User login
When to adjust the dosing of psychotropics in patients with renal impairment
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
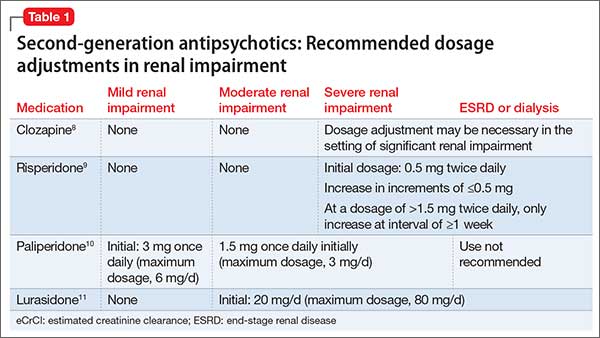
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
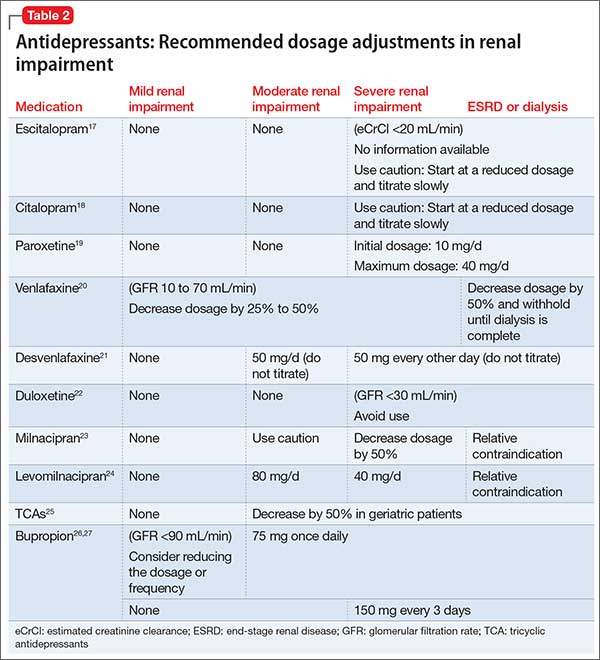
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
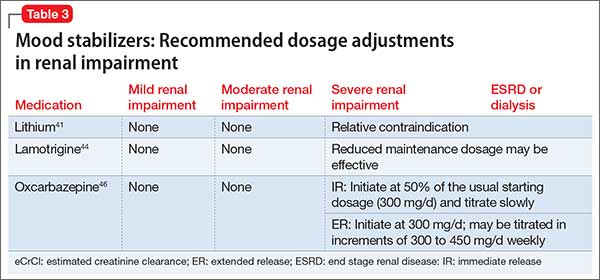
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
Renal disease can play a large role in altering the pharmacokinetics of medications, especially in elimination or clearance and plasma protein binding. Specifically, renal impairment decreases the plasma protein binding secondary to decreased albumin and retention of urea, which competes with medications to bind to the protein.1
Electrolyte shifts—which could lead to a fatal arrhythmia—are common among patients with renal impairment. The risk can be further increased in this population if a patient is taking a medication that can induce arrhythmia. If a drug is primarily excreted by the kidneys, elimination could be significantly altered, especially if the medication has active metabolites.1
Normal renal function is defined as an estimated creatinine clearance (eCrCl) of >80 mL/min. Renal impairment is classified as:
- mild: eCrCl, 51 to 80 mL/min
- moderate: eCrCl, 31 to 50 mL/min
- severe: eCrCl, ≤30 mL/min
- end-stage renal disease (ESRD): eCrCl, <10 mL/min.2
Overall, there is minimal information about the effects of renal disease on psychotropic therapy; our goal here is to summarize available data. We have created quick reference tables highlighting psychotropics that have renal dosing recommendations based on manufacturers’ package inserts.
Antipsychotics
First-generation antipsychotics (FGAs). Dosage adjustments based on renal function are not required for any FGA, according to manufacturers’ package inserts. Some of these antipsychotics are excreted in urine, but typically as inactive metabolites.
Although there are no dosage recommendations based on renal function provided by the labeling, there has been concern about the use of some FGAs in patients with renal impairment. Specifically, concerns center around the piperidine phenothiazines (thioridazine and mesoridazine) because of the increased risk of electrocardiographic changes and medication-induced arrhythmias in renal disease due to electrolyte imbalances.3,4 Additionally, there is case evidence5 that phenothiazine antipsychotics could increase a patient’s risk for hypotension in chronic renal failure. Haloperidol is considered safe in renal disease because <1% of the medication is excreted unchanged through urine.6
Second-generation antipsychotics (SGAs). Overall, SGAs are considered safe in patients with renal disease. Most SGAs undergo extensive hepatic metabolism before excretion, allowing them to be used safely in patients with renal disease.
Sheehan et al7 analyzed the metabolism and excretion of SGAs, evaluating 8 antipsychotics divided into 4 groups: (1) excretion primarily as an unchanged drug in urine, (2) changed drug in urine, (3) changed drug in feces, (4) and unchanged drug in feces.
- Paliperidone was found to be primarily excreted as an unchanged drug in urine.
- Clozapine, iloperidone, olanzapine, quetiapine, and risperidone all were found to be primarily excreted as a changed drug in urine.
- Aripiprazole and ziprasidone were found to be primarily excreted as a changed drug in feces.
The manufacturers’ package inserts for clozapine, paliperidone, risperidone, and lurasidone have recommended dosage adjustments based on renal function (Table 1).8-11
Ziprasidone. Although ziprasidone does not have a recommended renal dosage adjustment, caution is recommended because of the risk of electrocardiographic changes and potential for medication-induced arrhythmias in patients with electrolyte disturbances secondary to renal disease. A single-dosage study of ziprasidone by Aweeka et al12 demonstrated that the pharmacokinetics of ziprasidone are unchanged in patients with renal impairment.
Asenapine. A small study by Peeters et al13 evaluated the pharmacokinetics of asenapine in hepatic and renal impairment and found no clinically relevant changes in asenapine’s pharmacokinetics among patients with any level of renal impairment compared with patients with normal renal function.
Aripiprazole. Mallikaarjun et al14 completed a small study evaluating the pharmacokinetics of aripiprazole in patients with renal impairment. They found that the pharmacokinetics of aripiprazole in these patients is no different than it is in patients with normal renal function who are taking aripiprazole.
Quetiapine. Thyrum et al15 conducted a similar study with quetiapine, which showed no significant difference detected in the pharmacokinetics of quetiapine in patients with renal impairment. Additionally, quetiapine had no negative effect on patients’ creatinine clearance.
Lurasidone. During clinical trials of lurasidone in patients with mild, moderate, and severe renal impairment, the mean Cmax and area under the curve was higher compared with healthy patients, which led to recommended dosage adjustments in patients with renal impairment.11
As mentioned above, renal impairment decreases the protein binding percentage of medications. Hypothetically, the greater the protein binding, the lower the recommended dosage in patients with renal impairment because the free or unbound form correlates with efficacy and toxicity. Most FGAs and SGAs have the protein-binding characteristic of ≥90%.16 Although it seems this characteristic should result in recommendations to adjust dosage based on renal function, the various pharmacokinetic studies of antipsychotics have not shown this factor to play a role in the manufacturers’ recommendations.
Antidepressants
Comorbidity rates of depression in patients with renal disease range from 14% to 30%, making use of antidepressants in renal disease common.4 Antidepressants primarily are metabolized hepatically and excreted renally. Table 217-27 summarizes recommended dosing adjustments for antidepressants.
Selective serotonin reuptake inhibitors.Escitalopram is the (S)-enantiomer of the racemic antidepressant citalopram, both of which have been shown to decrease renal clearance in patients with mild or moderate renal impairment. However, according to the package insert, no dosage adjustments are needed.17 No extensive studies have been conducted on escitalopram or citalopram, but each should be initiated at a reduced dosage and the titration schedule should be prolonged in patients with severe renal impairment or ESRD.17,18
The plasma concentration of paroxetine has been noted to be elevated in patients with severe renal impairment, and the half-life can increase to nearly 50%.4 Paroxetine should be initiated at 10 mg/d, and then titrated slowly in patients with severe renal impairment.19,28
The pharmacokinetics of fluoxetine are unchanged in any stage of renal impairment. Patients in active renal dialysis report good tolerability and efficacy.4
Serotonin-norepinephrine reuptake inhibitors. Venlafaxine and its metabolite O-desmethylvenlafaxine (desvenlafaxine) are primarily excreted via renal elimination. Studies have shown that mild renal impairment can have an effect on plasma levels of the drug, and that moderate or severe impairment can increase the venlafaxine plasma concentration. According to the package insert, a dosage reduction of 50% is recommended for desvenlafaxine and venlafaxine.20,21
No significant pharmacokinetic changes with duloxetine have been noted in patients with mild or moderate renal impairment.22 However, duloxetine’s major metabolites, which are excreted renally, have been measured to be as much as 7 to 9 times higher in patients with ESRD compared with healthy subjects; therefore, it is recommended to avoid duloxetine in patients with severe renal disease.4,22 Our review of the literature produced limited recommendations on dosing milnacipran and its enantiomer levomilnacipran in renally impaired patients. The milnacipran package insert cautions its use in moderate renal impairment and recommends a 50% dosage reduction to 100 mg/d (50 mg twice daily) in patients with severe renal impairment.23 Dosage recommendations for levomilnacipran are 80 mg/d for moderate renal impairment and 40 mg/d for severe impairment. Both agents have relative contraindications for ESRD.23,24
Tricyclic antidepressants (TCAs) are predominantly metabolized hepatically, glucuronidated, and then eliminated renally. Desipramine, imipramine, and nortriptyline have nonspecific package insert recommendations for modified dosing in geriatric patients because of an age-related decrease in renal clearance.29-31 Review articles assert that elevated glucuronidated metabolites could increase patients’ sensitivity to side effects of TCAs. Because of concerns regarding elevated glucuronidated metabolites, it has been proposed to initiate TCAs at a low dosage, titrate slowly, and maintain the lowest effective dosage in patients with renal impairment.25
Monoamine oxidase inhibitors (MAOIs) and other antidepressants. The package inserts of the MAOIs isocarboxazid, phenelzine, selegiline, and tranylcypromine provide limited data and dosage recommendations for use in the context of renal impairment.32-36 Isocarboxazid should not be used in patients with severe renal impairment, according to the prescribing information.32 There are no dosing recommendations for transdermal selegiline in mild, moderate, or severe renal impairment.37 Extra vigilance is required when using MAOIs in patients with renal disease because of an increased risk of dialysis-induced hypotension (orthostatic hypotension is a common adverse effect of MAOIs).38
Bupropion is primarily metabolized hepatically to the active metabolite hydroxybupropion. Plasma levels of this metabolite at steady state are reported to be 10 times greater than bupropion’s concentration levels in healthy subjects; plasma levels are further increased in mild renal impairment.26 Hydroxybupropion is not dialyzable, which can increase the risk of toxicity with bupropion therapy in patients with renal impairment.3 If bupropion effectively treats depression in patients with declining renal function, specifically severe renal impairment and ESRD, then decreasing the dosage to 150 mg every 3 days is recommended to lessen the risk of toxicity. 27
Mood stabilizers
Lithium has the most published literature on dosing adjustments with renal impairment. Many providers are inclined to discontinue lithium use at the first sign of any change in renal function; however, monitoring, prevention, and treatment guidelines for lithium are well established after many years of research and clinical use.39 Lithium’s prescribing information recommends dosage adjustment in mild to moderate renal impairment and lists severe renal impairment and ESRD as relative contraindications.40
A recent study proposes more assertive use of lithium in patients with renal impairment of any severity. Rej et al41 compared continued lithium treatment to discontinuing treatment in geriatric patients with chronic renal failure, and reported (1) a statistically insignificant difference in renal function between groups at 2 years and (2) a “trending decrease” in renal function at 5 years in the lithium treatment group. With closely monitored plasma levels, lithium treatment is considered a workable treatment for patients with moderate renal impairment when mood stabilizer treatment has been effective.42
Lamotrigine and its main glucuronidated metabolite, lamotrigine-2N-glucuronide (L-2-N-G), are primarily excreted renally. In severe renal impairment and ESRD, the L-2-N-G levels are elevated but are not pharmacologically active and, therefore, do not affect plasma concentration or efficacy of lamotrigine.43 Although data are limited regarding the use of lamotrigine in severe renal impairment and ESRD, Kaufman44 reported a 17% to 20% decrease in concentration after dialysis—suggesting that post-dialysis titration might be needed in these patients.
Oxcarbazepine is metabolized by means of cytosolic enzymes in the liver to its primary pharmacologically active metabolite, 10-monohydroxy, which is further metabolized via glucuronidation and then renally excreted. There are no dosage adjustment recommendations for patients with an eCrCl >30 mL/min.45 Rouan et al46 suggest initiating oxcarbazepine at 50% of the recommended dosage and following a longer titration schedule in patients with an eCrCl 10 to 30 mL/min. No dosing suggestions for severe renal impairment and ESRD were provided because of study limitations; however, the general recommendation for psychotropic agents in patients in a severe stage of renal impairment is dosage reduction with close monitoring.46
Table 341,44,46 summarizes dosage adjustments for mood stabilizers in patients with renal impairment.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
1. Levy G. Pharmacokinetics in renal disease. Am J Med. 1977;62(4):461-465.
2. Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 3. Effects of renal and hepatic impairment. J Psychiatr Pract. 2012;18(6):430-437.
3. Cohen LM, Tessier EG, Germain MJ, et al. Update on psychotropic medication use in renal disease. Psychosomatics. 2004;45(1):34-48.
4. Baghdady NT, Banik S, Swartz SA, et al. Psychotropic drugs and renal failure: translating the evidence for clinical practice. Adv Ther. 2009;26(4):404-424.
5. Sheehan J, White A, Wilson R. Hazards of phenothiazines in chronic renal failure. Ir Med J. 1982;75(9):335.
6. Haloperidol [monograph]. In: Micromedex Drugdex [online database]. Greenwood Village, CO: Truven Health Analytics. Accessed December 17, 2014.
7. Sheehan JJ, Sliwa JK, Amatniek JC, et al. Atypical antipsychotic metabolism and excretion. Curr Drug Metab. 2010;11(6):516-525.
8. Clozaril [package insert]. East Hanover, NJ: Novartis Pharmaceuticals; 2014.
9. Risperdal [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
10. Invega [package insert]. Titusville, NJ: Janssen Pharmaceuticals; 2014.
11. Latuda [package insert]. Fort Lee, NJ: Sunovion Pharmaceuticals; 2013.
12. Aweeka F, Jayesekara D, Horton M, et al. The pharmacokinetics of ziprasidone in subjects with normal and impaired renal function. Br J Clin Pharmacol. 2004;49(suppl 1):27S-33S.
13. Peeters P, Bockbrader H, Spaans E, et al. Asenapine pharmacokinetics in hepatic and renal impairment. Clin Pharmacol. 2011;50(7):471-481.
14. Mallikaarjun S, Shoaf SE, Boulton DW, et al. Effects of hepatic or renal impairment on the pharmacokinetics of aripiprazole. Clin Pharmacokinet. 2008;47(8):533-542.
15. Thyrum PT, Wong YW, Yeh C. Single-dose pharmacokinetics of quetiapine in subjects with renal or hepatic impairment. Prog Neuropsychopharmacol Biol Psychiatry. 2000;24(4):521-533.
16. Lexi-Drugs. Lexicomp. Hudson, OH: Wolters Kluwer Health, Inc. http://online.lexi.com. Accessed May 28, 2015.
17. Lexapro [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
18. Celexa [package insert]. Forest Pharmaceuticals, Inc.: St. Louis, MO; 2014.
19. Paxil [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
20. Effexor [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2010.
21. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals Inc.; 2014.
22. Cymbalta [package insert]. Indianapolis, IN: Lilly USA, LLC; 2014.
23. Savella [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2013.
24. Fetzima [package insert]. St. Louis, MO: Forest Pharmaceuticals, Inc.; 2014.
25. Kurella M, Bennett WM, Chertow GM. Analgesia in patients with ESRD: a review of available evidence. Am J Kidney Dis. 2003;42(2):217-228.
26. Wellbutrin [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
27. Worrall SP, Almond MK, Dhillon S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron Clin Pract. 2004;97(3):c83-c89.
28. Nagler EV, Webster AC, Vanholder R, et al. Antidepressants for depression in stage 3-5 chronic kidney disease: a systematic review of pharmacokinetics, efficacy and safety with recommendations by European Renal Best Practice (ERBP). Nephrol Dial Transplant. 2012;27(10):3736-3745.
29. Norpramin. [package insert] Bridgewater, NJ: Sanofi-Aventis U.S. LLC; 2014.
30. Tofranil [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
31. Pamelor [package insert]. Hazelwood, MO: Mallinckrodt Inc.; 2014.
32. Marplan [package insert]. Parsippany, NJ: Validus Pharmaceuticals, LLC; 2012.
33. Nardil [package insert]. New York, NY: Parke-Davis Division of Pfizer Inc.; 2009.
34. EMSAM [package insert]. Morgantown, WV: Mylan Specialty, L.P.; 2014.
35. Eldepryl [package insert]. Morgantown, WV: Somerset Pharmaceuticals, Inc.; 2009.
36. Parnate [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2008.
37. Culpepper L. Reducing the burden of difficult-to-treat major depressive disorder: revisiting monoamine oxidase inhibitor therapy. Prim Care Companion CNS Disord. 2013;15(5). doi: 10.4088/PCC.13r01515.
38. Tossani E, Cassano P, Fava M. Depression and renal disease. Semin Dial. 2005;18(2):73-81.
39. Young AH, Hammond JM. Lithium in mood disorders: increasing evidence base, declining use? Br J Psychiatry. 2007;191:474-476.
40. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
41. Rej S, Looper K, Segal M. The effect of serum lithium levels on renal function in geriatric outpatients: a retrospective longitudinal study. Drugs Aging. 2013;30(6):409-415.
42. Malhi GS, Tanious M, Das P, et al. The science and practice of lithium therapy. Aust N Z J Psychiatry. 2012;46(3):192-211.
43. Lamictal [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2014.
44. Kaufman KR. Lamotrigine and hemodialysis in bipolar disorder: case analysis of dosing strategy with literature review. Bipolar Disord. 2010;12(4):446-449.
45. Trileptal [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2014.
46. Rouan MC, Lecaillon JB, Godbillon J, et al. The effect of renal impairment on the pharmacokinetics of oxcarbazepine and its metabolites. Eur J Clin Pharmacol. 1994;47(2):161-167.
Using lipid guidelines to manage metabolic syndrome for patients taking an antipsychotic
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?

Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
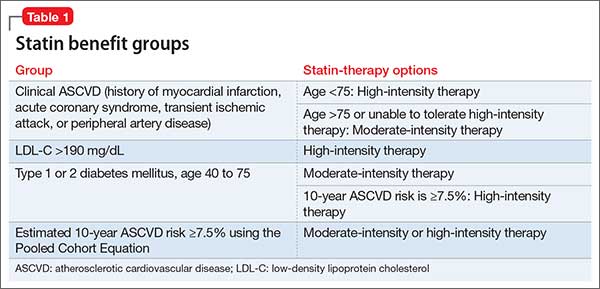
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
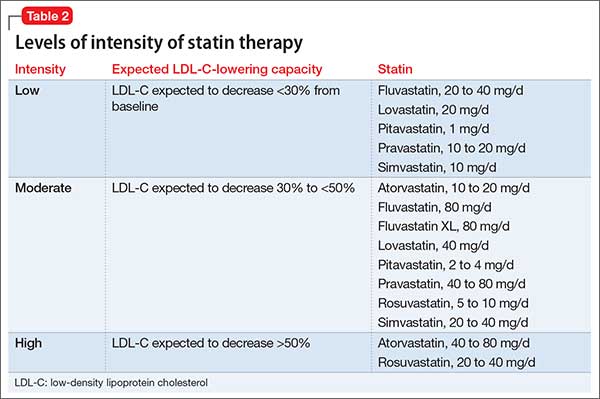
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
Your patient who has schizophrenia, Mr. W, age 48, requests that you switch him from olanzapine, 10 mg/d, to another antipsychotic because he gained 25 lb over 1 month taking the drug. He now weighs 275 lb. Mr. W reports smoking at least 2 packs of cigarettes a day and takes lisinopril, 20 mg/d, for hypertension. You decide to start risperidone, 1 mg/d. First, however, your initial work-up includes:
- high-density lipoprotein (HDL), 24 mg/dL
- total cholesterol, 220 mg/dL
- blood pressure, 154/80 mm Hgwaist circumference, 39 in
- body mass index (BMI), 29
- hemoglobin A1c, of 5.6%.
A prolactin level is pending.
How do you interpret these values?
Metabolic syndrome is defined as the cluster of central obesity, insulin resistance, hypertension, and dyslipidemia. Metabolic syndrome increases a patient's risk of diabetes 5-fold and cardiovascular disease 3-fold.1 Physical inactivity and eating high-fat foods typically precede weight gain and obesity that, in turn, develop into insulin resistance, hypertension, and dyslipidemia.1
Patients with severe psychiatric illness have an increased rate of mortality from cardiovascular disease, compared with the general population.2-4 The cause of this phenomenon is multifactorial: In general, patients with severe mental illness receive insufficient preventive health care, do not eat a balanced diet, and are more likely to smoke cigarettes than other people.2-4
Also, compared with the general population, the diet of men with schizophrenia contains less vegetables and grains and women with schizophrenia consume less grains. An estimated 70% of patients with schizophrenia smoke.4 As measured by BMI, 86% of women with schizophrenia and 70% of men with schizophrenia are overweight or obese.4
Antipsychotics used to treat severe mental illness also have been implicated in metabolic syndrome, specifically second-generation antipsychotics (SGAs).5 Several theories aim to explain how antipsychotics lead to metabolic alterations.
Oxidative stress. One theory centers on the production of oxidative stress and the consequent reactive oxygen species that form after SGA treatment.6
Mitochondrial function. Another theory assesses the impact of antipsychotic treatment on mitochondrial function. Mitochondrial dysfunction causes decreased fatty acid oxidation, leading to lipid accumulation.7
The culminating affect of severe mental illness alone as well as treatment-emergent side effects of antipsychotics raises the question of how to best treat the dyslipidemia component of metabolic syndrome. This article will:
- review which antipsychotics impact lipids the most
- provide an overview of the most recent lipid guidelines
- describe how to best manage patients to prevent and treat dyslipidemia.
Impact of antipsychotics on lipids
Antipsychotic treatment can lead to metabolic syndrome; SGAs are implicated in most cases.8 A study by Liao et al9 investigated the risk of developing type 2 diabetes mellitus, hypertension, and hyperlipidemia in patients with schizophrenia who received treatment with a first-generation antipsychotic (FGA) compared with patients who received a SGA. The significance-adjusted hazard ratio for the development of hyperlipidemia in patients treated with a SGA was statistically significant compared with the general population (1.41; 95% CI, 1.09-1.83). The risk of hyperlipidemia in patients treated with a FGA was not significant.
Studies have aimed to describe which SGAs carry the greatest risk of hyperlipidemia.10,11 To summarize findings, in 2004 the American Diabetes Association (ADA) and American Psychiatric Association released a consensus statement on the impact of antipsychotic medications on obesity and diabetes.12 The statement listed the following antipsychotics in order of greatest to least impact on hyperlipidemia:
- clozapine
- olanzapine
- quetiapine
- risperidone
- ziprasidone
- aripiprazole.
To evaluate newer SGAs, a systematic review and meta-analysis by De Hert et al13 aimed to assess the metabolic risks associated with asenapine, iloperidone, lurasidone, and paliperidone. In general, the studies included in the meta-analysis showed little or no clinically meaningful differences among these newer agents in terms of total cholesterol in short-term trials, except for asenapine and iloperidone.
Asenapine was found to increase the total cholesterol level in long-term trials (>12 weeks) by an average of 6.53 mg/dL. These trials also demonstrated a decrease in HDL cholesterol (−0.13 mg/dL) and a decrease in low-density lipoprotein cholesterol (LDL-C) (−1.72 mg/dL to −0.86 mg/dL). The impact of asenapine on these lab results does not appear to be clinically significant.13,14
Iloperidone. A study evaluating the impact iloperidone on lipid values showed a statistically significant increase in total cholesterol, HDL, and LDL-C levels after 12 weeks.13,15
Overview: Latest lipid guidelines
Current literature lacks information regarding statin use for overall prevention of metabolic syndrome. However, the most recent update to the American Heart Association's guideline on treating blood cholesterol to reduce atherosclerotic cardiovascular risk in adults describes the role of statin therapy to address dyslipidemia, which is one component of metabolic syndrome.16,17
Some of the greatest changes seen with the latest blood cholesterol guidelines include:
- focus on atherosclerotic cardiovascular disease (ASCVD) risk reduction to identify 4 statin benefit groups
- transition away from treating to a target LDL value
- use of the Pooled Cohort Equation to estimate 10-year ASCVD risk, rather than the Framingham Risk Score.
Placing patients in 1 of 4 statin benefit groups
Unlike the 2002 National Cholesterol Education Program Adult Treatment Panel III (ATP III) guidelines, the latest guidelines have identified 4 statin treatment benefit groups:
- patients with clinical ASCVD (including those who have had acute coronary syndrome, stroke, or myocardial infarction, or who have stable or unstable angina, transient ischemic attacks, or peripheral artery disease, or a combination of these findings)patients with LDL-C >190 mg/dL
- patients age 40 to 75 with type 1 or type 2 diabetes mellitus
- patients with an estimated 10-year ASCVD risk of ≥7.5% that was estimated using the Pooled Cohort Equation.16,17
Table 1 represents each statin benefit group and recommended treatment options.
Selected statin therapy for each statin benefit group is further delineated into low-, moderate-, and high-intensity therapy. Intensity of statin therapy represents the expected LDL lowering capacity of selected statins. Low-intensity statin therapy, on average, is expected to lower LDL-C by <30%. Moderate-intensity statin therapy is expected to lower LDL-C by 30% to <50%. High-intensity statin therapy is expected to lower LDL-C by >50%.
When selecting treatment for patients, it is important to first determine the statin benefit group that the patient falls under, and then select the appropriate statin intensity. The categorization of the different statins based on LDL-C lowering capacity is described in Table 2.
Whenever a patient is started on statin therapy, order a liver function test and lipid profile at baseline. Repeat these tests 4 to 12 weeks after statin initiation, then every 3 to 12 months.
Transition away from treating to a target LDL-C goal
ATP III guidelines suggested that elevated LDL was the leading cause of coronary heart disease and recommended therapy with LDL-lowering medications.18 The panel that developed the 2013 lipid guideline concluded that there was no evidence that showed benefit in treating to a designated LDL-C goal.16,17 Arguably, treating to a target may lead to overtreatment in some patients and under-treatment in others. Treatment is now recommended based on statin intensity.
Using the Pooled Cohort Equation
In moving away from the Framingham Risk Score, the latest lipid guidelines established a new calculation to assess cardiovascular disease. The Pooled Cohort Equation estimates the 10-year ASCVD risk for patients based on selected risk factors: age, sex, race, lipids, diabetes, smoking status, and blood pressure. Although other potential cardiovascular disease risk factors have been identified, the Pooled Cohort Equation focused on those risk factors that have been correlated with cardiovascular disease since the 1960s.16,17,19 The Pooled Cohort Equation is intended to (1) more accurately identify higher-risk patients and (2) assess who would best benefit from statin therapy.
Recommended lab tests and subsequent treatment
With the new lipid guidelines in place to direct dyslipidemia treatment and a better understanding of how certain antipsychotics impact lipid values, the next step is monitoring parameters for patients. Before initiating antipsychotic treatment and in accordance with the 2014 National Institute for Health and Care Excellence (NICE) guidelines, baseline measurements should include weight, waist circumference, pulse, blood pressure, fasting blood glucose, hemoglobin A1c, blood lipid profile, and, if risperidone or paliperidone is initiated, prolactin level.20 Additionally, patients should be assessed at baseline for any movement disorders as well as current nutritional status, diet, and level of physical activity.
Once treatment is selected on a patient-specific basis, weight should be measured weekly for the first 6 weeks, again at 12 weeks and 1 year, and then annually. Pulse and blood pressure should be obtained 12 weeks after treatment initiation and at 1 year. Fasting blood glucose, hemoglobin A1c, and blood lipid levels should be collected 12 weeks after treatment onset, then at the 1-year mark.20 These laboratory parameters should be measured annually while the patient is receiving antipsychotic treatment.
Alternately, you can follow the monitoring parameters in the more dated 2004 ADA consensus statement:
- baseline assessment to include BMI, waist circumference, blood pressure, fasting plasma glucose, fasting lipid profile, and personal and family history
- BMI measured again at 4 weeks, 8 weeks, 12 weeks, and then quarterly
- 12-week follow-up measurement of fasting plasma glucose, fasting lipids, and blood pressure
- annual measurement of fasting blood glucose, blood pressure, and waist circumference.12
In addition to the NICE guidelines and the ADA consensus statement, use of the current lipid guidelines and the Pooled Cohort Equation to assess 10-year ASCVD risk should be obtained at baseline and throughout antipsychotic treatment. If you identify an abnormality in the lipid profile, you have several options:
- Decrease the antipsychotic dosage
- Switch to an antipsychotic considered to be less risky
- Discontinue therapy
- Implement diet and exercise
- Refer the patient to a dietitian or other clinician skilled in managing overweight or obesity and hyperlipidemia.21
Furthermore, patients identified as being in 1 of the 4 statin benefit groups should be started on appropriate pharmacotherapy. Non-statin therapy as adjunct or in lieu of statin therapy is not considered to be first-line.16
CASE CONTINUED
After reviewing Mr. W's lab results, you calculate that he has a 24% ten-year ASCVD risk, using the Pooled Cohort Equation. Following the treatment algorithm for statin benefit groups, you see that Mr. W meets criteria for high-intensity statin therapy. You stop olanzapine, switch to risperidone, 1 mg/d, and initiate atorvastatin, 40 mg/d. You plan to assess Mr. W's weight weekly over the next 6 weeks and order a liver profile and lipid profile in 6 weeks.
Related Resource
- AHA/ACC 2013 Prevention Guidelines Tools CV Risk Calculator. https://professional.heart.org/professional/GuidelinesStatements/PreventionGuidelines/UCM_457698_Prevention-Guidelines.jsp.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Atorvastatin • Lipitor
Clozapine • Clozaril
Fluvastatin • Lescol
Iloperidone • Fanapt
Lovastatin • Mevacor
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Pitavastatin • Livalo
Pravastatin • Pravachol
Quetiapine • Seroquel
Risperidone • Risperdal
Rosuvastatin • Crestor
Simvastatin • Zocor
Ziprasidone • Geodon
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. This material is the result of work supported with resources and the use of facilities at the Chillicothe Veterans Affairs Medical Center in Chillicothe, Ohio.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
1. O’Neill S, O’Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1-12.
2. McCreadie RG; Scottish Schizophrenia Lifestyle Group. Diet, smoking and cardiovascular risk in people with schizophrenia: descriptive study. Br J Psychiatry. 2003;183:534-539.
3. Correll CU, Robinson DG, Schooler NR, et al. Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP Study. JAMA Psychiatry. 2014;7(12):1350-1363.
4. Nordentoft M, Wahlbeck K, Hällgren J, et al. Excess mortality, causes of death and life expectancy in 270,770 patients with recent onset of mental disorders in Denmark, Finland and Sweden. PLoS ONE. 2013;8(1):e55176. doi: 10.1371/journal.pone.0055176.
5. Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353-362.
6. Baig MR, Navaira E, Escamilla MA, et al. Clozapine treatment causes oxidation of proteins involved in energy metabolism in lymphoblastoid cells: a possible mechanism for antipsychotic-induced metabolic alterations. J Psychiatr Pract. 2010;16(5):325-333.
7. Schrauwen P, Schrauwen-Hinderling V, Hoeks J, et al. Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):266-271.
8. Watanabe J, Suzuki Y, Someya T. Lipid effects of psychiatric medications. Curr Atheroscler Rep. 2013;15(1):292.
9. Liao HH, Chang CS, Wei WC, et al. Schizophrenia patients at higher risk of diabetes, hypertension and hyperlipidemia: a population-based study. Schizophr Res. 2011;126(1-3):110-116.
10. Lidenmayer JP, Czobor P, Volavka J, et al. Changes in glucose and cholesterol levels in patients with schizophrenia treated with typical or atypical antipsychotics. Am J Psychiatry. 2003;160(2):290-296.
11. Olfson M, Marcus SC, Corey-Lisle P, et al. Hyperlipidemia following treatment with antipsychotic medications. Am J Psychiatry. 2006;163(10):1821-1825.
12. American Diabetes Association; American Psychiatric Association; American Association of Clinical Endocrinologists, et al. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
13. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone, and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
14. Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiary. 2014;75(3):238-245.
15. Cutler AJ, Kalali AH, Weiden PJ, et al. Four-week, double-blind, placebo-and ziprasidone-controlled trial of iloperidone in patients with acute exacerbations of schizophrenia. J Clin Psychopharmacol. 2008;28(2 suppl 1):S20-S28.
16. Stone NJ, Robinson J, Lichtenstein AH, et al. 2013 ACC/AHA Guideline on the Treatment of Blood Cholesterol to Reduce Atherosclerotic Cardiovascular Risk in Adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1-S45.
17. Goff DC Jr, Lloyd-Jones DM, Bennett G, et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S49-S72.
18. National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143-3421.
19. Ioannidis JP. More than a billion people taking statins? Potential implications of the new cardiovascular guidelines. JAMA. 2014;311(5):463-464.
20. National Collaborating Centre for Mental Health. Psychosis and schizophrenia in adults: treatment and management: the NICE Guideline on Treatment and Management. https://www.nice.org.uk/guidance/cg178/evidence/full-guideline-490503565. Published 2014. Accessed June 8, 2016.
21. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013;12(9):51-54.
Is the evidence compelling for using ketamine to treat resistant depression?
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.

as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
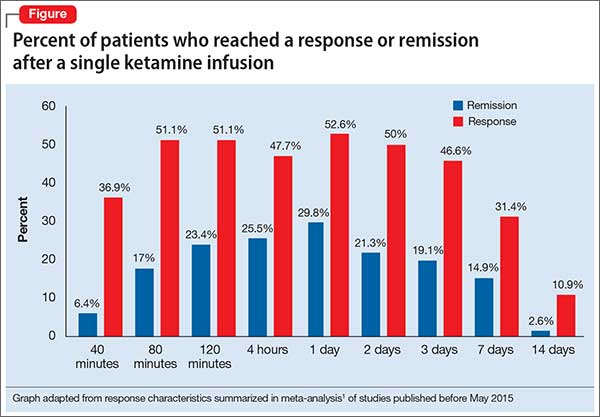
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.
as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
Ms. B, age 31, experienced her first depressive episode at age 24 during her second year of law school. These episodes are characterized by insomnia, sadness, guilt, suicidal ideation, and impaired concentration that affect her ability to function at work and interfere with her ability to maintain relationships. She has no history of mania, hypomania, or psychosis.
Ms. B has approximately 2 severe episodes a year, lasting 8 to 10 weeks. She has failed adequate (≥6 week) trials of sertraline, 200 mg/d; venlafaxine XR, 300 mg/d; bupropion XL, 450 mg/d; and vortioxetine, 20 mg/d. Adjunctive treatments were not well tolerated; lithium caused severe nausea and aripiprazole lead to intolerable akathisia. Psychotherapy was ineffective. A trial of electroconvulsive therapy relieved her depression but resulted in significant memory impairment.
Is ketamine a treatment option for Ms. B?
Ketamine, an N-methyl-D aspartate antagonist, was approved by the FDA in 1970.
as a dissociative anesthetic. It proved useful in military battlefield situations. The drug then became popular as a “club drug” and is used recreationally as a dissociative agent. It recently has been used clinically for treating post-operative pain and treatment-resistant depression (TRD). It has shown efficacy for several specific symptom clusters in depression, including anhedonia and suicidality.
Several small randomized, double-blind, placebo-controlled trials of ketamine—some of which studied TRD—have reported antidepressant effects after a single IV dose of 0.5 mg/kg in depressed patients.1,2 The response rate, defined as a 50% reduction in symptoms, is reported to be as high as 50% to 71% twenty-four hours after infusion, with significant improvements noted in some patients after just 40 minutes.1 These effects, peaking at 24 hours, last ≥72 hours in approximately 50% of patients, but gradually return to baseline over 1 to 2 weeks (Figure1). The most common post-infusion adverse effects include:
- dissociation
- dizziness
- blurred vision
- poor concentration
- nausea.
Transient sedation and psychotomimetic symptoms, such as hallucinations, abnormal sensations, and confusion, also have been noted, as well as a small but significant increase in blood pressure shortly after infusion.1
Use of repeated doses of ketamine also has been studied, although larger and extended-duration studies are lacking. Two groups3,4 examined thrice weekly infusions (N = 24) and 1 group5 studied twice weekly infusions of 0.5 mg/kg for 2 weeks (6 and 4 doses, respectively) (N = 10). With thrice weekly dosing, 79% to 90% of patients showed symptomatic response overall and 25% to 100% of patients saw improvement after the first dose.3,4 Of the 20 patients who responded, 65% were still reporting improved symptoms 2 weeks after the last infusion and 40% showed response for >28 days.3,4 With twice weekly dosing,5 the response rate was 80% in 10 patients, while 5 patients (50%) achieved remission, lasting at least 28 days in 2 patients.
The authors of a recent Cochrane review6 evaluated ketamine for treating depression and concluded that, although there is evidence for ketamine’s efficacy early in treatment, effects are less certain after 2 weeks post-treatment. The Canadian Agency for Drugs and Technologies in Health also conducted an appraisal7 of ketamine for treating a variety of mental illnesses and similarly noted that, despite evidence in acute studies, (1) the role of the drug in clinical practice is unclear and (2) further comparative studies, as well as longer-term studies, are needed.
Last, the American Psychiatric Association Council of Research Task Force on Novel Biomarkers and Treatments1 conducted a systematic review and meta-analysis, whose authors concluded that ketamine produces a rapid and robust antidepressant effect that appears to be transient. They warn that, although results are promising, “enthusiasm should be tempered” and suggest that “its use in the clinical setting warrants caution.”
Should you consider treating depression with ketamine?
Although evidence for using ketamine as a rapid treatment of TRD is promising and non-IV forms of ketamine are being researched (eg, intranasal esketamine), there are factors that limit clinical application:
- The short duration of effect noted in studies highlights the need for research on maintenance strategies to assess longer-term efficacy as well as safety. For example, long-term ketamine abuse has been associated with cases of ulcerative or hemorrhagic cystitis causing severe and persistent pain, requiring a partial cystectomy.8,9 Further, long-term ketamine use for pain has been associated with a transaminitis. Lastly, ketamine self-treatment for depression with escalating doses has also been associated with severe ketamine addiction and sequelae.10 The incidence and severity of these adverse effects at dosages and administration frequencies that might be required for maintenance treatment of depression is unclear and requires further investigation.
- Psychotomimetic and cardiovascular adverse effects of ketamine warrant monitoring in an acute clinical setting, until longer term safety and monitoring protocols are developed. Of note, the dosing regimen used in most studies requires anesthesia monitoring in many health care systems. Although acute adverse effects in studies to date are infrequent, both cardiovascular and gastrointestinal (vomiting) events requiring IV intervention have been reported,4 underscoring the importance of anesthesiologist involvement.
- Tolerance. It is unknown if patients develop tolerance to ketamine with recurring dosages and may present additional safety concerns with repeated, higher dosages. Lastly, patients on extended ketamine therapy could encounter drug interactions with agents commonly used to treat depression.
Although some authors1,6 advise caution with widespread ketamine use, patients with TRD want effective treatments and may discount these warnings. Even though longer-term studies are needed, ketamine “infusion clinics” are already being established. Before referring patients to such clinics, it is important to understand the current clinical and safety limitations and requirements for ketamine in TRD and to consider and discuss the risks and benefits carefully.
CASE CONTINUED
Because Ms. B has tried several antidepressants and adjunctive therapies without success, and her depression is severe enough to affect her functioning in several domains, it might be reasonable to discuss a trial of ketamine. However, Ms. B also should be presented non-ketamine alternatives, such as other adjunctive strategies (liothyronine, buspirone, cognitive-behavioral therapy) or a trial of nortriptyline or a monoamine oxidase inhibitor.
If ketamine is thought to be the best option for Ms. B, her provider needs to establish a clear expectation that the effects likely will be temporary. Monitoring should include applying a rating scale to assess depressive symptoms, suicidality, and psychotomimetic symptoms. During and shortly after infusion, anesthesia support should be provided and blood pressure and other vital signs should be monitored. Additional monitoring, such as telemetry, might be indicated.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
1. Newport DJ, Carpenter LL, McDonald WM, et al; APA Council of Research Task Force on Novel Biomarkers and Treatments. Ketamine and other NMDA antagonists: early clinical trials and possible mechanisms in depression. Am J Psychiatry. 2015;172(10):950-966.
2. Murrough JW, losifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
3. aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry. 2010;67(2):139-145.
4. Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disorder. 2014;155:123-129.
5. Ramussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27(5):444-450.
6. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;9:CD011612. doi: 10.1002/14651858.CD011612.pub2.
7. Canadian Agency for Drugs and Technologies in Health. Intravenous ketamine for the treatment of mental health disorders: a review of clinical effectiveness and guidelines. Ottawa, Ontario, Canada: Canadian Agency for Drugs and Technologies in Health; 2014. https://www.cadth.ca/media/pdf/htis/dec-2014/RC0572%20IV%20Ketamine%20Report%20final.pdf. Published August 20, 2014. Accessed April 13, 2016.
8. Jhang JF, Birder LA, Chancellor MB, et al. Patient characteristics for different therapeutic strategies in the management ketamine cystitis [published online March 21, 2016]. Neurourol Urodyn. doi: 10.1002/nau.22996.
9. Busse J, Phillips L, Schechter W. Long-term intravenous ketamine for analgesia in a child with severe chronic intestinal graft versus host disease. Case Rep Anesthesiol. 2015;2015:834168. doi:10.1155/2015/834168.
10. Bonnet U. Long-term ketamine self-injections in major depressive disorder: focus on tolerance in ketamine’s antidepressant response and the development of ketamine addiction. J Psychoactive Drugs. 2015;47(4):276-285.
When and why to initiate antipsychotic polypharmacy, and with which agents
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,

and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4

Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
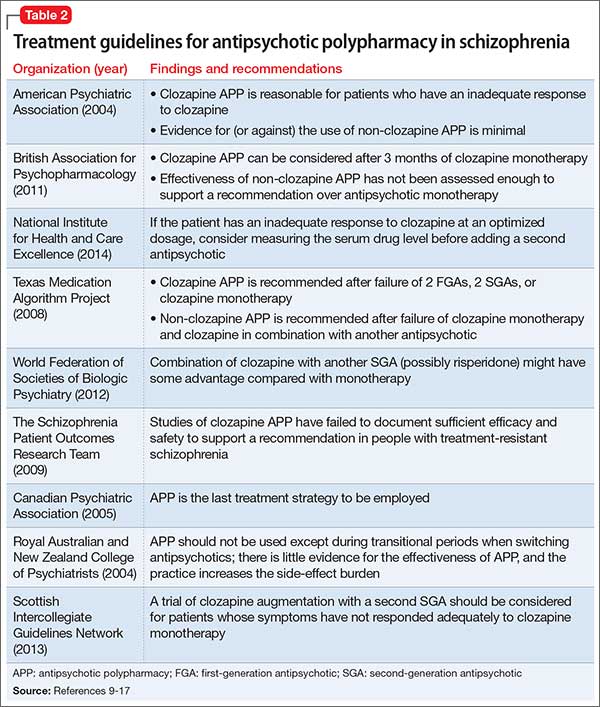

What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
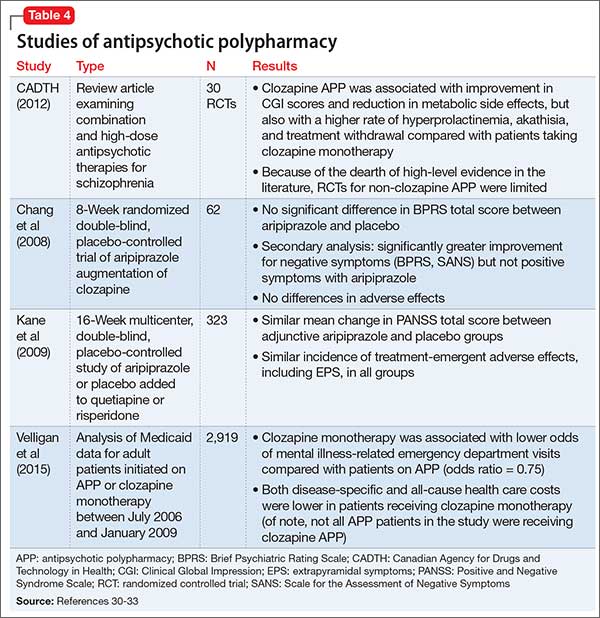
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
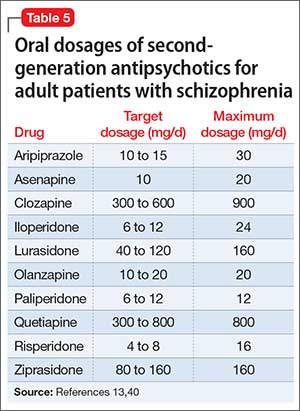
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,
and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4
Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
Mr. C, age 31, who has a 7-year history of schizophrenia and is currently on perphenazine, 24 mg twice a day, presents for psychiatric admission after experiencing paranoid delusions. Notable symptoms include delusions of reference and persecution, along with affective flattening and intermittent suicidal ideation. Perphenazine is tapered, and he is started on quetiapine, titrated to 600 mg/d.
Past antipsychotic trials include aripiprazole, olanzapine, paliperidone, haloperidol,
and ziprasidone. Because of his refractory symptoms and tolerability issues with other antipsychotics, Mr. C is switched to clozapine, 400 mg/d. His symptoms improve, but he experiences dose-limiting sialorrhea. Risperidone, 1 mg/d, is added to clozapine, which helps his psychosis and improves his functional status. Additionally, Mr. C develops enough insight to recognize his delusions and use skills learned in psychotherapy to cope with them.
Antipsychotic polypharmacy (APP), the concurrent use of ≥2 antipsychotics, is a topic of debate among mental health care providers. Studies indicate the prevalence of APP can reach upwards of 40%, with 1 systematic review citing more recent median APP prevalence in North America as 17%, an increase from a median of 12.7% in the 1980s.1 Other studies cite more recent figures as around 20%.2,3
The literature lists several reasons for use of long-term APP, including:
- incomplete cross-titration
- accidental continuation of APP that was intended to be temporary
- monotherapy failure
- mitigation or enhancement of effects of other antipsychotics (Table 1).1,4
Other factors include direct-to-consumer advertising, external pressures to decrease hospital stays, and low doctor-to-patient ratios.5 Although it can take as long as 16 weeks to see clinically significant improvement with an antipsychotic, prescribers might expect results after 4 weeks of treatment.6 Therefore, treatments could be labeled ineffective because trials did not last long enough, leading to premature use of polypharmacy. Combinations of a first- and second-generation antipsychotic (SGA) or 2 SGAs are most common.2,7,8
Treatment guidelines (Table 2)9-17 suggest APP could be considered after several failures of monotherapy, including clozapine monotherapy, although some guidelines do not address the issue or recommend against APP because of lack of efficacy and safety data. Additionally, APP poses safety concerns (Table 3).18-22 Recommendations for APP with combinations that do not include clozapine generally are not provided, because high-level evidence to support this strategy is lacking. Data on safety and efficacy of APP are mixed, with much of the literature dominated by case reports and uncontrolled studies.19
What to initiate
Clozapine. Higher-level evidence is available for clozapine APP. The combination of clozapine and risperidone is one of the most thoroughly studied and, therefore, is a reasonable first choice. Randomized controlled trials (RCTs) examining clozapine plus risperidone23-29 have yielded mixed results and have not provided conclusive information regarding benefit for positive vs negative symptoms.24-28
One RCT reported a significant change in Brief Psychiatric Rating Scale (BPRS) total and positive symptom scores.27 Other RCTs have shown a non-significant trend toward greater change in total, positive, and negative symptom scores with the clozapine-risperidone combination compared with clozapine monotherapy.25,28 In terms of cognition, this combination provided no additional benefit.23 Response, defined as ≥20% reduction in total BPRS or Positive and Negative Syndrome Scale (PANSS) scores, for clozapine plus risperidone range from 13% to 83%, compared with 8% to 29% for clozapine plus placebo.24,25,27,29
Data from 1 study27 suggest a number needed to treat of 4 to achieve at least a 20% improvement in BPRS scores with clozapine plus risperidone vs clozapine monotherapy. Across these studies, the average risperidone dosage was 4 mg/d, although using the lowest effective dosage is encouraged. A small number of RCTs and articles examining other APP combinations (Table 4)30-33 have yielded mixed results.
Overall, APP appears to be well-tolerated, although it is associated with an increased risk of adverse effects, including sedation, extrapyramidal symptoms, hyperprolactinemia, sexual dysfunction, cognitive impairment, anticholinergic effects, hyperlipidemia, and diabetes.23,24,34-36 Surprisingly, 1 literature review36 found no association between APP and increased risk of orthostasis. Increased occurrence of sedation, hyperprolactinemia, and an elevated fasting blood glucose level have been found for clozapine plus risperidone compared with clozapine monotherapy.24-26,28
Aripiprazole. Adjunctive aripiprazole, a dopamine partial agonist, could reduce elevated prolactin levels caused by other antipsychotics.32 In a study37 of 56 patients taking haloperidol who had hyperprolactinemia, prolactin levels normalized in 88.5% of patients taking adjunctive aripiprazole, 30 mg/d, compared with 3.6% of those with added placebo. Furthermore, results from 2 RCTs38,39 of patients taking clozapine or olanzapine suggest adjunctive aripiprazole could improve weight and metabolic profile. Therefore, adding aripiprazole to existing antipsychotic regimens is reasonable for patients with drug-induced symptomatic hyperprolactinemia or metabolic effects and who cannot be easily switched to another antipsychotic.
When to initiate
Most treatment guidelines9-17 recommend clozapine only after monotherapy with at least 2 other antipsychotics fails. It is reasonable to add an antipsychotic to clozapine in patients who have shown a partial response to clozapine after a minimum of 3 months. Non-clozapine APP should be considered when:
- a patient derives no benefit from clozapine
- refuses clozapine
- clozapine is contraindicated
- APP is initiated to mitigate side effects from another antipsychotic.
Antipsychotics could take up to 16 weeks to achieve full efficacy,6 therefore, an adequate trial period within the target dosage range is advised for all antipsychotics (Table 5).13,40
Why initiate
Based on available data, partial response to maximum recommended dosages of antipsychotic monotherapy, including clozapine, or inability to tolerate higher dosages, provides a reason for initiating APP. Non-clozapine APP generally should be considered only in patients who refuse, cannot tolerate, or do not respond to clozapine. Consider using validated rating scales to track treatment outcomes (ideally, a ≥20% symptomatic reduction on the BPRS or PANSS), although there is no formal guidance regarding their use or benefit in APP.
Summing up
APP is a fairly common prescribing practice, even though safety and efficacy data are mixed. The issue of APP has become prevalent enough that regulatory bodies are involved in its monitoring and documentation.41
Clozapine APP, especially with risperidone, has the most substantial evidence to support it. Although APP generally is well tolerated, the overall dearth of conclusive safety and efficacy data indicates that this practice should be reserved for patients who have not responded adequately to monotherapy, including clozapine. Adjunctive aripiprazole could be considered for addressing symptomatic hyperprolactinemia or other metabolic effects caused by other antipsychotics.
An adequate trial as long as 16 weeks is advised before assessing the efficacy of any antipsychotic regimen. If APP provides inadequate response, or if there is no clear indication for APP, consider switching the patient back to monotherapy.42-44
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
1. Gallego JA, Bonetti J, Zhang J, et al. Prevalence and correlates of antipsychotic polypharmacy: a systematic review and meta-regression of global and regional trends from the 1970s to 2009. Schizophr Res. 2012;138(1):18-28.
2. Gören JL, Meterko M, Williams S, et al. Antipsychotic prescribing pathways, polypharmacy, and clozapine use in treatment of schizophrenia. Psychiatr Serv. 2013;64(6):527-533.
3. Sun F, Stock EM, Copeland LA, et al. Polypharmacy with antipsychotic drugs in patients with schizophrenia: trends in multiple health care systems. Am J Health Syst Pharm. 2014;71(9):728-738.
4. Tapp A, Wood AE, Secrest L, et al. Combination antipsychotic therapy in clinical practice. Psychiatr Serv. 2003;54(1):55-59.
5. Ananth J, Parameswaran S, Gunatilake S. Antipsychotic polypharmacy. Curr Pharm Des. 2004;10(18):2231-2238.
6. Stahl SM. Antipsychotic polypharmacy: evidence based or eminence based? Acta Psychiatr Scand. 2002;106(5):321-322.
7. Botts S, Hines H, Littrell R. Antipsychotic polypharmacy in the ambulatory care setting, 1993-2000. Psychiatr Serv. 2003;54(8):1086.
8. Santone G, Bellantuono C, Rucci P, et al. Patient characteristics and process factors associated with antipsychotic polypharmacy in a nationwide sample of psychiatric inpatients in Italy. Pharmacoepidemiol Drug Saf. 2011;20(5):441-449.
9. American Psychiatric Association. Practice guideline for the treatment of patients with schizophrenia, second edition. http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/schizophrenia.pdf. Updated September 2009. Accessed September 20, 2014.
10. Barnes TRE; Schizophrenia Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. http://www.bap.org.uk/pdfs/Schizophrenia_Consensus_Guideline_Document.pdf. Updated 2011. Accessed September 20, 2014.
11. National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. http://www.nice.org.uk/guidance/cg178. Published February 2014. Accessed September 20, 2014.
12. Texas Medication Algorithm Project. Schizophrenia treatment algorithms. http://www.jpshealthnet.org/sites/default/files/tmapalgorithmforschizophrenia.pdf. Updated April 2008. Accessed September 20, 2014.
13. Hasan A, Falkai P, Wobrock T, et al; World Federation of Societies of Biological Psychiatry (WFSBP). World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and the management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-378.
14. Canadian Psychiatric Association. Clinical practice guidelines: treatment of schizophrenia. https://ww1.cpa-apc.org/Publications/Clinical_Guidelines/schizophrenia/november2005/index.asp. Updated November 2005. Accessed February 26, 2016.
15. Royal Australian and New Zealand College of Psychiatrists. Clinical practice guidelines for the treatment of schizophrenia and related disorders. http://www.ranzcp.org/Files/ranzcp-attachments/Resources/Publications/CPG/Clinician/CPG_Clinician_Full_Schizophrenia-pdf.aspx. Updated May 2005. Accessed February 26, 2016.
16. Scottish Intercollegiate Guidelines Network. Management of schizophrenia: a national clinical guideline. http://www.sign.ac.uk/guidelines/fulltext/131/index.html. Updated March 2013. Accessed September 20, 2014.
17. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71-93.
18. Correll CU, Gallego JA. Antipsychotic polypharmacy: a comprehensive evaluation of relevant correlates of a long-standing clinical practice. Psychiatr Clin North Am. 2012;35(3):661-681.
19. Tranulis C, Skalli L, Lalonde P, et al. Benefits and risks of antipsychotic polypharmacy: an evidence-based review of the literature. Drug Saf. 2008;31(1):7-20.
20. Barnes TR, Paton C. Antipsychotic polypharmacy in schizophrenia: benefits and risks. CNS Drugs. 2011;25(5):383-399.
21. Lochmann van Bennekom MW, Gijsman HJ, Zitman FG. Antipsychotic polypharmacy in psychotic disorders: a critical review of neurobiology, efficacy, tolerability and cost effectiveness. J Psychopharmacol. 2013;27(4):327-336.
22. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: systematic review. Schizophr Res. 2009;113(1):1-11.
23. Akdede BB, Anil Ya˘gcio˘glu AE, Alptekin K, et al. A double-blind study of combination of clozapine with risperidone in patients with schizophrenia: effects on cognition. J Clin Psychiatry. 2006;67(12):1912-1919.
24. Anil Ya˘gcio˘glu AE, Kivircik Akdede BB, Turgut TI, et al. A double-blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. J Clin Psychiatry. 2005;66(1):63-72.
25. Freudenreich O, Henderson DC, Walsh JP, et al. Risperidone augmentation for schizophrenia partially responsive to clozapine: a double-blind, placebo-controlled trial. Schizophr Res. 2007;92(1-3):90-94.
26. Honer WG, Thornton AE, Chen EY, et al; Clozapine and Risperidone Enhancement (CARE) Study Group. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. N Engl J Med. 2006;354(5):472-482.
27. Josiassen RC, Joseph A, Kohegyi E, et al. Clozapine augmented with risperidone in the treatment of schizophrenia: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2005;162(1):130-136.
28. Weiner E, Conley RR, Ball MP, et al. Adjunctive risperidone for partially responsive people with schizophrenia treated with clozapine. Neuropsychopharmacology. 2010;35(11):2274-2283.
29. Zink M, Kuwilsky A, Krumm B, et al. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomized controlled clinical trial. J Psychopharmacol. 2009;23(3):305-314.
30. Canadian Agency for Drugs and Technology in Health. Current utilization of antipsychotic agents for schizophrenia: combination and high-dose therapies. https://www.cadth.ca/sites/default/files/pdf/H0503_AAP-Current-Utilization-Report_e.pdf. Published August 2012. Accessed February 26, 2016.
31. Chang JS, Ahn YM, Park HJ, et al. Aripiprazole augmentation in clozapine treated patients with refractory schizophrenia: an 8-week, randomized, double blind, placebo-controlled trial. J Clin Psychiatry. 2008;69(5):720-731.
32. Kane JM, Correll CU, Goff DC, et al. A multicenter, randomized, double-blind, placebo-controlled, 16-week study of adjunctive aripiprazole for schizophrenia or schizoaffective disorder inadequately treated with quetiapine or risperidone monotherapy. J Clin Psychiatry. 2009;70(10):1348-1357.
33. Velligan DI, Carroll C, Lage MJ, et al. Outcomes of medicaid beneficiaries with schizophrenia receiving clozapine only or antipsychotic combinations. Psychiatr Serv. 2015;66(2):127-133.
34. Citrome L, Jaffe A, Levine J, et al. Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv. 2004;55(9):1006-1013.
35. Correll CU, Frederickson AM, Kane JM, et al. Does antipsychotic polypharmacy increase the risk for metabolic syndrome? Schizophr Res. 2007;89(1-3):91-100.
36. Gallego JA, Nielsen J, De Hert M, et al. Safety and tolerability of antipsychotic polypharmacy. Expert Opin Drug Saf. 2012;11(4):527-542.
37. Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404-1410.
38. Fan X, Borba CP, Copeland P, et al. Metabolic effects of adjunctive aripiprazole in clozapine-treated patients with schizophrenia. Acta Psychiatr Scand. 2013;127(3):217-226.
39. Henderson DC, Fan X, Copeland PM, et al. Aripiprazole added to overweight and obese olanzapine-treated schizophrenia patients. J Clin Psychopharmacol. 2009;26(2):165-169.
40. Drug Information Handbook, 22th ed. Hudson, OH: Lexi-Comp, Inc.; 2013:1143-1147.
41. The Joint Commission. Specifications Manual for Joint Commission National Quality Measures (v2013A1). https://manual.jointcommission.org/releases/TJC2013A/. Accessed on May 13, 2015.
42. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702-708.
43. Godleski LS, Kerler R, Barber JW, et al. Multiple versus single antipsychotic drug treatment in chronic psychosis. J Nerv Ment Dis. 1989;177(11):686-689.
44. Suzuki T, Uchida H, Tanaka KF, et al. Revising polypharmacy to a single antipsychotic regimen for patients with chronic schizophrenia. Int J Neuropsychopharmacol. 2004;7(2):133-142.
Urine drug screens: When might a test result be false-positive?
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.

However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.

There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
Mr. L, age 35, has an appointment at a mental health clinic for ongoing treatment of depression. His medication list includes atorvastatin, bupropion, lisinopril, and cranberry capsules for non-descriptive urinary issues. He has been treated for some time at a different outpatient facility; however he recently moved and changed clinics.
At this visit, his first, Mr. L receives a full physical exam, including a urine drug screen point-of-care (POC) test. He informs the nurse that he has an extensive history of drug abuse: “You name it, I’ve done it.” Although he experimented with many illicit substances, he acknowledges that “downers” were his favorite. He believes that his drug abuse could have caused his depression, but is proud to declare that he has been “clean” for 12 months and his depression is approaching remission.
However, the urine drug screen is positive for amphetamines. Mr. L vehemently swears that the test must be wrong, restating that he has been clean for 12 months. “Besides, I don’t even like ‘uppers’!” Because of Mr. L’s insistence, the clinician does a brief literature search about false-positive results in urine drug screening, which shows that, rarely, bupropion can trigger a false positive in the amphetamine immunoassay.
Could this be a false-positive result? Or is Mr. L not telling the truth?
Because no clinical lab test is perfect, any clinician who runs urine drug screens will encounter a false-positive result. (See the Box,1-3 for discussion of false negatives.) Understanding how each test works—and potential sources of error— can help you evaluate test results and determine the best course of action.
There are 2 main methods involved in urine drug testing: in-office (POC) urine testing and laboratory-based testing. This article describes the differences between these tests and summarizes the potential for false-positive results.
In-office urine testing
POC tests in urine drug screens use a technique called “immunoassay,” which is quantitative and generally will detect the agent in urine for only 3 to 7 days after ingestion.4 This test relies on the principle of competitive binding: If a parent drug or metabolite is present in urine, it will bind to a specific antibody site on the test strip and produce a positive result.5 Other compounds that are similarly “shaped” on a molecular level also can bind to these antibody sites when present in sufficient quantity, producing a “cross reaction,” also called a “false-positive” result. The Table6 lists agents that can cross-react with immunoassay tests. In addition to the cross-reaction, false positives also can occur because of technician or clerical error— making it important to review the process by which the specimen was obtained and tested if a false-positive result is suspected, as in the case described here.7
Different POC tests can have varying cross-reactivity patterns, based on the antibody used.8 In general, false positives in immunoassays are rare, but amphetamine and opiate false positives are more common than cocaine metabolite and cannabinoid false positives.9 The odds of a false positive vary, depending on the specificity of the immunoassay used and the substance under detection.6
A study that analyzed 10,000 POC urine drug screens found that 362 specimens tested positive for amphetamines, but that 128 of those did not test positive for amphetamines using more sensitive tests.10 Of these 128 false positives reported, 53 patients were taking bupropion at the time of the test.10 Therefore, clinicians should do a thorough patient medication review at the time of POC urine drug testing. In addition, consider identifying which type of test you are using at your practice site, and ask the manufacturer or lab to provide a list of known possible false positives.
Laboratory-based GC–MS testing
If a false positive is suspected on a POC immunoassay-based urine drug screen, results can be confirmed using gas chromatography–mass spectrometry (GC–MS). Although GC–MS is more accurate than an immunoassay, it also is more expensive and time-consuming.9
GC–MS breaks down a specimen into ionized fragments and separates them based on their mass–charge ratio. Because of this, GC–MS is able to identify the presence of a specific drug (eg, oxycodone) instead of a broad class (eg, opioid). The GC–MS method is a good tool to confirm initial positive screens when their integrity is in question because, unlike POC tests used during an office visit, GC–MS is not influenced by cross-reacting compounds.11-13
GC–MS is not error-free, however. For example, heroin and hydrocodone are metabolized into morphine and hydromorphone, respectively. Depending on when the specimen was collected, the metabolites, not the parents, might be the compounds identified, which might produce confusing results.
Clinical recommendations
When a POC drug screen is positive, confirming the result with GC–MS is good clinical practice. False positives can strain the relationship between patient and provider, thus compromising care. Examining the procedures that were used to obtain the specimen, as well as double-checking POC test results, is, when appropriate, good medicine.
CASE CONTINUED
Because Mr. L is adamant about his sobriety and the fact that his drugs of choice were sedatives, not stimulants, the clinician orders a second drug screen by GC–MS. The second screen is negative for substances of abuse; Mr. L’s clinician concludes that bupropion produced a false-positive result on the POC urine drug screen, confirming Mr. L’s assertions.
Related Resources
• Saitman A, Park HD, Fitzgerald RL. False-positive interferences of common urine drug screen immunoassays: a review. J Anal Toxicol. 2014;38(7):387-396.
• Tenore PL. Advanced urine toxicology testing. J Addict Dis. 2010;29(4):436-448.
Drug Brand Names
Amantadine • Symadine, Symmetrel
Amitriptyline • Elavil
Atorvastatin • Lipitor
Brompheniramine • Dimetane
Bupropion • Wellbutrin, Zyban
Carbamazepine • Carbatrol, Tegretol
Chlorpromazine • Thorazine
Clomipramine • Anafranil
Cyclobenzaprine • Amrix, Flexeril
Cyproheptadine • Periactin
Desipramine • Nopramin
Desoxyephedrine • Desoxyn
Dextromethorphan • Delsym, Robitussin
Dicyclomine • Bentyl, Dicyclocot
Diphenhydramine • Benadryl, Unisom
Doxylamine • Robitussin, NyQuil
Dronabinol • Marinol
Efavirenz • Sustiva
Ephedrine • Mistol, Va-Tro-Nol
Ergotamine • Ergomar, Cafergot
Hydrocodone • Vicodin
Hydromophone • Dilaudid, Palladone
Hydroxyzine • Atarax, Vistaril
Isometheptene • Amidrine, Migrend
Isoxsuprine • Vasodilan, Vasoprine
Ketoprofen • Orudis, Oruvail
Labetalol • Normodyne, Trandate
Lisinopril • Prinivil, Zestril
Meperidine • Demerol
Naproxen • Aleve, Naprosyn
Oxaprozin • Daypro
Oxycodone • Oxycontin, Percocet, Percodan, Roxicodone
Phentermine • Adipex, Phentrol
Phenylephrine • Sudafed PE, Neo-Synephrine
Piroxicam • Feldene
Promethazine • Phenergan
Pseudoephedrine • Sudafed, Dimetapp
Quetiapine • Seroquel
Ranitidine • Zantac
Rifampin • Rifadin, Rimactane
Selegiline • EMSAM
Sertraline • Zoloft
Sulindac • Clinoril
Sumatriptan • Imitrex
Thioridazine • Mellaril
Tolmetin • Tolectin
Trazodone • Desyrel, Oleptro
Trimethobenzamide • Benzacot, Tigan
Trimipramine • Surmontil
Verapamil • Calan, Isoptin
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.
1. Cobaugh DJ, Gainor C, Gaston CL, et al. The opioid abuse and misuse epidemic: implications for pharmacists in hospitals and health systems. Am J Health Syst Pharm. 2014;71(18):1539-1554.
2. Gilbert JW, Wheeler GR, Mick GE, et al. Importance of urine drug testing in the treatment of chronic noncancer pain: implications of recent medicare policy changes in Kentucky. Pain Physician. 2010;13(2):167-186.
3. Michna E, Jamison RN, Pham LD, et al. Urine toxicology screening among chronic pain patients on opioid therapy: frequency and predictability of abnormal findings. Clin J Pain. 2007;23(2):173-179.
4. U.S. Department of Justice. Fact sheet: drug testing in the criminal justice system. https://www.ncjrs.gov/pdffiles/dtest. pdf. Published March 1992. Accessed July 29, 2015.
5. Australian Diagnostic Services. Technical information: testing principle’s. http://www.australiandrugtesting. com/#!technical-info/c14h4. Accessed November 5, 2014.
6. University of Illinois at Chicago College of Pharmacy. What drugs are likely to interfere with urine drug screens? http://dig.pharm.uic.edu/faq/2011/Feb/faq1.aspx. Accessed November 5, 2014.
7. Wolff K, Farrell M, Marsden J, et al. A review of biological indicators of illicit drug use, practical considerations and clinical usefulness. Addiction. 1999;94(9):1279-1298.
8. Gourlay D, Heit H, Caplan YH. Urine drug testing in primary care – dispelling the myths & designing strategies. PharmaCom Group. http://www.mc.uky.edu/equip-4-pcps/documents/ section8/urine%20drug%20testing%20in%20clinical%20 practice.pdf. Accessed August 6, 2015.
9. Standridge JB, Adams SM, Zotos AP. Urine drug screen: a valuable office procedure. Am Fam Physician. 2010;81(5): 635-640.
10. Casey ER, Scott MG, Tang S, et al. Frequency of false positive amphetamine screens due to bupropion using the Syva EMIT II immunoassay. J Med Toxicol. 2011;7(2):105-108.
11. Casavant MJ. Urine drug screening in adolescents. Pediatr Clin N Am. 2002;49(2):317-327.
12. Shults TF. The medical review officer handbook. 7th ed. Chapel Hill, NC: Quadrangle Research; 1999.
13. Baden LR, Horowitz G, Jacoby H, et al. Quinolones and false-positive urine screening for opiates by immunoassay technology. JAMA. 2001;286(24):3115-3119.

Avoiding common drug−drug interactions
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight

hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. T, age 23, was given a diagnosis of bipolar disorder 1 year ago. After he experienced inadequate symptom relief with valproate, you switched him to extended-release lithium, 1,200 mg/d. Mr. T reported improved mood and stability with this medication adjustment. These positive changes led him to resume activities he enjoyed before onset of bipolar disorder, such as running, reading, and going out to dinner with friends.
Now, Mr. T’s mother calls your office to express concern about her son’s slight
hand tremor, which appeared after 2 days of gastrointestinal distress. She tells you that Mr. T sprained his ankle while running 1 week ago and has been taking over-the-counter ibuprofen for pain relief, which he did often in the past.
You suspect that Mr. T is experiencing lithium toxicity as a result of ibuprofen use.
Although mental health providers can easily recognize the drug−drug interaction between lithium and nonsteroidal anti-inflammatory drugs (NSAIDs) that Mr. T experienced, interpreting the safety of a medication regimen with respect to drug− drug interactions before prescribing often is more daunting. This article reviews the basics of drug−drug interactions, while briefly highlighting common examples in psychiatric medicine (Table 11-5). We also provide an outline of additional points to consider when reviewing your patients’ medication regimens and encountering unfamiliar drug−drug interactions.
Types of drug−drug interactions
Drug−drug interactions fall into 2 categories: pharmacodynamic (PD) and pharmacokinetic (PK):
• PD interactions are a result of the combined impact of medications on the body when there is no direct effect on absorption, distribution, metabolism, or excretion characteristics, such as 2 medications that act at the same receptor or lead to similar or opposing pharmacologic effects.
• PK interactions occur when a drug affects the absorption, distribution, metabolism, or excretion characteristics of another drug.
Although it is possible that drug−drug interactions will have no clinical effect, when the impact of a PD or PK drug−drug interaction is evident, it likely is the result of additive, synergistic, or antagonistic consequences on the medications’ intended impact or side-effect profile.
Pharmacodynamic interactions
Serotonin syndrome. The potential for serotonin syndrome occurs when medications that increase synaptic serotonin concentration are used concomitantly.1 This can occur through several mechanisms, including increased serotonin release, decreased reuptake, or decreased serotonin metabolism. A high serotonin concentration in the CNS and in the periphery overstimulates serotonin receptors, leading to signs and symptoms that can include diarrhea, fever, delirium, coma, and potentially death.
QT prolongation and anticholinergic toxicity are further examples of additive PD drug−drug interactions. Anticholinergic toxicity is possible when multiple medications contribute to inhibition of the neuro-transmitter acetylcholine at muscarinic receptors. This leads to adverse effects such as dry mouth, constipation, confusion, and urinary retention.
QT prolongation, which can lead to arrhythmia, occurs when a patient is taking several medications that can increase the QT interval. Consider close monitoring and using alternative agents with less potential to increase the QT interval in patients at risk of arrhythmias (geriatric patients, those with an increased QT interval at baseline, etc.).
Decreased seizure threshold. The increased risk of seizures with bupropion and other medications that lower the seizure threshold is another example of an additive PD drug interaction. Bupropion can increase the risk of seizures in a dose-dependent manner, which increases when bupropion is taken with other drugs that lower the seizure threshold.6 Seizure risk associated with alcohol or benzodiazepine withdrawal also may increase the risk for this interaction.
Of note, the increased risk of seizures with the combination of bupropion and alcohol in the absence of withdrawal is not well studied in humans, but positive correlation has been seen in an animal study.6
Decreased platelet function. Another example of a PD drug−drug interaction is increased risk of bleeding when a selective serotonin reuptake inhibitor is used with a NSAID or oral anticoagulant. The proposed mechanism for this interaction is that blocking serotonin reuptake on platelets leads to decreased platelet function and an increased risk for prolonged bleeding.7 This is somewhat controversial because, first, it has been noted that drugs with the highest degree of serotonin reuptake inhibition do not always cause the highest risk of bleeding and, second, most of the evidence for this interaction is from observational studies.7
This potential interaction could be most important for patients who need an antidepressant, are on chronic NSAID or anticoagulant therapy, and are at high risk of bleeding.
Pharmacokinetic interactions
PK interactions in psychiatry often are caused by interference of drug metabolizing enzymes. The cytochrome P450 (CYP450) family of metabolizing enzymes in particular is important to the breakdown of medications in the body. Many drug−drug interactions involve medications that can inhibit or induce metabolism of other drugs through their effect on the CYP450 system.
Inhibition interactions. When a drug’s metabolism is inhibited, the result is usually increased serum concentration of that medication (because of less breakdown) and a more potent impact on the primary mechanism of action or adverse effects. Sometimes, inhibiting metabolism can lead to decreased clinical effect. Tamoxifen (an oral agent used to treat breast cancer) and certain analgesics when used in combination with moderate or strong inhibitors of the CYP2D6 subfamily of CYP450 metabolizing enzymes are 2 examples of metabolism inhibition leading to decreased efficacy.8 Both tamoxifen and the analgesics listed in Table 11-5 are prodrugs; that is, they must be metabolized to be active. When the enzymes that metabolize these drugs into their active form are inhibited, the concentration of active drug decreases.
Induction interactions. Alternatively, there is an increased rate of drug breakdown and resulting decrease in effect when drugs that induce the activity of metabolizing enzymes are used with medications that are substrates of the same enzyme. Carbamazepine is commonly involved in this type of drug interaction because it is a strong inducer of CYP 1A2, 2B6, 2C19, 2C9, and 3A4, and the p-glycoprotein drug efflux pump.9 As a result of this rampant induction, carbamazepine can decrease the serum concentration of oral contraceptives below a reliably effective level. Therefore, it is recommended that women of childbearing potential use other contraceptive methods, such as a progestin implant or an intrauterine device.10
In addition, the polycyclic aromatic hydrocarbons found in cigarettes induce activity of CYP1A2. Patients who smoke and use medications metabolized by this enzyme, such as clozapine and olanzapine, may need a higher dosage.
Drug elimination interactions
The last drug−drug interaction discussed here returns the discussion to Mr. T and involves drug elimination.2 The NSAIDs Mr. T was using for pain likely caused decreased renal excretion of lithium. Because lithium is primarily excreted through the kidneys, Mr. T’s NSAID use, possibly in combination with dehydration caused by gastrointestinal distress, resulted in lithium toxicity. This class of analgesics should be avoided or used cautiously in patients taking lithium.
Clinical applications
The relatively common drug−drug interactions discussed here are just a fraction of the potential interactions mental health practitioners see on a daily basis. Understanding the basics of PD and PK interactions in the setting of patient-specific factors can help to clarify the information found in drug−drug interaction databases, such as Micromedex, Lexicomp, Facts and Comparisons, and Epocrates. Table 2 lists additional insights into drug interactions.
Related Resources
• CredibleMeds. Online resource on QT prolonging drugs. http://crediblemeds.org.
• Madhusoodanan S, Velama U, Parmar J, et al. A current review of cytochrome P450 interactions of psychotropic drugs. Ann Clin Psychiatry. 2014;26(2):120-138.
Drug Brand Names
Benztropine • Cogentin Olanzapine • Zyprexa
Bupropion • Wellbutrin Oxycodone • Oxycontin
Carbamazepine • Tegretol Paroxetine • Paxil
Clozapine • Clozaril Quetiapine • Seroquel
Diphenhydramine • Benadryl Sertraline • Zoloft
Duloxetine • Cymbalta Tamoxifen • Soltamox
Fluoxetine • Prozac Trazodone • Desyrel
Lithium • Eskalith, Lithobid Valproate • Divalproex
Haloperidol • Haldol Ziprasidone • Geodon
Hydrocodone • Vicodin
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
1. Buckley NA, Dawson AH, Isbister GK. Serotonin syndrome. BMJ. 2014;348:g1626. doi: 10.1136/bmj.g1626.
2. Eskalith [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2003.
3. Handler J. Lithium and antihypertensive medication: a potentially dangerous interaction. J Clin Hypertens (Greenwich). 2009;11(12):738-742.
4. Blanche P, Raynaud E, Kerob D, et al. Lithium intoxication in an elderly patient after combined treatment with losartan. Eur J Clin Pharmacol. 1997;52(6):501.
5. Atacand [package insert]. Wilmington, DE: AstraZeneca LP; 2013.
6. Silverstone PH, Williams R, McMahon L, et al. Alcohol significantly lowers the seizure threshold in mice when co-administered with bupropion hydrochloride. Ann Gen Psychiatry. 2008;7:11.
7. Spina E, Trifirò G, Caraci F. Clinically significant drug interactions with newer antidepressants. CNS Drugs. 2012;26(1):39-67.
8. Ereshefsky L, Sloan DM. Drug-drug interactions with the use of psychotropic medications. CNS Spectr. 2009;14(suppl Q and A forum 8):1-8.
9. Carbamazepine. Drug facts and comparisons database. St. Louis, MO: Wolters Kluwer Health Inc; November 2014.
10. Pennell PB. Pregnancy, epilepsy, and women’s issues. Continuum (Minneap Minn). 2013;19(3 Epilepsy):697-714.
Grapefruit juice and psychotropics: How to avoid potential interactions
Ms. H, age 42, was given a diagnosis of bipolar disorder 10 years ago and has been taking carbamazepine, 1,200 mg/d, and olanzapine, 10 mg/d, for the past 2 years. She has not experienced a mood episode while on this regimen, and her carbamazepine level was 9.2 μg/mL 6 months ago. The only adverse effect she experienced was weight gain of approximately 10 lb. Ms. H takes a calcium supplement, but no other medications.
Ms. H reports to her psychiatrist that, for the past few days, she has been feeling nauseated, fatigued, and dizzy, but has continued taking her medications as prescribed. Her carbamazepine level is found to be 13.1 μg/mL. Ms. H states she has not started any new medications or supplements; her serum creatinine and liver function test results are within normal limits.
Upon further questioning, Ms. H says that an upper respiratory infection has been “going around her office,” so she increased her vitamin C intake by drinking 2 glasses of grapefruit juice a day (she doesn’t like orange juice). She has heard grapefruit juice can cause problems with some drugs so she is careful not to drink it at the same time she takes her medications. Her psychiatrist recognizes there may be a drug interaction involved, and recommends Ms. H hold her carbamazepine for 1 day and not consume any more grapefruit juice. A few days later, she reports feeling much better during a follow-up call and she makes an appointment to have her carbamazepine level rechecked in a we

Although grapefruit products are high in vitamins and low in calories, they can be associated with potentially serious drug interactions. The interaction between grapefruit juice and the calcium channel blocker felodipine was discovered inadvertently >20 years ago; since that time, possible interactions with >85 medications have been identified.1 Interactions with grapefruit products are complicated because, although most result in increased drug exposure, reduced exposure of the medication also can occur. Additionally, the degree and clinical significance of the interaction varies among individuals and from one drug to another.
Mechanism of action
Most interactions with grapefruit products are thought to result from the inhibition of intestinal cytochrome P450 3A4 (CYP3A4). CYP3A4 is involved in the metabolism of numerous drugs, and is the most abundant cytochrome P450 enzyme in the liver and epithelial cells lining the intestine.2 Although hepatic CYP3A4 is thought to be minimally affected by grapefruit, inhibition of intestinal CYP3A4 can result in an overall increase in bioavailability of medications that are substrates and raise the risk of potential toxicity.3 Grapefruit contains various chemicals collectively known as furanocoumarins, which are largely responsible for inhibition of intestinal CYP3A4.4 Additionally, Seville oranges and the pomelo (a large, sweet grapefruit-like citrus fruit) also contain furanocoumarins and could have a similar effect, warranting caution with certain medications.5
Inhibition of CYP3A4 by furanocoumarins cannot be reversed, and new enzymes must be synthesized to return to the previous level of function.6 Therefore, drug interactions resulting from CYP3A4 inhibition can last for as long as 72 hours after ingesting grapefruit products.7 Separating consumption of grapefruit products and medication administration will not help manage this interaction.
Grapefruit products also could affect drug disposition through effects on various drug transporters. Decreased systemic exposure to certain medications could occur through grapefruit’s inhibition of organic anion-transporting polypeptides (OATPs). OATPs form a family of drug uptake transporters found in the intestine, liver, kidney, and brain.8 For drugs that are substrates of OATPs, grapefruit’s inhibition of this transporter can result in decreased absorption and a resulting decrease in efficacy. Flavanoids in grapefruit, such as naringin, inhibit OATPs, which is competitive in nature.9 Unlike the irreversible inhibition of CYP3A4 by furanocoumarins, flavanoids effects on OATPs have been shown to decrease within 4 hours.10
No psychotropic medications have been identified as being susceptible to this interaction, but for those medications affected—including fexofenadine and levothyroxine—separating consumption of grapefruit and medication administration by 4 hours could avoid this interaction.11 Additional data indicate that orange juice and apple juice could have similar effects on OATPs.12
Perhaps the most well-known drug transporter, P-glycoprotein is part of the multidrug-resistant subfamily of transporters. It is located throughout the body, including in the intestine, kidneys, liver, and blood-brain barrier. P-glycoprotein acts as an export pump to decrease the cellular concentration of many different drug substrates, and many agents can alter P-glycoprotein’s expression or function.
Small changes in P-glycoprotein’s activity can result in substantial changes in the disposition of substrates, which can include certain antineoplastics and antiretrovirals. Most reports have found grapefruit juice inhibits P-glycoprotein-mediated efflux; however, there also are reports of transporter activation.6 Additionally, P-glycoprotein and CYP3A4 share many substrates, so it can be difficult to isolate the contribution of P-glycoprotein to grapefruit−drug interactions.13 The effect of grapefruit on P-glycoprotein activity has been difficult to fully elucidate; more studies are needed.
Grapefruit consumption and its effect
Drug interactions can occur by consuming commercially produced grapefruit juice and juice from concentrate, as well as freshly squeezed juice and grapefruit segments.14 CYP3A4-inhibiting furanocoumarins also have been isolated in grapefruit peel; it is not known, however, whether items made from peel (marmalade, candied peel) contain concentrations high enough to pose a risk of a drug interaction.14 Contributing to the unpredictability of grapefruit-drug interactions, the amount or concentration of furanocoumarins can vary among grapefruit products and brands.15 This variability can be influenced by the variety or maturity of the fruit and the fruit’s exposure to environmental stress.4
The frequency of consuming a grapefruit product can influence the degree of a drug interaction. In general, consuming one 8-oz glass of grapefruit juice or the segments from a whole grapefruit is enough to alter a susceptible drug’s pharmacokinetics.14 Regular grapefruit product consumption, however, can result in an overall greater effect.16,17
Lilja et al16 conducted a randomized, 4-phase, crossover study to look at the effect of grapefruit juice dose on kinetics of triazolam. Grapefruit juice was found to increase the mean area under the concentration-time curve (AUC) of triazolam compared with water, but no difference was found between single glasses of normal-strength and double-strength grapefruit juice. However, repeated consumption of double-strength grapefruit juice (200 mL, 3 times/d for 3 days) increased triazolam’s mean AUC by 143%, compared with an increase of 49% with just a single 200-mL glass of double-strength juice.16 Recurrent consumption of grapefruit juice (8 oz, 3 times/d for 6 days) also was found to increase the kinetics of the antihypertensive felodipine more than a single glass of grapefruit juice.17
Clinical consequences of an interaction between a drug and grapefruit can be difficult to predict. Drug concentration changes caused by a grapefruit interaction could vary based on interindividual differences. The amount and activity of intestinal CYP3A4 can vary from person to person, and can be influenced by genetic polymorphisms in addition to race, age, and environmental variables.18 Interindividual sensitivity to a change in a drug’s concentration also will differ, and patient-specific factors, such as concomitant drugs or diseases, could influence the likelihood of harm.
Interactions with grapefruit products are not necessarily a “class effect,” and specific drugs within a therapeutic category can be affected (although others might not). Several drug-specific characteristics can help gauge the risk of a clinically relevant interaction with grapefruit, including:
• metabolism through CYP3A4
• low bioavailability
• oral administration
• a narrow therapeutic index.1
For drugs with low bioavailability because of first-pass metabolism, grapefruit’s inhibition of intestinal CYP3A4 can result in a greater relative increase in plasma concentrations compared with a drug with high bioavailability.19
For example, an increase in bioavailability from 5% to 10% will result in a much larger increase in AUC and overall clinical exposure compared with an increase from 85% to 90% even though both represent an absolute increase of 5%. Although a drug does not have to have low oral bioavailability for an interaction to occur, lower bioavailability means that a drug has a higher likelihood of causing a significant interaction because of altered pharmacokinetics. Of note, injectable medications will not interact with grapefruit because metabolism through intestinal CYP3A4 is bypassed and grapefruit does not significantly inhibit hepatic CYP3A4.
Although grapefruit products could alter the pharmacokinetics of susceptible drugs, those changes might not be associated with adverse effects. Therefore, a factor to consider in evaluating a potential interaction with grapefruit is the drug’s therapeutic index and its risk of serious adverse effects. Drugs with a narrow therapeutic index are of particular concern because a significant increase in therapeutic or adverse effects could result from a relatively small increase in the drug’s concentration.7
Which medications are affected?
Among medications identified as interacting with grapefruit, some cardiovascular agents and several of the HMG-CoA reductase inhibitors (statins) have garnered the most attention. However, grapefruit also can affect the metabolism of several psychotropic medications through inhibition of intestinal CYP3A4 (Table).16,20-35 Prescribing information for some drugs warns against consuming grapefruit while using the medication. Among CNS agents, buspirone, carbamazepine, lurasidone, pimozide, triazolam, and oral midazolam all have such warnings in their product labeling.
Buspirone currently is not recommended with “large quantities of grapefruit juice.”20 A randomized, 2-phase crossover study looking at the effects of grapefruit juice on buspirone’s pharmacokinetics found that double-strength grapefruit juice (200 mL, administered 3 times/d for 3 days) resulted in a 9.2-fold increase in mean AUC and a 4.3-fold increase in mean Cmax after a single 10-mg buspirone dose.22 Highlighting the wide interindividual variability seen with drug-grapefruit interactions, the increase found in buspirone’s AUC ranged from 3-fold to 20-fold among study participants.22
Carbamazepine product labeling lists grapefruit juice as a CYP3A4 inhibitor that is expected to or has been found to increase plasma levels of the drug.20 Carbamazepine’s bioavailability is influenced by intestinal CYP3A4 activity; in a randomized, 2-phase crossover study of 10 patients with epilepsy, grapefruit juice was found to increase AUC of carbamazepine by 41% and Cmax by 40%.23,36
Lurasidone and pimozide, although not specifically studied, have product labels that recommend avoiding grapefruit juice because it could inhibit metabolism of these agents by CYP3A4.20 Of particular concern is the potential for elevated levels of pimozide to increase the risk of adverse cardiovascular effects including QT interval prolongation.19
Midazolam. Although grapefruit juice does not affect the disposition of IV midazolam, pretreatment with grapefruit juice was found to increase the AUC and Cmax of oral midazolam by 52% and 56%, respectively.30
Other considerations in drug-grapefruit interactions
Cautionary statements about a possible interaction with grapefruit juice for many other psychotropics can be found in commonly used drug information references or online sources. If you are concerned about a possible interaction and avoiding grapefruit products is not feasible, consider a different medication in the same class.
However, you also should consider the level of evidence supporting any purported interaction. Several psychotropic agents do have studies or case reports supporting an interaction with grapefruit, but cautionary statements could be based on theoretical concerns because of a medication’s bioavailability, metabolic pathway, and concern for increased adverse events related to higher drug concentrations. Adding to the confusion, cautionary statements can be found about medications, such as clozapine, that have not been shown to have an interaction with grapefruit juice when studied.
With many of the drugs that have a reported or theoretical interaction with grapefruit, data are inconsistent as to whether the resulting interaction will be clinically relevant. A number of variables relating to the individual patient, grapefruit product, or particular drug can play a role in the significance of an interaction. Additionally, effects on drug disposition can last for a few days after consuming a grapefruit product.
Keep alert to situations of increased risk
Recall that the case patient, Ms. H, presented with an elevated carbamazepine level and suffered resulting adverse effects because of an interaction between the drug and grapefruit juice. Although Ms. H was careful to separate intake of grapefruit juice from carbamazepine administration, grapefruit’s inhibition of intestinal CYP3A4 still was present, leading to the interaction.
It is important for health care professionals to recognize this potential risk and to advise patients regarding possible interactions between medications and grapefruit products.
Related Resources
• U.S. Food and Drug Administration. Grapefruit juice and medicine may not mix. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm292276.htm.
• Hanley MJ, Cancalon P, Widmer WW, et al. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol. 2011;7(3):267-286.
• Andrade C. Fruit juice, organic anion transporting polypeptides, and drug interactions in psychiatry. J Clin Psychiatry. 2014;75(11):e1323-e1325.
Drug Brand Names
Alprazolam • Xanax Lurasidone • Latuda
Buspirone • BuSpar Midazolam • Versed
Carbamazepine • Tegretol Methadone • Dolophine
Clomipramine • Anafranil Nefazodone • Serzone
Clozapine • Clozaril Olanzapine • Zyprexa
Diazepam • Valium Pimozide • Orap
Felodipine • Plendil Quetiapine • Seroquel
Fexofenadine • Allegra Sertraline • Zoloft
Fluoxetine • Prozac Trazodone • Desyrel
Fluvoxamine • Luvox Triazolam • Halcion
Haloperidol • Haldol Ziprasidone • Geodon
Levothyroxine • Levoxyl, Synthroid
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bailey DG, Dresser G, Arnold JM. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? CMAJ. 2013;185(4):309-316.
2. Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21): 2211-2221.
3. Saito M, Hirata-Koizumi M, Matsumoto M, et al. Undesirable effects of citrus juice on the pharmacokinetics of drugs: focus on recent studies. Drug Saf. 2005;28(8):677- 694.
4. Cancalon PF, Barros SM, Haun C, et al. Effect of maturity, processing, and storage on the furanocoumarin composition of grapefruit and grapefruit juice. J Food Sci. 2011;76(4):C543-C548.
5. Pirmohamed M. Drug-grapefruit juice interactions: two mechanisms are clear but individual responses vary. BMJ. 2013;346:f1. doi: 10.1136/bmj.f1.
6. Dahan A, Altman H. Food-drug interaction: grapefruit juice augments drug bioavailability–mechanism, extent and relevance. Eur J Clin Nutr. 2004;58:1-9.
7. Stump AL, Mayo T, Blum A. Management of grapefruit-drug interactions. Am Fam Physician. 2006;74(4):605-608.
8. Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(suppl 2):1-5.
9. Bailey DG, Dressker GK, Leak BF, et al. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin Pharmacol Ther. 2007;81(4):495-502.
10. Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81(3):362-370.
11. Bailey DG. Fruit juice inhibition of uptake transport: a new type of food-drug interaction. Br J Clin Pharmacol. 2010;70(5): 645-655.
12. Dresser GK, Bailey DG, Leake BF, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther. 2002;71:11-20.
13. Seden K, Dickinson L, Khoo S, et al. Grapefruit-drug interactions. Drugs. 2010;70(18):2373-2407.
14. Bailey DG, Dresser GK, Kreeft JH, et al. Grapefruit-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther. 2000;68(5):468-477.
15. De Castro WV, Mertens-Talcott S, Rubner A, et al. Variation of flavonoids and furanocoumarins in grapefruit juices: a potential source of variability in grapefruit juice-drug interaction studies. J Agric Food Chem. 2006;54(1):249-255.
16. Lilja JJ, Kivistö KT, Backman JT, et al. Effect of grapefruit juice on grapefruit juice-triazolam interaction: repeated consumption prolongs triazolam half-life. Eur J Clin Pharmacol. 2000;56(5):411-415.
17. Lown KS, Bailey DG, Fontana RJ, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Invest. 1997;99(10):2545-2553.
18. Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535-567.
19. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000; 38(1):41-57.
20. U.S. Food and Drug Administration. Drugs@FDA. http://www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed July 14, 2014.
21. Yasui, N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;15(2):185-190.
22. Lilja JJ, Kivistö KT, Backman JT, et al. Grapefruit juice substantially increases plasma concentrations of buspirone. Clin Pharmacol Ther. 1998;64(6):655-660.
23. Garg SK, Kumar N, Bhargava VK, et al. Effect of grapefruit juice on carbamazepine bioavailability in patients with epilepsy. Clin Pharmacol Ther. 1998;64(3):286-288.
24. Oesterheld J, Kallepalli BR. Grapefruit juice and clomipramine: shifting metabolic ratios. J Clin Psychopharm. 1997;17(1):62-63.
25. Lane HY, Jann MW, Chang YC, et al. Repeated ingestion of grapefruit juice does not alter clozapine’s steady-state plasma levels, effectiveness, and tolerability. J Clin Psychiatry. 2001;62(10):812-817.
26. Ozdemir M, Aktan Y, Boydag BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59.
27. DeSilva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS. 2001;15(10):1281-1285.
28. Hori H, Yoshimura R, Ueda N, et al. Grapefruit juice-fluvoxamine interaction—is it risky or not? J Clin Psychopharmacol. 2003;23(4):422-424.
29. Yasui N, Kondo T, Suzuki A, et al. Lack of significant pharmacokinetic interaction between haloperidol and grapefruit juice. Int Clin Psychopharmacol. 1999;142(2):113-118.
30. Kupferschmidt HH, Ha HR, Ziegler WH, et al. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58(1):20-28.
31. Benmebarek M, Cevaud C, Gex-Fabry M, et al. Effects of grapefruit juice on the pharmacokinetics of the enantiomers of methadone. Clin Pharmacol Ther. 2004;76(1):55-63.
32. DeVane CL, Nemeroff CB. Clinical pharmacokinetics of quetiapine: an atypical antipsychotic. Clin Pharmacokinet. 2001;40(7):509-522.
33. Ueda N, Yoshimura R, Umene-Nakano W, et al. Grapefruit juice alters plasma sertraline levels after single ingestion of sertraline in healthy volunteers. World J Biol Psychiatry. 2009;10(4 pt 3):832-835.
34. Lee AJ, Chan WK, Harralson AF, et al. The effects of grapefruit juice on sertraline metabolism: an in vitro and in vivo study. Clin Ther. 1999;21(11):1890-1899.
35. Sugimoto K, Araki N, Ohmori M, et al. Interaction between grapefruit juice and hypnotic drugs: comparison of triazolam and quazepam. Eur J Clin Pharmacol. 2006;62(3):209-215.
36. Fagiolino P, Vazquez M, Olano I, et al. Systemic and presystemic conversion of carbamazepine to carbamazepine- 10-11-epoxide during long term treatment. Journal of Epilepsy and Clinical Neurophysiology. 2006;12(1):13-16.
Ms. H, age 42, was given a diagnosis of bipolar disorder 10 years ago and has been taking carbamazepine, 1,200 mg/d, and olanzapine, 10 mg/d, for the past 2 years. She has not experienced a mood episode while on this regimen, and her carbamazepine level was 9.2 μg/mL 6 months ago. The only adverse effect she experienced was weight gain of approximately 10 lb. Ms. H takes a calcium supplement, but no other medications.
Ms. H reports to her psychiatrist that, for the past few days, she has been feeling nauseated, fatigued, and dizzy, but has continued taking her medications as prescribed. Her carbamazepine level is found to be 13.1 μg/mL. Ms. H states she has not started any new medications or supplements; her serum creatinine and liver function test results are within normal limits.
Upon further questioning, Ms. H says that an upper respiratory infection has been “going around her office,” so she increased her vitamin C intake by drinking 2 glasses of grapefruit juice a day (she doesn’t like orange juice). She has heard grapefruit juice can cause problems with some drugs so she is careful not to drink it at the same time she takes her medications. Her psychiatrist recognizes there may be a drug interaction involved, and recommends Ms. H hold her carbamazepine for 1 day and not consume any more grapefruit juice. A few days later, she reports feeling much better during a follow-up call and she makes an appointment to have her carbamazepine level rechecked in a we
Although grapefruit products are high in vitamins and low in calories, they can be associated with potentially serious drug interactions. The interaction between grapefruit juice and the calcium channel blocker felodipine was discovered inadvertently >20 years ago; since that time, possible interactions with >85 medications have been identified.1 Interactions with grapefruit products are complicated because, although most result in increased drug exposure, reduced exposure of the medication also can occur. Additionally, the degree and clinical significance of the interaction varies among individuals and from one drug to another.
Mechanism of action
Most interactions with grapefruit products are thought to result from the inhibition of intestinal cytochrome P450 3A4 (CYP3A4). CYP3A4 is involved in the metabolism of numerous drugs, and is the most abundant cytochrome P450 enzyme in the liver and epithelial cells lining the intestine.2 Although hepatic CYP3A4 is thought to be minimally affected by grapefruit, inhibition of intestinal CYP3A4 can result in an overall increase in bioavailability of medications that are substrates and raise the risk of potential toxicity.3 Grapefruit contains various chemicals collectively known as furanocoumarins, which are largely responsible for inhibition of intestinal CYP3A4.4 Additionally, Seville oranges and the pomelo (a large, sweet grapefruit-like citrus fruit) also contain furanocoumarins and could have a similar effect, warranting caution with certain medications.5
Inhibition of CYP3A4 by furanocoumarins cannot be reversed, and new enzymes must be synthesized to return to the previous level of function.6 Therefore, drug interactions resulting from CYP3A4 inhibition can last for as long as 72 hours after ingesting grapefruit products.7 Separating consumption of grapefruit products and medication administration will not help manage this interaction.
Grapefruit products also could affect drug disposition through effects on various drug transporters. Decreased systemic exposure to certain medications could occur through grapefruit’s inhibition of organic anion-transporting polypeptides (OATPs). OATPs form a family of drug uptake transporters found in the intestine, liver, kidney, and brain.8 For drugs that are substrates of OATPs, grapefruit’s inhibition of this transporter can result in decreased absorption and a resulting decrease in efficacy. Flavanoids in grapefruit, such as naringin, inhibit OATPs, which is competitive in nature.9 Unlike the irreversible inhibition of CYP3A4 by furanocoumarins, flavanoids effects on OATPs have been shown to decrease within 4 hours.10
No psychotropic medications have been identified as being susceptible to this interaction, but for those medications affected—including fexofenadine and levothyroxine—separating consumption of grapefruit and medication administration by 4 hours could avoid this interaction.11 Additional data indicate that orange juice and apple juice could have similar effects on OATPs.12
Perhaps the most well-known drug transporter, P-glycoprotein is part of the multidrug-resistant subfamily of transporters. It is located throughout the body, including in the intestine, kidneys, liver, and blood-brain barrier. P-glycoprotein acts as an export pump to decrease the cellular concentration of many different drug substrates, and many agents can alter P-glycoprotein’s expression or function.
Small changes in P-glycoprotein’s activity can result in substantial changes in the disposition of substrates, which can include certain antineoplastics and antiretrovirals. Most reports have found grapefruit juice inhibits P-glycoprotein-mediated efflux; however, there also are reports of transporter activation.6 Additionally, P-glycoprotein and CYP3A4 share many substrates, so it can be difficult to isolate the contribution of P-glycoprotein to grapefruit−drug interactions.13 The effect of grapefruit on P-glycoprotein activity has been difficult to fully elucidate; more studies are needed.
Grapefruit consumption and its effect
Drug interactions can occur by consuming commercially produced grapefruit juice and juice from concentrate, as well as freshly squeezed juice and grapefruit segments.14 CYP3A4-inhibiting furanocoumarins also have been isolated in grapefruit peel; it is not known, however, whether items made from peel (marmalade, candied peel) contain concentrations high enough to pose a risk of a drug interaction.14 Contributing to the unpredictability of grapefruit-drug interactions, the amount or concentration of furanocoumarins can vary among grapefruit products and brands.15 This variability can be influenced by the variety or maturity of the fruit and the fruit’s exposure to environmental stress.4
The frequency of consuming a grapefruit product can influence the degree of a drug interaction. In general, consuming one 8-oz glass of grapefruit juice or the segments from a whole grapefruit is enough to alter a susceptible drug’s pharmacokinetics.14 Regular grapefruit product consumption, however, can result in an overall greater effect.16,17
Lilja et al16 conducted a randomized, 4-phase, crossover study to look at the effect of grapefruit juice dose on kinetics of triazolam. Grapefruit juice was found to increase the mean area under the concentration-time curve (AUC) of triazolam compared with water, but no difference was found between single glasses of normal-strength and double-strength grapefruit juice. However, repeated consumption of double-strength grapefruit juice (200 mL, 3 times/d for 3 days) increased triazolam’s mean AUC by 143%, compared with an increase of 49% with just a single 200-mL glass of double-strength juice.16 Recurrent consumption of grapefruit juice (8 oz, 3 times/d for 6 days) also was found to increase the kinetics of the antihypertensive felodipine more than a single glass of grapefruit juice.17
Clinical consequences of an interaction between a drug and grapefruit can be difficult to predict. Drug concentration changes caused by a grapefruit interaction could vary based on interindividual differences. The amount and activity of intestinal CYP3A4 can vary from person to person, and can be influenced by genetic polymorphisms in addition to race, age, and environmental variables.18 Interindividual sensitivity to a change in a drug’s concentration also will differ, and patient-specific factors, such as concomitant drugs or diseases, could influence the likelihood of harm.
Interactions with grapefruit products are not necessarily a “class effect,” and specific drugs within a therapeutic category can be affected (although others might not). Several drug-specific characteristics can help gauge the risk of a clinically relevant interaction with grapefruit, including:
• metabolism through CYP3A4
• low bioavailability
• oral administration
• a narrow therapeutic index.1
For drugs with low bioavailability because of first-pass metabolism, grapefruit’s inhibition of intestinal CYP3A4 can result in a greater relative increase in plasma concentrations compared with a drug with high bioavailability.19
For example, an increase in bioavailability from 5% to 10% will result in a much larger increase in AUC and overall clinical exposure compared with an increase from 85% to 90% even though both represent an absolute increase of 5%. Although a drug does not have to have low oral bioavailability for an interaction to occur, lower bioavailability means that a drug has a higher likelihood of causing a significant interaction because of altered pharmacokinetics. Of note, injectable medications will not interact with grapefruit because metabolism through intestinal CYP3A4 is bypassed and grapefruit does not significantly inhibit hepatic CYP3A4.
Although grapefruit products could alter the pharmacokinetics of susceptible drugs, those changes might not be associated with adverse effects. Therefore, a factor to consider in evaluating a potential interaction with grapefruit is the drug’s therapeutic index and its risk of serious adverse effects. Drugs with a narrow therapeutic index are of particular concern because a significant increase in therapeutic or adverse effects could result from a relatively small increase in the drug’s concentration.7
Which medications are affected?
Among medications identified as interacting with grapefruit, some cardiovascular agents and several of the HMG-CoA reductase inhibitors (statins) have garnered the most attention. However, grapefruit also can affect the metabolism of several psychotropic medications through inhibition of intestinal CYP3A4 (Table).16,20-35 Prescribing information for some drugs warns against consuming grapefruit while using the medication. Among CNS agents, buspirone, carbamazepine, lurasidone, pimozide, triazolam, and oral midazolam all have such warnings in their product labeling.
Buspirone currently is not recommended with “large quantities of grapefruit juice.”20 A randomized, 2-phase crossover study looking at the effects of grapefruit juice on buspirone’s pharmacokinetics found that double-strength grapefruit juice (200 mL, administered 3 times/d for 3 days) resulted in a 9.2-fold increase in mean AUC and a 4.3-fold increase in mean Cmax after a single 10-mg buspirone dose.22 Highlighting the wide interindividual variability seen with drug-grapefruit interactions, the increase found in buspirone’s AUC ranged from 3-fold to 20-fold among study participants.22
Carbamazepine product labeling lists grapefruit juice as a CYP3A4 inhibitor that is expected to or has been found to increase plasma levels of the drug.20 Carbamazepine’s bioavailability is influenced by intestinal CYP3A4 activity; in a randomized, 2-phase crossover study of 10 patients with epilepsy, grapefruit juice was found to increase AUC of carbamazepine by 41% and Cmax by 40%.23,36
Lurasidone and pimozide, although not specifically studied, have product labels that recommend avoiding grapefruit juice because it could inhibit metabolism of these agents by CYP3A4.20 Of particular concern is the potential for elevated levels of pimozide to increase the risk of adverse cardiovascular effects including QT interval prolongation.19
Midazolam. Although grapefruit juice does not affect the disposition of IV midazolam, pretreatment with grapefruit juice was found to increase the AUC and Cmax of oral midazolam by 52% and 56%, respectively.30
Other considerations in drug-grapefruit interactions
Cautionary statements about a possible interaction with grapefruit juice for many other psychotropics can be found in commonly used drug information references or online sources. If you are concerned about a possible interaction and avoiding grapefruit products is not feasible, consider a different medication in the same class.
However, you also should consider the level of evidence supporting any purported interaction. Several psychotropic agents do have studies or case reports supporting an interaction with grapefruit, but cautionary statements could be based on theoretical concerns because of a medication’s bioavailability, metabolic pathway, and concern for increased adverse events related to higher drug concentrations. Adding to the confusion, cautionary statements can be found about medications, such as clozapine, that have not been shown to have an interaction with grapefruit juice when studied.
With many of the drugs that have a reported or theoretical interaction with grapefruit, data are inconsistent as to whether the resulting interaction will be clinically relevant. A number of variables relating to the individual patient, grapefruit product, or particular drug can play a role in the significance of an interaction. Additionally, effects on drug disposition can last for a few days after consuming a grapefruit product.
Keep alert to situations of increased risk
Recall that the case patient, Ms. H, presented with an elevated carbamazepine level and suffered resulting adverse effects because of an interaction between the drug and grapefruit juice. Although Ms. H was careful to separate intake of grapefruit juice from carbamazepine administration, grapefruit’s inhibition of intestinal CYP3A4 still was present, leading to the interaction.
It is important for health care professionals to recognize this potential risk and to advise patients regarding possible interactions between medications and grapefruit products.
Related Resources
• U.S. Food and Drug Administration. Grapefruit juice and medicine may not mix. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm292276.htm.
• Hanley MJ, Cancalon P, Widmer WW, et al. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol. 2011;7(3):267-286.
• Andrade C. Fruit juice, organic anion transporting polypeptides, and drug interactions in psychiatry. J Clin Psychiatry. 2014;75(11):e1323-e1325.
Drug Brand Names
Alprazolam • Xanax Lurasidone • Latuda
Buspirone • BuSpar Midazolam • Versed
Carbamazepine • Tegretol Methadone • Dolophine
Clomipramine • Anafranil Nefazodone • Serzone
Clozapine • Clozaril Olanzapine • Zyprexa
Diazepam • Valium Pimozide • Orap
Felodipine • Plendil Quetiapine • Seroquel
Fexofenadine • Allegra Sertraline • Zoloft
Fluoxetine • Prozac Trazodone • Desyrel
Fluvoxamine • Luvox Triazolam • Halcion
Haloperidol • Haldol Ziprasidone • Geodon
Levothyroxine • Levoxyl, Synthroid
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. H, age 42, was given a diagnosis of bipolar disorder 10 years ago and has been taking carbamazepine, 1,200 mg/d, and olanzapine, 10 mg/d, for the past 2 years. She has not experienced a mood episode while on this regimen, and her carbamazepine level was 9.2 μg/mL 6 months ago. The only adverse effect she experienced was weight gain of approximately 10 lb. Ms. H takes a calcium supplement, but no other medications.
Ms. H reports to her psychiatrist that, for the past few days, she has been feeling nauseated, fatigued, and dizzy, but has continued taking her medications as prescribed. Her carbamazepine level is found to be 13.1 μg/mL. Ms. H states she has not started any new medications or supplements; her serum creatinine and liver function test results are within normal limits.
Upon further questioning, Ms. H says that an upper respiratory infection has been “going around her office,” so she increased her vitamin C intake by drinking 2 glasses of grapefruit juice a day (she doesn’t like orange juice). She has heard grapefruit juice can cause problems with some drugs so she is careful not to drink it at the same time she takes her medications. Her psychiatrist recognizes there may be a drug interaction involved, and recommends Ms. H hold her carbamazepine for 1 day and not consume any more grapefruit juice. A few days later, she reports feeling much better during a follow-up call and she makes an appointment to have her carbamazepine level rechecked in a we
Although grapefruit products are high in vitamins and low in calories, they can be associated with potentially serious drug interactions. The interaction between grapefruit juice and the calcium channel blocker felodipine was discovered inadvertently >20 years ago; since that time, possible interactions with >85 medications have been identified.1 Interactions with grapefruit products are complicated because, although most result in increased drug exposure, reduced exposure of the medication also can occur. Additionally, the degree and clinical significance of the interaction varies among individuals and from one drug to another.
Mechanism of action
Most interactions with grapefruit products are thought to result from the inhibition of intestinal cytochrome P450 3A4 (CYP3A4). CYP3A4 is involved in the metabolism of numerous drugs, and is the most abundant cytochrome P450 enzyme in the liver and epithelial cells lining the intestine.2 Although hepatic CYP3A4 is thought to be minimally affected by grapefruit, inhibition of intestinal CYP3A4 can result in an overall increase in bioavailability of medications that are substrates and raise the risk of potential toxicity.3 Grapefruit contains various chemicals collectively known as furanocoumarins, which are largely responsible for inhibition of intestinal CYP3A4.4 Additionally, Seville oranges and the pomelo (a large, sweet grapefruit-like citrus fruit) also contain furanocoumarins and could have a similar effect, warranting caution with certain medications.5
Inhibition of CYP3A4 by furanocoumarins cannot be reversed, and new enzymes must be synthesized to return to the previous level of function.6 Therefore, drug interactions resulting from CYP3A4 inhibition can last for as long as 72 hours after ingesting grapefruit products.7 Separating consumption of grapefruit products and medication administration will not help manage this interaction.
Grapefruit products also could affect drug disposition through effects on various drug transporters. Decreased systemic exposure to certain medications could occur through grapefruit’s inhibition of organic anion-transporting polypeptides (OATPs). OATPs form a family of drug uptake transporters found in the intestine, liver, kidney, and brain.8 For drugs that are substrates of OATPs, grapefruit’s inhibition of this transporter can result in decreased absorption and a resulting decrease in efficacy. Flavanoids in grapefruit, such as naringin, inhibit OATPs, which is competitive in nature.9 Unlike the irreversible inhibition of CYP3A4 by furanocoumarins, flavanoids effects on OATPs have been shown to decrease within 4 hours.10
No psychotropic medications have been identified as being susceptible to this interaction, but for those medications affected—including fexofenadine and levothyroxine—separating consumption of grapefruit and medication administration by 4 hours could avoid this interaction.11 Additional data indicate that orange juice and apple juice could have similar effects on OATPs.12
Perhaps the most well-known drug transporter, P-glycoprotein is part of the multidrug-resistant subfamily of transporters. It is located throughout the body, including in the intestine, kidneys, liver, and blood-brain barrier. P-glycoprotein acts as an export pump to decrease the cellular concentration of many different drug substrates, and many agents can alter P-glycoprotein’s expression or function.
Small changes in P-glycoprotein’s activity can result in substantial changes in the disposition of substrates, which can include certain antineoplastics and antiretrovirals. Most reports have found grapefruit juice inhibits P-glycoprotein-mediated efflux; however, there also are reports of transporter activation.6 Additionally, P-glycoprotein and CYP3A4 share many substrates, so it can be difficult to isolate the contribution of P-glycoprotein to grapefruit−drug interactions.13 The effect of grapefruit on P-glycoprotein activity has been difficult to fully elucidate; more studies are needed.
Grapefruit consumption and its effect
Drug interactions can occur by consuming commercially produced grapefruit juice and juice from concentrate, as well as freshly squeezed juice and grapefruit segments.14 CYP3A4-inhibiting furanocoumarins also have been isolated in grapefruit peel; it is not known, however, whether items made from peel (marmalade, candied peel) contain concentrations high enough to pose a risk of a drug interaction.14 Contributing to the unpredictability of grapefruit-drug interactions, the amount or concentration of furanocoumarins can vary among grapefruit products and brands.15 This variability can be influenced by the variety or maturity of the fruit and the fruit’s exposure to environmental stress.4
The frequency of consuming a grapefruit product can influence the degree of a drug interaction. In general, consuming one 8-oz glass of grapefruit juice or the segments from a whole grapefruit is enough to alter a susceptible drug’s pharmacokinetics.14 Regular grapefruit product consumption, however, can result in an overall greater effect.16,17
Lilja et al16 conducted a randomized, 4-phase, crossover study to look at the effect of grapefruit juice dose on kinetics of triazolam. Grapefruit juice was found to increase the mean area under the concentration-time curve (AUC) of triazolam compared with water, but no difference was found between single glasses of normal-strength and double-strength grapefruit juice. However, repeated consumption of double-strength grapefruit juice (200 mL, 3 times/d for 3 days) increased triazolam’s mean AUC by 143%, compared with an increase of 49% with just a single 200-mL glass of double-strength juice.16 Recurrent consumption of grapefruit juice (8 oz, 3 times/d for 6 days) also was found to increase the kinetics of the antihypertensive felodipine more than a single glass of grapefruit juice.17
Clinical consequences of an interaction between a drug and grapefruit can be difficult to predict. Drug concentration changes caused by a grapefruit interaction could vary based on interindividual differences. The amount and activity of intestinal CYP3A4 can vary from person to person, and can be influenced by genetic polymorphisms in addition to race, age, and environmental variables.18 Interindividual sensitivity to a change in a drug’s concentration also will differ, and patient-specific factors, such as concomitant drugs or diseases, could influence the likelihood of harm.
Interactions with grapefruit products are not necessarily a “class effect,” and specific drugs within a therapeutic category can be affected (although others might not). Several drug-specific characteristics can help gauge the risk of a clinically relevant interaction with grapefruit, including:
• metabolism through CYP3A4
• low bioavailability
• oral administration
• a narrow therapeutic index.1
For drugs with low bioavailability because of first-pass metabolism, grapefruit’s inhibition of intestinal CYP3A4 can result in a greater relative increase in plasma concentrations compared with a drug with high bioavailability.19
For example, an increase in bioavailability from 5% to 10% will result in a much larger increase in AUC and overall clinical exposure compared with an increase from 85% to 90% even though both represent an absolute increase of 5%. Although a drug does not have to have low oral bioavailability for an interaction to occur, lower bioavailability means that a drug has a higher likelihood of causing a significant interaction because of altered pharmacokinetics. Of note, injectable medications will not interact with grapefruit because metabolism through intestinal CYP3A4 is bypassed and grapefruit does not significantly inhibit hepatic CYP3A4.
Although grapefruit products could alter the pharmacokinetics of susceptible drugs, those changes might not be associated with adverse effects. Therefore, a factor to consider in evaluating a potential interaction with grapefruit is the drug’s therapeutic index and its risk of serious adverse effects. Drugs with a narrow therapeutic index are of particular concern because a significant increase in therapeutic or adverse effects could result from a relatively small increase in the drug’s concentration.7
Which medications are affected?
Among medications identified as interacting with grapefruit, some cardiovascular agents and several of the HMG-CoA reductase inhibitors (statins) have garnered the most attention. However, grapefruit also can affect the metabolism of several psychotropic medications through inhibition of intestinal CYP3A4 (Table).16,20-35 Prescribing information for some drugs warns against consuming grapefruit while using the medication. Among CNS agents, buspirone, carbamazepine, lurasidone, pimozide, triazolam, and oral midazolam all have such warnings in their product labeling.
Buspirone currently is not recommended with “large quantities of grapefruit juice.”20 A randomized, 2-phase crossover study looking at the effects of grapefruit juice on buspirone’s pharmacokinetics found that double-strength grapefruit juice (200 mL, administered 3 times/d for 3 days) resulted in a 9.2-fold increase in mean AUC and a 4.3-fold increase in mean Cmax after a single 10-mg buspirone dose.22 Highlighting the wide interindividual variability seen with drug-grapefruit interactions, the increase found in buspirone’s AUC ranged from 3-fold to 20-fold among study participants.22
Carbamazepine product labeling lists grapefruit juice as a CYP3A4 inhibitor that is expected to or has been found to increase plasma levels of the drug.20 Carbamazepine’s bioavailability is influenced by intestinal CYP3A4 activity; in a randomized, 2-phase crossover study of 10 patients with epilepsy, grapefruit juice was found to increase AUC of carbamazepine by 41% and Cmax by 40%.23,36
Lurasidone and pimozide, although not specifically studied, have product labels that recommend avoiding grapefruit juice because it could inhibit metabolism of these agents by CYP3A4.20 Of particular concern is the potential for elevated levels of pimozide to increase the risk of adverse cardiovascular effects including QT interval prolongation.19
Midazolam. Although grapefruit juice does not affect the disposition of IV midazolam, pretreatment with grapefruit juice was found to increase the AUC and Cmax of oral midazolam by 52% and 56%, respectively.30
Other considerations in drug-grapefruit interactions
Cautionary statements about a possible interaction with grapefruit juice for many other psychotropics can be found in commonly used drug information references or online sources. If you are concerned about a possible interaction and avoiding grapefruit products is not feasible, consider a different medication in the same class.
However, you also should consider the level of evidence supporting any purported interaction. Several psychotropic agents do have studies or case reports supporting an interaction with grapefruit, but cautionary statements could be based on theoretical concerns because of a medication’s bioavailability, metabolic pathway, and concern for increased adverse events related to higher drug concentrations. Adding to the confusion, cautionary statements can be found about medications, such as clozapine, that have not been shown to have an interaction with grapefruit juice when studied.
With many of the drugs that have a reported or theoretical interaction with grapefruit, data are inconsistent as to whether the resulting interaction will be clinically relevant. A number of variables relating to the individual patient, grapefruit product, or particular drug can play a role in the significance of an interaction. Additionally, effects on drug disposition can last for a few days after consuming a grapefruit product.
Keep alert to situations of increased risk
Recall that the case patient, Ms. H, presented with an elevated carbamazepine level and suffered resulting adverse effects because of an interaction between the drug and grapefruit juice. Although Ms. H was careful to separate intake of grapefruit juice from carbamazepine administration, grapefruit’s inhibition of intestinal CYP3A4 still was present, leading to the interaction.
It is important for health care professionals to recognize this potential risk and to advise patients regarding possible interactions between medications and grapefruit products.
Related Resources
• U.S. Food and Drug Administration. Grapefruit juice and medicine may not mix. http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm292276.htm.
• Hanley MJ, Cancalon P, Widmer WW, et al. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol. 2011;7(3):267-286.
• Andrade C. Fruit juice, organic anion transporting polypeptides, and drug interactions in psychiatry. J Clin Psychiatry. 2014;75(11):e1323-e1325.
Drug Brand Names
Alprazolam • Xanax Lurasidone • Latuda
Buspirone • BuSpar Midazolam • Versed
Carbamazepine • Tegretol Methadone • Dolophine
Clomipramine • Anafranil Nefazodone • Serzone
Clozapine • Clozaril Olanzapine • Zyprexa
Diazepam • Valium Pimozide • Orap
Felodipine • Plendil Quetiapine • Seroquel
Fexofenadine • Allegra Sertraline • Zoloft
Fluoxetine • Prozac Trazodone • Desyrel
Fluvoxamine • Luvox Triazolam • Halcion
Haloperidol • Haldol Ziprasidone • Geodon
Levothyroxine • Levoxyl, Synthroid
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Bailey DG, Dresser G, Arnold JM. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? CMAJ. 2013;185(4):309-316.
2. Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21): 2211-2221.
3. Saito M, Hirata-Koizumi M, Matsumoto M, et al. Undesirable effects of citrus juice on the pharmacokinetics of drugs: focus on recent studies. Drug Saf. 2005;28(8):677- 694.
4. Cancalon PF, Barros SM, Haun C, et al. Effect of maturity, processing, and storage on the furanocoumarin composition of grapefruit and grapefruit juice. J Food Sci. 2011;76(4):C543-C548.
5. Pirmohamed M. Drug-grapefruit juice interactions: two mechanisms are clear but individual responses vary. BMJ. 2013;346:f1. doi: 10.1136/bmj.f1.
6. Dahan A, Altman H. Food-drug interaction: grapefruit juice augments drug bioavailability–mechanism, extent and relevance. Eur J Clin Nutr. 2004;58:1-9.
7. Stump AL, Mayo T, Blum A. Management of grapefruit-drug interactions. Am Fam Physician. 2006;74(4):605-608.
8. Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(suppl 2):1-5.
9. Bailey DG, Dressker GK, Leak BF, et al. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin Pharmacol Ther. 2007;81(4):495-502.
10. Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81(3):362-370.
11. Bailey DG. Fruit juice inhibition of uptake transport: a new type of food-drug interaction. Br J Clin Pharmacol. 2010;70(5): 645-655.
12. Dresser GK, Bailey DG, Leake BF, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther. 2002;71:11-20.
13. Seden K, Dickinson L, Khoo S, et al. Grapefruit-drug interactions. Drugs. 2010;70(18):2373-2407.
14. Bailey DG, Dresser GK, Kreeft JH, et al. Grapefruit-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther. 2000;68(5):468-477.
15. De Castro WV, Mertens-Talcott S, Rubner A, et al. Variation of flavonoids and furanocoumarins in grapefruit juices: a potential source of variability in grapefruit juice-drug interaction studies. J Agric Food Chem. 2006;54(1):249-255.
16. Lilja JJ, Kivistö KT, Backman JT, et al. Effect of grapefruit juice on grapefruit juice-triazolam interaction: repeated consumption prolongs triazolam half-life. Eur J Clin Pharmacol. 2000;56(5):411-415.
17. Lown KS, Bailey DG, Fontana RJ, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Invest. 1997;99(10):2545-2553.
18. Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535-567.
19. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000; 38(1):41-57.
20. U.S. Food and Drug Administration. Drugs@FDA. http://www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed July 14, 2014.
21. Yasui, N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;15(2):185-190.
22. Lilja JJ, Kivistö KT, Backman JT, et al. Grapefruit juice substantially increases plasma concentrations of buspirone. Clin Pharmacol Ther. 1998;64(6):655-660.
23. Garg SK, Kumar N, Bhargava VK, et al. Effect of grapefruit juice on carbamazepine bioavailability in patients with epilepsy. Clin Pharmacol Ther. 1998;64(3):286-288.
24. Oesterheld J, Kallepalli BR. Grapefruit juice and clomipramine: shifting metabolic ratios. J Clin Psychopharm. 1997;17(1):62-63.
25. Lane HY, Jann MW, Chang YC, et al. Repeated ingestion of grapefruit juice does not alter clozapine’s steady-state plasma levels, effectiveness, and tolerability. J Clin Psychiatry. 2001;62(10):812-817.
26. Ozdemir M, Aktan Y, Boydag BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59.
27. DeSilva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS. 2001;15(10):1281-1285.
28. Hori H, Yoshimura R, Ueda N, et al. Grapefruit juice-fluvoxamine interaction—is it risky or not? J Clin Psychopharmacol. 2003;23(4):422-424.
29. Yasui N, Kondo T, Suzuki A, et al. Lack of significant pharmacokinetic interaction between haloperidol and grapefruit juice. Int Clin Psychopharmacol. 1999;142(2):113-118.
30. Kupferschmidt HH, Ha HR, Ziegler WH, et al. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58(1):20-28.
31. Benmebarek M, Cevaud C, Gex-Fabry M, et al. Effects of grapefruit juice on the pharmacokinetics of the enantiomers of methadone. Clin Pharmacol Ther. 2004;76(1):55-63.
32. DeVane CL, Nemeroff CB. Clinical pharmacokinetics of quetiapine: an atypical antipsychotic. Clin Pharmacokinet. 2001;40(7):509-522.
33. Ueda N, Yoshimura R, Umene-Nakano W, et al. Grapefruit juice alters plasma sertraline levels after single ingestion of sertraline in healthy volunteers. World J Biol Psychiatry. 2009;10(4 pt 3):832-835.
34. Lee AJ, Chan WK, Harralson AF, et al. The effects of grapefruit juice on sertraline metabolism: an in vitro and in vivo study. Clin Ther. 1999;21(11):1890-1899.
35. Sugimoto K, Araki N, Ohmori M, et al. Interaction between grapefruit juice and hypnotic drugs: comparison of triazolam and quazepam. Eur J Clin Pharmacol. 2006;62(3):209-215.
36. Fagiolino P, Vazquez M, Olano I, et al. Systemic and presystemic conversion of carbamazepine to carbamazepine- 10-11-epoxide during long term treatment. Journal of Epilepsy and Clinical Neurophysiology. 2006;12(1):13-16.
1. Bailey DG, Dresser G, Arnold JM. Grapefruit-medication interactions: forbidden fruit or avoidable consequences? CMAJ. 2013;185(4):309-316.
2. Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21): 2211-2221.
3. Saito M, Hirata-Koizumi M, Matsumoto M, et al. Undesirable effects of citrus juice on the pharmacokinetics of drugs: focus on recent studies. Drug Saf. 2005;28(8):677- 694.
4. Cancalon PF, Barros SM, Haun C, et al. Effect of maturity, processing, and storage on the furanocoumarin composition of grapefruit and grapefruit juice. J Food Sci. 2011;76(4):C543-C548.
5. Pirmohamed M. Drug-grapefruit juice interactions: two mechanisms are clear but individual responses vary. BMJ. 2013;346:f1. doi: 10.1136/bmj.f1.
6. Dahan A, Altman H. Food-drug interaction: grapefruit juice augments drug bioavailability–mechanism, extent and relevance. Eur J Clin Nutr. 2004;58:1-9.
7. Stump AL, Mayo T, Blum A. Management of grapefruit-drug interactions. Am Fam Physician. 2006;74(4):605-608.
8. Kim RB. Organic anion-transporting polypeptide (OATP) transporter family and drug disposition. Eur J Clin Invest. 2003;33(suppl 2):1-5.
9. Bailey DG, Dressker GK, Leak BF, et al. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin Pharmacol Ther. 2007;81(4):495-502.
10. Glaeser H, Bailey DG, Dresser GK, et al. Intestinal drug transporter expression and the impact of grapefruit juice in humans. Clin Pharmacol Ther. 2007;81(3):362-370.
11. Bailey DG. Fruit juice inhibition of uptake transport: a new type of food-drug interaction. Br J Clin Pharmacol. 2010;70(5): 645-655.
12. Dresser GK, Bailey DG, Leake BF, et al. Fruit juices inhibit organic anion transporting polypeptide-mediated drug uptake to decrease the oral availability of fexofenadine. Clin Pharmacol Ther. 2002;71:11-20.
13. Seden K, Dickinson L, Khoo S, et al. Grapefruit-drug interactions. Drugs. 2010;70(18):2373-2407.
14. Bailey DG, Dresser GK, Kreeft JH, et al. Grapefruit-felodipine interaction: effect of unprocessed fruit and probable active ingredients. Clin Pharmacol Ther. 2000;68(5):468-477.
15. De Castro WV, Mertens-Talcott S, Rubner A, et al. Variation of flavonoids and furanocoumarins in grapefruit juices: a potential source of variability in grapefruit juice-drug interaction studies. J Agric Food Chem. 2006;54(1):249-255.
16. Lilja JJ, Kivistö KT, Backman JT, et al. Effect of grapefruit juice on grapefruit juice-triazolam interaction: repeated consumption prolongs triazolam half-life. Eur J Clin Pharmacol. 2000;56(5):411-415.
17. Lown KS, Bailey DG, Fontana RJ, et al. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J Clin Invest. 1997;99(10):2545-2553.
18. Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535-567.
19. Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000; 38(1):41-57.
20. U.S. Food and Drug Administration. Drugs@FDA. http://www.accessdata.fda.gov/scripts/cder/drugsatfda. Accessed July 14, 2014.
21. Yasui, N, Kondo T, Furukori H, et al. Effects of repeated ingestion of grapefruit juice on the single and multiple oral-dose pharmacokinetics and pharmacodynamics of alprazolam. Psychopharmacology (Berl). 2000;15(2):185-190.
22. Lilja JJ, Kivistö KT, Backman JT, et al. Grapefruit juice substantially increases plasma concentrations of buspirone. Clin Pharmacol Ther. 1998;64(6):655-660.
23. Garg SK, Kumar N, Bhargava VK, et al. Effect of grapefruit juice on carbamazepine bioavailability in patients with epilepsy. Clin Pharmacol Ther. 1998;64(3):286-288.
24. Oesterheld J, Kallepalli BR. Grapefruit juice and clomipramine: shifting metabolic ratios. J Clin Psychopharm. 1997;17(1):62-63.
25. Lane HY, Jann MW, Chang YC, et al. Repeated ingestion of grapefruit juice does not alter clozapine’s steady-state plasma levels, effectiveness, and tolerability. J Clin Psychiatry. 2001;62(10):812-817.
26. Ozdemir M, Aktan Y, Boydag BS, et al. Interaction between grapefruit juice and diazepam in humans. Eur J Drug Metab Pharmacokinet. 1998;23(1):55-59.
27. DeSilva KE, Le Flore DB, Marston BJ, et al. Serotonin syndrome in HIV-infected individuals receiving antiretroviral therapy and fluoxetine. AIDS. 2001;15(10):1281-1285.
28. Hori H, Yoshimura R, Ueda N, et al. Grapefruit juice-fluvoxamine interaction—is it risky or not? J Clin Psychopharmacol. 2003;23(4):422-424.
29. Yasui N, Kondo T, Suzuki A, et al. Lack of significant pharmacokinetic interaction between haloperidol and grapefruit juice. Int Clin Psychopharmacol. 1999;142(2):113-118.
30. Kupferschmidt HH, Ha HR, Ziegler WH, et al. Interaction between grapefruit juice and midazolam in humans. Clin Pharmacol Ther. 1995;58(1):20-28.
31. Benmebarek M, Cevaud C, Gex-Fabry M, et al. Effects of grapefruit juice on the pharmacokinetics of the enantiomers of methadone. Clin Pharmacol Ther. 2004;76(1):55-63.
32. DeVane CL, Nemeroff CB. Clinical pharmacokinetics of quetiapine: an atypical antipsychotic. Clin Pharmacokinet. 2001;40(7):509-522.
33. Ueda N, Yoshimura R, Umene-Nakano W, et al. Grapefruit juice alters plasma sertraline levels after single ingestion of sertraline in healthy volunteers. World J Biol Psychiatry. 2009;10(4 pt 3):832-835.
34. Lee AJ, Chan WK, Harralson AF, et al. The effects of grapefruit juice on sertraline metabolism: an in vitro and in vivo study. Clin Ther. 1999;21(11):1890-1899.
35. Sugimoto K, Araki N, Ohmori M, et al. Interaction between grapefruit juice and hypnotic drugs: comparison of triazolam and quazepam. Eur J Clin Pharmacol. 2006;62(3):209-215.
36. Fagiolino P, Vazquez M, Olano I, et al. Systemic and presystemic conversion of carbamazepine to carbamazepine- 10-11-epoxide during long term treatment. Journal of Epilepsy and Clinical Neurophysiology. 2006;12(1):13-16.
What to tell your bipolar disorder patient who wants to breast-feed
Ms. K, age 35, soon will deliver her second child. She has a 12-year history of bipolar disorder, which was well controlled with lithium, 1,200 mg/d. During her first pregnancy 3 years ago, Ms. K stopped taking lithium because she was concerned about the risk of Ebstein’s anomaly. She experienced a bipolar relapse after her healthy baby was born, and developed postpartum psychosis that was treated by restarting lithium, 1,200 mg/d, and adding olanzapine, 10 mg/d.
Ms. K has continued these medications throughout her current pregnancy. She wants to breast-feed her infant and is concerned about the effects that psychotropics might have on her newborn.
Breast-feeding and medications
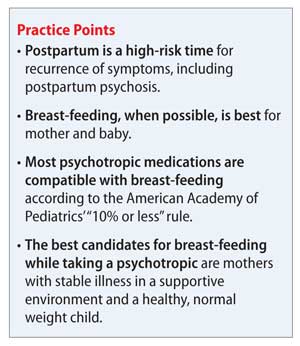
The benefits of breast-feeding for mother and infant are well-known. Despite this, some women with bipolar disorder are advised not to breast-feed or, worse, to discontinue their medications in order to breast-feed. Decisions about breast-feeding while taking medications should be based on evidence of benefits and risks to the infant, along with a discussion of the risks of untreated illness, which is high postpartum. The prescribing information for many of the medications used to treat bipolar disorder advise against breast-feeding, although there is little evidence of harm.
Drug dosages and levels in breast milk can be reported a few different ways:
• percentage of maternal dosage measured in the breast milk
• percentage weight-adjusted maternal dosage
• percentage of maternal plasma level, and milk-to-plasma ratio (M:P).
Daily infant dosage can be calculated by multiplying the average concentration of the drug in breast milk (mg/mL) by the average volume of milk the baby ingests in 24 hours (usually 150 mL).1 The relative infant dosage can be calculated as the percentage maternal dosage, which is the daily infant dosage (mg/kg/d) ÷ maternal dosage (mg/kg/d) × 100.1
According to the American Academy of Pediatrics, ≤10% of the maternal dosage is compatible with breast-feeding.1 Most psychotropics studied fall below this threshold. Keep in mind that all published research is for breast-feeding a full-term infant; exercise caution with premature or low birth weight infants. Infants born to mothers taking a psychotropic should be monitored for withdrawal symptoms, which might be associated with antidepressants and benzodiazepines, but otherwise are rare.
Lithium
Breast-feeding during lithium treatment has been considered contraindicated based on early reports that lithium was highly excreted in breast milk.2 A 2003 study2 of 11 women found that lithium was excreted in breast milk in amounts between zero and 30% of maternal dosage (mean, 12.2% ± 8.5%; median, 11.2%; 95% CI, 6.3% to 18.0%). Researchers measured serum concentrations in 2 infants and found that 1 received 17% to 20% of the maternal dosage, and the other showed 50%. None of the infants experienced adverse events. In a study of 10 mother-infant pairs, breast milk lithium concentration averaged 0.35 mEq/L (standard deviation [SD] = 0.10, range 0.19 to 0.48 mEq/L), with paired infant serum concentrations of 0.16 mEq/L (SD = 0.06, range 0.09 to 0.25 mEq/L).3 Some transient abnormalities were found in infant serum concentrations of thyroid-stimulating hormone (TSH), blood urea nitrogen, and creatinine; there were no adverse effects on development. The authors recommend monitoring for TSH abnormalities in infants.
Olanzapine
Olanzapine prescribing information cites a study reporting that 1.8% of the maternal dosage is transferred to breast milk.4 Yet the olanzapine prescribing information states, “It is recommended that women receiving olanzapine should not breast-feed.” Olanzapine use during breast-feeding has been studied more than many medications, in part because of a database maintained by the manufacturer. In a study using the manufacturer’s database (N = 102) adverse reactions were reported in 15.6% of the infants, with the most common being somnolence (3.9%), irritability (2%), tremor (2%), and insomnia (2%).5
Other second-generation antipsychotics
Aripiprazole. The only case report of aripiprazole excretion in human breast milk found a concentration of approximately 20% of the maternal plasma level and an M:P ratio of 0.18:0.2.6
Asenapine. According to asenapine prescribing information7 and a literature search, it is not known whether asenapine is excreted in breast milk of humans, although it is found in the milk of lactating rats.
Lurasidone. According to the lurasidone prescribing information8 and a literature search, it is not known whether lurasidone is excreted in human breast milk, although it is found in the milk of lactating rats.
Quetiapine. An initial study reported that 0.09% to 0.43% of the maternal dosage of quetiapine was excreted in breast milk.9 Further studies found excretion to be 0.09% of maternal dosage, with infant plasma levels reaching 6% of the maternal dosage.10 A case series found that one-third of babies exposed to quetiapine during breast-feeding showed some neurodevelopmental delay, although these mothers also were taking other psychotropics.11
Risperidone. A 2000 study12 of risperidone in lactation reported that 0.84% weight-adjusted maternal risperidone dosage and 3.46% of its metabolite 9-hydroxyrisperidone is transferred to the infant. A later study showed 2.3% to 4.7% of the maternal dosage is transferred, with no adverse events reported in infants.13 A case study reported no adverse events and normal neurodevelopment in a the child of a mother taking risperidone.14
Ziprasidone. According to the ziprasidone prescribing information15 and a literature search, is not known whether ziprasidone is excreted in human breast milk.
See the Table4,6-10,12,13,15 for a summary of the evidence levels of second-generation antipsychotics that are excreted in breast milk.

Other mood stabilizers
Carbamazepine has been measured in breast milk at 3.8% to 5.9% of the maternal dosage.16
Lamotrigine. In a study of 30 lactating women, the breast milk contained an average of 9.2% of the maternal dosage of lamotrigine.17 Mild thrombocytosis was detected in 7 of 8 infants; no other adverse effects were reported. A case study describes a woman who breast-fed while taking lamotrigine, 850 mg/d, and who experienced dizziness and visual disturbances. The infant had apnea episodes followed by a cyanotic crisis, which required resuscitation. The infant’s plasma lamotrigine level was 4.87 μg/mL. Symptoms disappeared when the mother stopped breast-feeding.18 Lamotrigine is considered to be moderately safe in breast-feeding patients with proper monitoring. The drug also has a known safety profile because of its use in children with epilepsy.
Valproic acid. Because of its high plasma protein binding, valproic acid does not pass readily into the breast milk. Newborns receive approximately 1.4% to 1.7% of the maternal dosage.16 Caution is advised, however, because of some reported adverse events. One case reported thrombocytopenic purpura and anemia in an infant.19 Valproic acid is considered to be compatible with breast-feeding with proper monitoring.
Benzodiazepines
Benzodiazepines can be helpful adjunctive medications to aid sleep, which is essential for the mother’s and infant’s health. In a prospectively recruited, retrospectively assessed cohort study that evaluated 124 women taking benzodiazepines while breast-feeding, adverse effects, specifically sedation, were noted in 1.6% of infants.20
Future developments in prescribing information
Under a 2008 FDA recommendation, the “Nursing Mothers” section of prescribing information would be replaced with a section entitled “Lactation.” This new heading would include the sub-headings Risk Summary, Clinical Considerations, and Data.1 It is expected that this new format will be more practical and will help clinicians and patients make informed decisions. The prescribing changes will be in effect on June 30, 2015.21
Related Resources
• Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
• LactMed. http://toxnet.nlm.nih.gov/newtoxnet/lactmed.htm.
• MOTHERISK. www.motherisk.org/women/ breastfeeding.jsp.
Drug Brand Names
Aripiprazole • Abilify Olanzapine • Zyprexa
Asenapine • Saphris Quetiapine • Seroquel
Carbamazepine • Tegretol Risperidone • Risperdal
Lamotrigine • Lamictal Valproic acid • Depakene
Lithium • Eskalith, Lithobid Ziprasidone • Geodon
Lurasidone • Latuda
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sach HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3);e796-e809.
2. Moretti ME, Koren G, Verjee Z, et al. Monitoring lithium in breast milk: an individualized approach for breast-feeding mothers. Ther Drug Monit. 2003;25(3):364-366.
3. Viguera AC, Newport DJ, Ritchie J, et al. Lithium in breast milk and nursing infants: clinical implications. Am J Psychiatry. 2007;164(2):342-345.
4. Xyprexa [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
5. Brunner E, Falk DM, Jones M, et al. Olanzapine in pregnancy and breastfeeding: a review of data from global safety surveillance. BMC Pharmacol Toxicol. 2013;14:38.
6. Schlotterbeck P, Leube D, Kircher T, et al. Aripiprazole in human milk. Int J Neuropsychopharmacol. 2007;10(3):433.
7. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2014.
8. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals; 2013.
9. Lee A, Giesbrecht E, Dunn E, et al. Excretion of quetiapine in breast milk. Am J Psychiatry. 2004;161(9):1715-1716.
10. Rampono J, Kristensen JH, Ilett KF, et al. Quetiapine and breastfeeding. Ann Pharmacother. 2007;41(4):711-714.
11. Misri S, Corral M, Wardrop AA, et al. Quetiapine augmentation in lactation: a series of case reports. J Clin Psychopharmacol. 2006;26(5):508-511.
12. Hill RC, McIvor RJ, Wojnar-Horton RE, et al. Risperidone distribution and excretion into human milk: case report and estimated infant exposure during breast-feeding. J Clin Psychopharmacol. 2000;20(2):285-286.
13. Ilett KF, Hackett LP, Kristensen JH, et al. Transfer of risperidone and 9-hydroxyrisperidone into human milk. Ann Pharmacother. 2004;38(2):273-276.
14. Aichhorn W, Stuppaek C, Whitworth AB. Risperidone and breast-feeding. J Psychopharmacol. 2005;19(2):211-213.
15. Geodon [package insert]. New York, NY: Pfizer; 2014.
16. Davanzo R, Dal Bo S, Bua J, et al. Antiepileptic drugs and breastfeeding. Ital J Pediatr. 2013;39:50.
17. Newport DJ, Pennell PB, Calamaras MR, et al. Lamotrigine in breast milk and nursing infants: determination of exposure. Pediatrics. 2008;122(1):e223-e231.
18. Nordmo E, Aronsen L, Wasland K, et al. Severe apnea in an infant exposed to lamotrigine in breast milk. Ann Pharmacother. 2009;43(11):1893-1897.
19. Stahl MM, Neiderud J, Vinge E. Thrombocytopenic purpura and anemia in a breast-fed infant whose mother was treated with valproic acid. J Pediatr. 1997;130(6):1001-1003.
20. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451.
21. U.S. Food and Drug Administration. FDA issues final rule on changes to pregnancy and lactation labeling information for prescription drug and biological products. http://www. fda.gov/NewsEvents/Newsroom/PressAnnouncements/ ucm425317.htm. Published December 3. 2014. Accessed March 4, 2015.
Ms. K, age 35, soon will deliver her second child. She has a 12-year history of bipolar disorder, which was well controlled with lithium, 1,200 mg/d. During her first pregnancy 3 years ago, Ms. K stopped taking lithium because she was concerned about the risk of Ebstein’s anomaly. She experienced a bipolar relapse after her healthy baby was born, and developed postpartum psychosis that was treated by restarting lithium, 1,200 mg/d, and adding olanzapine, 10 mg/d.
Ms. K has continued these medications throughout her current pregnancy. She wants to breast-feed her infant and is concerned about the effects that psychotropics might have on her newborn.
Breast-feeding and medications
The benefits of breast-feeding for mother and infant are well-known. Despite this, some women with bipolar disorder are advised not to breast-feed or, worse, to discontinue their medications in order to breast-feed. Decisions about breast-feeding while taking medications should be based on evidence of benefits and risks to the infant, along with a discussion of the risks of untreated illness, which is high postpartum. The prescribing information for many of the medications used to treat bipolar disorder advise against breast-feeding, although there is little evidence of harm.
Drug dosages and levels in breast milk can be reported a few different ways:
• percentage of maternal dosage measured in the breast milk
• percentage weight-adjusted maternal dosage
• percentage of maternal plasma level, and milk-to-plasma ratio (M:P).
Daily infant dosage can be calculated by multiplying the average concentration of the drug in breast milk (mg/mL) by the average volume of milk the baby ingests in 24 hours (usually 150 mL).1 The relative infant dosage can be calculated as the percentage maternal dosage, which is the daily infant dosage (mg/kg/d) ÷ maternal dosage (mg/kg/d) × 100.1
According to the American Academy of Pediatrics, ≤10% of the maternal dosage is compatible with breast-feeding.1 Most psychotropics studied fall below this threshold. Keep in mind that all published research is for breast-feeding a full-term infant; exercise caution with premature or low birth weight infants. Infants born to mothers taking a psychotropic should be monitored for withdrawal symptoms, which might be associated with antidepressants and benzodiazepines, but otherwise are rare.
Lithium
Breast-feeding during lithium treatment has been considered contraindicated based on early reports that lithium was highly excreted in breast milk.2 A 2003 study2 of 11 women found that lithium was excreted in breast milk in amounts between zero and 30% of maternal dosage (mean, 12.2% ± 8.5%; median, 11.2%; 95% CI, 6.3% to 18.0%). Researchers measured serum concentrations in 2 infants and found that 1 received 17% to 20% of the maternal dosage, and the other showed 50%. None of the infants experienced adverse events. In a study of 10 mother-infant pairs, breast milk lithium concentration averaged 0.35 mEq/L (standard deviation [SD] = 0.10, range 0.19 to 0.48 mEq/L), with paired infant serum concentrations of 0.16 mEq/L (SD = 0.06, range 0.09 to 0.25 mEq/L).3 Some transient abnormalities were found in infant serum concentrations of thyroid-stimulating hormone (TSH), blood urea nitrogen, and creatinine; there were no adverse effects on development. The authors recommend monitoring for TSH abnormalities in infants.
Olanzapine
Olanzapine prescribing information cites a study reporting that 1.8% of the maternal dosage is transferred to breast milk.4 Yet the olanzapine prescribing information states, “It is recommended that women receiving olanzapine should not breast-feed.” Olanzapine use during breast-feeding has been studied more than many medications, in part because of a database maintained by the manufacturer. In a study using the manufacturer’s database (N = 102) adverse reactions were reported in 15.6% of the infants, with the most common being somnolence (3.9%), irritability (2%), tremor (2%), and insomnia (2%).5
Other second-generation antipsychotics
Aripiprazole. The only case report of aripiprazole excretion in human breast milk found a concentration of approximately 20% of the maternal plasma level and an M:P ratio of 0.18:0.2.6
Asenapine. According to asenapine prescribing information7 and a literature search, it is not known whether asenapine is excreted in breast milk of humans, although it is found in the milk of lactating rats.
Lurasidone. According to the lurasidone prescribing information8 and a literature search, it is not known whether lurasidone is excreted in human breast milk, although it is found in the milk of lactating rats.
Quetiapine. An initial study reported that 0.09% to 0.43% of the maternal dosage of quetiapine was excreted in breast milk.9 Further studies found excretion to be 0.09% of maternal dosage, with infant plasma levels reaching 6% of the maternal dosage.10 A case series found that one-third of babies exposed to quetiapine during breast-feeding showed some neurodevelopmental delay, although these mothers also were taking other psychotropics.11
Risperidone. A 2000 study12 of risperidone in lactation reported that 0.84% weight-adjusted maternal risperidone dosage and 3.46% of its metabolite 9-hydroxyrisperidone is transferred to the infant. A later study showed 2.3% to 4.7% of the maternal dosage is transferred, with no adverse events reported in infants.13 A case study reported no adverse events and normal neurodevelopment in a the child of a mother taking risperidone.14
Ziprasidone. According to the ziprasidone prescribing information15 and a literature search, is not known whether ziprasidone is excreted in human breast milk.
See the Table4,6-10,12,13,15 for a summary of the evidence levels of second-generation antipsychotics that are excreted in breast milk.
Other mood stabilizers
Carbamazepine has been measured in breast milk at 3.8% to 5.9% of the maternal dosage.16
Lamotrigine. In a study of 30 lactating women, the breast milk contained an average of 9.2% of the maternal dosage of lamotrigine.17 Mild thrombocytosis was detected in 7 of 8 infants; no other adverse effects were reported. A case study describes a woman who breast-fed while taking lamotrigine, 850 mg/d, and who experienced dizziness and visual disturbances. The infant had apnea episodes followed by a cyanotic crisis, which required resuscitation. The infant’s plasma lamotrigine level was 4.87 μg/mL. Symptoms disappeared when the mother stopped breast-feeding.18 Lamotrigine is considered to be moderately safe in breast-feeding patients with proper monitoring. The drug also has a known safety profile because of its use in children with epilepsy.
Valproic acid. Because of its high plasma protein binding, valproic acid does not pass readily into the breast milk. Newborns receive approximately 1.4% to 1.7% of the maternal dosage.16 Caution is advised, however, because of some reported adverse events. One case reported thrombocytopenic purpura and anemia in an infant.19 Valproic acid is considered to be compatible with breast-feeding with proper monitoring.
Benzodiazepines
Benzodiazepines can be helpful adjunctive medications to aid sleep, which is essential for the mother’s and infant’s health. In a prospectively recruited, retrospectively assessed cohort study that evaluated 124 women taking benzodiazepines while breast-feeding, adverse effects, specifically sedation, were noted in 1.6% of infants.20
Future developments in prescribing information
Under a 2008 FDA recommendation, the “Nursing Mothers” section of prescribing information would be replaced with a section entitled “Lactation.” This new heading would include the sub-headings Risk Summary, Clinical Considerations, and Data.1 It is expected that this new format will be more practical and will help clinicians and patients make informed decisions. The prescribing changes will be in effect on June 30, 2015.21
Related Resources
• Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
• LactMed. http://toxnet.nlm.nih.gov/newtoxnet/lactmed.htm.
• MOTHERISK. www.motherisk.org/women/ breastfeeding.jsp.
Drug Brand Names
Aripiprazole • Abilify Olanzapine • Zyprexa
Asenapine • Saphris Quetiapine • Seroquel
Carbamazepine • Tegretol Risperidone • Risperdal
Lamotrigine • Lamictal Valproic acid • Depakene
Lithium • Eskalith, Lithobid Ziprasidone • Geodon
Lurasidone • Latuda
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. K, age 35, soon will deliver her second child. She has a 12-year history of bipolar disorder, which was well controlled with lithium, 1,200 mg/d. During her first pregnancy 3 years ago, Ms. K stopped taking lithium because she was concerned about the risk of Ebstein’s anomaly. She experienced a bipolar relapse after her healthy baby was born, and developed postpartum psychosis that was treated by restarting lithium, 1,200 mg/d, and adding olanzapine, 10 mg/d.
Ms. K has continued these medications throughout her current pregnancy. She wants to breast-feed her infant and is concerned about the effects that psychotropics might have on her newborn.
Breast-feeding and medications
The benefits of breast-feeding for mother and infant are well-known. Despite this, some women with bipolar disorder are advised not to breast-feed or, worse, to discontinue their medications in order to breast-feed. Decisions about breast-feeding while taking medications should be based on evidence of benefits and risks to the infant, along with a discussion of the risks of untreated illness, which is high postpartum. The prescribing information for many of the medications used to treat bipolar disorder advise against breast-feeding, although there is little evidence of harm.
Drug dosages and levels in breast milk can be reported a few different ways:
• percentage of maternal dosage measured in the breast milk
• percentage weight-adjusted maternal dosage
• percentage of maternal plasma level, and milk-to-plasma ratio (M:P).
Daily infant dosage can be calculated by multiplying the average concentration of the drug in breast milk (mg/mL) by the average volume of milk the baby ingests in 24 hours (usually 150 mL).1 The relative infant dosage can be calculated as the percentage maternal dosage, which is the daily infant dosage (mg/kg/d) ÷ maternal dosage (mg/kg/d) × 100.1
According to the American Academy of Pediatrics, ≤10% of the maternal dosage is compatible with breast-feeding.1 Most psychotropics studied fall below this threshold. Keep in mind that all published research is for breast-feeding a full-term infant; exercise caution with premature or low birth weight infants. Infants born to mothers taking a psychotropic should be monitored for withdrawal symptoms, which might be associated with antidepressants and benzodiazepines, but otherwise are rare.
Lithium
Breast-feeding during lithium treatment has been considered contraindicated based on early reports that lithium was highly excreted in breast milk.2 A 2003 study2 of 11 women found that lithium was excreted in breast milk in amounts between zero and 30% of maternal dosage (mean, 12.2% ± 8.5%; median, 11.2%; 95% CI, 6.3% to 18.0%). Researchers measured serum concentrations in 2 infants and found that 1 received 17% to 20% of the maternal dosage, and the other showed 50%. None of the infants experienced adverse events. In a study of 10 mother-infant pairs, breast milk lithium concentration averaged 0.35 mEq/L (standard deviation [SD] = 0.10, range 0.19 to 0.48 mEq/L), with paired infant serum concentrations of 0.16 mEq/L (SD = 0.06, range 0.09 to 0.25 mEq/L).3 Some transient abnormalities were found in infant serum concentrations of thyroid-stimulating hormone (TSH), blood urea nitrogen, and creatinine; there were no adverse effects on development. The authors recommend monitoring for TSH abnormalities in infants.
Olanzapine
Olanzapine prescribing information cites a study reporting that 1.8% of the maternal dosage is transferred to breast milk.4 Yet the olanzapine prescribing information states, “It is recommended that women receiving olanzapine should not breast-feed.” Olanzapine use during breast-feeding has been studied more than many medications, in part because of a database maintained by the manufacturer. In a study using the manufacturer’s database (N = 102) adverse reactions were reported in 15.6% of the infants, with the most common being somnolence (3.9%), irritability (2%), tremor (2%), and insomnia (2%).5
Other second-generation antipsychotics
Aripiprazole. The only case report of aripiprazole excretion in human breast milk found a concentration of approximately 20% of the maternal plasma level and an M:P ratio of 0.18:0.2.6
Asenapine. According to asenapine prescribing information7 and a literature search, it is not known whether asenapine is excreted in breast milk of humans, although it is found in the milk of lactating rats.
Lurasidone. According to the lurasidone prescribing information8 and a literature search, it is not known whether lurasidone is excreted in human breast milk, although it is found in the milk of lactating rats.
Quetiapine. An initial study reported that 0.09% to 0.43% of the maternal dosage of quetiapine was excreted in breast milk.9 Further studies found excretion to be 0.09% of maternal dosage, with infant plasma levels reaching 6% of the maternal dosage.10 A case series found that one-third of babies exposed to quetiapine during breast-feeding showed some neurodevelopmental delay, although these mothers also were taking other psychotropics.11
Risperidone. A 2000 study12 of risperidone in lactation reported that 0.84% weight-adjusted maternal risperidone dosage and 3.46% of its metabolite 9-hydroxyrisperidone is transferred to the infant. A later study showed 2.3% to 4.7% of the maternal dosage is transferred, with no adverse events reported in infants.13 A case study reported no adverse events and normal neurodevelopment in a the child of a mother taking risperidone.14
Ziprasidone. According to the ziprasidone prescribing information15 and a literature search, is not known whether ziprasidone is excreted in human breast milk.
See the Table4,6-10,12,13,15 for a summary of the evidence levels of second-generation antipsychotics that are excreted in breast milk.
Other mood stabilizers
Carbamazepine has been measured in breast milk at 3.8% to 5.9% of the maternal dosage.16
Lamotrigine. In a study of 30 lactating women, the breast milk contained an average of 9.2% of the maternal dosage of lamotrigine.17 Mild thrombocytosis was detected in 7 of 8 infants; no other adverse effects were reported. A case study describes a woman who breast-fed while taking lamotrigine, 850 mg/d, and who experienced dizziness and visual disturbances. The infant had apnea episodes followed by a cyanotic crisis, which required resuscitation. The infant’s plasma lamotrigine level was 4.87 μg/mL. Symptoms disappeared when the mother stopped breast-feeding.18 Lamotrigine is considered to be moderately safe in breast-feeding patients with proper monitoring. The drug also has a known safety profile because of its use in children with epilepsy.
Valproic acid. Because of its high plasma protein binding, valproic acid does not pass readily into the breast milk. Newborns receive approximately 1.4% to 1.7% of the maternal dosage.16 Caution is advised, however, because of some reported adverse events. One case reported thrombocytopenic purpura and anemia in an infant.19 Valproic acid is considered to be compatible with breast-feeding with proper monitoring.
Benzodiazepines
Benzodiazepines can be helpful adjunctive medications to aid sleep, which is essential for the mother’s and infant’s health. In a prospectively recruited, retrospectively assessed cohort study that evaluated 124 women taking benzodiazepines while breast-feeding, adverse effects, specifically sedation, were noted in 1.6% of infants.20
Future developments in prescribing information
Under a 2008 FDA recommendation, the “Nursing Mothers” section of prescribing information would be replaced with a section entitled “Lactation.” This new heading would include the sub-headings Risk Summary, Clinical Considerations, and Data.1 It is expected that this new format will be more practical and will help clinicians and patients make informed decisions. The prescribing changes will be in effect on June 30, 2015.21
Related Resources
• Massachusetts General Hospital Center for Women’s Mental Health. www.womensmentalhealth.org.
• LactMed. http://toxnet.nlm.nih.gov/newtoxnet/lactmed.htm.
• MOTHERISK. www.motherisk.org/women/ breastfeeding.jsp.
Drug Brand Names
Aripiprazole • Abilify Olanzapine • Zyprexa
Asenapine • Saphris Quetiapine • Seroquel
Carbamazepine • Tegretol Risperidone • Risperdal
Lamotrigine • Lamictal Valproic acid • Depakene
Lithium • Eskalith, Lithobid Ziprasidone • Geodon
Lurasidone • Latuda
Disclosure
The author reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Sach HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3);e796-e809.
2. Moretti ME, Koren G, Verjee Z, et al. Monitoring lithium in breast milk: an individualized approach for breast-feeding mothers. Ther Drug Monit. 2003;25(3):364-366.
3. Viguera AC, Newport DJ, Ritchie J, et al. Lithium in breast milk and nursing infants: clinical implications. Am J Psychiatry. 2007;164(2):342-345.
4. Xyprexa [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
5. Brunner E, Falk DM, Jones M, et al. Olanzapine in pregnancy and breastfeeding: a review of data from global safety surveillance. BMC Pharmacol Toxicol. 2013;14:38.
6. Schlotterbeck P, Leube D, Kircher T, et al. Aripiprazole in human milk. Int J Neuropsychopharmacol. 2007;10(3):433.
7. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2014.
8. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals; 2013.
9. Lee A, Giesbrecht E, Dunn E, et al. Excretion of quetiapine in breast milk. Am J Psychiatry. 2004;161(9):1715-1716.
10. Rampono J, Kristensen JH, Ilett KF, et al. Quetiapine and breastfeeding. Ann Pharmacother. 2007;41(4):711-714.
11. Misri S, Corral M, Wardrop AA, et al. Quetiapine augmentation in lactation: a series of case reports. J Clin Psychopharmacol. 2006;26(5):508-511.
12. Hill RC, McIvor RJ, Wojnar-Horton RE, et al. Risperidone distribution and excretion into human milk: case report and estimated infant exposure during breast-feeding. J Clin Psychopharmacol. 2000;20(2):285-286.
13. Ilett KF, Hackett LP, Kristensen JH, et al. Transfer of risperidone and 9-hydroxyrisperidone into human milk. Ann Pharmacother. 2004;38(2):273-276.
14. Aichhorn W, Stuppaek C, Whitworth AB. Risperidone and breast-feeding. J Psychopharmacol. 2005;19(2):211-213.
15. Geodon [package insert]. New York, NY: Pfizer; 2014.
16. Davanzo R, Dal Bo S, Bua J, et al. Antiepileptic drugs and breastfeeding. Ital J Pediatr. 2013;39:50.
17. Newport DJ, Pennell PB, Calamaras MR, et al. Lamotrigine in breast milk and nursing infants: determination of exposure. Pediatrics. 2008;122(1):e223-e231.
18. Nordmo E, Aronsen L, Wasland K, et al. Severe apnea in an infant exposed to lamotrigine in breast milk. Ann Pharmacother. 2009;43(11):1893-1897.
19. Stahl MM, Neiderud J, Vinge E. Thrombocytopenic purpura and anemia in a breast-fed infant whose mother was treated with valproic acid. J Pediatr. 1997;130(6):1001-1003.
20. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451.
21. U.S. Food and Drug Administration. FDA issues final rule on changes to pregnancy and lactation labeling information for prescription drug and biological products. http://www. fda.gov/NewsEvents/Newsroom/PressAnnouncements/ ucm425317.htm. Published December 3. 2014. Accessed March 4, 2015.
1. Sach HC; Committee on Drugs. The transfer of drugs and therapeutics into human breast milk: an update on selected topics. Pediatrics. 2013;132(3);e796-e809.
2. Moretti ME, Koren G, Verjee Z, et al. Monitoring lithium in breast milk: an individualized approach for breast-feeding mothers. Ther Drug Monit. 2003;25(3):364-366.
3. Viguera AC, Newport DJ, Ritchie J, et al. Lithium in breast milk and nursing infants: clinical implications. Am J Psychiatry. 2007;164(2):342-345.
4. Xyprexa [package insert]. Indianapolis, IN: Eli Lilly and Company; 2014.
5. Brunner E, Falk DM, Jones M, et al. Olanzapine in pregnancy and breastfeeding: a review of data from global safety surveillance. BMC Pharmacol Toxicol. 2013;14:38.
6. Schlotterbeck P, Leube D, Kircher T, et al. Aripiprazole in human milk. Int J Neuropsychopharmacol. 2007;10(3):433.
7. Saphris [package insert]. St. Louis, MO: Forest Pharmaceuticals; 2014.
8. Latuda [package insert]. Marlborough, MA: Sunovion Pharmaceuticals; 2013.
9. Lee A, Giesbrecht E, Dunn E, et al. Excretion of quetiapine in breast milk. Am J Psychiatry. 2004;161(9):1715-1716.
10. Rampono J, Kristensen JH, Ilett KF, et al. Quetiapine and breastfeeding. Ann Pharmacother. 2007;41(4):711-714.
11. Misri S, Corral M, Wardrop AA, et al. Quetiapine augmentation in lactation: a series of case reports. J Clin Psychopharmacol. 2006;26(5):508-511.
12. Hill RC, McIvor RJ, Wojnar-Horton RE, et al. Risperidone distribution and excretion into human milk: case report and estimated infant exposure during breast-feeding. J Clin Psychopharmacol. 2000;20(2):285-286.
13. Ilett KF, Hackett LP, Kristensen JH, et al. Transfer of risperidone and 9-hydroxyrisperidone into human milk. Ann Pharmacother. 2004;38(2):273-276.
14. Aichhorn W, Stuppaek C, Whitworth AB. Risperidone and breast-feeding. J Psychopharmacol. 2005;19(2):211-213.
15. Geodon [package insert]. New York, NY: Pfizer; 2014.
16. Davanzo R, Dal Bo S, Bua J, et al. Antiepileptic drugs and breastfeeding. Ital J Pediatr. 2013;39:50.
17. Newport DJ, Pennell PB, Calamaras MR, et al. Lamotrigine in breast milk and nursing infants: determination of exposure. Pediatrics. 2008;122(1):e223-e231.
18. Nordmo E, Aronsen L, Wasland K, et al. Severe apnea in an infant exposed to lamotrigine in breast milk. Ann Pharmacother. 2009;43(11):1893-1897.
19. Stahl MM, Neiderud J, Vinge E. Thrombocytopenic purpura and anemia in a breast-fed infant whose mother was treated with valproic acid. J Pediatr. 1997;130(6):1001-1003.
20. Kelly LE, Poon S, Madadi P, et al. Neonatal benzodiazepines exposure during breastfeeding. J Pediatr. 2012;161(3):448-451.
21. U.S. Food and Drug Administration. FDA issues final rule on changes to pregnancy and lactation labeling information for prescription drug and biological products. http://www. fda.gov/NewsEvents/Newsroom/PressAnnouncements/ ucm425317.htm. Published December 3. 2014. Accessed March 4, 2015.
Do glutamatergic drugs have a role in treating depression?
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1

Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period. 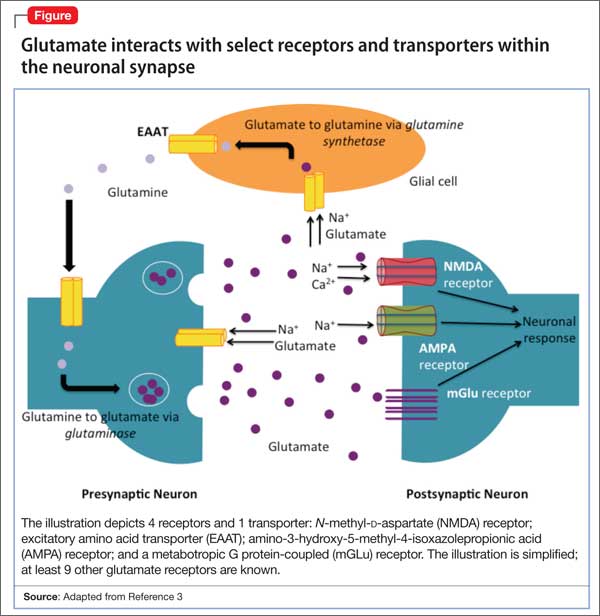
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1
Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period.
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7
Acamprosate, often used for treating alcohol abuse, is another a drug with glutamatergic activity that has been studied for possible use as an antidepressant.5
A review by Lapidus et al5 has a more extensive listing of current medications and investigational compounds that modulate glutamate transmission, and are of interest for their possible antidepressant activity. Given the relatively new “glutamatergic hypothesis” of depression, it is exciting that so many current and novel glutamatergic drug therapies are being evaluated.
Future of ketamine treatment
Glutamate has been shown to play an important part in the pathophysiology of depression. The rapid antidepressant efficacy of ketamine provides evidence that future medications with glutamate-modulating activity could be useful for patients who struggle to achieve symptom relief using available antidepressants. Several limitations exist regarding ketamine use, and more work in this important therapeutic area needs to be done. This last point is important to remember when speaking with patients such as Mrs. S. Although it is understandable for her to be excited about novel treatment options such as ketamine, stress to her that treating depression with ketamine at this time is strictly investigational, and that the drug needs to be thoroughly evaluated for safety and efficacy before it can be prescribed for this indication.
CASE CONTINUED
Mrs. S realizes that ketamine may not be the best next step for her, and she agrees to explore other approaches to treat her residual depressive symptoms.
Related Resources
• Machado-Vieira R, Ibrahim L, Henter ID, et al. Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol Biochem Behav. 2012;100(4):678-687.
• Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
Drug Brand Names
Acamprosate • Campral Duloxetine • Cymbalta
Aripiprazole • Abilify Ketamine • Ketalar
Bupropion • Wellbutrin, Zyban Riluzole • Rilutek
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in this article or with manufacturers of competing products.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
1. Niciu MJ, Ionescu DF, Richards EM, et al. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm. 2014;121(8):907-924.
2. Gaynes BN, Dusetzina SB, Ellis AR, et al. Treating depression after initial treatment failure: directly comparing switch and augmenting strategies in STAR*D. J Clin Psychopharmacol. 2012;32(1):114-119.
3. Curry SC, Mills KC, Ruha A, et al. Neurotransmitters and neuromodulators. In: Nelson LS, Lewin NA, Howland MA, et al, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York, NY: McGraw-Hill; 2011:189-220.
4. Zarate C Jr, Machado-Vieira R, Henter I, et al. Glutamatergic modulators: the future of treating mood disorders? Harv Rev Psychiatry. 2010;18(5):293-303.
5. Lapidus KA, Soleimani L, Murrough JW. Novel glutamatergic drugs for the treatment of mood disorders. Neuropsychiatr Dis Treat. 2013;9:1101-1112.
6. Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134-1142.
7. Ibrahim L, Diazgranados N, Franco-Chaves J, et al. Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology. 2012;37(6):1526-1533.
How to modify psychotropic therapy for patients who have liver dysfunction
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3

Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
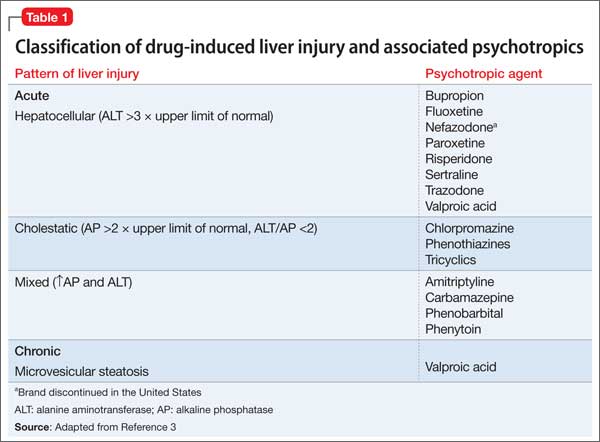
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5

When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Police bring Ms. R, age 35, to the psychiatric ER after they find her asleep in a park. She is awake but drowsy, and states that she has a history of bipolar disorder. She claims that she had been stable on valproic acid (VPA), 1,500 mg/d, bupropion XL, 300 mg/d, quetiapine, 400 mg/d, and trazodone, 100 mg/d, until 2 weeks ago, when her best friend died and she stopped taking her medications all together. The previous evening, feeling “alone, hopeless, and sad,” she attempted suicide by ingesting a handful of VPA and clonazepam, obtained from a friend, and 2 liters of vodka. She complains of nausea, vomiting, and abdominal pain. Elevated laboratory chemistries included aspartate aminotransferase (AST), 220 U/L; alanine aminotransferase (ALT), 182 U/L; alkaline phosphatase (AP), 75 U/L; γ-glutamyltransferase (GGT), 104 U/L; total bilirubin, 1.4 mg/dL; and an elevated VPA serum concentration of 152 μg/mL.
Drug-induced hepatotoxicity accounts for approximately 50% of acute liver failure cases, and almost 10% of liver transplants in some facilities.1 The incidence of drug-induced hepatotoxicity is between 0.001% and 0.1% in patients on standard medication doses.2 Drug-induced hepatotoxicity is characterized by:
• abnormalities in laboratory parameters (hepatocellular, cholestatic, or mixed)
• mechanisms of toxicity (direct, immune-mediated, idiosyncratic, mitochondrial toxicity)
• liver biopsy histology (steatosis, sinusoidal obstruction syndrome).3
Liver function test results of hepatocellular injury are characterized by ALT elevation and minimal AP elevation, whereas cholestatic injury manifests as high AP. Table 13 categorizes psychotropics based on type of liver injury and how each injury manifest in liver function tests. Delayed idiosyncratic reactions occur after taking the drug, whereas direct toxicities are dose-dependent and more predictable. By definition, a clinically significant hepatotoxicity is associated with an ALT >3 times the upper limit of normal.3
VPA-induced liver injury occurs in approximately 1 in 37,000 persons taking the drug.4 Patients at an increased risk of VPA-induced liver injury include:
• children
• patients with mitochondrial enzyme deficiencies
• Reye’s syndrome
• Friedreich’s ataxia
• polypharmacy patients
• patients with a sibling who has experienced VPA toxicity.4
Benign enzyme elevations occur in approximately 20% of patients taking VPA.5 In Ms. R’s case, concomitant VPA, clonazepam, and alcohol may have led to elevations in ALT, AST, and GGT. Her nausea, vomiting, and abdominal pain are consistent with hepatic dysfunction.
Carnitine is effective in increasing survival of patients with VPA-induced hepatotoxicity.4 Because Ms. R is symptomatic, discontinuing VPA and administering IV L-carnitine is warranted.5 L-carnitine can be initiated at 100 mg/kg as an IV bolus, followed by 50 mg/kg as an IV infusion every 8 hours, with a maximum dosage of 3,000 mg.6 Patients may require several days of therapy based on symptoms. L-carnitine should be continued until a patient shows clinical improvement, such as decreases in ALT and AST.
Ms. R experienced a VPA-induced hepatotoxic reaction. However, continuous monitoring is appropriate for all patients who are prescribed any potentially hepatotoxic psychotropic, especially after hepatic injuries resolve. This includes mood stabilizers, antipsychotics, benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), and serotonin-norepinephrine reuptake inhibitors, especially when given concomitantly with other hepatotoxic agents.
Table 2 lists dosing recommendations for commonly used psychotropics in patients with hepatic impairment. Among mood stabilizers, carbamazepine and VPA are associated with the highest incidence of hepatotoxicity.2 A follow-up study of more than 1,000,000 VPA prescriptions found 29 cases of fatal hepatotoxicity in a 7-year period.7 Although there are case reports of hepatotoxicity with oxcarbazepine, it may have a better liver safety profile than carbamazepine.2 Hepatotoxicity with lamotrigine is rare, although fatal cases have been reported.5
When initiating an antipsychotic, a temporary, benign increase in liver enzymes can be expected, but typically discontinuation is unnecessary.2 Phenothiazines in particular can cause increases in liver enzymes in 20% of patients.2 Hepatotoxicity with benzodiazepines is infrequent, with a few cases of cholestatic injury reported with diazepam, chlordiazepoxide, and flurazepam.2
SSRIs are relatively safe; incidents of hepatic injury are rare. Among SSRIs, paroxetine is most frequently associated with hepatotoxicity. Abnormal liver function tests have been observed with fluoxetine (0.5% of long-term recipients) and other SSRIs.1,2,4
Among antidepressants with dual serotonergic action, nefazodone carries a black-box warning for hepatotoxicity and is used rarely, whereas trazodone is not regarded as hepatotoxic.2 Antidepressants with dual norepinephrine and serotonin reuptake inhibitor properties carry a higher risk of liver injury, especially duloxetine. Hepatocellular, cholestatic, and mixed types of hepatotoxicity are associated with duloxetine-induced hepatotoxicity.2
Monitoring recommendations
Before prescribing potentially hepatotoxic medications, order baseline liver function tests. During therapy, periodic liver function monitoring is recommended. Elevated transaminase concentrations (>3 × the upper limit of normal), bilirubin (>2 × the upper limit of normal), and prolonged prothrombin times are indicators of hepatic injury.2 Caution should be taken to prevent polypharmacy with multiple hepatotoxic medications and over-the-counter use of hepatotoxic drugs and supplements.
When choosing a psychotropic, take into account patient-specific factors, such as underlying liver disease and alcohol consumption. Patients on potentially hepatotoxic medications should be counseled to recognize and report symptoms of liver dysfunction, including nausea, vomiting, jaundice, and lower-extremity edema.2 If liver injury occurs, modify therapy with the potential offending agent and check liver function periodically.
Related Resourcesa
• Bleibel W, Kim S, D’Silva K, et al. Drug-induced liver injury: review article. Dig Dis Sci. 2007;52(10):2463-2471.
• U.S. National Library of Medicine. LiverTox. National Institute of Health. www.livertox.nih.gov.
Drug Brand Names
Amitriptyline • Elavil Lurasidone • Latuda
Molindone • Moban Molindone • Moban
Aripiprazole • Abilify Nefazodone • Serzone
Asenapine • Saphris Nortriptyline • Pamelor
Bupropion XL • Wellbutrin XL Olanzapine • Zyprexa
Citalopram • Celexa Oxcarbazepine • Trileptal
Carbamazepine • Tegretol Paroxetine • Paxil
Chlordiazepoxide • Librium Perphenazine • Trilafon
Chlorpromazine • Thorazine Phenobarbital • Luminal
Clonazepam • Klonopin Phenytoin • Dilantin
Clozapine • Clozaril Quetiapine • Seroquel
Desvenlafaxine • Pristiq Risperidone • Risperdal
Diazepam • Valium Sertraline • Zoloft
Duloxetine • Cymbalta Thiothixene • Navane
Escitalopram • Lexapro Trazodone • Desyrel
Fluoxetine • Prozac Trifluoperazine • Stelazine
Fluphenazine • Prolixin Topiramate • Topamax
Flurazepam • Dalmane Valproic acid • Depakote
Haloperidol • Haldol Venlafaxine • Effexor
Iloperidone • Fanapt Ziprasidone • Geodon
Lamotrigine • Lamictal
Levocarnitine • L-carnitine
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.
1. Pugh AJ, Barve AJ, Falkner K, et al. Drug-induced hepatotoxicity or drug-induced liver injury. Clin Liver Dis. 2009;13(2):277-294.
2. Sedky K, Nazir R, Joshi A, et al. Which psychotropic medications induce hepatotoxicity? Gen Hosp Psychiatry. 2012;34(1):53-61.
3. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25(10):1135-1151.
4. Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22(2):169-183.
5. Murray KF, Hadzic N, Wirth S, et al. Drug-related hepatotoxicity and acute liver failure. J Pediatr Gastroenterol Nutr. 2008;47(4):395-405.
6. Perrott J, Murphy NG, Zed PJ. L-carnitine for acute valproic acid overdose: a systematic review of published cases. Ann Pharmacother. 2010;44(7-8):1287-1293.
7. Bryant AE 3rd, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology. 1996;46(2):465-469.