User login
Treating comorbid posttraumatic stress disorder and cardiovascular disease
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1

Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.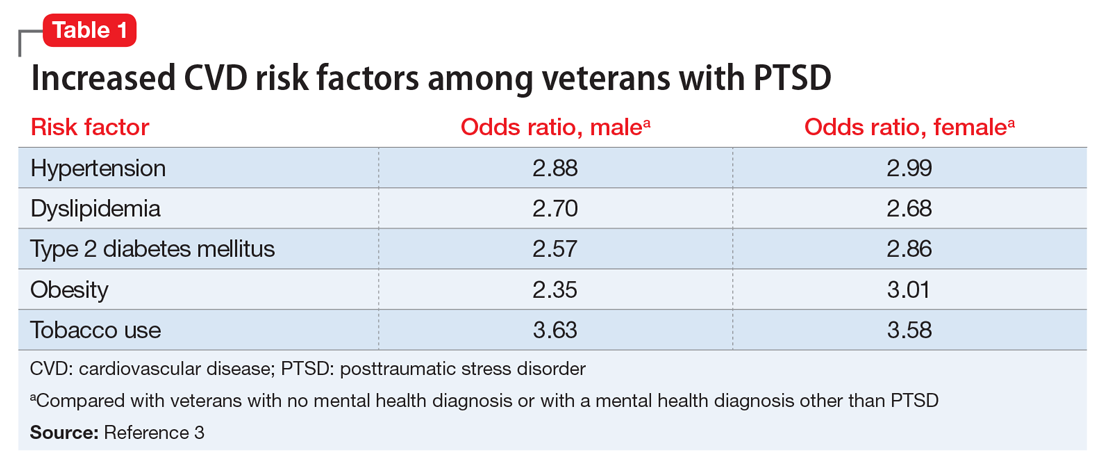
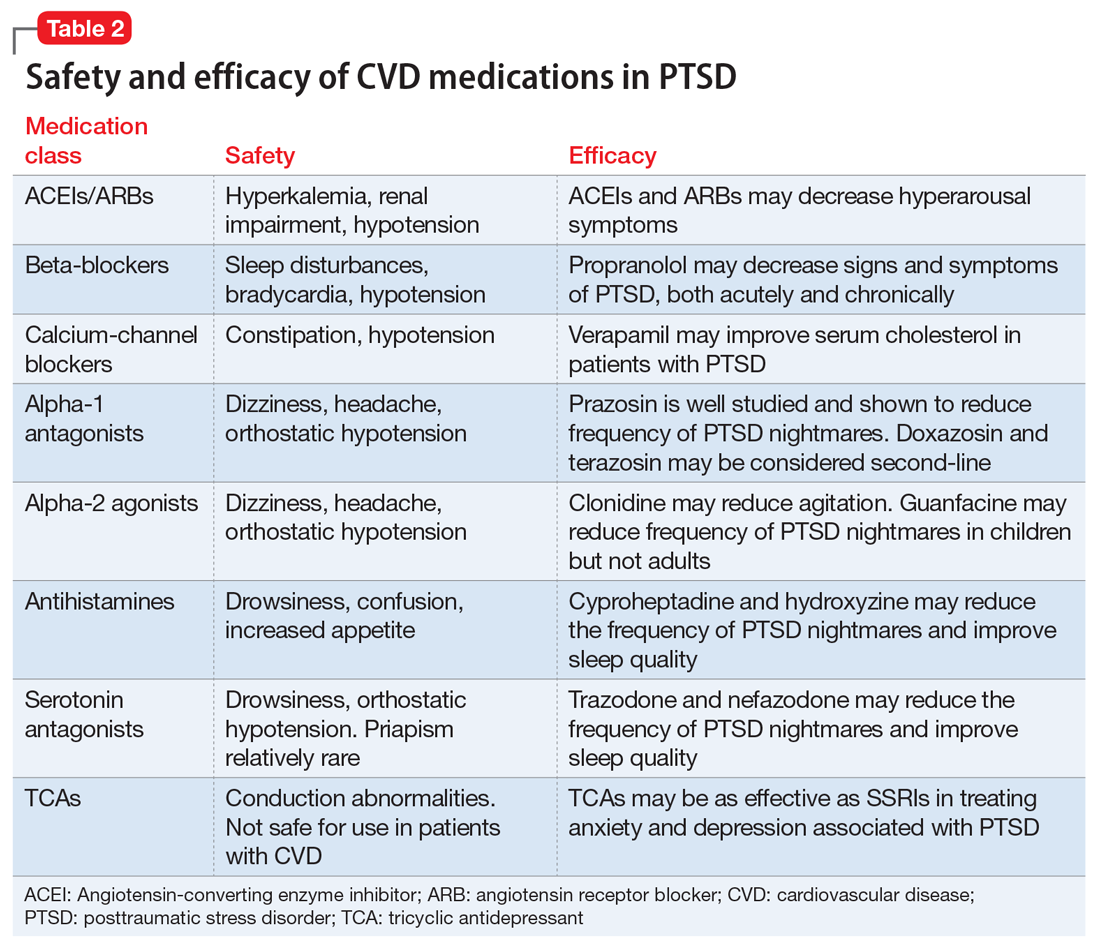
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15

Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1
Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15
Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
Mr. S, 64, has a history of posttraumatic stress disorder (PTSD), which has been well controlled for the past 15 years with cognitive-processing therapy and fluoxetine, 40 mg/d. However, over the past 6 weeks, Mr. S has experienced increased hypervigilance, nightmares, and flashbacks. He states that his primary care provider recommended an adjustment in pharmacotherapy to address this exacerbation of symptoms. Previous medication trials include sertraline, 200 mg/d, discontinued due to lack of perceived efficacy, and venlafaxine, 150 mg/d, discontinued due to increased blood pressure.
Mr. S’s medical history includes hypertension, dyslipidemia, and myocardial infarction (MI) 5 years ago. His family history includes sudden cardiac death (mother and father) and major depressive disorder (sister). His blood pressure is currently uncontrolled on lisinopril, 5 mg/d, and metoprolol succinate, 50 mg/d. Today, serial blood pressure readings measured approximately 180/90 mm Hg, with a pulse 50-60 beats per minute.
What is the next step in treating Mr. S’s hypertension and PTSD symptoms? Is there any evidence to support concomitant therapy?
PTSD is characterized by emotional and behavioral symptoms following exposure to a traumatic event. Its 12-month prevalence in the United States is estimated at 3.5%. Diagnostic criteria necessitate the presence of intrusive symptoms, persistent effortful avoidance of distressing trauma-related stimuli, negative cognitions or mood, and alterations in arousal and reactivity. PTSD negatively impacts social and occupational functioning.1
Studies have revealed a correlation between the presence of psychosocial factors, such as depression and anxiety, and the occurrence of cardiovascular events. The mechanism appears to consist of a behavioral component (eg, poor diet, tobacco use) and a direct pathophysiologic component (eg, excessive sympathetic nervous system activation) (Table 13).4 Management of concomitant PTSD and CVD presents a challenge to clinicians.
This article summarizes the evidence for the use of CVD medications in treating PTSD (Table 2) and how to apply these principles in patient care (Table 35-14).
ACEIs, ARBs, beta blockers, and calcium channel blockers
Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) inhibit the renin-angiotensin system: ACEIs prevent formation of angiotensin II, a potent vasoconstrictor, and ARBs prevent interaction between angiotensin II and its receptor. In one study, patients were recruited from a large public hospital serving primarily a highly traumatized, low-income population. Patients taking an ACEI or ARB who had experienced at least 1 traumatic event exhibited significantly decreased hyperarousal symptoms and decreased intrusive thoughts on the PTSD Symptom Scale and Clinician Administered PTSD Scale.5 Other studies have reported that blockade of angiotensin II AT1 receptors may result in decreased stress, anxiety, and inflammation.15
Evidence supports the use of the centrally acting, beta-adrenergic antagonist propranolol for decreasing the physiologic reactivity to acute trauma. Emotional arousal enhances the consolidation of emotional experiences into long-term memories via the adrenal stress hormones epinephrine and corticosterone. The amygdala mediates these stress hormones and releases norepinephrine, which subsequently activates noradrenergic receptors essential for memory enhancement. Several studies have reported that patients who received propranolol within several hours of a traumatic event experienced fewer physiologic signs of PTSD at follow-up 1 month later.16 Moreover, researchers have hypothesized that chronic treatment with propranolol may be effective in decreasing hyperarousal symptoms in patients with chronic PTSD by reducing tonically elevated norepinephrine signaling.6
Chronic elevation of noradrenergic activity may induce lipoprotein lipase and suppress low-density lipoprotein (LDL) receptor activity, which in turn elevates serum cholesterol levels. The results of one study suggested that verapamil, a non-dihydropyridine calcium channel blocker, significantly improves serum cholesterol levels in patients with PTSD by increasing LDL receptor activity and decreasing norepinephrine release.7
Alpha-1 and alpha-2 antagonists
Alpha-1 antagonists relax vascular smooth muscle by blocking norepinephrine stimulation at postsynaptic α-1-adrenergic receptors. They frequently are prescribed for hypertension and benign prostatic hypertrophy. One α-1 antagonist in particular, prazosin, appears especially useful in treating sleep disturbances, which occur in up to 90% of patients with PTSD.17 Because of its relatively greater lipophilicity, prazosin crosses the blood–brain barrier and acts centrally to reduce the fight-or-flight and hyperarousal reactions related to nightmares caused by PTSD.18 Common adverse effects include dizziness and orthostatic hypotension. These usually can be mitigated with titration to effective dose. In a study of active-duty soldiers who returned from Iraq and Afghanistan, Raskind et al8 found that prazosin doses up to 25 mg/d in men and 12 mg/d in women were tolerated with weekly adjustments and blood pressure monitoring.
Other α-1 antagonists have shown efficacy in a limited number of trials and may be considered second-line treatment of PTSD hyperarousal symptoms. Doxazosin has a longer half-life compared with prazosin (22 hours vs 3 hours) and may be useful in treating daytime hyperarousal with once-daily dosing. However, its hydrophilicity prevents it from crossing the blood–brain barrier to the same degree as prazosin.19 Terazosin also has a longer half-life (12 hours) and reaches peak plasma concentration in 1 hour. It undergoes minimal first-pass metabolism, leaving almost the entire circulating dose in the parent form, but clinical data are limited to only a small case report.10
Alpha-2 agonists inhibit sympathetic outflow in the CNS, which ultimately relaxes vascular smooth muscle like α-1 antagonists. Clonidine exhibits sedative properties, which derive from its nonspecific binding to α-2a-, -2b-, and -2c-adrenergic receptors. Several case studies have described a reduction in agitation in PTSD patients with the use of clonidine, likely through the induction of sleep and relaxation. Guanfacine, on the other hand, selectively binds to the α-2a-adrenergic receptor and therefore lacks the sedative properties of clonidine. Several placebo-controlled trials showed no alleviation of PTSD symptoms in adults with the use of guanfacine.11 However, case reports and open-label trials have suggested that guanfacine may reduce trauma-induced nightmares in pediatric patients. Further investigation is needed to clarify the potential use of guanfacine in pediatric PTSD.19
Antihistamines and antidepressants
Several second-line pharmacologic agents may be useful in patients with PTSD who are already taking cardiovascular medication. A limited number of studies have demonstrated reduced frequency of PTSD nightmares with the histamine-1 antagonists cyproheptadine and hydroxyzine, both of which exhibit minor anti-serotonergic properties.12,13 Likewise, the serotonin antagonists nefazodone and trazodone have been shown to reduce the frequency of PTSD nightmares, as well as improve overall sleep quality.14 Nefazodone should be considered an option only after treatment failure of multiple other medications, because it is associated with a small, but significant, risk of life-threatening hepatotoxicity.20
Tricyclic antidepressants (TCAs) may reduce anxiety and depression associated with PTSD to the same degree as SSRIs.21 However, their effect on PTSD-associated sleep disturbances is much less pronounced than other available medications.14 TCAs should be avoided in patients with CVD because they may exacerbate cardiac conduction abnormalities. This is especially true for those recovering from acute MI.22
CASE CONTINUED
Mr. S is started on prazosin, 1 mg at bedtime, titrated weekly to 6 mg at bedtime with regular blood pressure monitoring because of the risk of orthostatic hypotension. Although the frequency of his nightmares decreases to 1 or 2 per month, he still experiences flashbacks at the same frequency and intensity as before. Prazosin, 1 mg every morning, is added, titrated weekly to 4 mg every morning. This combination of morning and bedtime dosing leads to resolution of both nightmares and flashbacks along with a significant reduction in hyperarousal. Lisinopril is increased from 5 to 10 mg/d to address Mr. S’s uncontrolled hypertension; this change also could have contributed to the reduction in hyperarousal. CPT and fluoxetine are continued.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
1. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
2. Laslett LJ, Alagona P Jr, Clark BA 3rd, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues: a report from the American College of Cardiology. J Am Coll Cardiol. 2012;60(suppl 25):S1-S49.
3. Cohen BE, Marmar C, Ren L, et al. Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA. 2009;302(5):489-492.
4. Rozanski A, Blumenthal JA, Kaplan J. Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation. 1999;99(16):2192-2217.
5. Khoury NM, Marvar PJ, Gillespie CF, et al. The renin-angiotensin pathway in posttraumatic stress disorder: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers are associated with fewer traumatic stress symptoms. J Clin Psychiatry. 2012;73(6):849-855.
6. Giustino TF, Fitzgerald PJ, Maren S. Revisiting propranolol and PTSD: memory erasure or extinction enhancement? Neurobiol Learn Mem. 2016;130:26-33.
7. Ansari MA, Ahmed S. Calcium channel blocker verapamil: a new intervention for high cholesterol levels in patients with PTSD. Turk Jem. 2007;11:93-97.
8. Raskind MA, Peskind ER, Kanter ED, et al. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry. 2003;160(2):371-373.
9. De Jong J, Wauben P, Huijbrechts I, et al. Doxazosin treatment for posttraumatic stress disorder. J Clin Psychopharmacol. 2010;30(1):84-85.
10. Nirmalani-Gandhy A, Sanchez D, Catalano G. Terazosin for the treatment of trauma-related nightmares: a report of four cases. Clin Neuropharmacol. 2015;38(3):109-111.
11. Belkin MR, Schwartz TL. Alpha-2 receptor agonists for the treatment of posttraumatic stress disorder. Drugs Context. 2015;4:212286. doi: 10.7573/dic.212286.
12. Gupta S, Popli A, Bathurst E, et al. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Compr Psychiatry. 1998;39(3):160-164.
13. Ahmadpanah M, Sabzeiee P, Hosseini SM, et al. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology. 2014;69(4):235-242.
14. Maher MJ, Rego SA, Asnis GM. Sleep disturbances in patients with post-traumatic stress disorder: epidemiology, impact and approaches to management. CNS Drugs. 2006;20(7):567-590.
15. Saavedra JM, Sánchez-Lemus E, Benicky J. Blockade of brain angiotensin II AT1 receptors ameliorates stress, anxiety, brain inflammation, and ischemia: therapeutic implications. Psychoneuroendocrinology. 2011;36(1):1-18.
16. McGaugh JL. Making lasting memories: remembering the significant. Proc Natl Acad Sci U S A. 2013;110(suppl 2):10402-10407.
17. Writer BW, Meyer EG, Schillerstrom JE. Prazosin for military combat-related PTSD nightmares: a critical review. J Neuropsychiatry Clin Neurosci. 2014;26(1):24-33.
18. Kung S, Espinel Z, Lapid MI. Treatment of nightmares with prazosin: a systematic review. Mayo Clin Proc. 2012;87(9):890-900.
19. Arnsten AF, Raskind MA, Taylor FB, et al. The effects of stress exposure on prefrontal cortex: translating basic research into successful treatments for post-traumatic stress disorder. Neurobiol Stress. 2015;1:89-99.
20. Serzone [package insert]. Princeton, NJ: Bristol-Myers Squibb; 2003.
21. Puetz TW, Youngstedt SD, Herring MP. Effects of pharmacotherapy on combat-related PTSD, anxiety, and depression: a systematic review and meta-regression analysis. PLoS One. 2015;10(5):e0126529. doi: 10.1371/journal. pone.0126529.
22. Glassman AH. Cardiovascular effects of tricyclic antidepressants. Annu Rev Med. 1984;35:503-511.
Herb–drug interactions: Caution patients when changing supplements
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels.
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
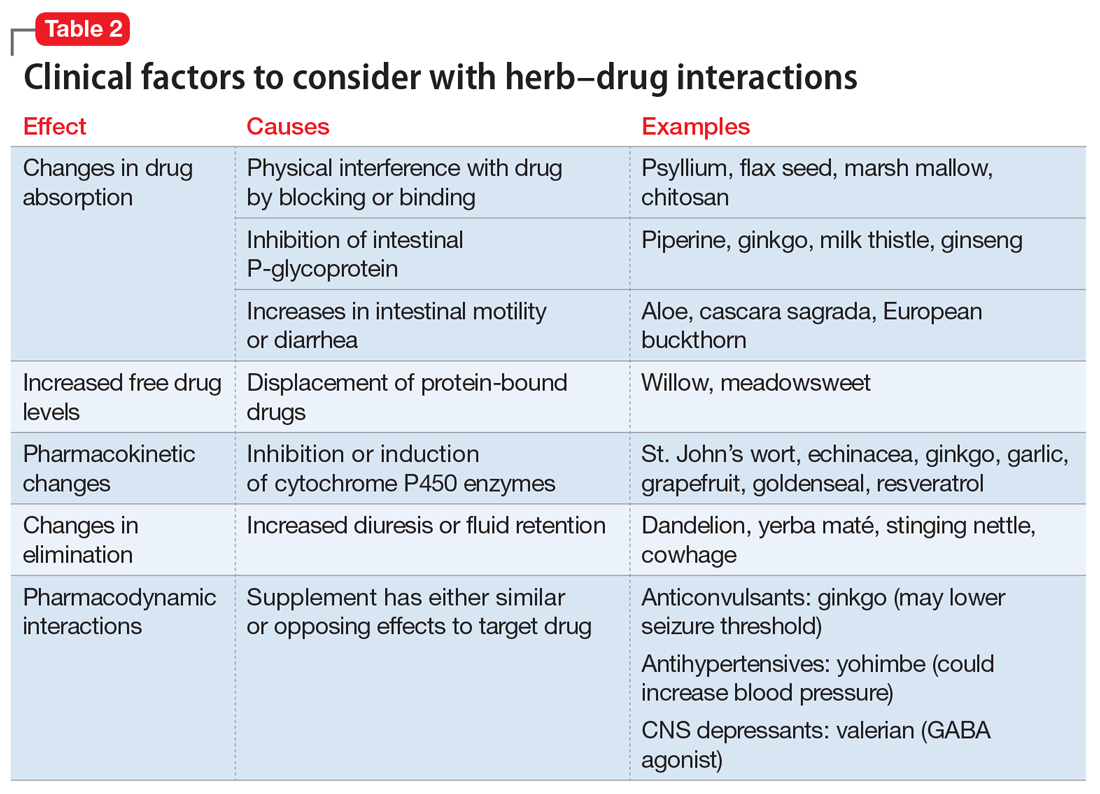
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels.
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
Ms. X, age 41, has a history of bipolar disorder and presents with extreme sleepiness, constipation with mild abdominal cramping, occasional dizziness, and “palpitations.” Although usually she is quite articulate, Ms. X seems to have trouble describing her symptoms and reports that they have been worsening over 4 to 6 days. She is worried because she is making mistakes at work and repeatedly misunderstanding directions.
Ms. X has a family history of hyperlipidemia, heart disease, and diabetes, and she has been employing a healthy diet, exercise, and use of supplements for cardiovascular health since her early 20s. Her medication regimen includes lithium, 600 mg, twice a day, quetiapine, 1,200 mg/d, a multivitamin and mineral tablet once a day, a brand name garlic supplement (garlic powder, 300 mg, vitamin C, 80 mg, vitamin E, 20 IU, vitamin A, 2,640 IU) twice a day, and fish oil, 2 g/d, at bedtime. Lithium levels consistently have been 0.8 to 0.9 mEq/L for the last 3 years.
Factors of drug–supplement interactions
Because an interaction is possible doesn’t always mean that a drug and an offending botanical cannot be used together. With awareness and planning, possible interactions can be safely managed (Table 1). Such was the case of Ms. X, who was stable on a higher-than-usual dosage of quetiapine (average target is 600 mg/d for bipolar disorder) because of presumed moderate enzyme induction by the brand name garlic supplement. Ms. X did not want to stop taking this supplement when she started quetiapine. Although garlic is listed as a possible moderate cytochrome P450 (CYP) 3A4 inducer, there is conflicting evidence.1 Ms. X’s clinician advised her to avoid changes in dosage, because it could affect her quetiapine levels. However, the change in the botanical preparation from dried, powdered garlic to garlic oil likely removed the CYP3A4 enzyme induction, leading to a lower rate of metabolism and accumulation of the drug to toxic levels.
Drug metabolism. Practitioners are increasingly aware that St. John’s wort can significantly affect concomitantly administered drug levels by induction of the CYP isoenzyme 3A4 and more resources are listing this same possible induction for garlic.1 However, what is less understood is the extent to which different preparations of the same plant possess different chemical profiles (Table 2).
Clinical studies with different garlic preparations—dried powder, aqueous extracts, deodorized preparations, oils—have demonstrated diverse and highly variable results in tests of effects on CYP isoenzymes and other metabolism activities.
Drug absorption. Small differences in amounts of vitamins in the supplement are unlikely to be clinically significant, but the addition of piperine could be affecting quetiapine absorption. Piperine, a constituent of black pepper and long pepper, is used in Ayurvedic medicine for:
- pain
- influenza
- rheumatoid arthritis
- asthma
- loss of appetite
- stimulating peristalsis.6
Animal studies have demonstrated anti-inflammatory, anticonvulsant, anticarcinogenic, and antioxidant effects, as well as stimulation of digestion via digestive enzyme secretion and increased gastromotility.3,6
Because piperine is known to increase intestinal absorption by various mechanisms, it often is added to botanical medicines to increase bioavailability of active components. BioPerine is a 95% piperine extract marketed to be included in vitamin and herbal supplements for that purpose.3 This allows use of lower dosages to achieve outcomes, which, for expensive botanicals, could be a cost savings for the manufacturer. Studies examining piperine’s influence on drug absorption have demonstrated significant increases in carbamazepine, rifampin, phenytoin, nevirapine, and many other drugs.
In addition to increased absorption, piperine seems to be a non-specific general inhibitor of CYP isoenzymes; IV phenytoin levels also were higher among test participants.6,8 Piperine reduces intestinal glucuronidation via uridine 5’-diphospho-glucuronosyltransferase inhibition, and the small or moderate effects on lithium levels seem to be the result of diuretic activities.3,7
Patients often are motivated to control at least 1 aspect of their medical treatment, such as the supplements they choose to take. Being open to patient use of non-harmful or low-risk supplements, even when they are unlikely to have any medicinal benefit, helps preserve a relationship in which patients are more likely to consider your recommendation to avoid a harmful or high-risk supplement.
Related Resources
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
1. Natural Medicines Database. Garlic monograph. http://naturaldatabase.therapeuticresearch.com. Accessed May 1, 2017.
2. Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (part 1). EXCLI J. 2014;13:347-391.
3. Colalto C. Herbal interactions on absorption of drugs: mechanism of action and clinical risk assessment. Pharmacol Res. 2010;62(3):207-227.
4. Gurley BJ, Gardner SF, Hubbard MA, et al. Clinical assessment of effects of botanical supplementation on cytochrome P450 phenotypes in the elderly: St. John’s wort, garlic oil, Panax ginseng and Ginkgo biloba. Drugs Aging. 2005;22(6):525-539.
5. Gallicano K, Foster B, Choudhri S. Effect of short-term administration of garlic supplements on single-dose ritonavir pharmacokinetics in healthy volunteers. Br J Clin Pharmacol. 2003;55(2):199-202.
6. Meghwal M, Goswami TK. Piper nigrum and piperine: an update. Phytother Res. 2013;27(8):1121-1130.
7. Natural Medicines Database. Black pepper monograph. https://www.naturalmedicines.therapeuticresearch.com. Accessed May 1, 2017.
8. Zhou S, Lim LY, Chowbay B. Herbal modulation of p-glycoprotein. Drug Metab Rev. 2004;36(1):57-104.
9. Chinta G, Syed B, Coumar MS, et al. Piperine: a comprehensive review of pre-clinical and clinical investigations. Curr Bioact Compd. 2015;11(3):156-169.
How you can simplify your patient’s medication regimen to enhance adherence
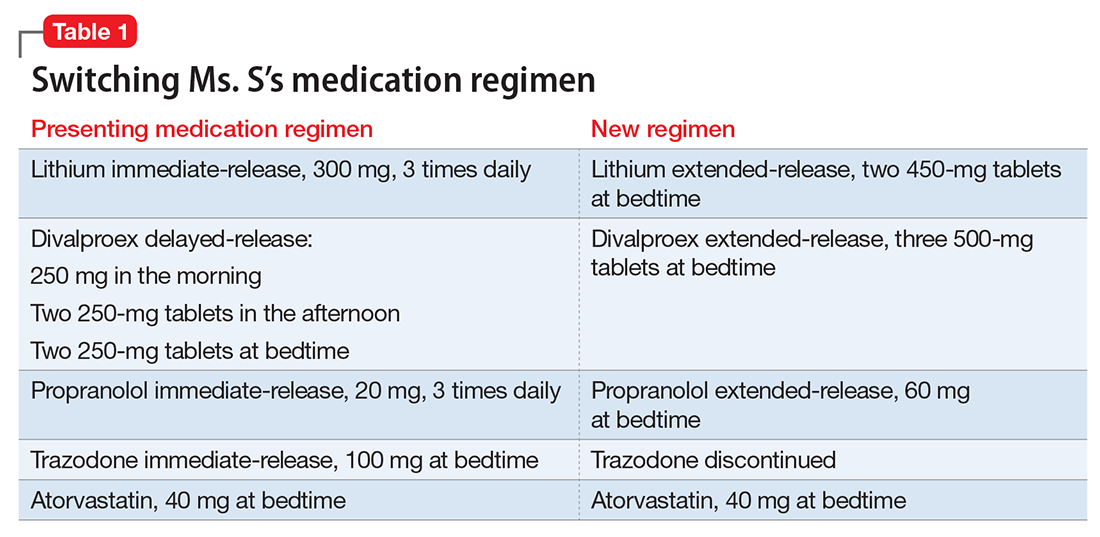
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2


Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
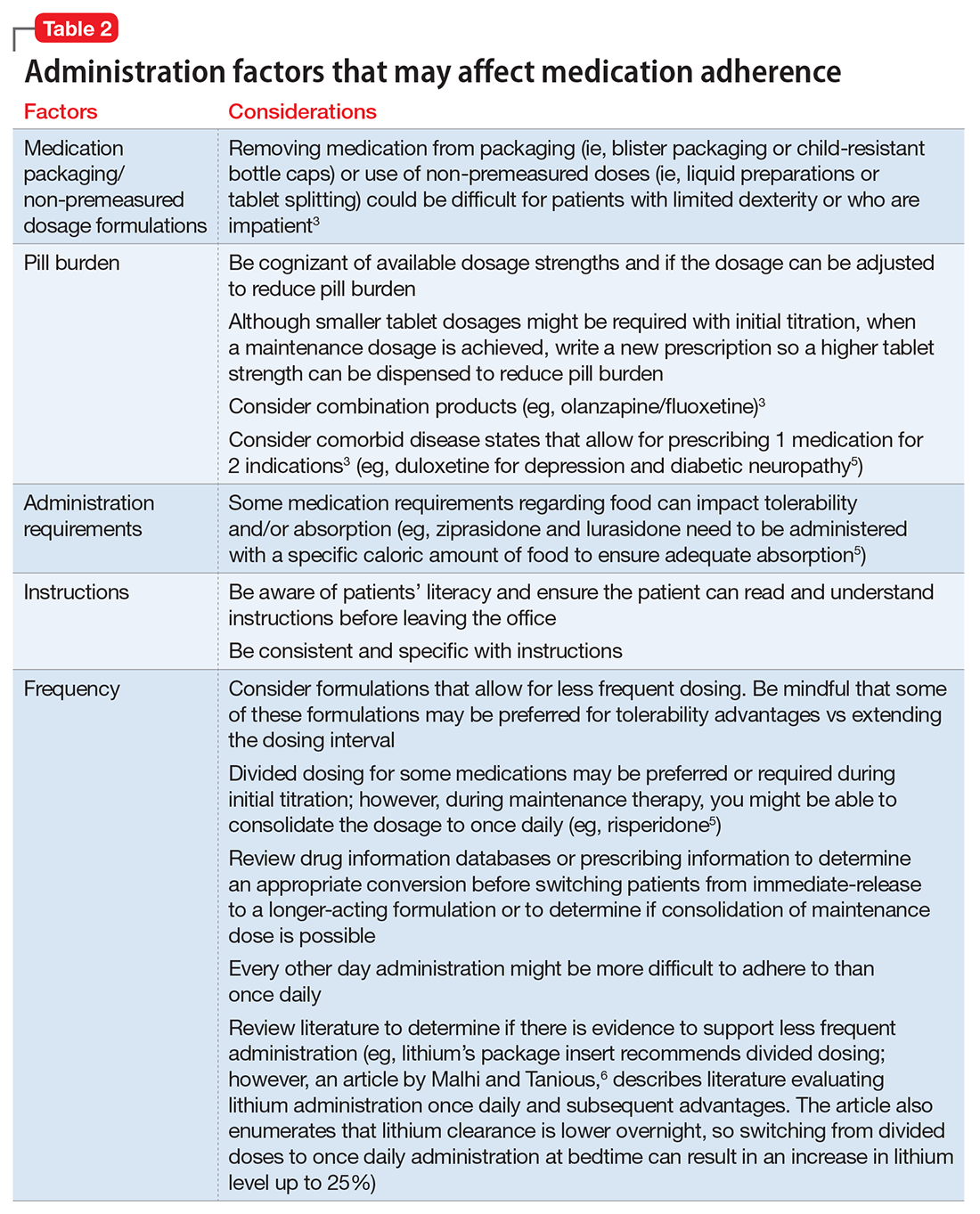
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
Ms. S, age 53, has bipolar disorder, dyslipidemia, and drug-induced tremor and presents to the clinic complaining of increasing depressive symptoms despite a history of response to her current medication regimen (Table 1). When informed that her lithium and divalproex levels are subtherapeutic, Ms. S admits that she doesn’t always take her medication. She understands her psychiatric and medical conditions and rationale for her current medications; however, she recently changed jobs, which has affected her ability to adhere to her regimen. Ms. S says the only thing preventing her from adhering to her medication is the frequency of administration.
Only approximately one-half of patients with chronic illness adhere to their medication regimen.1 Nonadherence has been reported in 20% to 72% of patients with schizophrenia, 20% to 50% of those with bipolar disorder, and 28% to 52% with major depressive disorder.2 Medication nonadherence can impact a patient’s health outcomes1 and could lead to increased hospitalizations, homelessness, substance use, decreased quality of life, and suicide; however, it is difficult to fully determine the extent of medication nonadherence due to lack of standard measurement methodology.2
Factors that affect medication adherence in patients with psychiatric diagnoses include:
- patient-related (ie, demographic factors)
- psychological (eg, lack of insight into illness, negative emotions toward medications)
- social and environmental (eg, therapeutic alliance with the physician, housing stability and support, and discharge planning)
- medication-related (eg, complex dosing schedule).2
Medication regimen tolerability, complexity, and cost; patient understanding of medication indications and onset of therapeutic effect; and patient’s view of benefits can impact adherence.1,3 Assessing medication adherence and identifying barriers specific to the patient is essential when developing a treatment plan. If complexity is a barrier, simplify the medication regimen.
Claxton et al4 found an inverse relationship between medication dosing and adherence. Reviewing data from 76 studies that used electronic monitoring (records the time and date of actual dosing events) the overall rate of medication adherence was 71% ± 17%. Adherence rates were significantly higher with once daily (79% ± 14%) vs 3 times daily (65% ± 16%) or 4 times daily (51% ± 20%), and twice daily (69% ± 15%) was significantly better than 4 times daily dosing. Adherence between once daily and twice daily or twice daily and 3 times daily did not result in a significant difference. The authors noted that electronic monitoring has limitations; patients could have opened the medication bottle but not ingested the drug.4
Consider these factors and strategies when developing a treatment plan (Table 2).3,5,6
Ease of administration
Medication packaging. Patients with limited dexterity might not be able to remove the medication from blister packaging or child-proof cap, measure non-unit dose liquid preparations, or split tablets in half.3 Patients with limited patience could get frustrated and skip medications that take longer to remove from packaging or have to be measured. Consult a pharmacist about medication packaging options or formulations that might be appropriate for some patients (ie, individuals with dysphagia), such as oral-disintegrating or sublingual tablets.
Assess pill burden. Although it might not be appropriate when titrating medications, consider adjusting the maintenance dosage to reduce the number of tablets (eg, a patient prescribed divalproex delayed-release, 2,750 mg/d, will take eleven 250-mg tablets vs taking divalproex delayed-release, 2,500 mg/d, which is five 500-mg tablets).
Keep in mind availability of combination medications (eg, olanzapine/fluoxetine) to reduce pill burden. Also, if possible, consider comorbid disease states that allow for prescribing 1 medication that can treat 2 conditions to reduce pill burden (eg, duloxetine for depression and diabetic neuropathy).3
Food recommendations. Review food requirements (ie, administration on an empty stomach vs the need for a specific caloric amount) and whether these are recommendations to improve tolerability or required to ensure adequate absorption. Nonadherence with dietary recommendations that can affect absorption may result in reduced effectiveness despite taking the medication.
Administration instructions
Keep administration instructions simple and be consistent with instructions and terminology.3 For example, if all medications are to be administered once daily in the morning, provide specific instructions (ie, “every morning”) because it may be confusing for patients if some medications are written for “once daily” and others for “every morning.” Some patients might prefer to have the medication indication noted in the administration instructions. Additionally, be aware of the patient’s literacy, and ensure the patient is able to read and understand instructions before leaving the office.
Administration frequency
Consider the required administration frequency and the patient’s self-reported ability to adhere to that frequency before initiating a new medication. Ask the patient what frequencies he (she) can best manage and evaluate his (her) regimen to determine if a less frequent schedule is possible. Consider formulations that may allow for less frequent dosing (eg, controlled-release, sustained-release, long-acting, or extended-release formulations) or consolidating divided doses to once daily if possible.3 Some of these formulations may be preferred for tolerability advantages vs extending the dosing interval (eg, regular-release and extended-release lithium tablets have the same half-life of approximately 18 to 36 hours; however, the extended-release formulation has a longer time to peak serum concentration, approximately 2 to 6 hours vs 0.5 to 3 hours, respectively. As a result, the extended-release formulation may offer improved tolerability in terms of peak-related side effects,5,7 which may be advantageous, especially when dosing lithium once daily). Keep in mind, for some patients every other day administration is more difficult to adhere to than once daily.
Review drug or prescribing information to determine an appropriate conversion before switching from an immediate-release to a longer-acting formulation. The switch may result in different drug serum concentrations (eg, propranolol sustained-release has different pharmacokinetics and produces lower blood levels than the immediate-release formulation). When switching between formulations, monitor patients to ensure the desired therapeutic effect is maintained.8
Consider collaborating with pharmacists, primary care providers, and other prescribers to simplify medical and psychiatric medications.
Other considerations
Lab monitoring requirements for drugs, such as clozapine, lithium, or divalproex, could affect a patient’s willingness to adhere. Use of weekly or monthly medication organizers, mobile apps, alarms (on cell phones or clocks), medication check-off sheets or calendars, and family or friend support could help improve medication adherence.
Case continued
After reviewing the medication regimen and consulting with a pharmacist, Ms. S’s regimen is simplified to once-daily administration, and pill burden is reduced by using extended-release formulations and consolidating doses at bedtime (Table 1). Additionally, trazodone is discontinued because divalproex, now taken once daily at bedtime, is sedating and aids in sleep.
For medications that require therapeutic blood monitoring such as lithium and divalproex, check drug levels when switching formulations. In the case of Ms. S, lithium, propranolol, and divalproex dosages were switched to extended-release preparations and consolidated to once daily at bedtime; the divalproex dosage was increased because an increase in total daily dose between 8% to 20% may be required to maintain similar serum concentrations.5 Lithium immediate-release was switched to the extended-release, which reduced the pill burden and could help tolerability if Ms. S experiences peak concentration-related side effects. Consolidating the lithium dosage from divided to once daily at bedtime can increase the lithium serum level by up to 25%.6
With a change in formulation, monitor tolerability and effectiveness of the medication regimen in regard to mood stabilization and tremor control, as well as check serum lithium and divalproex levels, creatinine, and sodium after 5 days, unless signs and symptoms of toxicity occur.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
1. World Health Organization. Adherence to long-term therapies: evidence for action. http://apps.who.int/iris/bitstream/10665/42682/1/9241545992.pdf. Published 2003. Accessed November 29, 2015.
2. Julius RJ, Novitsky MA, Dubin WR. Medication adherence: a review of the literature and implications for clinical practice. J Psychiatr Pract. 2009;15(1):34-44.
3. Atreja A, Bellam N, Levy SR. Strategies to enhance patient adherence: making it simple. MedGenMed. 2005;7(1):4.
4. Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther. 2001;23(8):1296-1310.
5. Lexicomp Online, Lexi-Drugs, Hudson, Ohio: Lexi-Comp, Inc.; February 28, 2016.
6. Malhi GS, Tanious M. Optimal frequency of lithium administration in the treatment of bipolar disorder: clinical and dosing considerations. CNS Drugs. 2011;25(4):289-298.
7. Jefferson JW, Greist JH, Ackerman DL, et al. Lithium: an overview. In: Lithium encyclopedia for clinical practice. 2nd ed. Washington, DC: American Psychiatric Press; 1987.
8. Inderal LA (propranolol extended release) [package insert]. Cranford, NJ: Akrimax Pharmaceuticals; November 2015.
Ruling out delirium: Therapeutic principles of withdrawing and changing medications
Ms. M, age 71, was diagnosed with Alzheimer’s disease several months ago and her clinical presentation and Mini-Mental Status Exam score of 22 indicates mild dementia. In addition to chronic medications for hypertension, Ms. M has been taking lorazepam, 1 mg, 3 times daily, for >15 years for unspecified anxiety.
Ms. M becomes more confused at home over the course of a few days, and her daughter brings her to her primary care physician for evaluation. Recognizing that benzodiazepines can contribute to delirium, the physician discontinues lorazepam. Three days later, Ms. M’s confusion worsens, and she develops nausea and a tremor. She is taken to the local emergency department where she is admitted for benzodiazepine withdrawal and diagnosed with a urinary tract infection.
Because dementia is a strong risk factor for developing delirium,1 withdrawing or changing
Consider withdrawing or replacing medications that are strongly implicated in causing delirium with another medication for the same indication with a lower potential for
In general, there are no firm rules for how to taper and discontinue potentially deliriogenic medications, as both the need to taper and the best strategy for doing so depends on a number of factors and requires clinical judgement. When determining how quickly to withdraw a potentially offending medication in a patient with suspected delirium, clinicians should consider:
Dosage and duration of treatment. Consider tapering and discontinuing benzodiazepines in a patient who is taking more than the minimal scheduled dosages for ≥2 weeks, especially after 8 weeks of scheduled treatment. Consider tapering opioids in a patient taking more than the minimal scheduled dosage for more than a few days. When attempting to rule out delirium, taper opioids as quickly and as safely possible, with a recommended reduction of ≤20% per day to prevent withdrawal symptoms. In general, potentially deliriogenic medications can be discontinued without tapering if they are taken on a non-daily, as-needed basis.
The half-life of a medication determines both the onset and duration of withdrawal symptoms. Withdrawal occurs earlier when discontinuing medications with short 
Nature of withdrawal symptoms. In patients with suspected delirium, tapering over weeks or
Care setting. When tapering and discontinuing a medication, regularly monitor patients for withdrawal symptoms; slow or temporarily stop the taper if withdrawal symptoms occur. Because close monitoring is easier in an inpatient vs an outpatient care setting, more aggressive tapering over 2 to 3 days generally can be considered, although more gradual tapering might be prudent to ensure safety of outpatients.
1. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
2. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
3. Clegg A, Young JB. Which medications to avoid in people at risk of delirium: a systematic review. Age Aging. 2010;40(1):23-29.
4. Han L, McCusker J, Cole M, et al. Use of medications with anticholinergic effect predicts clinical severity of delirium symptoms in older medical inpatients. Arch Intern Med. 2001;161(8):1099-1105.
5. Carnahan RM, Lund BC, Perry PJ, et al. The Anticholinergic Drug Scale as a measure of drug-related anticholinergic burden: associations with serum anticholinergic activity. J Clin Pharmacol. 2006;46(12):1481-1486.
6. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
Ms. M, age 71, was diagnosed with Alzheimer’s disease several months ago and her clinical presentation and Mini-Mental Status Exam score of 22 indicates mild dementia. In addition to chronic medications for hypertension, Ms. M has been taking lorazepam, 1 mg, 3 times daily, for >15 years for unspecified anxiety.
Ms. M becomes more confused at home over the course of a few days, and her daughter brings her to her primary care physician for evaluation. Recognizing that benzodiazepines can contribute to delirium, the physician discontinues lorazepam. Three days later, Ms. M’s confusion worsens, and she develops nausea and a tremor. She is taken to the local emergency department where she is admitted for benzodiazepine withdrawal and diagnosed with a urinary tract infection.
Because dementia is a strong risk factor for developing delirium,1 withdrawing or changing
Consider withdrawing or replacing medications that are strongly implicated in causing delirium with another medication for the same indication with a lower potential for
In general, there are no firm rules for how to taper and discontinue potentially deliriogenic medications, as both the need to taper and the best strategy for doing so depends on a number of factors and requires clinical judgement. When determining how quickly to withdraw a potentially offending medication in a patient with suspected delirium, clinicians should consider:
Dosage and duration of treatment. Consider tapering and discontinuing benzodiazepines in a patient who is taking more than the minimal scheduled dosages for ≥2 weeks, especially after 8 weeks of scheduled treatment. Consider tapering opioids in a patient taking more than the minimal scheduled dosage for more than a few days. When attempting to rule out delirium, taper opioids as quickly and as safely possible, with a recommended reduction of ≤20% per day to prevent withdrawal symptoms. In general, potentially deliriogenic medications can be discontinued without tapering if they are taken on a non-daily, as-needed basis.
The half-life of a medication determines both the onset and duration of withdrawal symptoms. Withdrawal occurs earlier when discontinuing medications with short
Nature of withdrawal symptoms. In patients with suspected delirium, tapering over weeks or
Care setting. When tapering and discontinuing a medication, regularly monitor patients for withdrawal symptoms; slow or temporarily stop the taper if withdrawal symptoms occur. Because close monitoring is easier in an inpatient vs an outpatient care setting, more aggressive tapering over 2 to 3 days generally can be considered, although more gradual tapering might be prudent to ensure safety of outpatients.
Ms. M, age 71, was diagnosed with Alzheimer’s disease several months ago and her clinical presentation and Mini-Mental Status Exam score of 22 indicates mild dementia. In addition to chronic medications for hypertension, Ms. M has been taking lorazepam, 1 mg, 3 times daily, for >15 years for unspecified anxiety.
Ms. M becomes more confused at home over the course of a few days, and her daughter brings her to her primary care physician for evaluation. Recognizing that benzodiazepines can contribute to delirium, the physician discontinues lorazepam. Three days later, Ms. M’s confusion worsens, and she develops nausea and a tremor. She is taken to the local emergency department where she is admitted for benzodiazepine withdrawal and diagnosed with a urinary tract infection.
Because dementia is a strong risk factor for developing delirium,1 withdrawing or changing
Consider withdrawing or replacing medications that are strongly implicated in causing delirium with another medication for the same indication with a lower potential for
In general, there are no firm rules for how to taper and discontinue potentially deliriogenic medications, as both the need to taper and the best strategy for doing so depends on a number of factors and requires clinical judgement. When determining how quickly to withdraw a potentially offending medication in a patient with suspected delirium, clinicians should consider:
Dosage and duration of treatment. Consider tapering and discontinuing benzodiazepines in a patient who is taking more than the minimal scheduled dosages for ≥2 weeks, especially after 8 weeks of scheduled treatment. Consider tapering opioids in a patient taking more than the minimal scheduled dosage for more than a few days. When attempting to rule out delirium, taper opioids as quickly and as safely possible, with a recommended reduction of ≤20% per day to prevent withdrawal symptoms. In general, potentially deliriogenic medications can be discontinued without tapering if they are taken on a non-daily, as-needed basis.
The half-life of a medication determines both the onset and duration of withdrawal symptoms. Withdrawal occurs earlier when discontinuing medications with short
Nature of withdrawal symptoms. In patients with suspected delirium, tapering over weeks or
Care setting. When tapering and discontinuing a medication, regularly monitor patients for withdrawal symptoms; slow or temporarily stop the taper if withdrawal symptoms occur. Because close monitoring is easier in an inpatient vs an outpatient care setting, more aggressive tapering over 2 to 3 days generally can be considered, although more gradual tapering might be prudent to ensure safety of outpatients.
1. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
2. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
3. Clegg A, Young JB. Which medications to avoid in people at risk of delirium: a systematic review. Age Aging. 2010;40(1):23-29.
4. Han L, McCusker J, Cole M, et al. Use of medications with anticholinergic effect predicts clinical severity of delirium symptoms in older medical inpatients. Arch Intern Med. 2001;161(8):1099-1105.
5. Carnahan RM, Lund BC, Perry PJ, et al. The Anticholinergic Drug Scale as a measure of drug-related anticholinergic burden: associations with serum anticholinergic activity. J Clin Pharmacol. 2006;46(12):1481-1486.
6. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
1. Inouye SK. Delirium in older persons. N Engl J Med. 2006;354(11):1157-1165.
2. Alagiakrishnan K, Wiens CA. An approach to drug induced delirium in the elderly. Postgrad Med J. 2004;80(945):388-393.
3. Clegg A, Young JB. Which medications to avoid in people at risk of delirium: a systematic review. Age Aging. 2010;40(1):23-29.
4. Han L, McCusker J, Cole M, et al. Use of medications with anticholinergic effect predicts clinical severity of delirium symptoms in older medical inpatients. Arch Intern Med. 2001;161(8):1099-1105.
5. Carnahan RM, Lund BC, Perry PJ, et al. The Anticholinergic Drug Scale as a measure of drug-related anticholinergic burden: associations with serum anticholinergic activity. J Clin Pharmacol. 2006;46(12):1481-1486.
6. American Geriatrics Society 2015 Beers Criteria Update Expert Panel. American Geriatrics Society 2015 Updated Beers Criteria for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc. 2015;63(11):2227-2246.
When to prescribe antidepressants to treat comorbid depression and pain disorders
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.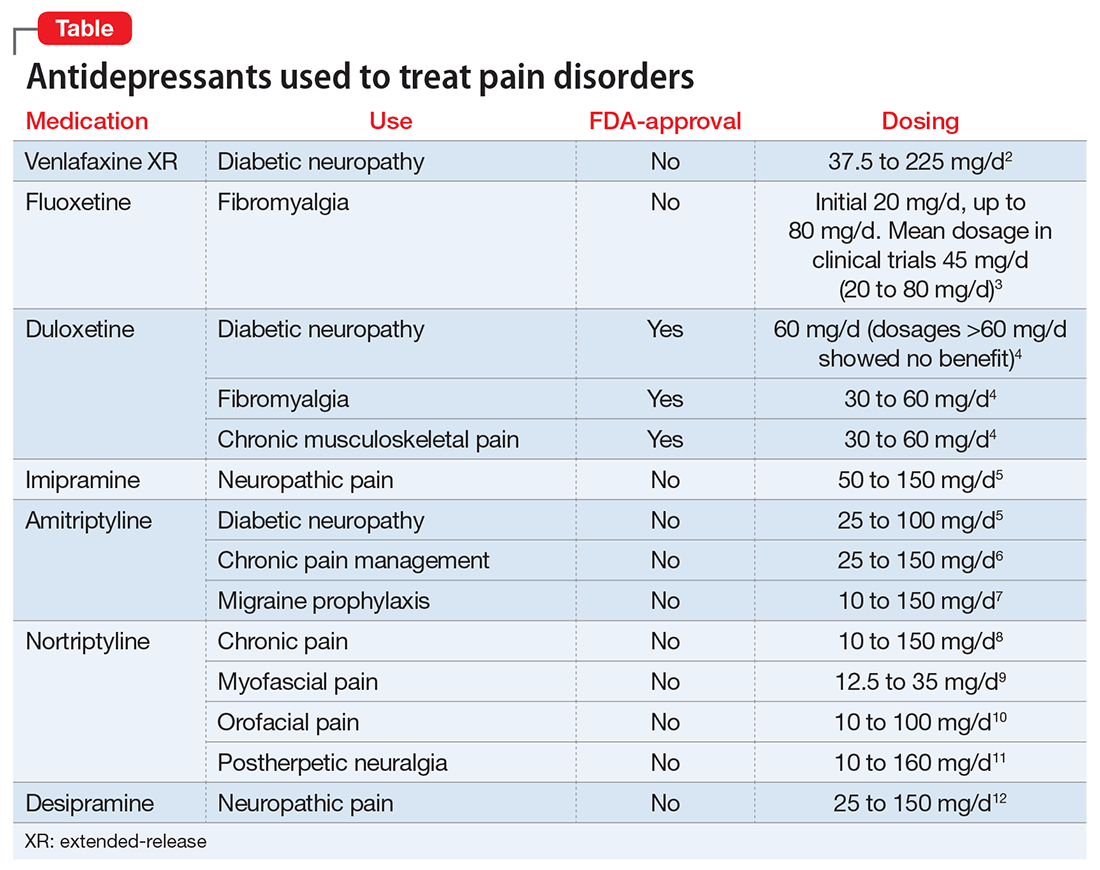
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
Ms. C, age 44, has a history of hypertension, chronic shoulder pain associated with a motor vehicle accident almost 2 decades ago, and major depressive disorder (MDD). Her medication regimen includes losartan, 100 mg/d; atenolol, 25 mg/d; gabapentin, 100 mg, 3 times a day; sertraline, 100 mg/d; and naproxen, 500 mg, twice a day as needed for pain. She does not take opioids for pain control because she had a poor response when used in the past. Ms. C denies muscle pain or tenderness but describes pain in nonspecific areas of her arm, shoulder, neck, and chest. Ms. C reports poor quality of sleep and early morning awakenings, which she attributes to her unmanaged pain. Her last appointment with a psychiatrist was “many, many months ago.”
A reciprocal relationship exists between depression and pain. A 2-year, population-based, prospective, observational study of 3,654 patients showed that pain at baseline was an independent predictor of depression and a depression diagnosis was a predictor of developing pain within 2 years.1 Patients with MDD might complain of physical symptoms, such as constipation, generalized aches, frequent headache, and fatigue, many of which overlap with chronic pain disorders. Therefore, a thorough symptom assessment and history is vital for an accurate diagnosis. To decrease polypharmacy and pill burden, optimal treatment should employ agents that treat both conditions.
Using antidepressants to treat pain disorders
Several antidepressants have been studied for managing pain disorders including:
- fibromyalgia
- diabetic neuropathy
- neuropathic pain
- postherpetic neuralgia
- migraine prophylaxis
- chronic musculoskeletal pain.
Antidepressants that treat both depression and chronic neuropathic pain include tricyclic antidepressants (TCAs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) (Table).2-12 Notably, most antidepressants studied for pain management are used off-label; duloxetine is the only medication with an FDA indication for MDD and pain disorders.
The hypothesized mechanism of action is dual serotonin and norepinephrine reuptake inhibition, based on the monoamine hypothesis of depression and pain signaling dysfunction in neuropathic pain. Antidepressants, such as TCAs and SNRIs, address pain by increasing the synaptic concentration of norepinephrine and/or serotonin in the dorsal horn, thereby inhibiting the release of excitatory neurotransmitters and blunting pain pathways.13
TCAs used to treat comorbid depression and pain conditions include amitriptyline, nortriptyline, imipramine, and desipramine.14 TCAs are cost-effective medications for managing neuropathy and headache; however, the dosages used for pain tend to be lower than those typically used for depression.
TCAs are not commonly prescribed for depression because of their side-effect profile and poor tolerability. TCAs are contraindicated in patients with cardiac conduction abnormalities, epilepsy, and narrow-angle glaucoma. Common adverse effects include dry mouth, sweating, dizziness, orthostatic hypotension, sedation, weight gain, urinary retention, and constipation. These adverse effects limit their use and have organizations, such as the American Geriatric Society, to caution against their use in geriatric patients.
SNRIs that have been studied for pain disorders include venlafaxine, duloxetine, and milnacipran.2 Of note, milnacipran is not FDA-approved for MDD, but its L-enantiomer, levomilnacipran, is. Unlike duloxetine and venlafaxine, both milnacipran and levomilnacipran are not available as a generic formulation, therefore they have a higher patient cost. The SNRI dosages used for pain management tend to be similar to those used for MDD, indicating that the target dosage may be effective for both depressive and pain symptoms.
Selective serotonin reuptake inhibitors (SSRIs). Compared with data available supporting the use of TCAs and SNRIs for pain management, the data for SSRI are sparse. Studies have evaluated fluoxetine, paroxetine, and citalopram for pain, with the most promising data supporting fluoxetine.2 Fluoxetine, 10 to 80 mg/d, has been evaluated in randomized, placebo-controlled trials for pain conditions, including fibromyalgia (n = 3), painful diabetic neuropathy (n = 1), and facial pain (n = 1). Fluoxetine was more effective than placebo at controlling pain in 2 fibromyalgia studies (dosage range, 10 to 80 mg/d) and 1 facial pain study (dosage, 20 mg/d).2
CASE CONTINUED
When evaluating potential treatment options, it is noted that Ms. C is prescribed sertraline, 200 mg/d, but has been taking a lower dosage. Ms. C states that she has been taking sertraline, 100 mg every morning, for months, and noticed some minor initial improvements in mood, but still has days when she don’t feel like doing anything. She fills out a depression rating scale classifying her current depression as moderately severe. Today she rates her pain as 7 out of 10. Suboptimal control of her depression may require a dosage increase; however, perhaps a change in therapy is warranted. It may be prudent to switch Ms. C to an SNRI, such as duloxetine, an agent that can address her depression and provide additional benefits of pain control.
Switching from a SSRI to duloxetine has been shown to be effective when targeting pain symptoms in patients with comorbid MDD. In addition, improvements in pain scores have been seen after a switch to duloxetine in patients with depression with nonresponse or partial response to a SSRI.15
Studies support the decision to change Ms. C’s medication from sertraline to duloxetine, despite an inadequate therapeutic trial of the SSRI.
Using pain medication to treat depression
Conversely, the use of pain medications to treat depression also has been studied. The most notable data supports the use of ketamine, an anesthetic. IV ketamine is well documented for treating pain and, in recent years, has been evaluated for MDD in several small studies. Results show that IV ketamine, 0.5 mg/kg, produced a rapid response in depressed patients.16 For pain conditions studies support the use of ketamine as an IV push, continuous infusion, intermittent infusion, as well as oral administration, for many conditions, including acute and postoperative pain, chronic regional pain, and neuropathic pain. However, there is little evidence evaluating ketamine’s effect on both pain scores and depression symptoms in patients such as Ms. C.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
1. Chou KL. Reciprocal relationship between pain and depression in older adults: evidence from the English Longitudinal Study of Ageing. J Affect Disord. 2007;102(1-3):115-123.
2. Lee YC, Chen PP. A review of SSRIs and SNRIs in neuropathic pain. Expert Opin Pharmacother. 2010;11(17):2813-2825.
3. Arnold LM, Hess EV, Hudson JI, et al. A randomized placebo-controlled, double-blind, flexible-dose study of fluoxetine in the treatment of women with fibromyalgia. Am J Med. 2002;112(3):191-197.
4. Cymbalta [package insert]. Indianapolis, IN: Eli Lily and Company; 2015.
5. Bril V, England J, Franklin GM, et al; American Academy of Neurology; American Association of Neuromuscular and Electrodiagnostic Medicine; American Academy of Physical Medicine and Rehabilitation. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology. 2011;76(20):1758-1765.
6. McQuay HJ, Carroll D, Glynn CJ. Low dose amitriptyline in the treatment of chronic pain. Anaesthesia. 1992;47(8):646-652.
7. Evers S, Afra J, Frese A, et al; European Federation of Neurological Societies. EFNS guideline on the drug treatment of migraine—revised report of an EFNS task force. Eur J Neurol. 2009;16(9):968-981.
8. Atkinson JH, Slater MA, Williams RA, et al. A placebo-controlled randomized clinical trial of nortriptyline for chronic low back pain. Pain. 1998;76(3):287-296.
9. Haviv Y, Rettman A, Aframian D, et al. Myofascial pain: an open study on the pharmacotherapeutic response to stepped treatment with tricyclic antidepressants and gabapentin. J Oral Facial Pain Headache. 2015;29(2):144-151.
10. Romero-Reyes M, Uyanik JM. Orofacial pain management: current perspectives. J Pain Res. 2014;7:99-115.
11. Raja SN, Haythornthwaite JA, Pappagallo M, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015-1021.
12. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
13. Argoff C. Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin. 2011;27(10):2019-2031.
14. Haanpää ML, Gourlay GK, Kent JL, et al. Treatment considerations for patients with neuropathic pain and other medical comorbidities. Mayo Clin Proc. 2010;85(suppl 3):S15-S25.
15. Perahia DGS, Quail D, Desaiah D, et al. Switching to duloxetine in selective serotonin reuptake inhibitor non- and partial-responders: effects on painful physical symptoms of depression. J Psychiatric Res. 2009;43(5):512-518.
16. Caddy C, Amit BH, McCloud TL, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database Syst Rev. 2015;(9):CD011612. doi: 10.1002/14651858.CD011612.pub2.
Managing clozapine-induced neutropenia and agranulocytosis
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to 
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
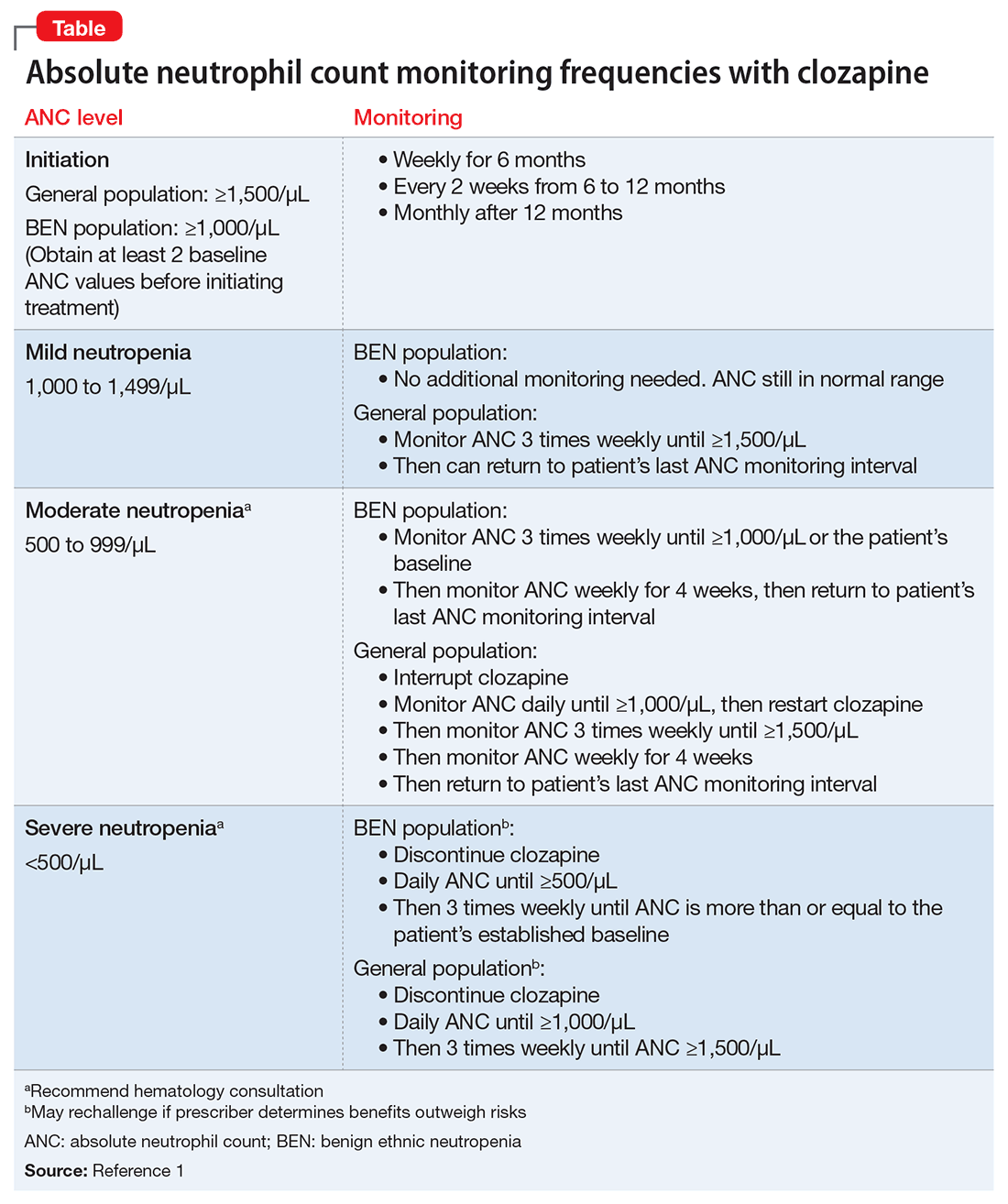
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
Mr. S, age 43, has schizophrenia and been stable on clozapine for 6 years after several other antipsychotic regimens failed. Mr. S also has a history of hypertension, dyslipidemia, and gastroesophageal reflux disorder. His medication regimen includes clozapine, 400 mg/d, lisinopril, 20 mg/d, atorvastatin, 40 mg/d, omeprazole, 40 mg/d, and a multivitamin. During routine blood monitoring, Mr. S shows a significant drop in absolute neutrophil count (ANC) (750/µL) (reference range, 1,500 to 8,000 µL). Mr. S , who is African American, has no history of benign ethnic neutropenia (BEN) or ANC <1,000/µL. While reviewing his chart, clinicians note that Mr. S had an ANC of 1,350/µL3 years earlier in 2013. Because a complete workup reveals no other cause for this lab abnormality, we determine that is clozapine-induced. Mr. S’s physician asks about treatment options that would allow him to stay on clozapine.
Because of clozapine’s efficacy in treatment-resistant schizophrenia, many psychiatrists aim to
Clozapine-induced neutropenia
Clozapine was approved in 1989 for managing treatment-resistant schizophrenia after demonstrating better efficacy than chlorpromazine.1 However, the adverse effects of neutropenia (white blood cell count [WBC] <3,000/μL) and agranulocytosis (ANC <500/μL3) leading to death were reported in later studies.2,3 One study in the United Kingdom and Ireland reported a prevalence of 2.9% for neutropenia and 0.8% for agranulocytosis among patients taking clozapine.3 Because of this risk, the FDA mandated WBC and ANC monitoring before initiating clozapine and periodically thereafter. In October 2015, the Risk Evaluation and Mitigation Strategies program for clozapine updated recommended ANC levels and eliminated WBC monitoring. ANC monitoring frequencies are summarized in the Table.1
The exact mechanism of clozapine-induced neutropenia is unknown, although it is possible it stems from the drug’s effect on white blood cell precursors.2 Neutropenia typically appears within 3 months of clozapine initiation; however, delayed cases have been reported. Additionally, the risk is higher in certain patient populations (African heritage, Yemenite, West Indians, and Arab). Patients with a lower ANC at clozapine initiation and advanced age appear to be at higher risk.2
Filgrastim
The use of granulocyte-colony stimulating factor, such as filgrastim, often is viewed as a “rescue” treatment. Filgrastim’s mechanism of action is related to neutrophil production and proliferation. Several articles from the 1990s reported efficacy in the short-term management of low WBC or ANC. However, few articles, mainly case reports, have looked at long-term use of these agents. One article examined 3 patients, average age 45, who developed neutropenia during clozapine treatment.4 Filgrastim at an average dosage of 0.6 to 0.9 mg/week was used successfully. The dosage was reduced to 0.3 mg/week in 1 patient, although neutropenia returned.
Because of the lack of literature regarding long-term therapy, it is recommended to consider short-term treatment with filgrastim to normalize ANC after a severe drop in a symptomatic patient. Physicians also must consider the potential barriers to filgrastim treatment including adverse effects, such as allergic reactions, bone pain, and thrombocytopenia, and high cost.
Adjunctive lithium
Lithium could cause leukocytosis, which could balance neutropenia induced by clozapine. One of the largest studies evaluating lithium therapy with clozapine-induced neutropenia and agranulocytosis studied 25 patients taking clozapine with a previous “red result” (WBC <3,000/μL, ANC <1,500/μL, or platelets <50,000/μL).3 Lithium treatment was started before or simultaneously with the reinitiation of clozapine in most patients; the remaining patients started treatment at a later date. Only 1 of 25 patients experienced a repeat “red result.” The average lithium level was 0.54 mEq/L.
It is important to remember that initiating adjunctive lithium carries risk. Adverse effects include gastrointestinal upset, tremors, polyuria, polydipsia, and nephrotoxicity.
Additionally, there is risk that lithium simply masks the preliminary states of neutropenia leading to a more severe agranulocytosis without warning.3 Again, the mechanism of action of clozapine-induced neutropenia is thought to be related to the drug’s effect on WBC precursors. The mechanism of lithium-induced leukocytosis is unknown, therefore it’s possible that lithium will not protect a patient from clozapine-induced neutropenia or agranulocytosis, and can lead to serious adverse events.
When deciding whether to rechallenge a patient on clozapine who had a prior episode of moderate or severe neutropenia or agranulocytosis, a risk vs benefit discussion is necessary. One study found that 20 of 53 patients (38%) experienced a repeat dyscrasia when rechallenged.5 Of these patients, most experienced a lower ANC that presented faster and took longer to resolve.5 If a patient has experienced true agranulocytosis, the recommendation is to not rechallenge clozapine.
Related Resources
• Clozapine REMS Program. www.clozapinerems.com.
• Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48,49.
• Whiskey E, Taylor D. Restarting clozapine after neutropenia: evaluating the possibilities and practicalities. CNS Drugs. 2007;21(1):25-35.
Drug Brand Names
Atorvastatin • Lipitor
Chlorpromazine • Thorazine
Clozapine • Clozaril
Fligrastim • Neupogen
Lisinopril • Prinivil
Lithium • Eskalith, Lithobid
Omeprazole • Prilosec
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.
1. Clozapine [package insert]. North Wales, PA: TEVA Pharmaceuticals USA; 2015.
2. Lundblad W, Azzam PN, Gopalan P, et al. Medical management of patients on clozapine: a guide for internists. J Hosp Med. 2015;8(8):537-543.
3. Kanaan RA, Kerwin RW. Lithium and clozapine rechallenge: a retrospective case analysis. J Clin Psychiatry. 2006;67(5):756-760.
4. Hägg S, Rosenius S, Spigset O. Long-term combination treatment with clozapine and filgrastim in patients with clozapine-induced agranulocytosis. Int Clin Psychopharmacol. 2003;18(3):173-174.
5. Dunk LR, Annan LJ, Andrews CD. Rechallenge with clozapine following leucopenia or neutropenia during previous therapy. Br J Psychiatry. 2006;188:255-263.