User login
Alvogen issues recall for mislabeled fentanyl patches
Alvogen has issued a voluntary recall of two lots of its Fentanyl Transdermal System 12-mcg/h transdermal patches because of a product mislabeling, according to the Food and Drug Administration.

The recall was issued because a small number of cartons labeled as containing 12-mcg/h patches contained 50-mcg/h patches. The 50-mcg/h patches were labeled as such within the package.
Application of a 50-mcg/h patch instead of a 12-mcg/h patch could result in serious, life-threatening, or fatal respiratory depression. Groups at potential risk for such adverse events include first-time users of the patch, children, and the elderly. No reports of serious adverse events have yet been reported.
“Pharmacies are requested not to dispense any product subject to this recall,” the FDA said in a press release. Patients who “have product subject to this recall should immediately remove any patch currently in use and contact their health care provider. Patients with unused product should return it to point of purchase for replacement.”
Find more information on the recall at the FDA website.
Alvogen has issued a voluntary recall of two lots of its Fentanyl Transdermal System 12-mcg/h transdermal patches because of a product mislabeling, according to the Food and Drug Administration.
The recall was issued because a small number of cartons labeled as containing 12-mcg/h patches contained 50-mcg/h patches. The 50-mcg/h patches were labeled as such within the package.
Application of a 50-mcg/h patch instead of a 12-mcg/h patch could result in serious, life-threatening, or fatal respiratory depression. Groups at potential risk for such adverse events include first-time users of the patch, children, and the elderly. No reports of serious adverse events have yet been reported.
“Pharmacies are requested not to dispense any product subject to this recall,” the FDA said in a press release. Patients who “have product subject to this recall should immediately remove any patch currently in use and contact their health care provider. Patients with unused product should return it to point of purchase for replacement.”
Find more information on the recall at the FDA website.
Alvogen has issued a voluntary recall of two lots of its Fentanyl Transdermal System 12-mcg/h transdermal patches because of a product mislabeling, according to the Food and Drug Administration.
The recall was issued because a small number of cartons labeled as containing 12-mcg/h patches contained 50-mcg/h patches. The 50-mcg/h patches were labeled as such within the package.
Application of a 50-mcg/h patch instead of a 12-mcg/h patch could result in serious, life-threatening, or fatal respiratory depression. Groups at potential risk for such adverse events include first-time users of the patch, children, and the elderly. No reports of serious adverse events have yet been reported.
“Pharmacies are requested not to dispense any product subject to this recall,” the FDA said in a press release. Patients who “have product subject to this recall should immediately remove any patch currently in use and contact their health care provider. Patients with unused product should return it to point of purchase for replacement.”
Find more information on the recall at the FDA website.
Restricting opioids after knee surgery did not increase refills
according to a study in the Journal of Arthroplasty.

Contrary to concerns that restrictive opioid prescribing might increase the number of patient call-ins and refill requests, one academic institution had significantly fewer call-ins and refills after it implemented a strict postoperative opioid prescribing protocol on Jan. 1, 2018.
“Orthopedic surgeons might be reluctant to change practice without evidence that new, more-restrictive practice will not impede patient care,” the researchers wrote. “As the current study demonstrates, there is room to significantly decrease postoperative opioid prescriptions in total joint arthroplasty. This places patients at lower risk of opioid abuse and diversion without significantly altering the risk of postoperative complications or compromising postoperative pain control.”
Opioid overuse is a major public health concern, and orthopedic surgeons may overprescribe opioids after surgery. The University of Iowa Hospitals and Clinics in Iowa City implemented strict postoperative opioid prescription guidelines that are based on the American Academy of Orthopedic Surgeons Clinical Practice Guidelines. As part of the protocol, patients receive a preoperative education session that emphasizes risks associated with opioid use. Before initiating this protocol, postoperative drug choice and quantity had not been standardized.
To examine changes in opioid prescriptions and the number of call-ins, postoperative complications, and prescription refill requests after the implementation of the restrictive opioid prescribing protocol, investigators at the institution conducted a retrospective study.
Andrew J. Holte, a researcher in the department of orthopedics and rehabilitation, and his colleagues reviewed cases from June 2017 to February 2018. Their analysis included 399 patients who underwent total hip arthroplasty or total knee arthroplasty.
In all, 282 patients underwent surgery before the restrictive protocol (the historical cohort) and 117 after (the restrictive cohort). In the historical cohort, about 48% of the patients underwent total knee arthroplasty. In the restrictive cohort, about 44% underwent total knee arthroplasty. Patients had an average age of about 61 years, and approximately 52% were women.
According to comparisons of morphine mg equivalents (MME), the historical cohort received significantly larger mean initial opioid prescriptions (752 MME vs. 387 MME), significantly more refills per patient (0.5 vs. 0.3), and significantly more medication through refills (253 MME vs. 84 MME).
“For reference, 50 pills of 5 mg oxycodone is equivalent to 300 MMEs,” the authors noted.
A multivariable model found that younger age and total knee arthroplasty, compared with total hip arthroplasty, were associated with increased likelihood of requests for refills and patient call-ins.
“Surprisingly, there were significantly more patient call-ins and requests for refills of opioids in the historical cohort,” Mr. Holte and his colleagues said. “Although this study did not collect direct data on patient pain scores, we believe that call-ins and requests for refills are sufficient surrogate markers for inadequate pain control.”
The study does not account for prescriptions from other providers or whether patients took none, some, or all of their filled prescriptions. Future studies are needed to assess how reduced opioid prescriptions affect pain and functional outcomes in the long term, the researchers said.
One or more study authors disclosed potential conflicts of interest. The disclosures can be found in Appendix A, Supplementary Data, at the end of the journal article.
SOURCE: Holte AJ et al. J Arthroplasty. 2019 Feb 20. doi: 10.1016/j.arth.2019.02.022.
according to a study in the Journal of Arthroplasty.
Contrary to concerns that restrictive opioid prescribing might increase the number of patient call-ins and refill requests, one academic institution had significantly fewer call-ins and refills after it implemented a strict postoperative opioid prescribing protocol on Jan. 1, 2018.
“Orthopedic surgeons might be reluctant to change practice without evidence that new, more-restrictive practice will not impede patient care,” the researchers wrote. “As the current study demonstrates, there is room to significantly decrease postoperative opioid prescriptions in total joint arthroplasty. This places patients at lower risk of opioid abuse and diversion without significantly altering the risk of postoperative complications or compromising postoperative pain control.”
Opioid overuse is a major public health concern, and orthopedic surgeons may overprescribe opioids after surgery. The University of Iowa Hospitals and Clinics in Iowa City implemented strict postoperative opioid prescription guidelines that are based on the American Academy of Orthopedic Surgeons Clinical Practice Guidelines. As part of the protocol, patients receive a preoperative education session that emphasizes risks associated with opioid use. Before initiating this protocol, postoperative drug choice and quantity had not been standardized.
To examine changes in opioid prescriptions and the number of call-ins, postoperative complications, and prescription refill requests after the implementation of the restrictive opioid prescribing protocol, investigators at the institution conducted a retrospective study.
Andrew J. Holte, a researcher in the department of orthopedics and rehabilitation, and his colleagues reviewed cases from June 2017 to February 2018. Their analysis included 399 patients who underwent total hip arthroplasty or total knee arthroplasty.
In all, 282 patients underwent surgery before the restrictive protocol (the historical cohort) and 117 after (the restrictive cohort). In the historical cohort, about 48% of the patients underwent total knee arthroplasty. In the restrictive cohort, about 44% underwent total knee arthroplasty. Patients had an average age of about 61 years, and approximately 52% were women.
According to comparisons of morphine mg equivalents (MME), the historical cohort received significantly larger mean initial opioid prescriptions (752 MME vs. 387 MME), significantly more refills per patient (0.5 vs. 0.3), and significantly more medication through refills (253 MME vs. 84 MME).
“For reference, 50 pills of 5 mg oxycodone is equivalent to 300 MMEs,” the authors noted.
A multivariable model found that younger age and total knee arthroplasty, compared with total hip arthroplasty, were associated with increased likelihood of requests for refills and patient call-ins.
“Surprisingly, there were significantly more patient call-ins and requests for refills of opioids in the historical cohort,” Mr. Holte and his colleagues said. “Although this study did not collect direct data on patient pain scores, we believe that call-ins and requests for refills are sufficient surrogate markers for inadequate pain control.”
The study does not account for prescriptions from other providers or whether patients took none, some, or all of their filled prescriptions. Future studies are needed to assess how reduced opioid prescriptions affect pain and functional outcomes in the long term, the researchers said.
One or more study authors disclosed potential conflicts of interest. The disclosures can be found in Appendix A, Supplementary Data, at the end of the journal article.
SOURCE: Holte AJ et al. J Arthroplasty. 2019 Feb 20. doi: 10.1016/j.arth.2019.02.022.
according to a study in the Journal of Arthroplasty.
Contrary to concerns that restrictive opioid prescribing might increase the number of patient call-ins and refill requests, one academic institution had significantly fewer call-ins and refills after it implemented a strict postoperative opioid prescribing protocol on Jan. 1, 2018.
“Orthopedic surgeons might be reluctant to change practice without evidence that new, more-restrictive practice will not impede patient care,” the researchers wrote. “As the current study demonstrates, there is room to significantly decrease postoperative opioid prescriptions in total joint arthroplasty. This places patients at lower risk of opioid abuse and diversion without significantly altering the risk of postoperative complications or compromising postoperative pain control.”
Opioid overuse is a major public health concern, and orthopedic surgeons may overprescribe opioids after surgery. The University of Iowa Hospitals and Clinics in Iowa City implemented strict postoperative opioid prescription guidelines that are based on the American Academy of Orthopedic Surgeons Clinical Practice Guidelines. As part of the protocol, patients receive a preoperative education session that emphasizes risks associated with opioid use. Before initiating this protocol, postoperative drug choice and quantity had not been standardized.
To examine changes in opioid prescriptions and the number of call-ins, postoperative complications, and prescription refill requests after the implementation of the restrictive opioid prescribing protocol, investigators at the institution conducted a retrospective study.
Andrew J. Holte, a researcher in the department of orthopedics and rehabilitation, and his colleagues reviewed cases from June 2017 to February 2018. Their analysis included 399 patients who underwent total hip arthroplasty or total knee arthroplasty.
In all, 282 patients underwent surgery before the restrictive protocol (the historical cohort) and 117 after (the restrictive cohort). In the historical cohort, about 48% of the patients underwent total knee arthroplasty. In the restrictive cohort, about 44% underwent total knee arthroplasty. Patients had an average age of about 61 years, and approximately 52% were women.
According to comparisons of morphine mg equivalents (MME), the historical cohort received significantly larger mean initial opioid prescriptions (752 MME vs. 387 MME), significantly more refills per patient (0.5 vs. 0.3), and significantly more medication through refills (253 MME vs. 84 MME).
“For reference, 50 pills of 5 mg oxycodone is equivalent to 300 MMEs,” the authors noted.
A multivariable model found that younger age and total knee arthroplasty, compared with total hip arthroplasty, were associated with increased likelihood of requests for refills and patient call-ins.
“Surprisingly, there were significantly more patient call-ins and requests for refills of opioids in the historical cohort,” Mr. Holte and his colleagues said. “Although this study did not collect direct data on patient pain scores, we believe that call-ins and requests for refills are sufficient surrogate markers for inadequate pain control.”
The study does not account for prescriptions from other providers or whether patients took none, some, or all of their filled prescriptions. Future studies are needed to assess how reduced opioid prescriptions affect pain and functional outcomes in the long term, the researchers said.
One or more study authors disclosed potential conflicts of interest. The disclosures can be found in Appendix A, Supplementary Data, at the end of the journal article.
SOURCE: Holte AJ et al. J Arthroplasty. 2019 Feb 20. doi: 10.1016/j.arth.2019.02.022.
FROM THE JOURNAL OF ARTHROPLASTY
FDA approves generic naloxone spray for opioid overdose treatment
The Food and Drug Administration on April 19 approved the first generic naloxone hydrochloride nasal spray (Narcan) as treatment for stopping or reversing an opioid overdose.

“In the wake of the opioid crisis, a number of efforts are underway to make this emergency overdose reversal treatment more readily available and more accessible,” said Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, in a press release. “In addition to this approval of the first generic naloxone nasal spray, moving forward, we will prioritize our review of generic drug applications for naloxone.”
The agency said the naloxone nasal spray does not need assembly and can be used by anyone, regardless of medical training. If the spray is administered quickly after the overdose begins, the effect of the opioid will be countered, often within minutes. However, patients should still seek immediate medical attention.
The FDA cautioned that, when used on a patient with an opioid dependence, naloxone can cause severe opioid withdrawal, characterized by symptoms such as body aches, diarrhea, tachycardia, fever, runny nose, sneezing, goose bumps, sweating, yawning, nausea or vomiting, nervousness, restlessness or irritability, shivering or trembling, abdominal cramps, weakness, and increased blood pressure.
Find the full press release on the FDA website.
The Food and Drug Administration on April 19 approved the first generic naloxone hydrochloride nasal spray (Narcan) as treatment for stopping or reversing an opioid overdose.
“In the wake of the opioid crisis, a number of efforts are underway to make this emergency overdose reversal treatment more readily available and more accessible,” said Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, in a press release. “In addition to this approval of the first generic naloxone nasal spray, moving forward, we will prioritize our review of generic drug applications for naloxone.”
The agency said the naloxone nasal spray does not need assembly and can be used by anyone, regardless of medical training. If the spray is administered quickly after the overdose begins, the effect of the opioid will be countered, often within minutes. However, patients should still seek immediate medical attention.
The FDA cautioned that, when used on a patient with an opioid dependence, naloxone can cause severe opioid withdrawal, characterized by symptoms such as body aches, diarrhea, tachycardia, fever, runny nose, sneezing, goose bumps, sweating, yawning, nausea or vomiting, nervousness, restlessness or irritability, shivering or trembling, abdominal cramps, weakness, and increased blood pressure.
Find the full press release on the FDA website.
The Food and Drug Administration on April 19 approved the first generic naloxone hydrochloride nasal spray (Narcan) as treatment for stopping or reversing an opioid overdose.
“In the wake of the opioid crisis, a number of efforts are underway to make this emergency overdose reversal treatment more readily available and more accessible,” said Douglas Throckmorton, MD, deputy center director for regulatory programs in the FDA’s Center for Drug Evaluation and Research, in a press release. “In addition to this approval of the first generic naloxone nasal spray, moving forward, we will prioritize our review of generic drug applications for naloxone.”
The agency said the naloxone nasal spray does not need assembly and can be used by anyone, regardless of medical training. If the spray is administered quickly after the overdose begins, the effect of the opioid will be countered, often within minutes. However, patients should still seek immediate medical attention.
The FDA cautioned that, when used on a patient with an opioid dependence, naloxone can cause severe opioid withdrawal, characterized by symptoms such as body aches, diarrhea, tachycardia, fever, runny nose, sneezing, goose bumps, sweating, yawning, nausea or vomiting, nervousness, restlessness or irritability, shivering or trembling, abdominal cramps, weakness, and increased blood pressure.
Find the full press release on the FDA website.
Calcium supplement use linked to cancer death
PHILADELPHIA – a nutrition specialist noted at the annual meeting of the American College of Physicians.

The report, published (Ann Intern Med. 2019 Apr 9. doi: 10.7326/M18-2478) just 2 days before the start of the Internal Medicine meeting, found no mortality benefits associated with any reported dietary supplement use among nearly 31,000 adults in the National Health and Nutrition Examination Survey.
On the contrary, they found that excess calcium consumption was associated with increased risk for cancer-related deaths. Calcium supplements were specifically implicated in the excess of mortality, according to the investigators, with a rate ratio of 1.53 (95% confidence interval, 1.04-2.25) for intakes of 1,000 mg/day versus no intake.
“It’s better to get all of your vitamins from your food, over supplements,” said Marijane Hynes, MD, director of weight management at George Washington University, Washington, in a meet-the-professor session at the conference.
The amount of calcium patients are getting from food can be estimated with one rule of thumb: Multiply the number of dairy servings per day by 300 mg, Dr. Hynes said, who added that a serving is 8 ounces of milk or 1 ounce of hard cheese. Dark green vegetables, breads, cereals, and some nuts can provide 100-200 mg of calcium per day.
Calcium carbonate can be taken with food to enhance calcium absorption, according to Dr. Hynes, while calcium citrate can be taken without food, and is preferred for patients taking acid reflux medications.
Because calcium absorption is reduced at higher doses, patients who need more than 600 mg/day should be taking divided doses, she said.
Bone health goes beyond the dairy aisle, Dr. Hynes added. High vitamin K intake was linked to reduced hip fracture risk among the Framingham Heart Study participants. To get the recommended amount of vitamin K in the diet, patients can consume one or more servings of broccoli, kale, collard greens, or dark green lettuce.
Dr. Hynes reported she that had no relationships with entities producing, marketing, reselling, or distributing health care goods or services consumed by, or used on, patients.
These are observational data. This is not saying we put someone on calcium, and they ended up with cancer, and when you look at this whole thing it’s amazing to me that nobody is discussing the benefits that were found in patients taking magnesium, vitamin K2, and other vitamins. The other thing I would like to point out is that, for at least a decade, it has been really well established that we shouldn’t be using more than 1,000 milligrams of calcium a day, especially from a supplements source. In this study, supplemental calcium intake of 1,000 mg/d or higher was associated with increased risk of cancer death, so what’s the big deal?
The big thing with calcium is calcium comes in 7 different forms. When you eat a variety of fruits and vegetables the source of calcium you get is mixed. The problem with supplements is you are using one or maybe two forms of calcium, but if your body doesn’t recognize that form of calcium then you aren’t getting calcium and it may not be beneficial to you.
What we need to do here, in my opinion, is we need to look at the whole picture. We know that dieting alone or exercising alone does not improve outcomes. It’s the combination of diet, exercise, hormone balance, nutrients from supplements, and emotional balance that makes you healthy. Similarly, you can’t say if you just take this one nutrient you are going to improve your quality of life.
With calcium and vitamin D, you have to take vitamin K2, because vitamin K2 activates osteocalcin, a protein that rebuilds the matrix of the bone. Without vitamin K2, you can’t deposit calcium in the bones. K2 also prevents the deposition of calcium in the blood vessels.
Magnesium is another tremendously important mineral, and magnesium deficiency is the most common mineral deficiency in the United States.
Probably one of the most common causes of magnesium deficiency is the use of acid blockers. I would be very curious to know how many people were taking proton pump inhibitors or acid blockers in general. I bet you most of them were.
Derrick DeSilva Jr., MD, is an internist, practicing in Edison, N.J. He made these comments in an interview. He reported serving as a consultant for Common Sense Supplements, a company that produces dietary supplements.
These are observational data. This is not saying we put someone on calcium, and they ended up with cancer, and when you look at this whole thing it’s amazing to me that nobody is discussing the benefits that were found in patients taking magnesium, vitamin K2, and other vitamins. The other thing I would like to point out is that, for at least a decade, it has been really well established that we shouldn’t be using more than 1,000 milligrams of calcium a day, especially from a supplements source. In this study, supplemental calcium intake of 1,000 mg/d or higher was associated with increased risk of cancer death, so what’s the big deal?
The big thing with calcium is calcium comes in 7 different forms. When you eat a variety of fruits and vegetables the source of calcium you get is mixed. The problem with supplements is you are using one or maybe two forms of calcium, but if your body doesn’t recognize that form of calcium then you aren’t getting calcium and it may not be beneficial to you.
What we need to do here, in my opinion, is we need to look at the whole picture. We know that dieting alone or exercising alone does not improve outcomes. It’s the combination of diet, exercise, hormone balance, nutrients from supplements, and emotional balance that makes you healthy. Similarly, you can’t say if you just take this one nutrient you are going to improve your quality of life.
With calcium and vitamin D, you have to take vitamin K2, because vitamin K2 activates osteocalcin, a protein that rebuilds the matrix of the bone. Without vitamin K2, you can’t deposit calcium in the bones. K2 also prevents the deposition of calcium in the blood vessels.
Magnesium is another tremendously important mineral, and magnesium deficiency is the most common mineral deficiency in the United States.
Probably one of the most common causes of magnesium deficiency is the use of acid blockers. I would be very curious to know how many people were taking proton pump inhibitors or acid blockers in general. I bet you most of them were.
Derrick DeSilva Jr., MD, is an internist, practicing in Edison, N.J. He made these comments in an interview. He reported serving as a consultant for Common Sense Supplements, a company that produces dietary supplements.
These are observational data. This is not saying we put someone on calcium, and they ended up with cancer, and when you look at this whole thing it’s amazing to me that nobody is discussing the benefits that were found in patients taking magnesium, vitamin K2, and other vitamins. The other thing I would like to point out is that, for at least a decade, it has been really well established that we shouldn’t be using more than 1,000 milligrams of calcium a day, especially from a supplements source. In this study, supplemental calcium intake of 1,000 mg/d or higher was associated with increased risk of cancer death, so what’s the big deal?
The big thing with calcium is calcium comes in 7 different forms. When you eat a variety of fruits and vegetables the source of calcium you get is mixed. The problem with supplements is you are using one or maybe two forms of calcium, but if your body doesn’t recognize that form of calcium then you aren’t getting calcium and it may not be beneficial to you.
What we need to do here, in my opinion, is we need to look at the whole picture. We know that dieting alone or exercising alone does not improve outcomes. It’s the combination of diet, exercise, hormone balance, nutrients from supplements, and emotional balance that makes you healthy. Similarly, you can’t say if you just take this one nutrient you are going to improve your quality of life.
With calcium and vitamin D, you have to take vitamin K2, because vitamin K2 activates osteocalcin, a protein that rebuilds the matrix of the bone. Without vitamin K2, you can’t deposit calcium in the bones. K2 also prevents the deposition of calcium in the blood vessels.
Magnesium is another tremendously important mineral, and magnesium deficiency is the most common mineral deficiency in the United States.
Probably one of the most common causes of magnesium deficiency is the use of acid blockers. I would be very curious to know how many people were taking proton pump inhibitors or acid blockers in general. I bet you most of them were.
Derrick DeSilva Jr., MD, is an internist, practicing in Edison, N.J. He made these comments in an interview. He reported serving as a consultant for Common Sense Supplements, a company that produces dietary supplements.
PHILADELPHIA – a nutrition specialist noted at the annual meeting of the American College of Physicians.
The report, published (Ann Intern Med. 2019 Apr 9. doi: 10.7326/M18-2478) just 2 days before the start of the Internal Medicine meeting, found no mortality benefits associated with any reported dietary supplement use among nearly 31,000 adults in the National Health and Nutrition Examination Survey.
On the contrary, they found that excess calcium consumption was associated with increased risk for cancer-related deaths. Calcium supplements were specifically implicated in the excess of mortality, according to the investigators, with a rate ratio of 1.53 (95% confidence interval, 1.04-2.25) for intakes of 1,000 mg/day versus no intake.
“It’s better to get all of your vitamins from your food, over supplements,” said Marijane Hynes, MD, director of weight management at George Washington University, Washington, in a meet-the-professor session at the conference.
The amount of calcium patients are getting from food can be estimated with one rule of thumb: Multiply the number of dairy servings per day by 300 mg, Dr. Hynes said, who added that a serving is 8 ounces of milk or 1 ounce of hard cheese. Dark green vegetables, breads, cereals, and some nuts can provide 100-200 mg of calcium per day.
Calcium carbonate can be taken with food to enhance calcium absorption, according to Dr. Hynes, while calcium citrate can be taken without food, and is preferred for patients taking acid reflux medications.
Because calcium absorption is reduced at higher doses, patients who need more than 600 mg/day should be taking divided doses, she said.
Bone health goes beyond the dairy aisle, Dr. Hynes added. High vitamin K intake was linked to reduced hip fracture risk among the Framingham Heart Study participants. To get the recommended amount of vitamin K in the diet, patients can consume one or more servings of broccoli, kale, collard greens, or dark green lettuce.
Dr. Hynes reported she that had no relationships with entities producing, marketing, reselling, or distributing health care goods or services consumed by, or used on, patients.
PHILADELPHIA – a nutrition specialist noted at the annual meeting of the American College of Physicians.
The report, published (Ann Intern Med. 2019 Apr 9. doi: 10.7326/M18-2478) just 2 days before the start of the Internal Medicine meeting, found no mortality benefits associated with any reported dietary supplement use among nearly 31,000 adults in the National Health and Nutrition Examination Survey.
On the contrary, they found that excess calcium consumption was associated with increased risk for cancer-related deaths. Calcium supplements were specifically implicated in the excess of mortality, according to the investigators, with a rate ratio of 1.53 (95% confidence interval, 1.04-2.25) for intakes of 1,000 mg/day versus no intake.
“It’s better to get all of your vitamins from your food, over supplements,” said Marijane Hynes, MD, director of weight management at George Washington University, Washington, in a meet-the-professor session at the conference.
The amount of calcium patients are getting from food can be estimated with one rule of thumb: Multiply the number of dairy servings per day by 300 mg, Dr. Hynes said, who added that a serving is 8 ounces of milk or 1 ounce of hard cheese. Dark green vegetables, breads, cereals, and some nuts can provide 100-200 mg of calcium per day.
Calcium carbonate can be taken with food to enhance calcium absorption, according to Dr. Hynes, while calcium citrate can be taken without food, and is preferred for patients taking acid reflux medications.
Because calcium absorption is reduced at higher doses, patients who need more than 600 mg/day should be taking divided doses, she said.
Bone health goes beyond the dairy aisle, Dr. Hynes added. High vitamin K intake was linked to reduced hip fracture risk among the Framingham Heart Study participants. To get the recommended amount of vitamin K in the diet, patients can consume one or more servings of broccoli, kale, collard greens, or dark green lettuce.
Dr. Hynes reported she that had no relationships with entities producing, marketing, reselling, or distributing health care goods or services consumed by, or used on, patients.
REPORTING FROM INTERNAL MEDICINE 2019
Clinical Pharmacist Credentialing and Privileging: A Process for Ensuring High-Quality Patient Care
The Red Lake Indian Health Service (IHS) health care facility is in north-central Minnesota within the Red Lake Nation. The facility supports primary care, emergency, urgent care, pharmacy, inpatient, optometry, dental, radiology, laboratory, physical therapy, and behavioral health services to about 10,000 Red Lake Band of Chippewa Indian patients. The Red Lake pharmacy provides inpatient and outpatient medication services and pharmacist-managed clinical patient care.
In 2013, the Red Lake IHS medical staff endorsed the implementation of comprehensive clinical pharmacy services to increase health care access and optimize clinical outcomes for patients. During the evolution of pharmacy-based patient-centric care, the clinical programs offered by Red Lake IHS pharmacy expanded from 1 anticoagulation clinic to multiple advanced-practice clinical pharmacy services. This included pharmacy primary care, medication-assisted therapy, naloxone, hepatitis C, and behavioral health medication management clinics.
The immense clinical growth of the pharmacy department demonstrated a need to assess and monitor pharmacist competency to ensure the delivery of quality patient care. Essential quality improvement processes were lacking. To fill these quality improvement gaps, a robust pharmacist credentialing and privileging program was implemented in 2015.
Patient Care
As efforts within health care establishments across the US focus on the delivery of efficient, high-quality, affordable health care, pharmacists have become increasingly instrumental in providing patient care within expanded clinical roles.1-8 Many clinical pharmacy models have evolved into interdisciplinary approaches to care.9 Within these models, abiding by state and federal laws, pharmacists practice under the indirect supervision of licensed independent practitioners (LIPs), such as physicians, nurse practitioners, and physician assistants.8 Under collaborative practice agreements (CPAs), patients are initially diagnosed by LIPs, then referred to clinical pharmacists for therapeutic management.5,7
Clinical pharmacist functions encompass comprehensive medication management (ie, prescribing, monitoring, and adjustment of medications), nonpharmacologic guidance, and coordination of care. Interdisciplinary collaboration allows pharmacists opportunities to provide direct patient care or consultations by telecommunication in many different clinical environments, including disease management, primary care, or specialty care. Pharmacists may manage chronic or acute illnesses associated with endocrine, cardiovascular, respiratory, gastrointestinal, or other systems.
Pharmacists may also provide comprehensive medication review services, such as medication therapy management (MTM), transitions of care, or chronic care management. Examples of specialized areas include psychiatric, opioid use disorder, palliative care, infectious disease, chronic pain, or oncology services. For hospitalized patients, pharmacists may monitor pharmacokinetics and adjust dosing, transition patients from IV to oral medications, or complete medication reconciliation.10 Within these clinical roles, pharmacists assist in providing patient care during shortages of other health care providers (HCPs), improve patient outcomes, decrease health care-associated costs by preventing emergency department and hospital admissions or readmissions, increase access to patient care, and increase revenue through pharmacist-managed clinics and services.11
Pharmacist Credentialing
With the advancement of modern clinical pharmacy practice, many pharmacists have undertaken responsibilities to fulfill the complex duties of clinical care and diverse patient situations, but with few or no requirements to prove initial or ongoing clinical competency.2 Traditionally, pharmacist credentialing is limited to a onetime or periodic review of education and licensure, with little to no involvement in privileging and ongoing monitoring of clinical proficiency.10 These quality assurance disparities can be met and satisfied through credentialing and privileging processes. Credentialing and privileging are systematic, evidence-based processes that provide validation to HCPs, employers, and patients that pharmacists are qualified to practice clinically. 2,9 According to the Council on Credentialing in Pharmacy, clinical pharmacists should be held accountable for demonstrating competency and providing quality care through credentialing and privileging, as required for other HCPs.2,12
Credentialing and recredentialing is a primary source verification process. These processes ensure that there are no license restrictions or revocations; certifications are current; mandatory courses, certificates, and continuing education are complete; training and orientation are satisfactory; and any disciplinary action, malpractice claims, or history of impairment is reported. Privileging is the review of credentials and evaluation of clinical training and competence by the Clinical Director and Medical Executive Committee to determine whether a clinical pharmacist is competent to practice within requested privileges.11
Credentialing and privileging processes are designed not only to initially confirm that a pharmacist is competent to practice clinically, but also monitor ongoing performance.2,13 Participation in professional practice evaluations, which includes peer reviews, ongoing professional practice evaluations, and focused professional practice evaluations, is required for all credentialed and privileged practitioners. These evaluations are used to identify, assess, and correct unsatisfactory trends. Individual practices, documentation, and processes are evaluated against existing department standards (eg, CPAs, policies, processes)11,13 The results of individual professional practice evaluations are reviewed with practitioners on a regular basis and performance improvement plans implemented as needed.
Since 2015, 17 pharmacists at the Red Lake IHS health care facility have been granted membership to the medical staff as credentialed and privileged practitioners. In a retrospective review of professional practice evaluations by the Red Lake IHS pharmacy clinical coordinator, 971 outpatient clinical peer reviews, including the evaluation of 21,526 peer-review elements were completed by pharmacists from fiscal year 2015 through 2018. Peer-review elements assessed
Conclusion
Pharmacists have become increasingly instrumental in providing effective, cost-efficient, and accessible clinical services by continuing to move toward expanding and evolving roles within comprehensive, patient-centered clinical pharmacy practice settings.5,6 Multifaceted clinical responsibilities associated with health care delivery necessitate assessment and monitoring of pharmacist performance. Credentialing and privileging is an established and trusted systematic process that assures HCPs, employers, and patients that pharmacists are qualified and competent to practice clinically.2,4,12 Implementation of professional practice evaluations suggest improved staff compliance with visit documentation, patient care standards, and clinic processes required by CPAs, policies, and department standards to ensure the delivery of safe, high-quality patient care.
1. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. https://www.accp.com/docs/positions/misc/Improving_Patient_and_Health_System_Outcomes.pdf. Published December 2011. Accessed March 15, 2019.
2. Rouse MJ, Vlasses PH, Webb CE; Council on Credentialing in Pharmacy. Credentialing and privileging of pharmacists: a resource paper from the Council on Credentialing in Pharmacy. Am J Health Syst Pharm. 2014;71(21):e109-e118.
3. Berwick DM, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff (Millwood). 2008;27(3):759-769.
4. Blair MM, Carmichael J, Young E, Thrasher K; Qualified Provider Model Ad Hoc Committee. Pharmacist privileging in a health system: report of the Qualified Provider Model Ad Hoc Committee. Am J Health Syst Pharm. 2007;64(22):2373-2381.
5. Claxton KI, Wojtal P. Design and implementation of a credentialing and privileging model for ambulatory care pharmacists. Am J Health Syst Pharm. 2006;63(17):1627-1632.
6. Jordan TA, Hennenfent JA, Lewin JJ III, Nesbit TW, Weber R. Elevating pharmacists’ scope of practice through a health-system clinical privileging process. Am J Health Syst Pharm. 2016;73(18):1395-1405.
7. Centers for Disease Control and Prevention. Collaborative practice agreements and pharmacists’ patient care services: a resource for doctors, nurses, physician assistants, and other providers. https://www.cdc.gov/dhdsp/pubs/docs/Translational_Tools_Providers.pdf. Published October 2013. Accessed March 18, 2019.
8. Council on Credentialing in Pharmacy, Albanese NP, Rouse MJ. Scope of contemporary pharmacy practice: roles, responsibilities, and functions of practitioners and pharmacy technicians. J Am Pharm Assoc (2003). 2010;50(2):e35-e69.
9. Philip B, Weber R. Enhancing pharmacy practice models through pharmacists’ privileging. Hosp Pharm. 2013; 48(2):160-165.
10. Galt KA. Credentialing and privileging of pharmacists. Am J Health Syst Pharm. 2004;61(7):661-670.
11. Smith ML, Gemelas MF; US Public Health Service; Indian Health Service. Indian Health Service medical staff credentialing and privileging guide. https://www.ihs.gov/riskmanagement/includes/themes/newihstheme/display_objects/documents/IHS-Medical-Staff-Credentialing-and-Privileging-Guide.pdf. Published September 2005. Accessed March 15, 2019.
12. US Department of Health and Human Services, Indian Health Service. Indian health manual: medical credentials and privileges review process. https://www.ihs.gov/ihm/pc/part-3/p3c1. Accessed March 15, 2019.
13. Holley SL, Ketel C. Ongoing professional practice evaluation and focused professional practice evaluation: an overview for advanced practice clinicians. J Midwifery Women Health. 2014;59(4):452-459.
The Red Lake Indian Health Service (IHS) health care facility is in north-central Minnesota within the Red Lake Nation. The facility supports primary care, emergency, urgent care, pharmacy, inpatient, optometry, dental, radiology, laboratory, physical therapy, and behavioral health services to about 10,000 Red Lake Band of Chippewa Indian patients. The Red Lake pharmacy provides inpatient and outpatient medication services and pharmacist-managed clinical patient care.
In 2013, the Red Lake IHS medical staff endorsed the implementation of comprehensive clinical pharmacy services to increase health care access and optimize clinical outcomes for patients. During the evolution of pharmacy-based patient-centric care, the clinical programs offered by Red Lake IHS pharmacy expanded from 1 anticoagulation clinic to multiple advanced-practice clinical pharmacy services. This included pharmacy primary care, medication-assisted therapy, naloxone, hepatitis C, and behavioral health medication management clinics.
The immense clinical growth of the pharmacy department demonstrated a need to assess and monitor pharmacist competency to ensure the delivery of quality patient care. Essential quality improvement processes were lacking. To fill these quality improvement gaps, a robust pharmacist credentialing and privileging program was implemented in 2015.
Patient Care
As efforts within health care establishments across the US focus on the delivery of efficient, high-quality, affordable health care, pharmacists have become increasingly instrumental in providing patient care within expanded clinical roles.1-8 Many clinical pharmacy models have evolved into interdisciplinary approaches to care.9 Within these models, abiding by state and federal laws, pharmacists practice under the indirect supervision of licensed independent practitioners (LIPs), such as physicians, nurse practitioners, and physician assistants.8 Under collaborative practice agreements (CPAs), patients are initially diagnosed by LIPs, then referred to clinical pharmacists for therapeutic management.5,7
Clinical pharmacist functions encompass comprehensive medication management (ie, prescribing, monitoring, and adjustment of medications), nonpharmacologic guidance, and coordination of care. Interdisciplinary collaboration allows pharmacists opportunities to provide direct patient care or consultations by telecommunication in many different clinical environments, including disease management, primary care, or specialty care. Pharmacists may manage chronic or acute illnesses associated with endocrine, cardiovascular, respiratory, gastrointestinal, or other systems.
Pharmacists may also provide comprehensive medication review services, such as medication therapy management (MTM), transitions of care, or chronic care management. Examples of specialized areas include psychiatric, opioid use disorder, palliative care, infectious disease, chronic pain, or oncology services. For hospitalized patients, pharmacists may monitor pharmacokinetics and adjust dosing, transition patients from IV to oral medications, or complete medication reconciliation.10 Within these clinical roles, pharmacists assist in providing patient care during shortages of other health care providers (HCPs), improve patient outcomes, decrease health care-associated costs by preventing emergency department and hospital admissions or readmissions, increase access to patient care, and increase revenue through pharmacist-managed clinics and services.11
Pharmacist Credentialing
With the advancement of modern clinical pharmacy practice, many pharmacists have undertaken responsibilities to fulfill the complex duties of clinical care and diverse patient situations, but with few or no requirements to prove initial or ongoing clinical competency.2 Traditionally, pharmacist credentialing is limited to a onetime or periodic review of education and licensure, with little to no involvement in privileging and ongoing monitoring of clinical proficiency.10 These quality assurance disparities can be met and satisfied through credentialing and privileging processes. Credentialing and privileging are systematic, evidence-based processes that provide validation to HCPs, employers, and patients that pharmacists are qualified to practice clinically. 2,9 According to the Council on Credentialing in Pharmacy, clinical pharmacists should be held accountable for demonstrating competency and providing quality care through credentialing and privileging, as required for other HCPs.2,12
Credentialing and recredentialing is a primary source verification process. These processes ensure that there are no license restrictions or revocations; certifications are current; mandatory courses, certificates, and continuing education are complete; training and orientation are satisfactory; and any disciplinary action, malpractice claims, or history of impairment is reported. Privileging is the review of credentials and evaluation of clinical training and competence by the Clinical Director and Medical Executive Committee to determine whether a clinical pharmacist is competent to practice within requested privileges.11
Credentialing and privileging processes are designed not only to initially confirm that a pharmacist is competent to practice clinically, but also monitor ongoing performance.2,13 Participation in professional practice evaluations, which includes peer reviews, ongoing professional practice evaluations, and focused professional practice evaluations, is required for all credentialed and privileged practitioners. These evaluations are used to identify, assess, and correct unsatisfactory trends. Individual practices, documentation, and processes are evaluated against existing department standards (eg, CPAs, policies, processes)11,13 The results of individual professional practice evaluations are reviewed with practitioners on a regular basis and performance improvement plans implemented as needed.
Since 2015, 17 pharmacists at the Red Lake IHS health care facility have been granted membership to the medical staff as credentialed and privileged practitioners. In a retrospective review of professional practice evaluations by the Red Lake IHS pharmacy clinical coordinator, 971 outpatient clinical peer reviews, including the evaluation of 21,526 peer-review elements were completed by pharmacists from fiscal year 2015 through 2018. Peer-review elements assessed
Conclusion
Pharmacists have become increasingly instrumental in providing effective, cost-efficient, and accessible clinical services by continuing to move toward expanding and evolving roles within comprehensive, patient-centered clinical pharmacy practice settings.5,6 Multifaceted clinical responsibilities associated with health care delivery necessitate assessment and monitoring of pharmacist performance. Credentialing and privileging is an established and trusted systematic process that assures HCPs, employers, and patients that pharmacists are qualified and competent to practice clinically.2,4,12 Implementation of professional practice evaluations suggest improved staff compliance with visit documentation, patient care standards, and clinic processes required by CPAs, policies, and department standards to ensure the delivery of safe, high-quality patient care.
The Red Lake Indian Health Service (IHS) health care facility is in north-central Minnesota within the Red Lake Nation. The facility supports primary care, emergency, urgent care, pharmacy, inpatient, optometry, dental, radiology, laboratory, physical therapy, and behavioral health services to about 10,000 Red Lake Band of Chippewa Indian patients. The Red Lake pharmacy provides inpatient and outpatient medication services and pharmacist-managed clinical patient care.
In 2013, the Red Lake IHS medical staff endorsed the implementation of comprehensive clinical pharmacy services to increase health care access and optimize clinical outcomes for patients. During the evolution of pharmacy-based patient-centric care, the clinical programs offered by Red Lake IHS pharmacy expanded from 1 anticoagulation clinic to multiple advanced-practice clinical pharmacy services. This included pharmacy primary care, medication-assisted therapy, naloxone, hepatitis C, and behavioral health medication management clinics.
The immense clinical growth of the pharmacy department demonstrated a need to assess and monitor pharmacist competency to ensure the delivery of quality patient care. Essential quality improvement processes were lacking. To fill these quality improvement gaps, a robust pharmacist credentialing and privileging program was implemented in 2015.
Patient Care
As efforts within health care establishments across the US focus on the delivery of efficient, high-quality, affordable health care, pharmacists have become increasingly instrumental in providing patient care within expanded clinical roles.1-8 Many clinical pharmacy models have evolved into interdisciplinary approaches to care.9 Within these models, abiding by state and federal laws, pharmacists practice under the indirect supervision of licensed independent practitioners (LIPs), such as physicians, nurse practitioners, and physician assistants.8 Under collaborative practice agreements (CPAs), patients are initially diagnosed by LIPs, then referred to clinical pharmacists for therapeutic management.5,7
Clinical pharmacist functions encompass comprehensive medication management (ie, prescribing, monitoring, and adjustment of medications), nonpharmacologic guidance, and coordination of care. Interdisciplinary collaboration allows pharmacists opportunities to provide direct patient care or consultations by telecommunication in many different clinical environments, including disease management, primary care, or specialty care. Pharmacists may manage chronic or acute illnesses associated with endocrine, cardiovascular, respiratory, gastrointestinal, or other systems.
Pharmacists may also provide comprehensive medication review services, such as medication therapy management (MTM), transitions of care, or chronic care management. Examples of specialized areas include psychiatric, opioid use disorder, palliative care, infectious disease, chronic pain, or oncology services. For hospitalized patients, pharmacists may monitor pharmacokinetics and adjust dosing, transition patients from IV to oral medications, or complete medication reconciliation.10 Within these clinical roles, pharmacists assist in providing patient care during shortages of other health care providers (HCPs), improve patient outcomes, decrease health care-associated costs by preventing emergency department and hospital admissions or readmissions, increase access to patient care, and increase revenue through pharmacist-managed clinics and services.11
Pharmacist Credentialing
With the advancement of modern clinical pharmacy practice, many pharmacists have undertaken responsibilities to fulfill the complex duties of clinical care and diverse patient situations, but with few or no requirements to prove initial or ongoing clinical competency.2 Traditionally, pharmacist credentialing is limited to a onetime or periodic review of education and licensure, with little to no involvement in privileging and ongoing monitoring of clinical proficiency.10 These quality assurance disparities can be met and satisfied through credentialing and privileging processes. Credentialing and privileging are systematic, evidence-based processes that provide validation to HCPs, employers, and patients that pharmacists are qualified to practice clinically. 2,9 According to the Council on Credentialing in Pharmacy, clinical pharmacists should be held accountable for demonstrating competency and providing quality care through credentialing and privileging, as required for other HCPs.2,12
Credentialing and recredentialing is a primary source verification process. These processes ensure that there are no license restrictions or revocations; certifications are current; mandatory courses, certificates, and continuing education are complete; training and orientation are satisfactory; and any disciplinary action, malpractice claims, or history of impairment is reported. Privileging is the review of credentials and evaluation of clinical training and competence by the Clinical Director and Medical Executive Committee to determine whether a clinical pharmacist is competent to practice within requested privileges.11
Credentialing and privileging processes are designed not only to initially confirm that a pharmacist is competent to practice clinically, but also monitor ongoing performance.2,13 Participation in professional practice evaluations, which includes peer reviews, ongoing professional practice evaluations, and focused professional practice evaluations, is required for all credentialed and privileged practitioners. These evaluations are used to identify, assess, and correct unsatisfactory trends. Individual practices, documentation, and processes are evaluated against existing department standards (eg, CPAs, policies, processes)11,13 The results of individual professional practice evaluations are reviewed with practitioners on a regular basis and performance improvement plans implemented as needed.
Since 2015, 17 pharmacists at the Red Lake IHS health care facility have been granted membership to the medical staff as credentialed and privileged practitioners. In a retrospective review of professional practice evaluations by the Red Lake IHS pharmacy clinical coordinator, 971 outpatient clinical peer reviews, including the evaluation of 21,526 peer-review elements were completed by pharmacists from fiscal year 2015 through 2018. Peer-review elements assessed
Conclusion
Pharmacists have become increasingly instrumental in providing effective, cost-efficient, and accessible clinical services by continuing to move toward expanding and evolving roles within comprehensive, patient-centered clinical pharmacy practice settings.5,6 Multifaceted clinical responsibilities associated with health care delivery necessitate assessment and monitoring of pharmacist performance. Credentialing and privileging is an established and trusted systematic process that assures HCPs, employers, and patients that pharmacists are qualified and competent to practice clinically.2,4,12 Implementation of professional practice evaluations suggest improved staff compliance with visit documentation, patient care standards, and clinic processes required by CPAs, policies, and department standards to ensure the delivery of safe, high-quality patient care.
1. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. https://www.accp.com/docs/positions/misc/Improving_Patient_and_Health_System_Outcomes.pdf. Published December 2011. Accessed March 15, 2019.
2. Rouse MJ, Vlasses PH, Webb CE; Council on Credentialing in Pharmacy. Credentialing and privileging of pharmacists: a resource paper from the Council on Credentialing in Pharmacy. Am J Health Syst Pharm. 2014;71(21):e109-e118.
3. Berwick DM, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff (Millwood). 2008;27(3):759-769.
4. Blair MM, Carmichael J, Young E, Thrasher K; Qualified Provider Model Ad Hoc Committee. Pharmacist privileging in a health system: report of the Qualified Provider Model Ad Hoc Committee. Am J Health Syst Pharm. 2007;64(22):2373-2381.
5. Claxton KI, Wojtal P. Design and implementation of a credentialing and privileging model for ambulatory care pharmacists. Am J Health Syst Pharm. 2006;63(17):1627-1632.
6. Jordan TA, Hennenfent JA, Lewin JJ III, Nesbit TW, Weber R. Elevating pharmacists’ scope of practice through a health-system clinical privileging process. Am J Health Syst Pharm. 2016;73(18):1395-1405.
7. Centers for Disease Control and Prevention. Collaborative practice agreements and pharmacists’ patient care services: a resource for doctors, nurses, physician assistants, and other providers. https://www.cdc.gov/dhdsp/pubs/docs/Translational_Tools_Providers.pdf. Published October 2013. Accessed March 18, 2019.
8. Council on Credentialing in Pharmacy, Albanese NP, Rouse MJ. Scope of contemporary pharmacy practice: roles, responsibilities, and functions of practitioners and pharmacy technicians. J Am Pharm Assoc (2003). 2010;50(2):e35-e69.
9. Philip B, Weber R. Enhancing pharmacy practice models through pharmacists’ privileging. Hosp Pharm. 2013; 48(2):160-165.
10. Galt KA. Credentialing and privileging of pharmacists. Am J Health Syst Pharm. 2004;61(7):661-670.
11. Smith ML, Gemelas MF; US Public Health Service; Indian Health Service. Indian Health Service medical staff credentialing and privileging guide. https://www.ihs.gov/riskmanagement/includes/themes/newihstheme/display_objects/documents/IHS-Medical-Staff-Credentialing-and-Privileging-Guide.pdf. Published September 2005. Accessed March 15, 2019.
12. US Department of Health and Human Services, Indian Health Service. Indian health manual: medical credentials and privileges review process. https://www.ihs.gov/ihm/pc/part-3/p3c1. Accessed March 15, 2019.
13. Holley SL, Ketel C. Ongoing professional practice evaluation and focused professional practice evaluation: an overview for advanced practice clinicians. J Midwifery Women Health. 2014;59(4):452-459.
1. Giberson S, Yoder S, Lee MP. Improving patient and health system outcomes through advanced pharmacy practice. https://www.accp.com/docs/positions/misc/Improving_Patient_and_Health_System_Outcomes.pdf. Published December 2011. Accessed March 15, 2019.
2. Rouse MJ, Vlasses PH, Webb CE; Council on Credentialing in Pharmacy. Credentialing and privileging of pharmacists: a resource paper from the Council on Credentialing in Pharmacy. Am J Health Syst Pharm. 2014;71(21):e109-e118.
3. Berwick DM, Nolan TW, Whittington J. The triple aim: care, health, and cost. Health Aff (Millwood). 2008;27(3):759-769.
4. Blair MM, Carmichael J, Young E, Thrasher K; Qualified Provider Model Ad Hoc Committee. Pharmacist privileging in a health system: report of the Qualified Provider Model Ad Hoc Committee. Am J Health Syst Pharm. 2007;64(22):2373-2381.
5. Claxton KI, Wojtal P. Design and implementation of a credentialing and privileging model for ambulatory care pharmacists. Am J Health Syst Pharm. 2006;63(17):1627-1632.
6. Jordan TA, Hennenfent JA, Lewin JJ III, Nesbit TW, Weber R. Elevating pharmacists’ scope of practice through a health-system clinical privileging process. Am J Health Syst Pharm. 2016;73(18):1395-1405.
7. Centers for Disease Control and Prevention. Collaborative practice agreements and pharmacists’ patient care services: a resource for doctors, nurses, physician assistants, and other providers. https://www.cdc.gov/dhdsp/pubs/docs/Translational_Tools_Providers.pdf. Published October 2013. Accessed March 18, 2019.
8. Council on Credentialing in Pharmacy, Albanese NP, Rouse MJ. Scope of contemporary pharmacy practice: roles, responsibilities, and functions of practitioners and pharmacy technicians. J Am Pharm Assoc (2003). 2010;50(2):e35-e69.
9. Philip B, Weber R. Enhancing pharmacy practice models through pharmacists’ privileging. Hosp Pharm. 2013; 48(2):160-165.
10. Galt KA. Credentialing and privileging of pharmacists. Am J Health Syst Pharm. 2004;61(7):661-670.
11. Smith ML, Gemelas MF; US Public Health Service; Indian Health Service. Indian Health Service medical staff credentialing and privileging guide. https://www.ihs.gov/riskmanagement/includes/themes/newihstheme/display_objects/documents/IHS-Medical-Staff-Credentialing-and-Privileging-Guide.pdf. Published September 2005. Accessed March 15, 2019.
12. US Department of Health and Human Services, Indian Health Service. Indian health manual: medical credentials and privileges review process. https://www.ihs.gov/ihm/pc/part-3/p3c1. Accessed March 15, 2019.
13. Holley SL, Ketel C. Ongoing professional practice evaluation and focused professional practice evaluation: an overview for advanced practice clinicians. J Midwifery Women Health. 2014;59(4):452-459.
Pharmacist Interventions to Reduce Modifiable Bleeding Risk Factors Using HAS-BLED in Patients Taking Warfarin (FULL)
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and is associated with a 5-fold increase in the risk of ischemic stroke and the risk increases with age.1-3 Oral anticoagulation (OAC) therapy effectively reduces the risk of ischemic stroke in patients with nonvalvular AF. However, OAC therapy carries a bleeding risk.4
Several bleeding risk scores have been developed and validated for patients with AF who are taking warfarin: HEMORR2HAGES (Hepatic or renal disease, Ethanol abuse, Malignancy, Older age, Reduced platelet count or function, Re-bleeding, Hypertension, Anemia, Genetic factors, Excessive fall risk, Stroke), ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation), and HAS-BLED (Hypertension, Abnormal renal and/or liver function, Stroke, Bleeding history or predisposition, Labile international normalized ratio [INR], Elderly, and Drugs and/or alcohol excess concomitantly).4,5 All 3 bleeding risk scores demonstrate only modest ability to predict clinically relevant bleeding in patients taking warfarin. The HAS-BLED score was superior to HEMORR2HAGES and ATRIA for predicting any clinically relevant bleeding and was the only bleeding risk score that demonstrated significant predictive performance for intracranial hemorrhage.5 Compared with HEMORR2HAGES, the HAS-BLED score is simpler to use and to assess risk factors that can be gathered from medical history or routinely tested in patients with AF.4 Unlike HAS-BLED, HEMORR2HAGES and ATRIA do not consider medications that could increase the risk of bleeding.
Despite the availability of validated bleeding risk scores, clinical application of these measures should not be used to exclude a patient from OAC therapy for patients who reach a threshold score. Rather, current guideline and expert consensus agree with the recommendation to use bleeding risk scores to identify risk factors and address those factors that are modifiable to reduce the risk of anticoagulant-associated major bleeding.6-8
The authors identified modifiable bleeding risk factors using the HAS-BLED score and evaluated pharmacist interventions to correct these factors in patients with nonvalvular AF who are taking warfarin. To the authors’ knowledge, there have been no published studies evaluating interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED score.
Methods
Clinical pharmacy specialists (CPSs) in the primary care (PC) clinics at the Clement J. Zablocki VAMC (CJZVAMC) in Milwaukee, Wisconsin, have prescriptive authority within their scope of practice to manage smoking cessation and diseases, including anticoagulation, diabetes mellitus, heart failure, hypertension, and dyslipidemia. Patients who are on OAC therapy, including warfarin, receive comprehensive anticoagulation management from PC CPSs, including prescribing OAC therapy, education, dosage adjustments, and laboratory monitoring.
Patients were included in the HAS-BLED risk scoring and intervention if their warfarin therapy was managed by a PC CPS, had an active warfarin prescription with a diagnosis of nonvalvular AF in their problem list, and had ≥ 1 modifiable risk factor(s) from the HAS-BLED risk score. Modifiable risk factors evaluated were systolic blood pressure (SBP) > 160 mm Hg, an active prescription for VA or non-VA (which generally indicates over-the-counter [OTC] medication use) aspirin, clopidogrel, or a nonsteroidal anti-inflammatory drug (NSAID). Excess alcohol consumption was not listed as a modifiable risk factor in this assessment because nearly all the anticoagulation patients already receive regular recommendations to minimize alcohol use from the PC CPSs.
Patients were excluded from analysis if they had an indication for warfarin use other than nonvalvular AF, such as atrial flutter, acute/chronic deep vein thrombosis or pulmonary embolism, history of venous thromboembolism, peripheral vascular disease, or aortic or mitral mechanical valve. Patients also were excluded if they were on antiplatelet therapy for unstable coronary artery disease (CAD), experienced acute coronary syndrome within the past 1 year, history of stent placement, carotid endarterectomy, carotid stenosis, or noncardioembolic stroke and no other modifiable risk factors. Last, patients were excluded if clinic SBP readings were > 160 mm Hg but there was documented white coat hypertension or home SBP readings < 160 mm Hg.
The following definitions or measurements were used for assessing the HAS-BLED bleeding risk score4:
- Uncontrolled hypertension: most recently charted SBP > 160 mm Hg;
- Abnormal renal function: dialysis, renal transplant, serum creatinine > 2.26 mg/dL;
- Abnormal liver function: chronic hepatic disease, biochemical evidence of significant hepatic derangement (bilirubin > 2 × upper limit of normal and/or AST/ALT/alkaline phosphatase > 3 × upper limit of normal);
- Stroke: including history of transient ischemic attack;
- Bleeding history or predisposition: history of major bleeding (intracranial and/or any bleeding requiring hospitalization and/or causing a decrease in hemoglobin (Hgb) level of > 2 g/dL and/or requiring blood transfusion), anemia (males: Hgb < 13 g/dL; females: Hgb < 12 g/dL);
- Labile INR: percentage of INRs in therapeutic range < 60% (using the CJZVAMC anticoagulation management tool, which calculates percentage of INRs in goal reported since the first visit);
- Geriatric: age > 65 years at initial assessment;
- Concomitant drug use (VA prescription or non-VA medication list): aspirin, clopidogrel, or NSAID; and
- Alcohol in excess: > 8 alcohol servings per week from chart documentation of the patient’s self-report.
In the HAS-BLED bleeding risk score, patients receive 1 point for each component for a maximum score of 9 points. The score is stratified into low (0 points), intermediate (1 to 2 points), and high (≥ 3 points) bleeding risk.4
The HAS-BLED risk factors were obtained from patient chart review, including problem list, laboratory results, and PC CPS anticoagulation notes. Interventions included primary care provider (PCP) notification of elevated BP and offer of BP management by a PC CPS, patient education and/or PCP contact to discontinue concurrent NSAID or addition of a proton pump inhibitor (PPI) based on bleeding risk factor if the NSAID was deemed necessary, and discontinuation of concomitant antiplatelet drug(s) or reduction in aspirin dosage in consultation with patient’s PCP and cardiologist.9 In order to complete the initial HAS-BLED assessment and implement interventions, a note template was developed and entered into the electronic health record (EHR) that identified the patient’s modifiable risk factors.
Once the PCP and cardiologist (if applicable) responded to the note, by either accepting or declining the PC CPS recommendation(s), the HAS-BLED score was recalculated and recorded. If the provider did not respond to the initial note, an attempt was made to follow up at 3 months and at 6 months if necessary. If the provider did not respond at 6 months, the nonresponse was documented. For patients whose PCP requested PC CPS management of BP, the HAS-BLED score was recalculated 6 months after response from the PCP.
The primary outcome was the proportion of patients whose HAS-BLED score was reduced by at least 1 point. Secondary outcomes included the proportion of patients whose HAS-BLED score was reduced from one category of bleeding risk to a lesser one, total number of pharmacist interventions completed, number of pharmacist interventions made of each type (BP management, NSAID use, or antiplatelet drug use), and PCP acceptance rate.
Results
A total of 897 patients taking warfarin received anticoagulation management by a PC CPS at CJZVAMC in 2015. Of these, 819 patients were excluded based on the exclusion criteria (eFigure).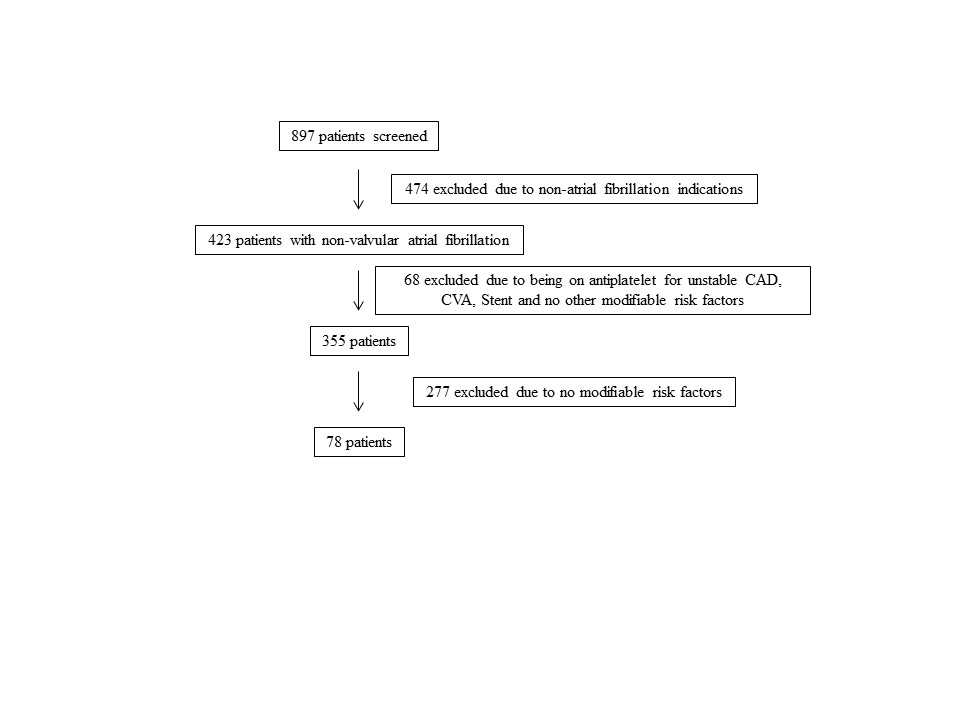
Seventy-eight patients were included in the assessment. Baseline HAS-BLED scores were calculated, and recommendations were made via an EHR progress note to the PCP and cardiologist (if applicable). Recommended interventions in the 78 patients resulted in 44 patients (56%) who experienced reduction in their HAS-BLED score by at least 1 point (Table 1). Twenty patients (25%) saw their HAS-BLED category reduced from a higher level of bleeding risk to a lower risk. The average HAS-BLED score in the 44 patients was 2.38 before intervention and 1.55 after the intervention. 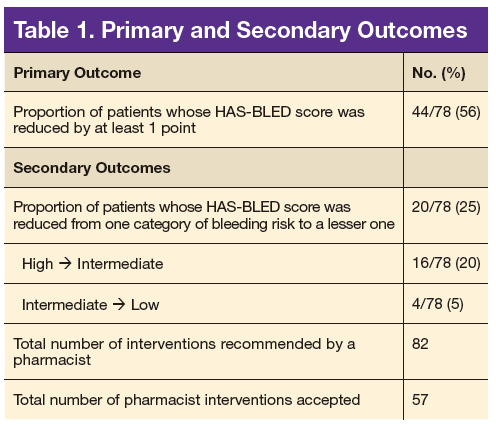
In 10 patients, the HAS-BLED score did not decrease despite accepted PC CPS intervention. Specifically, 7 patients were on both an antiplatelet agent and NSAID. As a result of the pharmacist intervention, the NSAID was discontinued, but the antiplatelet remained because of stent placement or carotid stenosis. In 1 patient, the aspirin dosage was decreased from 325 to 81 mg/d. In 2 patients where NSAID use was deemed necessary—meloxicam in both cases—a PPI was ordered based on bleeding risk.9
A total of 82 interventions were recommended; 57 interventions were accepted, resulting in a provider acceptance rate of 69.5% (Tables 2 and 3). Thirty-five of the accepted interventions (61%) involved discontinuing an antiplatelet (aspirin or clopidogrel) in consultation with the patient’s PCP and cardiologist. Twenty-seven of these patients had no documented CAD, and 8 of the patients had stable CAD. Seventeen (30%) of the accepted recommendations were for discontinuing NSAID therapy, 2 (4%) were for BP management by a PC CPS, 2 (4%) for addition of PPI with continued NSAID use (meloxicam), and 1 (1%) for decreasing aspirin dosage from 325 to 81 mg/d. The NSAIDs that were discontinued included ibuprofen, indomethacin, meloxicam, and naproxen.
Discussion
This project is the first, to the authors’ knowledge, to evaluate pharmacist interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED bleeding risk score. Most of the patients with nonvalvular AF in the PC clinics did not have modifiable bleeding risk factors. However, of the patients who received a recommendation to reduce a specific modifiable risk factor, most of the interventions were accepted by PCPs and
Most of the interventions recommended evaluating the use of antiplatelet agents, particularly aspirin. The benefits of antiplatelet therapy for secondary prevention of cardiovascular disease are well established. However, for AF patients on OAC therapy, the concomitant use of antiplatelet therapy significantly increases the risk of bleeding and should be reserved for high-risk patients.10 The 2012 American College of Chest Physicians guidelines support the use of OAC monotherapy in patients with AF with stable CAD, including patients with a myocardial infarction or percutaneous coronary intervention more than 1 year previously, which has been corroborated with guideline and expert consensus recommendations released in 2016.7,8,10,11
For patients taking warfarin for AF without CAD, the possible benefit of concomitant aspirin therapy for primary prevention is outweighed by the increased risk of major bleeding.12 Furthermore, warfarin monotherapy has been shown to be effective in primary prevention of CAD and seems to have cardiovascular benefit for secondary prevention but with increased bleeding.13,14 As a result, through the exclusion criteria this project aimed to evaluate warfarin patients with AF at low risk of cardiovascular events who might be taking unnecessary concurrent antiplatelet therapy. 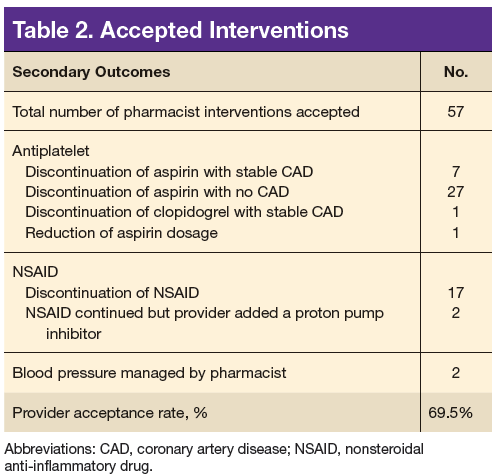
More than one-half of the interventions involved discontinuing antiplatelet therapy. Chart reviews revealed a lack of documentation for the indication and intended duration of antiplatelet therapy. In many patients, it is likely that aspirin use predated AF diagnosis and warfarin initiation. In some of these cases, it would have been appropriate to discontinue aspirin when starting warfarin use. Although there is guidance to support the use of OAC monotherapy in patients with AF with stable CAD, the patient’s provider and cardiologist made the decision to discontinue an antiplatelet agent after weighing benefits and risks. Regardless of the outcome, this analysis revealed the importance and need for routine review of antiplatelet therapy and documenting the rationale for antiplatelet use in addition to anticoagulation.
The second largest category of interventions accepted was for evaluation of NSAID use. A 2014 study by Lamberts and colleagues found that concomitant use of oral anticoagulants and NSAIDs conferred an independent risk for major bleeding and thromboembolism in patients with AF.15 The increase in serious bleeding (absolute risk difference of 2.5 events per 1,000 patients) was observed even with short-term NSAID exposure of 14 days across all NSAID types (selective COX-2 inhibitors or nonselective NSAIDs). In addition, there was an incremental increase in bleeding risk with high NSAID dosages. The risk of serious bleeding was even greater when an NSAID is added to OAC therapy and aspirin. Seven out of 17 warfarin patients (41%) who were taking an NSAID also were on an antiplatelet agent. As a result of the pharmacist interventions, NSAIDs were discontinued in all of these patients, but the antiplatelet remained because of stent placement or carotid stenosis.
This analysis captured only those patients with a documented active prescription or self-reported OTC use of an NSAID. It is unknown how many patients might take OTC NSAIDs occasionally but not report this use to a provider or pharmacist. Primary care CPSs educate patients to not use NSAIDs while taking warfarin during their initial visit and periodically thereafter; however, with the number of different NSAIDs available without a prescription and the various brand and generic names offered, it can be difficult for patients to understand what they should or should not take for minor pain or fever. Therefore, it is imperative that NSAID use is reviewed regularly at anticoagulant follow-up visits and patients are educated about alternative OTC agents for pain relief (eg, acetaminophen, topical agents, heating pad) when necessary. It also is equally important for PCPs to weigh the benefit vs risk for each patient before prescribing an NSAID if alternatives have been exhausted especially if the patient also is taking an OAC and antiplatelet agent.
The smallest number of interventions completed was for BP management. According to the HAS-BLED bleeding risk score, BP management was recommended only if the most recent clinic SBP was > 160 mm Hg, excluding patients with documented white-coat syndrome or home SBP readings < 160 mm Hg. One potential explanation for the small number of patients with SBP > 160 mm Hg is that for many of the patients taking warfarin, the PC CPSs at CJZVAMC have been involved in their BP management through earlier consultation by providers.
Limitations
A limitation of the BP component of the HAS-BLED score was that the assessment of BP for this project was only one point in time. In 3 cases, the SBP was > 160 mm Hg only during the most recent measurement, and these patients had normal BP readings on return to the clinic for follow-up. This category of recommendation also took more time for follow-up because a PC CPS would need to evaluate the patient in clinic, implement changes to BP medications, and follow-up at subsequent visits. Although some PCPs felt that the patient did not need pharmacist intervention, the elevated SBPs were brought to the provider’s attention, and some patients received further monitoring by the PCP or through a specialty clinic (eg, nephrology).
Another limitation of this project was that the bleeding risk evaluation occurred at only 1 visit. Patients’ medications and medical issues often change with time. Therefore, it is important to implement a process to regularly review (eg, annually) patients’ bleeding risk factors and to identify and act on modifiable risk factors. Another limitation was a lack of a comparator group and the time frame of the evaluation. As a result, the authors were unable to evaluate bleeding outcomes because of the small sample size and limited time frame. Future studies could consider evaluating bleeding events as an outcome, including additional modifiable risk factors, such as excess alcohol and labile INR, expanding the review to patients taking warfarin for indications other than AF, and review of patients on direct-acting oral anticoagulants (DOACs) with AF; keeping in mind that currently available bleeding risk calculators were developed for patients taking warfarin, not DOACs with AF. Patients could be counselled on reducing alcohol intake or switching to a DOAC if INR is labile despite adherence. 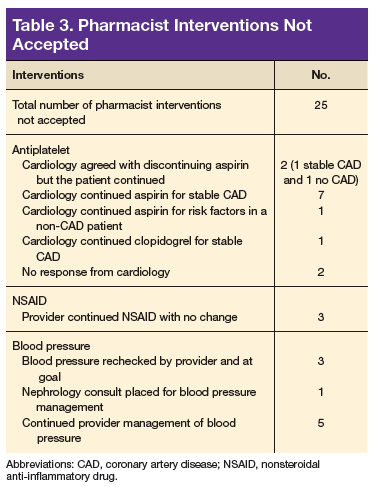
Conclusion
This quality improvement project successfully implemented use of the HAS-BLED bleeding risk score to identify and reduce modifiable bleeding risk factors in patients with AF taking warfarin. Pharmacist intervention resulted in a reduction of HAS-BLED scores and bleeding risk categories.
Click here to read the digital edition.
1. Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110(9):1042-1046.
2. Patel NJ, Deshmukh A, Pant S, et al. Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation. 2014;129(23):2371-2379.
3. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983-988.
4. Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100.
5. Apostolakis S, Lane DA, Guo Y, Buller H, Lip GY. Performance of the HEMORR2HAGES, ATRIA, and HAS-BLED bleeding risk-prediction scores in patients with atrial fibrillation undergoing anticoagulation: the AMADEUS (evaluating the use of SR34006 compared to warfarin or acenocoumarol in patients with atrial fibrillation) study. J Am Coll Cardiol. 2012;60(9):861-867.
6. Lane DA, Lip GY. Use of the CHA(2)DS(2)-VASc and HAS-BLED scores to aid decision making for thromboprophylaxis in nonvalvular atrial fibrillation. Circulation. 2012;126(7):860-865.
7. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962.
8. Ruff CT, Ansell JE, Becker RC, et al. North American thrombosis forum, AF action initiative consensus document. Am J Med. 2016;129(suppl 5):S1-S29.
9. Lanza FL, Chan FK, Quigley EM; Practice Parameters Committee of the American College of Gastroenterology. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104(3):728-738.
10. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation. Antithrombotic therapy and prevention of thrombosis, 9th ed. American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):e531S-e575S.
11. Macle L, Cairns J, Leblanc K, et al; CCS Atrial Fibrillation Guidelines Committee. 2016 focused update of the Canadian Cardiovascular Society guidelines for the management of atrial fibrillation. Can J Cardiol. 2016;32(10):1170-1185.
12. Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med. 2007;167(2):117-124.
13. The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet. 1998;351(9098):233-241.
14. Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
15. Lamberts M, Lip GY, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698.
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and is associated with a 5-fold increase in the risk of ischemic stroke and the risk increases with age.1-3 Oral anticoagulation (OAC) therapy effectively reduces the risk of ischemic stroke in patients with nonvalvular AF. However, OAC therapy carries a bleeding risk.4
Several bleeding risk scores have been developed and validated for patients with AF who are taking warfarin: HEMORR2HAGES (Hepatic or renal disease, Ethanol abuse, Malignancy, Older age, Reduced platelet count or function, Re-bleeding, Hypertension, Anemia, Genetic factors, Excessive fall risk, Stroke), ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation), and HAS-BLED (Hypertension, Abnormal renal and/or liver function, Stroke, Bleeding history or predisposition, Labile international normalized ratio [INR], Elderly, and Drugs and/or alcohol excess concomitantly).4,5 All 3 bleeding risk scores demonstrate only modest ability to predict clinically relevant bleeding in patients taking warfarin. The HAS-BLED score was superior to HEMORR2HAGES and ATRIA for predicting any clinically relevant bleeding and was the only bleeding risk score that demonstrated significant predictive performance for intracranial hemorrhage.5 Compared with HEMORR2HAGES, the HAS-BLED score is simpler to use and to assess risk factors that can be gathered from medical history or routinely tested in patients with AF.4 Unlike HAS-BLED, HEMORR2HAGES and ATRIA do not consider medications that could increase the risk of bleeding.
Despite the availability of validated bleeding risk scores, clinical application of these measures should not be used to exclude a patient from OAC therapy for patients who reach a threshold score. Rather, current guideline and expert consensus agree with the recommendation to use bleeding risk scores to identify risk factors and address those factors that are modifiable to reduce the risk of anticoagulant-associated major bleeding.6-8
The authors identified modifiable bleeding risk factors using the HAS-BLED score and evaluated pharmacist interventions to correct these factors in patients with nonvalvular AF who are taking warfarin. To the authors’ knowledge, there have been no published studies evaluating interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED score.
Methods
Clinical pharmacy specialists (CPSs) in the primary care (PC) clinics at the Clement J. Zablocki VAMC (CJZVAMC) in Milwaukee, Wisconsin, have prescriptive authority within their scope of practice to manage smoking cessation and diseases, including anticoagulation, diabetes mellitus, heart failure, hypertension, and dyslipidemia. Patients who are on OAC therapy, including warfarin, receive comprehensive anticoagulation management from PC CPSs, including prescribing OAC therapy, education, dosage adjustments, and laboratory monitoring.
Patients were included in the HAS-BLED risk scoring and intervention if their warfarin therapy was managed by a PC CPS, had an active warfarin prescription with a diagnosis of nonvalvular AF in their problem list, and had ≥ 1 modifiable risk factor(s) from the HAS-BLED risk score. Modifiable risk factors evaluated were systolic blood pressure (SBP) > 160 mm Hg, an active prescription for VA or non-VA (which generally indicates over-the-counter [OTC] medication use) aspirin, clopidogrel, or a nonsteroidal anti-inflammatory drug (NSAID). Excess alcohol consumption was not listed as a modifiable risk factor in this assessment because nearly all the anticoagulation patients already receive regular recommendations to minimize alcohol use from the PC CPSs.
Patients were excluded from analysis if they had an indication for warfarin use other than nonvalvular AF, such as atrial flutter, acute/chronic deep vein thrombosis or pulmonary embolism, history of venous thromboembolism, peripheral vascular disease, or aortic or mitral mechanical valve. Patients also were excluded if they were on antiplatelet therapy for unstable coronary artery disease (CAD), experienced acute coronary syndrome within the past 1 year, history of stent placement, carotid endarterectomy, carotid stenosis, or noncardioembolic stroke and no other modifiable risk factors. Last, patients were excluded if clinic SBP readings were > 160 mm Hg but there was documented white coat hypertension or home SBP readings < 160 mm Hg.
The following definitions or measurements were used for assessing the HAS-BLED bleeding risk score4:
- Uncontrolled hypertension: most recently charted SBP > 160 mm Hg;
- Abnormal renal function: dialysis, renal transplant, serum creatinine > 2.26 mg/dL;
- Abnormal liver function: chronic hepatic disease, biochemical evidence of significant hepatic derangement (bilirubin > 2 × upper limit of normal and/or AST/ALT/alkaline phosphatase > 3 × upper limit of normal);
- Stroke: including history of transient ischemic attack;
- Bleeding history or predisposition: history of major bleeding (intracranial and/or any bleeding requiring hospitalization and/or causing a decrease in hemoglobin (Hgb) level of > 2 g/dL and/or requiring blood transfusion), anemia (males: Hgb < 13 g/dL; females: Hgb < 12 g/dL);
- Labile INR: percentage of INRs in therapeutic range < 60% (using the CJZVAMC anticoagulation management tool, which calculates percentage of INRs in goal reported since the first visit);
- Geriatric: age > 65 years at initial assessment;
- Concomitant drug use (VA prescription or non-VA medication list): aspirin, clopidogrel, or NSAID; and
- Alcohol in excess: > 8 alcohol servings per week from chart documentation of the patient’s self-report.
In the HAS-BLED bleeding risk score, patients receive 1 point for each component for a maximum score of 9 points. The score is stratified into low (0 points), intermediate (1 to 2 points), and high (≥ 3 points) bleeding risk.4
The HAS-BLED risk factors were obtained from patient chart review, including problem list, laboratory results, and PC CPS anticoagulation notes. Interventions included primary care provider (PCP) notification of elevated BP and offer of BP management by a PC CPS, patient education and/or PCP contact to discontinue concurrent NSAID or addition of a proton pump inhibitor (PPI) based on bleeding risk factor if the NSAID was deemed necessary, and discontinuation of concomitant antiplatelet drug(s) or reduction in aspirin dosage in consultation with patient’s PCP and cardiologist.9 In order to complete the initial HAS-BLED assessment and implement interventions, a note template was developed and entered into the electronic health record (EHR) that identified the patient’s modifiable risk factors.
Once the PCP and cardiologist (if applicable) responded to the note, by either accepting or declining the PC CPS recommendation(s), the HAS-BLED score was recalculated and recorded. If the provider did not respond to the initial note, an attempt was made to follow up at 3 months and at 6 months if necessary. If the provider did not respond at 6 months, the nonresponse was documented. For patients whose PCP requested PC CPS management of BP, the HAS-BLED score was recalculated 6 months after response from the PCP.
The primary outcome was the proportion of patients whose HAS-BLED score was reduced by at least 1 point. Secondary outcomes included the proportion of patients whose HAS-BLED score was reduced from one category of bleeding risk to a lesser one, total number of pharmacist interventions completed, number of pharmacist interventions made of each type (BP management, NSAID use, or antiplatelet drug use), and PCP acceptance rate.
Results
A total of 897 patients taking warfarin received anticoagulation management by a PC CPS at CJZVAMC in 2015. Of these, 819 patients were excluded based on the exclusion criteria (eFigure).
Seventy-eight patients were included in the assessment. Baseline HAS-BLED scores were calculated, and recommendations were made via an EHR progress note to the PCP and cardiologist (if applicable). Recommended interventions in the 78 patients resulted in 44 patients (56%) who experienced reduction in their HAS-BLED score by at least 1 point (Table 1). Twenty patients (25%) saw their HAS-BLED category reduced from a higher level of bleeding risk to a lower risk. The average HAS-BLED score in the 44 patients was 2.38 before intervention and 1.55 after the intervention.
In 10 patients, the HAS-BLED score did not decrease despite accepted PC CPS intervention. Specifically, 7 patients were on both an antiplatelet agent and NSAID. As a result of the pharmacist intervention, the NSAID was discontinued, but the antiplatelet remained because of stent placement or carotid stenosis. In 1 patient, the aspirin dosage was decreased from 325 to 81 mg/d. In 2 patients where NSAID use was deemed necessary—meloxicam in both cases—a PPI was ordered based on bleeding risk.9
A total of 82 interventions were recommended; 57 interventions were accepted, resulting in a provider acceptance rate of 69.5% (Tables 2 and 3). Thirty-five of the accepted interventions (61%) involved discontinuing an antiplatelet (aspirin or clopidogrel) in consultation with the patient’s PCP and cardiologist. Twenty-seven of these patients had no documented CAD, and 8 of the patients had stable CAD. Seventeen (30%) of the accepted recommendations were for discontinuing NSAID therapy, 2 (4%) were for BP management by a PC CPS, 2 (4%) for addition of PPI with continued NSAID use (meloxicam), and 1 (1%) for decreasing aspirin dosage from 325 to 81 mg/d. The NSAIDs that were discontinued included ibuprofen, indomethacin, meloxicam, and naproxen.
Discussion
This project is the first, to the authors’ knowledge, to evaluate pharmacist interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED bleeding risk score. Most of the patients with nonvalvular AF in the PC clinics did not have modifiable bleeding risk factors. However, of the patients who received a recommendation to reduce a specific modifiable risk factor, most of the interventions were accepted by PCPs and
Most of the interventions recommended evaluating the use of antiplatelet agents, particularly aspirin. The benefits of antiplatelet therapy for secondary prevention of cardiovascular disease are well established. However, for AF patients on OAC therapy, the concomitant use of antiplatelet therapy significantly increases the risk of bleeding and should be reserved for high-risk patients.10 The 2012 American College of Chest Physicians guidelines support the use of OAC monotherapy in patients with AF with stable CAD, including patients with a myocardial infarction or percutaneous coronary intervention more than 1 year previously, which has been corroborated with guideline and expert consensus recommendations released in 2016.7,8,10,11
For patients taking warfarin for AF without CAD, the possible benefit of concomitant aspirin therapy for primary prevention is outweighed by the increased risk of major bleeding.12 Furthermore, warfarin monotherapy has been shown to be effective in primary prevention of CAD and seems to have cardiovascular benefit for secondary prevention but with increased bleeding.13,14 As a result, through the exclusion criteria this project aimed to evaluate warfarin patients with AF at low risk of cardiovascular events who might be taking unnecessary concurrent antiplatelet therapy.
More than one-half of the interventions involved discontinuing antiplatelet therapy. Chart reviews revealed a lack of documentation for the indication and intended duration of antiplatelet therapy. In many patients, it is likely that aspirin use predated AF diagnosis and warfarin initiation. In some of these cases, it would have been appropriate to discontinue aspirin when starting warfarin use. Although there is guidance to support the use of OAC monotherapy in patients with AF with stable CAD, the patient’s provider and cardiologist made the decision to discontinue an antiplatelet agent after weighing benefits and risks. Regardless of the outcome, this analysis revealed the importance and need for routine review of antiplatelet therapy and documenting the rationale for antiplatelet use in addition to anticoagulation.
The second largest category of interventions accepted was for evaluation of NSAID use. A 2014 study by Lamberts and colleagues found that concomitant use of oral anticoagulants and NSAIDs conferred an independent risk for major bleeding and thromboembolism in patients with AF.15 The increase in serious bleeding (absolute risk difference of 2.5 events per 1,000 patients) was observed even with short-term NSAID exposure of 14 days across all NSAID types (selective COX-2 inhibitors or nonselective NSAIDs). In addition, there was an incremental increase in bleeding risk with high NSAID dosages. The risk of serious bleeding was even greater when an NSAID is added to OAC therapy and aspirin. Seven out of 17 warfarin patients (41%) who were taking an NSAID also were on an antiplatelet agent. As a result of the pharmacist interventions, NSAIDs were discontinued in all of these patients, but the antiplatelet remained because of stent placement or carotid stenosis.
This analysis captured only those patients with a documented active prescription or self-reported OTC use of an NSAID. It is unknown how many patients might take OTC NSAIDs occasionally but not report this use to a provider or pharmacist. Primary care CPSs educate patients to not use NSAIDs while taking warfarin during their initial visit and periodically thereafter; however, with the number of different NSAIDs available without a prescription and the various brand and generic names offered, it can be difficult for patients to understand what they should or should not take for minor pain or fever. Therefore, it is imperative that NSAID use is reviewed regularly at anticoagulant follow-up visits and patients are educated about alternative OTC agents for pain relief (eg, acetaminophen, topical agents, heating pad) when necessary. It also is equally important for PCPs to weigh the benefit vs risk for each patient before prescribing an NSAID if alternatives have been exhausted especially if the patient also is taking an OAC and antiplatelet agent.
The smallest number of interventions completed was for BP management. According to the HAS-BLED bleeding risk score, BP management was recommended only if the most recent clinic SBP was > 160 mm Hg, excluding patients with documented white-coat syndrome or home SBP readings < 160 mm Hg. One potential explanation for the small number of patients with SBP > 160 mm Hg is that for many of the patients taking warfarin, the PC CPSs at CJZVAMC have been involved in their BP management through earlier consultation by providers.
Limitations
A limitation of the BP component of the HAS-BLED score was that the assessment of BP for this project was only one point in time. In 3 cases, the SBP was > 160 mm Hg only during the most recent measurement, and these patients had normal BP readings on return to the clinic for follow-up. This category of recommendation also took more time for follow-up because a PC CPS would need to evaluate the patient in clinic, implement changes to BP medications, and follow-up at subsequent visits. Although some PCPs felt that the patient did not need pharmacist intervention, the elevated SBPs were brought to the provider’s attention, and some patients received further monitoring by the PCP or through a specialty clinic (eg, nephrology).
Another limitation of this project was that the bleeding risk evaluation occurred at only 1 visit. Patients’ medications and medical issues often change with time. Therefore, it is important to implement a process to regularly review (eg, annually) patients’ bleeding risk factors and to identify and act on modifiable risk factors. Another limitation was a lack of a comparator group and the time frame of the evaluation. As a result, the authors were unable to evaluate bleeding outcomes because of the small sample size and limited time frame. Future studies could consider evaluating bleeding events as an outcome, including additional modifiable risk factors, such as excess alcohol and labile INR, expanding the review to patients taking warfarin for indications other than AF, and review of patients on direct-acting oral anticoagulants (DOACs) with AF; keeping in mind that currently available bleeding risk calculators were developed for patients taking warfarin, not DOACs with AF. Patients could be counselled on reducing alcohol intake or switching to a DOAC if INR is labile despite adherence.
Conclusion
This quality improvement project successfully implemented use of the HAS-BLED bleeding risk score to identify and reduce modifiable bleeding risk factors in patients with AF taking warfarin. Pharmacist intervention resulted in a reduction of HAS-BLED scores and bleeding risk categories.
Click here to read the digital edition.
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia and is associated with a 5-fold increase in the risk of ischemic stroke and the risk increases with age.1-3 Oral anticoagulation (OAC) therapy effectively reduces the risk of ischemic stroke in patients with nonvalvular AF. However, OAC therapy carries a bleeding risk.4
Several bleeding risk scores have been developed and validated for patients with AF who are taking warfarin: HEMORR2HAGES (Hepatic or renal disease, Ethanol abuse, Malignancy, Older age, Reduced platelet count or function, Re-bleeding, Hypertension, Anemia, Genetic factors, Excessive fall risk, Stroke), ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation), and HAS-BLED (Hypertension, Abnormal renal and/or liver function, Stroke, Bleeding history or predisposition, Labile international normalized ratio [INR], Elderly, and Drugs and/or alcohol excess concomitantly).4,5 All 3 bleeding risk scores demonstrate only modest ability to predict clinically relevant bleeding in patients taking warfarin. The HAS-BLED score was superior to HEMORR2HAGES and ATRIA for predicting any clinically relevant bleeding and was the only bleeding risk score that demonstrated significant predictive performance for intracranial hemorrhage.5 Compared with HEMORR2HAGES, the HAS-BLED score is simpler to use and to assess risk factors that can be gathered from medical history or routinely tested in patients with AF.4 Unlike HAS-BLED, HEMORR2HAGES and ATRIA do not consider medications that could increase the risk of bleeding.
Despite the availability of validated bleeding risk scores, clinical application of these measures should not be used to exclude a patient from OAC therapy for patients who reach a threshold score. Rather, current guideline and expert consensus agree with the recommendation to use bleeding risk scores to identify risk factors and address those factors that are modifiable to reduce the risk of anticoagulant-associated major bleeding.6-8
The authors identified modifiable bleeding risk factors using the HAS-BLED score and evaluated pharmacist interventions to correct these factors in patients with nonvalvular AF who are taking warfarin. To the authors’ knowledge, there have been no published studies evaluating interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED score.
Methods
Clinical pharmacy specialists (CPSs) in the primary care (PC) clinics at the Clement J. Zablocki VAMC (CJZVAMC) in Milwaukee, Wisconsin, have prescriptive authority within their scope of practice to manage smoking cessation and diseases, including anticoagulation, diabetes mellitus, heart failure, hypertension, and dyslipidemia. Patients who are on OAC therapy, including warfarin, receive comprehensive anticoagulation management from PC CPSs, including prescribing OAC therapy, education, dosage adjustments, and laboratory monitoring.
Patients were included in the HAS-BLED risk scoring and intervention if their warfarin therapy was managed by a PC CPS, had an active warfarin prescription with a diagnosis of nonvalvular AF in their problem list, and had ≥ 1 modifiable risk factor(s) from the HAS-BLED risk score. Modifiable risk factors evaluated were systolic blood pressure (SBP) > 160 mm Hg, an active prescription for VA or non-VA (which generally indicates over-the-counter [OTC] medication use) aspirin, clopidogrel, or a nonsteroidal anti-inflammatory drug (NSAID). Excess alcohol consumption was not listed as a modifiable risk factor in this assessment because nearly all the anticoagulation patients already receive regular recommendations to minimize alcohol use from the PC CPSs.
Patients were excluded from analysis if they had an indication for warfarin use other than nonvalvular AF, such as atrial flutter, acute/chronic deep vein thrombosis or pulmonary embolism, history of venous thromboembolism, peripheral vascular disease, or aortic or mitral mechanical valve. Patients also were excluded if they were on antiplatelet therapy for unstable coronary artery disease (CAD), experienced acute coronary syndrome within the past 1 year, history of stent placement, carotid endarterectomy, carotid stenosis, or noncardioembolic stroke and no other modifiable risk factors. Last, patients were excluded if clinic SBP readings were > 160 mm Hg but there was documented white coat hypertension or home SBP readings < 160 mm Hg.
The following definitions or measurements were used for assessing the HAS-BLED bleeding risk score4:
- Uncontrolled hypertension: most recently charted SBP > 160 mm Hg;
- Abnormal renal function: dialysis, renal transplant, serum creatinine > 2.26 mg/dL;
- Abnormal liver function: chronic hepatic disease, biochemical evidence of significant hepatic derangement (bilirubin > 2 × upper limit of normal and/or AST/ALT/alkaline phosphatase > 3 × upper limit of normal);
- Stroke: including history of transient ischemic attack;
- Bleeding history or predisposition: history of major bleeding (intracranial and/or any bleeding requiring hospitalization and/or causing a decrease in hemoglobin (Hgb) level of > 2 g/dL and/or requiring blood transfusion), anemia (males: Hgb < 13 g/dL; females: Hgb < 12 g/dL);
- Labile INR: percentage of INRs in therapeutic range < 60% (using the CJZVAMC anticoagulation management tool, which calculates percentage of INRs in goal reported since the first visit);
- Geriatric: age > 65 years at initial assessment;
- Concomitant drug use (VA prescription or non-VA medication list): aspirin, clopidogrel, or NSAID; and
- Alcohol in excess: > 8 alcohol servings per week from chart documentation of the patient’s self-report.
In the HAS-BLED bleeding risk score, patients receive 1 point for each component for a maximum score of 9 points. The score is stratified into low (0 points), intermediate (1 to 2 points), and high (≥ 3 points) bleeding risk.4
The HAS-BLED risk factors were obtained from patient chart review, including problem list, laboratory results, and PC CPS anticoagulation notes. Interventions included primary care provider (PCP) notification of elevated BP and offer of BP management by a PC CPS, patient education and/or PCP contact to discontinue concurrent NSAID or addition of a proton pump inhibitor (PPI) based on bleeding risk factor if the NSAID was deemed necessary, and discontinuation of concomitant antiplatelet drug(s) or reduction in aspirin dosage in consultation with patient’s PCP and cardiologist.9 In order to complete the initial HAS-BLED assessment and implement interventions, a note template was developed and entered into the electronic health record (EHR) that identified the patient’s modifiable risk factors.
Once the PCP and cardiologist (if applicable) responded to the note, by either accepting or declining the PC CPS recommendation(s), the HAS-BLED score was recalculated and recorded. If the provider did not respond to the initial note, an attempt was made to follow up at 3 months and at 6 months if necessary. If the provider did not respond at 6 months, the nonresponse was documented. For patients whose PCP requested PC CPS management of BP, the HAS-BLED score was recalculated 6 months after response from the PCP.
The primary outcome was the proportion of patients whose HAS-BLED score was reduced by at least 1 point. Secondary outcomes included the proportion of patients whose HAS-BLED score was reduced from one category of bleeding risk to a lesser one, total number of pharmacist interventions completed, number of pharmacist interventions made of each type (BP management, NSAID use, or antiplatelet drug use), and PCP acceptance rate.
Results
A total of 897 patients taking warfarin received anticoagulation management by a PC CPS at CJZVAMC in 2015. Of these, 819 patients were excluded based on the exclusion criteria (eFigure).
Seventy-eight patients were included in the assessment. Baseline HAS-BLED scores were calculated, and recommendations were made via an EHR progress note to the PCP and cardiologist (if applicable). Recommended interventions in the 78 patients resulted in 44 patients (56%) who experienced reduction in their HAS-BLED score by at least 1 point (Table 1). Twenty patients (25%) saw their HAS-BLED category reduced from a higher level of bleeding risk to a lower risk. The average HAS-BLED score in the 44 patients was 2.38 before intervention and 1.55 after the intervention.
In 10 patients, the HAS-BLED score did not decrease despite accepted PC CPS intervention. Specifically, 7 patients were on both an antiplatelet agent and NSAID. As a result of the pharmacist intervention, the NSAID was discontinued, but the antiplatelet remained because of stent placement or carotid stenosis. In 1 patient, the aspirin dosage was decreased from 325 to 81 mg/d. In 2 patients where NSAID use was deemed necessary—meloxicam in both cases—a PPI was ordered based on bleeding risk.9
A total of 82 interventions were recommended; 57 interventions were accepted, resulting in a provider acceptance rate of 69.5% (Tables 2 and 3). Thirty-five of the accepted interventions (61%) involved discontinuing an antiplatelet (aspirin or clopidogrel) in consultation with the patient’s PCP and cardiologist. Twenty-seven of these patients had no documented CAD, and 8 of the patients had stable CAD. Seventeen (30%) of the accepted recommendations were for discontinuing NSAID therapy, 2 (4%) were for BP management by a PC CPS, 2 (4%) for addition of PPI with continued NSAID use (meloxicam), and 1 (1%) for decreasing aspirin dosage from 325 to 81 mg/d. The NSAIDs that were discontinued included ibuprofen, indomethacin, meloxicam, and naproxen.
Discussion
This project is the first, to the authors’ knowledge, to evaluate pharmacist interventions to reduce modifiable bleeding risk factors identified by the HAS-BLED bleeding risk score. Most of the patients with nonvalvular AF in the PC clinics did not have modifiable bleeding risk factors. However, of the patients who received a recommendation to reduce a specific modifiable risk factor, most of the interventions were accepted by PCPs and
Most of the interventions recommended evaluating the use of antiplatelet agents, particularly aspirin. The benefits of antiplatelet therapy for secondary prevention of cardiovascular disease are well established. However, for AF patients on OAC therapy, the concomitant use of antiplatelet therapy significantly increases the risk of bleeding and should be reserved for high-risk patients.10 The 2012 American College of Chest Physicians guidelines support the use of OAC monotherapy in patients with AF with stable CAD, including patients with a myocardial infarction or percutaneous coronary intervention more than 1 year previously, which has been corroborated with guideline and expert consensus recommendations released in 2016.7,8,10,11
For patients taking warfarin for AF without CAD, the possible benefit of concomitant aspirin therapy for primary prevention is outweighed by the increased risk of major bleeding.12 Furthermore, warfarin monotherapy has been shown to be effective in primary prevention of CAD and seems to have cardiovascular benefit for secondary prevention but with increased bleeding.13,14 As a result, through the exclusion criteria this project aimed to evaluate warfarin patients with AF at low risk of cardiovascular events who might be taking unnecessary concurrent antiplatelet therapy.
More than one-half of the interventions involved discontinuing antiplatelet therapy. Chart reviews revealed a lack of documentation for the indication and intended duration of antiplatelet therapy. In many patients, it is likely that aspirin use predated AF diagnosis and warfarin initiation. In some of these cases, it would have been appropriate to discontinue aspirin when starting warfarin use. Although there is guidance to support the use of OAC monotherapy in patients with AF with stable CAD, the patient’s provider and cardiologist made the decision to discontinue an antiplatelet agent after weighing benefits and risks. Regardless of the outcome, this analysis revealed the importance and need for routine review of antiplatelet therapy and documenting the rationale for antiplatelet use in addition to anticoagulation.
The second largest category of interventions accepted was for evaluation of NSAID use. A 2014 study by Lamberts and colleagues found that concomitant use of oral anticoagulants and NSAIDs conferred an independent risk for major bleeding and thromboembolism in patients with AF.15 The increase in serious bleeding (absolute risk difference of 2.5 events per 1,000 patients) was observed even with short-term NSAID exposure of 14 days across all NSAID types (selective COX-2 inhibitors or nonselective NSAIDs). In addition, there was an incremental increase in bleeding risk with high NSAID dosages. The risk of serious bleeding was even greater when an NSAID is added to OAC therapy and aspirin. Seven out of 17 warfarin patients (41%) who were taking an NSAID also were on an antiplatelet agent. As a result of the pharmacist interventions, NSAIDs were discontinued in all of these patients, but the antiplatelet remained because of stent placement or carotid stenosis.
This analysis captured only those patients with a documented active prescription or self-reported OTC use of an NSAID. It is unknown how many patients might take OTC NSAIDs occasionally but not report this use to a provider or pharmacist. Primary care CPSs educate patients to not use NSAIDs while taking warfarin during their initial visit and periodically thereafter; however, with the number of different NSAIDs available without a prescription and the various brand and generic names offered, it can be difficult for patients to understand what they should or should not take for minor pain or fever. Therefore, it is imperative that NSAID use is reviewed regularly at anticoagulant follow-up visits and patients are educated about alternative OTC agents for pain relief (eg, acetaminophen, topical agents, heating pad) when necessary. It also is equally important for PCPs to weigh the benefit vs risk for each patient before prescribing an NSAID if alternatives have been exhausted especially if the patient also is taking an OAC and antiplatelet agent.
The smallest number of interventions completed was for BP management. According to the HAS-BLED bleeding risk score, BP management was recommended only if the most recent clinic SBP was > 160 mm Hg, excluding patients with documented white-coat syndrome or home SBP readings < 160 mm Hg. One potential explanation for the small number of patients with SBP > 160 mm Hg is that for many of the patients taking warfarin, the PC CPSs at CJZVAMC have been involved in their BP management through earlier consultation by providers.
Limitations
A limitation of the BP component of the HAS-BLED score was that the assessment of BP for this project was only one point in time. In 3 cases, the SBP was > 160 mm Hg only during the most recent measurement, and these patients had normal BP readings on return to the clinic for follow-up. This category of recommendation also took more time for follow-up because a PC CPS would need to evaluate the patient in clinic, implement changes to BP medications, and follow-up at subsequent visits. Although some PCPs felt that the patient did not need pharmacist intervention, the elevated SBPs were brought to the provider’s attention, and some patients received further monitoring by the PCP or through a specialty clinic (eg, nephrology).
Another limitation of this project was that the bleeding risk evaluation occurred at only 1 visit. Patients’ medications and medical issues often change with time. Therefore, it is important to implement a process to regularly review (eg, annually) patients’ bleeding risk factors and to identify and act on modifiable risk factors. Another limitation was a lack of a comparator group and the time frame of the evaluation. As a result, the authors were unable to evaluate bleeding outcomes because of the small sample size and limited time frame. Future studies could consider evaluating bleeding events as an outcome, including additional modifiable risk factors, such as excess alcohol and labile INR, expanding the review to patients taking warfarin for indications other than AF, and review of patients on direct-acting oral anticoagulants (DOACs) with AF; keeping in mind that currently available bleeding risk calculators were developed for patients taking warfarin, not DOACs with AF. Patients could be counselled on reducing alcohol intake or switching to a DOAC if INR is labile despite adherence.
Conclusion
This quality improvement project successfully implemented use of the HAS-BLED bleeding risk score to identify and reduce modifiable bleeding risk factors in patients with AF taking warfarin. Pharmacist intervention resulted in a reduction of HAS-BLED scores and bleeding risk categories.
Click here to read the digital edition.
1. Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110(9):1042-1046.
2. Patel NJ, Deshmukh A, Pant S, et al. Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation. 2014;129(23):2371-2379.
3. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983-988.
4. Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100.
5. Apostolakis S, Lane DA, Guo Y, Buller H, Lip GY. Performance of the HEMORR2HAGES, ATRIA, and HAS-BLED bleeding risk-prediction scores in patients with atrial fibrillation undergoing anticoagulation: the AMADEUS (evaluating the use of SR34006 compared to warfarin or acenocoumarol in patients with atrial fibrillation) study. J Am Coll Cardiol. 2012;60(9):861-867.
6. Lane DA, Lip GY. Use of the CHA(2)DS(2)-VASc and HAS-BLED scores to aid decision making for thromboprophylaxis in nonvalvular atrial fibrillation. Circulation. 2012;126(7):860-865.
7. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962.
8. Ruff CT, Ansell JE, Becker RC, et al. North American thrombosis forum, AF action initiative consensus document. Am J Med. 2016;129(suppl 5):S1-S29.
9. Lanza FL, Chan FK, Quigley EM; Practice Parameters Committee of the American College of Gastroenterology. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104(3):728-738.
10. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation. Antithrombotic therapy and prevention of thrombosis, 9th ed. American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):e531S-e575S.
11. Macle L, Cairns J, Leblanc K, et al; CCS Atrial Fibrillation Guidelines Committee. 2016 focused update of the Canadian Cardiovascular Society guidelines for the management of atrial fibrillation. Can J Cardiol. 2016;32(10):1170-1185.
12. Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med. 2007;167(2):117-124.
13. The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet. 1998;351(9098):233-241.
14. Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
15. Lamberts M, Lip GY, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698.
1. Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation. 2004;110(9):1042-1046.
2. Patel NJ, Deshmukh A, Pant S, et al. Contemporary trends of hospitalization for atrial fibrillation in the United States, 2000 through 2010: implications for healthcare planning. Circulation. 2014;129(23):2371-2379.
3. Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983-988.
4. Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093-1100.
5. Apostolakis S, Lane DA, Guo Y, Buller H, Lip GY. Performance of the HEMORR2HAGES, ATRIA, and HAS-BLED bleeding risk-prediction scores in patients with atrial fibrillation undergoing anticoagulation: the AMADEUS (evaluating the use of SR34006 compared to warfarin or acenocoumarol in patients with atrial fibrillation) study. J Am Coll Cardiol. 2012;60(9):861-867.
6. Lane DA, Lip GY. Use of the CHA(2)DS(2)-VASc and HAS-BLED scores to aid decision making for thromboprophylaxis in nonvalvular atrial fibrillation. Circulation. 2012;126(7):860-865.
7. Kirchhof P, Benussi S, Kotecha D, et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893-2962.
8. Ruff CT, Ansell JE, Becker RC, et al. North American thrombosis forum, AF action initiative consensus document. Am J Med. 2016;129(suppl 5):S1-S29.
9. Lanza FL, Chan FK, Quigley EM; Practice Parameters Committee of the American College of Gastroenterology. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104(3):728-738.
10. You JJ, Singer DE, Howard PA, et al. Antithrombotic therapy for atrial fibrillation. Antithrombotic therapy and prevention of thrombosis, 9th ed. American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2012;141(suppl 2):e531S-e575S.
11. Macle L, Cairns J, Leblanc K, et al; CCS Atrial Fibrillation Guidelines Committee. 2016 focused update of the Canadian Cardiovascular Society guidelines for the management of atrial fibrillation. Can J Cardiol. 2016;32(10):1170-1185.
12. Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med. 2007;167(2):117-124.
13. The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet. 1998;351(9098):233-241.
14. Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med. 2002;347(13):969-974.
15. Lamberts M, Lip GY, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698.
Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) Polypharmacy Clinic
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time. 
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit. 
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
An interprofessional polypharmacy clinic for intensive management of medication regimens helps high-risk patients manage their medications.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time.
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit.
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
In 2011, 5 VA medical centers (VAMCs) were selected by the Office of Academic Affiliations (OAA) to establish CoEPCE. Part of the VA New Models of Care initiative, the 5 Centers of Excellence (CoE) in Boise, Idaho; Cleveland, Ohio; San Francisco, California; Seattle, Washington; and West Haven, Connecticut, are utilizing VA primary care settings to develop and test innovative approaches to prepare physician residents and students, advanced practice nurse residents and undergraduate nursing students, and other professions of health trainees (eg, pharmacy, social work, psychology, physician assistants [PAs], physical therapists) for primary care practice in the 21st century. The CoEs are developing, implementing, and evaluating curricula designed to prepare learners from relevant professions to practice in patient-centered, interprofessional team-based primary care settings. The curricula at all CoEs must address 4 core domains (Table).
Health care professional education programs do not have many opportunities for workplace learning where trainees from different professions can learn and work together to provide care to patients in real time.
The VA Connecticut Healthcare System CoEPCE developed and implemented an education and practice-based immersion learning model with physician residents, nurse practitioner (NP) residents and NP students, pharmacy residents, postdoctorate psychology learners, and PA and physical therapy learners and faculty. This interprofessional, collaborative team model breaks from the traditional independent model of siloed primary care providers (PCPs) caring for a panel of patients.
Methods
In 2015, OAA evaluators reviewed background documents and conducted open-ended interviews with 12 West Haven CoEPCE staff, participating trainees, VA faculty, VA facility leadership, and affiliate faculty. Informants described their involvement, challenges encountered, and benefits of the Initiative to Minimize Pharmaceutical Risk in Older Veterans (IMPROVE) program to trainees, veterans, and the VA.
Lack of Clinical Approaches to Interprofessional Education and Care
Polypharmacy is a common problem among older adults with multiple chronic conditions, which places patients at higher risk for multiple negative health outcomes.2,3 The typical primary care visit rarely allows for a thorough review of a patient’s medications, much less the identification of strategies to reduce polypharmacy and improve medication management. Rather, the complexity inherent to polypharmacy makes it an ideal challenge for a team-based approach.
Team Approach to Medication Needs
A key CoEPCE program aim is to expand workplace learning instruction strategies and to create more clinical opportunities for CoEPCE trainees to work together as a team to anticipate and address the health care needs of veterans. To address this training need, the West Haven CoEPCE developed IMPROVE to focus on high-need patients and provides a venue in which trainees and supervisors from different professions can collaborate on a specific patient case, using a patient-centered framework. IMPROVE can be easily applied to a range of medication-related aims, such as reducing medications, managing medications and adherence, and addressing adverse effects (AEs). These goals are 2-fold: (1) implement a trainee-led performance improvement project that reduces polypharmacy in elderly veterans; and (2) develop a hands-on, experiential geriatrics training program that enhances trainee skills and knowledge related to safe prescribing.
Planning and Implementation
IMPROVE has its origins in a scholarly project developed by a West Haven CoE physician resident trainee. Development of the IMPROVE program involved VA health psychology, internal medicine faculty, geriatric medicine faculty, NP faculty, and geriatric pharmacy residents and faculty. Planning started in 2013 with a series of pilot clinics and became an official project of the West Haven CoE in September 2014. The intervention required no change in West Haven VAMC policy. However, the initiative required buy-in from West Haven CoE leadership and the director of the West Haven primary care clinic.
Curriculum
IMPROVE is an educational, workplace learning, and clinical activity that combines a 1-hour trainee teaching session, a 45-minute group visit, and a 60-minute individual clinic visit to address the complex problem of polypharmacy. It emphasizes the sharing of trainee and faculty backgrounds by serving as a venue for interprofessional trainees and providers to discuss pharmacologic and nonpharmacologic treatment in the elderly and brainstorm strategies to optimize treatment regimens, minimize risk, and execute medication plans with patients.
All CoEPCE trainees in West Haven are required to participate in IMPROVE and on average, each trainee presents and sees one of their patients at least 3 times per year in the program. Up to 5 trainees participate in each IMPROVE session. Trainees are responsible for reviewing their panels to identify patients who might benefit from participation, followed by inviting the patient to participate. Patients are instructed to bring their pill bottles to the visit. To prepare for the polypharmacy clinic, the trainees, the geriatrician, and the geriatric pharmacist perform an extensive medication chart review, using the medication review worksheet developed by West Haven VAMC providers.4 They also work with a protocol for medication discontinuation, which was compiled by West Haven VAMC clinicians. The teams use a variety of tools that guide appropriate prescribing in older adult populations.5,6 During a preclinic conference, trainees present their patients to the interprofessional team for discussion and participate in a short discussion led by a pharmacist, geriatrician, or health psychologist on a topic related to prescribing safety in older adults or nonpharmacologic treatments.
IMPROVE emphasizes a patient-centered approach to develop, execute, and monitor medication plans. Patients and their family members are invited by their trainee clinician to participate in a group visit. Typically, trainees invite patients aged ≥ 65 years who have ≥ 10 medications and are considered appropriate for a group visit.
The group visit process is based on health psychology strategies, which often incorporate group-based engagement with patients. The health psychologist can give advice to facilitate the visit and optimize participant involvement. There is a discussion facilitator guide that lists the education points to be covered by a designated trainee facilitator and sample questions to guide the discussion.7 A health psychology resident and other rotating trainees cofacilitate the group visit with a goal to reach out to each group member, including family members, and have them discuss perceptions and share concerns and treatment goals. There is shared responsibility among the trainees to address the educational material as well as involve their respective patients during the sessions.
Immediately following the group visit, trainees conduct a 1-hour clinic session that includes medication reconciliation, a review of an IMPROVE questionnaire, orthostatic vital signs, and the St. Louis University Mental Status (SLUMS) exam to assess changes in cognition.7,8 Discussion involved the patient’s medication list as well as possible changes that could be made to the list. Using shared decision-making techniques, this conversation considers the patients’ treatment goals, feelings about the medications, which medications they would like to stop, and AEs they may be experiencing. After the individual visit is completed, the trainee participates in a 10-minute interprofessional precepting session, which may include a geriatrician, a pharmacist, and a health psychologist. In the session they may discuss adjustments to medications and a safe follow-up plan, including appropriate referrals. Trainees discuss the plan with the patient and send a letter describing the plan shortly after the visit.
IMPROVE combines didactic teaching with experiential education. It embodies the 4 core domains that shape the CoEPCE curriculum. First, trainees learn interprofessional collaboration concepts, including highlighting the roles of each profession and working with an interprofessional team to solve problems. Second, CoEPCE trainees learn performance improvement under the supervision of faculty. Third, IMPROVE allows trainees to develop sustained relationships with other team members while improving the quality of the clinic experience as well as with patients through increased continuity of care. Trainees see patients on their panel and are responsible for outreach before and after the visit. Finally, with a focus on personalized patient goals, trainees have the opportunity to further develop skills in shared decision making (SDM).
Related: Reducing Benzodiazepine Prescribing in Older Veterans: A Direct-to-Consumer Educational Brochure
The IMPROVE model continues to evolve. The original curriculum involved an hour-long preclinic preparation session before the group visit in which trainees and faculty discussed the medication review for each patient scheduled that day. This preparation session was later shortened to 40 minutes, and a 20-minute didactic component was added to create the current preclinic session. The didactic component focused on a specific topic in appropriate prescribing for older patients. For example, one didactic lesson is on a particular class of medications, its common AEs, and practical prescribing and “deprescribing” strategies for that class. Initially, the oldest patients or patients who could be grouped thematically, such as those taking both narcotics and benzodiazepines, were invited to participate, but that limited the number of appropriate patients within the CoEPCE. Currently, trainees identify patients from their panels who might benefit, based on age, number of medications, or potential medication-related concerns, such as falls, cognitive impairment, or other concerns for adverse drug effects. These trainees have the unique opportunity to apply learned strategies to their patients to continue to optimize the medication regimen even after the IMPROVE visit. Another significant change was the inclusion of veterans who are comanaged with PCPs outside the VA, because we found that patients with multiple providers could benefit from improved coordination of care.
Faculty Role
CoE faculty and non-CoE VA faculty participate in supervisory, consulting, teaching and precepting roles. Some faculty members such as the health psychologists are already located in or near the VA primary care clinic, so they can assist in curriculum development and execution during their regular clinic duties. The geriatrician reviews the patients’ health records before the patients come into the clinic, participates in the group visit, and coprecepts during the 1:1 patient visits. Collaboration is inherent in IMPROVE. For example, the geriatrician works with the geriatric pharmacist to identify and teach an educational topic. IMPROVE is characterized by a strong faculty/trainee partnership, with trainees playing roles as both teacher and facilitator in addition to learning how to take a team approach to polypharmacy.
Resources
IMPROVE requires administrative and academic support, especially faculty and trainee preparation of education sessions. The CoEPCE internal medicine resident and the internal medicine chief resident work with the health technicians for each patient aligned care team (PACT) to enter the information into the VA medical scheduling system. Trainee clinic time is blocked for their group visits in advance. Patients are scheduled 1 to 3 weeks in advance. Trainees and faculty are expected to review the medication review worksheet and resources prior to the visit. One CoEPCE faculty member reviews patients prior to the preclinic session (about an hour of preparation per session). Sufficient space also is required: a room large enough to accommodate up to 10 people for both didactic lessons and preclinic sessions, a facility patient education conference room for the group visit, and up to 5 clinic exam rooms. CoEPCE staff developed a templated note in the VA Computerized Patient Record System (CPRS), the VA electronic health record system to guide trainees step-by-step through the clinic visit and allow them to directly enter information into the system.7
Monitoring and Assessment
CoEPCE staff are evaluating IMPROVE by building a database for patient-level and trainee-level outcomes, including changes in trainee knowledge and attitudes over time. The CoEPCE also validated the polypharmacy knowledge assessment tool for medicine and NP trainees.
Partnerships
IMPROVE has greatly benefited from partnerships with facility department leadership, particularly involvement of pharmacy staff. In addition, we have partnered with both the health psychology and pharmacy faculty and trainees to participate in the program. Geriatrics faculty and trainees also have contributed extensively to IMPROVE. Future goals include offering the program to non-COEPCE patients throughout primary care.
The Yale Primary Care Internal Medicine Residency program and the Yale Categorical Internal Medicine Residency Program are integral partners to the CoEPCE. IMPROVE supports their mandate to encourage interprofessional teamwork in primary care, meet the Accreditation Council for Graduate Medical Education interprofessional milestones, and promote individual trainee scholarship and performance improvement in areas of broad applicability. IMPROVE also is an opportunity to share ideas across institutions and stimulate new collaborations and dissemination of the model to other primary care settings outside the VA.
Challenges and Solutions
The demand for increased direct patient care pressures programs like IMPROVE, which is a time-intensive process with high impact on a few complex patients. The assumption is that managing medications will save money in the long run, but in the short-term, a strong case has to be made for securing resources, particularly blocking provider time and securing an education room for group visits and clinic exam rooms for individual visits. First, decision makers need to be convinced that polypharmacy is important and should be a training priority. The CoEPCE has tried different configurations to increase the number of patients being seen, such as having ≥ 1 IMPROVE session in an afternoon, but trainees found this to be labor intensive and stressful.
Second, patients with medications prescribed by providers outside the VA require additional communication and coordination to reduce medications. The CoEPCE initially excluded these patients, but after realizing that some of these patients needed the most help, it developed a process for reaching out to non-VA providers and coordinating care. Additionally, there is significant diversity in patient polypharmacy needs. These can range from adherence problems to the challenge of complex psychosocial needs that are more easily (but less effectively) addressed with medications. The issue of polypharmacy is further complicated by evolving understanding of medications’ relative risks and benefits in older adults with multiple chronic conditions. IMPROVE is an effective vehicle for synthesizing current science in medications and their management, especially in complex older patients with multiple chronic conditions.
Other challenges include developing a templated CPRS electronic note that interfaces with the VA information technology system. The process of creating a template, obtaining approval from the forms committee, and working with information technology personnel to implement the template was more time intensive than anticipated and required multiple iterations of proofreading and editing.
Factors for Success
The commitment to support new models of trainee education by West Haven CoEPCE faculty and leadership, and West Haven VAMC and primary care clinic leadership facilitated the implementation of IMPROVE. Additionally, there is strong CoEPCE collaboration at all levels—codirectors, faculty, and trainees—for the program. High interprofessional trainee interest, organizational insight, and an academic orientation were critical for developing and launching IMPROVE.
Additionally, there is synergy with other team-based professions. Geriatrics has a tradition of working in multidisciplinary teams as well as working with SDM concepts as part of care discussions. High interest and collaboration by a geriatrician and an experienced geriatric pharmacist has been key. The 2 specialties complement each other and address the complex health needs of participating veterans. Health psychologists transition patients to nonpharmacologic treatments, such as sleep hygiene education and cognitive behavioral therapy, in addition to exploring barriers to behavior change.
Another factor for success has been the CoEPCE framework and expertise in interprofessional education. While refining the model, program planners tapped into existing expertise in polypharmacy within the VA from the geriatrics, pharmacy, and clinical health psychology departments. The success of the individual components—the preparation session, the group visit, and the 1:1 patient visit—is in large part the result of a collective effort by CoEPCE staff and the integration of CoEPCE staff through coordination, communications, logistics, quality improvement, and faculty involvement from multiple professions.
The IMPROVE model is flexible and can accommodate diverse patient interests and issues. Model components are based on sound practices that have demonstrated success in other arenas, such as diabetes mellitus group visits. The model can also accommodate diverse trainee levels. Senior trainees can be more independent in developing their care plans, teaching the didactic topic, or precepting during the 1:1 patient exam.
Accomplishments and Benefits
Trainees are using team skills to provide patient-centered care. They are strengthening their clinical skills through exposure to patients in a group visit and 1:1 clinic visit. There have been significant improvements in the trainees’ provision of individual patient care. Key IMPROVE outcomes are outlined below.
Interprofessional Education
Unlike a traditional didactic, IMPROVE is an opportunity for health care professionals to work together to provide care in a clinic setting. It also expands CoEPCE interprofessional education capacity through colocation of different trainee and faculty professions during the conference session. This combination trains participants to work as a team and reflect on patients together, which has strengthened communications among professions. The model provides sufficient time and expertise to discuss the medications in detail and as a team, something that would not normally happen during a regular primary care visit.
CoEPCE trainees learn about medication management, its importance in preventing complications and improving patient health outcomes. Trainees of all professions learn to translate the skills they learn in IMPROVE to other patients, such as how to perform a complete medication reconciliation or lead a discussion using SDM. IMPROVE also provides techniques useful in other contexts, such as group visits and consideration of different medication options for patients who have been cared for by other (VA and non-VA) providers.
Interprofessional Collaboration
Understanding and leveraging the expertise of trainees and faculty from different professions is a primary goal of IMPROVE. Education sessions, the group visit, and precepting model are intentionally designed to break down silos and foster a team approach to care, which supports the PACT team model. Trainees and faculty all have their unique strengths and look at the issue from a different perspective, which increases the likelihood that the patient will hear a cohesive solution or strategy. The result is that trainees are more well rounded and become better practitioners who seek advice from other professions and work well in teams.
Trainees are expected to learn about other professions and their skill sets. For example, trainees learn early about the roles and scopes of practice of pharmacists and health psychologists for more effective referrals. Discussions during the session before the group visit may bring conditions like depression or dementia to the trainees’ attention. This is significant because issues like patient motivation may be better handled from a behavioral perspective.
Expanded Clinical Performance
IMPROVE is an opportunity for CoEPCE trainees to expand their clinical expertise. It provides exposure to a variety of patients and patient care needs and is an opportunity to present a high-risk patient to colleagues of various professions. As of December 2015, about 30 internal medicine residents and 6 NP residents have seen patients in the polypharmacy clinic. Each year, 4 NP residents, 2 health psychology residents, 4 clinical pharmacy residents, and 1 geriatric pharmacy resident participate in the IMPROVE clinic during their yearlong training program. During their 3-year training program, 17 to 19 internal medicine residents participate in IMPROVE.
A structured forum for discussing patients and their care options supports professionals’ utilization of the full scope of their practice. Trainees learn and apply team skills, such as communication and the warm handoff, which can be used in other clinic settings. A warm handoff is often described as an intervention in which “a clinician directly introduces a patient to another clinician at the time of the patient’s visit and often a brief encounter between the patient and the health care professional occurs.”9 An interprofessional care plan supports trainee clinical performance, providing a more robust approach to patient care than individual providers might on their own.
Patient Outcomes
IMPROVE is an enriched care plan informed by multiple professions with the potential to improve medication use and provide better care. Veterans also are receiving better medication education as well as access to a health psychologist who can help them with goal setting and effective behavioral interventions. On average, 5 patients participate each month. As of December 2015, 68 patients have participated in IMPROVE.
The group visit and the 1:1 patient visits focus exclusively on medication issues and solutions, which would be less common in a typical primary care visit with a complex patient who brings a list of agenda items. In addition to taking a thorough look at their medications and related problems, it also educates patients on related issues such as sleep hygiene. Participating veterans also are encouraged to share their concerns, experiences, and solutions with the group, which may increase the saliency of the message beyond what is offered in counseling from a provider.
To date, preliminary data suggest that in some patients, cognition (as measured with SLUMS after 6 months) has modestly improved after decreasing their medications. Other outcomes being monitored in follow-up are utilization of care, reported history of falls, number of medications, and vital signs at initial and follow-up visits.
Patients experience increased continuity of care because the patient now has a team focusing on his or her care. Team members have a shared understanding of the patient’s situation and are better able to establish therapeutic rapport with patients during the group visit. Moreover, CoEPCE trainees and faculty try to ensure that everyone knows about and concurs with medication changes, including outside providers and family members.
Satisfaction Questionnaire
Patients that are presented at IMPROVE can be particularly challenging, and there may be a psychological benefit to working with a team to develop a new care plan. Providers are able to get input and look at the patient in a new light.
Results of postvisit patient satisfaction questionnaires are encouraging and result in a high level of patient satisfaction and perception of clinical benefit. Patients identify an improvement in the understanding of their medications, feel they are able to safely decrease their medications, and are interested in participating again.
CoEPCE Benefits
IMPROVE expands the prevention and treatment options for populations at risk of hospitalization and adverse outcomes from medication complications, such as AEs and drug-drug or drug-disease interactions. Embedding the polypharmacy clinic within the primary care setting rather than in a separate specialty clinic results in an increased likelihood of implementation of pharmacist and geriatrician recommendations for polypharmacy and allows for direct interprofessional education and collaboration.
IMPROVE also combines key components of interprofessional education—an enriched clinical training model and knowledge of medications in an elderly population—into a training activity that complements other CoEPCE activities. The model not only has strengthened CoEPCE partnerships with other VA departments and specialties, but also revealed opportunities for collaboration with academic affiliates as a means to break down traditional silos among medicine, nursing, pharmacy, geriatrics, and psychology.
IMPROVE combines key components of interprofessional education, including all 4 CoEPCE core domains, to provide hands-on experience with knowledge learned in other aspects of the CoEPCE training program (eg, shared decision-making strategies for eliciting patient goals, weighing risks and benefits in complex clinical situations). Physician and NP trainees work together with trainees in pharmacy and health psychology in the complex approach to polypharmacy. IMPROVE provides the framework for an interprofessional clinic that could be used in the treatment of other complex or high-risk chronic conditions.
The Future
An opportunity for improvement and expansion includes increased patient involvement (as patients continue to learn they have a team working on their behalf). Opportunities exist to connect with patients who have several clinicians prescribing medications outside the CoEPCE to provide comprehensive care and decrease medication complexity.
The CoEPCE has been proactive in increasing the visibility of IMPROVE through multiple presentations at local and national meetings, facilitating collaborations and greater adoption in primary care. Individual and collective IMPROVE components can be adapted to other contexts. For example, the 20-minute geriatrics education session and the forms completed prior and during the patient visit can be readily applied to other complex patients that trainees meet in clinic. Under stage 2 of the CoEPCE program, the CoEPCE is developing an implementation kit that describes the training process and includes the medication worksheet, assessment tools, and directions for conducting the group visit.
It is hoped that working collaboratively with the West Haven COEPCE polypharmacy faculty, a similar model of education and training will be implemented at other health professional training sites at Yale University in New Haven, Connecticut. Additionally, the West Haven CoEPCE is planning to partner with the other original CoEPCE program sites to implement similar interprofessional polypharmacy clinics.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
1. US Department of Health and Human Services, Agency for Health Research and Quality. Transforming the organization and delivery of primary care. http://www.pcmh.ahrq .gov/. Accessed August 14, 2018.
2. Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in prescription drug use among adults in the United States from 1999-2012. JAMA. 2015;314(17):1818-1831.
3. Fried TR, O’Leary J, Towle V, Goldstein MK, Trentalange M, Martin DK. Health outcomes associated with polypharmacy in community-dwelling older adults: a systematic review. J Am Geriatr Soc. 2014;62(12):2261-2272.
4. Mecca M, Niehoff K, Grammas M. Medication review worksheet 2015. http://pogoe.org/productid/21872. Accessed August 14, 2018.
5. American Geriatrics Society 2015 Beers criteria update expert panel. American Geriatrics Society 2015 updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
6. O’Mahony D, O’Sullivan D, Byrne S, O’Connor MN, Ryan C, Gallagher P. STOPP/START criteria for potentially inappropriate prescribing in older people: version 2. Age Ageing. 2015;44(2):213-218.
7. Yale University. IMPROVE Polypharmacy Project. http://improvepolypharmacy.yale.edu. Accessed August 14, 2018.
8. Tariq SH, Tumosa N, Chibnall JT, Perry MH III, Morley JE. Comparison of the Saint Louis University mental status examination and the mini-mental state examination for detecting dementia and mild neurocognitive disorder—a pilot study. Am J Geriatr Psychiatry. 2006;14(11):900-910.
9. Cohen DJ, Balasubramanian BA, Davis M, et al. Understanding care integration from the ground up: Five organizing constructs that shape integrated practices. J Am Board Fam Med. 2015;28(suppl):S7-S20.
The Pharmacist’s Role in Medication Optimization for Patients With Chronic Heart Failure (FULL)
In the U.S., about 5.1 million people have clinically manifested heart failure (HF).1 The absolute mortality rate for HF is about 50% within 5 years of diagnosis, and 1 in 9 death certificates in the U.S. list HF as a cause of death.2 Heart failure is the primary diagnosis in more than 1 million hospitalizations annually.1 Patients with HF who are at risk for all-cause rehospitalization have a 1-month readmission rate of 25%, and their median survival time decreases with each hospitalization.3,4 Heart failure is the top reason for discharge of veterans treated within the VA health care system.5
Some medications decrease morbidity and mortality in patients with systolic dysfunction.6 These medications include angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and β-blockers (BBs). Studies have demonstrated effectiveness of these medications in patients with reduced ejection fraction (EF) of less or equal to 40%. Other medications with proven success include aldosterone antagonists, hydralazine in combination with a nitrate, and digoxin. The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines provide medication recommendations based on the ACCF/AHA stages of HF and the New York Heart Association (NYHA) functional classifications, designated as guideline-directed medical therapy (GDMT).6,7 Therapeutic interventions are aimed at reducing morbidity and mortality for ACCF/AHA stage C HF. The ACCF/AHA guidelines also recommend establishing multidisciplinary HF disease management programs for patients at high risk for hospital readmission to facilitate implementation of GDMT, address different barriers to behavior change, and reduce the risk of subsequent rehospitalization for HF.6
In October 2010, the Jesse Brown VAMC (JBVAMC) in Chicago, Illinois, opened its Heart Failure Disease Management Program (HFDMP) to prevent readmissions by targeting patients discharged after HF exacerbations and arranging follow-up in the HFDMP clinic. Enrollment in the clinic is initiated when an inpatient physician places a consultation. A cardiology nurse practitioner (NP) receives the consultation and schedules an in-clinic appointment for the patient within 1 week of discharge. The patient goes to the clinic on average every 2 weeks until he or she is on a stable, optimal medication regimen and is competent in self-management. After 3 months, the patient transitions to the general cardiology clinic. This process allows the HFDMP to see new patients in need of intense care and education for HF. The multidisciplinary HFDMP began with a NP and a cardiologist and 6 months later in April 2011 added a pharmacist.
After enrolling in HFDMP, the patient can be referred to the pharmacist for independent optimization of medication therapy in the Pharmacy Medication Titration Clinic (PMTC). The PMTC at JBVAMC is different from other HF clinics in that the pharmacist has prescribing authority and can interact face-to-face with patients to titrate medications. Once a patient is on an optimal medication regimen, he or she is referred to the NP and cardiologist. The PMTC is open 4 hours twice per month and offers 30-minute time slots. The authors conducted a study of the effectiveness of face-to-face PMTC appointments within the HFDMP.
Methods
This study, approved by the institutional review board at the University of Illinois at Chicago and the research and development service at JBVAMC, was a retrospective electronic chart review of patients enrolled in the HFDMP. Study patients were aged ≥ 18 years and were enrolled in the HFDMP between April 15, 2011 and April 15, 2013. Exclusion criteria included EF higher than 40%, 1 or no appointment attended, and enrollment before April 15, 2011. There were 2 study groups: HFDMP patients enrolled in PMTC (PMTC group) and HFDMP patients not enrolled in PMTC (no-PMTC group). For 1:1 comparison, the number of patients who met the criteria for the PMTC group was used to determine the number of patients to include in the no-PMTC group. Baseline date was the date of enrollment into either HFDMP or PMTC.
Data collected at baseline included demographics, NYHA class of HF, blood pressure (BP), heart rate, ejection fraction (EF), date of HF diagnosis, number of hospitalizations for HF within previous 6 months, serum creatinine level, height, weight, comorbidities, and HF medications. Data collected at the end date included NYHA class of HF; BP; heart rate; EF; HF medications; reason for not achieving target dose of medication or GDMT; ACEI, ARB, or BB adherence, defined as 80% of medication refills 6 months after date of discharge from group; readmission for HF within 30 days and 90 days; length of stay (LOS), including bed type if readmitted; emergency department (ED) visits for HF within 6 months of date of discharge from group; and death within 6 months of date of discharge from group. Clinical GDMT was defined as reaching the maximum tolerable or target dose of each HF medication for each patient depending on clinical presentation, as recommended by the ACCF/AHA guidelines for HF.6 It incorporated NYHA class of HF, contraindications, hypotension, bradycardia, dizziness, and hyperkalemia as well as the prescribing of aldosterone antagonists, hydralazine and isosorbide dinitrate, and digoxin.
Study Outcomes
There were 2 primary endpoints: difference in percentage of patients who achieved target ACEI or ARB doses and difference in percentage of patients who achieved target BB doses. Secondary endpoints were difference between PMTC and no-PMTC groups in percentage of patient achievement in clinical GDMT; percentage medication adherence; percentage of patients with change in NYHA class of HF; percentage of patients with change in EF, including mean change; percentage of patients with 30-day and 90-day readmissions for HF; mean LOS if readmitted within 30 days and 90 days; percentage of patients with ED visits for HF within 6 months after baseline; and percentage mortality within 6 months after baseline.
Statistical Analysis
A 2-tailed Fisher exact test was used for nominal data, and a Student t test for continuous data. Statistical significance was set at P < .05.
Results
Of the 228 HFDMP enrollees, 29 were seen in the PMTC during the study period, and 199 were not seen in the PMTC. Of the 29 patients seen in the PMTC, 24 met the criteria for the PMTC study group. Charts of 106 of the 199 patients not seen in the PMTC were randomly reviewed until 24 patients who met the study criteria were selected for the no-PMTC study group. Thus, the PMTC group and the no-PMTC group each had 24 patients for 1:1 comparison. Eighty-seven patients were excluded from the study: 22 with EF > 40%, 50 with 1 or no appointment attended, and 15 who were enrolled in HFDMP before April 15, 2011.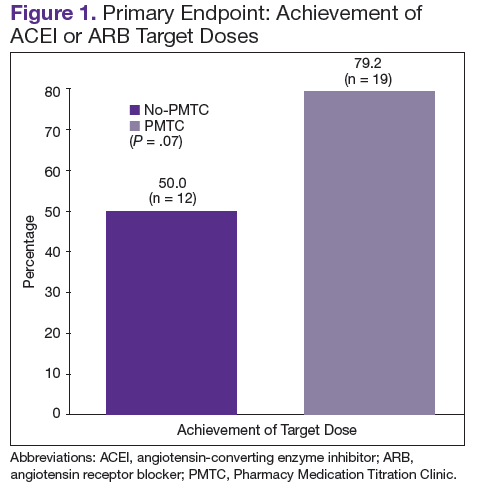
Mean age was 66 years. All patients were male, and most were African American. The baseline characteristics of the PMTC and no-PMTC groups were similar, except a higher percentage of patients in the PMTC group had NYHA class II or III of HF (Table 1). 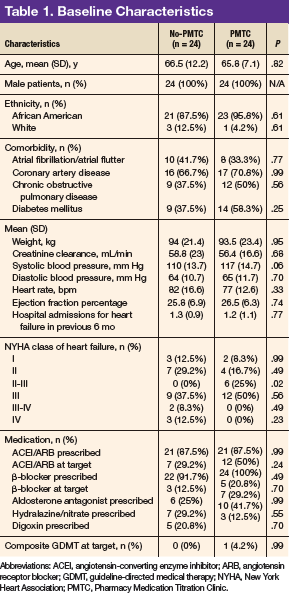
The ACEI or ARB target dose was achieved by a higher percentage of patients in the PMTC group, 79.2% (n = 19) vs 50% (n = 12), but the difference was not significant (P = .07) (Figure 1). However, BB target dose was achieved by a significantly higher (P = .01) percentage of patients in the PMTC group, 87.5% (n = 21) vs 20.8% (n = 5)(Figure 2). Furthermore, a significantly higher (P = .02) percentage of patients in the PMTC group, 62.5% (n = 15) vs 25% (n = 6), achieved composite clinical GDMT (Table 2). Last, there was not a statistically significant difference in 80% adherence with ACEI or ARB dosing or with BB dosing between the PMTC group (70.8%; n = 17) and the no-PMTC group (75%; n = 18). 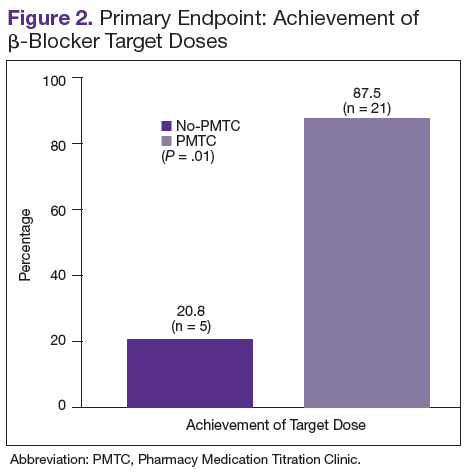
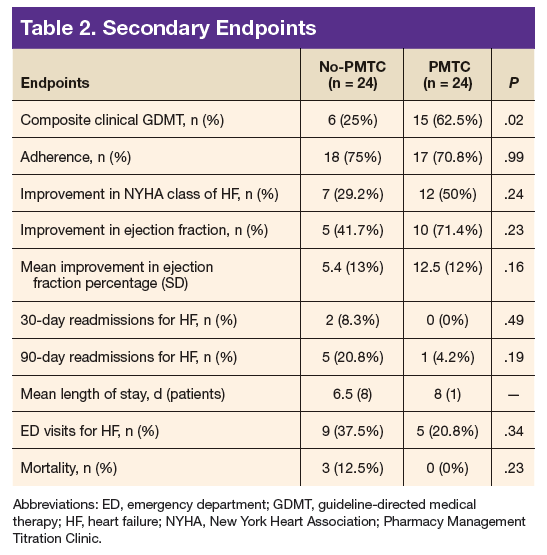
For a higher percentage of patients in the PMTC group, 50% (n = 12) vs 29.2% (n = 7), NYHA class of HF improved, but the difference was not significant (P = .24). In addition, EF improved in a higher percentage of patients in the PMTC group, 71.4% (n = 10) vs 41.7% (n = 5), and mean (SD) improvement in EF was higher in the PMTC group, 12.5% (12%) vs 5.4% (13%), but neither difference was significant (P = .23 and P = .16, respectively).
A higher percentage of patients in the no-PMTC group were readmitted for HF within 30 days, 8.3% (n = 2) vs 0%, but the difference was not significant. Likewise, a higher percentage of patients in the no-PMTC group were readmitted for HF within 90 days, 20.8% (n = 5) vs 4.2% (n = 1; P = .19). Mean LOS for these readmissions was longer in the PMTC group, 8 days (n = 1) vs 6.5 days (n = 8). There was a higher percentage of ED visits for HF in the no-PMTC group, 37.5% (n = 9) vs 20.8% (n = 5), but did not reach statistical significance (P = .34). Last, the no-PMTC group had a higher percentage of deaths within 6 months after baseline, 37.5% (n = 9) vs 20.8% (n = 9), which also was not significant.
Discussion
This study demonstrated that, within HFDMPs, there is a role for a pharmacist who has prescribing authority and interacts face-to-face with patients in the clinic. Significantly more patients in the PMTC group achieved target BB doses by the end of the study. Target doses of BBs have been found to decrease morbidity and mortality.8-10
The present study also found a positive trend toward achieving target ACEI or ARB doses. Reasons for not achieving target doses included contraindication to the medication, medication discontinuation during hospital admission, hypotension, hyperkalemia, and titration not complete by end of study period. Two of the many reasons for titration not being complete were clinic enrollment timing and nonadherence. Although achievement of target ACEI or ARB doses did not reach statistical significance, statistically significantly more patients achieved composite clinical GDMT.
As defined in the study, clinical GDMT captured the prescribing of hydralazine and isosorbide dinitrate in patients intolerant to ACEIs and ARBs. This study, the first known to evaluate achievement in GDMT, demonstrated a pharmacist’s ability to titrate more than just ACEI, ARB, and BB doses. This finding is clinically important in that appropriate pharmacologic therapy can reduce the number of hospitalizations for HF and improve survival, even though the study found that its PMTC group showed only trends toward fewer 30-day and 90-day readmissions for HF, fewer ED visits for HF, and less mortality.6 This finding may be attributable to the small number of readmissions for HF and deaths among the study groups.
One endpoint that did not show an expected difference with pharmacist intervention was medication adherence. However, medication nonadherence likely was a reason for patient referral to the PMTC. Baseline medication adherence was not determined, so improvement in adherence could not be assessed. Findings might have been different, too, if medication adherence had been evaluated with patient interviews and refill history, not just refill history.
In the PMTC group, LOS for readmissions for HF did not improve. However, the group had only 1 readmission, which may have skewed the result. No studies have linked outpatient pharmacist intervention to decreased LOS for readmission for HF. The endpoint was evaluated to assess whether medication stability leads to reduced LOS and to complete a limited cost analysis. Analysis of mean cost based on number of readmissions, bed type, and LOS revealed a cost savings of $167,556.82 for the PMTC group (Table 3). Other potential cost savings that are difficult to quantify and that were not accounted for include extended time between ED visits or readmissions for HF and increased quality of life and daily functioning. 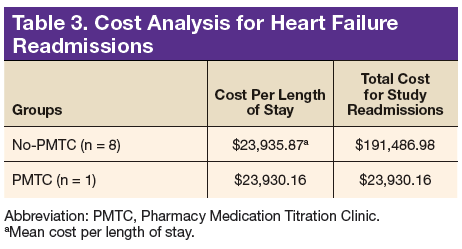
This is the first study known to evaluate a pharmacist who had prescribing authority and interacted face-to-face with patients. Other studies have evaluated the role of the pharmacist in the multidisciplinary management of patients with HF. In 1999, the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study reported the effect of direct HF-related patient care by a pharmacist who performed medication evaluations, provided patient education, and made medication recommendations to a physician.11
After a medication dosing change, the pharmacist provided telephone follow-up to assess for problems with the drug therapy and then, if any were identified, referred patients to the physician. Pharmacist intervention demonstrated a decrease in all-cause mortality and HF events and an increase in ACEI doses. Unlike the pharmacist in the present study, the pharmacist had to get recommendations approved and prescribed by a physician. The present PMTC allows for pharmacist intervention, including medication therapy changes and follow-up appointments without consultation with a physician. If needed, the HF cardiologist is available in the HFDMP clinic or by telephone.
Jain and colleagues evaluated a protocol-driven medication titration clinic staffed by nurse and pharmacist specialists.12 Although their study was limited by its descriptive nature, the authors concluded that the clinic increased the number of patients who achieved target ACEI/ARB or target BB doses. In the present study, the percentage of PMTC patients who achieved target doses increased during the study period, from 50% to 79.2% (ACEI/ARB) and from 20.8% to 87.5% (BB). Unlike other clinic pharmacists, however, the PMTC pharmacist titrates medications independently and does not follow a set clinic protocol.
Similar to the PHARM study, the Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) evaluated pharmacists who worked collaboratively with physicians to optimize HF therapy.13 As in the PMTC, patients and pharmacists had 30-minute appointments together. In HOOPS, however, physician agreement was needed before pharmacist recommendations were implemented. That study found that more patients with pharmacist intervention started an ACEI or ARB, had the medication titrated, and received recommended doses. More patients also either started a BB or increased its dose, but this did not increase the number of patients who received recommended BB doses. In addition, pharmacist intervention did not affect clinical outcomes. The authors acknowledged this finding might be attributable to the pharmacists’ lack of proper HF management training. Patients in the study were also more stable, whereas PMTC patients arrived after HF discharge and were followed until medication therapy was optimized. The HOOPS followed patients for only 3 or 4 visits, regardless of target dose achievement status.
In a study conducted within the VA health care system, Martinez and colleagues evaluated the use of pharmacists who had prescribing authority and were permitted to order laboratory tests under a scope of practice similar to that in the present study.14 However, their coordination agreement allowed them only to initiate and adjust doses of certain HF medications according to defined protocols. The pharmacist conducted monthly education classes and had medication titration appointments with individual patients by telephone over 2-week intervals. Face-to-face appointments were limited to medication reconciliations, whereas all appointments in the present study were face-to-face. In addition, Martinez and colleagues found that a higher percentage of patients who attended pharmacist appointments achieved target ACEI, ARB, and BB doses, whereas the present study found a higher percentage only of achieved target BB doses, not ACEI or ARB. However, the present study also found increased composite clinical GDMT achievement, which Martinez and colleagues did not evaluate. As mentioned, GDMT achievement may be a broader evaluation of optimal HF medical therapy, as it incorporates ACEI or ARB intolerance.
Martinez and colleagues acknowledged several study limitations different from those of the present study.14 Most members of their study population were white men, unlike this study’s population. Combining these 2 studies’ results may support use of pharmacist intervention for both white and African American men. In addition, the authors noted that patients often forgot their medications or were confused about doses, and concluded that forgetfulness and confusion may stem from having only telephone interviews and lacking written instructions for the interval between clinic appointments. By contrast, all PMTC patients were seen face-to-face, and handouts detailing any changes helped minimized confusion. Even with face-to-face appointments, however, patient nonadherence persisted, making it difficult to optimize HF therapy. Patients did not always follow instructions to bring HF medications (or medication lists) and daily weight measurements to clinic visits, which complicated medication reconciliations, interventions, and educational efforts regarding dose changes. Furthermore, patients sometimes missed face-to-face appointments, often because of transportation difficulties. In these situations, telephone appointments may be beneficial. The obstacle of transportation is removed, and, during the at-home telephone call, the patient has easy access to medications and measurements.
Limitations
This study had several limitations. It was retrospective, and its sample size was too small for conclusions regarding morbidity and mortality. As the population was predominantly African American males, results may not be applicable to other races and females. Furthermore, not evaluating HF causes at baseline could have confounded results, as disease progression, response to medications, and prognosis can vary, depending on etiology. Moreover, as patients are referred from the HFDMP to the PMTC, there may have been a bias in patient selection for the PMTC group. Patients in the PMTC group may have been more clinically stable yet had a larger knowledge deficit, an adherence issue, or a need for difficult, frequent titrations. Patients also may have been less likely to be seen during the first 30 days after discharge. In addition, it could have been beneficial to match patients on NYHA class of HF at baseline to ensure HF severity was balanced between groups. Last, the adherence analysis may not be accurate, as it relied on refill history, which may not reflect how medications were taken at home.
It would be beneficial to expand this initial study with a larger sample. Presumed HF causes and medication adherence should be captured at baseline. Additional endpoints, including quality of life and patient cognition, could enhance results. Furthermore, comparing the HFDMP with the general cardiology clinic may reveal other benefits of a focused HFDMP and its PMTC. Last, evaluating patients who are recently discharged from HF admission yet not enrolled in the HFDMP may provide more information regarding the utility of both the HFDMP and the PMTC.
Conclusion
For the PMTC group in this study, achievement of target BB doses and achievement in composite clinical GDMT were significant, but achievement of target ACEI/ARB doses were not.
Click here to read the digital edition.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6-e245.
2. Roger VL, Weston SA, Redfield MM, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292(3):344-350.
3. Krumholz HM, Merrill AR, Schone EM, et al. Patterns of hospital performance in acute myocardial infarction and heart failure 30-day mortality and readmission. Circ Cardiovasc Qual Outcomes. 2009;2(5):407-413.
4. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J. 2007;154(2):260-266.
5. U.S. Department of Veterans Affairs, VA Office of Research and Development, Executive Committee, Chronic Heart Failure Quality Enhancement Research Initiative (CHF-QUERI), Health Services Research and Development Service. Chronic heart failure [QUERI fact sheet]. https://www.queri.research.va.gov/about/factsheets/chf_factsheet.pdf. Published July 2014. Accessed October 9, 2017.
6. Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62(16):e147-e239.
7. New York Heart Association Criteria Committee. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9th ed. Boston, MA: Little Brown; 1994.
8. Parker M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334(21):1349-1355.
9. CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353(9146):9-13.
10. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353(9169):2001-2007.
11. Gattis WA, Hasselblad V, Whellan DJ, O’Connor CM. Reduction in heart failure events by the addition of a clinical pharmacist to the heart failure management team: results of the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study. Arch Intern Med. 1999;159(16):1939-1945.
12. Jain A, Mills P, Nunn LM, et al. Success of a multidisciplinary heart failure clinic for initiation and up-titration of key therapeutic agents. Eur J Heart Fail. 2005;7(3):405-410.
13. Lowrie R, Mair FS, Greenlaw N, et al; Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) Investigators. Pharmacist intervention in primary care to improve outcomes in patients with left ventricular systolic dysfunction. Eur Heart J. 2012;33(3):314-324.
14. Martinez AS, Saef J, Paszczuk A, Bhatt-Chugani H. Implementation of a pharmacist-managed heart failure medication titration clinic. Am J Health Syst Pharm. 2013;70(12):1070-1076.
In the U.S., about 5.1 million people have clinically manifested heart failure (HF).1 The absolute mortality rate for HF is about 50% within 5 years of diagnosis, and 1 in 9 death certificates in the U.S. list HF as a cause of death.2 Heart failure is the primary diagnosis in more than 1 million hospitalizations annually.1 Patients with HF who are at risk for all-cause rehospitalization have a 1-month readmission rate of 25%, and their median survival time decreases with each hospitalization.3,4 Heart failure is the top reason for discharge of veterans treated within the VA health care system.5
Some medications decrease morbidity and mortality in patients with systolic dysfunction.6 These medications include angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and β-blockers (BBs). Studies have demonstrated effectiveness of these medications in patients with reduced ejection fraction (EF) of less or equal to 40%. Other medications with proven success include aldosterone antagonists, hydralazine in combination with a nitrate, and digoxin. The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines provide medication recommendations based on the ACCF/AHA stages of HF and the New York Heart Association (NYHA) functional classifications, designated as guideline-directed medical therapy (GDMT).6,7 Therapeutic interventions are aimed at reducing morbidity and mortality for ACCF/AHA stage C HF. The ACCF/AHA guidelines also recommend establishing multidisciplinary HF disease management programs for patients at high risk for hospital readmission to facilitate implementation of GDMT, address different barriers to behavior change, and reduce the risk of subsequent rehospitalization for HF.6
In October 2010, the Jesse Brown VAMC (JBVAMC) in Chicago, Illinois, opened its Heart Failure Disease Management Program (HFDMP) to prevent readmissions by targeting patients discharged after HF exacerbations and arranging follow-up in the HFDMP clinic. Enrollment in the clinic is initiated when an inpatient physician places a consultation. A cardiology nurse practitioner (NP) receives the consultation and schedules an in-clinic appointment for the patient within 1 week of discharge. The patient goes to the clinic on average every 2 weeks until he or she is on a stable, optimal medication regimen and is competent in self-management. After 3 months, the patient transitions to the general cardiology clinic. This process allows the HFDMP to see new patients in need of intense care and education for HF. The multidisciplinary HFDMP began with a NP and a cardiologist and 6 months later in April 2011 added a pharmacist.
After enrolling in HFDMP, the patient can be referred to the pharmacist for independent optimization of medication therapy in the Pharmacy Medication Titration Clinic (PMTC). The PMTC at JBVAMC is different from other HF clinics in that the pharmacist has prescribing authority and can interact face-to-face with patients to titrate medications. Once a patient is on an optimal medication regimen, he or she is referred to the NP and cardiologist. The PMTC is open 4 hours twice per month and offers 30-minute time slots. The authors conducted a study of the effectiveness of face-to-face PMTC appointments within the HFDMP.
Methods
This study, approved by the institutional review board at the University of Illinois at Chicago and the research and development service at JBVAMC, was a retrospective electronic chart review of patients enrolled in the HFDMP. Study patients were aged ≥ 18 years and were enrolled in the HFDMP between April 15, 2011 and April 15, 2013. Exclusion criteria included EF higher than 40%, 1 or no appointment attended, and enrollment before April 15, 2011. There were 2 study groups: HFDMP patients enrolled in PMTC (PMTC group) and HFDMP patients not enrolled in PMTC (no-PMTC group). For 1:1 comparison, the number of patients who met the criteria for the PMTC group was used to determine the number of patients to include in the no-PMTC group. Baseline date was the date of enrollment into either HFDMP or PMTC.
Data collected at baseline included demographics, NYHA class of HF, blood pressure (BP), heart rate, ejection fraction (EF), date of HF diagnosis, number of hospitalizations for HF within previous 6 months, serum creatinine level, height, weight, comorbidities, and HF medications. Data collected at the end date included NYHA class of HF; BP; heart rate; EF; HF medications; reason for not achieving target dose of medication or GDMT; ACEI, ARB, or BB adherence, defined as 80% of medication refills 6 months after date of discharge from group; readmission for HF within 30 days and 90 days; length of stay (LOS), including bed type if readmitted; emergency department (ED) visits for HF within 6 months of date of discharge from group; and death within 6 months of date of discharge from group. Clinical GDMT was defined as reaching the maximum tolerable or target dose of each HF medication for each patient depending on clinical presentation, as recommended by the ACCF/AHA guidelines for HF.6 It incorporated NYHA class of HF, contraindications, hypotension, bradycardia, dizziness, and hyperkalemia as well as the prescribing of aldosterone antagonists, hydralazine and isosorbide dinitrate, and digoxin.
Study Outcomes
There were 2 primary endpoints: difference in percentage of patients who achieved target ACEI or ARB doses and difference in percentage of patients who achieved target BB doses. Secondary endpoints were difference between PMTC and no-PMTC groups in percentage of patient achievement in clinical GDMT; percentage medication adherence; percentage of patients with change in NYHA class of HF; percentage of patients with change in EF, including mean change; percentage of patients with 30-day and 90-day readmissions for HF; mean LOS if readmitted within 30 days and 90 days; percentage of patients with ED visits for HF within 6 months after baseline; and percentage mortality within 6 months after baseline.
Statistical Analysis
A 2-tailed Fisher exact test was used for nominal data, and a Student t test for continuous data. Statistical significance was set at P < .05.
Results
Of the 228 HFDMP enrollees, 29 were seen in the PMTC during the study period, and 199 were not seen in the PMTC. Of the 29 patients seen in the PMTC, 24 met the criteria for the PMTC study group. Charts of 106 of the 199 patients not seen in the PMTC were randomly reviewed until 24 patients who met the study criteria were selected for the no-PMTC study group. Thus, the PMTC group and the no-PMTC group each had 24 patients for 1:1 comparison. Eighty-seven patients were excluded from the study: 22 with EF > 40%, 50 with 1 or no appointment attended, and 15 who were enrolled in HFDMP before April 15, 2011.
Mean age was 66 years. All patients were male, and most were African American. The baseline characteristics of the PMTC and no-PMTC groups were similar, except a higher percentage of patients in the PMTC group had NYHA class II or III of HF (Table 1).
The ACEI or ARB target dose was achieved by a higher percentage of patients in the PMTC group, 79.2% (n = 19) vs 50% (n = 12), but the difference was not significant (P = .07) (Figure 1). However, BB target dose was achieved by a significantly higher (P = .01) percentage of patients in the PMTC group, 87.5% (n = 21) vs 20.8% (n = 5)(Figure 2). Furthermore, a significantly higher (P = .02) percentage of patients in the PMTC group, 62.5% (n = 15) vs 25% (n = 6), achieved composite clinical GDMT (Table 2). Last, there was not a statistically significant difference in 80% adherence with ACEI or ARB dosing or with BB dosing between the PMTC group (70.8%; n = 17) and the no-PMTC group (75%; n = 18).
For a higher percentage of patients in the PMTC group, 50% (n = 12) vs 29.2% (n = 7), NYHA class of HF improved, but the difference was not significant (P = .24). In addition, EF improved in a higher percentage of patients in the PMTC group, 71.4% (n = 10) vs 41.7% (n = 5), and mean (SD) improvement in EF was higher in the PMTC group, 12.5% (12%) vs 5.4% (13%), but neither difference was significant (P = .23 and P = .16, respectively).
A higher percentage of patients in the no-PMTC group were readmitted for HF within 30 days, 8.3% (n = 2) vs 0%, but the difference was not significant. Likewise, a higher percentage of patients in the no-PMTC group were readmitted for HF within 90 days, 20.8% (n = 5) vs 4.2% (n = 1; P = .19). Mean LOS for these readmissions was longer in the PMTC group, 8 days (n = 1) vs 6.5 days (n = 8). There was a higher percentage of ED visits for HF in the no-PMTC group, 37.5% (n = 9) vs 20.8% (n = 5), but did not reach statistical significance (P = .34). Last, the no-PMTC group had a higher percentage of deaths within 6 months after baseline, 37.5% (n = 9) vs 20.8% (n = 9), which also was not significant.
Discussion
This study demonstrated that, within HFDMPs, there is a role for a pharmacist who has prescribing authority and interacts face-to-face with patients in the clinic. Significantly more patients in the PMTC group achieved target BB doses by the end of the study. Target doses of BBs have been found to decrease morbidity and mortality.8-10
The present study also found a positive trend toward achieving target ACEI or ARB doses. Reasons for not achieving target doses included contraindication to the medication, medication discontinuation during hospital admission, hypotension, hyperkalemia, and titration not complete by end of study period. Two of the many reasons for titration not being complete were clinic enrollment timing and nonadherence. Although achievement of target ACEI or ARB doses did not reach statistical significance, statistically significantly more patients achieved composite clinical GDMT.
As defined in the study, clinical GDMT captured the prescribing of hydralazine and isosorbide dinitrate in patients intolerant to ACEIs and ARBs. This study, the first known to evaluate achievement in GDMT, demonstrated a pharmacist’s ability to titrate more than just ACEI, ARB, and BB doses. This finding is clinically important in that appropriate pharmacologic therapy can reduce the number of hospitalizations for HF and improve survival, even though the study found that its PMTC group showed only trends toward fewer 30-day and 90-day readmissions for HF, fewer ED visits for HF, and less mortality.6 This finding may be attributable to the small number of readmissions for HF and deaths among the study groups.
One endpoint that did not show an expected difference with pharmacist intervention was medication adherence. However, medication nonadherence likely was a reason for patient referral to the PMTC. Baseline medication adherence was not determined, so improvement in adherence could not be assessed. Findings might have been different, too, if medication adherence had been evaluated with patient interviews and refill history, not just refill history.
In the PMTC group, LOS for readmissions for HF did not improve. However, the group had only 1 readmission, which may have skewed the result. No studies have linked outpatient pharmacist intervention to decreased LOS for readmission for HF. The endpoint was evaluated to assess whether medication stability leads to reduced LOS and to complete a limited cost analysis. Analysis of mean cost based on number of readmissions, bed type, and LOS revealed a cost savings of $167,556.82 for the PMTC group (Table 3). Other potential cost savings that are difficult to quantify and that were not accounted for include extended time between ED visits or readmissions for HF and increased quality of life and daily functioning.
This is the first study known to evaluate a pharmacist who had prescribing authority and interacted face-to-face with patients. Other studies have evaluated the role of the pharmacist in the multidisciplinary management of patients with HF. In 1999, the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study reported the effect of direct HF-related patient care by a pharmacist who performed medication evaluations, provided patient education, and made medication recommendations to a physician.11
After a medication dosing change, the pharmacist provided telephone follow-up to assess for problems with the drug therapy and then, if any were identified, referred patients to the physician. Pharmacist intervention demonstrated a decrease in all-cause mortality and HF events and an increase in ACEI doses. Unlike the pharmacist in the present study, the pharmacist had to get recommendations approved and prescribed by a physician. The present PMTC allows for pharmacist intervention, including medication therapy changes and follow-up appointments without consultation with a physician. If needed, the HF cardiologist is available in the HFDMP clinic or by telephone.
Jain and colleagues evaluated a protocol-driven medication titration clinic staffed by nurse and pharmacist specialists.12 Although their study was limited by its descriptive nature, the authors concluded that the clinic increased the number of patients who achieved target ACEI/ARB or target BB doses. In the present study, the percentage of PMTC patients who achieved target doses increased during the study period, from 50% to 79.2% (ACEI/ARB) and from 20.8% to 87.5% (BB). Unlike other clinic pharmacists, however, the PMTC pharmacist titrates medications independently and does not follow a set clinic protocol.
Similar to the PHARM study, the Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) evaluated pharmacists who worked collaboratively with physicians to optimize HF therapy.13 As in the PMTC, patients and pharmacists had 30-minute appointments together. In HOOPS, however, physician agreement was needed before pharmacist recommendations were implemented. That study found that more patients with pharmacist intervention started an ACEI or ARB, had the medication titrated, and received recommended doses. More patients also either started a BB or increased its dose, but this did not increase the number of patients who received recommended BB doses. In addition, pharmacist intervention did not affect clinical outcomes. The authors acknowledged this finding might be attributable to the pharmacists’ lack of proper HF management training. Patients in the study were also more stable, whereas PMTC patients arrived after HF discharge and were followed until medication therapy was optimized. The HOOPS followed patients for only 3 or 4 visits, regardless of target dose achievement status.
In a study conducted within the VA health care system, Martinez and colleagues evaluated the use of pharmacists who had prescribing authority and were permitted to order laboratory tests under a scope of practice similar to that in the present study.14 However, their coordination agreement allowed them only to initiate and adjust doses of certain HF medications according to defined protocols. The pharmacist conducted monthly education classes and had medication titration appointments with individual patients by telephone over 2-week intervals. Face-to-face appointments were limited to medication reconciliations, whereas all appointments in the present study were face-to-face. In addition, Martinez and colleagues found that a higher percentage of patients who attended pharmacist appointments achieved target ACEI, ARB, and BB doses, whereas the present study found a higher percentage only of achieved target BB doses, not ACEI or ARB. However, the present study also found increased composite clinical GDMT achievement, which Martinez and colleagues did not evaluate. As mentioned, GDMT achievement may be a broader evaluation of optimal HF medical therapy, as it incorporates ACEI or ARB intolerance.
Martinez and colleagues acknowledged several study limitations different from those of the present study.14 Most members of their study population were white men, unlike this study’s population. Combining these 2 studies’ results may support use of pharmacist intervention for both white and African American men. In addition, the authors noted that patients often forgot their medications or were confused about doses, and concluded that forgetfulness and confusion may stem from having only telephone interviews and lacking written instructions for the interval between clinic appointments. By contrast, all PMTC patients were seen face-to-face, and handouts detailing any changes helped minimized confusion. Even with face-to-face appointments, however, patient nonadherence persisted, making it difficult to optimize HF therapy. Patients did not always follow instructions to bring HF medications (or medication lists) and daily weight measurements to clinic visits, which complicated medication reconciliations, interventions, and educational efforts regarding dose changes. Furthermore, patients sometimes missed face-to-face appointments, often because of transportation difficulties. In these situations, telephone appointments may be beneficial. The obstacle of transportation is removed, and, during the at-home telephone call, the patient has easy access to medications and measurements.
Limitations
This study had several limitations. It was retrospective, and its sample size was too small for conclusions regarding morbidity and mortality. As the population was predominantly African American males, results may not be applicable to other races and females. Furthermore, not evaluating HF causes at baseline could have confounded results, as disease progression, response to medications, and prognosis can vary, depending on etiology. Moreover, as patients are referred from the HFDMP to the PMTC, there may have been a bias in patient selection for the PMTC group. Patients in the PMTC group may have been more clinically stable yet had a larger knowledge deficit, an adherence issue, or a need for difficult, frequent titrations. Patients also may have been less likely to be seen during the first 30 days after discharge. In addition, it could have been beneficial to match patients on NYHA class of HF at baseline to ensure HF severity was balanced between groups. Last, the adherence analysis may not be accurate, as it relied on refill history, which may not reflect how medications were taken at home.
It would be beneficial to expand this initial study with a larger sample. Presumed HF causes and medication adherence should be captured at baseline. Additional endpoints, including quality of life and patient cognition, could enhance results. Furthermore, comparing the HFDMP with the general cardiology clinic may reveal other benefits of a focused HFDMP and its PMTC. Last, evaluating patients who are recently discharged from HF admission yet not enrolled in the HFDMP may provide more information regarding the utility of both the HFDMP and the PMTC.
Conclusion
For the PMTC group in this study, achievement of target BB doses and achievement in composite clinical GDMT were significant, but achievement of target ACEI/ARB doses were not.
Click here to read the digital edition.
In the U.S., about 5.1 million people have clinically manifested heart failure (HF).1 The absolute mortality rate for HF is about 50% within 5 years of diagnosis, and 1 in 9 death certificates in the U.S. list HF as a cause of death.2 Heart failure is the primary diagnosis in more than 1 million hospitalizations annually.1 Patients with HF who are at risk for all-cause rehospitalization have a 1-month readmission rate of 25%, and their median survival time decreases with each hospitalization.3,4 Heart failure is the top reason for discharge of veterans treated within the VA health care system.5
Some medications decrease morbidity and mortality in patients with systolic dysfunction.6 These medications include angiotensin-converting enzyme inhibitors (ACEIs), angiotensin receptor blockers (ARBs), and β-blockers (BBs). Studies have demonstrated effectiveness of these medications in patients with reduced ejection fraction (EF) of less or equal to 40%. Other medications with proven success include aldosterone antagonists, hydralazine in combination with a nitrate, and digoxin. The American College of Cardiology Foundation/American Heart Association (ACCF/AHA) guidelines provide medication recommendations based on the ACCF/AHA stages of HF and the New York Heart Association (NYHA) functional classifications, designated as guideline-directed medical therapy (GDMT).6,7 Therapeutic interventions are aimed at reducing morbidity and mortality for ACCF/AHA stage C HF. The ACCF/AHA guidelines also recommend establishing multidisciplinary HF disease management programs for patients at high risk for hospital readmission to facilitate implementation of GDMT, address different barriers to behavior change, and reduce the risk of subsequent rehospitalization for HF.6
In October 2010, the Jesse Brown VAMC (JBVAMC) in Chicago, Illinois, opened its Heart Failure Disease Management Program (HFDMP) to prevent readmissions by targeting patients discharged after HF exacerbations and arranging follow-up in the HFDMP clinic. Enrollment in the clinic is initiated when an inpatient physician places a consultation. A cardiology nurse practitioner (NP) receives the consultation and schedules an in-clinic appointment for the patient within 1 week of discharge. The patient goes to the clinic on average every 2 weeks until he or she is on a stable, optimal medication regimen and is competent in self-management. After 3 months, the patient transitions to the general cardiology clinic. This process allows the HFDMP to see new patients in need of intense care and education for HF. The multidisciplinary HFDMP began with a NP and a cardiologist and 6 months later in April 2011 added a pharmacist.
After enrolling in HFDMP, the patient can be referred to the pharmacist for independent optimization of medication therapy in the Pharmacy Medication Titration Clinic (PMTC). The PMTC at JBVAMC is different from other HF clinics in that the pharmacist has prescribing authority and can interact face-to-face with patients to titrate medications. Once a patient is on an optimal medication regimen, he or she is referred to the NP and cardiologist. The PMTC is open 4 hours twice per month and offers 30-minute time slots. The authors conducted a study of the effectiveness of face-to-face PMTC appointments within the HFDMP.
Methods
This study, approved by the institutional review board at the University of Illinois at Chicago and the research and development service at JBVAMC, was a retrospective electronic chart review of patients enrolled in the HFDMP. Study patients were aged ≥ 18 years and were enrolled in the HFDMP between April 15, 2011 and April 15, 2013. Exclusion criteria included EF higher than 40%, 1 or no appointment attended, and enrollment before April 15, 2011. There were 2 study groups: HFDMP patients enrolled in PMTC (PMTC group) and HFDMP patients not enrolled in PMTC (no-PMTC group). For 1:1 comparison, the number of patients who met the criteria for the PMTC group was used to determine the number of patients to include in the no-PMTC group. Baseline date was the date of enrollment into either HFDMP or PMTC.
Data collected at baseline included demographics, NYHA class of HF, blood pressure (BP), heart rate, ejection fraction (EF), date of HF diagnosis, number of hospitalizations for HF within previous 6 months, serum creatinine level, height, weight, comorbidities, and HF medications. Data collected at the end date included NYHA class of HF; BP; heart rate; EF; HF medications; reason for not achieving target dose of medication or GDMT; ACEI, ARB, or BB adherence, defined as 80% of medication refills 6 months after date of discharge from group; readmission for HF within 30 days and 90 days; length of stay (LOS), including bed type if readmitted; emergency department (ED) visits for HF within 6 months of date of discharge from group; and death within 6 months of date of discharge from group. Clinical GDMT was defined as reaching the maximum tolerable or target dose of each HF medication for each patient depending on clinical presentation, as recommended by the ACCF/AHA guidelines for HF.6 It incorporated NYHA class of HF, contraindications, hypotension, bradycardia, dizziness, and hyperkalemia as well as the prescribing of aldosterone antagonists, hydralazine and isosorbide dinitrate, and digoxin.
Study Outcomes
There were 2 primary endpoints: difference in percentage of patients who achieved target ACEI or ARB doses and difference in percentage of patients who achieved target BB doses. Secondary endpoints were difference between PMTC and no-PMTC groups in percentage of patient achievement in clinical GDMT; percentage medication adherence; percentage of patients with change in NYHA class of HF; percentage of patients with change in EF, including mean change; percentage of patients with 30-day and 90-day readmissions for HF; mean LOS if readmitted within 30 days and 90 days; percentage of patients with ED visits for HF within 6 months after baseline; and percentage mortality within 6 months after baseline.
Statistical Analysis
A 2-tailed Fisher exact test was used for nominal data, and a Student t test for continuous data. Statistical significance was set at P < .05.
Results
Of the 228 HFDMP enrollees, 29 were seen in the PMTC during the study period, and 199 were not seen in the PMTC. Of the 29 patients seen in the PMTC, 24 met the criteria for the PMTC study group. Charts of 106 of the 199 patients not seen in the PMTC were randomly reviewed until 24 patients who met the study criteria were selected for the no-PMTC study group. Thus, the PMTC group and the no-PMTC group each had 24 patients for 1:1 comparison. Eighty-seven patients were excluded from the study: 22 with EF > 40%, 50 with 1 or no appointment attended, and 15 who were enrolled in HFDMP before April 15, 2011.
Mean age was 66 years. All patients were male, and most were African American. The baseline characteristics of the PMTC and no-PMTC groups were similar, except a higher percentage of patients in the PMTC group had NYHA class II or III of HF (Table 1).
The ACEI or ARB target dose was achieved by a higher percentage of patients in the PMTC group, 79.2% (n = 19) vs 50% (n = 12), but the difference was not significant (P = .07) (Figure 1). However, BB target dose was achieved by a significantly higher (P = .01) percentage of patients in the PMTC group, 87.5% (n = 21) vs 20.8% (n = 5)(Figure 2). Furthermore, a significantly higher (P = .02) percentage of patients in the PMTC group, 62.5% (n = 15) vs 25% (n = 6), achieved composite clinical GDMT (Table 2). Last, there was not a statistically significant difference in 80% adherence with ACEI or ARB dosing or with BB dosing between the PMTC group (70.8%; n = 17) and the no-PMTC group (75%; n = 18).
For a higher percentage of patients in the PMTC group, 50% (n = 12) vs 29.2% (n = 7), NYHA class of HF improved, but the difference was not significant (P = .24). In addition, EF improved in a higher percentage of patients in the PMTC group, 71.4% (n = 10) vs 41.7% (n = 5), and mean (SD) improvement in EF was higher in the PMTC group, 12.5% (12%) vs 5.4% (13%), but neither difference was significant (P = .23 and P = .16, respectively).
A higher percentage of patients in the no-PMTC group were readmitted for HF within 30 days, 8.3% (n = 2) vs 0%, but the difference was not significant. Likewise, a higher percentage of patients in the no-PMTC group were readmitted for HF within 90 days, 20.8% (n = 5) vs 4.2% (n = 1; P = .19). Mean LOS for these readmissions was longer in the PMTC group, 8 days (n = 1) vs 6.5 days (n = 8). There was a higher percentage of ED visits for HF in the no-PMTC group, 37.5% (n = 9) vs 20.8% (n = 5), but did not reach statistical significance (P = .34). Last, the no-PMTC group had a higher percentage of deaths within 6 months after baseline, 37.5% (n = 9) vs 20.8% (n = 9), which also was not significant.
Discussion
This study demonstrated that, within HFDMPs, there is a role for a pharmacist who has prescribing authority and interacts face-to-face with patients in the clinic. Significantly more patients in the PMTC group achieved target BB doses by the end of the study. Target doses of BBs have been found to decrease morbidity and mortality.8-10
The present study also found a positive trend toward achieving target ACEI or ARB doses. Reasons for not achieving target doses included contraindication to the medication, medication discontinuation during hospital admission, hypotension, hyperkalemia, and titration not complete by end of study period. Two of the many reasons for titration not being complete were clinic enrollment timing and nonadherence. Although achievement of target ACEI or ARB doses did not reach statistical significance, statistically significantly more patients achieved composite clinical GDMT.
As defined in the study, clinical GDMT captured the prescribing of hydralazine and isosorbide dinitrate in patients intolerant to ACEIs and ARBs. This study, the first known to evaluate achievement in GDMT, demonstrated a pharmacist’s ability to titrate more than just ACEI, ARB, and BB doses. This finding is clinically important in that appropriate pharmacologic therapy can reduce the number of hospitalizations for HF and improve survival, even though the study found that its PMTC group showed only trends toward fewer 30-day and 90-day readmissions for HF, fewer ED visits for HF, and less mortality.6 This finding may be attributable to the small number of readmissions for HF and deaths among the study groups.
One endpoint that did not show an expected difference with pharmacist intervention was medication adherence. However, medication nonadherence likely was a reason for patient referral to the PMTC. Baseline medication adherence was not determined, so improvement in adherence could not be assessed. Findings might have been different, too, if medication adherence had been evaluated with patient interviews and refill history, not just refill history.
In the PMTC group, LOS for readmissions for HF did not improve. However, the group had only 1 readmission, which may have skewed the result. No studies have linked outpatient pharmacist intervention to decreased LOS for readmission for HF. The endpoint was evaluated to assess whether medication stability leads to reduced LOS and to complete a limited cost analysis. Analysis of mean cost based on number of readmissions, bed type, and LOS revealed a cost savings of $167,556.82 for the PMTC group (Table 3). Other potential cost savings that are difficult to quantify and that were not accounted for include extended time between ED visits or readmissions for HF and increased quality of life and daily functioning.
This is the first study known to evaluate a pharmacist who had prescribing authority and interacted face-to-face with patients. Other studies have evaluated the role of the pharmacist in the multidisciplinary management of patients with HF. In 1999, the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study reported the effect of direct HF-related patient care by a pharmacist who performed medication evaluations, provided patient education, and made medication recommendations to a physician.11
After a medication dosing change, the pharmacist provided telephone follow-up to assess for problems with the drug therapy and then, if any were identified, referred patients to the physician. Pharmacist intervention demonstrated a decrease in all-cause mortality and HF events and an increase in ACEI doses. Unlike the pharmacist in the present study, the pharmacist had to get recommendations approved and prescribed by a physician. The present PMTC allows for pharmacist intervention, including medication therapy changes and follow-up appointments without consultation with a physician. If needed, the HF cardiologist is available in the HFDMP clinic or by telephone.
Jain and colleagues evaluated a protocol-driven medication titration clinic staffed by nurse and pharmacist specialists.12 Although their study was limited by its descriptive nature, the authors concluded that the clinic increased the number of patients who achieved target ACEI/ARB or target BB doses. In the present study, the percentage of PMTC patients who achieved target doses increased during the study period, from 50% to 79.2% (ACEI/ARB) and from 20.8% to 87.5% (BB). Unlike other clinic pharmacists, however, the PMTC pharmacist titrates medications independently and does not follow a set clinic protocol.
Similar to the PHARM study, the Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) evaluated pharmacists who worked collaboratively with physicians to optimize HF therapy.13 As in the PMTC, patients and pharmacists had 30-minute appointments together. In HOOPS, however, physician agreement was needed before pharmacist recommendations were implemented. That study found that more patients with pharmacist intervention started an ACEI or ARB, had the medication titrated, and received recommended doses. More patients also either started a BB or increased its dose, but this did not increase the number of patients who received recommended BB doses. In addition, pharmacist intervention did not affect clinical outcomes. The authors acknowledged this finding might be attributable to the pharmacists’ lack of proper HF management training. Patients in the study were also more stable, whereas PMTC patients arrived after HF discharge and were followed until medication therapy was optimized. The HOOPS followed patients for only 3 or 4 visits, regardless of target dose achievement status.
In a study conducted within the VA health care system, Martinez and colleagues evaluated the use of pharmacists who had prescribing authority and were permitted to order laboratory tests under a scope of practice similar to that in the present study.14 However, their coordination agreement allowed them only to initiate and adjust doses of certain HF medications according to defined protocols. The pharmacist conducted monthly education classes and had medication titration appointments with individual patients by telephone over 2-week intervals. Face-to-face appointments were limited to medication reconciliations, whereas all appointments in the present study were face-to-face. In addition, Martinez and colleagues found that a higher percentage of patients who attended pharmacist appointments achieved target ACEI, ARB, and BB doses, whereas the present study found a higher percentage only of achieved target BB doses, not ACEI or ARB. However, the present study also found increased composite clinical GDMT achievement, which Martinez and colleagues did not evaluate. As mentioned, GDMT achievement may be a broader evaluation of optimal HF medical therapy, as it incorporates ACEI or ARB intolerance.
Martinez and colleagues acknowledged several study limitations different from those of the present study.14 Most members of their study population were white men, unlike this study’s population. Combining these 2 studies’ results may support use of pharmacist intervention for both white and African American men. In addition, the authors noted that patients often forgot their medications or were confused about doses, and concluded that forgetfulness and confusion may stem from having only telephone interviews and lacking written instructions for the interval between clinic appointments. By contrast, all PMTC patients were seen face-to-face, and handouts detailing any changes helped minimized confusion. Even with face-to-face appointments, however, patient nonadherence persisted, making it difficult to optimize HF therapy. Patients did not always follow instructions to bring HF medications (or medication lists) and daily weight measurements to clinic visits, which complicated medication reconciliations, interventions, and educational efforts regarding dose changes. Furthermore, patients sometimes missed face-to-face appointments, often because of transportation difficulties. In these situations, telephone appointments may be beneficial. The obstacle of transportation is removed, and, during the at-home telephone call, the patient has easy access to medications and measurements.
Limitations
This study had several limitations. It was retrospective, and its sample size was too small for conclusions regarding morbidity and mortality. As the population was predominantly African American males, results may not be applicable to other races and females. Furthermore, not evaluating HF causes at baseline could have confounded results, as disease progression, response to medications, and prognosis can vary, depending on etiology. Moreover, as patients are referred from the HFDMP to the PMTC, there may have been a bias in patient selection for the PMTC group. Patients in the PMTC group may have been more clinically stable yet had a larger knowledge deficit, an adherence issue, or a need for difficult, frequent titrations. Patients also may have been less likely to be seen during the first 30 days after discharge. In addition, it could have been beneficial to match patients on NYHA class of HF at baseline to ensure HF severity was balanced between groups. Last, the adherence analysis may not be accurate, as it relied on refill history, which may not reflect how medications were taken at home.
It would be beneficial to expand this initial study with a larger sample. Presumed HF causes and medication adherence should be captured at baseline. Additional endpoints, including quality of life and patient cognition, could enhance results. Furthermore, comparing the HFDMP with the general cardiology clinic may reveal other benefits of a focused HFDMP and its PMTC. Last, evaluating patients who are recently discharged from HF admission yet not enrolled in the HFDMP may provide more information regarding the utility of both the HFDMP and the PMTC.
Conclusion
For the PMTC group in this study, achievement of target BB doses and achievement in composite clinical GDMT were significant, but achievement of target ACEI/ARB doses were not.
Click here to read the digital edition.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6-e245.
2. Roger VL, Weston SA, Redfield MM, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292(3):344-350.
3. Krumholz HM, Merrill AR, Schone EM, et al. Patterns of hospital performance in acute myocardial infarction and heart failure 30-day mortality and readmission. Circ Cardiovasc Qual Outcomes. 2009;2(5):407-413.
4. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J. 2007;154(2):260-266.
5. U.S. Department of Veterans Affairs, VA Office of Research and Development, Executive Committee, Chronic Heart Failure Quality Enhancement Research Initiative (CHF-QUERI), Health Services Research and Development Service. Chronic heart failure [QUERI fact sheet]. https://www.queri.research.va.gov/about/factsheets/chf_factsheet.pdf. Published July 2014. Accessed October 9, 2017.
6. Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62(16):e147-e239.
7. New York Heart Association Criteria Committee. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9th ed. Boston, MA: Little Brown; 1994.
8. Parker M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334(21):1349-1355.
9. CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353(9146):9-13.
10. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353(9169):2001-2007.
11. Gattis WA, Hasselblad V, Whellan DJ, O’Connor CM. Reduction in heart failure events by the addition of a clinical pharmacist to the heart failure management team: results of the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study. Arch Intern Med. 1999;159(16):1939-1945.
12. Jain A, Mills P, Nunn LM, et al. Success of a multidisciplinary heart failure clinic for initiation and up-titration of key therapeutic agents. Eur J Heart Fail. 2005;7(3):405-410.
13. Lowrie R, Mair FS, Greenlaw N, et al; Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) Investigators. Pharmacist intervention in primary care to improve outcomes in patients with left ventricular systolic dysfunction. Eur Heart J. 2012;33(3):314-324.
14. Martinez AS, Saef J, Paszczuk A, Bhatt-Chugani H. Implementation of a pharmacist-managed heart failure medication titration clinic. Am J Health Syst Pharm. 2013;70(12):1070-1076.
1. Go AS, Mozaffarian D, Roger VL, et al; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6-e245.
2. Roger VL, Weston SA, Redfield MM, et al. Trends in heart failure incidence and survival in a community-based population. JAMA. 2004;292(3):344-350.
3. Krumholz HM, Merrill AR, Schone EM, et al. Patterns of hospital performance in acute myocardial infarction and heart failure 30-day mortality and readmission. Circ Cardiovasc Qual Outcomes. 2009;2(5):407-413.
4. Setoguchi S, Stevenson LW, Schneeweiss S. Repeated hospitalizations predict mortality in the community population with heart failure. Am Heart J. 2007;154(2):260-266.
5. U.S. Department of Veterans Affairs, VA Office of Research and Development, Executive Committee, Chronic Heart Failure Quality Enhancement Research Initiative (CHF-QUERI), Health Services Research and Development Service. Chronic heart failure [QUERI fact sheet]. https://www.queri.research.va.gov/about/factsheets/chf_factsheet.pdf. Published July 2014. Accessed October 9, 2017.
6. Yancy CW, Jessup M, Bozkurt B, et al; American College of Cardiology Foundation; American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62(16):e147-e239.
7. New York Heart Association Criteria Committee. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9th ed. Boston, MA: Little Brown; 1994.
8. Parker M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med. 1996;334(21):1349-1355.
9. CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353(9146):9-13.
10. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353(9169):2001-2007.
11. Gattis WA, Hasselblad V, Whellan DJ, O’Connor CM. Reduction in heart failure events by the addition of a clinical pharmacist to the heart failure management team: results of the Pharmacist in Heart Failure Assessment Recommendation and Monitoring (PHARM) study. Arch Intern Med. 1999;159(16):1939-1945.
12. Jain A, Mills P, Nunn LM, et al. Success of a multidisciplinary heart failure clinic for initiation and up-titration of key therapeutic agents. Eur J Heart Fail. 2005;7(3):405-410.
13. Lowrie R, Mair FS, Greenlaw N, et al; Heart Failure Optimal Outcomes From Pharmacy Study (HOOPS) Investigators. Pharmacist intervention in primary care to improve outcomes in patients with left ventricular systolic dysfunction. Eur Heart J. 2012;33(3):314-324.
14. Martinez AS, Saef J, Paszczuk A, Bhatt-Chugani H. Implementation of a pharmacist-managed heart failure medication titration clinic. Am J Health Syst Pharm. 2013;70(12):1070-1076.
FDA Boxed Warnings Updates: October 2018
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
The FDA’s MedWatch program safety labeling changes for boxed warnings are compiled quarterly for drugs and therapeutic biologics where important changes have been made to the safety information. These and other label changes are searchable in the Drug Safety Labeling Changes (SLC) database, where data are available to the public in downloadable and searchable formats. Boxed warnings are ordinarily used to highlight either adverse reactions so serious in proportion to the potential benefi t from the drug that it is essential that it be considered in assessing the risks and benefi ts of using the drug; or serious adverse reactions that can be prevented/reduced in frequency or severity by appropriate use of the drug; or FDA approved the drug with restrictions to ensure safe use because FDA concluded that the drug can be safely used only if distribution or use is restricted. For complete FDA Drug Safety Labeling changes, please visit http://www.accessdata.fda.gov/scripts/cder/safetylabelingchanges.
DESOGEN (DESOGESTREL AND ETHINYL ESTRADIOL TABLETS
- Edited boxed warning, June 2018
WARNING: CIGARETTE SMOKING AND SERIOUS CARDIOVASCULAR EVENTS
Cigarette smoking increases the risk of serious cardiovascular events from combination oral contraceptive (COC) use. This risk increases with age, particularly in women over 35 years of age, and with the number of cigarettes smoked. For this reason, COCs are contraindicated in women who are over 35 years of age, and smoke.
TRIZIVIR (ABACAVIR, LAMIVUDINE, AND ZIDOVUDINE TABLETS)
- Edited boxed warning, April 2018
Lactic Acidosis and Severe Hepatomegaly with Steatosis: Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues, including abacavir, lamivudine, and zidovudine (components of TRIZIVIR). Discontinue TRIZIVIR if clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity occur [see Warnings and Precautions (5.4)].
ERBITUX (CETUXIMAB)
- Edited boxed warning, June 2018
WARNING: INFUSION REACTIONS and CARDIOPULMONARY ARREST
Infusion Reactions: ERBITUX can cause serious and fatal infusion reactions [see Warnings and Precautions (5.1), Adverse Reactions (6)]. Immediately interrupt and permanently discontinue ERBITUX for serious infusion reactions [see Dosage and Administration (2.4)].
Cardiopulmonary Arrest: Cardiopulmonary arrest or sudden death occurred in patients with squamous cell carcinoma of the head and neck receiving ERBITUX with radiation therapy or a cetuximab product with platinum-based therapy and fluorouracil. Monitor serum electrolytes, including serum magnesium, potassium, and calcium, during and after ERBITUX administration [see Warnings and Precautions (5.2, 5.6)].
AUSTEDO (DEUTETRABENAZINE)
- Edited boxed warning, June 2018
WARNING: DEPRESSION AND SUICIDALITY IN PATIENTS WITH HUNTINGTON’S DISEASE
AUSTEDO can increase the risk of depression …
EXJADE (DEFERASIROX)
- Edited boxed warning, May 2018
Renal Failure: EXJADE can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders. Evaluate baseline renal function prior to starting or increasing Exjade dosing in all patients. Exjade is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with pre-existing renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation.
TUXARIN ER (CODEINE PHOSPHATE AND CHLORPHENIRAMINE MALEATE)
- Edited boxed warning, June 2018
WARNING: ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; ULTRA-RAPID METABOLISM OF CODEINE AND OTHER RISK FACTORS FOR LIFE-THREATENING RESPIRATORY DEPRESSION IN CHILDREN; MEDICATION ERRORS; INTERACTIONS WITH DRUGS AFFECTING CYTOCHROME P450 ISOENZYMES; CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS; NEONATAL OPIOID WITHDRAWAL SYNDROME.
Addiction, Abuse, and Misuse: TUXARIN ER exposes patients and other users to the risks of opioid addiction, abuse, and misuse,
which can lead to overdose and death. Reserve TUXARIN ER for use in adult patients for whom the benefits of cough suppression
are expected to outweigh the risks, and in whom an adequate assessment of the etiology of the cough has been made. Assess each
patient’s risk prior to prescribing TUXARIN ER, prescribe TUXARIN ER for the shortest duration that is consistent with individual patient
treatment goals, monitor all patients regularly for the development of addition or abuse, and refill only after reevaluation of the need for continued treatment. [see Warnings and Precautions (5.1)]
Life-Threatening Respiratory Depression: Serious, life-threatening, or fatal respiratory depression may occur with use of TUXARIN ER.
Monitor for respiratory depression, especially during initiation of TUXARIN ER therapy or when used in patients at higher risk [see Warnings and Precautions (5.2)].
Accidental Ingestion: Accidental ingestion of even one dose of TUXARIN ER, especially by children, can result in a fatal overdose of codeine [see Warnings and Precautions (5.2)].
Ultra-Rapid Metabolism of Codeine and Other Risk Factors for Life-Threatening Respiratory Depression in Children: Life threatening
respiratory depression and death have occurred in children who received codeine. Most of the reported cases occurred following tonsillectomy and/or adenoidectomy, and many of the children had evidence of being an ultra-rapid metabolizer of codeine due to a
CYP2D6 polymorphism. [see Warnings and Precautions (5.3)]. TUXARIN ER is contraindicated in children younger than 12 years of age and
in children younger than 18 years of age following tonsillectomy and/or adenoidectomy [see Contraindications (4)]. Avoid the use of TUXARIN ER in adolescents 12 to 18 years of age who have other risk factors that may increase their sensitivity to the respiratory depressant effects of codeine. [see Warnings and Precautions (5.1)].
Interactions with Drugs Affecting Cytochrome P450 Isoenzymes: The effects of concomitant use or discontinuation of cytochrome P450 3A4 inducers, 3A4 inhibitors, or 2D6 inhibitors with codeine are complex, requiring careful consideration of the effects on the parent drug, codeine, and the active metabolite, morphine. Avoid the use of TUXARIN ER in patients who are taking a CYP3A4 inhibitor, CYP3A4 inducer, or 2D6 inhibitor [see Warnings and Precautions (5.8), Drug Interactions (7.1,7.2, 7.4)]
Risks from Concomitant Use with Benzodiazepines, CNS Depressants: Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Avoid use of TUXARIN ER in patients taking benzodiazepines, other CNS depressants, or alcohol. [see Warning and Precautions (5.9) Drug Interactions (7.5)].
Neonatal Opioid Withdrawal Syndrome: TUXARIN ER is not recommended for use in pregnant women [see Use in Specific Populations
(8.1)]. Prolonged use of TUXARIN ER during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If TUXARIN ER is used for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.15)].
PROGRAF (TACROLIMUS)
- Edited boxed warning, May 2018
Increased risk for developing serious infections and malignancies with PROGRAF or other immunosuppressants that may lead to hospitalization or death [(see Warnings and Precautions 5.1,5.2)].
SAMSCA (TOLVAPTAN)
- Edited boxed warning, April 2018
Addition of the following:
WARNING: NOT FOR USE FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE (ADPKD)
Because of the risk of hepatoxicity, tolvaptan should not be used for ADPKD outside of the FDA-approved REMS
GLUCOPHAGE/GLUCOPHAGE XR (METFORMIN HYDROCHLORIDE)
- Edited boxed warning, May 2018
PLR conversion; Lactic Acidosis warning is highlighted as boxed warning.
Tedizolid Use in Immunocompromised Patients
Immunocompromised patients are often susceptible to opportunistic infections, including those caused by multidrug-resistant organisms (MDROs). Transplant recipients are at high risk for developing infections due to lifelong immunosuppressive therapy.1,2 Additionally, patients receiving chemotherapy and those with HIV and AIDS are in an immunocompromised state.3-8
Regardless of the etiology for immunosuppression, decreased absolute neutrophil and platelet counts are seen in this condition. Although immunosuppressed individuals may be at increased risk of Gram-negative or Gram-positive infections, this review focuses on the treatment of Gram-positive bacterial infections. Of particular concern are opportunistic infections caused by Gram-positive MDROs, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus species (VRE), penicillin-resistant Streptococcus pneumoniae, and Nocardia species. Treatment of infections in the immunocompromised patient population warrants careful antimicrobial selection to ensure that a patient’s immune system is not further compromised due to adverse effects (AEs) secondary to therapy. As such, clinicians are exploring alternative antimicrobials, such as tedizolid, to treat various opportunistic infections.
Recently, requests at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin, for off-label use of tedizolid have increased despite having other cheaper alternatives with comparable Gram-positive coverage. This review examines available literature regarding off-label use of tedizolid with a focus on use in immunocompromised patients.
Tedizolid phosphate (Sivextro) is an oxazolidinone antibiotic prodrug that joined linezolid as the second in its class in 2014. Oxazolidinones inhibit bacterial protein synthesis by binding to the 50S subunit of bacterial ribosomes in Gram-positive bacteria and are often used to treat MRSA and VRE infections.9 In vitro, oxazolidinones have shown bacteriostatic activity against Enterococcus and Staphylococcus species while exhibiting bactericidal activity against most Streptococcus species.10 Tedizolid has a US Food and Drug Administration (FDA) -approved, simplified dosing profile of 200 mg daily for 6 days compared with linezolid 400 to 600 mg twice daily for 10 to 14 days. Both medications are highly bioavailable with direct IV to oral conversion.11,12 Potential, expanded use of tedizolid against Gram-positive MDROs rests on a more favorable AE profile than does its linezolid predecessor. Tedizolid has been associated with less antibiotic-induced myelosuppression, which could prove valuable for immunocompromised patients.13
Tedizolid is approved for the sole indication of acute bacterial skin and skin structure infections (ABSSSI), whereas its predecessor has many approved indications and has been used extensively for off-label indications (Table). 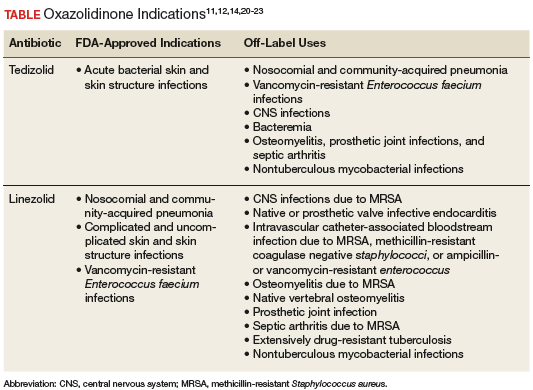
Adverse reactions, as determined by 2 phase 2 and 2 phase 3 clinical trials evaluating 1,050 patients treated with tedizolid and 662 patients treated with linezolid, were similar between the oxazolidinones. Nausea was the most common AE and was reported in 8% and 12% of patients taking tedizolid and linezolid, respectively. Other common AEs (1%-6%) reported for both agents included vomiting, diarrhea, headache, and dizziness.11 Myelosuppression, peripheral neuropathy, and optic nerve disorders were the most common severe AEs reported with oxazolidinones. Tedizolid demonstrated a significantly decreased incidence of neutropenia (3%), defined by absolute neutrophil count 9/L compared with that of linezolid (7%) (P = .024).13 Evaluation of peripheral neuropathy and optic nerve disorders within the tedizolid and linezolid groups revealed similar incidences (peripheral neuropathy 1.2% vs 0.6%; optic nerve disorders 0.3% vs 0.2%, respectively).11
There is one preclinical trial that described the use of tedizolid in a murine model. A murine model study compared the antistaphylococcal killing effect of doses of tedizolid equivalent to human exposures ranging from 200 to 3,200 mg/d in both granulocytopenic and normal mice. The mice were evaluated at 24, 48, and 72 hours after therapy initiation. The presence of granulocytes had a dramatic effect on the antimicrobial effect of tedizolid. Dose response, demonstrated by the ratio of the area under the curve over the minimum inhibitory concentration, was on average > 25-fold for nonneutropenic vs neutropenic models. Near maximal effect of the nonneutropenic group, irrespective of duration of therapy, was achieved at the lowest dose tested (an exposure of about 200-mg tedizolid phosphate per day in humans).This study suggests that immunocompromised patients may warrant higher doses of tedizolid than the currently FDA-approved dose due to a decreased number of granulocytes available for modulating bacterial infections.15
Use of tedizolid doses higher than that which is FDA-approved may negate the favorable AE profile. A phase 1 clinical study was conducted to evaluate the safety, tolerability, and pharmacokinetics of tedizolid compared with those of linezolid in 40 healthy volunteers in a 21-day multiple ascending dose study.16 Subjects were stratified into 5 treatment cohorts: 200-, 300-, or 400-mg tedizolid orally once a day, 600-mg linezolid orally twice a day, and placebo. Tedizolid given at 200 mg had a hematologic safety profile similar to that of placebo. However, mean platelet counts decreased over time in a dose-dependent manner for tedizolid, with the 400-mg tedizolid and linezolid groups reporting similar reductions in platelet counts.16
Some evidence is available examining linezolid in neutropenic patients. Rafailidis and colleagues reviewed available literature regarding linezolid in neutropenic patients with Gram-positive infections. Evaluation of linezolid administration at usual doses to 438 neutropenic patients from 2 prospective comparative studies, a prospective cohort study, 2 retrospective studies, and 8 case reports was performed. Results of the evaluation revealed a clinical cure rate between 57% and 87% in the intention-to-treat population of the prospective studies.17 Given the similarities in bacterial spectrum of activity between linezolid and tedizolid, it may be reasonable to infer that tedizolid’s decreased myelosuppression profile would make it useful in the setting of neutropenia in immunocompromised patients.
There is little evidence regarding the use of tedizolid in immunocompromised patients, as only 2 case reports were found. The first described a 60-year-old male postrenal transplant complicated with VRE bacteremia, rhabdomyolysis, and thrombocytopenia. This patient was treated with prolonged tedizolid 200 mg daily due to multiple contraindications for treatment with other antibiotics. The patient was cured with a 14-day course of tedizolid without any noted AEs.18
The second identified case report described the use of tedizolid for the treatment of central nervous system (CNS) manifestations secondary to nocardiosis. Effective treatment of CNS nocardiosis requires high concentrations and prolonged duration of antimicrobial exposure. This case report described a 68-year-old, chronically immunocompromised female patient with multiple myeloma who was hospitalized for 3 months for the treatment of a CNS nocardiosis infection. After discharge, the patient was treated with an oral regimen of 200-mg tedizolid daily in combination with sulfamethoxazole/trimethoprim (800 mg/160 mg) 3 times daily. After 6 months of combination therapy, magnetic resonance imaging revealed complete resolution of nocardiosis-related central lesions. Although the patient’s malignancy advanced during combination antibiotic therapy, the patient’s absolute neutrophil count remained stable and showed an increase in absolute CD4+ cell counts with no other documented AEs.19
Tedizolid is the latest FDA-approved oxazolidinone antibiotic for susceptible Gram-positive acute bacterial skin and skin structure infections. It has a simplified and shorter duration of treatment and imparts similar AEs at improved rates compared with that of linezolid, most notably in relation to hematologic AEs. Due to the lack of established literature and an agreed-upon dosing strategy for the use of tedizolid in immunocompromised patients, tedizolid therapy for Gram-positive infections in immunocompromised patients should be reserved for salvage therapy when more established Gram-positive antibiotic agents lack efficacy or when patient contraindications to their use exist.
1. Fishman JA, Issa NC. Infection in organ transplantation: risk factors and evolving patterns of infection. Infect Dis Clin North Am. 2010;24(2):273-283.
2. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614.
3. Nightingale SD, Byrd LT, Southern PM, Jockusch JD, Cal SX, Wynne BA. Incidence of mycobacterium avium-intracellulare complex bacteremia in human immunodeficiency virus-positive patients. J Infect Dis. 1992;165(6):1082-1085.
4. O’Brien S, Kantarjian H, Beran M, et al. Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood. 1993;82(6):1695-1700.
5. Anaissie E, Kontoyiannis DP, Kantarjian H, Elting L, Robertson LE, Keating M. Listeriosis in patients with chronic lymphocytic leukemia who were treated with fludarabine and prednisone. Ann Intern Med. 1992;117(6):466-469.
6. Morrison VA, Rai KR, Peterson BL, et al. Impact of therapy with chlorambucil, fludarabine, or fludarabine plus chlorambucil on infections in patients with chronic lymphocytic leukemia: Intergroup Study Cancer and Leukemia Group B 9011. J Clin Oncol. 2001;19(16):3611-3621.
7. Nucci M, Anaissie E. Infections in patients with multiple myeloma in the era of high-dose therapy and novel agents. Clin Infect Dis. 2009;49(8):1211-1225.
8. Naseer M, Dailey FE, Juboori AA, Samiullah S, Tahan V. Epidemiology, determinants, and managements of AIDS cholangiopathy: a review. World J Gastroenterol. 2018;24(7):767-774.
9. Radunz S, Juntermanns B, Kaiser GM, et al. Efficacy and safety of linezolid in liver transplant patients. Transpl Infect Dis. 2011;13(4):353-358.
10. Roger C, Roberts JA, Muller L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clin Pharmacokinet. 2018;57(5):559-575.
11. Sivextro [package insert]. Whitehouse Station, NJ: Merck & Co Inc; 2016.
12. Zyvox [package insert]. New York, NY: Pfizer Inc; 2018.
13. Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomized, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696-705.
14. Liu C, Bayer A, Cosgrove S, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
15. Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300-5305.
16. Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother. 2016;71(9):2553-2558.
17. Rafailidis PI, Kouranos VD, Christodoulou C, Falagas ME. Linezolid for patients with neutropenia: are bacteriostatic agents appropriate? Expert Rev Anti Infect Ther. 2009;7(4):415-422.
18. Sudhindra P, Lee L, Wang G, Dhand A. Tedizolid for treatment of enterococcal bacteremia. Open Forum Infect Dis. 2016;3(suppl 1):1344.
19. Matin A, Sharma S, Mathur P, Apewokin SK. Myelosuppression-sparing treatment of central nervous system nocardiosis in a multiple myeloma patient utilizing a tedizolid-based regimen: a case report. Int J Antimicrob Agents. 2017;49(4):488-492.
20. Dryden MS. Alternative clinical indications for novel antibiotics licensed for skin and soft tissue infections? Curr Opin Infect Dis. 2015;28(2):117-124.
21. Milstein M, Brzezinski A, Varaine F, Mitnick CD. (Re)moving the needle: prospects for all-oral treatment for multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2016;20(12):18-23.
22. Winthrop KL, Ku JH, Marras TK, et al. The tolerability of linezolid in the treatment of nontuberculous mycobacterial disease. Eur Respir J. 2015;45(4):1177-1179.
23. Yuste JR, Bertó J, Del Pozo JL, Leiva J. Prolonged use of tedizolid in a pulmonary non-tuberculosis mycobacterial infection after linezlid-induced toxicity. J Antimicrob Chemother. 2017;72(2):625-628.
Immunocompromised patients are often susceptible to opportunistic infections, including those caused by multidrug-resistant organisms (MDROs). Transplant recipients are at high risk for developing infections due to lifelong immunosuppressive therapy.1,2 Additionally, patients receiving chemotherapy and those with HIV and AIDS are in an immunocompromised state.3-8
Regardless of the etiology for immunosuppression, decreased absolute neutrophil and platelet counts are seen in this condition. Although immunosuppressed individuals may be at increased risk of Gram-negative or Gram-positive infections, this review focuses on the treatment of Gram-positive bacterial infections. Of particular concern are opportunistic infections caused by Gram-positive MDROs, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus species (VRE), penicillin-resistant Streptococcus pneumoniae, and Nocardia species. Treatment of infections in the immunocompromised patient population warrants careful antimicrobial selection to ensure that a patient’s immune system is not further compromised due to adverse effects (AEs) secondary to therapy. As such, clinicians are exploring alternative antimicrobials, such as tedizolid, to treat various opportunistic infections.
Recently, requests at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin, for off-label use of tedizolid have increased despite having other cheaper alternatives with comparable Gram-positive coverage. This review examines available literature regarding off-label use of tedizolid with a focus on use in immunocompromised patients.
Tedizolid phosphate (Sivextro) is an oxazolidinone antibiotic prodrug that joined linezolid as the second in its class in 2014. Oxazolidinones inhibit bacterial protein synthesis by binding to the 50S subunit of bacterial ribosomes in Gram-positive bacteria and are often used to treat MRSA and VRE infections.9 In vitro, oxazolidinones have shown bacteriostatic activity against Enterococcus and Staphylococcus species while exhibiting bactericidal activity against most Streptococcus species.10 Tedizolid has a US Food and Drug Administration (FDA) -approved, simplified dosing profile of 200 mg daily for 6 days compared with linezolid 400 to 600 mg twice daily for 10 to 14 days. Both medications are highly bioavailable with direct IV to oral conversion.11,12 Potential, expanded use of tedizolid against Gram-positive MDROs rests on a more favorable AE profile than does its linezolid predecessor. Tedizolid has been associated with less antibiotic-induced myelosuppression, which could prove valuable for immunocompromised patients.13
Tedizolid is approved for the sole indication of acute bacterial skin and skin structure infections (ABSSSI), whereas its predecessor has many approved indications and has been used extensively for off-label indications (Table).
Adverse reactions, as determined by 2 phase 2 and 2 phase 3 clinical trials evaluating 1,050 patients treated with tedizolid and 662 patients treated with linezolid, were similar between the oxazolidinones. Nausea was the most common AE and was reported in 8% and 12% of patients taking tedizolid and linezolid, respectively. Other common AEs (1%-6%) reported for both agents included vomiting, diarrhea, headache, and dizziness.11 Myelosuppression, peripheral neuropathy, and optic nerve disorders were the most common severe AEs reported with oxazolidinones. Tedizolid demonstrated a significantly decreased incidence of neutropenia (3%), defined by absolute neutrophil count 9/L compared with that of linezolid (7%) (P = .024).13 Evaluation of peripheral neuropathy and optic nerve disorders within the tedizolid and linezolid groups revealed similar incidences (peripheral neuropathy 1.2% vs 0.6%; optic nerve disorders 0.3% vs 0.2%, respectively).11
There is one preclinical trial that described the use of tedizolid in a murine model. A murine model study compared the antistaphylococcal killing effect of doses of tedizolid equivalent to human exposures ranging from 200 to 3,200 mg/d in both granulocytopenic and normal mice. The mice were evaluated at 24, 48, and 72 hours after therapy initiation. The presence of granulocytes had a dramatic effect on the antimicrobial effect of tedizolid. Dose response, demonstrated by the ratio of the area under the curve over the minimum inhibitory concentration, was on average > 25-fold for nonneutropenic vs neutropenic models. Near maximal effect of the nonneutropenic group, irrespective of duration of therapy, was achieved at the lowest dose tested (an exposure of about 200-mg tedizolid phosphate per day in humans).This study suggests that immunocompromised patients may warrant higher doses of tedizolid than the currently FDA-approved dose due to a decreased number of granulocytes available for modulating bacterial infections.15
Use of tedizolid doses higher than that which is FDA-approved may negate the favorable AE profile. A phase 1 clinical study was conducted to evaluate the safety, tolerability, and pharmacokinetics of tedizolid compared with those of linezolid in 40 healthy volunteers in a 21-day multiple ascending dose study.16 Subjects were stratified into 5 treatment cohorts: 200-, 300-, or 400-mg tedizolid orally once a day, 600-mg linezolid orally twice a day, and placebo. Tedizolid given at 200 mg had a hematologic safety profile similar to that of placebo. However, mean platelet counts decreased over time in a dose-dependent manner for tedizolid, with the 400-mg tedizolid and linezolid groups reporting similar reductions in platelet counts.16
Some evidence is available examining linezolid in neutropenic patients. Rafailidis and colleagues reviewed available literature regarding linezolid in neutropenic patients with Gram-positive infections. Evaluation of linezolid administration at usual doses to 438 neutropenic patients from 2 prospective comparative studies, a prospective cohort study, 2 retrospective studies, and 8 case reports was performed. Results of the evaluation revealed a clinical cure rate between 57% and 87% in the intention-to-treat population of the prospective studies.17 Given the similarities in bacterial spectrum of activity between linezolid and tedizolid, it may be reasonable to infer that tedizolid’s decreased myelosuppression profile would make it useful in the setting of neutropenia in immunocompromised patients.
There is little evidence regarding the use of tedizolid in immunocompromised patients, as only 2 case reports were found. The first described a 60-year-old male postrenal transplant complicated with VRE bacteremia, rhabdomyolysis, and thrombocytopenia. This patient was treated with prolonged tedizolid 200 mg daily due to multiple contraindications for treatment with other antibiotics. The patient was cured with a 14-day course of tedizolid without any noted AEs.18
The second identified case report described the use of tedizolid for the treatment of central nervous system (CNS) manifestations secondary to nocardiosis. Effective treatment of CNS nocardiosis requires high concentrations and prolonged duration of antimicrobial exposure. This case report described a 68-year-old, chronically immunocompromised female patient with multiple myeloma who was hospitalized for 3 months for the treatment of a CNS nocardiosis infection. After discharge, the patient was treated with an oral regimen of 200-mg tedizolid daily in combination with sulfamethoxazole/trimethoprim (800 mg/160 mg) 3 times daily. After 6 months of combination therapy, magnetic resonance imaging revealed complete resolution of nocardiosis-related central lesions. Although the patient’s malignancy advanced during combination antibiotic therapy, the patient’s absolute neutrophil count remained stable and showed an increase in absolute CD4+ cell counts with no other documented AEs.19
Tedizolid is the latest FDA-approved oxazolidinone antibiotic for susceptible Gram-positive acute bacterial skin and skin structure infections. It has a simplified and shorter duration of treatment and imparts similar AEs at improved rates compared with that of linezolid, most notably in relation to hematologic AEs. Due to the lack of established literature and an agreed-upon dosing strategy for the use of tedizolid in immunocompromised patients, tedizolid therapy for Gram-positive infections in immunocompromised patients should be reserved for salvage therapy when more established Gram-positive antibiotic agents lack efficacy or when patient contraindications to their use exist.
Immunocompromised patients are often susceptible to opportunistic infections, including those caused by multidrug-resistant organisms (MDROs). Transplant recipients are at high risk for developing infections due to lifelong immunosuppressive therapy.1,2 Additionally, patients receiving chemotherapy and those with HIV and AIDS are in an immunocompromised state.3-8
Regardless of the etiology for immunosuppression, decreased absolute neutrophil and platelet counts are seen in this condition. Although immunosuppressed individuals may be at increased risk of Gram-negative or Gram-positive infections, this review focuses on the treatment of Gram-positive bacterial infections. Of particular concern are opportunistic infections caused by Gram-positive MDROs, including methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus species (VRE), penicillin-resistant Streptococcus pneumoniae, and Nocardia species. Treatment of infections in the immunocompromised patient population warrants careful antimicrobial selection to ensure that a patient’s immune system is not further compromised due to adverse effects (AEs) secondary to therapy. As such, clinicians are exploring alternative antimicrobials, such as tedizolid, to treat various opportunistic infections.
Recently, requests at the William S. Middleton Memorial Veterans Hospital in Madison, Wisconsin, for off-label use of tedizolid have increased despite having other cheaper alternatives with comparable Gram-positive coverage. This review examines available literature regarding off-label use of tedizolid with a focus on use in immunocompromised patients.
Tedizolid phosphate (Sivextro) is an oxazolidinone antibiotic prodrug that joined linezolid as the second in its class in 2014. Oxazolidinones inhibit bacterial protein synthesis by binding to the 50S subunit of bacterial ribosomes in Gram-positive bacteria and are often used to treat MRSA and VRE infections.9 In vitro, oxazolidinones have shown bacteriostatic activity against Enterococcus and Staphylococcus species while exhibiting bactericidal activity against most Streptococcus species.10 Tedizolid has a US Food and Drug Administration (FDA) -approved, simplified dosing profile of 200 mg daily for 6 days compared with linezolid 400 to 600 mg twice daily for 10 to 14 days. Both medications are highly bioavailable with direct IV to oral conversion.11,12 Potential, expanded use of tedizolid against Gram-positive MDROs rests on a more favorable AE profile than does its linezolid predecessor. Tedizolid has been associated with less antibiotic-induced myelosuppression, which could prove valuable for immunocompromised patients.13
Tedizolid is approved for the sole indication of acute bacterial skin and skin structure infections (ABSSSI), whereas its predecessor has many approved indications and has been used extensively for off-label indications (Table).
Adverse reactions, as determined by 2 phase 2 and 2 phase 3 clinical trials evaluating 1,050 patients treated with tedizolid and 662 patients treated with linezolid, were similar between the oxazolidinones. Nausea was the most common AE and was reported in 8% and 12% of patients taking tedizolid and linezolid, respectively. Other common AEs (1%-6%) reported for both agents included vomiting, diarrhea, headache, and dizziness.11 Myelosuppression, peripheral neuropathy, and optic nerve disorders were the most common severe AEs reported with oxazolidinones. Tedizolid demonstrated a significantly decreased incidence of neutropenia (3%), defined by absolute neutrophil count 9/L compared with that of linezolid (7%) (P = .024).13 Evaluation of peripheral neuropathy and optic nerve disorders within the tedizolid and linezolid groups revealed similar incidences (peripheral neuropathy 1.2% vs 0.6%; optic nerve disorders 0.3% vs 0.2%, respectively).11
There is one preclinical trial that described the use of tedizolid in a murine model. A murine model study compared the antistaphylococcal killing effect of doses of tedizolid equivalent to human exposures ranging from 200 to 3,200 mg/d in both granulocytopenic and normal mice. The mice were evaluated at 24, 48, and 72 hours after therapy initiation. The presence of granulocytes had a dramatic effect on the antimicrobial effect of tedizolid. Dose response, demonstrated by the ratio of the area under the curve over the minimum inhibitory concentration, was on average > 25-fold for nonneutropenic vs neutropenic models. Near maximal effect of the nonneutropenic group, irrespective of duration of therapy, was achieved at the lowest dose tested (an exposure of about 200-mg tedizolid phosphate per day in humans).This study suggests that immunocompromised patients may warrant higher doses of tedizolid than the currently FDA-approved dose due to a decreased number of granulocytes available for modulating bacterial infections.15
Use of tedizolid doses higher than that which is FDA-approved may negate the favorable AE profile. A phase 1 clinical study was conducted to evaluate the safety, tolerability, and pharmacokinetics of tedizolid compared with those of linezolid in 40 healthy volunteers in a 21-day multiple ascending dose study.16 Subjects were stratified into 5 treatment cohorts: 200-, 300-, or 400-mg tedizolid orally once a day, 600-mg linezolid orally twice a day, and placebo. Tedizolid given at 200 mg had a hematologic safety profile similar to that of placebo. However, mean platelet counts decreased over time in a dose-dependent manner for tedizolid, with the 400-mg tedizolid and linezolid groups reporting similar reductions in platelet counts.16
Some evidence is available examining linezolid in neutropenic patients. Rafailidis and colleagues reviewed available literature regarding linezolid in neutropenic patients with Gram-positive infections. Evaluation of linezolid administration at usual doses to 438 neutropenic patients from 2 prospective comparative studies, a prospective cohort study, 2 retrospective studies, and 8 case reports was performed. Results of the evaluation revealed a clinical cure rate between 57% and 87% in the intention-to-treat population of the prospective studies.17 Given the similarities in bacterial spectrum of activity between linezolid and tedizolid, it may be reasonable to infer that tedizolid’s decreased myelosuppression profile would make it useful in the setting of neutropenia in immunocompromised patients.
There is little evidence regarding the use of tedizolid in immunocompromised patients, as only 2 case reports were found. The first described a 60-year-old male postrenal transplant complicated with VRE bacteremia, rhabdomyolysis, and thrombocytopenia. This patient was treated with prolonged tedizolid 200 mg daily due to multiple contraindications for treatment with other antibiotics. The patient was cured with a 14-day course of tedizolid without any noted AEs.18
The second identified case report described the use of tedizolid for the treatment of central nervous system (CNS) manifestations secondary to nocardiosis. Effective treatment of CNS nocardiosis requires high concentrations and prolonged duration of antimicrobial exposure. This case report described a 68-year-old, chronically immunocompromised female patient with multiple myeloma who was hospitalized for 3 months for the treatment of a CNS nocardiosis infection. After discharge, the patient was treated with an oral regimen of 200-mg tedizolid daily in combination with sulfamethoxazole/trimethoprim (800 mg/160 mg) 3 times daily. After 6 months of combination therapy, magnetic resonance imaging revealed complete resolution of nocardiosis-related central lesions. Although the patient’s malignancy advanced during combination antibiotic therapy, the patient’s absolute neutrophil count remained stable and showed an increase in absolute CD4+ cell counts with no other documented AEs.19
Tedizolid is the latest FDA-approved oxazolidinone antibiotic for susceptible Gram-positive acute bacterial skin and skin structure infections. It has a simplified and shorter duration of treatment and imparts similar AEs at improved rates compared with that of linezolid, most notably in relation to hematologic AEs. Due to the lack of established literature and an agreed-upon dosing strategy for the use of tedizolid in immunocompromised patients, tedizolid therapy for Gram-positive infections in immunocompromised patients should be reserved for salvage therapy when more established Gram-positive antibiotic agents lack efficacy or when patient contraindications to their use exist.
1. Fishman JA, Issa NC. Infection in organ transplantation: risk factors and evolving patterns of infection. Infect Dis Clin North Am. 2010;24(2):273-283.
2. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614.
3. Nightingale SD, Byrd LT, Southern PM, Jockusch JD, Cal SX, Wynne BA. Incidence of mycobacterium avium-intracellulare complex bacteremia in human immunodeficiency virus-positive patients. J Infect Dis. 1992;165(6):1082-1085.
4. O’Brien S, Kantarjian H, Beran M, et al. Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood. 1993;82(6):1695-1700.
5. Anaissie E, Kontoyiannis DP, Kantarjian H, Elting L, Robertson LE, Keating M. Listeriosis in patients with chronic lymphocytic leukemia who were treated with fludarabine and prednisone. Ann Intern Med. 1992;117(6):466-469.
6. Morrison VA, Rai KR, Peterson BL, et al. Impact of therapy with chlorambucil, fludarabine, or fludarabine plus chlorambucil on infections in patients with chronic lymphocytic leukemia: Intergroup Study Cancer and Leukemia Group B 9011. J Clin Oncol. 2001;19(16):3611-3621.
7. Nucci M, Anaissie E. Infections in patients with multiple myeloma in the era of high-dose therapy and novel agents. Clin Infect Dis. 2009;49(8):1211-1225.
8. Naseer M, Dailey FE, Juboori AA, Samiullah S, Tahan V. Epidemiology, determinants, and managements of AIDS cholangiopathy: a review. World J Gastroenterol. 2018;24(7):767-774.
9. Radunz S, Juntermanns B, Kaiser GM, et al. Efficacy and safety of linezolid in liver transplant patients. Transpl Infect Dis. 2011;13(4):353-358.
10. Roger C, Roberts JA, Muller L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clin Pharmacokinet. 2018;57(5):559-575.
11. Sivextro [package insert]. Whitehouse Station, NJ: Merck & Co Inc; 2016.
12. Zyvox [package insert]. New York, NY: Pfizer Inc; 2018.
13. Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomized, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696-705.
14. Liu C, Bayer A, Cosgrove S, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
15. Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300-5305.
16. Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother. 2016;71(9):2553-2558.
17. Rafailidis PI, Kouranos VD, Christodoulou C, Falagas ME. Linezolid for patients with neutropenia: are bacteriostatic agents appropriate? Expert Rev Anti Infect Ther. 2009;7(4):415-422.
18. Sudhindra P, Lee L, Wang G, Dhand A. Tedizolid for treatment of enterococcal bacteremia. Open Forum Infect Dis. 2016;3(suppl 1):1344.
19. Matin A, Sharma S, Mathur P, Apewokin SK. Myelosuppression-sparing treatment of central nervous system nocardiosis in a multiple myeloma patient utilizing a tedizolid-based regimen: a case report. Int J Antimicrob Agents. 2017;49(4):488-492.
20. Dryden MS. Alternative clinical indications for novel antibiotics licensed for skin and soft tissue infections? Curr Opin Infect Dis. 2015;28(2):117-124.
21. Milstein M, Brzezinski A, Varaine F, Mitnick CD. (Re)moving the needle: prospects for all-oral treatment for multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2016;20(12):18-23.
22. Winthrop KL, Ku JH, Marras TK, et al. The tolerability of linezolid in the treatment of nontuberculous mycobacterial disease. Eur Respir J. 2015;45(4):1177-1179.
23. Yuste JR, Bertó J, Del Pozo JL, Leiva J. Prolonged use of tedizolid in a pulmonary non-tuberculosis mycobacterial infection after linezlid-induced toxicity. J Antimicrob Chemother. 2017;72(2):625-628.
1. Fishman JA, Issa NC. Infection in organ transplantation: risk factors and evolving patterns of infection. Infect Dis Clin North Am. 2010;24(2):273-283.
2. Fishman JA. Infection in solid-organ transplant recipients. N Engl J Med. 2007;357(25):2601-2614.
3. Nightingale SD, Byrd LT, Southern PM, Jockusch JD, Cal SX, Wynne BA. Incidence of mycobacterium avium-intracellulare complex bacteremia in human immunodeficiency virus-positive patients. J Infect Dis. 1992;165(6):1082-1085.
4. O’Brien S, Kantarjian H, Beran M, et al. Results of fludarabine and prednisone therapy in 264 patients with chronic lymphocytic leukemia with multivariate analysis-derived prognostic model for response to treatment. Blood. 1993;82(6):1695-1700.
5. Anaissie E, Kontoyiannis DP, Kantarjian H, Elting L, Robertson LE, Keating M. Listeriosis in patients with chronic lymphocytic leukemia who were treated with fludarabine and prednisone. Ann Intern Med. 1992;117(6):466-469.
6. Morrison VA, Rai KR, Peterson BL, et al. Impact of therapy with chlorambucil, fludarabine, or fludarabine plus chlorambucil on infections in patients with chronic lymphocytic leukemia: Intergroup Study Cancer and Leukemia Group B 9011. J Clin Oncol. 2001;19(16):3611-3621.
7. Nucci M, Anaissie E. Infections in patients with multiple myeloma in the era of high-dose therapy and novel agents. Clin Infect Dis. 2009;49(8):1211-1225.
8. Naseer M, Dailey FE, Juboori AA, Samiullah S, Tahan V. Epidemiology, determinants, and managements of AIDS cholangiopathy: a review. World J Gastroenterol. 2018;24(7):767-774.
9. Radunz S, Juntermanns B, Kaiser GM, et al. Efficacy and safety of linezolid in liver transplant patients. Transpl Infect Dis. 2011;13(4):353-358.
10. Roger C, Roberts JA, Muller L. Clinical pharmacokinetics and pharmacodynamics of oxazolidinones. Clin Pharmacokinet. 2018;57(5):559-575.
11. Sivextro [package insert]. Whitehouse Station, NJ: Merck & Co Inc; 2016.
12. Zyvox [package insert]. New York, NY: Pfizer Inc; 2018.
13. Moran GJ, Fang E, Corey GR, Das AF, De Anda C, Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomized, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696-705.
14. Liu C, Bayer A, Cosgrove S, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18-e55.
15. Drusano GL, Liu W, Kulawy R, Louie A. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300-5305.
16. Lodise TP, Bidell MR, Flanagan SD, Zasowski EJ, Minassian SL, Prokocimer P. Characterization of the haematological profile of 21 days of tedizolid in healthy subjects. J Antimicrob Chemother. 2016;71(9):2553-2558.
17. Rafailidis PI, Kouranos VD, Christodoulou C, Falagas ME. Linezolid for patients with neutropenia: are bacteriostatic agents appropriate? Expert Rev Anti Infect Ther. 2009;7(4):415-422.
18. Sudhindra P, Lee L, Wang G, Dhand A. Tedizolid for treatment of enterococcal bacteremia. Open Forum Infect Dis. 2016;3(suppl 1):1344.
19. Matin A, Sharma S, Mathur P, Apewokin SK. Myelosuppression-sparing treatment of central nervous system nocardiosis in a multiple myeloma patient utilizing a tedizolid-based regimen: a case report. Int J Antimicrob Agents. 2017;49(4):488-492.
20. Dryden MS. Alternative clinical indications for novel antibiotics licensed for skin and soft tissue infections? Curr Opin Infect Dis. 2015;28(2):117-124.
21. Milstein M, Brzezinski A, Varaine F, Mitnick CD. (Re)moving the needle: prospects for all-oral treatment for multidrug-resistant tuberculosis. Int J Tuberc Lung Dis. 2016;20(12):18-23.
22. Winthrop KL, Ku JH, Marras TK, et al. The tolerability of linezolid in the treatment of nontuberculous mycobacterial disease. Eur Respir J. 2015;45(4):1177-1179.
23. Yuste JR, Bertó J, Del Pozo JL, Leiva J. Prolonged use of tedizolid in a pulmonary non-tuberculosis mycobacterial infection after linezlid-induced toxicity. J Antimicrob Chemother. 2017;72(2):625-628.