User login
How should you use the lab to monitor patients taking a mood stabilizer?
Ms. W, age 27, presents with a chief concern of “depression.” She describes a history of several hypomanic episodes as well as the current depressive episode, prompting a bipolar II disorder diagnosis. She is naïve to all psychotropics. You plan to initiate a mood-stabilizing agent. What would you include in your initial workup before starting treatment and how would you monitor her as she continues treatment?
Mood stabilizers are employed to treat bipolar spectrum disorders (bipolar I, bipolar II, and cyclothymic disorder) and schizoaffective disorder, bipolar type. Some evidence suggests that mood stabilizers also can be used for treatment-resistant depressive disorders and borderline personality disorder.1 Mood stabilizers include lithium, valproate, carbamazepine, oxcarbazepine, and lamotrigine.2-5
This review focuses on applications and monitoring of mood stabilizers for bipolar I and II disorders. We also will briefly review atypical antipsychotics because they also are used to treat bipolar spectrum disorders (see the September 2013 issue of Current Psychiatry at CurrentPsychiatry.com for a more detailed article on monitoring of antipsychotics).6
There are several well-researched guidelines used to guide clinical practice.2-5 Many guidelines recommend baseline and routine monitoring parameters based on the characteristics of the agent used. However, the International Society for Bipolar Disorders (ISBD) guidelines highlight the importance of monitoring medical comorbidities, which are common among patients with bipolar disorder and can affect pharmacotherapy and clinical outcomes. These recommendations are similar to metabolic monitoring guidelines for antipsychotics.5
Reviews of therapeutic monitoring show that only one-third to one-half of patien

taking a mood stabilizer are appropriately monitored. Poor adherence to guideline recommendations often is observed because of patients’ lack of insight or medication adherence and because psychiatric care generally is segregated from other medical care.7-9
Baseline testing
The ISBD guidelines recommend an initial workup for all patients that includes:
• waist circumference or body mass index (BMI), or both
• blood pressure
• complete blood count (CBC)
• electrolytes
• blood urea nitrogen (BUN) and creatinine
• liver function tests (LFTs)
• fasting glucose
• fasting lipid profile.
In addition, medical history, cigarette smoking status, alcohol intake, and family history of cardiovascular disease, cerebrovascular disease, hypertension, dyslipidemia, and diabetes mellitus should be documented. Rule out pregnancy in women of childbearing potential.2 The Figure describes monitoring parameters based on selected agent.
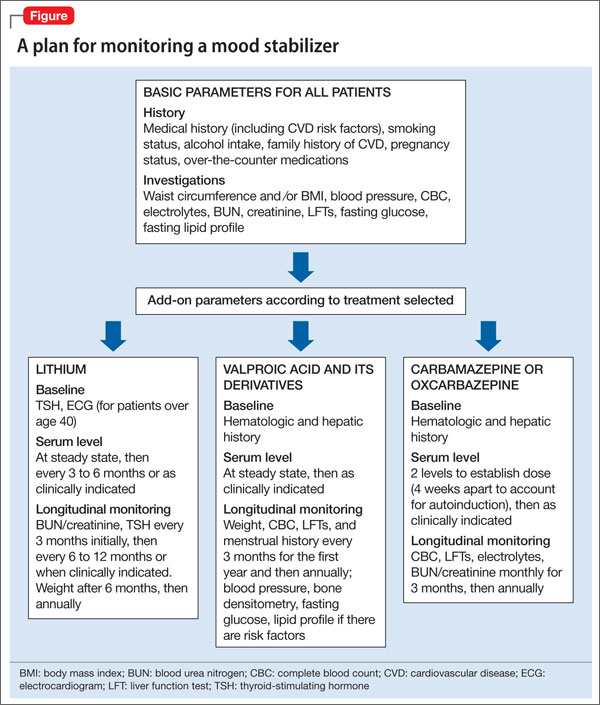
Agent-specific monitoring
Lithium. Patients beginning lithium therapy should undergo thyroid function testing and, for patients age >40, ECG monitoring. Educate patients about potential side effects of lithium, signs and symptoms of lithium toxicity, and the importance of avoiding dehydration. Adding or changing certain medications could elevate the serum lithium level (eg, diuretics, angiotensin-converting enzyme [ACE]-inhibitors, nonsteroidal anti-inflammatory drugs [NSAIDs], COX-2 inhibitors).
Lithium can cause weight gain and adverse effects in several organ systems, including:
• gastrointestinal (GI) (nausea, vomiting, abdominal pain, loss of appetite, diarrhea)
• renal (nephrogenic diabetes insipidus, tubulointerstitial renal disease)
• neurologic (tremors, cognitive dulling, raised intracranial pressure)
• endocrine (thyroid and parathyroid dysfunction)
• cardiac (benign electrocardiographic changes, conduction abnormalities)
• dermatologic (acne, psoriasis, hair loss)
• hematologic (benign leukocytosis).
Lithium has a narrow therapeutic index (0.5 to 1.2 mEq/L), which means that small changes in the serum level can result in therapeutic inefficacy or toxicity. Lithium toxicity can cause irreversible organ damage or death. Serum lithium levels, symptomatic response, emergence and evolution of adverse drug reactions (ADRs), and the recognition of patient risk factors for toxicity can help guide dosing. From a safety monitoring viewpoint, lithium toxicity, renal and endocrine adverse effects, and potential drug interactions are foremost concerns.
Lithium usually is started at a low, divided dosages to minimize side effects, and titrated according to response. Check lithium levels before and after each dose increase. Serum levels reach steady state 5 days after dosage adjustment, but might need to be checked sooner if a rapid increase is necessary, such as when treating acute mania, or if you suspect toxicity.
If the patient has renal insufficiency, it may take longer for the lithium to reach steady state; therefore, delaying a blood level beyond 5 days may be necessary to gauge a true steady state. Also, anytime a medication that interferes with lithium renal elimination, such as diuretics, ACE inhibitors, NSAIDs, COX-2 inhibitors, is added or the dosage is changed, a new lithium level will need to be obtained to reassess the level in 5 days, assuming adequate renal function. In general, renal function and thyroid function should be evaluated once or twice during the first 6 months of lithium treatment.
Subsequently, renal and thyroid function can be checked every 6 months to 1 year in stable patients or when clinically indicated. Check a patient’s weight after 6 months of therapy, then at least annually.2
Valproic acid (VPA) and its derivatives. The most important initial monitoring for VPA therapy includes LFTs and CBC. Before initiating VPA treatment, take a medical history, with special attention to hepatic, hematologic, and bleeding abnormalities. Therapeutic blood monitoring can be conducted once steady state is achieved and as clinically necessary thereafter.
VPA can be administered at an initial starting dosage of 20 to 30 mg/kg/d in inpatients. In outpatients it is given in low, divided doses or as once-daily dosing using an extended-release formulation to minimize GI and neurologic toxicity and titrated every few days. Target serum level is 50 to 125 μg/mL.
Side effects of VPA include GI distress (eg, anorexia, nausea, dyspepsia, vomiting, diarrhea), hematologic effects (reversible leukopenia, thrombocytopenia), hair loss, weight gain, tremor, hepatic effects (benign LFT elevations, hepatotoxicity), osteoporosis, and sedation. Patients with prior or current hepatic disease may be at greater risk for hepatotoxicity. There is an association between VPA and polycystic ovarian syndrome. Rare, idiosyncratic, but potentially fatal adverse events with valproate include irreversible hepatic failure, hemorrhagic pancreatitis, and agranulocytosis.
Older monitoring standards indicated taking LFTs and CBC every 6 months and serum VPA level as clinically indicated. According to ISBD guidelines, weight, CBC, LFTs, and menstrual history should be monitored every 3 months for the first year and then annually; blood pressure, bone status (densitometry), fasting glucose, and fasting lipids should be monitored only in patients with related risk factors. Routine ammonia levels are not recommended but might be indicated if a patient has sudden mental status changes or change in condition.2
Carbamazepine and oxcarbazepine. The most important initial monitoring for carbamazepine therapy includes LFTs, renal function, electrolytes, and CBC. Before treatment, take a medical history, with special emphasis on history of blood dyscrasias or liver disease. After initiating carbamazepine, CBC, LFTs, electrolytes, and renal function should be done monthly for 3 months, then repeated annually.
Carbamazepine is a substrate and an inducer of the cytochrome P450 (CYP) system, so it can reduce levels of many other drugs including other antiepileptics, warfarin, and oral contraceptives. Serum level of carbamazepine can be measured at trough after 5 days, with a target level of 4 to 12 μg/mL. Two levels should be drawn, 4 weeks apart, to establish therapeutic dosage secondary to autoinduction of the CYP450 system.2
As many as one-half of patients experience side effects with carbamazepine. The most common side effects include fatigue, nausea, and neurologic symptoms (diplopia, blurred vision, and ataxia). Less frequent side effects include skin rashes, leukopenia, liver enzyme elevations, thrombocytopenia, hyponatremia, and hypo-osmolality. Rare, potentially fatal side effects include agranulocytosis, aplastic anemia, thrombocytopenia, hepatic failure, and exfoliative dermatitis (eg, Stevens-Johnson syndrome).
Patients of Asian descent who are taking carbamazepine should undergo genetic testing for the HLA-B*1502 enzyme because persons with this allele are at higher risk of developing Stevens-Johnson syndrome. Also, patients should be educated about the signs and symptoms of these rare adverse reactions so that medical treatment is not delayed should these adverse events present.
Lamotrigine does not require further laboratory monitoring beyond the initial recommended workup. The most important variables to consider are interactions with other medications (especially other antiepileptics, such as VPA and carbamazepine) and observing for rash. Titration takes several weeks to minimize risk of developing a rash.2 Similar to carbamazepine, the patient should be educated on the signs and symptoms of exfoliative dermatitis (eg, Stevens-Johnson syndrome) so that medical treatment is sought out should this reaction occur.
Atypical antipsychotics. Baseline workup includes the general monitoring parameters described above. Atypical antipsychotics have a lower incidence of extrapyramidal side effects than typical antipsychotics, but are associated with an increased risk of metabolic complications. Other major ADRs to consider are cardiac effects and hyperprolactinemia; clinicians should therefore inquire about a personal or family history of cardiac problems, including congenital long QT syndrome. Patients should be screened for any medications that can prolong the QTc interval or interact with the metabolism of medications known to cause QTc prolongation.
Measure weight monthly for the first 3 months, then every 3 months to monitor for metabolic side effects during ongoing treatment. Obtain blood pressure and fasting glucose every 3 months for the first year, then annually. Repeat a fasting lipid profile 3 months after initiating treatment, then annually. Cardiac effects and prolactin levels can be monitored as needed if clinically indicated.2
CASE CONTINUED
You discuss with Ms. W choices of a mood stabilizing agent to treat her bipolar II disorder; she agrees to start lithium. Before initiating treatment, you obtain her weight (and calculate her BMI), blood pressure, CBC, electrolyte levels, BUN and creatinine levels, liver function tests, fasting glucose, fasting lipid profile, and thyroid panel. You also review her medical history, lifestyle factors (cigarette smoking status, alcohol intake), and family history. A urine pregnancy screen is negative. The pharmacist assists in screening for potential drug-drug interactions, including over-the-counter medications that Ms. W occasionally takes as needed. She is counseled on the use of NSAIDS because these drugs can increase the lithium level.
Ms. W tolerates and responds well to lithium. No further dosing recommendations are made, based on clinical response. You measure her weight at 6 months, then annually. Renal function and thyroid function are monitored at 3 and 6 months after lithium is initiated, and then annually. One year after starting lithium, she continues to tolerate the medication and has minimal metabolic side effects.
Related Resources
• McInnis MG. Lithium for bipolar disorder: A re-emerging treatment for mood instability. Current Psychiatry. 2014; 13(6):38-44.
• Stahl SM. Stahl’s illustrated mood stabilizers. New York, NY: Cambridge University Press; 2009.
Drug Brand Names
Carbamazepine • Tegretol Valproic acid • Depacon, Depakote
Lamotrigine • Lamictal Warfarin • Coumadin
Lithium • Lithobid, Eskalith
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Maglione M, Ruelaz Maher A, Hu J, et al. Off-label use of atypical antipsychotics: an update. Comparative Effectiveness Review No. 43. Rockville, MD: Agency for Healthcare Research and Quality; 2011. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/778/CER43_Off-LabelAntipsychotics_20110928.pdf. Published September 2011. Accessed June 6, 2014.
2. American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry. 2002;159(suppl 4):1-50.
3. Ng F, Mammen OK, Wilting I, et al; International Society for Bipolar Disorders. The International Society for Bipolar Disorders (ISBD) consensus guidelines for the safety monitoring of bipolar disorder treatments. Bipolar Disord. 2009;11(6):559-595.
4. National Institute for Health and Clinical Excellence. Bipolar disorder (CG38). The management of bipolar disorder in adults, children and adolescents, in primary and secondary care. http://www.nice.org.uk/CG038. Updated February 13, 2014. Accessed June 6, 2014.
5. Yatham LN, Kennedy SH, O’Donovan C, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007. Bipolar Disord. 2006;8(6):721-739.
6. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013; 12(9):51-54.
7. Krishnan KR. Psychiatric and medical comorbidities of bipolar disorder. Psychosom Med. 2005;67(1):1-8.
8. Kilbourne AM, Post EP, Bauer MS, et al. Therapeutic drug and cardiovascular disease risk monitoring in patients with bipolar disorder. J Affect Disord. 2007;102(1-3):145-151.
9. Marcus SC, Olfson M, Pincus HA, et al. Therapeutic drug monitoring of mood stabilizers in Medicaid patients with bipolar disorder. Am J Psychiatry. 1999;156(7):1014-1018.
Ms. W, age 27, presents with a chief concern of “depression.” She describes a history of several hypomanic episodes as well as the current depressive episode, prompting a bipolar II disorder diagnosis. She is naïve to all psychotropics. You plan to initiate a mood-stabilizing agent. What would you include in your initial workup before starting treatment and how would you monitor her as she continues treatment?
Mood stabilizers are employed to treat bipolar spectrum disorders (bipolar I, bipolar II, and cyclothymic disorder) and schizoaffective disorder, bipolar type. Some evidence suggests that mood stabilizers also can be used for treatment-resistant depressive disorders and borderline personality disorder.1 Mood stabilizers include lithium, valproate, carbamazepine, oxcarbazepine, and lamotrigine.2-5
This review focuses on applications and monitoring of mood stabilizers for bipolar I and II disorders. We also will briefly review atypical antipsychotics because they also are used to treat bipolar spectrum disorders (see the September 2013 issue of Current Psychiatry at CurrentPsychiatry.com for a more detailed article on monitoring of antipsychotics).6
There are several well-researched guidelines used to guide clinical practice.2-5 Many guidelines recommend baseline and routine monitoring parameters based on the characteristics of the agent used. However, the International Society for Bipolar Disorders (ISBD) guidelines highlight the importance of monitoring medical comorbidities, which are common among patients with bipolar disorder and can affect pharmacotherapy and clinical outcomes. These recommendations are similar to metabolic monitoring guidelines for antipsychotics.5
Reviews of therapeutic monitoring show that only one-third to one-half of patien
taking a mood stabilizer are appropriately monitored. Poor adherence to guideline recommendations often is observed because of patients’ lack of insight or medication adherence and because psychiatric care generally is segregated from other medical care.7-9
Baseline testing
The ISBD guidelines recommend an initial workup for all patients that includes:
• waist circumference or body mass index (BMI), or both
• blood pressure
• complete blood count (CBC)
• electrolytes
• blood urea nitrogen (BUN) and creatinine
• liver function tests (LFTs)
• fasting glucose
• fasting lipid profile.
In addition, medical history, cigarette smoking status, alcohol intake, and family history of cardiovascular disease, cerebrovascular disease, hypertension, dyslipidemia, and diabetes mellitus should be documented. Rule out pregnancy in women of childbearing potential.2 The Figure describes monitoring parameters based on selected agent.
Agent-specific monitoring
Lithium. Patients beginning lithium therapy should undergo thyroid function testing and, for patients age >40, ECG monitoring. Educate patients about potential side effects of lithium, signs and symptoms of lithium toxicity, and the importance of avoiding dehydration. Adding or changing certain medications could elevate the serum lithium level (eg, diuretics, angiotensin-converting enzyme [ACE]-inhibitors, nonsteroidal anti-inflammatory drugs [NSAIDs], COX-2 inhibitors).
Lithium can cause weight gain and adverse effects in several organ systems, including:
• gastrointestinal (GI) (nausea, vomiting, abdominal pain, loss of appetite, diarrhea)
• renal (nephrogenic diabetes insipidus, tubulointerstitial renal disease)
• neurologic (tremors, cognitive dulling, raised intracranial pressure)
• endocrine (thyroid and parathyroid dysfunction)
• cardiac (benign electrocardiographic changes, conduction abnormalities)
• dermatologic (acne, psoriasis, hair loss)
• hematologic (benign leukocytosis).
Lithium has a narrow therapeutic index (0.5 to 1.2 mEq/L), which means that small changes in the serum level can result in therapeutic inefficacy or toxicity. Lithium toxicity can cause irreversible organ damage or death. Serum lithium levels, symptomatic response, emergence and evolution of adverse drug reactions (ADRs), and the recognition of patient risk factors for toxicity can help guide dosing. From a safety monitoring viewpoint, lithium toxicity, renal and endocrine adverse effects, and potential drug interactions are foremost concerns.
Lithium usually is started at a low, divided dosages to minimize side effects, and titrated according to response. Check lithium levels before and after each dose increase. Serum levels reach steady state 5 days after dosage adjustment, but might need to be checked sooner if a rapid increase is necessary, such as when treating acute mania, or if you suspect toxicity.
If the patient has renal insufficiency, it may take longer for the lithium to reach steady state; therefore, delaying a blood level beyond 5 days may be necessary to gauge a true steady state. Also, anytime a medication that interferes with lithium renal elimination, such as diuretics, ACE inhibitors, NSAIDs, COX-2 inhibitors, is added or the dosage is changed, a new lithium level will need to be obtained to reassess the level in 5 days, assuming adequate renal function. In general, renal function and thyroid function should be evaluated once or twice during the first 6 months of lithium treatment.
Subsequently, renal and thyroid function can be checked every 6 months to 1 year in stable patients or when clinically indicated. Check a patient’s weight after 6 months of therapy, then at least annually.2
Valproic acid (VPA) and its derivatives. The most important initial monitoring for VPA therapy includes LFTs and CBC. Before initiating VPA treatment, take a medical history, with special attention to hepatic, hematologic, and bleeding abnormalities. Therapeutic blood monitoring can be conducted once steady state is achieved and as clinically necessary thereafter.
VPA can be administered at an initial starting dosage of 20 to 30 mg/kg/d in inpatients. In outpatients it is given in low, divided doses or as once-daily dosing using an extended-release formulation to minimize GI and neurologic toxicity and titrated every few days. Target serum level is 50 to 125 μg/mL.
Side effects of VPA include GI distress (eg, anorexia, nausea, dyspepsia, vomiting, diarrhea), hematologic effects (reversible leukopenia, thrombocytopenia), hair loss, weight gain, tremor, hepatic effects (benign LFT elevations, hepatotoxicity), osteoporosis, and sedation. Patients with prior or current hepatic disease may be at greater risk for hepatotoxicity. There is an association between VPA and polycystic ovarian syndrome. Rare, idiosyncratic, but potentially fatal adverse events with valproate include irreversible hepatic failure, hemorrhagic pancreatitis, and agranulocytosis.
Older monitoring standards indicated taking LFTs and CBC every 6 months and serum VPA level as clinically indicated. According to ISBD guidelines, weight, CBC, LFTs, and menstrual history should be monitored every 3 months for the first year and then annually; blood pressure, bone status (densitometry), fasting glucose, and fasting lipids should be monitored only in patients with related risk factors. Routine ammonia levels are not recommended but might be indicated if a patient has sudden mental status changes or change in condition.2
Carbamazepine and oxcarbazepine. The most important initial monitoring for carbamazepine therapy includes LFTs, renal function, electrolytes, and CBC. Before treatment, take a medical history, with special emphasis on history of blood dyscrasias or liver disease. After initiating carbamazepine, CBC, LFTs, electrolytes, and renal function should be done monthly for 3 months, then repeated annually.
Carbamazepine is a substrate and an inducer of the cytochrome P450 (CYP) system, so it can reduce levels of many other drugs including other antiepileptics, warfarin, and oral contraceptives. Serum level of carbamazepine can be measured at trough after 5 days, with a target level of 4 to 12 μg/mL. Two levels should be drawn, 4 weeks apart, to establish therapeutic dosage secondary to autoinduction of the CYP450 system.2
As many as one-half of patients experience side effects with carbamazepine. The most common side effects include fatigue, nausea, and neurologic symptoms (diplopia, blurred vision, and ataxia). Less frequent side effects include skin rashes, leukopenia, liver enzyme elevations, thrombocytopenia, hyponatremia, and hypo-osmolality. Rare, potentially fatal side effects include agranulocytosis, aplastic anemia, thrombocytopenia, hepatic failure, and exfoliative dermatitis (eg, Stevens-Johnson syndrome).
Patients of Asian descent who are taking carbamazepine should undergo genetic testing for the HLA-B*1502 enzyme because persons with this allele are at higher risk of developing Stevens-Johnson syndrome. Also, patients should be educated about the signs and symptoms of these rare adverse reactions so that medical treatment is not delayed should these adverse events present.
Lamotrigine does not require further laboratory monitoring beyond the initial recommended workup. The most important variables to consider are interactions with other medications (especially other antiepileptics, such as VPA and carbamazepine) and observing for rash. Titration takes several weeks to minimize risk of developing a rash.2 Similar to carbamazepine, the patient should be educated on the signs and symptoms of exfoliative dermatitis (eg, Stevens-Johnson syndrome) so that medical treatment is sought out should this reaction occur.
Atypical antipsychotics. Baseline workup includes the general monitoring parameters described above. Atypical antipsychotics have a lower incidence of extrapyramidal side effects than typical antipsychotics, but are associated with an increased risk of metabolic complications. Other major ADRs to consider are cardiac effects and hyperprolactinemia; clinicians should therefore inquire about a personal or family history of cardiac problems, including congenital long QT syndrome. Patients should be screened for any medications that can prolong the QTc interval or interact with the metabolism of medications known to cause QTc prolongation.
Measure weight monthly for the first 3 months, then every 3 months to monitor for metabolic side effects during ongoing treatment. Obtain blood pressure and fasting glucose every 3 months for the first year, then annually. Repeat a fasting lipid profile 3 months after initiating treatment, then annually. Cardiac effects and prolactin levels can be monitored as needed if clinically indicated.2
CASE CONTINUED
You discuss with Ms. W choices of a mood stabilizing agent to treat her bipolar II disorder; she agrees to start lithium. Before initiating treatment, you obtain her weight (and calculate her BMI), blood pressure, CBC, electrolyte levels, BUN and creatinine levels, liver function tests, fasting glucose, fasting lipid profile, and thyroid panel. You also review her medical history, lifestyle factors (cigarette smoking status, alcohol intake), and family history. A urine pregnancy screen is negative. The pharmacist assists in screening for potential drug-drug interactions, including over-the-counter medications that Ms. W occasionally takes as needed. She is counseled on the use of NSAIDS because these drugs can increase the lithium level.
Ms. W tolerates and responds well to lithium. No further dosing recommendations are made, based on clinical response. You measure her weight at 6 months, then annually. Renal function and thyroid function are monitored at 3 and 6 months after lithium is initiated, and then annually. One year after starting lithium, she continues to tolerate the medication and has minimal metabolic side effects.
Related Resources
• McInnis MG. Lithium for bipolar disorder: A re-emerging treatment for mood instability. Current Psychiatry. 2014; 13(6):38-44.
• Stahl SM. Stahl’s illustrated mood stabilizers. New York, NY: Cambridge University Press; 2009.
Drug Brand Names
Carbamazepine • Tegretol Valproic acid • Depacon, Depakote
Lamotrigine • Lamictal Warfarin • Coumadin
Lithium • Lithobid, Eskalith
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. W, age 27, presents with a chief concern of “depression.” She describes a history of several hypomanic episodes as well as the current depressive episode, prompting a bipolar II disorder diagnosis. She is naïve to all psychotropics. You plan to initiate a mood-stabilizing agent. What would you include in your initial workup before starting treatment and how would you monitor her as she continues treatment?
Mood stabilizers are employed to treat bipolar spectrum disorders (bipolar I, bipolar II, and cyclothymic disorder) and schizoaffective disorder, bipolar type. Some evidence suggests that mood stabilizers also can be used for treatment-resistant depressive disorders and borderline personality disorder.1 Mood stabilizers include lithium, valproate, carbamazepine, oxcarbazepine, and lamotrigine.2-5
This review focuses on applications and monitoring of mood stabilizers for bipolar I and II disorders. We also will briefly review atypical antipsychotics because they also are used to treat bipolar spectrum disorders (see the September 2013 issue of Current Psychiatry at CurrentPsychiatry.com for a more detailed article on monitoring of antipsychotics).6
There are several well-researched guidelines used to guide clinical practice.2-5 Many guidelines recommend baseline and routine monitoring parameters based on the characteristics of the agent used. However, the International Society for Bipolar Disorders (ISBD) guidelines highlight the importance of monitoring medical comorbidities, which are common among patients with bipolar disorder and can affect pharmacotherapy and clinical outcomes. These recommendations are similar to metabolic monitoring guidelines for antipsychotics.5
Reviews of therapeutic monitoring show that only one-third to one-half of patien
taking a mood stabilizer are appropriately monitored. Poor adherence to guideline recommendations often is observed because of patients’ lack of insight or medication adherence and because psychiatric care generally is segregated from other medical care.7-9
Baseline testing
The ISBD guidelines recommend an initial workup for all patients that includes:
• waist circumference or body mass index (BMI), or both
• blood pressure
• complete blood count (CBC)
• electrolytes
• blood urea nitrogen (BUN) and creatinine
• liver function tests (LFTs)
• fasting glucose
• fasting lipid profile.
In addition, medical history, cigarette smoking status, alcohol intake, and family history of cardiovascular disease, cerebrovascular disease, hypertension, dyslipidemia, and diabetes mellitus should be documented. Rule out pregnancy in women of childbearing potential.2 The Figure describes monitoring parameters based on selected agent.
Agent-specific monitoring
Lithium. Patients beginning lithium therapy should undergo thyroid function testing and, for patients age >40, ECG monitoring. Educate patients about potential side effects of lithium, signs and symptoms of lithium toxicity, and the importance of avoiding dehydration. Adding or changing certain medications could elevate the serum lithium level (eg, diuretics, angiotensin-converting enzyme [ACE]-inhibitors, nonsteroidal anti-inflammatory drugs [NSAIDs], COX-2 inhibitors).
Lithium can cause weight gain and adverse effects in several organ systems, including:
• gastrointestinal (GI) (nausea, vomiting, abdominal pain, loss of appetite, diarrhea)
• renal (nephrogenic diabetes insipidus, tubulointerstitial renal disease)
• neurologic (tremors, cognitive dulling, raised intracranial pressure)
• endocrine (thyroid and parathyroid dysfunction)
• cardiac (benign electrocardiographic changes, conduction abnormalities)
• dermatologic (acne, psoriasis, hair loss)
• hematologic (benign leukocytosis).
Lithium has a narrow therapeutic index (0.5 to 1.2 mEq/L), which means that small changes in the serum level can result in therapeutic inefficacy or toxicity. Lithium toxicity can cause irreversible organ damage or death. Serum lithium levels, symptomatic response, emergence and evolution of adverse drug reactions (ADRs), and the recognition of patient risk factors for toxicity can help guide dosing. From a safety monitoring viewpoint, lithium toxicity, renal and endocrine adverse effects, and potential drug interactions are foremost concerns.
Lithium usually is started at a low, divided dosages to minimize side effects, and titrated according to response. Check lithium levels before and after each dose increase. Serum levels reach steady state 5 days after dosage adjustment, but might need to be checked sooner if a rapid increase is necessary, such as when treating acute mania, or if you suspect toxicity.
If the patient has renal insufficiency, it may take longer for the lithium to reach steady state; therefore, delaying a blood level beyond 5 days may be necessary to gauge a true steady state. Also, anytime a medication that interferes with lithium renal elimination, such as diuretics, ACE inhibitors, NSAIDs, COX-2 inhibitors, is added or the dosage is changed, a new lithium level will need to be obtained to reassess the level in 5 days, assuming adequate renal function. In general, renal function and thyroid function should be evaluated once or twice during the first 6 months of lithium treatment.
Subsequently, renal and thyroid function can be checked every 6 months to 1 year in stable patients or when clinically indicated. Check a patient’s weight after 6 months of therapy, then at least annually.2
Valproic acid (VPA) and its derivatives. The most important initial monitoring for VPA therapy includes LFTs and CBC. Before initiating VPA treatment, take a medical history, with special attention to hepatic, hematologic, and bleeding abnormalities. Therapeutic blood monitoring can be conducted once steady state is achieved and as clinically necessary thereafter.
VPA can be administered at an initial starting dosage of 20 to 30 mg/kg/d in inpatients. In outpatients it is given in low, divided doses or as once-daily dosing using an extended-release formulation to minimize GI and neurologic toxicity and titrated every few days. Target serum level is 50 to 125 μg/mL.
Side effects of VPA include GI distress (eg, anorexia, nausea, dyspepsia, vomiting, diarrhea), hematologic effects (reversible leukopenia, thrombocytopenia), hair loss, weight gain, tremor, hepatic effects (benign LFT elevations, hepatotoxicity), osteoporosis, and sedation. Patients with prior or current hepatic disease may be at greater risk for hepatotoxicity. There is an association between VPA and polycystic ovarian syndrome. Rare, idiosyncratic, but potentially fatal adverse events with valproate include irreversible hepatic failure, hemorrhagic pancreatitis, and agranulocytosis.
Older monitoring standards indicated taking LFTs and CBC every 6 months and serum VPA level as clinically indicated. According to ISBD guidelines, weight, CBC, LFTs, and menstrual history should be monitored every 3 months for the first year and then annually; blood pressure, bone status (densitometry), fasting glucose, and fasting lipids should be monitored only in patients with related risk factors. Routine ammonia levels are not recommended but might be indicated if a patient has sudden mental status changes or change in condition.2
Carbamazepine and oxcarbazepine. The most important initial monitoring for carbamazepine therapy includes LFTs, renal function, electrolytes, and CBC. Before treatment, take a medical history, with special emphasis on history of blood dyscrasias or liver disease. After initiating carbamazepine, CBC, LFTs, electrolytes, and renal function should be done monthly for 3 months, then repeated annually.
Carbamazepine is a substrate and an inducer of the cytochrome P450 (CYP) system, so it can reduce levels of many other drugs including other antiepileptics, warfarin, and oral contraceptives. Serum level of carbamazepine can be measured at trough after 5 days, with a target level of 4 to 12 μg/mL. Two levels should be drawn, 4 weeks apart, to establish therapeutic dosage secondary to autoinduction of the CYP450 system.2
As many as one-half of patients experience side effects with carbamazepine. The most common side effects include fatigue, nausea, and neurologic symptoms (diplopia, blurred vision, and ataxia). Less frequent side effects include skin rashes, leukopenia, liver enzyme elevations, thrombocytopenia, hyponatremia, and hypo-osmolality. Rare, potentially fatal side effects include agranulocytosis, aplastic anemia, thrombocytopenia, hepatic failure, and exfoliative dermatitis (eg, Stevens-Johnson syndrome).
Patients of Asian descent who are taking carbamazepine should undergo genetic testing for the HLA-B*1502 enzyme because persons with this allele are at higher risk of developing Stevens-Johnson syndrome. Also, patients should be educated about the signs and symptoms of these rare adverse reactions so that medical treatment is not delayed should these adverse events present.
Lamotrigine does not require further laboratory monitoring beyond the initial recommended workup. The most important variables to consider are interactions with other medications (especially other antiepileptics, such as VPA and carbamazepine) and observing for rash. Titration takes several weeks to minimize risk of developing a rash.2 Similar to carbamazepine, the patient should be educated on the signs and symptoms of exfoliative dermatitis (eg, Stevens-Johnson syndrome) so that medical treatment is sought out should this reaction occur.
Atypical antipsychotics. Baseline workup includes the general monitoring parameters described above. Atypical antipsychotics have a lower incidence of extrapyramidal side effects than typical antipsychotics, but are associated with an increased risk of metabolic complications. Other major ADRs to consider are cardiac effects and hyperprolactinemia; clinicians should therefore inquire about a personal or family history of cardiac problems, including congenital long QT syndrome. Patients should be screened for any medications that can prolong the QTc interval or interact with the metabolism of medications known to cause QTc prolongation.
Measure weight monthly for the first 3 months, then every 3 months to monitor for metabolic side effects during ongoing treatment. Obtain blood pressure and fasting glucose every 3 months for the first year, then annually. Repeat a fasting lipid profile 3 months after initiating treatment, then annually. Cardiac effects and prolactin levels can be monitored as needed if clinically indicated.2
CASE CONTINUED
You discuss with Ms. W choices of a mood stabilizing agent to treat her bipolar II disorder; she agrees to start lithium. Before initiating treatment, you obtain her weight (and calculate her BMI), blood pressure, CBC, electrolyte levels, BUN and creatinine levels, liver function tests, fasting glucose, fasting lipid profile, and thyroid panel. You also review her medical history, lifestyle factors (cigarette smoking status, alcohol intake), and family history. A urine pregnancy screen is negative. The pharmacist assists in screening for potential drug-drug interactions, including over-the-counter medications that Ms. W occasionally takes as needed. She is counseled on the use of NSAIDS because these drugs can increase the lithium level.
Ms. W tolerates and responds well to lithium. No further dosing recommendations are made, based on clinical response. You measure her weight at 6 months, then annually. Renal function and thyroid function are monitored at 3 and 6 months after lithium is initiated, and then annually. One year after starting lithium, she continues to tolerate the medication and has minimal metabolic side effects.
Related Resources
• McInnis MG. Lithium for bipolar disorder: A re-emerging treatment for mood instability. Current Psychiatry. 2014; 13(6):38-44.
• Stahl SM. Stahl’s illustrated mood stabilizers. New York, NY: Cambridge University Press; 2009.
Drug Brand Names
Carbamazepine • Tegretol Valproic acid • Depacon, Depakote
Lamotrigine • Lamictal Warfarin • Coumadin
Lithium • Lithobid, Eskalith
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Maglione M, Ruelaz Maher A, Hu J, et al. Off-label use of atypical antipsychotics: an update. Comparative Effectiveness Review No. 43. Rockville, MD: Agency for Healthcare Research and Quality; 2011. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/778/CER43_Off-LabelAntipsychotics_20110928.pdf. Published September 2011. Accessed June 6, 2014.
2. American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry. 2002;159(suppl 4):1-50.
3. Ng F, Mammen OK, Wilting I, et al; International Society for Bipolar Disorders. The International Society for Bipolar Disorders (ISBD) consensus guidelines for the safety monitoring of bipolar disorder treatments. Bipolar Disord. 2009;11(6):559-595.
4. National Institute for Health and Clinical Excellence. Bipolar disorder (CG38). The management of bipolar disorder in adults, children and adolescents, in primary and secondary care. http://www.nice.org.uk/CG038. Updated February 13, 2014. Accessed June 6, 2014.
5. Yatham LN, Kennedy SH, O’Donovan C, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007. Bipolar Disord. 2006;8(6):721-739.
6. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013; 12(9):51-54.
7. Krishnan KR. Psychiatric and medical comorbidities of bipolar disorder. Psychosom Med. 2005;67(1):1-8.
8. Kilbourne AM, Post EP, Bauer MS, et al. Therapeutic drug and cardiovascular disease risk monitoring in patients with bipolar disorder. J Affect Disord. 2007;102(1-3):145-151.
9. Marcus SC, Olfson M, Pincus HA, et al. Therapeutic drug monitoring of mood stabilizers in Medicaid patients with bipolar disorder. Am J Psychiatry. 1999;156(7):1014-1018.
1. Maglione M, Ruelaz Maher A, Hu J, et al. Off-label use of atypical antipsychotics: an update. Comparative Effectiveness Review No. 43. Rockville, MD: Agency for Healthcare Research and Quality; 2011. http://www.effectivehealthcare.ahrq.gov/ehc/products/150/778/CER43_Off-LabelAntipsychotics_20110928.pdf. Published September 2011. Accessed June 6, 2014.
2. American Psychiatric Association. Practice guideline for the treatment of patients with bipolar disorder (revision). Am J Psychiatry. 2002;159(suppl 4):1-50.
3. Ng F, Mammen OK, Wilting I, et al; International Society for Bipolar Disorders. The International Society for Bipolar Disorders (ISBD) consensus guidelines for the safety monitoring of bipolar disorder treatments. Bipolar Disord. 2009;11(6):559-595.
4. National Institute for Health and Clinical Excellence. Bipolar disorder (CG38). The management of bipolar disorder in adults, children and adolescents, in primary and secondary care. http://www.nice.org.uk/CG038. Updated February 13, 2014. Accessed June 6, 2014.
5. Yatham LN, Kennedy SH, O’Donovan C, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) guidelines for the management of patients with bipolar disorder: update 2007. Bipolar Disord. 2006;8(6):721-739.
6. Zeier K, Connell R, Resch W, et al. Recommendations for lab monitoring of atypical antipsychotics. Current Psychiatry. 2013; 12(9):51-54.
7. Krishnan KR. Psychiatric and medical comorbidities of bipolar disorder. Psychosom Med. 2005;67(1):1-8.
8. Kilbourne AM, Post EP, Bauer MS, et al. Therapeutic drug and cardiovascular disease risk monitoring in patients with bipolar disorder. J Affect Disord. 2007;102(1-3):145-151.
9. Marcus SC, Olfson M, Pincus HA, et al. Therapeutic drug monitoring of mood stabilizers in Medicaid patients with bipolar disorder. Am J Psychiatry. 1999;156(7):1014-1018.
What to do when your patient who takes clozapine enters a smoke-free facility
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence

Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4

Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.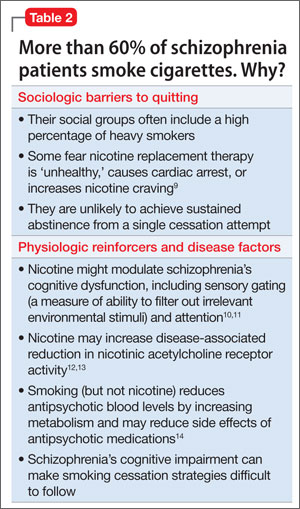
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence
Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4
Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. D, age 30, has a 12-year history of schizophrenia and is experiencing worsening auditory hallucinations despite reported medication adherence. He has been taking clozapine, maintenance dosages 500 to 700 mg/d, for 4 years and smokes 2 packs of cigarettes a day. When Mr. D is admitted to a nonsmoking inpatient psychiatric facility, he receives nicotine transdermal patches, 21 mg/d, for nicotine withdrawal. Mr. D’s most recent outpatient clozapine dosage, 700 mg/d, is resumed. All laboratory tests, including complete blood count with differential, are within normal limits at admission.
Five days later Mr. D is tachycardic with a heart rate of 109 beats per minute. When assessing Mr. D, we notice he has alogia and that, when he does speak, his speech is slowed with a 4 to 5 second delay in response. He also appears sedated. We observe occasional mild jerking of his shoulder and lower legs.
Mr. D reports that his auditory hallucinations have lessened since his admission, but complains of difficulty remembering information and feeling tired during the day. The treatment team suspects clozapine toxicity; his trough clozapine level is 1,350 ng/mL (therapeutic range, 350 to 1,000 ng/mL).
It is well documented that cigarette smoke can induce cytochrome P450 (CYP) isoenzymes, specifically CYP1A1, CYP1A2, and CYP2E1. Because clozapine is primarily metabolized by CYP1A2 (approximately 70%), smoking can induce clozapine metabolism and abruptly stopping smoking can increase clozapine levels.1 The polycyclic aromatic hydrocarbons, not the nicotine, found in cigarettes are thought to be responsible for CYP1A2 induction; therefore, use of a nicotine replacement product did not prevent the increase in Mr. D’s clozapine levels.
Examining the evidence
Meyer1 evaluated clozapine levels before and after implementation of a hospital-wide smoking ban (N = 11). Clozapine dosages were not adjusted at the time of the smoking ban, which resulted in a mean 72% increase in clozapine levels after a minimum of 2 weeks as nonsmokers. Even after eliminating 2 outliers, the mean increase in clozapine levels was 36.1%. Murayama-Sung et al2 reported a statistically significant increase in the level of clozapine (46%, P = .004) and the level of norclozapine (23%, P = .02) after a hospital-wide smoking ban was instituted (N = 14). However, the pre-change and post-change in the ratio of clozapine to norclozapine level was not found to be statistically significant. Haslemo et al3 found that smoking as few as 7 to 12 cigarettes a day was sufficient for maximum induction of CYP1A2. Because Mr. D was smoking 2 packs of cigarettes a day (40 cigarettes) with an clozapine dosage 700 mg/d as an outpatient, he likely experienced significant induction of clozapine metabolism through CYP1A2, which was no longer present when he stopped smoking.
Therapeutic clozapine concentrations are typically above 350 and 420 ng/mL.4 Concentrations >700 ng/mL are associated with increased adverse effects, but generally are not associated with a higher response; levels >900 ng/mL have been associated with toxicity.4 Clozapine-treated patients on a stable dosage who smoke can experience clozapine-related adverse effects after admission to a smoke-free facility secondary to an increase in the clozapine concentration (Table 1).4
Five days after admission to the facility, Mr. D was noted to have myoclonus, somnolence, and tachycardia, with a clozapine level of 1,350 ng/mL. Additional adverse effects that can be seen include orthostatic hypotension, sialorrhea, worsening psychiatric symptoms (eg, hallucinations), and seizures.5 Although there is variability in the timing of the decrease in CYP1A2 activity after smoking cessation, practitioners should begin to monitor for clozapine-related adverse effects 1 or 2 days after smoking cessation.6
Treatment recommendations
Monitoring of the clozapine concentration and adjustment of the dosage might be needed to account for the fluctuation seen with smoking cessation to maintain efficacy and minimize adverse effects. However, a test of the clozapine level may not be available at all facilities, often requiring that the specimen be sent to an outside laboratory, taking 3 to 7 days to receive results.
Faber and Fuhr6 recommended reducing the dosage of a CYP1A2 substrate medication, such as clozapine, olanzapine, or theophylline, by 10% each day until the dosage has been reduced by 40% in patients who stop smoking. Lowe and Ackman5 proposed reducing the clozapine dosage by 30% to 40% to achieve a pre-cessation serum concentration at 1 week. For Mr. D, this would mean decreasing the clozapine dosage to 425 to 500 mg/d.
Assuming that Mr. D’s clozapine dosage is decreased during his hospitalization and that he resumes smoking after discharge, it is likely the dosage will need to be increased. It may take several weeks to see maximal induction, because new CYP enzymes need to be synthesized when the patient resumes smoking.7 One recommendation is to increase the clozapine dosage by a factor of 1.5 over 2 to 4 weeks, with close monitoring of the clozapine concentration and adverse effects because this increase is approximate.7 Depending on when Mr. D’s follow-up appointment is scheduled, the practitioner may need to plan a dosage adjustment to prevent a decrease in his clozapine level caused by smoking to prevent a worsening of symptoms and rehospitalization.
This case emphasizes the importance of asking clozapine-treated patients about their smoking history when they are admitted to a smoke-free facility. For several reasons, >60% of patients with schizophrenia smoke cigarettes8 (Table 2).9-14 Patients who smoke and are on a stable dosage of clozapine might require a dosage reduction when they are admitted to a smoke-free facility to avoid adverse effects. If the dosage is not adjusted, a patient may experience clozapine-induced adverse effects, such as tachycardia, sedation, and seizures. It is likely that patients such as Mr. D will experience fluctuation in the clozapine level and possibly changes in efficacy and tolerability transitioning between inpatient and outpatient settings if the dosage is not adjusted.
Related Resources
• Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
• Fankhauser MP. Drug interactions with tobacco smoke: Implications for patient care. Current Psychiatry. 2013; 12(1):12-16.
• Greenwood-Smith C, Lubman DI, Castle DJ. Serum clozapine levels: a review of their clinical utility. J Psychopharmacol. 2003;17(2):234-248.
• Olesen OV, Thomsen K, Jensen PN, et al. Clozapine serum levels and side effects during steady state treatment of schizophrenic patients: a cross sectional study. Psychopharmacology (Berl). 1995;117(3):371-378.
Drug Brand Names
Clozapine • Clozaril Theophylline • Theo-Dur
Olanzapine • Zyprexa
Disclosure
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
1. Meyer JM. Individual changes in clozapine levels after smoking cessation: results and a predictive model. J Clin Psychopharmacol. 2001;21(6):569-574.
2. Murayama-Sung L, Ahmed I, Goebert D, et al. The impact of hospital smoking ban on clozapine and norclozapine levels. J Clin Psychopharmacol. 2011;31(1):124-126.
3. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Psychopharmacol. 2006;62(12): 1049-1053.
4. Nielsen J, Damkier P, Lublin H, et al. Optimizing clozapine treatment. Acta Psychiatr Scand. 2011;123(6):411-422.
5. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine and olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
6. Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
7. de Leon J. Atypical antipsychotic dosing: the effect of smoking and caffeine. Psychiatr Serv. 2004;55(5):491-493.
8. Dickerson F, Stallings CR, Origoni AE, et al. Cigarette smoking among persons with schizophrenia or bipolar disorder in routine clinical settings, 1999-2011. Psychiatr Serv. 2013;64(1):44-50.
9. Esterberg ML, Compton MT. Smoking behavior in persons with a schizophrenia-spectrum disorder: a qualitative investigation of the transtheoretical model. Soc Sci Med. 2005;61(2):293-303.
10. Barr RS, Culhane MA, Jubelt LE, et al. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008; 33(3):480-490.
11. Adler LE, Hoffer LD, Wiser A, et al. Normalization of auditory physiology by cigarette smoking in schizophrenic patients. Am J Psychiatry. 1993;150(12):1856-1861.
12. Sallette J, Pons S, Devillers-Thiery A, et al. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46(4):595-607.
13. Breese CR, Lee MJ, Adams CE, et al. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23(4):351-364.
14. Miller DD, Kelly MW, Perry PJ, et al. The influence of cigarette smoking on haloperidol pharmacokinetics. J Clin Psychiatry. 1990;28(6):529-231.
Should you use an anticonvulsant to treat impulsivity and aggression?
Mr. V, age 29, is a US Army veteran who presents to the psychiatric emergency department because of increasing aggression. He recently returned from deployment overseas and lives with his parents. Mr. V’s mother reports that he has been increasingly “unstable” and describes an incident during which he punched a hole in his bedroom window after a temporary slow-down in the home’s Internet connection.
The workup and review of the history rules out substance abuse, posttraumatic stress disorder, bipolar disorder, seizure disorder, and personality disorders. He is currently taking only omeprazole, 40 mg/d, for acid reflux. The psychiatrist considers prescribing an antiepileptic medication to treat the agitation. Why this choice of agent?
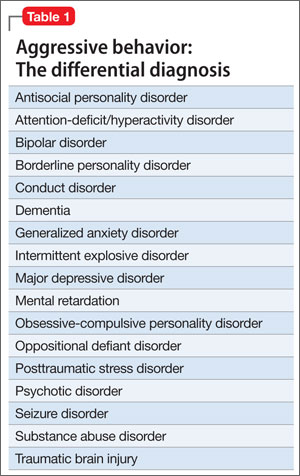
According to DSM-5, patients who have repeated episodes of aggression can be given a diagnosis of intermittent explosive disorder, but such behavior can occur secondary to other psychiatric diagnoses (Table 1). No medications are FDA approved for aggression.1
Aggression and associated verbal and physical acts fall into 2 subtypes: impulsive type and premeditated (predatory) type. Impulsive aggression generally is described as an emotionally charged aggressive response characterized by a loss of behavioral control.
Premeditated aggression

Pharmacotherapy is directed primarily at treating impulsive aggression because this subtype is thought to be caused by neurologic deficits that can affect a person’s ability to process, and react appropriately to, external stimuli. Agitation can result from neuronal hyperactivity.2 Agents such as antiepileptic drugs (AEDs) have the potential to reduce the intensity and frequency of such behaviors.2
In this article, we focus on the use of AEDs for treating impulsive aggression in adults.
Reviewing the evidence for AEDs
The neurobiology of aggression involves multiple neurotransmitters, intracellular pathways, and ion channels.3 AEDs have several mechanisms of action, however; primary mechanisms include action on sodium and calcium channels and modulation of γ-aminobutyric acid (GABA), glutamate, and carbonic anhydrase.2,3 Agent-specific mechanisms of actions are listed in Table 2.

Phenytoin. Several double-blind, placebo-controlled trials have found a statistically significant difference between phenytoin and placebo for treating impulsive aggression, as measured by the Overt Aggression Scale (OAS)a or a modified version (MOAS/ OAS-M).1,2,4 Researchers found that phenytoin, 300 mg/d, but not 100 mg/d, decreased impulsive aggression.4
a Studies generally used the OAS, or one of its modifications, to evaluate aggressive behavior.2,4
Valproate. Trials of valproate for decreasing aggressive behaviors have produced mixed results with regard to primary outcome when used at standard dosages and within the therapeutic range measured by serum concentration.2,3 In a pooled analysis of studies that met stringent criteria (randomized, controlled trial, aggressive behavior as primary outcome, patients free of organic illness or neurologic illness), Jones and colleagues1 reported that valproate/divalproex did not produce statistically significant results compared with placebo for treating impulsive aggression.
Carbamazepine and oxcarbazepine. Double-blind, placebo-controlled trials and case studies of carbamazepine have shown mixed results. In contrast, oxcarbazepine has been found to significantly decrease aggressive behavior, measured by OAS/MOAS/ OAS-M scores.2,3 Total daily dosages of oxcarbazepine ranged from 1,500 to 2,400 mg.2-4 It has been speculated that oxcarbazepine might be a useful option for treating impulsive aggression because of its therapeutic value in temporal lobe seizures—a subtype of seizure disorder that involves the limbic system, which also modulates aggressiveness.5
Additionally, when compared with carbamazepine, oxcarbazepine has a lower risk of cardiotoxicity, neurotoxicity, and blood dyscrasia. Oxcarbazepine has fewer drug-drug interactions because of a lower degree of hepatic enzyme induction.
Topiramate. Several studies have confirmed the efficacy of topiramate for aggressive behavior.2,3 However, there have been reports that topiramate can induce or exacerbate aggression in some patients, an effect that might be dose-related. Aggression might respond better to a higher, short-term dosage (eg, 400 mg/d) than to lower (100 to 300 mg/d) dosages, which might exacerbate aggression.3
Gabapentin. Research on using gabapentin for aggression is limited. Speculation is that the combined activity of gabapentin on GABA and glutamate give the drug its antiaggressive effect.3 No randomized, double-blind, placebo-controlled trials are underway comparing gabapentin and placebo or other active medication for impulsive aggression.
Some case reports and small-scale, open-label studies report a decrease in aggression with gabapentin. As is the case with topiramate, a lower dosage (200 mg to 400 mg) has been reported to result in increased aggression—whereas a higher dosages (800 mg) decreases aggressive behavior.2,3
Lamotrigine. The results of several studies, including double-blind, placebo-controlled trials, support the use of lamotrigine for aggressive behavior. A number of these studies, however, used scales other than OAS (or its modifications) to determine this outcome. One trial showed increased aggression in several patients on lower-dosage lamotrigine (100 mg/d) that resolved when the dosage was increased.2,3
Treatment recommendations
Although all AEDs have some documented efficacy against aggression, choosing the appropriate agent depends on patient-specific variables. Avoiding divalproex in patients with liver dysfunction, for example, or carbamazepine in those with a preexisting cardiac conduction abnormality will improve outcomes by avoiding complications.
It is important to rule out all other causes of aggression before selecting a treatment. The presence of one or more of the diagnoses listed in Table 1 could lead to selection of an alternate class of medication. Nondrug therapies, such as cognitive-behavioral therapy, also should be considered.
Related Resources
• Coccaro EF. Aggression. Psychiatric assessment and treatment. Chicago, IL: Marcel Dekker, Inc.; 2003.
• Citrome LL. Aggression. http://emedicine.medscape.com/article/288689-overview. Updated June 18, 2012. Accessed February 28, 2014.
Drug Brand Names
Carbamazepine • Tegretol Phenytoin • Dilantin
Gabapentin • Neurontin Topiramate • Topamax
Lamotrigine • Lamictal Valproate/Divalproex
Omeprazole • Prilosec • Depakote
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jones RM, Arlidge J, Gilham R, et al. Efficacy of mood stabilizers in the treatment of impulsive or repetitive aggression: systemic review and meta-analysis. Br J Psychiatry. 2011;198(2):93-98.
2. Stanford MS, Anderson NE, Lake SL, et al. Pharmacologic treatment of impulsive aggression with antiepileptic drugs. Curr Treat Options Neurol. 2009;11(5):383-390.
3. Comai S, Tau M, Pavlovic Z, et al. The psychopharmacology of aggressive behavior: a translational approach: part 2: clinical studies using atypical antipsychotics, anticonvulsants, and lithium. J Clin Psychopharmacol. 2012;32(2):237-260.
4. Huband N, Ferriter M, Nathan R, et al. Antiepileptics for aggression and associated impulsivity. Cochrane Database Sys Rev. 2010;2:CD003499.
5. Mattes JA. Medications for aggressiveness in prison: focus on oxcarbazepine. J Am Acad Psychiatry Law. 2012;40(2):234-238.
Mr. V, age 29, is a US Army veteran who presents to the psychiatric emergency department because of increasing aggression. He recently returned from deployment overseas and lives with his parents. Mr. V’s mother reports that he has been increasingly “unstable” and describes an incident during which he punched a hole in his bedroom window after a temporary slow-down in the home’s Internet connection.
The workup and review of the history rules out substance abuse, posttraumatic stress disorder, bipolar disorder, seizure disorder, and personality disorders. He is currently taking only omeprazole, 40 mg/d, for acid reflux. The psychiatrist considers prescribing an antiepileptic medication to treat the agitation. Why this choice of agent?
According to DSM-5, patients who have repeated episodes of aggression can be given a diagnosis of intermittent explosive disorder, but such behavior can occur secondary to other psychiatric diagnoses (Table 1). No medications are FDA approved for aggression.1
Aggression and associated verbal and physical acts fall into 2 subtypes: impulsive type and premeditated (predatory) type. Impulsive aggression generally is described as an emotionally charged aggressive response characterized by a loss of behavioral control.
Premeditated aggression
Pharmacotherapy is directed primarily at treating impulsive aggression because this subtype is thought to be caused by neurologic deficits that can affect a person’s ability to process, and react appropriately to, external stimuli. Agitation can result from neuronal hyperactivity.2 Agents such as antiepileptic drugs (AEDs) have the potential to reduce the intensity and frequency of such behaviors.2
In this article, we focus on the use of AEDs for treating impulsive aggression in adults.
Reviewing the evidence for AEDs
The neurobiology of aggression involves multiple neurotransmitters, intracellular pathways, and ion channels.3 AEDs have several mechanisms of action, however; primary mechanisms include action on sodium and calcium channels and modulation of γ-aminobutyric acid (GABA), glutamate, and carbonic anhydrase.2,3 Agent-specific mechanisms of actions are listed in Table 2.
Phenytoin. Several double-blind, placebo-controlled trials have found a statistically significant difference between phenytoin and placebo for treating impulsive aggression, as measured by the Overt Aggression Scale (OAS)a or a modified version (MOAS/ OAS-M).1,2,4 Researchers found that phenytoin, 300 mg/d, but not 100 mg/d, decreased impulsive aggression.4
a Studies generally used the OAS, or one of its modifications, to evaluate aggressive behavior.2,4
Valproate. Trials of valproate for decreasing aggressive behaviors have produced mixed results with regard to primary outcome when used at standard dosages and within the therapeutic range measured by serum concentration.2,3 In a pooled analysis of studies that met stringent criteria (randomized, controlled trial, aggressive behavior as primary outcome, patients free of organic illness or neurologic illness), Jones and colleagues1 reported that valproate/divalproex did not produce statistically significant results compared with placebo for treating impulsive aggression.
Carbamazepine and oxcarbazepine. Double-blind, placebo-controlled trials and case studies of carbamazepine have shown mixed results. In contrast, oxcarbazepine has been found to significantly decrease aggressive behavior, measured by OAS/MOAS/ OAS-M scores.2,3 Total daily dosages of oxcarbazepine ranged from 1,500 to 2,400 mg.2-4 It has been speculated that oxcarbazepine might be a useful option for treating impulsive aggression because of its therapeutic value in temporal lobe seizures—a subtype of seizure disorder that involves the limbic system, which also modulates aggressiveness.5
Additionally, when compared with carbamazepine, oxcarbazepine has a lower risk of cardiotoxicity, neurotoxicity, and blood dyscrasia. Oxcarbazepine has fewer drug-drug interactions because of a lower degree of hepatic enzyme induction.
Topiramate. Several studies have confirmed the efficacy of topiramate for aggressive behavior.2,3 However, there have been reports that topiramate can induce or exacerbate aggression in some patients, an effect that might be dose-related. Aggression might respond better to a higher, short-term dosage (eg, 400 mg/d) than to lower (100 to 300 mg/d) dosages, which might exacerbate aggression.3
Gabapentin. Research on using gabapentin for aggression is limited. Speculation is that the combined activity of gabapentin on GABA and glutamate give the drug its antiaggressive effect.3 No randomized, double-blind, placebo-controlled trials are underway comparing gabapentin and placebo or other active medication for impulsive aggression.
Some case reports and small-scale, open-label studies report a decrease in aggression with gabapentin. As is the case with topiramate, a lower dosage (200 mg to 400 mg) has been reported to result in increased aggression—whereas a higher dosages (800 mg) decreases aggressive behavior.2,3
Lamotrigine. The results of several studies, including double-blind, placebo-controlled trials, support the use of lamotrigine for aggressive behavior. A number of these studies, however, used scales other than OAS (or its modifications) to determine this outcome. One trial showed increased aggression in several patients on lower-dosage lamotrigine (100 mg/d) that resolved when the dosage was increased.2,3
Treatment recommendations
Although all AEDs have some documented efficacy against aggression, choosing the appropriate agent depends on patient-specific variables. Avoiding divalproex in patients with liver dysfunction, for example, or carbamazepine in those with a preexisting cardiac conduction abnormality will improve outcomes by avoiding complications.
It is important to rule out all other causes of aggression before selecting a treatment. The presence of one or more of the diagnoses listed in Table 1 could lead to selection of an alternate class of medication. Nondrug therapies, such as cognitive-behavioral therapy, also should be considered.
Related Resources
• Coccaro EF. Aggression. Psychiatric assessment and treatment. Chicago, IL: Marcel Dekker, Inc.; 2003.
• Citrome LL. Aggression. http://emedicine.medscape.com/article/288689-overview. Updated June 18, 2012. Accessed February 28, 2014.
Drug Brand Names
Carbamazepine • Tegretol Phenytoin • Dilantin
Gabapentin • Neurontin Topiramate • Topamax
Lamotrigine • Lamictal Valproate/Divalproex
Omeprazole • Prilosec • Depakote
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. V, age 29, is a US Army veteran who presents to the psychiatric emergency department because of increasing aggression. He recently returned from deployment overseas and lives with his parents. Mr. V’s mother reports that he has been increasingly “unstable” and describes an incident during which he punched a hole in his bedroom window after a temporary slow-down in the home’s Internet connection.
The workup and review of the history rules out substance abuse, posttraumatic stress disorder, bipolar disorder, seizure disorder, and personality disorders. He is currently taking only omeprazole, 40 mg/d, for acid reflux. The psychiatrist considers prescribing an antiepileptic medication to treat the agitation. Why this choice of agent?
According to DSM-5, patients who have repeated episodes of aggression can be given a diagnosis of intermittent explosive disorder, but such behavior can occur secondary to other psychiatric diagnoses (Table 1). No medications are FDA approved for aggression.1
Aggression and associated verbal and physical acts fall into 2 subtypes: impulsive type and premeditated (predatory) type. Impulsive aggression generally is described as an emotionally charged aggressive response characterized by a loss of behavioral control.
Premeditated aggression
Pharmacotherapy is directed primarily at treating impulsive aggression because this subtype is thought to be caused by neurologic deficits that can affect a person’s ability to process, and react appropriately to, external stimuli. Agitation can result from neuronal hyperactivity.2 Agents such as antiepileptic drugs (AEDs) have the potential to reduce the intensity and frequency of such behaviors.2
In this article, we focus on the use of AEDs for treating impulsive aggression in adults.
Reviewing the evidence for AEDs
The neurobiology of aggression involves multiple neurotransmitters, intracellular pathways, and ion channels.3 AEDs have several mechanisms of action, however; primary mechanisms include action on sodium and calcium channels and modulation of γ-aminobutyric acid (GABA), glutamate, and carbonic anhydrase.2,3 Agent-specific mechanisms of actions are listed in Table 2.
Phenytoin. Several double-blind, placebo-controlled trials have found a statistically significant difference between phenytoin and placebo for treating impulsive aggression, as measured by the Overt Aggression Scale (OAS)a or a modified version (MOAS/ OAS-M).1,2,4 Researchers found that phenytoin, 300 mg/d, but not 100 mg/d, decreased impulsive aggression.4
a Studies generally used the OAS, or one of its modifications, to evaluate aggressive behavior.2,4
Valproate. Trials of valproate for decreasing aggressive behaviors have produced mixed results with regard to primary outcome when used at standard dosages and within the therapeutic range measured by serum concentration.2,3 In a pooled analysis of studies that met stringent criteria (randomized, controlled trial, aggressive behavior as primary outcome, patients free of organic illness or neurologic illness), Jones and colleagues1 reported that valproate/divalproex did not produce statistically significant results compared with placebo for treating impulsive aggression.
Carbamazepine and oxcarbazepine. Double-blind, placebo-controlled trials and case studies of carbamazepine have shown mixed results. In contrast, oxcarbazepine has been found to significantly decrease aggressive behavior, measured by OAS/MOAS/ OAS-M scores.2,3 Total daily dosages of oxcarbazepine ranged from 1,500 to 2,400 mg.2-4 It has been speculated that oxcarbazepine might be a useful option for treating impulsive aggression because of its therapeutic value in temporal lobe seizures—a subtype of seizure disorder that involves the limbic system, which also modulates aggressiveness.5
Additionally, when compared with carbamazepine, oxcarbazepine has a lower risk of cardiotoxicity, neurotoxicity, and blood dyscrasia. Oxcarbazepine has fewer drug-drug interactions because of a lower degree of hepatic enzyme induction.
Topiramate. Several studies have confirmed the efficacy of topiramate for aggressive behavior.2,3 However, there have been reports that topiramate can induce or exacerbate aggression in some patients, an effect that might be dose-related. Aggression might respond better to a higher, short-term dosage (eg, 400 mg/d) than to lower (100 to 300 mg/d) dosages, which might exacerbate aggression.3
Gabapentin. Research on using gabapentin for aggression is limited. Speculation is that the combined activity of gabapentin on GABA and glutamate give the drug its antiaggressive effect.3 No randomized, double-blind, placebo-controlled trials are underway comparing gabapentin and placebo or other active medication for impulsive aggression.
Some case reports and small-scale, open-label studies report a decrease in aggression with gabapentin. As is the case with topiramate, a lower dosage (200 mg to 400 mg) has been reported to result in increased aggression—whereas a higher dosages (800 mg) decreases aggressive behavior.2,3
Lamotrigine. The results of several studies, including double-blind, placebo-controlled trials, support the use of lamotrigine for aggressive behavior. A number of these studies, however, used scales other than OAS (or its modifications) to determine this outcome. One trial showed increased aggression in several patients on lower-dosage lamotrigine (100 mg/d) that resolved when the dosage was increased.2,3
Treatment recommendations
Although all AEDs have some documented efficacy against aggression, choosing the appropriate agent depends on patient-specific variables. Avoiding divalproex in patients with liver dysfunction, for example, or carbamazepine in those with a preexisting cardiac conduction abnormality will improve outcomes by avoiding complications.
It is important to rule out all other causes of aggression before selecting a treatment. The presence of one or more of the diagnoses listed in Table 1 could lead to selection of an alternate class of medication. Nondrug therapies, such as cognitive-behavioral therapy, also should be considered.
Related Resources
• Coccaro EF. Aggression. Psychiatric assessment and treatment. Chicago, IL: Marcel Dekker, Inc.; 2003.
• Citrome LL. Aggression. http://emedicine.medscape.com/article/288689-overview. Updated June 18, 2012. Accessed February 28, 2014.
Drug Brand Names
Carbamazepine • Tegretol Phenytoin • Dilantin
Gabapentin • Neurontin Topiramate • Topamax
Lamotrigine • Lamictal Valproate/Divalproex
Omeprazole • Prilosec • Depakote
Oxcarbazepine • Trileptal
Disclosure
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Jones RM, Arlidge J, Gilham R, et al. Efficacy of mood stabilizers in the treatment of impulsive or repetitive aggression: systemic review and meta-analysis. Br J Psychiatry. 2011;198(2):93-98.
2. Stanford MS, Anderson NE, Lake SL, et al. Pharmacologic treatment of impulsive aggression with antiepileptic drugs. Curr Treat Options Neurol. 2009;11(5):383-390.
3. Comai S, Tau M, Pavlovic Z, et al. The psychopharmacology of aggressive behavior: a translational approach: part 2: clinical studies using atypical antipsychotics, anticonvulsants, and lithium. J Clin Psychopharmacol. 2012;32(2):237-260.
4. Huband N, Ferriter M, Nathan R, et al. Antiepileptics for aggression and associated impulsivity. Cochrane Database Sys Rev. 2010;2:CD003499.
5. Mattes JA. Medications for aggressiveness in prison: focus on oxcarbazepine. J Am Acad Psychiatry Law. 2012;40(2):234-238.
1. Jones RM, Arlidge J, Gilham R, et al. Efficacy of mood stabilizers in the treatment of impulsive or repetitive aggression: systemic review and meta-analysis. Br J Psychiatry. 2011;198(2):93-98.
2. Stanford MS, Anderson NE, Lake SL, et al. Pharmacologic treatment of impulsive aggression with antiepileptic drugs. Curr Treat Options Neurol. 2009;11(5):383-390.
3. Comai S, Tau M, Pavlovic Z, et al. The psychopharmacology of aggressive behavior: a translational approach: part 2: clinical studies using atypical antipsychotics, anticonvulsants, and lithium. J Clin Psychopharmacol. 2012;32(2):237-260.
4. Huband N, Ferriter M, Nathan R, et al. Antiepileptics for aggression and associated impulsivity. Cochrane Database Sys Rev. 2010;2:CD003499.
5. Mattes JA. Medications for aggressiveness in prison: focus on oxcarbazepine. J Am Acad Psychiatry Law. 2012;40(2):234-238.
What is the relevance of a 2-week response to an antipsychotic?
Mr. M, age 28, was given a diagnosis of schizophrenia 6 years ago after experiencing a psychotic break involving auditory hallucinations and paranoia. Olanzapine, 10 mg/d, relieved his symptoms, but he stopped taking the drug after gaining 40 pounds and developing diabetes mellitus. He had 2 other hospital admissions for acute psychosis and has taken at least 1 other medication, the name of which he can’t recall. Recently, Mr. M was involuntarily admitted to the psychiatric ward of his local hospital. His psychiatrist started aripiprazole, 10 mg/d, which was titrated to 30 mg/d. After 2 weeks he reports only a slight decrease in hallucinations. His mother is growing concerned about the effectiveness of this medication and wants to know if it’s time to consider another drug.
Time to onset of action of antipsychotic agents has been debated since at least 1970.1 Supporters of the delayed-onset hypothesis assert that antipsychotics take weeks or months to show significant improvement of symptoms because of the need for depolarization block for efficacy.2 Trials of 4 to 6 weeks often are recommended for patients before failure is declared,3,4 and trials of this length or longer have proved useful for first-episode patients.5-7 Recent studies suggest, however, that response is cumulative for chronically ill triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic âpatients with most improvement occurring during weeks 1 and 2.1,8
Two meta-analyses found the greatest rate of cumulative improvement in symptoms during the first 2 weeks.1,8 These analyses included chronically ill patients with mean duration of illness of 15.5 and 10.4 years, respectively. Patients reported 21.9% and 20.5% reductions in symptoms from baseline at 2 weeks, with total responses between 30% at 4 weeks and 40% at 1 year, respectively. These meta-analyses indicate that most of the benefit from antipsychotics in this patient population occurs in the first 2 weeks, which supports the early-response hypothesis.
These observations led to questions about the predictive value of early response and minimum time to determine treatment failure. This article discusses the significance of early response and non-response to antipsychotics and their impact on treating patients with schizophrenia.
What are the predictive factors? How can they guide treatment?
Of the 8 studies in our literature review, only 2 reported early response rates >50%.9,10 (see this article at CurrentPsychiatry.com for a Box describing the literature review.) Positive predictive value (PPV) ranged from 0.51 to 0.81, meaning that 51% to 81% of early responders continued to respond. Six of the 8 studies reported PPV of 50% to 70%. 9,11-15 This appears to be true for chronic and first-episode patients, suggesting that 30% to 50% of early responders will fail to have a sustained response (Table 1,9-16Table 2,9-16 and Figure).

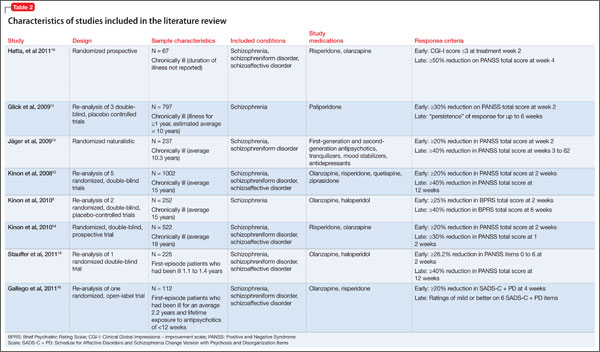
Compared with early response, early non-response is a more consistent predictor of final non-response. In every study of chronically ill patients, negative predictive value (NPV) was greater than PPV (Table 1).9-16 NPVs in the literature suggest that 58% to 91% of early non-responders will continue to be non-responders. This seems to be true of chronically ill patients for whom NPVs consistently were between 75% and 85%. By comparison, in first-episode patients NPVs of 58% and 66% were calculated (Table 19-16 and Figure).14,15
These observations suggest that reassessing drug therapy is indicated early in treatment for early non-responders, particularly in chronically ill patients. However, early non-response in a first-episode patient is not as strong a predictor of eventual treatment failure, supporting the idea that first-episode patients may experience a delayed response to therapy. Researchers studying onset of antipsychotic effect report that median time to response onset in first-episode patients may be ≥8 weeks.6,8 In patients who do not achieve modest early response, assess dose, adherence, substance abuse, and psychosocial stressors.3 For patients without dose, adherence, substance use, or stress issues, switching drug therapy in chronically ill early non-responders is reasonable because the probability of a late response is small.
Individual patient characteristics determine how much these data aid clinical decision-making. If a patient has a good response to an antipsychotic in the first 2 weeks, continue the drug, but observe the patient closely because response may not be sustained. In first-episode patients who fail to respond within 2 weeks of starting an antipsychotic, it is reasonable to continue the drug for several weeks because these patients may be more likely to respond later in therapy.
Clinicians treating chronically ill patients who have failed several antipsychotics and demonstrate a poor response after 2 weeks of an appropriate antipsychotic dose are justified in changing medications because later significant response is unlikely. If a patient has a poor early response but has failed several other antipsychotics with few remaining alternatives, it is reasonable to continue the maximum tolerated dose of the current therapy because the patient may be a late responder. However, early non-response predicts future non-response in many patients.
Case continued
In the case described here, Mr. M is failing his current treatment regimen with a reasonable antipsychotic dose after 2 weeks. Because Mr. M has been on 2 antipsychotics and demonstrated a good response to olanzapine, changing medications should be considered.
Related Resource
- Correll CU, Hauser M. The year in psychosis and bipolar disorder: predicting response to schizophrenia treatment. www.medscape.com/viewarticle/735211_7.
Drug Brand Names
Aripiprazole • Abilify Quetiapine • Seroquel
Haloperidol • Haldol Risperidone • Risperdal
Olanzapine • Zyprexa Ziprasidone • Geodon
Paliperidone • Invega
Disclosures
Dr. Straley owns stock in Johnson & Johnson. Dr. Webster reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Agid O, Kapur S, Arenovich T, et al. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry. 2003;60(12):1228-1235.
2. Grace AA, Bunney BS, Moore H, et al. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20(1):31-37.
3. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
4. Meltzer HY, Bobo WV, Heckers SH, et al. Chapter 16. Schizophrenia. In: Ebert MH, Loosen PT, Nurcombe B, Leckman JF, eds. CURRENT Diagnosis & Treatment: Psychiatry. 2nd ed. New York: McGraw-Hill; 2008. http://www.accessmedicine.com/content.aspx?aID=3284037. Accessed December 5, 2013.
5. Robinson DG, Woerner MG, Alvir JM, et al. Predictors of treatment response from a first episode of schizophrenia or schizoaffective disorder. Am J Psychiatry. 1999;156(4):544-549.
6. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
7. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry. 2005;57(12):1543-1549.
9. Kinon BJ, Chen L, Stauffer VL, et al. Early onset of antipsychotic action in schizophrenia: evaluating the possibility of shorter acute efficacy trials. J Clin Psychopharmacol. 2010;30(3):286-289.
10. Hatta K, Otachi T, Sudo Y, et al. Difference in early prediction of antipsychotic non-response between risperidone and olanzapine in the treatment of acute-phase schizophrenia. Schizophr Res. 2011;128(1-3):127-135.
11. Glick ID, Bossie CA, Alphs L, et al. Onset and persistence of antipsychotic response in patients with schizophrenia. J Clin Psychopharmacol. 2009;29(6):542-547.
12. Kinon BJ, Chen L, Ascher-Svanum H, et al. Predicting response to atypical antipsychotics based on early response in the treatment of schizophrenia. Schizophr Res. 2008;102(1-3):230-240.
13. Jäger M, Schmauss M, Laux G, et al. Early improvement as a predictor of remission and response in schizophrenia: results from a naturalistic study. Eur Psychiatry. 2009;24(8):501-506.
14. Kinon BJ, Chen L, Ascher-Svanum H, et al. Early response to antipsychotic drug therapy as a clinical marker of subsequent response in the treatment of schizophrenia. Neuropsychopharmacology. 2010;35(2):581-590.
15. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
16. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
Mr. M, age 28, was given a diagnosis of schizophrenia 6 years ago after experiencing a psychotic break involving auditory hallucinations and paranoia. Olanzapine, 10 mg/d, relieved his symptoms, but he stopped taking the drug after gaining 40 pounds and developing diabetes mellitus. He had 2 other hospital admissions for acute psychosis and has taken at least 1 other medication, the name of which he can’t recall. Recently, Mr. M was involuntarily admitted to the psychiatric ward of his local hospital. His psychiatrist started aripiprazole, 10 mg/d, which was titrated to 30 mg/d. After 2 weeks he reports only a slight decrease in hallucinations. His mother is growing concerned about the effectiveness of this medication and wants to know if it’s time to consider another drug.
Time to onset of action of antipsychotic agents has been debated since at least 1970.1 Supporters of the delayed-onset hypothesis assert that antipsychotics take weeks or months to show significant improvement of symptoms because of the need for depolarization block for efficacy.2 Trials of 4 to 6 weeks often are recommended for patients before failure is declared,3,4 and trials of this length or longer have proved useful for first-episode patients.5-7 Recent studies suggest, however, that response is cumulative for chronically ill triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic âpatients with most improvement occurring during weeks 1 and 2.1,8
Two meta-analyses found the greatest rate of cumulative improvement in symptoms during the first 2 weeks.1,8 These analyses included chronically ill patients with mean duration of illness of 15.5 and 10.4 years, respectively. Patients reported 21.9% and 20.5% reductions in symptoms from baseline at 2 weeks, with total responses between 30% at 4 weeks and 40% at 1 year, respectively. These meta-analyses indicate that most of the benefit from antipsychotics in this patient population occurs in the first 2 weeks, which supports the early-response hypothesis.
These observations led to questions about the predictive value of early response and minimum time to determine treatment failure. This article discusses the significance of early response and non-response to antipsychotics and their impact on treating patients with schizophrenia.
What are the predictive factors? How can they guide treatment?
Of the 8 studies in our literature review, only 2 reported early response rates >50%.9,10 (see this article at CurrentPsychiatry.com for a Box describing the literature review.) Positive predictive value (PPV) ranged from 0.51 to 0.81, meaning that 51% to 81% of early responders continued to respond. Six of the 8 studies reported PPV of 50% to 70%. 9,11-15 This appears to be true for chronic and first-episode patients, suggesting that 30% to 50% of early responders will fail to have a sustained response (Table 1,9-16Table 2,9-16 and Figure).
Compared with early response, early non-response is a more consistent predictor of final non-response. In every study of chronically ill patients, negative predictive value (NPV) was greater than PPV (Table 1).9-16 NPVs in the literature suggest that 58% to 91% of early non-responders will continue to be non-responders. This seems to be true of chronically ill patients for whom NPVs consistently were between 75% and 85%. By comparison, in first-episode patients NPVs of 58% and 66% were calculated (Table 19-16 and Figure).14,15
These observations suggest that reassessing drug therapy is indicated early in treatment for early non-responders, particularly in chronically ill patients. However, early non-response in a first-episode patient is not as strong a predictor of eventual treatment failure, supporting the idea that first-episode patients may experience a delayed response to therapy. Researchers studying onset of antipsychotic effect report that median time to response onset in first-episode patients may be ≥8 weeks.6,8 In patients who do not achieve modest early response, assess dose, adherence, substance abuse, and psychosocial stressors.3 For patients without dose, adherence, substance use, or stress issues, switching drug therapy in chronically ill early non-responders is reasonable because the probability of a late response is small.
Individual patient characteristics determine how much these data aid clinical decision-making. If a patient has a good response to an antipsychotic in the first 2 weeks, continue the drug, but observe the patient closely because response may not be sustained. In first-episode patients who fail to respond within 2 weeks of starting an antipsychotic, it is reasonable to continue the drug for several weeks because these patients may be more likely to respond later in therapy.
Clinicians treating chronically ill patients who have failed several antipsychotics and demonstrate a poor response after 2 weeks of an appropriate antipsychotic dose are justified in changing medications because later significant response is unlikely. If a patient has a poor early response but has failed several other antipsychotics with few remaining alternatives, it is reasonable to continue the maximum tolerated dose of the current therapy because the patient may be a late responder. However, early non-response predicts future non-response in many patients.
Case continued
In the case described here, Mr. M is failing his current treatment regimen with a reasonable antipsychotic dose after 2 weeks. Because Mr. M has been on 2 antipsychotics and demonstrated a good response to olanzapine, changing medications should be considered.
Related Resource
- Correll CU, Hauser M. The year in psychosis and bipolar disorder: predicting response to schizophrenia treatment. www.medscape.com/viewarticle/735211_7.
Drug Brand Names
Aripiprazole • Abilify Quetiapine • Seroquel
Haloperidol • Haldol Risperidone • Risperdal
Olanzapine • Zyprexa Ziprasidone • Geodon
Paliperidone • Invega
Disclosures
Dr. Straley owns stock in Johnson & Johnson. Dr. Webster reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Mr. M, age 28, was given a diagnosis of schizophrenia 6 years ago after experiencing a psychotic break involving auditory hallucinations and paranoia. Olanzapine, 10 mg/d, relieved his symptoms, but he stopped taking the drug after gaining 40 pounds and developing diabetes mellitus. He had 2 other hospital admissions for acute psychosis and has taken at least 1 other medication, the name of which he can’t recall. Recently, Mr. M was involuntarily admitted to the psychiatric ward of his local hospital. His psychiatrist started aripiprazole, 10 mg/d, which was titrated to 30 mg/d. After 2 weeks he reports only a slight decrease in hallucinations. His mother is growing concerned about the effectiveness of this medication and wants to know if it’s time to consider another drug.
Time to onset of action of antipsychotic agents has been debated since at least 1970.1 Supporters of the delayed-onset hypothesis assert that antipsychotics take weeks or months to show significant improvement of symptoms because of the need for depolarization block for efficacy.2 Trials of 4 to 6 weeks often are recommended for patients before failure is declared,3,4 and trials of this length or longer have proved useful for first-episode patients.5-7 Recent studies suggest, however, that response is cumulative for chronically ill triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic âpatients with most improvement occurring during weeks 1 and 2.1,8
Two meta-analyses found the greatest rate of cumulative improvement in symptoms during the first 2 weeks.1,8 These analyses included chronically ill patients with mean duration of illness of 15.5 and 10.4 years, respectively. Patients reported 21.9% and 20.5% reductions in symptoms from baseline at 2 weeks, with total responses between 30% at 4 weeks and 40% at 1 year, respectively. These meta-analyses indicate that most of the benefit from antipsychotics in this patient population occurs in the first 2 weeks, which supports the early-response hypothesis.
These observations led to questions about the predictive value of early response and minimum time to determine treatment failure. This article discusses the significance of early response and non-response to antipsychotics and their impact on treating patients with schizophrenia.
What are the predictive factors? How can they guide treatment?
Of the 8 studies in our literature review, only 2 reported early response rates >50%.9,10 (see this article at CurrentPsychiatry.com for a Box describing the literature review.) Positive predictive value (PPV) ranged from 0.51 to 0.81, meaning that 51% to 81% of early responders continued to respond. Six of the 8 studies reported PPV of 50% to 70%. 9,11-15 This appears to be true for chronic and first-episode patients, suggesting that 30% to 50% of early responders will fail to have a sustained response (Table 1,9-16Table 2,9-16 and Figure).
Compared with early response, early non-response is a more consistent predictor of final non-response. In every study of chronically ill patients, negative predictive value (NPV) was greater than PPV (Table 1).9-16 NPVs in the literature suggest that 58% to 91% of early non-responders will continue to be non-responders. This seems to be true of chronically ill patients for whom NPVs consistently were between 75% and 85%. By comparison, in first-episode patients NPVs of 58% and 66% were calculated (Table 19-16 and Figure).14,15
These observations suggest that reassessing drug therapy is indicated early in treatment for early non-responders, particularly in chronically ill patients. However, early non-response in a first-episode patient is not as strong a predictor of eventual treatment failure, supporting the idea that first-episode patients may experience a delayed response to therapy. Researchers studying onset of antipsychotic effect report that median time to response onset in first-episode patients may be ≥8 weeks.6,8 In patients who do not achieve modest early response, assess dose, adherence, substance abuse, and psychosocial stressors.3 For patients without dose, adherence, substance use, or stress issues, switching drug therapy in chronically ill early non-responders is reasonable because the probability of a late response is small.
Individual patient characteristics determine how much these data aid clinical decision-making. If a patient has a good response to an antipsychotic in the first 2 weeks, continue the drug, but observe the patient closely because response may not be sustained. In first-episode patients who fail to respond within 2 weeks of starting an antipsychotic, it is reasonable to continue the drug for several weeks because these patients may be more likely to respond later in therapy.
Clinicians treating chronically ill patients who have failed several antipsychotics and demonstrate a poor response after 2 weeks of an appropriate antipsychotic dose are justified in changing medications because later significant response is unlikely. If a patient has a poor early response but has failed several other antipsychotics with few remaining alternatives, it is reasonable to continue the maximum tolerated dose of the current therapy because the patient may be a late responder. However, early non-response predicts future non-response in many patients.
Case continued
In the case described here, Mr. M is failing his current treatment regimen with a reasonable antipsychotic dose after 2 weeks. Because Mr. M has been on 2 antipsychotics and demonstrated a good response to olanzapine, changing medications should be considered.
Related Resource
- Correll CU, Hauser M. The year in psychosis and bipolar disorder: predicting response to schizophrenia treatment. www.medscape.com/viewarticle/735211_7.
Drug Brand Names
Aripiprazole • Abilify Quetiapine • Seroquel
Haloperidol • Haldol Risperidone • Risperdal
Olanzapine • Zyprexa Ziprasidone • Geodon
Paliperidone • Invega
Disclosures
Dr. Straley owns stock in Johnson & Johnson. Dr. Webster reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Agid O, Kapur S, Arenovich T, et al. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry. 2003;60(12):1228-1235.
2. Grace AA, Bunney BS, Moore H, et al. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20(1):31-37.
3. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
4. Meltzer HY, Bobo WV, Heckers SH, et al. Chapter 16. Schizophrenia. In: Ebert MH, Loosen PT, Nurcombe B, Leckman JF, eds. CURRENT Diagnosis & Treatment: Psychiatry. 2nd ed. New York: McGraw-Hill; 2008. http://www.accessmedicine.com/content.aspx?aID=3284037. Accessed December 5, 2013.
5. Robinson DG, Woerner MG, Alvir JM, et al. Predictors of treatment response from a first episode of schizophrenia or schizoaffective disorder. Am J Psychiatry. 1999;156(4):544-549.
6. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
7. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry. 2005;57(12):1543-1549.
9. Kinon BJ, Chen L, Stauffer VL, et al. Early onset of antipsychotic action in schizophrenia: evaluating the possibility of shorter acute efficacy trials. J Clin Psychopharmacol. 2010;30(3):286-289.
10. Hatta K, Otachi T, Sudo Y, et al. Difference in early prediction of antipsychotic non-response between risperidone and olanzapine in the treatment of acute-phase schizophrenia. Schizophr Res. 2011;128(1-3):127-135.
11. Glick ID, Bossie CA, Alphs L, et al. Onset and persistence of antipsychotic response in patients with schizophrenia. J Clin Psychopharmacol. 2009;29(6):542-547.
12. Kinon BJ, Chen L, Ascher-Svanum H, et al. Predicting response to atypical antipsychotics based on early response in the treatment of schizophrenia. Schizophr Res. 2008;102(1-3):230-240.
13. Jäger M, Schmauss M, Laux G, et al. Early improvement as a predictor of remission and response in schizophrenia: results from a naturalistic study. Eur Psychiatry. 2009;24(8):501-506.
14. Kinon BJ, Chen L, Ascher-Svanum H, et al. Early response to antipsychotic drug therapy as a clinical marker of subsequent response in the treatment of schizophrenia. Neuropsychopharmacology. 2010;35(2):581-590.
15. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
16. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
1. Agid O, Kapur S, Arenovich T, et al. Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry. 2003;60(12):1228-1235.
2. Grace AA, Bunney BS, Moore H, et al. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20(1):31-37.
3. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association Steering Committee on Practice Guidelines et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
4. Meltzer HY, Bobo WV, Heckers SH, et al. Chapter 16. Schizophrenia. In: Ebert MH, Loosen PT, Nurcombe B, Leckman JF, eds. CURRENT Diagnosis & Treatment: Psychiatry. 2nd ed. New York: McGraw-Hill; 2008. http://www.accessmedicine.com/content.aspx?aID=3284037. Accessed December 5, 2013.
5. Robinson DG, Woerner MG, Alvir JM, et al. Predictors of treatment response from a first episode of schizophrenia or schizoaffective disorder. Am J Psychiatry. 1999;156(4):544-549.
6. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743-745.
7. Lieberman JA, Phillips M, Gu H, et al. Atypical and conventional antipsychotic drugs in treatment-naive first-episode schizophrenia: a 52-week randomized trial of clozapine vs chlorpromazine. Neuropsychopharmacology. 2003;28(5):995-1003.
8. Leucht S, Busch R, Hamann J, et al. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry. 2005;57(12):1543-1549.
9. Kinon BJ, Chen L, Stauffer VL, et al. Early onset of antipsychotic action in schizophrenia: evaluating the possibility of shorter acute efficacy trials. J Clin Psychopharmacol. 2010;30(3):286-289.
10. Hatta K, Otachi T, Sudo Y, et al. Difference in early prediction of antipsychotic non-response between risperidone and olanzapine in the treatment of acute-phase schizophrenia. Schizophr Res. 2011;128(1-3):127-135.
11. Glick ID, Bossie CA, Alphs L, et al. Onset and persistence of antipsychotic response in patients with schizophrenia. J Clin Psychopharmacol. 2009;29(6):542-547.
12. Kinon BJ, Chen L, Ascher-Svanum H, et al. Predicting response to atypical antipsychotics based on early response in the treatment of schizophrenia. Schizophr Res. 2008;102(1-3):230-240.
13. Jäger M, Schmauss M, Laux G, et al. Early improvement as a predictor of remission and response in schizophrenia: results from a naturalistic study. Eur Psychiatry. 2009;24(8):501-506.
14. Kinon BJ, Chen L, Ascher-Svanum H, et al. Early response to antipsychotic drug therapy as a clinical marker of subsequent response in the treatment of schizophrenia. Neuropsychopharmacology. 2010;35(2):581-590.
15. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691-1696.
16. Stauffer VL, Case M, Kinon BJ, et al. Early response to antipsychotic therapy as a clinical marker of subsequent response in the treatment of patients with first-episode psychosis. Psychiatry Res. 2011;187(1-2):42-48.
SSRIs in pregnancy: What should you tell your depressed patient?
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.


Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.
Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
Mrs. D is a 28-year-old married woman who became depressed after her first pregnancy. The depression was treated successfully with paroxetine, 20 mg/d. Before beginning treatment, she reported low mood, spent most of the day in bed, was unable to care for herself, and confessed to thoughts of harming her child.
Mrs. D presents to your clinic asking whether she should continue her selective serotonin reuptake inhibitor (SSRI) because she and her husband are thinking about having a second child. Recently, she tells you, she saw a news article suggesting that antidepressants show little benefit, and she is concerned that her baby might have a heart defect if she continues paroxetine.
Mrs. D wants to discontinue her medication, but her husband thought she should discuss doing so with you first. During this visit she takes a pregnancy test, which is positive. She wants to know what to do.
women experience depression; 3.8% of pregnant women receive an SSRI.1 SSRIs are the most commonly prescribed antidepressants during pregnancy, but their use remains controversial. There is disagreement about the maternal and neonatal risks of untreated depression and SSRI exposure.2-10 Media reports of studies demonstrating adverse effects associated with SSRIs may generate fear among women, possibly prompting them to self-discontinue medication.
Evidence of risks and benefits
Clinicians should be aware of possible adverse effects of SSRI use and untreated depression (Table).2-10 The available data precludes definitive associations between untreated depression and poor outcomes (Box). Studies of SSRI use during pregnancy have shown conflicting results for all potential outcomes. Absolute risk, with the exception of neonatal adaptation syndrome, is estimated to be small. Neonatal adaptation syndrome—which is characterized by jitteriness, poor muscle tone, weak cries, respiratory distress, hypoglycemia, low Apgar scores, and seizures—occurs in 15% to 30% of infants born to mothers taking SSRIs, but it is transient and resolves during the first weeks of life.
Treatment recommendations
Given the conflicting nature of the evidence, treatment plans should be individualized, weighing the risks and benefits of treatment and the patient’s beliefs and psychiatric history. Consider severity of symptoms and history, including effective therapy and history of relapse. For women with mild or moderate depression, cognitive-behavioral therapy might be an appropriate first-line therapy. However, non-pharmacotherapeutic interventions might not relieve severe depression or be available to all women. When discontinuing an SSRI before pregnancy, counsel the patient to not discontinue the medication abruptly and provide an appropriate taper schedule. See Related Resources for detailed recommendations from the American Psychiatric Association and the American College of Obstetricians and Gynecologists.
Reviewing the SSRI literature regarding pregnancy
Sertraline, paroxetine, citalopram, and fluoxetine are the most studied SSRIs during pregnancy; little information is available on escitalopram and fluvoxamine.11 Prescribing preference generally is given to the medications with the most evidence; paroxetine may be an exception. In 2005, the FDA requested a change in paroxetine’s pregnancy category from C to D, indicating that adequate studies demonstrated a risk of congenital cardiac malformations.11 Additional studies have been conducted, and the teratogenicity of paroxetine is debatable. A recent review reports 8 studies that suggest a malformation risk, compared with 15 studies that show no risk.12
The American Academy of Pediatrics considers SSRIs to be compatible with breast-feeding.13 The best-studied drugs include sertraline and paroxetine. Fluoxetine should be avoided when possible because a long elimination half-life can cause the drug to accumulate in the newborn, increasing the risk of irritability, hypertonia, sedation, and poor suckle.7
There is no best SSRI for all pregnant women. Risks and benefits, including previous treatment success and failure, should be taken into account before starting or switching therapy. Whenever possible, consider monotherapy to avoid compounding the risk of harm.
Related Resources
- Yonkers KA, Wisner KL, Stewart DE, et al. The management of depression during pregnancy: a report from the American Psychiatric Association and the American College of Obstetricians and Gynecologists. Gen Hosp Psychiatry. 2009;31:403-413.
- MGH Center for Women’s Mental Health. www.womensmentalhealth.org.
Drug Brand Names
Citalopram • Celexa Escitalopram • Lexapro Fluoxetine • Prozac
Fluvoxamine • Luvox Paroxetine • Paxil Sertraline • Zoloft
Disclosures
Dr. Leino reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. Dr. Ellingrod receives grant support from the National Institute of Mental Health.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
1. Alwan S, Reefhuis J, Rasmussen SA, et al. Patterns of antidepressant medication use among pregnant women in a United States population. J Clin Pharmacol. 2011;51(2):264-270.
2. Domar AD, Moragianni VA, Ryley DA, et al. The risks of selective serotonin reuptake inhibitor use in infertile women: a review of the impact on fertility, pregnancy, neonatal health and beyond. Hum Reprod. 20113;28(1):160-171.
3. Davalos DB, Yadon CA, Tregellas HC. Untreated prenatal maternal depression and the potential risks to offspring: a review. Arch Womens Ment Health. 2012;15(1):1-14.
4. Spinelli M. Antidepressant treatment during pregnancy. Am J Psychiatry. 2012;169(2):121-124.
5. Oyebode F, Rastogi A, Berrisford G, et al. Psychotropics in pregnancy: safety and other considerations. Pharmacol Ther. 2012;135(1):71-77.
6. Byatt N, Deligiannidis KM, Freeman MP. Antidepressant use in pregnancy: a critical review focused on risks and controversies. Acta Psychiatr Scand. 2013;127(2):94-114.
7. Sie SD, Wennink JM, van Driel JJ, et al. Maternal use of SSRIs, SNRIs and NaSSAs: practical recommendations during pregnancy and lactation. Arch Dis Child Fetal Neonatal Ed. 2012;97(6):F472-476.
8. Jimenez-Solem E, Andersen JT, Petersen M, et al. SSRI use during pregnancy and risk of stillbirth and neonatal mortality. Am J Psychiatry. 2013;170(3):299-304.
9. Nikfar S, Rahimi R, Hendoiee N, et al. Increasing the risk of spontaneous abortion and major malformations in newborns following use of serotonin reuptake inhibitors during pregnancy: a systematic review and updated meta-analysis. Daru. 2012;20(1):75.
10. Stephansson O, Kieler H, Haglund B, et al. Selective serotonin reuptake inhibitors during pregnancy and risk of stillbirth and infant mortality. JAMA. 2013;309(1):48-54.
11. U.S. Food and Drug Administration. Public health advisory: paroxetine. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/Public HealthAdvisories/ucm051731.htm. Published December 8, 2005. Accessed September 27, 2013.
12. Koren G, Nordeng H. Antidepressant use during pregnancy: the benefit-risk ratio. Am J Obstet Gynecol. 2012;207(3):157-163.
13. American Academy of Pediatrics Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics. 2001;108:776-789.
Recommendations for lab monitoring of atypical antipsychotics
Mr. H, age 31, is admitted to an acute psychiatric unit with major depressive disorder, substance dependence, insomnia, and generalized anxiety. In the past, he was treated unsuccessfully with sertraline, fluoxetine, clonazepam, venlafaxine, and lithium. The treatment team starts Mr. H on quetiapine, titrated to 150 mg at bedtime, to address suspected bipolar II disorder.
At baseline, Mr. H is 68 inches tall and slightly overweight at 176 lbs (body mass index [BMI] 26.8 kg/m2). The laboratory reports his glycated hemoglobin (HbA1c) at 5.4%; low-density lipoprotein (LDL), 60 mg/dL; total cholesterol, 122 mg/dL; triglycerides, 141 mg/dL; and high-density lipoprotein (HDL), 34 mg/dL.
Within 1 month, Mr. H experiences a 16% increase in body weight. HbA1c increases to 5.6%; LDL, to 93 mg/dL. These metabolic changes are not addressed, and he continues quetiapine for another 5 months. At the end of 6 months, Mr. H weighs 223.8 lbs (BMI 34 kg/m2)—a 27% increase from baseline. HbA1c is in the prediabetic range, at 5.9%, and LDL is 120 mg/dL.1 The treatment team discusses the risks of further metabolic effects, cardiovascular disease, and diabetes with Mr. H. He agrees to a change in therapy.
An increase in weight is thought to be associated with the actions of antipsychotics on H1and 5-HT2c receptors.7 Clozapine and olanzapine pose the highest risk of weight gain. Quetiapine and risperidone are considered of intermediate risk; aripiprazole and ziprasidone present the lowest risk(Table 1).5,7
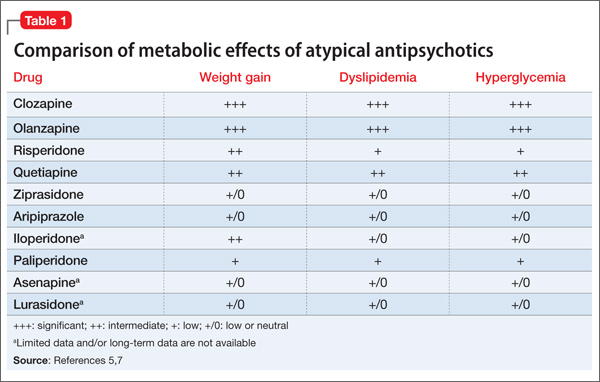
Patients taking an atypical antipsychotic may experience an elevation of blood glucose, serum triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic â-cells.7 Clozapine and olanzapine pose the highest risk of dyslipidemia. Quetiapine and risperidone are considered of intermediate risk; the risk associated with quetiapine is closer to that of olanzpine.8,9 Aripiprazole and ziprasidone present a lower risk of dyslipidemia and glucose elevations.5
Newer atypical antipsychotics, such as asenapine, iloperidone, paliperidone, and lurasidone, seem to have a lower metabolic risk profile, similar to those seen with aripiprazole and ziprasidone.5 Patients enrolled in initial clinical trials might not be antipsychotic naïve, however, and may have been taking a high metabolic risk antipsychotic. When these patients are switched to an antipsychotic that carries less of a metabolic risk, it might appear that they are experiencing a decrease in metabolic adverse events.
Metabolic data on newer atypical antipsychotics are limited; most have not been subject to long-term study. Routine monitoring of metabolic side effects is recommended for all atypical antipsychotics, regardless of risk profile.
Recommended monitoring
Because of the known metabolic side effects that occur in patients taking an atypical antipsychotic, baseline and periodic monitoring is recommended (Table 2).2,10 BMI and waist circumference should be recorded at baseline and tracked throughout treatment. Ideally, obtain measurements monthly for the first 3 months of therapy, or after any medication adjustments, then at 6 months, and annually thereafter. Encourage patients to track their own weight.
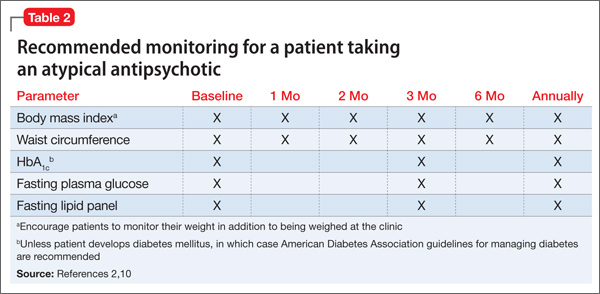
HbA1c and fasting plasma glucose levels should be measured at baseline and throughout the course of treatment. Obtain another set of measurements at 3 months, then annually thereafter, unless the patient develops type 2 diabetes mellitus.2
Obtaining a fasting lipid panel at baseline and periodically throughout the course of treatment is recommended. After baseline measurement, another panel should be taken at 3 months and annually thereafter. Guidelines of the American Diabetes Association recommend a fasting lipid panel every 5 years—however, good clinical practice dictates obtaining a lipid panel annually.
Managing metabolic side effects
Assess whether the patient can benefit from a lower dosage of current medication, switching to an antipsychotic with less of a risk of metabolic disturbance, or from discontinuation of therapy. In most cases, aim to use monotherapy because polypharmacy contributes to an increased risk of side effects.10
Weight management. Recommend nutrition counseling and physical activity for all patients who are overweight. Referral to a health care professional or to a program with expertise in weight management also might be beneficial.2 Include family members and significant others in the patient’s education when possible.
Impaired fasting glucose. Encourage a low-carbohydrate, high-protein diet with high intake of vegetables. Patients should obtain at least 30 minutes of physical activity, five times a week. Referral to a diabetes self-management class also is appropriate. Consider referral to a primary care physician or a clinician with expertise in diabetes.2
Impaired fasting lipids. Encourage your patients to adhere to a heart-healthy diet that is low in saturated fats and to get adequate physical activity. Referral to a dietician and primary care provider for medical management of dyslipidemia might be appropriate.2
Related Resources
- American Diabetes Association. Guide to living with diabetes. www.diabetes.org/living-with-diabetes.
- MOVE! Weight Management Program for Veterans. www. move.va.gov.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Clonazepam • Klonopin
Clozapine • Clozaril
Fluoxetine • Prozac
Iloperidone • Fanapt
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosure
The authors report no financial relationships with any of the manufacturers mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association. Executive summary: standards of medical care in diabetes—2010. Diabetes Care. 2010;33:
S4-S10.
2. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and the North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
3. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
4. Tarricone I, Ferrari Gozzi B, Serretti A, et al. Weight gain in antipsychotic-naive patients: a review and meta-analysis. Psychol Med. 2010;40(2):187-200.
5. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
6. De Hert M, Dobbelaere M, Sheridan EM, et al. Metabolic and endocrine adverse effects of second-generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice. Eur Psychiatry. 2011;26(3):144-158.
7. Stahl SM. Stahl’s essential psychopharmacology, neuroscientific basis and practical applications. Oxford, United Kingdom: Cambridge University Press; 2008.
8. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):
1209-1223.
9. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
10. Gothefors D, Adolfsson R, Attvall S, et al; Swedish Psychiatric Association. Swedish clinical guidelines – prevention and management of metabolic risk in patients with severe psychiatric disorders. Nord J Psychiatry. 2010;64(5):294-302.
11. Schneiderhan ME, Batscha CL, Rosen C. Assessment of a point-of-care metabolic risk screening program in outpatients receiving antipsychotic agents. Pharmacotherapy. 2009;29(8): 975-987.
Mr. H, age 31, is admitted to an acute psychiatric unit with major depressive disorder, substance dependence, insomnia, and generalized anxiety. In the past, he was treated unsuccessfully with sertraline, fluoxetine, clonazepam, venlafaxine, and lithium. The treatment team starts Mr. H on quetiapine, titrated to 150 mg at bedtime, to address suspected bipolar II disorder.
At baseline, Mr. H is 68 inches tall and slightly overweight at 176 lbs (body mass index [BMI] 26.8 kg/m2). The laboratory reports his glycated hemoglobin (HbA1c) at 5.4%; low-density lipoprotein (LDL), 60 mg/dL; total cholesterol, 122 mg/dL; triglycerides, 141 mg/dL; and high-density lipoprotein (HDL), 34 mg/dL.
Within 1 month, Mr. H experiences a 16% increase in body weight. HbA1c increases to 5.6%; LDL, to 93 mg/dL. These metabolic changes are not addressed, and he continues quetiapine for another 5 months. At the end of 6 months, Mr. H weighs 223.8 lbs (BMI 34 kg/m2)—a 27% increase from baseline. HbA1c is in the prediabetic range, at 5.9%, and LDL is 120 mg/dL.1 The treatment team discusses the risks of further metabolic effects, cardiovascular disease, and diabetes with Mr. H. He agrees to a change in therapy.
An increase in weight is thought to be associated with the actions of antipsychotics on H1and 5-HT2c receptors.7 Clozapine and olanzapine pose the highest risk of weight gain. Quetiapine and risperidone are considered of intermediate risk; aripiprazole and ziprasidone present the lowest risk(Table 1).5,7
Patients taking an atypical antipsychotic may experience an elevation of blood glucose, serum triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic â-cells.7 Clozapine and olanzapine pose the highest risk of dyslipidemia. Quetiapine and risperidone are considered of intermediate risk; the risk associated with quetiapine is closer to that of olanzpine.8,9 Aripiprazole and ziprasidone present a lower risk of dyslipidemia and glucose elevations.5
Newer atypical antipsychotics, such as asenapine, iloperidone, paliperidone, and lurasidone, seem to have a lower metabolic risk profile, similar to those seen with aripiprazole and ziprasidone.5 Patients enrolled in initial clinical trials might not be antipsychotic naïve, however, and may have been taking a high metabolic risk antipsychotic. When these patients are switched to an antipsychotic that carries less of a metabolic risk, it might appear that they are experiencing a decrease in metabolic adverse events.
Metabolic data on newer atypical antipsychotics are limited; most have not been subject to long-term study. Routine monitoring of metabolic side effects is recommended for all atypical antipsychotics, regardless of risk profile.
Recommended monitoring
Because of the known metabolic side effects that occur in patients taking an atypical antipsychotic, baseline and periodic monitoring is recommended (Table 2).2,10 BMI and waist circumference should be recorded at baseline and tracked throughout treatment. Ideally, obtain measurements monthly for the first 3 months of therapy, or after any medication adjustments, then at 6 months, and annually thereafter. Encourage patients to track their own weight.
HbA1c and fasting plasma glucose levels should be measured at baseline and throughout the course of treatment. Obtain another set of measurements at 3 months, then annually thereafter, unless the patient develops type 2 diabetes mellitus.2
Obtaining a fasting lipid panel at baseline and periodically throughout the course of treatment is recommended. After baseline measurement, another panel should be taken at 3 months and annually thereafter. Guidelines of the American Diabetes Association recommend a fasting lipid panel every 5 years—however, good clinical practice dictates obtaining a lipid panel annually.
Managing metabolic side effects
Assess whether the patient can benefit from a lower dosage of current medication, switching to an antipsychotic with less of a risk of metabolic disturbance, or from discontinuation of therapy. In most cases, aim to use monotherapy because polypharmacy contributes to an increased risk of side effects.10
Weight management. Recommend nutrition counseling and physical activity for all patients who are overweight. Referral to a health care professional or to a program with expertise in weight management also might be beneficial.2 Include family members and significant others in the patient’s education when possible.
Impaired fasting glucose. Encourage a low-carbohydrate, high-protein diet with high intake of vegetables. Patients should obtain at least 30 minutes of physical activity, five times a week. Referral to a diabetes self-management class also is appropriate. Consider referral to a primary care physician or a clinician with expertise in diabetes.2
Impaired fasting lipids. Encourage your patients to adhere to a heart-healthy diet that is low in saturated fats and to get adequate physical activity. Referral to a dietician and primary care provider for medical management of dyslipidemia might be appropriate.2
Related Resources
- American Diabetes Association. Guide to living with diabetes. www.diabetes.org/living-with-diabetes.
- MOVE! Weight Management Program for Veterans. www. move.va.gov.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Clonazepam • Klonopin
Clozapine • Clozaril
Fluoxetine • Prozac
Iloperidone • Fanapt
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosure
The authors report no financial relationships with any of the manufacturers mentioned in this article or with manufacturers of competing products.
Mr. H, age 31, is admitted to an acute psychiatric unit with major depressive disorder, substance dependence, insomnia, and generalized anxiety. In the past, he was treated unsuccessfully with sertraline, fluoxetine, clonazepam, venlafaxine, and lithium. The treatment team starts Mr. H on quetiapine, titrated to 150 mg at bedtime, to address suspected bipolar II disorder.
At baseline, Mr. H is 68 inches tall and slightly overweight at 176 lbs (body mass index [BMI] 26.8 kg/m2). The laboratory reports his glycated hemoglobin (HbA1c) at 5.4%; low-density lipoprotein (LDL), 60 mg/dL; total cholesterol, 122 mg/dL; triglycerides, 141 mg/dL; and high-density lipoprotein (HDL), 34 mg/dL.
Within 1 month, Mr. H experiences a 16% increase in body weight. HbA1c increases to 5.6%; LDL, to 93 mg/dL. These metabolic changes are not addressed, and he continues quetiapine for another 5 months. At the end of 6 months, Mr. H weighs 223.8 lbs (BMI 34 kg/m2)—a 27% increase from baseline. HbA1c is in the prediabetic range, at 5.9%, and LDL is 120 mg/dL.1 The treatment team discusses the risks of further metabolic effects, cardiovascular disease, and diabetes with Mr. H. He agrees to a change in therapy.
An increase in weight is thought to be associated with the actions of antipsychotics on H1and 5-HT2c receptors.7 Clozapine and olanzapine pose the highest risk of weight gain. Quetiapine and risperidone are considered of intermediate risk; aripiprazole and ziprasidone present the lowest risk(Table 1).5,7
Patients taking an atypical antipsychotic may experience an elevation of blood glucose, serum triglyceride, and LDL levels, and a decrease in the HDL level.2 These effects may be seen without an increase in BMI, and should be considered a direct effect of the antipsychotic.5 Although the mechanism by which dyslipidemia occurs is poorly understood, an increase in the blood glucose level is thought to be, in part, mediated by antagonism of M3 muscarinic receptors on pancreatic â-cells.7 Clozapine and olanzapine pose the highest risk of dyslipidemia. Quetiapine and risperidone are considered of intermediate risk; the risk associated with quetiapine is closer to that of olanzpine.8,9 Aripiprazole and ziprasidone present a lower risk of dyslipidemia and glucose elevations.5
Newer atypical antipsychotics, such as asenapine, iloperidone, paliperidone, and lurasidone, seem to have a lower metabolic risk profile, similar to those seen with aripiprazole and ziprasidone.5 Patients enrolled in initial clinical trials might not be antipsychotic naïve, however, and may have been taking a high metabolic risk antipsychotic. When these patients are switched to an antipsychotic that carries less of a metabolic risk, it might appear that they are experiencing a decrease in metabolic adverse events.
Metabolic data on newer atypical antipsychotics are limited; most have not been subject to long-term study. Routine monitoring of metabolic side effects is recommended for all atypical antipsychotics, regardless of risk profile.
Recommended monitoring
Because of the known metabolic side effects that occur in patients taking an atypical antipsychotic, baseline and periodic monitoring is recommended (Table 2).2,10 BMI and waist circumference should be recorded at baseline and tracked throughout treatment. Ideally, obtain measurements monthly for the first 3 months of therapy, or after any medication adjustments, then at 6 months, and annually thereafter. Encourage patients to track their own weight.
HbA1c and fasting plasma glucose levels should be measured at baseline and throughout the course of treatment. Obtain another set of measurements at 3 months, then annually thereafter, unless the patient develops type 2 diabetes mellitus.2
Obtaining a fasting lipid panel at baseline and periodically throughout the course of treatment is recommended. After baseline measurement, another panel should be taken at 3 months and annually thereafter. Guidelines of the American Diabetes Association recommend a fasting lipid panel every 5 years—however, good clinical practice dictates obtaining a lipid panel annually.
Managing metabolic side effects
Assess whether the patient can benefit from a lower dosage of current medication, switching to an antipsychotic with less of a risk of metabolic disturbance, or from discontinuation of therapy. In most cases, aim to use monotherapy because polypharmacy contributes to an increased risk of side effects.10
Weight management. Recommend nutrition counseling and physical activity for all patients who are overweight. Referral to a health care professional or to a program with expertise in weight management also might be beneficial.2 Include family members and significant others in the patient’s education when possible.
Impaired fasting glucose. Encourage a low-carbohydrate, high-protein diet with high intake of vegetables. Patients should obtain at least 30 minutes of physical activity, five times a week. Referral to a diabetes self-management class also is appropriate. Consider referral to a primary care physician or a clinician with expertise in diabetes.2
Impaired fasting lipids. Encourage your patients to adhere to a heart-healthy diet that is low in saturated fats and to get adequate physical activity. Referral to a dietician and primary care provider for medical management of dyslipidemia might be appropriate.2
Related Resources
- American Diabetes Association. Guide to living with diabetes. www.diabetes.org/living-with-diabetes.
- MOVE! Weight Management Program for Veterans. www. move.va.gov.
Drug Brand Names
Aripiprazole • Abilify
Asenapine • Saphris
Clonazepam • Klonopin
Clozapine • Clozaril
Fluoxetine • Prozac
Iloperidone • Fanapt
Lithium • Eskalith, Lithobid
Lurasidone • Latuda
Olanzapine • Zyprexa
Paliperidone • Invega
Quetiapine • Seroquel
Risperidone • Risperdal
Sertraline • Zoloft
Venlafaxine • Effexor
Ziprasidone • Geodon
Disclosure
The authors report no financial relationships with any of the manufacturers mentioned in this article or with manufacturers of competing products.
1. American Diabetes Association. Executive summary: standards of medical care in diabetes—2010. Diabetes Care. 2010;33:
S4-S10.
2. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and the North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
3. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
4. Tarricone I, Ferrari Gozzi B, Serretti A, et al. Weight gain in antipsychotic-naive patients: a review and meta-analysis. Psychol Med. 2010;40(2):187-200.
5. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
6. De Hert M, Dobbelaere M, Sheridan EM, et al. Metabolic and endocrine adverse effects of second-generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice. Eur Psychiatry. 2011;26(3):144-158.
7. Stahl SM. Stahl’s essential psychopharmacology, neuroscientific basis and practical applications. Oxford, United Kingdom: Cambridge University Press; 2008.
8. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):
1209-1223.
9. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
10. Gothefors D, Adolfsson R, Attvall S, et al; Swedish Psychiatric Association. Swedish clinical guidelines – prevention and management of metabolic risk in patients with severe psychiatric disorders. Nord J Psychiatry. 2010;64(5):294-302.
11. Schneiderhan ME, Batscha CL, Rosen C. Assessment of a point-of-care metabolic risk screening program in outpatients receiving antipsychotic agents. Pharmacotherapy. 2009;29(8): 975-987.
1. American Diabetes Association. Executive summary: standards of medical care in diabetes—2010. Diabetes Care. 2010;33:
S4-S10.
2. American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and the North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596-601.
3. Kahn RS, Fleischhacker WW, Boter H, et al; EUFEST study group. Effectiveness of antipsychotic drugs in first-episode schizophrenia and schizophreniform disorder: an open randomised clinical trial. Lancet. 2008;371(9618):1085-1097.
4. Tarricone I, Ferrari Gozzi B, Serretti A, et al. Weight gain in antipsychotic-naive patients: a review and meta-analysis. Psychol Med. 2010;40(2):187-200.
5. De Hert M, Yu W, Detraux J, et al. Body weight and metabolic adverse effects of asenapine, iloperidone, lurasidone and paliperidone in the treatment of schizophrenia and bipolar disorder: a systematic review and exploratory meta-analysis. CNS Drugs. 2012;26(9):733-759.
6. De Hert M, Dobbelaere M, Sheridan EM, et al. Metabolic and endocrine adverse effects of second-generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice. Eur Psychiatry. 2011;26(3):144-158.
7. Stahl SM. Stahl’s essential psychopharmacology, neuroscientific basis and practical applications. Oxford, United Kingdom: Cambridge University Press; 2008.
8. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):
1209-1223.
9. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765-1773.
10. Gothefors D, Adolfsson R, Attvall S, et al; Swedish Psychiatric Association. Swedish clinical guidelines – prevention and management of metabolic risk in patients with severe psychiatric disorders. Nord J Psychiatry. 2010;64(5):294-302.
11. Schneiderhan ME, Batscha CL, Rosen C. Assessment of a point-of-care metabolic risk screening program in outpatients receiving antipsychotic agents. Pharmacotherapy. 2009;29(8): 975-987.
Lithium-induced diabetes insipidus: Prevention and management
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
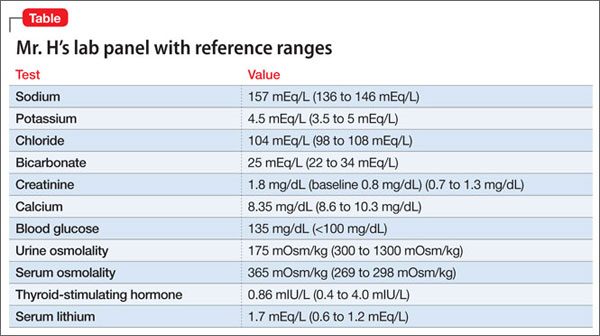
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
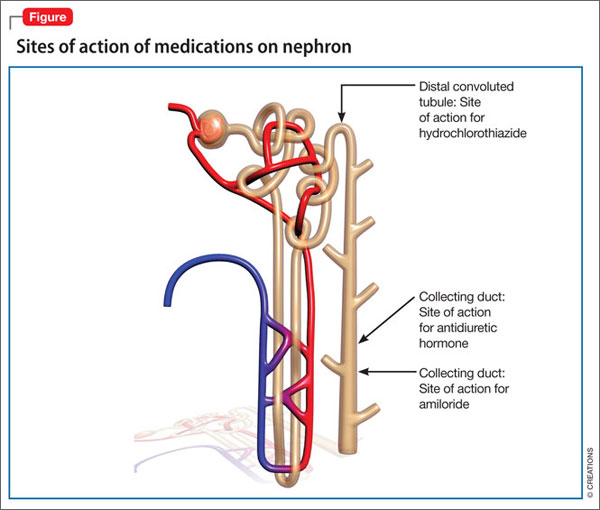
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
Mr. H, age 33, was diagnosed with bipolar I disorder 9 years ago. For the past year, his mood symptoms have been well controlled with lithium 300 mg, 3 times a day, and olanzapine, 20 mg/d. He presents to the outpatient clinic for a routine visit complaining of insomnia, daytime sleepiness, and increased thirst. He also notes that his tremor has become more prominent over the last few weeks. Concerned about his symptoms, Mr. H’s clinician orders a comprehensive laboratory panel (Table).
Upon further questioning, Mr. H’s physician determines that his insomnia is caused by nocturnal urination, which is consistent with fluid and electrolyte imbalances seen in Mr. H’s laboratory panel. Mr. H is diagnosed with lithium-induced diabetes insipidus.
Although lithium’s exact mechanism of action is unknown, it is known that lithium can negatively affect the kidneys.1,2 Typically, antidiuretic hormone (ADH) regulates water permeability in the collecting duct of the nephron, allowing water to be reabsorbed through simple diffusion in the kidney’s collecting duct (Figure).3 Chronic lithium use reduces or desensitizes the kidney’s ability to respond to ADH. Resistance to ADH occurs when lithium accumulates in the cells of the collecting duct and inhibits ADH’s ability to increase water permeability. This inhibition can cause some of Mr. H’s symptoms, such as polydipsia and polyuria, and is estimated to occur in approximately 40% of patients receiving long-term lithium therapy.4,5
Diagnosis
Diagnosing lithium-induced nephrogenic diabetes insipidus (NDI) begins with a history of the patient’s symptoms and ordering lab tests.5 The next step involves a water restriction test, also known as a thirst test, to measure the patient’s ability to concentrate his or her urine. Baseline serum osmolality and electrolytes are compared with new values obtained after completing the water restriction test. Healthy people will have a 2-to-4-fold increase in urine osmolality compared with patients who have NDI. The last step includes administering desmopressin and differentiates between central diabetes insipidus and NDI.6
After desmopressin use, patients who have central diabetes insipidus will have a >50% increase in urine osmolality, whereas patients who have NDI will have <10% increase in urine osmolality. This distinction is important because patients with central diabetes insipidus might have more severe disease and might not benefit from measures commonly used for lithium-induced NDI.7
Prevention and management
Lithium-induced NDI is thought to be dose-dependent and may be prevented by using the lowest effective dose of lithium for an individual patient. It is important that patients taking lithium receive basic electrolyte, hematologic, liver function, renal function, and thyroid function tests at baseline and every 6 to 12 months after the lithium regimen is stable. Additionally, lithium levels should be monitored frequently. The frequency of these tests may range from twice weekly to every 3 to 4 months or longer, depending on the patient’s condition. This monitoring allows the prescriber to quickly identify emerging adverse effects.
Patients with impaired renal function and those with a urine output >3 liters a day are more susceptible to NDI and require monitoring every 3 months. Also, instruct patients to monitor their urine output and educate them about the dangers of fluid and electrolyte imbalances and the signs and symptoms of NDI, such as excessive thirst and urination.1,2
When a patient experiences lithium-induced NDI, re-evaluate treatment and dosage, including simplifying the dosing regimen or switching to once-daily dosing, usually at bedtime. Once-daily dosing results in a lower overall lithium trough, which might allow the kidneys more “drug-free” time.4,5 Additionally, 12-hour lithium levels are approximately 20% higher with once-daily monitoring; continued monitoring is needed during this switch. Patients who have a moderate or severe form of lithium-induced NDI may need to discontinue lithium altogether. There are several options for treating lithium-induced NDI in patients who need to take lithium. Closely monitor kidney function and lithium routinely with these strategies.
Amiloride. This potassium-sparing diuretic minimizes accumulation of lithium by inhibiting collecting duct sodium channels. Studies have shown that amiloride can decrease mean urine volume, increase urine osmolality, and improve the kidneys’ ability to respond to exogenous arginine vasopressin.8
Thiazide diuretics produce mild sodium depletion, which decreases the distal tubule delivery of sodium, therefore increasing water reabsorption in the collecting duct. Hydrochlorothiazide has been shown to reduce urine output by >50% in patients with NDI on a sodium-restricted diet. Hydrochlorothiazide use requires careful monitoring of potassium and lithium levels. Use of a thiazide diuretic also might warrant decreasing the lithium dose by as much as 50% to prevent toxicity.9,10
Low-sodium diet plus hydrochlorothiazide. This route provides another option to decrease urine output during lithium-induced NDI. A reduction in urine output has been shown to be directly proportional to a decrease in salt intake and excretion. Restricting sodium to <2.3 g/d is an appropriate goal for many patients to prevent reoccurring symptoms, which is more than the 3 g/d average that most Americans consume. Potassium and lithium levels must be monitored closely.9
Nonsteroidal anti-inflammatory drugs (NSAIDs). These drugs’ ability to inhibit prostaglandin synthesis prevents prostaglandins from antagonizing actions of ADH in the kidney. The result is increased urine concentration via the actions of ADH. Indomethacin has a greater effect than ibuprofen in increasing ADH’s actions on the kidney. Use of concomitant NSAIDs with lithium requires close monitoring of renal function tests.11
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.
1. Ecelbarger CA. Lithium treatment and remodeling of the collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F37-38.
2. Christensen BM, Kim YH, Kwon TH, et al. Lithium treatment induces a marked proliferation of primarily principal cells in rat kidney inner medullary collecting duct. Am J Physiol Renal Physiol. 2006;291(1):F39-48.
3. Francis SG, Gardner DG. Basic and clinical endocrinology. 7th ed. New York, NY: McGraw Hill; 2003:154-158.
4. Stone KA. Lithium-induced nephrogenic diabetes insipidus. J Am Board Fam Pract. 1999;12(1):43-47.
5. Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270-276.
6. Wesche D, Deen PM, Knoers NV. Congenital nephrogenic diabetes insipidus: the current state of affairs. Pediatr Nephrol. 2012;27(12):2183-2204.
7. Rose BD, Post TW. Clinical physiology of acid-base and electrolyte disorders. 5th ed. New York, NY: McGraw-Hill; 2001:754-759,782-783.
8. Batlle DC, von Riotte AB, Gaviria M, et al. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408-414.
9. Earley LE, Orloff J. The mechanism of antidiuresis associated with the administration of hydrochlorothiazide to patients with vasopressin-resistant diabetes insipidus. J Clin Invest. 1962;41(11):1988-1997.
10. Kim GH, Lee JW, Oh YK, et al. Antidiuretic effect of hydrochlorothiazide in lithium-induced nephrogenic diabetes insipidus is associated with upregulation of aquaporin-2, Na-Cl co-transporter, and epithelial sodium channel. J Am Soc Nephrol. 2004;15(11):2836-2843.
11. Libber S, Harrison H, Spector D. Treatment of nephrogenic diabetes insipidus with prostaglandin synthesis inhibitors. J Pediatr. 1986;108(2):305-311.
Sildenafil for SSRI-induced sexual dysfunction in women
- Sexual dysfunction can arise from environmental, social, medical, or drug effects and requires a multifaceted approach to treatment.
- When possible, take a baseline sexual dysfunction measurement to assess if selective serotonin reuptake inhibitor use is correlated with onset or worsening of sexual dysfunction.
- Nonpharmacologic options should be considered before and during pharmacotherapy.
- Sildenafil may be useful for treating anorgasmia in women taking serotonergic antidepressants.
- Phosphodiesterase type 5 inhibitors are not FDA-approved for sexual dysfunction in women.
Mrs. L, age 27, has a history of major depressive disorder with symptoms of anxiety. She was managed successfully for 2 years with bupropion XL, 300 mg/d, but was switched to venlafaxine, titrated to 225 mg/d, after she developed seizures secondary to a head injury sustained in a car accident. After the switch, Mrs. L’s mood deteriorated and she was hospitalized. Since then, she’s received several medication trials, including paroxetine, 30 mg/d, a selective serotonin reuptake inhibitor (SSRI), and the tricyclic antidepressant (TCA) nortriptyline, 75 mg/d, but she could not tolerate these medications because of severe xerostomia.
After taking sertraline, 150 mg/d, for 8 weeks, Mrs. L improves and has a Patient Health Questionnaire score of 6, indicating mild depression. Her initial complaints of diarrhea and nausea have resolved, but Mrs. L now reports that she and her husband are having marital difficulties because she cannot achieve orgasm during sexual intercourse. She did not have this problem when she was taking bupropion. Her husband occasionally takes the phosphodiesterase type 5 (PDE5) inhibitor sildenafil before intercourse, and Mrs. L asks you if this medication will help her achieve orgasm.
DSM-IV-TR defines sexual dysfunction as disturbances in sexual desire and/or in the sexual response cycle (excitement, plateau, orgasm, and resolution) that result in marked distress and interpersonal difficulty.1 Sexual dysfunction can occur with the use of any antidepressant with serotonergic activity; it affects an estimated 50% to 70% of patients who take SSRIs.2 Sexual dysfunction can occur with all SSRIs; however, higher rates of sexual dysfunction are found with citalopram, fluoxetine, paroxetine, and sertraline.3 Studies have suggested there may be a dose-side effect relationship with SSRI-induced sexual dysfunction.4
Several factors can increase a patient’s risk of sexual dysfunction and should be considered before prescribing an antidepressant or when a patient presents with new or worsening sexual dysfunction (Table 1).5 In general, nonserotonergic agents such as bupropion, mirtazapine, and nefazodone are associated with lower rates of sexual dysfunction. The pharmacology of these agents explains their decreased propensity to cause sexual dysfunction. These agents increase levels of dopamine in the mesolimbic dopaminergic system either by blocking reuptake (bupropion) or antagonizing the serotonin subtype-2 receptor and facilitating disinhibition of decreased dopamine downstream (nefazodone and mirtazapine).
Table 1
Risk factors for sexual dysfunction
| Sex | Risk factors |
|---|---|
| Women | History of sexual, physical, or emotional abuse, physical inactivity |
| Men | Severe hyperprolactinemia, smoking |
| Both sexes | Poor to fair health, genitourinary disease, diabetes mellitus, cardiovascular disease, hypertension, increasing age, psychiatric disorders, relationship difficulties |
| Source: Reference 5 | |
One option for treating antidepressant-induced sexual dysfunction in women is PDE5 inhibitors, which are used to treat erectile dysfunction (ED). These medications ameliorate ED by inhibiting degradation of cyclic guanosine monophosphate by PDE5, which increases blood flow to the penis during sexual stimulation. Although these medications are not FDA-approved for treating sexual dysfunction in women, adjunctive PDE5 inhibitor treatment may be beneficial for sexual dysfunction in females because similar mediators, such as nitric oxide and cyclic guanosine monophosphate, involved in the nonadrenergic-noncholinergic signaling that controls sexual stimulation in men also are found in female genital tissue.6
When treating a woman with SSRI-induced sexual dysfunction, consider nonpharmacologic treatments both before and during pharmacotherapy (Table 2).7,8 See Table 3 for a comparison of pharmacokinetics, side effects, and drug interactions of the 4 FDA-approved PDE5 inhibitors—avanafil, sildenafil, tadalafil, and vardenafil.
Table 2
Management strategies for SSRI-induced sexual dysfunction
| Intervention | Comments |
|---|---|
| Nonpharmacologic | |
| Lifestyle modifications | Encourage healthy eating, weight loss, smoking cessation, substance abuse treatment, or minimizing alcohol intake to improve patient self-image and overall health |
| Cognitive-behavioral therapy | Patients can identify coping strategies for reducing symptom severity and preventing worsening sexual dysfunction |
| Sex therapy | May benefit patients with relationship difficulties |
| ‘Watch and wait’ | Spontaneous resolving (or ‘adaptation’) of sexual dysfunction with antidepressants can take ≥6 months. Studies have found adaptation rates generally are low (~10%) |
| Pharmacologic | |
| Drug holiday | May be an option for patients taking antidepressants with shorter half-lives and patients taking lower doses. Be cautious of empowering patients to stop their own medications as needed |
| Dosage reduction | Serotonergic antidepressant-induced sexual dysfunction may be related to dose. Little research has been conducted on this method and the patient’s clinical status must be considered |
| Dose timing | Instructing a patient to take the antidepressant after his or her usual time of sexual activity (eg, patients who engage in sexual activity at night should take the antidepressant before falling asleep). This may allow the drug level to be lowest during sexual activity |
| Switching medications | Case reports, retrospective studies, and RCTs suggest switching to a different antidepressant with less serotonergic activity may be appropriate, particularly if the patient has not responded to the current antidepressant |
| Adjunctive therapy | RCTs support adjunctive bupropion (≥300 mg/d) or olanzapine (5 mg/d) as treatment for SSRI-induced sexual dysfunction in women Studies have found no improvement in sexual functioning with adjunctive buspirone, granisetron, amantadine, mirtazapine, yohimbine, ephedrine, or ginkgo biloba in women |
| RCTs: randomized controlled trials; SSRI: selective serotonin reuptake inhibitor Source: Reference 7,8 | |
Table 3
Phosphodiesterase type 5 inhibitors: A comparison
| Medication | Dose rangea | Pharmacokinetics | Side effects | Significant drug interactions |
|---|---|---|---|---|
| Avanafil | 50 to 200 mg, 30 minutes before sexual activity | Bioavailability: N/A (high-fat meal delays Tmax by 60 minutes and reduces Cmax by 24% to 39%; clinically insignificant) Half life: 5 hours Metabolism: CYP3A4 | Headache, flushing, nasal congestion, nasopharyngitis, backache | Strong CYP3A4 inhibitors (increased avanafil levels) Contraindicated within 12 hours of nitrate use (eg, nitroglycerin) |
| Sildenafil | 25 to 100 mg, 1 to 2 hours before sexual activity | Bioavailability: 41% (food/high-fat meal delays Tmax by 60 minutes and reduces Cmax by 29%) Half life: 4 hours Metabolism: CYP3A4 | Headache, flushing, erythema, indigestion, insomnia, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased sildenafil levels) Contraindicated within 24 hours of nitrate use |
| Tadalafil | 10 to 20 mg, 30 minutes before sexual activity | Bioavailability: N/A (not affected by food) Half life: 17.5 hours (duration of action up to 36 hours) Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, myalgia, and back pain | Strong CYP3A4 inhibitors (increased tadalafil levels) Contraindicated within 48 hours of nitrate use |
| Vardenafil | 5 to 20 mg, 30 minutes to 2 hours before sexual activity | Bioavailability: 15% for film-coated tablet (high-fat meal reduces Cmax by 18% to 50%) Half life: 4 to 5 hours Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased vardenafil levels) Contraindicated within 24 hours of nitrate use |
| aTypical dose range for treatment of erectile dysfunction Cmax: maximum concentration; CYP: cytochrome P450; Tmax: time to maximum concentration Source: Micromedex® Healthcare Series [Internet database]. Greenwood Village, CO: Thomson Healthcare. Accessed October 10, 2012 | ||||
Limited evidence for sildenafil
Case reports, a few small open-label trials, and 1 prospective, randomized controlled trial (RCT) have evaluated sildenafil as an adjunctive treatment for serotonergic antidepressant-associated sexual dysfunction in women.6,9 Nurnberg et al6 examined the efficacy of adjunctive sildenafil in women with SSRI-induced sexual dysfunction. This 8-week, placebo-controlled, double-blind, RCT used a flexible dose (50 or 100 mg), intention-to-treat design to assess the effect of sildenafil on 98 premenopausal women whose depression was in remission. Ten patients were taking the serotonin-norepinephrine inhibitor venlafaxine, 1 was taking the TCA clomipramine, and 87 were receiving an SSRI. Patients were instructed to take sildenafil or placebo 1 to 2 hours before sexual activity. The primary outcome was mean change from baseline on the Clinical Global Impression-Sexual Function (CGI-SF) scale.
Women taking sildenafil showed significant improvement compared with those taking placebo, with a treatment difference between groups of 0.8 (95% CI, 0.6 to 1.0; =.001). Additionally, 23% of sildenafil-treated patients reported no improvement with the intervention, compared with 73% of patients receiving placebo. Secondary outcomes using 3 validated scales that evaluated specific phases of sexual function found that patients’ orgasmic function significantly benefited from sildenafil treatment, while desire, arousal, and overall satisfaction were not significantly different.
Although these findings seem to support sildenafil for treating serotonergic antidepressant-associated sexual dysfunction in women, the study had a relatively small treatment effect in a well-defined patient population; therefore, replication in future trials and different patient populations is warranted. Overall, sildenafil was well tolerated, despite patient reports of headaches, flushing, visual disturbances, dyspepsia, nasal congestion, and palpitations. Finally, cost vs benefit should be considered; PDE5 inhibitors may not be covered by insurance or may require prior authorization.
CASE CONTINUED: Symptoms resolve
Bupropion is not an appropriate choice for Mrs. L because of her seizure risk. Mirtazapine is ruled out because in the past she experienced excessive somnolence that impaired her ability to function. You are not comfortable prescribing nefazodone because of its risk of hepatotoxicity or suggesting that Mrs. L take a “drug holiday” (stop taking any antidepressants for a short period) because of the risk of depressive relapse. You suggest that Mrs. L continue to take sertraline because sometimes antidepressant-induced sexual dysfunction resolves after ≥6 months of treatment with the same agent, but she is adamant that her relationship with her husband will deteriorate if she waits that long. She also declines cognitive-behavioral therapy because her job doesn’t allow the time or flexibility to commit to the sessions.
You prescribe sildenafil, 50 mg, and instruct Mrs. L to take 1 tablet 1 to 2 hours before sexual activity. This treatment improves her ability to achieve orgasm. She tolerates the drug well and after 8 weeks of treatment her CGI-SF score improves from 6 at baseline, indicating extreme dysfunction, to 2, indicating normal function. Ten months into her sertraline treatment, Mrs. L discovers she no longer requires sildenafil to achieve orgasm.
Related Resources
- Nurnberg HG. An evidence-based review updating the various treatment and management approaches to serotonin reuptake inhibitor-associated sexual dysfunction. Drugs Today (Barc). 2008;44(2):147-168.
- NIH Medline Plus. Sexual problems in women. www.nlm.nih.gov/medlineplus/sexualproblemsinwomen.html.
- Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Pract. 2002;51(8):681.
Drug Brand Names
- Amantadine • Symadine, Symmetrel
- Avanafil • Stendra
- Bupropion • Wellbutrin, Zyban
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Fluoxetine • Prozac
- Granisetron • Kytril
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nitroglycerin • Nitrostat
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
- Tadalafil • Cialis
- Vardenafil • Levitra
- Venlafaxine • Effexor
Disclosures
Dr. Burghardt receives grant or research support from the University of Michigan Depression Center.
Ms. Gardner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Montejo AL, Llorca G, Izquierdo JA, et al. Incidence of sexual dysfunction associated with antidepressant agents: a prospective multicenter study of 1022 outpatients. Spanish Working Group for the Study of Psychotropic-Related Sexual Dysfunction. J Clin Psychiatry. 2001;62(suppl 3):10-21.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a meta-analysis. J Clin Psychopharmacol. 2009;29(3):259-266.
4. Clayton AH, Pradko JF, Croft HA, et al. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63(4):357-366.
5. Lewis RW, Fugl-Meyer KS, Bosch R, et al. Epidemiology/risk factors of sexual dysfunction. J Sex Med. 2004;1(1):35-39.
6. Nurnberg HG, Hensley PL, Heiman JR, et al. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA. 2008;300(4):395-404.
7. Taylor MJ, Rudkin L, Hawton K. Strategies for managing antidepressant-induced sexual dysfunction: systematic review of randomised controlled trials. J Affect Disord. 2005;88(3):241-254.
8. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509.
9. Brown DA, Kyle JA, Ferrill MJ. Assessing the clinical efficacy of sildenafil for the treatment of female sexual dysfunction. Ann Pharmacother. 2009;43(7):1275-1285.
Kyle J. Burghardt, PharmD
Psychiatric Pharmacogenomics Research Fellow, University of Michigan College of Pharmacy, Ann Arbor, MI
Kristen N. Gardner, BS
PharmD candidate, University of Michigan College of Pharmacy, Ann Arbor, MI
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Kyle J. Burghardt, PharmD
Psychiatric Pharmacogenomics Research Fellow, University of Michigan College of Pharmacy, Ann Arbor, MI
Kristen N. Gardner, BS
PharmD candidate, University of Michigan College of Pharmacy, Ann Arbor, MI
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Kyle J. Burghardt, PharmD
Psychiatric Pharmacogenomics Research Fellow, University of Michigan College of Pharmacy, Ann Arbor, MI
Kristen N. Gardner, BS
PharmD candidate, University of Michigan College of Pharmacy, Ann Arbor, MI
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
- Sexual dysfunction can arise from environmental, social, medical, or drug effects and requires a multifaceted approach to treatment.
- When possible, take a baseline sexual dysfunction measurement to assess if selective serotonin reuptake inhibitor use is correlated with onset or worsening of sexual dysfunction.
- Nonpharmacologic options should be considered before and during pharmacotherapy.
- Sildenafil may be useful for treating anorgasmia in women taking serotonergic antidepressants.
- Phosphodiesterase type 5 inhibitors are not FDA-approved for sexual dysfunction in women.
Mrs. L, age 27, has a history of major depressive disorder with symptoms of anxiety. She was managed successfully for 2 years with bupropion XL, 300 mg/d, but was switched to venlafaxine, titrated to 225 mg/d, after she developed seizures secondary to a head injury sustained in a car accident. After the switch, Mrs. L’s mood deteriorated and she was hospitalized. Since then, she’s received several medication trials, including paroxetine, 30 mg/d, a selective serotonin reuptake inhibitor (SSRI), and the tricyclic antidepressant (TCA) nortriptyline, 75 mg/d, but she could not tolerate these medications because of severe xerostomia.
After taking sertraline, 150 mg/d, for 8 weeks, Mrs. L improves and has a Patient Health Questionnaire score of 6, indicating mild depression. Her initial complaints of diarrhea and nausea have resolved, but Mrs. L now reports that she and her husband are having marital difficulties because she cannot achieve orgasm during sexual intercourse. She did not have this problem when she was taking bupropion. Her husband occasionally takes the phosphodiesterase type 5 (PDE5) inhibitor sildenafil before intercourse, and Mrs. L asks you if this medication will help her achieve orgasm.
DSM-IV-TR defines sexual dysfunction as disturbances in sexual desire and/or in the sexual response cycle (excitement, plateau, orgasm, and resolution) that result in marked distress and interpersonal difficulty.1 Sexual dysfunction can occur with the use of any antidepressant with serotonergic activity; it affects an estimated 50% to 70% of patients who take SSRIs.2 Sexual dysfunction can occur with all SSRIs; however, higher rates of sexual dysfunction are found with citalopram, fluoxetine, paroxetine, and sertraline.3 Studies have suggested there may be a dose-side effect relationship with SSRI-induced sexual dysfunction.4
Several factors can increase a patient’s risk of sexual dysfunction and should be considered before prescribing an antidepressant or when a patient presents with new or worsening sexual dysfunction (Table 1).5 In general, nonserotonergic agents such as bupropion, mirtazapine, and nefazodone are associated with lower rates of sexual dysfunction. The pharmacology of these agents explains their decreased propensity to cause sexual dysfunction. These agents increase levels of dopamine in the mesolimbic dopaminergic system either by blocking reuptake (bupropion) or antagonizing the serotonin subtype-2 receptor and facilitating disinhibition of decreased dopamine downstream (nefazodone and mirtazapine).
Table 1
Risk factors for sexual dysfunction
| Sex | Risk factors |
|---|---|
| Women | History of sexual, physical, or emotional abuse, physical inactivity |
| Men | Severe hyperprolactinemia, smoking |
| Both sexes | Poor to fair health, genitourinary disease, diabetes mellitus, cardiovascular disease, hypertension, increasing age, psychiatric disorders, relationship difficulties |
| Source: Reference 5 | |
One option for treating antidepressant-induced sexual dysfunction in women is PDE5 inhibitors, which are used to treat erectile dysfunction (ED). These medications ameliorate ED by inhibiting degradation of cyclic guanosine monophosphate by PDE5, which increases blood flow to the penis during sexual stimulation. Although these medications are not FDA-approved for treating sexual dysfunction in women, adjunctive PDE5 inhibitor treatment may be beneficial for sexual dysfunction in females because similar mediators, such as nitric oxide and cyclic guanosine monophosphate, involved in the nonadrenergic-noncholinergic signaling that controls sexual stimulation in men also are found in female genital tissue.6
When treating a woman with SSRI-induced sexual dysfunction, consider nonpharmacologic treatments both before and during pharmacotherapy (Table 2).7,8 See Table 3 for a comparison of pharmacokinetics, side effects, and drug interactions of the 4 FDA-approved PDE5 inhibitors—avanafil, sildenafil, tadalafil, and vardenafil.
Table 2
Management strategies for SSRI-induced sexual dysfunction
| Intervention | Comments |
|---|---|
| Nonpharmacologic | |
| Lifestyle modifications | Encourage healthy eating, weight loss, smoking cessation, substance abuse treatment, or minimizing alcohol intake to improve patient self-image and overall health |
| Cognitive-behavioral therapy | Patients can identify coping strategies for reducing symptom severity and preventing worsening sexual dysfunction |
| Sex therapy | May benefit patients with relationship difficulties |
| ‘Watch and wait’ | Spontaneous resolving (or ‘adaptation’) of sexual dysfunction with antidepressants can take ≥6 months. Studies have found adaptation rates generally are low (~10%) |
| Pharmacologic | |
| Drug holiday | May be an option for patients taking antidepressants with shorter half-lives and patients taking lower doses. Be cautious of empowering patients to stop their own medications as needed |
| Dosage reduction | Serotonergic antidepressant-induced sexual dysfunction may be related to dose. Little research has been conducted on this method and the patient’s clinical status must be considered |
| Dose timing | Instructing a patient to take the antidepressant after his or her usual time of sexual activity (eg, patients who engage in sexual activity at night should take the antidepressant before falling asleep). This may allow the drug level to be lowest during sexual activity |
| Switching medications | Case reports, retrospective studies, and RCTs suggest switching to a different antidepressant with less serotonergic activity may be appropriate, particularly if the patient has not responded to the current antidepressant |
| Adjunctive therapy | RCTs support adjunctive bupropion (≥300 mg/d) or olanzapine (5 mg/d) as treatment for SSRI-induced sexual dysfunction in women Studies have found no improvement in sexual functioning with adjunctive buspirone, granisetron, amantadine, mirtazapine, yohimbine, ephedrine, or ginkgo biloba in women |
| RCTs: randomized controlled trials; SSRI: selective serotonin reuptake inhibitor Source: Reference 7,8 | |
Table 3
Phosphodiesterase type 5 inhibitors: A comparison
| Medication | Dose rangea | Pharmacokinetics | Side effects | Significant drug interactions |
|---|---|---|---|---|
| Avanafil | 50 to 200 mg, 30 minutes before sexual activity | Bioavailability: N/A (high-fat meal delays Tmax by 60 minutes and reduces Cmax by 24% to 39%; clinically insignificant) Half life: 5 hours Metabolism: CYP3A4 | Headache, flushing, nasal congestion, nasopharyngitis, backache | Strong CYP3A4 inhibitors (increased avanafil levels) Contraindicated within 12 hours of nitrate use (eg, nitroglycerin) |
| Sildenafil | 25 to 100 mg, 1 to 2 hours before sexual activity | Bioavailability: 41% (food/high-fat meal delays Tmax by 60 minutes and reduces Cmax by 29%) Half life: 4 hours Metabolism: CYP3A4 | Headache, flushing, erythema, indigestion, insomnia, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased sildenafil levels) Contraindicated within 24 hours of nitrate use |
| Tadalafil | 10 to 20 mg, 30 minutes before sexual activity | Bioavailability: N/A (not affected by food) Half life: 17.5 hours (duration of action up to 36 hours) Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, myalgia, and back pain | Strong CYP3A4 inhibitors (increased tadalafil levels) Contraindicated within 48 hours of nitrate use |
| Vardenafil | 5 to 20 mg, 30 minutes to 2 hours before sexual activity | Bioavailability: 15% for film-coated tablet (high-fat meal reduces Cmax by 18% to 50%) Half life: 4 to 5 hours Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased vardenafil levels) Contraindicated within 24 hours of nitrate use |
| aTypical dose range for treatment of erectile dysfunction Cmax: maximum concentration; CYP: cytochrome P450; Tmax: time to maximum concentration Source: Micromedex® Healthcare Series [Internet database]. Greenwood Village, CO: Thomson Healthcare. Accessed October 10, 2012 | ||||
Limited evidence for sildenafil
Case reports, a few small open-label trials, and 1 prospective, randomized controlled trial (RCT) have evaluated sildenafil as an adjunctive treatment for serotonergic antidepressant-associated sexual dysfunction in women.6,9 Nurnberg et al6 examined the efficacy of adjunctive sildenafil in women with SSRI-induced sexual dysfunction. This 8-week, placebo-controlled, double-blind, RCT used a flexible dose (50 or 100 mg), intention-to-treat design to assess the effect of sildenafil on 98 premenopausal women whose depression was in remission. Ten patients were taking the serotonin-norepinephrine inhibitor venlafaxine, 1 was taking the TCA clomipramine, and 87 were receiving an SSRI. Patients were instructed to take sildenafil or placebo 1 to 2 hours before sexual activity. The primary outcome was mean change from baseline on the Clinical Global Impression-Sexual Function (CGI-SF) scale.
Women taking sildenafil showed significant improvement compared with those taking placebo, with a treatment difference between groups of 0.8 (95% CI, 0.6 to 1.0; =.001). Additionally, 23% of sildenafil-treated patients reported no improvement with the intervention, compared with 73% of patients receiving placebo. Secondary outcomes using 3 validated scales that evaluated specific phases of sexual function found that patients’ orgasmic function significantly benefited from sildenafil treatment, while desire, arousal, and overall satisfaction were not significantly different.
Although these findings seem to support sildenafil for treating serotonergic antidepressant-associated sexual dysfunction in women, the study had a relatively small treatment effect in a well-defined patient population; therefore, replication in future trials and different patient populations is warranted. Overall, sildenafil was well tolerated, despite patient reports of headaches, flushing, visual disturbances, dyspepsia, nasal congestion, and palpitations. Finally, cost vs benefit should be considered; PDE5 inhibitors may not be covered by insurance or may require prior authorization.
CASE CONTINUED: Symptoms resolve
Bupropion is not an appropriate choice for Mrs. L because of her seizure risk. Mirtazapine is ruled out because in the past she experienced excessive somnolence that impaired her ability to function. You are not comfortable prescribing nefazodone because of its risk of hepatotoxicity or suggesting that Mrs. L take a “drug holiday” (stop taking any antidepressants for a short period) because of the risk of depressive relapse. You suggest that Mrs. L continue to take sertraline because sometimes antidepressant-induced sexual dysfunction resolves after ≥6 months of treatment with the same agent, but she is adamant that her relationship with her husband will deteriorate if she waits that long. She also declines cognitive-behavioral therapy because her job doesn’t allow the time or flexibility to commit to the sessions.
You prescribe sildenafil, 50 mg, and instruct Mrs. L to take 1 tablet 1 to 2 hours before sexual activity. This treatment improves her ability to achieve orgasm. She tolerates the drug well and after 8 weeks of treatment her CGI-SF score improves from 6 at baseline, indicating extreme dysfunction, to 2, indicating normal function. Ten months into her sertraline treatment, Mrs. L discovers she no longer requires sildenafil to achieve orgasm.
Related Resources
- Nurnberg HG. An evidence-based review updating the various treatment and management approaches to serotonin reuptake inhibitor-associated sexual dysfunction. Drugs Today (Barc). 2008;44(2):147-168.
- NIH Medline Plus. Sexual problems in women. www.nlm.nih.gov/medlineplus/sexualproblemsinwomen.html.
- Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Pract. 2002;51(8):681.
Drug Brand Names
- Amantadine • Symadine, Symmetrel
- Avanafil • Stendra
- Bupropion • Wellbutrin, Zyban
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Fluoxetine • Prozac
- Granisetron • Kytril
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nitroglycerin • Nitrostat
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
- Tadalafil • Cialis
- Vardenafil • Levitra
- Venlafaxine • Effexor
Disclosures
Dr. Burghardt receives grant or research support from the University of Michigan Depression Center.
Ms. Gardner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
- Sexual dysfunction can arise from environmental, social, medical, or drug effects and requires a multifaceted approach to treatment.
- When possible, take a baseline sexual dysfunction measurement to assess if selective serotonin reuptake inhibitor use is correlated with onset or worsening of sexual dysfunction.
- Nonpharmacologic options should be considered before and during pharmacotherapy.
- Sildenafil may be useful for treating anorgasmia in women taking serotonergic antidepressants.
- Phosphodiesterase type 5 inhibitors are not FDA-approved for sexual dysfunction in women.
Mrs. L, age 27, has a history of major depressive disorder with symptoms of anxiety. She was managed successfully for 2 years with bupropion XL, 300 mg/d, but was switched to venlafaxine, titrated to 225 mg/d, after she developed seizures secondary to a head injury sustained in a car accident. After the switch, Mrs. L’s mood deteriorated and she was hospitalized. Since then, she’s received several medication trials, including paroxetine, 30 mg/d, a selective serotonin reuptake inhibitor (SSRI), and the tricyclic antidepressant (TCA) nortriptyline, 75 mg/d, but she could not tolerate these medications because of severe xerostomia.
After taking sertraline, 150 mg/d, for 8 weeks, Mrs. L improves and has a Patient Health Questionnaire score of 6, indicating mild depression. Her initial complaints of diarrhea and nausea have resolved, but Mrs. L now reports that she and her husband are having marital difficulties because she cannot achieve orgasm during sexual intercourse. She did not have this problem when she was taking bupropion. Her husband occasionally takes the phosphodiesterase type 5 (PDE5) inhibitor sildenafil before intercourse, and Mrs. L asks you if this medication will help her achieve orgasm.
DSM-IV-TR defines sexual dysfunction as disturbances in sexual desire and/or in the sexual response cycle (excitement, plateau, orgasm, and resolution) that result in marked distress and interpersonal difficulty.1 Sexual dysfunction can occur with the use of any antidepressant with serotonergic activity; it affects an estimated 50% to 70% of patients who take SSRIs.2 Sexual dysfunction can occur with all SSRIs; however, higher rates of sexual dysfunction are found with citalopram, fluoxetine, paroxetine, and sertraline.3 Studies have suggested there may be a dose-side effect relationship with SSRI-induced sexual dysfunction.4
Several factors can increase a patient’s risk of sexual dysfunction and should be considered before prescribing an antidepressant or when a patient presents with new or worsening sexual dysfunction (Table 1).5 In general, nonserotonergic agents such as bupropion, mirtazapine, and nefazodone are associated with lower rates of sexual dysfunction. The pharmacology of these agents explains their decreased propensity to cause sexual dysfunction. These agents increase levels of dopamine in the mesolimbic dopaminergic system either by blocking reuptake (bupropion) or antagonizing the serotonin subtype-2 receptor and facilitating disinhibition of decreased dopamine downstream (nefazodone and mirtazapine).
Table 1
Risk factors for sexual dysfunction
| Sex | Risk factors |
|---|---|
| Women | History of sexual, physical, or emotional abuse, physical inactivity |
| Men | Severe hyperprolactinemia, smoking |
| Both sexes | Poor to fair health, genitourinary disease, diabetes mellitus, cardiovascular disease, hypertension, increasing age, psychiatric disorders, relationship difficulties |
| Source: Reference 5 | |
One option for treating antidepressant-induced sexual dysfunction in women is PDE5 inhibitors, which are used to treat erectile dysfunction (ED). These medications ameliorate ED by inhibiting degradation of cyclic guanosine monophosphate by PDE5, which increases blood flow to the penis during sexual stimulation. Although these medications are not FDA-approved for treating sexual dysfunction in women, adjunctive PDE5 inhibitor treatment may be beneficial for sexual dysfunction in females because similar mediators, such as nitric oxide and cyclic guanosine monophosphate, involved in the nonadrenergic-noncholinergic signaling that controls sexual stimulation in men also are found in female genital tissue.6
When treating a woman with SSRI-induced sexual dysfunction, consider nonpharmacologic treatments both before and during pharmacotherapy (Table 2).7,8 See Table 3 for a comparison of pharmacokinetics, side effects, and drug interactions of the 4 FDA-approved PDE5 inhibitors—avanafil, sildenafil, tadalafil, and vardenafil.
Table 2
Management strategies for SSRI-induced sexual dysfunction
| Intervention | Comments |
|---|---|
| Nonpharmacologic | |
| Lifestyle modifications | Encourage healthy eating, weight loss, smoking cessation, substance abuse treatment, or minimizing alcohol intake to improve patient self-image and overall health |
| Cognitive-behavioral therapy | Patients can identify coping strategies for reducing symptom severity and preventing worsening sexual dysfunction |
| Sex therapy | May benefit patients with relationship difficulties |
| ‘Watch and wait’ | Spontaneous resolving (or ‘adaptation’) of sexual dysfunction with antidepressants can take ≥6 months. Studies have found adaptation rates generally are low (~10%) |
| Pharmacologic | |
| Drug holiday | May be an option for patients taking antidepressants with shorter half-lives and patients taking lower doses. Be cautious of empowering patients to stop their own medications as needed |
| Dosage reduction | Serotonergic antidepressant-induced sexual dysfunction may be related to dose. Little research has been conducted on this method and the patient’s clinical status must be considered |
| Dose timing | Instructing a patient to take the antidepressant after his or her usual time of sexual activity (eg, patients who engage in sexual activity at night should take the antidepressant before falling asleep). This may allow the drug level to be lowest during sexual activity |
| Switching medications | Case reports, retrospective studies, and RCTs suggest switching to a different antidepressant with less serotonergic activity may be appropriate, particularly if the patient has not responded to the current antidepressant |
| Adjunctive therapy | RCTs support adjunctive bupropion (≥300 mg/d) or olanzapine (5 mg/d) as treatment for SSRI-induced sexual dysfunction in women Studies have found no improvement in sexual functioning with adjunctive buspirone, granisetron, amantadine, mirtazapine, yohimbine, ephedrine, or ginkgo biloba in women |
| RCTs: randomized controlled trials; SSRI: selective serotonin reuptake inhibitor Source: Reference 7,8 | |
Table 3
Phosphodiesterase type 5 inhibitors: A comparison
| Medication | Dose rangea | Pharmacokinetics | Side effects | Significant drug interactions |
|---|---|---|---|---|
| Avanafil | 50 to 200 mg, 30 minutes before sexual activity | Bioavailability: N/A (high-fat meal delays Tmax by 60 minutes and reduces Cmax by 24% to 39%; clinically insignificant) Half life: 5 hours Metabolism: CYP3A4 | Headache, flushing, nasal congestion, nasopharyngitis, backache | Strong CYP3A4 inhibitors (increased avanafil levels) Contraindicated within 12 hours of nitrate use (eg, nitroglycerin) |
| Sildenafil | 25 to 100 mg, 1 to 2 hours before sexual activity | Bioavailability: 41% (food/high-fat meal delays Tmax by 60 minutes and reduces Cmax by 29%) Half life: 4 hours Metabolism: CYP3A4 | Headache, flushing, erythema, indigestion, insomnia, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased sildenafil levels) Contraindicated within 24 hours of nitrate use |
| Tadalafil | 10 to 20 mg, 30 minutes before sexual activity | Bioavailability: N/A (not affected by food) Half life: 17.5 hours (duration of action up to 36 hours) Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, myalgia, and back pain | Strong CYP3A4 inhibitors (increased tadalafil levels) Contraindicated within 48 hours of nitrate use |
| Vardenafil | 5 to 20 mg, 30 minutes to 2 hours before sexual activity | Bioavailability: 15% for film-coated tablet (high-fat meal reduces Cmax by 18% to 50%) Half life: 4 to 5 hours Metabolism: CYP3A4 | Headache, flushing, indigestion, nasal congestion, dizziness, visual disturbances (blue vision) | Strong CYP3A4 inhibitors (increased vardenafil levels) Contraindicated within 24 hours of nitrate use |
| aTypical dose range for treatment of erectile dysfunction Cmax: maximum concentration; CYP: cytochrome P450; Tmax: time to maximum concentration Source: Micromedex® Healthcare Series [Internet database]. Greenwood Village, CO: Thomson Healthcare. Accessed October 10, 2012 | ||||
Limited evidence for sildenafil
Case reports, a few small open-label trials, and 1 prospective, randomized controlled trial (RCT) have evaluated sildenafil as an adjunctive treatment for serotonergic antidepressant-associated sexual dysfunction in women.6,9 Nurnberg et al6 examined the efficacy of adjunctive sildenafil in women with SSRI-induced sexual dysfunction. This 8-week, placebo-controlled, double-blind, RCT used a flexible dose (50 or 100 mg), intention-to-treat design to assess the effect of sildenafil on 98 premenopausal women whose depression was in remission. Ten patients were taking the serotonin-norepinephrine inhibitor venlafaxine, 1 was taking the TCA clomipramine, and 87 were receiving an SSRI. Patients were instructed to take sildenafil or placebo 1 to 2 hours before sexual activity. The primary outcome was mean change from baseline on the Clinical Global Impression-Sexual Function (CGI-SF) scale.
Women taking sildenafil showed significant improvement compared with those taking placebo, with a treatment difference between groups of 0.8 (95% CI, 0.6 to 1.0; =.001). Additionally, 23% of sildenafil-treated patients reported no improvement with the intervention, compared with 73% of patients receiving placebo. Secondary outcomes using 3 validated scales that evaluated specific phases of sexual function found that patients’ orgasmic function significantly benefited from sildenafil treatment, while desire, arousal, and overall satisfaction were not significantly different.
Although these findings seem to support sildenafil for treating serotonergic antidepressant-associated sexual dysfunction in women, the study had a relatively small treatment effect in a well-defined patient population; therefore, replication in future trials and different patient populations is warranted. Overall, sildenafil was well tolerated, despite patient reports of headaches, flushing, visual disturbances, dyspepsia, nasal congestion, and palpitations. Finally, cost vs benefit should be considered; PDE5 inhibitors may not be covered by insurance or may require prior authorization.
CASE CONTINUED: Symptoms resolve
Bupropion is not an appropriate choice for Mrs. L because of her seizure risk. Mirtazapine is ruled out because in the past she experienced excessive somnolence that impaired her ability to function. You are not comfortable prescribing nefazodone because of its risk of hepatotoxicity or suggesting that Mrs. L take a “drug holiday” (stop taking any antidepressants for a short period) because of the risk of depressive relapse. You suggest that Mrs. L continue to take sertraline because sometimes antidepressant-induced sexual dysfunction resolves after ≥6 months of treatment with the same agent, but she is adamant that her relationship with her husband will deteriorate if she waits that long. She also declines cognitive-behavioral therapy because her job doesn’t allow the time or flexibility to commit to the sessions.
You prescribe sildenafil, 50 mg, and instruct Mrs. L to take 1 tablet 1 to 2 hours before sexual activity. This treatment improves her ability to achieve orgasm. She tolerates the drug well and after 8 weeks of treatment her CGI-SF score improves from 6 at baseline, indicating extreme dysfunction, to 2, indicating normal function. Ten months into her sertraline treatment, Mrs. L discovers she no longer requires sildenafil to achieve orgasm.
Related Resources
- Nurnberg HG. An evidence-based review updating the various treatment and management approaches to serotonin reuptake inhibitor-associated sexual dysfunction. Drugs Today (Barc). 2008;44(2):147-168.
- NIH Medline Plus. Sexual problems in women. www.nlm.nih.gov/medlineplus/sexualproblemsinwomen.html.
- Sturpe DA, Mertens MK, Scoville C. What are the treatment options for SSRI-related sexual dysfunction? J Fam Pract. 2002;51(8):681.
Drug Brand Names
- Amantadine • Symadine, Symmetrel
- Avanafil • Stendra
- Bupropion • Wellbutrin, Zyban
- Buspirone • BuSpar
- Citalopram • Celexa
- Clomipramine • Anafranil
- Fluoxetine • Prozac
- Granisetron • Kytril
- Mirtazapine • Remeron
- Nefazodone • Serzone
- Nitroglycerin • Nitrostat
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Sertraline • Zoloft
- Sildenafil • Viagra
- Tadalafil • Cialis
- Vardenafil • Levitra
- Venlafaxine • Effexor
Disclosures
Dr. Burghardt receives grant or research support from the University of Michigan Depression Center.
Ms. Gardner reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Montejo AL, Llorca G, Izquierdo JA, et al. Incidence of sexual dysfunction associated with antidepressant agents: a prospective multicenter study of 1022 outpatients. Spanish Working Group for the Study of Psychotropic-Related Sexual Dysfunction. J Clin Psychiatry. 2001;62(suppl 3):10-21.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a meta-analysis. J Clin Psychopharmacol. 2009;29(3):259-266.
4. Clayton AH, Pradko JF, Croft HA, et al. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63(4):357-366.
5. Lewis RW, Fugl-Meyer KS, Bosch R, et al. Epidemiology/risk factors of sexual dysfunction. J Sex Med. 2004;1(1):35-39.
6. Nurnberg HG, Hensley PL, Heiman JR, et al. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA. 2008;300(4):395-404.
7. Taylor MJ, Rudkin L, Hawton K. Strategies for managing antidepressant-induced sexual dysfunction: systematic review of randomised controlled trials. J Affect Disord. 2005;88(3):241-254.
8. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509.
9. Brown DA, Kyle JA, Ferrill MJ. Assessing the clinical efficacy of sildenafil for the treatment of female sexual dysfunction. Ann Pharmacother. 2009;43(7):1275-1285.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Montejo AL, Llorca G, Izquierdo JA, et al. Incidence of sexual dysfunction associated with antidepressant agents: a prospective multicenter study of 1022 outpatients. Spanish Working Group for the Study of Psychotropic-Related Sexual Dysfunction. J Clin Psychiatry. 2001;62(suppl 3):10-21.
3. Serretti A, Chiesa A. Treatment-emergent sexual dysfunction related to antidepressants: a meta-analysis. J Clin Psychopharmacol. 2009;29(3):259-266.
4. Clayton AH, Pradko JF, Croft HA, et al. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63(4):357-366.
5. Lewis RW, Fugl-Meyer KS, Bosch R, et al. Epidemiology/risk factors of sexual dysfunction. J Sex Med. 2004;1(1):35-39.
6. Nurnberg HG, Hensley PL, Heiman JR, et al. Sildenafil treatment of women with antidepressant-associated sexual dysfunction: a randomized controlled trial. JAMA. 2008;300(4):395-404.
7. Taylor MJ, Rudkin L, Hawton K. Strategies for managing antidepressant-induced sexual dysfunction: systematic review of randomised controlled trials. J Affect Disord. 2005;88(3):241-254.
8. Balon R. SSRI-associated sexual dysfunction. Am J Psychiatry. 2006;163(9):1504-1509.
9. Brown DA, Kyle JA, Ferrill MJ. Assessing the clinical efficacy of sildenafil for the treatment of female sexual dysfunction. Ann Pharmacother. 2009;43(7):1275-1285.
Drug interactions with tobacco smoke: Implications for patient care
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Martha P. Fankhauser, MS Pharm, FASHP, BCPP
Clinical Professor, Department of Pharmacy Practice and Science, College of Pharmacy and Pharmacotherapy Specialist, Arizona Smokers' Helpline, Mel and Enid Zuckerman College of Public Health, University of Arizona, Tucson, AZ
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
- Tobacco smokers often are treated with medications that are metabolized by hepatic cytochrome (CYP) 1A2 enzymes. Starting or stopping tobacco smoking may cause drug interactions because polycyclic aromatic hydrocarbons in cigarette smoke induce CYP1A2 enzymes.
- Drugs that are significantly metabolized by CYP1A2 (major substrates) are more likely to be impacted by changes in tobacco smoking compared with minor substrates.
- Induction of hepatic CYP1A2 enzymes may be greater in heavy or moderate smokers compared with light smokers (eg, <10 cigarettes per day).
- Evidence-based approaches for treating tobacco use in health care settings should address the risk of CYP1A2 drug interactions in tobacco smokers and how this impacts their clinical care.
Mrs. C, age 51, experiences exacerbated asthma and difficulty breathing and is admitted to a non-smoking hospital. She also has chronic obstructive pulmonary disease, type 2 diabetes mellitus, hypertension, hypercholesterolemia, hypothyroidism, gastroesophageal reflux disease, overactive bladder, muscle spasms, fibromyalgia, bipolar disorder, insomnia, and nicotine and caffeine dependence. She takes 19 prescribed and over-the-counter medications, drinks up to 8 cups of coffee per day, and smokes 20 to 30 cigarettes per day. In the emergency room, she receives albuterol/ipratropium inhalation therapy to help her breathing and a 21-mg nicotine replacement patch to avoid nicotine withdrawal.
In the United States, 19% of adults smoke cigarettes.1 Heavy tobacco smoking and nicotine dependence are common among psychiatric patients and contribute to higher rates of tobacco-related morbidity and mortality.2 When smokers stop smoking or are admitted to smoke-free facilities and are forced to abstain, nicotine withdrawal symptoms and changes in drug metabolism can develop over several days.3-5
Smokers such as Mrs. C are at risk for cytochrome (CYP) P450 drug interactions when they are admitted to or discharged from a smoke-free facility. Nine of Mrs. C’s medications are substrates of CYP1A2 (acetaminophen, caffeine, cyclobenzaprine, diazepam, duloxetine, melatonin, olanzapine, ondansetron, and zolpidem). When Mrs. C stops smoking while in the hospital, she could experience higher serum concentrations and adverse effects of these medications. If Mrs. C resumes smoking after bring discharged, metabolism and clearance of any medications started while she was hospitalized that are substrates of CYP1A2 enzymes could be increased, which could lead to reduced efficacy and poor clinical outcomes.
Pharmacokinetic effects
Polycyclic aromatic hydrocarbons in tobacco smoke induce hepatic CYP1A1, 1A2, and possibly 2E1 isoenzymes.6-12 CYP1A2 is a hepatic enzyme responsible for metabolizing and eliminating several classes of substrates (eg, drugs, hormones, endogenous compounds, and procarcinogens).6,13 Genetic, epigenetic, and environmental factors such as smoking impact the expression and activity of CYP1A2 and result in large interpatient variability in pharmacokinetic drug interactions.6,12,13 CYP1A2 enzymes can be induced or inhibited by drugs and substances, which can result in decreased or increased serum concentrations of substrates, respectively. When individuals stop smoking and switch to other nicotine products or devices, CYP1A2 induction of hepatic enzymes will revert to normal metabolism over several weeks to a month.10 Besides tobacco smoke, other CYP1A2 inducers include charbroiled food, carbamazepine, omeprazole, phenobarbital, primidone, and rifampin.4,5 Nicotine replacement products—such as gum, inhalers, lozenges, patches, and nasal spray—and nicotine delivery devices such as electronic cigarettes do not induce hepatic CYP1A2 enzymes or cause the same drug interactions as cigarette smoking.
Table 13-11 and Table 23-11 list commonly prescribed CYP1A2 substrates that could be affected by tobacco smoke. There are no specific guidelines for how to assess, monitor, or manage pharmacokinetic drug interactions with tobacco smoke.6-13 Induction of hepatic CYP1A2 enzymes by cigarette smoke may require increased dosages of some psychotropics—such as tricyclic antidepressants, duloxetine, mirtazapine, and some first- and second-generation antipsychotics (SGAs)—to achieve serum concentrations adequate for clinical efficacy. Serum concentrations may increase to toxic levels and result in adverse effects when a person quits smoking cigarettes or if a CYP1A2 inhibitor, such as amlodipine, cimetidine, ciprofloxacin, diclofenac, fluoxetine, fluvoxamine, or nifedipine, is added.5
Table 1
Common major cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Alosetron3,5,6 | Irritable bowel syndrome: serotonin 3 antagonist |
| Aminophylline3,5 | Bronchodilator: theophylline derivative |
| Betaxolol3,5 | β-1 selective adrenergic receptor blocking agent |
| Caffeine3-9 | Stimulant |
| Clomipramine3-11 | Tricyclic antidepressant |
| Clozapine3-10 | Second-generation antipsychotic |
| Cyclobenzaprine3-7 | Skeletal muscle relaxant |
| Doxepin3,7,10,11 | Tricyclic antidepressant |
| Duloxetine3-6 | Serotonin-norepinephrine reuptake inhibitor |
| Estradiol3,5-8 | Estrogen (active) |
| Estrogens: conjugated and estropipate3,5; estrone3,7 | Estrogen (derivatives) |
| Fluvoxamine3,8,9 | Selective serotonin reuptake inhibitor |
| Guanabenz3,5-7 | α-2 adrenergic agonist |
| Mirtazapine3-7 | Antidepressant: α-2 antagonist/serotonin 2A, 2C antagonist |
| Olanzapine3-11 | Second-generation antipsychotic |
| Pimozide3,5,7 | First-generation antipsychotic |
| Propranolol3-11 | β-adrenergic blocker |
| Ramelteon3,5,10 | Melatonin receptor agonist |
| Rasagiline3,5 | Antiparkinson: type B monoamine oxidase inhibitor |
| Riluzole3-7,10 | Glutamate inhibitor |
| Ropinirole3,5-7 | Antiparkinson: dopamine agonist |
| Theophylline3-6,8-11 | Bronchodilator: methylxanthine |
| Thiothixene3,5 | First-generation antipsychotic |
| Trifluoperazine3,5,9 | First-generation antipsychotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral, and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
Table 2
Common minor cytochrome P450 (CYP) 1A2 substrates
| Drug | Class |
|---|---|
| Acetaminophen3-9 | Analgesic |
| Almotriptan6 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Amitriptyline3-7,9-11 | Tricyclic antidepressant |
| Asenapine9 | Second-generation antipsychotic |
| Carvedilol5-7 | β and α adrenergic blocking activity |
| Chlorpromazine3,4,7-9,11 | First-generation antipsychotic |
| Chlorzoxazone4,7 | Skeletal muscle relaxant |
| Clopidogrel5 | Antiplatelet |
| Desipramine4,7,10,11 | Tricyclic antidepressant |
| Diazepam4,7,9,10 | Benzodiazepine |
| Diclofenac5,7 | Nonsteroidal anti-inflammatory drug |
| Diphenhydramine6 | Antihistamine |
| Febuxostat5 | Xanthine oxidase inhibitor |
| Fluphenazine3,9 | First-generation antipsychotic |
| Frovatriptan3 | Antimigraine: serotonin 1 agonist |
| Haloperidol3,4,6,8,9 | First-generation antipsychotic |
| Imipramine3,4,6-11 | Tricyclic antidepressant |
| Maprotiline6 | Tetracyclic antidepressant |
| Melatonin3,4,6,7 | Sleep-regulating hormone |
| Metoclopramide3 | Antiemetic: prokinetic gastrointestinal agent |
| Nabumetone6 | Nonsteroidal anti-inflammatory drug |
| Naproxen3,4,6,7 | Nonsteroidal anti-inflammatory drug |
| Naratriptan10 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Nicardipine3,7 | Calcium channel blocker |
| Nortriptyline4,6,7,9-11 | Tricyclic antidepressant |
| Ondansetron3,4,6,7 | Antiemetic: serotonin 3 antagonist |
| Palonosetron5 | Antiemetic: serotonin 3 antagonist |
| Perphenazine3,7 | First-generation antipsychotic |
| Progesterone5,7 | Progestin |
| Propofol4,6,7 | General anesthetic |
| Ranitidine5,7 | H2 antagonist |
| Rivastigmine10 | Acetylcholinesterase inhibitor |
| Selegiline6,7 | Antiparkinson: type B monoamine oxidase inhibitor |
| Thioridazine3,4,6 | First-generation antipsychotic |
| Tizanidine3-6 | Skeletal muscle relaxant: α-2 adrenergic agonist |
| Trazodone6,9 | Serotonin reuptake inhibitor and antagonist |
| Triamterene6 | Diuretic: potassium sparing |
| Verapamil3,4,6,7,10 | Calcium channel blocker |
| Warfarin3,4,6-10 | Anticoagulant: coumarin derivative |
| Zileuton3,4,6,7 | Asthma agent: 5-lipoxygenase inhibitor |
| Ziprasidone3,4 | Second-generation antipsychotic |
| Zolmitriptan3,6,7 | Antimigraine: serotonin 1B, 1D receptor agonist |
| Zolpidem4,6,7 | Nonbenzodiazepine hypnotic |
| Several classes of CYP1A2 substrates are not included and may cause toxicity with smoking cessation or require dosage increases in tobacco smokers (eg, antiarrhythmic, antifungal, antimalarial, antineoplastic, antiretroviral and anthelmintic agents and the antibiotic quinolone). Clinicians should be most concerned about drugs with a narrow therapeutic index and those that may be toxic with smoking cessation (eg, bleeding from warfarin and clopidogrel; high serum concentrations of caffeine, clozapine, olanzapine, propranolol, and theophylline) | |
SGA such as clozapine and olanzapine are major substrates of CYP1A2 and dosages may need to be adjusted when smoking status changes, depending on clinical efficacy and adverse effects.10,14,15 Maximum induction of clozapine and olanzapine metabolism may occur with 7 to 12 cigarettes per day and smokers may have 40% to 50% lower serum concentrations compared with nonsmokers.14 When a patient stops smoking, clozapine and olanzapine dosages may need to be reduced by 30% to 40% (eg, a stepwise 10% reduction in daily dose until day 4) to avoid elevated serum concentrations and risk of toxicity symptoms.15
Tobacco smokers can tolerate high daily intake of caffeinated beverages because of increased metabolism and clearance of caffeine, a major substrate of CYP1A2.11 When patients stop smoking, increased caffeine serum concentrations may cause anxiety, irritability, restlessness, insomnia, tremors, palpitations, and tachycardia. Caffeine toxicity also can mimic symptoms of nicotine withdrawal; therefore, smokers should be advised to reduce their caffeine intake by half to avoid adverse effects when they stop smoking.10,11
Adjusting dosing
Factors such as the amount and frequency of tobacco smoking, how quickly CYP1A2 enzymes change when starting and stopping smoking, exposure to secondhand smoke, and other concomitant drugs contribute to variability in pharmacokinetic drug interactions. Heavy smokers (≥30 cigarettes per day) should be closely monitored because variations in drug serum concentrations may be affected significantly by changes in smoking status.4,9,11 Dosage reductions of potentially toxic drugs should be done immediately when a heavy tobacco user stops smoking.10 For CYP1A2 substrates with a narrow therapeutic range, a conservative approach is to reduce the daily dose by 10% per day for several days after smoking cessation.11,16 The impact on drug metabolism may continue for weeks to a month after the person stops smoking; therefore, there may be a delay until CYP1A2 enzymes return to normal hepatic metabolism.4,8,9,15 In most situations, smoking cessation reverses induction of hepatic CYP1A2 enzymes back to normal metabolism and causes serum drug concentrations to increase.10 Because secondhand smoke induces hepatic CYP1A2 enzymes, those exposed to smoke may have changes in drug metabolism due to environmental smoke exposure.11
Tobacco smokers who take medications and hormones that are metabolized by CYP1A2 enzymes should be closely monitored because dosage adjustments may be necessary when they start or stop smoking cigarettes. An assessment of CYP drug interactions and routine monitoring of efficacy and/or toxicity should be done to avoid potential adverse effects from medications and to determine if changes in dosages and disease state management are required.
Related Resources
- Rx for Change. Drug interactions with smoking. http://smokingcessationleadership.ucsf.edu/interactions.pdf.
- Fiore MC, Baker TB. Treating smokers in the health care setting. N Engl J Med. 2011;365(13):1222-1231.
Drug Brand Names
- Albuterol/ipratropium • Combivent
- Almotriptan • Axert
- Alosetron • Lotronex
- Aminophylline • Phyllocontin, Truphylline
- Amitriptyline • Elavil
- Amlodipine • Norvasc
- Asenapine • Saphris
- Betaxolol • Kerlone
- Carbamazepine • Carbatrol, Tegretol
- Carvedilol • Coreg
- Chlorpromazine • Thorazine
- Chlorzoxazone • Parafon Forte
- Cimetidine • Tagamet
- Ciprofloxacin • Cipro
- Clomipramine • Anafranil
- Clopidogrel • Plavix
- Clozapine • Clozaril
- Cyclobenzaprine • Flexeril
- Desipramine • Norpramin
- Diazepam • Valium
- Diclofenac • Voltaren
- Diphenhydramine • Benadryl
- Doxepin • Silenor, Sinequan
- Duloxetine • Cymbalta
- Estradiol • Estrace
- Estrogens (conjugated) • Cenestin, Premarin
- Estropipate • Ogen
- Febuxostat • Uloric
- Fluoxetine • Prozac
- Fluphenazine • Prolixin
- Fluvoxamine • Luvox
- Frovatriptan • Frova
- Guanabenz • Wytensin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Maprotiline • Ludiomil
- Metoclopramide • Reglan
- Mirtazapine • Remeron
- Nabumetone • Relafen
- Naratriptan • Amerge
- Nicardipine • Cardene
- Nifedipine • Adalat, Procardia
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Omeprazole • Prilosec
- Ondansetron • Zofran
- Palonosetron • Aloxi
- Perphenazine • Trilafon
- Pimozide • Orap
- Primidone • Mysoline
- Progesterone • Prometrium
- Propofol • Diprivan
- Propranolol • Inderal
- Ramelteon • Rozerem
- Ranitidine • Zantac
- Rasagiline • Azilect
- Rifampin • Rifadin, Rimactane
- Riluzole • Rilutek
- Rivastigmine • Exelon
- Ropinirole • Requip
- Selegiline • Eldepryl, EMSAM, others
- Theophylline • Elixophyllin
- Thioridazine • Mellaril
- Thiothixene • Navane
- Tizanidine • Zanaflex
- Trazodone • Desyrel, Oleptro
- Triamterene • Dyrenium
- Trifluoperazine • Stelazine
- Verapamil • Calan, Verelan
- Warfarin • Coumadin, Jantoven
- Zileuton • Zyflo
- Ziprasidone • Geodon
- Zolmitriptan • Zomig
- Zolpidem • Ambien, Edluar
Disclosure
Ms. Fankhauser reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
1. Centers for Disease Control and Prevention (CDC). Vital signs: current cigarette smoking among adults aged ≥18 years—United States 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60(35):1207-1212.
2. Ziedonis D, Hitsman B, Beckham JC, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10(12):1691-1715.
3. Choe JY. Drug actions and interactions. New York NY: McGraw-Hill Medical; 2011.
4. Tatro DS. Drug interaction facts. St. Louis MO: Wolters Kluwer Health; 2011.
5. Lacy CF, Armstrong LL, Goldman MP, et al. eds. Drug information handbook, 20th ed. Hudson, OH: Lexicomp; 2011.
6. Zhou SF, Yang LP, Zhou ZW, et al. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481-494.
7. Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83-448.
8. Zevin S, Benowitz NL. Drug interactions with tobacco smoking. An update. Clin Pharmacokinet. 1999;36(6):425-438.
9. Desai HD, Seabolt J, Jann MW. Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs. 2001;15(6):469-494.
10. Schaffer SD, Yoon S, Zadezensky I. A review of smoking cessation: potentially risky effects on prescribed medications. J Clin Nurs. 2009;18(11):1533-1540.
11. Kroon LA. Drug interactions with smoking. Am J Health Syst Pharm. 2007;64(18):1917-1921.
12. Plowchalk DR, Yeo KR. Prediction of drug clearance in a smoking population: modeling the impact of variable cigarette consumption on the induction of CYP1A2. Eur J Pharmacol. 2012;68(6):951-960.
13. Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why how, and when? Basic Clin Pharmacol Toxicol. 2005;97(3):125-134.
14. Haslemo T, Eikeseth PH, Tanum L, et al. The effect of variable cigarette consumption on the interaction with clozapine and olanzapine. Eur J Clin Pharmacol. 2006;62(12):1049-1053.
15. Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010;44(4):727-732.
16. Faber MS, Fuhr U. Time response of cytochrome P4501A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004;76(2):178-184.
Which psychotropics carry the greatest risk of QTc prolongation?
- Screen patients for risk factors for prolonged QTc interval, such as congenital long QT syndrome, family history of cardiac conduction abnormalities, and previous occurrences of medication-mediated QTc prolongation.
- Obtain baseline and steady state ECG when initiating high-risk agents, particularly when administering combination therapy.
- Use the lowest effective dose of antidepressants and antipsychotics and monitor symptoms closely.
Mrs. A, age 68, has a 40-year history of schizoaffective disorder with comorbid anxiety disorder not otherwise specified, type 2 diabetes mellitus, and hypertension. She takes furosemide, 40 mg/d, lisinopril, 20 mg/d, and metformin, 2,000 mg/d, for hypertension and diabetes; lorazepam, 1.5 mg/d, and paroxetine, 40 mg/d, for anxiety; and quetiapine extended release, 800 mg/d, for psychotic features and mood dysregulation with schizoaffective disorder. Mrs. A’s husband died 5 years ago and she lives alone in a senior care facility. Mrs. A uses a weekly pill reminder box because her residential facility does not monitor medication adherence. She sees her psychiatrist once a month and her primary care provider every 3 months. She has no history of illicit drug, alcohol, or tobacco use.
Two weeks ago, Mrs. A was found leaning against the wall in a hallway, complaining of dizziness and disorientation, and unable to find her way back to her apartment. In the emergency department, her serum potassium is low (3.0 mEq/L; normal range: 3.5 to 5.0), fasting glucose is elevated (110 mg/dL; range: 65 to 99), and ECG reveals a prolonged QTc interval of 530 milliseconds. Before this episode, Mrs. A had been medically stable without mood or psychotic symptoms, although her daughter reported medication self-administration was becoming difficult.
Exposure to psychotropics carries a risk of QTc prolongation. The QT interval is an ECG measure of ventricular depolarization and repolarization. The QTc designation indicates a correction for heart rate with increasing heart rate correlating with a shorter QT interval. Readings of 440 milliseconds are considered normal.1 QTc prolongation is defined as >450 milliseconds for men and >470 milliseconds for women.2 An increase in the QT interval is a predictor of serious cardiac events.3
Antidepressants and antipsychotics have been associated with QTc prolongation. When identifying agents that could disrupt cardiac conduction, clinicians need to consider whether the drug’s molecular structure, receptor affinity, or pharmacologic effects are most critical.2 Although these may be important, patient-specific variables that increase the risk of QTc prolongation may have greater impact. These include:
- age >65
- female sex
- electrolyte imbalances (specifically low serum potassium and magnesium levels)
- high or toxic serum levels of the suspected drug
- preexisting cardiovascular impairment, such as bradycardia.4,5
Other risk factors include concurrent use of an agent with similar cardiovascular effects or one that competes for metabolism (either enzymatic or at the binding site), physiologic limitations such as renal insufficiency, and medication changes that may increase or decrease psychotropic clearance.4,6 Geriatric patients with dementia have an increased risk for cardiovascular-related death.7,8
Antidepressants
Among tricyclic antidepressants, most reports of QTc prolongation involve amitriptyline and maprotiline.9 Risk factors include demographics (eg, female sex, age), personal or family history (congenital long QT syndrome, cardiovascular disease), and concurrent conditions or drug use, particularly those associated with QTc prolongation.3 Desipramine and nortriptyline also have been identified as high-risk agents.10
QTc prolongation has been reported with all selective serotonin reuptake inhibitors at plasma concentrations above the therapeutic level.11 Fluoxetine-associated QTc prolongation was limited to cases of overdose or when additional risk factors were reported.4 QTc prolongation from psychotropics could increase the risk of torsades de pointes, according to an analysis of the FDA Adverse Event Reporting System.12 In 2011, the FDA reported an increased risk of abnormal heart rhythms—including QTc prolongation—with citalopram doses >40 mg/d.13 Although cases of QTc prolongation with paroxetine have not been reported,11 the Arizona Center for Education and Research on Therapeutics lists paroxetine with other agents that may increase the risk for QTc prolongation with concurrent use of medications that may prolong QTc interval.14 Venlafaxine doses >300 mg/d may require additional cardiac monitoring.5,12 Data from venlafaxine poisoning case reports found a positive correlation between dose and QTc prolongation.15 In a review of toxicology database information, Wenzel-Seifert et al4 found extended QT interval with citalopram, fluoxetine, and venlafaxine at toxic doses or in the presence of additional risk factors such as sex, older age, or personal or family history of congenital long QT syndrome or cardiovascular disease.
Antipsychotics
Case reports, case series, and research trials have evaluated the risk of QTc prolongation with antipsychotics (Table).1,2,4,16,17 The first-generation antipsychotics thioridazine,4,16,18 mesoridazine,16,18 chlorpromazine,19 and haloperidol3 warrant cardiac monitoring. The QTc prolongation effects of thioridazine and its active metabolite mesoridazine are well-documented and thioridazine-mediated QTc prolongation increases are dose-dependent.4,18 ECG monitoring is recommended with IV haloperidol, which is used for delirium in adults.20 QTc prolongation has been associated with long-term ziprasidone use more often than with risperidone, olanzapine, or quetiapine.19 Ziprasidone prolongs the QTc interval an average of 20 milliseconds,21 which could represent a clinically significant change. QTc prolongation for iloperidone is comparable to ziprasidone and haloperidol.22 There is some evidence that aripiprazole may shorten, rather than prolong, the QTc interval.4,17
Cardiovascular adverse effects associated with clozapine—including QTc prolongation—are dose-dependent.3 Olanzapine prolongs QTc interval, although the mean change is less than with other agents unless other variables were present, such as:
- concomitant use of medications that may prolong QTc interval (ie, amantadine, hydroxyzine, or tamoxifen2)
- preexisting cardiovascular conduction disorders
- higher doses (>40 mg/d).3,23
In 17 case reports of cardiac changes associated with quetiapine use, doses ranged from 100 mg/d24 to an overdose of 36 g/d.25 Only 1 patient death was reported secondary to overdose and preexisting dysrhythmia and hypertension.26 QTc prolongation associated with risperidone was minor1 based on oral doses in the normal therapeutic range and incidences of overdose.10 Paliperidone27 and lurasidone28 are associated with clinically insignificant QTc prolongation. Changes in QTc interval were positively correlated with asenapine dose, although at the highest dose of 40 mg/d, the increase was <5 milliseconds.29
Mrs. A presents with a number of risk factors for QTc prolongation, including older age, female sex, and psychiatric and medical comorbidities that require medication. A pill count revealed that she was taking more than the prescribed daily doses of her medications. During the interview, Mrs. A said that if she missed her medication time, she would take them when she remembered. If she could not remember if she took her pills, she would take them again. Her physicians will explore strategies to increase medication adherence.
Table
Examples of QTc prolongation associated with select antipsychoticsa
| Antipsychotic | Approximate QTc interval prolongation in millisecondsb |
|---|---|
| Aripiprazole4,17 | -1 to -4 |
| Clozapine4 | 10 |
| Haloperidol1,2 | 7 to 15 |
| Mesoridazine16 | 39 to 53 |
| Olanzapine1 | 2 to 6.5 |
| Paliperidone4 | 2 to 4 |
| Pimozide2 | 19 |
| Quetiapine1,2 | 6 to 15 |
| Risperidone1,2 | 3.5 to 10 |
| Sertindole1 | 30 |
| Thioridazine2,16 | 33 to 41 |
| Ziprasidone1,2 | 16 to 21 |
| aList is not comprehensive. Other antipsychotics may be associated with QTc prolongation bQTc prolongation interval may depend on the route of administration | |
Related Resources
- De Hert M, Detraux J, van Winkel R, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2011;8(2):114-126.
- Vieweg WV, Wood MA, Fernandez A, et al. Proarrhythmic risk with antipsychotic and antidepressant drugs: implications in the elderly. Drugs Aging. 2009;26(12):997-1012.
- Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Asenapine • Saphris
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Furosemide • Lasix
- Haloperidol • Haldol
- Hydroxyzine • Atarax, Vistaril
- Iloperidone • Fanapt
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Lurasidone • Latuda
- Maprotiline • Ludiomil
- Mesoridazine • Serentil
- Metformin • Glucophage
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tamoxifen • Nolvadex, Soltamox
- Thioridazine • Mellaril
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. No similar work by the authors is under review or in press. No funding was requested or received in conjunction with this manuscript.
1. Muscatello MR, Bruno A, Pandolfo G, et al. Emerging treatments in the management of schizophrenia - focus on sertindole. Drug Des Devel Ther. 2010;4:187-201.
2. Taylor DM. Antipsychotics and QT prolongation. Acta Psychiatr Scand. 2003;107(2):85-95.
3. Alvarez PA, Pahissa J. QT alterations in psychopharmacology: proven candidates and suspects. Curr Drug Saf. 2010;5(1):97-104.
4. Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of torsade de pointes. Dtsch Arztebl Int. 2011;108(41):687-693.
5. Vieweg WV. New generation antipsychotic drugs and QTc interval prolongation. Prim Care Companion J Clin Psychiatry. 2003;5(5):205-215.
6. Nielsen J, Graff C, Kanters JK, et al. Assessing QT interval prolongation and its associated risks with antipsychotics. CNS Drugs. 2011;25(6):473-490.
7. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
8. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
9. Vieweg WV, Wood MA. Tricyclic antidepressants QT interval prolongation, and torsade de pointes. Psychosomatics. 2004;45(5):371-377.
10. Jeon SH, Jaekal J, Lee SH, et al. Effects of nortriptyline on QT prolongation: a safety pharmacology study. Hum Exp Toxicol. 2011;30(10):1649-1656.
11. Wenzel-Seifert K, Wittmann M, Haen E. Torsade de pointes episodes under treatment with selective serotonin reuptake inhibitors. Pharmacopsychiatry. 2010;43(7):279-281.
12. Poluzzi E, Raschi E, Moretti U, et al. Drug-induced torsades de pointes: data mining of the public version of the FDA Adverse Event Reporting System (AERS). Pharmacoepidemiol Drug Saf. 2009;18(6):512-518.
13. U.S. Food and Drug Administration. FDA drug safety communication: revised recommendations for Celexa (citalopram hydrobromide) related to a potential risk of abnormal heart rhythms with high doses. http://www.fda.gov/Drugs/DrugSafety/ucm297391.htm. Published March 28, 2012. Accessed June 26, 2012.
14. Arizona CERT-QT Center for Education and Research on Therapeutics. QT drug lists by risk groups. http://www.azcert.org/medical-pros/drug-lists/drug-lists.cfm. Accessed June 26 2012.
15. Howell C, Wilson AD, Waring WS. Cardiovascular toxicity due to venlafaxine poisoning in adults: a review of 235 consecutive cases. Br J Clin Pharmacol. 2007;64(2):192-197.
16. Salih IS, Thanacoody RH, McKay GA, et al. Comparison of the effects of thioridazine and mesoridazine on the QT interval in healthy adults after single oral doses. Clin Pharmacol Ther. 2007;82(5):548-554.
17. Goodnick PJ, Jerry J, Parra F. Psychotropic drugs and the ECG: focus on the QTc interval. Expert Opin Pharmacother. 2002;3(5):479-498.
18. Dallaire S. Thioridazine (Mellaril) and mesoridazine (Serentil): prolongation of the QTc interval. CMAJ. 2001;164(1):91,95.-
19. Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation torsade de pointes and sudden death. Drugs. 2002;62(11):1649-1671.
20. Shapiro BA, Warren J, Egol AB, et al. Practice parameters for intravenous analgesia and sedation for adult patients in the intensive care unit: an executive summary. Crit Care Med. 1995;23(9):1596-1600.
21. Vieweg WV, Hasnain M. Question regarding ziprasidone and QTc interval prolongation in the ZODIAC Study. Am J Psychiatry. 2011;168(6):650-651.
22. Caccia S, Pasina L, Nobili A. New atypical antipsychotics for schizophrenia: iloperidone. Drug Des Devel Ther. 2010;4:33-48.
23. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics. 2003;44(2):174-175.
24. Vieweg WV, Schneider RK, Wood MA. Torsade de pointes in a patient with complex medical and psychiatric conditions receiving low-dose quetiapine. Acta Psychiatr Scand. 2005;112(4):318-322.
25. Capuano A, Ruggiero S, Vestini F, et al. Survival from coma induced by an intentional 36-g overdose of extended-release quetiapine. Drug Chem Toxicol. 2011;34(4):475-477.
26. Fernandes PP, Marcil WA. Death associated with quetiapine overdose. Am J Psychiatry. 2002;159(12):2114.-
27. Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once-monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-50.
28. Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189-210.
29. Chapel S, Hutmacher MM, Haig G, et al. Exposure-response analysis in patients with schizophrenia to assess the effect of asenapine on QTc prolongation. J Clin Pharmacol. 2009;49(11):1297-1308.
Nicole B. Washington, DO
Dr. Washington is Assistant Professor, Department of Psychiatry, School of Community Medicine, University of Oklahoma, Tulsa, OK
Nancy C. Brahm, PharmD, MS, BCPP, CGP
Dr. Brahm is Clinical Professor, College of Pharmacy, University of Oklahoma, Tulsa, OK
Julie Kissack, PharmD, BCPP
Dr. Kissack is Professor and Chair, Department of Pharmacy Practice, Harding University College of Pharmacy, Searcy, AR
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Nicole B. Washington, DO
Dr. Washington is Assistant Professor, Department of Psychiatry, School of Community Medicine, University of Oklahoma, Tulsa, OK
Nancy C. Brahm, PharmD, MS, BCPP, CGP
Dr. Brahm is Clinical Professor, College of Pharmacy, University of Oklahoma, Tulsa, OK
Julie Kissack, PharmD, BCPP
Dr. Kissack is Professor and Chair, Department of Pharmacy Practice, Harding University College of Pharmacy, Searcy, AR
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
Nicole B. Washington, DO
Dr. Washington is Assistant Professor, Department of Psychiatry, School of Community Medicine, University of Oklahoma, Tulsa, OK
Nancy C. Brahm, PharmD, MS, BCPP, CGP
Dr. Brahm is Clinical Professor, College of Pharmacy, University of Oklahoma, Tulsa, OK
Julie Kissack, PharmD, BCPP
Dr. Kissack is Professor and Chair, Department of Pharmacy Practice, Harding University College of Pharmacy, Searcy, AR
Vicki L. Ellingrod, PharmD, BCPP, FCCP
Series Editor
- Screen patients for risk factors for prolonged QTc interval, such as congenital long QT syndrome, family history of cardiac conduction abnormalities, and previous occurrences of medication-mediated QTc prolongation.
- Obtain baseline and steady state ECG when initiating high-risk agents, particularly when administering combination therapy.
- Use the lowest effective dose of antidepressants and antipsychotics and monitor symptoms closely.
Mrs. A, age 68, has a 40-year history of schizoaffective disorder with comorbid anxiety disorder not otherwise specified, type 2 diabetes mellitus, and hypertension. She takes furosemide, 40 mg/d, lisinopril, 20 mg/d, and metformin, 2,000 mg/d, for hypertension and diabetes; lorazepam, 1.5 mg/d, and paroxetine, 40 mg/d, for anxiety; and quetiapine extended release, 800 mg/d, for psychotic features and mood dysregulation with schizoaffective disorder. Mrs. A’s husband died 5 years ago and she lives alone in a senior care facility. Mrs. A uses a weekly pill reminder box because her residential facility does not monitor medication adherence. She sees her psychiatrist once a month and her primary care provider every 3 months. She has no history of illicit drug, alcohol, or tobacco use.
Two weeks ago, Mrs. A was found leaning against the wall in a hallway, complaining of dizziness and disorientation, and unable to find her way back to her apartment. In the emergency department, her serum potassium is low (3.0 mEq/L; normal range: 3.5 to 5.0), fasting glucose is elevated (110 mg/dL; range: 65 to 99), and ECG reveals a prolonged QTc interval of 530 milliseconds. Before this episode, Mrs. A had been medically stable without mood or psychotic symptoms, although her daughter reported medication self-administration was becoming difficult.
Exposure to psychotropics carries a risk of QTc prolongation. The QT interval is an ECG measure of ventricular depolarization and repolarization. The QTc designation indicates a correction for heart rate with increasing heart rate correlating with a shorter QT interval. Readings of 440 milliseconds are considered normal.1 QTc prolongation is defined as >450 milliseconds for men and >470 milliseconds for women.2 An increase in the QT interval is a predictor of serious cardiac events.3
Antidepressants and antipsychotics have been associated with QTc prolongation. When identifying agents that could disrupt cardiac conduction, clinicians need to consider whether the drug’s molecular structure, receptor affinity, or pharmacologic effects are most critical.2 Although these may be important, patient-specific variables that increase the risk of QTc prolongation may have greater impact. These include:
- age >65
- female sex
- electrolyte imbalances (specifically low serum potassium and magnesium levels)
- high or toxic serum levels of the suspected drug
- preexisting cardiovascular impairment, such as bradycardia.4,5
Other risk factors include concurrent use of an agent with similar cardiovascular effects or one that competes for metabolism (either enzymatic or at the binding site), physiologic limitations such as renal insufficiency, and medication changes that may increase or decrease psychotropic clearance.4,6 Geriatric patients with dementia have an increased risk for cardiovascular-related death.7,8
Antidepressants
Among tricyclic antidepressants, most reports of QTc prolongation involve amitriptyline and maprotiline.9 Risk factors include demographics (eg, female sex, age), personal or family history (congenital long QT syndrome, cardiovascular disease), and concurrent conditions or drug use, particularly those associated with QTc prolongation.3 Desipramine and nortriptyline also have been identified as high-risk agents.10
QTc prolongation has been reported with all selective serotonin reuptake inhibitors at plasma concentrations above the therapeutic level.11 Fluoxetine-associated QTc prolongation was limited to cases of overdose or when additional risk factors were reported.4 QTc prolongation from psychotropics could increase the risk of torsades de pointes, according to an analysis of the FDA Adverse Event Reporting System.12 In 2011, the FDA reported an increased risk of abnormal heart rhythms—including QTc prolongation—with citalopram doses >40 mg/d.13 Although cases of QTc prolongation with paroxetine have not been reported,11 the Arizona Center for Education and Research on Therapeutics lists paroxetine with other agents that may increase the risk for QTc prolongation with concurrent use of medications that may prolong QTc interval.14 Venlafaxine doses >300 mg/d may require additional cardiac monitoring.5,12 Data from venlafaxine poisoning case reports found a positive correlation between dose and QTc prolongation.15 In a review of toxicology database information, Wenzel-Seifert et al4 found extended QT interval with citalopram, fluoxetine, and venlafaxine at toxic doses or in the presence of additional risk factors such as sex, older age, or personal or family history of congenital long QT syndrome or cardiovascular disease.
Antipsychotics
Case reports, case series, and research trials have evaluated the risk of QTc prolongation with antipsychotics (Table).1,2,4,16,17 The first-generation antipsychotics thioridazine,4,16,18 mesoridazine,16,18 chlorpromazine,19 and haloperidol3 warrant cardiac monitoring. The QTc prolongation effects of thioridazine and its active metabolite mesoridazine are well-documented and thioridazine-mediated QTc prolongation increases are dose-dependent.4,18 ECG monitoring is recommended with IV haloperidol, which is used for delirium in adults.20 QTc prolongation has been associated with long-term ziprasidone use more often than with risperidone, olanzapine, or quetiapine.19 Ziprasidone prolongs the QTc interval an average of 20 milliseconds,21 which could represent a clinically significant change. QTc prolongation for iloperidone is comparable to ziprasidone and haloperidol.22 There is some evidence that aripiprazole may shorten, rather than prolong, the QTc interval.4,17
Cardiovascular adverse effects associated with clozapine—including QTc prolongation—are dose-dependent.3 Olanzapine prolongs QTc interval, although the mean change is less than with other agents unless other variables were present, such as:
- concomitant use of medications that may prolong QTc interval (ie, amantadine, hydroxyzine, or tamoxifen2)
- preexisting cardiovascular conduction disorders
- higher doses (>40 mg/d).3,23
In 17 case reports of cardiac changes associated with quetiapine use, doses ranged from 100 mg/d24 to an overdose of 36 g/d.25 Only 1 patient death was reported secondary to overdose and preexisting dysrhythmia and hypertension.26 QTc prolongation associated with risperidone was minor1 based on oral doses in the normal therapeutic range and incidences of overdose.10 Paliperidone27 and lurasidone28 are associated with clinically insignificant QTc prolongation. Changes in QTc interval were positively correlated with asenapine dose, although at the highest dose of 40 mg/d, the increase was <5 milliseconds.29
Mrs. A presents with a number of risk factors for QTc prolongation, including older age, female sex, and psychiatric and medical comorbidities that require medication. A pill count revealed that she was taking more than the prescribed daily doses of her medications. During the interview, Mrs. A said that if she missed her medication time, she would take them when she remembered. If she could not remember if she took her pills, she would take them again. Her physicians will explore strategies to increase medication adherence.
Table
Examples of QTc prolongation associated with select antipsychoticsa
| Antipsychotic | Approximate QTc interval prolongation in millisecondsb |
|---|---|
| Aripiprazole4,17 | -1 to -4 |
| Clozapine4 | 10 |
| Haloperidol1,2 | 7 to 15 |
| Mesoridazine16 | 39 to 53 |
| Olanzapine1 | 2 to 6.5 |
| Paliperidone4 | 2 to 4 |
| Pimozide2 | 19 |
| Quetiapine1,2 | 6 to 15 |
| Risperidone1,2 | 3.5 to 10 |
| Sertindole1 | 30 |
| Thioridazine2,16 | 33 to 41 |
| Ziprasidone1,2 | 16 to 21 |
| aList is not comprehensive. Other antipsychotics may be associated with QTc prolongation bQTc prolongation interval may depend on the route of administration | |
Related Resources
- De Hert M, Detraux J, van Winkel R, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2011;8(2):114-126.
- Vieweg WV, Wood MA, Fernandez A, et al. Proarrhythmic risk with antipsychotic and antidepressant drugs: implications in the elderly. Drugs Aging. 2009;26(12):997-1012.
- Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Asenapine • Saphris
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Furosemide • Lasix
- Haloperidol • Haldol
- Hydroxyzine • Atarax, Vistaril
- Iloperidone • Fanapt
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Lurasidone • Latuda
- Maprotiline • Ludiomil
- Mesoridazine • Serentil
- Metformin • Glucophage
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tamoxifen • Nolvadex, Soltamox
- Thioridazine • Mellaril
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. No similar work by the authors is under review or in press. No funding was requested or received in conjunction with this manuscript.
- Screen patients for risk factors for prolonged QTc interval, such as congenital long QT syndrome, family history of cardiac conduction abnormalities, and previous occurrences of medication-mediated QTc prolongation.
- Obtain baseline and steady state ECG when initiating high-risk agents, particularly when administering combination therapy.
- Use the lowest effective dose of antidepressants and antipsychotics and monitor symptoms closely.
Mrs. A, age 68, has a 40-year history of schizoaffective disorder with comorbid anxiety disorder not otherwise specified, type 2 diabetes mellitus, and hypertension. She takes furosemide, 40 mg/d, lisinopril, 20 mg/d, and metformin, 2,000 mg/d, for hypertension and diabetes; lorazepam, 1.5 mg/d, and paroxetine, 40 mg/d, for anxiety; and quetiapine extended release, 800 mg/d, for psychotic features and mood dysregulation with schizoaffective disorder. Mrs. A’s husband died 5 years ago and she lives alone in a senior care facility. Mrs. A uses a weekly pill reminder box because her residential facility does not monitor medication adherence. She sees her psychiatrist once a month and her primary care provider every 3 months. She has no history of illicit drug, alcohol, or tobacco use.
Two weeks ago, Mrs. A was found leaning against the wall in a hallway, complaining of dizziness and disorientation, and unable to find her way back to her apartment. In the emergency department, her serum potassium is low (3.0 mEq/L; normal range: 3.5 to 5.0), fasting glucose is elevated (110 mg/dL; range: 65 to 99), and ECG reveals a prolonged QTc interval of 530 milliseconds. Before this episode, Mrs. A had been medically stable without mood or psychotic symptoms, although her daughter reported medication self-administration was becoming difficult.
Exposure to psychotropics carries a risk of QTc prolongation. The QT interval is an ECG measure of ventricular depolarization and repolarization. The QTc designation indicates a correction for heart rate with increasing heart rate correlating with a shorter QT interval. Readings of 440 milliseconds are considered normal.1 QTc prolongation is defined as >450 milliseconds for men and >470 milliseconds for women.2 An increase in the QT interval is a predictor of serious cardiac events.3
Antidepressants and antipsychotics have been associated with QTc prolongation. When identifying agents that could disrupt cardiac conduction, clinicians need to consider whether the drug’s molecular structure, receptor affinity, or pharmacologic effects are most critical.2 Although these may be important, patient-specific variables that increase the risk of QTc prolongation may have greater impact. These include:
- age >65
- female sex
- electrolyte imbalances (specifically low serum potassium and magnesium levels)
- high or toxic serum levels of the suspected drug
- preexisting cardiovascular impairment, such as bradycardia.4,5
Other risk factors include concurrent use of an agent with similar cardiovascular effects or one that competes for metabolism (either enzymatic or at the binding site), physiologic limitations such as renal insufficiency, and medication changes that may increase or decrease psychotropic clearance.4,6 Geriatric patients with dementia have an increased risk for cardiovascular-related death.7,8
Antidepressants
Among tricyclic antidepressants, most reports of QTc prolongation involve amitriptyline and maprotiline.9 Risk factors include demographics (eg, female sex, age), personal or family history (congenital long QT syndrome, cardiovascular disease), and concurrent conditions or drug use, particularly those associated with QTc prolongation.3 Desipramine and nortriptyline also have been identified as high-risk agents.10
QTc prolongation has been reported with all selective serotonin reuptake inhibitors at plasma concentrations above the therapeutic level.11 Fluoxetine-associated QTc prolongation was limited to cases of overdose or when additional risk factors were reported.4 QTc prolongation from psychotropics could increase the risk of torsades de pointes, according to an analysis of the FDA Adverse Event Reporting System.12 In 2011, the FDA reported an increased risk of abnormal heart rhythms—including QTc prolongation—with citalopram doses >40 mg/d.13 Although cases of QTc prolongation with paroxetine have not been reported,11 the Arizona Center for Education and Research on Therapeutics lists paroxetine with other agents that may increase the risk for QTc prolongation with concurrent use of medications that may prolong QTc interval.14 Venlafaxine doses >300 mg/d may require additional cardiac monitoring.5,12 Data from venlafaxine poisoning case reports found a positive correlation between dose and QTc prolongation.15 In a review of toxicology database information, Wenzel-Seifert et al4 found extended QT interval with citalopram, fluoxetine, and venlafaxine at toxic doses or in the presence of additional risk factors such as sex, older age, or personal or family history of congenital long QT syndrome or cardiovascular disease.
Antipsychotics
Case reports, case series, and research trials have evaluated the risk of QTc prolongation with antipsychotics (Table).1,2,4,16,17 The first-generation antipsychotics thioridazine,4,16,18 mesoridazine,16,18 chlorpromazine,19 and haloperidol3 warrant cardiac monitoring. The QTc prolongation effects of thioridazine and its active metabolite mesoridazine are well-documented and thioridazine-mediated QTc prolongation increases are dose-dependent.4,18 ECG monitoring is recommended with IV haloperidol, which is used for delirium in adults.20 QTc prolongation has been associated with long-term ziprasidone use more often than with risperidone, olanzapine, or quetiapine.19 Ziprasidone prolongs the QTc interval an average of 20 milliseconds,21 which could represent a clinically significant change. QTc prolongation for iloperidone is comparable to ziprasidone and haloperidol.22 There is some evidence that aripiprazole may shorten, rather than prolong, the QTc interval.4,17
Cardiovascular adverse effects associated with clozapine—including QTc prolongation—are dose-dependent.3 Olanzapine prolongs QTc interval, although the mean change is less than with other agents unless other variables were present, such as:
- concomitant use of medications that may prolong QTc interval (ie, amantadine, hydroxyzine, or tamoxifen2)
- preexisting cardiovascular conduction disorders
- higher doses (>40 mg/d).3,23
In 17 case reports of cardiac changes associated with quetiapine use, doses ranged from 100 mg/d24 to an overdose of 36 g/d.25 Only 1 patient death was reported secondary to overdose and preexisting dysrhythmia and hypertension.26 QTc prolongation associated with risperidone was minor1 based on oral doses in the normal therapeutic range and incidences of overdose.10 Paliperidone27 and lurasidone28 are associated with clinically insignificant QTc prolongation. Changes in QTc interval were positively correlated with asenapine dose, although at the highest dose of 40 mg/d, the increase was <5 milliseconds.29
Mrs. A presents with a number of risk factors for QTc prolongation, including older age, female sex, and psychiatric and medical comorbidities that require medication. A pill count revealed that she was taking more than the prescribed daily doses of her medications. During the interview, Mrs. A said that if she missed her medication time, she would take them when she remembered. If she could not remember if she took her pills, she would take them again. Her physicians will explore strategies to increase medication adherence.
Table
Examples of QTc prolongation associated with select antipsychoticsa
| Antipsychotic | Approximate QTc interval prolongation in millisecondsb |
|---|---|
| Aripiprazole4,17 | -1 to -4 |
| Clozapine4 | 10 |
| Haloperidol1,2 | 7 to 15 |
| Mesoridazine16 | 39 to 53 |
| Olanzapine1 | 2 to 6.5 |
| Paliperidone4 | 2 to 4 |
| Pimozide2 | 19 |
| Quetiapine1,2 | 6 to 15 |
| Risperidone1,2 | 3.5 to 10 |
| Sertindole1 | 30 |
| Thioridazine2,16 | 33 to 41 |
| Ziprasidone1,2 | 16 to 21 |
| aList is not comprehensive. Other antipsychotics may be associated with QTc prolongation bQTc prolongation interval may depend on the route of administration | |
Related Resources
- De Hert M, Detraux J, van Winkel R, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2011;8(2):114-126.
- Vieweg WV, Wood MA, Fernandez A, et al. Proarrhythmic risk with antipsychotic and antidepressant drugs: implications in the elderly. Drugs Aging. 2009;26(12):997-1012.
- Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
Drug Brand Names
- Amantadine • Symmetrel
- Amitriptyline • Elavil
- Aripiprazole • Abilify
- Asenapine • Saphris
- Chlorpromazine • Thorazine
- Citalopram • Celexa
- Clozapine • Clozaril
- Desipramine • Norpramin
- Fluoxetine • Prozac
- Furosemide • Lasix
- Haloperidol • Haldol
- Hydroxyzine • Atarax, Vistaril
- Iloperidone • Fanapt
- Lisinopril • Prinivil, Zestril
- Lorazepam • Ativan
- Lurasidone • Latuda
- Maprotiline • Ludiomil
- Mesoridazine • Serentil
- Metformin • Glucophage
- Nortriptyline • Pamelor
- Olanzapine • Zyprexa
- Paliperidone • Invega
- Paroxetine • Paxil
- Pimozide • Orap
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Tamoxifen • Nolvadex, Soltamox
- Thioridazine • Mellaril
- Venlafaxine • Effexor
- Ziprasidone • Geodon
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products. No similar work by the authors is under review or in press. No funding was requested or received in conjunction with this manuscript.
1. Muscatello MR, Bruno A, Pandolfo G, et al. Emerging treatments in the management of schizophrenia - focus on sertindole. Drug Des Devel Ther. 2010;4:187-201.
2. Taylor DM. Antipsychotics and QT prolongation. Acta Psychiatr Scand. 2003;107(2):85-95.
3. Alvarez PA, Pahissa J. QT alterations in psychopharmacology: proven candidates and suspects. Curr Drug Saf. 2010;5(1):97-104.
4. Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of torsade de pointes. Dtsch Arztebl Int. 2011;108(41):687-693.
5. Vieweg WV. New generation antipsychotic drugs and QTc interval prolongation. Prim Care Companion J Clin Psychiatry. 2003;5(5):205-215.
6. Nielsen J, Graff C, Kanters JK, et al. Assessing QT interval prolongation and its associated risks with antipsychotics. CNS Drugs. 2011;25(6):473-490.
7. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
8. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
9. Vieweg WV, Wood MA. Tricyclic antidepressants QT interval prolongation, and torsade de pointes. Psychosomatics. 2004;45(5):371-377.
10. Jeon SH, Jaekal J, Lee SH, et al. Effects of nortriptyline on QT prolongation: a safety pharmacology study. Hum Exp Toxicol. 2011;30(10):1649-1656.
11. Wenzel-Seifert K, Wittmann M, Haen E. Torsade de pointes episodes under treatment with selective serotonin reuptake inhibitors. Pharmacopsychiatry. 2010;43(7):279-281.
12. Poluzzi E, Raschi E, Moretti U, et al. Drug-induced torsades de pointes: data mining of the public version of the FDA Adverse Event Reporting System (AERS). Pharmacoepidemiol Drug Saf. 2009;18(6):512-518.
13. U.S. Food and Drug Administration. FDA drug safety communication: revised recommendations for Celexa (citalopram hydrobromide) related to a potential risk of abnormal heart rhythms with high doses. http://www.fda.gov/Drugs/DrugSafety/ucm297391.htm. Published March 28, 2012. Accessed June 26, 2012.
14. Arizona CERT-QT Center for Education and Research on Therapeutics. QT drug lists by risk groups. http://www.azcert.org/medical-pros/drug-lists/drug-lists.cfm. Accessed June 26 2012.
15. Howell C, Wilson AD, Waring WS. Cardiovascular toxicity due to venlafaxine poisoning in adults: a review of 235 consecutive cases. Br J Clin Pharmacol. 2007;64(2):192-197.
16. Salih IS, Thanacoody RH, McKay GA, et al. Comparison of the effects of thioridazine and mesoridazine on the QT interval in healthy adults after single oral doses. Clin Pharmacol Ther. 2007;82(5):548-554.
17. Goodnick PJ, Jerry J, Parra F. Psychotropic drugs and the ECG: focus on the QTc interval. Expert Opin Pharmacother. 2002;3(5):479-498.
18. Dallaire S. Thioridazine (Mellaril) and mesoridazine (Serentil): prolongation of the QTc interval. CMAJ. 2001;164(1):91,95.-
19. Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation torsade de pointes and sudden death. Drugs. 2002;62(11):1649-1671.
20. Shapiro BA, Warren J, Egol AB, et al. Practice parameters for intravenous analgesia and sedation for adult patients in the intensive care unit: an executive summary. Crit Care Med. 1995;23(9):1596-1600.
21. Vieweg WV, Hasnain M. Question regarding ziprasidone and QTc interval prolongation in the ZODIAC Study. Am J Psychiatry. 2011;168(6):650-651.
22. Caccia S, Pasina L, Nobili A. New atypical antipsychotics for schizophrenia: iloperidone. Drug Des Devel Ther. 2010;4:33-48.
23. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics. 2003;44(2):174-175.
24. Vieweg WV, Schneider RK, Wood MA. Torsade de pointes in a patient with complex medical and psychiatric conditions receiving low-dose quetiapine. Acta Psychiatr Scand. 2005;112(4):318-322.
25. Capuano A, Ruggiero S, Vestini F, et al. Survival from coma induced by an intentional 36-g overdose of extended-release quetiapine. Drug Chem Toxicol. 2011;34(4):475-477.
26. Fernandes PP, Marcil WA. Death associated with quetiapine overdose. Am J Psychiatry. 2002;159(12):2114.-
27. Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once-monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-50.
28. Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189-210.
29. Chapel S, Hutmacher MM, Haig G, et al. Exposure-response analysis in patients with schizophrenia to assess the effect of asenapine on QTc prolongation. J Clin Pharmacol. 2009;49(11):1297-1308.
1. Muscatello MR, Bruno A, Pandolfo G, et al. Emerging treatments in the management of schizophrenia - focus on sertindole. Drug Des Devel Ther. 2010;4:187-201.
2. Taylor DM. Antipsychotics and QT prolongation. Acta Psychiatr Scand. 2003;107(2):85-95.
3. Alvarez PA, Pahissa J. QT alterations in psychopharmacology: proven candidates and suspects. Curr Drug Saf. 2010;5(1):97-104.
4. Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of torsade de pointes. Dtsch Arztebl Int. 2011;108(41):687-693.
5. Vieweg WV. New generation antipsychotic drugs and QTc interval prolongation. Prim Care Companion J Clin Psychiatry. 2003;5(5):205-215.
6. Nielsen J, Graff C, Kanters JK, et al. Assessing QT interval prolongation and its associated risks with antipsychotics. CNS Drugs. 2011;25(6):473-490.
7. Gill SS, Bronskill SE, Normand SL, et al. Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med. 2007;146(11):775-786.
8. Schneeweiss S, Setoguchi S, Brookhart A, et al. Risk of death associated with the use of conventional versus atypical antipsychotic drugs among elderly patients. CMAJ. 2007;176(5):627-632.
9. Vieweg WV, Wood MA. Tricyclic antidepressants QT interval prolongation, and torsade de pointes. Psychosomatics. 2004;45(5):371-377.
10. Jeon SH, Jaekal J, Lee SH, et al. Effects of nortriptyline on QT prolongation: a safety pharmacology study. Hum Exp Toxicol. 2011;30(10):1649-1656.
11. Wenzel-Seifert K, Wittmann M, Haen E. Torsade de pointes episodes under treatment with selective serotonin reuptake inhibitors. Pharmacopsychiatry. 2010;43(7):279-281.
12. Poluzzi E, Raschi E, Moretti U, et al. Drug-induced torsades de pointes: data mining of the public version of the FDA Adverse Event Reporting System (AERS). Pharmacoepidemiol Drug Saf. 2009;18(6):512-518.
13. U.S. Food and Drug Administration. FDA drug safety communication: revised recommendations for Celexa (citalopram hydrobromide) related to a potential risk of abnormal heart rhythms with high doses. http://www.fda.gov/Drugs/DrugSafety/ucm297391.htm. Published March 28, 2012. Accessed June 26, 2012.
14. Arizona CERT-QT Center for Education and Research on Therapeutics. QT drug lists by risk groups. http://www.azcert.org/medical-pros/drug-lists/drug-lists.cfm. Accessed June 26 2012.
15. Howell C, Wilson AD, Waring WS. Cardiovascular toxicity due to venlafaxine poisoning in adults: a review of 235 consecutive cases. Br J Clin Pharmacol. 2007;64(2):192-197.
16. Salih IS, Thanacoody RH, McKay GA, et al. Comparison of the effects of thioridazine and mesoridazine on the QT interval in healthy adults after single oral doses. Clin Pharmacol Ther. 2007;82(5):548-554.
17. Goodnick PJ, Jerry J, Parra F. Psychotropic drugs and the ECG: focus on the QTc interval. Expert Opin Pharmacother. 2002;3(5):479-498.
18. Dallaire S. Thioridazine (Mellaril) and mesoridazine (Serentil): prolongation of the QTc interval. CMAJ. 2001;164(1):91,95.-
19. Haddad PM, Anderson IM. Antipsychotic-related QTc prolongation torsade de pointes and sudden death. Drugs. 2002;62(11):1649-1671.
20. Shapiro BA, Warren J, Egol AB, et al. Practice parameters for intravenous analgesia and sedation for adult patients in the intensive care unit: an executive summary. Crit Care Med. 1995;23(9):1596-1600.
21. Vieweg WV, Hasnain M. Question regarding ziprasidone and QTc interval prolongation in the ZODIAC Study. Am J Psychiatry. 2011;168(6):650-651.
22. Caccia S, Pasina L, Nobili A. New atypical antipsychotics for schizophrenia: iloperidone. Drug Des Devel Ther. 2010;4:33-48.
23. Dineen S, Withrow K, Voronovitch L, et al. QTc prolongation and high-dose olanzapine. Psychosomatics. 2003;44(2):174-175.
24. Vieweg WV, Schneider RK, Wood MA. Torsade de pointes in a patient with complex medical and psychiatric conditions receiving low-dose quetiapine. Acta Psychiatr Scand. 2005;112(4):318-322.
25. Capuano A, Ruggiero S, Vestini F, et al. Survival from coma induced by an intentional 36-g overdose of extended-release quetiapine. Drug Chem Toxicol. 2011;34(4):475-477.
26. Fernandes PP, Marcil WA. Death associated with quetiapine overdose. Am J Psychiatry. 2002;159(12):2114.-
27. Sedky K, Nazir R, Lindenmayer JP, et al. Paliperidone palmitate: once-monthly treatment option for schizophrenia. Current Psychiatry. 2010;9(3):48-50.
28. Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189-210.
29. Chapel S, Hutmacher MM, Haig G, et al. Exposure-response analysis in patients with schizophrenia to assess the effect of asenapine on QTc prolongation. J Clin Pharmacol. 2009;49(11):1297-1308.