User login
Cost of imatinib still high despite generic options, team says

The availability of generic imatinib has had limited effects on costs of the drug, according to research published in Health Affairs.
Data suggest the cost of Gleevec in the US has more than doubled since the drug was approved in 2001, and the introduction of generic imatinib has reduced costs only slightly.
Two years after generic imatinib hit the market, a month’s supply of Gleevec cost about $9000, and the cost for generic imatinib was about $8000.
“Patients and providers have all looked forward to generic entry, expecting major price reductions,” said study author Stacie Dusetzina, PhD, of Vanderbilt University School of Medicine in Nashville, Tennessee.
“Unfortunately, we don’t see prices drop as quickly and as low as we would hope when generics are available.”
For this study, Dr Dusetzina and a colleague analyzed data from the MarketScan Commercial Research Database. The database contained records of 139,233 prescription fills for imatinib, which were made by 7201 patients from May 2001 through September 2017.
The researchers noted that Gleevec was priced at nearly $4000 for a 1-month (400 mg) supply when it came on the market in 2001. That price escalated to nearly $10,000 by 2015 before a generic competitor entered the market.
However, prices for Gleevec and generic imatinib remained high 2 years later. In 2017, a month’s supply of Gleevec cost about $9000, and the cost of generic imatinib was about $8000.
The researchers said the Gleevec case demonstrates several potential barriers to effective generic price competition, including shifts in prescribing toward more expensive brand-name treatments and smaller-than-expected price reductions.
Twenty-four percent of imatinib (Gleevec) prescriptions claims were for “dispense as written,” according to the researchers. This suggests that patients or providers specifically wanted to stay on the brand-name drug instead of switching to the generic.
“The more than doubling of the drug price over time and the lack of price reductions observed with nearly 2 years of generic drug competition is concerning,” Dr Dusetzina said.
“It begs the question whether we can rely on generic entry as a primary approach to address drug pricing for high-priced specialty medications. We need robust competition to move prices in this space.”
The availability of generic imatinib has had limited effects on costs of the drug, according to research published in Health Affairs.
Data suggest the cost of Gleevec in the US has more than doubled since the drug was approved in 2001, and the introduction of generic imatinib has reduced costs only slightly.
Two years after generic imatinib hit the market, a month’s supply of Gleevec cost about $9000, and the cost for generic imatinib was about $8000.
“Patients and providers have all looked forward to generic entry, expecting major price reductions,” said study author Stacie Dusetzina, PhD, of Vanderbilt University School of Medicine in Nashville, Tennessee.
“Unfortunately, we don’t see prices drop as quickly and as low as we would hope when generics are available.”
For this study, Dr Dusetzina and a colleague analyzed data from the MarketScan Commercial Research Database. The database contained records of 139,233 prescription fills for imatinib, which were made by 7201 patients from May 2001 through September 2017.
The researchers noted that Gleevec was priced at nearly $4000 for a 1-month (400 mg) supply when it came on the market in 2001. That price escalated to nearly $10,000 by 2015 before a generic competitor entered the market.
However, prices for Gleevec and generic imatinib remained high 2 years later. In 2017, a month’s supply of Gleevec cost about $9000, and the cost of generic imatinib was about $8000.
The researchers said the Gleevec case demonstrates several potential barriers to effective generic price competition, including shifts in prescribing toward more expensive brand-name treatments and smaller-than-expected price reductions.
Twenty-four percent of imatinib (Gleevec) prescriptions claims were for “dispense as written,” according to the researchers. This suggests that patients or providers specifically wanted to stay on the brand-name drug instead of switching to the generic.
“The more than doubling of the drug price over time and the lack of price reductions observed with nearly 2 years of generic drug competition is concerning,” Dr Dusetzina said.
“It begs the question whether we can rely on generic entry as a primary approach to address drug pricing for high-priced specialty medications. We need robust competition to move prices in this space.”
The availability of generic imatinib has had limited effects on costs of the drug, according to research published in Health Affairs.
Data suggest the cost of Gleevec in the US has more than doubled since the drug was approved in 2001, and the introduction of generic imatinib has reduced costs only slightly.
Two years after generic imatinib hit the market, a month’s supply of Gleevec cost about $9000, and the cost for generic imatinib was about $8000.
“Patients and providers have all looked forward to generic entry, expecting major price reductions,” said study author Stacie Dusetzina, PhD, of Vanderbilt University School of Medicine in Nashville, Tennessee.
“Unfortunately, we don’t see prices drop as quickly and as low as we would hope when generics are available.”
For this study, Dr Dusetzina and a colleague analyzed data from the MarketScan Commercial Research Database. The database contained records of 139,233 prescription fills for imatinib, which were made by 7201 patients from May 2001 through September 2017.
The researchers noted that Gleevec was priced at nearly $4000 for a 1-month (400 mg) supply when it came on the market in 2001. That price escalated to nearly $10,000 by 2015 before a generic competitor entered the market.
However, prices for Gleevec and generic imatinib remained high 2 years later. In 2017, a month’s supply of Gleevec cost about $9000, and the cost of generic imatinib was about $8000.
The researchers said the Gleevec case demonstrates several potential barriers to effective generic price competition, including shifts in prescribing toward more expensive brand-name treatments and smaller-than-expected price reductions.
Twenty-four percent of imatinib (Gleevec) prescriptions claims were for “dispense as written,” according to the researchers. This suggests that patients or providers specifically wanted to stay on the brand-name drug instead of switching to the generic.
“The more than doubling of the drug price over time and the lack of price reductions observed with nearly 2 years of generic drug competition is concerning,” Dr Dusetzina said.
“It begs the question whether we can rely on generic entry as a primary approach to address drug pricing for high-priced specialty medications. We need robust competition to move prices in this space.”
CHMP backs approval of dasatinib for kids

The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended changes to the marketing authorization for dasatinib (Sprycel).
The CHMP is recommending approval for dasatinib as a treatment for pediatric patients with newly diagnosed, Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase (CP) or Ph+ CML-CP that is resistant or intolerant to prior therapy, including imatinib.
The CHMP has also recommended approval of a new formulation of dasatinib—a powder for oral suspension (PFOS)—for use in pediatric patients.
Dasatinib is already approved in the European Union to treat adults with:
- Newly diagnosed Ph+ CML-CP
- Chronic, accelerated, or blast phase CML with resistance or intolerance to prior therapy, including imatinib
- Ph+ acute lymphoblastic leukemia and lymphoid blast CML with resistance or intolerance to prior therapy.
The CHMP’s opinion on dasatinib for pediatric patients will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s opinion on dasatinib for pediatric patients is supported by 2 studies. Results from the phase 1 study (NCT00306202) were published in the Journal of Clinical Oncology in 2013. Phase 2 (NCT00777036) results were published in the same journal this year.
Phase 1
The phase 1 trial included 17 patients with CML-CP, all of whom had received prior imatinib.
Eleven patients received dasatinib at a starting dose of 60 mg/m2 once daily, and 6 received the drug at a starting dose of 80 mg/m2 once daily. Dose escalation was allowed based on tolerance and response. The median duration of treatment was 24.1 months (range, 2.3 to 50.6 months).
The 60 mg/m2 starting dose appeared more tolerable than 80 mg/m2 dose.
Drug-related adverse events (AEs) occurring in at least 20% of patients included neutropenia (82.4%), anemia (70.6%), thrombocytopenia (64.7%), nausea (29.4%), headache (35.3%), diarrhea (23.5%), and pain in extremity (23.5%). Grade 3-4 AEs included neutropenia (23.5%), thrombocytopenia (11.8%), and headache (5.9%). There were no drug-related deaths.
Ninety-four percent of patients achieved a complete hematologic response (CHR), 88% had a major cytogenetic response (MCyR), 82% had a complete cytogenetic response (CCyR), 47% had a major molecular response (MMR), and 24% had a complete molecular response (CMR).
Patients who received the lower starting dose of dasatinib had lower rates of cumulative CCyR (72.7% vs 100%) and CHR (90.9% vs 100%) but higher rates of cumulative MMR (54.5% vs 33.3%) and CMR (27.3% vs 16.7).
The median progression-free survival (PFS) and overall survival (OS) had not been reached at last follow-up. At 24 months, the estimated PFS was 61%, and the estimated OS was 88%.
Phase 2
The phase 2 trial included 29 patients with imatinib-resistant/intolerant CML-CP and 84 with newly diagnosed CML-CP.
The previously treated patients received dasatinib tablets. Newly diagnosed patients were treated with dasatinib tablets (n=51) or PFOS (n=33). Patients who started on PFOS could switch to tablets after receiving PFOS for at least 1 year. Sixty-seven percent of patients on PFOS switched to tablets due to patient preference.
The average daily dose of dasatinib was 58.18 mg/m2 in the previously treated patients and 59.84 mg/m2 in the newly diagnosed patients (for both tablets and PFOS). The median duration of treatment was 49.91 months (range, 1.9 to 90.2) and 42.30 months (range, 0.1 to 75.2), respectively.
Rates of confirmed CHR (at any time) were 93% in the previously treated patients and 96% in the newly diagnosed patients.
At 12 months, previously treated patients had an MMR rate of 41% and a CMR rate of 7%. In newly diagnosed patients, MMR was 52%, and CMR was 8%.
At 24 months, previously treated patients had an MMR rate of 55% and a CMR rate of 17%. In the newly diagnosed patients, MMR was 70%, and CMR was 21%.
The rate of MCyR at any time was 89.7% in all previously treated patients and 90% when the researchers excluded patients with MCyR or unknown cytogenetic status at baseline.
The rate of CCyR at any time was 94% in all newly diagnosed patients and 93.9% when the researchers excluded patients with CCyR or unknown cytogenetic status at baseline.
The median PFS and OS had not been reached at last follow-up. The estimated 48-month PFS was 78% in the previously treated patients and 93% in the newly diagnosed patients. The estimated 48-month OS was 96% and 100%, respectively.
Dasatinib-related AEs occurring in at least 10% of the previously treated patients included nausea/vomiting (31%), myalgia/arthralgia (17%), fatigue (14%), rash (14%), diarrhea (14%), hemorrhage (10%), bone growth and development events (10%), and shortness of breath (10%).
Dasatinib-related AEs occurring in at least 10% of the newly diagnosed patients included nausea/vomiting (20%), myalgia/arthralgia (10%), fatigue (11%), rash (19%), diarrhea (18%), and hemorrhage (10%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended changes to the marketing authorization for dasatinib (Sprycel).
The CHMP is recommending approval for dasatinib as a treatment for pediatric patients with newly diagnosed, Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase (CP) or Ph+ CML-CP that is resistant or intolerant to prior therapy, including imatinib.
The CHMP has also recommended approval of a new formulation of dasatinib—a powder for oral suspension (PFOS)—for use in pediatric patients.
Dasatinib is already approved in the European Union to treat adults with:
- Newly diagnosed Ph+ CML-CP
- Chronic, accelerated, or blast phase CML with resistance or intolerance to prior therapy, including imatinib
- Ph+ acute lymphoblastic leukemia and lymphoid blast CML with resistance or intolerance to prior therapy.
The CHMP’s opinion on dasatinib for pediatric patients will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s opinion on dasatinib for pediatric patients is supported by 2 studies. Results from the phase 1 study (NCT00306202) were published in the Journal of Clinical Oncology in 2013. Phase 2 (NCT00777036) results were published in the same journal this year.
Phase 1
The phase 1 trial included 17 patients with CML-CP, all of whom had received prior imatinib.
Eleven patients received dasatinib at a starting dose of 60 mg/m2 once daily, and 6 received the drug at a starting dose of 80 mg/m2 once daily. Dose escalation was allowed based on tolerance and response. The median duration of treatment was 24.1 months (range, 2.3 to 50.6 months).
The 60 mg/m2 starting dose appeared more tolerable than 80 mg/m2 dose.
Drug-related adverse events (AEs) occurring in at least 20% of patients included neutropenia (82.4%), anemia (70.6%), thrombocytopenia (64.7%), nausea (29.4%), headache (35.3%), diarrhea (23.5%), and pain in extremity (23.5%). Grade 3-4 AEs included neutropenia (23.5%), thrombocytopenia (11.8%), and headache (5.9%). There were no drug-related deaths.
Ninety-four percent of patients achieved a complete hematologic response (CHR), 88% had a major cytogenetic response (MCyR), 82% had a complete cytogenetic response (CCyR), 47% had a major molecular response (MMR), and 24% had a complete molecular response (CMR).
Patients who received the lower starting dose of dasatinib had lower rates of cumulative CCyR (72.7% vs 100%) and CHR (90.9% vs 100%) but higher rates of cumulative MMR (54.5% vs 33.3%) and CMR (27.3% vs 16.7).
The median progression-free survival (PFS) and overall survival (OS) had not been reached at last follow-up. At 24 months, the estimated PFS was 61%, and the estimated OS was 88%.
Phase 2
The phase 2 trial included 29 patients with imatinib-resistant/intolerant CML-CP and 84 with newly diagnosed CML-CP.
The previously treated patients received dasatinib tablets. Newly diagnosed patients were treated with dasatinib tablets (n=51) or PFOS (n=33). Patients who started on PFOS could switch to tablets after receiving PFOS for at least 1 year. Sixty-seven percent of patients on PFOS switched to tablets due to patient preference.
The average daily dose of dasatinib was 58.18 mg/m2 in the previously treated patients and 59.84 mg/m2 in the newly diagnosed patients (for both tablets and PFOS). The median duration of treatment was 49.91 months (range, 1.9 to 90.2) and 42.30 months (range, 0.1 to 75.2), respectively.
Rates of confirmed CHR (at any time) were 93% in the previously treated patients and 96% in the newly diagnosed patients.
At 12 months, previously treated patients had an MMR rate of 41% and a CMR rate of 7%. In newly diagnosed patients, MMR was 52%, and CMR was 8%.
At 24 months, previously treated patients had an MMR rate of 55% and a CMR rate of 17%. In the newly diagnosed patients, MMR was 70%, and CMR was 21%.
The rate of MCyR at any time was 89.7% in all previously treated patients and 90% when the researchers excluded patients with MCyR or unknown cytogenetic status at baseline.
The rate of CCyR at any time was 94% in all newly diagnosed patients and 93.9% when the researchers excluded patients with CCyR or unknown cytogenetic status at baseline.
The median PFS and OS had not been reached at last follow-up. The estimated 48-month PFS was 78% in the previously treated patients and 93% in the newly diagnosed patients. The estimated 48-month OS was 96% and 100%, respectively.
Dasatinib-related AEs occurring in at least 10% of the previously treated patients included nausea/vomiting (31%), myalgia/arthralgia (17%), fatigue (14%), rash (14%), diarrhea (14%), hemorrhage (10%), bone growth and development events (10%), and shortness of breath (10%).
Dasatinib-related AEs occurring in at least 10% of the newly diagnosed patients included nausea/vomiting (20%), myalgia/arthralgia (10%), fatigue (11%), rash (19%), diarrhea (18%), and hemorrhage (10%).
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended changes to the marketing authorization for dasatinib (Sprycel).
The CHMP is recommending approval for dasatinib as a treatment for pediatric patients with newly diagnosed, Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase (CP) or Ph+ CML-CP that is resistant or intolerant to prior therapy, including imatinib.
The CHMP has also recommended approval of a new formulation of dasatinib—a powder for oral suspension (PFOS)—for use in pediatric patients.
Dasatinib is already approved in the European Union to treat adults with:
- Newly diagnosed Ph+ CML-CP
- Chronic, accelerated, or blast phase CML with resistance or intolerance to prior therapy, including imatinib
- Ph+ acute lymphoblastic leukemia and lymphoid blast CML with resistance or intolerance to prior therapy.
The CHMP’s opinion on dasatinib for pediatric patients will be reviewed by the European Commission (EC).
If the EC agrees with the CHMP, the commission will grant a centralized marketing authorization that will be valid in the European Union. Norway, Iceland, and Liechtenstein will make corresponding decisions on the basis of the EC’s decision.
The EC typically makes a decision within 67 days of the CHMP’s recommendation.
The CHMP’s opinion on dasatinib for pediatric patients is supported by 2 studies. Results from the phase 1 study (NCT00306202) were published in the Journal of Clinical Oncology in 2013. Phase 2 (NCT00777036) results were published in the same journal this year.
Phase 1
The phase 1 trial included 17 patients with CML-CP, all of whom had received prior imatinib.
Eleven patients received dasatinib at a starting dose of 60 mg/m2 once daily, and 6 received the drug at a starting dose of 80 mg/m2 once daily. Dose escalation was allowed based on tolerance and response. The median duration of treatment was 24.1 months (range, 2.3 to 50.6 months).
The 60 mg/m2 starting dose appeared more tolerable than 80 mg/m2 dose.
Drug-related adverse events (AEs) occurring in at least 20% of patients included neutropenia (82.4%), anemia (70.6%), thrombocytopenia (64.7%), nausea (29.4%), headache (35.3%), diarrhea (23.5%), and pain in extremity (23.5%). Grade 3-4 AEs included neutropenia (23.5%), thrombocytopenia (11.8%), and headache (5.9%). There were no drug-related deaths.
Ninety-four percent of patients achieved a complete hematologic response (CHR), 88% had a major cytogenetic response (MCyR), 82% had a complete cytogenetic response (CCyR), 47% had a major molecular response (MMR), and 24% had a complete molecular response (CMR).
Patients who received the lower starting dose of dasatinib had lower rates of cumulative CCyR (72.7% vs 100%) and CHR (90.9% vs 100%) but higher rates of cumulative MMR (54.5% vs 33.3%) and CMR (27.3% vs 16.7).
The median progression-free survival (PFS) and overall survival (OS) had not been reached at last follow-up. At 24 months, the estimated PFS was 61%, and the estimated OS was 88%.
Phase 2
The phase 2 trial included 29 patients with imatinib-resistant/intolerant CML-CP and 84 with newly diagnosed CML-CP.
The previously treated patients received dasatinib tablets. Newly diagnosed patients were treated with dasatinib tablets (n=51) or PFOS (n=33). Patients who started on PFOS could switch to tablets after receiving PFOS for at least 1 year. Sixty-seven percent of patients on PFOS switched to tablets due to patient preference.
The average daily dose of dasatinib was 58.18 mg/m2 in the previously treated patients and 59.84 mg/m2 in the newly diagnosed patients (for both tablets and PFOS). The median duration of treatment was 49.91 months (range, 1.9 to 90.2) and 42.30 months (range, 0.1 to 75.2), respectively.
Rates of confirmed CHR (at any time) were 93% in the previously treated patients and 96% in the newly diagnosed patients.
At 12 months, previously treated patients had an MMR rate of 41% and a CMR rate of 7%. In newly diagnosed patients, MMR was 52%, and CMR was 8%.
At 24 months, previously treated patients had an MMR rate of 55% and a CMR rate of 17%. In the newly diagnosed patients, MMR was 70%, and CMR was 21%.
The rate of MCyR at any time was 89.7% in all previously treated patients and 90% when the researchers excluded patients with MCyR or unknown cytogenetic status at baseline.
The rate of CCyR at any time was 94% in all newly diagnosed patients and 93.9% when the researchers excluded patients with CCyR or unknown cytogenetic status at baseline.
The median PFS and OS had not been reached at last follow-up. The estimated 48-month PFS was 78% in the previously treated patients and 93% in the newly diagnosed patients. The estimated 48-month OS was 96% and 100%, respectively.
Dasatinib-related AEs occurring in at least 10% of the previously treated patients included nausea/vomiting (31%), myalgia/arthralgia (17%), fatigue (14%), rash (14%), diarrhea (14%), hemorrhage (10%), bone growth and development events (10%), and shortness of breath (10%).
Dasatinib-related AEs occurring in at least 10% of the newly diagnosed patients included nausea/vomiting (20%), myalgia/arthralgia (10%), fatigue (11%), rash (19%), diarrhea (18%), and hemorrhage (10%).
Do industry payments increase prescribing for some targeted therapies?
Physicians receiving general payments from the company marketing a targeted cancer therapy were more likely to prescribe it in three out of six drugs evaluated, researchers reported.
Prescribing of sunitinib, dasatinib, and nilotinib was increased for physicians receiving such payments versus not receiving them, while prescribing of imatinib, sorafenib, and pazopanib were not, according to the analysis by Aaron P. Mitchell, MD, of the Lineberger Comprehensive Cancer Center, UNC School of Medicine, University of North Carolina at Chapel Hill, and his coauthors.
In previous studies, pharmaceutical industry payments to physicians have been associated with “higher-cost, brand-name pharmaceutical prescribing,” Dr. Mitchell and his colleagues wrote. The report was published in JAMA Internal Medicine.
“Whether industry payments are associated with physician treatment choice in oncology is uncertain,” they said.
To evaluate the association between payments to oncologists and drug selection, Dr. Mitchell and his colleagues linked Open Payments data from the Centers for Medicare & Medicaid Services to data from Medicare Part D Prescriber Public Use File for the years 2013-2014.
The primary variable in the study was payments received during 2013, according to investigators, and the primary outcome of the analysis was prescriptions filled during 2014.
Open Payments reported in 2013 had a total dollar value of $4.08 billion, including $1.20 billion paid to physicians, according to CMS data.
The researchers focused on targeted therapies for two therapeutic areas: metastatic renal cell carcinoma (RCC), including sorafenib, sunitinib, and pazopanib; and chronic myeloid leukemia (CML), including imatinib, dasatinib, and nilotinib.
They limited their analysis to physicians listed as oncologists who filled at least 20 prescriptions for each of the three drugs in metastatic RCC (n = 354) or in CML (n = 2,225).
Receiving payments categorized as “general,” such as gifts, speaker fees, meals, and travel, increased the odds of prescribing drugs for both metastatic RCC (odds ratio, 2.05; 95% confidence interval, 1.34-3.14; P = .001) and for CML (odds ratio, 1.29; 95% CI, 1.13-1.47; P less than .001).
By contrast, research payments did not increase the odds of prescribing those drugs, the investigators reported.
Looking at specific drugs, they found that receipt of general payments from a drug’s manufacturer was associated with increased prescribing of sunitinib (50.5% versus 34.4%, P = .01), dasatinib (13.8% versus 11.4%, P = .02), and nilotinib (15.4% vs 12.5%, P = .01).
However, no such association was found for sorafenib or pazopanib.
For imatinib, by contrast, investigators said industry payments were associated with a prescribing decrease.
“This may reflect a strategy by the manufacturer of imatinib, which also produces nilotinib, to promote switching to nilotinib before the patent expiration of imatinib in 2015,” the researchers wrote.
Dr. Mitchell and his coauthors reported no conflict of interest disclosures related to the study.
SOURCE: Mitchell AP, et al. JAMA Intern Med. 2018 Apr 9. doi: 0.1001/jamainternmed.2018.0776.
Physicians receiving general payments from the company marketing a targeted cancer therapy were more likely to prescribe it in three out of six drugs evaluated, researchers reported.
Prescribing of sunitinib, dasatinib, and nilotinib was increased for physicians receiving such payments versus not receiving them, while prescribing of imatinib, sorafenib, and pazopanib were not, according to the analysis by Aaron P. Mitchell, MD, of the Lineberger Comprehensive Cancer Center, UNC School of Medicine, University of North Carolina at Chapel Hill, and his coauthors.
In previous studies, pharmaceutical industry payments to physicians have been associated with “higher-cost, brand-name pharmaceutical prescribing,” Dr. Mitchell and his colleagues wrote. The report was published in JAMA Internal Medicine.
“Whether industry payments are associated with physician treatment choice in oncology is uncertain,” they said.
To evaluate the association between payments to oncologists and drug selection, Dr. Mitchell and his colleagues linked Open Payments data from the Centers for Medicare & Medicaid Services to data from Medicare Part D Prescriber Public Use File for the years 2013-2014.
The primary variable in the study was payments received during 2013, according to investigators, and the primary outcome of the analysis was prescriptions filled during 2014.
Open Payments reported in 2013 had a total dollar value of $4.08 billion, including $1.20 billion paid to physicians, according to CMS data.
The researchers focused on targeted therapies for two therapeutic areas: metastatic renal cell carcinoma (RCC), including sorafenib, sunitinib, and pazopanib; and chronic myeloid leukemia (CML), including imatinib, dasatinib, and nilotinib.
They limited their analysis to physicians listed as oncologists who filled at least 20 prescriptions for each of the three drugs in metastatic RCC (n = 354) or in CML (n = 2,225).
Receiving payments categorized as “general,” such as gifts, speaker fees, meals, and travel, increased the odds of prescribing drugs for both metastatic RCC (odds ratio, 2.05; 95% confidence interval, 1.34-3.14; P = .001) and for CML (odds ratio, 1.29; 95% CI, 1.13-1.47; P less than .001).
By contrast, research payments did not increase the odds of prescribing those drugs, the investigators reported.
Looking at specific drugs, they found that receipt of general payments from a drug’s manufacturer was associated with increased prescribing of sunitinib (50.5% versus 34.4%, P = .01), dasatinib (13.8% versus 11.4%, P = .02), and nilotinib (15.4% vs 12.5%, P = .01).
However, no such association was found for sorafenib or pazopanib.
For imatinib, by contrast, investigators said industry payments were associated with a prescribing decrease.
“This may reflect a strategy by the manufacturer of imatinib, which also produces nilotinib, to promote switching to nilotinib before the patent expiration of imatinib in 2015,” the researchers wrote.
Dr. Mitchell and his coauthors reported no conflict of interest disclosures related to the study.
SOURCE: Mitchell AP, et al. JAMA Intern Med. 2018 Apr 9. doi: 0.1001/jamainternmed.2018.0776.
Physicians receiving general payments from the company marketing a targeted cancer therapy were more likely to prescribe it in three out of six drugs evaluated, researchers reported.
Prescribing of sunitinib, dasatinib, and nilotinib was increased for physicians receiving such payments versus not receiving them, while prescribing of imatinib, sorafenib, and pazopanib were not, according to the analysis by Aaron P. Mitchell, MD, of the Lineberger Comprehensive Cancer Center, UNC School of Medicine, University of North Carolina at Chapel Hill, and his coauthors.
In previous studies, pharmaceutical industry payments to physicians have been associated with “higher-cost, brand-name pharmaceutical prescribing,” Dr. Mitchell and his colleagues wrote. The report was published in JAMA Internal Medicine.
“Whether industry payments are associated with physician treatment choice in oncology is uncertain,” they said.
To evaluate the association between payments to oncologists and drug selection, Dr. Mitchell and his colleagues linked Open Payments data from the Centers for Medicare & Medicaid Services to data from Medicare Part D Prescriber Public Use File for the years 2013-2014.
The primary variable in the study was payments received during 2013, according to investigators, and the primary outcome of the analysis was prescriptions filled during 2014.
Open Payments reported in 2013 had a total dollar value of $4.08 billion, including $1.20 billion paid to physicians, according to CMS data.
The researchers focused on targeted therapies for two therapeutic areas: metastatic renal cell carcinoma (RCC), including sorafenib, sunitinib, and pazopanib; and chronic myeloid leukemia (CML), including imatinib, dasatinib, and nilotinib.
They limited their analysis to physicians listed as oncologists who filled at least 20 prescriptions for each of the three drugs in metastatic RCC (n = 354) or in CML (n = 2,225).
Receiving payments categorized as “general,” such as gifts, speaker fees, meals, and travel, increased the odds of prescribing drugs for both metastatic RCC (odds ratio, 2.05; 95% confidence interval, 1.34-3.14; P = .001) and for CML (odds ratio, 1.29; 95% CI, 1.13-1.47; P less than .001).
By contrast, research payments did not increase the odds of prescribing those drugs, the investigators reported.
Looking at specific drugs, they found that receipt of general payments from a drug’s manufacturer was associated with increased prescribing of sunitinib (50.5% versus 34.4%, P = .01), dasatinib (13.8% versus 11.4%, P = .02), and nilotinib (15.4% vs 12.5%, P = .01).
However, no such association was found for sorafenib or pazopanib.
For imatinib, by contrast, investigators said industry payments were associated with a prescribing decrease.
“This may reflect a strategy by the manufacturer of imatinib, which also produces nilotinib, to promote switching to nilotinib before the patent expiration of imatinib in 2015,” the researchers wrote.
Dr. Mitchell and his coauthors reported no conflict of interest disclosures related to the study.
SOURCE: Mitchell AP, et al. JAMA Intern Med. 2018 Apr 9. doi: 0.1001/jamainternmed.2018.0776.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Oncologists receiving general payments from the company marketing a cancer drug were more likely to prescribe it in three out of six drugs evaluated.
Major finding: Prescribing was significantly increased for sunitinib (50.5% versus 34.4%, P = .01), dasatinib (13.8% versus 11.4%, P = .02), and nilotinib (15.4% vs. 12.5%, P = .01), but not for imatinib, sorafenib, or pazopanib.
Study details: An analysis of Centers for Medicare & Medicaid Services Open Payments data and Medicare Part D Prescriber Public Use File for the years 2013 to 2014.
Disclosures: The authors reported no conflict of interest disclosures related to the study.
Source: Mitchell AP et al. JAMA Intern Med. 2018 Apr 9. doi: 0.1001/jamainternmed.2018.0776.
Agent exhibits activity in leukemias, MDS

The experimental agent prexigebersen (formerly BP1001) was considered well-tolerated and demonstrated early evidence of activity against relapsed/refractory hematologic disorders in a phase 1/1b trial.
The drug reduced blasts in the bone marrow and peripheral blood for patients with acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and myelodysplastic syndrome (MDS).
When given in combination with low-dose cytarabine, prexigebersen produced complete responses (CRs) in patients with AML.
Researchers said that, overall, the toxic effects of prexigebersen were manageable.
There was 1 patient who had dose-limiting toxicities, 1 who discontinued treatment due to possible drug-related toxic effects, and 1 treatment-related death.
Still, the maximum tolerated dose of prexigebersen was not established.
These results were published in The Lancet Haematology. The study was sponsored by Bio-Path Holdings, Inc., the company developing prexigebersen.
Prexigebersen is an anti-sense oligodeoxynucleotide developed to block Grb2 expression and function. Researchers tested the drug in a single-center, dose-escalation, phase 1/1b trial that enrolled and treated 39 patients.
In the phase 1 portion of the trial, patients received prexigebersen monotherapy. In the phase 1b portion, they received the drug in combination with low-dose cytarabine.
There were 32 patients in the phase 1 portion of the trial. Most (n=23) had AML, 5 had CML in blast phase, and 4 had MDS. The patients’ median age was 63 (range, 56-73), and they had received a median of 4 prior therapies.
All 7 patients in the phase 1b portion had AML. They had a median age of 72 (range, 70-76) and had all received 1 prior therapy.
For phase 1, prexigebersen was administered intravenously, twice weekly for 28 days at doses of 5 mg/m² in cohort 1 (n=13), 10 mg/m² in cohort 2 (n=6), 20 mg/m² in cohort 3 (n=3), 40 mg/m² in cohort 4 (n=3), 60 mg/m² in cohort 5 (n=3), and 90 mg/m² in cohort 6 (n=4).
In the phase 1b portion, patients received prexigebersen at 60 mg/m² (n=4) or 90 mg/m² (n=3) in combination with 20 mg of cytarabine (twice-daily subcutaneous injections).
Safety
Twenty-seven patients were evaluable for dose-limiting toxicity—21 from phase 1 and 6 from 1b.
One patient in cohort 1 developed mucositis and hand-foot syndrome, which were considered possibly related to prexigebersen and deemed dose-limiting toxicities. The patient was also receiving hydroxyurea (3 g/day) for CML and had a history of hydroxyurea-induced mucositis.
There were no other dose-limiting toxicities, and the researchers did not identify a maximum tolerated dose of prexigebersen.
The most common grade 3-4 adverse events (AEs) were cardiopulmonary disorders and fevers (including neutropenic fevers and infections).
In the monotherapy group, 17% of patients had grade 3-4 cardiopulmonary AEs, and 11% had fevers. In the prexigebersen-cytarabine combination group, 8% had grade 3-4 cardiopulmonary AEs, and 6% had fevers.
There were 5 grade 5 AEs in 4 patients, all of whom received monotherapy. These included cardiopulmonary disorders (n=2), fevers (n=2), and multi-organ failure (n=1). One patient had both fever (sepsis) and multi-organ failure.
Efficacy
According to the researchers’ assessments, 22% of phase 1 patients (7/32) benefited from prexigebersen monotherapy and therefore received more than 1 cycle of treatment. Five of these patients had AML, and 2 had MDS.
Single-agent activity was observed in other patients as well.
Thirty-three percent (9/27) of patients who had peripheral blood blasts at baseline saw their blasts reduced by 50% or more while receiving monotherapy. One of these patients had CML, and the rest had AML.
Ten percent (3/29) of patients with bone marrow blasts at baseline had a reduction in blasts of 50% or more while receiving monotherapy. Two of these patients had AML, and 1 had MDS.
Of the 7 patients receiving prexigebersen with cytarabine, 2 achieved a CR, and 1 had a CR with incomplete hematological recovery.
Two of the patients had stable disease, and the remaining 2 patients progressed. One of the patients with progressive disease withdrew from the study, and the other died.
Deaths
There were a total of 8 deaths.
One death was considered treatment-related. This patient had progressive CML in blast phase and died of multiple organ failure. This was the first patient treated on the trial, who also had the only dose-limiting toxicities.
Two patients with AML and 1 with MDS died of disease progression. Three AML patients died of sepsis, pneumonia, and cardiac arrest. And a CML patient died of respiratory distress.
The experimental agent prexigebersen (formerly BP1001) was considered well-tolerated and demonstrated early evidence of activity against relapsed/refractory hematologic disorders in a phase 1/1b trial.
The drug reduced blasts in the bone marrow and peripheral blood for patients with acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and myelodysplastic syndrome (MDS).
When given in combination with low-dose cytarabine, prexigebersen produced complete responses (CRs) in patients with AML.
Researchers said that, overall, the toxic effects of prexigebersen were manageable.
There was 1 patient who had dose-limiting toxicities, 1 who discontinued treatment due to possible drug-related toxic effects, and 1 treatment-related death.
Still, the maximum tolerated dose of prexigebersen was not established.
These results were published in The Lancet Haematology. The study was sponsored by Bio-Path Holdings, Inc., the company developing prexigebersen.
Prexigebersen is an anti-sense oligodeoxynucleotide developed to block Grb2 expression and function. Researchers tested the drug in a single-center, dose-escalation, phase 1/1b trial that enrolled and treated 39 patients.
In the phase 1 portion of the trial, patients received prexigebersen monotherapy. In the phase 1b portion, they received the drug in combination with low-dose cytarabine.
There were 32 patients in the phase 1 portion of the trial. Most (n=23) had AML, 5 had CML in blast phase, and 4 had MDS. The patients’ median age was 63 (range, 56-73), and they had received a median of 4 prior therapies.
All 7 patients in the phase 1b portion had AML. They had a median age of 72 (range, 70-76) and had all received 1 prior therapy.
For phase 1, prexigebersen was administered intravenously, twice weekly for 28 days at doses of 5 mg/m² in cohort 1 (n=13), 10 mg/m² in cohort 2 (n=6), 20 mg/m² in cohort 3 (n=3), 40 mg/m² in cohort 4 (n=3), 60 mg/m² in cohort 5 (n=3), and 90 mg/m² in cohort 6 (n=4).
In the phase 1b portion, patients received prexigebersen at 60 mg/m² (n=4) or 90 mg/m² (n=3) in combination with 20 mg of cytarabine (twice-daily subcutaneous injections).
Safety
Twenty-seven patients were evaluable for dose-limiting toxicity—21 from phase 1 and 6 from 1b.
One patient in cohort 1 developed mucositis and hand-foot syndrome, which were considered possibly related to prexigebersen and deemed dose-limiting toxicities. The patient was also receiving hydroxyurea (3 g/day) for CML and had a history of hydroxyurea-induced mucositis.
There were no other dose-limiting toxicities, and the researchers did not identify a maximum tolerated dose of prexigebersen.
The most common grade 3-4 adverse events (AEs) were cardiopulmonary disorders and fevers (including neutropenic fevers and infections).
In the monotherapy group, 17% of patients had grade 3-4 cardiopulmonary AEs, and 11% had fevers. In the prexigebersen-cytarabine combination group, 8% had grade 3-4 cardiopulmonary AEs, and 6% had fevers.
There were 5 grade 5 AEs in 4 patients, all of whom received monotherapy. These included cardiopulmonary disorders (n=2), fevers (n=2), and multi-organ failure (n=1). One patient had both fever (sepsis) and multi-organ failure.
Efficacy
According to the researchers’ assessments, 22% of phase 1 patients (7/32) benefited from prexigebersen monotherapy and therefore received more than 1 cycle of treatment. Five of these patients had AML, and 2 had MDS.
Single-agent activity was observed in other patients as well.
Thirty-three percent (9/27) of patients who had peripheral blood blasts at baseline saw their blasts reduced by 50% or more while receiving monotherapy. One of these patients had CML, and the rest had AML.
Ten percent (3/29) of patients with bone marrow blasts at baseline had a reduction in blasts of 50% or more while receiving monotherapy. Two of these patients had AML, and 1 had MDS.
Of the 7 patients receiving prexigebersen with cytarabine, 2 achieved a CR, and 1 had a CR with incomplete hematological recovery.
Two of the patients had stable disease, and the remaining 2 patients progressed. One of the patients with progressive disease withdrew from the study, and the other died.
Deaths
There were a total of 8 deaths.
One death was considered treatment-related. This patient had progressive CML in blast phase and died of multiple organ failure. This was the first patient treated on the trial, who also had the only dose-limiting toxicities.
Two patients with AML and 1 with MDS died of disease progression. Three AML patients died of sepsis, pneumonia, and cardiac arrest. And a CML patient died of respiratory distress.
The experimental agent prexigebersen (formerly BP1001) was considered well-tolerated and demonstrated early evidence of activity against relapsed/refractory hematologic disorders in a phase 1/1b trial.
The drug reduced blasts in the bone marrow and peripheral blood for patients with acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and myelodysplastic syndrome (MDS).
When given in combination with low-dose cytarabine, prexigebersen produced complete responses (CRs) in patients with AML.
Researchers said that, overall, the toxic effects of prexigebersen were manageable.
There was 1 patient who had dose-limiting toxicities, 1 who discontinued treatment due to possible drug-related toxic effects, and 1 treatment-related death.
Still, the maximum tolerated dose of prexigebersen was not established.
These results were published in The Lancet Haematology. The study was sponsored by Bio-Path Holdings, Inc., the company developing prexigebersen.
Prexigebersen is an anti-sense oligodeoxynucleotide developed to block Grb2 expression and function. Researchers tested the drug in a single-center, dose-escalation, phase 1/1b trial that enrolled and treated 39 patients.
In the phase 1 portion of the trial, patients received prexigebersen monotherapy. In the phase 1b portion, they received the drug in combination with low-dose cytarabine.
There were 32 patients in the phase 1 portion of the trial. Most (n=23) had AML, 5 had CML in blast phase, and 4 had MDS. The patients’ median age was 63 (range, 56-73), and they had received a median of 4 prior therapies.
All 7 patients in the phase 1b portion had AML. They had a median age of 72 (range, 70-76) and had all received 1 prior therapy.
For phase 1, prexigebersen was administered intravenously, twice weekly for 28 days at doses of 5 mg/m² in cohort 1 (n=13), 10 mg/m² in cohort 2 (n=6), 20 mg/m² in cohort 3 (n=3), 40 mg/m² in cohort 4 (n=3), 60 mg/m² in cohort 5 (n=3), and 90 mg/m² in cohort 6 (n=4).
In the phase 1b portion, patients received prexigebersen at 60 mg/m² (n=4) or 90 mg/m² (n=3) in combination with 20 mg of cytarabine (twice-daily subcutaneous injections).
Safety
Twenty-seven patients were evaluable for dose-limiting toxicity—21 from phase 1 and 6 from 1b.
One patient in cohort 1 developed mucositis and hand-foot syndrome, which were considered possibly related to prexigebersen and deemed dose-limiting toxicities. The patient was also receiving hydroxyurea (3 g/day) for CML and had a history of hydroxyurea-induced mucositis.
There were no other dose-limiting toxicities, and the researchers did not identify a maximum tolerated dose of prexigebersen.
The most common grade 3-4 adverse events (AEs) were cardiopulmonary disorders and fevers (including neutropenic fevers and infections).
In the monotherapy group, 17% of patients had grade 3-4 cardiopulmonary AEs, and 11% had fevers. In the prexigebersen-cytarabine combination group, 8% had grade 3-4 cardiopulmonary AEs, and 6% had fevers.
There were 5 grade 5 AEs in 4 patients, all of whom received monotherapy. These included cardiopulmonary disorders (n=2), fevers (n=2), and multi-organ failure (n=1). One patient had both fever (sepsis) and multi-organ failure.
Efficacy
According to the researchers’ assessments, 22% of phase 1 patients (7/32) benefited from prexigebersen monotherapy and therefore received more than 1 cycle of treatment. Five of these patients had AML, and 2 had MDS.
Single-agent activity was observed in other patients as well.
Thirty-three percent (9/27) of patients who had peripheral blood blasts at baseline saw their blasts reduced by 50% or more while receiving monotherapy. One of these patients had CML, and the rest had AML.
Ten percent (3/29) of patients with bone marrow blasts at baseline had a reduction in blasts of 50% or more while receiving monotherapy. Two of these patients had AML, and 1 had MDS.
Of the 7 patients receiving prexigebersen with cytarabine, 2 achieved a CR, and 1 had a CR with incomplete hematological recovery.
Two of the patients had stable disease, and the remaining 2 patients progressed. One of the patients with progressive disease withdrew from the study, and the other died.
Deaths
There were a total of 8 deaths.
One death was considered treatment-related. This patient had progressive CML in blast phase and died of multiple organ failure. This was the first patient treated on the trial, who also had the only dose-limiting toxicities.
Two patients with AML and 1 with MDS died of disease progression. Three AML patients died of sepsis, pneumonia, and cardiac arrest. And a CML patient died of respiratory distress.
FDA approves nilotinib for children with CML
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
Nilotinib is now approved for use by children aged 1 year and older with Philadelphia chromosome–positive chronic myeloid leukemia (Ph+ CML) in the chronic phase.
The Food and Drug Administration expanded the drug’s indication to include use as first- and second-line treatment in children.
Adverse events in the pediatric studies were similar to those observed in adults. However, children experienced hyperbilirubinemia (grade 3/4: 13%) and transaminase elevation (AST grade 3/4: 1%; ALT grade 3/4: 9%). Additionally, one previously treated pediatric patient progressed with advance phase/blast crisis after about 10 months of treatment.
Nilotinib (Tasigna) was already approved in adults with newly diagnosed Ph+ CML in the chronic phase and adults with chronic phase and accelerated phase Ph+ CML resistant or intolerant to prior therapy.
FDA approves nilotinib for kids with CML
The US Food and Drug Administration (FDA) has expanded the approved indication for nilotinib (Tasigna®) to include the treatment of children.
The drug is now approved to treat patients age 1 and older who have newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.
Nilotinib is also approved to treat pediatric patients age 1 and older who have chronic phase, Ph+ CML that is resistant or intolerant to prior tyrosine kinase inhibitor (TKI) therapy, as well as adults with Ph+ CML in chronic phase and accelerated phase that is resistant or intolerant to prior therapy including imatinib.
The new pediatric indications for nilotinib, granted under the FDA’s priority review designation, are based on results from 2 studies of the drug—a phase 1 and phase 2.
According to Novartis, the studies included 69 CML patients who ranged in age from 2 to 17. They had either newly diagnosed, chronic phase, Ph+ CML or chronic phase, Ph+ CML with resistance or intolerance to prior TKI therapy.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation.
There was 1 patient with resistant/intolerant CML who progressed to advance phase/blast crisis after about 10 months on nilotinib.
The US Food and Drug Administration (FDA) has expanded the approved indication for nilotinib (Tasigna®) to include the treatment of children.
The drug is now approved to treat patients age 1 and older who have newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.
Nilotinib is also approved to treat pediatric patients age 1 and older who have chronic phase, Ph+ CML that is resistant or intolerant to prior tyrosine kinase inhibitor (TKI) therapy, as well as adults with Ph+ CML in chronic phase and accelerated phase that is resistant or intolerant to prior therapy including imatinib.
The new pediatric indications for nilotinib, granted under the FDA’s priority review designation, are based on results from 2 studies of the drug—a phase 1 and phase 2.
According to Novartis, the studies included 69 CML patients who ranged in age from 2 to 17. They had either newly diagnosed, chronic phase, Ph+ CML or chronic phase, Ph+ CML with resistance or intolerance to prior TKI therapy.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation.
There was 1 patient with resistant/intolerant CML who progressed to advance phase/blast crisis after about 10 months on nilotinib.
The US Food and Drug Administration (FDA) has expanded the approved indication for nilotinib (Tasigna®) to include the treatment of children.
The drug is now approved to treat patients age 1 and older who have newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.
Nilotinib is also approved to treat pediatric patients age 1 and older who have chronic phase, Ph+ CML that is resistant or intolerant to prior tyrosine kinase inhibitor (TKI) therapy, as well as adults with Ph+ CML in chronic phase and accelerated phase that is resistant or intolerant to prior therapy including imatinib.
The new pediatric indications for nilotinib, granted under the FDA’s priority review designation, are based on results from 2 studies of the drug—a phase 1 and phase 2.
According to Novartis, the studies included 69 CML patients who ranged in age from 2 to 17. They had either newly diagnosed, chronic phase, Ph+ CML or chronic phase, Ph+ CML with resistance or intolerance to prior TKI therapy.
In the newly diagnosed patients, the major molecular response (MMR) rate was 60.0% at 12 cycles, with 15 patients achieving MMR.
In patients with resistance or intolerance to prior therapy, the MMR rate was 40.9% at 12 cycles, with 18 patients being in MMR.
In newly diagnosed patients, the cumulative MMR rate was 64.0% by cycle 12. In patients with resistance or intolerance to prior therapy, the cumulative MMR rate was 47.7% by cycle 12.
Adverse events were generally consistent with those observed in adults, with the exception of hyperbilirubinemia and transaminase elevation, which were reported at a higher frequency than in adults.
The rate of grade 3/4 hyperbilirubinemia was 13.0%, the rate of grade 3/4 AST elevation was 1.4%, and the rate of grade 3/4 ALT elevation was 8.7%.
There were no deaths on treatment or after treatment discontinuation.
There was 1 patient with resistant/intolerant CML who progressed to advance phase/blast crisis after about 10 months on nilotinib.
Ponatinib efficacy maintained despite dose reductions
Final results from the phase 2 PACE trial suggest dose reductions did not have an effect on the efficacy of ponatinib in patients with chronic phase (CP) chronic myeloid leukemia (CML).
Trial investigators began reducing ponatinib doses after the drug was linked to arterial occlusive events (AOEs).
Most patients who achieved a major molecular response (MMR) or major cytogenetic response (MCyR) on higher doses of ponatinib maintained those responses after dose reductions.
These results were published in Blood. The trial was sponsored by Ariad Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Earlier results from the PACE trial indicated that ponatinib increased a patient’s risk of vascular occlusive events (VOEs), including AOEs.
Therefore, in October 2013, a phase 3 trial of ponatinib was discontinued, and all other ponatinib trials were put on partial clinical hold. Trial enrollment was halted temporarily, and investigators began reducing ponatinib doses.
Then, the US Food and Drug Administration suspended sales and marketing of ponatinib, pending results of a safety evaluation.
However, in December 2013, the agency decided ponatinib could return to the market if new safety measures were implemented. In January 2014, the drug was back on the market.
Patients
The PACE trial included 449 patients with CML or acute lymphoblastic leukemia, but the results published in Blood focused only on patients with CP-CML. All of these patients were resistant or intolerant to dasatinib or nilotinib, or they had the BCR-ABLT315I mutation.
There were 270 patients with CP-CML. They had a median age of 60 (range, 18-94), and 47% were female.
Ninety-three percent had received at least 2 prior approved tyrosine kinase inhibitors (TKIs), and 57% had received at least 3. The median duration of prior TKI treatment was 5.4 years (range, 0.4-13.3).
Eighty percent of patients were resistant to dasatinib or nilotinib, 14% were intolerant to either drug, and 19% were both resistant and intolerant. Twenty-four percent of patients had the T315I mutation.
Treatment
The starting dose of ponatinib was 45 mg once daily. Doses were reduced to 30 mg or 15 mg once daily to manage adverse events (AEs), or reductions were implemented proactively (starting on October 10, 2013) due to concerns about VOEs.
Unless a benefit-risk analysis justified use of a higher dose, the recommendation for CP-CML patients was a 15 mg daily dose for those with an MCyR and a 30 mg daily dose for those without an MCyR.
Patients received ponatinib until disease progression, intolerance, or the patient or investigator decided to stop treatment.
The median duration of treatment was 32.1 months (range, 0.1-73.0), and the median follow-up was 56.8 months (range, 0.1-73.1).
Efficacy
Of the 267 evaluable patients, 60% achieved an MCyR at any time, with 54% achieving a complete cytogenetic response. Forty-eight percent of patients were still in MCyR at the last response assessment, and 82% of patients who achieved an MCyR were estimated to remain in MCyR at 5 years.
There were 69 patients in MCyR as of October 10, 2013, when pre-emptive dose reduction began, who had their dose reduced. Ninety-six percent of these patients (n=66) maintained MCyR after dose reduction. Of the 34 patients in MCyR who did not have pre-emptive dose reductions, 94% (n=32) maintained MCyR.
Forty percent of patients attained a major molecular response (MMR) at any time during the study, with 30% achieving MR4 and 24% achieving MR4.5. Thirty percent of patients were in MMR at the last response assessment, and 59% of patients who achieved MMR were estimated to remain in MMR at 5 years.
There were 52 patients in MMR as of October 10, 2013, who had their dose reduced. Ninety percent (n=47) of these patients maintained MMR following dose reduction. Of the 19 patients in MMR who did not have pre-emptive dose reductions, 95% (n=18) maintained MMR.
“The PACE trial is among the longest and largest studies of patients with CP-CML who have received 2 or 3 prior TKIs, and the findings provide treating physicians with important updated information . . . ,” said study author Jorge Eduardo Cortes, MD, of MD Anderson Cancer Center in Houston, Texas.
“These final PACE results demonstrate that [ponatinib] provides lasting clinically meaningful responses, irrespective of dose reductions, in this population.”
At 5 years, the progression-free survival rate was 53%, and the overall survival rate was 73%.
AOEs
The incidence of AOEs was 31%, and the incidence of serious AOEs was 26%. This included cardiovascular AOEs (16%, 12% serious), cerebrovascular (13%, 10% serious), and peripheral vascular AOEs (14%, 11% serious). The exposure-adjusted incidence of AOEs was 14.1 per 100 patient-years.
Among patients without AOEs prior to October 2013 when pre-emptive dose reductions began, the incidence of AOEs was 19% in patients who had dose reductions and 18% in patients who did not.
The cumulative incidence of AOEs increased over time, but the exposure-adjusted incidence of new AOEs did not. It was 15.8 per 100 patient-years in year 1 and 4.9 per 100 patient-years in year 5.
The investigators said the lack of increase in exposure-adjusted AOE incidence could be due to the natural history or etiology of AOEs, dose reductions, or a change in the patient population (ie, an enrichment of patients who may have a lower risk of vascular events).
Therefore, it is unclear whether lower doses of ponatinib reduce the risk of AOEs in patients with risk factors. However, AOEs appear to be dose-related and modified by pre-existing cardiovascular disease and other risk factors.
Venous thromboembolism (VTE), on the other hand, does not seem to be dose-related. The investigators said the rate of VTE in this study was consistent with rates typically observed in cancer patients.
The incidence of VTE was 6%, and 5% of patients had serious VTEs. The exposure-adjusted incidence of VTEs was 2.1 per 100 patient-years.
Other AEs
The most common any-grade treatment-emergent AEs (≥40%) were rash (47%), abdominal pain (46%), thrombocytopenia (46%), headache (43%), dry skin (42%), and constipation (41%).
The most common grade 3/4 treatment-emergent AEs (≥10%) were thrombocytopenia (35%), neutropenia (17%), hypertension (14%), increased lipase (13%), abdominal pain (10%), and anemia (10%).
Serious AEs (≥5%) included pancreatitis (7%), atrial fibrillation (6%), pneumonia (6%), and angina pectoris (5%).
There were 12 deaths (4%) that occurred on study or within 30 days of the end of study treatment. Two deaths were considered possibly or probably related to ponatinib, 1 due to pneumonia and 1 due to acute myocardial infarction.
Final results from the phase 2 PACE trial suggest dose reductions did not have an effect on the efficacy of ponatinib in patients with chronic phase (CP) chronic myeloid leukemia (CML).
Trial investigators began reducing ponatinib doses after the drug was linked to arterial occlusive events (AOEs).
Most patients who achieved a major molecular response (MMR) or major cytogenetic response (MCyR) on higher doses of ponatinib maintained those responses after dose reductions.
These results were published in Blood. The trial was sponsored by Ariad Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Earlier results from the PACE trial indicated that ponatinib increased a patient’s risk of vascular occlusive events (VOEs), including AOEs.
Therefore, in October 2013, a phase 3 trial of ponatinib was discontinued, and all other ponatinib trials were put on partial clinical hold. Trial enrollment was halted temporarily, and investigators began reducing ponatinib doses.
Then, the US Food and Drug Administration suspended sales and marketing of ponatinib, pending results of a safety evaluation.
However, in December 2013, the agency decided ponatinib could return to the market if new safety measures were implemented. In January 2014, the drug was back on the market.
Patients
The PACE trial included 449 patients with CML or acute lymphoblastic leukemia, but the results published in Blood focused only on patients with CP-CML. All of these patients were resistant or intolerant to dasatinib or nilotinib, or they had the BCR-ABLT315I mutation.
There were 270 patients with CP-CML. They had a median age of 60 (range, 18-94), and 47% were female.
Ninety-three percent had received at least 2 prior approved tyrosine kinase inhibitors (TKIs), and 57% had received at least 3. The median duration of prior TKI treatment was 5.4 years (range, 0.4-13.3).
Eighty percent of patients were resistant to dasatinib or nilotinib, 14% were intolerant to either drug, and 19% were both resistant and intolerant. Twenty-four percent of patients had the T315I mutation.
Treatment
The starting dose of ponatinib was 45 mg once daily. Doses were reduced to 30 mg or 15 mg once daily to manage adverse events (AEs), or reductions were implemented proactively (starting on October 10, 2013) due to concerns about VOEs.
Unless a benefit-risk analysis justified use of a higher dose, the recommendation for CP-CML patients was a 15 mg daily dose for those with an MCyR and a 30 mg daily dose for those without an MCyR.
Patients received ponatinib until disease progression, intolerance, or the patient or investigator decided to stop treatment.
The median duration of treatment was 32.1 months (range, 0.1-73.0), and the median follow-up was 56.8 months (range, 0.1-73.1).
Efficacy
Of the 267 evaluable patients, 60% achieved an MCyR at any time, with 54% achieving a complete cytogenetic response. Forty-eight percent of patients were still in MCyR at the last response assessment, and 82% of patients who achieved an MCyR were estimated to remain in MCyR at 5 years.
There were 69 patients in MCyR as of October 10, 2013, when pre-emptive dose reduction began, who had their dose reduced. Ninety-six percent of these patients (n=66) maintained MCyR after dose reduction. Of the 34 patients in MCyR who did not have pre-emptive dose reductions, 94% (n=32) maintained MCyR.
Forty percent of patients attained a major molecular response (MMR) at any time during the study, with 30% achieving MR4 and 24% achieving MR4.5. Thirty percent of patients were in MMR at the last response assessment, and 59% of patients who achieved MMR were estimated to remain in MMR at 5 years.
There were 52 patients in MMR as of October 10, 2013, who had their dose reduced. Ninety percent (n=47) of these patients maintained MMR following dose reduction. Of the 19 patients in MMR who did not have pre-emptive dose reductions, 95% (n=18) maintained MMR.
“The PACE trial is among the longest and largest studies of patients with CP-CML who have received 2 or 3 prior TKIs, and the findings provide treating physicians with important updated information . . . ,” said study author Jorge Eduardo Cortes, MD, of MD Anderson Cancer Center in Houston, Texas.
“These final PACE results demonstrate that [ponatinib] provides lasting clinically meaningful responses, irrespective of dose reductions, in this population.”
At 5 years, the progression-free survival rate was 53%, and the overall survival rate was 73%.
AOEs
The incidence of AOEs was 31%, and the incidence of serious AOEs was 26%. This included cardiovascular AOEs (16%, 12% serious), cerebrovascular (13%, 10% serious), and peripheral vascular AOEs (14%, 11% serious). The exposure-adjusted incidence of AOEs was 14.1 per 100 patient-years.
Among patients without AOEs prior to October 2013 when pre-emptive dose reductions began, the incidence of AOEs was 19% in patients who had dose reductions and 18% in patients who did not.
The cumulative incidence of AOEs increased over time, but the exposure-adjusted incidence of new AOEs did not. It was 15.8 per 100 patient-years in year 1 and 4.9 per 100 patient-years in year 5.
The investigators said the lack of increase in exposure-adjusted AOE incidence could be due to the natural history or etiology of AOEs, dose reductions, or a change in the patient population (ie, an enrichment of patients who may have a lower risk of vascular events).
Therefore, it is unclear whether lower doses of ponatinib reduce the risk of AOEs in patients with risk factors. However, AOEs appear to be dose-related and modified by pre-existing cardiovascular disease and other risk factors.
Venous thromboembolism (VTE), on the other hand, does not seem to be dose-related. The investigators said the rate of VTE in this study was consistent with rates typically observed in cancer patients.
The incidence of VTE was 6%, and 5% of patients had serious VTEs. The exposure-adjusted incidence of VTEs was 2.1 per 100 patient-years.
Other AEs
The most common any-grade treatment-emergent AEs (≥40%) were rash (47%), abdominal pain (46%), thrombocytopenia (46%), headache (43%), dry skin (42%), and constipation (41%).
The most common grade 3/4 treatment-emergent AEs (≥10%) were thrombocytopenia (35%), neutropenia (17%), hypertension (14%), increased lipase (13%), abdominal pain (10%), and anemia (10%).
Serious AEs (≥5%) included pancreatitis (7%), atrial fibrillation (6%), pneumonia (6%), and angina pectoris (5%).
There were 12 deaths (4%) that occurred on study or within 30 days of the end of study treatment. Two deaths were considered possibly or probably related to ponatinib, 1 due to pneumonia and 1 due to acute myocardial infarction.
Final results from the phase 2 PACE trial suggest dose reductions did not have an effect on the efficacy of ponatinib in patients with chronic phase (CP) chronic myeloid leukemia (CML).
Trial investigators began reducing ponatinib doses after the drug was linked to arterial occlusive events (AOEs).
Most patients who achieved a major molecular response (MMR) or major cytogenetic response (MCyR) on higher doses of ponatinib maintained those responses after dose reductions.
These results were published in Blood. The trial was sponsored by Ariad Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Earlier results from the PACE trial indicated that ponatinib increased a patient’s risk of vascular occlusive events (VOEs), including AOEs.
Therefore, in October 2013, a phase 3 trial of ponatinib was discontinued, and all other ponatinib trials were put on partial clinical hold. Trial enrollment was halted temporarily, and investigators began reducing ponatinib doses.
Then, the US Food and Drug Administration suspended sales and marketing of ponatinib, pending results of a safety evaluation.
However, in December 2013, the agency decided ponatinib could return to the market if new safety measures were implemented. In January 2014, the drug was back on the market.
Patients
The PACE trial included 449 patients with CML or acute lymphoblastic leukemia, but the results published in Blood focused only on patients with CP-CML. All of these patients were resistant or intolerant to dasatinib or nilotinib, or they had the BCR-ABLT315I mutation.
There were 270 patients with CP-CML. They had a median age of 60 (range, 18-94), and 47% were female.
Ninety-three percent had received at least 2 prior approved tyrosine kinase inhibitors (TKIs), and 57% had received at least 3. The median duration of prior TKI treatment was 5.4 years (range, 0.4-13.3).
Eighty percent of patients were resistant to dasatinib or nilotinib, 14% were intolerant to either drug, and 19% were both resistant and intolerant. Twenty-four percent of patients had the T315I mutation.
Treatment
The starting dose of ponatinib was 45 mg once daily. Doses were reduced to 30 mg or 15 mg once daily to manage adverse events (AEs), or reductions were implemented proactively (starting on October 10, 2013) due to concerns about VOEs.
Unless a benefit-risk analysis justified use of a higher dose, the recommendation for CP-CML patients was a 15 mg daily dose for those with an MCyR and a 30 mg daily dose for those without an MCyR.
Patients received ponatinib until disease progression, intolerance, or the patient or investigator decided to stop treatment.
The median duration of treatment was 32.1 months (range, 0.1-73.0), and the median follow-up was 56.8 months (range, 0.1-73.1).
Efficacy
Of the 267 evaluable patients, 60% achieved an MCyR at any time, with 54% achieving a complete cytogenetic response. Forty-eight percent of patients were still in MCyR at the last response assessment, and 82% of patients who achieved an MCyR were estimated to remain in MCyR at 5 years.
There were 69 patients in MCyR as of October 10, 2013, when pre-emptive dose reduction began, who had their dose reduced. Ninety-six percent of these patients (n=66) maintained MCyR after dose reduction. Of the 34 patients in MCyR who did not have pre-emptive dose reductions, 94% (n=32) maintained MCyR.
Forty percent of patients attained a major molecular response (MMR) at any time during the study, with 30% achieving MR4 and 24% achieving MR4.5. Thirty percent of patients were in MMR at the last response assessment, and 59% of patients who achieved MMR were estimated to remain in MMR at 5 years.
There were 52 patients in MMR as of October 10, 2013, who had their dose reduced. Ninety percent (n=47) of these patients maintained MMR following dose reduction. Of the 19 patients in MMR who did not have pre-emptive dose reductions, 95% (n=18) maintained MMR.
“The PACE trial is among the longest and largest studies of patients with CP-CML who have received 2 or 3 prior TKIs, and the findings provide treating physicians with important updated information . . . ,” said study author Jorge Eduardo Cortes, MD, of MD Anderson Cancer Center in Houston, Texas.
“These final PACE results demonstrate that [ponatinib] provides lasting clinically meaningful responses, irrespective of dose reductions, in this population.”
At 5 years, the progression-free survival rate was 53%, and the overall survival rate was 73%.
AOEs
The incidence of AOEs was 31%, and the incidence of serious AOEs was 26%. This included cardiovascular AOEs (16%, 12% serious), cerebrovascular (13%, 10% serious), and peripheral vascular AOEs (14%, 11% serious). The exposure-adjusted incidence of AOEs was 14.1 per 100 patient-years.
Among patients without AOEs prior to October 2013 when pre-emptive dose reductions began, the incidence of AOEs was 19% in patients who had dose reductions and 18% in patients who did not.
The cumulative incidence of AOEs increased over time, but the exposure-adjusted incidence of new AOEs did not. It was 15.8 per 100 patient-years in year 1 and 4.9 per 100 patient-years in year 5.
The investigators said the lack of increase in exposure-adjusted AOE incidence could be due to the natural history or etiology of AOEs, dose reductions, or a change in the patient population (ie, an enrichment of patients who may have a lower risk of vascular events).
Therefore, it is unclear whether lower doses of ponatinib reduce the risk of AOEs in patients with risk factors. However, AOEs appear to be dose-related and modified by pre-existing cardiovascular disease and other risk factors.
Venous thromboembolism (VTE), on the other hand, does not seem to be dose-related. The investigators said the rate of VTE in this study was consistent with rates typically observed in cancer patients.
The incidence of VTE was 6%, and 5% of patients had serious VTEs. The exposure-adjusted incidence of VTEs was 2.1 per 100 patient-years.
Other AEs
The most common any-grade treatment-emergent AEs (≥40%) were rash (47%), abdominal pain (46%), thrombocytopenia (46%), headache (43%), dry skin (42%), and constipation (41%).
The most common grade 3/4 treatment-emergent AEs (≥10%) were thrombocytopenia (35%), neutropenia (17%), hypertension (14%), increased lipase (13%), abdominal pain (10%), and anemia (10%).
Serious AEs (≥5%) included pancreatitis (7%), atrial fibrillation (6%), pneumonia (6%), and angina pectoris (5%).
There were 12 deaths (4%) that occurred on study or within 30 days of the end of study treatment. Two deaths were considered possibly or probably related to ponatinib, 1 due to pneumonia and 1 due to acute myocardial infarction.
A global snapshot of leukemia incidence
, according to an analysis of World Health Organization cancer databases.
Incidence also is generally higher in males, with a global male to female ratio of 1.4. For men, the highest regional leukemia rate – estimated at 11.3 per 100,000 population for 2012 – was found in Australia and New Zealand, with northern America (the United States and Canada) next at 10.5 per 100,000. Australia/New Zealand and northern America had the highest rate for women at 7.2 per 100,000, followed by western Europe and northern Europe at 6.0 per 100,000, reported Adalberto Miranda-Filho, PhD, of the WHO’s International Agency for Research on Cancer in Lyon, France, and his associates.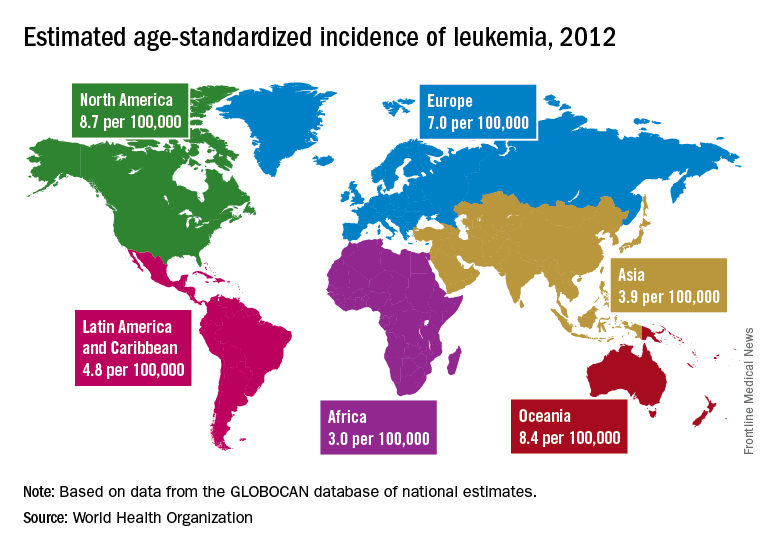
The lowest regional rates for women were found in western Africa (1.2 per 100,000), middle Africa (1.8), and Micronesia/Polynesia (2.1). For men, leukemia incidence was lowest in western Africa (1.4 per 100,000), middle Africa (2.6), and south-central Asia (3.4), according to data from the WHO’s GLOBOCAN database. The report was published in The Lancet Haematology.
Estimates for leukemia subtypes in 2003-2007 – calculated for 54 countries, not regions – also showed a great deal of variation. For acute lymphoblastic leukemia, Ecuador had the highest rates for both males (2.8 per 100,000) and females (3.3), with high rates seen in Costa Rica, Columbia, and Cyprus. Rates in the United States were near the top: 2.1 for males and 1.6 for females. Rates were lowest for men in Jamaica (0.4) and Serbia (0.6) and for women in India (0.5) and Serbia and Cuba (0.6), Dr. Miranda-Filho and his associates said.
Incidence rates for acute myeloid leukemia were highest in Australia for men (2.8 per 100,000) and Austria for women (2.2), with the United States near the top for both men (2.6) and women (1.9). The lowest rates occurred in Cuba and Egypt for men (0.9 per 100,000) and Cuba for women (0.4), data from the WHO’s Cancer Incidence in Five Continents Volume X show.
Chronic lymphocytic leukemia incidence was highest for men in Canada (4.5 per 100,000), Ireland and Lithuania (4.4), and Slovakia (4.3). The incidence was highest for women in Lithuania (2.5), Canada (2.3), and Slovakia and Denmark (2.1). Incidence in the United States was 3.5 for men and 1.8 for women. At the other end of the scale, the lowest rates for both men and women were in Japan and Malaysia (0.1), the investigators’ analysis showed.
Chronic myeloid leukemia rates were the lowest of the subtypes, and Tunisia was the lowest for men at 0.4 per 100,000 and tied for lowest with Serbia, Slovenia, and Puerto Rico for women at 0.3. Incidence was highest for men in Australia at 1.8 per 100,000 and highest for women in Uruguay at 1.1. Rates in the United States were 1.3 for men and 0.8 for women, Dr. Miranda-Filho and his associates said.
“The higher incidence of acute lymphoblastic leukaemia in parts of South America, as well as of chronic lymphocytic leukaemia in populations across North America and Oceania, alongside a lower incidence in Asia, might be important markers for further epidemiological study, and a means to better understand the underlying factors to support future cancer prevention strategies,” the investigators wrote.
SOURCE: Miranda-Filho A et al. Lancet Haematol. 2018;5:e14-24.
, according to an analysis of World Health Organization cancer databases.
Incidence also is generally higher in males, with a global male to female ratio of 1.4. For men, the highest regional leukemia rate – estimated at 11.3 per 100,000 population for 2012 – was found in Australia and New Zealand, with northern America (the United States and Canada) next at 10.5 per 100,000. Australia/New Zealand and northern America had the highest rate for women at 7.2 per 100,000, followed by western Europe and northern Europe at 6.0 per 100,000, reported Adalberto Miranda-Filho, PhD, of the WHO’s International Agency for Research on Cancer in Lyon, France, and his associates.
The lowest regional rates for women were found in western Africa (1.2 per 100,000), middle Africa (1.8), and Micronesia/Polynesia (2.1). For men, leukemia incidence was lowest in western Africa (1.4 per 100,000), middle Africa (2.6), and south-central Asia (3.4), according to data from the WHO’s GLOBOCAN database. The report was published in The Lancet Haematology.
Estimates for leukemia subtypes in 2003-2007 – calculated for 54 countries, not regions – also showed a great deal of variation. For acute lymphoblastic leukemia, Ecuador had the highest rates for both males (2.8 per 100,000) and females (3.3), with high rates seen in Costa Rica, Columbia, and Cyprus. Rates in the United States were near the top: 2.1 for males and 1.6 for females. Rates were lowest for men in Jamaica (0.4) and Serbia (0.6) and for women in India (0.5) and Serbia and Cuba (0.6), Dr. Miranda-Filho and his associates said.
Incidence rates for acute myeloid leukemia were highest in Australia for men (2.8 per 100,000) and Austria for women (2.2), with the United States near the top for both men (2.6) and women (1.9). The lowest rates occurred in Cuba and Egypt for men (0.9 per 100,000) and Cuba for women (0.4), data from the WHO’s Cancer Incidence in Five Continents Volume X show.
Chronic lymphocytic leukemia incidence was highest for men in Canada (4.5 per 100,000), Ireland and Lithuania (4.4), and Slovakia (4.3). The incidence was highest for women in Lithuania (2.5), Canada (2.3), and Slovakia and Denmark (2.1). Incidence in the United States was 3.5 for men and 1.8 for women. At the other end of the scale, the lowest rates for both men and women were in Japan and Malaysia (0.1), the investigators’ analysis showed.
Chronic myeloid leukemia rates were the lowest of the subtypes, and Tunisia was the lowest for men at 0.4 per 100,000 and tied for lowest with Serbia, Slovenia, and Puerto Rico for women at 0.3. Incidence was highest for men in Australia at 1.8 per 100,000 and highest for women in Uruguay at 1.1. Rates in the United States were 1.3 for men and 0.8 for women, Dr. Miranda-Filho and his associates said.
“The higher incidence of acute lymphoblastic leukaemia in parts of South America, as well as of chronic lymphocytic leukaemia in populations across North America and Oceania, alongside a lower incidence in Asia, might be important markers for further epidemiological study, and a means to better understand the underlying factors to support future cancer prevention strategies,” the investigators wrote.
SOURCE: Miranda-Filho A et al. Lancet Haematol. 2018;5:e14-24.
, according to an analysis of World Health Organization cancer databases.
Incidence also is generally higher in males, with a global male to female ratio of 1.4. For men, the highest regional leukemia rate – estimated at 11.3 per 100,000 population for 2012 – was found in Australia and New Zealand, with northern America (the United States and Canada) next at 10.5 per 100,000. Australia/New Zealand and northern America had the highest rate for women at 7.2 per 100,000, followed by western Europe and northern Europe at 6.0 per 100,000, reported Adalberto Miranda-Filho, PhD, of the WHO’s International Agency for Research on Cancer in Lyon, France, and his associates.
The lowest regional rates for women were found in western Africa (1.2 per 100,000), middle Africa (1.8), and Micronesia/Polynesia (2.1). For men, leukemia incidence was lowest in western Africa (1.4 per 100,000), middle Africa (2.6), and south-central Asia (3.4), according to data from the WHO’s GLOBOCAN database. The report was published in The Lancet Haematology.
Estimates for leukemia subtypes in 2003-2007 – calculated for 54 countries, not regions – also showed a great deal of variation. For acute lymphoblastic leukemia, Ecuador had the highest rates for both males (2.8 per 100,000) and females (3.3), with high rates seen in Costa Rica, Columbia, and Cyprus. Rates in the United States were near the top: 2.1 for males and 1.6 for females. Rates were lowest for men in Jamaica (0.4) and Serbia (0.6) and for women in India (0.5) and Serbia and Cuba (0.6), Dr. Miranda-Filho and his associates said.
Incidence rates for acute myeloid leukemia were highest in Australia for men (2.8 per 100,000) and Austria for women (2.2), with the United States near the top for both men (2.6) and women (1.9). The lowest rates occurred in Cuba and Egypt for men (0.9 per 100,000) and Cuba for women (0.4), data from the WHO’s Cancer Incidence in Five Continents Volume X show.
Chronic lymphocytic leukemia incidence was highest for men in Canada (4.5 per 100,000), Ireland and Lithuania (4.4), and Slovakia (4.3). The incidence was highest for women in Lithuania (2.5), Canada (2.3), and Slovakia and Denmark (2.1). Incidence in the United States was 3.5 for men and 1.8 for women. At the other end of the scale, the lowest rates for both men and women were in Japan and Malaysia (0.1), the investigators’ analysis showed.
Chronic myeloid leukemia rates were the lowest of the subtypes, and Tunisia was the lowest for men at 0.4 per 100,000 and tied for lowest with Serbia, Slovenia, and Puerto Rico for women at 0.3. Incidence was highest for men in Australia at 1.8 per 100,000 and highest for women in Uruguay at 1.1. Rates in the United States were 1.3 for men and 0.8 for women, Dr. Miranda-Filho and his associates said.
“The higher incidence of acute lymphoblastic leukaemia in parts of South America, as well as of chronic lymphocytic leukaemia in populations across North America and Oceania, alongside a lower incidence in Asia, might be important markers for further epidemiological study, and a means to better understand the underlying factors to support future cancer prevention strategies,” the investigators wrote.
SOURCE: Miranda-Filho A et al. Lancet Haematol. 2018;5:e14-24.
FROM THE LANCET HAEMATOLOGY
TFR achievable with second-line nilotinib for chronic CML
Second-line nilotinib may lead to maintained molecular response and treatment-free remission that can last 48 weeks or longer for patients with chronic myeloid leukemia (CML), findings from a phase 2 study suggest.
Treatment-free remission (TFR) is an emerging treatment goal for patients with CML in the chronic phase, according to François-Xavier Mahon, MD, PhD, of the University of Bordeaux (France) and his colleagues. “Potential motivators and benefits of achieving TFR may include relief of treatment side effects, reduced risk for long-term [tyrosine kinase inhibitor] toxicity, and the ability to plan a family,” they wrote. “When TFR is a treatment goal, achievement of [deep molecular response] is a key prerequisite.”
Established molecular response benchmarks include major molecular response (MMR), MR4 and MR4.5, according to the researchers.
In an open-label phase 2 study, the researchers enrolled patients with Philadelphia chromosome-positive CML who received nilotinib for 2 years or longer after having received imatinib for longer than 4 weeks. The other key criterion for enrollment was achieving MR4.5 during treatment with nilotinib, according to the study, published in Annals of Internal Medicine.
In total, 163 patients were enrolled and entered the 1-year consolidation phase. Of those patients, 126 were eligible for the TFR phase during which nilotinib treatment was stopped.
Dr. Mahon and his colleagues reported that 73 (58%) patients in the TFR phase maintained TFR at 48 weeks, 67 of whom had MR4.5. Of the seven patients who had a loss of MR4.5, four did not have a loss of MMR or confirmed loss of MR4, according to the researchers.
While the primary endpoint was TFR at 48 weeks, the researchers reported that 53% of patients maintained TFR at 96 weeks. Some patients had reinitiated nilotinib by the 96-week cutoff. Of those patients, the study showed that 93% regained MR4 and MR4.5.
The researchers noted that the safety findings were consistent with previously published data of nilotinib. “Improvements in quality of life have been cited as a motivator for stopping treatment,” they wrote. “Minimal changes in quality of life were seen with treatment cessation, possibly because the patients in this study already had a relatively high quality of life, given that they had tolerated at least 3 years of nilotinib therapy before stopping treatment.”
Novartis Pharmaceuticals funded the study. Dr. Mahon and other researchers reported financial ties to several pharmaceutical companies, including Novartis.
SOURCE: Mahon FX et al. Ann Intern Med. 2018 Feb 20. doi: 10.7326/M17-1094.
Second-line nilotinib may lead to maintained molecular response and treatment-free remission that can last 48 weeks or longer for patients with chronic myeloid leukemia (CML), findings from a phase 2 study suggest.
Treatment-free remission (TFR) is an emerging treatment goal for patients with CML in the chronic phase, according to François-Xavier Mahon, MD, PhD, of the University of Bordeaux (France) and his colleagues. “Potential motivators and benefits of achieving TFR may include relief of treatment side effects, reduced risk for long-term [tyrosine kinase inhibitor] toxicity, and the ability to plan a family,” they wrote. “When TFR is a treatment goal, achievement of [deep molecular response] is a key prerequisite.”
Established molecular response benchmarks include major molecular response (MMR), MR4 and MR4.5, according to the researchers.
In an open-label phase 2 study, the researchers enrolled patients with Philadelphia chromosome-positive CML who received nilotinib for 2 years or longer after having received imatinib for longer than 4 weeks. The other key criterion for enrollment was achieving MR4.5 during treatment with nilotinib, according to the study, published in Annals of Internal Medicine.
In total, 163 patients were enrolled and entered the 1-year consolidation phase. Of those patients, 126 were eligible for the TFR phase during which nilotinib treatment was stopped.
Dr. Mahon and his colleagues reported that 73 (58%) patients in the TFR phase maintained TFR at 48 weeks, 67 of whom had MR4.5. Of the seven patients who had a loss of MR4.5, four did not have a loss of MMR or confirmed loss of MR4, according to the researchers.
While the primary endpoint was TFR at 48 weeks, the researchers reported that 53% of patients maintained TFR at 96 weeks. Some patients had reinitiated nilotinib by the 96-week cutoff. Of those patients, the study showed that 93% regained MR4 and MR4.5.
The researchers noted that the safety findings were consistent with previously published data of nilotinib. “Improvements in quality of life have been cited as a motivator for stopping treatment,” they wrote. “Minimal changes in quality of life were seen with treatment cessation, possibly because the patients in this study already had a relatively high quality of life, given that they had tolerated at least 3 years of nilotinib therapy before stopping treatment.”
Novartis Pharmaceuticals funded the study. Dr. Mahon and other researchers reported financial ties to several pharmaceutical companies, including Novartis.
SOURCE: Mahon FX et al. Ann Intern Med. 2018 Feb 20. doi: 10.7326/M17-1094.
Second-line nilotinib may lead to maintained molecular response and treatment-free remission that can last 48 weeks or longer for patients with chronic myeloid leukemia (CML), findings from a phase 2 study suggest.
Treatment-free remission (TFR) is an emerging treatment goal for patients with CML in the chronic phase, according to François-Xavier Mahon, MD, PhD, of the University of Bordeaux (France) and his colleagues. “Potential motivators and benefits of achieving TFR may include relief of treatment side effects, reduced risk for long-term [tyrosine kinase inhibitor] toxicity, and the ability to plan a family,” they wrote. “When TFR is a treatment goal, achievement of [deep molecular response] is a key prerequisite.”
Established molecular response benchmarks include major molecular response (MMR), MR4 and MR4.5, according to the researchers.
In an open-label phase 2 study, the researchers enrolled patients with Philadelphia chromosome-positive CML who received nilotinib for 2 years or longer after having received imatinib for longer than 4 weeks. The other key criterion for enrollment was achieving MR4.5 during treatment with nilotinib, according to the study, published in Annals of Internal Medicine.
In total, 163 patients were enrolled and entered the 1-year consolidation phase. Of those patients, 126 were eligible for the TFR phase during which nilotinib treatment was stopped.
Dr. Mahon and his colleagues reported that 73 (58%) patients in the TFR phase maintained TFR at 48 weeks, 67 of whom had MR4.5. Of the seven patients who had a loss of MR4.5, four did not have a loss of MMR or confirmed loss of MR4, according to the researchers.
While the primary endpoint was TFR at 48 weeks, the researchers reported that 53% of patients maintained TFR at 96 weeks. Some patients had reinitiated nilotinib by the 96-week cutoff. Of those patients, the study showed that 93% regained MR4 and MR4.5.
The researchers noted that the safety findings were consistent with previously published data of nilotinib. “Improvements in quality of life have been cited as a motivator for stopping treatment,” they wrote. “Minimal changes in quality of life were seen with treatment cessation, possibly because the patients in this study already had a relatively high quality of life, given that they had tolerated at least 3 years of nilotinib therapy before stopping treatment.”
Novartis Pharmaceuticals funded the study. Dr. Mahon and other researchers reported financial ties to several pharmaceutical companies, including Novartis.
SOURCE: Mahon FX et al. Ann Intern Med. 2018 Feb 20. doi: 10.7326/M17-1094.
FROM ANNALS OF INTERNAL MEDICINE
Key clinical point:
Major finding: In total, 58% of patients who switched to nilotinib experienced treatment-free remission at 48 weeks.
Study details: A single-group, open-label phase 2 study.
Disclosures: Novartis Pharmaceuticals funded the study. The researchers reported financial ties to Novartis and other pharmaceutical companies.
Source: Mahon FX et al. Ann Intern Med. 2018 Feb 20. doi: 10.7326/M17-1094.
Drug could improve treatment of CML, team says

A microRNA-targeting drug could improve the effectiveness of tyrosine kinase inhibitors (TKIs) against chronic myelogenous leukemia (CML), according to preclinical research published in Nature Medicine.
The drug, miristen, targets miR-126, a microRNA expressed in leukemia stem cells (LSCs).
Researchers found that miristen “enhanced the anti-leukemic effects of TKI treatment” in mouse models of CML and “strongly diminished LSC leukemia-initiating capacity.”
“This could be a major breakthrough for people who are in remission for CML because there is always a concern that the disease will come back if TKI treatment is stopped,” said study author Bin Zhang, PhD, of City of Hope Medical Center in Duarte, California.
“Miristen could be the drug that sends the disease into permanent remission.”
For this study, Dr Zhang and her colleagues tested miristen alone and in combination with the TKI nilotinib in mouse models of CML.
The best results were seen in mice treated with miristen and nilotinib. Transplantation of bone marrow cells collected from mice treated with miristen and nilotinib resulted in no sign of leukemia in the healthy recipient mice, meaning all LSCs were eliminated.
The researchers believe miristen simply makes TKIs more effective in killing LSCs. The team also thinks they have discovered the key to the treatment’s success.
The researchers found that endothelial cells in the blood vessels of the bone marrow contain high levels of miR-126. These endothelial cells transfer miR-126 to LSCs, essentially feeding the leukemia what it needs to survive and grow.
The team hypothesized that to eliminate CML, miristen had to lower miR-126 in both the LSCs and the endothelial cells. Testing proved this theory correct.
“What we have discovered is how the microenvironment surrounding the leukemia stem cells supports them and how you need to target miR-126 in the leukemia stem cells and the microenvironment to completely eradicate the disease,” said study author Guido Marcucci, MD, of City of Hope.
“Our current study showed these findings may also apply to other types of leukemia.”
A microRNA-targeting drug could improve the effectiveness of tyrosine kinase inhibitors (TKIs) against chronic myelogenous leukemia (CML), according to preclinical research published in Nature Medicine.
The drug, miristen, targets miR-126, a microRNA expressed in leukemia stem cells (LSCs).
Researchers found that miristen “enhanced the anti-leukemic effects of TKI treatment” in mouse models of CML and “strongly diminished LSC leukemia-initiating capacity.”
“This could be a major breakthrough for people who are in remission for CML because there is always a concern that the disease will come back if TKI treatment is stopped,” said study author Bin Zhang, PhD, of City of Hope Medical Center in Duarte, California.
“Miristen could be the drug that sends the disease into permanent remission.”
For this study, Dr Zhang and her colleagues tested miristen alone and in combination with the TKI nilotinib in mouse models of CML.
The best results were seen in mice treated with miristen and nilotinib. Transplantation of bone marrow cells collected from mice treated with miristen and nilotinib resulted in no sign of leukemia in the healthy recipient mice, meaning all LSCs were eliminated.
The researchers believe miristen simply makes TKIs more effective in killing LSCs. The team also thinks they have discovered the key to the treatment’s success.
The researchers found that endothelial cells in the blood vessels of the bone marrow contain high levels of miR-126. These endothelial cells transfer miR-126 to LSCs, essentially feeding the leukemia what it needs to survive and grow.
The team hypothesized that to eliminate CML, miristen had to lower miR-126 in both the LSCs and the endothelial cells. Testing proved this theory correct.
“What we have discovered is how the microenvironment surrounding the leukemia stem cells supports them and how you need to target miR-126 in the leukemia stem cells and the microenvironment to completely eradicate the disease,” said study author Guido Marcucci, MD, of City of Hope.
“Our current study showed these findings may also apply to other types of leukemia.”
A microRNA-targeting drug could improve the effectiveness of tyrosine kinase inhibitors (TKIs) against chronic myelogenous leukemia (CML), according to preclinical research published in Nature Medicine.
The drug, miristen, targets miR-126, a microRNA expressed in leukemia stem cells (LSCs).
Researchers found that miristen “enhanced the anti-leukemic effects of TKI treatment” in mouse models of CML and “strongly diminished LSC leukemia-initiating capacity.”
“This could be a major breakthrough for people who are in remission for CML because there is always a concern that the disease will come back if TKI treatment is stopped,” said study author Bin Zhang, PhD, of City of Hope Medical Center in Duarte, California.
“Miristen could be the drug that sends the disease into permanent remission.”
For this study, Dr Zhang and her colleagues tested miristen alone and in combination with the TKI nilotinib in mouse models of CML.
The best results were seen in mice treated with miristen and nilotinib. Transplantation of bone marrow cells collected from mice treated with miristen and nilotinib resulted in no sign of leukemia in the healthy recipient mice, meaning all LSCs were eliminated.
The researchers believe miristen simply makes TKIs more effective in killing LSCs. The team also thinks they have discovered the key to the treatment’s success.
The researchers found that endothelial cells in the blood vessels of the bone marrow contain high levels of miR-126. These endothelial cells transfer miR-126 to LSCs, essentially feeding the leukemia what it needs to survive and grow.
The team hypothesized that to eliminate CML, miristen had to lower miR-126 in both the LSCs and the endothelial cells. Testing proved this theory correct.
“What we have discovered is how the microenvironment surrounding the leukemia stem cells supports them and how you need to target miR-126 in the leukemia stem cells and the microenvironment to completely eradicate the disease,” said study author Guido Marcucci, MD, of City of Hope.
“Our current study showed these findings may also apply to other types of leukemia.”