamblers. Acta Psychiatr Scand 2003;108(3):203-7.
20. Ladouceur R, Dube D, Bujold A. Prevalence of pathological gambling and related problems among college students in the Quebec metropolitan area. Can J Psychiatry 1994;39(5):289-93.
21. Ladouceur R, Boisvert JM, Dumont J. Cognitive-behavioral treatment for adolescent pathological gamblers. Behav Modif 1994;18(2):230-42.
22. Stewart RM, Brown RI. An outcome study of Gamblers Anonymous. Br J Psychiatry 1988;152:284-8.
23. Grant JE, Kim SW. Pharmacotherapy of pathological gambling. Psychiatr Ann 2002;32:186-91.
24. Carlton PL, Manowitz P, McBride H, et al. Attention deficit disorder and pathological gambling. J Clin Psychiatry 1987;48(12):487-8.
25. Langhinrichsen-Rohling J, Rohde P, Seeley JR, Rohling ML. Individual, family, and peer correlates of adolescent gambling. J Gambl Stud 2004;20(1):23-46.
1. Gupta R, Derevensky JL. Adolescents with gambling problems: from research to treatment. J Gambl Stud 2000;16(2-3):315-42.
2. Evans RI. Some theoretical models and constructs generic to substance abuse prevention programs for adolescents: possible relevance and limitations for problem gambling. J Gambl Stud 2003;19(3):287-302.
3. Crockford DN, Goodyear B, Edwards J, et al. Cue-induced brain activity in pathological gamblers. Biol Psychiatry 2005;58(10):787-95.
4. Gupta R, Derevensky JL. Adolescent gambling behavior: A prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;14(4):319-45.
5. Derevensky JL, Gupta R, Winters K. Prevalence rates of youth gambling problems: are the current rates inflated? J Gambl Stud 2003;19(4):405-25.
6. Griffiths M. Adolescent gambling. London: Routledge; 1995.
7. Pietrzak RH, Ladd GT, Petry NM. Disordered gambling in adolescents: epidemiology, diagnosis, and treatment. Paediatr Drugs 2003;5(9):583-95.
8. Slutske WS, Jackson KM, Sher KJ. The natural history of problem gambling from age 18 to 29. J Abnorm Psychol 2003;112(2):263-74.
9. Goudriaan AE, Oosterlaan J, de Beurs E, Van den Brink W. Pathological gambling: a comprehensive review of biobehavioral findings. Neurosci Biobehav Rev 2004;28(2):123-41.
10. Johnson EE, Hamer RM, Nora RM. The Lie/Bet Questionnaire for screening pathological gamblers: a follow-up study. Psychol Rep 1998;83(3 Pt 2):1219-24.
11. Wiebe JM, Cox BJ, Mehmel BG. The South Oaks Gambling Screen Revised for Adolescents (SOGS-RA): further psychometric findings from a community sample. J Gambl Stud 2000;16(2-3):275-8.
12. Ursua MP, Uribelarrea LL. 20 questions of Gamblers Anonymous: a psychometric study with population of Spain. J Gambl Stud 1998;14:3-15.
13. Ladouceur R, Dube D. Gambling among primary school students in the Quebec Metropolitan area. J Gambl Stud 1994;10:363-70.
14. Shaffer HJ, Korn DA. Gambling and related mental disorders: a public health analysis. Annu Rev Public Health 2002;23:171-212.
15. Winters KC, Anderson N. Gambling involvement and drug use among adolescents. J Gambl Stud 2000;16(2-3):175-98.
16. Proimos J, DuRant RH, Pierce JD, Goodman E. Gambling and other risk behaviors among 8th- to 12th-grade students. Pediatrics 1998;102(2):e23.-
17. Gupta R, Derevensky JL. Adolescent gambling behavior: a prevalence study and examination of the correlates associated with problem gambling. J Gambl Stud 1998;(4):319-45.
18. Kaminer Y, Burleson JA, Jadamec A. Gambling behavior in adolescent substance abuse. Subst Abus 2002;23(3):191-8.
19. Grant JE, Kim SW. Comorbidity of impulse control disorders in pathological gamblers. Acta Psychiatr Scand 2003;108(3):203-7.
20. Ladouceur R, Dube D, Bujold A. Prevalence of pathological gambling and related problems among college students in the Quebec metropolitan area. Can J Psychiatry 1994;39(5):289-93.
21. Ladouceur R, Boisvert JM, Dumont J. Cognitive-behavioral treatment for adolescent pathological gamblers. Behav Modif 1994;18(2):230-42.
22. Stewart RM, Brown RI. An outcome study of Gamblers Anonymous. Br J Psychiatry 1988;152:284-8.
23. Grant JE, Kim SW. Pharmacotherapy of pathological gambling. Psychiatr Ann 2002;32:186-91.
24. Carlton PL, Manowitz P, McBride H, et al. Attention deficit disorder and pathological gambling. J Clin Psychiatry 1987;48(12):487-8.
25. Langhinrichsen-Rohling J, Rohde P, Seeley JR, Rohling ML. Individual, family, and peer correlates of adolescent gambling. J Gambl Stud 2004;20(1):23-46.
Corticosteroid-induced mania: Prepare for the unpredictable
Can corticosteroids “unlock” hidden potential for mania, or are steroid-induced mood symptoms a temporary reaction? And when these mood symptoms occur, what is the best way to treat them?
Psychiatric symptoms develop in 5% to 18% of patients treated with corticosteroids. These effects—most often mania or depression—emerge within days to weeks of starting steroids. To help you head off manic and mixed mood symptoms, this paper examines how to:
- treat steroid-induced mania or mixed bipolar symptoms
- reduce the risk of a mood episode in patients who require sustained corticosteroid therapy.
‘Steroid psychosis’
Jane Pauley, NBC’s Today Show broadcaster, described in her autobiography how hypomania developed within weeks after she started corticosteroids for idiopathic urticaria edema: “I was so energized that I didn’t just walk down the hall, I felt like I was motoring down the hall. I was suddenly the equal of my high-energy friends who move fast and talk fast and loud. I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”1
Pauley’s hypomania led to a manic episode and eventually to depression. She was started on antidepressants, which triggered another manic episode. Pauley—who had no history of bipolar disorder—spent 3 weeks in a New York psychiatric hospital.1
Diagnostic symptoms. Corticosteroids’ psychiatric effects—cognitive, mood, anxiety, and psychotic symptoms—were first described as “steroid psychosis.” Psychosis can occur, but mood symptoms are more common:
- Among 122 patients, 40% experienced depression, followed by mania (28%), psychosis (14%), delirium (10%), and mixed mood episodes (8%).2
- Among 130 patients, mania was most prevalent (35%), followed by depression (28%), mixed mood episodes (12%), delirium (13%), and psychosis (11%).3
- Corticosteroids caused 54% of organic mania cases on a hospital psychiatric consult service.4
- In a prospective study of 50 patients treated with corticosteroids, 13 developed hypomania and 5 developed depression.5
Steroid-induced symptoms emerge from 3 to 4 days to a median of 11 days after a patient starts corticosteroid therapy. After steroids are discontinued, depressive symptoms persist approximately 4 weeks, mania 3 weeks, and delirium a few days. Approximately one-half of patients with steroid psychosis improve in 4 days and one-half within 2 weeks.2,6
Continue to: Who is at risk?
Who is at risk?
Corticosteroids include the steroids produced in the adrenal gland (such as corticosterone) and their synthetic—and often more potent—analogues (such as prednisone).7 Because of their glucocorticoid, immunosuppressant, mineralocorticoid, and anti-inflammatory properties, steroids are used as replacement therapy and to treat a wide variety of illnesses (Table 1).
Table 1
Medical conditions for which corticosteroids are commonly used
| Disorder | Indications for corticosteroids |
|---|---|
| Acute adrenal insufficiency | Acute; replacement therapy |
| Addison’s disease | Chronic; replacement therapy |
| Asthma | Acute and chronic; anti-inflammatory |
| Inflammatory bowel disease | Acute; anti-inflammatory |
| Multiple sclerosis | Acute; exacerbations, immunosuppressant |
| Organ transplant | Chronic; immunosuppressant |
| Rheumatoid arthritis | Chronic; anti-inflammatory |
| Systemic lupus erythematosus | Acute; severe exacerbation, immunosuppressant (high doses are used) |
Age and gender. Patient age appears unrelated to development of psychiatric symptoms after corticosteroid use.2 One study suggested women are twice as likely as men to develop psychiatric symptoms (77 versus 38 cases in 115 patients),3 but many illnesses that require corticosteroid treatment occur more frequently in women. Other researchers found a slight female predominance (58% versus 42% of cases) when they excluded patients with systemic lupus erythematosus and rheumatoid arthritis, which are more common in women than in men.2
Dosage. Higher corticosteroid dosages increase the risk of psychiatric symptoms. In patients taking prednisone, the Boston Collaborative Drug Surveillance Project8 found the incidence of psychiatric side effects to be:
- 1.3% in patients taking
- 4.6% in those taking 41 to 80 mg
- 18.4% in those taking >80 mg.
Psychiatric history. Past psychiatric illness does not seem to be a risk factor for psychiatric side effects of corticosteroids,9 although patients with a history of posttraumatic stress disorder are more likely to suffer depression while taking corticosteroids.10
Corticosteroid exposure. Patients who did not experience psychiatric side effects with corticosteroids in the past appear not to be protected if corticosteroids are used again. One report examined 17 cases of steroid-induced psychiatric illness in patients with previous exposure to corticosteroid therapy. Six patients had previous psychiatric side effects while taking corticosteroids, and 11 did not.2
Bipolar trigger?
Do corticosteroids’ acute psychiatric side effects have long-term sequelae? Longitudinal evidence is scarce, but a few reports suggest corticosteroids could play a role in the onset of primary bipolar I disorder:
- A 28-year-old woman with no known mood symptoms before a short course of prednisone experienced six episodes of mania and depression when not taking corticosteroids during the subsequent 18 months.11
- Among 16 patients with first-onset mood symptoms after corticosteroid use, a retrospective chart review found 7 had recurrent manic and depressive symptoms unrelated to additional corticosteroid use.12
Continue to: ... case reports are inconclusive...
Although intriguing, these case reports are inconclusive. Because bipolar type I incidence in the general population is 1.5%,13 many persons with bipolar disorder undergo corticosteroid treatment. Nevertheless, these results—especially from the retrospective review12—suggest that corticosteroid use may contribute to the onset of bipolar I illness.
Symptomatic treatment
Corticosteroid-induced side effects are usually managed by tapering off the steroids and treating the psychiatric symptoms.2,3 Simply tapering off the steroids—without additional treatments—led to recovery in 33 of 36 patients.2 Stopping corticosteroids is not always possible or desirable, however, especially in many medically complicated cases seen by psychiatric consult services.
In a recent case, I was asked to see a man, age 69, on the oncology service who was receiving corticosteroids every 2 weeks as part of his chemotherapy. The patient was admitted to the hospital for acute mental status changes 2 days after his last corticosteroid dose. He had pressured speech, grandiosity, and had not slept in 2 days. We started risperidone, 1 mg bid, and most of his manic symptoms resolved within 2 days. His chemotherapy was continued without corticosteroids. If this had not been not possible, I would have recommended continuing risperidone prophylactically.
No double-blind, placebo-controlled studies have examined prevention or treatment of steroid-induced mania or other psychiatric symptoms. Uncontrolled trials and case reports suggest benefit from some symptomatic and preventive treatments (Table 2).
Table 2
Mood stabilizers with evidence of benefit in treating corticosteroid-induced mania
| Indication | Medication | Dosage/blood level | Evidence |
|---|---|---|---|
| Preventing psychiatric effects in patients requiring long-term corticosteroids | Lithium | 0.8 to 1.2 mEq/L | Prospective trial (27 with multiple sclerosis)24 |
| Preventing recurrence of manic symptoms in patients requiring additional steroid pulses | Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case report16 |
| Gabapentin | 300 mg tid | Case report26 | |
| Treating steroid-induced manic symptoms | Olanzapine | Initially 2.5 mg/d, titrated to 20 mg/d | Open-label trial (12 patients)14 |
| Lithium | 0.7 mEq/L | Case report15 | |
| Quetiapine | 25 mg qhs and 12.5 mg bid prn | Case report17 | |
| Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case reports12,16 | |
| Haloperidol | 2 to 20 mg/d* | Case reports12,16 | |
| Treating steroid-induced depressive symptoms | Fluoxetine | 20 mg/d | Case report18 |
| Amitriptyline | 30 mg/d (usual effective range is 50 to 300 mg/d)* | Case report12 | |
| Lamotrigine | Up to 400 mg/d | Case report19 | |
| Lithium | 0.1 to 0.8 mEq/L | Case reports20,21 | |
| Treating steroid-induced psychotic symptoms | Haloperidol | 5 mg IV on day 1, then 2 mg po bid | Case report22 |
| Risperidone | 1.5 mg/d | Case report23 | |
| *Dosage not included in published report; recommendation based on experience or anecdotal information | |||
Treating manic and mixed mood symptoms. Twelve outpatients with manic or mixed symptoms from corticosteroid use received olanzapine in a 5-week, open-label trial. Flexible dosing started at 2.5 mg/d and was increased as needed (maximum 20 mg/d). One patient dropped out for lack of efficacy. For the others, manic and mixed symptoms improved significantly, as indicated by scores on the Young Mania Rating Scale, Hamilton Rating Scale for Depression, and Brief Psychotic Rating Scale.14 Patient weight, blood glucose, and involuntary movements did not change significantly.
Evidence from case reports indicates that lithium,15 carbamazepine,12,16 haloperidol,12,16 or quetiapine17 also can successfully treat steroid-induced manic symptoms.
Continue to: Treating other psychiatric symptoms
Treating other psychiatric symptoms. Case reports support electroconvulsive therapy,2,15 fluoxetine,18 amitriptyline,12 lamotrigine,19 or lithium20,21 for steroid-induced depression, and haloperidol22 or risperidone23 for steroid-induced psychosis.
In four cases,6 tricyclic antidepressants appeared to worsen corticosteroids’ psychiatric side effects. These case patients might have had steroid-induced delirium instead of mood disorders or psychosis, however, and the tricyclics’ anticholinergic effects could have worsened the delirium.9
Preventing steroid-induced symptoms
Although clear guidelines on when to start preventive treatments do not exist, potential candidates for pretreatment with lithium or other agents include patients who:
- have developed psychiatric symptoms multiple times after repeated corticosteroid use
- are at high risk if psychiatric side effects occur.
Lithium. Prophylactic lithium was given to 27 patients with multiple sclerosis and taking corticosteroids for acute exacerbations. None developed psychiatric symptoms.24 At the same clinic, 6 of 44 patients with multiple sclerosis or retrobulbar neuritis developed psychiatric side effects after using corticosteroids without lithium.
Be cautious when using prophylactic lithium because some conditions treated with corticosteroids—such as systemic lupus erythematosus—can impair renal function.20 Corticosteroids also can affect sodium balance and increase the risk of lithium intoxication.25
Check renal function before and during lithium titration, and initiate corticosteroid therapy when lithium is at effective blood levels (0.8 to 1.2 mEq/L). Monitor lithium levels and renal function frequently during steroid treatment.
Other mood stabilizers. Two case reports describe patients who repeatedly developed manic symptoms after multiple corticosteroid doses. Carbamazepine, 600 mg qd,16 and gabapentin, 300 mg tid,26 prevented manic symptoms after additional corticosteroid pulses.
Related resources
- Brown ES, Chandler PA. Mood and cognitive changes during systemic corticosteroid therapy. Prim Care Companion J Clin Psychiatry 2001;3(1):17-21. www.psychiatrist.com/pcc/abstracts/pcc030103.htm.
- Merrill W. Case 35-1998: use of lithium to prevent corticosteroid-induced mania. N Engl J Med 1999;340:1123.
Drug brand names
- Amitriptyline • Elavil
- Carbamazepine • Tegretol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
1. Pauley J. Skywriting: a life out of the blue. New York: Random House; 2004.
2. Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord 1983;5:319-32.
3. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry 2003;25:27-33.
4. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci 1989;1:398-400.
5. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days’ corticosteroid treatment. A prospective study. Psychoneuroendocrinology 1996;21:25-31.
6. Hall R, Popkin M, Stickney S, Gardner E. Presentation of the steroid psychoses. J Nerv Ment Dis 1979;167:229-36.
7. Schimmer B, Parker K. Adenohypophyseal hormones and their hypothalamic releasing factors. In: Hardman J, Limbird L, Gilman A (eds). Goodman and Gilman’s the pharmacological basis of therapeutics, 9th ed. New York: McGraw-Hill;1996:1459-86.
8. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther 1972;13:694-8.
9. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf 2000;22:111-22.
10. Brown ES, Suppes T, Khan DA, Carmody TJ, 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol 2002;22:55-61.
11. Pies R. Persistent bipolar illness after steroid administration. Arch Intern Med 1981;141:1087.-
12. Wada K, Yamada N, Suzuki K, et al. Recurrent cases of corticosteroid-induced mood disorder: a clinical characteristics and treatment. J Clin Psychiatry 2000;61:261-7.
13. Narrow WE, Rae DS, Robins LN, Regier DA. Revised prevalence estimates of mental disorders in the United States: using a clinical significance criterion to reconcile 2 surveys’ estimates. Arch Gen Psychiatry 2002;59:115-23.
14. Brown ES, Chamberlain W, Dhanani N, et al. An open-label trial of olanzapine for corticosteroid-induced mood symptoms. J Affect Disord 2004;83:277-81.
15. Blazer DG, 2nd, Petrie WM, Wilson WP. Affective psychosis following renal transplant. Dis Nerv Syst 1976;37:663-7.
16. Wada K, Yamada N, Yamauchi Y, Kuroda S. Carbamazepine treatment of corticosteroid-induced mood disorder. J Affect Disord 2001;65:315-7.
17. Siddiqui Z, Ramaswamy S, Petty F. Quetiapine therapy for corticosteroid-induced mania. Can J Psychiatry 2005;50:77-8.
18. Wyszynski AA, Wyszynski B. Treatment of depression with fluoxetine in corticosteroid-dependent central nervous system Sjogren’s syndrome. Psychosomatics 1993;34:173-7.
19. Brown ES, Frol A, Bobadilla L, et al. Effect of lamotrigine on mood and cognition in patients receiving chronic exogenous corticosteroids. Psychosomatics 2003;44:204-8.
20. Terao T, Mizuki T, Ohji T, Abe K. Antidepressant effect of lithium in patients with systemic lupus erythematosus and cerebral infarction, treated with corticosteroid. Br J Psychiatry 1994;164:109-11.
21. Terao T, Yoshimura R, Shiratuchi T, Abe K. Effects of lithium on steroid-induced depression. Biol Psychiatry 1997;41:1225-6.
22. Ahmad M, Rasul FM. Steroid-induced psychosis treated with haloperidol in a patient with active chronic obstructive pulmonary disease [letter]. Am J Emerg Med 1999;17:735.-
23. DeSilva CC, Nurse MC, Vokey K. Steroid-induced psychosis treated with risperidone. Can J Psychiatry 2002;47:388-9.
24. Falk WE, Mahnke MW, Poskanzer DC. Lithium prophylaxis of corticotropin-induced psychosis. JAMA 1979;241:1011-2.
25. Saklad SR. Management of corticosteroid-induced psychosis with lithium. Clin Pharm 1987;6:186.-
26. Ginsberg DL, Sussman N. Gabapentin as prophylaxis against steroid-induced mania. Can J Psychiatry 2001;46:455-6.
Can corticosteroids “unlock” hidden potential for mania, or are steroid-induced mood symptoms a temporary reaction? And when these mood symptoms occur, what is the best way to treat them?
Psychiatric symptoms develop in 5% to 18% of patients treated with corticosteroids. These effects—most often mania or depression—emerge within days to weeks of starting steroids. To help you head off manic and mixed mood symptoms, this paper examines how to:
- treat steroid-induced mania or mixed bipolar symptoms
- reduce the risk of a mood episode in patients who require sustained corticosteroid therapy.
‘Steroid psychosis’
Jane Pauley, NBC’s Today Show broadcaster, described in her autobiography how hypomania developed within weeks after she started corticosteroids for idiopathic urticaria edema: “I was so energized that I didn’t just walk down the hall, I felt like I was motoring down the hall. I was suddenly the equal of my high-energy friends who move fast and talk fast and loud. I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”1
Pauley’s hypomania led to a manic episode and eventually to depression. She was started on antidepressants, which triggered another manic episode. Pauley—who had no history of bipolar disorder—spent 3 weeks in a New York psychiatric hospital.1
Diagnostic symptoms. Corticosteroids’ psychiatric effects—cognitive, mood, anxiety, and psychotic symptoms—were first described as “steroid psychosis.” Psychosis can occur, but mood symptoms are more common:
- Among 122 patients, 40% experienced depression, followed by mania (28%), psychosis (14%), delirium (10%), and mixed mood episodes (8%).2
- Among 130 patients, mania was most prevalent (35%), followed by depression (28%), mixed mood episodes (12%), delirium (13%), and psychosis (11%).3
- Corticosteroids caused 54% of organic mania cases on a hospital psychiatric consult service.4
- In a prospective study of 50 patients treated with corticosteroids, 13 developed hypomania and 5 developed depression.5
Steroid-induced symptoms emerge from 3 to 4 days to a median of 11 days after a patient starts corticosteroid therapy. After steroids are discontinued, depressive symptoms persist approximately 4 weeks, mania 3 weeks, and delirium a few days. Approximately one-half of patients with steroid psychosis improve in 4 days and one-half within 2 weeks.2,6
Continue to: Who is at risk?
Who is at risk?
Corticosteroids include the steroids produced in the adrenal gland (such as corticosterone) and their synthetic—and often more potent—analogues (such as prednisone).7 Because of their glucocorticoid, immunosuppressant, mineralocorticoid, and anti-inflammatory properties, steroids are used as replacement therapy and to treat a wide variety of illnesses (Table 1).
Table 1
Medical conditions for which corticosteroids are commonly used
| Disorder | Indications for corticosteroids |
|---|---|
| Acute adrenal insufficiency | Acute; replacement therapy |
| Addison’s disease | Chronic; replacement therapy |
| Asthma | Acute and chronic; anti-inflammatory |
| Inflammatory bowel disease | Acute; anti-inflammatory |
| Multiple sclerosis | Acute; exacerbations, immunosuppressant |
| Organ transplant | Chronic; immunosuppressant |
| Rheumatoid arthritis | Chronic; anti-inflammatory |
| Systemic lupus erythematosus | Acute; severe exacerbation, immunosuppressant (high doses are used) |
Age and gender. Patient age appears unrelated to development of psychiatric symptoms after corticosteroid use.2 One study suggested women are twice as likely as men to develop psychiatric symptoms (77 versus 38 cases in 115 patients),3 but many illnesses that require corticosteroid treatment occur more frequently in women. Other researchers found a slight female predominance (58% versus 42% of cases) when they excluded patients with systemic lupus erythematosus and rheumatoid arthritis, which are more common in women than in men.2
Dosage. Higher corticosteroid dosages increase the risk of psychiatric symptoms. In patients taking prednisone, the Boston Collaborative Drug Surveillance Project8 found the incidence of psychiatric side effects to be:
- 1.3% in patients taking
- 4.6% in those taking 41 to 80 mg
- 18.4% in those taking >80 mg.
Psychiatric history. Past psychiatric illness does not seem to be a risk factor for psychiatric side effects of corticosteroids,9 although patients with a history of posttraumatic stress disorder are more likely to suffer depression while taking corticosteroids.10
Corticosteroid exposure. Patients who did not experience psychiatric side effects with corticosteroids in the past appear not to be protected if corticosteroids are used again. One report examined 17 cases of steroid-induced psychiatric illness in patients with previous exposure to corticosteroid therapy. Six patients had previous psychiatric side effects while taking corticosteroids, and 11 did not.2
Bipolar trigger?
Do corticosteroids’ acute psychiatric side effects have long-term sequelae? Longitudinal evidence is scarce, but a few reports suggest corticosteroids could play a role in the onset of primary bipolar I disorder:
- A 28-year-old woman with no known mood symptoms before a short course of prednisone experienced six episodes of mania and depression when not taking corticosteroids during the subsequent 18 months.11
- Among 16 patients with first-onset mood symptoms after corticosteroid use, a retrospective chart review found 7 had recurrent manic and depressive symptoms unrelated to additional corticosteroid use.12
Continue to: ... case reports are inconclusive...
Although intriguing, these case reports are inconclusive. Because bipolar type I incidence in the general population is 1.5%,13 many persons with bipolar disorder undergo corticosteroid treatment. Nevertheless, these results—especially from the retrospective review12—suggest that corticosteroid use may contribute to the onset of bipolar I illness.
Symptomatic treatment
Corticosteroid-induced side effects are usually managed by tapering off the steroids and treating the psychiatric symptoms.2,3 Simply tapering off the steroids—without additional treatments—led to recovery in 33 of 36 patients.2 Stopping corticosteroids is not always possible or desirable, however, especially in many medically complicated cases seen by psychiatric consult services.
In a recent case, I was asked to see a man, age 69, on the oncology service who was receiving corticosteroids every 2 weeks as part of his chemotherapy. The patient was admitted to the hospital for acute mental status changes 2 days after his last corticosteroid dose. He had pressured speech, grandiosity, and had not slept in 2 days. We started risperidone, 1 mg bid, and most of his manic symptoms resolved within 2 days. His chemotherapy was continued without corticosteroids. If this had not been not possible, I would have recommended continuing risperidone prophylactically.
No double-blind, placebo-controlled studies have examined prevention or treatment of steroid-induced mania or other psychiatric symptoms. Uncontrolled trials and case reports suggest benefit from some symptomatic and preventive treatments (Table 2).
Table 2
Mood stabilizers with evidence of benefit in treating corticosteroid-induced mania
| Indication | Medication | Dosage/blood level | Evidence |
|---|---|---|---|
| Preventing psychiatric effects in patients requiring long-term corticosteroids | Lithium | 0.8 to 1.2 mEq/L | Prospective trial (27 with multiple sclerosis)24 |
| Preventing recurrence of manic symptoms in patients requiring additional steroid pulses | Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case report16 |
| Gabapentin | 300 mg tid | Case report26 | |
| Treating steroid-induced manic symptoms | Olanzapine | Initially 2.5 mg/d, titrated to 20 mg/d | Open-label trial (12 patients)14 |
| Lithium | 0.7 mEq/L | Case report15 | |
| Quetiapine | 25 mg qhs and 12.5 mg bid prn | Case report17 | |
| Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case reports12,16 | |
| Haloperidol | 2 to 20 mg/d* | Case reports12,16 | |
| Treating steroid-induced depressive symptoms | Fluoxetine | 20 mg/d | Case report18 |
| Amitriptyline | 30 mg/d (usual effective range is 50 to 300 mg/d)* | Case report12 | |
| Lamotrigine | Up to 400 mg/d | Case report19 | |
| Lithium | 0.1 to 0.8 mEq/L | Case reports20,21 | |
| Treating steroid-induced psychotic symptoms | Haloperidol | 5 mg IV on day 1, then 2 mg po bid | Case report22 |
| Risperidone | 1.5 mg/d | Case report23 | |
| *Dosage not included in published report; recommendation based on experience or anecdotal information | |||
Treating manic and mixed mood symptoms. Twelve outpatients with manic or mixed symptoms from corticosteroid use received olanzapine in a 5-week, open-label trial. Flexible dosing started at 2.5 mg/d and was increased as needed (maximum 20 mg/d). One patient dropped out for lack of efficacy. For the others, manic and mixed symptoms improved significantly, as indicated by scores on the Young Mania Rating Scale, Hamilton Rating Scale for Depression, and Brief Psychotic Rating Scale.14 Patient weight, blood glucose, and involuntary movements did not change significantly.
Evidence from case reports indicates that lithium,15 carbamazepine,12,16 haloperidol,12,16 or quetiapine17 also can successfully treat steroid-induced manic symptoms.
Continue to: Treating other psychiatric symptoms
Treating other psychiatric symptoms. Case reports support electroconvulsive therapy,2,15 fluoxetine,18 amitriptyline,12 lamotrigine,19 or lithium20,21 for steroid-induced depression, and haloperidol22 or risperidone23 for steroid-induced psychosis.
In four cases,6 tricyclic antidepressants appeared to worsen corticosteroids’ psychiatric side effects. These case patients might have had steroid-induced delirium instead of mood disorders or psychosis, however, and the tricyclics’ anticholinergic effects could have worsened the delirium.9
Preventing steroid-induced symptoms
Although clear guidelines on when to start preventive treatments do not exist, potential candidates for pretreatment with lithium or other agents include patients who:
- have developed psychiatric symptoms multiple times after repeated corticosteroid use
- are at high risk if psychiatric side effects occur.
Lithium. Prophylactic lithium was given to 27 patients with multiple sclerosis and taking corticosteroids for acute exacerbations. None developed psychiatric symptoms.24 At the same clinic, 6 of 44 patients with multiple sclerosis or retrobulbar neuritis developed psychiatric side effects after using corticosteroids without lithium.
Be cautious when using prophylactic lithium because some conditions treated with corticosteroids—such as systemic lupus erythematosus—can impair renal function.20 Corticosteroids also can affect sodium balance and increase the risk of lithium intoxication.25
Check renal function before and during lithium titration, and initiate corticosteroid therapy when lithium is at effective blood levels (0.8 to 1.2 mEq/L). Monitor lithium levels and renal function frequently during steroid treatment.
Other mood stabilizers. Two case reports describe patients who repeatedly developed manic symptoms after multiple corticosteroid doses. Carbamazepine, 600 mg qd,16 and gabapentin, 300 mg tid,26 prevented manic symptoms after additional corticosteroid pulses.
Related resources
- Brown ES, Chandler PA. Mood and cognitive changes during systemic corticosteroid therapy. Prim Care Companion J Clin Psychiatry 2001;3(1):17-21. www.psychiatrist.com/pcc/abstracts/pcc030103.htm.
- Merrill W. Case 35-1998: use of lithium to prevent corticosteroid-induced mania. N Engl J Med 1999;340:1123.
Drug brand names
- Amitriptyline • Elavil
- Carbamazepine • Tegretol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Can corticosteroids “unlock” hidden potential for mania, or are steroid-induced mood symptoms a temporary reaction? And when these mood symptoms occur, what is the best way to treat them?
Psychiatric symptoms develop in 5% to 18% of patients treated with corticosteroids. These effects—most often mania or depression—emerge within days to weeks of starting steroids. To help you head off manic and mixed mood symptoms, this paper examines how to:
- treat steroid-induced mania or mixed bipolar symptoms
- reduce the risk of a mood episode in patients who require sustained corticosteroid therapy.
‘Steroid psychosis’
Jane Pauley, NBC’s Today Show broadcaster, described in her autobiography how hypomania developed within weeks after she started corticosteroids for idiopathic urticaria edema: “I was so energized that I didn’t just walk down the hall, I felt like I was motoring down the hall. I was suddenly the equal of my high-energy friends who move fast and talk fast and loud. I told everyone that I could understand why men felt like they could run the world, because I felt like that. This was a new me, and I liked her!”1
Pauley’s hypomania led to a manic episode and eventually to depression. She was started on antidepressants, which triggered another manic episode. Pauley—who had no history of bipolar disorder—spent 3 weeks in a New York psychiatric hospital.1
Diagnostic symptoms. Corticosteroids’ psychiatric effects—cognitive, mood, anxiety, and psychotic symptoms—were first described as “steroid psychosis.” Psychosis can occur, but mood symptoms are more common:
- Among 122 patients, 40% experienced depression, followed by mania (28%), psychosis (14%), delirium (10%), and mixed mood episodes (8%).2
- Among 130 patients, mania was most prevalent (35%), followed by depression (28%), mixed mood episodes (12%), delirium (13%), and psychosis (11%).3
- Corticosteroids caused 54% of organic mania cases on a hospital psychiatric consult service.4
- In a prospective study of 50 patients treated with corticosteroids, 13 developed hypomania and 5 developed depression.5
Steroid-induced symptoms emerge from 3 to 4 days to a median of 11 days after a patient starts corticosteroid therapy. After steroids are discontinued, depressive symptoms persist approximately 4 weeks, mania 3 weeks, and delirium a few days. Approximately one-half of patients with steroid psychosis improve in 4 days and one-half within 2 weeks.2,6
Continue to: Who is at risk?
Who is at risk?
Corticosteroids include the steroids produced in the adrenal gland (such as corticosterone) and their synthetic—and often more potent—analogues (such as prednisone).7 Because of their glucocorticoid, immunosuppressant, mineralocorticoid, and anti-inflammatory properties, steroids are used as replacement therapy and to treat a wide variety of illnesses (Table 1).
Table 1
Medical conditions for which corticosteroids are commonly used
| Disorder | Indications for corticosteroids |
|---|---|
| Acute adrenal insufficiency | Acute; replacement therapy |
| Addison’s disease | Chronic; replacement therapy |
| Asthma | Acute and chronic; anti-inflammatory |
| Inflammatory bowel disease | Acute; anti-inflammatory |
| Multiple sclerosis | Acute; exacerbations, immunosuppressant |
| Organ transplant | Chronic; immunosuppressant |
| Rheumatoid arthritis | Chronic; anti-inflammatory |
| Systemic lupus erythematosus | Acute; severe exacerbation, immunosuppressant (high doses are used) |
Age and gender. Patient age appears unrelated to development of psychiatric symptoms after corticosteroid use.2 One study suggested women are twice as likely as men to develop psychiatric symptoms (77 versus 38 cases in 115 patients),3 but many illnesses that require corticosteroid treatment occur more frequently in women. Other researchers found a slight female predominance (58% versus 42% of cases) when they excluded patients with systemic lupus erythematosus and rheumatoid arthritis, which are more common in women than in men.2
Dosage. Higher corticosteroid dosages increase the risk of psychiatric symptoms. In patients taking prednisone, the Boston Collaborative Drug Surveillance Project8 found the incidence of psychiatric side effects to be:
- 1.3% in patients taking
- 4.6% in those taking 41 to 80 mg
- 18.4% in those taking >80 mg.
Psychiatric history. Past psychiatric illness does not seem to be a risk factor for psychiatric side effects of corticosteroids,9 although patients with a history of posttraumatic stress disorder are more likely to suffer depression while taking corticosteroids.10
Corticosteroid exposure. Patients who did not experience psychiatric side effects with corticosteroids in the past appear not to be protected if corticosteroids are used again. One report examined 17 cases of steroid-induced psychiatric illness in patients with previous exposure to corticosteroid therapy. Six patients had previous psychiatric side effects while taking corticosteroids, and 11 did not.2
Bipolar trigger?
Do corticosteroids’ acute psychiatric side effects have long-term sequelae? Longitudinal evidence is scarce, but a few reports suggest corticosteroids could play a role in the onset of primary bipolar I disorder:
- A 28-year-old woman with no known mood symptoms before a short course of prednisone experienced six episodes of mania and depression when not taking corticosteroids during the subsequent 18 months.11
- Among 16 patients with first-onset mood symptoms after corticosteroid use, a retrospective chart review found 7 had recurrent manic and depressive symptoms unrelated to additional corticosteroid use.12
Continue to: ... case reports are inconclusive...
Although intriguing, these case reports are inconclusive. Because bipolar type I incidence in the general population is 1.5%,13 many persons with bipolar disorder undergo corticosteroid treatment. Nevertheless, these results—especially from the retrospective review12—suggest that corticosteroid use may contribute to the onset of bipolar I illness.
Symptomatic treatment
Corticosteroid-induced side effects are usually managed by tapering off the steroids and treating the psychiatric symptoms.2,3 Simply tapering off the steroids—without additional treatments—led to recovery in 33 of 36 patients.2 Stopping corticosteroids is not always possible or desirable, however, especially in many medically complicated cases seen by psychiatric consult services.
In a recent case, I was asked to see a man, age 69, on the oncology service who was receiving corticosteroids every 2 weeks as part of his chemotherapy. The patient was admitted to the hospital for acute mental status changes 2 days after his last corticosteroid dose. He had pressured speech, grandiosity, and had not slept in 2 days. We started risperidone, 1 mg bid, and most of his manic symptoms resolved within 2 days. His chemotherapy was continued without corticosteroids. If this had not been not possible, I would have recommended continuing risperidone prophylactically.
No double-blind, placebo-controlled studies have examined prevention or treatment of steroid-induced mania or other psychiatric symptoms. Uncontrolled trials and case reports suggest benefit from some symptomatic and preventive treatments (Table 2).
Table 2
Mood stabilizers with evidence of benefit in treating corticosteroid-induced mania
| Indication | Medication | Dosage/blood level | Evidence |
|---|---|---|---|
| Preventing psychiatric effects in patients requiring long-term corticosteroids | Lithium | 0.8 to 1.2 mEq/L | Prospective trial (27 with multiple sclerosis)24 |
| Preventing recurrence of manic symptoms in patients requiring additional steroid pulses | Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case report16 |
| Gabapentin | 300 mg tid | Case report26 | |
| Treating steroid-induced manic symptoms | Olanzapine | Initially 2.5 mg/d, titrated to 20 mg/d | Open-label trial (12 patients)14 |
| Lithium | 0.7 mEq/L | Case report15 | |
| Quetiapine | 25 mg qhs and 12.5 mg bid prn | Case report17 | |
| Carbamazepine | 600 mg qd (to therapeutic range of 8 to 12 μg/mL)* | Case reports12,16 | |
| Haloperidol | 2 to 20 mg/d* | Case reports12,16 | |
| Treating steroid-induced depressive symptoms | Fluoxetine | 20 mg/d | Case report18 |
| Amitriptyline | 30 mg/d (usual effective range is 50 to 300 mg/d)* | Case report12 | |
| Lamotrigine | Up to 400 mg/d | Case report19 | |
| Lithium | 0.1 to 0.8 mEq/L | Case reports20,21 | |
| Treating steroid-induced psychotic symptoms | Haloperidol | 5 mg IV on day 1, then 2 mg po bid | Case report22 |
| Risperidone | 1.5 mg/d | Case report23 | |
| *Dosage not included in published report; recommendation based on experience or anecdotal information | |||
Treating manic and mixed mood symptoms. Twelve outpatients with manic or mixed symptoms from corticosteroid use received olanzapine in a 5-week, open-label trial. Flexible dosing started at 2.5 mg/d and was increased as needed (maximum 20 mg/d). One patient dropped out for lack of efficacy. For the others, manic and mixed symptoms improved significantly, as indicated by scores on the Young Mania Rating Scale, Hamilton Rating Scale for Depression, and Brief Psychotic Rating Scale.14 Patient weight, blood glucose, and involuntary movements did not change significantly.
Evidence from case reports indicates that lithium,15 carbamazepine,12,16 haloperidol,12,16 or quetiapine17 also can successfully treat steroid-induced manic symptoms.
Continue to: Treating other psychiatric symptoms
Treating other psychiatric symptoms. Case reports support electroconvulsive therapy,2,15 fluoxetine,18 amitriptyline,12 lamotrigine,19 or lithium20,21 for steroid-induced depression, and haloperidol22 or risperidone23 for steroid-induced psychosis.
In four cases,6 tricyclic antidepressants appeared to worsen corticosteroids’ psychiatric side effects. These case patients might have had steroid-induced delirium instead of mood disorders or psychosis, however, and the tricyclics’ anticholinergic effects could have worsened the delirium.9
Preventing steroid-induced symptoms
Although clear guidelines on when to start preventive treatments do not exist, potential candidates for pretreatment with lithium or other agents include patients who:
- have developed psychiatric symptoms multiple times after repeated corticosteroid use
- are at high risk if psychiatric side effects occur.
Lithium. Prophylactic lithium was given to 27 patients with multiple sclerosis and taking corticosteroids for acute exacerbations. None developed psychiatric symptoms.24 At the same clinic, 6 of 44 patients with multiple sclerosis or retrobulbar neuritis developed psychiatric side effects after using corticosteroids without lithium.
Be cautious when using prophylactic lithium because some conditions treated with corticosteroids—such as systemic lupus erythematosus—can impair renal function.20 Corticosteroids also can affect sodium balance and increase the risk of lithium intoxication.25
Check renal function before and during lithium titration, and initiate corticosteroid therapy when lithium is at effective blood levels (0.8 to 1.2 mEq/L). Monitor lithium levels and renal function frequently during steroid treatment.
Other mood stabilizers. Two case reports describe patients who repeatedly developed manic symptoms after multiple corticosteroid doses. Carbamazepine, 600 mg qd,16 and gabapentin, 300 mg tid,26 prevented manic symptoms after additional corticosteroid pulses.
Related resources
- Brown ES, Chandler PA. Mood and cognitive changes during systemic corticosteroid therapy. Prim Care Companion J Clin Psychiatry 2001;3(1):17-21. www.psychiatrist.com/pcc/abstracts/pcc030103.htm.
- Merrill W. Case 35-1998: use of lithium to prevent corticosteroid-induced mania. N Engl J Med 1999;340:1123.
Drug brand names
- Amitriptyline • Elavil
- Carbamazepine • Tegretol
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Lithium • Eskalith, others
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
1. Pauley J. Skywriting: a life out of the blue. New York: Random House; 2004.
2. Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord 1983;5:319-32.
3. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry 2003;25:27-33.
4. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci 1989;1:398-400.
5. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days’ corticosteroid treatment. A prospective study. Psychoneuroendocrinology 1996;21:25-31.
6. Hall R, Popkin M, Stickney S, Gardner E. Presentation of the steroid psychoses. J Nerv Ment Dis 1979;167:229-36.
7. Schimmer B, Parker K. Adenohypophyseal hormones and their hypothalamic releasing factors. In: Hardman J, Limbird L, Gilman A (eds). Goodman and Gilman’s the pharmacological basis of therapeutics, 9th ed. New York: McGraw-Hill;1996:1459-86.
8. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther 1972;13:694-8.
9. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf 2000;22:111-22.
10. Brown ES, Suppes T, Khan DA, Carmody TJ, 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol 2002;22:55-61.
11. Pies R. Persistent bipolar illness after steroid administration. Arch Intern Med 1981;141:1087.-
12. Wada K, Yamada N, Suzuki K, et al. Recurrent cases of corticosteroid-induced mood disorder: a clinical characteristics and treatment. J Clin Psychiatry 2000;61:261-7.
13. Narrow WE, Rae DS, Robins LN, Regier DA. Revised prevalence estimates of mental disorders in the United States: using a clinical significance criterion to reconcile 2 surveys’ estimates. Arch Gen Psychiatry 2002;59:115-23.
14. Brown ES, Chamberlain W, Dhanani N, et al. An open-label trial of olanzapine for corticosteroid-induced mood symptoms. J Affect Disord 2004;83:277-81.
15. Blazer DG, 2nd, Petrie WM, Wilson WP. Affective psychosis following renal transplant. Dis Nerv Syst 1976;37:663-7.
16. Wada K, Yamada N, Yamauchi Y, Kuroda S. Carbamazepine treatment of corticosteroid-induced mood disorder. J Affect Disord 2001;65:315-7.
17. Siddiqui Z, Ramaswamy S, Petty F. Quetiapine therapy for corticosteroid-induced mania. Can J Psychiatry 2005;50:77-8.
18. Wyszynski AA, Wyszynski B. Treatment of depression with fluoxetine in corticosteroid-dependent central nervous system Sjogren’s syndrome. Psychosomatics 1993;34:173-7.
19. Brown ES, Frol A, Bobadilla L, et al. Effect of lamotrigine on mood and cognition in patients receiving chronic exogenous corticosteroids. Psychosomatics 2003;44:204-8.
20. Terao T, Mizuki T, Ohji T, Abe K. Antidepressant effect of lithium in patients with systemic lupus erythematosus and cerebral infarction, treated with corticosteroid. Br J Psychiatry 1994;164:109-11.
21. Terao T, Yoshimura R, Shiratuchi T, Abe K. Effects of lithium on steroid-induced depression. Biol Psychiatry 1997;41:1225-6.
22. Ahmad M, Rasul FM. Steroid-induced psychosis treated with haloperidol in a patient with active chronic obstructive pulmonary disease [letter]. Am J Emerg Med 1999;17:735.-
23. DeSilva CC, Nurse MC, Vokey K. Steroid-induced psychosis treated with risperidone. Can J Psychiatry 2002;47:388-9.
24. Falk WE, Mahnke MW, Poskanzer DC. Lithium prophylaxis of corticotropin-induced psychosis. JAMA 1979;241:1011-2.
25. Saklad SR. Management of corticosteroid-induced psychosis with lithium. Clin Pharm 1987;6:186.-
26. Ginsberg DL, Sussman N. Gabapentin as prophylaxis against steroid-induced mania. Can J Psychiatry 2001;46:455-6.
1. Pauley J. Skywriting: a life out of the blue. New York: Random House; 2004.
2. Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord 1983;5:319-32.
3. Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry 2003;25:27-33.
4. Rundell JR, Wise MG. Causes of organic mood disorder. J Neuropsychiatry Clin Neurosci 1989;1:398-400.
5. Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days’ corticosteroid treatment. A prospective study. Psychoneuroendocrinology 1996;21:25-31.
6. Hall R, Popkin M, Stickney S, Gardner E. Presentation of the steroid psychoses. J Nerv Ment Dis 1979;167:229-36.
7. Schimmer B, Parker K. Adenohypophyseal hormones and their hypothalamic releasing factors. In: Hardman J, Limbird L, Gilman A (eds). Goodman and Gilman’s the pharmacological basis of therapeutics, 9th ed. New York: McGraw-Hill;1996:1459-86.
8. The Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther 1972;13:694-8.
9. Patten SB, Neutel CI. Corticosteroid-induced adverse psychiatric effects: incidence, diagnosis and management. Drug Saf 2000;22:111-22.
10. Brown ES, Suppes T, Khan DA, Carmody TJ, 3rd. Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol 2002;22:55-61.
11. Pies R. Persistent bipolar illness after steroid administration. Arch Intern Med 1981;141:1087.-
12. Wada K, Yamada N, Suzuki K, et al. Recurrent cases of corticosteroid-induced mood disorder: a clinical characteristics and treatment. J Clin Psychiatry 2000;61:261-7.
13. Narrow WE, Rae DS, Robins LN, Regier DA. Revised prevalence estimates of mental disorders in the United States: using a clinical significance criterion to reconcile 2 surveys’ estimates. Arch Gen Psychiatry 2002;59:115-23.
14. Brown ES, Chamberlain W, Dhanani N, et al. An open-label trial of olanzapine for corticosteroid-induced mood symptoms. J Affect Disord 2004;83:277-81.
15. Blazer DG, 2nd, Petrie WM, Wilson WP. Affective psychosis following renal transplant. Dis Nerv Syst 1976;37:663-7.
16. Wada K, Yamada N, Yamauchi Y, Kuroda S. Carbamazepine treatment of corticosteroid-induced mood disorder. J Affect Disord 2001;65:315-7.
17. Siddiqui Z, Ramaswamy S, Petty F. Quetiapine therapy for corticosteroid-induced mania. Can J Psychiatry 2005;50:77-8.
18. Wyszynski AA, Wyszynski B. Treatment of depression with fluoxetine in corticosteroid-dependent central nervous system Sjogren’s syndrome. Psychosomatics 1993;34:173-7.
19. Brown ES, Frol A, Bobadilla L, et al. Effect of lamotrigine on mood and cognition in patients receiving chronic exogenous corticosteroids. Psychosomatics 2003;44:204-8.
20. Terao T, Mizuki T, Ohji T, Abe K. Antidepressant effect of lithium in patients with systemic lupus erythematosus and cerebral infarction, treated with corticosteroid. Br J Psychiatry 1994;164:109-11.
21. Terao T, Yoshimura R, Shiratuchi T, Abe K. Effects of lithium on steroid-induced depression. Biol Psychiatry 1997;41:1225-6.
22. Ahmad M, Rasul FM. Steroid-induced psychosis treated with haloperidol in a patient with active chronic obstructive pulmonary disease [letter]. Am J Emerg Med 1999;17:735.-
23. DeSilva CC, Nurse MC, Vokey K. Steroid-induced psychosis treated with risperidone. Can J Psychiatry 2002;47:388-9.
24. Falk WE, Mahnke MW, Poskanzer DC. Lithium prophylaxis of corticotropin-induced psychosis. JAMA 1979;241:1011-2.
25. Saklad SR. Management of corticosteroid-induced psychosis with lithium. Clin Pharm 1987;6:186.-
26. Ginsberg DL, Sussman N. Gabapentin as prophylaxis against steroid-induced mania. Can J Psychiatry 2001;46:455-6.
Is this patient competent to stand trial?
Mr. P, age 39, attacks a convenience store clerk with a knife and is charged with aggravated assault. The judge grants the defense attorney’s request that Mr. P’s competency to stand trial be evaluated. Mr. P’s medical records show paranoid schizophrenia diagnosed at age 22, multiple psychiatric hospitalizations, and chronic medication noncompliance.
You may be called on to determine capacity—such as whether a patient can provide informed consent for a medical procedure. Judges or juries make decisions about competency—often based on a psychiatrist’s opinion about a person’s capacity.
This article describes how to prepare a report stating your opinion about whether a defendant such as Mr. P is competent to stand trial.
What is competency?
The defendant’s attorney usually raises the question of whether a defendant is competent to stand trial, but a judge or prosecuting attorney also may suggest an evaluation. Defense attorneys question their clients’ competence to stand trial in approximately 8% to 15% of felony cases, and up to 50,000 defendants are referred for competency evaluations each year.1-4 Competency may be questioned when the defendant:
- is obviously mentally ill or has a history of mental illness
- appears to be making irrational decisions
- has difficulty interacting with the court or defense counsel.5
“Competency” and “sanity” are often used together in discussions of criminal prosecution of mentally-ill defendants. This article describes evaluating competence to stand trial; we will discuss how to evaluate sanity in a future issue of Current Psychiatry.
Competency is dynamic; the law defines many types, each with a legal definition and requisite capacity. A person may be competent in one area but incompetent in another. He may be incompetent to make a decision about psychiatric hospitalization, for example, yet retain competency to give or withhold informed consent for treatment.
Evaluating competency also is dynamic, depending on the patient’s present state:
- She might be incapable of giving informed consent for surgery while delirious but capable to make a competent decision about treatment after sensorium clears.
- A psychotic defendant may be incompetent to stand trial initially but may be restored to competency after treatment.
The ‘dusky standard’
Courts have long recognized that the mentally ill may be incapable of defending themselves against criminal charges (Box).6 The U.S. Supreme Court in 1960 established in Dusky v. United States that the legal standard for competence to stand trial is “whether [the person] has sufficient present ability to consult with his lawyer with a reasonable degree of rational understanding—and whether he has a rational as well as factual understanding of the proceedings against him.” This standard has been adopted in principle by all states and the federal jurisdiction.7
The “Dusky standard” indicates that a defendant is incompetent to stand trial if, because of a mental illness or other condition, he is unable to:
- understand the nature and objectives of the court proceedings
- or assist in his defense (Table 1).
Judges ultimately determine defendants’ competence to stand trial, but psychiatrists’ opinions are adopted in 90% of cases.8,9
The concept of competence to stand trial originated in 13th-century England. Persons charged with a crime were required to enter a plea in the King’s Court. Defendants who refused to enter a plea were either:
- confined and starved (“prison forte et dure”)
- or slowly crushed under the weight of stones (“peine forte et dure”).2
Before this punishment was exacted, the reason the alleged criminals did not enter a plea had to be determined. Defendants deemed mute by malice (intentionally withholding a plea) were subjected to the aforementioned cruelties. A defendant deemed mute by visitation of God (unable to comprehend that he was required to enter a plea because of mental illness/retardation) was spared, and a plea of not guilty was entered for him.
In the United States, a person’s right to be competent in legal proceedings is implicitly guaranteed by two constitutional amendments:
- right to counsel (Sixth Amendment)
- right to due process (Fourteenth Amendment).
The ‘Dusky standard’ of competence
| A defendant is incompetent to stand trial if he is: |
|
| ‘Nature and objectives’ of a trial include: |
|
| ‘Assisting in own defense’ includes ability to: |
|
| Source: Dusky v US (1960) |
How to assess competency
If a judge asks you to evaluate a defendant’s competency, you need to know the standard governing competence to stand trial in the judge’s jurisdiction. All courts in the United States use the Dusky standard, but the wording varies.
Review the defendant’s case records, including court papers (with a list of charges), medical records, and psychiatric records. Then interview the defendant to thoroughly evaluate his mental status and collect a detailed psychiatric history.
If the defendant has a mental disorder, it must impair his ability to understand the proceedings or participate in his defense to result in incompetency. Sources who know the defendant (spouse, family, or friends) may provide useful collateral information.
Assessment tools. Some argue that tools designed to help determine competency can assess understanding of facts related to the trial but not ability to reason. The MacArthur Competency Assessment Tool—Criminal Adjudication is thought to assess both decisional competency and factual understanding.10 Another new tool, the Evaluation of Competency to Stand Trial-revised (ECST-R), is beginning to be used more frequently to evaluate possible malingering and case-specific information.
These tools can be purchased online through vendors such as www3.parinc.com. Though useful, these tools serve as adjuncts to the clinical interview.
In your report to the court, include relevant information from the mental status evaluation, the diagnosis, and—most important—a clear, concise opinion of the defendant’s current competence to stand trial.
Case: does Mr. P meet the standard?
During your interview, Mr. P endorses chronic auditory hallucinations telling him to harm others. He is alert and oriented to location, date, and current events. He can adequately describe courtroom proceedings and each individual’s role, noting that he had been to court before on a drug possession charge, for which he received probation.
When you ask Mr. P about his attorney, he leans in and whispers, “My attorney and my mother have a secret plan to send me to prison for the rest of my life.” He contends his attorney is telling him to claim he is “crazy” to make him “look bad” in court.
In a separate interview, you question the corrections officer who accompanied Mr. P to the evaluation. He says Mr. P refuses to see his mother and his attorney when they come to visit him in jail and takes his medications only sporadically.
Understanding court proceedings. A defendant such as Mr. P must be able to understand the charges against him, that he is on trial for those charges, and the severity of the charges. He must be able to understand the pleas he can offer (guilty, not guilty, not guilty by reason of insanity, or no contest).
The defendant also must be aware of the roles of trial participants, including defense attorney, prosecutor, witnesses, judge, and jury. He must appreciate the trial’s adversarial nature, that his attorney is acting in his best interests and defending him, and that the prosecutor is trying to convict him.
Ability to assist in defense. A defendant must be able to have logical, coherent discussions with his attorney and be free of paranoid beliefs about the attorney. He must recognize his role as the defendant and maintain no delusions that he is somehow immune to prosecution.
In cooperation with his attorney, he must be able to evaluate the evidence against him and predict the trial’s probable outcome. He must help his attorney formulate a plan for his defense and make reasonable decisions about that plan. If relevant, he must be willing to consider using a mental illness defense at trial; therefore, he must possess a reasonable amount of insight into his mental illness. He also must:
- be able to participate with his attorney in plea bargaining and grasp the meaning and outcome of this process
- have sufficient memory and concentration to understand the trial proceedings.
Finally, a defendant must be motivated to assist in his defense and free of self-defeating behavior. For example, severely depressed patients seeking to punish themselves by causing an unfavorable trial outcome could be considered incompetent.
Mentally ill and incompetent
Mr. P has a clear history of mental illness, the first criterion for a defendant to be considered incompetent to stand trial. He has psychotic symptoms, but these alone are insufficient to consider him incompetent. Also, having competently stood trial in the past does not necessarily mean he is competent now.
Based on the beginning of the interview, Mr. P appears to understand the nature and objectives of court proceedings. His delusions about his attorney, however, clearly would impair his ability to assist in his defense.
Reporting to the court. A forensic evaluator’s report to the court might say: “It is my opinion, with reasonable medical certainty, that although Mr. P understands the nature and objectives of the court proceedings against him, he has a significant thought disorder that currently impairs his ability to assist in his own defense. In particular, Mr. P maintains delusions (a fixed, false belief held despite evidence to the contrary) that his attorney is plotting against him.”
Treatment to restore competency. Approximately 30% of evaluated defendants are adjudicated incompetent for a variety of reasons (Table 2).11 They often are committed to a forensic mental hospital for treatment to restore competency, which occurs in up to 90% of cases.12
Mr. P will likely be committed to restore competency, which may be achieved by treating his schizophrenia.
Defendants with disorders such as dementia or mental retardation may be considered unable to be restored to competency, and their charges are dismissed or held in abeyance. They may then be involuntarily hospitalized if committed through civil proceedings.
Table 2
7 common reasons defendants are found incompetent to stand trial
|
| Source: Reference 5 |
- Grisso T. Evaluating competencies: forensic assessments and instruments, 2nd ed. New York: Springer; 2002.
- Melton GB, Petrila J, Poythress G, Slobogin C. Psychological evaluations for the courts: a handbook for mental health professionals and lawyers, 2nd ed. New York: Guilford Press; 1997.
- American Academy of Psychiatry and the Law. www.aapl.org.
1. Hoge SK, Bonnie RJ, Poythress N, Monahan J. Attorney-client decision making in criminal cases: Client competence and participation as perceived by their attorneys. Behav Sci Law 1992;10:385-94.
2. Poythress NG, Bonnie RJ, Hoge SK, et al. Client abilities to assist counsel and make decisions in criminal cases: Findings from three studies. Law Hum Behav 1994;18(4):437-52.
3. Hoge SK, Bonnie RJ, Poythress N, et al. The MacArthur Adjudicative Competency Study: development and validation of a research instrument. Law Hum Behav 1997;21(2):141-79.
4. Skeem JL, Golding SL, Cohn NB, et al. Logic and reliability of evaluations of competence to stand trial. Law Hum Behav 1998;22(5):519-47.
5. Resnick PJ, Noffsinger SG. Competence to stand trial and the insanity defense. In: Simon RL, Gold LH, eds. Textbook of forensic psychiatry: the clinicians guide to assessment Arlington, VA; American Psychiatric Publishing, 2003;329-47.
6. Grubin D. Fitness to plead in England and Wales. East Sussex, UK: Psychology Press; 1996.
7. Dusky v United States, 362, 402 (US 1960).
8. Freckelton I. Rationality and flexibility in assessment of fitness to stand trial. Int J Law Psychiatry 1986;19:39-59.
9. Reich J, Tookey L. Disagreements between court and psychiatrist on competency to stand trial. J Clin Psychiatry 1986;47:29-30.
10. Bonnie R. The competence of criminal defendants: beyond Dusky and Drop. University of Miami Law Review 1993;47:539-601.
11. Nicholson R, Kugler K. Competent and incompetent criminal defendants: a quantitative review of comparative research. Psychol Bull 1991;109:355-70.
12. Noffsinger SG. Restoration to competency practice guidelines. Int J Offender Ther Comparative Criminology 2001;45(2):356-62.
Mr. P, age 39, attacks a convenience store clerk with a knife and is charged with aggravated assault. The judge grants the defense attorney’s request that Mr. P’s competency to stand trial be evaluated. Mr. P’s medical records show paranoid schizophrenia diagnosed at age 22, multiple psychiatric hospitalizations, and chronic medication noncompliance.
You may be called on to determine capacity—such as whether a patient can provide informed consent for a medical procedure. Judges or juries make decisions about competency—often based on a psychiatrist’s opinion about a person’s capacity.
This article describes how to prepare a report stating your opinion about whether a defendant such as Mr. P is competent to stand trial.
What is competency?
The defendant’s attorney usually raises the question of whether a defendant is competent to stand trial, but a judge or prosecuting attorney also may suggest an evaluation. Defense attorneys question their clients’ competence to stand trial in approximately 8% to 15% of felony cases, and up to 50,000 defendants are referred for competency evaluations each year.1-4 Competency may be questioned when the defendant:
- is obviously mentally ill or has a history of mental illness
- appears to be making irrational decisions
- has difficulty interacting with the court or defense counsel.5
“Competency” and “sanity” are often used together in discussions of criminal prosecution of mentally-ill defendants. This article describes evaluating competence to stand trial; we will discuss how to evaluate sanity in a future issue of Current Psychiatry.
Competency is dynamic; the law defines many types, each with a legal definition and requisite capacity. A person may be competent in one area but incompetent in another. He may be incompetent to make a decision about psychiatric hospitalization, for example, yet retain competency to give or withhold informed consent for treatment.
Evaluating competency also is dynamic, depending on the patient’s present state:
- She might be incapable of giving informed consent for surgery while delirious but capable to make a competent decision about treatment after sensorium clears.
- A psychotic defendant may be incompetent to stand trial initially but may be restored to competency after treatment.
The ‘dusky standard’
Courts have long recognized that the mentally ill may be incapable of defending themselves against criminal charges (Box).6 The U.S. Supreme Court in 1960 established in Dusky v. United States that the legal standard for competence to stand trial is “whether [the person] has sufficient present ability to consult with his lawyer with a reasonable degree of rational understanding—and whether he has a rational as well as factual understanding of the proceedings against him.” This standard has been adopted in principle by all states and the federal jurisdiction.7
The “Dusky standard” indicates that a defendant is incompetent to stand trial if, because of a mental illness or other condition, he is unable to:
- understand the nature and objectives of the court proceedings
- or assist in his defense (Table 1).
Judges ultimately determine defendants’ competence to stand trial, but psychiatrists’ opinions are adopted in 90% of cases.8,9
The concept of competence to stand trial originated in 13th-century England. Persons charged with a crime were required to enter a plea in the King’s Court. Defendants who refused to enter a plea were either:
- confined and starved (“prison forte et dure”)
- or slowly crushed under the weight of stones (“peine forte et dure”).2
Before this punishment was exacted, the reason the alleged criminals did not enter a plea had to be determined. Defendants deemed mute by malice (intentionally withholding a plea) were subjected to the aforementioned cruelties. A defendant deemed mute by visitation of God (unable to comprehend that he was required to enter a plea because of mental illness/retardation) was spared, and a plea of not guilty was entered for him.
In the United States, a person’s right to be competent in legal proceedings is implicitly guaranteed by two constitutional amendments:
- right to counsel (Sixth Amendment)
- right to due process (Fourteenth Amendment).
The ‘Dusky standard’ of competence
| A defendant is incompetent to stand trial if he is: |
|
| ‘Nature and objectives’ of a trial include: |
|
| ‘Assisting in own defense’ includes ability to: |
|
| Source: Dusky v US (1960) |
How to assess competency
If a judge asks you to evaluate a defendant’s competency, you need to know the standard governing competence to stand trial in the judge’s jurisdiction. All courts in the United States use the Dusky standard, but the wording varies.
Review the defendant’s case records, including court papers (with a list of charges), medical records, and psychiatric records. Then interview the defendant to thoroughly evaluate his mental status and collect a detailed psychiatric history.
If the defendant has a mental disorder, it must impair his ability to understand the proceedings or participate in his defense to result in incompetency. Sources who know the defendant (spouse, family, or friends) may provide useful collateral information.
Assessment tools. Some argue that tools designed to help determine competency can assess understanding of facts related to the trial but not ability to reason. The MacArthur Competency Assessment Tool—Criminal Adjudication is thought to assess both decisional competency and factual understanding.10 Another new tool, the Evaluation of Competency to Stand Trial-revised (ECST-R), is beginning to be used more frequently to evaluate possible malingering and case-specific information.
These tools can be purchased online through vendors such as www3.parinc.com. Though useful, these tools serve as adjuncts to the clinical interview.
In your report to the court, include relevant information from the mental status evaluation, the diagnosis, and—most important—a clear, concise opinion of the defendant’s current competence to stand trial.
Case: does Mr. P meet the standard?
During your interview, Mr. P endorses chronic auditory hallucinations telling him to harm others. He is alert and oriented to location, date, and current events. He can adequately describe courtroom proceedings and each individual’s role, noting that he had been to court before on a drug possession charge, for which he received probation.
When you ask Mr. P about his attorney, he leans in and whispers, “My attorney and my mother have a secret plan to send me to prison for the rest of my life.” He contends his attorney is telling him to claim he is “crazy” to make him “look bad” in court.
In a separate interview, you question the corrections officer who accompanied Mr. P to the evaluation. He says Mr. P refuses to see his mother and his attorney when they come to visit him in jail and takes his medications only sporadically.
Understanding court proceedings. A defendant such as Mr. P must be able to understand the charges against him, that he is on trial for those charges, and the severity of the charges. He must be able to understand the pleas he can offer (guilty, not guilty, not guilty by reason of insanity, or no contest).
The defendant also must be aware of the roles of trial participants, including defense attorney, prosecutor, witnesses, judge, and jury. He must appreciate the trial’s adversarial nature, that his attorney is acting in his best interests and defending him, and that the prosecutor is trying to convict him.
Ability to assist in defense. A defendant must be able to have logical, coherent discussions with his attorney and be free of paranoid beliefs about the attorney. He must recognize his role as the defendant and maintain no delusions that he is somehow immune to prosecution.
In cooperation with his attorney, he must be able to evaluate the evidence against him and predict the trial’s probable outcome. He must help his attorney formulate a plan for his defense and make reasonable decisions about that plan. If relevant, he must be willing to consider using a mental illness defense at trial; therefore, he must possess a reasonable amount of insight into his mental illness. He also must:
- be able to participate with his attorney in plea bargaining and grasp the meaning and outcome of this process
- have sufficient memory and concentration to understand the trial proceedings.
Finally, a defendant must be motivated to assist in his defense and free of self-defeating behavior. For example, severely depressed patients seeking to punish themselves by causing an unfavorable trial outcome could be considered incompetent.
Mentally ill and incompetent
Mr. P has a clear history of mental illness, the first criterion for a defendant to be considered incompetent to stand trial. He has psychotic symptoms, but these alone are insufficient to consider him incompetent. Also, having competently stood trial in the past does not necessarily mean he is competent now.
Based on the beginning of the interview, Mr. P appears to understand the nature and objectives of court proceedings. His delusions about his attorney, however, clearly would impair his ability to assist in his defense.
Reporting to the court. A forensic evaluator’s report to the court might say: “It is my opinion, with reasonable medical certainty, that although Mr. P understands the nature and objectives of the court proceedings against him, he has a significant thought disorder that currently impairs his ability to assist in his own defense. In particular, Mr. P maintains delusions (a fixed, false belief held despite evidence to the contrary) that his attorney is plotting against him.”
Treatment to restore competency. Approximately 30% of evaluated defendants are adjudicated incompetent for a variety of reasons (Table 2).11 They often are committed to a forensic mental hospital for treatment to restore competency, which occurs in up to 90% of cases.12
Mr. P will likely be committed to restore competency, which may be achieved by treating his schizophrenia.
Defendants with disorders such as dementia or mental retardation may be considered unable to be restored to competency, and their charges are dismissed or held in abeyance. They may then be involuntarily hospitalized if committed through civil proceedings.
Table 2
7 common reasons defendants are found incompetent to stand trial
|
| Source: Reference 5 |
- Grisso T. Evaluating competencies: forensic assessments and instruments, 2nd ed. New York: Springer; 2002.
- Melton GB, Petrila J, Poythress G, Slobogin C. Psychological evaluations for the courts: a handbook for mental health professionals and lawyers, 2nd ed. New York: Guilford Press; 1997.
- American Academy of Psychiatry and the Law. www.aapl.org.
Mr. P, age 39, attacks a convenience store clerk with a knife and is charged with aggravated assault. The judge grants the defense attorney’s request that Mr. P’s competency to stand trial be evaluated. Mr. P’s medical records show paranoid schizophrenia diagnosed at age 22, multiple psychiatric hospitalizations, and chronic medication noncompliance.
You may be called on to determine capacity—such as whether a patient can provide informed consent for a medical procedure. Judges or juries make decisions about competency—often based on a psychiatrist’s opinion about a person’s capacity.
This article describes how to prepare a report stating your opinion about whether a defendant such as Mr. P is competent to stand trial.
What is competency?
The defendant’s attorney usually raises the question of whether a defendant is competent to stand trial, but a judge or prosecuting attorney also may suggest an evaluation. Defense attorneys question their clients’ competence to stand trial in approximately 8% to 15% of felony cases, and up to 50,000 defendants are referred for competency evaluations each year.1-4 Competency may be questioned when the defendant:
- is obviously mentally ill or has a history of mental illness
- appears to be making irrational decisions
- has difficulty interacting with the court or defense counsel.5
“Competency” and “sanity” are often used together in discussions of criminal prosecution of mentally-ill defendants. This article describes evaluating competence to stand trial; we will discuss how to evaluate sanity in a future issue of Current Psychiatry.
Competency is dynamic; the law defines many types, each with a legal definition and requisite capacity. A person may be competent in one area but incompetent in another. He may be incompetent to make a decision about psychiatric hospitalization, for example, yet retain competency to give or withhold informed consent for treatment.
Evaluating competency also is dynamic, depending on the patient’s present state:
- She might be incapable of giving informed consent for surgery while delirious but capable to make a competent decision about treatment after sensorium clears.
- A psychotic defendant may be incompetent to stand trial initially but may be restored to competency after treatment.
The ‘dusky standard’
Courts have long recognized that the mentally ill may be incapable of defending themselves against criminal charges (Box).6 The U.S. Supreme Court in 1960 established in Dusky v. United States that the legal standard for competence to stand trial is “whether [the person] has sufficient present ability to consult with his lawyer with a reasonable degree of rational understanding—and whether he has a rational as well as factual understanding of the proceedings against him.” This standard has been adopted in principle by all states and the federal jurisdiction.7
The “Dusky standard” indicates that a defendant is incompetent to stand trial if, because of a mental illness or other condition, he is unable to:
- understand the nature and objectives of the court proceedings
- or assist in his defense (Table 1).
Judges ultimately determine defendants’ competence to stand trial, but psychiatrists’ opinions are adopted in 90% of cases.8,9
The concept of competence to stand trial originated in 13th-century England. Persons charged with a crime were required to enter a plea in the King’s Court. Defendants who refused to enter a plea were either:
- confined and starved (“prison forte et dure”)
- or slowly crushed under the weight of stones (“peine forte et dure”).2
Before this punishment was exacted, the reason the alleged criminals did not enter a plea had to be determined. Defendants deemed mute by malice (intentionally withholding a plea) were subjected to the aforementioned cruelties. A defendant deemed mute by visitation of God (unable to comprehend that he was required to enter a plea because of mental illness/retardation) was spared, and a plea of not guilty was entered for him.
In the United States, a person’s right to be competent in legal proceedings is implicitly guaranteed by two constitutional amendments:
- right to counsel (Sixth Amendment)
- right to due process (Fourteenth Amendment).
The ‘Dusky standard’ of competence
| A defendant is incompetent to stand trial if he is: |
|
| ‘Nature and objectives’ of a trial include: |
|
| ‘Assisting in own defense’ includes ability to: |
|
| Source: Dusky v US (1960) |
How to assess competency
If a judge asks you to evaluate a defendant’s competency, you need to know the standard governing competence to stand trial in the judge’s jurisdiction. All courts in the United States use the Dusky standard, but the wording varies.
Review the defendant’s case records, including court papers (with a list of charges), medical records, and psychiatric records. Then interview the defendant to thoroughly evaluate his mental status and collect a detailed psychiatric history.
If the defendant has a mental disorder, it must impair his ability to understand the proceedings or participate in his defense to result in incompetency. Sources who know the defendant (spouse, family, or friends) may provide useful collateral information.
Assessment tools. Some argue that tools designed to help determine competency can assess understanding of facts related to the trial but not ability to reason. The MacArthur Competency Assessment Tool—Criminal Adjudication is thought to assess both decisional competency and factual understanding.10 Another new tool, the Evaluation of Competency to Stand Trial-revised (ECST-R), is beginning to be used more frequently to evaluate possible malingering and case-specific information.
These tools can be purchased online through vendors such as www3.parinc.com. Though useful, these tools serve as adjuncts to the clinical interview.
In your report to the court, include relevant information from the mental status evaluation, the diagnosis, and—most important—a clear, concise opinion of the defendant’s current competence to stand trial.
Case: does Mr. P meet the standard?
During your interview, Mr. P endorses chronic auditory hallucinations telling him to harm others. He is alert and oriented to location, date, and current events. He can adequately describe courtroom proceedings and each individual’s role, noting that he had been to court before on a drug possession charge, for which he received probation.
When you ask Mr. P about his attorney, he leans in and whispers, “My attorney and my mother have a secret plan to send me to prison for the rest of my life.” He contends his attorney is telling him to claim he is “crazy” to make him “look bad” in court.
In a separate interview, you question the corrections officer who accompanied Mr. P to the evaluation. He says Mr. P refuses to see his mother and his attorney when they come to visit him in jail and takes his medications only sporadically.
Understanding court proceedings. A defendant such as Mr. P must be able to understand the charges against him, that he is on trial for those charges, and the severity of the charges. He must be able to understand the pleas he can offer (guilty, not guilty, not guilty by reason of insanity, or no contest).
The defendant also must be aware of the roles of trial participants, including defense attorney, prosecutor, witnesses, judge, and jury. He must appreciate the trial’s adversarial nature, that his attorney is acting in his best interests and defending him, and that the prosecutor is trying to convict him.
Ability to assist in defense. A defendant must be able to have logical, coherent discussions with his attorney and be free of paranoid beliefs about the attorney. He must recognize his role as the defendant and maintain no delusions that he is somehow immune to prosecution.
In cooperation with his attorney, he must be able to evaluate the evidence against him and predict the trial’s probable outcome. He must help his attorney formulate a plan for his defense and make reasonable decisions about that plan. If relevant, he must be willing to consider using a mental illness defense at trial; therefore, he must possess a reasonable amount of insight into his mental illness. He also must:
- be able to participate with his attorney in plea bargaining and grasp the meaning and outcome of this process
- have sufficient memory and concentration to understand the trial proceedings.
Finally, a defendant must be motivated to assist in his defense and free of self-defeating behavior. For example, severely depressed patients seeking to punish themselves by causing an unfavorable trial outcome could be considered incompetent.
Mentally ill and incompetent
Mr. P has a clear history of mental illness, the first criterion for a defendant to be considered incompetent to stand trial. He has psychotic symptoms, but these alone are insufficient to consider him incompetent. Also, having competently stood trial in the past does not necessarily mean he is competent now.
Based on the beginning of the interview, Mr. P appears to understand the nature and objectives of court proceedings. His delusions about his attorney, however, clearly would impair his ability to assist in his defense.
Reporting to the court. A forensic evaluator’s report to the court might say: “It is my opinion, with reasonable medical certainty, that although Mr. P understands the nature and objectives of the court proceedings against him, he has a significant thought disorder that currently impairs his ability to assist in his own defense. In particular, Mr. P maintains delusions (a fixed, false belief held despite evidence to the contrary) that his attorney is plotting against him.”
Treatment to restore competency. Approximately 30% of evaluated defendants are adjudicated incompetent for a variety of reasons (Table 2).11 They often are committed to a forensic mental hospital for treatment to restore competency, which occurs in up to 90% of cases.12
Mr. P will likely be committed to restore competency, which may be achieved by treating his schizophrenia.
Defendants with disorders such as dementia or mental retardation may be considered unable to be restored to competency, and their charges are dismissed or held in abeyance. They may then be involuntarily hospitalized if committed through civil proceedings.
Table 2
7 common reasons defendants are found incompetent to stand trial
|
| Source: Reference 5 |
- Grisso T. Evaluating competencies: forensic assessments and instruments, 2nd ed. New York: Springer; 2002.
- Melton GB, Petrila J, Poythress G, Slobogin C. Psychological evaluations for the courts: a handbook for mental health professionals and lawyers, 2nd ed. New York: Guilford Press; 1997.
- American Academy of Psychiatry and the Law. www.aapl.org.
1. Hoge SK, Bonnie RJ, Poythress N, Monahan J. Attorney-client decision making in criminal cases: Client competence and participation as perceived by their attorneys. Behav Sci Law 1992;10:385-94.
2. Poythress NG, Bonnie RJ, Hoge SK, et al. Client abilities to assist counsel and make decisions in criminal cases: Findings from three studies. Law Hum Behav 1994;18(4):437-52.
3. Hoge SK, Bonnie RJ, Poythress N, et al. The MacArthur Adjudicative Competency Study: development and validation of a research instrument. Law Hum Behav 1997;21(2):141-79.
4. Skeem JL, Golding SL, Cohn NB, et al. Logic and reliability of evaluations of competence to stand trial. Law Hum Behav 1998;22(5):519-47.
5. Resnick PJ, Noffsinger SG. Competence to stand trial and the insanity defense. In: Simon RL, Gold LH, eds. Textbook of forensic psychiatry: the clinicians guide to assessment Arlington, VA; American Psychiatric Publishing, 2003;329-47.
6. Grubin D. Fitness to plead in England and Wales. East Sussex, UK: Psychology Press; 1996.
7. Dusky v United States, 362, 402 (US 1960).
8. Freckelton I. Rationality and flexibility in assessment of fitness to stand trial. Int J Law Psychiatry 1986;19:39-59.
9. Reich J, Tookey L. Disagreements between court and psychiatrist on competency to stand trial. J Clin Psychiatry 1986;47:29-30.
10. Bonnie R. The competence of criminal defendants: beyond Dusky and Drop. University of Miami Law Review 1993;47:539-601.
11. Nicholson R, Kugler K. Competent and incompetent criminal defendants: a quantitative review of comparative research. Psychol Bull 1991;109:355-70.
12. Noffsinger SG. Restoration to competency practice guidelines. Int J Offender Ther Comparative Criminology 2001;45(2):356-62.
1. Hoge SK, Bonnie RJ, Poythress N, Monahan J. Attorney-client decision making in criminal cases: Client competence and participation as perceived by their attorneys. Behav Sci Law 1992;10:385-94.
2. Poythress NG, Bonnie RJ, Hoge SK, et al. Client abilities to assist counsel and make decisions in criminal cases: Findings from three studies. Law Hum Behav 1994;18(4):437-52.
3. Hoge SK, Bonnie RJ, Poythress N, et al. The MacArthur Adjudicative Competency Study: development and validation of a research instrument. Law Hum Behav 1997;21(2):141-79.
4. Skeem JL, Golding SL, Cohn NB, et al. Logic and reliability of evaluations of competence to stand trial. Law Hum Behav 1998;22(5):519-47.
5. Resnick PJ, Noffsinger SG. Competence to stand trial and the insanity defense. In: Simon RL, Gold LH, eds. Textbook of forensic psychiatry: the clinicians guide to assessment Arlington, VA; American Psychiatric Publishing, 2003;329-47.
6. Grubin D. Fitness to plead in England and Wales. East Sussex, UK: Psychology Press; 1996.
7. Dusky v United States, 362, 402 (US 1960).
8. Freckelton I. Rationality and flexibility in assessment of fitness to stand trial. Int J Law Psychiatry 1986;19:39-59.
9. Reich J, Tookey L. Disagreements between court and psychiatrist on competency to stand trial. J Clin Psychiatry 1986;47:29-30.
10. Bonnie R. The competence of criminal defendants: beyond Dusky and Drop. University of Miami Law Review 1993;47:539-601.
11. Nicholson R, Kugler K. Competent and incompetent criminal defendants: a quantitative review of comparative research. Psychol Bull 1991;109:355-70.
12. Noffsinger SG. Restoration to competency practice guidelines. Int J Offender Ther Comparative Criminology 2001;45(2):356-62.
Beating obesity: Help patients control binge eating disorder and night eating syndrome
Say “eating disorders,” and young, thin, Caucasian women with anorexia or bulimia nervosa come to mind. Psychiatry outpatients, however, are more likely to have binge eating disorder (BED) or night eating syndrome (NES) and to be middle-aged, obese, male, or African-American.
Like anorexia and bulimia, BED and NES cause distress, impairment, and medical morbidity. But BED and NES are different because you can manage many patients without referring them to eating disorder treatment centers. You can improve patients’ function and quality of life by:
- correcting eating disorder behaviors and thoughts
- identifying and managing psychiatric comorbidity
- identifying and treating associated medical problems (usually obesity complications such as diabetes mellitus, hypertension, and dyslipidemia)
- helping them achieve and maintain a healthy (but realistic) body weight.
Characteristics of BED and NES
BED and NES are coded as eating disorder, not otherwise specified in DSM-IV-TR, and their diagnostic criteria are provisional. Research criteria for BED are listed in Appendix B of DSM-IV (Box 1); diagnostic criteria for NES are being developed (Box 2).
- Recurrent episodes of binge eating. An episode of binge eating is characterized by both of the following:
- Eating, in a discrete period of time (eg, within any 2-hour period), an amount of food that is definitely larger than most people would eat in a similar period of time under similar circumstances
- A sense of lack of control over eating during the episode (eg, a feeling that one cannot stop eating or control what or how much one is eating)
- The binge-eating episodes are associated with three (or more) of the following:
- Eating much more rapidly than normal
- Eating until feeling uncomfortably full
- Eating large amounts of food when not feeling physically hungry
- Eating alone because of being embarrassed by how much one is eating
- Feeling disgusted with oneself, depressed, or very guilty after overeating
- Marked distress regarding binge eating is present.
- The binge eating occurs, on average, at least 2 days a week for 6 months.
- Binge eating is not associated with the regular use of inappropriate compensatory behaviors (eg, purging, fasting, excessive exercise) and does not occur exclusively during the course of anorexia nervosa or bulimia nervosa.
Source: American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
- Morning anorexia, even if the patient eats breakfast
- Evening hyperphagia, in which ≥50% of daily energy intake is consumed after the evening meal
- Awakening at least once a night and eating snacks
- Duration of at least 3 months
- Patient does not meet criteria for bulimia nervosa or binge eating disorder
Source: Birketvedt GS, Florholmen J, Sundsfjord J, et al. Behavioral and neuroendocrine characteristics of the night-eating syndrome. JAMA 1999;282:657-63.
Prevalence. How common are these eating disorders? Two small studies examined BED and NES prevalence in outpatient psychiatric populations. A European study found 4% of 234 psychiatry clinic patients met criteria for BED,1 whereas 12% in 399 patients in two U.S. clinics met criteria for NES (with possibly higher rates in patients who took atypical antipsychotics).2
Demographics. Men experience BED and NES nearly as often as women, and distribution among women is similar across age groups.3 Binge eating may be more common among African-Americans than Caucasians.4
Obesity. One-half or more of persons with BED or NES are obese, with body mass index (BMI) ≥30.5,6 Obesity prevalence increases over time—from 22% at baseline to 39% 5 years later in one study of BED.7
Psychiatric comorbidity. Overweight or obesity increase the risk for early mortality and impaired quality of life.8 Persons with obesity plus BED have poorer physical and psychosocial function and lower quality of life than do obese persons without BED.9
Structured clinical interviews of 128 obese subjects found higher rates of psychiatric disorders in those with BED. Obesity with comorbid binge eating increased lifetime relative risk:
- >6-fold for major depression
- >8-fold for panic disorder
- >13-fold for borderline personality disorder, compared with obesity alone.10
Similarly, overweight patients with NES have more depression, lower self-esteem, and more difficulty losing weight than those without NES.11 They meet criteria significantly more often for major depressive disorder, anxiety disorders, and substance use disorders.12 Most NES patients view their nocturnal eating as shameful,13 and distress and guilt are among the diagnostic criteria for BED.
Fortunately, successful treatment of BED or NES almost always improves comorbid medical and psychiatric conditions as well. Ongoing treatment is critical for sustaining weight loss.14
Diagnosis and evaluation
Start by asking overweight patients if they binge eat or do most of their eating at night. Follow up with questions to assess whether they meet provisional diagnostic criteria for BED or NES and to rule out other disorders in the differential diagnosis (Box 3). These include bulimia and sleep-related eating disorder, which is generally regarded as a parasomnia.
Obtain a history of the patient’s eating disorder and weight, calculate BMI, and assess for psychiatric comorbidity.15 Make sure blood pressure and fasting lipids and glucose are monitored in patients who are overweight (BMI ≥27) or obese (BMI ≥30).16 Question patients with night eating about sleep disorder symptoms and use of hypnotics—especially short-acting benzodiazepines and zolpidem, which have been associated with sleep-related eating disorder.
| Disorder | Bulimia nervosa | Binge eating disorder | Night-eating syndrome | Sleep-related eating disorder |
|---|---|---|---|---|
| Morning anorexia | No | No | Yes | Yes |
| Evening hyperphagia | No | No | Yes | No |
| Eating pattern | Binges | Binges | Snacks | Snacks, unusual items |
| Compensatory behavior | Yes | No | No | No |
| Awareness of eating | Yes | Yes | Yes | No |
| Polysomnography | Normal | Normal | Low sleep efficiency | Sleep disorder |
| Treatment | CBT, SSRIs | CBT, SSRIs | Sertraline, relaxation | Treat sleep disorder; dopamine agonists |
| CBT: cognitive-behavioral therapy | ||||
| SSRIs: selective serotonin reuptake inhibitors | ||||
Controlling binge eating
Cognitive-behavioral therapy (CBT), interpersonal therapy (IPT), dialectical behavior therapy (DBT), and medications have treated BED effectively in randomized, controlled trials:
- The psychotherapies are equally effective in decreasing bingeing but have little impact on weight.
- Medications are less effective in reducing bingeing but are associated with modest weight loss.
Psychotherapy. The most-studied intervention for BED is CBT, which leads to remission (abstinence from bingeing ≥28 days) in 50% to 60% of patients.17 CBT techniques for BED adapt readily to self-help programs (Box 4).
In one study patients worked with a self-help manual while meeting biweekly with therapists for 15 to 20 minutes in individual sessions. They were randomly assigned to CBT, behavioral weight loss, or control (self-monitoring only) groups. At 12 weeks, remission rates were:
- 46% with CBT
- 18.4% with behavioral weight loss
- 13.3% for controls.
Patients in the intervention groups lost some weight, but no group showed significant changes in BMI.18 The manual used in this study is available in bookstores and online (see Related resources for patients and clinicians).
Although somewhat less effective than therapist-led CBT, guided self-help is easy to implement in a general psychiatric practice.
A randomized, controlled trial compared CBT with IPT in 20 weekly group sessions. Posttreatment remission rates were equivalent—79% for CBT versus 73% for IPT—and weight in both groups was essentially unchanged.19
Abstinence rates after group DBT were 89% in a randomized, controlled trial of 44 women with BED. Binge eating improved significantly more in those assigned to DBT, compared with wait-listed controls. Differences in weight and mood were not significant, and abstinence rates slipped to 56% 6 months after DBT ended.20
Self-monitor
- Keep detailed records of all dietary intake
- Look for patterns in timing, type, and amount of food eaten
- Note antecedents and consequences of binges
Eat regularly
- Have 3 planned meals and 2 snacks per day
- Reduce cues to eat at other times
Substitute other behaviors for bingeing
- List pleasant alternate activities
- Recognize urges to binge
- Choose a substitute activity
- Review efficacy of substitute behaviors in preventing binges
Revise erroneous thinking patterns
- Reduce unrealistic expectations (especially about weight loss)
- Minimize self-criticism in response to lapses
- Change polarized thinking (“I’ve blown my diet; I may as well binge.”)
Limit vulnerabilities to relapse
- Reduce concerns about weight and shape
- Address problems with self-esteem, depression, or anxiety
- Maintain realistic expectations
Source: Fairburn CG. Overcoming binge eating. New York: Guilford Press; 1995.
Medications evaluated for BED in randomized, placebo-controlled trials include selective serotonin reuptake inhibitors (SSRIs) and a tricyclic, obesity management agents (sibutramine and orlistat), and topiramate (Box 5). Binge eating remission rates were highest with antidepressants, and patients lost the most weight with orlistat and sibutramine.
| Medication | Dosage (mg/d) | Duration (weeks) | N | BED remission (%) | Weight loss (kg)* | |
|---|---|---|---|---|---|---|
| Drug | Placebo | |||||
| Citalopram | 20 to 60 | 6 | 38 | 47 | 21 | 2.3 |
| Desipramine | 100 to 300 | 8 | 23 | 60 | 15 | 2.3 |
| Fluoxetine | 20 to 80 | 6 | 60 | 45 | 21 | 4.6 |
| Fluvoxamine | 50 to 300 | 9 | 85 | 38 | 26 | 1.7 |
| Orlistat | 120 tid | 24 | 89 | 23 | 29 | 5.1 |
| Sertraline | 50 to 200 | 6 | 34 | 47 | 14 | 4.4 |
| Sibutramine† | 15 | 12 | 60 | Not reported | 8.8 | |
| Topiramate | 50 to 600 | 14 | 58 | 64 | 30 | 4.8 |
| * Difference between weight lost with drug and weight lost with placebo | ||||||
| † Sibutramine is a controlled substance (schedule IV) and is recommended only for obese patients with BMI ≥30 (≥27 if cardiac risk factors are present). Do not use with monoamine oxidase inhibitors or serotonergic agents, and monitor blood pressure. | ||||||
| Source: Carter WP, Hudson JI, Lalonde JK, et al. Pharmacologic treatment of binge eating disorder. Int J Eat Disord 2003;34:S74-S88 | ||||||
Combining CBT with medications or exercise has also been evaluated for BED in randomized, controlled trials:21
- Group CBT and fluoxetine, 60 mg/d, were compared with placebo in 108 patients. After 16 weeks, intent-to-treat remission rates were 22% (fluoxetine), 26% (placebo), 50% (CBT + fluoxetine), and 61% (CBT + placebo). Weight loss did not differ significantly among treatments but was associated with binge eating remission.
- Guided self-help CBT combined with orlistat, 120 mg tid, or placebo were compared in 50 patients. After 12 weeks, intent-to-treat remission rates were significantly higher with orlistat (64% versus 36%) but not 3 months later (52% each). Weight loss of ≥5% was seen in 36% of those taking orlistat and in 8% taking placebo.
- Binge eating abstinence doubled when exercise (45 minutes. 3 times/week) was added to CBT; weight loss and mood also improved.
Little is known about appropriate dosages and durations for treating BED. Based on bulimia studies, most experts recommend higher-than-usual SSRI dosing (such as fluoxetine, 60 mg/d) and continuing treatment at least 6 months.22
Behavioral weight-loss programs have not been evaluated for BED in randomized, controlled trials. Obese persons with BED experience weight loss equivalent to that of those without BED, however, and more than one-half of persons with BED stop bingeing.9
Most programs combine reduced-calorie diets, increased activity, and behavior modification. Obese patients typically experience a 10% weight loss across 4 months to 1 year, but without continued intervention their weight returns to baseline.23 Weight Watchers is one behavioral weight-loss program with documented efficacy in controlled trials.24
Advocating calorie restriction for binge-eating patients has been controversial because dieting plays a role in triggering and maintaining bulimia nervosa. Recent evidence suggests, however, that binge eating disorder can be safely managed with dieting. In a randomized, controlled trial, 123 obese women without BED were randomly assigned to 3 groups:
- 1,000 kcal/d liquid meal replacement
- 1,200 to 1,500 kcal/d diet of conventional food
- a non-dieting approach to weight control.
Weight and depressive symptoms declined significantly among women in the two dieting groups but not in non-dieters. More episodes of binge eating were observed in subjects on the liquid diet at week 28, but no differences were seen at weeks 40 and 65, and no subjects in any group developed bulimia or binge eating disorder.25
Surprisingly, a 2003 review found that weight loss treatment that ignores bingeing is as effective in reducing bingeing as treatment that focuses solely on that symptom.22
Recommendations. A variety of treatments may be effective for BED, but no guidelines exist to help you choose among them. CBT is considered the treatment of choice, but most overweight BED patients require adjunctive exercise, medication, or behavioral weight-loss treatment.
We recommend that you base each patient’s treatment on five factors:
- treatment availability and cost
- past treatment response
- patient preference
- psychiatric and medical comorbidities
- BMI and past weight-loss experience.
For example, self-help CBT plus exercise or orlistat might benefit an obese man with bipolar disorder who was unable to tolerate adjunctive topiramate. An overweight depressed woman who needs weight-loss support could be given sertraline and encouraged to attend Weight Watchers.
Educate patients about realistic weight loss goals. A reasonable expectation is to lose 0.5 to 2 lbs/week, for a 10% loss across 6 months. Refer to guidelines for obesity risk assessment and treatment23 when advising patients about exercise and weight loss.
Treating night eating syndrome
Research into NES is just beginning, and one small, randomized trial has been published. Twenty patients with NES were randomly assigned to sit quietly or practice progressive muscle relaxation 20 minutes/day for 1 week. Muscle relaxation was associated with improved stress, anxiety, and depression scores, along with trends toward reduced nocturnal eating.26
This study supports a role for stress and anxiety in NES and suggests a potentially effective treatment. These results need to be replicated, however. In other preliminary work:
- After 12 weeks of sertraline therapy (average 188 mg/d), 17 obese patients with NES were eating less often at night, taking in fewer calories after the evening meal, and awakening less often. Five patients (29%) experienced remission, with an average weight loss of 4.8 kg.27
- One of two NES patients treated with topiramate (mean dose 218 mg at night) experienced remission and the other a marked response. Sleep improved, and average weight loss was 11 kg across 8 months.28
- One woman, age 51, with NES and nonseasonal depression experienced remission of depression and NES after 14 phototherapy sessions. NES returned when light therapy was discontinued.29
Recommendations. Suggest that NES patients start progressive muscle relaxation (see Related resources for instructions, or patients can purchase audiotapes). If benefits are insufficient, consider adjunctive sertraline, topiramate, or phototherapy. The efficacy of self-help for NES has not been evaluated, although a manual is available (see Related resources).
For clinicians
- Devlin MJ, Yanovski SZ, Wilson GT. Obesity: What mental health professionals need to know. Am J Psychiatry 2000;157:854-66.
- Instructions for progressive muscle relaxation. www.webmd.com/hw/health_guide_atoz/ta4146.asp?navbar=hw153409.
For patients and clinicians
- Anorexia and related eating disorders. www.anred.com (information about BED and NES).
- Self-help manuals available at bookstores or at Gürze Books (www.gurze.com):
- Fairburn CG. Overcoming binge eating. New York: Guilford Press, 1995.
- Allison KC, Stunkard AJ, Thier SL. Overcoming night eating syndrome: A step-by-step guide to breaking the cycle. Oakland, CA: New Harbinger Publications; 2004.
- Weight Control Information Network (WIN). National Institute of Diabetes and Digestive and Kidney Diseases. http://win.niddk.nih.gov
Drug brand names
- Citalopram • Celexa
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Orlistat • Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax
Disclosures
Dr. Cloak owns Pfizer Inc. stock but otherwise reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Powers reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Taraldsen KW, Eriksen L, Gotestam KG. Prevalence of eating disorders among Norwegian women and men in a psychiatric outpatient unit. Int J Eat Disord 1996;20:185-90.
2. Lundgren JD, Allison KC, Crow S, et al. Prevalence of the night-eating syndrome in a psychiatric population. Am J Psychiatry 2006;163:156-8.
3. Streigel-Moore RH, Franko DL. Epidemiology of binge eating disorder. Int J Eating Disord 2003;34:S19-S29.
4. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9:83-7.
5. Marshall HM, Allison KC, O’Reardon JP, et al. Night eating syndrome among nonobese persons. Int J Eat Disord 2004;35:217-22.
6. Spitzer RL, Yanovski S, Wadden T, et al. Binge eating disorder: its further validation in a multisite study. Int J Eat Disord 1993;13:137-53.
7. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;37:659-65.
8. Fontaine KR, Redden DT, Wang C, et al. Years of life lost due to obesity. JAMA 2003;289:187-93.
9. Rieger E, Wilfley DE, Stein RI, et al. A comparison of quality of life in obese individuals with and without binge eating disorder. Int J Eat Disord 2005;37:234-40.
10. Yanovski SZ, Nelson JE, Dubbert BK, Spitzer RL. Association of binge eating disorder and psychiatric co-morbidity in obese subjects. Am J Psychiatry 1993;150:1472-9.
11. Gluck ME, Geliebter A, Satov T. Night eating syndrome is associated with depression, low self-esteem, reduced daytime hunger, and less weight loss in obese outpatients. Obes Res 2001;9:264-7.
12. Stunkard AJ, Allison KC. Two forms of disordered eating in obesity: Binge eating and night eating. Int J Obes Relat Metab Disord 2003;7:1-12.
13. O’Reardon JP, Peshek A, Allison K. Night eating syndrome: Diagnosis, epidemiology, and management. CNS Drugs 2005;19:997-1008.
14. Agras WS, Teich CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge-eating disorder. J Consult Clin Psychol 1997;65:343-7.
15. Cloak NL, Powers PS. Are undiagnosed eating disorders keeping your patients sick? Current Psychiatry 2005;4(12):65-75.
16. Kushner RF, Roth JL. Medical evaluation of the obese individual. Psychiatr Clin North Am 2005;28:89-103.
17. Wonderlich SA, de Zwaan M, Mitchell JE, et al. Psychological and dietary treatments of binge eating disorder: conceptual implications. Int J Eat Disord 2003;34:S58-S73.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge-eating disorder. Behav Res Ther 2005;43:1509-25.
19. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal therapy for the treatment of overweight individuals with binge eating disorder. Arch Gen Psychiatry 2002;59:713-21.
20. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
21. Pendleton VR, Goodrick CK, Poston WS, et al. Exercise augments the effects of cognitive-behavioral therapy in the treatment of binge eating. Int J Eat Disord 2002;31:172-84.
22. Agras WS. Pharmacotherapy of bulimia nervosa and binge eating disorder: longer-term outcomes. Psychopharmacol Bull 1997;33:433-6.
23. Clinical guidelines on the identification evaluation and treatment of obesity in adults Executive summary, 1998. Bethesda, MD: National Heart, Lung, and Blood Institute. Available at: http://www.nhlbi.nih.gov/guidelines/obesity. Accessed April 18, 2006.
24. Tsai AG, Wadden TA, Womble LG, Byrne KJ. Commercial and self-help programs for weight control. Psychiatr Clin North Am 2005;28:171-92.
25. Wadden TA, Foster GD, Sarwer DB, et al. Dieting and the development of eating disorders in obese women: Results of a randomized controlled trial. Am J Clin Nutr 2004;80:560-8.
26. Pawlow LA, O’Neil PM, Malcolm RJ. Night eating syndrome: Effects of brief relaxation training on stress, mood, hunger, and eating patterns. Int J Obes Relat Metab Disord 2003;27:970-8.
27. O’Reardon JP, Stunkard AJ, Allison KC. A clinical trial of sertraline in the treatment of night eating syndrome. Int J Eat Disord 2004;35:16-26.
28. Winkelman JW. Treatment of nocturnal eating syndrome and sleep-related eating disorder with topiramate. Sleep Med 2003;4(3):243-6.
29. Friedman S, Even C, Dardennes R, Guelfi JD. Light therapy, obesity, and night-eating syndrome. Am J Psychiatry 2002;159:875-6.
Say “eating disorders,” and young, thin, Caucasian women with anorexia or bulimia nervosa come to mind. Psychiatry outpatients, however, are more likely to have binge eating disorder (BED) or night eating syndrome (NES) and to be middle-aged, obese, male, or African-American.
Like anorexia and bulimia, BED and NES cause distress, impairment, and medical morbidity. But BED and NES are different because you can manage many patients without referring them to eating disorder treatment centers. You can improve patients’ function and quality of life by:
- correcting eating disorder behaviors and thoughts
- identifying and managing psychiatric comorbidity
- identifying and treating associated medical problems (usually obesity complications such as diabetes mellitus, hypertension, and dyslipidemia)
- helping them achieve and maintain a healthy (but realistic) body weight.
Characteristics of BED and NES
BED and NES are coded as eating disorder, not otherwise specified in DSM-IV-TR, and their diagnostic criteria are provisional. Research criteria for BED are listed in Appendix B of DSM-IV (Box 1); diagnostic criteria for NES are being developed (Box 2).
- Recurrent episodes of binge eating. An episode of binge eating is characterized by both of the following:
- Eating, in a discrete period of time (eg, within any 2-hour period), an amount of food that is definitely larger than most people would eat in a similar period of time under similar circumstances
- A sense of lack of control over eating during the episode (eg, a feeling that one cannot stop eating or control what or how much one is eating)
- The binge-eating episodes are associated with three (or more) of the following:
- Eating much more rapidly than normal
- Eating until feeling uncomfortably full
- Eating large amounts of food when not feeling physically hungry
- Eating alone because of being embarrassed by how much one is eating
- Feeling disgusted with oneself, depressed, or very guilty after overeating
- Marked distress regarding binge eating is present.
- The binge eating occurs, on average, at least 2 days a week for 6 months.
- Binge eating is not associated with the regular use of inappropriate compensatory behaviors (eg, purging, fasting, excessive exercise) and does not occur exclusively during the course of anorexia nervosa or bulimia nervosa.
Source: American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
- Morning anorexia, even if the patient eats breakfast
- Evening hyperphagia, in which ≥50% of daily energy intake is consumed after the evening meal
- Awakening at least once a night and eating snacks
- Duration of at least 3 months
- Patient does not meet criteria for bulimia nervosa or binge eating disorder
Source: Birketvedt GS, Florholmen J, Sundsfjord J, et al. Behavioral and neuroendocrine characteristics of the night-eating syndrome. JAMA 1999;282:657-63.
Prevalence. How common are these eating disorders? Two small studies examined BED and NES prevalence in outpatient psychiatric populations. A European study found 4% of 234 psychiatry clinic patients met criteria for BED,1 whereas 12% in 399 patients in two U.S. clinics met criteria for NES (with possibly higher rates in patients who took atypical antipsychotics).2
Demographics. Men experience BED and NES nearly as often as women, and distribution among women is similar across age groups.3 Binge eating may be more common among African-Americans than Caucasians.4
Obesity. One-half or more of persons with BED or NES are obese, with body mass index (BMI) ≥30.5,6 Obesity prevalence increases over time—from 22% at baseline to 39% 5 years later in one study of BED.7
Psychiatric comorbidity. Overweight or obesity increase the risk for early mortality and impaired quality of life.8 Persons with obesity plus BED have poorer physical and psychosocial function and lower quality of life than do obese persons without BED.9
Structured clinical interviews of 128 obese subjects found higher rates of psychiatric disorders in those with BED. Obesity with comorbid binge eating increased lifetime relative risk:
- >6-fold for major depression
- >8-fold for panic disorder
- >13-fold for borderline personality disorder, compared with obesity alone.10
Similarly, overweight patients with NES have more depression, lower self-esteem, and more difficulty losing weight than those without NES.11 They meet criteria significantly more often for major depressive disorder, anxiety disorders, and substance use disorders.12 Most NES patients view their nocturnal eating as shameful,13 and distress and guilt are among the diagnostic criteria for BED.
Fortunately, successful treatment of BED or NES almost always improves comorbid medical and psychiatric conditions as well. Ongoing treatment is critical for sustaining weight loss.14
Diagnosis and evaluation
Start by asking overweight patients if they binge eat or do most of their eating at night. Follow up with questions to assess whether they meet provisional diagnostic criteria for BED or NES and to rule out other disorders in the differential diagnosis (Box 3). These include bulimia and sleep-related eating disorder, which is generally regarded as a parasomnia.
Obtain a history of the patient’s eating disorder and weight, calculate BMI, and assess for psychiatric comorbidity.15 Make sure blood pressure and fasting lipids and glucose are monitored in patients who are overweight (BMI ≥27) or obese (BMI ≥30).16 Question patients with night eating about sleep disorder symptoms and use of hypnotics—especially short-acting benzodiazepines and zolpidem, which have been associated with sleep-related eating disorder.
| Disorder | Bulimia nervosa | Binge eating disorder | Night-eating syndrome | Sleep-related eating disorder |
|---|---|---|---|---|
| Morning anorexia | No | No | Yes | Yes |
| Evening hyperphagia | No | No | Yes | No |
| Eating pattern | Binges | Binges | Snacks | Snacks, unusual items |
| Compensatory behavior | Yes | No | No | No |
| Awareness of eating | Yes | Yes | Yes | No |
| Polysomnography | Normal | Normal | Low sleep efficiency | Sleep disorder |
| Treatment | CBT, SSRIs | CBT, SSRIs | Sertraline, relaxation | Treat sleep disorder; dopamine agonists |
| CBT: cognitive-behavioral therapy | ||||
| SSRIs: selective serotonin reuptake inhibitors | ||||
Controlling binge eating
Cognitive-behavioral therapy (CBT), interpersonal therapy (IPT), dialectical behavior therapy (DBT), and medications have treated BED effectively in randomized, controlled trials:
- The psychotherapies are equally effective in decreasing bingeing but have little impact on weight.
- Medications are less effective in reducing bingeing but are associated with modest weight loss.
Psychotherapy. The most-studied intervention for BED is CBT, which leads to remission (abstinence from bingeing ≥28 days) in 50% to 60% of patients.17 CBT techniques for BED adapt readily to self-help programs (Box 4).
In one study patients worked with a self-help manual while meeting biweekly with therapists for 15 to 20 minutes in individual sessions. They were randomly assigned to CBT, behavioral weight loss, or control (self-monitoring only) groups. At 12 weeks, remission rates were:
- 46% with CBT
- 18.4% with behavioral weight loss
- 13.3% for controls.
Patients in the intervention groups lost some weight, but no group showed significant changes in BMI.18 The manual used in this study is available in bookstores and online (see Related resources for patients and clinicians).
Although somewhat less effective than therapist-led CBT, guided self-help is easy to implement in a general psychiatric practice.
A randomized, controlled trial compared CBT with IPT in 20 weekly group sessions. Posttreatment remission rates were equivalent—79% for CBT versus 73% for IPT—and weight in both groups was essentially unchanged.19
Abstinence rates after group DBT were 89% in a randomized, controlled trial of 44 women with BED. Binge eating improved significantly more in those assigned to DBT, compared with wait-listed controls. Differences in weight and mood were not significant, and abstinence rates slipped to 56% 6 months after DBT ended.20
Self-monitor
- Keep detailed records of all dietary intake
- Look for patterns in timing, type, and amount of food eaten
- Note antecedents and consequences of binges
Eat regularly
- Have 3 planned meals and 2 snacks per day
- Reduce cues to eat at other times
Substitute other behaviors for bingeing
- List pleasant alternate activities
- Recognize urges to binge
- Choose a substitute activity
- Review efficacy of substitute behaviors in preventing binges
Revise erroneous thinking patterns
- Reduce unrealistic expectations (especially about weight loss)
- Minimize self-criticism in response to lapses
- Change polarized thinking (“I’ve blown my diet; I may as well binge.”)
Limit vulnerabilities to relapse
- Reduce concerns about weight and shape
- Address problems with self-esteem, depression, or anxiety
- Maintain realistic expectations
Source: Fairburn CG. Overcoming binge eating. New York: Guilford Press; 1995.
Medications evaluated for BED in randomized, placebo-controlled trials include selective serotonin reuptake inhibitors (SSRIs) and a tricyclic, obesity management agents (sibutramine and orlistat), and topiramate (Box 5). Binge eating remission rates were highest with antidepressants, and patients lost the most weight with orlistat and sibutramine.
| Medication | Dosage (mg/d) | Duration (weeks) | N | BED remission (%) | Weight loss (kg)* | |
|---|---|---|---|---|---|---|
| Drug | Placebo | |||||
| Citalopram | 20 to 60 | 6 | 38 | 47 | 21 | 2.3 |
| Desipramine | 100 to 300 | 8 | 23 | 60 | 15 | 2.3 |
| Fluoxetine | 20 to 80 | 6 | 60 | 45 | 21 | 4.6 |
| Fluvoxamine | 50 to 300 | 9 | 85 | 38 | 26 | 1.7 |
| Orlistat | 120 tid | 24 | 89 | 23 | 29 | 5.1 |
| Sertraline | 50 to 200 | 6 | 34 | 47 | 14 | 4.4 |
| Sibutramine† | 15 | 12 | 60 | Not reported | 8.8 | |
| Topiramate | 50 to 600 | 14 | 58 | 64 | 30 | 4.8 |
| * Difference between weight lost with drug and weight lost with placebo | ||||||
| † Sibutramine is a controlled substance (schedule IV) and is recommended only for obese patients with BMI ≥30 (≥27 if cardiac risk factors are present). Do not use with monoamine oxidase inhibitors or serotonergic agents, and monitor blood pressure. | ||||||
| Source: Carter WP, Hudson JI, Lalonde JK, et al. Pharmacologic treatment of binge eating disorder. Int J Eat Disord 2003;34:S74-S88 | ||||||
Combining CBT with medications or exercise has also been evaluated for BED in randomized, controlled trials:21
- Group CBT and fluoxetine, 60 mg/d, were compared with placebo in 108 patients. After 16 weeks, intent-to-treat remission rates were 22% (fluoxetine), 26% (placebo), 50% (CBT + fluoxetine), and 61% (CBT + placebo). Weight loss did not differ significantly among treatments but was associated with binge eating remission.
- Guided self-help CBT combined with orlistat, 120 mg tid, or placebo were compared in 50 patients. After 12 weeks, intent-to-treat remission rates were significantly higher with orlistat (64% versus 36%) but not 3 months later (52% each). Weight loss of ≥5% was seen in 36% of those taking orlistat and in 8% taking placebo.
- Binge eating abstinence doubled when exercise (45 minutes. 3 times/week) was added to CBT; weight loss and mood also improved.
Little is known about appropriate dosages and durations for treating BED. Based on bulimia studies, most experts recommend higher-than-usual SSRI dosing (such as fluoxetine, 60 mg/d) and continuing treatment at least 6 months.22
Behavioral weight-loss programs have not been evaluated for BED in randomized, controlled trials. Obese persons with BED experience weight loss equivalent to that of those without BED, however, and more than one-half of persons with BED stop bingeing.9
Most programs combine reduced-calorie diets, increased activity, and behavior modification. Obese patients typically experience a 10% weight loss across 4 months to 1 year, but without continued intervention their weight returns to baseline.23 Weight Watchers is one behavioral weight-loss program with documented efficacy in controlled trials.24
Advocating calorie restriction for binge-eating patients has been controversial because dieting plays a role in triggering and maintaining bulimia nervosa. Recent evidence suggests, however, that binge eating disorder can be safely managed with dieting. In a randomized, controlled trial, 123 obese women without BED were randomly assigned to 3 groups:
- 1,000 kcal/d liquid meal replacement
- 1,200 to 1,500 kcal/d diet of conventional food
- a non-dieting approach to weight control.
Weight and depressive symptoms declined significantly among women in the two dieting groups but not in non-dieters. More episodes of binge eating were observed in subjects on the liquid diet at week 28, but no differences were seen at weeks 40 and 65, and no subjects in any group developed bulimia or binge eating disorder.25
Surprisingly, a 2003 review found that weight loss treatment that ignores bingeing is as effective in reducing bingeing as treatment that focuses solely on that symptom.22
Recommendations. A variety of treatments may be effective for BED, but no guidelines exist to help you choose among them. CBT is considered the treatment of choice, but most overweight BED patients require adjunctive exercise, medication, or behavioral weight-loss treatment.
We recommend that you base each patient’s treatment on five factors:
- treatment availability and cost
- past treatment response
- patient preference
- psychiatric and medical comorbidities
- BMI and past weight-loss experience.
For example, self-help CBT plus exercise or orlistat might benefit an obese man with bipolar disorder who was unable to tolerate adjunctive topiramate. An overweight depressed woman who needs weight-loss support could be given sertraline and encouraged to attend Weight Watchers.
Educate patients about realistic weight loss goals. A reasonable expectation is to lose 0.5 to 2 lbs/week, for a 10% loss across 6 months. Refer to guidelines for obesity risk assessment and treatment23 when advising patients about exercise and weight loss.
Treating night eating syndrome
Research into NES is just beginning, and one small, randomized trial has been published. Twenty patients with NES were randomly assigned to sit quietly or practice progressive muscle relaxation 20 minutes/day for 1 week. Muscle relaxation was associated with improved stress, anxiety, and depression scores, along with trends toward reduced nocturnal eating.26
This study supports a role for stress and anxiety in NES and suggests a potentially effective treatment. These results need to be replicated, however. In other preliminary work:
- After 12 weeks of sertraline therapy (average 188 mg/d), 17 obese patients with NES were eating less often at night, taking in fewer calories after the evening meal, and awakening less often. Five patients (29%) experienced remission, with an average weight loss of 4.8 kg.27
- One of two NES patients treated with topiramate (mean dose 218 mg at night) experienced remission and the other a marked response. Sleep improved, and average weight loss was 11 kg across 8 months.28
- One woman, age 51, with NES and nonseasonal depression experienced remission of depression and NES after 14 phototherapy sessions. NES returned when light therapy was discontinued.29
Recommendations. Suggest that NES patients start progressive muscle relaxation (see Related resources for instructions, or patients can purchase audiotapes). If benefits are insufficient, consider adjunctive sertraline, topiramate, or phototherapy. The efficacy of self-help for NES has not been evaluated, although a manual is available (see Related resources).
For clinicians
- Devlin MJ, Yanovski SZ, Wilson GT. Obesity: What mental health professionals need to know. Am J Psychiatry 2000;157:854-66.
- Instructions for progressive muscle relaxation. www.webmd.com/hw/health_guide_atoz/ta4146.asp?navbar=hw153409.
For patients and clinicians
- Anorexia and related eating disorders. www.anred.com (information about BED and NES).
- Self-help manuals available at bookstores or at Gürze Books (www.gurze.com):
- Fairburn CG. Overcoming binge eating. New York: Guilford Press, 1995.
- Allison KC, Stunkard AJ, Thier SL. Overcoming night eating syndrome: A step-by-step guide to breaking the cycle. Oakland, CA: New Harbinger Publications; 2004.
- Weight Control Information Network (WIN). National Institute of Diabetes and Digestive and Kidney Diseases. http://win.niddk.nih.gov
Drug brand names
- Citalopram • Celexa
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Orlistat • Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax
Disclosures
Dr. Cloak owns Pfizer Inc. stock but otherwise reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Powers reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Say “eating disorders,” and young, thin, Caucasian women with anorexia or bulimia nervosa come to mind. Psychiatry outpatients, however, are more likely to have binge eating disorder (BED) or night eating syndrome (NES) and to be middle-aged, obese, male, or African-American.
Like anorexia and bulimia, BED and NES cause distress, impairment, and medical morbidity. But BED and NES are different because you can manage many patients without referring them to eating disorder treatment centers. You can improve patients’ function and quality of life by:
- correcting eating disorder behaviors and thoughts
- identifying and managing psychiatric comorbidity
- identifying and treating associated medical problems (usually obesity complications such as diabetes mellitus, hypertension, and dyslipidemia)
- helping them achieve and maintain a healthy (but realistic) body weight.
Characteristics of BED and NES
BED and NES are coded as eating disorder, not otherwise specified in DSM-IV-TR, and their diagnostic criteria are provisional. Research criteria for BED are listed in Appendix B of DSM-IV (Box 1); diagnostic criteria for NES are being developed (Box 2).
- Recurrent episodes of binge eating. An episode of binge eating is characterized by both of the following:
- Eating, in a discrete period of time (eg, within any 2-hour period), an amount of food that is definitely larger than most people would eat in a similar period of time under similar circumstances
- A sense of lack of control over eating during the episode (eg, a feeling that one cannot stop eating or control what or how much one is eating)
- The binge-eating episodes are associated with three (or more) of the following:
- Eating much more rapidly than normal
- Eating until feeling uncomfortably full
- Eating large amounts of food when not feeling physically hungry
- Eating alone because of being embarrassed by how much one is eating
- Feeling disgusted with oneself, depressed, or very guilty after overeating
- Marked distress regarding binge eating is present.
- The binge eating occurs, on average, at least 2 days a week for 6 months.
- Binge eating is not associated with the regular use of inappropriate compensatory behaviors (eg, purging, fasting, excessive exercise) and does not occur exclusively during the course of anorexia nervosa or bulimia nervosa.
Source: American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000.
- Morning anorexia, even if the patient eats breakfast
- Evening hyperphagia, in which ≥50% of daily energy intake is consumed after the evening meal
- Awakening at least once a night and eating snacks
- Duration of at least 3 months
- Patient does not meet criteria for bulimia nervosa or binge eating disorder
Source: Birketvedt GS, Florholmen J, Sundsfjord J, et al. Behavioral and neuroendocrine characteristics of the night-eating syndrome. JAMA 1999;282:657-63.
Prevalence. How common are these eating disorders? Two small studies examined BED and NES prevalence in outpatient psychiatric populations. A European study found 4% of 234 psychiatry clinic patients met criteria for BED,1 whereas 12% in 399 patients in two U.S. clinics met criteria for NES (with possibly higher rates in patients who took atypical antipsychotics).2
Demographics. Men experience BED and NES nearly as often as women, and distribution among women is similar across age groups.3 Binge eating may be more common among African-Americans than Caucasians.4
Obesity. One-half or more of persons with BED or NES are obese, with body mass index (BMI) ≥30.5,6 Obesity prevalence increases over time—from 22% at baseline to 39% 5 years later in one study of BED.7
Psychiatric comorbidity. Overweight or obesity increase the risk for early mortality and impaired quality of life.8 Persons with obesity plus BED have poorer physical and psychosocial function and lower quality of life than do obese persons without BED.9
Structured clinical interviews of 128 obese subjects found higher rates of psychiatric disorders in those with BED. Obesity with comorbid binge eating increased lifetime relative risk:
- >6-fold for major depression
- >8-fold for panic disorder
- >13-fold for borderline personality disorder, compared with obesity alone.10
Similarly, overweight patients with NES have more depression, lower self-esteem, and more difficulty losing weight than those without NES.11 They meet criteria significantly more often for major depressive disorder, anxiety disorders, and substance use disorders.12 Most NES patients view their nocturnal eating as shameful,13 and distress and guilt are among the diagnostic criteria for BED.
Fortunately, successful treatment of BED or NES almost always improves comorbid medical and psychiatric conditions as well. Ongoing treatment is critical for sustaining weight loss.14
Diagnosis and evaluation
Start by asking overweight patients if they binge eat or do most of their eating at night. Follow up with questions to assess whether they meet provisional diagnostic criteria for BED or NES and to rule out other disorders in the differential diagnosis (Box 3). These include bulimia and sleep-related eating disorder, which is generally regarded as a parasomnia.
Obtain a history of the patient’s eating disorder and weight, calculate BMI, and assess for psychiatric comorbidity.15 Make sure blood pressure and fasting lipids and glucose are monitored in patients who are overweight (BMI ≥27) or obese (BMI ≥30).16 Question patients with night eating about sleep disorder symptoms and use of hypnotics—especially short-acting benzodiazepines and zolpidem, which have been associated with sleep-related eating disorder.
| Disorder | Bulimia nervosa | Binge eating disorder | Night-eating syndrome | Sleep-related eating disorder |
|---|---|---|---|---|
| Morning anorexia | No | No | Yes | Yes |
| Evening hyperphagia | No | No | Yes | No |
| Eating pattern | Binges | Binges | Snacks | Snacks, unusual items |
| Compensatory behavior | Yes | No | No | No |
| Awareness of eating | Yes | Yes | Yes | No |
| Polysomnography | Normal | Normal | Low sleep efficiency | Sleep disorder |
| Treatment | CBT, SSRIs | CBT, SSRIs | Sertraline, relaxation | Treat sleep disorder; dopamine agonists |
| CBT: cognitive-behavioral therapy | ||||
| SSRIs: selective serotonin reuptake inhibitors | ||||
Controlling binge eating
Cognitive-behavioral therapy (CBT), interpersonal therapy (IPT), dialectical behavior therapy (DBT), and medications have treated BED effectively in randomized, controlled trials:
- The psychotherapies are equally effective in decreasing bingeing but have little impact on weight.
- Medications are less effective in reducing bingeing but are associated with modest weight loss.
Psychotherapy. The most-studied intervention for BED is CBT, which leads to remission (abstinence from bingeing ≥28 days) in 50% to 60% of patients.17 CBT techniques for BED adapt readily to self-help programs (Box 4).
In one study patients worked with a self-help manual while meeting biweekly with therapists for 15 to 20 minutes in individual sessions. They were randomly assigned to CBT, behavioral weight loss, or control (self-monitoring only) groups. At 12 weeks, remission rates were:
- 46% with CBT
- 18.4% with behavioral weight loss
- 13.3% for controls.
Patients in the intervention groups lost some weight, but no group showed significant changes in BMI.18 The manual used in this study is available in bookstores and online (see Related resources for patients and clinicians).
Although somewhat less effective than therapist-led CBT, guided self-help is easy to implement in a general psychiatric practice.
A randomized, controlled trial compared CBT with IPT in 20 weekly group sessions. Posttreatment remission rates were equivalent—79% for CBT versus 73% for IPT—and weight in both groups was essentially unchanged.19
Abstinence rates after group DBT were 89% in a randomized, controlled trial of 44 women with BED. Binge eating improved significantly more in those assigned to DBT, compared with wait-listed controls. Differences in weight and mood were not significant, and abstinence rates slipped to 56% 6 months after DBT ended.20
Self-monitor
- Keep detailed records of all dietary intake
- Look for patterns in timing, type, and amount of food eaten
- Note antecedents and consequences of binges
Eat regularly
- Have 3 planned meals and 2 snacks per day
- Reduce cues to eat at other times
Substitute other behaviors for bingeing
- List pleasant alternate activities
- Recognize urges to binge
- Choose a substitute activity
- Review efficacy of substitute behaviors in preventing binges
Revise erroneous thinking patterns
- Reduce unrealistic expectations (especially about weight loss)
- Minimize self-criticism in response to lapses
- Change polarized thinking (“I’ve blown my diet; I may as well binge.”)
Limit vulnerabilities to relapse
- Reduce concerns about weight and shape
- Address problems with self-esteem, depression, or anxiety
- Maintain realistic expectations
Source: Fairburn CG. Overcoming binge eating. New York: Guilford Press; 1995.
Medications evaluated for BED in randomized, placebo-controlled trials include selective serotonin reuptake inhibitors (SSRIs) and a tricyclic, obesity management agents (sibutramine and orlistat), and topiramate (Box 5). Binge eating remission rates were highest with antidepressants, and patients lost the most weight with orlistat and sibutramine.
| Medication | Dosage (mg/d) | Duration (weeks) | N | BED remission (%) | Weight loss (kg)* | |
|---|---|---|---|---|---|---|
| Drug | Placebo | |||||
| Citalopram | 20 to 60 | 6 | 38 | 47 | 21 | 2.3 |
| Desipramine | 100 to 300 | 8 | 23 | 60 | 15 | 2.3 |
| Fluoxetine | 20 to 80 | 6 | 60 | 45 | 21 | 4.6 |
| Fluvoxamine | 50 to 300 | 9 | 85 | 38 | 26 | 1.7 |
| Orlistat | 120 tid | 24 | 89 | 23 | 29 | 5.1 |
| Sertraline | 50 to 200 | 6 | 34 | 47 | 14 | 4.4 |
| Sibutramine† | 15 | 12 | 60 | Not reported | 8.8 | |
| Topiramate | 50 to 600 | 14 | 58 | 64 | 30 | 4.8 |
| * Difference between weight lost with drug and weight lost with placebo | ||||||
| † Sibutramine is a controlled substance (schedule IV) and is recommended only for obese patients with BMI ≥30 (≥27 if cardiac risk factors are present). Do not use with monoamine oxidase inhibitors or serotonergic agents, and monitor blood pressure. | ||||||
| Source: Carter WP, Hudson JI, Lalonde JK, et al. Pharmacologic treatment of binge eating disorder. Int J Eat Disord 2003;34:S74-S88 | ||||||
Combining CBT with medications or exercise has also been evaluated for BED in randomized, controlled trials:21
- Group CBT and fluoxetine, 60 mg/d, were compared with placebo in 108 patients. After 16 weeks, intent-to-treat remission rates were 22% (fluoxetine), 26% (placebo), 50% (CBT + fluoxetine), and 61% (CBT + placebo). Weight loss did not differ significantly among treatments but was associated with binge eating remission.
- Guided self-help CBT combined with orlistat, 120 mg tid, or placebo were compared in 50 patients. After 12 weeks, intent-to-treat remission rates were significantly higher with orlistat (64% versus 36%) but not 3 months later (52% each). Weight loss of ≥5% was seen in 36% of those taking orlistat and in 8% taking placebo.
- Binge eating abstinence doubled when exercise (45 minutes. 3 times/week) was added to CBT; weight loss and mood also improved.
Little is known about appropriate dosages and durations for treating BED. Based on bulimia studies, most experts recommend higher-than-usual SSRI dosing (such as fluoxetine, 60 mg/d) and continuing treatment at least 6 months.22
Behavioral weight-loss programs have not been evaluated for BED in randomized, controlled trials. Obese persons with BED experience weight loss equivalent to that of those without BED, however, and more than one-half of persons with BED stop bingeing.9
Most programs combine reduced-calorie diets, increased activity, and behavior modification. Obese patients typically experience a 10% weight loss across 4 months to 1 year, but without continued intervention their weight returns to baseline.23 Weight Watchers is one behavioral weight-loss program with documented efficacy in controlled trials.24
Advocating calorie restriction for binge-eating patients has been controversial because dieting plays a role in triggering and maintaining bulimia nervosa. Recent evidence suggests, however, that binge eating disorder can be safely managed with dieting. In a randomized, controlled trial, 123 obese women without BED were randomly assigned to 3 groups:
- 1,000 kcal/d liquid meal replacement
- 1,200 to 1,500 kcal/d diet of conventional food
- a non-dieting approach to weight control.
Weight and depressive symptoms declined significantly among women in the two dieting groups but not in non-dieters. More episodes of binge eating were observed in subjects on the liquid diet at week 28, but no differences were seen at weeks 40 and 65, and no subjects in any group developed bulimia or binge eating disorder.25
Surprisingly, a 2003 review found that weight loss treatment that ignores bingeing is as effective in reducing bingeing as treatment that focuses solely on that symptom.22
Recommendations. A variety of treatments may be effective for BED, but no guidelines exist to help you choose among them. CBT is considered the treatment of choice, but most overweight BED patients require adjunctive exercise, medication, or behavioral weight-loss treatment.
We recommend that you base each patient’s treatment on five factors:
- treatment availability and cost
- past treatment response
- patient preference
- psychiatric and medical comorbidities
- BMI and past weight-loss experience.
For example, self-help CBT plus exercise or orlistat might benefit an obese man with bipolar disorder who was unable to tolerate adjunctive topiramate. An overweight depressed woman who needs weight-loss support could be given sertraline and encouraged to attend Weight Watchers.
Educate patients about realistic weight loss goals. A reasonable expectation is to lose 0.5 to 2 lbs/week, for a 10% loss across 6 months. Refer to guidelines for obesity risk assessment and treatment23 when advising patients about exercise and weight loss.
Treating night eating syndrome
Research into NES is just beginning, and one small, randomized trial has been published. Twenty patients with NES were randomly assigned to sit quietly or practice progressive muscle relaxation 20 minutes/day for 1 week. Muscle relaxation was associated with improved stress, anxiety, and depression scores, along with trends toward reduced nocturnal eating.26
This study supports a role for stress and anxiety in NES and suggests a potentially effective treatment. These results need to be replicated, however. In other preliminary work:
- After 12 weeks of sertraline therapy (average 188 mg/d), 17 obese patients with NES were eating less often at night, taking in fewer calories after the evening meal, and awakening less often. Five patients (29%) experienced remission, with an average weight loss of 4.8 kg.27
- One of two NES patients treated with topiramate (mean dose 218 mg at night) experienced remission and the other a marked response. Sleep improved, and average weight loss was 11 kg across 8 months.28
- One woman, age 51, with NES and nonseasonal depression experienced remission of depression and NES after 14 phototherapy sessions. NES returned when light therapy was discontinued.29
Recommendations. Suggest that NES patients start progressive muscle relaxation (see Related resources for instructions, or patients can purchase audiotapes). If benefits are insufficient, consider adjunctive sertraline, topiramate, or phototherapy. The efficacy of self-help for NES has not been evaluated, although a manual is available (see Related resources).
For clinicians
- Devlin MJ, Yanovski SZ, Wilson GT. Obesity: What mental health professionals need to know. Am J Psychiatry 2000;157:854-66.
- Instructions for progressive muscle relaxation. www.webmd.com/hw/health_guide_atoz/ta4146.asp?navbar=hw153409.
For patients and clinicians
- Anorexia and related eating disorders. www.anred.com (information about BED and NES).
- Self-help manuals available at bookstores or at Gürze Books (www.gurze.com):
- Fairburn CG. Overcoming binge eating. New York: Guilford Press, 1995.
- Allison KC, Stunkard AJ, Thier SL. Overcoming night eating syndrome: A step-by-step guide to breaking the cycle. Oakland, CA: New Harbinger Publications; 2004.
- Weight Control Information Network (WIN). National Institute of Diabetes and Digestive and Kidney Diseases. http://win.niddk.nih.gov
Drug brand names
- Citalopram • Celexa
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Orlistat • Xenical
- Sertraline • Zoloft
- Sibutramine • Meridia
- Topiramate • Topamax
Disclosures
Dr. Cloak owns Pfizer Inc. stock but otherwise reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Powers reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Taraldsen KW, Eriksen L, Gotestam KG. Prevalence of eating disorders among Norwegian women and men in a psychiatric outpatient unit. Int J Eat Disord 1996;20:185-90.
2. Lundgren JD, Allison KC, Crow S, et al. Prevalence of the night-eating syndrome in a psychiatric population. Am J Psychiatry 2006;163:156-8.
3. Streigel-Moore RH, Franko DL. Epidemiology of binge eating disorder. Int J Eating Disord 2003;34:S19-S29.
4. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9:83-7.
5. Marshall HM, Allison KC, O’Reardon JP, et al. Night eating syndrome among nonobese persons. Int J Eat Disord 2004;35:217-22.
6. Spitzer RL, Yanovski S, Wadden T, et al. Binge eating disorder: its further validation in a multisite study. Int J Eat Disord 1993;13:137-53.
7. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;37:659-65.
8. Fontaine KR, Redden DT, Wang C, et al. Years of life lost due to obesity. JAMA 2003;289:187-93.
9. Rieger E, Wilfley DE, Stein RI, et al. A comparison of quality of life in obese individuals with and without binge eating disorder. Int J Eat Disord 2005;37:234-40.
10. Yanovski SZ, Nelson JE, Dubbert BK, Spitzer RL. Association of binge eating disorder and psychiatric co-morbidity in obese subjects. Am J Psychiatry 1993;150:1472-9.
11. Gluck ME, Geliebter A, Satov T. Night eating syndrome is associated with depression, low self-esteem, reduced daytime hunger, and less weight loss in obese outpatients. Obes Res 2001;9:264-7.
12. Stunkard AJ, Allison KC. Two forms of disordered eating in obesity: Binge eating and night eating. Int J Obes Relat Metab Disord 2003;7:1-12.
13. O’Reardon JP, Peshek A, Allison K. Night eating syndrome: Diagnosis, epidemiology, and management. CNS Drugs 2005;19:997-1008.
14. Agras WS, Teich CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge-eating disorder. J Consult Clin Psychol 1997;65:343-7.
15. Cloak NL, Powers PS. Are undiagnosed eating disorders keeping your patients sick? Current Psychiatry 2005;4(12):65-75.
16. Kushner RF, Roth JL. Medical evaluation of the obese individual. Psychiatr Clin North Am 2005;28:89-103.
17. Wonderlich SA, de Zwaan M, Mitchell JE, et al. Psychological and dietary treatments of binge eating disorder: conceptual implications. Int J Eat Disord 2003;34:S58-S73.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge-eating disorder. Behav Res Ther 2005;43:1509-25.
19. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal therapy for the treatment of overweight individuals with binge eating disorder. Arch Gen Psychiatry 2002;59:713-21.
20. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
21. Pendleton VR, Goodrick CK, Poston WS, et al. Exercise augments the effects of cognitive-behavioral therapy in the treatment of binge eating. Int J Eat Disord 2002;31:172-84.
22. Agras WS. Pharmacotherapy of bulimia nervosa and binge eating disorder: longer-term outcomes. Psychopharmacol Bull 1997;33:433-6.
23. Clinical guidelines on the identification evaluation and treatment of obesity in adults Executive summary, 1998. Bethesda, MD: National Heart, Lung, and Blood Institute. Available at: http://www.nhlbi.nih.gov/guidelines/obesity. Accessed April 18, 2006.
24. Tsai AG, Wadden TA, Womble LG, Byrne KJ. Commercial and self-help programs for weight control. Psychiatr Clin North Am 2005;28:171-92.
25. Wadden TA, Foster GD, Sarwer DB, et al. Dieting and the development of eating disorders in obese women: Results of a randomized controlled trial. Am J Clin Nutr 2004;80:560-8.
26. Pawlow LA, O’Neil PM, Malcolm RJ. Night eating syndrome: Effects of brief relaxation training on stress, mood, hunger, and eating patterns. Int J Obes Relat Metab Disord 2003;27:970-8.
27. O’Reardon JP, Stunkard AJ, Allison KC. A clinical trial of sertraline in the treatment of night eating syndrome. Int J Eat Disord 2004;35:16-26.
28. Winkelman JW. Treatment of nocturnal eating syndrome and sleep-related eating disorder with topiramate. Sleep Med 2003;4(3):243-6.
29. Friedman S, Even C, Dardennes R, Guelfi JD. Light therapy, obesity, and night-eating syndrome. Am J Psychiatry 2002;159:875-6.
1. Taraldsen KW, Eriksen L, Gotestam KG. Prevalence of eating disorders among Norwegian women and men in a psychiatric outpatient unit. Int J Eat Disord 1996;20:185-90.
2. Lundgren JD, Allison KC, Crow S, et al. Prevalence of the night-eating syndrome in a psychiatric population. Am J Psychiatry 2006;163:156-8.
3. Streigel-Moore RH, Franko DL. Epidemiology of binge eating disorder. Int J Eating Disord 2003;34:S19-S29.
4. Striegel-Moore RH, Wilfley DE, Pike KM, et al. Recurrent binge eating in black American women. Arch Fam Med 2000;9:83-7.
5. Marshall HM, Allison KC, O’Reardon JP, et al. Night eating syndrome among nonobese persons. Int J Eat Disord 2004;35:217-22.
6. Spitzer RL, Yanovski S, Wadden T, et al. Binge eating disorder: its further validation in a multisite study. Int J Eat Disord 1993;13:137-53.
7. Fairburn CG, Cooper Z, Doll HA, et al. The natural course of bulimia nervosa and binge eating disorder in young women. Arch Gen Psychiatry 2000;37:659-65.
8. Fontaine KR, Redden DT, Wang C, et al. Years of life lost due to obesity. JAMA 2003;289:187-93.
9. Rieger E, Wilfley DE, Stein RI, et al. A comparison of quality of life in obese individuals with and without binge eating disorder. Int J Eat Disord 2005;37:234-40.
10. Yanovski SZ, Nelson JE, Dubbert BK, Spitzer RL. Association of binge eating disorder and psychiatric co-morbidity in obese subjects. Am J Psychiatry 1993;150:1472-9.
11. Gluck ME, Geliebter A, Satov T. Night eating syndrome is associated with depression, low self-esteem, reduced daytime hunger, and less weight loss in obese outpatients. Obes Res 2001;9:264-7.
12. Stunkard AJ, Allison KC. Two forms of disordered eating in obesity: Binge eating and night eating. Int J Obes Relat Metab Disord 2003;7:1-12.
13. O’Reardon JP, Peshek A, Allison K. Night eating syndrome: Diagnosis, epidemiology, and management. CNS Drugs 2005;19:997-1008.
14. Agras WS, Teich CF, Arnow B, et al. One-year follow-up of cognitive-behavioral therapy for obese individuals with binge-eating disorder. J Consult Clin Psychol 1997;65:343-7.
15. Cloak NL, Powers PS. Are undiagnosed eating disorders keeping your patients sick? Current Psychiatry 2005;4(12):65-75.
16. Kushner RF, Roth JL. Medical evaluation of the obese individual. Psychiatr Clin North Am 2005;28:89-103.
17. Wonderlich SA, de Zwaan M, Mitchell JE, et al. Psychological and dietary treatments of binge eating disorder: conceptual implications. Int J Eat Disord 2003;34:S58-S73.
18. Grilo CM, Masheb RM. A randomized controlled comparison of guided self-help cognitive behavioral therapy and behavioral weight loss for binge-eating disorder. Behav Res Ther 2005;43:1509-25.
19. Wilfley DE, Welch RR, Stein RI, et al. A randomized comparison of group cognitive-behavioral therapy and group interpersonal therapy for the treatment of overweight individuals with binge eating disorder. Arch Gen Psychiatry 2002;59:713-21.
20. Telch CF, Agras WS, Linehan MM. Dialectical behavior therapy for binge eating disorder. J Consult Clin Psychol 2001;69:1061-5.
21. Pendleton VR, Goodrick CK, Poston WS, et al. Exercise augments the effects of cognitive-behavioral therapy in the treatment of binge eating. Int J Eat Disord 2002;31:172-84.
22. Agras WS. Pharmacotherapy of bulimia nervosa and binge eating disorder: longer-term outcomes. Psychopharmacol Bull 1997;33:433-6.
23. Clinical guidelines on the identification evaluation and treatment of obesity in adults Executive summary, 1998. Bethesda, MD: National Heart, Lung, and Blood Institute. Available at: http://www.nhlbi.nih.gov/guidelines/obesity. Accessed April 18, 2006.
24. Tsai AG, Wadden TA, Womble LG, Byrne KJ. Commercial and self-help programs for weight control. Psychiatr Clin North Am 2005;28:171-92.
25. Wadden TA, Foster GD, Sarwer DB, et al. Dieting and the development of eating disorders in obese women: Results of a randomized controlled trial. Am J Clin Nutr 2004;80:560-8.
26. Pawlow LA, O’Neil PM, Malcolm RJ. Night eating syndrome: Effects of brief relaxation training on stress, mood, hunger, and eating patterns. Int J Obes Relat Metab Disord 2003;27:970-8.
27. O’Reardon JP, Stunkard AJ, Allison KC. A clinical trial of sertraline in the treatment of night eating syndrome. Int J Eat Disord 2004;35:16-26.
28. Winkelman JW. Treatment of nocturnal eating syndrome and sleep-related eating disorder with topiramate. Sleep Med 2003;4(3):243-6.
29. Friedman S, Even C, Dardennes R, Guelfi JD. Light therapy, obesity, and night-eating syndrome. Am J Psychiatry 2002;159:875-6.
Learning from lab-rat love
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
Get creative to manage dementia-related behaviors
Mrs. A, age 82, has advanced Alzheimer’s disease and has resided in a nursing home for 2 years. She does not recognize that she lives in a nursing home and waits by the door for her son to take her home. She spends her days weeping, telling visitors and staff she has been abandoned and must go home to care for her children.
Recently she has been wandering from the facility. When staff attempt to direct her away from the door, she resists, becomes physically aggressive, and hollers loudly. Her behavior bothers visitors and other patients, who frequently complain.
Her primary care physician prescribes a trial of olanzapine, 10 mg/d, but she becomes confused and suffers a fall. Staff report that Mrs. A is sleeping poorly and losing weight.
Deciding how to manage agitation, aggression, or psychotic symptoms of dementia is dicey at best. You can try an atypical antipsychotic despite the FDA’s black-box warning (Risks of using vs. not using atypical antipsychotics. Current Psychiatry 2005;4(8):14-28.
- Carbamazepine • Carbatrol
- Donepezil • Aricept
- Lorazepam • Ativan
- Memantine • Namenda
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Trazodone • Desyrel
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series: Treatment of dementia and its behavioral disturbances. Minneapolis, MN: McGraw-Hill; 2005.
2. Schneider LS, Dagerman LS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: Meta-analysis of randomized placebo-controlled trials. JAMA 2005;294:1934-43.
3. FDA Talk Paper. FDA issues public health advisory for antipsychotic drugs used for treatment of behavioral disorders in elderly patients. U.S. Food and Drug Administration. Available at: http://www.fda.gov/bbs/topics/ANSWERS/2005/ANS01350.html. Accessed March 10, 2006.
4. Treatment of agitation in older persons with dementia. The Expert Consensus Panel for Agitation in Dementia. Postgrad Med 1998;SPEC NO:1-88.
5. Sutor B, Rummans TA, Smith GE. Assessment and management of behavioral disturbances in nursing home patients with dementia. Mayo Clin Proc 2001;76:540-50.
6. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia in Lewy bodies: Third report of the DLB consortium. Neurology 2005;65:1863-72.
7. Feldman H, Lyketsos LD, Steinberg QM, et al. A 24-week, randomized, double-blind study of donepezil in moderate to severe Alzheimer’s disease. Arch Gen Psychiatry 2003;60:737-46.
8. Sink KM, Holden KF, Yaffe K. Pharmacologic treatment of neuropsychiatric symptoms of dementia: A review of the evidence. JAMA 2005;293:596-608.
9. Gauthier S, Wirth Y, Mobius HJ. Effects of memantine on behavioral symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomized controlled studies. Int J Geriatr Psychiatry 2005;20:259-62.
10. Porsteinsson AP, Tariot PN, Jakimovich LJ, et al. Valproate therapy for agitation in dementia: Open-label extension of a double-blind trial. Am J Geriatr Psychiatry 2003;11:434-40.
11. Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer’s disease complicated by agitation: A randomized controlled trial. Am J Geriatr Psychiatry 2005;13:942-9.
Mrs. A, age 82, has advanced Alzheimer’s disease and has resided in a nursing home for 2 years. She does not recognize that she lives in a nursing home and waits by the door for her son to take her home. She spends her days weeping, telling visitors and staff she has been abandoned and must go home to care for her children.
Recently she has been wandering from the facility. When staff attempt to direct her away from the door, she resists, becomes physically aggressive, and hollers loudly. Her behavior bothers visitors and other patients, who frequently complain.
Her primary care physician prescribes a trial of olanzapine, 10 mg/d, but she becomes confused and suffers a fall. Staff report that Mrs. A is sleeping poorly and losing weight.
Deciding how to manage agitation, aggression, or psychotic symptoms of dementia is dicey at best. You can try an atypical antipsychotic despite the FDA’s black-box warning (Risks of using vs. not using atypical antipsychotics. Current Psychiatry 2005;4(8):14-28.
- Carbamazepine • Carbatrol
- Donepezil • Aricept
- Lorazepam • Ativan
- Memantine • Namenda
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Trazodone • Desyrel
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mrs. A, age 82, has advanced Alzheimer’s disease and has resided in a nursing home for 2 years. She does not recognize that she lives in a nursing home and waits by the door for her son to take her home. She spends her days weeping, telling visitors and staff she has been abandoned and must go home to care for her children.
Recently she has been wandering from the facility. When staff attempt to direct her away from the door, she resists, becomes physically aggressive, and hollers loudly. Her behavior bothers visitors and other patients, who frequently complain.
Her primary care physician prescribes a trial of olanzapine, 10 mg/d, but she becomes confused and suffers a fall. Staff report that Mrs. A is sleeping poorly and losing weight.
Deciding how to manage agitation, aggression, or psychotic symptoms of dementia is dicey at best. You can try an atypical antipsychotic despite the FDA’s black-box warning (Risks of using vs. not using atypical antipsychotics. Current Psychiatry 2005;4(8):14-28.
- Carbamazepine • Carbatrol
- Donepezil • Aricept
- Lorazepam • Ativan
- Memantine • Namenda
- Mirtazapine • Remeron
- Olanzapine • Zyprexa
- Oxazepam • Serax
- Trazodone • Desyrel
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series: Treatment of dementia and its behavioral disturbances. Minneapolis, MN: McGraw-Hill; 2005.
2. Schneider LS, Dagerman LS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: Meta-analysis of randomized placebo-controlled trials. JAMA 2005;294:1934-43.
3. FDA Talk Paper. FDA issues public health advisory for antipsychotic drugs used for treatment of behavioral disorders in elderly patients. U.S. Food and Drug Administration. Available at: http://www.fda.gov/bbs/topics/ANSWERS/2005/ANS01350.html. Accessed March 10, 2006.
4. Treatment of agitation in older persons with dementia. The Expert Consensus Panel for Agitation in Dementia. Postgrad Med 1998;SPEC NO:1-88.
5. Sutor B, Rummans TA, Smith GE. Assessment and management of behavioral disturbances in nursing home patients with dementia. Mayo Clin Proc 2001;76:540-50.
6. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia in Lewy bodies: Third report of the DLB consortium. Neurology 2005;65:1863-72.
7. Feldman H, Lyketsos LD, Steinberg QM, et al. A 24-week, randomized, double-blind study of donepezil in moderate to severe Alzheimer’s disease. Arch Gen Psychiatry 2003;60:737-46.
8. Sink KM, Holden KF, Yaffe K. Pharmacologic treatment of neuropsychiatric symptoms of dementia: A review of the evidence. JAMA 2005;293:596-608.
9. Gauthier S, Wirth Y, Mobius HJ. Effects of memantine on behavioral symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomized controlled studies. Int J Geriatr Psychiatry 2005;20:259-62.
10. Porsteinsson AP, Tariot PN, Jakimovich LJ, et al. Valproate therapy for agitation in dementia: Open-label extension of a double-blind trial. Am J Geriatr Psychiatry 2003;11:434-40.
11. Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer’s disease complicated by agitation: A randomized controlled trial. Am J Geriatr Psychiatry 2005;13:942-9.
1. Alexopoulos GS, Jeste DV, Chung H, et al. The expert consensus guideline series: Treatment of dementia and its behavioral disturbances. Minneapolis, MN: McGraw-Hill; 2005.
2. Schneider LS, Dagerman LS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: Meta-analysis of randomized placebo-controlled trials. JAMA 2005;294:1934-43.
3. FDA Talk Paper. FDA issues public health advisory for antipsychotic drugs used for treatment of behavioral disorders in elderly patients. U.S. Food and Drug Administration. Available at: http://www.fda.gov/bbs/topics/ANSWERS/2005/ANS01350.html. Accessed March 10, 2006.
4. Treatment of agitation in older persons with dementia. The Expert Consensus Panel for Agitation in Dementia. Postgrad Med 1998;SPEC NO:1-88.
5. Sutor B, Rummans TA, Smith GE. Assessment and management of behavioral disturbances in nursing home patients with dementia. Mayo Clin Proc 2001;76:540-50.
6. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia in Lewy bodies: Third report of the DLB consortium. Neurology 2005;65:1863-72.
7. Feldman H, Lyketsos LD, Steinberg QM, et al. A 24-week, randomized, double-blind study of donepezil in moderate to severe Alzheimer’s disease. Arch Gen Psychiatry 2003;60:737-46.
8. Sink KM, Holden KF, Yaffe K. Pharmacologic treatment of neuropsychiatric symptoms of dementia: A review of the evidence. JAMA 2005;293:596-608.
9. Gauthier S, Wirth Y, Mobius HJ. Effects of memantine on behavioral symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomized controlled studies. Int J Geriatr Psychiatry 2005;20:259-62.
10. Porsteinsson AP, Tariot PN, Jakimovich LJ, et al. Valproate therapy for agitation in dementia: Open-label extension of a double-blind trial. Am J Geriatr Psychiatry 2003;11:434-40.
11. Tariot PN, Raman R, Jakimovich L, et al. Divalproex sodium in nursing home residents with possible or probable Alzheimer’s disease complicated by agitation: A randomized controlled trial. Am J Geriatr Psychiatry 2005;13:942-9.
Traumatic brain injury: Choosing drugs to assist recovery
Choosing medications for patients with traumatic brain injury (TBI) requires caution; some drugs slow their recovery, and no standard post-TBI treatment exists.
As consulting psychiatrist on a TBI rehabilitation team, I am asked to manage enduring cognitive and emotional problems—aggression, apathy, learning disabilities, dementia—in patients with moderate to severe head injuries. This article describes how we apply available evidence to treat neurobehavioral symptoms in these patients.
Case: An iraq war casualty
The physical medicine and rehabilitation service asks for help in managing agitation, anxiety, and nightmares in Mr. N, age 20, a U.S. combat soldier. While on patrol 2 months ago in Iraq, he suffered a penetrating right frontoparietal brain injury from an improvised explosive device.
Mr. N has undergone a right temporoparietal craniectomy with debridement, ventriculostomy placement, and scalp flap closure. He has had seizures and then pancreatitis—thought to be caused by divalproex prescribed to treat the seizures. Divalproex was replaced with phenytoin at our hospital, and the pancreatitis resolved.
How serious an injury?
TBI ranges from self-limited concussion to devastating, permanent CNS impairment and life-long disability. Brain injuries from sudden impact—from assaults, falls, motor vehicle accidents, combat, or sports—can cause diffuse axonal injury and confusion or unconsciousness, even without radiographic evidence of cerebral bleeding, edema, or mass effect.
No hierarchy or nomenclature is universally accepted for TBI. The term “concussion” is generally used for milder injury and TBI for more-severe injuries.
Concussion. The American Academy of Neurology defines concussion as a trauma-induced alteration in mental status that may or may not involve loss of consciousness. Confusion and amnesia—the hallmarks of concussion—may occur immediately after the head trauma or several minutes later.1 This definition recognizes three concussion grades:
- Grade 1: confusion lasts
- Grade 2: confusion persists >15 minutes but without LOC
- Grade 3: concussion with LOC. The confusional state is marked by disorientation, delayed verbal and motor responses, inattention, incoordination, emotional lability, and slurred or incoherent speech.
- Mild TBI: GCS 13 to 15, LOC 1,3
- Moderate TBI: GCS 9 to 12, LOC 30 minutes to 7 days, and PTA 24 hours to 7 days.
- Severe TBI: GCS ≤8, LOC, and PTA >7 days,4 or any focal neuroimaging abnormalities.3
Using Glasgow Coma Scale scores to evaluate brain injury severity
| Component | Response | Score |
|---|---|---|
| Best eye response | No eye opening | 1 |
| Eye opening to pain | 2 | |
| Eye opening to verbal command | 3 | |
| Eyes open spontaneously | 4 | |
| Best verbal response | No verbal response | 1 |
| Incomprehensible sounds | 2 | |
| Inappropriate words | 3 | |
| Confused | 4 | |
| Oriented | 5 | |
| Best motor response | No motor response | 1 |
| Extension to pain | 2 | |
| Flexion to pain | 3 | |
| Withdrawal from pain | 4 | |
| Localizing pain | 5 | |
| Obeys commands | 6 | |
| GCS total score ≥12 is mild injury, 9 to 11 is moderate, and ≤8 is severe (90% of patients with scores ≤8 are in a coma). Coma is defined as not opening eyes, not obeying commands, and not saying understandable words. Composite scores with eye, verbal, and motor responses (such as E3V3M5) are clinically more useful than totals. | ||
| Source: Reference 2. | ||
Case continued: ‘They’re hurting me’
Mr. N meets criteria for severe TBI. He is periodically agitated and aggressive and refuses to return to physical therapy, complaining that rehabilitation nurses are intentionally hurting him. He occasionally hits the staff and throws things. His medications include:
- phenytoin, 100 mg every 6 hours for seizure prophylaxis
- lamotrigine, 50 mg bid for seizure prophylaxis
- zolpidem, 5 mg as needed at bedtime for pain
- methadone, 10 mg/d for pain
- oxycodone, 5 mg every 4 hours as needed for breakthrough pain.
Assessing progress
For patients such as Mr. N, TBI recovery progress is measured with the Rancho Los Amigos Scale.
The original Rancho scale—developed in 1972 by staff at the Rancho Los Amigos rehabilitation hospital in Downey, CA—described eight levels of cognitive and adaptive functioning, from coma and total care through normal cognition and independence. A 1997 revised version separates the highest cognitive functioning level (VIII, purposeful, appropriate function) into three parts, expanding the scale to 10 levels (Table 2).5
Of course, not all TBI patients begin recovery at Rancho level I, and unfortunately not all achieve level X. Some experience dementia caused by head trauma, with persistent memory impairment and cognitive deficits in language, apraxia, agnosia, or executive function.6
Most patients recover as predicted by the initial injury’s severity. Others experience diffuse cerebral swelling with sudden, rapid deterioration after what appeared to be a grade 1 or grade 2 concussion. Diffuse cerebral swelling is sometimes considered a “second-impact syndrome,” but it can also occur after a single impact.7 A second TBI is not universally believed to cause the precipitous decline, but animal studies suggest an additive effect of rapid sequential TBI.8
Table 2
10-level Rancho Los Amigos Scale for assessing TBI recovery
| Level | Cognitive and adaptive function | Assistance required |
|---|---|---|
| I | No response | Total assistance |
| II | Generalized response | Total assistance |
| III | Localized response | Total assistance |
| IV | Confused/agitated | Maximal assistance |
| V | Confused, inappropriate non-agitated | Maximal assistance |
| VI | Confused, appropriate | Moderate assistance |
| VII | Automatic, appropriate | Minimal assistance |
| VIII | Purposeful, appropriate | Stand-by assistance |
| IX | Purposeful, appropriate | Stand-by assistanceon request |
| X | Purposeful, appropriate | Modified independent |
| Source: Traumatic Brain Injury Resource Guide. www.neuroskills.com. | ||
Recovery for a patient such as Mr. N with Rancho level IV to V TBI may be complicated by marked mood lability, spontaneous aggression, psychomotor agitation, extremely short attention with marked distractibility, little to no short-term memory, and noncooperation with treatment and care. Patients may also show disorders of diminished motivation, characterized by normal consciousness but decreased goal-directed behavior and affective flattening.9
Case continued: Calling in reinforcements
Besides combat nightmares, Mr. N is experiencing other signs of posttraumatic stress disorder (PTSD): intrusive memories of dead comrades, anhedonia, insomnia, irritability, and hypervigilance. We recommend a trial of citalopram, 10 mg/d, but within 1 week he becomes more irritable, agitated, and aggressive, with worsening sleep. We arrange a meeting to obtain collateral information from Mr. N’s aunt, mother, and clinical psychologist. We learn that a first-degree relative had bipolar disorder, and Mr. N lived with various relatives during childhood.
As a child, Mr. N was easily angered, hyperactive, unpredictably aggressive with peers, and impulsive. He was diagnosed with “explosive disorder” at age 8. A psychiatrist prescribed methylphenidate (which helped) and paroxetine (which worsened his behavior and aggression). Based on this history, we make a presumptive diagnosis of comorbid bipolar disorder.
Treating psychopathology
Comorbidities. Adolescents and adults with pre-existing attention-deficit/hyperactivity disorder or bipolar disorder may be predisposed to carelessness or risk taking that lead to accidents and TBI. Likewise, alcoholism and substance use disorders are risk factors for head injuries. These pre-existing conditions will complicate the post-TBI course and must be treated concurrently.
Depression and PTSD may follow a head injury and complicate recovery. In fact, post-TBI symptoms—poor sleep, poor memory and concentration, and irritability—are common to both depression and PTSD.
A team approach. Regardless of its severity or recovery stage, TBI requires multidisciplinary treatment. Physical, occupational, and speech therapies are essential initially. As recovery progresses, vocational rehabilitation may need to be added. Throughout rehabilitation, supportive individual and family therapy can help patients reintegrate into the community. Psychologists, neuropsychologists, and clinical social workers are indispensable to the treatment team.
Medication precautions
Using medications to manage post-TBI syndromes is difficult and controversial. No standard regimen exists, and few clinical trials guide treatment. Small, uncontrolled studies (human and animal) suggest commonly prescribed drugs may worsen outcomes (Table 3).10,11 For example:
- Cognitive function improved in three TBI patients after thioridazine was discontinued in two and haloperidol in one.12
- Haloperidol given to 11 patients with TBI made no difference in rehabilitation outcomes when compared with 15 patients who did not receive the antipsychotic. Those receiving haloperidol also had longer post-trauma amnesia (5 to 30 weeks), compared with the untreated group (1 to 18 weeks).13
- In animal studies of TBI, motor recovery was slowed with haloperidol but not olanzapine,14,15 and with clonidine,16 phenytoin,17 and trazodone.18 Phenobarbitol.19 and diazepam20 have been associated with delayed behavioral recovery and chronic behavior problems, respectively, in rats with TBI. How these agents might affect human patients is speculative.
Medications with potential to impede TBI recovery*
| Class | Medications |
|---|---|
| Alpha-2 agonist | Clonidine |
| Antidepressant | Trazodone |
| Antiepileptic | Phenytoin, phenobarbital |
| Benzodiazepine | Diazepam |
| Neuroleptic | Haloperidol, thioridazine |
| *Suggested by animal or clinical studies | |
| Source: References 11-20 | |
- Psychostimulants have improved recovery of motor function in animal trials if given before physical therapy.14
- Stimulants and dopaminergic agonists such as bromocriptine and amantadine might help disorders of diminished motivation.22
- Dextroamphetamine and methylphenidate have improved impulsivity, memory, and concentration in a patient with TBI.23
Table 4
Drugs considered safe and effective
for TBI neurobehavioral symptoms
| Target symptom(s) | Drug | Usual daily dosage* |
|---|---|---|
| Apathy | Amantadine | 100 to 400 mg |
| Bromocriptine | 1.25 to 100 mg | |
| Cognition | Donepezil | |
| Inattention | Dextroamphetamine | 5 to 60 mg |
| Methylphenidate | 10 to 60 mg | |
| Depression, PTSD symptoms | Fluoxetine | 20 to 80 mg |
| Agitation, mood stabilization | Anticonvulsants | |
| Lamotrigine | 25 to 200 mg | |
| Divalproex sodium | 10 to 15 mg/kg/day† | |
| Carbamazepine | 400 to 1,600 mg‡ | |
| Atypical antipsychotics | ||
| Olanzapine | 2.5 to 20 mg | |
| Quetiapine | 50 to 800 mg | |
| Risperidone | 0.5 to 6 mg | |
| Ziprasidone | 20 to 160 mg | |
| Beta blocker | ||
| Propranolol | 20 to 480 mg | |
| PTSD: posttraumatic stress disorder | ||
| * Dosage may be divided; see full prescribing information. | ||
| † Adjust dosage to achieve serum level of 50 to 100 mcg/mL. | ||
| ‡ Adjust dosage to achieve serum level of 4 to 12 mcg/mL. | ||
Small studies of anticonvulsants for post-TBI agitation report:
- valproic acid might improve behavioral control and decrease aggression, and it did not worsen performance on neuropsychological testing
- carbamazepine reduced agitation in seven TBI patients and reduced anger outbursts in 8 of 10 others
- gabapentin caused paradoxical effects in two TBI patients25
- lamotrigine improved agitation in one TBI patient.26
- propranolol, 420 to 520 mg/d
- pindolol, 60 mg/d
- metoprolol, 200 mg/d.21
Dosing atypical antipsychotics
for agitation and aggression in TBI
| Drug | Initial daily dosage* | Maximum daily dosage* |
|---|---|---|
| Aripiprazole | 2.5 to 5 mg | 30 mg |
| Olanzapine | 2.5 mg | 20 mg |
| Quetiapine | 12.5 to 50 mg | 800 mg |
| Risperidone | 0.25 mg | 8 mg |
| Ziprasidone | 20 mg | 160 mg |
| *Daily dosages may be divided | ||
Depression and PTSD in TBI patients are considered indications for selective serotonin reuptake inhibitors (SSRIs). Animal data suggest that fluoxetine is safe for patients with TBI,27 though no human data have been published.
For PTSD with bipolar depression, we usually prescribe lamotrigine or combine an atypical antipsychotic with an SSRI. Lithium would be second-line therapy. PTSD with bipolar mania is more difficult to treat because little evidence guides medication choices. As with depression and PTSD, we usually combine an atypical antipsychotic with an SSRI. We try to control manic and psychotic symptoms first, then add the SSRI for anxiety after the mood becomes more stable.
Cognitive impairment. A dozen published studies and case reports indicate that donepezil improves cognition in subacute and chronic TBI. For example:
- An open-label trial showed subjective improvement in cognitive functions in 8 of 10 patients given donepezil.28
- In a double-blind, placebo-controlled, crossover trial, short-term memory and attention improved with donepezil in 18 patients with post-acute TBI, as shown by neuropsychological test scores.29
- A retrospective case-control study showed no significant difference in cognitive outcome between controls and 18 patients prescribed donepezil but did suggest that cognition improved more rapidly when patients started donepezil earlier in recovery.30
Case continued: Back to rehab
We replace Mr. N’s phenytoin with carbamazepine, 700 mg/d (serum level about 12 mcg/mL), discontinue citalopram, and start him on quetiapine as a mood stabilizer, titrating the dosage to 600 mg/d over 3 weeks. We select quetiapine based on experience using it as a mood stabilizer and carbamazepine for additional mood stabilization and seizure prophylaxis.
We continue methadone and oxycodone at the same dosages for pain management, with good results. We eventually switch him from zolpidem to trazodone, 50 mg as needed at bedtime. We discontinue lamotrigine because he is no longer having seizures.
Mr. N tolerates quetiapine and carbamazepine well. The nursing staff reports he is much less irritable and aggressive and his sleep has improved, but he is not oversedated. He returns to and participates in physical, occupational, and speech therapies.
Tips for using medications
Many TBI patients are unusually sensitive to or intolerant of medication side effects. Because no randomized, controlled clinical trials support using any medication in these patients, be cautious. The following recommendations can help:
- Use psychotropics with a low risk of complications.
- Start with low dosages and increase gradually to assess side effects and efficacy of medication trials.
- Give full trials and adequate dosing before you decide a medication has not improved symptoms sufficiently.
- Monitor closely for side effects.
- Seek information from family members to evaluate a medication’s effectiveness, as patients’ cognitive deficits may limit their ability to reliably report symptoms.
Related resources
- Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press, 2005.
- Traumatic Brain Injury Resource Guide. www.neuroskills.com
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonidine • Catapres
- Dextroamphetamine • Dexedrine
- Diazepam • Valium
- Divalproex sodium • Depakote
- Donepezil • Aricept
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Methadone • Dolophine
- Methylphenidate • Ritalin
- Metoprolol • Lopressor
- Olanzapine • Zyprexa
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Pindolol • Visken
- Propranolol • Inderal
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Ziprasidone • Geodon
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Academy of Neurology. Practice parameter: The management of concussion in sports. Neurology 1997;48:581-5.
2. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;2(7872):81-4.
3. Alexander MP. Mild traumatic brain injury: Pathophysiology, natural history, and clinical management. Neurology 1995;45:1253-60.
4. Arlinghaus KA, Shoaib AM, Price TRP. Neuropsychiatric assessment. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press; 2005:59-78.
5. Hagen C, Malkmus D, Durham P. Communication Disorders Service, Rancho Los Amigos Rehabilitation Hospital, Downey, CA, 1972 (rev. 1997).
6. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 2000.
7. McCrory P. Does second impact syndrome exist? Clin J Sport Med 2001;11:144-9.
8. Vagnozzi R, Signoretti S, Tavazzi B, et al. Hypothesis of the postconcussive vulnerable brain: experimental evidence of its metabolic occurrence. Neurosurgery 2005;57:164-71.
9. Marin RS, Chakravorty S. Disorders of diminished motivation. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA; American Psychiatric Press; 2005:337-52.
10. Goldstein LB. Prescribing of potentially harmful drugs to patients admitted to hospital after head injury. J Neurol Neurosurg Psychiatry 1995;58:753-5.
11. Phillips JP, Devier DJ, Feeney DM. Rehabilitation pharmacology bridging laboratory work to clinical application. J Head Trauma Rehabil 2003;18:342-56.
12. Stanislaw SL. Cognitive effects of antipsychotic agents in persons with traumatic brain injury. Brain Injury 1997;11:335-41.
13. Rao N, Jellinek HM, Woolston DC. Agitation in closed head injury: haloperidol effects on rehabilitation outcome. Arch Phys Med Rehabil 1985;66:30-4.
14. Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science 1982;217:855-7.
15. Wilson MS, Gibson CL, Hamm RJ. Haloperidol, but not olanzapine, impairs cognitive performance after traumatic brain injury in rats. Am J Phys Med Rehabil 2003;82:871-9.
16. Goldstein LB, Davis JN. Clonidine impairs recovery of beamwalking after a sensorimotor cortex lesion in the rat. Brain Research 1990;508:305-9.
17. Brailowsky S, Knight RT, Efron R. Phenytoin increases the severity of cortical hemiplegia in rats. Brain Research 1986;376:71-7.
18. Boyeson MG, Harmon RL. Effects of trazodone and desipramine on motor recovery in brain-injured rats. Am J Phys Med Rehabil 1993;72:286-93.
19. Hernandez TD, Holling LC. Disruption of behavioral recovery by the anticonvulsant phenobarbital. Brain Research 1994;635:300-6.
20. Schallert T, Hernandez TD, Barth TM. Recovery of function after brain damage: severe and chronic disruption by diazepam. Brain Research 1986;379:104-11.
21. Deb S, Crownshaw T. The role of pharmacotherapy in the management of behavior disorders in traumatic brain injury patients. Brain Injury 2004;18:1-31.
22. Campbell JJ, Duffy JD. Treatment strategies in amotivated patients. Psychiatric Annals 1997;27(1):44-9.
23. Evans RW, Gualtieri CT, Patterson D. Treatment of chronic closed head injury with psychostimulant drugs: a controlled case study and an appropriate evaluation procedure. J Nerv Ment Dis 1987;175:106-10.
24. Elovic EP, Lansang R, Li Y, Ricker JH. The use of atypical antipsychotics in traumatic brain injury. J Head Trauma Rehabil 2003;18:177-95.
25. Lombard LA, Zafonte RD. Agitation after traumatic brain injury: considerations and treatment options. Am J Phys Med Rehabil 2005;84:797-812.
26. Pachet A, Friesen S, Winkelaar D, Gray S. Beneficial behavioural effects of lamotrigine in traumatic brain injury. Brain Injury 2003;17:715-22.
27. Boyeson MG, Harmon RL, Jones JL. Comparative effects of fluoxetine, amitriptyline, and serotonin on functional motor recovery after sensorimotor cortex injury. Am J Phys Med Rehabil 1994;73:76-83.
28. Khateb A, Ammann J, Annoni JM, Diserens K. Cognition enhancing effects of onepezil in traumatic brain injury (abstract). Eur Neurol 2005;54:39-45.
29. Zhang L, Plotkin RC, Wang G, et al. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004;85:1005-55.
30. Walker W, Seel R, Gibellato M, et al. The effects of donepezil on traumatic brain injury acute rehabilitation outcomes. Brain Inj 2004;18:739-50.
Choosing medications for patients with traumatic brain injury (TBI) requires caution; some drugs slow their recovery, and no standard post-TBI treatment exists.
As consulting psychiatrist on a TBI rehabilitation team, I am asked to manage enduring cognitive and emotional problems—aggression, apathy, learning disabilities, dementia—in patients with moderate to severe head injuries. This article describes how we apply available evidence to treat neurobehavioral symptoms in these patients.
Case: An iraq war casualty
The physical medicine and rehabilitation service asks for help in managing agitation, anxiety, and nightmares in Mr. N, age 20, a U.S. combat soldier. While on patrol 2 months ago in Iraq, he suffered a penetrating right frontoparietal brain injury from an improvised explosive device.
Mr. N has undergone a right temporoparietal craniectomy with debridement, ventriculostomy placement, and scalp flap closure. He has had seizures and then pancreatitis—thought to be caused by divalproex prescribed to treat the seizures. Divalproex was replaced with phenytoin at our hospital, and the pancreatitis resolved.
How serious an injury?
TBI ranges from self-limited concussion to devastating, permanent CNS impairment and life-long disability. Brain injuries from sudden impact—from assaults, falls, motor vehicle accidents, combat, or sports—can cause diffuse axonal injury and confusion or unconsciousness, even without radiographic evidence of cerebral bleeding, edema, or mass effect.
No hierarchy or nomenclature is universally accepted for TBI. The term “concussion” is generally used for milder injury and TBI for more-severe injuries.
Concussion. The American Academy of Neurology defines concussion as a trauma-induced alteration in mental status that may or may not involve loss of consciousness. Confusion and amnesia—the hallmarks of concussion—may occur immediately after the head trauma or several minutes later.1 This definition recognizes three concussion grades:
- Grade 1: confusion lasts
- Grade 2: confusion persists >15 minutes but without LOC
- Grade 3: concussion with LOC. The confusional state is marked by disorientation, delayed verbal and motor responses, inattention, incoordination, emotional lability, and slurred or incoherent speech.
- Mild TBI: GCS 13 to 15, LOC 1,3
- Moderate TBI: GCS 9 to 12, LOC 30 minutes to 7 days, and PTA 24 hours to 7 days.
- Severe TBI: GCS ≤8, LOC, and PTA >7 days,4 or any focal neuroimaging abnormalities.3
Using Glasgow Coma Scale scores to evaluate brain injury severity
| Component | Response | Score |
|---|---|---|
| Best eye response | No eye opening | 1 |
| Eye opening to pain | 2 | |
| Eye opening to verbal command | 3 | |
| Eyes open spontaneously | 4 | |
| Best verbal response | No verbal response | 1 |
| Incomprehensible sounds | 2 | |
| Inappropriate words | 3 | |
| Confused | 4 | |
| Oriented | 5 | |
| Best motor response | No motor response | 1 |
| Extension to pain | 2 | |
| Flexion to pain | 3 | |
| Withdrawal from pain | 4 | |
| Localizing pain | 5 | |
| Obeys commands | 6 | |
| GCS total score ≥12 is mild injury, 9 to 11 is moderate, and ≤8 is severe (90% of patients with scores ≤8 are in a coma). Coma is defined as not opening eyes, not obeying commands, and not saying understandable words. Composite scores with eye, verbal, and motor responses (such as E3V3M5) are clinically more useful than totals. | ||
| Source: Reference 2. | ||
Case continued: ‘They’re hurting me’
Mr. N meets criteria for severe TBI. He is periodically agitated and aggressive and refuses to return to physical therapy, complaining that rehabilitation nurses are intentionally hurting him. He occasionally hits the staff and throws things. His medications include:
- phenytoin, 100 mg every 6 hours for seizure prophylaxis
- lamotrigine, 50 mg bid for seizure prophylaxis
- zolpidem, 5 mg as needed at bedtime for pain
- methadone, 10 mg/d for pain
- oxycodone, 5 mg every 4 hours as needed for breakthrough pain.
Assessing progress
For patients such as Mr. N, TBI recovery progress is measured with the Rancho Los Amigos Scale.
The original Rancho scale—developed in 1972 by staff at the Rancho Los Amigos rehabilitation hospital in Downey, CA—described eight levels of cognitive and adaptive functioning, from coma and total care through normal cognition and independence. A 1997 revised version separates the highest cognitive functioning level (VIII, purposeful, appropriate function) into three parts, expanding the scale to 10 levels (Table 2).5
Of course, not all TBI patients begin recovery at Rancho level I, and unfortunately not all achieve level X. Some experience dementia caused by head trauma, with persistent memory impairment and cognitive deficits in language, apraxia, agnosia, or executive function.6
Most patients recover as predicted by the initial injury’s severity. Others experience diffuse cerebral swelling with sudden, rapid deterioration after what appeared to be a grade 1 or grade 2 concussion. Diffuse cerebral swelling is sometimes considered a “second-impact syndrome,” but it can also occur after a single impact.7 A second TBI is not universally believed to cause the precipitous decline, but animal studies suggest an additive effect of rapid sequential TBI.8
Table 2
10-level Rancho Los Amigos Scale for assessing TBI recovery
| Level | Cognitive and adaptive function | Assistance required |
|---|---|---|
| I | No response | Total assistance |
| II | Generalized response | Total assistance |
| III | Localized response | Total assistance |
| IV | Confused/agitated | Maximal assistance |
| V | Confused, inappropriate non-agitated | Maximal assistance |
| VI | Confused, appropriate | Moderate assistance |
| VII | Automatic, appropriate | Minimal assistance |
| VIII | Purposeful, appropriate | Stand-by assistance |
| IX | Purposeful, appropriate | Stand-by assistanceon request |
| X | Purposeful, appropriate | Modified independent |
| Source: Traumatic Brain Injury Resource Guide. www.neuroskills.com. | ||
Recovery for a patient such as Mr. N with Rancho level IV to V TBI may be complicated by marked mood lability, spontaneous aggression, psychomotor agitation, extremely short attention with marked distractibility, little to no short-term memory, and noncooperation with treatment and care. Patients may also show disorders of diminished motivation, characterized by normal consciousness but decreased goal-directed behavior and affective flattening.9
Case continued: Calling in reinforcements
Besides combat nightmares, Mr. N is experiencing other signs of posttraumatic stress disorder (PTSD): intrusive memories of dead comrades, anhedonia, insomnia, irritability, and hypervigilance. We recommend a trial of citalopram, 10 mg/d, but within 1 week he becomes more irritable, agitated, and aggressive, with worsening sleep. We arrange a meeting to obtain collateral information from Mr. N’s aunt, mother, and clinical psychologist. We learn that a first-degree relative had bipolar disorder, and Mr. N lived with various relatives during childhood.
As a child, Mr. N was easily angered, hyperactive, unpredictably aggressive with peers, and impulsive. He was diagnosed with “explosive disorder” at age 8. A psychiatrist prescribed methylphenidate (which helped) and paroxetine (which worsened his behavior and aggression). Based on this history, we make a presumptive diagnosis of comorbid bipolar disorder.
Treating psychopathology
Comorbidities. Adolescents and adults with pre-existing attention-deficit/hyperactivity disorder or bipolar disorder may be predisposed to carelessness or risk taking that lead to accidents and TBI. Likewise, alcoholism and substance use disorders are risk factors for head injuries. These pre-existing conditions will complicate the post-TBI course and must be treated concurrently.
Depression and PTSD may follow a head injury and complicate recovery. In fact, post-TBI symptoms—poor sleep, poor memory and concentration, and irritability—are common to both depression and PTSD.
A team approach. Regardless of its severity or recovery stage, TBI requires multidisciplinary treatment. Physical, occupational, and speech therapies are essential initially. As recovery progresses, vocational rehabilitation may need to be added. Throughout rehabilitation, supportive individual and family therapy can help patients reintegrate into the community. Psychologists, neuropsychologists, and clinical social workers are indispensable to the treatment team.
Medication precautions
Using medications to manage post-TBI syndromes is difficult and controversial. No standard regimen exists, and few clinical trials guide treatment. Small, uncontrolled studies (human and animal) suggest commonly prescribed drugs may worsen outcomes (Table 3).10,11 For example:
- Cognitive function improved in three TBI patients after thioridazine was discontinued in two and haloperidol in one.12
- Haloperidol given to 11 patients with TBI made no difference in rehabilitation outcomes when compared with 15 patients who did not receive the antipsychotic. Those receiving haloperidol also had longer post-trauma amnesia (5 to 30 weeks), compared with the untreated group (1 to 18 weeks).13
- In animal studies of TBI, motor recovery was slowed with haloperidol but not olanzapine,14,15 and with clonidine,16 phenytoin,17 and trazodone.18 Phenobarbitol.19 and diazepam20 have been associated with delayed behavioral recovery and chronic behavior problems, respectively, in rats with TBI. How these agents might affect human patients is speculative.
Medications with potential to impede TBI recovery*
| Class | Medications |
|---|---|
| Alpha-2 agonist | Clonidine |
| Antidepressant | Trazodone |
| Antiepileptic | Phenytoin, phenobarbital |
| Benzodiazepine | Diazepam |
| Neuroleptic | Haloperidol, thioridazine |
| *Suggested by animal or clinical studies | |
| Source: References 11-20 | |
- Psychostimulants have improved recovery of motor function in animal trials if given before physical therapy.14
- Stimulants and dopaminergic agonists such as bromocriptine and amantadine might help disorders of diminished motivation.22
- Dextroamphetamine and methylphenidate have improved impulsivity, memory, and concentration in a patient with TBI.23
Table 4
Drugs considered safe and effective
for TBI neurobehavioral symptoms
| Target symptom(s) | Drug | Usual daily dosage* |
|---|---|---|
| Apathy | Amantadine | 100 to 400 mg |
| Bromocriptine | 1.25 to 100 mg | |
| Cognition | Donepezil | |
| Inattention | Dextroamphetamine | 5 to 60 mg |
| Methylphenidate | 10 to 60 mg | |
| Depression, PTSD symptoms | Fluoxetine | 20 to 80 mg |
| Agitation, mood stabilization | Anticonvulsants | |
| Lamotrigine | 25 to 200 mg | |
| Divalproex sodium | 10 to 15 mg/kg/day† | |
| Carbamazepine | 400 to 1,600 mg‡ | |
| Atypical antipsychotics | ||
| Olanzapine | 2.5 to 20 mg | |
| Quetiapine | 50 to 800 mg | |
| Risperidone | 0.5 to 6 mg | |
| Ziprasidone | 20 to 160 mg | |
| Beta blocker | ||
| Propranolol | 20 to 480 mg | |
| PTSD: posttraumatic stress disorder | ||
| * Dosage may be divided; see full prescribing information. | ||
| † Adjust dosage to achieve serum level of 50 to 100 mcg/mL. | ||
| ‡ Adjust dosage to achieve serum level of 4 to 12 mcg/mL. | ||
Small studies of anticonvulsants for post-TBI agitation report:
- valproic acid might improve behavioral control and decrease aggression, and it did not worsen performance on neuropsychological testing
- carbamazepine reduced agitation in seven TBI patients and reduced anger outbursts in 8 of 10 others
- gabapentin caused paradoxical effects in two TBI patients25
- lamotrigine improved agitation in one TBI patient.26
- propranolol, 420 to 520 mg/d
- pindolol, 60 mg/d
- metoprolol, 200 mg/d.21
Dosing atypical antipsychotics
for agitation and aggression in TBI
| Drug | Initial daily dosage* | Maximum daily dosage* |
|---|---|---|
| Aripiprazole | 2.5 to 5 mg | 30 mg |
| Olanzapine | 2.5 mg | 20 mg |
| Quetiapine | 12.5 to 50 mg | 800 mg |
| Risperidone | 0.25 mg | 8 mg |
| Ziprasidone | 20 mg | 160 mg |
| *Daily dosages may be divided | ||
Depression and PTSD in TBI patients are considered indications for selective serotonin reuptake inhibitors (SSRIs). Animal data suggest that fluoxetine is safe for patients with TBI,27 though no human data have been published.
For PTSD with bipolar depression, we usually prescribe lamotrigine or combine an atypical antipsychotic with an SSRI. Lithium would be second-line therapy. PTSD with bipolar mania is more difficult to treat because little evidence guides medication choices. As with depression and PTSD, we usually combine an atypical antipsychotic with an SSRI. We try to control manic and psychotic symptoms first, then add the SSRI for anxiety after the mood becomes more stable.
Cognitive impairment. A dozen published studies and case reports indicate that donepezil improves cognition in subacute and chronic TBI. For example:
- An open-label trial showed subjective improvement in cognitive functions in 8 of 10 patients given donepezil.28
- In a double-blind, placebo-controlled, crossover trial, short-term memory and attention improved with donepezil in 18 patients with post-acute TBI, as shown by neuropsychological test scores.29
- A retrospective case-control study showed no significant difference in cognitive outcome between controls and 18 patients prescribed donepezil but did suggest that cognition improved more rapidly when patients started donepezil earlier in recovery.30
Case continued: Back to rehab
We replace Mr. N’s phenytoin with carbamazepine, 700 mg/d (serum level about 12 mcg/mL), discontinue citalopram, and start him on quetiapine as a mood stabilizer, titrating the dosage to 600 mg/d over 3 weeks. We select quetiapine based on experience using it as a mood stabilizer and carbamazepine for additional mood stabilization and seizure prophylaxis.
We continue methadone and oxycodone at the same dosages for pain management, with good results. We eventually switch him from zolpidem to trazodone, 50 mg as needed at bedtime. We discontinue lamotrigine because he is no longer having seizures.
Mr. N tolerates quetiapine and carbamazepine well. The nursing staff reports he is much less irritable and aggressive and his sleep has improved, but he is not oversedated. He returns to and participates in physical, occupational, and speech therapies.
Tips for using medications
Many TBI patients are unusually sensitive to or intolerant of medication side effects. Because no randomized, controlled clinical trials support using any medication in these patients, be cautious. The following recommendations can help:
- Use psychotropics with a low risk of complications.
- Start with low dosages and increase gradually to assess side effects and efficacy of medication trials.
- Give full trials and adequate dosing before you decide a medication has not improved symptoms sufficiently.
- Monitor closely for side effects.
- Seek information from family members to evaluate a medication’s effectiveness, as patients’ cognitive deficits may limit their ability to reliably report symptoms.
Related resources
- Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press, 2005.
- Traumatic Brain Injury Resource Guide. www.neuroskills.com
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonidine • Catapres
- Dextroamphetamine • Dexedrine
- Diazepam • Valium
- Divalproex sodium • Depakote
- Donepezil • Aricept
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Methadone • Dolophine
- Methylphenidate • Ritalin
- Metoprolol • Lopressor
- Olanzapine • Zyprexa
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Pindolol • Visken
- Propranolol • Inderal
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Ziprasidone • Geodon
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Choosing medications for patients with traumatic brain injury (TBI) requires caution; some drugs slow their recovery, and no standard post-TBI treatment exists.
As consulting psychiatrist on a TBI rehabilitation team, I am asked to manage enduring cognitive and emotional problems—aggression, apathy, learning disabilities, dementia—in patients with moderate to severe head injuries. This article describes how we apply available evidence to treat neurobehavioral symptoms in these patients.
Case: An iraq war casualty
The physical medicine and rehabilitation service asks for help in managing agitation, anxiety, and nightmares in Mr. N, age 20, a U.S. combat soldier. While on patrol 2 months ago in Iraq, he suffered a penetrating right frontoparietal brain injury from an improvised explosive device.
Mr. N has undergone a right temporoparietal craniectomy with debridement, ventriculostomy placement, and scalp flap closure. He has had seizures and then pancreatitis—thought to be caused by divalproex prescribed to treat the seizures. Divalproex was replaced with phenytoin at our hospital, and the pancreatitis resolved.
How serious an injury?
TBI ranges from self-limited concussion to devastating, permanent CNS impairment and life-long disability. Brain injuries from sudden impact—from assaults, falls, motor vehicle accidents, combat, or sports—can cause diffuse axonal injury and confusion or unconsciousness, even without radiographic evidence of cerebral bleeding, edema, or mass effect.
No hierarchy or nomenclature is universally accepted for TBI. The term “concussion” is generally used for milder injury and TBI for more-severe injuries.
Concussion. The American Academy of Neurology defines concussion as a trauma-induced alteration in mental status that may or may not involve loss of consciousness. Confusion and amnesia—the hallmarks of concussion—may occur immediately after the head trauma or several minutes later.1 This definition recognizes three concussion grades:
- Grade 1: confusion lasts
- Grade 2: confusion persists >15 minutes but without LOC
- Grade 3: concussion with LOC. The confusional state is marked by disorientation, delayed verbal and motor responses, inattention, incoordination, emotional lability, and slurred or incoherent speech.
- Mild TBI: GCS 13 to 15, LOC 1,3
- Moderate TBI: GCS 9 to 12, LOC 30 minutes to 7 days, and PTA 24 hours to 7 days.
- Severe TBI: GCS ≤8, LOC, and PTA >7 days,4 or any focal neuroimaging abnormalities.3
Using Glasgow Coma Scale scores to evaluate brain injury severity
| Component | Response | Score |
|---|---|---|
| Best eye response | No eye opening | 1 |
| Eye opening to pain | 2 | |
| Eye opening to verbal command | 3 | |
| Eyes open spontaneously | 4 | |
| Best verbal response | No verbal response | 1 |
| Incomprehensible sounds | 2 | |
| Inappropriate words | 3 | |
| Confused | 4 | |
| Oriented | 5 | |
| Best motor response | No motor response | 1 |
| Extension to pain | 2 | |
| Flexion to pain | 3 | |
| Withdrawal from pain | 4 | |
| Localizing pain | 5 | |
| Obeys commands | 6 | |
| GCS total score ≥12 is mild injury, 9 to 11 is moderate, and ≤8 is severe (90% of patients with scores ≤8 are in a coma). Coma is defined as not opening eyes, not obeying commands, and not saying understandable words. Composite scores with eye, verbal, and motor responses (such as E3V3M5) are clinically more useful than totals. | ||
| Source: Reference 2. | ||
Case continued: ‘They’re hurting me’
Mr. N meets criteria for severe TBI. He is periodically agitated and aggressive and refuses to return to physical therapy, complaining that rehabilitation nurses are intentionally hurting him. He occasionally hits the staff and throws things. His medications include:
- phenytoin, 100 mg every 6 hours for seizure prophylaxis
- lamotrigine, 50 mg bid for seizure prophylaxis
- zolpidem, 5 mg as needed at bedtime for pain
- methadone, 10 mg/d for pain
- oxycodone, 5 mg every 4 hours as needed for breakthrough pain.
Assessing progress
For patients such as Mr. N, TBI recovery progress is measured with the Rancho Los Amigos Scale.
The original Rancho scale—developed in 1972 by staff at the Rancho Los Amigos rehabilitation hospital in Downey, CA—described eight levels of cognitive and adaptive functioning, from coma and total care through normal cognition and independence. A 1997 revised version separates the highest cognitive functioning level (VIII, purposeful, appropriate function) into three parts, expanding the scale to 10 levels (Table 2).5
Of course, not all TBI patients begin recovery at Rancho level I, and unfortunately not all achieve level X. Some experience dementia caused by head trauma, with persistent memory impairment and cognitive deficits in language, apraxia, agnosia, or executive function.6
Most patients recover as predicted by the initial injury’s severity. Others experience diffuse cerebral swelling with sudden, rapid deterioration after what appeared to be a grade 1 or grade 2 concussion. Diffuse cerebral swelling is sometimes considered a “second-impact syndrome,” but it can also occur after a single impact.7 A second TBI is not universally believed to cause the precipitous decline, but animal studies suggest an additive effect of rapid sequential TBI.8
Table 2
10-level Rancho Los Amigos Scale for assessing TBI recovery
| Level | Cognitive and adaptive function | Assistance required |
|---|---|---|
| I | No response | Total assistance |
| II | Generalized response | Total assistance |
| III | Localized response | Total assistance |
| IV | Confused/agitated | Maximal assistance |
| V | Confused, inappropriate non-agitated | Maximal assistance |
| VI | Confused, appropriate | Moderate assistance |
| VII | Automatic, appropriate | Minimal assistance |
| VIII | Purposeful, appropriate | Stand-by assistance |
| IX | Purposeful, appropriate | Stand-by assistanceon request |
| X | Purposeful, appropriate | Modified independent |
| Source: Traumatic Brain Injury Resource Guide. www.neuroskills.com. | ||
Recovery for a patient such as Mr. N with Rancho level IV to V TBI may be complicated by marked mood lability, spontaneous aggression, psychomotor agitation, extremely short attention with marked distractibility, little to no short-term memory, and noncooperation with treatment and care. Patients may also show disorders of diminished motivation, characterized by normal consciousness but decreased goal-directed behavior and affective flattening.9
Case continued: Calling in reinforcements
Besides combat nightmares, Mr. N is experiencing other signs of posttraumatic stress disorder (PTSD): intrusive memories of dead comrades, anhedonia, insomnia, irritability, and hypervigilance. We recommend a trial of citalopram, 10 mg/d, but within 1 week he becomes more irritable, agitated, and aggressive, with worsening sleep. We arrange a meeting to obtain collateral information from Mr. N’s aunt, mother, and clinical psychologist. We learn that a first-degree relative had bipolar disorder, and Mr. N lived with various relatives during childhood.
As a child, Mr. N was easily angered, hyperactive, unpredictably aggressive with peers, and impulsive. He was diagnosed with “explosive disorder” at age 8. A psychiatrist prescribed methylphenidate (which helped) and paroxetine (which worsened his behavior and aggression). Based on this history, we make a presumptive diagnosis of comorbid bipolar disorder.
Treating psychopathology
Comorbidities. Adolescents and adults with pre-existing attention-deficit/hyperactivity disorder or bipolar disorder may be predisposed to carelessness or risk taking that lead to accidents and TBI. Likewise, alcoholism and substance use disorders are risk factors for head injuries. These pre-existing conditions will complicate the post-TBI course and must be treated concurrently.
Depression and PTSD may follow a head injury and complicate recovery. In fact, post-TBI symptoms—poor sleep, poor memory and concentration, and irritability—are common to both depression and PTSD.
A team approach. Regardless of its severity or recovery stage, TBI requires multidisciplinary treatment. Physical, occupational, and speech therapies are essential initially. As recovery progresses, vocational rehabilitation may need to be added. Throughout rehabilitation, supportive individual and family therapy can help patients reintegrate into the community. Psychologists, neuropsychologists, and clinical social workers are indispensable to the treatment team.
Medication precautions
Using medications to manage post-TBI syndromes is difficult and controversial. No standard regimen exists, and few clinical trials guide treatment. Small, uncontrolled studies (human and animal) suggest commonly prescribed drugs may worsen outcomes (Table 3).10,11 For example:
- Cognitive function improved in three TBI patients after thioridazine was discontinued in two and haloperidol in one.12
- Haloperidol given to 11 patients with TBI made no difference in rehabilitation outcomes when compared with 15 patients who did not receive the antipsychotic. Those receiving haloperidol also had longer post-trauma amnesia (5 to 30 weeks), compared with the untreated group (1 to 18 weeks).13
- In animal studies of TBI, motor recovery was slowed with haloperidol but not olanzapine,14,15 and with clonidine,16 phenytoin,17 and trazodone.18 Phenobarbitol.19 and diazepam20 have been associated with delayed behavioral recovery and chronic behavior problems, respectively, in rats with TBI. How these agents might affect human patients is speculative.
Medications with potential to impede TBI recovery*
| Class | Medications |
|---|---|
| Alpha-2 agonist | Clonidine |
| Antidepressant | Trazodone |
| Antiepileptic | Phenytoin, phenobarbital |
| Benzodiazepine | Diazepam |
| Neuroleptic | Haloperidol, thioridazine |
| *Suggested by animal or clinical studies | |
| Source: References 11-20 | |
- Psychostimulants have improved recovery of motor function in animal trials if given before physical therapy.14
- Stimulants and dopaminergic agonists such as bromocriptine and amantadine might help disorders of diminished motivation.22
- Dextroamphetamine and methylphenidate have improved impulsivity, memory, and concentration in a patient with TBI.23
Table 4
Drugs considered safe and effective
for TBI neurobehavioral symptoms
| Target symptom(s) | Drug | Usual daily dosage* |
|---|---|---|
| Apathy | Amantadine | 100 to 400 mg |
| Bromocriptine | 1.25 to 100 mg | |
| Cognition | Donepezil | |
| Inattention | Dextroamphetamine | 5 to 60 mg |
| Methylphenidate | 10 to 60 mg | |
| Depression, PTSD symptoms | Fluoxetine | 20 to 80 mg |
| Agitation, mood stabilization | Anticonvulsants | |
| Lamotrigine | 25 to 200 mg | |
| Divalproex sodium | 10 to 15 mg/kg/day† | |
| Carbamazepine | 400 to 1,600 mg‡ | |
| Atypical antipsychotics | ||
| Olanzapine | 2.5 to 20 mg | |
| Quetiapine | 50 to 800 mg | |
| Risperidone | 0.5 to 6 mg | |
| Ziprasidone | 20 to 160 mg | |
| Beta blocker | ||
| Propranolol | 20 to 480 mg | |
| PTSD: posttraumatic stress disorder | ||
| * Dosage may be divided; see full prescribing information. | ||
| † Adjust dosage to achieve serum level of 50 to 100 mcg/mL. | ||
| ‡ Adjust dosage to achieve serum level of 4 to 12 mcg/mL. | ||
Small studies of anticonvulsants for post-TBI agitation report:
- valproic acid might improve behavioral control and decrease aggression, and it did not worsen performance on neuropsychological testing
- carbamazepine reduced agitation in seven TBI patients and reduced anger outbursts in 8 of 10 others
- gabapentin caused paradoxical effects in two TBI patients25
- lamotrigine improved agitation in one TBI patient.26
- propranolol, 420 to 520 mg/d
- pindolol, 60 mg/d
- metoprolol, 200 mg/d.21
Dosing atypical antipsychotics
for agitation and aggression in TBI
| Drug | Initial daily dosage* | Maximum daily dosage* |
|---|---|---|
| Aripiprazole | 2.5 to 5 mg | 30 mg |
| Olanzapine | 2.5 mg | 20 mg |
| Quetiapine | 12.5 to 50 mg | 800 mg |
| Risperidone | 0.25 mg | 8 mg |
| Ziprasidone | 20 mg | 160 mg |
| *Daily dosages may be divided | ||
Depression and PTSD in TBI patients are considered indications for selective serotonin reuptake inhibitors (SSRIs). Animal data suggest that fluoxetine is safe for patients with TBI,27 though no human data have been published.
For PTSD with bipolar depression, we usually prescribe lamotrigine or combine an atypical antipsychotic with an SSRI. Lithium would be second-line therapy. PTSD with bipolar mania is more difficult to treat because little evidence guides medication choices. As with depression and PTSD, we usually combine an atypical antipsychotic with an SSRI. We try to control manic and psychotic symptoms first, then add the SSRI for anxiety after the mood becomes more stable.
Cognitive impairment. A dozen published studies and case reports indicate that donepezil improves cognition in subacute and chronic TBI. For example:
- An open-label trial showed subjective improvement in cognitive functions in 8 of 10 patients given donepezil.28
- In a double-blind, placebo-controlled, crossover trial, short-term memory and attention improved with donepezil in 18 patients with post-acute TBI, as shown by neuropsychological test scores.29
- A retrospective case-control study showed no significant difference in cognitive outcome between controls and 18 patients prescribed donepezil but did suggest that cognition improved more rapidly when patients started donepezil earlier in recovery.30
Case continued: Back to rehab
We replace Mr. N’s phenytoin with carbamazepine, 700 mg/d (serum level about 12 mcg/mL), discontinue citalopram, and start him on quetiapine as a mood stabilizer, titrating the dosage to 600 mg/d over 3 weeks. We select quetiapine based on experience using it as a mood stabilizer and carbamazepine for additional mood stabilization and seizure prophylaxis.
We continue methadone and oxycodone at the same dosages for pain management, with good results. We eventually switch him from zolpidem to trazodone, 50 mg as needed at bedtime. We discontinue lamotrigine because he is no longer having seizures.
Mr. N tolerates quetiapine and carbamazepine well. The nursing staff reports he is much less irritable and aggressive and his sleep has improved, but he is not oversedated. He returns to and participates in physical, occupational, and speech therapies.
Tips for using medications
Many TBI patients are unusually sensitive to or intolerant of medication side effects. Because no randomized, controlled clinical trials support using any medication in these patients, be cautious. The following recommendations can help:
- Use psychotropics with a low risk of complications.
- Start with low dosages and increase gradually to assess side effects and efficacy of medication trials.
- Give full trials and adequate dosing before you decide a medication has not improved symptoms sufficiently.
- Monitor closely for side effects.
- Seek information from family members to evaluate a medication’s effectiveness, as patients’ cognitive deficits may limit their ability to reliably report symptoms.
Related resources
- Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press, 2005.
- Traumatic Brain Injury Resource Guide. www.neuroskills.com
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Carbamazepine • Tegretol
- Citalopram • Celexa
- Clonidine • Catapres
- Dextroamphetamine • Dexedrine
- Diazepam • Valium
- Divalproex sodium • Depakote
- Donepezil • Aricept
- Fluoxetine • Prozac
- Gabapentin • Neurontin
- Haloperidol • Haldol
- Lamotrigine • Lamictal
- Methadone • Dolophine
- Methylphenidate • Ritalin
- Metoprolol • Lopressor
- Olanzapine • Zyprexa
- Oxycodone • Oxycontin
- Paroxetine • Paxil
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Pindolol • Visken
- Propranolol • Inderal
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Thioridazine • Mellaril
- Trazodone • Desyrel
- Ziprasidone • Geodon
- Zolpidem • Ambien
The author reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. American Academy of Neurology. Practice parameter: The management of concussion in sports. Neurology 1997;48:581-5.
2. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;2(7872):81-4.
3. Alexander MP. Mild traumatic brain injury: Pathophysiology, natural history, and clinical management. Neurology 1995;45:1253-60.
4. Arlinghaus KA, Shoaib AM, Price TRP. Neuropsychiatric assessment. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press; 2005:59-78.
5. Hagen C, Malkmus D, Durham P. Communication Disorders Service, Rancho Los Amigos Rehabilitation Hospital, Downey, CA, 1972 (rev. 1997).
6. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 2000.
7. McCrory P. Does second impact syndrome exist? Clin J Sport Med 2001;11:144-9.
8. Vagnozzi R, Signoretti S, Tavazzi B, et al. Hypothesis of the postconcussive vulnerable brain: experimental evidence of its metabolic occurrence. Neurosurgery 2005;57:164-71.
9. Marin RS, Chakravorty S. Disorders of diminished motivation. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA; American Psychiatric Press; 2005:337-52.
10. Goldstein LB. Prescribing of potentially harmful drugs to patients admitted to hospital after head injury. J Neurol Neurosurg Psychiatry 1995;58:753-5.
11. Phillips JP, Devier DJ, Feeney DM. Rehabilitation pharmacology bridging laboratory work to clinical application. J Head Trauma Rehabil 2003;18:342-56.
12. Stanislaw SL. Cognitive effects of antipsychotic agents in persons with traumatic brain injury. Brain Injury 1997;11:335-41.
13. Rao N, Jellinek HM, Woolston DC. Agitation in closed head injury: haloperidol effects on rehabilitation outcome. Arch Phys Med Rehabil 1985;66:30-4.
14. Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science 1982;217:855-7.
15. Wilson MS, Gibson CL, Hamm RJ. Haloperidol, but not olanzapine, impairs cognitive performance after traumatic brain injury in rats. Am J Phys Med Rehabil 2003;82:871-9.
16. Goldstein LB, Davis JN. Clonidine impairs recovery of beamwalking after a sensorimotor cortex lesion in the rat. Brain Research 1990;508:305-9.
17. Brailowsky S, Knight RT, Efron R. Phenytoin increases the severity of cortical hemiplegia in rats. Brain Research 1986;376:71-7.
18. Boyeson MG, Harmon RL. Effects of trazodone and desipramine on motor recovery in brain-injured rats. Am J Phys Med Rehabil 1993;72:286-93.
19. Hernandez TD, Holling LC. Disruption of behavioral recovery by the anticonvulsant phenobarbital. Brain Research 1994;635:300-6.
20. Schallert T, Hernandez TD, Barth TM. Recovery of function after brain damage: severe and chronic disruption by diazepam. Brain Research 1986;379:104-11.
21. Deb S, Crownshaw T. The role of pharmacotherapy in the management of behavior disorders in traumatic brain injury patients. Brain Injury 2004;18:1-31.
22. Campbell JJ, Duffy JD. Treatment strategies in amotivated patients. Psychiatric Annals 1997;27(1):44-9.
23. Evans RW, Gualtieri CT, Patterson D. Treatment of chronic closed head injury with psychostimulant drugs: a controlled case study and an appropriate evaluation procedure. J Nerv Ment Dis 1987;175:106-10.
24. Elovic EP, Lansang R, Li Y, Ricker JH. The use of atypical antipsychotics in traumatic brain injury. J Head Trauma Rehabil 2003;18:177-95.
25. Lombard LA, Zafonte RD. Agitation after traumatic brain injury: considerations and treatment options. Am J Phys Med Rehabil 2005;84:797-812.
26. Pachet A, Friesen S, Winkelaar D, Gray S. Beneficial behavioural effects of lamotrigine in traumatic brain injury. Brain Injury 2003;17:715-22.
27. Boyeson MG, Harmon RL, Jones JL. Comparative effects of fluoxetine, amitriptyline, and serotonin on functional motor recovery after sensorimotor cortex injury. Am J Phys Med Rehabil 1994;73:76-83.
28. Khateb A, Ammann J, Annoni JM, Diserens K. Cognition enhancing effects of onepezil in traumatic brain injury (abstract). Eur Neurol 2005;54:39-45.
29. Zhang L, Plotkin RC, Wang G, et al. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004;85:1005-55.
30. Walker W, Seel R, Gibellato M, et al. The effects of donepezil on traumatic brain injury acute rehabilitation outcomes. Brain Inj 2004;18:739-50.
1. American Academy of Neurology. Practice parameter: The management of concussion in sports. Neurology 1997;48:581-5.
2. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet 1974;2(7872):81-4.
3. Alexander MP. Mild traumatic brain injury: Pathophysiology, natural history, and clinical management. Neurology 1995;45:1253-60.
4. Arlinghaus KA, Shoaib AM, Price TRP. Neuropsychiatric assessment. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA: American Psychiatric Press; 2005:59-78.
5. Hagen C, Malkmus D, Durham P. Communication Disorders Service, Rancho Los Amigos Rehabilitation Hospital, Downey, CA, 1972 (rev. 1997).
6. Diagnostic and statistical manual of mental disorders (4th ed, text rev). Washington, DC: American Psychiatric Association; 2000.
7. McCrory P. Does second impact syndrome exist? Clin J Sport Med 2001;11:144-9.
8. Vagnozzi R, Signoretti S, Tavazzi B, et al. Hypothesis of the postconcussive vulnerable brain: experimental evidence of its metabolic occurrence. Neurosurgery 2005;57:164-71.
9. Marin RS, Chakravorty S. Disorders of diminished motivation. In: Silver JM, McAllister TW, Yudofsky SC (eds). Textbook of traumatic brain injury. Arlington, VA; American Psychiatric Press; 2005:337-52.
10. Goldstein LB. Prescribing of potentially harmful drugs to patients admitted to hospital after head injury. J Neurol Neurosurg Psychiatry 1995;58:753-5.
11. Phillips JP, Devier DJ, Feeney DM. Rehabilitation pharmacology bridging laboratory work to clinical application. J Head Trauma Rehabil 2003;18:342-56.
12. Stanislaw SL. Cognitive effects of antipsychotic agents in persons with traumatic brain injury. Brain Injury 1997;11:335-41.
13. Rao N, Jellinek HM, Woolston DC. Agitation in closed head injury: haloperidol effects on rehabilitation outcome. Arch Phys Med Rehabil 1985;66:30-4.
14. Feeney DM, Gonzalez A, Law WA. Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science 1982;217:855-7.
15. Wilson MS, Gibson CL, Hamm RJ. Haloperidol, but not olanzapine, impairs cognitive performance after traumatic brain injury in rats. Am J Phys Med Rehabil 2003;82:871-9.
16. Goldstein LB, Davis JN. Clonidine impairs recovery of beamwalking after a sensorimotor cortex lesion in the rat. Brain Research 1990;508:305-9.
17. Brailowsky S, Knight RT, Efron R. Phenytoin increases the severity of cortical hemiplegia in rats. Brain Research 1986;376:71-7.
18. Boyeson MG, Harmon RL. Effects of trazodone and desipramine on motor recovery in brain-injured rats. Am J Phys Med Rehabil 1993;72:286-93.
19. Hernandez TD, Holling LC. Disruption of behavioral recovery by the anticonvulsant phenobarbital. Brain Research 1994;635:300-6.
20. Schallert T, Hernandez TD, Barth TM. Recovery of function after brain damage: severe and chronic disruption by diazepam. Brain Research 1986;379:104-11.
21. Deb S, Crownshaw T. The role of pharmacotherapy in the management of behavior disorders in traumatic brain injury patients. Brain Injury 2004;18:1-31.
22. Campbell JJ, Duffy JD. Treatment strategies in amotivated patients. Psychiatric Annals 1997;27(1):44-9.
23. Evans RW, Gualtieri CT, Patterson D. Treatment of chronic closed head injury with psychostimulant drugs: a controlled case study and an appropriate evaluation procedure. J Nerv Ment Dis 1987;175:106-10.
24. Elovic EP, Lansang R, Li Y, Ricker JH. The use of atypical antipsychotics in traumatic brain injury. J Head Trauma Rehabil 2003;18:177-95.
25. Lombard LA, Zafonte RD. Agitation after traumatic brain injury: considerations and treatment options. Am J Phys Med Rehabil 2005;84:797-812.
26. Pachet A, Friesen S, Winkelaar D, Gray S. Beneficial behavioural effects of lamotrigine in traumatic brain injury. Brain Injury 2003;17:715-22.
27. Boyeson MG, Harmon RL, Jones JL. Comparative effects of fluoxetine, amitriptyline, and serotonin on functional motor recovery after sensorimotor cortex injury. Am J Phys Med Rehabil 1994;73:76-83.
28. Khateb A, Ammann J, Annoni JM, Diserens K. Cognition enhancing effects of onepezil in traumatic brain injury (abstract). Eur Neurol 2005;54:39-45.
29. Zhang L, Plotkin RC, Wang G, et al. Cholinergic augmentation with donepezil enhances recovery in short-term memory and sustained attention after traumatic brain injury. Arch Phys Med Rehabil 2004;85:1005-55.
30. Walker W, Seel R, Gibellato M, et al. The effects of donepezil on traumatic brain injury acute rehabilitation outcomes. Brain Inj 2004;18:739-50.
Protect against drug-drug interactions with anxiolytics
Patients with anxiety disorders are at risk for drug-drug interactions (DDIs) with anxiolytics because they often take medications for comorbid medical or psychiatric illnesses.1-3 Prescribing anxiolytics for them without contemplating both physiology and chemistry leads to what Osler called “popgun pharmacy, hitting now the malady and again the patient,” while “not knowing which.”4
To help you “hit” the anxiety instead of the patient,1 we explain the pharmacokinetics and pharmacodynamics of benzodiazepines, buspirone, and propranolol. Practical tables provide information at a glance about which combinations to avoid and which have potential clinical effects (Box 1) you could use to your patients’ advantage.
Clinical effect=Affinity for site of action (pharmacodynamics)×Concentration at site of action (pharmacokinetics)×Patient’s biology (genetics, age, disease, internal environment)
Pharmacodynamics
What a drug does to the body (actions that mediate its efficacy and adverse effects)
Pharmacokinetics
What the body does to a drug (absorption, distribution, metabolism, elimination) that determines its concentration at the site of action
Patient’s biology
Why patients respond differently to the same dose of the same medication (internal environment includes what patients consume, such as foods and co-prescribed drugs)
Benzodiazepines
Benzodiazepines provide an anxiolytic effect by increasing the relative efficiency of the gamma-aminobutyric acid (GABA) receptor when it is stimulated by GABA.5 As a class, benzodiazepines are efficacious for treating panic disorder, social anxiety disorder, generalized anxiety disorder, alcohol withdrawal, and situational anxiety.
Oxidative metabolism. Some benzodiazepines require bio-transformation in the liver by oxidative metabolism; others—such as lorazepam, oxazepam, and emazepam—undergo only glucuronidation reactions and do not have active metabolites (Table 1).6-8
Table 1
Benzodiazepines: How metabolized and half-lives
| Benzodiazepine | Metabolism | Half-life (includes metabolites) |
|---|---|---|
| Alprazolam | Oxidation 3A3/4 | 8 to 12 hrs |
| Chlordiazepoxide | Oxidation 3A3/4 | 10 to 20 hrs |
| Clonazepam | Oxidation 3A3/4 | 18 to 50 hrs |
| Clorazepate | Oxidation 3A3/4 | 40 to 100 hrs |
| Diazepam | Oxidation 1A2, 2C8/9, 2C19, 3A3/4 | 20 to 70 hrs |
| Lorazepam | Conjugation | 10 to 20 hrs |
| Oxazepam | Conjugation | 5 to 15 hrs |
| Source: References 5-7. | ||
Benzodiazepines that undergo oxidative metabolism are more likely than those that do not to be influenced by old age, liver disease, or co-administration of other drugs that increase or decrease hepatic CYP enzyme function. Some (midazolam and triazolam) have high first-pass metabolism before reaching systemic circulation.
Pharmacodynamic DDIs. Giving benzodiazepines with other CNS depressants—such as barbiturates, tricyclics and tetracyclics, dopamine receptor antagonists, opioids, or antihistamines, or alcohol—can cause potentially serious oversedation and respiratory depression (Table 2).
Table 2
Clinical effects of drug-drug interactions with benzodiazepines
| Pharmacodynamic |
| Respiratory depression with alcohol, barbiturates, tricyclic and tetracyclic drugs, dopamine receptor antagonists, opioids, antihistamines |
| With mirtazapine ↑ sedation |
| With lithium, antipsychotics, and clonazepam → ataxia and dysarthria |
| With clozapine → delirium |
| Pharmacokinetic |
| Cimetidine, disulfiram, isoniazid, estrogen, oral contraceptives ↑ diazepam, chlordiazepoxide plasma concentrations |
| Nefazodone and fluvoxamine ↑ plasma concentration of triazolam, alprazolam |
| Carbamazepine ↓ alprazolam plasma concentration |
| Food, antacids ↓ benzodiazepine plasma concentrations |
| Cigarette smoking ↑ benzodiazepine metabolism |
| Benzodiazepines ↑ plasma concentrations of digoxin, phenytoin |
Using benzodiazepines with lithium or antipsychotics may cause ataxia and dysarthria, and benzodiazepines with clozapine can cause delirium.
At-risk patients. Benzodiazepine use is a significant predictor of falling, especially in elderly persons taking more than one sedative. In a controlled study of hospitalized older patients, 84 (46%) of 181 who fell were taking one or more benzodiazepine, compared with 48 (27%) of 181 age-matched controls who did not fall.10 The message: seek an alternative to benzodiazepines to sedate older patients, especially those taking another CNS depressant.
Alprazolam and DDIs. Alprazolam is commonly prescribed, despite its high potential for abuse and association with dangerous DDIs:
- A study of 172 deaths involving oxycodone showed that 117 patients died from combined drug toxicity. Benzodiazepines (detected in 96 cases) were the most common co-intoxicants and were led by alprazolam.11
- Benzodiazepine abuse is common among clients at methadone maintenance clinics and was reported in 3 fatal drug overdoses caused by co-ingestion of methadone and alprazolam.12
- Cocaine and methadone were the most common co-intoxicants with alprazolam in a study of 87 deaths attributed to combined drug toxicity.13
- In a study of patients who overdosed with benzodiazepines, 22% of those who took alprazolam required ICU admission. This was twice the rate of ICU admission after overdose with other benzodiazepines.14
These studies indicate that alprazolam may be more toxic than other benzodiazepines in overdose and when used with other drugs. We recommend that you exercise great care when prescribing alprazolam, particularly for patients who may be at risk of deliberate self-poisoning and lethal DDIs.
Pharmacokinetic DDIs. Diazepam and chlordiazepoxide plasma concentrations increase in combination with drugs that inhibit CYP enzymes, including cimetidine, disulfiram, isoniazid, estrogen, and oral contraceptives.15
Nefazodone—a CYP 3A3/4 inhibitor—can increase plasma concentrations of triazolam and alprazolam to potentially toxic levels. Nefazodone’s manufacturer recommends lowering triazolam dosages by 75% and alprazolam dosages by 50% when used with nefazodone.3
Carbamazepine—a CYP 3A3/4 inducer—induces both its own and other drugs’ metabolism. It can lower plasma concentrations of alprazolam, clonazepam, midazolam, and triazolam, which are metabolized by 3A3/4. Smoking, food, and antacids also may decrease benzodiazepine plasma concentrations.
As perpetuator drugs, benzodiazepines might increase digoxin plasma concentration, probably because of reduced digoxin renal clearance.16 Diazepam may inhibit CYP 2C9 and/or 2C19 by being an alternate substrate for enzymebinding sites,15,17 increasing the concentration of other drugs such as phenytoin.
Buspirone: Complicated pharmacology
One of buspirone’s major clinical advantages is that it does not pharmacodynamically or pharmacokinetically affect benzodiazepines. Buspirone, the only azaspirodecanedione marketed in the United States, has complex central 5-HT effects.18,19 Because it is a partial 5-HT1A agonist, buspirone’s net effect depends on 5-HT concentration at the receptor:
- If 5-HT concentration is low, buspirone will act as an agonist.
- If 5-HT concentration is high, buspirone—being a partial agonist—will antagonize the effect of excessive 5-HT.
Buspirone’s pharmacology is further complicated by its conversion via oxidative metabolism into an active metabolite—1-pheyl-piperazine (1-PP). Buspirone is a CYP 3A3/4 enzyme substrate, so it is extensively metabolized as it crosses the duodenum and passes through the liver. As a result, the parent drug has low bioavailability and is principally converted into 1-PP before entering systemic circulation.6
1-PP works differently than the parent drug. As an alpha-2-adrenergic antagonist, 1-PP increases the firing rate of adrenergic neurons in the locus ceruleus by blocking a receptor in presynaptic feedback system.
Which traits of buspirone and its active metabolite produce the drug’s anxiolytic effect? It might be one of these, all of them, or some other unknown trait.
Pharmacodynamic DDIs. Presumably because of its effects on serotonin release at 5-HT1A receptors, buspirone may cause hypertensive episodes when used with monoamine oxidase inhibitors (MAOIs) (Table 3). This is why a 2-week washout is recommended between discontinuing an MAOIs and starting buspirone.21
Table 3
Clinical effects of drug-drug interactions with buspirone
| Pharmacodynamic |
| DO NOT use buspirone with monoamine oxidase inhibitors (MAOIs); allow 2-week washout after stopping an MAOI before starting buspirone |
| Pharmacokinetic |
| Food ↑ buspirone Cmax and AUC 2-fold |
| Renal impairment ↑ buspirone plasma concentration 2-fold |
| Hepatic impairment ↑ buspirone Cmax and AUC 15-fold and ↑ half-life 2-fold |
| Verapamil, diltiazem, erythromycin, itraconazole ↑ buspirone plasma concentration |
| Rifampicin ↓ buspirone plasma concentration 10-fold |
| Buspirone ↑ haloperidol plasma concentration |
| Erythromycin, itraconazole, nefazodone, grapefruit juice ↑ buspirone plasma concentration |
| Cmax: maximum drug concentration |
| AUC: area under the curve (mathematical calculation of the body’s total exposure to a drug over time) |
- buspirone affects 5-HT mechanisms
- drugs that affect serotonin reuptake inhibition, 5HT1A receptors, and 5HT2 receptors may have synergy.20
Some SSRIs—such as high-dose fluoxetine and usual doses of fluvoxamine—may increase buspirone serum concentration by inhibiting CYP 3A4.6 Consider this clinical effect before you combine an SSRI with buspirone. Using buspirone with fluoxetine, paroxetine, or bupropion also increases serum 1-PP. This increase, which occurs when CYP 2D6 slows 1-PP clearance, could cause euphoria, mania, or seizures.20
Coadministering rifampin can lower buspirone plasma concentrations almost 10-fold because rifampin induces CYP 3A3/4.22
As a perpetuator, buspirone can increase haloperidol plasma concentrations, but probably not to a clinically important extent. In an open trial, Goff23 added buspirone, mean dosage 23.8 mg/d, to a stable regimen of haloperidol in 7 patients with schizophrenia. Although haloperidol’s mean plasma concentration increased by 26% after 6 weeks, this modest change would be difficult to detect in clinical practice.
Huang et al24 found no clinically significant pharmacokinetic interaction between buspirone, 10 mg tid, and haloperidol, 10 to 40 mg/d, during 6 weeks of coadministration in 27 patients with schizophrenia.
Propranolol: Beta-blocking anxiolytic
Propranolol is prescribed off-label for anxiety disorders more often than other beta blockers. It may help patients with situational or performance anxiety.
Beta-adrenergic blockers competitively antagonize norepinephrine and epinephrine at the beta-adrenergic receptor. These cardiovascular agents can reduce many of anxiety’s peripheral manifestations, such as tachycardia, diaphoresis, trembling, and blushing. All beta blockers share this pharmacologic effect, but their pharmacokinetics differ greatly. Some depend on a single CYP enzyme for clearance (metoprolol, by CYP 2D6), whereas others, such as propranolol, are metabolized by multiple CYP enzymes.
Pharmacodynamic DDIs. Drugs that block alpha-1 adrenergic receptors potentiate beta blockers’ blood pressure-lowering effects and increase the risk of orthostatic hypotension. This is probably why haloperidol can potentiate propranolol’s hypotensive effects.6 Other alpha-1 adrenergic antagonists—though not normally classified as such—include some tertiary amine tricyclic antidepressants (amitriptyline and imipramine) and some antipsychotics (quetiapine).
Reports have associated hypertensive crises and bradycardia with coadministration of beta blockers and MAOIs.21 Depressed myocardial contractility and A-V nodal conduction may occur when beta blockers are combined with calcium channel inhibitors.21 Beta blockers also can decrease IV anesthetic dose requirements because they decrease cardiac output.25
In patients using insulin for diabetes mellitus, propranolol inhibits recovery from insulin-induced hypoglycemia and may cause hypertension and bradycardia. Beta blockers also can mask the tachycardia that usually accompanies insulin-induced hypoglycemia.
Pharmacokinetic DDIs. Propranolol has an extensive first-pass effect, being etabolized in the liver to active and inactive compounds that interact with CYP enzymes 1A2, 2C18, 2C19 and 2D6.6
Coadministering strong CYP 2D6 inhibitors such as bupropion, fluoxetine, or paroxetine can reduce propanolol clearance, increasing its effect and risking cardiac toxicity6 (Table 4). CYP 1A2 inhibitors such as amiodarone and fluoroquinolones or CYP 2C19 inhibitors such as fluvoxamine also increase serum concentrations of propranolol.
Table 4
How to avoid drug interactions with three common anxiolytics*
| When prescribing benzodiazepines… | |
| DO | DO NOT |
| Advise patients not to combine benzodiazepines with alcohol | Use with other CNS depressants or nefazodone |
| Talk to patients about potential for abuse/dependency, and monitor benzodiazepine use | Use in elderly patients or in patients with high potential for substance abuse |
| When prescribing buspirone… | |
| DO | DO NOT |
| Allow a 2-week washout between discontinuing an MAOI and starting buspirone | Use with MAOIs, verapamil, diltiazem, erythromycin, or itraconazole |
| Consider adding buspirone when SSRI monotherapy has not adequately helped patients with anxiety | Co-administer with rifampin |
| Combine with benzodiazepines, if needed | |
| When prescribing propranolol… | |
| DO | DO NOT |
| Educate patients using insulin for diabetes mellitus that propranolol may inhibit recovery from insulin-induced hypoglycemia, cause bradycardia, or mask tachycardia | Combine with medications with strong hypotensive effects |
| Coadminister with strong CYP 2D6 or 1A2 inhibitors | |
| Recheck anticonvulsant plasma concentrations after starting propranolol | Add to calcium inhibitors for patients with ↓ myocardial contractility and A-V nodal conduction |
| * Before prescribing any anxiolytic, review all co-prescribed medications for potential DDIs | |
| DDI: drug-drug interaction | |
| MAOI: monoamine oxidase inhibitor | |
| SSRI: selective serotonin reuptake inhibitor | |
As a perpetuator, propranolol produces small increases in diazepam concentration, suggesting that the beta-blocker inhibits diazepam metabolism. This interaction can impair kinetic visual acuity, which is correlated with the ability to discriminate moving objects in space.26
Propranolol increases plasma concentrations of antipsychotics, anticonvulsants, theophylline, and levothyroxine (Table 5)—possibly because of the beta blocker’s negative inotropic effects (decreased cardiac output reduces hepatic and renal blood flow).
Table 5
Clinical effects of drug-drug interactions with propranolol
| Pharmacodynamic |
| With MAO inhibitors → hypertensive crisis and bradycardia |
| With calcium channel inhibitors → ↓ myocardial contractility and A-V nodal conduction |
| ↓ intravenous anesthetic dose requirements |
| ↓ diazepam metabolism |
| ↓ median effective dosage of valproate and diazepam; might improve antiepileptic potential of valproate |
| Pharmacokinetic |
| ↑ plasma concentration of antipsychotics, anticonvulsants, theophylline, levothyroxine |
| Barbiturates, phenytoin, and cigarette smoking ↑ propranolol elimination |
- Cytochrome P450 interactions. www.drug-interaction.com.
- FDA. Food and drug interactions. http://vm.cfsan.fda.gov/~lrd/fdinter.html.
- Psychiatric drug interactions. www.preskorn.com.
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Carbamazepine • Carbatrol, others
- Chlordiazepoxide • Librium
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Clozapine • Clozaril
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Lorazepam • Ativan
- Midazolam • Versed
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenytoin • Dilantin
- Propranolol • Inderal
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Triazolam • Halcion
- Valproate • various
- Verapamil • Calan, Isoptin
Drs. Ramadan and Werder report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has received grants or has been a consultant or speaker for Abbott Laboratories, AstraZeneca Pharmaceuticals, Boehringer-Ingelheim, Bristol-Myers Squibb Co., Merck & Co., Eisai, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Johnson & Johnson, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceutical, Pfizer, Solvay Pharmaceuticals, Sanofi-Aventis, and Wyeth.
1. Preskorn S, Flockhart D. Psychiatric drug interactions guide. New York: MBL Communications.; 2004.
2. Bruce SE, Yonkers KA, Otto MW, et al. Influence of psychiatric comorbidity on recovery and recurrence in generalized anxiety disorder, social phobia, and panic disorder 12-year prospective study. Am J Psychiatry 2005;162:1179-87.
3. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(Suppl 8):13-21.
4. Bean RB, Bean WB. Sir William Osler: Aphorisms from his bedside teaching and writing. Springfield, IL: Charles C. Thomas; 1961:53.
5. Tasman A, Kay J, Lieberman JA. Psychiatry Therapeutics. 2nd ed. West Sussex, UK: John Wiley & Sons; 2003:347.
6. Fuller M, Sajatovic M. Drug information for mental health. 3rd ed. Hudson, OH: Lexi-Comp; 2001.
7. Stahl SM. Essential psychopharmacology: Neuroscientific basis and practical applications. 2nd ed. New York: Cambridge University Press; 2000.
8. Janicak PG, Davis JM, Preskorn SH, Ayad FJ. Principles and practice of psychopharmacology. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2001.
9. Tanaka E. Toxicological interactions involving psychiatric drugs and alcohol: an update. J Clin Pharm Ther 2003;28(2):81-95.
10. Frels C, Williams P, Narayanan S, Gariballa SE. Iatrogenic causes of falls in hospitalised elderly patients: a case-control study. Postgrad Med 2002;78(922):487-9.
11. Wolf BC, Lavezzi WA, Sullivan LM, Flannagan LM. One hundred seventy two deaths involving the use of oxycodone in Palm Beach County. J Forensic Sci 2005;50(1):192-5.
12. Rogers WO, Hall MA, Brissie RM, Robinson CA. Detection of alprazolam in three cases of methadone/benzodiazepine overdose. J Forensic Sci 1997;42(1):155-6.
13. Wolf BC, Lavezzi WA, Sullivan LM, et al. Alprazolam-related deaths in Palm Beach County. Am J Forensic Med Pathol 2005;26(1):24-7.
14. Isbister GK, O’Regan L, Sibbritt D, et al. Alprazolam is relatively more toxic than other benzodiazepines in overdose. Br J Clin Pharmacol 2004;58(1):88-95.
15. Sadock BJ, Sadock VA. Kaplan and Sadock’s pocket handbook of psychiatric drug treatment. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001.
16. Tollefson G, Lesar T, Grothe D, et al. Alprazolam-related digoxin toxicity. Am J Psychiatry 1984;141(12):1612-3.
17. Murphy A, Wilbur K. Phenytoin-diazepam interaction. Ann Pharmacother 2003;37(5):659-3.
18. Sharp T, McQuade R, Bramwell S, et al. Effect of acute and repeated administration of 5-HT1A receptor agonists on 5-HT release in rat brain in vivo. Naunhyn Schmiedebergs Arch Pharmacol 1993;348(4):339-46.
19. Van den Hooff P, Galvan M. Actions of 5-hydroxytryptamine and 5-HT1A receptor ligands on rat dorso-lateral septal neurons in vitro. Br J Pharmacol 1992;106(4):893-9.
20. Preskorn SH Do you believe in magic? Journal of Practical Psychiatry and Behavioral Health March 1997;99-103
21. Physicians’ Desk Reference. 59th ed. Montvale, NJ: Thomson PDR; 2005.
22. Mahmood I, Sahajwalla C. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin Pharmacokinet 1999;36(4):277-87.
23. Goff DC, Midha KK, Brotman AW, et al. An open trial of buspirone added to neuroleptics in schizophrenic patients. J Clin Psychopharmacol 1991;11(3):193-7.
24. Huang HF, Jann MW, Wei FC, et al. Lack of pharmacokinetic interaction between buspirone and haloperidol in patients with schizophrenia. J Clin Pharmacol 1996;36(10):963-9.
25. Avram MJ, Krejcie TC, Henthorn TK, et al. Beta-adrenergic blockade affects initial drug distribution due to decreased cardiac output and altered blood flow distribution. J Pharmacol Exp Ther 2004;311(2):617-24.
26. Hawksworth G, Betts T, Crowe A, et al. Diazepam/beta-adrenoceptor antagonist interactions. Br J Clin Pharmacol 1984;17(Suppl 1):69S-76S.
27. Hallengren B, Nilsson OR, Karlberg BE, et al. Influence of hyperthyroidism on the kinetics of methimazole, propranolol, metoprolol and atenolol. Eur J Clin Pharmacol 1982;21(5):379-84.
Patients with anxiety disorders are at risk for drug-drug interactions (DDIs) with anxiolytics because they often take medications for comorbid medical or psychiatric illnesses.1-3 Prescribing anxiolytics for them without contemplating both physiology and chemistry leads to what Osler called “popgun pharmacy, hitting now the malady and again the patient,” while “not knowing which.”4
To help you “hit” the anxiety instead of the patient,1 we explain the pharmacokinetics and pharmacodynamics of benzodiazepines, buspirone, and propranolol. Practical tables provide information at a glance about which combinations to avoid and which have potential clinical effects (Box 1) you could use to your patients’ advantage.
Clinical effect=Affinity for site of action (pharmacodynamics)×Concentration at site of action (pharmacokinetics)×Patient’s biology (genetics, age, disease, internal environment)
Pharmacodynamics
What a drug does to the body (actions that mediate its efficacy and adverse effects)
Pharmacokinetics
What the body does to a drug (absorption, distribution, metabolism, elimination) that determines its concentration at the site of action
Patient’s biology
Why patients respond differently to the same dose of the same medication (internal environment includes what patients consume, such as foods and co-prescribed drugs)
Benzodiazepines
Benzodiazepines provide an anxiolytic effect by increasing the relative efficiency of the gamma-aminobutyric acid (GABA) receptor when it is stimulated by GABA.5 As a class, benzodiazepines are efficacious for treating panic disorder, social anxiety disorder, generalized anxiety disorder, alcohol withdrawal, and situational anxiety.
Oxidative metabolism. Some benzodiazepines require bio-transformation in the liver by oxidative metabolism; others—such as lorazepam, oxazepam, and emazepam—undergo only glucuronidation reactions and do not have active metabolites (Table 1).6-8
Table 1
Benzodiazepines: How metabolized and half-lives
| Benzodiazepine | Metabolism | Half-life (includes metabolites) |
|---|---|---|
| Alprazolam | Oxidation 3A3/4 | 8 to 12 hrs |
| Chlordiazepoxide | Oxidation 3A3/4 | 10 to 20 hrs |
| Clonazepam | Oxidation 3A3/4 | 18 to 50 hrs |
| Clorazepate | Oxidation 3A3/4 | 40 to 100 hrs |
| Diazepam | Oxidation 1A2, 2C8/9, 2C19, 3A3/4 | 20 to 70 hrs |
| Lorazepam | Conjugation | 10 to 20 hrs |
| Oxazepam | Conjugation | 5 to 15 hrs |
| Source: References 5-7. | ||
Benzodiazepines that undergo oxidative metabolism are more likely than those that do not to be influenced by old age, liver disease, or co-administration of other drugs that increase or decrease hepatic CYP enzyme function. Some (midazolam and triazolam) have high first-pass metabolism before reaching systemic circulation.
Pharmacodynamic DDIs. Giving benzodiazepines with other CNS depressants—such as barbiturates, tricyclics and tetracyclics, dopamine receptor antagonists, opioids, or antihistamines, or alcohol—can cause potentially serious oversedation and respiratory depression (Table 2).
Table 2
Clinical effects of drug-drug interactions with benzodiazepines
| Pharmacodynamic |
| Respiratory depression with alcohol, barbiturates, tricyclic and tetracyclic drugs, dopamine receptor antagonists, opioids, antihistamines |
| With mirtazapine ↑ sedation |
| With lithium, antipsychotics, and clonazepam → ataxia and dysarthria |
| With clozapine → delirium |
| Pharmacokinetic |
| Cimetidine, disulfiram, isoniazid, estrogen, oral contraceptives ↑ diazepam, chlordiazepoxide plasma concentrations |
| Nefazodone and fluvoxamine ↑ plasma concentration of triazolam, alprazolam |
| Carbamazepine ↓ alprazolam plasma concentration |
| Food, antacids ↓ benzodiazepine plasma concentrations |
| Cigarette smoking ↑ benzodiazepine metabolism |
| Benzodiazepines ↑ plasma concentrations of digoxin, phenytoin |
Using benzodiazepines with lithium or antipsychotics may cause ataxia and dysarthria, and benzodiazepines with clozapine can cause delirium.
At-risk patients. Benzodiazepine use is a significant predictor of falling, especially in elderly persons taking more than one sedative. In a controlled study of hospitalized older patients, 84 (46%) of 181 who fell were taking one or more benzodiazepine, compared with 48 (27%) of 181 age-matched controls who did not fall.10 The message: seek an alternative to benzodiazepines to sedate older patients, especially those taking another CNS depressant.
Alprazolam and DDIs. Alprazolam is commonly prescribed, despite its high potential for abuse and association with dangerous DDIs:
- A study of 172 deaths involving oxycodone showed that 117 patients died from combined drug toxicity. Benzodiazepines (detected in 96 cases) were the most common co-intoxicants and were led by alprazolam.11
- Benzodiazepine abuse is common among clients at methadone maintenance clinics and was reported in 3 fatal drug overdoses caused by co-ingestion of methadone and alprazolam.12
- Cocaine and methadone were the most common co-intoxicants with alprazolam in a study of 87 deaths attributed to combined drug toxicity.13
- In a study of patients who overdosed with benzodiazepines, 22% of those who took alprazolam required ICU admission. This was twice the rate of ICU admission after overdose with other benzodiazepines.14
These studies indicate that alprazolam may be more toxic than other benzodiazepines in overdose and when used with other drugs. We recommend that you exercise great care when prescribing alprazolam, particularly for patients who may be at risk of deliberate self-poisoning and lethal DDIs.
Pharmacokinetic DDIs. Diazepam and chlordiazepoxide plasma concentrations increase in combination with drugs that inhibit CYP enzymes, including cimetidine, disulfiram, isoniazid, estrogen, and oral contraceptives.15
Nefazodone—a CYP 3A3/4 inhibitor—can increase plasma concentrations of triazolam and alprazolam to potentially toxic levels. Nefazodone’s manufacturer recommends lowering triazolam dosages by 75% and alprazolam dosages by 50% when used with nefazodone.3
Carbamazepine—a CYP 3A3/4 inducer—induces both its own and other drugs’ metabolism. It can lower plasma concentrations of alprazolam, clonazepam, midazolam, and triazolam, which are metabolized by 3A3/4. Smoking, food, and antacids also may decrease benzodiazepine plasma concentrations.
As perpetuator drugs, benzodiazepines might increase digoxin plasma concentration, probably because of reduced digoxin renal clearance.16 Diazepam may inhibit CYP 2C9 and/or 2C19 by being an alternate substrate for enzymebinding sites,15,17 increasing the concentration of other drugs such as phenytoin.
Buspirone: Complicated pharmacology
One of buspirone’s major clinical advantages is that it does not pharmacodynamically or pharmacokinetically affect benzodiazepines. Buspirone, the only azaspirodecanedione marketed in the United States, has complex central 5-HT effects.18,19 Because it is a partial 5-HT1A agonist, buspirone’s net effect depends on 5-HT concentration at the receptor:
- If 5-HT concentration is low, buspirone will act as an agonist.
- If 5-HT concentration is high, buspirone—being a partial agonist—will antagonize the effect of excessive 5-HT.
Buspirone’s pharmacology is further complicated by its conversion via oxidative metabolism into an active metabolite—1-pheyl-piperazine (1-PP). Buspirone is a CYP 3A3/4 enzyme substrate, so it is extensively metabolized as it crosses the duodenum and passes through the liver. As a result, the parent drug has low bioavailability and is principally converted into 1-PP before entering systemic circulation.6
1-PP works differently than the parent drug. As an alpha-2-adrenergic antagonist, 1-PP increases the firing rate of adrenergic neurons in the locus ceruleus by blocking a receptor in presynaptic feedback system.
Which traits of buspirone and its active metabolite produce the drug’s anxiolytic effect? It might be one of these, all of them, or some other unknown trait.
Pharmacodynamic DDIs. Presumably because of its effects on serotonin release at 5-HT1A receptors, buspirone may cause hypertensive episodes when used with monoamine oxidase inhibitors (MAOIs) (Table 3). This is why a 2-week washout is recommended between discontinuing an MAOIs and starting buspirone.21
Table 3
Clinical effects of drug-drug interactions with buspirone
| Pharmacodynamic |
| DO NOT use buspirone with monoamine oxidase inhibitors (MAOIs); allow 2-week washout after stopping an MAOI before starting buspirone |
| Pharmacokinetic |
| Food ↑ buspirone Cmax and AUC 2-fold |
| Renal impairment ↑ buspirone plasma concentration 2-fold |
| Hepatic impairment ↑ buspirone Cmax and AUC 15-fold and ↑ half-life 2-fold |
| Verapamil, diltiazem, erythromycin, itraconazole ↑ buspirone plasma concentration |
| Rifampicin ↓ buspirone plasma concentration 10-fold |
| Buspirone ↑ haloperidol plasma concentration |
| Erythromycin, itraconazole, nefazodone, grapefruit juice ↑ buspirone plasma concentration |
| Cmax: maximum drug concentration |
| AUC: area under the curve (mathematical calculation of the body’s total exposure to a drug over time) |
- buspirone affects 5-HT mechanisms
- drugs that affect serotonin reuptake inhibition, 5HT1A receptors, and 5HT2 receptors may have synergy.20
Some SSRIs—such as high-dose fluoxetine and usual doses of fluvoxamine—may increase buspirone serum concentration by inhibiting CYP 3A4.6 Consider this clinical effect before you combine an SSRI with buspirone. Using buspirone with fluoxetine, paroxetine, or bupropion also increases serum 1-PP. This increase, which occurs when CYP 2D6 slows 1-PP clearance, could cause euphoria, mania, or seizures.20
Coadministering rifampin can lower buspirone plasma concentrations almost 10-fold because rifampin induces CYP 3A3/4.22
As a perpetuator, buspirone can increase haloperidol plasma concentrations, but probably not to a clinically important extent. In an open trial, Goff23 added buspirone, mean dosage 23.8 mg/d, to a stable regimen of haloperidol in 7 patients with schizophrenia. Although haloperidol’s mean plasma concentration increased by 26% after 6 weeks, this modest change would be difficult to detect in clinical practice.
Huang et al24 found no clinically significant pharmacokinetic interaction between buspirone, 10 mg tid, and haloperidol, 10 to 40 mg/d, during 6 weeks of coadministration in 27 patients with schizophrenia.
Propranolol: Beta-blocking anxiolytic
Propranolol is prescribed off-label for anxiety disorders more often than other beta blockers. It may help patients with situational or performance anxiety.
Beta-adrenergic blockers competitively antagonize norepinephrine and epinephrine at the beta-adrenergic receptor. These cardiovascular agents can reduce many of anxiety’s peripheral manifestations, such as tachycardia, diaphoresis, trembling, and blushing. All beta blockers share this pharmacologic effect, but their pharmacokinetics differ greatly. Some depend on a single CYP enzyme for clearance (metoprolol, by CYP 2D6), whereas others, such as propranolol, are metabolized by multiple CYP enzymes.
Pharmacodynamic DDIs. Drugs that block alpha-1 adrenergic receptors potentiate beta blockers’ blood pressure-lowering effects and increase the risk of orthostatic hypotension. This is probably why haloperidol can potentiate propranolol’s hypotensive effects.6 Other alpha-1 adrenergic antagonists—though not normally classified as such—include some tertiary amine tricyclic antidepressants (amitriptyline and imipramine) and some antipsychotics (quetiapine).
Reports have associated hypertensive crises and bradycardia with coadministration of beta blockers and MAOIs.21 Depressed myocardial contractility and A-V nodal conduction may occur when beta blockers are combined with calcium channel inhibitors.21 Beta blockers also can decrease IV anesthetic dose requirements because they decrease cardiac output.25
In patients using insulin for diabetes mellitus, propranolol inhibits recovery from insulin-induced hypoglycemia and may cause hypertension and bradycardia. Beta blockers also can mask the tachycardia that usually accompanies insulin-induced hypoglycemia.
Pharmacokinetic DDIs. Propranolol has an extensive first-pass effect, being etabolized in the liver to active and inactive compounds that interact with CYP enzymes 1A2, 2C18, 2C19 and 2D6.6
Coadministering strong CYP 2D6 inhibitors such as bupropion, fluoxetine, or paroxetine can reduce propanolol clearance, increasing its effect and risking cardiac toxicity6 (Table 4). CYP 1A2 inhibitors such as amiodarone and fluoroquinolones or CYP 2C19 inhibitors such as fluvoxamine also increase serum concentrations of propranolol.
Table 4
How to avoid drug interactions with three common anxiolytics*
| When prescribing benzodiazepines… | |
| DO | DO NOT |
| Advise patients not to combine benzodiazepines with alcohol | Use with other CNS depressants or nefazodone |
| Talk to patients about potential for abuse/dependency, and monitor benzodiazepine use | Use in elderly patients or in patients with high potential for substance abuse |
| When prescribing buspirone… | |
| DO | DO NOT |
| Allow a 2-week washout between discontinuing an MAOI and starting buspirone | Use with MAOIs, verapamil, diltiazem, erythromycin, or itraconazole |
| Consider adding buspirone when SSRI monotherapy has not adequately helped patients with anxiety | Co-administer with rifampin |
| Combine with benzodiazepines, if needed | |
| When prescribing propranolol… | |
| DO | DO NOT |
| Educate patients using insulin for diabetes mellitus that propranolol may inhibit recovery from insulin-induced hypoglycemia, cause bradycardia, or mask tachycardia | Combine with medications with strong hypotensive effects |
| Coadminister with strong CYP 2D6 or 1A2 inhibitors | |
| Recheck anticonvulsant plasma concentrations after starting propranolol | Add to calcium inhibitors for patients with ↓ myocardial contractility and A-V nodal conduction |
| * Before prescribing any anxiolytic, review all co-prescribed medications for potential DDIs | |
| DDI: drug-drug interaction | |
| MAOI: monoamine oxidase inhibitor | |
| SSRI: selective serotonin reuptake inhibitor | |
As a perpetuator, propranolol produces small increases in diazepam concentration, suggesting that the beta-blocker inhibits diazepam metabolism. This interaction can impair kinetic visual acuity, which is correlated with the ability to discriminate moving objects in space.26
Propranolol increases plasma concentrations of antipsychotics, anticonvulsants, theophylline, and levothyroxine (Table 5)—possibly because of the beta blocker’s negative inotropic effects (decreased cardiac output reduces hepatic and renal blood flow).
Table 5
Clinical effects of drug-drug interactions with propranolol
| Pharmacodynamic |
| With MAO inhibitors → hypertensive crisis and bradycardia |
| With calcium channel inhibitors → ↓ myocardial contractility and A-V nodal conduction |
| ↓ intravenous anesthetic dose requirements |
| ↓ diazepam metabolism |
| ↓ median effective dosage of valproate and diazepam; might improve antiepileptic potential of valproate |
| Pharmacokinetic |
| ↑ plasma concentration of antipsychotics, anticonvulsants, theophylline, levothyroxine |
| Barbiturates, phenytoin, and cigarette smoking ↑ propranolol elimination |
- Cytochrome P450 interactions. www.drug-interaction.com.
- FDA. Food and drug interactions. http://vm.cfsan.fda.gov/~lrd/fdinter.html.
- Psychiatric drug interactions. www.preskorn.com.
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Carbamazepine • Carbatrol, others
- Chlordiazepoxide • Librium
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Clozapine • Clozaril
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Lorazepam • Ativan
- Midazolam • Versed
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenytoin • Dilantin
- Propranolol • Inderal
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Triazolam • Halcion
- Valproate • various
- Verapamil • Calan, Isoptin
Drs. Ramadan and Werder report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has received grants or has been a consultant or speaker for Abbott Laboratories, AstraZeneca Pharmaceuticals, Boehringer-Ingelheim, Bristol-Myers Squibb Co., Merck & Co., Eisai, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Johnson & Johnson, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceutical, Pfizer, Solvay Pharmaceuticals, Sanofi-Aventis, and Wyeth.
Patients with anxiety disorders are at risk for drug-drug interactions (DDIs) with anxiolytics because they often take medications for comorbid medical or psychiatric illnesses.1-3 Prescribing anxiolytics for them without contemplating both physiology and chemistry leads to what Osler called “popgun pharmacy, hitting now the malady and again the patient,” while “not knowing which.”4
To help you “hit” the anxiety instead of the patient,1 we explain the pharmacokinetics and pharmacodynamics of benzodiazepines, buspirone, and propranolol. Practical tables provide information at a glance about which combinations to avoid and which have potential clinical effects (Box 1) you could use to your patients’ advantage.
Clinical effect=Affinity for site of action (pharmacodynamics)×Concentration at site of action (pharmacokinetics)×Patient’s biology (genetics, age, disease, internal environment)
Pharmacodynamics
What a drug does to the body (actions that mediate its efficacy and adverse effects)
Pharmacokinetics
What the body does to a drug (absorption, distribution, metabolism, elimination) that determines its concentration at the site of action
Patient’s biology
Why patients respond differently to the same dose of the same medication (internal environment includes what patients consume, such as foods and co-prescribed drugs)
Benzodiazepines
Benzodiazepines provide an anxiolytic effect by increasing the relative efficiency of the gamma-aminobutyric acid (GABA) receptor when it is stimulated by GABA.5 As a class, benzodiazepines are efficacious for treating panic disorder, social anxiety disorder, generalized anxiety disorder, alcohol withdrawal, and situational anxiety.
Oxidative metabolism. Some benzodiazepines require bio-transformation in the liver by oxidative metabolism; others—such as lorazepam, oxazepam, and emazepam—undergo only glucuronidation reactions and do not have active metabolites (Table 1).6-8
Table 1
Benzodiazepines: How metabolized and half-lives
| Benzodiazepine | Metabolism | Half-life (includes metabolites) |
|---|---|---|
| Alprazolam | Oxidation 3A3/4 | 8 to 12 hrs |
| Chlordiazepoxide | Oxidation 3A3/4 | 10 to 20 hrs |
| Clonazepam | Oxidation 3A3/4 | 18 to 50 hrs |
| Clorazepate | Oxidation 3A3/4 | 40 to 100 hrs |
| Diazepam | Oxidation 1A2, 2C8/9, 2C19, 3A3/4 | 20 to 70 hrs |
| Lorazepam | Conjugation | 10 to 20 hrs |
| Oxazepam | Conjugation | 5 to 15 hrs |
| Source: References 5-7. | ||
Benzodiazepines that undergo oxidative metabolism are more likely than those that do not to be influenced by old age, liver disease, or co-administration of other drugs that increase or decrease hepatic CYP enzyme function. Some (midazolam and triazolam) have high first-pass metabolism before reaching systemic circulation.
Pharmacodynamic DDIs. Giving benzodiazepines with other CNS depressants—such as barbiturates, tricyclics and tetracyclics, dopamine receptor antagonists, opioids, or antihistamines, or alcohol—can cause potentially serious oversedation and respiratory depression (Table 2).
Table 2
Clinical effects of drug-drug interactions with benzodiazepines
| Pharmacodynamic |
| Respiratory depression with alcohol, barbiturates, tricyclic and tetracyclic drugs, dopamine receptor antagonists, opioids, antihistamines |
| With mirtazapine ↑ sedation |
| With lithium, antipsychotics, and clonazepam → ataxia and dysarthria |
| With clozapine → delirium |
| Pharmacokinetic |
| Cimetidine, disulfiram, isoniazid, estrogen, oral contraceptives ↑ diazepam, chlordiazepoxide plasma concentrations |
| Nefazodone and fluvoxamine ↑ plasma concentration of triazolam, alprazolam |
| Carbamazepine ↓ alprazolam plasma concentration |
| Food, antacids ↓ benzodiazepine plasma concentrations |
| Cigarette smoking ↑ benzodiazepine metabolism |
| Benzodiazepines ↑ plasma concentrations of digoxin, phenytoin |
Using benzodiazepines with lithium or antipsychotics may cause ataxia and dysarthria, and benzodiazepines with clozapine can cause delirium.
At-risk patients. Benzodiazepine use is a significant predictor of falling, especially in elderly persons taking more than one sedative. In a controlled study of hospitalized older patients, 84 (46%) of 181 who fell were taking one or more benzodiazepine, compared with 48 (27%) of 181 age-matched controls who did not fall.10 The message: seek an alternative to benzodiazepines to sedate older patients, especially those taking another CNS depressant.
Alprazolam and DDIs. Alprazolam is commonly prescribed, despite its high potential for abuse and association with dangerous DDIs:
- A study of 172 deaths involving oxycodone showed that 117 patients died from combined drug toxicity. Benzodiazepines (detected in 96 cases) were the most common co-intoxicants and were led by alprazolam.11
- Benzodiazepine abuse is common among clients at methadone maintenance clinics and was reported in 3 fatal drug overdoses caused by co-ingestion of methadone and alprazolam.12
- Cocaine and methadone were the most common co-intoxicants with alprazolam in a study of 87 deaths attributed to combined drug toxicity.13
- In a study of patients who overdosed with benzodiazepines, 22% of those who took alprazolam required ICU admission. This was twice the rate of ICU admission after overdose with other benzodiazepines.14
These studies indicate that alprazolam may be more toxic than other benzodiazepines in overdose and when used with other drugs. We recommend that you exercise great care when prescribing alprazolam, particularly for patients who may be at risk of deliberate self-poisoning and lethal DDIs.
Pharmacokinetic DDIs. Diazepam and chlordiazepoxide plasma concentrations increase in combination with drugs that inhibit CYP enzymes, including cimetidine, disulfiram, isoniazid, estrogen, and oral contraceptives.15
Nefazodone—a CYP 3A3/4 inhibitor—can increase plasma concentrations of triazolam and alprazolam to potentially toxic levels. Nefazodone’s manufacturer recommends lowering triazolam dosages by 75% and alprazolam dosages by 50% when used with nefazodone.3
Carbamazepine—a CYP 3A3/4 inducer—induces both its own and other drugs’ metabolism. It can lower plasma concentrations of alprazolam, clonazepam, midazolam, and triazolam, which are metabolized by 3A3/4. Smoking, food, and antacids also may decrease benzodiazepine plasma concentrations.
As perpetuator drugs, benzodiazepines might increase digoxin plasma concentration, probably because of reduced digoxin renal clearance.16 Diazepam may inhibit CYP 2C9 and/or 2C19 by being an alternate substrate for enzymebinding sites,15,17 increasing the concentration of other drugs such as phenytoin.
Buspirone: Complicated pharmacology
One of buspirone’s major clinical advantages is that it does not pharmacodynamically or pharmacokinetically affect benzodiazepines. Buspirone, the only azaspirodecanedione marketed in the United States, has complex central 5-HT effects.18,19 Because it is a partial 5-HT1A agonist, buspirone’s net effect depends on 5-HT concentration at the receptor:
- If 5-HT concentration is low, buspirone will act as an agonist.
- If 5-HT concentration is high, buspirone—being a partial agonist—will antagonize the effect of excessive 5-HT.
Buspirone’s pharmacology is further complicated by its conversion via oxidative metabolism into an active metabolite—1-pheyl-piperazine (1-PP). Buspirone is a CYP 3A3/4 enzyme substrate, so it is extensively metabolized as it crosses the duodenum and passes through the liver. As a result, the parent drug has low bioavailability and is principally converted into 1-PP before entering systemic circulation.6
1-PP works differently than the parent drug. As an alpha-2-adrenergic antagonist, 1-PP increases the firing rate of adrenergic neurons in the locus ceruleus by blocking a receptor in presynaptic feedback system.
Which traits of buspirone and its active metabolite produce the drug’s anxiolytic effect? It might be one of these, all of them, or some other unknown trait.
Pharmacodynamic DDIs. Presumably because of its effects on serotonin release at 5-HT1A receptors, buspirone may cause hypertensive episodes when used with monoamine oxidase inhibitors (MAOIs) (Table 3). This is why a 2-week washout is recommended between discontinuing an MAOIs and starting buspirone.21
Table 3
Clinical effects of drug-drug interactions with buspirone
| Pharmacodynamic |
| DO NOT use buspirone with monoamine oxidase inhibitors (MAOIs); allow 2-week washout after stopping an MAOI before starting buspirone |
| Pharmacokinetic |
| Food ↑ buspirone Cmax and AUC 2-fold |
| Renal impairment ↑ buspirone plasma concentration 2-fold |
| Hepatic impairment ↑ buspirone Cmax and AUC 15-fold and ↑ half-life 2-fold |
| Verapamil, diltiazem, erythromycin, itraconazole ↑ buspirone plasma concentration |
| Rifampicin ↓ buspirone plasma concentration 10-fold |
| Buspirone ↑ haloperidol plasma concentration |
| Erythromycin, itraconazole, nefazodone, grapefruit juice ↑ buspirone plasma concentration |
| Cmax: maximum drug concentration |
| AUC: area under the curve (mathematical calculation of the body’s total exposure to a drug over time) |
- buspirone affects 5-HT mechanisms
- drugs that affect serotonin reuptake inhibition, 5HT1A receptors, and 5HT2 receptors may have synergy.20
Some SSRIs—such as high-dose fluoxetine and usual doses of fluvoxamine—may increase buspirone serum concentration by inhibiting CYP 3A4.6 Consider this clinical effect before you combine an SSRI with buspirone. Using buspirone with fluoxetine, paroxetine, or bupropion also increases serum 1-PP. This increase, which occurs when CYP 2D6 slows 1-PP clearance, could cause euphoria, mania, or seizures.20
Coadministering rifampin can lower buspirone plasma concentrations almost 10-fold because rifampin induces CYP 3A3/4.22
As a perpetuator, buspirone can increase haloperidol plasma concentrations, but probably not to a clinically important extent. In an open trial, Goff23 added buspirone, mean dosage 23.8 mg/d, to a stable regimen of haloperidol in 7 patients with schizophrenia. Although haloperidol’s mean plasma concentration increased by 26% after 6 weeks, this modest change would be difficult to detect in clinical practice.
Huang et al24 found no clinically significant pharmacokinetic interaction between buspirone, 10 mg tid, and haloperidol, 10 to 40 mg/d, during 6 weeks of coadministration in 27 patients with schizophrenia.
Propranolol: Beta-blocking anxiolytic
Propranolol is prescribed off-label for anxiety disorders more often than other beta blockers. It may help patients with situational or performance anxiety.
Beta-adrenergic blockers competitively antagonize norepinephrine and epinephrine at the beta-adrenergic receptor. These cardiovascular agents can reduce many of anxiety’s peripheral manifestations, such as tachycardia, diaphoresis, trembling, and blushing. All beta blockers share this pharmacologic effect, but their pharmacokinetics differ greatly. Some depend on a single CYP enzyme for clearance (metoprolol, by CYP 2D6), whereas others, such as propranolol, are metabolized by multiple CYP enzymes.
Pharmacodynamic DDIs. Drugs that block alpha-1 adrenergic receptors potentiate beta blockers’ blood pressure-lowering effects and increase the risk of orthostatic hypotension. This is probably why haloperidol can potentiate propranolol’s hypotensive effects.6 Other alpha-1 adrenergic antagonists—though not normally classified as such—include some tertiary amine tricyclic antidepressants (amitriptyline and imipramine) and some antipsychotics (quetiapine).
Reports have associated hypertensive crises and bradycardia with coadministration of beta blockers and MAOIs.21 Depressed myocardial contractility and A-V nodal conduction may occur when beta blockers are combined with calcium channel inhibitors.21 Beta blockers also can decrease IV anesthetic dose requirements because they decrease cardiac output.25
In patients using insulin for diabetes mellitus, propranolol inhibits recovery from insulin-induced hypoglycemia and may cause hypertension and bradycardia. Beta blockers also can mask the tachycardia that usually accompanies insulin-induced hypoglycemia.
Pharmacokinetic DDIs. Propranolol has an extensive first-pass effect, being etabolized in the liver to active and inactive compounds that interact with CYP enzymes 1A2, 2C18, 2C19 and 2D6.6
Coadministering strong CYP 2D6 inhibitors such as bupropion, fluoxetine, or paroxetine can reduce propanolol clearance, increasing its effect and risking cardiac toxicity6 (Table 4). CYP 1A2 inhibitors such as amiodarone and fluoroquinolones or CYP 2C19 inhibitors such as fluvoxamine also increase serum concentrations of propranolol.
Table 4
How to avoid drug interactions with three common anxiolytics*
| When prescribing benzodiazepines… | |
| DO | DO NOT |
| Advise patients not to combine benzodiazepines with alcohol | Use with other CNS depressants or nefazodone |
| Talk to patients about potential for abuse/dependency, and monitor benzodiazepine use | Use in elderly patients or in patients with high potential for substance abuse |
| When prescribing buspirone… | |
| DO | DO NOT |
| Allow a 2-week washout between discontinuing an MAOI and starting buspirone | Use with MAOIs, verapamil, diltiazem, erythromycin, or itraconazole |
| Consider adding buspirone when SSRI monotherapy has not adequately helped patients with anxiety | Co-administer with rifampin |
| Combine with benzodiazepines, if needed | |
| When prescribing propranolol… | |
| DO | DO NOT |
| Educate patients using insulin for diabetes mellitus that propranolol may inhibit recovery from insulin-induced hypoglycemia, cause bradycardia, or mask tachycardia | Combine with medications with strong hypotensive effects |
| Coadminister with strong CYP 2D6 or 1A2 inhibitors | |
| Recheck anticonvulsant plasma concentrations after starting propranolol | Add to calcium inhibitors for patients with ↓ myocardial contractility and A-V nodal conduction |
| * Before prescribing any anxiolytic, review all co-prescribed medications for potential DDIs | |
| DDI: drug-drug interaction | |
| MAOI: monoamine oxidase inhibitor | |
| SSRI: selective serotonin reuptake inhibitor | |
As a perpetuator, propranolol produces small increases in diazepam concentration, suggesting that the beta-blocker inhibits diazepam metabolism. This interaction can impair kinetic visual acuity, which is correlated with the ability to discriminate moving objects in space.26
Propranolol increases plasma concentrations of antipsychotics, anticonvulsants, theophylline, and levothyroxine (Table 5)—possibly because of the beta blocker’s negative inotropic effects (decreased cardiac output reduces hepatic and renal blood flow).
Table 5
Clinical effects of drug-drug interactions with propranolol
| Pharmacodynamic |
| With MAO inhibitors → hypertensive crisis and bradycardia |
| With calcium channel inhibitors → ↓ myocardial contractility and A-V nodal conduction |
| ↓ intravenous anesthetic dose requirements |
| ↓ diazepam metabolism |
| ↓ median effective dosage of valproate and diazepam; might improve antiepileptic potential of valproate |
| Pharmacokinetic |
| ↑ plasma concentration of antipsychotics, anticonvulsants, theophylline, levothyroxine |
| Barbiturates, phenytoin, and cigarette smoking ↑ propranolol elimination |
- Cytochrome P450 interactions. www.drug-interaction.com.
- FDA. Food and drug interactions. http://vm.cfsan.fda.gov/~lrd/fdinter.html.
- Psychiatric drug interactions. www.preskorn.com.
- Alprazolam • Xanax
- Bupropion • Wellbutrin
- Buspirone • BuSpar
- Carbamazepine • Carbatrol, others
- Chlordiazepoxide • Librium
- Cimetidine • Tagamet
- Clonazepam • Klonopin
- Clorazepate • Tranxene
- Clozapine • Clozaril
- Diazepam • Valium
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Haloperidol • Haldol
- Itraconazole • Sporanox
- Lorazepam • Ativan
- Midazolam • Versed
- Mirtazapine • Remeron
- Oxazepam • Serax
- Paroxetine • Paxil
- Phenytoin • Dilantin
- Propranolol • Inderal
- Quetiapine • Seroquel
- Rifampin • Rifadin, Rimactane
- Triazolam • Halcion
- Valproate • various
- Verapamil • Calan, Isoptin
Drs. Ramadan and Werder report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Preskorn has received grants or has been a consultant or speaker for Abbott Laboratories, AstraZeneca Pharmaceuticals, Boehringer-Ingelheim, Bristol-Myers Squibb Co., Merck & Co., Eisai, Eli Lilly and Co., GlaxoSmithKline, Janssen Pharmaceutica, Johnson & Johnson, Novartis Pharmaceuticals Corp., Organon, Otsuka America Pharmaceutical, Pfizer, Solvay Pharmaceuticals, Sanofi-Aventis, and Wyeth.
1. Preskorn S, Flockhart D. Psychiatric drug interactions guide. New York: MBL Communications.; 2004.
2. Bruce SE, Yonkers KA, Otto MW, et al. Influence of psychiatric comorbidity on recovery and recurrence in generalized anxiety disorder, social phobia, and panic disorder 12-year prospective study. Am J Psychiatry 2005;162:1179-87.
3. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(Suppl 8):13-21.
4. Bean RB, Bean WB. Sir William Osler: Aphorisms from his bedside teaching and writing. Springfield, IL: Charles C. Thomas; 1961:53.
5. Tasman A, Kay J, Lieberman JA. Psychiatry Therapeutics. 2nd ed. West Sussex, UK: John Wiley & Sons; 2003:347.
6. Fuller M, Sajatovic M. Drug information for mental health. 3rd ed. Hudson, OH: Lexi-Comp; 2001.
7. Stahl SM. Essential psychopharmacology: Neuroscientific basis and practical applications. 2nd ed. New York: Cambridge University Press; 2000.
8. Janicak PG, Davis JM, Preskorn SH, Ayad FJ. Principles and practice of psychopharmacology. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2001.
9. Tanaka E. Toxicological interactions involving psychiatric drugs and alcohol: an update. J Clin Pharm Ther 2003;28(2):81-95.
10. Frels C, Williams P, Narayanan S, Gariballa SE. Iatrogenic causes of falls in hospitalised elderly patients: a case-control study. Postgrad Med 2002;78(922):487-9.
11. Wolf BC, Lavezzi WA, Sullivan LM, Flannagan LM. One hundred seventy two deaths involving the use of oxycodone in Palm Beach County. J Forensic Sci 2005;50(1):192-5.
12. Rogers WO, Hall MA, Brissie RM, Robinson CA. Detection of alprazolam in three cases of methadone/benzodiazepine overdose. J Forensic Sci 1997;42(1):155-6.
13. Wolf BC, Lavezzi WA, Sullivan LM, et al. Alprazolam-related deaths in Palm Beach County. Am J Forensic Med Pathol 2005;26(1):24-7.
14. Isbister GK, O’Regan L, Sibbritt D, et al. Alprazolam is relatively more toxic than other benzodiazepines in overdose. Br J Clin Pharmacol 2004;58(1):88-95.
15. Sadock BJ, Sadock VA. Kaplan and Sadock’s pocket handbook of psychiatric drug treatment. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001.
16. Tollefson G, Lesar T, Grothe D, et al. Alprazolam-related digoxin toxicity. Am J Psychiatry 1984;141(12):1612-3.
17. Murphy A, Wilbur K. Phenytoin-diazepam interaction. Ann Pharmacother 2003;37(5):659-3.
18. Sharp T, McQuade R, Bramwell S, et al. Effect of acute and repeated administration of 5-HT1A receptor agonists on 5-HT release in rat brain in vivo. Naunhyn Schmiedebergs Arch Pharmacol 1993;348(4):339-46.
19. Van den Hooff P, Galvan M. Actions of 5-hydroxytryptamine and 5-HT1A receptor ligands on rat dorso-lateral septal neurons in vitro. Br J Pharmacol 1992;106(4):893-9.
20. Preskorn SH Do you believe in magic? Journal of Practical Psychiatry and Behavioral Health March 1997;99-103
21. Physicians’ Desk Reference. 59th ed. Montvale, NJ: Thomson PDR; 2005.
22. Mahmood I, Sahajwalla C. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin Pharmacokinet 1999;36(4):277-87.
23. Goff DC, Midha KK, Brotman AW, et al. An open trial of buspirone added to neuroleptics in schizophrenic patients. J Clin Psychopharmacol 1991;11(3):193-7.
24. Huang HF, Jann MW, Wei FC, et al. Lack of pharmacokinetic interaction between buspirone and haloperidol in patients with schizophrenia. J Clin Pharmacol 1996;36(10):963-9.
25. Avram MJ, Krejcie TC, Henthorn TK, et al. Beta-adrenergic blockade affects initial drug distribution due to decreased cardiac output and altered blood flow distribution. J Pharmacol Exp Ther 2004;311(2):617-24.
26. Hawksworth G, Betts T, Crowe A, et al. Diazepam/beta-adrenoceptor antagonist interactions. Br J Clin Pharmacol 1984;17(Suppl 1):69S-76S.
27. Hallengren B, Nilsson OR, Karlberg BE, et al. Influence of hyperthyroidism on the kinetics of methimazole, propranolol, metoprolol and atenolol. Eur J Clin Pharmacol 1982;21(5):379-84.
1. Preskorn S, Flockhart D. Psychiatric drug interactions guide. New York: MBL Communications.; 2004.
2. Bruce SE, Yonkers KA, Otto MW, et al. Influence of psychiatric comorbidity on recovery and recurrence in generalized anxiety disorder, social phobia, and panic disorder 12-year prospective study. Am J Psychiatry 2005;162:1179-87.
3. Nemeroff CB. Use of atypical antipsychotics in refractory depression and anxiety. J Clin Psychiatry 2005;66(Suppl 8):13-21.
4. Bean RB, Bean WB. Sir William Osler: Aphorisms from his bedside teaching and writing. Springfield, IL: Charles C. Thomas; 1961:53.
5. Tasman A, Kay J, Lieberman JA. Psychiatry Therapeutics. 2nd ed. West Sussex, UK: John Wiley & Sons; 2003:347.
6. Fuller M, Sajatovic M. Drug information for mental health. 3rd ed. Hudson, OH: Lexi-Comp; 2001.
7. Stahl SM. Essential psychopharmacology: Neuroscientific basis and practical applications. 2nd ed. New York: Cambridge University Press; 2000.
8. Janicak PG, Davis JM, Preskorn SH, Ayad FJ. Principles and practice of psychopharmacology. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2001.
9. Tanaka E. Toxicological interactions involving psychiatric drugs and alcohol: an update. J Clin Pharm Ther 2003;28(2):81-95.
10. Frels C, Williams P, Narayanan S, Gariballa SE. Iatrogenic causes of falls in hospitalised elderly patients: a case-control study. Postgrad Med 2002;78(922):487-9.
11. Wolf BC, Lavezzi WA, Sullivan LM, Flannagan LM. One hundred seventy two deaths involving the use of oxycodone in Palm Beach County. J Forensic Sci 2005;50(1):192-5.
12. Rogers WO, Hall MA, Brissie RM, Robinson CA. Detection of alprazolam in three cases of methadone/benzodiazepine overdose. J Forensic Sci 1997;42(1):155-6.
13. Wolf BC, Lavezzi WA, Sullivan LM, et al. Alprazolam-related deaths in Palm Beach County. Am J Forensic Med Pathol 2005;26(1):24-7.
14. Isbister GK, O’Regan L, Sibbritt D, et al. Alprazolam is relatively more toxic than other benzodiazepines in overdose. Br J Clin Pharmacol 2004;58(1):88-95.
15. Sadock BJ, Sadock VA. Kaplan and Sadock’s pocket handbook of psychiatric drug treatment. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001.
16. Tollefson G, Lesar T, Grothe D, et al. Alprazolam-related digoxin toxicity. Am J Psychiatry 1984;141(12):1612-3.
17. Murphy A, Wilbur K. Phenytoin-diazepam interaction. Ann Pharmacother 2003;37(5):659-3.
18. Sharp T, McQuade R, Bramwell S, et al. Effect of acute and repeated administration of 5-HT1A receptor agonists on 5-HT release in rat brain in vivo. Naunhyn Schmiedebergs Arch Pharmacol 1993;348(4):339-46.
19. Van den Hooff P, Galvan M. Actions of 5-hydroxytryptamine and 5-HT1A receptor ligands on rat dorso-lateral septal neurons in vitro. Br J Pharmacol 1992;106(4):893-9.
20. Preskorn SH Do you believe in magic? Journal of Practical Psychiatry and Behavioral Health March 1997;99-103
21. Physicians’ Desk Reference. 59th ed. Montvale, NJ: Thomson PDR; 2005.
22. Mahmood I, Sahajwalla C. Clinical pharmacokinetics and pharmacodynamics of buspirone, an anxiolytic drug. Clin Pharmacokinet 1999;36(4):277-87.
23. Goff DC, Midha KK, Brotman AW, et al. An open trial of buspirone added to neuroleptics in schizophrenic patients. J Clin Psychopharmacol 1991;11(3):193-7.
24. Huang HF, Jann MW, Wei FC, et al. Lack of pharmacokinetic interaction between buspirone and haloperidol in patients with schizophrenia. J Clin Pharmacol 1996;36(10):963-9.
25. Avram MJ, Krejcie TC, Henthorn TK, et al. Beta-adrenergic blockade affects initial drug distribution due to decreased cardiac output and altered blood flow distribution. J Pharmacol Exp Ther 2004;311(2):617-24.
26. Hawksworth G, Betts T, Crowe A, et al. Diazepam/beta-adrenoceptor antagonist interactions. Br J Clin Pharmacol 1984;17(Suppl 1):69S-76S.
27. Hallengren B, Nilsson OR, Karlberg BE, et al. Influence of hyperthyroidism on the kinetics of methimazole, propranolol, metoprolol and atenolol. Eur J Clin Pharmacol 1982;21(5):379-84.