User login
Team links telomere degeneration and MDS
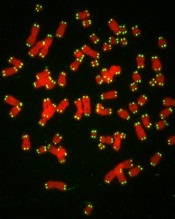
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
telomeres in green
Image by Claus Azzalin
New research has revealed a direct link between telomere degeneration and myelodysplastic syndromes (MDS).
“MDS risk correlates with advancing age, therapy-induced DNA damage, and/or shorter telomeres, but whether telomere erosion directly causes MDS is unknown,” said Simona Colla, PhD, of the MD Anderson Cancer Center in Houston, Texas.
“Our study provided genetic evidence that DNA damage caused by telomere loss is linked to this disorder.”
Dr Colla and her colleagues described this study in Cancer Cell.
The team’s in vitro and in vivo work showed that DNA damage caused by dysfunctional telomeres resulted in repressed expression of the gene SRSF2.
SRSF2 is an RNA splicing gene that plays a role in cellular processes. This change impacted common myeloid progenitors (CMPs), affecting their ability to differentiate or fully mature.
“This study established an intimate link across telomere biology, aberrant RNA splicing, and CMP differentiation,” said Ron DiPinho, MD, also of the MD Anderson Cancer Center.
“This may suggest that strategies to mitigate this DNA damage may be useful for preventing and/or treating MDS.”
Dr Colla added that the researchers’ findings “were consistent with long-standing observations that poor prognosis in MDS correlates strongly with short telomeres and elevated DNA damage in CMP cells.”
“This improved understanding should provide highly specific risk biomarkers for preventing and treating this incurable disease,” she said. ![]()
New method to treat EPO-resistant anemias

Researchers believe they have found a new way to treat anemias that are resistant to erythropoietin (EPO).
The team identified a pair of drugs, one that is already approved for use in the US, that can activate the cell receptor PPAR-α and synergize with low amounts of
glucocorticoids to increase red blood cell (RBC) production.
Their research is serving as the foundation for an upcoming clinical trial in patients with Diamond-Blackfan anemia (DBA).
Harvey Lodish, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts, and his colleagues described the research in a letter to Nature.
The team noted that certain anemias, such as DBA, cannot be treated with EPO. EPO controls RBC production by causing colony-forming-unit erythroids (CFU-Es) to divide and differentiate into RBCs. In DBA, the CFU-Es die before they can make RBCs, and patients have too few CFU-Es to make EPO treatment effective.
In 2010, Dr Lodish and his colleagues determined that glucocorticoids increase RBCs in EPO-resistant anemias by acting on burst-forming-unit erythroids (BFU-Es).
Glucocorticoids increase the likelihood that, when BFU-Es divide, one or both of the resulting cells remains a BFU-E instead of differentiating into CFU-Es. Patients treated with glucocorticoids have more BFU-Es, which, in turn, produce more CFU-Es and, ultimately, more RBCs.
With this in mind, the researchers screened for drugs that could interact with and boost glucocorticoids’ activity.
Glucocorticoids act by binding to a receptor in the cytoplasm that migrates into the nucleus and affects the expression of multiple genes. So the team screened for drugs that inhibit or activate other nuclear receptors.
They found that two drugs used to treat lipid disorders worked with glucocorticoids to increase RBC production in vitro. Both of these drugs, GW7647 and fenofibrate, activate PPARα. Fenofibrate was approved by the US Food and Drug Administration in 2001 and has been used to treat high cholesterol in adults and children.
When the researchers studied the mechanism of action of glucocorticoids and GW7647/fenofibrate, they found the glucocorticoid receptor binds to approximately 1000 sites in the DNA and turns on a large number of genes.
Fenofibrate and GW7647 activate the PPARα receptor, which subsequently binds adjacent to the glucocorticoid receptor on the DNA. The receptors modulate genes that are critical for BFU-E cell self-renewal and ultimately produce more RBCs.
When combined with dexamethasone, treatment with either GW7647 or fenofibrate led to a 150-fold increase in erythroblast production, which is 3- to 5-fold greater than the increase observed with dexamethasone alone.
These results have led the researchers to begin a clinical trial to test the effectiveness of a glucocorticoid/fenofibrate treatment in children with DBA.
Beyond the treatment of DBA, the researchers are optimistic about the impact that glucocorticoid/fenofibrate treatment might have on seemingly unrelated conditions.
“Glucocorticoids represent one of the most prescribed classes of drugs,” said Xiaofei Gao, PhD, a researcher in the Lodish lab.
“Basically, if PPARα is important in a disease that is currently treated by glucocorticoids, we may have a new way to treat those diseases that reduces the harmful side effects of glucocorticoids. That could affect a lot of patients.” ![]()
Researchers believe they have found a new way to treat anemias that are resistant to erythropoietin (EPO).
The team identified a pair of drugs, one that is already approved for use in the US, that can activate the cell receptor PPAR-α and synergize with low amounts of
glucocorticoids to increase red blood cell (RBC) production.
Their research is serving as the foundation for an upcoming clinical trial in patients with Diamond-Blackfan anemia (DBA).
Harvey Lodish, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts, and his colleagues described the research in a letter to Nature.
The team noted that certain anemias, such as DBA, cannot be treated with EPO. EPO controls RBC production by causing colony-forming-unit erythroids (CFU-Es) to divide and differentiate into RBCs. In DBA, the CFU-Es die before they can make RBCs, and patients have too few CFU-Es to make EPO treatment effective.
In 2010, Dr Lodish and his colleagues determined that glucocorticoids increase RBCs in EPO-resistant anemias by acting on burst-forming-unit erythroids (BFU-Es).
Glucocorticoids increase the likelihood that, when BFU-Es divide, one or both of the resulting cells remains a BFU-E instead of differentiating into CFU-Es. Patients treated with glucocorticoids have more BFU-Es, which, in turn, produce more CFU-Es and, ultimately, more RBCs.
With this in mind, the researchers screened for drugs that could interact with and boost glucocorticoids’ activity.
Glucocorticoids act by binding to a receptor in the cytoplasm that migrates into the nucleus and affects the expression of multiple genes. So the team screened for drugs that inhibit or activate other nuclear receptors.
They found that two drugs used to treat lipid disorders worked with glucocorticoids to increase RBC production in vitro. Both of these drugs, GW7647 and fenofibrate, activate PPARα. Fenofibrate was approved by the US Food and Drug Administration in 2001 and has been used to treat high cholesterol in adults and children.
When the researchers studied the mechanism of action of glucocorticoids and GW7647/fenofibrate, they found the glucocorticoid receptor binds to approximately 1000 sites in the DNA and turns on a large number of genes.
Fenofibrate and GW7647 activate the PPARα receptor, which subsequently binds adjacent to the glucocorticoid receptor on the DNA. The receptors modulate genes that are critical for BFU-E cell self-renewal and ultimately produce more RBCs.
When combined with dexamethasone, treatment with either GW7647 or fenofibrate led to a 150-fold increase in erythroblast production, which is 3- to 5-fold greater than the increase observed with dexamethasone alone.
These results have led the researchers to begin a clinical trial to test the effectiveness of a glucocorticoid/fenofibrate treatment in children with DBA.
Beyond the treatment of DBA, the researchers are optimistic about the impact that glucocorticoid/fenofibrate treatment might have on seemingly unrelated conditions.
“Glucocorticoids represent one of the most prescribed classes of drugs,” said Xiaofei Gao, PhD, a researcher in the Lodish lab.
“Basically, if PPARα is important in a disease that is currently treated by glucocorticoids, we may have a new way to treat those diseases that reduces the harmful side effects of glucocorticoids. That could affect a lot of patients.” ![]()
Researchers believe they have found a new way to treat anemias that are resistant to erythropoietin (EPO).
The team identified a pair of drugs, one that is already approved for use in the US, that can activate the cell receptor PPAR-α and synergize with low amounts of
glucocorticoids to increase red blood cell (RBC) production.
Their research is serving as the foundation for an upcoming clinical trial in patients with Diamond-Blackfan anemia (DBA).
Harvey Lodish, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts, and his colleagues described the research in a letter to Nature.
The team noted that certain anemias, such as DBA, cannot be treated with EPO. EPO controls RBC production by causing colony-forming-unit erythroids (CFU-Es) to divide and differentiate into RBCs. In DBA, the CFU-Es die before they can make RBCs, and patients have too few CFU-Es to make EPO treatment effective.
In 2010, Dr Lodish and his colleagues determined that glucocorticoids increase RBCs in EPO-resistant anemias by acting on burst-forming-unit erythroids (BFU-Es).
Glucocorticoids increase the likelihood that, when BFU-Es divide, one or both of the resulting cells remains a BFU-E instead of differentiating into CFU-Es. Patients treated with glucocorticoids have more BFU-Es, which, in turn, produce more CFU-Es and, ultimately, more RBCs.
With this in mind, the researchers screened for drugs that could interact with and boost glucocorticoids’ activity.
Glucocorticoids act by binding to a receptor in the cytoplasm that migrates into the nucleus and affects the expression of multiple genes. So the team screened for drugs that inhibit or activate other nuclear receptors.
They found that two drugs used to treat lipid disorders worked with glucocorticoids to increase RBC production in vitro. Both of these drugs, GW7647 and fenofibrate, activate PPARα. Fenofibrate was approved by the US Food and Drug Administration in 2001 and has been used to treat high cholesterol in adults and children.
When the researchers studied the mechanism of action of glucocorticoids and GW7647/fenofibrate, they found the glucocorticoid receptor binds to approximately 1000 sites in the DNA and turns on a large number of genes.
Fenofibrate and GW7647 activate the PPARα receptor, which subsequently binds adjacent to the glucocorticoid receptor on the DNA. The receptors modulate genes that are critical for BFU-E cell self-renewal and ultimately produce more RBCs.
When combined with dexamethasone, treatment with either GW7647 or fenofibrate led to a 150-fold increase in erythroblast production, which is 3- to 5-fold greater than the increase observed with dexamethasone alone.
These results have led the researchers to begin a clinical trial to test the effectiveness of a glucocorticoid/fenofibrate treatment in children with DBA.
Beyond the treatment of DBA, the researchers are optimistic about the impact that glucocorticoid/fenofibrate treatment might have on seemingly unrelated conditions.
“Glucocorticoids represent one of the most prescribed classes of drugs,” said Xiaofei Gao, PhD, a researcher in the Lodish lab.
“Basically, if PPARα is important in a disease that is currently treated by glucocorticoids, we may have a new way to treat those diseases that reduces the harmful side effects of glucocorticoids. That could affect a lot of patients.” ![]()
Drug can alleviate transfusion dependence in non-del-5q MDS

Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Photo courtesy of Celgene
WASHINGTON, DC—Results of a phase 3 trial support the use of lenalidomide in patients with lower-risk myelodysplastic syndromes (MDS) without 5q deletion who are unresponsive or refractory to erythropoiesis-stimulating agents (ESAs), according to researchers.
About 27% of patients who received lenalidomide achieved transfusion independence for 8 weeks or more, and about 18% were transfusion-independent for 24 weeks or more.
Valeria Santini, MD, of AOU Careggi in Florence, Italy, and her colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 115). The trial, MDS-005, was supported by Celgene Corporation, the company developing lenalidomide.
The trial was a comparison of lenalidomide and placebo in 239 patients with non-del-5q MDS who had failed treatment with ESAs. The patients were transfusion-dependent and had low- or intermediate-1-risk disease according to the International Prognostic Scoring System.
Patients were randomized 2:1 to receive oral lenalidomide at 10 mg once daily on days 1 to 28 of 28-day cycles (5 mg for patients with creatinine clearance 40 to 60 mL/min) or placebo.
Significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 8 weeks or more—26.9% vs 2.5% (P<0.001).
Likewise, significantly more patients in the lenalidomide arm than in the placebo arm achieved transfusion independence for 24 weeks or more—17.5% vs 0% (P<0.001).
Ninety percent of the lenalidomide-treated patients who achieved transfusion independence for 8 weeks or more responded within 4 cycles of treatment. The median duration of response was 32.9 weeks.
The median follow-up was 1.6 years (range, 0-3.6) in the lenalidomide arm and 1.3 years (range, 0-4.0) in the placebo arm. Within these time periods, patients in the placebo arm were more likely than those who received lenalidomide to progress to acute myeloid leukemia (AML) or to develop second primary malignancies (SPMs).
The AML incidence rate per 100 person-years was 1.91 in the lenalidomide arm and 2.46 in the placebo arm. And the SPM incidence rate per 100 person-years was 2.19 in the lenalidomide arm and 2.27 in the placebo arm.
As expected, treatment-emergent adverse events (AEs) were more common in the lenalidomide arm than in the placebo arm. AEs included neutropenia (64.4% vs 12.7%), thrombocytopenia (39.4% vs 7.6%), diarrhea (42.5% vs 22.8%), constipation (22.5% vs 12.7%), infections (51.9% vs 43%), hemorrhage (20.6% vs 10.1%), hepatic disorders (14.4% vs 5.1%), cardiac arrhythmia (11.3% vs 8.9%), and cutaneous reactions (10% vs 1.3%).
Grade 3-4 AEs in the lenalidomide arm included neutropenia (61.9%), thrombocytopenia (35.6%), infections (14.4%), hepatic disorders (5%), diarrhea (2.5%), hemorrhage (1.9%), deep vein thrombosis (1.9%), cardiac arrhythmia (1.3%), and cutaneous reactions (1.3%).
Based on the results of this trial, Celgene plans to submit a regulatory filing with the US Food and Drug Administration in the second half of 2015. Lenalidomide is not approved in the US to treat patients with non-del-5q MDS. ![]()
Inhibitor improves OS in poor-prognosis MDS

WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
WASHINGTON, DC—A small-molecule inhibitor can improve overall survival (OS) in certain patients with previously treated myelodysplastic syndromes (MDS), results of a phase 3 trial suggest.
Overall, patients who received the dual PI3K/PLK pathway inhibitor rigosertib along with best supportive care (BSC) did not see a significant improvement in OS compared to patients who received BSC alone.
However, rigosertib did improve OS in patients with poor prognosis.
Guillermo Garcia-Manero, MD, of the MD Anderson Cancer Center in Houston, Texas, and his colleagues presented these results at the 13th International Symposium on Myelodysplastic Syndromes (abstract 112).
The trial, known as ONTIME, was sponsored by Onconova Therapeutics, Inc., the company developing rigosertib.
The trial included 299 higher-risk MDS patients with excess blasts (5% to 30% bone marrow blasts) who had failed to respond to (25%), progressed on (37%), or relapsed after (38%) treatment with hypomethylating agents (HMAs).
Patients were randomized 2:1 to receive rigosertib plus BSC or BSC alone. Patients treated with rigosertib received 1800 mg every 24 hours for 72 hours as a continuous, intravenous, ambulatory infusion, every 2 weeks for the first 16 weeks, then every 4 weeks.
The treatment arms were generally balanced in terms of baseline characteristics. The majority of patients were male (66%) and white (82%). The median age was 74 years. Most patients (85%) had an Eastern Cooperative Oncology Group score of 0 or 1.
The median duration of the last HMA therapy was 8.8 months for patients in the rigosertib arm and 10.3 months for patients in the BSC arm.
The researchers found no significant difference in OS between the treatment arms. The median OS was 8.4 months in the rigosertib arm and 5.9 months in the BSC arm, and the 12-month OS was 35% and 25%, respectively (hazard ratio[HR]=0.87, P=0.31).
On the other hand, certain patients did see a significant improvement in OS with rigosertib. Among patients with primary HMA failure (those who failed to respond to or progressed during HMA therapy), the median OS was 8.6 months in the rigosertib arm and 5.3 months in the BSC arm (HR=0.69, P=0.040).
For patients who received HMAs for less than 9 months, the median OS was 7.7 months in the rigosertib arm and 4.5 months in the BSC arm (HR=0.55, P=0.003). Among patients younger than 75 years of age, the median OS was 9.7 months in the rigosertib arm and 4.1 months in the BSC arm (HR=0.52, P=0.0004).
And for patients with very high-risk disease according to the Revised International Prognostic Scoring System, the median OS was 7.6 months in the rigosertib arm and 3.2 months in the BSC arm (HR=0.56, P=0.005).
The researchers said there were no obvious differences between the treatment arms with regard to overall adverse events (AEs) or grade 3 or higher AEs.
Overall, 99% of patients in the rigosertib arm and 85% in the BSC arm experienced treatment-emergent AEs. The incidence of grade 3 or higher AEs was 79% and 68%, respectively.
Treatment-emergent AEs of all grades—occurring in the rigosertib and BSC arms, respectively—included nausea (35% vs 18%), diarrhea (33% vs 20%), constipation (31% vs 11%), fatigue (30% vs 18%), pyrexia (27% vs 21%), anemia (23% vs 9%), peripheral edema (21% vs 16%), and thrombocytopenia (21% vs 8%).
Considering the study results together, Dr Garcia-Manero and his colleagues concluded that rigosertib is likely most effective in high-risk MDS patients with the worst prognosis, and these patients can safely receive the drug. ![]()
Drug shows promise for lower-risk MDS

WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
WASHINGTON, DC—An investigational drug can increase hemoglobin levels and eliminate transfusion dependence in patients with lower-risk myelodysplastic syndromes (MDS), results of a phase 2 trial suggest.
The drug, luspatercept, is a modified activin receptor type IIB fusion protein that acts as a ligand trap for members in the TGF-β superfamily involved in the late stages of erythropoiesis.
Luspatercept regulates late-stage erythrocyte precursor differentiation and maturation.
Uwe Platzbecker, MD, of the University Hospital in Dresden, Germany, presented results from an ongoing phase 2 study of luspatercept at the 13th International Symposium on Myelodysplastic Syndromes (abstract 53).
The trial is supported by Acceleron Pharma Inc. and Celgene Corporation, the companies developing luspatercept.
“We are excited by the results in lower-risk MDS patients, which confirm and extend our previous findings,” Dr Platzbecker said. “Luspatercept may be useful early in the treatment of lower-risk MDS patients, either as the initial treatment for anemia or in patients who do not respond or become refractory to treatment with erythropoiesis-stimulating agents.”
Patient and dosing details
The researchers enrolled 58 patients in this study. Twenty-seven have completed treatment as part of the dose-escalation cohort. These patients received luspatercept at 7 doses ranging from 0.125 mg/kg to 1.75 mg/kg.
Thirty-one patients are still receiving treatment in the expansion cohort. The starting dose in this cohort is 1.0 mg/kg, and patients are receiving individual dose titration up to 1.75 mg/kg. Seventeen patients from this cohort received at least 4 cycles of treatment or discontinued early and were included in the analysis presented at the meeting.
In all, Dr Platzbecker presented results in 44 patients. Their median age was 71 (range, 27-88), and 57% were male. The median time since diagnosis was 2.5 years (range, 0.2-13.6 years). Sixty-one percent of patients had received prior treatment with erythropoiesis-stimulating agents, and 21% had received lenalidomide.
Fifteen patients had a low transfusion burden (LTB), as they received less than 4 units of red blood cells (RBCs) over 8 weeks. For these patients, the median hemoglobin at baseline was 9.0 g/dL (range, 6.8-10.1), and the median number of RBCs transfused over 8 weeks was 2 (range, 2-2).
Twenty-nine patients had a high transfusion burden (HTB) and received 4 or more RBC units over 8 weeks. The median number of RBCs transfused in this group was 6 (range, 4-14).
Fifty percent of patients had low-risk MDS according to IPSS, 46% had intermediate-1-risk disease, and 4% had intermediate-2-risk MDS. Eighty-one percent of patients were positive for ring sideroblasts, and 58% had the SF3B1 splicing mutation.
Efficacy and safety
The study’s primary efficacy endpoint was an increase in hemoglobin and/or a reduction in transfusion use. For LTB patients, the endpoint was a hemoglobin increase of 1.5 g/dL or more for 2 weeks or longer. For HTB patients, it was decrease in transfusion of 4 or more RBC units or a 50% or greater reduction in transfusion over 8 weeks.
Among the 9 patients who received lower doses of luspatercept (0.125-0.5 mg/kg), 33% met the primary efficacy endpoint. And 63% of the 35 patients in the higher dose group (0.75-1.75 mg/kg) achieved the primary efficacy endpoint.
Twenty-two percent of patients in the lower dose group achieved the International Working Group (IWG) hematologic improvement-erythroid (HI-E) threshold of efficacy, as did 54% of patients in the higher dose group.
Fourteen percent of patients in the lower dose group achieved transfusion independence, as did 36% of patients in the higher dose group. In the higher dose group, this included 4 of 6 patients with LTB and 6 of 22 patients with HTB.
Among patients who were ring-sideroblast-positive and received higher doses of luspatercept, 39% achieved transfusion independence, and 63% achieved IWG HI-E.
The majority of adverse events (AEs) were mild to moderate (grade 1 or 2). AEs included nasopharyngitis (14%), diarrhea (14%), myalgia (11%), bone pain (9%), bronchitis (9%), headache (9%), and muscle spasms (9%).
There were 2 serious AEs—grade 3 muscle pain and grade 3 worsening of general condition—that were considered possibly related to treatment. One non-serious grade 3 AE of blast cell count increase was considered possibly treatment-related as well.
In closing, Dr Platzbecker said luspatercept was generally safe and well-tolerated, in addition to providing “robust hematologic improvement.” And these results support further study of the drug in patients with lower-risk MDS. ![]()
Analysis helps quantify HU use in SCA

Photo by Zak Hubbard
Results of a retrospective study reinforce the idea that hydroxyurea (HU) is underused in US patients with sickle cell anemia (SCA).
Researchers analyzed data from an insurance claims database and identified 570 patients with probable SCA who were available for follow-up and likely would have benefitted from receiving HU.
Less than a quarter of those patients actually received the drug within a year of their third visit to a hospital seeking treatment for pain crises.
The researchers noted that these data may not be representative of the US population because the analysis does not include uninsured patients or publicly insured patients.
Nicolas Stettler, MD, of the Lewin Group in Falls Church, Virginia, and his colleagues conducted this research and reported the results in a letter to JAMA.
The researchers noted that the 2014 National Heart, Lung, and Blood Institute guidelines recommend using HU to treat all adults with SCA who experience 3 or more moderate-to-severe pain crises within a year. This is a “strong” recommendation based on high-quality evidence reviewed in 2008.
Despite this recommendation, it is thought that HU is underused, although the extent of its use has been unclear.
With that in mind, Dr Stettler and his colleagues examined the use of HU when indicated for SCA in the Optum Normative Health Informatics database. This database is a nationwide sample of commercial health and pharmacy claims from more than 36 million residents in all 50 states and Washington, DC.
The researchers identified adults ages 18 and older with 1 or more inpatient or outpatient claims for SCA between January 2009 and June 2013.
The team selected patients when they had 3 or more hospitalizations, emergency department visits, or both within 12 months that included 1 of the 5 most frequent diagnosis codes used for patients with SCA and pain crises.
Treatment was defined as filling 1 or more HU prescriptions during the 3, 6, or 12 months of continued enrollment following the third episode.
Of the enrolled population (n=26,631,901), the researchers identified 2086 adults with probable SCA. Of these patients, 677 had at least 3 pain-related hospitalizations or emergency department visits within 12 months, and 570 had at least 3 months of coverage after the third episode.
Among those 570 patients, 86 (15%) were treated with HU within 3 months of their third encounter. The percentage of treated patients increased slightly to 18% at 6 months and to 23% at 12 months.
The researchers noted that there are several barriers to HU treatment, including fear of adverse events, lack of clinician training, and failure to engage in shared decision-making.
The team also pointed out that their data do not include the uninsured or publicly insured population, which may have more limited access to healthcare or awareness of treatment options than the patients studied. So these findings may not be representative of the entire US population with SCA and may be a conservative estimate of the HU treatment gap.
To address this gap, it may be necessary to enhance patient outreach and clinician training and develop healthcare quality measures aimed at increasing the use of HU for all patients who would benefit, the researchers said. ![]()
Photo by Zak Hubbard
Results of a retrospective study reinforce the idea that hydroxyurea (HU) is underused in US patients with sickle cell anemia (SCA).
Researchers analyzed data from an insurance claims database and identified 570 patients with probable SCA who were available for follow-up and likely would have benefitted from receiving HU.
Less than a quarter of those patients actually received the drug within a year of their third visit to a hospital seeking treatment for pain crises.
The researchers noted that these data may not be representative of the US population because the analysis does not include uninsured patients or publicly insured patients.
Nicolas Stettler, MD, of the Lewin Group in Falls Church, Virginia, and his colleagues conducted this research and reported the results in a letter to JAMA.
The researchers noted that the 2014 National Heart, Lung, and Blood Institute guidelines recommend using HU to treat all adults with SCA who experience 3 or more moderate-to-severe pain crises within a year. This is a “strong” recommendation based on high-quality evidence reviewed in 2008.
Despite this recommendation, it is thought that HU is underused, although the extent of its use has been unclear.
With that in mind, Dr Stettler and his colleagues examined the use of HU when indicated for SCA in the Optum Normative Health Informatics database. This database is a nationwide sample of commercial health and pharmacy claims from more than 36 million residents in all 50 states and Washington, DC.
The researchers identified adults ages 18 and older with 1 or more inpatient or outpatient claims for SCA between January 2009 and June 2013.
The team selected patients when they had 3 or more hospitalizations, emergency department visits, or both within 12 months that included 1 of the 5 most frequent diagnosis codes used for patients with SCA and pain crises.
Treatment was defined as filling 1 or more HU prescriptions during the 3, 6, or 12 months of continued enrollment following the third episode.
Of the enrolled population (n=26,631,901), the researchers identified 2086 adults with probable SCA. Of these patients, 677 had at least 3 pain-related hospitalizations or emergency department visits within 12 months, and 570 had at least 3 months of coverage after the third episode.
Among those 570 patients, 86 (15%) were treated with HU within 3 months of their third encounter. The percentage of treated patients increased slightly to 18% at 6 months and to 23% at 12 months.
The researchers noted that there are several barriers to HU treatment, including fear of adverse events, lack of clinician training, and failure to engage in shared decision-making.
The team also pointed out that their data do not include the uninsured or publicly insured population, which may have more limited access to healthcare or awareness of treatment options than the patients studied. So these findings may not be representative of the entire US population with SCA and may be a conservative estimate of the HU treatment gap.
To address this gap, it may be necessary to enhance patient outreach and clinician training and develop healthcare quality measures aimed at increasing the use of HU for all patients who would benefit, the researchers said. ![]()
Photo by Zak Hubbard
Results of a retrospective study reinforce the idea that hydroxyurea (HU) is underused in US patients with sickle cell anemia (SCA).
Researchers analyzed data from an insurance claims database and identified 570 patients with probable SCA who were available for follow-up and likely would have benefitted from receiving HU.
Less than a quarter of those patients actually received the drug within a year of their third visit to a hospital seeking treatment for pain crises.
The researchers noted that these data may not be representative of the US population because the analysis does not include uninsured patients or publicly insured patients.
Nicolas Stettler, MD, of the Lewin Group in Falls Church, Virginia, and his colleagues conducted this research and reported the results in a letter to JAMA.
The researchers noted that the 2014 National Heart, Lung, and Blood Institute guidelines recommend using HU to treat all adults with SCA who experience 3 or more moderate-to-severe pain crises within a year. This is a “strong” recommendation based on high-quality evidence reviewed in 2008.
Despite this recommendation, it is thought that HU is underused, although the extent of its use has been unclear.
With that in mind, Dr Stettler and his colleagues examined the use of HU when indicated for SCA in the Optum Normative Health Informatics database. This database is a nationwide sample of commercial health and pharmacy claims from more than 36 million residents in all 50 states and Washington, DC.
The researchers identified adults ages 18 and older with 1 or more inpatient or outpatient claims for SCA between January 2009 and June 2013.
The team selected patients when they had 3 or more hospitalizations, emergency department visits, or both within 12 months that included 1 of the 5 most frequent diagnosis codes used for patients with SCA and pain crises.
Treatment was defined as filling 1 or more HU prescriptions during the 3, 6, or 12 months of continued enrollment following the third episode.
Of the enrolled population (n=26,631,901), the researchers identified 2086 adults with probable SCA. Of these patients, 677 had at least 3 pain-related hospitalizations or emergency department visits within 12 months, and 570 had at least 3 months of coverage after the third episode.
Among those 570 patients, 86 (15%) were treated with HU within 3 months of their third encounter. The percentage of treated patients increased slightly to 18% at 6 months and to 23% at 12 months.
The researchers noted that there are several barriers to HU treatment, including fear of adverse events, lack of clinician training, and failure to engage in shared decision-making.
The team also pointed out that their data do not include the uninsured or publicly insured population, which may have more limited access to healthcare or awareness of treatment options than the patients studied. So these findings may not be representative of the entire US population with SCA and may be a conservative estimate of the HU treatment gap.
To address this gap, it may be necessary to enhance patient outreach and clinician training and develop healthcare quality measures aimed at increasing the use of HU for all patients who would benefit, the researchers said. ![]()
More than 75% with sickle cell crises don’t get hydroxyurea
More than 75% of adults with sickle cell anemia who have frequent pain crises fail to receive hydroxyurea therapy as strongly recommended in National Heart, Lung, and Blood Institute clinical guidelines, according to a Research Letter to the Editor published online April 28 in JAMA.
Despite proven benefits in decreasing pain crises, hospitalizations, blood transfusions, and possibly mortality, hydroxyurea, a “safe and inexpensive drug,” is thought to be underused. To document the actual use of the drug when indicated, investigators analyzed information in a nationwide insurance claims database covering nearly 27 million patients per year. They focused on the records of 570 adults hospitalized or treated in an emergency department for a sickle cell pain crisis at least three times during a 1-year period, said Dr. Nicolas Stettler of the Lewin Group, a health care consulting firm in Falls Church, Va., and his associates.
Only 15.1% of these patients received hydroxyurea within 3 months of their third crisis, only 18.2% received the agent within 6 months, and only 22.7% received it within 12 months. These figures likely represent a conservative estimate of the hydroxyurea treatment gap, since the study didn’t include the large uninsured and publicly insured populations who have more limited access to health care, Dr. Stettler and his associates noted (JAMA 2015;313:1671-2).
Several barriers to this treatment have been identified in previous research, including fear of adverse events, lack of clinician training, and failure to use shared decision making. “To address this gap, it may be necessary to enhance patient outreach and clinician training and develop health care quality measures aimed at increasing the use of hydroxyurea for all patients who would benefit,” they added.
More than 75% of adults with sickle cell anemia who have frequent pain crises fail to receive hydroxyurea therapy as strongly recommended in National Heart, Lung, and Blood Institute clinical guidelines, according to a Research Letter to the Editor published online April 28 in JAMA.
Despite proven benefits in decreasing pain crises, hospitalizations, blood transfusions, and possibly mortality, hydroxyurea, a “safe and inexpensive drug,” is thought to be underused. To document the actual use of the drug when indicated, investigators analyzed information in a nationwide insurance claims database covering nearly 27 million patients per year. They focused on the records of 570 adults hospitalized or treated in an emergency department for a sickle cell pain crisis at least three times during a 1-year period, said Dr. Nicolas Stettler of the Lewin Group, a health care consulting firm in Falls Church, Va., and his associates.
Only 15.1% of these patients received hydroxyurea within 3 months of their third crisis, only 18.2% received the agent within 6 months, and only 22.7% received it within 12 months. These figures likely represent a conservative estimate of the hydroxyurea treatment gap, since the study didn’t include the large uninsured and publicly insured populations who have more limited access to health care, Dr. Stettler and his associates noted (JAMA 2015;313:1671-2).
Several barriers to this treatment have been identified in previous research, including fear of adverse events, lack of clinician training, and failure to use shared decision making. “To address this gap, it may be necessary to enhance patient outreach and clinician training and develop health care quality measures aimed at increasing the use of hydroxyurea for all patients who would benefit,” they added.
More than 75% of adults with sickle cell anemia who have frequent pain crises fail to receive hydroxyurea therapy as strongly recommended in National Heart, Lung, and Blood Institute clinical guidelines, according to a Research Letter to the Editor published online April 28 in JAMA.
Despite proven benefits in decreasing pain crises, hospitalizations, blood transfusions, and possibly mortality, hydroxyurea, a “safe and inexpensive drug,” is thought to be underused. To document the actual use of the drug when indicated, investigators analyzed information in a nationwide insurance claims database covering nearly 27 million patients per year. They focused on the records of 570 adults hospitalized or treated in an emergency department for a sickle cell pain crisis at least three times during a 1-year period, said Dr. Nicolas Stettler of the Lewin Group, a health care consulting firm in Falls Church, Va., and his associates.
Only 15.1% of these patients received hydroxyurea within 3 months of their third crisis, only 18.2% received the agent within 6 months, and only 22.7% received it within 12 months. These figures likely represent a conservative estimate of the hydroxyurea treatment gap, since the study didn’t include the large uninsured and publicly insured populations who have more limited access to health care, Dr. Stettler and his associates noted (JAMA 2015;313:1671-2).
Several barriers to this treatment have been identified in previous research, including fear of adverse events, lack of clinician training, and failure to use shared decision making. “To address this gap, it may be necessary to enhance patient outreach and clinician training and develop health care quality measures aimed at increasing the use of hydroxyurea for all patients who would benefit,” they added.
FROM JAMA
Key clinical point: More than 75% of adults with sickle cell anemia who have frequent pain crises fail to get hydroxyurea therapy as recommended.
Major finding: Only 15.1% of adults with sickle cell anemia received hydroxyurea within 3 months of their third pain crisis, only 18.2% received the agent within 6 months, and only 22.7% received it within 12 months.
Data source: An analysis of information in a large nationwide insurance claims database involving 570 adults with frequent hospitalizations for sickle cell pain crises.
Disclosures: This study was funded by the Lewin Group, a health care consulting firm. Dr. Stettler and his associates reported having no relevant financial disclosures.
Gene therapy appears effective against WAS
![]()
Photo by Chad McNeeley
Results of a small study suggest gene therapy can lead to clinical improvements in children and teens with Wiskott-Aldrich syndrome (WAS).
The gene therapy—autologous, gene-corrected hematopoietic stem cells (HSCs) given along with chemotherapy—improved infectious complications, severe eczema, and symptoms of autoimmunity in 6 of the 7 patients studied.
The therapy also reduced patients’ use of blood products and the amount of time they spent in the hospital.
Marina Cavazzana, MD, PhD, of Necker Children’s Hospital in Paris, France, and colleagues reported these results in JAMA. The study was sponsored by Genethon.
The researchers noted that WAS is caused by loss-of-function mutations in the WAS gene. The condition is characterized by thrombocytopenia, eczema, and recurring infections. In the absence of definitive treatment, patients with classic WAS generally do not survive beyond their second or third decade of life.
Partially HLA-matched, allogeneic HSC transplant is often curative, but it is associated with a high incidence of complications. Dr Cavazzana and colleagues speculated that transplanting autologous, gene-corrected HSCs may be an effective and potentially safer alternative.
So the team assessed the outcomes and safety of autologous HSC gene therapy in 7 patients (age range, 0.8-15.5 years) with severe WAS who lacked HLA antigen-matched related or unrelated HSC donors.
Patients were enrolled in France and England and treated between December 2010 and January 2014. Follow-up ranged from 9 months to 42 months.
The treatment involved collecting mutated HSCs from patients and correcting the cells in the lab by introducing a healthy WAS gene using a lentiviral vector developed and produced by Genethon. The corrected cells were reinjected into patients who, in parallel, received chemotherapy to suppress their defective stem cells and autoimmune cells to make room for new, corrected cells.
Six of the 7 patients saw clinical improvements after this treatment. One patient died of pre-existing, treatment-refractory infectious disease.
In the 6 surviving patients, infectious complications resolved after gene therapy, and prophylactic antibiotic therapy was successfully discontinued in 3 cases. Severe eczema resolved in all affected patients, as did signs and symptoms of autoimmunity.
There were no severe bleeding episodes after treatment. And, at last follow-up, none of the 6 surviving patients required blood product support.
The median number of hospitalization days decreased from 25 during the 2 years before treatment to 0 during the 2 years after treatment.
“[T]he patients showed a significant clinical improvement due to the re-expression of the protein WASp in the cells of the immune system,” Dr
Cavazzana said.
However, the researchers also noted that the interpretation of these results is constrained by the small number of patients studied. So the team said they could not draw conclusions on long-term outcomes and safety without further follow-up and additional trials. ![]()
![]()
Photo by Chad McNeeley
Results of a small study suggest gene therapy can lead to clinical improvements in children and teens with Wiskott-Aldrich syndrome (WAS).
The gene therapy—autologous, gene-corrected hematopoietic stem cells (HSCs) given along with chemotherapy—improved infectious complications, severe eczema, and symptoms of autoimmunity in 6 of the 7 patients studied.
The therapy also reduced patients’ use of blood products and the amount of time they spent in the hospital.
Marina Cavazzana, MD, PhD, of Necker Children’s Hospital in Paris, France, and colleagues reported these results in JAMA. The study was sponsored by Genethon.
The researchers noted that WAS is caused by loss-of-function mutations in the WAS gene. The condition is characterized by thrombocytopenia, eczema, and recurring infections. In the absence of definitive treatment, patients with classic WAS generally do not survive beyond their second or third decade of life.
Partially HLA-matched, allogeneic HSC transplant is often curative, but it is associated with a high incidence of complications. Dr Cavazzana and colleagues speculated that transplanting autologous, gene-corrected HSCs may be an effective and potentially safer alternative.
So the team assessed the outcomes and safety of autologous HSC gene therapy in 7 patients (age range, 0.8-15.5 years) with severe WAS who lacked HLA antigen-matched related or unrelated HSC donors.
Patients were enrolled in France and England and treated between December 2010 and January 2014. Follow-up ranged from 9 months to 42 months.
The treatment involved collecting mutated HSCs from patients and correcting the cells in the lab by introducing a healthy WAS gene using a lentiviral vector developed and produced by Genethon. The corrected cells were reinjected into patients who, in parallel, received chemotherapy to suppress their defective stem cells and autoimmune cells to make room for new, corrected cells.
Six of the 7 patients saw clinical improvements after this treatment. One patient died of pre-existing, treatment-refractory infectious disease.
In the 6 surviving patients, infectious complications resolved after gene therapy, and prophylactic antibiotic therapy was successfully discontinued in 3 cases. Severe eczema resolved in all affected patients, as did signs and symptoms of autoimmunity.
There were no severe bleeding episodes after treatment. And, at last follow-up, none of the 6 surviving patients required blood product support.
The median number of hospitalization days decreased from 25 during the 2 years before treatment to 0 during the 2 years after treatment.
“[T]he patients showed a significant clinical improvement due to the re-expression of the protein WASp in the cells of the immune system,” Dr
Cavazzana said.
However, the researchers also noted that the interpretation of these results is constrained by the small number of patients studied. So the team said they could not draw conclusions on long-term outcomes and safety without further follow-up and additional trials. ![]()
![]()
Photo by Chad McNeeley
Results of a small study suggest gene therapy can lead to clinical improvements in children and teens with Wiskott-Aldrich syndrome (WAS).
The gene therapy—autologous, gene-corrected hematopoietic stem cells (HSCs) given along with chemotherapy—improved infectious complications, severe eczema, and symptoms of autoimmunity in 6 of the 7 patients studied.
The therapy also reduced patients’ use of blood products and the amount of time they spent in the hospital.
Marina Cavazzana, MD, PhD, of Necker Children’s Hospital in Paris, France, and colleagues reported these results in JAMA. The study was sponsored by Genethon.
The researchers noted that WAS is caused by loss-of-function mutations in the WAS gene. The condition is characterized by thrombocytopenia, eczema, and recurring infections. In the absence of definitive treatment, patients with classic WAS generally do not survive beyond their second or third decade of life.
Partially HLA-matched, allogeneic HSC transplant is often curative, but it is associated with a high incidence of complications. Dr Cavazzana and colleagues speculated that transplanting autologous, gene-corrected HSCs may be an effective and potentially safer alternative.
So the team assessed the outcomes and safety of autologous HSC gene therapy in 7 patients (age range, 0.8-15.5 years) with severe WAS who lacked HLA antigen-matched related or unrelated HSC donors.
Patients were enrolled in France and England and treated between December 2010 and January 2014. Follow-up ranged from 9 months to 42 months.
The treatment involved collecting mutated HSCs from patients and correcting the cells in the lab by introducing a healthy WAS gene using a lentiviral vector developed and produced by Genethon. The corrected cells were reinjected into patients who, in parallel, received chemotherapy to suppress their defective stem cells and autoimmune cells to make room for new, corrected cells.
Six of the 7 patients saw clinical improvements after this treatment. One patient died of pre-existing, treatment-refractory infectious disease.
In the 6 surviving patients, infectious complications resolved after gene therapy, and prophylactic antibiotic therapy was successfully discontinued in 3 cases. Severe eczema resolved in all affected patients, as did signs and symptoms of autoimmunity.
There were no severe bleeding episodes after treatment. And, at last follow-up, none of the 6 surviving patients required blood product support.
The median number of hospitalization days decreased from 25 during the 2 years before treatment to 0 during the 2 years after treatment.
“[T]he patients showed a significant clinical improvement due to the re-expression of the protein WASp in the cells of the immune system,” Dr
Cavazzana said.
However, the researchers also noted that the interpretation of these results is constrained by the small number of patients studied. So the team said they could not draw conclusions on long-term outcomes and safety without further follow-up and additional trials.
FDA grants drug orphan designation for SCD
Image by Graham Beards
The US Food and Drug Administration (FDA) has granted orphan drug designation for the bovine PEGylated carboxyhemoglobin product Sanguinate to treat sickle cell disease (SCD).
Through its anti-vaso-constrictive properties, Sanguinate facilitates the transfer of oxygen to oxygen-deprived cells and tissues.
By correcting oxygen levels and downregulating inflammation, the drug could potentially treat many of the comorbidities associated with SCD.
Trials of Sanguinate
The company developing Sanguinate, Prolong Pharmaceuticals, has several clinical studies underway to determine the safety and efficacy of the drug in SCD and other diseases caused by the effects of oxygen deprivation.
In a phase 1 trial, Sanguinate proved safe and well-tolerated in healthy volunteers. Three cohorts of 8 subjects received single, ascending doses of Sanguinate at 80 mg/kg, 120 mg/kg, or 160 mg/kg. Two volunteers in each cohort were control subjects who received saline.
There were no serious adverse events reported with Sanguinate. Subjects experienced decreases in serum haptoglobin, but this did not appear to be dose-related. Sanguinate’s half-life was dose-dependent and ranged from 7.9 hours to 13.8 hours.
A phase 1 study of Sanguinate in SCD patients has been completed, and researchers are now conducting a phase 2 study testing the drug for the reduction or prevention of delayed cerebral ischemia following subarachnoid hemorrhage.
Phase 2 trials are also planned for vaso-occlusive crisis and leg ulcers secondary to SCD, as well as for preventing delayed graft function following kidney transplant. Sanguinate is also being evaluated for the treatment of beta-thalassemia.
About orphan designation
The FDA grants orphan designation to diseases affecting fewer than 200,000 people in the US.
Orphan designation provides the company developing a drug with certain benefits and incentives, including a 7-year period of marketing exclusivity upon regulatory approval, potential tax credits for certain activities, eligibility for orphan drug grants, and the waiver of certain administrative fees.
Image by Graham Beards
The US Food and Drug Administration (FDA) has granted orphan drug designation for the bovine PEGylated carboxyhemoglobin product Sanguinate to treat sickle cell disease (SCD).
Through its anti-vaso-constrictive properties, Sanguinate facilitates the transfer of oxygen to oxygen-deprived cells and tissues.
By correcting oxygen levels and downregulating inflammation, the drug could potentially treat many of the comorbidities associated with SCD.
Trials of Sanguinate
The company developing Sanguinate, Prolong Pharmaceuticals, has several clinical studies underway to determine the safety and efficacy of the drug in SCD and other diseases caused by the effects of oxygen deprivation.
In a phase 1 trial, Sanguinate proved safe and well-tolerated in healthy volunteers. Three cohorts of 8 subjects received single, ascending doses of Sanguinate at 80 mg/kg, 120 mg/kg, or 160 mg/kg. Two volunteers in each cohort were control subjects who received saline.
There were no serious adverse events reported with Sanguinate. Subjects experienced decreases in serum haptoglobin, but this did not appear to be dose-related. Sanguinate’s half-life was dose-dependent and ranged from 7.9 hours to 13.8 hours.
A phase 1 study of Sanguinate in SCD patients has been completed, and researchers are now conducting a phase 2 study testing the drug for the reduction or prevention of delayed cerebral ischemia following subarachnoid hemorrhage.
Phase 2 trials are also planned for vaso-occlusive crisis and leg ulcers secondary to SCD, as well as for preventing delayed graft function following kidney transplant. Sanguinate is also being evaluated for the treatment of beta-thalassemia.
About orphan designation
The FDA grants orphan designation to diseases affecting fewer than 200,000 people in the US.
Orphan designation provides the company developing a drug with certain benefits and incentives, including a 7-year period of marketing exclusivity upon regulatory approval, potential tax credits for certain activities, eligibility for orphan drug grants, and the waiver of certain administrative fees.
Image by Graham Beards
The US Food and Drug Administration (FDA) has granted orphan drug designation for the bovine PEGylated carboxyhemoglobin product Sanguinate to treat sickle cell disease (SCD).
Through its anti-vaso-constrictive properties, Sanguinate facilitates the transfer of oxygen to oxygen-deprived cells and tissues.
By correcting oxygen levels and downregulating inflammation, the drug could potentially treat many of the comorbidities associated with SCD.
Trials of Sanguinate
The company developing Sanguinate, Prolong Pharmaceuticals, has several clinical studies underway to determine the safety and efficacy of the drug in SCD and other diseases caused by the effects of oxygen deprivation.
In a phase 1 trial, Sanguinate proved safe and well-tolerated in healthy volunteers. Three cohorts of 8 subjects received single, ascending doses of Sanguinate at 80 mg/kg, 120 mg/kg, or 160 mg/kg. Two volunteers in each cohort were control subjects who received saline.
There were no serious adverse events reported with Sanguinate. Subjects experienced decreases in serum haptoglobin, but this did not appear to be dose-related. Sanguinate’s half-life was dose-dependent and ranged from 7.9 hours to 13.8 hours.
A phase 1 study of Sanguinate in SCD patients has been completed, and researchers are now conducting a phase 2 study testing the drug for the reduction or prevention of delayed cerebral ischemia following subarachnoid hemorrhage.
Phase 2 trials are also planned for vaso-occlusive crisis and leg ulcers secondary to SCD, as well as for preventing delayed graft function following kidney transplant. Sanguinate is also being evaluated for the treatment of beta-thalassemia.
About orphan designation
The FDA grants orphan designation to diseases affecting fewer than 200,000 people in the US.
Orphan designation provides the company developing a drug with certain benefits and incentives, including a 7-year period of marketing exclusivity upon regulatory approval, potential tax credits for certain activities, eligibility for orphan drug grants, and the waiver of certain administrative fees.
FDA approves new formulation of iron overload drug
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com.
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com.
The US Food and Drug Administration (FDA) has granted accelerated approval for Jadenu, a new oral formulation of Exjade (deferasirox).
Jadenu is now approved to treat patients 2 years of age and older who have chronic iron overload resulting from blood transfusions. The drug is also approved to treat chronic iron overload in patients 10 years of age and older who have non-transfusion-dependent thalassemia.
Jadenu can be swallowed whole and taken with or without a light meal. Exjade is a dispersible tablet that must be mixed in liquid and taken on an empty stomach.
Jadenu has been approved with a boxed warning, which states that the drug may cause serious and fatal renal toxicity (including failure), hepatic toxicity (including failure), and gastrointestinal hemorrhage. Therefore, treatment with Jadenu requires close patient monitoring, including laboratory tests of renal and hepatic function.
The FDA has granted Jadenu accelerated approval based on the drug showing a reduction of liver iron concentrations and serum ferritin levels. Continued FDA approval for Jadenu may be contingent upon verification and description of clinical benefit in confirmatory trials.
Jadenu has been evaluated in trials of healthy volunteers, but there are no clinical data showing the effects of Jadenu in patients with chronic iron overload.
Exjade, on the other hand, has been evaluated in several trials of patients with chronic iron overload resulting from transfusions and patients with non-transfusion-dependent thalassemia who have chronic iron overload.
Data from these trials can be found in the prescribing information for Jadenu, available at www.jadenu.com.