User login
Venetoclax gets accelerated approval in older AML patients
(AML).

The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
(AML).
The BCL-2 inhibitor is now approved for use in combination with azacitidine, decitabine, or low-dose cytarabine to treat newly diagnosed adults with AML who are aged 75 and older or who are ineligible for intensive chemotherapy.
The FDA grants accelerated approval based on a surrogate or intermediate endpoint that is reasonably likely to predict clinical benefit. Continued approval of venetoclax in AML may be contingent upon verification of clinical benefit in confirmatory trials.
The approval is based on data from two studies – the phase 1b M14-358 trial ( NCT02203773 ) and the phase 1/2 M14-387 trial ( NCT02287233 ).
In M14-358, newly diagnosed AML patients received venetoclax in combination with azacitidine (n=84) or decitabine (n=31). There were 67 patients in the azacitidine arm and 13 in the decitabine arm who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via a daily ramp-up to a final dose of 400 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
They received azacitidine at 75 mg/m 2 on days 1-7 of each 28-day cycle or decitabine at 20 mg/m 2 on days 1-5 of each cycle. Patients continued treatment until disease progression or unacceptable toxicity.
The median follow-up was 7.9 months for the azacitidine arm and 11 months for the decitabine arm.
The complete response (CR) rate was 37% (25/67) in the azacitidine arm and 54% (7/13) in the decitabine arm. The rates of CR with partial hematologic recovery were 24% (16/67) and 7.7% (1/13), respectively. The most common adverse events (AEs) – occurring in at least 30% of patients in both arms – were nausea, diarrhea, constipation, neutropenia, thrombocytopenia, hemorrhage, peripheral edema, vomiting, fatigue, febrile neutropenia, rash, and anemia. The incidence of serious AEs was 75% overall. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, pneumonia (excluding fungal), sepsis (excluding fungal), respiratory failure, and multiple organ dysfunction syndrome. The incidence of fatal AEs was 1.5% within 30 days of treatment initiation. The M14-387 trial included 82 AML patients who received venetoclax plus low-dose cytarabine. Patients were newly diagnosed with AML, but some had previous exposure to a hypomethylating agent for an antecedent hematologic disorder.
There were 61 patients who were aged 75 or older or were ineligible for intensive induction chemotherapy.
Patients received venetoclax via daily ramp-up to a final dose of 600 mg once daily. They received prophylaxis for tumor lysis syndrome and were hospitalized for monitoring during the ramp-up.
Cytarabine was given at 20 mg/m 2 on days 1-10 of each 28-day cycle. Patients continued to receive treatment until disease progression or unacceptable toxicity.
At a median follow-up of 6.5 months, the CR rate was 21% (13/61), and the rate of CR with partial hematologic recovery was 21% (13/61).
The most common AEs (occurring in at least 30% of patients) were nausea, thrombocytopenia, hemorrhage, febrile neutropenia, neutropenia, diarrhea, fatigue, constipation, and dyspnea. The incidence of serious AEs was 95%. The most frequent serious AEs (occurring in at least 5% of patients) were febrile neutropenia, sepsis (excluding fungal), hemorrhage, pneumonia (excluding fungal), and device-related infection. The incidence of fatal AEs was 4.9% within 30 days of treatment initiation.
Additional details from the M14-358 and M14-387 trials are available in the prescribing information for venetoclax.
Venetoclax is being developed by AbbVie and Roche.
FDA approves generic drugs for APL
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
The U.S. Food and Drug Administration (FDA) has now approved three generic arsenic trioxide products for use in patients with acute promyelocytic leukemia (APL).
Two of the products—from Zydus Cadila and Amring Pharmaceuticals—were approved on November 13.
The third—from Fresenius Kabi—was approved in August and launched in the United States last month.
All three injectable arsenic trioxide products (1 mg/mL) are generic versions of Teva’s Trisenox.
Since 2000, Trisenox has been FDA-approved to induce remission and as consolidation therapy for patients with APL who are refractory to, or have relapsed after, retinoid and anthracycline chemotherapy, and whose APL is characterized by presence of the t(15;17) translocation or PML/RAR-alpha gene expression.
In January, the FDA approved Trisenox for use in combination with tretinoin to treat adults with newly diagnosed, low-risk APL with the t(15;17) translocation or PML/RAR-alpha gene expression.
Azacitidine-nivolumab combo 'encouraging' in AML
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months. However, the researchers identified factors associated with improved response and survival that could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment related.
Naval Daver, MD, of the University of Texas MD Anderson Cancer Center, Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 years. More than half of the patients (56%) had de novo AML, and 44% had secondary AML. The median number of prior therapies was two; 64% of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was three. Patients had a median time on study of 3.5 months and reasons for discontinuation included primary refractory disease, relapse after initial response, proceeding to SCT, patient preference, and death.
The most common treatment-related, nonhematologic AEs were constipation, diarrhea, pneumonitis, nausea, and lung infection. The rate of immune-related AEs was 25% (n = 18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4); 14 responded to steroids and were safely rechallenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) because of AEs. Two patients died of AEs that were considered possibly related to treatment. One death was caused by progressive pneumonia/pneumonitis, and one was caused by hemophagocytic lymphohistiocytosis.
The overall response rate was 33% (n = 23), with 4 patients achieving a complete response (CR) and 11 achieving a CR with incomplete count recovery (CRi). One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. Additionally, a higher frequency of pretherapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said in a statement.
At a median follow-up of 21.4 months, 81% of patients (n = 57) had died; 16 died on study treatment and 41 died after discontinuation. The median OS overall was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in nonresponders (P less than .0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P less than .001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver said.
This research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Individual researchers also reported financial relationships with Bristol-Myers Squibb.
SOURCE: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
FROM CANCER DISCOVERY
Key clinical point:
Major finding: The overall response rate was 33%.
Study details: This phase 2 trial included 70 patients with relapsed/refractory acute myeloid leukemia.
Disclosures: The research was supported by Bristol-Myers Squibb, the University of Texas MD Anderson Cancer Center, and the Dick Clark Immunotherapy Research Fund. Researchers reported financial relationships with Bristol-Myers Squibb.
Source: Daver N et al. Cancer Discov. 2018 Nov 8. doi: 10.1158/2159-8290.CD-18-0774.
AP-1 plays key role in various AML subtypes, team says
The AP-1 transcription factor family is of “major importance” in acute myeloid leukemia (AML), according to researchers.
The team said they identified transcription factor networks specific to AML subtypes, which showed that leukemic growth is dependent upon certain transcription factors, and “the global activation of signaling pathways parallels a growth dependence on AP-1 activity in multiple types of AML.”
Constanze Bonifer, PhD, of the University of Birmingham in the U.K., and her colleagues conducted this research and detailed their findings in Nature Genetics.
The researchers noted that previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA methylation patterns.
“Our work now defines these networks in detail and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” Dr. Bonifer and her colleagues wrote.
The researchers combined data obtained via several analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from AML patients with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the team reported.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, the researchers transduced AML cells with a doxycycline-inducible version of a dominant-negative (dn) FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB, and JDP families of transcription factors,” the researchers wrote. “[T]hus, it is challenging to target by defined RNA interference approaches.”
Results of the in vitro study showed that induction of dnFOS, mediated by doxycycline, inhibited proliferation of t(8;21)+ Kasumi-1 cells and FLT3-ITD-expressing MV4-11 cells.
Induction of dnFOS also inhibited the colony-forming ability of primary CD34+ FLT3-ITD cells but not CD34+ hematopoietic stem and progenitor cells.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, the researchers transplanted either of two cell lines—Kasumi-1 or MV4-11—expressing inducible dnFOS in immunodeficient mice.
With Kasumi-1, granulosarcomas developed in six of seven untreated control mice and two mice treated with doxycycline, neither of which expressed the inducible protein.
With MV4-11, doxycycline inhibited leukemia development, and untreated mice rapidly developed tumors.
The researchers declared no competing interests related to this work, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship, and an MRC/Leuka Clinical Training Fellowship.
The AP-1 transcription factor family is of “major importance” in acute myeloid leukemia (AML), according to researchers.
The team said they identified transcription factor networks specific to AML subtypes, which showed that leukemic growth is dependent upon certain transcription factors, and “the global activation of signaling pathways parallels a growth dependence on AP-1 activity in multiple types of AML.”
Constanze Bonifer, PhD, of the University of Birmingham in the U.K., and her colleagues conducted this research and detailed their findings in Nature Genetics.
The researchers noted that previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA methylation patterns.
“Our work now defines these networks in detail and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” Dr. Bonifer and her colleagues wrote.
The researchers combined data obtained via several analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from AML patients with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the team reported.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, the researchers transduced AML cells with a doxycycline-inducible version of a dominant-negative (dn) FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB, and JDP families of transcription factors,” the researchers wrote. “[T]hus, it is challenging to target by defined RNA interference approaches.”
Results of the in vitro study showed that induction of dnFOS, mediated by doxycycline, inhibited proliferation of t(8;21)+ Kasumi-1 cells and FLT3-ITD-expressing MV4-11 cells.
Induction of dnFOS also inhibited the colony-forming ability of primary CD34+ FLT3-ITD cells but not CD34+ hematopoietic stem and progenitor cells.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, the researchers transplanted either of two cell lines—Kasumi-1 or MV4-11—expressing inducible dnFOS in immunodeficient mice.
With Kasumi-1, granulosarcomas developed in six of seven untreated control mice and two mice treated with doxycycline, neither of which expressed the inducible protein.
With MV4-11, doxycycline inhibited leukemia development, and untreated mice rapidly developed tumors.
The researchers declared no competing interests related to this work, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship, and an MRC/Leuka Clinical Training Fellowship.
The AP-1 transcription factor family is of “major importance” in acute myeloid leukemia (AML), according to researchers.
The team said they identified transcription factor networks specific to AML subtypes, which showed that leukemic growth is dependent upon certain transcription factors, and “the global activation of signaling pathways parallels a growth dependence on AP-1 activity in multiple types of AML.”
Constanze Bonifer, PhD, of the University of Birmingham in the U.K., and her colleagues conducted this research and detailed their findings in Nature Genetics.
The researchers noted that previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA methylation patterns.
“Our work now defines these networks in detail and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” Dr. Bonifer and her colleagues wrote.
The researchers combined data obtained via several analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from AML patients with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the team reported.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, the researchers transduced AML cells with a doxycycline-inducible version of a dominant-negative (dn) FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB, and JDP families of transcription factors,” the researchers wrote. “[T]hus, it is challenging to target by defined RNA interference approaches.”
Results of the in vitro study showed that induction of dnFOS, mediated by doxycycline, inhibited proliferation of t(8;21)+ Kasumi-1 cells and FLT3-ITD-expressing MV4-11 cells.
Induction of dnFOS also inhibited the colony-forming ability of primary CD34+ FLT3-ITD cells but not CD34+ hematopoietic stem and progenitor cells.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, the researchers transplanted either of two cell lines—Kasumi-1 or MV4-11—expressing inducible dnFOS in immunodeficient mice.
With Kasumi-1, granulosarcomas developed in six of seven untreated control mice and two mice treated with doxycycline, neither of which expressed the inducible protein.
With MV4-11, doxycycline inhibited leukemia development, and untreated mice rapidly developed tumors.
The researchers declared no competing interests related to this work, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship, and an MRC/Leuka Clinical Training Fellowship.
Transcription factor plays key role in AML gene regulatory networks
The AP-1 transcription factor family, important in many tumor types, plays a major role in acute myeloid leukemia, according to researchers who conducted a comprehensive global analysis of gene regulatory networks involved in this disease.

This observation suggests new opportunities for targeted treatment of AML, according to the researchers, led by Peter N. Cockerill, PhD, and Constanze Bonifer, PhD, with the Institute of Cancer and Genomic Sciences, University of Birmingham, England.
“Induced and aberrantly expressed transcription factors are not bystanders, but are important for network maintenance and leukemic growth,” the investigators wrote in Nature Genetics.
Investigators combined data obtained via several different analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from specific subgroups of subjects with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the investigators reported.
Previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA-methylation patterns.
“Our work now defines these networks in detail, and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” the authors said in their report.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, investigators transduced AML cells with a doxycycline-inducible version of a dominant negative FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB and JDP families of transcription factors, thus it is challenging to target by defined RNA interference approaches,” the investigators explained.
Results of the in vitro study showed that induction of that protein, mediated by doxycycline, inhibited proliferation and colony-forming ability in AML cell lines.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, they transplanted two different types of cells expressing inducible dominant negative FOS protein in immunodeficient mice.
For the first cell type, granulosarcomas developed in six out of seven mice in a control group, but in only two mice treated with doxycycline, neither of which expressed the inducible protein, suggesting that the transgene was silenced, according to the investigators. For the second cell type, doxycycline inhibited leukemia development, while untreated mice rapidly developed tumors.
“Taken together, these findings demonstrate the importance of AP-1 for several AML subtypes and emphasize the potential of transcriptional network analyses to predict transcription factors crucial for malignant propagation,” the investigators wrote.
They declared no competing interests related to their research, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship and a MRC/Leuka Clinical Training Fellowship.
SOURCE: Assi SA et al. Nat Genet. 2018 Nov 12. doi: 10.1038/s41588-018-0270-1.
The AP-1 transcription factor family, important in many tumor types, plays a major role in acute myeloid leukemia, according to researchers who conducted a comprehensive global analysis of gene regulatory networks involved in this disease.
This observation suggests new opportunities for targeted treatment of AML, according to the researchers, led by Peter N. Cockerill, PhD, and Constanze Bonifer, PhD, with the Institute of Cancer and Genomic Sciences, University of Birmingham, England.
“Induced and aberrantly expressed transcription factors are not bystanders, but are important for network maintenance and leukemic growth,” the investigators wrote in Nature Genetics.
Investigators combined data obtained via several different analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from specific subgroups of subjects with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the investigators reported.
Previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA-methylation patterns.
“Our work now defines these networks in detail, and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” the authors said in their report.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, investigators transduced AML cells with a doxycycline-inducible version of a dominant negative FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB and JDP families of transcription factors, thus it is challenging to target by defined RNA interference approaches,” the investigators explained.
Results of the in vitro study showed that induction of that protein, mediated by doxycycline, inhibited proliferation and colony-forming ability in AML cell lines.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, they transplanted two different types of cells expressing inducible dominant negative FOS protein in immunodeficient mice.
For the first cell type, granulosarcomas developed in six out of seven mice in a control group, but in only two mice treated with doxycycline, neither of which expressed the inducible protein, suggesting that the transgene was silenced, according to the investigators. For the second cell type, doxycycline inhibited leukemia development, while untreated mice rapidly developed tumors.
“Taken together, these findings demonstrate the importance of AP-1 for several AML subtypes and emphasize the potential of transcriptional network analyses to predict transcription factors crucial for malignant propagation,” the investigators wrote.
They declared no competing interests related to their research, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship and a MRC/Leuka Clinical Training Fellowship.
SOURCE: Assi SA et al. Nat Genet. 2018 Nov 12. doi: 10.1038/s41588-018-0270-1.
The AP-1 transcription factor family, important in many tumor types, plays a major role in acute myeloid leukemia, according to researchers who conducted a comprehensive global analysis of gene regulatory networks involved in this disease.
This observation suggests new opportunities for targeted treatment of AML, according to the researchers, led by Peter N. Cockerill, PhD, and Constanze Bonifer, PhD, with the Institute of Cancer and Genomic Sciences, University of Birmingham, England.
“Induced and aberrantly expressed transcription factors are not bystanders, but are important for network maintenance and leukemic growth,” the investigators wrote in Nature Genetics.
Investigators combined data obtained via several different analytic techniques to construct transcription factor networks in normal CD34+ cells and cells from specific subgroups of subjects with defined mutations, including RUNX1 mutations, t(8;21) translocations, mutations of both alleles of the CEBPA gene, and FLT3-ITD with or without NPM1 mutation.
The AP-1 family network was of “high regulatory relevance” for all AML subtypes evaluated, the investigators reported.
Previous work revealed the existence of gene regulatory networks in different types of AML classified by gene expression and DNA-methylation patterns.
“Our work now defines these networks in detail, and shows that leukemic drivers determine the regulatory phenotype by establishing and maintaining specific gene regulatory and signaling networks that are distinct from those in normal cells,” the authors said in their report.
Follow-up in vitro and in vivo studies confirmed the importance of AP-1 for different AML subtypes.
In the in vitro study, investigators transduced AML cells with a doxycycline-inducible version of a dominant negative FOS protein.
“AP-1 is a heterodimer formed by members of the FOS, JUN, ATF, CREB and JDP families of transcription factors, thus it is challenging to target by defined RNA interference approaches,” the investigators explained.
Results of the in vitro study showed that induction of that protein, mediated by doxycycline, inhibited proliferation and colony-forming ability in AML cell lines.
To evaluate the relevance of AP-1 for leukemia propagation in vivo, they transplanted two different types of cells expressing inducible dominant negative FOS protein in immunodeficient mice.
For the first cell type, granulosarcomas developed in six out of seven mice in a control group, but in only two mice treated with doxycycline, neither of which expressed the inducible protein, suggesting that the transgene was silenced, according to the investigators. For the second cell type, doxycycline inhibited leukemia development, while untreated mice rapidly developed tumors.
“Taken together, these findings demonstrate the importance of AP-1 for several AML subtypes and emphasize the potential of transcriptional network analyses to predict transcription factors crucial for malignant propagation,” the investigators wrote.
They declared no competing interests related to their research, which was funded by Bloodwise, Cancer Research UK, a Kay Kendall Clinical Training Fellowship and a MRC/Leuka Clinical Training Fellowship.
SOURCE: Assi SA et al. Nat Genet. 2018 Nov 12. doi: 10.1038/s41588-018-0270-1.
FROM NATURE GENETICS
Key clinical point:
Major finding: The AP-1 factor family gene regulatory network was of high regulatory relevance in multiple subtypes of AML with defined mutations.
Study details: Analysis of normal CD34+ cells and cells from AML subjects.
Disclosures: Funding came from Bloodwise and Cancer Research UK, among other sources. The researchers reported having no competing financial interests.
Source: Assi SA et al. Nat Genet. 2018 Nov 12. doi: 10.1038/s41588-018-0270-1.
‘Compelling’ new target found for monocytic AML

Efforts to determine why immune checkpoint blockade is not successful in treating leukemia have resulted in a “compelling” new target to treat monocytic acute myeloid leukemia (AML), according to researchers.
They discovered that leukocyte immunoglobulin-like receptor B4 (LILRB4), a marker of monocytic leukemia, creates an immunosuppressive microenvironment by mediating T-cell suppression.
Using a mouse model and human cells, the research team showed that LILRB4 supports tumor infiltration into tissues and suppresses T-cell activity through a signaling pathway involving APOE, LILRB4, SHP-2, uPAR, and ARG1.
Senior author Chengcheng “Alec” Zhang, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported their findings in Nature.
The team first compared surface expression of LILRB4 on normal monocytes and neoplastic monocytes from 105 AML patient samples. They observed that LILRB4 levels were higher on monocytic AML cells than on normal monocytes.
The researchers then tested whether LILRB4 expressed on AML cells suppressed T cells. They cultured LILRB4-positive leukemia cells, LILRB4-negative leukemia cells, and normal hematopoietic cells with either autologous T cells or T cells from healthy donors. The team determined that only LILRB4-positive monocytic AML cells substantially suppressed T-cell proliferation.
When the researchers knocked out LILRB4, the ability of AML cells to suppress T cells was reduced and could be restored with forced expression of wild-type LILRB4. Additionally, LILRB4-mediated T-cell inhibition could be reversed by LILRB4-blocking antibodies.
The team then used a humanized mouse xenograft model and an immunocompetent mouse model to investigate LILRB4 function in immune checkpoint blockade. Blocking LILRB4 lowered tumor burden and prolonged survival in the mice.
The researchers performed numerous in vitro and in vivo experiments and observed that antibody blockade of LILRB4 shrank tumors and decreased leukemic infiltration into internal organs, including the bone marrow, liver, and brain.
And so the team hypothesized that LILRB4 promotes leukemia infiltration in addition to inhibiting T cells.
To test the hypothesis, they performed transendothelial migration and homing assays and monitored leukemia infiltration relative to LILRB4 expression on leukemia cells. They observed that LILRB4-mediated migration enhanced extramedullary infiltration of monocytic AML cells, thus contributing to immune evasion.
The researchers also found that APOE protein activated the immune inhibitory receptor LILRB4.
To ascertain whether suppression of T cells by LILRB4 depends on APOE, the team co-cultured T cells with control or human AML cells with APOE knocked out.
Through a series of experiments, they determined that APOE is an extracellular binding protein of LILRB4 and that APOE activates LILRB4 to support T-cell proliferation and AML cell migration.
The researchers believe that targeting LILRB4 may have minimal toxicity. This is because LILRB4 expression on normal monocytic cells is limited, LILRB4 signaling may differ in leukemia cells, and LILRB4 blockade did not significantly interfere with normal hematopoietic function.
Dr. Zhang anticipates that if the preclinical studies go well, clinical trials could begin as early as next year.
The University of Texas System has exclusively licensed LILRB4-related patent applications to California-based Immune-Onc Therapeutics Inc., which contributed to the research and is conducting preclinical studies.
Dr. Zhang and another author are scientific advisory board members with Immune-Onc Therapeutics. Two other authors are employees of and hold equities in Immune-Onc Therapeutics.
The researchers received additional funding for this work from the National Cancer Institute, Leukemia & Lymphoma Society, the March of Dimes, the Cancer Prevention and Research Institute of Texas, the Robert A. Welch Foundation, the National Natural Science Foundation of China, the National Basic Research Program of China, and the China Scholarship Council.
Efforts to determine why immune checkpoint blockade is not successful in treating leukemia have resulted in a “compelling” new target to treat monocytic acute myeloid leukemia (AML), according to researchers.
They discovered that leukocyte immunoglobulin-like receptor B4 (LILRB4), a marker of monocytic leukemia, creates an immunosuppressive microenvironment by mediating T-cell suppression.
Using a mouse model and human cells, the research team showed that LILRB4 supports tumor infiltration into tissues and suppresses T-cell activity through a signaling pathway involving APOE, LILRB4, SHP-2, uPAR, and ARG1.
Senior author Chengcheng “Alec” Zhang, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported their findings in Nature.
The team first compared surface expression of LILRB4 on normal monocytes and neoplastic monocytes from 105 AML patient samples. They observed that LILRB4 levels were higher on monocytic AML cells than on normal monocytes.
The researchers then tested whether LILRB4 expressed on AML cells suppressed T cells. They cultured LILRB4-positive leukemia cells, LILRB4-negative leukemia cells, and normal hematopoietic cells with either autologous T cells or T cells from healthy donors. The team determined that only LILRB4-positive monocytic AML cells substantially suppressed T-cell proliferation.
When the researchers knocked out LILRB4, the ability of AML cells to suppress T cells was reduced and could be restored with forced expression of wild-type LILRB4. Additionally, LILRB4-mediated T-cell inhibition could be reversed by LILRB4-blocking antibodies.
The team then used a humanized mouse xenograft model and an immunocompetent mouse model to investigate LILRB4 function in immune checkpoint blockade. Blocking LILRB4 lowered tumor burden and prolonged survival in the mice.
The researchers performed numerous in vitro and in vivo experiments and observed that antibody blockade of LILRB4 shrank tumors and decreased leukemic infiltration into internal organs, including the bone marrow, liver, and brain.
And so the team hypothesized that LILRB4 promotes leukemia infiltration in addition to inhibiting T cells.
To test the hypothesis, they performed transendothelial migration and homing assays and monitored leukemia infiltration relative to LILRB4 expression on leukemia cells. They observed that LILRB4-mediated migration enhanced extramedullary infiltration of monocytic AML cells, thus contributing to immune evasion.
The researchers also found that APOE protein activated the immune inhibitory receptor LILRB4.
To ascertain whether suppression of T cells by LILRB4 depends on APOE, the team co-cultured T cells with control or human AML cells with APOE knocked out.
Through a series of experiments, they determined that APOE is an extracellular binding protein of LILRB4 and that APOE activates LILRB4 to support T-cell proliferation and AML cell migration.
The researchers believe that targeting LILRB4 may have minimal toxicity. This is because LILRB4 expression on normal monocytic cells is limited, LILRB4 signaling may differ in leukemia cells, and LILRB4 blockade did not significantly interfere with normal hematopoietic function.
Dr. Zhang anticipates that if the preclinical studies go well, clinical trials could begin as early as next year.
The University of Texas System has exclusively licensed LILRB4-related patent applications to California-based Immune-Onc Therapeutics Inc., which contributed to the research and is conducting preclinical studies.
Dr. Zhang and another author are scientific advisory board members with Immune-Onc Therapeutics. Two other authors are employees of and hold equities in Immune-Onc Therapeutics.
The researchers received additional funding for this work from the National Cancer Institute, Leukemia & Lymphoma Society, the March of Dimes, the Cancer Prevention and Research Institute of Texas, the Robert A. Welch Foundation, the National Natural Science Foundation of China, the National Basic Research Program of China, and the China Scholarship Council.
Efforts to determine why immune checkpoint blockade is not successful in treating leukemia have resulted in a “compelling” new target to treat monocytic acute myeloid leukemia (AML), according to researchers.
They discovered that leukocyte immunoglobulin-like receptor B4 (LILRB4), a marker of monocytic leukemia, creates an immunosuppressive microenvironment by mediating T-cell suppression.
Using a mouse model and human cells, the research team showed that LILRB4 supports tumor infiltration into tissues and suppresses T-cell activity through a signaling pathway involving APOE, LILRB4, SHP-2, uPAR, and ARG1.
Senior author Chengcheng “Alec” Zhang, PhD, of the University of Texas Southwestern Medical Center in Dallas, and his colleagues reported their findings in Nature.
The team first compared surface expression of LILRB4 on normal monocytes and neoplastic monocytes from 105 AML patient samples. They observed that LILRB4 levels were higher on monocytic AML cells than on normal monocytes.
The researchers then tested whether LILRB4 expressed on AML cells suppressed T cells. They cultured LILRB4-positive leukemia cells, LILRB4-negative leukemia cells, and normal hematopoietic cells with either autologous T cells or T cells from healthy donors. The team determined that only LILRB4-positive monocytic AML cells substantially suppressed T-cell proliferation.
When the researchers knocked out LILRB4, the ability of AML cells to suppress T cells was reduced and could be restored with forced expression of wild-type LILRB4. Additionally, LILRB4-mediated T-cell inhibition could be reversed by LILRB4-blocking antibodies.
The team then used a humanized mouse xenograft model and an immunocompetent mouse model to investigate LILRB4 function in immune checkpoint blockade. Blocking LILRB4 lowered tumor burden and prolonged survival in the mice.
The researchers performed numerous in vitro and in vivo experiments and observed that antibody blockade of LILRB4 shrank tumors and decreased leukemic infiltration into internal organs, including the bone marrow, liver, and brain.
And so the team hypothesized that LILRB4 promotes leukemia infiltration in addition to inhibiting T cells.
To test the hypothesis, they performed transendothelial migration and homing assays and monitored leukemia infiltration relative to LILRB4 expression on leukemia cells. They observed that LILRB4-mediated migration enhanced extramedullary infiltration of monocytic AML cells, thus contributing to immune evasion.
The researchers also found that APOE protein activated the immune inhibitory receptor LILRB4.
To ascertain whether suppression of T cells by LILRB4 depends on APOE, the team co-cultured T cells with control or human AML cells with APOE knocked out.
Through a series of experiments, they determined that APOE is an extracellular binding protein of LILRB4 and that APOE activates LILRB4 to support T-cell proliferation and AML cell migration.
The researchers believe that targeting LILRB4 may have minimal toxicity. This is because LILRB4 expression on normal monocytic cells is limited, LILRB4 signaling may differ in leukemia cells, and LILRB4 blockade did not significantly interfere with normal hematopoietic function.
Dr. Zhang anticipates that if the preclinical studies go well, clinical trials could begin as early as next year.
The University of Texas System has exclusively licensed LILRB4-related patent applications to California-based Immune-Onc Therapeutics Inc., which contributed to the research and is conducting preclinical studies.
Dr. Zhang and another author are scientific advisory board members with Immune-Onc Therapeutics. Two other authors are employees of and hold equities in Immune-Onc Therapeutics.
The researchers received additional funding for this work from the National Cancer Institute, Leukemia & Lymphoma Society, the March of Dimes, the Cancer Prevention and Research Institute of Texas, the Robert A. Welch Foundation, the National Natural Science Foundation of China, the National Basic Research Program of China, and the China Scholarship Council.
‘Encouraging’ phase 2 results in rel/ref AML

The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months.
However, the researchers identified factors associated with improved response and survival that they believe could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment-related.
Naval Daver, MD, of The University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 (range, 22-90). Fifty-six percent had de novo AML, and 44% had secondary AML.
The median number of prior therapies was 2 (range, 1 to 7). Sixty-four percent of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was 3 (range, 1 to 25).
Patients had a median time on study of 3.5 months (range, 0.3 to 26.3 months). Reasons for discontinuation included primary refractory disease (n=27), relapse after initial response (n=19), death (n=16), proceeding to SCT (n=3), and patient preference (n=3).
Safety
The most common treatment-related, non-hematologic AEs were constipation (26%), diarrhea (20%), pneumonitis (13%), nausea (11%), and lung infection (11%).
The rate of immune-related AEs was 25% (n=18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4). Fourteen of these patients responded to steroids and were safely re-challenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) due to AEs—pneumonitis (n=7), cytokine release syndrome (n=1), and immune nephritis (n=1).
Two patients died of AEs that were considered possibly related to treatment. One death was due to progressive pneumonia/pneumonitis, and one was due to hemophagocytosis lymphohistiocytosis.
Response
The overall response rate was 33% (n=23). Four patients had a complete response (CR), and 11 had a CR with incomplete count recovery (CRi).
One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. In addition, a higher frequency of pre-therapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said.
Survival
At a median follow-up of 21.4 months, 81% of patients (n=57) had died. Sixteen patients died on study treatment, and 41 died after discontinuation.
The median OS was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in non-responders (P<0.0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P<0.001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver noted.
This research was supported by Bristol-Myers Squibb, MD Anderson, and the Dick Clark Immunotherapy Research Fund. In addition, individual researchers reported financial relationships with Bristol-Myers Squibb.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months.
However, the researchers identified factors associated with improved response and survival that they believe could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment-related.
Naval Daver, MD, of The University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 (range, 22-90). Fifty-six percent had de novo AML, and 44% had secondary AML.
The median number of prior therapies was 2 (range, 1 to 7). Sixty-four percent of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was 3 (range, 1 to 25).
Patients had a median time on study of 3.5 months (range, 0.3 to 26.3 months). Reasons for discontinuation included primary refractory disease (n=27), relapse after initial response (n=19), death (n=16), proceeding to SCT (n=3), and patient preference (n=3).
Safety
The most common treatment-related, non-hematologic AEs were constipation (26%), diarrhea (20%), pneumonitis (13%), nausea (11%), and lung infection (11%).
The rate of immune-related AEs was 25% (n=18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4). Fourteen of these patients responded to steroids and were safely re-challenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) due to AEs—pneumonitis (n=7), cytokine release syndrome (n=1), and immune nephritis (n=1).
Two patients died of AEs that were considered possibly related to treatment. One death was due to progressive pneumonia/pneumonitis, and one was due to hemophagocytosis lymphohistiocytosis.
Response
The overall response rate was 33% (n=23). Four patients had a complete response (CR), and 11 had a CR with incomplete count recovery (CRi).
One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. In addition, a higher frequency of pre-therapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said.
Survival
At a median follow-up of 21.4 months, 81% of patients (n=57) had died. Sixteen patients died on study treatment, and 41 died after discontinuation.
The median OS was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in non-responders (P<0.0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P<0.001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver noted.
This research was supported by Bristol-Myers Squibb, MD Anderson, and the Dick Clark Immunotherapy Research Fund. In addition, individual researchers reported financial relationships with Bristol-Myers Squibb.
The combination of azacitidine and nivolumab produced “encouraging” results in a phase 2 trial of patients with relapsed or refractory acute myeloid leukemia (AML), according to researchers.
The overall response rate was 33%, and the median overall survival (OS) was 6.3 months.
However, the researchers identified factors associated with improved response and survival that they believe could be used to select patients for this treatment.
A quarter of patients on this trial had immune-related adverse events (AEs) that were considered related to treatment, and two patients died of AEs that may have been treatment-related.
Naval Daver, MD, of The University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer Discovery.
The trial included 70 patients with a median age of 70 (range, 22-90). Fifty-six percent had de novo AML, and 44% had secondary AML.
The median number of prior therapies was 2 (range, 1 to 7). Sixty-four percent of patients had received hypomethylating agents, 47% had received targeted therapies, and 19% had received allogeneic stem cell transplant (SCT).
For this trial, patients received azacitidine at 75 mg/m2 on days 1 to 7 and nivolumab at 3 mg/kg on days 1 and 14 of each cycle. The median number of cycles was 3 (range, 1 to 25).
Patients had a median time on study of 3.5 months (range, 0.3 to 26.3 months). Reasons for discontinuation included primary refractory disease (n=27), relapse after initial response (n=19), death (n=16), proceeding to SCT (n=3), and patient preference (n=3).
Safety
The most common treatment-related, non-hematologic AEs were constipation (26%), diarrhea (20%), pneumonitis (13%), nausea (11%), and lung infection (11%).
The rate of immune-related AEs was 25% (n=18), with grade 2-4 immune-related AEs occurring in 16 patients (8 with grade 3-4). Fourteen of these patients responded to steroids and were safely re-challenged with nivolumab, according to the researchers.
Nine patients (13%) discontinued nivolumab (but continued with azacitidine) due to AEs—pneumonitis (n=7), cytokine release syndrome (n=1), and immune nephritis (n=1).
Two patients died of AEs that were considered possibly related to treatment. One death was due to progressive pneumonia/pneumonitis, and one was due to hemophagocytosis lymphohistiocytosis.
Response
The overall response rate was 33% (n=23). Four patients had a complete response (CR), and 11 had a CR with incomplete count recovery (CRi).
One patient had a partial response, and seven had hematologic improvement in one or more parameter maintained for more than 6 months. Six patients had stable disease lasting more than 6 months.
The researchers noted that the response rate was higher among patients who had not received prior treatment with hypomethylating agents. In addition, a higher frequency of pre-therapy CD3 and CD8 cells in the bone marrow or peripheral blood appeared to predict response.
“In particular, CD3 appeared to have a high sensitivity and specificity rate for predicting response, indicating it might serve as a reliable biomarker for selecting patients for this combination therapy,” Dr. Daver said.
Survival
At a median follow-up of 21.4 months, 81% of patients (n=57) had died. Sixteen patients died on study treatment, and 41 died after discontinuation.
The median OS was 6.3 months, and the median event-free survival was 4.5 months.
The median OS was 16.1 months in patients with CR/CRi, partial response, hematologic improvement, or stable disease and 4.1 months in non-responders (P<0.0001). This difference was still significant after the researchers censored the three patients who had gone on to SCT in CR/CRi (P<0.001).
The researchers also found that being in first salvage was associated with improved OS in a univariate analysis and in a comparison with historical controls.
Dr. Daver and his colleagues concluded that azacitidine and nivolumab “produced an encouraging response rate and overall survival” in patients with relapsed/refractory AML.
“We believe that implementation of clinical and immune biomarkers to select patients are likely to yield further improved outcomes with these types of therapies in AML,” Dr. Daver noted.
This research was supported by Bristol-Myers Squibb, MD Anderson, and the Dick Clark Immunotherapy Research Fund. In addition, individual researchers reported financial relationships with Bristol-Myers Squibb.
Report details financial burden of blood cancers
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
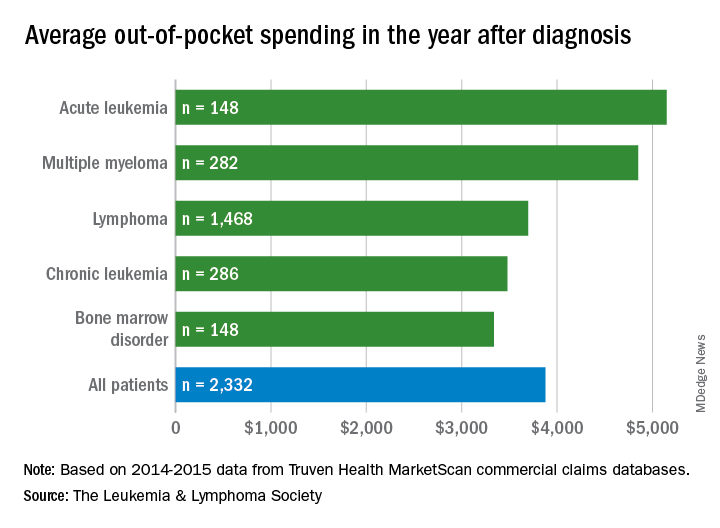
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
with costs for acute leukemia almost tripling that amount, according to a new report from the Leukemia & Lymphoma Society (LLS).
Total allowed cost – the average amount paid by the insurer and patient combined – for acute leukemia was more than $463,000 for the 12 months after initial diagnosis. Averages for the other four cancers included in the analysis came in at $214,000 for multiple myeloma, $134,000 for bone marrow disorders, $131,000 for lymphoma, and $89,000 for chronic leukemia, the LLS said.
The cost figures are drawn from claims data for 2,332 patients diagnosed in 2014.
Differences in out-of-pocket (OOP) costs were smaller, with the average for all patients at almost $3,900 in the year after diagnosis and acute leukemia coming in the highest at $5,100. Over time, however, OOP costs for multiple myeloma patients became the highest, totaling $9,100 for the 3 years after diagnosis, compared with $8,800 for acute leukemia and an average of less than $7,800 for the other blood cancers, the LLS said in the report, which was prepared by the actuarial firm Milliman.
OOP costs also varied by the type of plan. Patients in high-deductible plans averaged nearly $5,400 for the first year after diagnosis, compared with $3,300 for those with traditional insurance, the LLS noted. For acute leukemia, the OOP costs of high-deductible plans were more than twice as high as those of traditional plans.
The study was based on data for adults aged 18-64 years from the Truven Health MarketScan commercial claims databases for the years from 2013 to 2016. The LLS received support for the study from Pfizer, Genentech, and Amgen.
Quizartinib receives accelerated assessment for AML

The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
Changes related to AML relapse may be reversible

New research suggests relapse of acute myeloid leukemia (AML) after allogeneic hematopoietic stem cell transplant (HSCT) is related to changes in immune-related gene expression that may be reversible.
Researchers observed downregulation of major histocompatibility complex (MHC) class II genes in samples from patients who relapsed after HSCT.
However, interferon-gamma “rapidly reversed this phenotype” in vitro, according to the researchers.
Matthew J. Christopher, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri, and his colleagues reported these findings in The New England Journal of Medicine.
The researchers set out to determine how genetic and epigenetic changes after HSCT may allow leukemic cells to avoid the graft-vs-leukemia effect and to see whether immune-related genes are affected by HSCT.
The team analyzed paired samples obtained at diagnosis and relapse from 15 AML patients who relapsed after HSCT and 20 AML patients who relapsed after chemotherapy. The team also analyzed additional samples from patients who relapsed after HSCT to validate initial findings.
Methods of analysis included enhanced exome sequencing, RNA sequencing, flow cytometry, and immunohistochemical analysis.
Findings
The researchers first looked for relapse-specific mutations but found no driver mutations associated with relapse after HSCT.
The mutations seen post-HSCT relapse were generally similar to those seen both before treatment and after relapse in patients who had undergone chemotherapy, and the researchers could not identify any patterns of mutations related to relapse.
They then looked for, but did not find, relapse-specific mutations in genes involved in modulation of immune checkpoints, antigen presentation, or cytokine signaling.
The researchers did, however, find evidence of epigenetic changes that were more common in the samples from patients with post-transplant relapses.
RNA sequencing showed that MHC class II genes (HLA-DPA1, HLA-DPB1, HLA-DQB1, and HLA-DRB1) were downregulated three- to 12-fold after transplant.
Flow cytometry and immunohistochemical analysis confirmed that MHC class II expression was decreased at relapse after HSCT in 17 of 34 samples evaluated.
The researchers said there was no association between the downregulation of MHC class II and donor type or use of immunosuppression.
To see whether the downregulation of MHC class II genes was reversible, the researchers treated three post-HSCT relapse samples with interferon-gamma, which is known to upregulate MHC class II on certain cells.
Culturing patient cells with interferon-gamma “rapidly induced MHC class II protein expression on leukemic blasts,” the researchers said. They observed “essentially full restoration of MHC class II protein expression in nearly all AML blasts after 72 hours.”
This study was supported by the National Institutes of Health, Leukemia and Lymphoma Society, and the Barnes-Jewish Hospital Foundation.
Several study authors reported personal fees and/or research support from industry outside the submitted work.
New research suggests relapse of acute myeloid leukemia (AML) after allogeneic hematopoietic stem cell transplant (HSCT) is related to changes in immune-related gene expression that may be reversible.
Researchers observed downregulation of major histocompatibility complex (MHC) class II genes in samples from patients who relapsed after HSCT.
However, interferon-gamma “rapidly reversed this phenotype” in vitro, according to the researchers.
Matthew J. Christopher, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri, and his colleagues reported these findings in The New England Journal of Medicine.
The researchers set out to determine how genetic and epigenetic changes after HSCT may allow leukemic cells to avoid the graft-vs-leukemia effect and to see whether immune-related genes are affected by HSCT.
The team analyzed paired samples obtained at diagnosis and relapse from 15 AML patients who relapsed after HSCT and 20 AML patients who relapsed after chemotherapy. The team also analyzed additional samples from patients who relapsed after HSCT to validate initial findings.
Methods of analysis included enhanced exome sequencing, RNA sequencing, flow cytometry, and immunohistochemical analysis.
Findings
The researchers first looked for relapse-specific mutations but found no driver mutations associated with relapse after HSCT.
The mutations seen post-HSCT relapse were generally similar to those seen both before treatment and after relapse in patients who had undergone chemotherapy, and the researchers could not identify any patterns of mutations related to relapse.
They then looked for, but did not find, relapse-specific mutations in genes involved in modulation of immune checkpoints, antigen presentation, or cytokine signaling.
The researchers did, however, find evidence of epigenetic changes that were more common in the samples from patients with post-transplant relapses.
RNA sequencing showed that MHC class II genes (HLA-DPA1, HLA-DPB1, HLA-DQB1, and HLA-DRB1) were downregulated three- to 12-fold after transplant.
Flow cytometry and immunohistochemical analysis confirmed that MHC class II expression was decreased at relapse after HSCT in 17 of 34 samples evaluated.
The researchers said there was no association between the downregulation of MHC class II and donor type or use of immunosuppression.
To see whether the downregulation of MHC class II genes was reversible, the researchers treated three post-HSCT relapse samples with interferon-gamma, which is known to upregulate MHC class II on certain cells.
Culturing patient cells with interferon-gamma “rapidly induced MHC class II protein expression on leukemic blasts,” the researchers said. They observed “essentially full restoration of MHC class II protein expression in nearly all AML blasts after 72 hours.”
This study was supported by the National Institutes of Health, Leukemia and Lymphoma Society, and the Barnes-Jewish Hospital Foundation.
Several study authors reported personal fees and/or research support from industry outside the submitted work.
New research suggests relapse of acute myeloid leukemia (AML) after allogeneic hematopoietic stem cell transplant (HSCT) is related to changes in immune-related gene expression that may be reversible.
Researchers observed downregulation of major histocompatibility complex (MHC) class II genes in samples from patients who relapsed after HSCT.
However, interferon-gamma “rapidly reversed this phenotype” in vitro, according to the researchers.
Matthew J. Christopher, MD, PhD, of Washington University School of Medicine in St. Louis, Missouri, and his colleagues reported these findings in The New England Journal of Medicine.
The researchers set out to determine how genetic and epigenetic changes after HSCT may allow leukemic cells to avoid the graft-vs-leukemia effect and to see whether immune-related genes are affected by HSCT.
The team analyzed paired samples obtained at diagnosis and relapse from 15 AML patients who relapsed after HSCT and 20 AML patients who relapsed after chemotherapy. The team also analyzed additional samples from patients who relapsed after HSCT to validate initial findings.
Methods of analysis included enhanced exome sequencing, RNA sequencing, flow cytometry, and immunohistochemical analysis.
Findings
The researchers first looked for relapse-specific mutations but found no driver mutations associated with relapse after HSCT.
The mutations seen post-HSCT relapse were generally similar to those seen both before treatment and after relapse in patients who had undergone chemotherapy, and the researchers could not identify any patterns of mutations related to relapse.
They then looked for, but did not find, relapse-specific mutations in genes involved in modulation of immune checkpoints, antigen presentation, or cytokine signaling.
The researchers did, however, find evidence of epigenetic changes that were more common in the samples from patients with post-transplant relapses.
RNA sequencing showed that MHC class II genes (HLA-DPA1, HLA-DPB1, HLA-DQB1, and HLA-DRB1) were downregulated three- to 12-fold after transplant.
Flow cytometry and immunohistochemical analysis confirmed that MHC class II expression was decreased at relapse after HSCT in 17 of 34 samples evaluated.
The researchers said there was no association between the downregulation of MHC class II and donor type or use of immunosuppression.
To see whether the downregulation of MHC class II genes was reversible, the researchers treated three post-HSCT relapse samples with interferon-gamma, which is known to upregulate MHC class II on certain cells.
Culturing patient cells with interferon-gamma “rapidly induced MHC class II protein expression on leukemic blasts,” the researchers said. They observed “essentially full restoration of MHC class II protein expression in nearly all AML blasts after 72 hours.”
This study was supported by the National Institutes of Health, Leukemia and Lymphoma Society, and the Barnes-Jewish Hospital Foundation.
Several study authors reported personal fees and/or research support from industry outside the submitted work.