User login
Turoctocog alfa prevented, treated bleeds in previously untreated pediatric patients with severe hemophilia A
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.

Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.
Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
The recombinant factor VIII turoctocog alfa effectively prevented and stopped bleeds among previously untreated children with hemophilia A enrolled in a multicenter trial, investigators have reported.
Nearly 86% of patients who developed inhibitors and started immune tolerance induction with turoctocog alfa went on to have a negative inhibitor titer, according to investigators in guardian 4, an international, nonrandomized, phase 3 trial.
The treatment was well tolerated in these pediatric patients, and the rate of inhibitor development in the study was “within the expected range” for previously untreated patients with severe hemophilia A, wrote the investigators, led by Hassan M. Yaish, MD, of the University of Utah, Salt Lake City.
Turoctocog alfa, a recombinant, B domain–truncated human coagulation factor VIII (FVIII), is known to be safe and effective in previously treated patients with hemophilia A based on results of the guardian 1, 2, and 3 clinical trials, according to Dr. Yaish and coauthors.
“This is the first trial within the guardian clinical trial program to evaluate turoctocog alfa in previously untreated patients, a patient population that is generally at an increased risk of developing antibodies to foreign protein such as exogenous FVIII,” wrote Dr. Yaish and coauthors in their report on guardian 4, which appears in Haemophilia.
The guardian 4 trial included a total of 60 patients aged less than 6 years with severe hemophilia A treated at 1 of 40 sites internationally, including the United States. The investigators noted that 58 of those patients received turoctocog alfa as prophylaxis, and 49 completed the study.
The primary endpoint of the study was the incidence of FVIII inhibitors within the first 50 exposure days, the time period during which inhibitors most frequently occur, investigators wrote.
In all, 25 of the 58 prophylactically treated patients (43.1%) developed FVIII inhibitors in that period, according to Dr. Yaish and colleagues.
The inhibitor incidence rate for other recombinant FVIII products in key studies of previously untreated patients range from 24.4% to 44.5%, they wrote in a discussion of that result.
In the present study, guardian 4, the estimated mean annualized bleed rate was 4.26 bleeds per patient per year, the investigators also reported.
The success rate for treatment of bleeds during prophylaxis was 88.5%. Of 402 bleeds, 227 (56.5%) were controlled with a single injection of turoctocog alfa, according to the report, while 22.6% and 7.5% were controlled with two and three injections, respectively.
Of 21 patients with inhibitors who started immune tolerance induction with turoctocog alfa, 18 (85.7%) had a negative inhibitor titer at the end of the trial, while treatment was withdrawn in 3 (14.3%) because insufficient decline in titer or persistent positivity for titers following 2 years of the therapy, according to the report.
Novo Nordisk sponsored the guardian 4 study. Dr. Yaish reported disclosures related to Novo Nordisk, Shire, Bayer, and Octapharma.
SOURCE: Yaish HM et al. Haemophilia. 2019 Dec 9. doi: 10.1111/hae.13883.
FROM HAEMOPHILIA
Age, sex, and other factors linked to risk of intracranial hemorrhage in ITP
ORLANDO – A large, retrospective study suggests several factors are associated with an increased risk of intracranial hemorrhage in patients with immune thrombocytopenia.

Data on more than 300,000 immune thrombocytopenia (ITP) hospitalizations indicated that older age, male sex, not having private insurance, having a gastrointestinal or “other” bleed, and receiving treatment at a hospital in the western United States, a medium- or large-sized hospital, or an urban teaching hospital were all associated with an increased risk of intracranial hemorrhage (ICH).
Mayank Sharma, of the University of Miami, detailed these findings at the annual meeting of the American Society of Hematology.
Mr. Sharma and colleagues analyzed data from the National Inpatient Sample database from 2007 to 2016. Of the 348,906 ITP hospitalizations included, there were 3,408 (0.98%) cases of ICH.
The overall incidence of ICH was low and remained stable over time, “which is reassuring,” Mr. Sharma said. However, the mortality rate was higher among patients with ICH than among those without it – 26.7% and 3.2%, respectively.
A multivariate analysis showed that female patients had a decreased likelihood of ICH, with an odds ratio of 0.81 (95% confidence interval, 0.68-0.97). Patients with private insurance had a decreased likelihood of ICH as well, with an OR of 0.81 (95% CI, 0.61-1.08).
Conversely, older patients had an increased likelihood of ICH. The OR was 2.23 (95% CI, 1.51-3.31) for patients aged 25-64 years, and the OR was 3.69 (95% CI, 2.34-5.84) for patients aged 65 years and older.
Patients with a gastrointestinal bleed or an other bleed (not including hematuria or epistaxis) had an increased likelihood of ICH. The ORs were 1.60 (95% CI, 1.18-2.16) and 1.69 (95% CI, 1.19-2.42), respectively.
Patients hospitalized in the western United States (OR, 1.62; 95% CI, 1.26-2.08), at a medium-sized hospital (OR, 1.64; 95% CI, 1.08-2.47), at a large hospital (OR, 2.42; 95% CI, 1.65-3.55), or at an urban teaching hospital (OR, 2.73; 95% CI, 1.80-4.13) all had an increased likelihood of ICH.
“Our second objective was to study the factors associated with mortality in ITP patients with ICH,” Mr. Sharma said. “We found female gender and Medicaid, private, or self-pay as primary payers to be associated with a lower mortality in ITP with ICH.
“[A]ge of 25-64 and 65 years and above, coexistence of a GI bleed or other bleed, and admission to a large or urban teaching hospital were associated with a higher mortality,” he added.
Mr. Sharma said the study’s strengths are that it is the most recent study on trends in ITP/ICH hospitalizations, and that it’s a longitudinal assessment of data from a nationally representative database.
The study’s limitations include its retrospective nature and the use of ICD codes, which could lead to inaccuracies. Data on prior therapies and long-term outcomes were not available, and the researchers were unable to differentiate between acute and chronic ITP.
Mr. Sharma said he had no relevant conflicts of interest.
SOURCE: Sharma M et al. ASH 2019, Abstract 55.
ORLANDO – A large, retrospective study suggests several factors are associated with an increased risk of intracranial hemorrhage in patients with immune thrombocytopenia.
Data on more than 300,000 immune thrombocytopenia (ITP) hospitalizations indicated that older age, male sex, not having private insurance, having a gastrointestinal or “other” bleed, and receiving treatment at a hospital in the western United States, a medium- or large-sized hospital, or an urban teaching hospital were all associated with an increased risk of intracranial hemorrhage (ICH).
Mayank Sharma, of the University of Miami, detailed these findings at the annual meeting of the American Society of Hematology.
Mr. Sharma and colleagues analyzed data from the National Inpatient Sample database from 2007 to 2016. Of the 348,906 ITP hospitalizations included, there were 3,408 (0.98%) cases of ICH.
The overall incidence of ICH was low and remained stable over time, “which is reassuring,” Mr. Sharma said. However, the mortality rate was higher among patients with ICH than among those without it – 26.7% and 3.2%, respectively.
A multivariate analysis showed that female patients had a decreased likelihood of ICH, with an odds ratio of 0.81 (95% confidence interval, 0.68-0.97). Patients with private insurance had a decreased likelihood of ICH as well, with an OR of 0.81 (95% CI, 0.61-1.08).
Conversely, older patients had an increased likelihood of ICH. The OR was 2.23 (95% CI, 1.51-3.31) for patients aged 25-64 years, and the OR was 3.69 (95% CI, 2.34-5.84) for patients aged 65 years and older.
Patients with a gastrointestinal bleed or an other bleed (not including hematuria or epistaxis) had an increased likelihood of ICH. The ORs were 1.60 (95% CI, 1.18-2.16) and 1.69 (95% CI, 1.19-2.42), respectively.
Patients hospitalized in the western United States (OR, 1.62; 95% CI, 1.26-2.08), at a medium-sized hospital (OR, 1.64; 95% CI, 1.08-2.47), at a large hospital (OR, 2.42; 95% CI, 1.65-3.55), or at an urban teaching hospital (OR, 2.73; 95% CI, 1.80-4.13) all had an increased likelihood of ICH.
“Our second objective was to study the factors associated with mortality in ITP patients with ICH,” Mr. Sharma said. “We found female gender and Medicaid, private, or self-pay as primary payers to be associated with a lower mortality in ITP with ICH.
“[A]ge of 25-64 and 65 years and above, coexistence of a GI bleed or other bleed, and admission to a large or urban teaching hospital were associated with a higher mortality,” he added.
Mr. Sharma said the study’s strengths are that it is the most recent study on trends in ITP/ICH hospitalizations, and that it’s a longitudinal assessment of data from a nationally representative database.
The study’s limitations include its retrospective nature and the use of ICD codes, which could lead to inaccuracies. Data on prior therapies and long-term outcomes were not available, and the researchers were unable to differentiate between acute and chronic ITP.
Mr. Sharma said he had no relevant conflicts of interest.
SOURCE: Sharma M et al. ASH 2019, Abstract 55.
ORLANDO – A large, retrospective study suggests several factors are associated with an increased risk of intracranial hemorrhage in patients with immune thrombocytopenia.
Data on more than 300,000 immune thrombocytopenia (ITP) hospitalizations indicated that older age, male sex, not having private insurance, having a gastrointestinal or “other” bleed, and receiving treatment at a hospital in the western United States, a medium- or large-sized hospital, or an urban teaching hospital were all associated with an increased risk of intracranial hemorrhage (ICH).
Mayank Sharma, of the University of Miami, detailed these findings at the annual meeting of the American Society of Hematology.
Mr. Sharma and colleagues analyzed data from the National Inpatient Sample database from 2007 to 2016. Of the 348,906 ITP hospitalizations included, there were 3,408 (0.98%) cases of ICH.
The overall incidence of ICH was low and remained stable over time, “which is reassuring,” Mr. Sharma said. However, the mortality rate was higher among patients with ICH than among those without it – 26.7% and 3.2%, respectively.
A multivariate analysis showed that female patients had a decreased likelihood of ICH, with an odds ratio of 0.81 (95% confidence interval, 0.68-0.97). Patients with private insurance had a decreased likelihood of ICH as well, with an OR of 0.81 (95% CI, 0.61-1.08).
Conversely, older patients had an increased likelihood of ICH. The OR was 2.23 (95% CI, 1.51-3.31) for patients aged 25-64 years, and the OR was 3.69 (95% CI, 2.34-5.84) for patients aged 65 years and older.
Patients with a gastrointestinal bleed or an other bleed (not including hematuria or epistaxis) had an increased likelihood of ICH. The ORs were 1.60 (95% CI, 1.18-2.16) and 1.69 (95% CI, 1.19-2.42), respectively.
Patients hospitalized in the western United States (OR, 1.62; 95% CI, 1.26-2.08), at a medium-sized hospital (OR, 1.64; 95% CI, 1.08-2.47), at a large hospital (OR, 2.42; 95% CI, 1.65-3.55), or at an urban teaching hospital (OR, 2.73; 95% CI, 1.80-4.13) all had an increased likelihood of ICH.
“Our second objective was to study the factors associated with mortality in ITP patients with ICH,” Mr. Sharma said. “We found female gender and Medicaid, private, or self-pay as primary payers to be associated with a lower mortality in ITP with ICH.
“[A]ge of 25-64 and 65 years and above, coexistence of a GI bleed or other bleed, and admission to a large or urban teaching hospital were associated with a higher mortality,” he added.
Mr. Sharma said the study’s strengths are that it is the most recent study on trends in ITP/ICH hospitalizations, and that it’s a longitudinal assessment of data from a nationally representative database.
The study’s limitations include its retrospective nature and the use of ICD codes, which could lead to inaccuracies. Data on prior therapies and long-term outcomes were not available, and the researchers were unable to differentiate between acute and chronic ITP.
Mr. Sharma said he had no relevant conflicts of interest.
SOURCE: Sharma M et al. ASH 2019, Abstract 55.
REPORTING FROM ASH 2019
Phase 2 studies show potential of FcRn blockade in primary ITP
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
ORLANDO – Treatments targeted to the neonatal Fc receptor are showing promise in phase 2 studies in primary immune thrombocytopenia, investigators reported at the annual meeting of the American Society of Hematology.
Encouraging outcomes support the continued phase 3 development of these agents, which are designed to block the neonatal Fc receptor (FcRn) in patients with this IgG-mediated disease.
Blocking FcRN is intended to prevent recycling of IgG, resulting in IgG degradation, according to the authors of studies evaluating rozanolixizumab, a subcutaneously administered monoclonal antibody, and efgartigimod, an intravenously administered antibody fragment, in primary immune thrombocytopenia (ITP).
Rozanolixizumab
Results of the phase 2 study of rozanolixizumab demonstrated that this agent reduced IgG levels and improved platelet counts at all doses tested, according to the investigators, led by Tadeusz Robak, MD, of the department of hematology at the Medical University of Lodz (Poland).
Efficacy endpoints were seen more quickly – by day 8 of treatment – with single subcutaneous infusions at higher doses, according to the researchers.
Headaches of mild to moderate severity were noted at higher doses, and no patients left the study because of adverse events, they reported.
“These safety, tolerability, and efficacy data support phase 3 development of rozanolixizumab in patients with primary ITP,” wrote Dr. Robak and coauthors in the abstract for their study.
A total of 66 adult patients with primary ITP were enrolled and treated with single or multiple subcutaneous doses of rozanolixizumab administered at 1-week intervals.
Baseline characteristics suggested a “difficult-to-treat” patient cohort that had a median ITP duration of nearly 6 years and a median of four prior therapies, including thrombopoietin receptor agonists in about one-third of patients, according to the investigators.
Platelet counts of at least 50 x 109/L were achieved by day 8 in more than half of patients who received single doses of rozanolixizumab at higher dose levels of 15 mg/kg (58.3%) and 20 mg/kg (54.5%), Dr. Robak and colleagues reported.
Mild to moderate headaches were seen in about 40% of patients over an 8-week observation period. There were no serious infections and, of four serious adverse events occurring during the study, none were deemed to be treatment related, according to the investigators.
“People who have primary ITP may experience low platelet count that can put them at risk for severe bleeding, and there are limited options that provide a rapid increase in platelet count to reduce this risk,” Dr. Robak said in an interview. “These data build on the growing body of evidence that suggest targeting the FcRn pathway could have the potential to transform the treatment experience for people with rare IgG autoantibody–mediated diseases such as primary ITP.”
Efgartigimod
Substantial reductions in IgG levels and clinically relevant increases in platelet counts were seen following a 3-week treatment cycle with efgartigimod in patients with treatment-refractory ITP, according to investigator Adrian C. Newland, MB, BCh, of the Royal London Hospital and coinvestigators.
The human IgG1 antibody Fc-fragment, a natural ligand of FcRN, is engineered to have increased affinity to FcRn, while preserving its pH‐dependent binding, according to the investigators.
Efgartigimod treatment was well tolerated and reduced the proportion of patients with bleeding in the phase 2 study presented at ASH 2019.
“This suggests that targeted IgG reduction with efgartigimod is a potential new treatment modality in primary ITP, and warrants evaluation of longer-term treatment in a larger phase 3 study,” the investigators reported in the abstract for their study.
The report described 38 patients randomized to four weekly intravenous infusions of placebo or efgartigimod at one of two dosing levels. Patients had long-standing ITP, with a median 4.8 years disease duration, and all had either failed splenectomy or had inadequate response to prior treatment.
Efgartigimod treatment rapidly reduced total IgG in all patients who received it, with a mean change from baseline of up to 63.7%, according to investigators.
Platelet counts favored the investigational treatment over placebo by several measures. Platelet counts of at least 50 x 109/L on two or more occasions were seen in 46% of efgartigimod-treated patients and 25% of the placebo group; that platelet count was achieved for 10 or more days in 38% and 0% of the efgartigimod and placebo groups, respectively.
Treatment was well tolerated, according to the investigators, who said there were “no dose-related safety observations.” Full results of the phase 2 investigation were published in the American Journal of Hematology, concurrent with the meeting (2019 Dec 10. doi.org/10.1002/ajh.25680).
The study of rozanolixizumab was supported by UCB; Dr. Robak reported disclosures related to UCB (honoraria, research funding), as well as Takeda, Janssen, Amgen, Roche, AbbVie, Gilead, BeiGene, Acerta, and MorphoSys. The study of efgartigimod was supported by argenx; Dr. Newland reported disclosures related to argenx, Novartis, Angle, Amgen, Ono Pharmaceutical, Shionogi, Rigel, and Dova Pharmaceuticals.
SOURCEs: Robak T et al. ASH 2019, Abstract 897; Newland AC et al. ASH 2019, Abstract 895.
REPORTING FROM ASH 2019
Think twice: Choosing Wisely recommendations on testing to avoid in pediatric hematology
ORLANDO – There’s with some exceptions.
![]()
The list, which was produced by an expert panel with representatives from the American Society of Hematology and the American Society of Pediatric Hematology/Oncology (ASPHO), includes five tests or procedures that are considered unnecessary. The recommendations were released at the annual meeting of the American Society of Hematology.
The five recommendations are:
- Don’t perform routine preoperative hemostatic testing in an otherwise healthy child with no prior personal or family history of bleeding.
- Don’t transfuse platelets in a nonbleeding pediatric patient with a platelet count greater than 10,000/mcL, unless other signs of bleeding are present, or if the patient is set to undergo an invasive procedure.
- Don’t order thrombophilia testing on children with venous access-associated thrombosis in the absence of a positive family history.
- Don’t transfuse packed RBCs for iron-deficiency anemia in asymptomatic pediatric patients when there is no evidence of hemodynamic instability or active bleeding.
- Don’t routinely administer granulocyte colony–stimulating factor (G-CSF) for empiric treatment of pediatric patients with asymptomatic autoimmune neutropenia in the absence of recurrent or severe bacterial and/or fungal infections.
This is the third Choosing Wisely list produced by ASH. The group released the first list in 2013 and the second in 2014. But officials at both ASH and ASPHO have received feedback over the years that there should also be a pediatric-focused list in hematology, said Sarah O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and cochair of the expert panel that put together the recommendations.
Hemostatic testing
The panel recommended against preoperative hemostatic screening in healthy children with no personal or family history of excessive bleeding because the test does not effectively predict who will have unexpected surgical bleeding. The testing could instead identify artifacts or disorders unrelated to bleeding risk, such as factor XII deficiency or an infection-associated, transient lupus anticoagulant, according to Veronica H. Flood, MD, of the Medical College of Wisconsin, Milwaukee, and a member of the expert panel.
Performing this type of testing also adds cost and stress for families, and often delays surgery.
A look at the current literature reveals that there is little evidence to support coagulation testing in healthy children undergoing surgery. “Despite all this evidence, there remain practitioners who perform such screening on a regular basis,” Dr. Flood said.
For physicians concerned about bleeding risk, Dr. Flood said that existing guidelines support taking a bleeding history in preoperative patients. “This may take a little more time, but in the end will result in better results and less expense.”
Platelet transfusion
The panel recommended against platelet transfusion in nonbleeding pediatric patients with hypoproliferative thrombocytopenia and a platelet count greater than 10,000/mcL. The caveats for this recommendation are that it does not apply if there are other signs or symptoms of bleeding, if the patient is undergoing an invasive procedure, if the patient is aged 1 year or younger, or if the patient has immune-mediated thrombocytopenia, according to Rachel Bercovitz, MD, of the Ann & Robert H. Lurie Children’s Hospital of Chicago and a member of the expert panel.
Previous studies on the platelet transfusions in patients with hematologic malignancies have shown that 10,000/mcL is the appropriate threshold, with no difference in bleeding above that number and increased bleeding below it, Dr. Bercovitz said.
Additionally, while platelet transfusion is a safe procedure, Dr. Bercovitz said, it is not without acute and long-term risks.
Cost is also a factor. “Platelets are a limited and expensive resource,” she said.
Thrombophilia testing
Thrombophilia testing in children with a central venous catheter-associated thrombosis was once common practice but should be avoided, explained Leslie J. Raffini, MD, of the Children’s Hospital of Philadelphia and a member of the expert panel.
Thrombophilia does not influence the initial management of a first episode of provoked venous thrombosis, it does not inform the intensity of duration of anticoagulant therapy, and it does not predict recurrence of venous thrombosis in children, Dr. Raffini said.
In the 2013 Choosing Wisely list, ASH made the same recommendation against testing in adult patients with venous thromboembolism occurring in the setting of major transient risk factors. Thrombophilia testing is also expensive, often has to be repeated, and can be misinterpreted, Dr. Raffini said.
Packed RBC transfusion
The panel recommended against transfusion with packed RBCs for children with iron-deficiency anemia who have no symptoms and no evidence of hemodynamic instability or active bleeding. Transfusion is appropriate if children are symptomatic or are hemodynamically unstable, said Patrick T. McGann, MD, of Cincinnati Children’s Hospital and a member of the expert panel.
Rather than jump to transfusion, Dr. McGann said this group of asymptomatic and hemodynamically stable children should be treated for their iron deficiency through oral or intravenous iron. “This is not about ignoring iron deficiency.”
Both are effective treatments with multiple options available, he said. But sending a child to the hospital for transfusion is a costly option that is stressful for families and only provides a temporary solution to the issue, since treatment of the underlying iron deficiency still needs to be addressed, Dr. McGann said.
G-CSF treatment
The panel also recommended against routine administration of G-CSF in children with asymptomatic autoimmune neutropenia. Peter E. Newburger, MD, of Boston Children’s Hospital and a member of the expert guideline panel, said that there is limited evidence available and no published guidelines in this area, so the panel was guided by expert opinion.
In most cases, G-CSF is not necessary because autoimmune neutropenia resolves spontaneously by age 4-5 years and the risk of serious infection is extremely low. Appropriate management includes antibiotics for acute bacterial infection, good dental hygiene, and continued immunizations, Dr. Newburger said.
G-CSF may be appropriate in limited cases to improve quality of life, but it should be started at a low dose of 1-2 mcg/kg.
In cases of serious infection, Dr. Newburger said physicians should consider alternative diagnoses, such as congenital neutropenia or myelodysplastic syndromes.
ORLANDO – There’s with some exceptions.
![]()
The list, which was produced by an expert panel with representatives from the American Society of Hematology and the American Society of Pediatric Hematology/Oncology (ASPHO), includes five tests or procedures that are considered unnecessary. The recommendations were released at the annual meeting of the American Society of Hematology.
The five recommendations are:
- Don’t perform routine preoperative hemostatic testing in an otherwise healthy child with no prior personal or family history of bleeding.
- Don’t transfuse platelets in a nonbleeding pediatric patient with a platelet count greater than 10,000/mcL, unless other signs of bleeding are present, or if the patient is set to undergo an invasive procedure.
- Don’t order thrombophilia testing on children with venous access-associated thrombosis in the absence of a positive family history.
- Don’t transfuse packed RBCs for iron-deficiency anemia in asymptomatic pediatric patients when there is no evidence of hemodynamic instability or active bleeding.
- Don’t routinely administer granulocyte colony–stimulating factor (G-CSF) for empiric treatment of pediatric patients with asymptomatic autoimmune neutropenia in the absence of recurrent or severe bacterial and/or fungal infections.
This is the third Choosing Wisely list produced by ASH. The group released the first list in 2013 and the second in 2014. But officials at both ASH and ASPHO have received feedback over the years that there should also be a pediatric-focused list in hematology, said Sarah O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and cochair of the expert panel that put together the recommendations.
Hemostatic testing
The panel recommended against preoperative hemostatic screening in healthy children with no personal or family history of excessive bleeding because the test does not effectively predict who will have unexpected surgical bleeding. The testing could instead identify artifacts or disorders unrelated to bleeding risk, such as factor XII deficiency or an infection-associated, transient lupus anticoagulant, according to Veronica H. Flood, MD, of the Medical College of Wisconsin, Milwaukee, and a member of the expert panel.
Performing this type of testing also adds cost and stress for families, and often delays surgery.
A look at the current literature reveals that there is little evidence to support coagulation testing in healthy children undergoing surgery. “Despite all this evidence, there remain practitioners who perform such screening on a regular basis,” Dr. Flood said.
For physicians concerned about bleeding risk, Dr. Flood said that existing guidelines support taking a bleeding history in preoperative patients. “This may take a little more time, but in the end will result in better results and less expense.”
Platelet transfusion
The panel recommended against platelet transfusion in nonbleeding pediatric patients with hypoproliferative thrombocytopenia and a platelet count greater than 10,000/mcL. The caveats for this recommendation are that it does not apply if there are other signs or symptoms of bleeding, if the patient is undergoing an invasive procedure, if the patient is aged 1 year or younger, or if the patient has immune-mediated thrombocytopenia, according to Rachel Bercovitz, MD, of the Ann & Robert H. Lurie Children’s Hospital of Chicago and a member of the expert panel.
Previous studies on the platelet transfusions in patients with hematologic malignancies have shown that 10,000/mcL is the appropriate threshold, with no difference in bleeding above that number and increased bleeding below it, Dr. Bercovitz said.
Additionally, while platelet transfusion is a safe procedure, Dr. Bercovitz said, it is not without acute and long-term risks.
Cost is also a factor. “Platelets are a limited and expensive resource,” she said.
Thrombophilia testing
Thrombophilia testing in children with a central venous catheter-associated thrombosis was once common practice but should be avoided, explained Leslie J. Raffini, MD, of the Children’s Hospital of Philadelphia and a member of the expert panel.
Thrombophilia does not influence the initial management of a first episode of provoked venous thrombosis, it does not inform the intensity of duration of anticoagulant therapy, and it does not predict recurrence of venous thrombosis in children, Dr. Raffini said.
In the 2013 Choosing Wisely list, ASH made the same recommendation against testing in adult patients with venous thromboembolism occurring in the setting of major transient risk factors. Thrombophilia testing is also expensive, often has to be repeated, and can be misinterpreted, Dr. Raffini said.
Packed RBC transfusion
The panel recommended against transfusion with packed RBCs for children with iron-deficiency anemia who have no symptoms and no evidence of hemodynamic instability or active bleeding. Transfusion is appropriate if children are symptomatic or are hemodynamically unstable, said Patrick T. McGann, MD, of Cincinnati Children’s Hospital and a member of the expert panel.
Rather than jump to transfusion, Dr. McGann said this group of asymptomatic and hemodynamically stable children should be treated for their iron deficiency through oral or intravenous iron. “This is not about ignoring iron deficiency.”
Both are effective treatments with multiple options available, he said. But sending a child to the hospital for transfusion is a costly option that is stressful for families and only provides a temporary solution to the issue, since treatment of the underlying iron deficiency still needs to be addressed, Dr. McGann said.
G-CSF treatment
The panel also recommended against routine administration of G-CSF in children with asymptomatic autoimmune neutropenia. Peter E. Newburger, MD, of Boston Children’s Hospital and a member of the expert guideline panel, said that there is limited evidence available and no published guidelines in this area, so the panel was guided by expert opinion.
In most cases, G-CSF is not necessary because autoimmune neutropenia resolves spontaneously by age 4-5 years and the risk of serious infection is extremely low. Appropriate management includes antibiotics for acute bacterial infection, good dental hygiene, and continued immunizations, Dr. Newburger said.
G-CSF may be appropriate in limited cases to improve quality of life, but it should be started at a low dose of 1-2 mcg/kg.
In cases of serious infection, Dr. Newburger said physicians should consider alternative diagnoses, such as congenital neutropenia or myelodysplastic syndromes.
ORLANDO – There’s with some exceptions.
![]()
The list, which was produced by an expert panel with representatives from the American Society of Hematology and the American Society of Pediatric Hematology/Oncology (ASPHO), includes five tests or procedures that are considered unnecessary. The recommendations were released at the annual meeting of the American Society of Hematology.
The five recommendations are:
- Don’t perform routine preoperative hemostatic testing in an otherwise healthy child with no prior personal or family history of bleeding.
- Don’t transfuse platelets in a nonbleeding pediatric patient with a platelet count greater than 10,000/mcL, unless other signs of bleeding are present, or if the patient is set to undergo an invasive procedure.
- Don’t order thrombophilia testing on children with venous access-associated thrombosis in the absence of a positive family history.
- Don’t transfuse packed RBCs for iron-deficiency anemia in asymptomatic pediatric patients when there is no evidence of hemodynamic instability or active bleeding.
- Don’t routinely administer granulocyte colony–stimulating factor (G-CSF) for empiric treatment of pediatric patients with asymptomatic autoimmune neutropenia in the absence of recurrent or severe bacterial and/or fungal infections.
This is the third Choosing Wisely list produced by ASH. The group released the first list in 2013 and the second in 2014. But officials at both ASH and ASPHO have received feedback over the years that there should also be a pediatric-focused list in hematology, said Sarah O’Brien, MD, of Nationwide Children’s Hospital in Columbus, Ohio, and cochair of the expert panel that put together the recommendations.
Hemostatic testing
The panel recommended against preoperative hemostatic screening in healthy children with no personal or family history of excessive bleeding because the test does not effectively predict who will have unexpected surgical bleeding. The testing could instead identify artifacts or disorders unrelated to bleeding risk, such as factor XII deficiency or an infection-associated, transient lupus anticoagulant, according to Veronica H. Flood, MD, of the Medical College of Wisconsin, Milwaukee, and a member of the expert panel.
Performing this type of testing also adds cost and stress for families, and often delays surgery.
A look at the current literature reveals that there is little evidence to support coagulation testing in healthy children undergoing surgery. “Despite all this evidence, there remain practitioners who perform such screening on a regular basis,” Dr. Flood said.
For physicians concerned about bleeding risk, Dr. Flood said that existing guidelines support taking a bleeding history in preoperative patients. “This may take a little more time, but in the end will result in better results and less expense.”
Platelet transfusion
The panel recommended against platelet transfusion in nonbleeding pediatric patients with hypoproliferative thrombocytopenia and a platelet count greater than 10,000/mcL. The caveats for this recommendation are that it does not apply if there are other signs or symptoms of bleeding, if the patient is undergoing an invasive procedure, if the patient is aged 1 year or younger, or if the patient has immune-mediated thrombocytopenia, according to Rachel Bercovitz, MD, of the Ann & Robert H. Lurie Children’s Hospital of Chicago and a member of the expert panel.
Previous studies on the platelet transfusions in patients with hematologic malignancies have shown that 10,000/mcL is the appropriate threshold, with no difference in bleeding above that number and increased bleeding below it, Dr. Bercovitz said.
Additionally, while platelet transfusion is a safe procedure, Dr. Bercovitz said, it is not without acute and long-term risks.
Cost is also a factor. “Platelets are a limited and expensive resource,” she said.
Thrombophilia testing
Thrombophilia testing in children with a central venous catheter-associated thrombosis was once common practice but should be avoided, explained Leslie J. Raffini, MD, of the Children’s Hospital of Philadelphia and a member of the expert panel.
Thrombophilia does not influence the initial management of a first episode of provoked venous thrombosis, it does not inform the intensity of duration of anticoagulant therapy, and it does not predict recurrence of venous thrombosis in children, Dr. Raffini said.
In the 2013 Choosing Wisely list, ASH made the same recommendation against testing in adult patients with venous thromboembolism occurring in the setting of major transient risk factors. Thrombophilia testing is also expensive, often has to be repeated, and can be misinterpreted, Dr. Raffini said.
Packed RBC transfusion
The panel recommended against transfusion with packed RBCs for children with iron-deficiency anemia who have no symptoms and no evidence of hemodynamic instability or active bleeding. Transfusion is appropriate if children are symptomatic or are hemodynamically unstable, said Patrick T. McGann, MD, of Cincinnati Children’s Hospital and a member of the expert panel.
Rather than jump to transfusion, Dr. McGann said this group of asymptomatic and hemodynamically stable children should be treated for their iron deficiency through oral or intravenous iron. “This is not about ignoring iron deficiency.”
Both are effective treatments with multiple options available, he said. But sending a child to the hospital for transfusion is a costly option that is stressful for families and only provides a temporary solution to the issue, since treatment of the underlying iron deficiency still needs to be addressed, Dr. McGann said.
G-CSF treatment
The panel also recommended against routine administration of G-CSF in children with asymptomatic autoimmune neutropenia. Peter E. Newburger, MD, of Boston Children’s Hospital and a member of the expert guideline panel, said that there is limited evidence available and no published guidelines in this area, so the panel was guided by expert opinion.
In most cases, G-CSF is not necessary because autoimmune neutropenia resolves spontaneously by age 4-5 years and the risk of serious infection is extremely low. Appropriate management includes antibiotics for acute bacterial infection, good dental hygiene, and continued immunizations, Dr. Newburger said.
G-CSF may be appropriate in limited cases to improve quality of life, but it should be started at a low dose of 1-2 mcg/kg.
In cases of serious infection, Dr. Newburger said physicians should consider alternative diagnoses, such as congenital neutropenia or myelodysplastic syndromes.
REPORTING FROM ASH 2019
Best practice alerts really can work
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
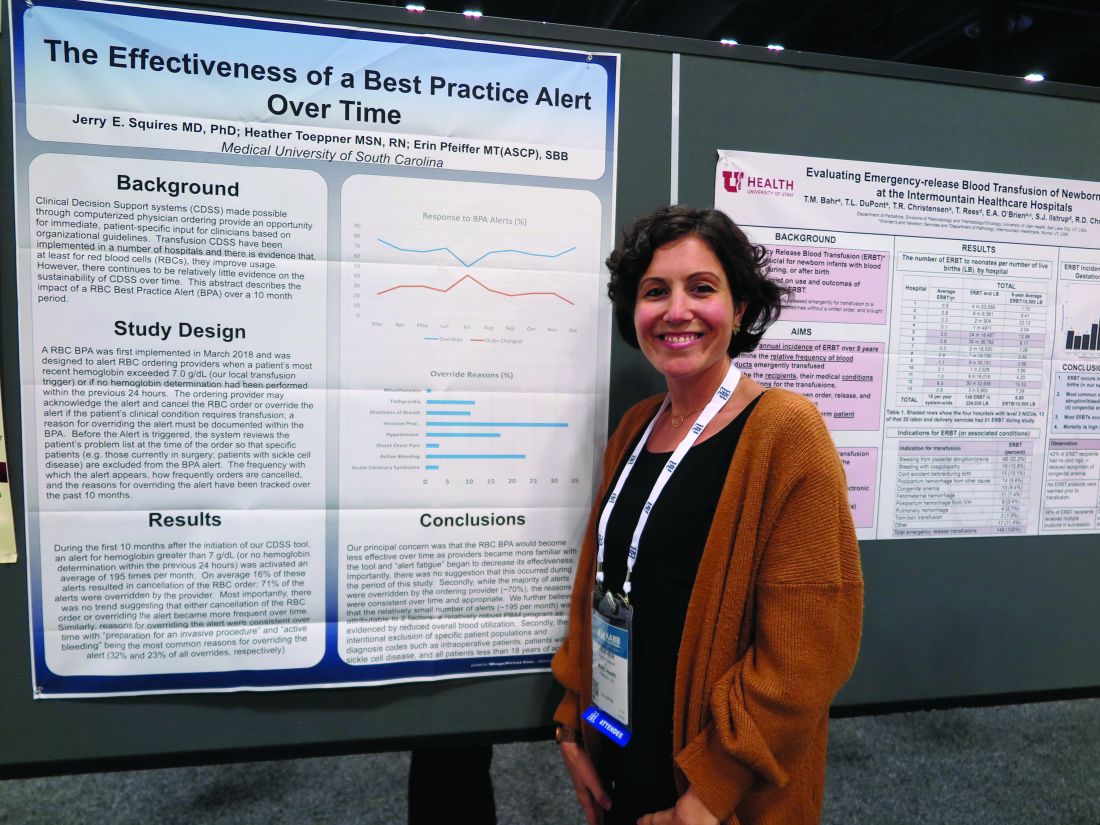
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
SAN ANTONIO – Clinicians don’t appear to mind too much when their red blood cell orders are flagged for review by a best practice alert system, and alert fatigue doesn’t seem to hamper patient blood management efforts, investigators in a single-center study reported.
At the Medical University of South Carolina, Charleston (MUSC), if clinicians order RBC transfusions for patients with hemoglobin levels over 7.0 g/dL or for patients who did not have a hemoglobin determination over the past 24 hours, they receive a best practice alert. They must acknowledge it and cancel the order, or override it and document a reason in the medical record.
Although approximately 70% of alerts were overridden, the reasons for the overrides “were consistent over time and appropriate,” reported Jerry E. Squires, MD, PhD, and colleagues from MUSC in a poster presentation at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The goal of the study was to find out if the effectiveness of the alert was wearing out after months of active use by clinicians. “Is it true that they’re clicking too much and they’re inundated with other [best practice alerts], and are they even paying attention?” said coauthor Heather Toeppner, RN, also from MUSC, in an interview. “All in all, we found that the alert is making a lasting impression in our institution,” she said.
Transfusion clinical decision support systems that produce automated alerts for clinicians can improve usage and reduce waste of RBCs, but whether the effect is sustained over time was unknown, Ms. Toeppner said, prompting the investigators to study the effect of the RBC best practice alert over 10 months.
As noted, the alert is triggered when providers order RBCs for patients with hemoglobin levels over 7.0 g/dL or when there is no record of a hemoglobin test in the chart within the past 24 hours. Before the alert is triggered, however, the system reviews the record and excludes alerts for patients with specific conditions, such as concurrent surgery or sickle cell disease.
The authors found that the alert was triggered an average of 195 times per month over the 10 months studied. On average, 16% of the alerts resulted in a cancellation of the RBC order, and 71% of alerts were overridden.
“Most importantly, there was no trend suggesting that either cancellation of the RBC order or overriding the alert became more frequent over time,” the investigators wrote. “Similarly, reasons for overriding the alert were consistent over time, with ‘preparation for an invasive procedure’ and ‘active bleeding’ being the most common reasons for overriding the alert (32% and 23% of all overrides, respectively).”
Other common reasons for overrides included tachycardia, shortness of breath, hypotension, onset of chest pain, and acute coronary syndrome.
Interestingly, but perhaps not surprisingly, they found that overrides dropped sharply and changed orders rose by the same magnitude in July, when new residents started their rotations.
The investigators wrote that the relatively small number of alerts may be attributable to their institution’s robust patient blood management program and the intentional exclusion of orders for patients with specific diagnostic codes, including intraoperative patients, those with sickle cell disease, and all patients aged younger than 18 years.
The study was internally funded. The authors reported having no conflicts of interest.
REPORTING FROM AABB 2019
Fresh RBCs offer no benefit over older cells in pediatric ICU
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.

Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
SAN ANTONIO – Fresh red cells were no better than conventional stored red cells when transfused into critically ill children, and there was some evidence in the ABC PICU trial suggesting that fresh red cells could be associated with a higher incidence of posttransfusion organ dysfunction.
Among 1,461 children randomly assigned to receive RBC transfusions with either fresh cells (stored for 7 days or less) or standard-issue cells (stored anywhere from 2-42 days), there were no differences in the primary endpoint of new or progressive multiple organ dysfunction syndrome (NPMODS), reported Philip Spinella, MD from Washington University, St. Louis.
“Our results do not support current blood management policies that recommend providing fresh red cell units to certain populations of children,” he said at the annual meeting of AABB, the group formerly known as the American Association of Blood Banks.
The study findings support those of a systematic review (Transfus Med Rev. 2018;32:77-88), whose authors found that “transfusion of fresher RBCs is not associated with decreased risk of death but is associated with higher rates of transfusion reactions and possibly infection.” The authors of the review concluded that “the current evidence does not support a change from current usual transfusion practice.”
Is fresh really better?
The launch of the ABC PICU trial was motivated by laboratory and observational evidence suggesting that older RBCs may be less safe or efficacious than fresh RBCs, especially in vulnerable populations such as critically ill children.
Although physician and institutional practice has been to transfuse fresh RBCs to some pediatric patients, the standard practice among blood banks has been to deliver the oldest stored units first, in an effort to prevent product wastage.
Dr. Spinella and colleagues across 50 centers in the United States, Canada, France, Italy, and Israel enrolled patients who were admitted to a pediatric ICU who received their first RBC transfusion within 7 days of admission and had an expected length of stay after transfusion of more than 24 hours.
The median patient age was 1.8 years for those who received fresh cells, and 1.9 years for those who received usual care.
There were 1,630 transfusions of fresh RBCs stored for a median of 5 days and 1,533 transfusions of standard RBCs stored for a median of 18 days. The median volume of red cell units transfused was 17.5 mL/kg in the fresh group and 16.6 mL/kg in the standard group.
The incidence of NPMODS was 20.2% for fresh-RBC recipients and 18.2% for standard-product recipients. The absolute difference of 2.0% was not statistically significant.
There were also no significant differences in the timing of NPMODS occurrence between the groups, and no significant differences by patient age (28 days or younger, 29-365 days old, or older than 1 year).
Similarly, there were no differences in NPMODS incidence between the groups by country, although in Canada there was a trend toward a higher incidence of organ dysfunction in the group that received fresh RBCs, Dr. Spinella noted.
Additionally, there were no significant differences between the groups by admission to the ICU by medical, surgical, cardiac, or trauma services; no differences by quartile of red cell volume transfused; and no differences in mortality rates either in the ICU or the main hospital, or at 28 or 90 days after discharge.
Why no difference?
Seeking explanations for why fresh RBCs did not perform better than older stored cells, Dr. Spinella suggested that changes such as storage lesions that occur over time may not be as clinically relevant as previously supposed.
“Another possibility is that these study patients didn’t need red cells to begin with to improve oxygen delivery,” he said.
Other potential explanations include the possibility that exposure to fresh red cells may be associated with immune suppression because viable white cells may also be present in the product, and that the chronological age of a stored red cell unit may not equate to its biologic or metabolic age or performance, he added.
ABC PICU was supported by Washington University; the National Heart, Lung, and Blood Institute; the Canadian and French governments; and other groups. Dr. Spinella reported having no relevant conflicts of interest.
REPORTING FROM AABB 2019
Emicizumab effective in children with hemophilia A and inhibitors
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
Emicizumab (Hemlibra) is well tolerated and has substantial, clinically meaningful efficacy in pediatric patients with hemophilia A and factor VIII inhibitors, according to an analysis of data from the HAVEN 2 trial in this bispecific humanized monoclonal antibody.
Among those receiving once-weekly emicizumab prophylaxis, 77% had no treated bleeding events and 100% of evaluable target joints resolved in the HAVEN 2 study, which investigators say is the largest prospective study so far of bleed prevention in pediatric patients with hemophilia A and inhibitors.
Moreover, emicizumab resulted in a 99% reduction in bleeding rate versus previous bypassing agent prophylaxis, subsequent, according to an intraindividual comparison described in the report.
Based on these results, emicizumab stands to become the “next-generation standard of care” for pediatric patients with hemophilia A and factor VIII inhibitors, reported Guy Young, MD, of Children’s Hospital Los Angeles and University of Southern California Keck School of Medicine, and his coinvestigators.
“Despite a rapidly evolving treatment landscape in hemophilia, children have been largely excluded from recent trials of novel agents,” they said in the report, which appears in the journal Blood.
Before emicizumab, current treatment options for pediatric patients with factor VIII inhibitors were limited to immune tolerance induction, or use of bypassing agents with efficacy that “can be suboptimal and unpredictable,” said Dr. Young and colleagues.
“More effective prophylactic options with reduced treatment burden are needed,” they said.
Emicizumab works in hemophilia A by bridging activated factor IX and factor X, restoring the function of missing factor VIIIa, according to the report.
A total of 88 male pediatric patients with congenital hemophilia A were enrolled in HAVEN 2, an ongoing phase 3 multicenter study that is nonrandomized and open label. The median age of patients was 7 years (range, 1-15 years). Most participants (97%) had severe hemophilia A, 72% had previously undergone immune tolerance induction, and 75% were receiving treatment with prophylactic bypassing agents.
Most participants received 4 once-weekly loading doses of 3 mg/kg body weight of emicizumab subcutaneously, followed by a maintenance regimen of 1.5 mg/kg weekly.
The annualized bleed rate was 0.3, with 77% of participants having zero bleeding events, for the 65 participants in the trial who were under 12 and received emicizumab 1.5 mg/kg weekly, according to the report.
In the subset of those patients with target joints, the mean annualized bleed rate was 3.3 for the first 24 weeks, and 0 for the next 24 weeks; these findings suggest that bleed rates decrease over time with continuing emicizumab treatment “even in patients with a more severe phenotype,” the investigators wrote.
For 15 patients younger than 12 years of age receiving bypassing agent prophylaxis, emicizumab 1.5 mg/kg once weekly resulted in an annualized bleed rate of 0.3, compared to 21.1 for the prior bypassing agent, which translates into a 99% reduction in bleeding risk, according to investigators.
Annualized bleed rates for patients receiving emicizumab 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks were 0.2 and 2.2, respectively; hover, the numerically higher annualized bleed rate in the 6 mg/kg every-4-weeks group was based on 2 out of the 10 patients treated with that maintenance regimen, including one who had 6 target joint bleeds in the 24 weeks before enrolling in the study, and another who developed antidrug antibodies within the first 8 weeks of emicizumab treatment.
Out of 23 patients who had target joints and received emicizumab prophylaxis for at least 52 weeks, 100% (45 of 45) target joints resolved, according to the report.
“Notably, this is the first report of a treatment resolving target joints in an inhibitor population, which until now has only been reported when using factor VIII products in patients without inhibitors,” Dr. Young and colleagues said.
The most common of the 721 adverse events reported in HAVEN 2 were nasopharyngitis and injection-site reactions. Of 21 serious adverse events, only 1 (antidrug antibodies with neutralizing potential) was thought by investigators to be related to emicizumab.
The Food and Drug Administration (FDA) initially approved emicizumab in November 2017 on the basis of the HAVEN 2 pediatric trial, and on HAVEN 1, a randomized, multicenter, open-label, phase 3 trial including 109 adult and adolescent males with hemophilia A and FVIII inhibitors. The indication for emicizumab was expanded to include patients without inhibitors in October 2018, on the basis of the HAVEN 3 and HAVEN 4 randomized phase 3 trials.
Dr. Young reported disclosures related to Alnylam, Bayer, Bioverativ, CSL Behring, Genentech/Roche, Grifols, Kedrion, Novo Nordisk, Shire, Spark, and uniQure.
SOURCE: Young G, et al. Blood. 2019 Oct 10. doi: 10.1182/blood.2019001869.
FROM Blood
Fibrinogen concentrate effective, safe for postop bleeding
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.

Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
SAN ANTONIO – Fibrinogen concentrate was noninferior to cryoprecipitate for controlling bleeding following cardiac surgery in the randomized FIBRES trial, Canadian investigators reported.
Among 827 patients undergoing cardiopulmonary bypass, there were no significant differences in the use of allogenenic transfusion products within 24 hours of surgery for patients assigned to receive fibrinogen concentrate for control of bleeding, compared with patients who received cryoprecipitate, reported Jeannie Callum, MD, from Sunnybrook Health Sciences Centre in Toronto, on behalf of coinvestigators in the FIBRES trial.
Fibrinogen concentrate, commonly used to control postoperative bleeding in Europe, was associated with numerically, but not statistically, lower incidence of both adverse events and serious adverse events than cryoprecipitate, the current standard of care in North America.
“Given its safety and logistical advantages, fibrinogen concentrate may be considered in bleeding patients with acquired hypofibrinogenemia,” Dr. Callum said at the annual meeting of the AABB, the group formerly known as the American Association of Blood Banks.
Results of the FIBRES trial were published simultaneously in JAMA (2019 Oct 21. doi: 10.1001/jama.2019.17312).
Acquired hypofibrinogenemia, defined as a fibrinogen level below the range of 1.5-2.0 g/L, is a major cause of excess bleeding after cardiac surgery. European guidelines on the management of bleeding following trauma or cardiac surgery recommend the use of either cryoprecipitate or fibrinogen concentrate to control excessive bleeding in patients with acquired hypofibrinogenemia, Dr. Callum noted.
Cryoprecipitate is a pooled plasma–derived product that contains fibrinogen, but also fibronectin, platelet microparticles, coagulation factors VIII and XIII, and von Willebrand factor.
Additionally, fibrinogen levels in cryoprecipitate can range from as low as 3 g/L to as high as 30 g/L, and the product is normally kept and shipped frozen, and is then thawed for use and pooled prior to administration, with a shelf life of just 4-6 hours.
In contrast, fibrinogen concentrates “are pathogen-reduced and purified; have standardized fibrinogen content (20 g/L); are lyophilized, allowing for easy storage, reconstitution, and administration; and have longer shelf life after reconstitution (up to 24 hours),” Dr. Callum and her colleagues reported.
Despite the North American preference for cryoprecipitate and the European preference for fibrinogen concentrate, there have been few studies directly comparing the two products, which prompted the FIBRES investigators to design a head-to-head trial.
The randomized trial was conducted in 11 Canadian hospitals with adults undergoing cardiac surgery with cardiopulmonary bypass for whom fibrinogen supplementation was ordered in accordance with accepted clinical standards.
Patients were randomly assigned to received either 4 g of fibrinogen concentrate or 10 units of cryoprecipitate for 24 hours, with all patients receiving additional cryoprecipitate as needed after the first day.
Of 15,412 cardiac patients treated at the participating sites, 827 patients met the trial criteria and were randomized. Because the trial met the prespecified stopping criterion for noninferiority of fibrinogen at the interim analysis, the trial was halted, leaving the 827 patients as the final analysis population.
The mean number of allogeneic blood component units administered – the primary outcome – was 16.3 units in the fibrinogen concentrate group and 17.0 units in the cryoprecipitate group (mean ratio, 0.96; P for noninferiority less than .001; P for superiority = .50).
Fibrinogen was also noninferior for the secondary outcomes of individual 24-hour and cumulative 7-day blood component transfusions, and in a post-hoc analysis of cumulative transfusions measured from product administration to 24 hours after termination of cardiopulmonary bypass. These endpoints should be interpreted with caution, however, because they were not corrected for type 1 error, the investigators noted.
Fibrinogen concentrate also appeared to be noninferior for all defined subgroups, except for patients who underwent nonelective procedures, which included all patients in critical state before surgery.
Adverse events (AEs) of any kind occurred in 66.7% of patients with fibrinogen concentrate vs. 72.7% of those on cryoprecipitate. Serious AEs occurred in 31.5% vs. 34.7%, respectively.
Thromboembolic events – stroke or transient ischemic attack, amaurosis fugax (temporary vision loss), myocardial infarction, deep-vein thrombosis, pulmonary embolism, other-vessel thrombosis, disseminated intravascular coagulation, or thrombophlebitis – occurred in 7% vs. 9.6%, respectively.
The investigators acknowledged that the study was limited by the inability to blind the clinical team to the product used, by the adult-only population, and by the likelihood of variable dosing in the cryoprecipitate group.
Advantages of fibrinogen concentrate over cryoprecipitate are that the former is pathogen reduced and is easier to deliver, the investigators said.
“One important consideration is the cost differential that currently favors cryoprecipitate, but this varies across regions, and the most recent economic analysis failed to include the costs of future emerging pathogens and did not include comprehensive activity-based costing,” the investigators wrote in JAMA.
The trial was sponsored by Octapharma AG, which also provided fibrinogen concentrate. Cryoprecipitate was provided by the Canadian Blood Services and Héma-Québec. Dr. Callum reported receiving grants from Canadian Blood Services, Octapharma, and CSL Behring during the conduct of the study. Multiple coauthors had similar disclosures.
SOURCE: Callum J et al. JAMA. 2019 Oct 21. doi:10.1001/jama.2019.17312.
REPORTING FROM AABB 2019
Updated international consensus recommendations on management of acute upper GI bleeding
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
These updated consensus guidelines provide a rigorous review of evidence on managing nonvariceal upper gastrointestinal bleeding. The recommendations for patients on anticoagulant or antiplatelet therapy will be particularly helpful because of increasing use of these medications. The advice on proton pump inhibitor therapy in patients on these drugs who have had previous ulcer bleeding can help allay concerns about possible integrations between PPIs and clopidogrel.
While the guidelines recommend using the Glasgow Blatchford scale to guide hospitalization decisions, prognostic scores are not commonly used in the emergency department, and many patients present with a Glasgow Blatchford score greater than 1, so this tool may have little impact on hospitalization rates. More studies are needed to compare clinical judgment with these prognostic scores.
Angel Lanas, MD, is from the University Clinic Hospital at the University of Zaragoza (Spain). These comments are adapted from an accompanying editorial (Ann Intern Med 2019, October 22. doi: 10.7326/M19-2789). Dr. Lanas declared unrelated personal fees from the pharmaceutical sector.
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
Guidelines on the management of acute upper gastrointestinal bleeding have been updated, including recommendations on managing patients on antiplatelet or anticoagulant therapy and on use of endoscopy and new therapeutic approaches.
Writing in Annals of Internal Medicine, an international group of experts published an update to the 2010 International Consensus Recommendations on the Management of Patients With Nonvariceal Upper Gastrointestinal Bleeding, with a focus on resuscitation and risk assessment; pre-endoscopic, endoscopic, and pharmacologic management; and secondary prophylaxis.
Alan N. Barkun, MDCM, MSc, from McGill University, Montreal, and coauthors first recommended that fluid resuscitation should be initiated in patients with acute upper gastrointestinal bleeding and hemodynamic instability to avoid hemorrhagic shock and restore end-organ perfusion and tissue oxygenation while the bleeding is brought under control.
They acknowledged the uncertainty around whether colloid or crystalloid fluid should be used, but suggested routine use of colloids was not justified because they were more expensive and did not appear to increase survival.
On the question of whether the resuscitation should be aggressive or restrictive in its timing and rate, the group said there was not enough evidence to support a recommendation on this. “The important issue in patients with hemorrhagic shock due to trauma or UGIB [upper gastrointestinal bleeding] is to stop the bleeding while minimizing hemodynamic compromise,” they wrote.
They also advised blood transfusions in patients with a hemoglobin level below 80 g/L who did not have underlying cardiovascular disease, but suggested a higher hemoglobin threshold for those with underlying cardiovascular disease.
The second recommendation was that patients with a Glasgow Blatchford score of 1 or less were at very low risk for rebleeding and mortality, and these patients may therefore not need hospitalization or inpatient endoscopy. They advised against using the AIMS65 prognostic score for this purpose because it was designed to identify patients at high risk of death, not those at low risk for safe discharge.
In regard to endoscopic management, they advocated that all patients with acute upper gastrointestinal bleeding – whether low or high risk – undergo endoscopy within 24 hours of presentation. This was even more urgent in patients being treated with anticoagulants. “Because of the recognized benefits of early endoscopy, coagulopathy should be treated as necessary but endoscopy should not be delayed,” they wrote.
Patients with acutely bleeding ulcers with high-risk stigmata should undergo endoscopic therapy preferably with thermocoagulation or sclerosant injection, or with hemoclips depending on the bleeding location and patient characteristics.
The group also included two conditional recommendations, based on very-low-quality evidence, that patients with actively bleeding ulcers receive TC-325 hemostatic powder as temporizing therapy to stop the bleeding if conventional endoscopic therapies aren’t available or fail. However, they stressed that TC-325 should not be used as a single therapeutic strategy.
Because of a lack of efficacy data and low availability of expertise in the technology, the authors said they could not make a recommendation for or against using a Doppler endoscopic probe (DEP) to assess the need for further endoscopic therapy.
“The group generally agreed that although making a recommendation for or against using DEP to manage UGIB is premature, DEP has the potential to alter the usual approach to visually assessing bleeding lesion risk when evaluating the need for, and adequacy of, endoscopic hemostasis.”
The guidelines also addressed the issue of pharmacologic management of acute upper gastrointestinal bleeding. They strongly recommended that patients with bleeding ulcers and high-risk stigmata who have undergone successful endoscopic therapy should then receive an intravenous loading dose of proton pump inhibitor (PPI) therapy, followed by continuous intravenous infusion.
“Cost-effectiveness studies have suggested that high-dose intravenous PPIs after successful endoscopic hemostasis improve outcomes at a modest cost increase relative to non–high-dose intravenous or oral PPI strategies,” they wrote.
A second conditional recommendation, based on very-low-quality evidence, was that patients with a bleeding ulcer who were at high risk for rebleeding be also treated twice-daily with oral PPIs for 2 weeks, then once-daily. They also recommended patients on cardiovascular prophylaxis with single or dual antiplatelet therapy or anticoagulant therapy be given PPIs.
“The consensus group concluded that, for high-risk patients with an ongoing need for anticoagulants, the evidence suggests that the benefits of secondary prophylaxis outweigh the risks.”
The group was supported by a grant from CIHR Institute of Nutrition, Metabolism and Diabetes and from the Saudi Gastroenterology Association. Nine authors declared grants, personal fees, honoraria and other funding from the pharmaceutical and medical device sector outside the submitted work. No other conflicts of interest were declared.
SOURCE: Barkun A et al. Ann Intern Med 2019, October 22. doi: 10.7326/M19-1795.
FROM ANNALS OF INTERNAL MEDICINE

ISTH releases draft guideline for TTP diagnosis, treatment
A new draft guideline for the diagnosis and management of thrombocytopenic purpura (TTP) was recently released by the International Society on Thrombosis and Hemostasis (ISTH).
According to the panel of experts involved, the ISTH guideline takes into account the latest TTP findings, offering a more up-to-date resource for clinicians than the two previously published guidelines in 2012 from the British Committee for Standards in Haematology and in 2017 from the TTP group of Japan’s Blood Coagulation Abnormalities Research Team.
“Since the publication of these guidelines, there have been significant developments in the diagnosis and treatment of TTP, and an increase in published data on how management strategies affect objective health outcomes,” the panel members wrote in the guideline, which is available at ISTH.org.
Despite these advancements, TTP remains a challenging condition for both clinicians and patients for a variety of reasons, the panel noted, which was led by clinical cochair X. Long Zheng, MD, PhD, of the University of Alabama in Birmingham and method cochair Sara K. Vesely, PhD, of the University of Oklahoma, Oklahoma City.
The ISTH guideline provides recommendations for adult patients with either immune or hereditary TTP, from acute events through remission, including diagnostic steps to determine if a case of thrombotic microangiopathy is in fact TTP.
Nearly all the recommendations are based on very-low-certainty evidence. Some of the key treatment recommendations include the following:
- For patients with immune TTP experiencing a first acute event, add corticosteroids to therapeutic plasma exchange (TPE), rather than treating with TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a first acute event, add rituximab to corticosteroids and TPE, rather than corticosteroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing a relapse, add corticosteroids to TPE, rather than TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a relapse, add rituximab to corticosteroids and TPE, rather than steroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing an acute event – either a first event or a relapse – use caplacizumab. This is a conditional recommendation based on moderate-certainty evidence.
- For patients with immune TTP who are in remission and have low plasma ADAMTS13 activity but no other symptoms of TMA, use rituximab for prophylaxis. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, plasma infusion or a watch-and-wait strategy is recommended. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, do not use factor VIII concentrate infusions. A watch-and-wait strategy is advised. This is a conditional recommendation.
- For patients with immune TTP who are pregnant and have decreased plasma ADAMTS13 activity but no symptoms of TMA, use prophylactic treatment. This is a strong recommendation.
- For patients with hereditary TTP who are pregnant, use prophylactic treatment. This is a strong recommendation. The panel further recommended treatment with plasma infusion rather than factor VIII products, which was a conditional recommendation.
The multidisciplinary expert panel included hematologists and pathologists with expertise in TTP, neurologists, nephrologists, intensive care specialists, and patient representatives. The panel followed the GRADE approach and the Population, Intervention, Comparison, Outcome (PICO) framework, and adhered to standards set forth by the Health and Medicine Division (HMD) of the National Academies of Sciences, Engineering, and Medicine and the GIN-McMaster Guideline Development Checklist.
Even with an experienced group of physicians and a structured plan, however, the creation of guidelines for rare diseases like TTP presents a unique set of obstacles, according to the panel members. Challenges include a small body of relevant evidence that is often inconsistent and lacking in high certainty and studies that do not address outcomes important to patients. These shortcomings can make it difficult for guideline developers to issue strong recommendations.
“However, well-developed clinical practice guidelines are vital in rare diseases; these conditions are, by their nature, encountered very infrequently by individual clinicians, who may feel unprepared to address their diagnosis and treatment,” the panelists wrote. “Well-synthesized evidence and clear recommendations play an important role in supporting clinical decision making. Systematically created guidelines can also highlight areas where evidence is uncertain, clinical judgement is required, and future research in the area is warranted.”
The guideline was supported by ISTH. Fifty percent of the panel members had no or minimal conflict of interest; those with conflicts of interest abstained from voting on recommendations relevant to their conflicts.
A new draft guideline for the diagnosis and management of thrombocytopenic purpura (TTP) was recently released by the International Society on Thrombosis and Hemostasis (ISTH).
According to the panel of experts involved, the ISTH guideline takes into account the latest TTP findings, offering a more up-to-date resource for clinicians than the two previously published guidelines in 2012 from the British Committee for Standards in Haematology and in 2017 from the TTP group of Japan’s Blood Coagulation Abnormalities Research Team.
“Since the publication of these guidelines, there have been significant developments in the diagnosis and treatment of TTP, and an increase in published data on how management strategies affect objective health outcomes,” the panel members wrote in the guideline, which is available at ISTH.org.
Despite these advancements, TTP remains a challenging condition for both clinicians and patients for a variety of reasons, the panel noted, which was led by clinical cochair X. Long Zheng, MD, PhD, of the University of Alabama in Birmingham and method cochair Sara K. Vesely, PhD, of the University of Oklahoma, Oklahoma City.
The ISTH guideline provides recommendations for adult patients with either immune or hereditary TTP, from acute events through remission, including diagnostic steps to determine if a case of thrombotic microangiopathy is in fact TTP.
Nearly all the recommendations are based on very-low-certainty evidence. Some of the key treatment recommendations include the following:
- For patients with immune TTP experiencing a first acute event, add corticosteroids to therapeutic plasma exchange (TPE), rather than treating with TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a first acute event, add rituximab to corticosteroids and TPE, rather than corticosteroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing a relapse, add corticosteroids to TPE, rather than TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a relapse, add rituximab to corticosteroids and TPE, rather than steroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing an acute event – either a first event or a relapse – use caplacizumab. This is a conditional recommendation based on moderate-certainty evidence.
- For patients with immune TTP who are in remission and have low plasma ADAMTS13 activity but no other symptoms of TMA, use rituximab for prophylaxis. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, plasma infusion or a watch-and-wait strategy is recommended. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, do not use factor VIII concentrate infusions. A watch-and-wait strategy is advised. This is a conditional recommendation.
- For patients with immune TTP who are pregnant and have decreased plasma ADAMTS13 activity but no symptoms of TMA, use prophylactic treatment. This is a strong recommendation.
- For patients with hereditary TTP who are pregnant, use prophylactic treatment. This is a strong recommendation. The panel further recommended treatment with plasma infusion rather than factor VIII products, which was a conditional recommendation.
The multidisciplinary expert panel included hematologists and pathologists with expertise in TTP, neurologists, nephrologists, intensive care specialists, and patient representatives. The panel followed the GRADE approach and the Population, Intervention, Comparison, Outcome (PICO) framework, and adhered to standards set forth by the Health and Medicine Division (HMD) of the National Academies of Sciences, Engineering, and Medicine and the GIN-McMaster Guideline Development Checklist.
Even with an experienced group of physicians and a structured plan, however, the creation of guidelines for rare diseases like TTP presents a unique set of obstacles, according to the panel members. Challenges include a small body of relevant evidence that is often inconsistent and lacking in high certainty and studies that do not address outcomes important to patients. These shortcomings can make it difficult for guideline developers to issue strong recommendations.
“However, well-developed clinical practice guidelines are vital in rare diseases; these conditions are, by their nature, encountered very infrequently by individual clinicians, who may feel unprepared to address their diagnosis and treatment,” the panelists wrote. “Well-synthesized evidence and clear recommendations play an important role in supporting clinical decision making. Systematically created guidelines can also highlight areas where evidence is uncertain, clinical judgement is required, and future research in the area is warranted.”
The guideline was supported by ISTH. Fifty percent of the panel members had no or minimal conflict of interest; those with conflicts of interest abstained from voting on recommendations relevant to their conflicts.
A new draft guideline for the diagnosis and management of thrombocytopenic purpura (TTP) was recently released by the International Society on Thrombosis and Hemostasis (ISTH).
According to the panel of experts involved, the ISTH guideline takes into account the latest TTP findings, offering a more up-to-date resource for clinicians than the two previously published guidelines in 2012 from the British Committee for Standards in Haematology and in 2017 from the TTP group of Japan’s Blood Coagulation Abnormalities Research Team.
“Since the publication of these guidelines, there have been significant developments in the diagnosis and treatment of TTP, and an increase in published data on how management strategies affect objective health outcomes,” the panel members wrote in the guideline, which is available at ISTH.org.
Despite these advancements, TTP remains a challenging condition for both clinicians and patients for a variety of reasons, the panel noted, which was led by clinical cochair X. Long Zheng, MD, PhD, of the University of Alabama in Birmingham and method cochair Sara K. Vesely, PhD, of the University of Oklahoma, Oklahoma City.
The ISTH guideline provides recommendations for adult patients with either immune or hereditary TTP, from acute events through remission, including diagnostic steps to determine if a case of thrombotic microangiopathy is in fact TTP.
Nearly all the recommendations are based on very-low-certainty evidence. Some of the key treatment recommendations include the following:
- For patients with immune TTP experiencing a first acute event, add corticosteroids to therapeutic plasma exchange (TPE), rather than treating with TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a first acute event, add rituximab to corticosteroids and TPE, rather than corticosteroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing a relapse, add corticosteroids to TPE, rather than TPE alone. This is a strong recommendation.
- For patients with immune TTP experiencing a relapse, add rituximab to corticosteroids and TPE, rather than steroids and TPE alone. This is a conditional recommendation.
- For patients with immune TTP experiencing an acute event – either a first event or a relapse – use caplacizumab. This is a conditional recommendation based on moderate-certainty evidence.
- For patients with immune TTP who are in remission and have low plasma ADAMTS13 activity but no other symptoms of TMA, use rituximab for prophylaxis. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, plasma infusion or a watch-and-wait strategy is recommended. This is a conditional recommendation.
- For patients with hereditary TTP who are in remission, do not use factor VIII concentrate infusions. A watch-and-wait strategy is advised. This is a conditional recommendation.
- For patients with immune TTP who are pregnant and have decreased plasma ADAMTS13 activity but no symptoms of TMA, use prophylactic treatment. This is a strong recommendation.
- For patients with hereditary TTP who are pregnant, use prophylactic treatment. This is a strong recommendation. The panel further recommended treatment with plasma infusion rather than factor VIII products, which was a conditional recommendation.
The multidisciplinary expert panel included hematologists and pathologists with expertise in TTP, neurologists, nephrologists, intensive care specialists, and patient representatives. The panel followed the GRADE approach and the Population, Intervention, Comparison, Outcome (PICO) framework, and adhered to standards set forth by the Health and Medicine Division (HMD) of the National Academies of Sciences, Engineering, and Medicine and the GIN-McMaster Guideline Development Checklist.
Even with an experienced group of physicians and a structured plan, however, the creation of guidelines for rare diseases like TTP presents a unique set of obstacles, according to the panel members. Challenges include a small body of relevant evidence that is often inconsistent and lacking in high certainty and studies that do not address outcomes important to patients. These shortcomings can make it difficult for guideline developers to issue strong recommendations.
“However, well-developed clinical practice guidelines are vital in rare diseases; these conditions are, by their nature, encountered very infrequently by individual clinicians, who may feel unprepared to address their diagnosis and treatment,” the panelists wrote. “Well-synthesized evidence and clear recommendations play an important role in supporting clinical decision making. Systematically created guidelines can also highlight areas where evidence is uncertain, clinical judgement is required, and future research in the area is warranted.”
The guideline was supported by ISTH. Fifty percent of the panel members had no or minimal conflict of interest; those with conflicts of interest abstained from voting on recommendations relevant to their conflicts.