User login
Addressing the rarity and complexities of sarcomas
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
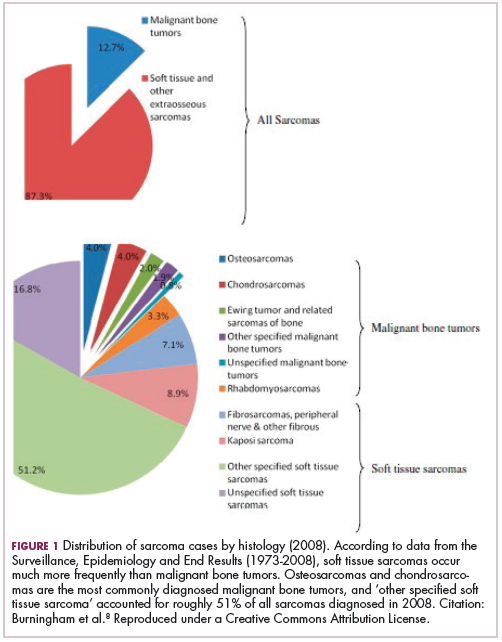
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3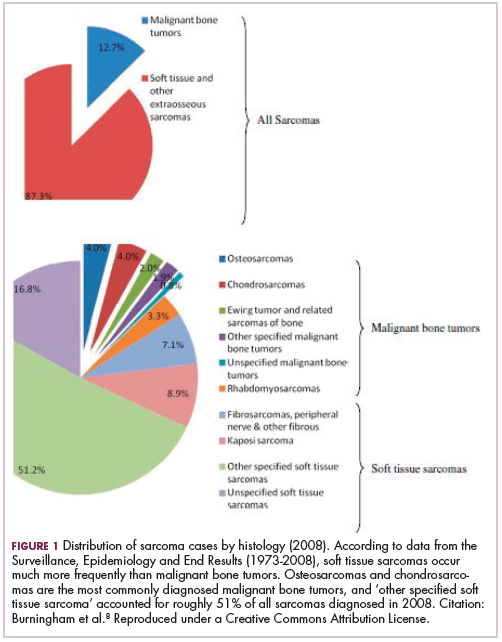
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).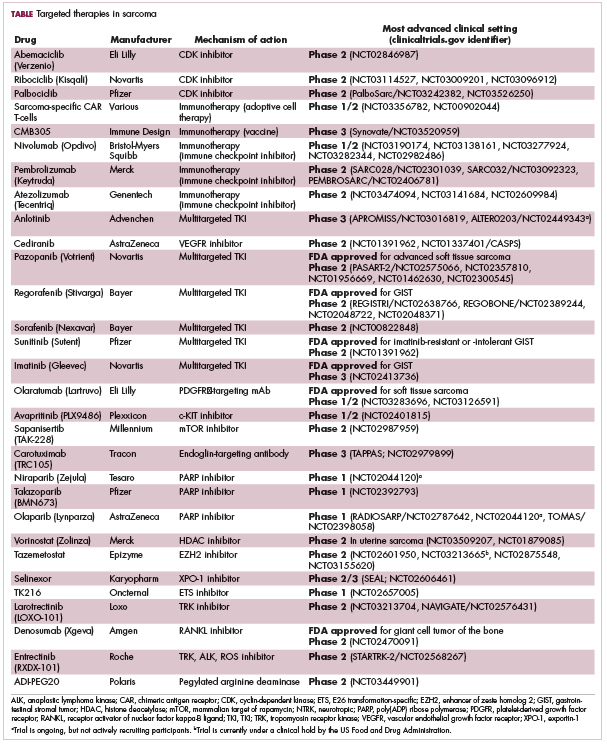
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33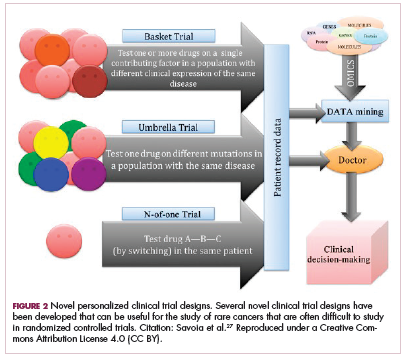
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
The rarity and complexities of bone and soft tissue sarcomas pose a major challenge to effective treatment. Historically, there has been a blanket approach to treatment, but more recently that has begun to change thanks to genome profiling studies and novel clinical trial strategies. Here, we discuss the resulting enrichment of the therapeutic armamentarium with molecularly targeted and immune therapies.
A challenging tumor type
Sarcomas are a large group of histologically diverse cancers that arise in the mesenchymal cells. They can be broadly divided into bone and soft tissue sarcomas (STS) but are further subdivided according to the type of cell from which they derive; osteosarcomas in the bone, rhabdomyosarcomas in the skeletal muscle, liposarcomas in the fat tissues, leiomyosarcomas in the smooth muscle, and chondrosarcomas in the cartilaginous tissue, for example.
Each sarcoma subtype itself encompasses a range of different cancers with unique biology. Under the umbrella of liposarcoma, for example, are well/dedifferentiated liposarcomas and myxoid liposarcomas, which have very different pathologies and clinical courses.
As a whole, sarcomas are extremely rare tumors, accounting for less than 1% of all adult cancers, although they disproportionately affect children and young adults, with a prevalence closer to 15%.1,2 Certain sarcoma subtypes are exceptionally rare, with only a few cases diagnosed worldwide each year, whereas liposarcomas are at the other end of the spectrum, comprising the most common form of STS (Figure 1).3
In the early stages, sarcomas are generally highly treatable with a combination of surgical resection, chemotherapy, and radiation therapy. However, many patients develop advanced, metastatic disease, which presents much more of a challenge.4,5
Magic bullet for GIST
Despite their clear heterogeneity and complexity, sarcomas have tended to be treated as a single entity. Chemotherapy has played a central role in the treatment of advanced sarcomas and continues to do so, with 2 newer drugs approved by the United States Food and Drug Administration (FDA) in the past several years.6,7
The development of targeted therapy, on the other hand, for the most part proved unsuccessful. In general, studies examining the somatic mutation landscape in sarcomas found very few that were highly recurrent. The exception was gastrointestinal stromal tumors (GIST), which represent around 8% of STS.8 Frequent mutations in several highly targetable tyrosine kinases, notably KIT, which is mutated in around 85% of cases,9 and platelet-derived growth factor receptor alpha (PDGFRα) were identified in these tumors.10This prompted the development of tyrosine kinase inhibitors (TKIs), targeting these and other kinases, for the treatment of patients with GIST, and culminated in the approval of imatinib for this indication in 2002. This revolutionized the treatment of GIST, which had a poor prognosis and were resistant to chemotherapy, extending median overall survival in patients with metastatic disease almost to 5 years.11-13
Imatinib was also shown to benefit patients with surgically resectable disease and was subsequently approved in the adjuvant setting in 2008. A recent trial demonstrated that 3-year continuation of adjuvant imatinib resulted in a significantly longer progression-free survival (PFS) compared with 1 year of adjuvant imatinib, and even longer time periods are now being evaluated.14,15 The TKIs sunitinib and regorafenib have also been approved for the treatment of patients who become resistant to imatinib.16,17 Avapritinib, a newer, more specific inhibitor of KIT is also being evaluated in patients with GIST (Table).
Long-sought success for STS
Sunitinib and regorafenib include PDGFRα and the vascular endothelial growth factor receptors (VEGFRs) among their targets, receptors that play crucial roles in the formation of new blood vessels (angiogenesis). Many types of non-GIST sarcomas have been shown to be highly vascularized and express high levels of both of those receptors and other angiogenic proteins, which sparked interest in the development of multitargeted TKIs and other anti-angiogenic drugs in patients with STS.18
In 2012, pazopanib became the first FDA-approved molecularly targeted therapy for the treatment of non-GIST sarcomas. Approval in the second-line setting was based on the demonstration of a 3-month improvement in PFS compared with placebo.19 Four years later, the monoclonal antibody olaratumab, a more specific inhibitor of PDGFRα, was approved in combination with doxorubicin, marking the first front-line approval for more than 4 decades.20Numerous other anti-angiogenic drugs continue to be evaluated for the treatment of advanced STS. Among them, anlotinib is being tested in phase 3 clinical trials, and results from the ALTER0203 trial were presented at the 2018 annual meeting of the American Society of Clinical Oncology (ASCO).21 After failure of chemotherapy, 223 patients were randomly assigned to receive either anlotinib or placebo. Anlotinib significantly improved median PFS across all patients, compared with placebo (6.27 vs 1.4 months, respectively; hazard ratio [HR], 0.33; P < .0001), but was especially effective in patients with alveolar soft part sarcoma (ASPS; mPFS: 18.2 vs 3 months) and was well tolerated.21
Sarcoma secrets revealed
Advancements in genome sequencing technologies have made it possible to interrogate the molecular underpinnings of sarcomas in greater detail. However, their rarity presents a significant technical challenge, with a dearth of samples available for genomic testing. Large-scale worldwide collaborative efforts have facilitated the collection of sufficiently large patient populations to provide statistically robust data in many cases. The Cancer Genome Atlas has established a rare tumor characterization project to facilitate the genomic sequencing of rare cancer types like sarcomas.
Genome sequencing studies have revealed 2 types of sarcomas: those with relatively stable genomes and few molecular alterations, exemplified by Ewing sarcoma, which has a mutational load of 0.15 mutations/Megabase (Mb); and those that are much more complex with frequent somatic mutations, the prime example being leiomyosarcoma. The latter are characterized by mutations in the TP53 gene, dubbed the “guardian of the genome” for its essential role in genome stability.
The 2 types are likely to require very different therapeutic strategies. Although genomically complex tumors offer up lots of potential targets for therapy, they also display significant heterogeneity and it can be challenging to find a shared target across different tumor samples. The p53 protein would make a logical target but, to date, tumor suppressor proteins are not readily druggable.
The most common type of molecular alterations in sarcomas are chromosomal translocations, where part of a chromosome breaks off and becomes reattached to another chromosome. This can result in the formation of a gene fusion when parts of 2 different genes are brought together in a way in which the genetic code can still be read, leading to the formation of a fusion protein with altered activity.22-25
In sarcomas, these chromosomal translocations predominantly involve genes encoding transcription factors and the gene fusion results in their aberrant expression and activation of the transcriptional programs that they regulate.
Ewing sarcoma is a prime example of a sarcoma that is defined by chromosomal translocations. Most often, the resulting gene fusions occur between members of theten-eleven translocation (TET) family of RNA-binding proteins and the E26 transformation-specific (ETS) family of transcription factors. The most common fusion is between the EWSR1 and FLI1 genes, observed in between 85% and 90% of cases.
Significant efforts have been made to target EWSR1-FLI1. Since direct targeting of transcription factors is challenging, those efforts focused on targeting the aberrant transcriptional programs that they initiate. A major downstream target is the insulin-like growth factor receptor 1 (IGF1R) and numerous IGF1R inhibitors were developed and tested in patients with Ewing sarcoma, but unfortunately success was limited. Attention turned to the mammalian target of rapamycin (mTOR) as a potential mechanism of resistance to IGF1R inhibitors and explanation for the limited responses. Clinical trials combining mTOR and IGF1R inhibitors also proved unsuccessful.26
Although overall these trials were deemed failures, they were notable for the dramatic responses that were seen in 1 or 2 patients. Researchers are probing these “exceptional responses” using novel N-of-1 clinical trial designs that focus on a single patient (Figure 2).27-30 More recently, the first drug to specifically target the EWSR1-FLI1 fusion protein was developed. TK216 binds to the fusion protein and prevents it from binding to RNA helicase A, thereby blocking its function.31
Another type of gene fusion, involving the neurotrophic tropomyosin receptor kinase (NTRK) genes, has recently come into the spotlight for the treatment of lung cancer. According to a recent study, NTRK fusions may also be common in sarcomas. They were observed in 8% of patients with breast sarcomas, 5% with fibrosarcomas, and 5% with stomach or small intestine sarcomas.32
The NTRK genes encode TRK proteins and several small molecule inhibitors of TRK have been developed to treat patients with NTRK fusion-positive cancers. Another novel clinical trial design – the basket trial – is being used to test these inhibitors. This type of trial uses a tumor-agnostic approach, recruiting patients with all different histological subtypes of cancer that are unified by the shared presence of a specific molecular alteration.33
Repurposing gynecologic cancer drugs
More recently, a third group of sarcomas was categorized, with intermediate genomic complexity. These tumors, including well/dedifferentiated liposarcomas, were characterized by amplifications of chromosome 12, involving genes such as cyclin-dependent kinase 4 (CDK4). In fact, more than 90% of patients with well/dedifferentiated sarcomas display CDK4 amplification, making it a logical therapeutic target.36
CDK4 encodes CDK4 protein, a cell cycle-associated protein that regulates the transition from G1-S phase, known as the restriction point, beyond which the cell commits to undergoing mitosis. Aberrant expression of CDK4 in cancer drives the hallmark process of unchecked cellular proliferation.
Some small molecule CDK4/6 inhibitors have been developed and have shown significant promise in the treatment of breast cancer. They are also being evaluatedin patients with sarcoma whose tumors display CDK4 overexpression. In a recently published phase 2 trial of palbociclib in 60 patients with well/dedifferentiated liposarcomas, there was 1 CR.37
Another group of drugs that has advanced the treatment of gynecologic cancers comprises the poly (ADP-ribose) polymerase (PARP) inhibitors. In this context, PARP inhibitors are used in patients with mutations in the breast cancer susceptibility genes, BRCA1/2. The BRCA and PARP proteins are both involved in DNA repair pathways and the inhibition of PARP in patients who already have a defective BRCA pathway renders a lethal double blow to the cancer cell. According to the Broad Institute Cancer Cell Line Encyclopedia, Ewing sarcomas express high levels of the PARP1 enzyme, which could render them sensitive to PARP inhibition. Preclinical studies seemed to confirm that sensitivity, however, so far this has yet to translate into success in clinical trials, with no objective responses observed as yet.38
Expanding the field
Other treatment strategies being tested in patients with sarcoma are moving the field beyond conventional targeted therapies. There has been substantial focus in recent years on epigenetic alterations and their potential role in the development of cancer. Epigenetics is the secondary layer of regulation that acts on the genome and directs the spatial and temporal expression of genes.
Both DNA and the histone proteins they are packaged up with to form chromatin in nondividing cells can be modified by the attachment of chemical groups, such as acetyl and methyl groups, which can alter access to the DNA for transcription.
EZH2 is an enzyme that participates in histone methylation and thereby regulates transcriptional repression. Some types of sarcoma are characterized by a loss of expression of the INI1 gene, also known as SMARCB1. The INI1 protein is part of a chromatin remodeling complex that relieves transcriptional repression and when INI1 is lost, cells become dependent upon EZH2.39Clinical trials of the EZH2 inhibitor tazemetostat are ongoing in several types of sarcoma. Results from a phase 2 study in adults with INI1-negative tumors were presented at ASCO in 2017. Among 31 patients treated with 800 mg tazemetostat in continuous 28-day cycles, mPFS was 5.7 months, disease control rate was 10%, and confirmed overall response rate was 13%. The FDA has granted tazemetostat orphan drug designation in this indication.40A pediatric basket trial of tazemetostat is also ongoing, but the FDA recently placed it under a clinical hold as a result of a safety update from the trial in which a pediatric patient with advanced poorly differentiated chordoma developed a secondary T-cell lymphoma.41
Targeting the unique metabolism of sarcomas may offer a promising therapeutic strategy, although this is in the preliminary stages of evaluation. A recent study showed that the expression of the argininosuccinate synthase 1 enzyme, which is involved in the generation of arginine through the urea cycle, was lost in up to 90% of STS. A pegylated arginine deaminase (ADI-PEG20), is being evaluated in a phase 2 clinical trial.42
Finally, the concept of using immunotherapy to boost the anti-tumor immune response is also being examined in sarcomas. A significant number of cases of STS, osteosarcoma and GIST have been shown to express programmed cell death protein-ligand 1, therefore the use of immune checkpoint inhibitors that block this ligand or its receptor and help to reactive tumor-infiltrating T cells, could be a beneficial strategy.
Limited activity has been observed in studies conducted to date, however combination therapies, especially with inhibitors of the indoleamine 2,3-dioxygenase (IDO) enzyme, which plays a key role in immunosuppression, could help to harness the power of these drugs. Studies have suggested that sarcomas may be infiltrated by immunosuppressive macrophages that express IDO.43
It is generally believed that immunotherapy is most effective in tumors that are highly mutated because that allows a large number of cancer antigens to provoke an anti-tumor immune response. However, a single highly expressed antigen can also be strongly immunogenic. Synovial sarcomas have a relatively low mutational burden but they do express high levels of the cancer testis antigen NY-ESO-1.
NY-ESO-1 has provided a useful target for the development of adoptive cell therapies and vaccines for the treatment of sarcomas. CMB305 is an NY-ESO-1 vaccine that also incorporates a toll-like receptor 4 agonist. It is being evaluated in the phase 3 Synovate study as maintenance monotherapy in patients with locally advanced, unresectable or metastatic synovial sarcoma. In a phase 1 study, at a median follow-up of just under 18 months, the median OS for all 25 patients was 23.7 months.44
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29.
2. Toro JR, Travis LB, Wu HJ, Zhu K, Fletcher CD, Devesa SS. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int J Cancer. 2006;119(12):2922-2930.
3. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res. 2012;2(1):14.
4. Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma. Cancer. 2011;117(5):1049-1054.
5. Savina M, Le Cesne A, Blay JY, et al. Patterns of care and outcomes of patients with METAstatic soft tissue SARComa in a real-life setting: the METASARC observational study. BMC Med. 2017;15(1):78.
6. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786-793.
7. Schöffski P, Chawla S, Maki RG, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet. 2016;387(10028):1629-1637.
8. Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260(3):416-421; discussion 421-412.
9. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349.
10. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299(5607):708-710.
11. Dagher R, Cohen M, Williams G, et al. Approval summary. Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8(10):3034-3038.
12. Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626-632.
13. Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127-1134.
14. Zhao R, Wang Y, Huang Y, et al. Adjuvant imatinib for patients with high-risk gastrointestinal stromal tumors: a retrospective cohort study. Scientific Reports. 2017;7:16834.
15. Raut C, Espat N, Maki R, Araujo D, Williams T, Wolff J. Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. J Clin Oncol. 2017;35(15_suppl):11009.
16. Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295-302.
17. Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329-1338.
18. Versleijen-Jonkers YM, Vlenterie M, van de Luijtgaarden AC, van der Graaf WT. Anti-angiogenic therapy, a new player in the field of sarcoma treatment. Crit Rev Oncol Hematol. 2014;91(2):172-185.
19. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879-1886.
20. Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488-497.
21. Chi Y, Yao Y, Wang S, et al. Anlotinib for metastatic soft tissue sarcoma: A randomized, double-blind, placebo-controlled and multi-centered clinical trial. J Clin Oncol. 2018;36(suppl):abstr 11503.
22. Brohl AS, Shah HR, Wang Y-C, Kasarskis A, Maki RG. The somatic mutational landscape in soft tissue sarcoma: Early results from TCGA data. J Clin Oncol. 2015;33(15_suppl):10508-10508.
23. Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326-1341.
24. Jour G, Scarborough JD, Jones RL, et al. Molecular profiling of soft tissue sarcomas using next-generation sequencing: a pilot study toward precision therapeutics. Hum Pathol. 2014;45(8):1563-1571.
25. Yang J-L. Investigation of osteosarcoma genomics and its impact on targeted therapy: an international collaboration to conquer human osteosarcoma. Chin J Cancer. 2014;33(12):575-580.
26. Cidre-Aranaz F, Alonso J. EWS/FLI1 target genes and therapeutic opportunities in Ewing sarcoma. Front Oncol. 2015;5:162.
27. Savoia C, Volpe M, Grassi G, Borghi C, Agabiti Rosei E, Touyz RM. Personalized medicine-a modern approach for the diagnosis and management of hypertension. Clin Sci (Lond). 2017;131(22):2671-2685.
28. Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop. 2016;7(9):527-538.
29. van Maldegem AM, Bovée JVMG, Peterse EFP, Hogendoorn PCW, Gelderblom H. Ewing sarcoma: the clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur J Cancer. 2016;53:171-180.
30. Subbiah V, Hess KR, Khawaja MR, et al. Evaluation of novel targeted therapies in aggressive biology sarcoma patients after progression from US FDA approved therapies. Sci Rep. 2016;6:35448.
31. Jessen K, Moseley E, Chung EYL, et al. TK216, a novel, small molecule inhibitor of the ETS-family of transcription factors, displays anti-tumor activity in AML and DLBCL. Blood. 2016;128(22):4035-4035.
32. Sankhala K, Potts S, Christiansen J, et al. Immunohistochemistry screening to increase the efficacy of next-generation sequencing for detection of NTRK, ROS1, and ALK gene rearrangements (fusions) in sarcoma patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 9-12, 2016, 2016; Lisbon, Portugal.
33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett. 2017;387:121-126.
34. DuBois S, Laetsch T, Federman N, et al. The use of larotrectinib in the management of locally advanced pediatric NTRK-fusion sarcoma. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
35. Multani P, Manavel E, Hornby Z. Preliminary evidence of clinical response to entrectinib in three sarcome patients. Paper presented at: Connective Tissue Oncology Society Annual Meeting; November 8-11, 2017; Maui, Hawaii.
36. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42(8):715-721.
37. Dickson MA, Schwartz GK, Keohan ML, et al. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: a phase 2 clinical trial. JAMA Oncol. 2016;2(7):937-940.
38. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-607.
39. Kenichi K, Yoshinao O. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Science. 2017;108(4):547-552.
40. US Food and Drug Administration. Orphan drug designations and approvals. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/detailedIndex.cfm?cfgridkey=544416. Designated date September 28, 2017. Accessed July 4, 2018.
41. Press release. Epizyme provides update regarding tazemetostat clinical program. https://globenewswire.com/news-release/2018/04/23/1485765/0/en/Epizyme-Provides-Update-Regarding-Tazemetostat-Clinical-Program.html. Released April 23, 2018. Accessed July 4, 2018.
42. Bean GR, Kremer JC, Prudner BC, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death &Amp; Disease. 2016;7:e2406.
43. Bourcier K, Italiano A. Newer therapeutic strategies for soft-tissue sarcomas. Pharmacol Ther. 2018;188:118-123.
44. Somaiah N, Chawla SP, Block MS, et al. Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS). J Clin Oncol. 2017;35(15_suppl):11006-11006.
Meeting the potential of immunotherapy: new targets provide rational combinations
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.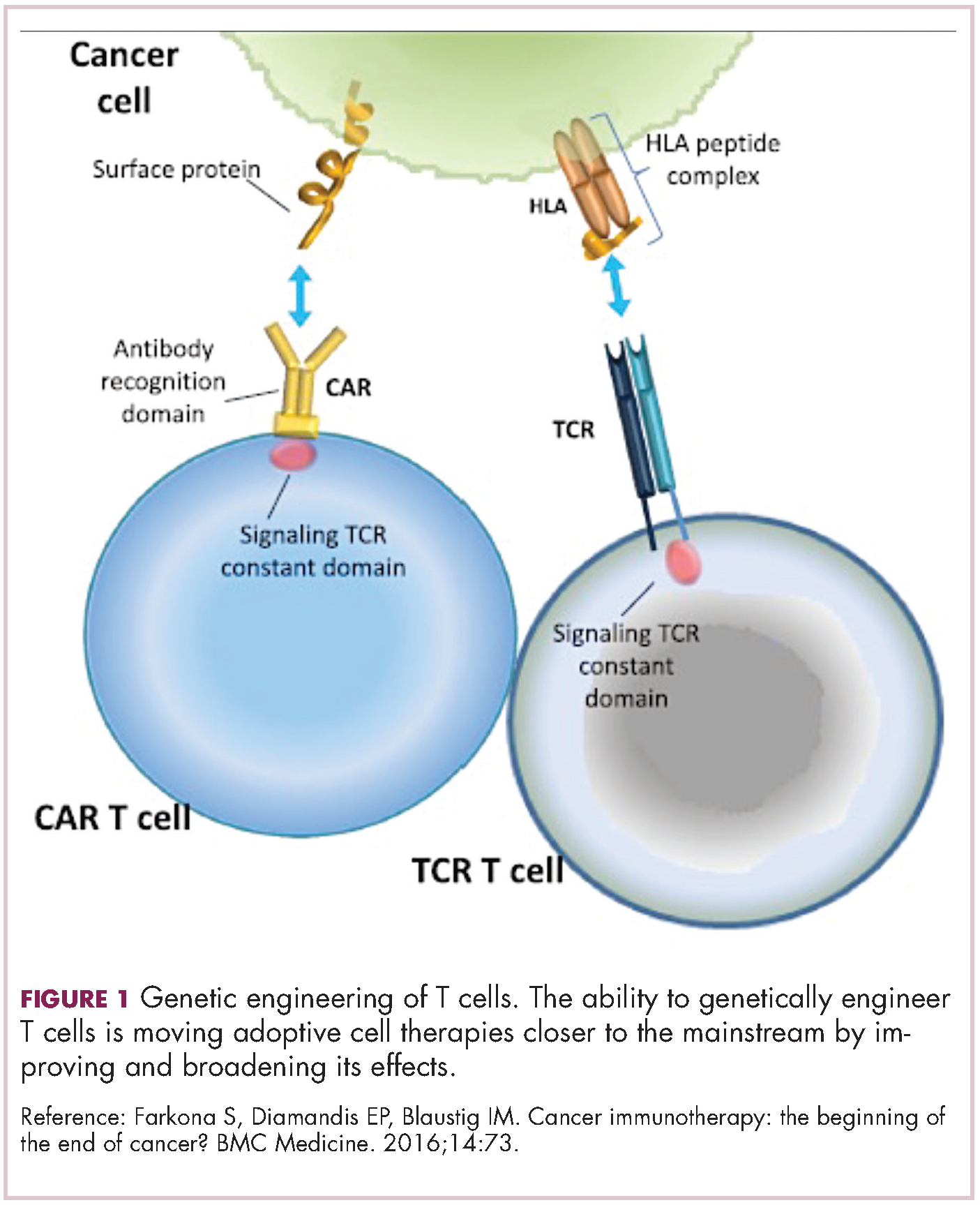
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17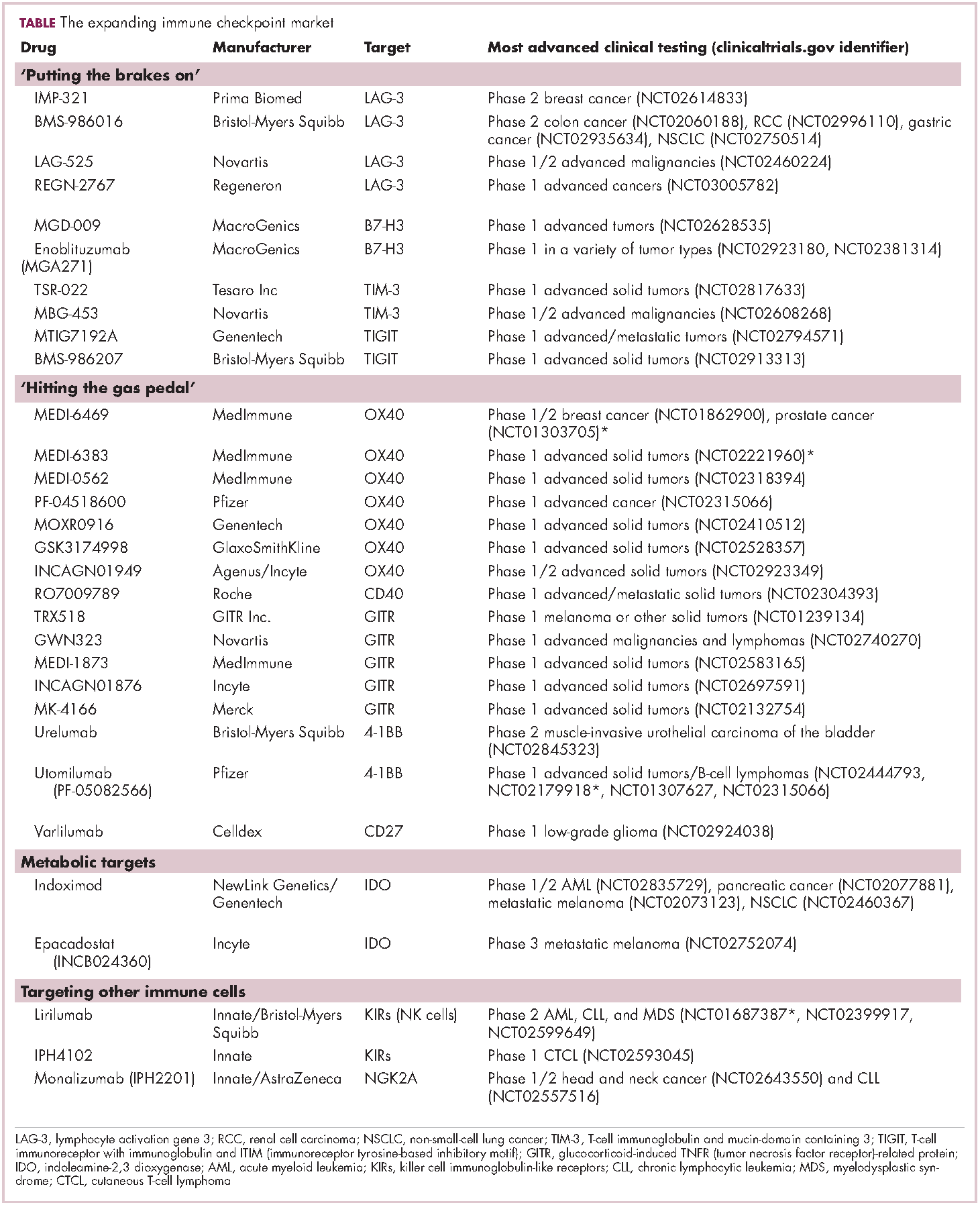
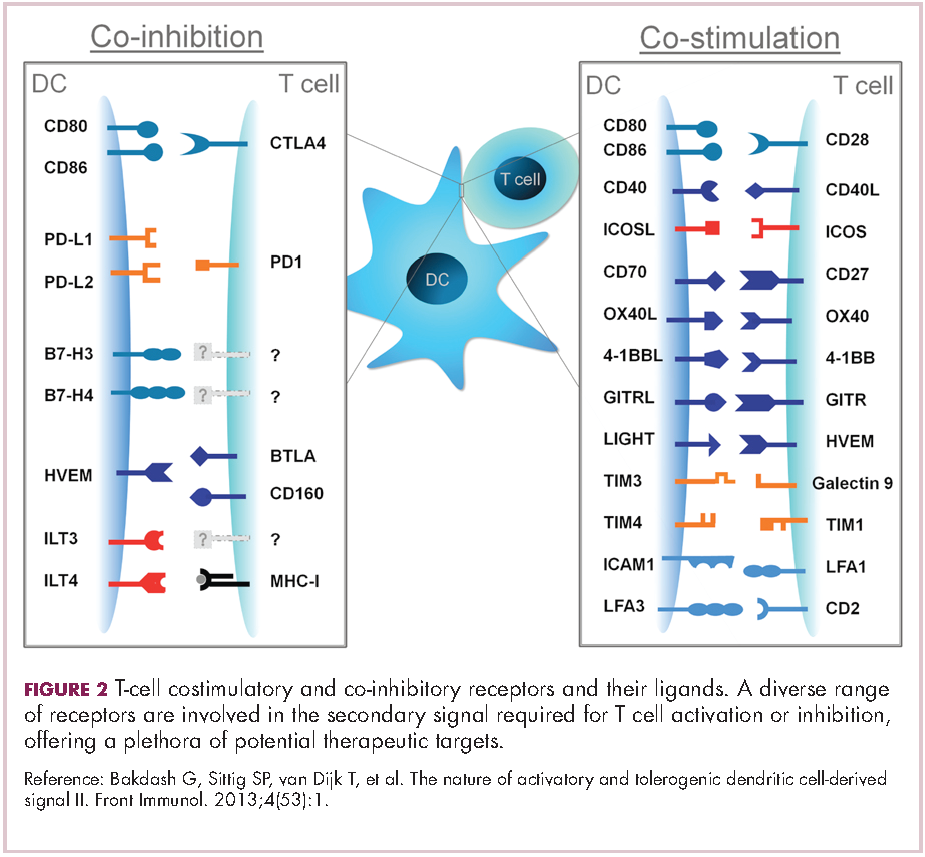
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).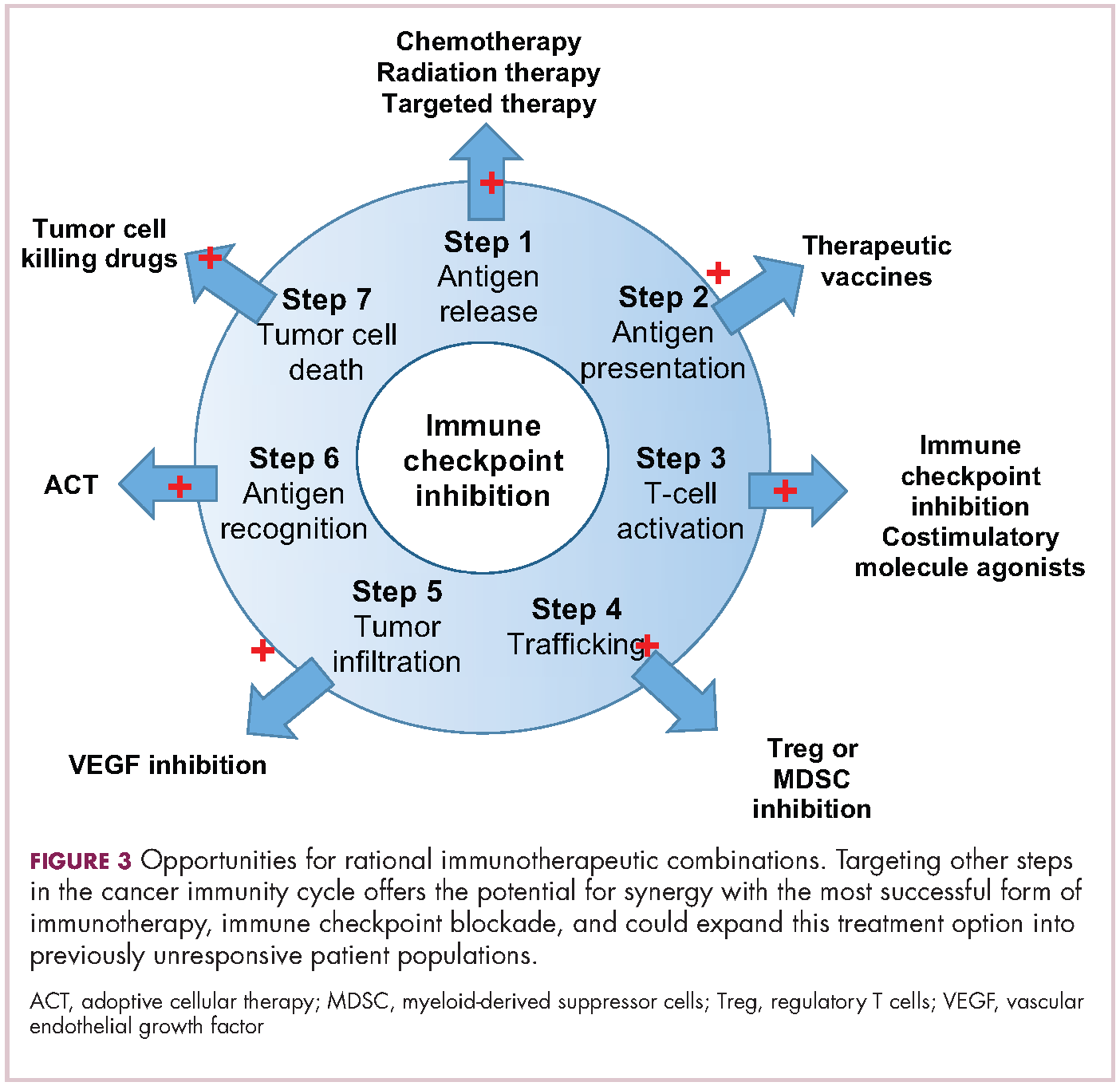
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
Immunotherapies shape the treatment landscape for hematologic malignancies
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.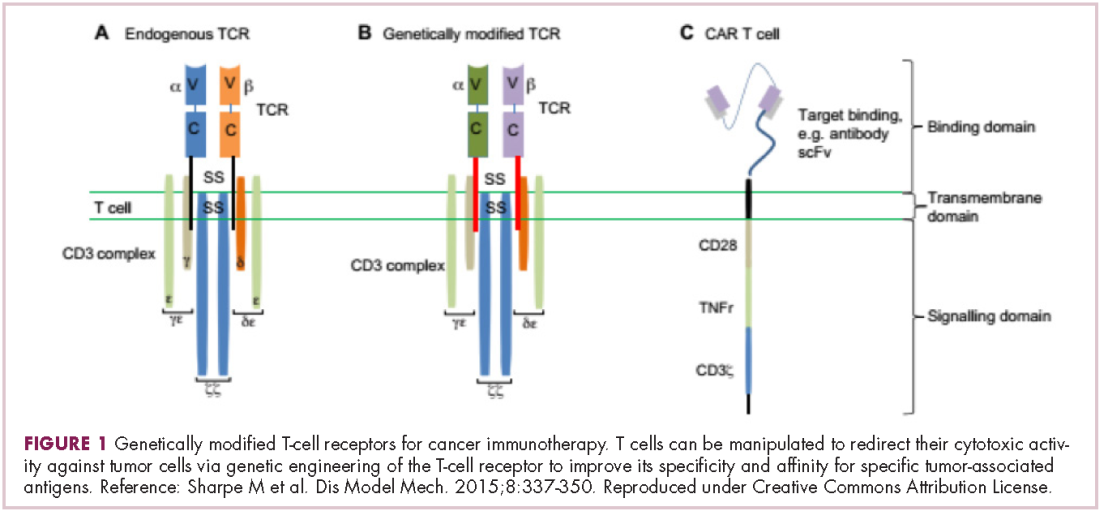
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5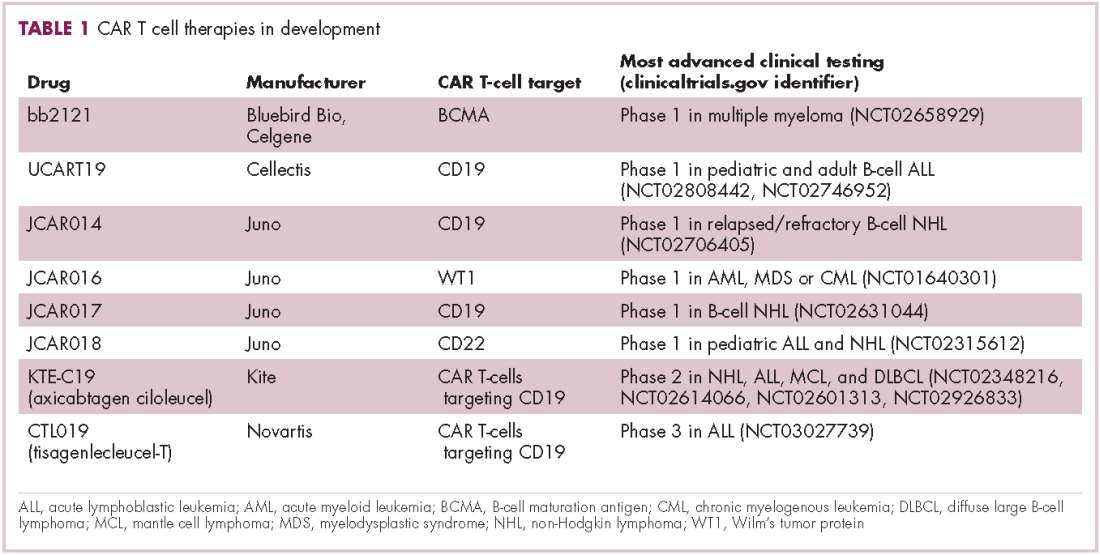
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12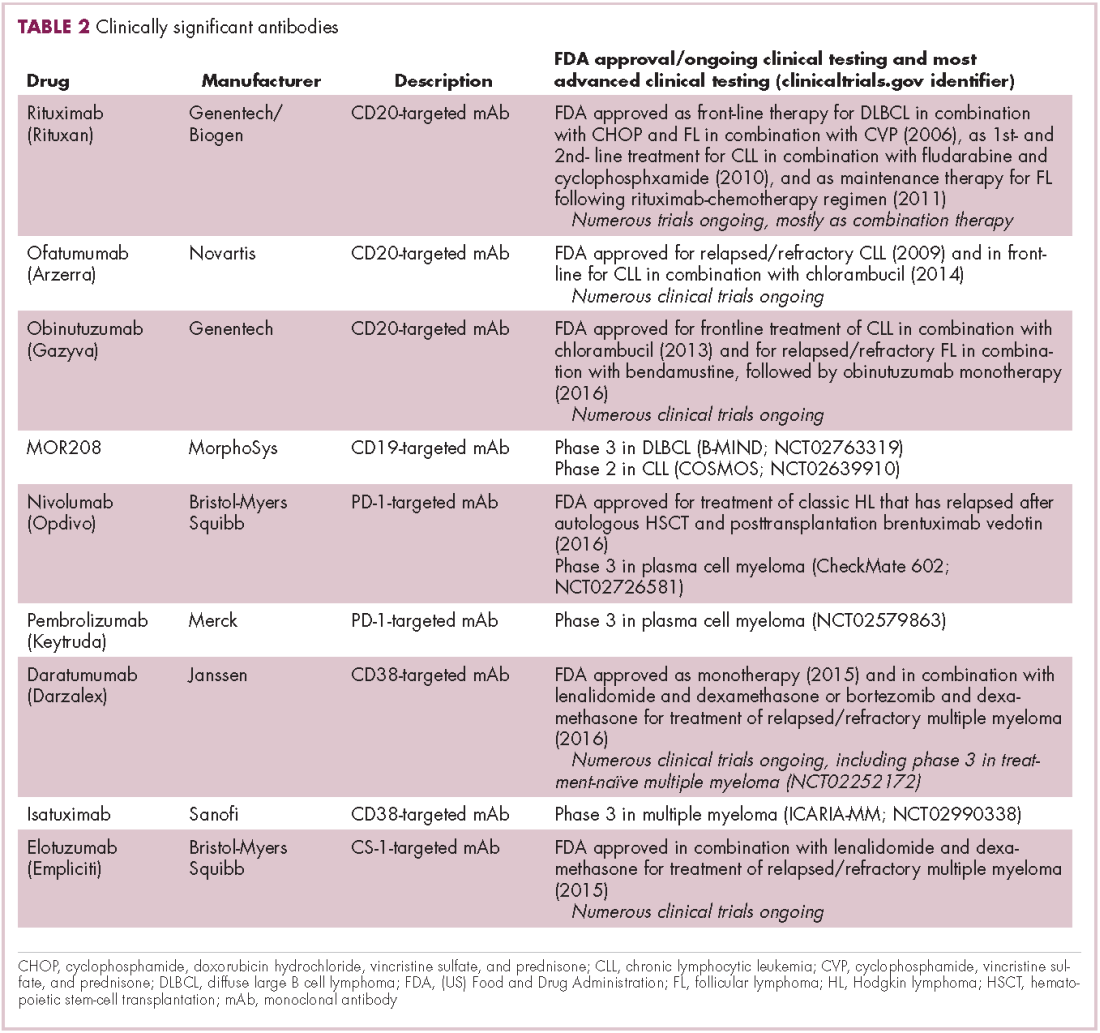
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20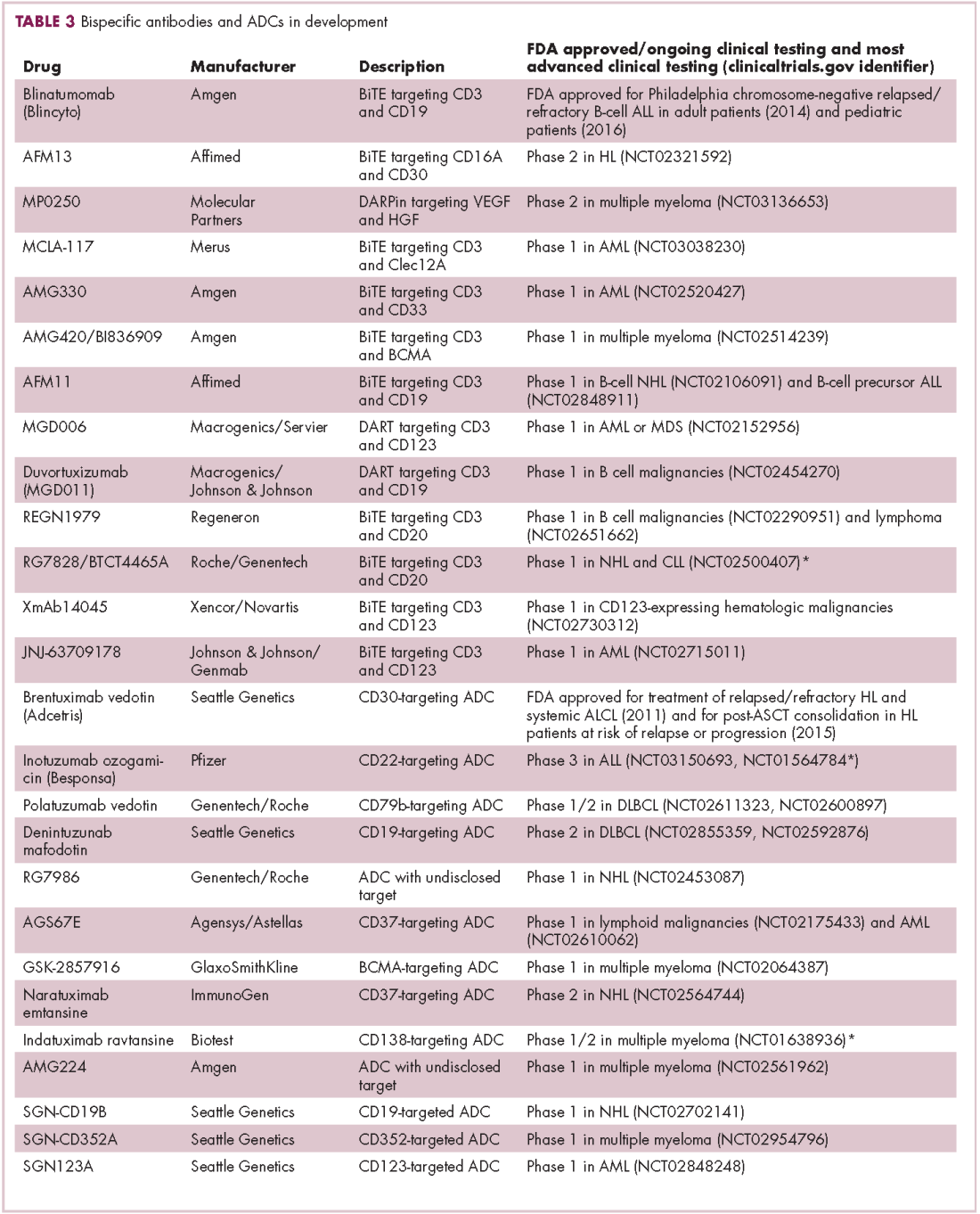
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
The treatment landscape for hematologic malignancies is evolving faster than ever before, with a range of available therapeutic options that is now almost as diverse as this group of tumors. Immunotherapy in particular is front and center in the battle to control these diseases. Here, we describe the latest promising developments.
Exploiting T cells
The treatment landscape for hematologic malignancies is diverse, but one particular type of therapy has led the charge in improving patient outcomes. Several features of hematologic malignancies may make them particularly amenable to immunotherapy, including the fact that they are derived from corrupt immune cells and come into constant contact with other immune cells within the hematopoietic environment in which they reside. One of the oldest forms of immunotherapy, hematopoietic stem-cell transplantation (HSCT), remains the only curative option for many patients with hematologic malignancies.1,2
Given the central role of T lymphocytes in antitumor immunity, research efforts have focused on harnessing their activity for cancer treatment. One example of this is adoptive cellular therapy (ACT), in which T cells are collected from a patient, grown outside the body to increase their number and then reinfused back to the patient. Allogeneic HSCT, in which the stem cells are collected from a matching donor and transplanted into the patient, is a crude example of ACT. The graft-versus-tumor effect is driven by donor cells present in the transplant, but is limited by the development of graft-versus-host disease (GvHD), whereby the donor T cells attack healthy host tissue.
Other types of ACT have been developed in an effort to capitalize on the anti-tumor effects of the patients own T cells and thus avoid the potentially fatal complication of GvHD. Tumor-infiltrating lymphocyte (TIL) therapy was developed to exploit the presence of tumor-specific T cells in the tumor microenvironment. To date, the efficacy of TIL therapy has been predominantly limited to melanoma.1,3,4
Most recently, there has been a substantial buzz around the idea of genetically engineering T cells before they are reintroduced into the patient, to increase their anti-tumor efficacy and minimize damage to healthy tissue. This is achieved either by manipulating the antigen binding portion of the T-cell receptor to alter its specificity (TCR T cells) or by generating artificial fusion receptors known as chimeric antigen receptors (CAR T cells; Figure 1). The former is limited by the need for the TCR to be genetically matched to the patient’s immune type, whereas the latter is more flexible in this regard and has proved most successful.
CARs are formed by fusing part of the single-chain variable fragment of a monoclonal antibody to part of the TCR and one or more costimulatory molecules. In this way, the T cell is guided to the tumor through antibody recognition of a particular tumor-associated antigen, whereupon its effector functions are activated by engagement of the TCR and costimulatory signal.5
Headlining advancements with CAR T cells
CAR T cells directed against the CD19 antigen, found on the surface of many hematologic malignancies, are the most clinically advanced in this rapidly evolving field (Table 1). Durable remissions have been demonstrated in patients with relapsed and refractory hematologic malignancies, including non-Hodgkin lymphoma (NHL), chronic lymphocytic leukemia (CLL), and acute lymphoblastic lymphoma (ALL), with efficacy in both the pre- and posttransplant setting and in patients with chemotherapy-refractory disease.4,5
CTL019, a CD19-targeted CAR-T cell therapy, also known as tisagenlecleucel-T, has received breakthrough therapy designation from the US Food and Drug Administration (FDA) for the treatment of pediatric and adult patients with relapsed/refractory B-cell ALL and, more recently, for the treatment of adult patients with relapsed/refractory diffuse large B cell lymphoma.6
It is edging closer to FDA approval for the ALL indication, having been granted priority review in March on the basis of the phase 2 ELIANA trial, in which 50 patients received a single infusion of CTL019. Data presented at the American Society of Hematology annual meeting in December 2016 showed that 82% of patients achieved either complete remission (CR) or CR with incomplete blood count recovery (CRi) 3 months after treatment.7
Meanwhile, Kite Pharma has a rolling submission with the FDA for KTE-C19 (axicabtagene ciloleucel) for the treatment of patients with relapsed/refractory B-cell NHL who are ineligible for HSCT. In the ZUMA-1 trial, this therapy demonstrated an overall response rate (ORR) of 71%.8 Juno Therapeutics is developing several CAR T-cell therapies, including JCAR017, which elicited CR in 60% of patients with relapsed/refractory NHL.9
Target antigens other than CD19 are being explored, but these are mostly in the early stages of clinical development. While the focus has predominantly been on the treatment of lymphoma and leukemia, a presentation at the American Society for Clinical Oncology annual meeting in June reported the efficacy of a CAR-T cell therapy targeting the B-cell maturation antigen in patients with multiple myeloma. Results from 19 patients enrolled in an ongoing phase 1 trial in China showed that 14 had achieved stringent CR, 1 partial remission (PR) and 4 very good partial remission (VGPR).10
Antibodies evolve
Another type of immunotherapy that has revolutionized the treatment of hematologic malignancies is monoclonal antibodies (mAbs), targeting antigens on the surface of malignant B and T cells, in particular CD20. The approval of CD20-targeting mAb rituximab in 1997 was the first coup for the development of immunotherapy for the treatment of hematologic malignancies. It has become part of the standard treatment regimen for B-cell malignancies, including NHL and CLL, in combination with various types of chemotherapy.
Several other CD20-targeting antibodies have been developed (Table 2), some of which work in the same way as rituximab (eg, ofatumumab) and some that have a slightly different mechanism of action (eg, obinutuzumab).11 Both types of antibody have proved highly effective; ofatumumab is FDA approved for the treatment of advanced CLL and is being evaluated in phase 3 trials in other hematologic malignancies, while obinutuzumab has received regulatory approval for the first-line treatment of CLL, replacing the standard rituximab-containing regimen.12
The use of ofatumumab as maintenance therapy is supported by the results of the phase 3 PROLONG study in which 474 patients were randomly assigned to ofatumumab maintenance for 2 years or observation. Over a median follow-up of close to 20 months, ofatumumab-treated patients experienced improved progression-free survival (PFS; median PFS: 29.4 months vs 15.2 months; hazard ratio [HR], 0.50; P < .0001).13 Obinutuzumab’s new indication is based on data from the phase 3 GADOLIN trial, in which the obinutuzumab arm showed improved 3-year PFS compared with rituximab.14Until recently, multiple myeloma had proven relatively resistant to mAb therapy, but two new drug targets have dramatically altered the treatment landscape for this type of hematologic malignancy. CD2 subset 1 (CS1), also known as signaling lymphocytic activation molecule 7 (SLAMF7), and CD38 are glycoproteins expressed highly and nearly uniformly on the surface of multiple myeloma cells and only at low levels on other lymphoid and myeloid cells.15
Several antibodies directed at these targets are in clinical development, but daratumumab and elotuzumab, targeting CD38 and CS1, respectively, are both newly approved by the FDA for relapsed/refractory disease, daratumumab as monotherapy and elotuzumab in combination with lenalidomide and dexamethasone.
The indication for daratumumab was subsequently expanded to include its use in combination with lenalidomide plus dexamethasone or bortezomib plus dexamethasone. Support for this new indication came from 2 pivotal phase 3 trials. In the CASTOR trial, the combination of daratumumab with bortezomib–dexamethasone reduced the risk of disease progression or death by 61%, compared with bortezomib–dexamethasone alone, whereas daratumumab with lenalidomide–dexamethasone reduced the risk of disease progression or death by 63% in the POLLUX trial.16,17
Numerous clinical trials for both drugs are ongoing, including in the front-line setting in multiple myeloma, as well as trials in other types of B-cell malignancy, and several other CD38-targeting mAbs are also in development, including isatuximab, which has reached the phase 3 stage (NCT02990338).
Innovative design
Newer drug designs, which have sought to take mAb therapy to the next level, have also shown significant efficacy in hematologic malignancies. Antibody-drug conjugates (ADCs) combine the cytotoxic efficacy of chemotherapeutic agents with the specificity of a mAb targeting a tumor-specific antigen. This essentially creates a targeted payload that improves upon the efficacy of mAb monotherapy but mitigates some of the side effects of chemotherapy related to their indiscriminate killing of both cancerous and healthy cells.
The development of ADCs has been somewhat of a rollercoaster ride, with the approval and subsequent withdrawal of the first-in-class drug gemtuzumab ozogamicin in 2010, but the field was reinvigorated with the successful development of brentuximab vedotin, which targets the CD30 antigen and is approved for the treatment of multiple different hematologic malignancies, including, most recently, for posttransplant consolidation therapy in patients with Hodgkin lymphoma at high risk of relapse or progression.18
Brentuximab vedotin may soon be joined by another FDA-approved ADC, this one targeting CD22. Inotuzumab ozogamicin was recently granted priority review for the treatment of relapsed/refractory ALL. The FDA is reviewing data from the phase 3 INO-VATE study in which inotuzumab ozogamicin reduced the risk of disease progression or death by 55% compared with standard therapy, and a decision is expected by August.19 Other ADC targets being investigated in clinical trials include CD138, CD19, and CD33 (Table 3). Meanwhile, a meta-analysis of randomized trials suggested that the withdrawal of gemtuzumab ozogamicin may have been premature, indicating that it does improve long-term overall survival (OS) and reduces the risk of relapse.20
Bispecific antibodies that link natural killer (NK) cells to tumor cells, by targeting the NK-cell receptor CD16, known as BiKEs, are also in development in an attempt to harness the power of the innate immune response.
B-cell signaling a ripe target
Beyond immunotherapy, molecularly targeted drugs directed against key drivers of hematologic malignancies are also showing great promise. In particular, the B-cell receptor (BCR) signaling pathway, a central regulator of B-cell function, and its constituent kinases that are frequently dysregulated in B cell malignancies, has emerged as an exciting therapeutic avenue.
A variety of small molecule inhibitors targeting different nodes of the BCR pathway have been developed (Table 4), but the greatest success to date has been achieved with drugs targeting Bruton’s tyrosine kinase (BTK). Their clinical development culminated in the approval of ibrutinib for the treatment of patients with mantle cell lymphoma in 2013 and subsequently for patients with CLL, Waldenström macroglobulinemia, and most recently for patients with marginal zone lymphoma.
More than 100 clinical trials of ibrutinib are ongoing in an effort to further clarify its role in a variety of different disease settings. Furthermore, in an effort to address some of the toxicity concerns with ibrutinib, more specific BTK inhibitors are also being developed.
Other kinases that orchestrate the BCR pathway, including phosphatidylinositol-3-kinase (PI3K) and SYK, are also being targeted. The delta isoform of PI3K is expressed exclusively in hematopoietic cells and a number of PI3K delta inhibitors have been developed. Idelalisib received regulatory approval for the treatment of patients with CLL in combination with rituximab, and for patients with follicular lymphoma and small lymphocytic leukemia.
As with ibrutinib, a plethora of clinical trials are ongoing, however a major setback was suffered in the frontline setting when Gilead Sciences halted 6 clinical trials due to reports of increased rates of adverse events, including deaths.26 Meanwhile, SYK inhibitors have lagged behind somewhat in their development, but one such offering, entospletinib, is showing promise in patients with AML.27
Finally, there has been some success in targeting one of the downstream targets of the BCR signaling pathway, the Bcl2 protein that is involved in the regulation of apoptosis. Venetoclax was approved last year for the treatment of patients with relapsed/refractory CLL in patients who have a chromosome 17p deletion, based on the demonstration of impressive, durable responses.28
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
1. Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haemato- logical malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201-215.
2. Im A, Pavletic SZ. Immunotherapy in hematologic malignancies: past, present, and future. J Hematol Oncol. 2017;10(1):94.
3. Gill S. Planes, trains, and automobiles: perspectives on CAR T cells and other cellular therapies for hematologic malignancies. Curr Hematol Malig Rep. 2016;11(4):318-325.
4. Ye B, Stary CM, Gao Q, et al. Genetically modified T-cell-based adoptive immunotherapy in hematological malignancies. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5237740/. Published January 2, 2017. Accessed July 22, 2017.
5. Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
6. Novartis. Novartis personalized cell therapy CTL019 receives FDA breakthrough therapy designation. https://www.novartis.com/news/media-releases/novartis-personalized-cell-therapy-ctl019-receivesfda-breakthrough-therapy. Published July 7, 2014. Accessed June 19,
2017.
7. Novartis. Novartis presents results from first global registration trial of CTL019 in pediatric and young adult patients with r/r B-ALL. https://www.novartis.com/news/media-releases/novartis-presentsresults-first-global-registration-trial-ctl019-pediatric-and. Published December 4, 2016. Accessed June 19, 2017.
8. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA1: a multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285-295.
9. Abramson JS, Palomba L, Gordon L. Transcend NHL 001: immunotherapy with the CD19-Directd CAR T-cell product JCAR017 results in high complete response rates in relapsed or refractory B-cell non-Hodgkin lymphoma. Paper presented at 58th American Society of Hematology Annual Meeting; December 3-6, 2016; San Diego, CA.
10. Fan F, Zhao W, Liu J, et al. Durable remissions with BCMA-specific chimeric antigen receptor (CAR)-modified T cells in patients with refractory/relapsed multiple myeloma. J Clin Oncol. 2017;35(suppl;):Abstr LBA3001.
11. Okroj M, Osterborg A, Blom AM. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat Rev. 2013;39(6):632-639.
12. Safdari Y, Ahmadzadeh V, Farajnia S. CD20-targeting in B-cell malignancies: novel prospects for antibodies and combination therapies. Invest New Drugs. 2016;34(4):497-512.
13. van Oers MH, Kuliczkowski K, Smolej L, et al. Ofatumumab maintenance versus observation in relapsed chronic lymphocytic leukaemia (PROLONG): an open-label, multicentre, randomised phase 3 study. Lancet Oncol. 2015;16(13):1370-1379.
14. Sehn LH, Chua N, Mayer J, et al. Obinutuzumab plus bendamustine versus bendamustine monotherapy in patients with rituximab-refractory indolent non-Hodgkin lymphoma (GADOLIN): a randomised, controlled, open-label, multicentre, phase 3 trial. Lancet Oncol. 2016;17(8):1081-1093.
15. Touzeau C, Moreau P, Dumontet C. Monoclonal antibody therapy in multiple myeloma. Leukemia. 2017;31(5):1039-1047.
16. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754-766.
17. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319-1331.
18. Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315-337.
19. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740-753.
20. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986-996.
21. Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93(3):290-296.
22. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836-847.
23. Koehrer S, Burger JA. B-cell receptor signaling in chronic lymphocytic leukemia and other B-cell malignancies. Clin Adv Hematol Oncol. 2016;14(1):55-65.
24. Seda V, Mraz M. B-cell receptor signalling and its crosstalk with other pathways in normal and malignant cells. Eur J Haematol. 2015;94(3):193-205.
25. Bojarczuk K, Bobrowicz M, Dwojak M, et al. B-cell receptor signaling in the pathogenesis of lymphoid malignancies. Blood Cells Mol Dis. 2015;55(3):255-265.
26. Medscape Medical News. Gilead stops six trials adding idelalisib to other drugs. http://www.medscape.com/viewarticle/860372. Published March 14, 2016. Accessed June 19, 2017.
27. Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157-170.
28. Food and Drug Administration. FDA approves new drug for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm495253.htm. Released April 11, 2016. Accessed June 19, 2017.
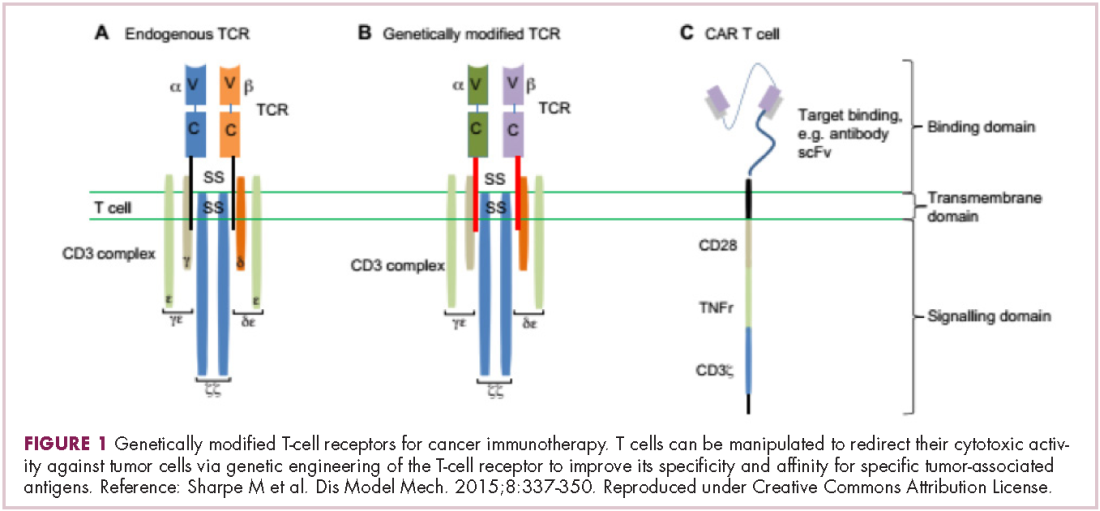
Tumor heterogeneity: a central foe in the war on cancer
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.

Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).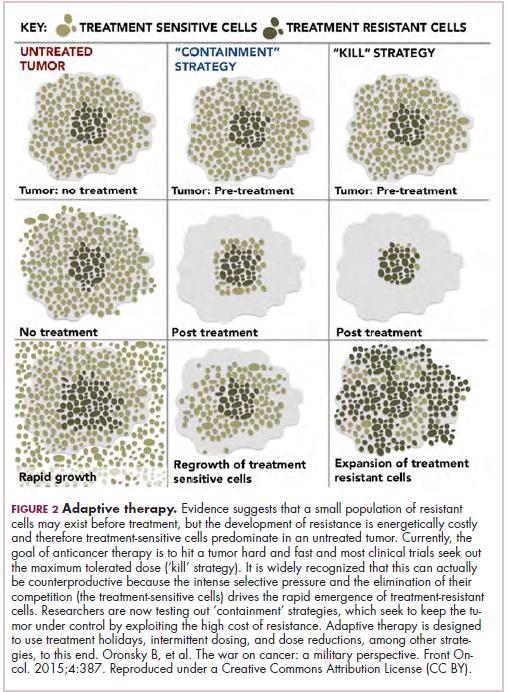
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22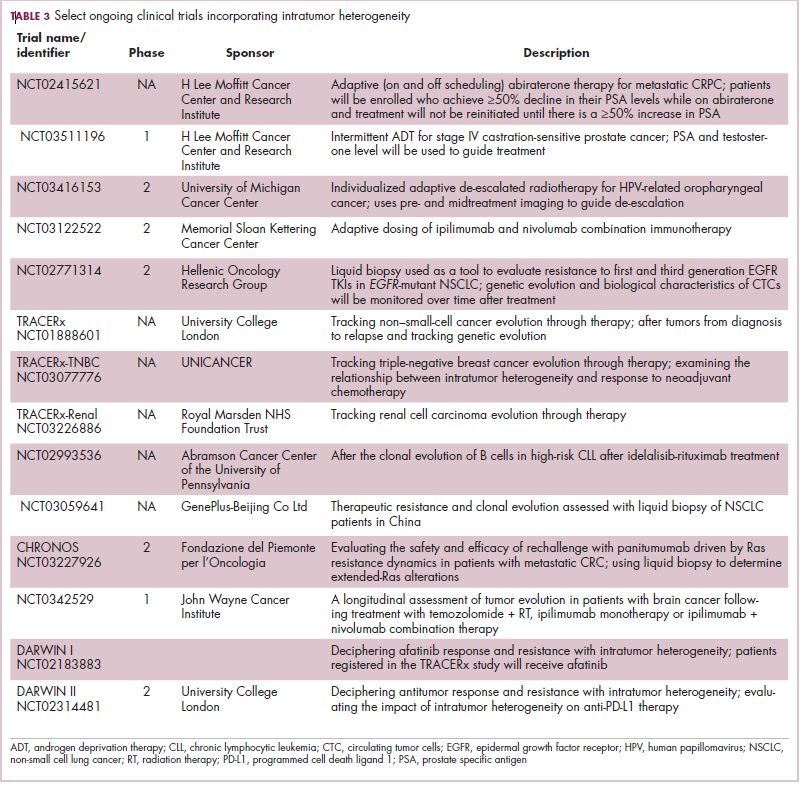
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.
Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
A major challenge to effective cancer treatment is the astounding level of heterogeneity that tumors display on many different fronts. Here, we discuss how a deeper appreciation of this heterogeneity and its impact is driving research efforts to better understand and tackle it and a radical rethink of treatment paradigms.
A complex and dynamic disease
The nonuniformity of cancer has long been appreciated, reflected most visibly in the variation of response to the same treatment across patients with the same type of tumor (inter-tumor heterogeneity). The extent of tumor heterogeneity is being fully realized only now, with the advent of next-generation sequencing technologies. Even within the same tumor, there can be significant heterogeneity from cell to cell (intra-tumor heterogeneity), yielding substantial complexity in cancer.
Heterogeneity reveals itself on many different levels. Histologically speaking, tumors are composed of a nonhomogenous mass of cells that vary in type and number. In terms of their molecular make-up, there is substantial variation in the types of molecular alterations observed, all the way down to the single cell level. In even more abstract terms, beyond the cancer itself, the microenvironment in which it resides can be highly heterogeneous, composed of a plethora of different supportive and tumor-infiltrating normal cells.
Heterogeneity can manifest spatially, reflecting differences in the composition of the primary tumor and tumors at secondary sites or across regions of the same tumor mass and temporally, at different time points across a tumor’s natural history. Evocative of the second law of thermodynamics, cancers generally become more diverse and complex over time.1-3
A tale of 2 models
It is widely accepted that the transformation of a normal cell into a malignant one occurs with the acquisition of certain “hallmark” abilities, but there are myriad ways in which these can be attained.
The clonal evolution model
As cells divide, they randomly acquire mutations as a result of DNA damage. The clonal evolution model posits that cancer develops as the result of a multistep accumulation of a series of “driver” mutations that confer a promalignant advantage to the cell and ultimately fuel a cancerous hallmark.
This evolution can occur in a linear fashion, whereby the emergence of a new driver mutation conveys such a potent evolutionary advantage that it outcompetes all previous clones. There is limited evidence for linear evolution in most advanced human cancers; instead, they are thought to evolve predominantly through a process of branching evolution, in which multiple clones can diverge in parallel from a common ancestor through the acquisition of different driver mutations. This results in common clonal mutations that form the trunk of the cancer’s evolutionary tree and are shared by all cells and subclonal mutations, which make up the branches and differ from cell to cell.
More recently, several other mechanisms of clonal evolution have been proposed, including neutral evolution, a type of branching evolution in which there are no selective pressures and evolution occurs by random mutations occurring over time that lead to genetic drift, and punctuated evolution, in which there are short evolutionary bursts of hypermutation.4,5
The CSC model
This model posits that the ability to form and sustain a cancer is restricted to a single cell type – the cancer stem cells – which have the unique capacity for self-renewal and differentiation. Although the forces of evolution are still involved in this model, they act on a hierarchy of cells, with stem cells sitting at the top. A tumor is derived from a single stem cell that has acquired a mutation, and the heterogeneity observed results both from the differentiation and the accumulation of mutations in CSCs.
Accumulated experimental evidence suggests that these models are not mutually exclusive and that they can all contribute to heterogeneity in varied amounts across different tumor types. What is clear is that heterogeneity and evolution are intricately intertwined in cancer development.1,2,6
An unstable genome
Heterogeneity and evolution are fueled by genomic alterations and the genome instability that they foster. This genome instability can range from single base pair substitutions to a doubling of the entire genome and results from both exposure to exogenous mutagens (eg, chemicals and ultraviolet radiation) and genomic alterations that have an impact on important cellular processes (eg, DNA repair or replication).
Among the most common causes of genome instability are mutations in the DNA mismatch repair pathway proteins or in the proofreading polymerase enzymes. Genome instability is often associated with unique mutational signatures – characteristic combinations of mutations that arose as the result of the specific biological processes underlying them.7
Genome-wide analyses have begun to reveal these mutational signatures across the spectrum of human cancers. The Wellcome Sanger Institute’s Catalogue of Somatic Mutations in Cancer (COSMIC) database has generated a set of 30 mutational signatures based on analysis of almost 11,000 exomes and more than 1,000 whole genomes spanning 40 different cancer types, some of which have been linked with specific mutagenic processes, such as tobacco, UV radiation, and DNA repair deficiency (Table 1).8
Fueling resistance
Arguably, heterogeneity presents one of the most significant barriers to effective cancer therapy, and this has become increasingly true in the era of personalized medicine in which targeted therapies take aim at specific molecular abnormalities.
It is vital that drugs target the truncal alterations that are present in all cancer cells to ensure that the entire cancer is eradicated. However, it is not always possible to target these alterations, for example, at the present time tumor suppressor proteins like p53 are not druggable.
Even when truncal alterations have been targeted successfully, such as epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) chromosomal rearrangements in non–small-cell lung cancer (NSCLC) and BRAF mutations in melanoma, the long-term efficacy of these drugs is almost invariably limited by the development of resistance.
Tumor heterogeneity and the clonal evolution it fuels are central drivers of resistance. Because tumors are dynamic and continue to evolve, anticancer treatments can act as a strong selective pressure and drive the emergence of drug-resistant subclones that allow the tumor to persist. In fact, study findings have revealed that small populations of resistant cells may be present before treatment. Thus, resistance may also occur as a result of the outgrowth of preexisting treatment-resistant cells that suddenly find that they acquire a survival advantage in the presence of a drug.1,6
Tackling heterogeneity
Despite extensive clinical documentation of the existence of heterogeneity and its underlying mechanisms across a range of tumor types, the development of novel clinical trial designs and therapeutic strategies that account for its effects have only recently begun to be explored.
For the most part, this was because of a lack of effective methods for evaluating intratumor heterogeneity. Multiregion biopsies, in which tissue derived from multiple different regions of a single tumor mass or from distinct cancerous lesions within the same patient, give a snapshot of tumor heterogeneity at a single point in time. The repeated longitudinal sampling required to gain a deeper appreciation of tumor heterogeneity over the course of tumor evolution is often not possible because of the morbidity associated with repeated surgical procedures.
Liquid biopsies, in which DNA sequencing can be performed on tumor components that are found circulating in the blood of cancer patients (including circulating tumor cells and cell-free circulating tumor DNA) have rapidly gained traction in the past several decades and offer an unprecedented opportunity for real-time assessment of evolving tumor heterogeneity.
They have proved to be highly sensitive and specific, with a high degree of concordance with tissue biopsy, they can identify both clonal and subclonal mutations, and they can detect resistance substantially earlier than radiographic imaging, which could permit earlier intervention.10,11 The first liquid biopsy-based companion diagnostic test was approved by the US Food and Drug Administration in 2016, for the detection of EGFR mutations associated with NSCLC.
Yet, even liquid biopsy alone is not able to fully dissect the extent of tumor heterogeneity, especially because it is limited in its ability to assess spatial heterogeneity. Truly effective assessment of tumor heterogeneity is likely to require a combination of liquid biopsy, carefully selected tumor tissue biopsies, imaging diagnostics, and biomarkers.
The ongoing TRACERx (Tracking cancer evolution through therapy [Rx]) trials are evaluating a combination of approaches to follow tumor evolution across the course of treatment. The study in NSCLC began in 2014 with a target enrollment of 842 patients and will follow patients over 6 years. Preliminary data from the first 100 patients were recently published and demonstrated that increased intratumor heterogeneity correlated with increased risk of recurrence or death.12
If patients consent, the TRACERx trials also feed into the PEACE (Posthumous evaluation of advanced cancer environment) trials, which are collecting postmortem biopsies to further evaluate tumor heterogeneity and evolution. TRACERx trials in several other cancer types are now also underway.
Cutting off the source
The main therapeutic strategies for overcoming tumor heterogeneity are focused on the mechanisms of resistance that it drives. It is becoming increasingly apparent that rationally designed combinations of drugs are likely to be required and might need to be administered early in the course of disease to prevent resistance.
However, according to mathematical modeling studies, combinations of at least 3 drugs may be necessary.13 In many cases, this is unlikely to be feasible owing to the unavailability of drugs for certain targets and issues of toxicity, as well as the high cost.
An alternative strategy is to use immunotherapy, because a single treatment can target multiple neoantigens simultaneously. Although immunotherapy has proved to be a highly effective treatment paradigm in multiple tumor types, resistance still arises through varied mechanisms with tumor heterogeneity at their core.14,15
A promising avenue for drug development is to cut off the source of tumor heterogeneity – genomic instability and the mutagenic processes that foster it (Table 2). This is exemplified by the success of poly(ADP-ribose) polymerase (PARP) inhibitors in patients with breast cancer susceptibility (BRCA1/2) gene mutations.
Both germline and somatic mutations in the BRCA1/2 genes are observed in 10% to 15% of patients with ovarian cancer and a substantial number of patients with other types of cancer, including breast, pancreatic, and prostate cancers.16,17
These genes play a central role in the homologous recombination (HR) pathway of DNA repair, which repairs double-strand breaks in DNA. PARP inhibitors target a different DNA repair pathway, base excision repair, which repairs single-strand breaks. The use of PARP inhibitors in patients with BRCA1/2 mutations is designed to create irreparable damage to the DNA repair processes and drive an unsustainable level of genome instability that leads to cell death, whereas normal cells without HR deficiency can survive.18
A growing number of PARP inhibitors are now approved for use in the United States for the treatment of ovarian cancer. In January, olaparib became the first PARP inhibitor approved for patients with BRCA1/2-mutant breast cancer, based on data from the OlympiAD trial in which 302 patients were randomized to receive olaparib 300 mg twice daily or physician’s choice of chemotherapy. Olaparib improved progression-free survival from 4.2 months to 7.0 months (hazard ratio, 0.58; P = .0009), and the most common adverse events included anemia, nausea, fatigue, and vomiting.19
Tumors with other defects in HR have also shown susceptibility to PARP inhibition, shifting interest toward identifying and treating these tumors as a group, independent of histology – about a quarter of all tumors display HR deficiency.20 This novel strategy of targeting mutational processes across a range of tumor types has also been exploited in the development of immunotherapies.
Patients with defects in the mismatch repair (MMR) pathway and microsatellite instability (MSI) – multiple alterations in the length of microsatellite markers within the DNA – are more sensitive to immunotherapy, likely because they are predisposed to a high level of somatic mutations that can serve as neoantigens to provoke a strong anti-tumor immune response.
In 2017, 2 immune checkpoint inhibitors were approved for use in patients with MSI-high or defective MMR (dMMR) cancers. The indication for pembrolizumab (Keytruda) was independent of tumor histology, the first approval of its kind. It was based on the results of 5 clinical trials in which 149 patients with MSI-H or dMMR cancers were given pembrolizumab 200 mg every 3 weeks or 10 mg/kg every 2 weeks for a maximum of 24 months. The overall response rate was 39.6%, including 11 complete responses and 48 partial responses.21
A new paradigm
Treatment of a tumor is one of the major selective pressures that shapes its evolution and recent evidence has emerged that these selective pressures can be highly dynamic. Study findings have shown that there is a cost associated with evolution of resistant subclones and, if the selective pressure of therapy is removed, that cost may become too high, such that resistant subclones are then outcompeted by drug-sensitive ones. There have been reports of reversal of drug resistance when drug treatment is interrupted.
The current treatment paradigm is to try to eliminate tumors by hitting them hard and fast with the maximum tolerated dose (MTD) of a drug. However, there is increasing appreciation that this may be inadvertently fostering more rapid disease progression because it selects for the emergence of resistant cells and eliminates all their competitors (Figure 2).
This is driving a potential paradigm shift, in which researchers are applying concepts from evolutionary biology and the control of invasive species to the treatment of cancer. Instead of completely eliminating a cancer, a strategy of adaptive therapy could be used to set up competition between different subclones and keep tumor growth in check by exploiting the high cost of resistance.22
Adaptive therapy involves the use of treatment holidays, intermittent dosing schedules or reduced drug doses, rather than using the MTD. Adaptive therapy was tested recently in mice with triple-negative and estrogen receptor-positive breast cancer. The standard maximum dose of chemotherapy was compared with adaptive therapy with either reduced doses or skipped doses as the tumor responded. Tumor growth initially decreased with all 3 treatment scenarios, but then regrew when chemotherapy was stopped or doses were skipped. However, adaptive therapy with lower doses resulted in long-term stabilization of the tumor where treatment was eventually able to be withdrawn.23 Clinical trials of several different types of adaptive therapy strategies are ongoing (Table 3).
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
1. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81-94.
2. Dzobo K, Senthebane DA, Thomford NE, Rowe A, Dandara C, Parker MI. Not everyone fits the mold: intratumor and intertumor heterogeneity and innovative cancer drug design and development. OMICS. 2018;22(1):17-34.
3. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168(4):613-628.
4. Davis A, Gao R, Navin N. Tumor evolution: linear, branching, neutral or punctuated? Biochim Biophys Acta. 2017;1867(2):151-161.
5. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. Published online first July 20, 2017. Accessed May 23, 2018. doi: 10.1158/2159-8290.CD-17-0343
6. Wu D, Wang DC, Cheng Y, et al. Roles of tumor heterogeneity in the development of drug resistance: a call for precision therapy. Semin Cancer Biol. 2017;42:13-19.
7. Ferguson LR, Chen H, Collins AR, et al. Genomic instability in human cancer: molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin Cancer Biol. 2015;35(suppl):S5-S24.
8. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805-811.
9. Rosenthal R, McGranahan N, Herrero J, Swanton C. Deciphering genetic intratumor heterogeneity and its impact on cancer evolution. Ann Rev Cancer Biol. 2017;1(1):223-240.
10. Esposito A, Criscitiello C, Locatelli M, Milano M, Curigliano G. Liquid biopsies for solid tumors: understanding tumor heterogeneity and real time monitoring of early resistance to targeted therapies. Pharmacol Ther. 2016;157:120-124.
11. Venesio T, Siravegna G, Bardelli A, Sapino A. Liquid biopsies for monitoring temporal genomic heterogeneity in breast and colon cancers. Pathobiology. 2018;85(1-2):146-154.
12. Jamal-Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non–small-cell lung cancer. New Engl J Med. 2017;376(22):2109-2121.
13. Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747.
14. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551-1566.
15. Ventola CL. Cancer immunotherapy, Part 3: challenges and future trends. PT. 2017;42(8):514-521.
16. Cavanagh H, Rogers KMA. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin Pract. 2015;13:16.
17. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
18. Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20-37.
19. Robson M, Im S-A, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. New England Journal of Medicine. 2017;377(6):523-533.
20. Williers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer: molecular targets and clinical applications. In: Kelley M, Fishel M, eds. DNA repair in cancer therapy. 2nd ed: Academic Press; 2016:119-160.
21. U.S. Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2017; https://www.fda.gov/Drugs/InformationOnDrugs/ ApprovedDrugs/ucm560040.htm. Accessed May 1st,, 2018.
22. Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Adaptive Therapy For Heterogeneous Cancer: Exploiting Space And Trade-Offs In Drug Scheduling. bioRxiv. 2017.
23. Enriquez-Navas PM, Kam Y, Das T, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8(327):327ra24.
Heart Transplantation Outcomes in Patients With Hepatitis C Virus Infection: Potential Impact of Newer Antiviral Treatments After Transplantation (FULL)
More than 185 million people worldwide, including more than 4 million in the U.S., are infected with the hepatitis C virus (HCV).1 Because of the indolent nature of the disease, actual prevalence is underestimated.2,3 Detection of HCV in people already infected is estimated to continue to increase over the next decade.4 Although primary manifestations of the disease are the result of liver damage, HCV infection is a systemic illness. In a study of more than 19,000 patients, HCV infection was identified as an independent risk factor for development of heart failure.5 In the U.S., prevalence of HCV infection in patients with heart failure is reported to be as high as 15%, much higher than the general population prevalence of 1.8%.6 When first identified in 1989, HCV infection was considered incurable. Clinical trials have since found a steady improvement in outcome, and now the disease is considered curable in up to 90% of cases.7
Clinical outcomes of heart transplantation (HTx) historically have been inferior in patients with HCV infection.8,9 The authors hypothesized that the literature on HTx outcomes has not accounted for the improvements in HCV infection treatment options that have occurred since the 1990s. In the study reported here, United Network of Organ Sharing (UNOS) data on adult HTx was used to evaluate clinical outcomes of HCV infection over 4 treatment eras.
Material and Methods
The authors analyzed UNOS data on adult HTx from January 1991 to March 2014. Two groups were created: patients with HCV infection (HC+) and noninfected patients (HC–). Eligible patients were aged > 18 years. Hepatitis C virus status was defined with antibody testing at time of HTx. Patients with multiorgan transplantation or with hepatitis B virus or HIV infection were excluded. For comparison of post-HTx survival, the 23-year study period was divided into 4 eras reflecting the evolution in HCV infection treatment options in the U.S. (Table 1). The first medication was interferon α (IFN-α), which was used alone (first era, 1991-1997) and then with the newly introduced ribavirin (second era, 1998-2000). The combination of IFN-α and ribavirin increased sustained virologic response rates, but the rates of adverse effects (AEs), such as cytopenia and depression, were high, and many patients could not tolerate the extended (48-week) regimen.10,11

Peginterferon, a long-acting IFN introduced in 2001, significantly increased adherence to 2-drug treatment for HCV infection, and its use in combination with ribavirin marked the third era (2001-2010). The fourth era (2011-2014) began with the introduction of direct-acting antiviral agents and their remarkable results. Since 2014, direct protease inhibitors without IFN found a dramatic impact on HCV treatment: fewer AEs, shorter treatment (24 weeks), and high (> 90%) sustained virologic response.7,12,13
Statistical Analysis
Categoric variables were analyzed with the χ2 test or the Fisher exact test and are reported as percentages. Continuous variables were analyzed with the Student t test or the Wilcoxon rank sum test and are reported as means, medians, and SDs. Statistical significance was set at P < .05. Survival curves were plotted with the Kaplan-Meier method, and comparisons made with log-rank tests. Analysis was performed with SAS Version 9.3 (Cary, NC).
Results
Between January 1991 and March 2014, adult HTx was performed 36,589 times, including 778 times (2.1%) in patients with HCV infection. There was no significant difference in percentage of HC+ patients who underwent HTx over the 4 treatment eras (first, 2.1%; second, 2.9%; third, 2.1%; fourth, 1.6%) (Table 2). Mean patient age for the HC+ and HC– groups was comparable. Percentage of African American patients was higher in the HC+ group than in the HC– group (18.9% vs 15.0%), as was percentage of patients of other race (11.2% vs 9.2%; P = .0008).
Regarding indications for HTx, ischemic (and nonischemic) cardiomyopathy was similar in prevalence between the 2 groups, but the “other” heart failure etiologies (congenital heart disease, valvular heart disease, postpartum cardiomyopathy, restrictive heart disease) were more prevalent in the HC+ group (12.9% vs 9.7%; P = .013). The HC+ group also had higher rates of tobacco use history (42.2% vs 40.1%; P = .002) and hypertension (23.1% vs 20.9%; P = .014). Mean (SD) bilirubin level at time of transplantation for the HC+ and HC– groups was comparable: 1.12 (1) mg/dL and 1.11 (1) mg/dL, respectively (P = .707). Of the heart donor variables (Table 3), only tobacco use history was significantly higher in the HC+ group (23.5% vs 19.8%; P = .008).
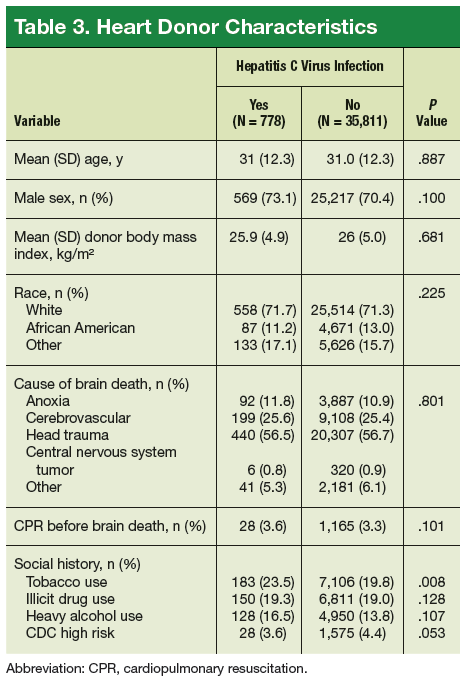
Survival Data
Mean (SD) overall follow-up was 6.2 (5.3) years (median, 5 years; range, 0-23.3 years) for all patients; 5.6 (4.3) years (median, 5.05 years; range, 0-23.2 years) for HC+ patients; and 6.2 (5.3) years (median, 6.1 years; range, 0-23.2 years) for HC– patients.
HC+ patients’ survival rates were 82.5% (1 year), 64.4% (5 years), and 42.1% (10 years), and HC– patients’ rates were 87.2% (1 year), 73.4% (5 years), and 54.7% (10 years). The HC+ group’s inferior survival at 1, 5, and 10 years was statistically significant (P < .0002) (Table 4).
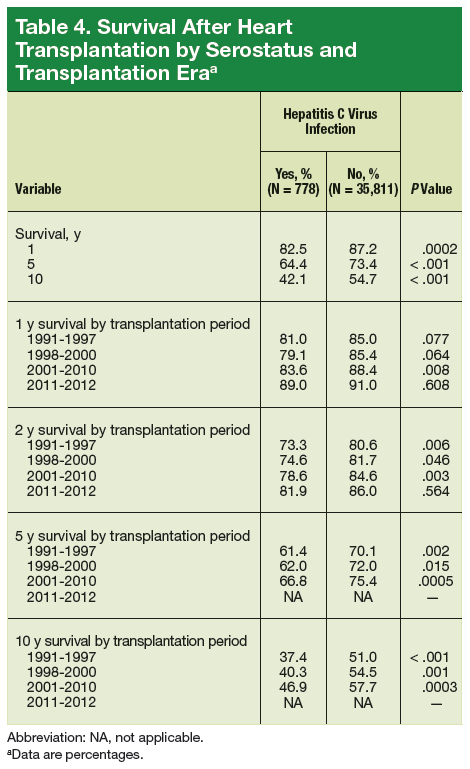
During the first era (1991-1997), HC+ patients’ survival rates were 81.0% (1 year), 73.3% (2 years), and 61.4% (5 years), and HC– patients’ rates were 85.0% (1 year), 80.6% (2 years), and 70.3% (5 years) (P < .05). During the second era (1998-2000), HC+ patients’ rates were 79.1% (1 year), 74.6% (2 years), and 62.0% (5 years), and HC– patients’ rates were 85.4% (1 year), 81.7% (2 years), and 72.0% (5 years) (P < .05). During the third era (2001-2010), HC+ patients’ rates were 83.6% (1 year), 78.6% (2 years), and 66.8% (5 years), and HC– patients’ rates were 88.4% (1 year), 84.6% (2 years), and 75.4% (5 years) (P < .05).
Survival data for the fourth treatment era (2011-2014) were available only for 1 and 2 years. HC+ patients’ survival rates were 89.01% (1 year) and 81.89% (2 years), and HC– patients’ rates were 91.00% (1 year) and 86.00% (2 years). The inferior survival found for the HC+ group during the 3 preceding eras was not found this era, during which HC+ and HC– patients had comparable rates of survival at 1 year (Figures 1 and 2).
Survival 1 year after HTx was compared between the HC+ and HC– groups over the 4 treatment eras (Figures 3 and 4). The HC+ patients’ survival after HTx improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91% (P = .9).
Hepatic decompensation leading to death was uncommon, but the rate was significantly higher (P = .0001) in the HC+ group (2.8%) than in the HC– group (0.6%).
Discussion
HCV infection is a risk factor for the development of cardiovascular illness and advanced heart failure. Given the worldwide prevalence of HCV infection, more HC+ patients will be evaluated for HTx in the future.2-4 Significant progress has been made in HCV infection treatment since the virus was first described. What was once incurable now has up to a 90% cure rate with newer treatment options.7,12,13 The present study findings showed consistent improvement in HC+ patients’ post-HTx survival during each treatment era. During the latest era, HC+ and HC– patients’ post-HTx survival was statistically similar.
It is possible that HC+ patients’ improvement in post-HTx survival could have resulted from improvement in overall post-HTx survival.14 Over the 23-year study period, the survival rates of both groups (HC+, HC–) improved, likely secondary to improved immunosuppression and perioperative care, but the magnitude of improvement was more pronounced in the HC+ group. HC+ patients’ post-HTx survival improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91%. The improvement in HC+ patients’ short-term survival over the study period was substantial but did not reach statistical significance (P = .9).
Over the study period, the percentage of HC+ patients who underwent HTx remained low, ranging from 2.9% during the second era (1998-2000) to 1.7% during the fourth era (2011-2014). Overall, only 2.2% of study patients were HC+ at time of transplantation—a rate similar to previously reported rates.8,15 This rate likely represents a selection bias for HTx listing, in which patients with nearly normal liver function were selected for HTx, as evident by normal bilirubin levels in both groups. In the present study, a high proportion of HC+ patients were not white. This distribution also was noted in epidemiologic studies of HCV infection by ethnicity.6 In the U.S., the highest prevalence of HCV infection was noted in African Americans and the lowest in whites. According to the U.S. census report, African Americans constitute 12% of the total U.S. population,16 whereas 22% of HC+ patients are African American.1
It has been postulated that the immunosuppression that accompanies the post-HTx state accelerates HCV disease progression and shortens HC+ patients’ survival.17,18 These concerns were not validated in the most recent studies of kidney, liver, and heart transplantation in HC+ patients.15,19,20 Furthermore, where post-HTx cause of death was examined in HC+ patients, death was attributed primarily to post-HTx malignancy and bacterial sepsis but seldom directly to hepatic failure. In a study in which liver function was serially monitored after HTx in 11 HC+ patients, immunosuppression did not affect HCV disease progression, and there was no liver function impairment.15 In the same study, the 3 more recent HTx patients (of the 11) had their serial HCV viral load monitored. Viral load remained steady in 2 of the patients and decreased in the third. Similarly, there has been a theoretical concern that heightened immunologic status in HC+ patients might lead to more frequent rejection episodes. However, this concern has not been substantiated in reported studies.21,22 In the present study, the rate of graft failure as the cause of death was 11.2% in HC+ patients and 13% in HC– patients, though the difference was not statistically significant. Thus, concerns about HCV reactivation during immunocompromise or during increased allograft rejection were not substantiated.
Vasculopathy is a leading cause of morbidity and mortality after HTx and seems to be influenced by HC+. In a study of HC+ donor hearts transplanted to HC– patients, the prevalence of HCV infection in the recipients was 75% at a mean follow-up of 4.2 years.23 Serial angiogram showed coronary vasculopathy in 46% of HC+ patients and 24% of HC– patients 3.2 years after HTx.23 Similar concerns were raised in another small study, in which 2 of 4 HC+ patient deaths at 3.7-year follow-up were attributed to cardiovascular causes with features similar to those of transplantation vasculopathy.24 Those findings contrast with the present findings of cardiovascular deaths in 17.6% of HC+ patients and 17.2% of HC– patients. Outcomes similar to the present outcomes were reported by Lee and colleagues: Post-HTx cardiovascular deaths occurred in 16.4% of HC+ patients and 15.2% of HC– patients.8
Survival data on HC+ patients who undergo HTx are mixed, with some studies finding similar shortterm and midterm post-HTx survival21,22 and others finding decreased survival.8,9 It is difficult to interpret survival results from these studies, as some have included HCV infection that developed after HTx,9 and others have excluded early postoperative deaths from analysis.21 In addition, in the larger of these studies, which spanned 15 years, propensity matching was used for survival analysis. 8
It is possible that the selection and treatment of HC+ patients who were awaiting or underwent HTx changed over the study period. Thus, it might not be accurate to compare HTx outcomes of patients without considering the significant progress that has been made in the management of HCV infection. Although the present study’s aggregate (23-year) post-HTx survival results at 1, 5, and 10 years were similar to those reported in large series of HC+ patients who underwent HTx, the present early and intermediate survival results showed consistent improvements over time.8,9 This improvement in survival of HC+ patients was not examined in previous studies.
In the present study, the distribution of causes of post-HTx deaths was diverse (Table 5). Although the leading causes of death were cardiovascular or were related to sepsis or multi-organ failure, deaths attributed to liver failure were uncommon. Only 2.8% of deaths in the HC+ group and 0.6% of deaths in the HC– group were attributed to liver failure (P ≤ .001). These findings are similar to the cause-specific mortality reported in the literature.8,9,15,22 It is possible that these mortality results may be secondary to selection of patients with preserved liver function.
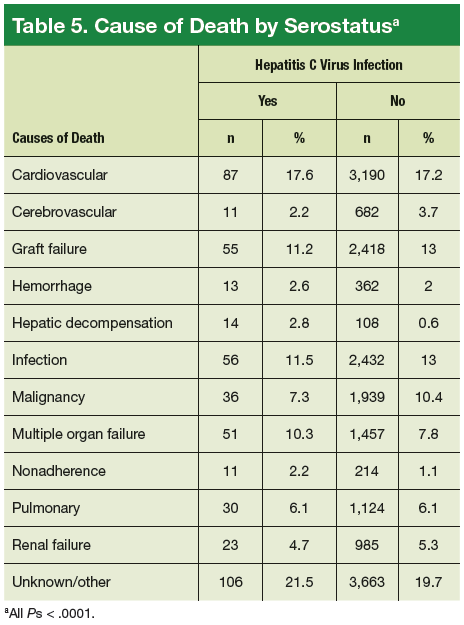
Future of HCV Infection and Heart Transplantation
Modern diagnostic methods will be used to accurately assess HC+ patients for HCV disease burden, and treatment will be provided before HTx is performed. Historically, the major limitations to treating HC+ patients awaiting HTx have been the long (48 week) duration of therapy and the anemia that can exacerbate heart failure symptoms and shorten the safe HTx waiting time.25 Most of the HCV treatment AEs have been attributed to use of IFN-α. Newer HCV treatments (direct-acting protease inhibitors without IFN) are of shorter duration (24 weeks) and have fewer AEs and higher cure rates. Most important, these treatments obtain similar cure rates irrespective of viral load, viral genotype, patient race, and previous HCV response status.7,12,13
Authors researching other solid-organ transplantation (eg, liver, kidney) have studied HCV pretreatment and found sustained virologic response before and after transplantation.26,27 Although these studies were conducted before the advent of direct-acting protease inhibitors, the feasibility of treating HCV before transplantation has been demonstrated.
Limitations
The limitations of retrospective data analysis are applicable to the present findings. Although HTx is infrequently performed in HC+ patients, the authors used a large national database and a 23-year study period and thus were able to gather a significant number of patients and perform meaningful statistical analysis. HCV disease burden, which influences disease progression, is much better quantified with recent quantitative viral loads, but these were not available in the UNOS database. It should be noted that UNOS does not gather information on HCV genotypes or forms of treatment received, both of which influence treatment response and prognosis. It is therefore not possible to elucidate, from the UNOS database, the influences of virus genotype and treatment response on post-HTx outcomes.
Conclusion
The treatment of HCV infection has significantly evolved since the virus was identified in 1989. At first the disease was considered incurable, but now a > 90% cure rate is possible with newer treatment regimens. This study found significant improvements in post-HTx outcomes in HC+ patients between 1991 and 2014. Both HC+ and HC– patients have had similar post-HTx clinical outcomes in recent years. The noted improvements in post-HTx survival in HC+ patients may be secondary to better patient selection or more effective antiviral treatments. Future studies will provide the answers.
Click here to read the digital edition.
1. Razavi H, Waked I, Sarrazin C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21(suppl 1):34-59.
2. Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31(4):1014-1018.
3. Tong MJ, el-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusionassociated hepatitis C. N Engl J Med. 1995;332(22):1463-1466.
4. Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):513-521.
5. Younossi ZM, Stepanova M, Nader F, Younossi Z, Elsheikh E. Associations of chronic hepatitis C with metabolic and cardiac outcomes. Aliment Pharmacol Ther. 2013;37(6):647-652.
6. Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556-562.
7. Ferenci P, Bernstein D, Lalezari J, et al; PEARL-III Study; PEARL-IV Study. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983-1992.
8. Lee I, Localio R, Brensinger CM, et al. Decreased post-transplant survival among heart transplant recipients with pre-transplant hepatitis C virus positivity. J Heart Lung Transplant. 2011;30(11):1266-1274.
9. Fong TL, Hou L, Hutchinson IV, Cicciarelli JC, Cho YW. Impact of hepatitis C infection on outcomes after heart transplantation. Transplantation. 2009;88(9):1137-1141.
10. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975-982.
11. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958-965.
12. Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973-1982.
13. Zeuzem S, Dusheiko GM, Salupere R, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370(21):1993-2001.
14. Lund LH, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant. 2013;32(10):951-964.
15. Cano O, Almenar L, Martínez-Dolz L, et al. Course of patients with chronic hepatitis C virus infection undergoing heart transplantation. Transplant Proc. 2007;39(7):2353-2354.
16. Hobbs F, Stoops N. Demographic Trends in the 20th Century. Washington, DC: U.S. Government Printing Office; 2002. U.S. Census Bureau, Census 2000 Special Reports, Series CENSR-4. https://www.census.gov/prod/2002pubs/censr-4.pdf. Issued November 2002. Accessed February 7, 2017.
17. Pol S, Samuel D, Cadranel J, et al. Hepatitis and solid organ transplantation. Transplant Proc. 2000;32(2):454-457.
18. Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364(25):2429-2438.
19. Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int. 1994;45(1):238-244.
20. Garcia-Saenz-de-Sicilia M, Olivera-Martinez MA, Grant WJ, et al. Impact of anti-thymocyte globulin during immunosuppression induction in patients with hepatitis C after liver transplantation. Dig Dis Sci. 2014;59(11): 2804-2812.
21. Lake KD, Smith CI, Milfred-La Forest SK, Pritzker MR, Emery RW. Outcomes of hepatitis C positive (HCV+) heart transplant recipients. Transplant Proc. 1997;29(1-2):581-582.
22. Shafii AE, Su JW, Smedira NG, et al. The effect of recipient hepatitis C virus infection on outcomes following heart transplantation. Transplant Proc. 2010;42(5):1784-1787.
23. Haji SA, Starling RC, Avery RK, et al. Donor hepatitis-C seropositivity is an independent risk factor for the development of accelerated coronary vasculopathy and predicts outcome after cardiac transplantation. J Heart Lung Transplant. 2004;23(3):277-283.
24. Castella M, Tenderich G, Koerner MM, et al. Outcome of heart transplantation in patients previously infected with hepatitis C virus. J Heart Lung Transplant. 2001;20(5):595-598.
25. Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med. 2006;355(23):2444-2451.
26. Thomas RM, Brems JJ, Guzman-Hartman G, Yong S, Cavaliere P, Van Thiel DH. Infection with chronic hepatitis C virus and liver transplantation: a role for interferon therapy before transplantation. Liver Transpl. 2003;9(9):905-915.
27. Everson GT, Terrault NA, Lok AS, et al. A randomized controlled trial of pretransplant antiviral therapy to prevent recurrence of hepatitis C after liver transplantation. Hepatology. 2013;57(5):1752-1762.
More than 185 million people worldwide, including more than 4 million in the U.S., are infected with the hepatitis C virus (HCV).1 Because of the indolent nature of the disease, actual prevalence is underestimated.2,3 Detection of HCV in people already infected is estimated to continue to increase over the next decade.4 Although primary manifestations of the disease are the result of liver damage, HCV infection is a systemic illness. In a study of more than 19,000 patients, HCV infection was identified as an independent risk factor for development of heart failure.5 In the U.S., prevalence of HCV infection in patients with heart failure is reported to be as high as 15%, much higher than the general population prevalence of 1.8%.6 When first identified in 1989, HCV infection was considered incurable. Clinical trials have since found a steady improvement in outcome, and now the disease is considered curable in up to 90% of cases.7
Clinical outcomes of heart transplantation (HTx) historically have been inferior in patients with HCV infection.8,9 The authors hypothesized that the literature on HTx outcomes has not accounted for the improvements in HCV infection treatment options that have occurred since the 1990s. In the study reported here, United Network of Organ Sharing (UNOS) data on adult HTx was used to evaluate clinical outcomes of HCV infection over 4 treatment eras.
Material and Methods
The authors analyzed UNOS data on adult HTx from January 1991 to March 2014. Two groups were created: patients with HCV infection (HC+) and noninfected patients (HC–). Eligible patients were aged > 18 years. Hepatitis C virus status was defined with antibody testing at time of HTx. Patients with multiorgan transplantation or with hepatitis B virus or HIV infection were excluded. For comparison of post-HTx survival, the 23-year study period was divided into 4 eras reflecting the evolution in HCV infection treatment options in the U.S. (Table 1). The first medication was interferon α (IFN-α), which was used alone (first era, 1991-1997) and then with the newly introduced ribavirin (second era, 1998-2000). The combination of IFN-α and ribavirin increased sustained virologic response rates, but the rates of adverse effects (AEs), such as cytopenia and depression, were high, and many patients could not tolerate the extended (48-week) regimen.10,11
Peginterferon, a long-acting IFN introduced in 2001, significantly increased adherence to 2-drug treatment for HCV infection, and its use in combination with ribavirin marked the third era (2001-2010). The fourth era (2011-2014) began with the introduction of direct-acting antiviral agents and their remarkable results. Since 2014, direct protease inhibitors without IFN found a dramatic impact on HCV treatment: fewer AEs, shorter treatment (24 weeks), and high (> 90%) sustained virologic response.7,12,13
Statistical Analysis
Categoric variables were analyzed with the χ2 test or the Fisher exact test and are reported as percentages. Continuous variables were analyzed with the Student t test or the Wilcoxon rank sum test and are reported as means, medians, and SDs. Statistical significance was set at P < .05. Survival curves were plotted with the Kaplan-Meier method, and comparisons made with log-rank tests. Analysis was performed with SAS Version 9.3 (Cary, NC).
Results
Between January 1991 and March 2014, adult HTx was performed 36,589 times, including 778 times (2.1%) in patients with HCV infection. There was no significant difference in percentage of HC+ patients who underwent HTx over the 4 treatment eras (first, 2.1%; second, 2.9%; third, 2.1%; fourth, 1.6%) (Table 2). Mean patient age for the HC+ and HC– groups was comparable. Percentage of African American patients was higher in the HC+ group than in the HC– group (18.9% vs 15.0%), as was percentage of patients of other race (11.2% vs 9.2%; P = .0008).
Regarding indications for HTx, ischemic (and nonischemic) cardiomyopathy was similar in prevalence between the 2 groups, but the “other” heart failure etiologies (congenital heart disease, valvular heart disease, postpartum cardiomyopathy, restrictive heart disease) were more prevalent in the HC+ group (12.9% vs 9.7%; P = .013). The HC+ group also had higher rates of tobacco use history (42.2% vs 40.1%; P = .002) and hypertension (23.1% vs 20.9%; P = .014). Mean (SD) bilirubin level at time of transplantation for the HC+ and HC– groups was comparable: 1.12 (1) mg/dL and 1.11 (1) mg/dL, respectively (P = .707). Of the heart donor variables (Table 3), only tobacco use history was significantly higher in the HC+ group (23.5% vs 19.8%; P = .008).
Survival Data
Mean (SD) overall follow-up was 6.2 (5.3) years (median, 5 years; range, 0-23.3 years) for all patients; 5.6 (4.3) years (median, 5.05 years; range, 0-23.2 years) for HC+ patients; and 6.2 (5.3) years (median, 6.1 years; range, 0-23.2 years) for HC– patients.
HC+ patients’ survival rates were 82.5% (1 year), 64.4% (5 years), and 42.1% (10 years), and HC– patients’ rates were 87.2% (1 year), 73.4% (5 years), and 54.7% (10 years). The HC+ group’s inferior survival at 1, 5, and 10 years was statistically significant (P < .0002) (Table 4).
During the first era (1991-1997), HC+ patients’ survival rates were 81.0% (1 year), 73.3% (2 years), and 61.4% (5 years), and HC– patients’ rates were 85.0% (1 year), 80.6% (2 years), and 70.3% (5 years) (P < .05). During the second era (1998-2000), HC+ patients’ rates were 79.1% (1 year), 74.6% (2 years), and 62.0% (5 years), and HC– patients’ rates were 85.4% (1 year), 81.7% (2 years), and 72.0% (5 years) (P < .05). During the third era (2001-2010), HC+ patients’ rates were 83.6% (1 year), 78.6% (2 years), and 66.8% (5 years), and HC– patients’ rates were 88.4% (1 year), 84.6% (2 years), and 75.4% (5 years) (P < .05).
Survival data for the fourth treatment era (2011-2014) were available only for 1 and 2 years. HC+ patients’ survival rates were 89.01% (1 year) and 81.89% (2 years), and HC– patients’ rates were 91.00% (1 year) and 86.00% (2 years). The inferior survival found for the HC+ group during the 3 preceding eras was not found this era, during which HC+ and HC– patients had comparable rates of survival at 1 year (Figures 1 and 2).
Survival 1 year after HTx was compared between the HC+ and HC– groups over the 4 treatment eras (Figures 3 and 4). The HC+ patients’ survival after HTx improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91% (P = .9).
Hepatic decompensation leading to death was uncommon, but the rate was significantly higher (P = .0001) in the HC+ group (2.8%) than in the HC– group (0.6%).
Discussion
HCV infection is a risk factor for the development of cardiovascular illness and advanced heart failure. Given the worldwide prevalence of HCV infection, more HC+ patients will be evaluated for HTx in the future.2-4 Significant progress has been made in HCV infection treatment since the virus was first described. What was once incurable now has up to a 90% cure rate with newer treatment options.7,12,13 The present study findings showed consistent improvement in HC+ patients’ post-HTx survival during each treatment era. During the latest era, HC+ and HC– patients’ post-HTx survival was statistically similar.
It is possible that HC+ patients’ improvement in post-HTx survival could have resulted from improvement in overall post-HTx survival.14 Over the 23-year study period, the survival rates of both groups (HC+, HC–) improved, likely secondary to improved immunosuppression and perioperative care, but the magnitude of improvement was more pronounced in the HC+ group. HC+ patients’ post-HTx survival improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91%. The improvement in HC+ patients’ short-term survival over the study period was substantial but did not reach statistical significance (P = .9).
Over the study period, the percentage of HC+ patients who underwent HTx remained low, ranging from 2.9% during the second era (1998-2000) to 1.7% during the fourth era (2011-2014). Overall, only 2.2% of study patients were HC+ at time of transplantation—a rate similar to previously reported rates.8,15 This rate likely represents a selection bias for HTx listing, in which patients with nearly normal liver function were selected for HTx, as evident by normal bilirubin levels in both groups. In the present study, a high proportion of HC+ patients were not white. This distribution also was noted in epidemiologic studies of HCV infection by ethnicity.6 In the U.S., the highest prevalence of HCV infection was noted in African Americans and the lowest in whites. According to the U.S. census report, African Americans constitute 12% of the total U.S. population,16 whereas 22% of HC+ patients are African American.1
It has been postulated that the immunosuppression that accompanies the post-HTx state accelerates HCV disease progression and shortens HC+ patients’ survival.17,18 These concerns were not validated in the most recent studies of kidney, liver, and heart transplantation in HC+ patients.15,19,20 Furthermore, where post-HTx cause of death was examined in HC+ patients, death was attributed primarily to post-HTx malignancy and bacterial sepsis but seldom directly to hepatic failure. In a study in which liver function was serially monitored after HTx in 11 HC+ patients, immunosuppression did not affect HCV disease progression, and there was no liver function impairment.15 In the same study, the 3 more recent HTx patients (of the 11) had their serial HCV viral load monitored. Viral load remained steady in 2 of the patients and decreased in the third. Similarly, there has been a theoretical concern that heightened immunologic status in HC+ patients might lead to more frequent rejection episodes. However, this concern has not been substantiated in reported studies.21,22 In the present study, the rate of graft failure as the cause of death was 11.2% in HC+ patients and 13% in HC– patients, though the difference was not statistically significant. Thus, concerns about HCV reactivation during immunocompromise or during increased allograft rejection were not substantiated.
Vasculopathy is a leading cause of morbidity and mortality after HTx and seems to be influenced by HC+. In a study of HC+ donor hearts transplanted to HC– patients, the prevalence of HCV infection in the recipients was 75% at a mean follow-up of 4.2 years.23 Serial angiogram showed coronary vasculopathy in 46% of HC+ patients and 24% of HC– patients 3.2 years after HTx.23 Similar concerns were raised in another small study, in which 2 of 4 HC+ patient deaths at 3.7-year follow-up were attributed to cardiovascular causes with features similar to those of transplantation vasculopathy.24 Those findings contrast with the present findings of cardiovascular deaths in 17.6% of HC+ patients and 17.2% of HC– patients. Outcomes similar to the present outcomes were reported by Lee and colleagues: Post-HTx cardiovascular deaths occurred in 16.4% of HC+ patients and 15.2% of HC– patients.8
Survival data on HC+ patients who undergo HTx are mixed, with some studies finding similar shortterm and midterm post-HTx survival21,22 and others finding decreased survival.8,9 It is difficult to interpret survival results from these studies, as some have included HCV infection that developed after HTx,9 and others have excluded early postoperative deaths from analysis.21 In addition, in the larger of these studies, which spanned 15 years, propensity matching was used for survival analysis. 8
It is possible that the selection and treatment of HC+ patients who were awaiting or underwent HTx changed over the study period. Thus, it might not be accurate to compare HTx outcomes of patients without considering the significant progress that has been made in the management of HCV infection. Although the present study’s aggregate (23-year) post-HTx survival results at 1, 5, and 10 years were similar to those reported in large series of HC+ patients who underwent HTx, the present early and intermediate survival results showed consistent improvements over time.8,9 This improvement in survival of HC+ patients was not examined in previous studies.
In the present study, the distribution of causes of post-HTx deaths was diverse (Table 5). Although the leading causes of death were cardiovascular or were related to sepsis or multi-organ failure, deaths attributed to liver failure were uncommon. Only 2.8% of deaths in the HC+ group and 0.6% of deaths in the HC– group were attributed to liver failure (P ≤ .001). These findings are similar to the cause-specific mortality reported in the literature.8,9,15,22 It is possible that these mortality results may be secondary to selection of patients with preserved liver function.
Future of HCV Infection and Heart Transplantation
Modern diagnostic methods will be used to accurately assess HC+ patients for HCV disease burden, and treatment will be provided before HTx is performed. Historically, the major limitations to treating HC+ patients awaiting HTx have been the long (48 week) duration of therapy and the anemia that can exacerbate heart failure symptoms and shorten the safe HTx waiting time.25 Most of the HCV treatment AEs have been attributed to use of IFN-α. Newer HCV treatments (direct-acting protease inhibitors without IFN) are of shorter duration (24 weeks) and have fewer AEs and higher cure rates. Most important, these treatments obtain similar cure rates irrespective of viral load, viral genotype, patient race, and previous HCV response status.7,12,13
Authors researching other solid-organ transplantation (eg, liver, kidney) have studied HCV pretreatment and found sustained virologic response before and after transplantation.26,27 Although these studies were conducted before the advent of direct-acting protease inhibitors, the feasibility of treating HCV before transplantation has been demonstrated.
Limitations
The limitations of retrospective data analysis are applicable to the present findings. Although HTx is infrequently performed in HC+ patients, the authors used a large national database and a 23-year study period and thus were able to gather a significant number of patients and perform meaningful statistical analysis. HCV disease burden, which influences disease progression, is much better quantified with recent quantitative viral loads, but these were not available in the UNOS database. It should be noted that UNOS does not gather information on HCV genotypes or forms of treatment received, both of which influence treatment response and prognosis. It is therefore not possible to elucidate, from the UNOS database, the influences of virus genotype and treatment response on post-HTx outcomes.
Conclusion
The treatment of HCV infection has significantly evolved since the virus was identified in 1989. At first the disease was considered incurable, but now a > 90% cure rate is possible with newer treatment regimens. This study found significant improvements in post-HTx outcomes in HC+ patients between 1991 and 2014. Both HC+ and HC– patients have had similar post-HTx clinical outcomes in recent years. The noted improvements in post-HTx survival in HC+ patients may be secondary to better patient selection or more effective antiviral treatments. Future studies will provide the answers.
Click here to read the digital edition.
More than 185 million people worldwide, including more than 4 million in the U.S., are infected with the hepatitis C virus (HCV).1 Because of the indolent nature of the disease, actual prevalence is underestimated.2,3 Detection of HCV in people already infected is estimated to continue to increase over the next decade.4 Although primary manifestations of the disease are the result of liver damage, HCV infection is a systemic illness. In a study of more than 19,000 patients, HCV infection was identified as an independent risk factor for development of heart failure.5 In the U.S., prevalence of HCV infection in patients with heart failure is reported to be as high as 15%, much higher than the general population prevalence of 1.8%.6 When first identified in 1989, HCV infection was considered incurable. Clinical trials have since found a steady improvement in outcome, and now the disease is considered curable in up to 90% of cases.7
Clinical outcomes of heart transplantation (HTx) historically have been inferior in patients with HCV infection.8,9 The authors hypothesized that the literature on HTx outcomes has not accounted for the improvements in HCV infection treatment options that have occurred since the 1990s. In the study reported here, United Network of Organ Sharing (UNOS) data on adult HTx was used to evaluate clinical outcomes of HCV infection over 4 treatment eras.
Material and Methods
The authors analyzed UNOS data on adult HTx from January 1991 to March 2014. Two groups were created: patients with HCV infection (HC+) and noninfected patients (HC–). Eligible patients were aged > 18 years. Hepatitis C virus status was defined with antibody testing at time of HTx. Patients with multiorgan transplantation or with hepatitis B virus or HIV infection were excluded. For comparison of post-HTx survival, the 23-year study period was divided into 4 eras reflecting the evolution in HCV infection treatment options in the U.S. (Table 1). The first medication was interferon α (IFN-α), which was used alone (first era, 1991-1997) and then with the newly introduced ribavirin (second era, 1998-2000). The combination of IFN-α and ribavirin increased sustained virologic response rates, but the rates of adverse effects (AEs), such as cytopenia and depression, were high, and many patients could not tolerate the extended (48-week) regimen.10,11
Peginterferon, a long-acting IFN introduced in 2001, significantly increased adherence to 2-drug treatment for HCV infection, and its use in combination with ribavirin marked the third era (2001-2010). The fourth era (2011-2014) began with the introduction of direct-acting antiviral agents and their remarkable results. Since 2014, direct protease inhibitors without IFN found a dramatic impact on HCV treatment: fewer AEs, shorter treatment (24 weeks), and high (> 90%) sustained virologic response.7,12,13
Statistical Analysis
Categoric variables were analyzed with the χ2 test or the Fisher exact test and are reported as percentages. Continuous variables were analyzed with the Student t test or the Wilcoxon rank sum test and are reported as means, medians, and SDs. Statistical significance was set at P < .05. Survival curves were plotted with the Kaplan-Meier method, and comparisons made with log-rank tests. Analysis was performed with SAS Version 9.3 (Cary, NC).
Results
Between January 1991 and March 2014, adult HTx was performed 36,589 times, including 778 times (2.1%) in patients with HCV infection. There was no significant difference in percentage of HC+ patients who underwent HTx over the 4 treatment eras (first, 2.1%; second, 2.9%; third, 2.1%; fourth, 1.6%) (Table 2). Mean patient age for the HC+ and HC– groups was comparable. Percentage of African American patients was higher in the HC+ group than in the HC– group (18.9% vs 15.0%), as was percentage of patients of other race (11.2% vs 9.2%; P = .0008).
Regarding indications for HTx, ischemic (and nonischemic) cardiomyopathy was similar in prevalence between the 2 groups, but the “other” heart failure etiologies (congenital heart disease, valvular heart disease, postpartum cardiomyopathy, restrictive heart disease) were more prevalent in the HC+ group (12.9% vs 9.7%; P = .013). The HC+ group also had higher rates of tobacco use history (42.2% vs 40.1%; P = .002) and hypertension (23.1% vs 20.9%; P = .014). Mean (SD) bilirubin level at time of transplantation for the HC+ and HC– groups was comparable: 1.12 (1) mg/dL and 1.11 (1) mg/dL, respectively (P = .707). Of the heart donor variables (Table 3), only tobacco use history was significantly higher in the HC+ group (23.5% vs 19.8%; P = .008).
Survival Data
Mean (SD) overall follow-up was 6.2 (5.3) years (median, 5 years; range, 0-23.3 years) for all patients; 5.6 (4.3) years (median, 5.05 years; range, 0-23.2 years) for HC+ patients; and 6.2 (5.3) years (median, 6.1 years; range, 0-23.2 years) for HC– patients.
HC+ patients’ survival rates were 82.5% (1 year), 64.4% (5 years), and 42.1% (10 years), and HC– patients’ rates were 87.2% (1 year), 73.4% (5 years), and 54.7% (10 years). The HC+ group’s inferior survival at 1, 5, and 10 years was statistically significant (P < .0002) (Table 4).
During the first era (1991-1997), HC+ patients’ survival rates were 81.0% (1 year), 73.3% (2 years), and 61.4% (5 years), and HC– patients’ rates were 85.0% (1 year), 80.6% (2 years), and 70.3% (5 years) (P < .05). During the second era (1998-2000), HC+ patients’ rates were 79.1% (1 year), 74.6% (2 years), and 62.0% (5 years), and HC– patients’ rates were 85.4% (1 year), 81.7% (2 years), and 72.0% (5 years) (P < .05). During the third era (2001-2010), HC+ patients’ rates were 83.6% (1 year), 78.6% (2 years), and 66.8% (5 years), and HC– patients’ rates were 88.4% (1 year), 84.6% (2 years), and 75.4% (5 years) (P < .05).
Survival data for the fourth treatment era (2011-2014) were available only for 1 and 2 years. HC+ patients’ survival rates were 89.01% (1 year) and 81.89% (2 years), and HC– patients’ rates were 91.00% (1 year) and 86.00% (2 years). The inferior survival found for the HC+ group during the 3 preceding eras was not found this era, during which HC+ and HC– patients had comparable rates of survival at 1 year (Figures 1 and 2).
Survival 1 year after HTx was compared between the HC+ and HC– groups over the 4 treatment eras (Figures 3 and 4). The HC+ patients’ survival after HTx improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91% (P = .9).
Hepatic decompensation leading to death was uncommon, but the rate was significantly higher (P = .0001) in the HC+ group (2.8%) than in the HC– group (0.6%).
Discussion
HCV infection is a risk factor for the development of cardiovascular illness and advanced heart failure. Given the worldwide prevalence of HCV infection, more HC+ patients will be evaluated for HTx in the future.2-4 Significant progress has been made in HCV infection treatment since the virus was first described. What was once incurable now has up to a 90% cure rate with newer treatment options.7,12,13 The present study findings showed consistent improvement in HC+ patients’ post-HTx survival during each treatment era. During the latest era, HC+ and HC– patients’ post-HTx survival was statistically similar.
It is possible that HC+ patients’ improvement in post-HTx survival could have resulted from improvement in overall post-HTx survival.14 Over the 23-year study period, the survival rates of both groups (HC+, HC–) improved, likely secondary to improved immunosuppression and perioperative care, but the magnitude of improvement was more pronounced in the HC+ group. HC+ patients’ post-HTx survival improved from 81% during the earliest era (1991-1997) to 89% during the latest era (2011-2014), whereas HC– patients’ survival improved from 85% to 91%. The improvement in HC+ patients’ short-term survival over the study period was substantial but did not reach statistical significance (P = .9).
Over the study period, the percentage of HC+ patients who underwent HTx remained low, ranging from 2.9% during the second era (1998-2000) to 1.7% during the fourth era (2011-2014). Overall, only 2.2% of study patients were HC+ at time of transplantation—a rate similar to previously reported rates.8,15 This rate likely represents a selection bias for HTx listing, in which patients with nearly normal liver function were selected for HTx, as evident by normal bilirubin levels in both groups. In the present study, a high proportion of HC+ patients were not white. This distribution also was noted in epidemiologic studies of HCV infection by ethnicity.6 In the U.S., the highest prevalence of HCV infection was noted in African Americans and the lowest in whites. According to the U.S. census report, African Americans constitute 12% of the total U.S. population,16 whereas 22% of HC+ patients are African American.1
It has been postulated that the immunosuppression that accompanies the post-HTx state accelerates HCV disease progression and shortens HC+ patients’ survival.17,18 These concerns were not validated in the most recent studies of kidney, liver, and heart transplantation in HC+ patients.15,19,20 Furthermore, where post-HTx cause of death was examined in HC+ patients, death was attributed primarily to post-HTx malignancy and bacterial sepsis but seldom directly to hepatic failure. In a study in which liver function was serially monitored after HTx in 11 HC+ patients, immunosuppression did not affect HCV disease progression, and there was no liver function impairment.15 In the same study, the 3 more recent HTx patients (of the 11) had their serial HCV viral load monitored. Viral load remained steady in 2 of the patients and decreased in the third. Similarly, there has been a theoretical concern that heightened immunologic status in HC+ patients might lead to more frequent rejection episodes. However, this concern has not been substantiated in reported studies.21,22 In the present study, the rate of graft failure as the cause of death was 11.2% in HC+ patients and 13% in HC– patients, though the difference was not statistically significant. Thus, concerns about HCV reactivation during immunocompromise or during increased allograft rejection were not substantiated.
Vasculopathy is a leading cause of morbidity and mortality after HTx and seems to be influenced by HC+. In a study of HC+ donor hearts transplanted to HC– patients, the prevalence of HCV infection in the recipients was 75% at a mean follow-up of 4.2 years.23 Serial angiogram showed coronary vasculopathy in 46% of HC+ patients and 24% of HC– patients 3.2 years after HTx.23 Similar concerns were raised in another small study, in which 2 of 4 HC+ patient deaths at 3.7-year follow-up were attributed to cardiovascular causes with features similar to those of transplantation vasculopathy.24 Those findings contrast with the present findings of cardiovascular deaths in 17.6% of HC+ patients and 17.2% of HC– patients. Outcomes similar to the present outcomes were reported by Lee and colleagues: Post-HTx cardiovascular deaths occurred in 16.4% of HC+ patients and 15.2% of HC– patients.8
Survival data on HC+ patients who undergo HTx are mixed, with some studies finding similar shortterm and midterm post-HTx survival21,22 and others finding decreased survival.8,9 It is difficult to interpret survival results from these studies, as some have included HCV infection that developed after HTx,9 and others have excluded early postoperative deaths from analysis.21 In addition, in the larger of these studies, which spanned 15 years, propensity matching was used for survival analysis. 8
It is possible that the selection and treatment of HC+ patients who were awaiting or underwent HTx changed over the study period. Thus, it might not be accurate to compare HTx outcomes of patients without considering the significant progress that has been made in the management of HCV infection. Although the present study’s aggregate (23-year) post-HTx survival results at 1, 5, and 10 years were similar to those reported in large series of HC+ patients who underwent HTx, the present early and intermediate survival results showed consistent improvements over time.8,9 This improvement in survival of HC+ patients was not examined in previous studies.
In the present study, the distribution of causes of post-HTx deaths was diverse (Table 5). Although the leading causes of death were cardiovascular or were related to sepsis or multi-organ failure, deaths attributed to liver failure were uncommon. Only 2.8% of deaths in the HC+ group and 0.6% of deaths in the HC– group were attributed to liver failure (P ≤ .001). These findings are similar to the cause-specific mortality reported in the literature.8,9,15,22 It is possible that these mortality results may be secondary to selection of patients with preserved liver function.
Future of HCV Infection and Heart Transplantation
Modern diagnostic methods will be used to accurately assess HC+ patients for HCV disease burden, and treatment will be provided before HTx is performed. Historically, the major limitations to treating HC+ patients awaiting HTx have been the long (48 week) duration of therapy and the anemia that can exacerbate heart failure symptoms and shorten the safe HTx waiting time.25 Most of the HCV treatment AEs have been attributed to use of IFN-α. Newer HCV treatments (direct-acting protease inhibitors without IFN) are of shorter duration (24 weeks) and have fewer AEs and higher cure rates. Most important, these treatments obtain similar cure rates irrespective of viral load, viral genotype, patient race, and previous HCV response status.7,12,13
Authors researching other solid-organ transplantation (eg, liver, kidney) have studied HCV pretreatment and found sustained virologic response before and after transplantation.26,27 Although these studies were conducted before the advent of direct-acting protease inhibitors, the feasibility of treating HCV before transplantation has been demonstrated.
Limitations
The limitations of retrospective data analysis are applicable to the present findings. Although HTx is infrequently performed in HC+ patients, the authors used a large national database and a 23-year study period and thus were able to gather a significant number of patients and perform meaningful statistical analysis. HCV disease burden, which influences disease progression, is much better quantified with recent quantitative viral loads, but these were not available in the UNOS database. It should be noted that UNOS does not gather information on HCV genotypes or forms of treatment received, both of which influence treatment response and prognosis. It is therefore not possible to elucidate, from the UNOS database, the influences of virus genotype and treatment response on post-HTx outcomes.
Conclusion
The treatment of HCV infection has significantly evolved since the virus was identified in 1989. At first the disease was considered incurable, but now a > 90% cure rate is possible with newer treatment regimens. This study found significant improvements in post-HTx outcomes in HC+ patients between 1991 and 2014. Both HC+ and HC– patients have had similar post-HTx clinical outcomes in recent years. The noted improvements in post-HTx survival in HC+ patients may be secondary to better patient selection or more effective antiviral treatments. Future studies will provide the answers.
Click here to read the digital edition.
1. Razavi H, Waked I, Sarrazin C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21(suppl 1):34-59.
2. Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31(4):1014-1018.
3. Tong MJ, el-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusionassociated hepatitis C. N Engl J Med. 1995;332(22):1463-1466.
4. Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):513-521.
5. Younossi ZM, Stepanova M, Nader F, Younossi Z, Elsheikh E. Associations of chronic hepatitis C with metabolic and cardiac outcomes. Aliment Pharmacol Ther. 2013;37(6):647-652.
6. Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556-562.
7. Ferenci P, Bernstein D, Lalezari J, et al; PEARL-III Study; PEARL-IV Study. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983-1992.
8. Lee I, Localio R, Brensinger CM, et al. Decreased post-transplant survival among heart transplant recipients with pre-transplant hepatitis C virus positivity. J Heart Lung Transplant. 2011;30(11):1266-1274.
9. Fong TL, Hou L, Hutchinson IV, Cicciarelli JC, Cho YW. Impact of hepatitis C infection on outcomes after heart transplantation. Transplantation. 2009;88(9):1137-1141.
10. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975-982.
11. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958-965.
12. Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973-1982.
13. Zeuzem S, Dusheiko GM, Salupere R, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370(21):1993-2001.
14. Lund LH, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant. 2013;32(10):951-964.
15. Cano O, Almenar L, Martínez-Dolz L, et al. Course of patients with chronic hepatitis C virus infection undergoing heart transplantation. Transplant Proc. 2007;39(7):2353-2354.
16. Hobbs F, Stoops N. Demographic Trends in the 20th Century. Washington, DC: U.S. Government Printing Office; 2002. U.S. Census Bureau, Census 2000 Special Reports, Series CENSR-4. https://www.census.gov/prod/2002pubs/censr-4.pdf. Issued November 2002. Accessed February 7, 2017.
17. Pol S, Samuel D, Cadranel J, et al. Hepatitis and solid organ transplantation. Transplant Proc. 2000;32(2):454-457.
18. Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364(25):2429-2438.
19. Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int. 1994;45(1):238-244.
20. Garcia-Saenz-de-Sicilia M, Olivera-Martinez MA, Grant WJ, et al. Impact of anti-thymocyte globulin during immunosuppression induction in patients with hepatitis C after liver transplantation. Dig Dis Sci. 2014;59(11): 2804-2812.
21. Lake KD, Smith CI, Milfred-La Forest SK, Pritzker MR, Emery RW. Outcomes of hepatitis C positive (HCV+) heart transplant recipients. Transplant Proc. 1997;29(1-2):581-582.
22. Shafii AE, Su JW, Smedira NG, et al. The effect of recipient hepatitis C virus infection on outcomes following heart transplantation. Transplant Proc. 2010;42(5):1784-1787.
23. Haji SA, Starling RC, Avery RK, et al. Donor hepatitis-C seropositivity is an independent risk factor for the development of accelerated coronary vasculopathy and predicts outcome after cardiac transplantation. J Heart Lung Transplant. 2004;23(3):277-283.
24. Castella M, Tenderich G, Koerner MM, et al. Outcome of heart transplantation in patients previously infected with hepatitis C virus. J Heart Lung Transplant. 2001;20(5):595-598.
25. Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med. 2006;355(23):2444-2451.
26. Thomas RM, Brems JJ, Guzman-Hartman G, Yong S, Cavaliere P, Van Thiel DH. Infection with chronic hepatitis C virus and liver transplantation: a role for interferon therapy before transplantation. Liver Transpl. 2003;9(9):905-915.
27. Everson GT, Terrault NA, Lok AS, et al. A randomized controlled trial of pretransplant antiviral therapy to prevent recurrence of hepatitis C after liver transplantation. Hepatology. 2013;57(5):1752-1762.
1. Razavi H, Waked I, Sarrazin C, et al. The present and future disease burden of hepatitis C virus (HCV) infection with today’s treatment paradigm. J Viral Hepat. 2014;21(suppl 1):34-59.
2. Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31(4):1014-1018.
3. Tong MJ, el-Farra NS, Reikes AR, Co RL. Clinical outcomes after transfusionassociated hepatitis C. N Engl J Med. 1995;332(22):1463-1466.
4. Davis GL, Alter MJ, El-Serag H, Poynard T, Jennings LW. Aging of hepatitis C virus (HCV)-infected persons in the United States: a multiple cohort model of HCV prevalence and disease progression. Gastroenterology. 2010;138(2):513-521.
5. Younossi ZM, Stepanova M, Nader F, Younossi Z, Elsheikh E. Associations of chronic hepatitis C with metabolic and cardiac outcomes. Aliment Pharmacol Ther. 2013;37(6):647-652.
6. Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556-562.
7. Ferenci P, Bernstein D, Lalezari J, et al; PEARL-III Study; PEARL-IV Study. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983-1992.
8. Lee I, Localio R, Brensinger CM, et al. Decreased post-transplant survival among heart transplant recipients with pre-transplant hepatitis C virus positivity. J Heart Lung Transplant. 2011;30(11):1266-1274.
9. Fong TL, Hou L, Hutchinson IV, Cicciarelli JC, Cho YW. Impact of hepatitis C infection on outcomes after heart transplantation. Transplantation. 2009;88(9):1137-1141.
10. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975-982.
11. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958-965.
12. Poordad F, Hezode C, Trinh R, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973-1982.
13. Zeuzem S, Dusheiko GM, Salupere R, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med. 2014;370(21):1993-2001.
14. Lund LH, Edwards LB, Kucheryavaya AY, et al. The registry of the International Society for Heart and Lung Transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant. 2013;32(10):951-964.
15. Cano O, Almenar L, Martínez-Dolz L, et al. Course of patients with chronic hepatitis C virus infection undergoing heart transplantation. Transplant Proc. 2007;39(7):2353-2354.
16. Hobbs F, Stoops N. Demographic Trends in the 20th Century. Washington, DC: U.S. Government Printing Office; 2002. U.S. Census Bureau, Census 2000 Special Reports, Series CENSR-4. https://www.census.gov/prod/2002pubs/censr-4.pdf. Issued November 2002. Accessed February 7, 2017.
17. Pol S, Samuel D, Cadranel J, et al. Hepatitis and solid organ transplantation. Transplant Proc. 2000;32(2):454-457.
18. Rosen HR. Clinical practice. Chronic hepatitis C infection. N Engl J Med. 2011;364(25):2429-2438.
19. Roth D, Zucker K, Cirocco R, et al. The impact of hepatitis C virus infection on renal allograft recipients. Kidney Int. 1994;45(1):238-244.
20. Garcia-Saenz-de-Sicilia M, Olivera-Martinez MA, Grant WJ, et al. Impact of anti-thymocyte globulin during immunosuppression induction in patients with hepatitis C after liver transplantation. Dig Dis Sci. 2014;59(11): 2804-2812.
21. Lake KD, Smith CI, Milfred-La Forest SK, Pritzker MR, Emery RW. Outcomes of hepatitis C positive (HCV+) heart transplant recipients. Transplant Proc. 1997;29(1-2):581-582.
22. Shafii AE, Su JW, Smedira NG, et al. The effect of recipient hepatitis C virus infection on outcomes following heart transplantation. Transplant Proc. 2010;42(5):1784-1787.
23. Haji SA, Starling RC, Avery RK, et al. Donor hepatitis-C seropositivity is an independent risk factor for the development of accelerated coronary vasculopathy and predicts outcome after cardiac transplantation. J Heart Lung Transplant. 2004;23(3):277-283.
24. Castella M, Tenderich G, Koerner MM, et al. Outcome of heart transplantation in patients previously infected with hepatitis C virus. J Heart Lung Transplant. 2001;20(5):595-598.
25. Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med. 2006;355(23):2444-2451.
26. Thomas RM, Brems JJ, Guzman-Hartman G, Yong S, Cavaliere P, Van Thiel DH. Infection with chronic hepatitis C virus and liver transplantation: a role for interferon therapy before transplantation. Liver Transpl. 2003;9(9):905-915.
27. Everson GT, Terrault NA, Lok AS, et al. A randomized controlled trial of pretransplant antiviral therapy to prevent recurrence of hepatitis C after liver transplantation. Hepatology. 2013;57(5):1752-1762.
Neuromodulation for Treatment-Refractory PTSD (FULL)
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
Failure of fear extinction is a core feature of posttraumatic stress disorder (PTSD).1 Recently, it was confirmed that the amygdala and the orbitofrontal cortex are crucial for both fear acquisition and fear extinction.2 The amygdala was found to have neurons active only during fear acquisition, and other neurons active only during fear extinction.3 In essence, the balance of activity between these 2 neuronal populations determines whether if an incoming stimulus is feared or not feared. This balance is under the influence of several cognitive domains, including memory, reward, and executive function.
In PTSD, the equilibrium is shifted heavily toward fear acquisition. The majority of patients spontaneously regain the capacity for fear extinction over time4 or with the help of treatment.5,6 Nonetheless, some patients with severe PTSD seem unable to recover the ability of fear extinction and remain refractory to both standard and novel psychotherapeutic or psychopharmacologic treatments.7 For these patients, direct modulation of the neural activity in the amygdala may permit fear extinction. This article describes the rationale for using deep brain stimulation (DBS) and initial results from the first-ever clinical trial.
Deep Brain Stimulation
Deep brain stimulation involves inserting electrodes in precise cerebral targets and then connecting the leads to a pulse generator (similar to a pacemaker) inserted in a subclavicular pocket. The generator controls the electrical signal (amplitude, pulse width, pulse frequency) delivered to the brain target and can be adjusted with use of a noninvasive programmer. In 1997, the FDA approved DBS for patients with Parkinson disease or essential tremor. Since then, its efficacy in these movement disorders has been confirmed in several studies.8,9
The mechanism by which the small electrical pulses of DBS influence activity is not clear. Clinically, DBS functionally inhibits the activity of local neurons.10 One theory describes “frequency jamming,” a concept similar to cardiac overdrive pacing in which the resultant high-frequency neuronal signal is meaningless and discounted by the rest of the brain.11
Over the years, DBS has demonstrated a strong safety profile.12 The risks of electrode insertion are mitigated with targeting based on high-quality magnetic resonance imaging (MRI) and computed tomography (Figure). Unlike a destructive lesion, DBS is reversible, and the implanted system can be removed in its entirety. Histologic analyses have shown only a small amount of scarring around the electrode tip.13 In movement disorder treatment, clinical experience has shown that stimulation-related adverse effects (AEs) are reversible with readjustment of stimulation parameters by external programmer.14
Novel Applications of DBS
The advantageous safety profile of DBS has permitted its evaluation in the treatment of other conditions thought to have malfunctioning networks at their core—such as intractable epilepsy (in resective surgery noncandidates).15,16 Although several trials have shown promising results of using DBS for treatment-resistant depression,17 the results of pivotal sham-controlled trials have been mixed.18,19 Obsessive-compulsive disorder, on the other hand, received the FDA humanitarian device exemption designation on the basis of positive long-term results.20 In treatment-resistant depression and obsessive-compulsive disorder, functional neuroimaging has identified DBS targets.21,22 Functional MRI or positron emission tomography (PET) images can be compared at resting state, at symptomatic state, and after treatment response. Nodes hyperactive during a symptomatic state and less active after successful treatment can be targeted with high-frequency DBS to directly reduce the hyperactivity and indirectly modulate or normalize the overall function of the circuit.23
Given the functional MRI and O15 (oxygen-15) PET evidence of amygdala hyperactivity in patients with PTSD having core symptoms,24-26 the authors hypothesized that high-frequency DBS targeting of the amygdala would improve PTSD-associated hyperarousal and reexperiencing symptoms in treatment-refractory patients. Indirect data supporting this hypothesis include a correlation between amygdala hyperactivity of increased intensity and symptom severity measured with the Clinician-Administered PTSD Scale (CAPS),27 and a correlation between reduced pretreatment amygdala hyperactivity and successful cognitive-behavioral treatment.28,29
Preclinical Work
Using a rodent model in which a novel object serves as a cue reminder of foot shocks (traumatic event), the authors tested the hypothesis that amygdala DBS would reduce PTSD-like symptoms.30 When untreated rats were presented with the object in their cage a week after the initial exposure, they immediately buried the object under bedding to avoid being reminded of the shocks. In contrast, rats treated with DBS did not bury the object. In most cases, in fact, they played with it.
The authors also replicated their results but with the addition of rats treated with paroxetine.31 Using the same rodent model, they found DBS superior to paroxetine in treating PTSD-like symptoms. This study had a crossover design: DBS and sham DBS. Briefly, 20 rats received an electrode in the amygdala and were exposed to inescapable shocks in the presence of the cue object. The rats were then randomly assigned to a DBS group (10 rats) or a sham-DBS group (10 rats). After 1 week, behavioral testing showed fear extinction in the DBS group and no improvement in the sham-DBS group. Then the groups were switched: The rats originally treated with DBS received no treatment, and the rats that were originally sham-treated underwent DBS. One week later, behavioral testing showed acquisition of fear extinction in all the rats. These results suggested DBS can be effective even when delayed after establishment of fear persistence and PTSD symptoms. These results also showed that DBS effects persist even after therapy discontinuation.
Similarly, other investigators have reported that the role of the amygdala is not limited to fear acquisition; it extends to fear expression. A lesion in the amygdala can prevent fear expression even if the disruption is performed subsequent to fear-conditioning training.32 This finding is important for humans, as DBS would be initiated during the chronic phase of the disorder, after failure of less invasive treatment options, such as pharmacotherapy and psychotherapy.
Early Clinical Experience
The authors have initiated the first ever clinical trial (NCT02091843) evaluating use of DBS for PTSD and are now recruiting patients. Enrollment is limited to 6 combat veterans with disabling PTSD that has not responded to pharmacotherapy and psychotherapy. This VA-funded single-site study, being conducted at the VA Greater Los Angeles Healthcare System (VAGLAHS), was approved by the VAGLAHS Institutional Review Board and the FDA. The authors have published the 2-year trial’s protocol, which includes an active-versus-sham stimulation phase; continuous electroencephalogram monitoring; baseline and posttreatment 18FDG (fluorodeoxyglucose) PET performed during a resting state vs during investigator-guided exposure to trauma reminders; and extensive psychological and neuropsychological assessments.33 The literature includes only 1 case report on amygdala DBS.34 The authors of that report used DBS of the basolateral nucleus of the amygdala to treat a teenaged boy with severe autism and found that the therapy was safe.
As of this writing, the authors have recruited and implanted 1 patient and reported on his clinical results (including baseline PET) over the first 8 months of stimulation35 and on the electrophysiologic findings over the first year.36 After experiencing extremely severe combat PTSD refractory to pharmacotherapy and psychotherapy treatments for more than 20 years, the patient treated with DBS is now experiencing substantial symptom relief, and his CAPS score (primary outcome measure) has improved by about 40%. He has tolerated continuous stimulation without any serious DBS-related AEs for up to 16 months. Notably, he has not had a single severe combat nightmare in a year—in stark contrast to nightly combat nightmares during the 20-year period leading to the trial. Furthermore, he has not been having any episodes of severe dissociation, which had been a common disabling problem before the trial. He has taken a second trip out of the country, improved his relationships with family, and made strides (albeit limited) in pursuing additional social interactions.
Avoidance remains a major problem. He recently left his job after 7 years, because he prefers a more nature-oriented rather than people-oriented environment. In addition, his interest in intensive psychotherapy has increased, and he has been considering options for spending more time working on his therapy.
Over 15 months of treatment, the patient’s CAPS total and subscale scores have decreased—his symptoms have improved (Table).21 He has had rapid and substantial reductions in recurrence and hyperarousal symptoms but slower improvement in avoidance. Improvements in emotional reactivity would be expected to occur before any change in behavior (eg, avoidance). Patients likely must first recognize changes in emotional reactivity to events before they can engage in a cognitive process to modify learned behavioral responses to those events.
After about 9 months of treatment, all of the study patient’s symptoms were somewhat stabilized, and the authors began making gradual stimulation adjustments to the latest parameters—3.5 V, 60 µs, and 160 Hz for the right electrode and 1.5 V, 60 µs, and 160 Hz for the left electrode—using the contacts in the basolateral nucleus of the amygdala, per postoperative neuroimaging.3
After 15 to 18 months, when improvement peaked at 48% symptom reduction from baseline, the patient experienced psychiatric decompensation (depression, suicide gesture) not attributable to changes in stimulation settings and not associated with exacerbation of PTSD symptoms. Treatment team members and independent psychiatric consultants attributed the decompensation to the patient’s difficulty in changing a long-standing avoidant behavior routine, owing to severe recurrence and hyperarousal symptoms in the past. His persistent inability to overcome avoidance and isolation, despite core PTSD symptom improvement, had left him feeling worthless.
The patient remains in the study but also is participating in other medication and psychotherapy trials and is making a career change. Periodic decompensations may be part of the treatment course as patients reach a more complex and volatile phase of improvement that requires more intensive cognitive reprocessing. If this proves to be the case with other patients enrolling in the study, intensive psychotherapy that addresses cognitive and emotional PTSD symptoms may be needed once there is improvement in intrusive and hyperarousal symptoms.
Conclusion
Deep brain stimulation has been successful in treating Parkinson disease and essential tremor. Physiologically, DBS seems to inhibit specific brain regions’ dysfunctional activity stemming from a disease process. Deep brain stimulation-induced inhibition of a dysfunctional node improves clinical outcomes in movement disorders.
Given the reversibility and positive safety profile of DBS, new applications are being studied. The authors propose that DBS may benefit patients with severe treatment-refractory PTSD. Their first patient’s core PTSD symptoms have improved significantly, as expected, but as in other psychiatric DBS cases, the seriousness and chronicity of his illness may be complicating the course of recovery. The authors plan to recruit 6 patients for this early-phase safety trial.
Click here to read the digital edition.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
1. Milad MR, Pitman RK, Ellis CB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66(12):1075-1082.
2. Marin MF, Song H, VanElzakker MB, et al. Association of resting metabolism in the fear neural network with extinction recall activations and clinical measures in trauma-exposed individuals. Am J Psychiatry. 2016;173(9):930-938.
3. Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454(7204):600-606.
4. Morina N, Wicherts JM, Lobbrecht J, Priebe S. Remission from post-traumatic stress disorder in adults: a systematic review and meta-analysis of long term outcome studies. Clin Psychol Rev. 2014;34(3):249-255.
5. Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489-500.
6. Hoskins M, Pearce J, Bethell A, et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br J Psychiatry. 2015;206(2):93-100.
7. Koek RJ, Schwartz HN, Scully S, et al. Treatment-refractory posttraumatic stress disorder (TRPTSD): a review and framework for the future. Prog Neuropsychopharmacol Biol Psychiatry. 2016;70:170-218.
8. Wagle Shukla A, Okun MS. State of the art for deep brain stimulation therapy in movement disorders: a clinical and technological perspective. IEEE Rev Biomed Eng. 2016;9:219-233.
9. Weaver FM, Follett K, Stern M, et al; CSP 468 Study Group. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63-73.
10. Benabid AL, Benazzouz A, Hoffmann D, Limousin P, Krack P, Pollack P. Long-term electrical inhibition of deep brain targets in movement disorders. Mov Disord. 1998;13(suppl 3):119-125.
11. Benabid AL, Wallace B, Mitrofanis J, et al. A putative generalized model of the effects and mechanism of action of high frequency electrical stimulation of the central nervous system. Acta Neurol Belg. 2005;105(3):149-157.
12. Fenoy AJ, Simpson RK Jr. Risks of common complications in deep brain stimulation surgery: management and avoidance. J Neurosurg. 2014;120(1):132-139.
13. DiLorenzo DJ, Jankovic J, Simpson RK, Takei H, Powell SZ. Neurohistopathological findings at the electrode–tissue interface in long-term deep brain stimulation: systematic literature review, case report, and assessment of stimulation threshold safety. Neuromodulation. 2014;17(5):405-418.
14. Revell MA. Deep brain stimulation for movement disorders. Nurs Clin North Am. 2015;50(4):691-701.
15. Fisher R, Salanova V, Witt T, et al; SANTE Study Group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
16. Salanova V, Witt T, Worth R, et al; SANTE Study Group. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. 2015;84(10):1017-1025.
17. Berlim MT, McGirr A, Van den Eynde F, Fleck MP, Giacobbe P. Effectiveness and acceptability of deep brain stimulation (DBS) of the subgenual cingulate cortex for treatment-resistant depression: a systematic review and exploratory meta-analysis. J Affect Disord. 2014;159:31-38.
18. Dougherty DD, Rezai AR, Carpenter LL, et al. A randomized sham-controlled trial of deep brain stimulation of the ventral capsule/ventral striatum for chronic treatment-resistant depression. Biol Psychiatry. 2015;78(4):240-248.
19. Bergfeld IO, Mantione M, Hoogendoorn ML, et al. Deep brain stimulation of the ventral anterior limb of the internal capsule for treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2016;73(5):456-464.
20. Greenberg BD, Malone DA, Friehs GM, et al. Three-year outcomes in deep brain stimulation for highly resistant obsessive-compulsive disorder. Neuropsychopharmacology. 2006;31(11):2384-2393.
21. Mayber HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675-682.
22. Rauch SL, Jenike MA, Alpert NM, et al. Regional cerebral blood flow measured during symptom provocation in obsessive-compulsive disorder using oxygen 15-labeled carbon dioxide and positron emission tomography. Arch Gen Psychiatry. 1994;51(1):62-70.
23. Williams NR, Taylor JJ, Lamb K, Hanlon CA, Short EB, George MS. Role of functional imaging in the development and refinement of invasive neuromodulation for psychiatric disorders. World J Radiol. 2014;6(10):756-778.
24. Francati V, Vermetten E, Bremner JD. Functional neuroimaging studies in posttraumatic stress disorder: review of current methods and findings. Depress Anxiety. 2007;24(3):202-218.
25. Shin LM, Orr SP, Carson MA, et al. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61(2):168-176.
26. Armony JL, Corbo V, Clément MH, Brunet A. Amygdala response in patients with acute PTSD to masked and unmasked emotional facial expressions. Am J Psychiatry. 2005;162(10):1961-1963.
27. Blake DD, Weathers FW, Nagy LM, et al. The development of a Clinician-Administered PTSD Scale. J Trauma Stress. 1995;8(1):75-90.
28. Felmingham K, Kemp A, Williams L, et al. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol Sci. 2007;18(2):127-129.
29. Peres JF, Newberg AB, Mercante JP, et al. Cerebral blood flow changes during retrieval of traumatic memories before and after psychotherapy: a SPECT study. Psychol Med. 2007;37(10):1481-1491.
30. Langevin JP, De Salles AA, Kosoyan HP, Krahl SE. Deep brain stimulation of the amygdala alleviates post-traumatic stress disorder symptoms in a rat model. J Psychiatr Res. 2010;44(16):1241-1245.
31. Stidd DA, Vogelsang K, Krahl SE, Langevin JP, Fellous JM. Amygdala deep brain stimulation is superior to paroxetine treatment in a rat model of posttraumatic stress disorder. Brain Stimul. 2013;6(6):837-844.
32. Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25(42):9680-9685.
33. Koek RJ, Langevin JP, Krahl SE, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory combat post-traumatic stress disorder (PTSD): study protocol for a pilot randomized controlled trial with blinded, staggered onset of stimulation. Trials. 2014;15:356.
34. Sturm V, Fricke O, Bührle CP, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2013;6:341.
35. Langevin JP, Koek RJ, Schwartz HN, et al. Deep brain stimulation of the basolateral amygdala for treatment-refractory posttraumatic stress disorder. Biol Psychiatry. 2016;79(10):e82-e84.
36. Langevin JP, Chen JW, Koek RJ, et al. Deep brain stimulation of the basolateral amygdala: targeting technique and electrodiagnostic findings. Brain Sci. 2016;6(3):E28.
Expanding treatment options for diverse neuroendocrine tumors
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).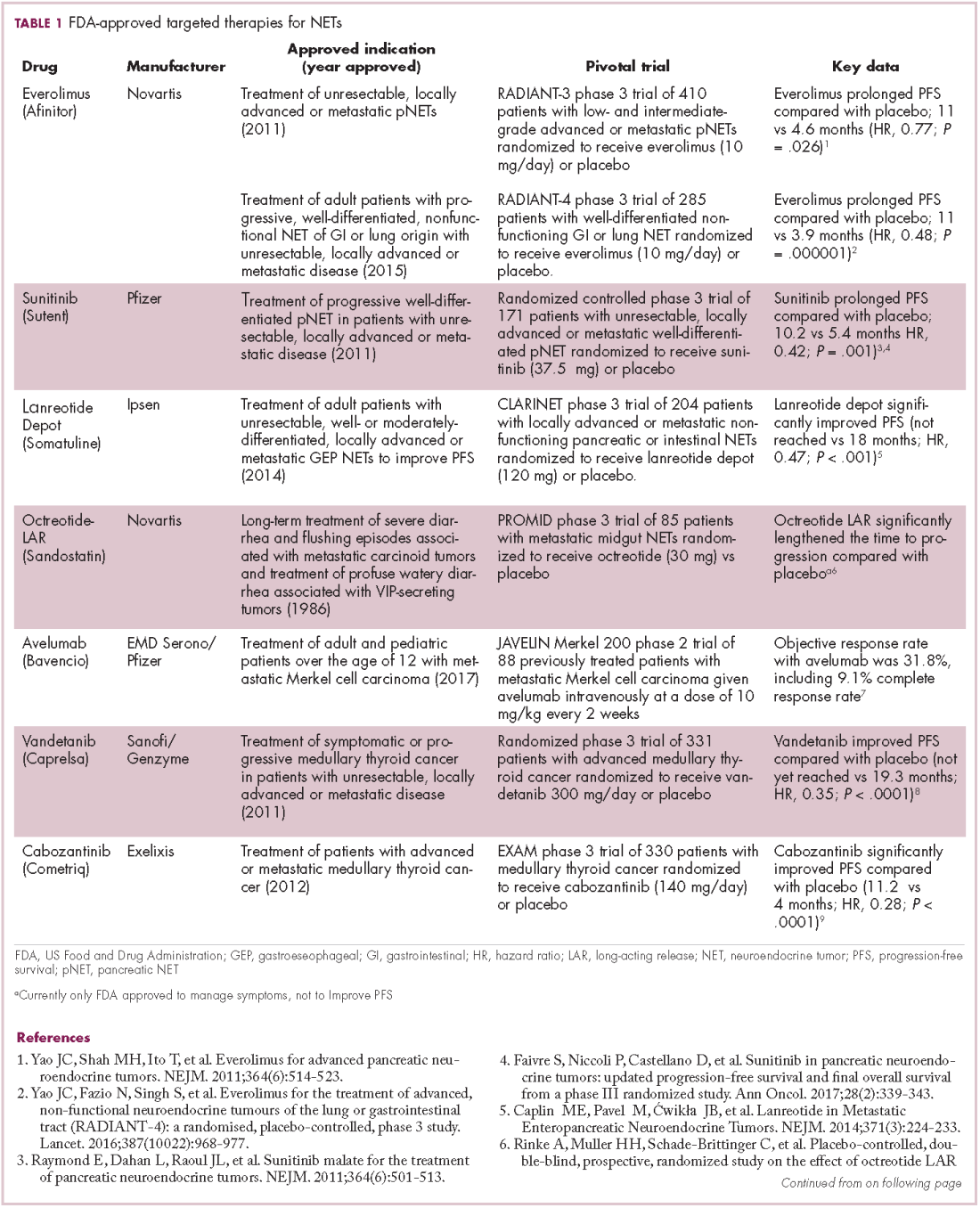

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24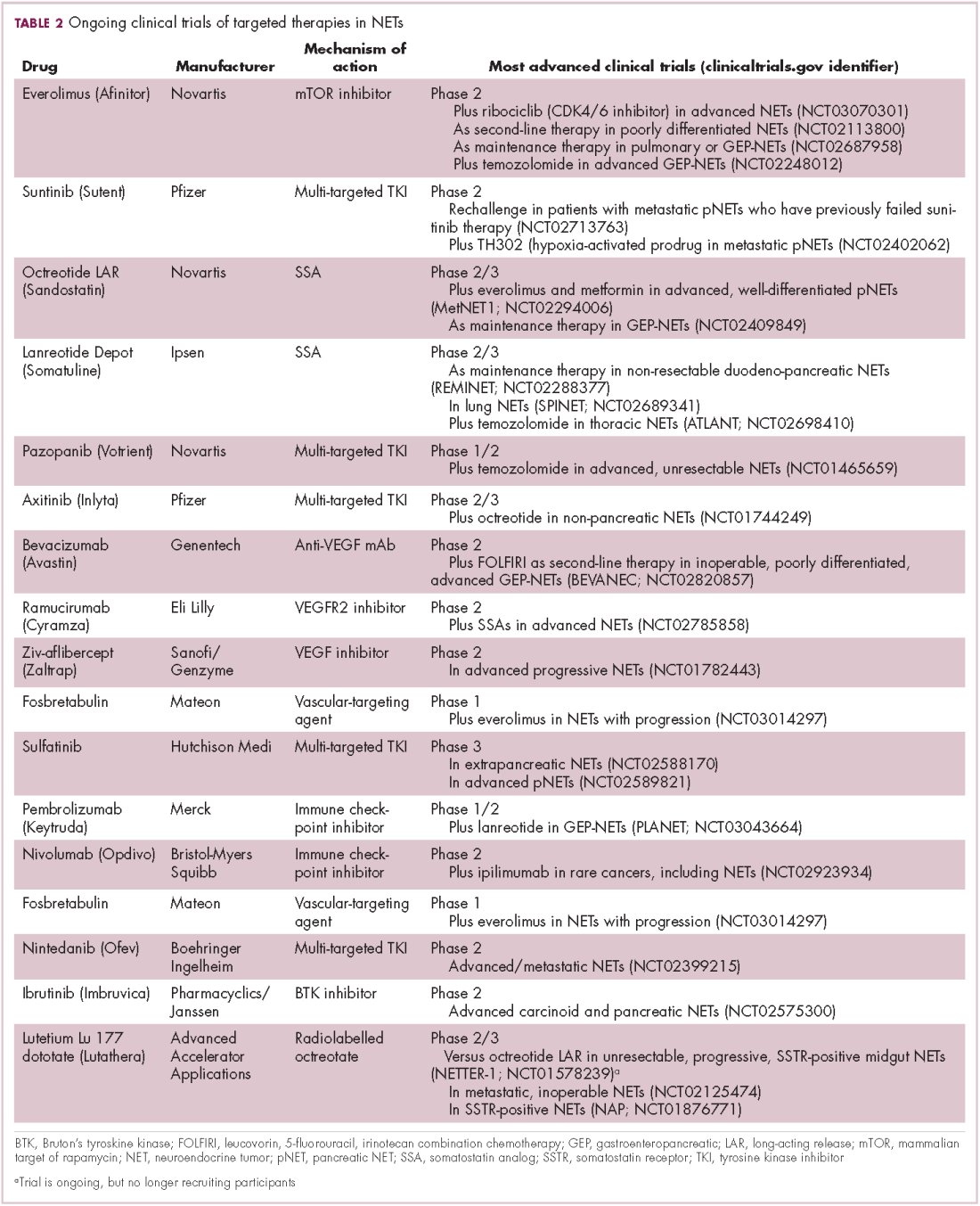
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.
More stable synthetic analogs of the somatostatin hormone (somatostatin analogs [SSAs]), which has a very short half-life in the circulation, have been developed that act as SSTR agonists. Two long-acting SSAs, octreotide (Sandostatin LAR Depot) and lanreotide (Somatuline Depot), which bind SSTR2 and SSTR5, have been approved by the United States Food and Drug Administration (FDA), but were primarily used for the alleviation of the symptoms associated with NETs resulting from carcinoid syndrome.
In recent years, evidence has begun to emerge that SSAs also have an anti-tumor effect, which is thought to be both direct and indirect in nature. Direct effects result from the interaction between the SSA and SSTRs expressed on tumor cells, blocking the protumor cellular effects of SSTR signaling that are poorly understood but thought to involve the mitogen-activated protein kinase (MAPK) pathway. Indirect effects are fortuitous side effects mediated through off-target effects, such as the suppression of other cellular activities of SSTRs and the other growth factors that they bind to, which can impact processes such as angiogenesis and immune modulation.7,12
Several clinical trials have been designed to test the anti-tumor effects of NETs, including the PROMID trial of octreotide and the CLARINET trial of lanreotide, the latter leading to the 2014 approval of lanreotide for the improvement of progression-free survival (PFS) in patients with advanced GI- and pNETs.
The randomized phase 3 study compared lanreotide 120 mg with placebo in 204 patients with locally advanced or metastatic nonfunctioning pancreatic or intestinal NETs. Lanreotide treatment resulted in a significant improvement in PFS (Not yet reached vs 18 months for placebo; hazard ratio [HR], 0.47; P < .001).13
Meanwhile, the PROMID trial compared octreotide 30 mg with placebo in 85 patients with advanced midgut NETs and demonstrated that octreotide increased time to progression (TTP; 14.3 months vs 6 months for placebo; P = .000072) with no significant difference in side effects.14
Pasireotide is a second-generation SSA with improved binding affinity to SSTR1, 3, and 5. Despite its improved specificity, pasireotide has not proved more effective than other SSAs and its development for the treatment of NETs has been discontinued.
Coupling radioisotopes to SSAs provides another promising therapeutic option for NETs, known as peptide receptor radionuclide therapy, or PRRT, which uses SSAs to deliver therapeutic radiation directly to the tumor cells. Several variations have been studied with different radioactive isotopes, but most promising is lutetium-177 (177Lu). A 177Lu-labelled octreotide (177Lu-Dotatate) recently demonstrated significant efficacy in the phase 3 NETTER-1 clinical trial in patients with advanced stage NETs of the small bowel. The trial randomly assigned 229 patients who were progressing on an SSA to either 177Lu-Dotatate or high-dose octreotide LAR (long-acting release). There was a significant increase in PFS in the 177Lu-Dotatate arm (Not yet reached vs 8.4 months; P < .0001). There was also a trend toward improved overall survival (OS), and longer follow-up is eagerly anticipated for confirmation. 177Lu-Dotatate has been granted priority review by the FDA, and a decision on its approval is expected in the next few months.11,15-17
Molecularly and immune-targeted therapies continue to take aim
The mammalian target of rapamycin, or mTOR, is a serine/threonine kinase that sits at the confluence of a number of different upstream signaling pathways and mediates key cellular processes including cell proliferation and survival (Figure 1).
Alterations in nearly all members of the mTOR pathway, including upstream activators and downstream effectors, have been observed in NETs, in both sporadic disease and the genetic syndromes associated with the development of NETs.18
The involvement of the mTOR pathway in the pathogenesis of NETs first came into focus in pNETs and the mTOR inhibitor, everolimus (Afinitor) has been extensively studied in this indication, culminating in its regulatory approval in 2011. In the pivotal trial (RADIANT-3), everolimus monotherapy was compared with placebo in 410 patients with low- and intermediate-grade pNETs. There was a statistically significant improvement in PFS from 4.6 months to 11 months (HR, 0.77; P = .026).19 The final OS analysis for this trial also revealed a benefit of more than 6 months in the everolimus arm, although this was not statistically significant, which the study authors attribute to the high rate of crossover from the placebo arm after progression.20
More recently, the results of the RADIANT-4 trial, in which everolimus was compare with placebo in patients with advanced, well-differentiated, nonfunctioning NETs of the GI tract and lung, led to a new approved indication for the mTOR inhibitor and the first approved targeted therapy for advanced lung NETs. In the overall study population (n = 285), everolimus prolonged PFS by more than 7 months (11 months vs 3.9 months for placebo; HR, 0.48; P = .000001), corresponding to a 52% reduction in the risk of disease progression or death.21,22
Everolimus continues to be evaluated, with a particular focus on combination therapy to overcome the resistance that commonly occurs after treatment with molecularly targeted drugs (Table 2). For example, preclinical studies suggested that mTOR inhibitors and SSAs may have synergistic activity owing to combined inhibition of the mTOR and insulin-like growth factor pathways. In a phase 1 study, the combination of pasireotide and everolimus was found to be safe and to have preliminary anti-tumor activity. However, the subsequent phase 2 COOPERATE-2 study failed to show improved PFS.23,24
The observation that NETs are highly vascularized and frequently express vascular endothelial growth factor (VEGF) and its receptor (VEGFR), which play a key role in coordinating angiogenesis, led to the pursuit of anti-angiogenic therapies in NETs. Both the anti-VEGF monoclonal antibody bevacizumab and small molecule tyrosine kinase inhibitors that include among their targets VEGFRs and other receptors involved in angiogenesis, such as platelet-derived growth factor receptor, have been tested.
Sunitinib was approved for the treatment of pNETs in 2011, making it a banner year for this tumor type. Approval was granted on the basis of significantly improved PFS in the sunitinib arm of a phase 3 randomized trial, but long-term follow-up suggested that sunitinib also improved OS by 10 months. Like everolimus, the OS benefit was not statistically significant, and again this was thought to be the result of extensive crossover.
Two other multikinase inhibitors have received regulatory approval for a much rarer form of NET, medullary thyroid cancer. Vandetanib and cabozantinib were approved for this indication in 2011 and 2012, respectively. Early in 2017, the results of a single-arm phase 2 trial of cabozantinib suggested that this drug may also have significant activity in other types of NET. In patients with advanced carcinoid and pNETs who received cabozantinib at 60 mg/day orally, partial responses were observed in 15% of patients and the median PFS was 21.8 months in the pNET cohort and >30 months in the carcinoid tumor cohort.25 Confirmatory phase 3 trials are planned but not currently underway.
Sulfatinib is a novel kinase inhibitor that targets the VEGFRs and fibroblast growth factor receptor 1. It has recently shown significant promise in the treatment of patients with advanced NETs. According to data presented at this year’s annual conference of the European Neuroendocrine Tumor Society in Barcelona, sulfatinib demonstrated an overall response rate of 17.1% in pancreatic NETs and 15% in extra-pancreatic NETs, with an overall disease control rate of 91.4%, and was well tolerated.26 Based on these and other promising phase 1 and 2 data, 2 phase 3 trials are ongoing.
Meanwhile, earlier this year, Mateon Therapeutics presented data from a phase 2 trial of a different kind of anti-angiogenic drug in patients with GI- or pNETs. Fosbretabulin is a vascular disrupting agent that targets the existing tumor vasculature rather than preventing the formation of new blood vessels. They do this via a number of different mechanisms, in the case of fosbretabulin it specifically targets endothelial cells and inhibits the assembly of microtubules and, hence, blocks mitosis. In 18 patients, fosbretabulin treatment resulted in 1 partial response and 7 patients who had stable disease; more than half of the patients reported improved quality of life.27 Fosbretabulin continues to be studied in NETs in combination with everolimus.
Finally, researchers are beginning to make a foray into the immunotherapy field that has revolutionized the treatment of many other tumor types. The immune checkpoint inhibitors nivolumab and pembrolizumab are being evaluated in ongoing phase 1 and 2 trials, while avelumab (Bavencio) was very recently approved by the FDA for the treatment of Merkel cell carcinoma.28,29
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.
1. Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255-1269.
2. Rorstad O. Prognostic indicators for carcinoid neuroendocrine tumors of the gastrointestinal tract. J Surg Oncol. 2005;89(3):151-160.
3. The NET Alliance. Characterizing a challenging cancer. http://www.thenetalliance.com/hcp/facts-about-net/characterization/. Publishing date not provided. Accessed October 18, 2017.
4. Yao JC, Hassan M, Phan A, et al. One hundred years after ‘carcinoid’: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063-3072.
5. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer. 2003;97(4):934-959.
6. Spada F, Valente M. Review of recent advances in medical treatment for neuroendocrine neoplasms: somatostatin analogs and chemotherapy. J Cancer Metastasis Treat. 2016;2(8):313-320.
7. Kelgiorgi D, Dervenis C. Pancreatic neuroendocrine tumors: the basics, the gray zone, and the target. F1000Research. 2017;6:663.
8. Viudez A, De Jesus-Acosta A, Carvalho FL, Vera R, Martin-Algarra S, Ramirez N. Pancreatic neuroendocrine tumors: Challenges in an underestimated disease. Crit Rev Oncol Hematol. 2016;101:193-206.
9. World Health Organization, International Agency for Research on Cancer. Bosman FT, Carneiro F, Hruban RH, Theise ND (eds). WHO classification of tumours of the digestive system. 2010, 4th ed (vol 3).
10. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors. J Thorac Oncol. 2015;10(9):1243-1260.
11. Lee A, Chan DL, Wong MH, et al. Systematic review of the role of targeted therapy in metastatic neuroendocrine tumors. Neuroendocrinology. 2017;104(3):209-222.
12. Theodoropoulou M, Stalla GK. Somatostatin receptors: from signaling to clinical practice. Front Neuroendocrinol. 2013;34(3):228-252.
13. Caplin ME, Pavel M, Cwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224-233.
14. Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656-4663.
15. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125-135.
16. Falconi M, Partelli S. Neuroendocrine tumours in 2016: defining rules for increasingly personalized treatments. Nat Rev Clin Oncol. 2017;14(2):80-82.
17. Hutchinson L. Targeted therapies: widening the treatment NET. Nat Rev Clin Oncol. 2017;14(1):2-3.
18. Cingarlini S, Bonomi M, Corbo V, Scarpa A, Tortora G. Profiling mTOR pathway in neuroendocrine tumors. Target Oncol. 2012;7(3):183-188.
19. Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514-523.
20. Yao JC, Pavel M, Lombard-Bohas C, et al. Everolimus for the treatment of advanced pancreatic neuroendocrine tumors: overall survival and circulating biomarkers from the randomized, phase III RADIANT-3 study. J Clin Oncol. http://ascopubs.org/ doi/abs/10.1200/JCO.2016.68.0702?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmed. September 12, 2016. E-pub ahead of print.
21. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968-977.
22. Gajate P, Martínez-Sáez O, Alonso-Gordoa T, Grande E. Emerging use of everolimus in the treatment of neuroendocrine tumors. Cancer Manage Res. 2017;9:215-224.
23. Chan JA, Ryan DP, Zhu AX, et al. Phase I study of pasireotide (SOM 230) and everolimus (RAD001) in advanced neuroendocrine tumors. Endocr Relat Cancer. 2012;19(5):615-623.
24. Kulke MH, Ruszniewski P, Van Cutsem E, et al. A randomized, open-label, phase 2 study of everolimus in combination with pasireotide LAR or everolimus alone in advanced, well-dierentiated, progressive pancreatic neuroendocrine tumors: COOPERATE-2 trial. Ann Oncol. 2017;28(6):1309-1315.
25. Chan JA, Faris JE, Murphy JE, et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J Clin Oncol. 2017;35(4 suppl):228-228.
26. Xu J, Li J, Bai CM, et al. An open-label phase Ib/II study of sulfatinib in patients with advanced neuroendocrine tumors (NCT02267967). Paper presented at the 14th Annual European Neuroendocrine Tumor Society Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease; March 8-10, 2017, Barcelona, Spain.
27. Libutti SK, Anthony LB, Chaplin DJ, Sosa JA. A phase II study of combretastatin A4-phosphate (CA4P) in the treatment of well-differentiated, low- to intermediate-grade, unresectable, recurrent, or metastatic pancreatic, or GI neuroendocrine tumors/carcinoid (GI-NETs/PNETs) with elevated biomarkers. J Clin Oncol. 2017;35(4 suppl):432-432.
28. Cordes LM, Gulley JL. Avelumab for the treatment of metastatic Merkel cell carcinoma. Drugs Today (Barc). 2017;53(7):377-383.
29. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374-1385.